 Yezhe Cheng†Xiaoxi Yuan†Qiang Tian†Xiuying Huang†Yang Chen†Yuzhi PuHu LongMingyu XuYafei Ji
Yezhe Cheng†Xiaoxi Yuan†Qiang Tian†Xiuying Huang†Yang Chen†Yuzhi PuHu LongMingyu XuYafei Ji Jia XieYuping Tan
Jia XieYuping Tan Xi Zhao*Hongmei Song*
Xi Zhao*Hongmei Song*- Center of Translational Medicine, Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd., Chengdu, China
Purpose: The aim of this study was to improve the intratumoral accumulation of an antibody–drug conjugate (ADC) and minimize its off-target toxicity, SKB264, a novel anti-trophoblast antigen 2 (TROP2) ADC that was developed using 2-methylsulfonyl pyrimidine as the linker to conjugate its payload (KL610023), a belotecan-derivative topoisomerase I inhibitor. The preclinical pharmacologic profiles of SKB264 were assessed in this study.
Methods: The in vitro and in vivo pharmacologic profiles of SKB264, including efficacy, pharmacokinetics–pharmacodynamics (PK-PD), safety, and tissue distribution, were investigated using TROP2-positive cell lines, cell-derived xenograft (CDX), patient-derived xenograft (PDX) models, and cynomolgus monkeys. Moreover, some profiles were compared with IMMU-132.
Results: In vitro, SKB264 and SKB264 monoclonal antibody (mAb) had similar internalization abilities and binding affinities to TROP2. After cellular internalization, KL610023 was released and inhibited tumor cell survival. In vivo, SKB264 significantly inhibited tumor growth in a dose-dependent manner in both CDX and PDX models. After SKB264 administration, the serum or plasma concentration/exposure of SKB264 (conjugated ADC, number of payload units ≥1), total antibody (Tab, unconjugated and conjugated mAb regardless of the number of the payload units), and KL610023 in cynomolgus monkeys increased proportionally with increasing dosage from 1 to 10 mg/kg. The linker stability of SKB264 was significantly enhanced as shown by prolonged payload half-life in vivo (SKB264 vs. IMMU-132, 56.3 h vs. 15.5 h). At the same dose, SKB264’s exposure in tumor tissue was 4.6-fold higher than that of IMMU-132.
Conclusions: Compared with IMMU-132, the longer half-life of SKB264 had a stronger targeting effect and better antitumor activity, suggesting the better therapeutic potential of SKB264 for treating TROP2-positive tumors.
Introduction
TROP2, a trophoblast cell-surface antigen, also known as tumor-associated calcium signal transductor (TACSTD2) is overexpressed in a variety of human epithelial cancers, including breast, lung, gastric, colorectal, pancreatic, prostatic, cervical, head-and-neck, and ovarian carcinomas (1, 2). Elevated TROP2 expression is associated with tumor invasion, aggression, progression, and metastasis (3–5). Preclinical and clinical studies have demonstrated the efficacy of anti-TROP2 antibody–drug conjugates (ADCs), such as sacituzumab govitecan (IMMU-132), an anti-TROP2 humanized antibody-SN-38 conjugate for cancer treatment (6, 7). The U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) approved IMMU-132 as a first-in-class ADC for cancer treatment. The clinical results for the use of IMMU-132 to treat refractory solid tumors were quite encouraging (8). The objective response rate (ORR) of IMMU-132 in drug-resistant triple-negative breast cancer (TNBC) patients reached 33% and definitive efficacy was demonstrated in patients after immunosuppressive or chemotherapy (9, 10). The antibody–drug linker is an important feature of IMMU-132, because payload release depends on the acid-mediated cleavage of the carbonate linkage between the linker and SN-38 (11), by which SN-38 is continually released at the targeted tumor site (12). However, the half-life of IMMU-132 in serum is relatively short (~1 day), because of the less stable thiol–maleimide coupling due to the known retro-Michael addition (13). Breakage of the linker–payload conjugate from the maleimide-based ADC can occur through thiol exchange under physiological conditions, thus leading to off-target effects. Accordingly, IMMU-132 may have relatively high off-target effects.
SKB264 is a TROP2-targeting ADC that shares the same monoclonal antibody (mAb) as IMMU-132. The mAb is conjugated with seven to eight molecules of novel payload via the coupling sites on the reduced inter-chain disulfide bonds. TL033, a novel drug-linker intermediate, contains the toxic payload KL610023 (also named T030), which is a topoisomerase I inhibitor belotecan derivative with a bystander effect, and can arrest cell cycle at the G2/S stage after its internalization, leading to cell death (14). Moreover, the 2-methylsulfonyl pyrimidine has been used as a novel ADC coupling group between the linker and mAb through irreversible nucleophilic aromatic substitution, achieving higher ADC stability (compared to IMMU-132) due to the lack of retro-Michael reaction in vivo (15). Our previous studies showed that the structure of both the payload and linker contributed to the increased ADC stability, thus maintaining ADC bioactivity. The half-life of SKB264 was around 57 h in the tumor-bearing mice model, while that of IMMU-132 was 14 h in the same model (10). Comparing the molecular structure of linker between SKB264 and IMMU-132, they have the same benzyl carbonate site, which is considered a pH-mediated cleavage site for releasing the payload from the linker in the tumor microenvironment. With a longer half-life than IMMU-132, SKB264 might achieve a different and more effective balance of extracellular stability and intracellular rupture of the ADC, resulting in a better efficacy. Preclinical studies of SKB264 have demonstrated its excellent ability to inhibit tumor growth, such as mammary, lung, stomach, and colorectal cancers, in a variety of CDX and PDX models. However, the characteristics of SKB264 remain incompletely understood. Therefore, extensive studies on SKB264 are needed to achieve a better therapeutic index through improved dynamics of tissue distribution and payload release.
The present study evaluated the preclinical profiles of SKB264, related to efficacy, PK-PD, and safety, compared with those of IMMU-132. The results showed that SKB264 had a stronger targeting effect and excellent antitumor activity with favorable ADC stability and safety profiles, suggesting the potential clinical utility of SKB264 for treating cancers with TROP2 overexpression.
Materials and methods
Antibodies and ADCs
SKB264 mAb is a humanized anti-TROP2 mAb with reference to the same amino acid sequence as IMMU-132. SKB264 is a novel anti-TROP2 ADC, in which SKB264 mAb is conjugated with seven to eight molecules of novel payload named KL610023 via the coupling sites on the reduced inter-chain disulfide bonds. Human IgG1 (hIgG1), KL610023, and the linker of SKB264 are developed by Kelun-Biotech (Sichuan Chengdu, China). IMMU-132, a reference ADC used in this study, was purchased from Immunomedics, Inc. (Morris Plains, NJ).
Cell lines
NCI-N87 (human gastric carcinoma), BxPC-3 (human pancreas adenocarcinoma), and Calu-3 (human non-small cell lung cancer) were purchased from ATCC (American Type Culture Collection). Both HCC1806 (human breast adenocarcinoma) and NCI-H23 (human non-small cell lung cancer) were purchased from CoBioer (Nanjing, China). The TROP2-overexpressing NCI-H23 (TROP2+) line, which is a subtype of NCI-H23 and constructed by Kelun-Biotech, and other cell lines, including NCI-N87, BxPC-3, HCC1806, and CI-H23 (parental), were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS). Calu-3 cells were cultured in Minimum Essential Medium (MEM) supplemented with 10% FBS and non-essential amino acids. All cell lines were maintained at 37°C in 5% CO2. All the cell lines were examined by short tandem repeat authentication via Genegle (Chengdu, China) and tested for mycoplasma using a mycoplasma detection kit (Yise, Shanghai, China).
TROP2 expression in tumor cell lines
Flow cytometry (FCM) was used to detect TROP2 expression in tumor cell lines. First, cells were incubated with the following antibodies on ice for 30 min: 1 μg/ml phycoerythrin (PE)-conjugated mouse IgG2a, k isotype control, or PE-conjugated anti-human TROP2 antibody (eBioscience, San Diego, CA). After rinsing, the labeled cells were analyzed by FCM (BD Accuri C6). The following gating strategy was used for FCM. In the FSC/SSC plot, cells were selected using a gate for high FSC cells except for cell debris. Then, single cells were selected using a gate for a single slope in the FSS-A/FSC-H plot. In the PE-A plot, nonspecific binding was excluded by isotype using a gate with little PE-A cells.
Binding and dynamic affinity
An enzyme-linked immunosorbent assay (ELISA) was used to evaluate the binding and dynamics affinity. Human TROP2 protein (1 μg/ml) (Sino Biological Inc, Wayne, PA) was coated on a 96-well plate overnight at 4°C. The plate was then blocked by Super block reagent (ScyTek Inc. Logan UT) at room temperature for 1 h. Diluted SKB264, SKB264 mAb, or isotype control (human IgG1 antibody) was added for incubation at 37°C for 1 h. After washing, the diluted (1:10,000) goat anti-human IgG Fc-horseradish peroxidase (HRP) (Abcam, Waltham, MA) was added for further incubation for 45 min at 37°C. Finally, TMB solution was added for absorbance measurement at 450 nm using a SpectraMax Plus384 Microplate reader (Molecular Devices, USA).
Surface plasmon resonance (SPR) was an alternative method to evaluate dynamic affinity, in which human TROP2 protein was captured with goat anti-human IgG Ab immobilized onto a CM5 sensor chip (Biacore Inc., Raleigh, NC). Serial titrations of SKB264 or IMMU-132 ranging from 0.0823 to 2.22 nM and running buffer were individually injected over the surface, which was captured human TROP2 for an association phase of 120 s, followed by a 600-s dissociation time. Kinetic parameters were calculated using the BIA evaluation software with the average values of two datasets collected on separate days. Association (Ka) and dissociation (Kd) rate constant values were calculated by fitting analysis assuming a Langmuir binding model (16) and a stoichiometry of (1:1) to determine KD (equilibrium dissociation constant) values.
Cytotoxic assay
Tumor cells were seeded on 96-well plates at the following concentrations: HCC1806 (3,000 cells per well), NCI-N87 (5,000 cells per well), BxPC-3 (2,000 cells per well), Calu-3 (8,000 cells per well), NCI-H23 (TROP2+, 3,000 cells per well), and NCI-H23 (parental, 3,000 cells per well). After overnight incubation, the diluted testing items were added respectively. After 72 h, cell viability was evaluated using a CellTiter-Glo Luminescent Cell Viability Assay (Promega Corp. Madison, WI), following the manufacturer’s instruction.
CDX models
All animal experiments in this study were conducted in the laboratory fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International. The contents and procedures related to animal experiments in the study complied with the relevant laws and regulations in the experimental animal use and management and the relevant requirements of the Institutional Animal Care and Use Committee (IACUC). The study protocol was approved by IACUC (IACUC Serial Number: IACUC-A2018086-T014-01).
The CDX models of HCC1806 (breast cancer) and NCI-N87 (gastric carcinoma) cells were developed by subcutaneously engrafting tumor cells into female BALB/c nude mice (GemPharma, Nanjing, China). When the average tumor volume reached about 120 mm3, mice were randomized into five groups (n = 8) and subsequently administered by intravenous (i.v.) injection with the testing item twice a week for six times. SKB264 treatments were performed at doses of 1, 3, and 10 mg/kg in the HCC1806 xenograft model and 0.3, 1, and 3 mg/kg in the NCI-N87 xenograft model. Irinotecan (30 mg/kg) treatment was an arm of the standard of care in the CDX models. The percentage of tumor growth inhibition (TGI) was defined as %TGI = [1 − (Ti − T0)/(Vi − V0)] × 100%, to evaluate the antitumor efficacy in vivo, where T0 and Ti were the mean tumor volume of the treatment group on the days of randomization and measurement, respectively, and V0 and Vi were the mean tumor volume of the control group on the days of randomization and measurement, respectively. Body weight changes and mortality were recorded to evaluate the tolerability of the testing items.
PDX models
The HuPrime® breast cancer xenograft model, which utilized primary BR1282 breast cancer derived from a female patient, was provided by Crown Bioscience Inc. (Cambridge, MA), and four gastric cancer PDX models, namely, 0406022, A11068, 0501116, and A19058, were provided by Personal Oncology (Nanjing, China). The BR1282 PDX model was confirmed to be TROP2-positive (high expression) by pathological examination. The four gastric cancer PDX models (0406022, A11068, 0501116, and A19058) were also confirmed to have high, moderate, low, and negative TROP2 expression, respectively. Immunohistochemistry (IHC) analysis of the paraffin-embedded PDX tissue section was performed with anti-TROP2 antibody (Abcam, Waltham, MA), following the set procedure of the Bond RX Fully Automated Research Stainer (Leica Biosystems Inc., Buffalo Grove, IL). NDP2.0-HT, a next-generation viewer software (Hamamatsu), was used for the image analysis. After primary tumor tissue harvested in “stock mice”, the tumor fragments (2 to 3 mm in length) were implanted subcutaneously into the right flank of BALB/c nude (BR1282) or NCG (four gastric cancer PDX models) mice purchased from GemPharma (Nanjing, China). Mice were randomized in five groups when the average tumor volume reached about 80–120 mm3 (n = 8 or 6). The tumor-bearing mice were treated with SKB264 via i.v. injection at doses of 0.5, 1.5, and 5 mg/kg for BR1282 and 1, 3, and 10 mg/kg for gastric cancer PDX models twice a week for six times. Irinotecan, paclitaxel, or paclitaxel (albumin-bound form) were the standard of care in the PDX models. Tumor volume and body weight were measured twice a week. Tumor weight was measured at study termination. Body weight changes and mortality were recorded to evaluate the tolerability of the testing items.
PK properties of SKB264 and KL610023 in cynomolgus monkeys
The PK properties of SKB264 and Tab were determined by ELISA, for which the lower limit of quantitation was 7.813 ng/ml. The serum concentration of KL610023 was measured by a validated liquid chromatography tandem mass spectrometry (LC/MS-MS), for which the lower limit of quantitation was 0.100 ng/ml. A total of 24 cynomolgus monkeys were randomized into four groups (n = 3/sex/group). Groups #1, #2, and #3 were treated with 1, 3, and 10 mg/kg of SKB264, respectively, via 15 min i.v. infusion once. Group #4 received 0.24 mg/kg of unconjugated KL610023 via i.v. infusion once. The serum concentrations of SKB264, Tab, and KL610023 were continually measured up to 672 h post-dose.
Tissue distribution of SKB264 after single i.v. injection in tumor-bearing nude mice
A total of 96 female nude mice bearing human HCC1806 tumor cells were divided into 16 groups (n = 6 mice/group) according to body weights and tumor volumes. After receiving a single i.v. injection of [3H]KL610023, at 0.083, 4, 24, and 72 h, as well as a single i.v. injection of [3H-KL610023]SKB264, [3H]SKB264-mAb, or [3H-mAb]SKB264 at 1, 24, 168, and 336 h post-dosing, blood, tumor tissue, and 16 types of normal tissues were collected from the euthanized mice.
Plasma samples were directly quantified by liquid scintillation counting (LSC) without further manipulation; however, the blood and tissue samples were digested with 1N of KOH at 90°C prior to quantification by LSC. The tissue residue approach (TRA) for blood, plasma, and tissues was converted to nanogram equivalent of [3H]KL610023 or TRA (ng Eq./g).
Toxicity assessment in cynomolgus monkeys
Forty cynomolgus monkeys were randomized into four groups (n = 5 animals/sex/group). The experimental animals were administered with 0.9% NaCl injection (negative control) or SKB264 (25, 50, or 75 mg/kg) via i.v. infusion once every 2 weeks for four doses (Table 1). The i.v. infusion volume of the SKB264 or the control was 10 ml/kg. Toxicity assessments were as follows: mortality/moribundity, clinical signs, body weights, food consumption, body temperature, ophthalmic examinations, local tolerance (injection site) evaluations, safety pharmacology [12-lead electrocardiogram (ECG), blood pressure, heart rate, and respiration examinations], clinical pathology (hematology, coagulation, and serum chemistry), urinalysis, cytokine levels [interleukin-2 (IL-2), interleukin-6 (IL-6), interleukin-8 (IL-8), tumor necrosis factor-alpha (TNF-α), and interferon-gamma (IFN-γ)], bone marrow smear evaluations, toxicokinetics (TK), immunology, anti-drug antibody (ADA) analysis, organ weights, and macroscopic and microscopic examinations.

Table 1 Summary results of repeated SKB264 administration in cynomolgus monkeys.
Comparison of in vivo efficacy and PK-PD between SKB264 and IMMU-132
Female BALB/c nude mice (Charles River, Beijing, China) were inoculated with 2×106 HCC1806 cells in the right front flank. To evaluate in vivo efficacy of ADC, when the average tumor size reached about 120 mm3, mice were randomized into 7 groups (n = 6 mice/group). The mice were treated with SKB264 or IMMU-132 at doses of 1, 3, and 10 mg/kg twice a week for six times via i.v. injection. To evaluate the PK-PD of ADC, when the average tumor size reached approximately 270 mm3, mice were randomized into three groups (n = 5/time point). The mice received a single i.v. injection of control hIgG1, SKB264, or IMMU-132 (3 mg/kg), respectively. Blood samples (from five mice) were collected at 1 h, 4 h, 8 h, 24 h, 48 h, 96 h, 192 h, or 336 h post-dosing to detect the concentration of payload at each time point. The rate of TGI (n = 5 mice, measured at endpoint, i.e., 336 h) was determined to evaluate the antitumor efficacy of ADC. Body weight changes and mortality were also recorded to evaluate the tolerability of the testing items.
In vitro stability of SKB264 and IMMU-132 in monkey and human plasma
To evaluate the in vitro stability of SKB264 and IMMU-132 in monkey and human plasma, the release rate of payload from SKB264 and IMMU-132 at the concentration of 50 μg/ml at 37°C up to 144 h was examined by measurement of payload concentration in plasma using LC/MS-MS, with a lower limit of quantitation was 2.00 ng/ml.
Statistical analysis
Phoenix WinNonlin 8.1 was used to calculate the PK parameters in the non-compartment model, including t1/2, Tmax, Cmax, AUC0-t, and AUC0-∞. The in vitro data were analyzed and displayed using software PRISM 5.0 (GraphPad). The half maximal effective concentration (EC50) and half maximal inhibitory concentration (IC50) were calculated using a nonlinear regression model with sigmoidal fitting. The in vitro antitumor activities of drugs were evaluated by a two-tailed paired t-test.
Results
Structure of SKB264
The structure of SKB264 is shown in Figure 1A. SKB264 is a TROP2-targeting ADC, which consists of a recombinant humanized anti-TROP2 antibody and a topoisomerase I inhibitor derived from Belotecan (KL610023), integrated together via a sulfonyl pyrimidine-CL2A-carbonate linker. The payload is conjugated with the antibody through irreversible nucleophilic aromatic substitution by tris (2-carboxyethyl) phosphine hydrochloride (TCEP HCl), a reducing agent that can reduce the interchain disulfide bounds. The drug-to-antibody ratio (DAR) of SKB264 was approximately 7.4, which was determined by the reversed-phase chromatography (RPC), and might be the theoretical maximum drug loading number for conventional interchain cysteine conjugation (Figure 1B). SKB264 was designed to release payload KL610023 after internalization by TROP2-expressing tumor cells. The inhibitory effect of KL610023 on topoisomerase I was almost similar to that of SN-38, as measured by a topoisomerase I-mediated DNA relaxation assay (Figure 1C).
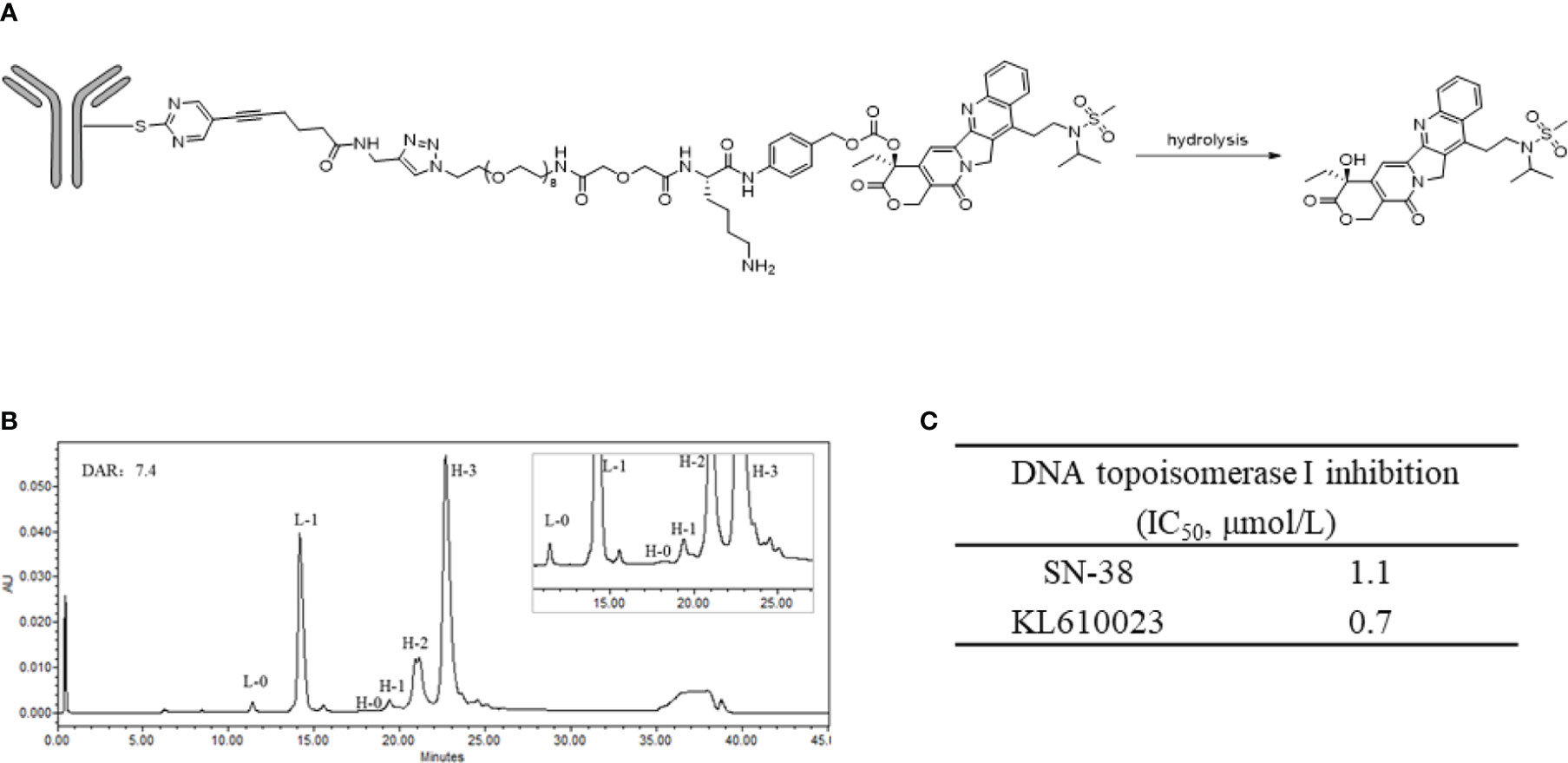
Figure 1 Structure and characterization of SKB264. (A) The chemical structure of the linkage of SKB264. (B) The distribution of the conjugated drug determined by the reverse-phase chromatography analysis (L-0: Unconjugated light chain, L-1: Light chain conjugated with one payload, H-0: Unconjugated heavy chain, H-1: Heavy chain conjugated with one payload, H-2: Heavy chain conjugated with two payloads, H-3: Heavy chain conjugated with three payloads). (C) Comparison of the inhibitory activity of cell-free DNA topoisomerase I between SN-38 and KL610023.
Action mechanism of SKB264
To examine whether the anti-TROP2 mAb plays a targeting role via specifically recognizing the highly expressed TROP2 on the surface of tumor cells, the following experiments were conducted (17). At the protein level, both SKB264 and SKB264 mAb had similar affinity to TROP2 protein, with an EC50 of 2.787 ng/ml and 2.731 ng/ml, respectively (Figure 2A), indicating that coupling payload KL610023 does not affect the affinity of mAb terminal. Compared to SKB264 mAb, the cell-level affinity of SKB264 showed a similar binding ability to TROP2-positive cells HCC1806 and NCI-N87. The EC50 of SKB264 was 11.21 nM and 6.213 nM, respectively, in two cell lines as mentioned above, while the EC50 of SKB264 mAb in them was 16.14 nM and 8.094 nM, respectively (Figure 2B). Those results suggest that the targeting role of SKB264 is mainly dependent on anti-TROP2 antibody, which can specifically bind to TROP2-positive cells, as KL610023 did not affect mAb affinity with TROP2. The results of the endocytosis experiment also showed that the percentage of endocytosis of SKB264 and SKB264 mAb was increased over time in TROP2-positive cells (HCC1806 and NCI-N87, Figure 2C). Taken together, these data imply that SKB264 specifically binds to TROP2-positive cells by targeting the TROP2 protein expressed on the cell surface and thereby inducing endocytosis.
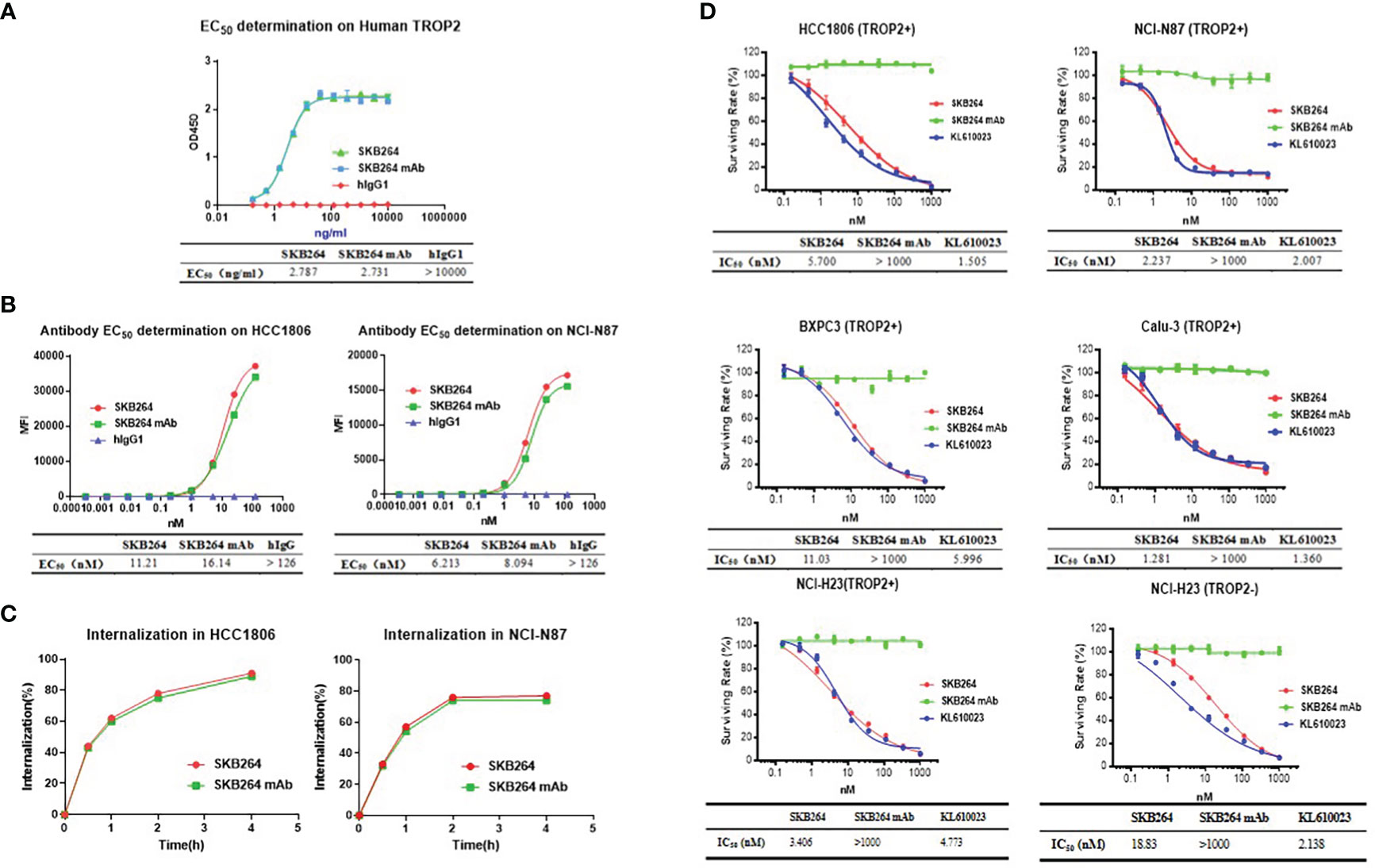
Figure 2 In vitro activities of SKB264. (A) Binding to human TROP2 estimated by ELISA (duplicates, mean ± SEM). (B) SKB264 binding to HCC1806 and NCI-N87 estimated by FACS. (C) Internalization of SKB264 or SKB264 mAb in HCC1806 and NCI-N87 cells. (D) In vitro cell growth inhibitory activity (triplicates, mean ± SEM) in HCC1806, NCI-N87, BxPC-3, Calu-3, NCI-H23 (TROP2+), and NCI-H23 (parental) cells.
In vitro antitumor activity of SKB264
To evaluate the effect of SKB264 on tumor cell proliferation, in vitro growth inhibition of cancer cell lines was determined by CellTiter-Glo Luminescent Cell Viability Assay. Cancer cell lines, including HCC1806, NCI-N87, BxPC3, Calu3, and NCI-H23 (TROP2+), highly expressed TROP2, while NCI-H23 (parental) was TROP2-negative (Figure 3). As shown in Figure 2D, unlike SKB264 mAb alone that has no inhibitory effect on tumor proliferation, both SKB264 and KL610023 could inhibit tumor cell growth, with IC50 values of 1.281–18.83 nM (SKB264) and 1.360–5.996 nM (KL610023). NCI-H23 (TROP2+) cells were more sensitive to SKB264 than parental cells (TROP2−). These results indicate that SKB264 exhibits tumor suppression ability mainly through its payload terminal. Taken together, after SKB264 endocytosed through its interaction with TROP2 on cell surface, the releasing free KL610023 can inhibit tumor cell proliferation.
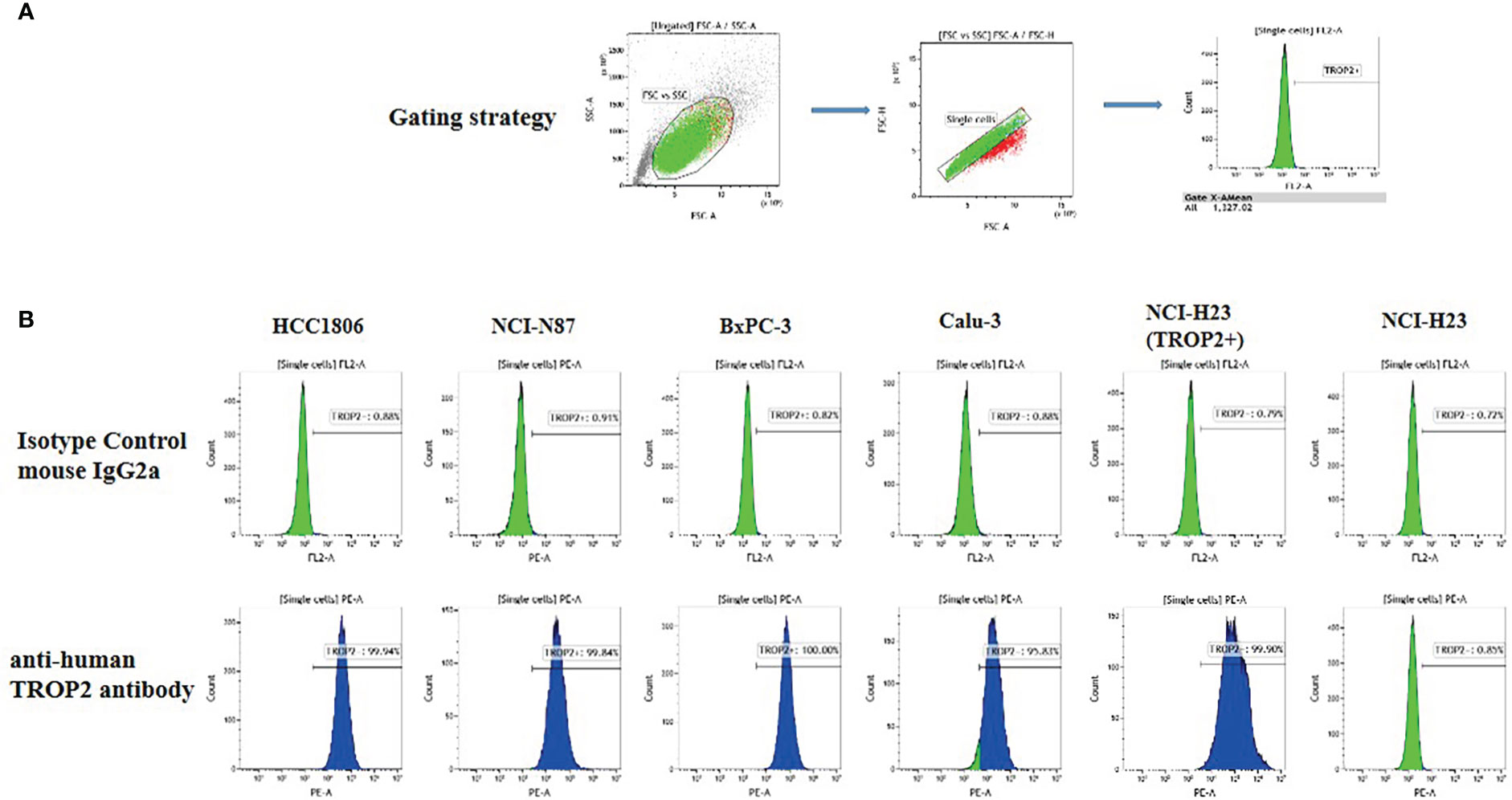
Figure 3 TROP2 expression on the cell surface. (A) Gating strategy. (B) FACS measurement results for six cell lines, namely, HCC1806, NCI-N87, BxPC-3, Calu-3, NCI-H23 (TROP2+), and NCI-H23 (parental).
In vivo antitumor activity of SKB264
In vivo antitumor activity of SKB264 was evaluated using both the CDX model and the PDX model. In the xenograft tumor model of NCI-N87 cells (Figure 4A), on the 24th day after first dose, the percentage of TGI of SKB264 (0.3, 1, and 3 mg/kg) was 78.4%, 139.2%, and 151.2%, respectively, suggesting the high efficacy of SKB264 in the inhibition of tumor growth in vivo (in a dose-dependent manner, p-values in three dose groups were <0.001, as compared to the control group). Similar to the NCI-N87 xenograft model, in the HCC1806 xenograft model (Figure 4B), the percentage of TGI of SKB264 (1, 3, and 10 mg/kg) was 75.6%, 98.5%, and 105.0% (p-values all < 0.001), respectively, on the 24th day after first dose. Furthermore, high TROP2 expression in BR1282 tumor xenograft (derived from the tumor tissue of a breast cancer patient) was found (Figure 4C). As shown in Figure 4D, in the BR1282 PDX model, on the 24th day after the first dose, the percentage of TGI of SKB264 (0.5, 1.5, and 5 mg/kg) was 44.0%, 92.6%, and 104.8%, respectively (p-values in the dose groups of 1.5 and 5 mg/kg were <0.001). Overall, our study demonstrated high efficacy (in dose-dependent manner) of the in vivo antitumor activity of SKB264 in both CDX and PDX models.
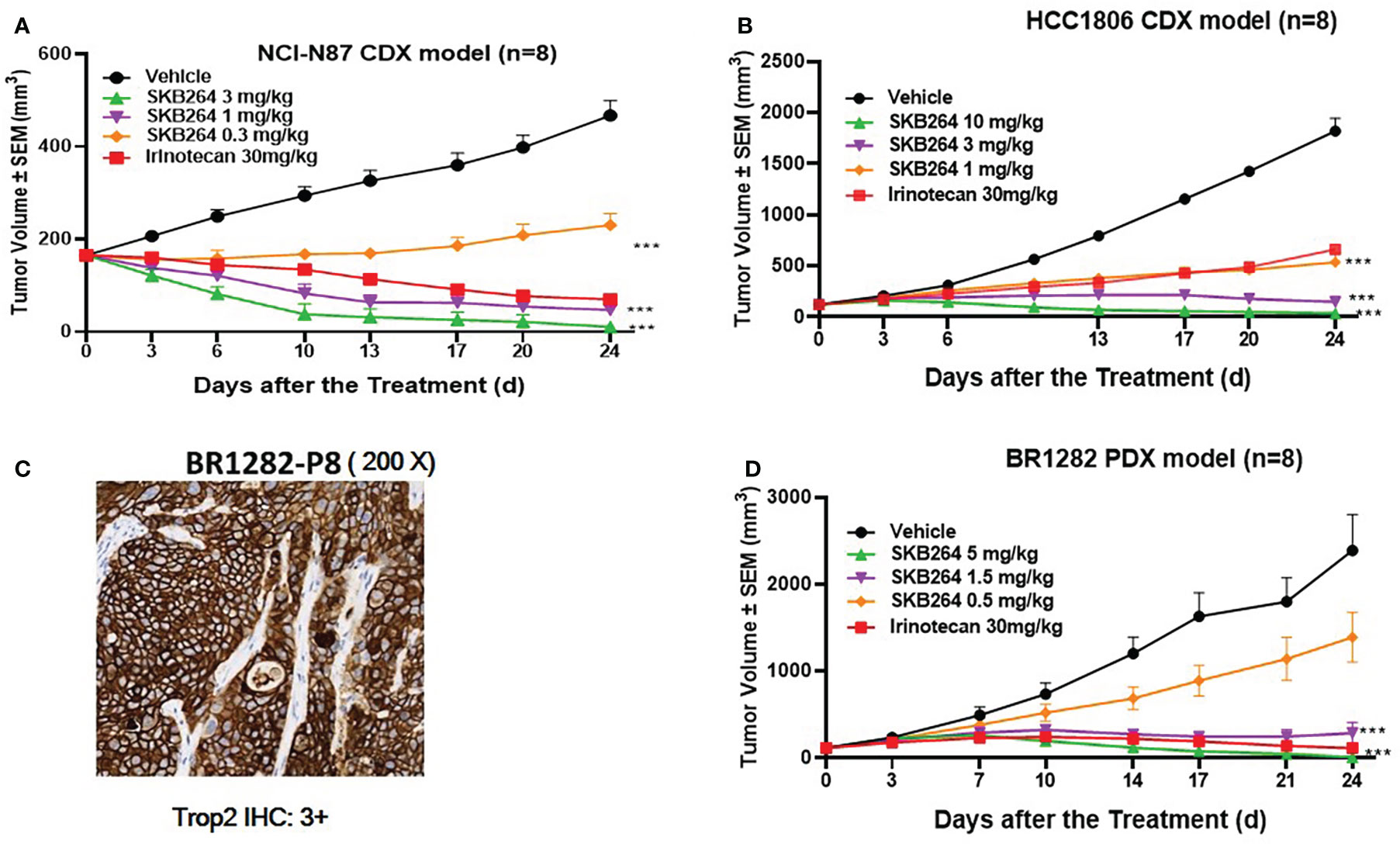
Figure 4 In vivo efficacy of the SKB264 treatment. Two-tailed paired t-test, ***p < 0.001 (vs. vehicle). (A) Antitumor efficacy of SKB264 in the HCC1806 xenograft model. The tumor-bearing mice were intravenously administered with SKB264 (3, 1, and 0.3 mg/kg, respectively), irinotecan, and vehicle control, n = 8. (B) Antitumor efficacy of SKB264 in the HCC1806 xenograft model. The tumor-bearing mice were intravenously administered with SKB264 (10, 3, and 1 mg/kg) and vehicle control, n = 8. (C) IHC analysis of TROP2 expression in the BR1282 xenografted tumor. IHC staining intensity: 3+ (high expression): >10% of tumor cells showed strong cell membrane staining. (D) In vivo antitumor efficacy of SKB264 in BR1282 xenograft model (n = 8).
To explore a minimum level of TROP2 to exert an antitumoral activity, four gastric cancer PDX models with different TROP2 expression levels were transplanted into mice. As shown in Figure 5, the gastric cancer PDX models (0406022, A11068, 0501116, and A19058) expressed high (IHC 3+), moderate (IHC 2+), low (IHC 1+), and negative (IHC 0) levels of TROP2, respectively. SKB264 exhibited a significant antitumor effect in three TROP2-positive gastric PDX models (0406022, A11068, and 0501116), with all TGI% more than 100% at 3 mg/kg, while SKB264 did not show an antitumor effect in the TROP2-negative PDX model (A19058) at 3 mg/kg. Taken together, these results indicate that SKB264 was effective for treating TROP2-positive PDX (TROP2 IHC ≥1+). There was no significant weight loss or mortality related to treatment in any of the CDX and PDX models.
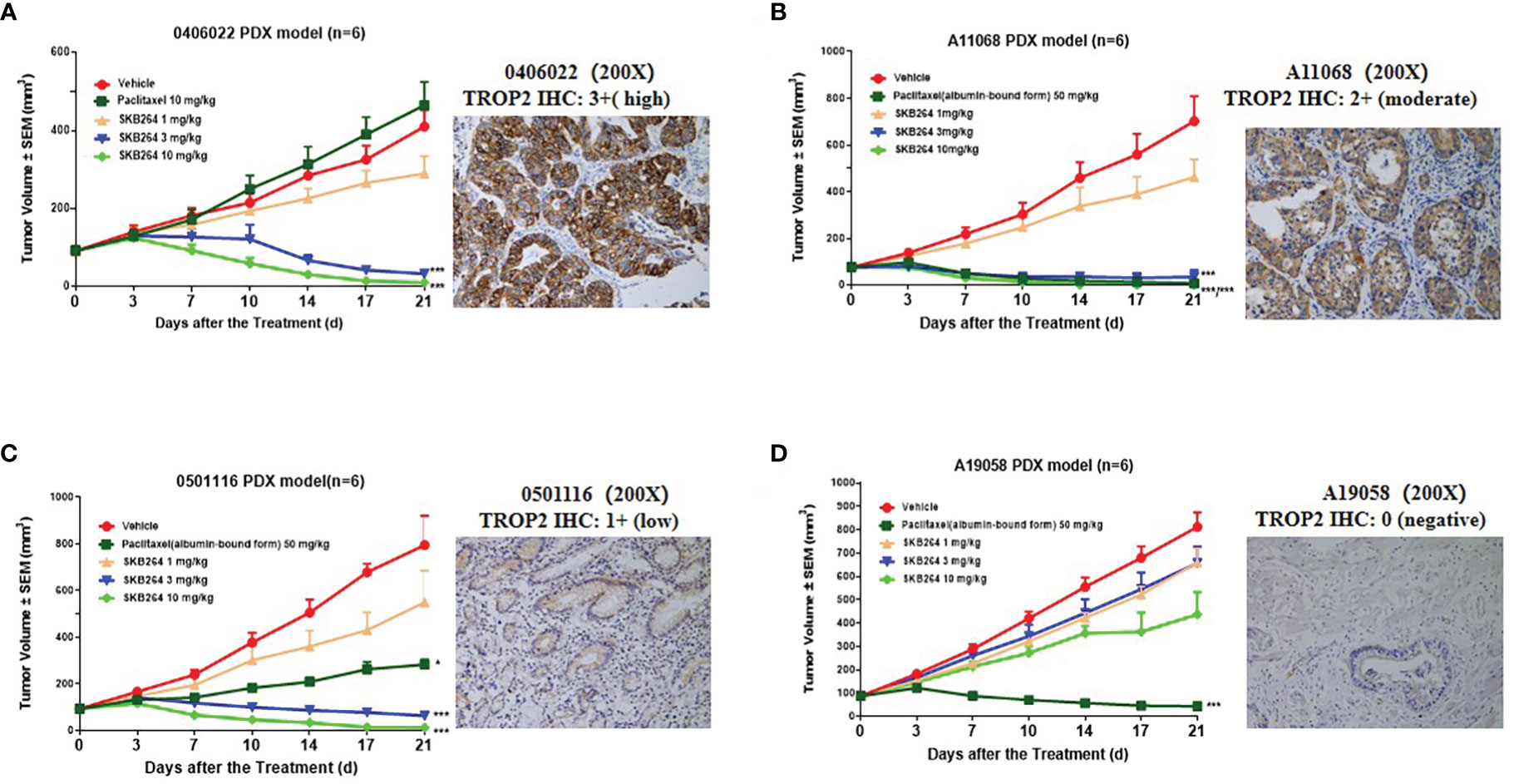
Figure 5 In vivo treatment efficacy of the SKB264 in gastric carcinoma PDX models. IHC staining intensity: 0 (Negative): no staining or ≤10% tumor cells showing any intensity of cell membrane staining; 1+ (low expression): >10% tumor cells showing weak cell membrane staining; 2+ (moderate expression): >10% tumor cells showing moderate intensity of cell membrane staining; and 3+ (high expression): >10% of tumor cells showing strong cell membrane staining. The tumor-bearing mice were intravenously treated with SKB264 (1, 3, and 10 mg/kg, respectively), vehicle control, and paclitaxel/paclitaxel (albumin-bound form) (n = 6 per group). On two-tailed paired t-test, ***p < 0.001 and *p < 0.05 (vs. vehicle). (A) 0406022; (B) A11068; (C) 0501116; and (D) A19058.
Pharmacokinetics in cynomolgus monkeys
Firstly, the affinities of SKB264 and SKB264 mAb to TROP2 of different species were evaluated by ELISA. The EC50 of the affinity of SKB264 and SKB264 mAb with monkey TROP2 was 4.363 ng/ml and 3.399 ng/ml, respectively. These results correlated with the affinity of SKB264 and SKB264 mAb with human TROP2 protein. However, SKB264 and SKB264 mAb bound to rat and mouse TROP2 protein with low affinity (EC50 > 5,000 ng/ml), indicating that cynomolgus monkey was an appropriate species for preclinical pharmacokinetic and toxicological analysis of SKB264 because of the similar binding affinity to human TROP2. Therefore, we performed the pharmacokinetic experiments in cynomolgus monkeys.
The effect of the increased exposure of SKB264, Tab, and KL610023 in cynomolgus monkeys was roughly proportional to that of the increasing dose of a single intravenous administration of SKB264 (1 mg/kg, 3 mg/kg, and 10 mg/kg, respectively). The clearance of SKB264 was slow after a single injection (t1/2: 24–57 h) (Figure 6A; Table 2), while the KL610023 was cleared rapidly (t1/2: 5.7 h) after a single intravenous administration at a dose of 0.24 mg/kg (Figure 6B; Table 3), indicating that SKB264 can significantly prolong the half-life of the payload (KL610023). Although KL610023 would be released in the plasma following the injection of SKB264, it accounted for <0.010% of systemic exposure of SKB264, which benefited from the unique design of SKB264 to ensure its stability in vivo.
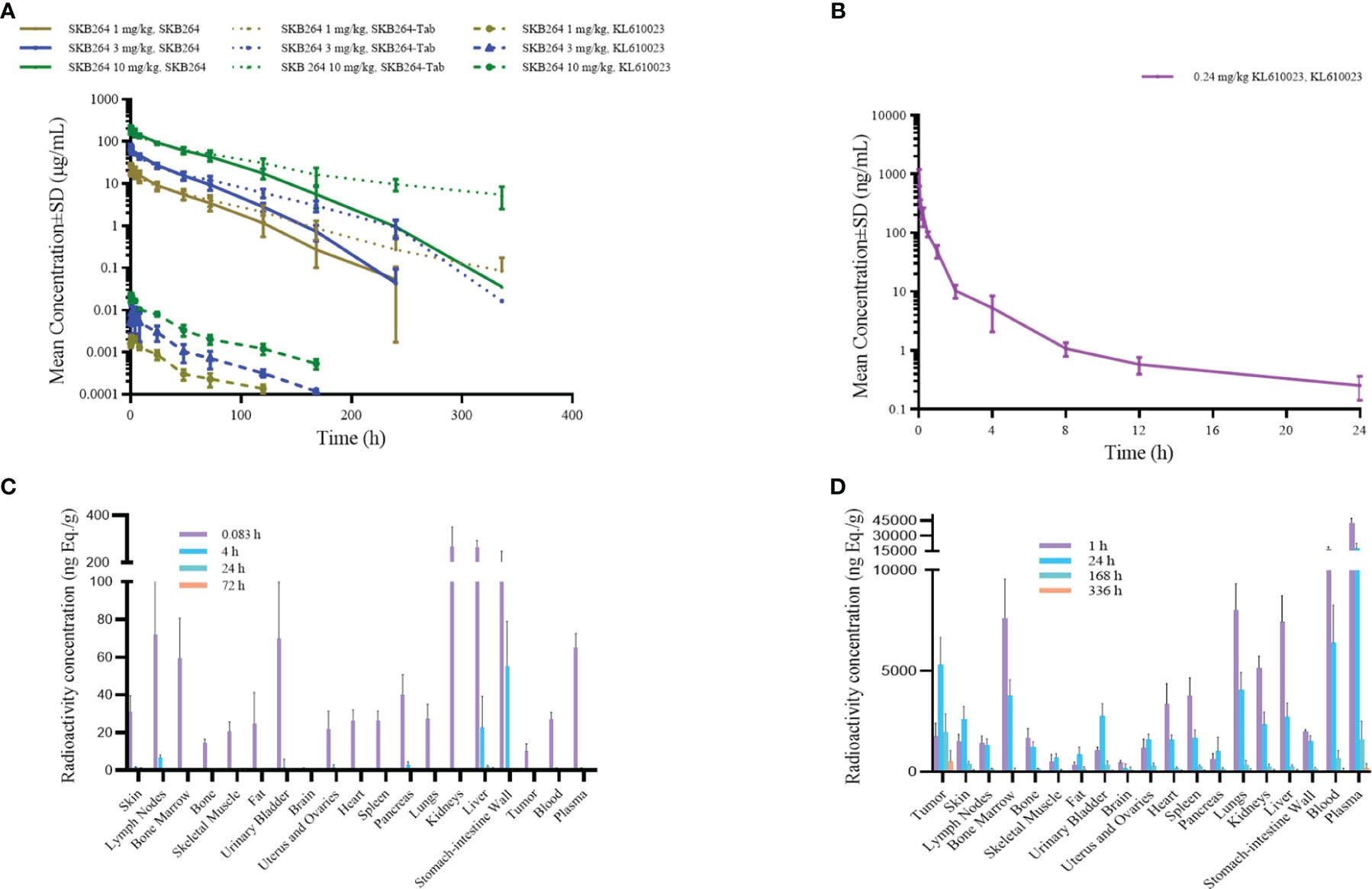
Figure 6 (A) Mean serum concentration–time curve of SKB264 (at different doses), Tab, and KL610023 following single administration (i.v.) of SKB264 in cynomolgus monkeys (n = 6/group). (B) Mean serum concentration–time curve of KL610023 following single administration (i.v.) of KL610023 in cynomolgus monkeys (n = 6). (C) Total radioactivity (TRA) in blood, plasma, and normal tissues following a single intravenous dose of [3H]KL610023 at 0.1 mg/100 µCi/kg in xenografted nude mice (n = 6). (D) TRA in blood, plasma, and normal tissues following a single intravenous dose of [3H-KL610023]SKB264 at 3 mg/100 µCi/kg in xenografted nude mice (n = 6).
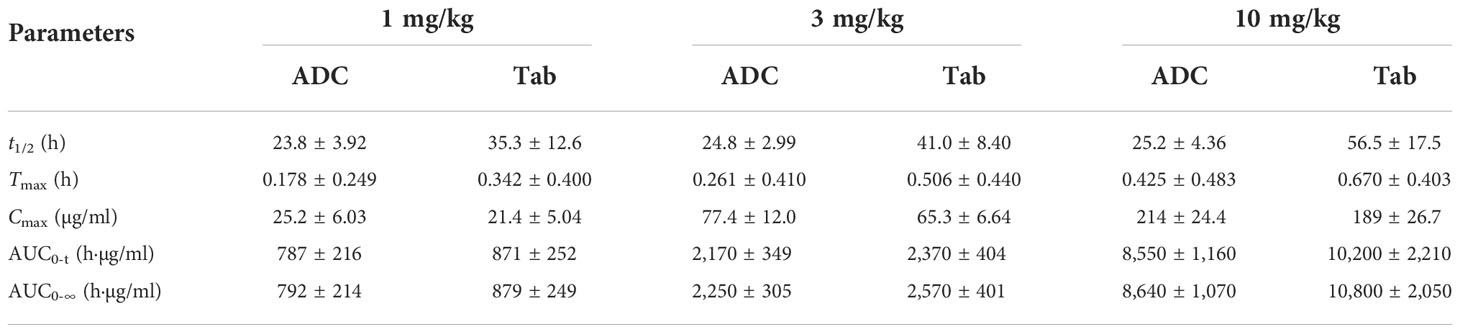
Table 2 Serum pharmacokinetic parameters of ADC and Tab after administration of SKB264 in cynomolgus monkeys (mean ± SD).
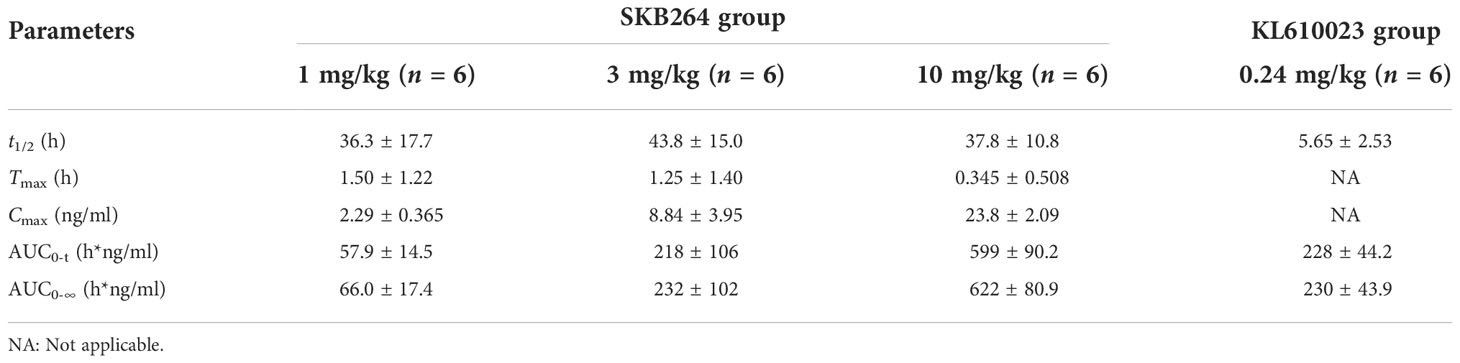
Table 3 Serum pharmacokinetic parameters of the payload after administration of SKB264 and KL610023 in cynomolgus monkeys (mean ± SD).
Tissue distribution of SKB264 in tumor-bearing mice
After a single intravenous administration of [3H-mAb]SKB264 or [3H]SKB264-mAb in the tumor-bearing mice, the distribution and elimination trends of total radioactivity of the isotope-labeled mAb or ADC were similar, indicating that the targeting and distribution behavior of SKB264 mAb remained without remarkable change after attaching payload. After a single intravenous administration of the payload, i.e., 0.1 mg/100 µCi/kg [3H]KL610023, its radioactivity was widely distributed in the tissues and organs that had more abundant blood flow (such as lymph nodes, bone marrow, plasma, intestine, stomach, lung, liver, and kidney) and would be quickly cleared (Figure 6C). However, the relative abundance of drug was higher in tumor tissue after administration of [3H]KL610023]SKB264, compared with [3H]KL610023 (Figure 6D). The elimination of total radioactivity in tumor tissue was also slower. These results indicate that SKB264 has abilities of target-specific uptake and retention in the tumors with high TROP2 expression.
Safety profile of SKB264
The in vivo safety study was performed in cynomolgus monkeys, in which the repeated intravenous dosing of SKB264 (every 2 weeks for four doses) was conducted. In detail, 40 cynomolgus monkeys were randomized into four groups to receive saline or different doses of SKB264 (25, 50, and 75 mg/kg) via i.v. infusion. The first dosing day was defined as Day 1. Five monkeys (two females and three males) in the group of SKB264 (75 mg/kg) treatment were euthanized due to their status of moribundity on Days 10–24. The cause of the moribundity appeared to be the deteriorating condition of the animal, the symptoms of which included loss of body weight, reduced food consumption, bone marrow toxicity, and intestinal toxicity. Histopathological examination found several abnormalities in in the group of SKB264 (75 mg/kg), including intestinal villi atrophy, chronic inflammation, intestinal gland dilatation, thymic cortex lymphopenia and cysts, bone marrow hematopoietic cell reduction, skin hair follicle atrophy, pigmentation and other morbid conditions, and changes in the corresponding clinic pathology indicators. In the SKB264 (50 mg/kg) group, there were also similar changes, but its incidence or severity was less or low. However, in the SKB264 (25 mg/kg) group, only the lesions in the skin and thymus were observed, most of which could be recovered or unobserved at the end of the recovery phase, except for the lesions in thymus. Although gastrointestinal toxicity and bone marrow toxicity are typical dose-limiting factors related to the clinical application of topoisomerase I inhibitors, in our study, the observed SKB264 effect on the intestines was very slight, even in the SKB264 (50 mg/kg) group. The dramatic decrease of side effects was presumable due to less off-target toxicity. TROP2-negative cells and the PDX model were less sensitive to SKB264, probably because it mainly targets and releases payload in TROP2-positive cells or tissues (Figures 2D, 5). The bone marrow toxicity, accompanied by a decreased reticulocyte ratio, was observed only in the SKB264 (50 mg/kg) group. In the thymus of monkeys in the SKB264 (25 mg/kg) group, the decreased cortical lymphocytes had no toxicological significance as shown in Table 1. No accumulation was found after the four repeated dosing of SKB264 (75 mg/kg). There was no apparent gender difference of the AUC and Cmax of SKB264 after the first dosing in the SKB264 (75 mg/kg) group, but they were higher in females than the males after the last dosing. Under the experimental conditions, the highest non-seriously toxic dose (HNSTD) of SKB264 was 50 mg/kg in monkeys following the repeated administration. The SKB264 HNSTD in monkeys provides evidence of preclinical safety of this ADC, which meets the requirement of further clinical trial. Therefore, those data are acceptable for entry into human clinical trials.
Comparison of the efficacy between SKB264 and IMMU-132
Both SKB264 and IMMU-132 are the TROP2-targeting ADCs, and their antibodies share the same amino acid sequence. SPR study revealed that both SKB264 and IMMU-132 could bind to the recombinant human TROP2 with Kd values of 0.3083 nM and 0.2935 nM, respectively, demonstrating that SKB264 and IMMU-132 have similar affinity for TROP2 (Figures 7A, B). Although the antibodies are similar, the coupled payload of SKB264 is KL610023, a derivative of belotecan, while that of IMMU-132 is SN-38, an active metabolite of irinotecan. Nevertheless, those two coupled payloads were potent to inhibit the proliferation of HCC1806 cells; as shown in Figure 7C, the IC50 of KL610023 and SN-38 was 1.816 nM and 2.610 nM, respectively. An in vitro study showed that there was no statistically significant difference in pharmacological activity between those two payloads. However, the difference in in vivo efficacy between SKB264 and IMMU-132 was found in our study, which might be due to their different linker and payload. In the HCC1806 xenograft tumor model, SKB264 could significantly inhibit tumor growth at doses of 1, 3, and 10 mg/kg, and the TGI was 81.06%, 189.46%, and 188.86%, respectively. In the same xenograft tumor model, the TGI of the IMMU-132 treatment (1, 3, and 10 mg/kg) was 48.12%, 70.64%, and 153.27%, respectively (Figure 7D). On the other hand, there was no significant change in the body weight of the experimental mice in all treatment groups (Figure 7E), indicating that SKB264 had a stronger ability to inhibit tumor growth than IMMU-132 at the same dose, so that the low dose of SKB264 could reach the same efficacy of the high dose of IMMU-132, avoiding stronger adverse reaction.
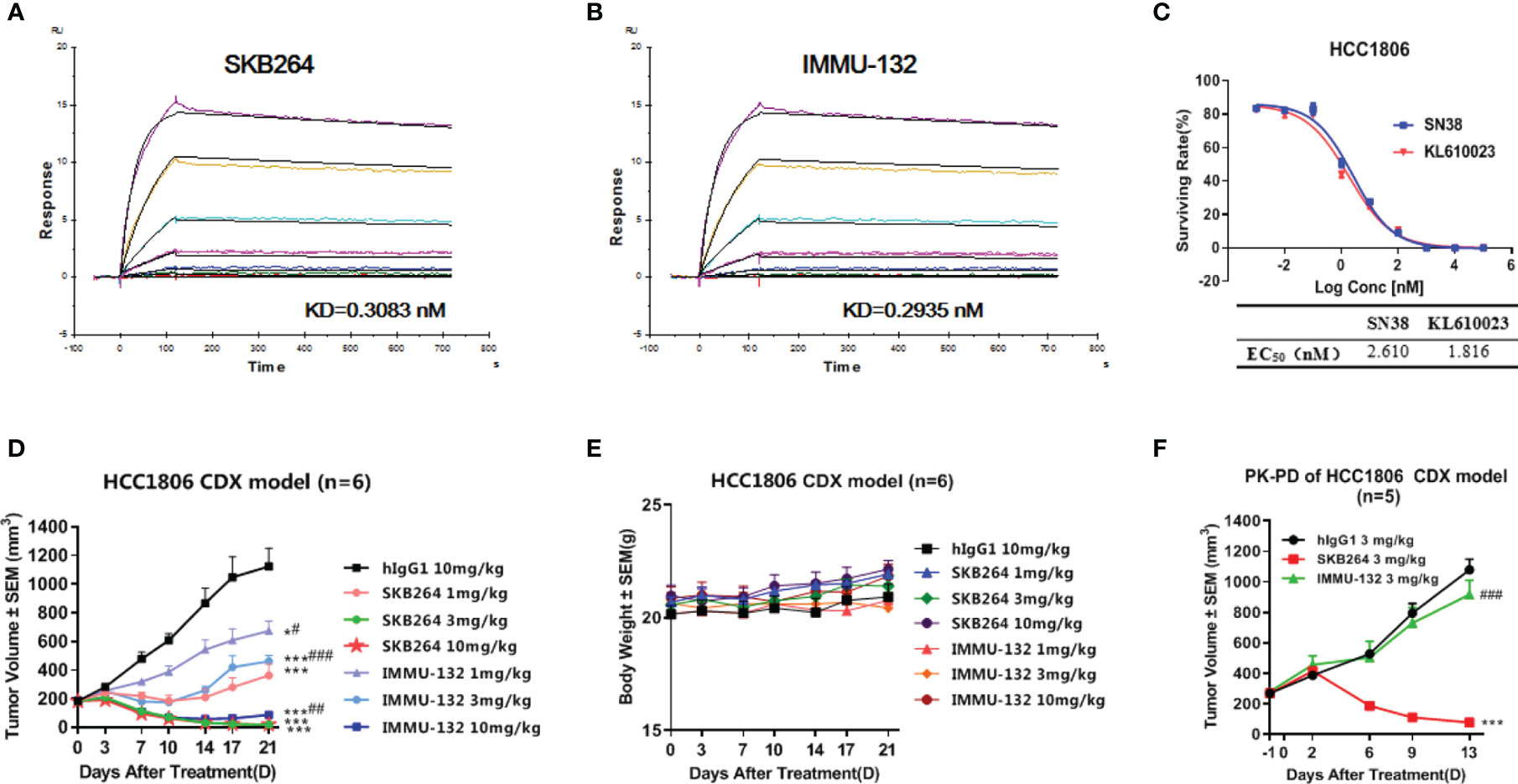
Figure 7 (A) Dynamic affinity of SKB264 to human TROP2. (B) Dynamic affinity of IMMU-132 to human TROP2. (C) In vitro inhibitory effect of KL610023 and SN-38 on HCC1806 cell growth (duplicates, mean ± SEM). (D) Antitumor efficacy of SKB264 and IMMU-132 in the HCC1806 xenograft model. The tumor-bearing mice were intravenously treated with SKB264 or IMMU-132 at dosages of 10, 3, and 1 mg/kg twice weekly, n = 6. Two-tailed paired t-test, *p < 0.05; ***p < 0.001 (vs. hIgG1); #p < 0.05; ##p < 0.01; ###p < 0.001 (vs. the same dose of SKB264). (E) Body weight of the tumor-bearing mice (n = 6). (F) Antitumor efficacy of single-dose SKB264 and IMMU-132 in the HCC1806 xenograft model in PK-PD (n = 5). Two-tailed paired t-test, ***p < 0.001 (vs. hIgG1); ###p < 0.001 (vs. SKB264).
The PK-PD study using the HCC1806 xenograft tumor model showed that the TGI of SKB264 (3 mg/kg) or IMMU-132 (3 mg/kg) after a 13-day single administration was 123.47% and 21.05%, respectively, indicating that the inhibitory effect of SKB264 on tumor growth was stronger than that of IMMU-132 after a single administration at the same dose (Figure 7F). Meanwhile, a PK study also found that the mouse plasma payload exposure of both SKB264 and IMMU-132 was similar, but the intratumor payload exposure of SKB264 was about 4.6 times that of IMMU-132 (Table 4). The larger intratumor exposure of SKB264 accounted for the better TGI, implying its greater effectiveness in cancer treatment.
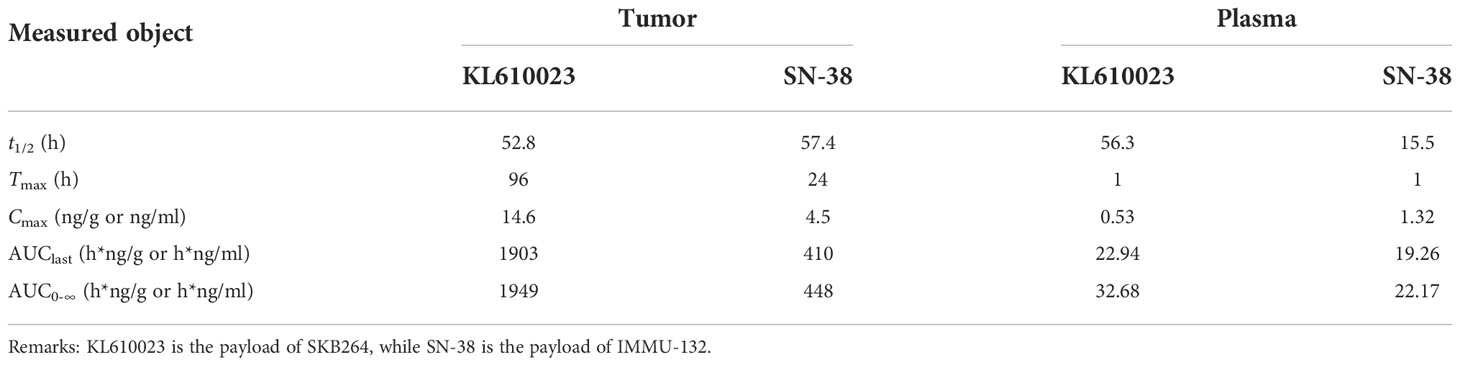
Table 4 Payload pharmacokinetic parameters in tumor and plasma after administration of either SKB264 or IMMU-132 in tumor-bearing mice (mean, n = 5).
In vitro stability of SKB264 and IMMU-132 in plasma
The release rates of the payload from IMMU-132 in cynomolgus monkeys and human plasma were almost 100% after 48-h incubation, while only 70% payload was released from SKB264 after 144-h incubation (Figure 8). These results indicate that SKB264 is more stable and releases payload more slowly in cynomolgus monkeys and human plasma than IMMU-132.
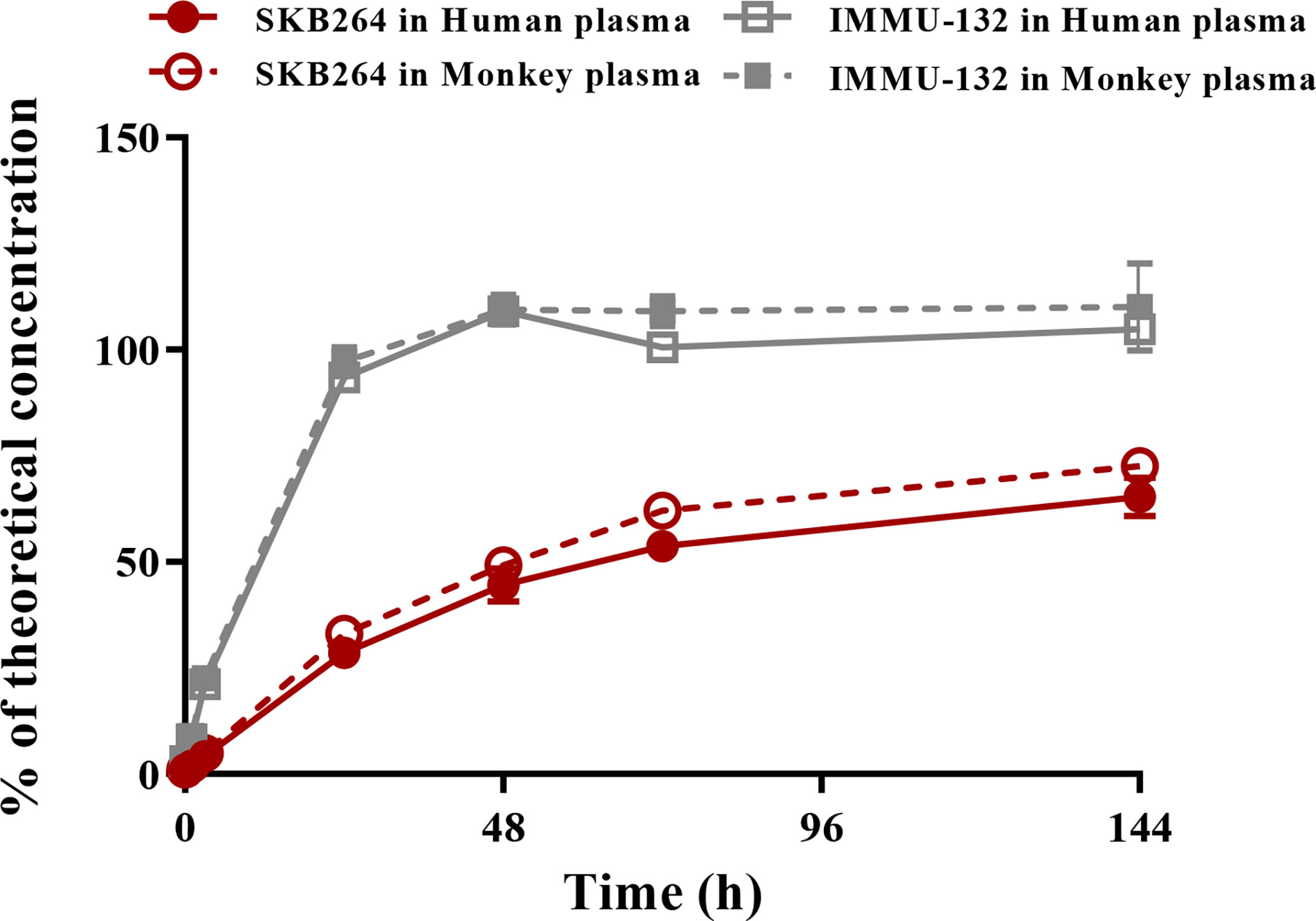
Figure 8 Percent of free payload in total available payload during the incubation of SKB264 or IMMU-132 with plasma of monkey and human.
Discussion
Cancer is characterized by the uncontrollable growth of abnormal cells, causing the disease that seriously affects human health and quality of life (18). It is reported that about one-sixth of all deaths are caused by cancer (19). TROP2 is a protein that is highly expressed in many epithelial tumors, including breast, lung, gastric, colorectal, pancreatic, prostatic, cervical, head-and-neck, and ovarian carcinomas, making it a potential and important target for ADCs (20, 21). As shown in Figure 9, based on the specificity of mAbs to recognize the cell-surface antigens of cancer, ADCs can deliver toxic payload to the tumor site, thereby enhancing the cytotoxicity in the tumor cells and reducing the toxic effect of drug in normal tissues (8). In April 2020, sacituzumab govitecan (IMMU-132), the first anti-TROP2 ADC, was approved in the US for the treatment of metastatic TNBC (22), spurring more research on the TROP2 biology and the development of anti-TROP2 ADC. Moreover, IMMU-132 was also used to treat advanced solid tumors in clinical studies (23).
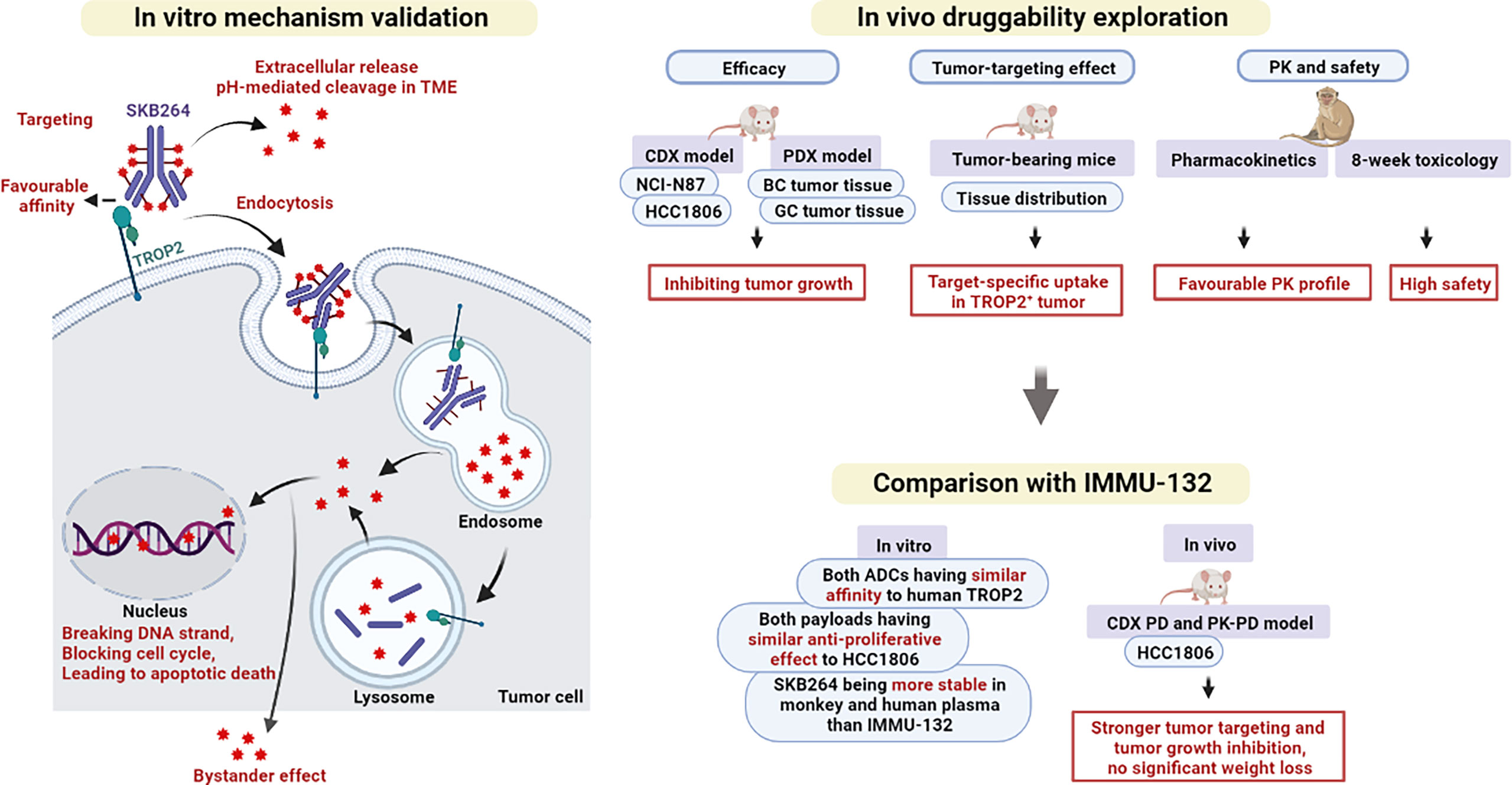
Figure 9 Graphical summary of the main findings in this study.
Since TROP2 is also expressed in multiple normal tissues, concerns have been raised on the specificity of anti-TROP2 ADC (1). Anti-TROP2-ADC may produce strong systemic toxicity in normal organs before reaching the effective level. Like any other treatment method, the therapeutic window is the key factor to tap the potential of ADC drugs. It has been reported that IMMU-132 development is successful because of lower payload toxicity than other ADCs, such as T-DM1 (1). However, IMMU-132 is not perfect, because the breakage of the linker–payload conjugate from this ADC may occur through thiol exchange under physiological conditions, making it relatively unstable in the circulation (24). The shedding of payload (SN-38) in the circulatory system will cause off-target toxicity, such as neutropenia, diarrhea, vomiting, and nausea (FDA Blackbox Warning) (25, 26). To minimize the off-target toxicity of ADCs like IMMU-132, SKB264 is developed with a novel DNA topoisomerase I inhibitor (KL610023) and a linker that confers an optimized conjugation method. Unlike the reversible addition of maleimide to cysteine in IMMU-132, methylsulfonyl pyrimidine is coupled with the linker of SKB264, via an irreversible covalent binding with disulfide reduced cysteine, by which the problem that the payload falls off easily from ADC can be fixed. Moreover, the use of toxic payload KL610023 further increases the stability of SKB264 as well. We hope to enhance the targeting ability of SKB264, reduce its off-target toxicity, and obtain a broader therapeutic window through the innovative design of SKB264.
To verify the targeting and antitumor effects of SKB264, both in vivo and in vitro experiments were conducted in the present study (Figure 9). The results of comparison showed that there was no significant difference between SKB264 and SKB264 mAb, in respect of their affinity (in vitro) to TROP2 protein and their ability to bind to TROP2 expressed on HCC1806 and NCI-N87 cells, followed by being internalized, suggesting that KL610023 conjugation cannot affect the affinity of SKB264 mAb. On the other hand, the ability of tumor targeting and internalization of SKB264 was mediated by SKB264 mAb. Furthermore, the results of cytotoxic assay showed that the antitumor effect of either SKB264 or KL610023 was almost the same, but the SKB264 mAb did not affect the survival of tumor cells, suggesting that KL610023 is the main cytotoxic factor to kill tumor cells. In in vivo experiments, SKB264 could significantly improve the TGI rate in both CDX and PDX models in a dose-dependent manner and also manifest better antitumor effect in vivo, as compared to that of IMMU-132 at the same dose. As to the mechanism of its action, SKB264 targets TROP2-positive tumor cells and mediates endocytosis via SKB264 mAb. After internalization, SKB264 releases its payload (KL610023) to kill tumor cells. However, there is variation in the process of SKB264 internalization and KL610023 release, which might be due to the difference in the factors related to intracellular trafficking pathways, for example, clathrin- or caveolin-mediated endocytosis, or lysosomal enzyme activities (27). Recycling and turnover of the TROP2 protein could also affect the KL610023 release rate (28). Therefore, further analysis is necessary to clarify the detailed dynamics of SKB264 in tumor cells.
PK experiments for SKB264 were conducted in cynomolgus monkeys, and the results showed that the affinity of both SKB264 and SKB264 mAb to TROP2 protein of cynomolgus monkeys was similar to that of human, suggesting that the cynomolgus monkey is the most appropriate and related species to human for the preclinical study of SKB264. We found that the concentrations of SKB264, Tab, and KL610023 in the serum of cynomolgus monkeys were roughly proportional to the dosage after a single intravenous administration, indicating that their metabolic process was stable in vivo (29). Since the metabolism of ADC is different from that of the toxic payload (30, 31), the linkage of SKB264 mAb can dramatically prolong the half-life of KL610023 as compared to KL610023 alone. Nevertheless, after being released from SKB264, KL610023 could be rapidly eliminated. Its proven short half-life (1.6 h) in cynomolgus monkeys leads to less toxicity or side reactions in normal tissues (Table 5) (32). In addition, only a low level of KL610023 could be detected after a single intravenous administration of SKB264, suggesting the high stability of SKB264. These results indicate that the modification of SKB264 mAb can reduce the exposure of KL610023 in the circulatory system, and the drug-mediated damage in normal tissues could be significantly reduced (33). Moreover, the isotope labeling experiments revealed that the linkage of SKB264 mAb was associated with a slow clearance of KL610023 and showed its enrichment in the tumor tissues, suggesting that this linkage may effectively improve the antitumor effect of KL610023. Overall, the proprietary technology used in our study to construct SKB264 could ensure a more precise delivery process.

Table 5 Payload KL610023 pharmacokinetic parameters in plasma after administrated in cynomolgus monkey (n=4, half male and half female).
Similar to IMMU-132 (10), the off-target toxicity of SKB264 observed in this study mainly occurred in the TROP2 highly expressed tissues (34), including the gastrointestinal, hematopoietic, and immune systems. In addition, the metabolic related experiments showed that unlike the metabolism of SN-38, KL6100023 had no hepatic circulation. In contrast, SN-38G can be converted into SN-38 in the intestine via its hepatic circulation (35). Therefore, as compared to IMMU-132, KL6100023 has less gastrointestinal adverse reactions, such as diarrhea (11). Moreover, within the dose range of 25–75 mg/kg, there was no significant gender difference in respect of the exposure/concentration of SKB264 in monkey serum; thus, the increased serum concentration of SKB264 was basically proportional to dose escalation. Under the experimental conditions, the HNSTD of SKB264 was 50 mg/kg, which was far greater than the pharmacodynamics dose of the model mice (3 mg/kg), indicating that the treatment window of SKB264 was wide enough, because of its stable linker leading to less payload release in circulation. This advantage enables less hematotoxicity and gastrointestinal toxicity, providing the opportunity to a possible combination therapy of SKB264.
It is noteworthy that there was difference in the in vivo behavior between SKB264 and IMMU-132, due to the features of their different linkers. The release rate of the payload of IMMU-132 was almost 100% in the plasma of both cynomolgus monkey and human, after 48-h incubation, while only 70% payload was released from SKB264 after 144-h incubation. The payload releasing curve of IMMU-132 was closer to the curve of the exponential growth before reaching the peak, while that of SKB264 was closer to the curve of the linear growth, indicating that SKB264 was more stable in plasma than IMMU-132. Furthermore, in case of the same dose, the payload exposure of SKB264 in tumor tissue is about 4.6 times that of IMMU-132 found in the PK-PD study. The intratumoral accumulation of KL6100023 is associated with the better stability of SKB264 in plasma and its longer circulation time in vivo (11). Taken together, the findings of our study can explain why the anticancer effect of SKB264 is better than that of IMMU-132 at the same dose.
The potentially lower hematotoxicity and gastrointestinal toxicity feature of SKB264 might provide the opportunity of a possible combination therapy with other drug modalities. For example, the combination therapy with both Topo I inhibitor and poly(ADP-ribose) polymerase (PARP) inhibitor is an attractive therapeutic approach, because both of them target the DNA damage response (DDR) pathway with promising results from preclinical studies (36, 37). However, this kind of combination therapy is still challenged by the overlap of the hematological side effects (38–40). Relying on its unique feature of less myelosuppressive side effects, SKB264 could offer improved clinical outcomes in the combination therapy with PARP inhibitors. Another attractive feature of the combination therapy with SKB264 might be immunotherapy, because Topo I inhibitors can improve the antitumor effects of immune checkpoint inhibitors (ICIs) (41–43). In the preclinical syngeneic mouse tumor model, it has been demonstrated that trastuzumab deruxtecan could sensitize tumors to ICI therapy by the enhanced antitumor immunity, which is activated by the delivered DNA topoisomerase I inhibitor payload (44–46). Therefore, the similar combination therapy with SKB264 and ICIs is expected and its preclinical evaluation is in progress.
In summary, we developed SKB264, a novel TROP2-targeting ADC using several patented technologies. Our study showed the remarkable efficacy of SKB264 in the nonclinical TROP2-expressing xenograft models, with an acceptable safety profile and an excellent therapeutic window in animal studies, suggesting that SKB264 treatment may be a promising therapeutic therapy for treating gastric carcinoma and breast cancer in the clinical setting. With its highly promising results in preclinical studies, SKB264 has been applied in clinical trials, hopefully to provide clinical benefit for patients with gastric carcinoma, lung cancer, and breast cancer.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee (IACUC).
Author contributions
YZC: Conceptualization, formal analysis, investigation, methodology, and writing—original draft. XY, QT, XH and YC: Conceptualization, methodology, and writing—original draft. YP and HL: Formal analysis, methodology, and writing—review and editing. MX, YJ, JX and YT: Investigation and writing—review and editing. XZ and HS: Project management and writing—review and editing. All authors contributed to the article and approved the submitted version.
Funding
The study was funded by Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd.
Conflict of interest
All authors were employed by Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Stepan LP, Trueblood ES, Hale K, Babcook J, Borges L, Sutherland CL. Expression of Trop2 cell surface glycoprotein in normal and tumor tissues: potential implications as a cancer therapeutic target. J Histochem Cytochem (2011) 59:701–10. doi: 10.1369/0022155411410430
2. Guo X, Zhu X, Zhao L, Li X, Cheng D, Feng K. Tumor-associated calcium signal transducer 2 regulates neovascularization of non-small-cell lung cancer via activating ERK1/2 signaling pathway. Tumour Biol (2017) 39:1010428317694324. doi: 10.1177/1010428317694324
3. Wu B, Yu C, Zhou B, Huang T, Gao L, Liu T, et al. Overexpression of TROP2 promotes proliferation and invasion of ovarian cancer cells. Exp Ther Med (2017) 14:1947–52. doi: 10.3892/etm.2017.4788
4. Ohmachi T, Tanaka F, Mimori K, Inoue H, Yanaga K, Mori M. Clinical significance of TROP2 expression in colorectal cancer. Clin Cancer Res (2006) 12:3057–63. doi: 10.1158/1078-0432.ccr-05-1961
5. Lenárt S, Lenárt P, Šmarda J, Remšík J, Souček K, Beneš P. Trop2: Jack of all trades, master of none. Cancers (Basel) (2020) 12:7. doi: 10.3390/cancers12113328
6. Sharkey RM, McBride WJ, Cardillo TM, Govindan SV, Wang Y, Rossi EA, et al. Enhanced delivery of SN-38 to human tumor xenografts with an anti-Trop-2-SN-38 antibody conjugate (Sacituzumab govitecan). Clin Cancer Res (2015) 21:5131–8. doi: 10.1158/1078-0432.ccr-15-0670
7. Cardillo TM, Govindan SV, Sharkey RM, Trisal P, Arrojo R, Liu D, et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2/SN-38 antibody-drug conjugate: Characterization and efficacy in pancreatic, gastric, and other cancers. Bioconjug Chem (2015) 26:919–31. doi: 10.1021/acs.bioconjchem.5b00223
8. Bardia A, Messersmith WA, Kio EA, Berlin JD, Vahdat L, Masters GA, et al. Sacituzumab govitecan, a trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann Oncol (2021) 32:746–56. doi: 10.1016/j.annonc.2021.03.005
9. Bardia A, Mayer IA, Diamond JR, Moroose RL, Isakoff SJ, Starodub AN, et al. Efficacy and safety of anti-Trop-2 antibody drug conjugate sacituzumab govitecan (IMMU-132) in heavily pretreated patients with metastatic triple-negative breast cancer. J Clin Oncol (2017) 35:2141–8. doi: 10.1200/jco.2016.70.8297
10. Starodub AN, Ocean AJ, Shah MA, Guarino MJ, Picozzi VJ Jr., Vahdat LT, et al. First-in-Human trial of a novel anti-Trop-2 antibody-SN-38 conjugate, sacituzumab govitecan, for the treatment of diverse metastatic solid tumors. Clin Cancer Res (2015) 21:3870–8. doi: 10.1158/1078-0432.ccr-14-3321
11. Goldenberg DM, Cardillo TM, Govindan SV, Rossi EA, Sharkey RM. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC). Oncotarget (2015) 6:22496–512. doi: 10.18632/oncotarget.4318
12. Trail PA, Dubowchik GM, Lowinger TB. Antibody drug conjugates for treatment of breast cancer: Novel targets and diverse approaches in ADC design. Pharmacol Ther (2018) 181:126–42. doi: 10.1016/j.pharmthera.2017.07.013
13. Fontaine SD, Reid R, Robinson L, Ashley GW, Santi DV. Long-term stabilization of maleimide-thiol conjugates. Bioconjug Chem (2015) 26:145–52. doi: 10.1021/bc5005262
14. Lee JH, Lee JM, Kim JK, Ahn SK, Lee SJ, Kim MY, et al. Antitumor activity of 7-[2-(N-isopropylamino)ethyl]-(20S)-camptothecin, CKD602, as a potent DNA topoisomerase I inhibitor. Arch Pharm Res (1998) 21:581–90. doi: 10.1007/bf02975379
15. Christie RJ, Tiberghien AC, Du Q, Bezabeh B, Fleming R, Shannon A, et al. Pyrrolobenzodiazepine antibody-drug conjugates designed for stable thiol conjugation. Antibodies (Basel) (2017) 6:2–3. doi: 10.3390/antib6040020
16. Kulschewski T, Pleiss J. Binding of solvent molecules to a protein surface in binary mixtures follows a competitive langmuir model. Langmuir (2016) 32:8960–8. doi: 10.1021/acs.langmuir.6b02546
17. McDougall AR, Tolcos M, Hooper SB, Cole TJ, Wallace MJ. Trop2: from development to disease. Dev Dyn. (2015) 244:99–109. doi: 10.1002/dvdy.24242
18. Roy PS, Saikia BJ. Cancer and cure: A critical analysis. Indian J Cancer (2016) 53:441–2. doi: 10.4103/0019-509x.200658
19. Abdollahpour-Alitappeh M, Lotfinia M, Bagheri N, Sineh Sepehr K, Habibi-Anbouhi M, Kobarfard F, et al. Trastuzumab-monomethyl auristatin e conjugate exhibits potent cytotoxic activity in vitro against HER2-positive human breast cancer. J Cell Physiol (2019) 234:2693–704. doi: 10.1002/jcp.27085
20. Cubas R, Li M, Chen C, Yao Q. Trop2: a possible therapeutic target for late stage epithelial carcinomas. Biochim Biophys Acta (2009) 1796:309–14. doi: 10.1016/j.bbcan.2009.08.001
21. Kobayashi H, Minami Y, Anami Y, Kondou Y, Iijima T, Kano J, et al. Expression of the GA733 gene family and its relationship to prognosis in pulmonary adenocarcinoma. Virchows Arch (2010) 457:69–76. doi: 10.1007/s00428-010-0930-8
22. Hafeez U, Parakh S, Gan HK, Scott AM. Antibody-drug conjugates for cancer therapy. Molecules (2020) 25:4764. doi: 10.3390/molecules25204764
23. Sands H, Jones PL. Methods for the study of the metabolism of radiolabeled monoclonal antibodies by liver and tumor. J Nucl Med (1987) 28:390–8.
24. Kubizek F, Eggenreich B, Spadiut O. Status quo in antibody-drug conjugates - can glyco- enzymes solve the current challenges? Protein Pept Lett (2017) 24:686–95. doi: 10.2174/0929866524666170724105211
25. Ocean AJ, Starodub AN, Bardia A, Vahdat LT, Isakoff SJ, Guarino M, et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: Safety and pharmacokinetics. Cancer (2017) 123:3843–54. doi: 10.1002/cncr.30789
26. Pandey R, Gruslova A, Chiou J, Brenner AJ, Tiziani S. Stable isotope dilution LC-HRMS assay to determine free SN-38, total SN-38, and SN-38G in a tumor xenograft model after intravenous administration of antibody-drug conjugate (Sacituzumab govitecan). Anal Chem (2020) 92:1260–7. doi: 10.1021/acs.analchem.9b04419
27. Lucas AT, Moody A, Schorzman AN, Zamboni WC. Importance and considerations of antibody engineering in antibody-drug conjugates development from a clinical pharmacologist's perspective. Antibodies (Basel) (2021) 10:10. doi: 10.3390/antib10030030
28. Hammood M, Craig AW, Leyton JV. Impact of endocytosis mechanisms for the receptors targeted by the currently approved antibody-drug conjugates (ADCs)-a necessity for future ADC research and development. Pharm (Basel) (2021) 14:14–15. doi: 10.3390/ph14070674
29. Kirchner GI, Meier-Wiedenbach I, Manns MP. Clinical pharmacokinetics of everolimus. Clin Pharmacokinet (2004) 43:83–95. doi: 10.2165/00003088-200443020-00002
30. Andersson S, Antonsson M, Elebring M, Jansson-Löfmark R, Weidolf L. Drug metabolism and pharmacokinetic strategies for oligonucleotide- and mRNA-based drug development. Drug Discovery Today (2018) 23:1733–45. doi: 10.1016/j.drudis.2018.05.030
31. Sun X, Ponte JF, Yoder NC, Laleau R, Coccia J, Lanieri L, et al. Effects of drug-antibody ratio on pharmacokinetics, biodistribution, efficacy, and tolerability of antibody-maytansinoid conjugates. Bioconjug Chem (2017) 28:1371–81. doi: 10.1021/acs.bioconjchem.7b00062
32. Nagai Y, Oitate M, Shiozawa H, Ando O. Comprehensive preclinical pharmacokinetic evaluations of trastuzumab deruxtecan (DS-8201a), a HER2-targeting antibody-drug conjugate, in cynomolgus monkeys. Xenobiotica (2019) 49:1086–96. doi: 10.1080/00498254.2018.1531158
33. Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs (2016) 8:659–71. doi: 10.1080/19420862.2016.1156829
34. Shvartsur A, Bonavida B. Trop2 and its overexpression in cancers: regulation and clinical/therapeutic implications. Genes Cancer (2015) 6:84–105. doi: 10.18632/genesandcancer.40
35. Stein A, Voigt W, Jordan K. Chemotherapy-induced diarrhea: pathophysiology, frequency and guideline-based management. Ther Adv Med Oncol (2010) 2:51–63. doi: 10.1177/1758834009355164
36. Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, et al. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther (2014) 349:408–16. doi: 10.1124/jpet.113.210146
37. Cardillo TM, Sharkey RM, Rossi DL, Arrojo R, Mostafa AA, Goldenberg DM. Synthetic lethality exploitation by an anti-Trop-2-SN-38 antibody-drug conjugate, IMMU-132, plus PARP inhibitors in BRCA1/2-wild-type triple-negative breast cancer. Clin Cancer Res (2017) 23:3405–15. doi: 10.1158/1078-0432.ccr-16-2401
38. Thomas A, Pommier Y. Targeting topoisomerase I in the era of precision medicine. Clin Cancer Res (2019) 25:6581–9. doi: 10.1158/1078-0432.ccr-19-1089
39. Chen EX, Jonker DJ, Siu LL, McKeever K, Keller D, Wells J, et al. A phase I study of olaparib and irinotecan in patients with colorectal cancer: Canadian cancer trials group IND 187. Invest New Drugs (2016) 34:450–7. doi: 10.1007/s10637-016-0351-x
40. Wahner Hendrickson AE, Menefee ME, Hartmann LC, Long HJ, Northfelt DW, Reid JM, et al. A phase I clinical trial of the Poly(ADP-ribose) polymerase inhibitor veliparib and weekly topotecan in patients with solid tumors. Clin Cancer Res (2018) 24:744–52. doi: 10.1158/1078-0432.ccr-17-1590
41. McKenzie JA, Mbofung RM, Malu S, Zhang M, Ashkin E, Devi S, et al. The effect of topoisomerase I inhibitors on the efficacy of T-Cell-Based cancer immunotherapy. J Natl Cancer Inst (2018) 110:777–86. doi: 10.1093/jnci/djx257
42. Chang CH, Wang Y, Li R, Rossi DL, Liu D, Rossi EA, et al. Combination therapy with bispecific antibodies and PD-1 blockade enhances the antitumor potency of T cells. Cancer Res (2017) 77:5384–94. doi: 10.1158/0008-5472.can-16-3431
43. Bednova O, Leyton JV. Targeted molecular therapeutics for bladder cancer-a new option beyond the mixed fortunes of immune checkpoint inhibitors? Int J Mol Sci (2020) 21:12. doi: 10.3390/ijms21197268
44. Iwata TN, Ishii C, Ishida S, Ogitani Y, Wada T, Agatsuma T. A HER2-targeting antibody-drug conjugate, trastuzumab deruxtecan (DS-8201a), enhances antitumor immunity in a mouse model. Mol Cancer Ther (2018) 17:1494–503. doi: 10.1158/1535-7163.mct-17-0749
45. Haratani K, Yonesaka K, Takamura S, Maenishi O, Kato R, Takegawa N, et al. U3-1402 sensitizes HER3-expressing tumors to PD-1 blockade by immune activation. J Clin Invest. (2020) 130:374–88. doi: 10.1172/jci126598
Keywords: antibody drug conjugate, SKB264, TROP2, PK-PD, solid tumor
Citation: Cheng Y, Yuan X, Tian Q, Huang X, Chen Y, Pu Y, Long H, Xu M, Ji Y, Xie J, Tan Y, Zhao X and Song H (2022) Preclinical profiles of SKB264, a novel anti-TROP2 antibody conjugated to topoisomerase inhibitor, demonstrated promising antitumor efficacy compared to IMMU-132. Front. Oncol. 12:951589. doi: 10.3389/fonc.2022.951589
Received: 24 May 2022; Accepted: 29 November 2022;
Published: 23 December 2022.
Edited by:
Tristan M. Sissung, National Institutes of Health (NIH), United StatesReviewed by:
Sihem Ait-Oudhia, Merck and Co., Inc., United StatesEnrique J. Arenas, Vall d’Hebron Institute of Oncology (VHIO), Spain
Copyright © 2022 Cheng, Yuan, Tian, Huang, Chen, Pu, Long, Xu, Ji, Xie, Tan, Zhao and Song. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongmei Song, c29uZ2hvbmdtZWlAa2VsdW4uY29t; Xi Zhao, emhhb3hpQGtlbHVuLmNvbQ==
†These authors have contributed equally to this work and share first authorship