 Jingchang Zhang
Jingchang Zhang Renfeng Li*
Renfeng Li*- The First Affiliated Hospital of Zhengzhou University, Zhengzhou University, Zhengzhou, China
Pancreatic cancer has the seventh highest death rate of all cancers. The absence of any serious symptoms, coupled with a lack of early prognostic and diagnostic markers, makes the disease untreatable in most cases. This leads to a delay in diagnosis and the disease progresses so there is no cure. Only about 20% of cases are diagnosed early. Surgical removal is the preferred treatment for cancer, but chemotherapy is standard for advanced cancer, although patients can eventually develop drug resistance and serious side effects. Chemoresistance is multifactorial because of the interaction among pancreatic cancer cells, cancer stem cells, and the tumor microenvironment (TME). Nevertheless, more pancreatic cancer patients will benefit from precision treatment and targeted drugs. This review focuses on the immune-related components of TME and the interactions between tumor cells and TME during the development and progression of pancreatic cancer, including immunosuppression, tumor dormancy and escape. Finally, we discussed a variety of immune components-oriented immunotargeting drugs in TME from a clinical perspective.
Introduction
Pancreatic cancer is one of the deadliest malignancies. Despite substantial improvements in the survival rates for other major cancer forms, pancreatic cancer survival rates have remained relatively unchanged since the 1960s. Pancreatic cancer is usually detected at an advanced stage and most treatment regimens are ineffective, contributing to the poor overall prognosis (1). There have been great advances in the diagnosis and treatment of pancreatic cancer in recent years, but clinical data show that only 4% of patients survive five years (2). Pancreatic cancer has been categorized into several types based on their site of origin, difference in pathogenesis, and molecular biology. These are, namely, pancreatic ductal adenocarcinoma (PDAC), pancreatic neuroendocrine neoplasm (PanNEN), acinar cell carcinoma, pancreatoblastoma, and solid pseudo-papillary neoplasm (SPN) (3). PDAC is the most common malignant neoplasm of the pancreas, the conventional type of PDAC which is a tubular adenocarcinoma accounts for 80%-90% of pancreatic cancers, with 60%-70% occurring at the head of the pancreas, mostly in men (4). PDAC is characterized by an immunosuppressive TME accompanied by the major expression of myeloid-derived suppressor cells (MDSCs) and M2 tumor-associated macrophages (TAMs). In contrast, the expression of CD8+ T cells is significantly low. Immunotherapeutic agents target the immunity mediators and empower them to suppress the tumor and effectively treat PDAC. Different targets are studied and exploited to induce an antitumor immune response in PDAC patients (5). The PDAC microenvironment consists of cancer cells, stromal cells, and extracellular components. Stromal cells that contribute to cancer progression are mainly pancreatic stellate cells (PSCs), regulatory T cells (Tregs), MDSCs, cancer-associated fibroblasts (CAFs) and TAMs. These cells and tumor cells can secrete extracellular components, such as extracellular matrix (ECM), matrix metalloproteinase (MMP), growth factors, and transforming growth factor-β (TGF-β), to maintain the TME (6). Recent studies have demonstrated that the TME plays a critical role in PDAC progression (7). Tumor immunotherapy is a therapeutic method to control and eliminate tumors by restarting and maintaining the tumor-immune cycle and restoring the body’s normal anti-tumor immune response, including monoclonal antibody immune checkpoint inhibitors, therapeutic antibodies, cancer vaccines, cell therapy and small molecule inhibitors, etc. In recent years, the good news of tumor immunotherapy has been continuously, and it has demonstrated strong antitumor activity in the treatment of pancreatic cancer (8–10). Tumor immunotherapies can enhance the immune system of the body, increase the specific recognition and memory of tumor cells, reduce the toxic and side effects on the body, and then achieve durable cure (10). Immunotherapy has become a powerful clinical strategy for treating cancer. The number of immunotherapy drug approvals has been increasing, with numerous treatments in clinical and preclinical development (8).
Immunosuppressive associated cells during the progression of PDAC
Tregs
Tregs, called regulatory T cells, are typically CD4 and CD25 positive, a subtype of T cell. The main function of these cells is to inhibit the proliferation and induction of effector T cells and to maintain autoantigen tolerance (11). Tregs infiltration constitute a prominent feature of PDAC. Tregs is an important part of tumor interstitial immune infiltration, and their presence is associated with poor clinical outcomes in many cancer types. Treg cells are the most potent antitumor immunosuppressants known, inhibiting the activity of CD4+, CD8+ and NK cells. Various mechanisms of Treg cell-mediated immunosuppression include the direct elimination of effector T cells or the acquisition of antigen-presenting cells in competition with effector T cells (12). Tregs release of TGFβ and interleukin-10 (IL-10) have also been suggested as a possible mechanism leading to this type of cellular immunosuppression. However, the exact role of Treg cells in pancreatic tumorigenesis remains largely unknown (13). Jan et al. demonstrated that Treg can confer an immunosuppressive property to their critical target, CD11c+ DC, which suppress immunity against cancer cells. Furthermore, the adhesion molecule L1CAM (CD171) is upregulated in pancreatic duct epithelium during the progression of PDAC and is associated with the accumulation of immunosuppressive T cells in tumor stroma (14). From a clinical perspective, the total number of circulating white blood cells and platelets may have a prognostic impact on patients with PDAC. These patients often have a poor prognosis associated with reduced lymphocyte counts, increased platelet count and polymorphonuclear cell counts (12). To understand the functional role of Tregs in PDAC development, we adopted an in situ implantation model. Pancreatic duct epithelial cells (Kras G12D-PDECs) originally representing KrasG12D were injected into the pancreas of C57BL/6 wild-type (WT) mice with the same gene by GFP labeling (GFP-KRAS G12D-PDECS). This model Outlines histologically the preinvasive stage of pancreatic intraepithelial neoplasia (PanIN) that develops and induces a similar intrapancreatic immune response (15). These data indicate that the development of PanIN is accompanied by the progressive accumulation of activated Tregs (14, 15). The expression of Foxp3 and CTLA-4 mRNA in peripheral blood Tregs of patients with advanced and advanced PDAC is higher, and there should be further positive correlation between IL-10 or TGF-β level and PDAC progression (16). Tregs can inhibit the body’s original anti-tumor effects, that is, tumor immunity, by combining multiple cytokines and pathways. Tregs secrete a range of inhibitory cytokines and molecules, including IL-10 and TGF-β, which, as shown in clinical studies, inhibit effector T cell function and result in lysis and inactivation (17). The process of Treg induced effector T cell lysis and inactivation is also involved in granzyme B (18, 19), TRAIL pathway (20) and galectin-1 (21), etc. Studies have shown that PDAC cells can recruit Tregs through the TRAIL pathway and promote tumorigenesis and progression (22). Furthermore, Tregs competitively bind with IL-2 to inhibit proliferation of effector cells involved in antitumor immunity (23). CTLA-4 expressed by Tregs upregulates indoleamine 2, 3-dioxygenase (IDO) pathways in dendritic cells and effector T cells and leads to their dysfunction (24, 25). Tregs also regulated lipid metabolism in M2-like TAMs. Liu et al. found that Tregs inhibited the secretion of interferon-γ (IFN-γ) by CD8+T cells, thereby blocking the activation of fatty acid synthesis mediated by sterol regulatory element binding protein-1 (SREBP1) in M2 TAMs. Thus, Tregs indirectly but selectively maintained m2-like TAM metabolic activity, mitochondrial integrity, and survival. Therefore, there are a positive feedback loop between TAMs and Tregs, which further enhance their immunosuppressive effect in TME (26) (Figure 1).
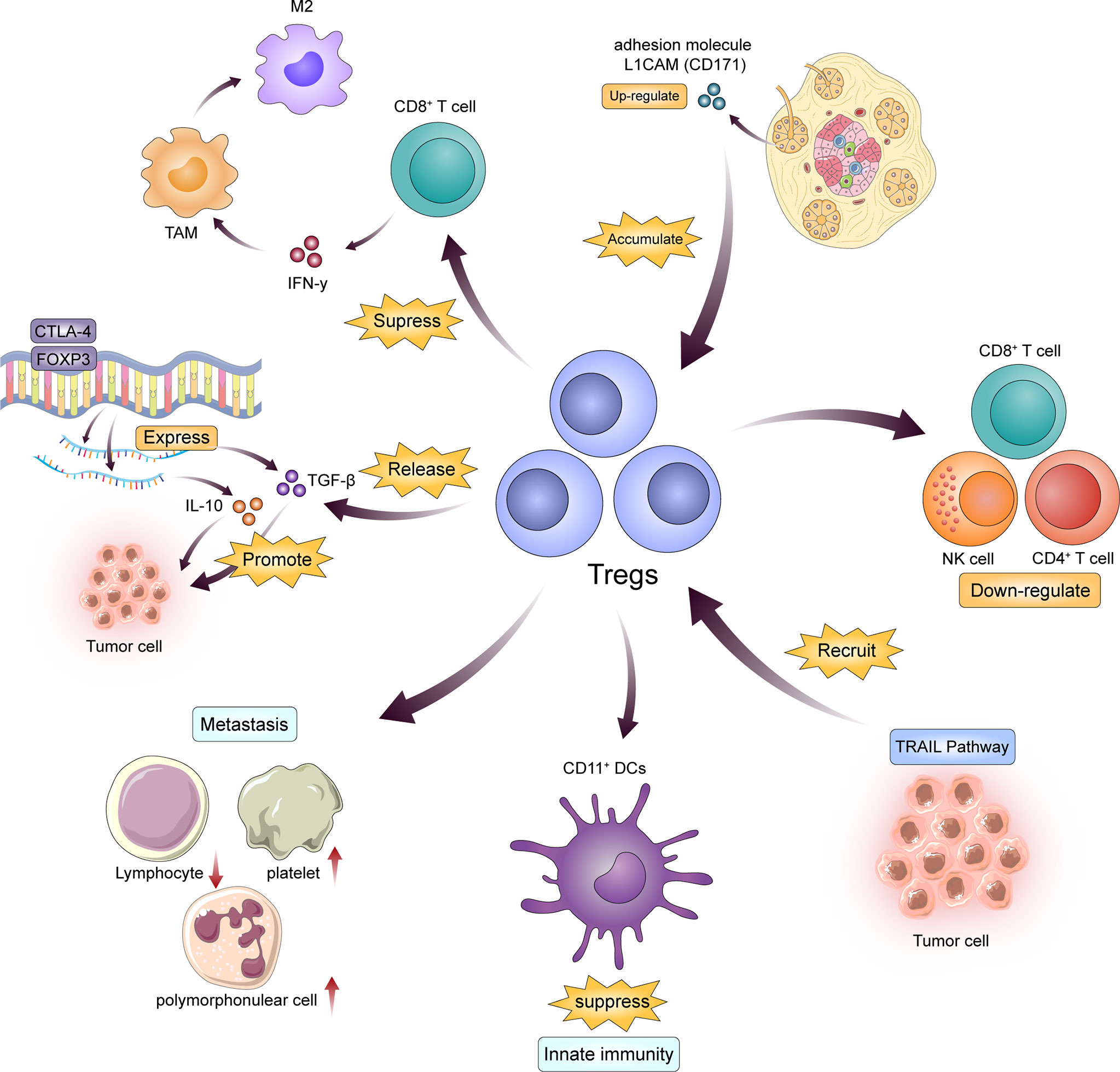
Figure 1 Tregs are the key to immunosuppression in the progression of PDAC. On the one hand, PDAC cells recruit Tregs in a variety of ways; on the other hand, Tregs change the blood environment and maintain the activity of M2-TAMS and DCs by releasing cytokines such as TGF and IL, thus achieving the effect of inhibiting effector T cells.
TAMs
Macrophages derived from monocytes are phagocytes involved in the innate immune system. Due to their plasticity, macrophages are composed of heterogeneous populations of cells with different functional and phenotypic characteristics (27). Based on activation mechanisms, macrophages are classified as M1 (activated by IFN-γ and TLR ligands, expressing higher levels of IL-12, IL-23, MHC II and inducible nitric oxide synthase, with tumoricidal effects) or M2 (activated by IL-4 and IL-13, expressing higher levels of IL-10 and TGF-β, and promote tumor progression) (28, 29). Studies have shown that the M2 phenotype of TAMs have obvious immunosuppressive effect (30). The accumulation of TAMs is associated with the progression of malignant tumor and treatment resistance, especially the immunosuppression of PDAC (31). Overall survival was shorter in patients with high density M2 macrophage infiltration than in patients with low density M2 macrophage infiltration (30). The M2 phenotype of TAMs can contribute to tumor progression by producing multiple mediators that maintain TME. These mediators mainly include growth factors and cytokines which support the proliferation of tumor cells. NF-κ B-mediated protective factors of apoptosis (e.g., IL-1β, IL-6, tumor necrosis factor (TNF)-α, C-C motif chemokine (CCL)2, C-X-C motif chemokine (CXCL)8 and CXCL10) (32, 33); Angiogenic growth factors, such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), TGF-β and fibroblast growth factor (FGF) (34–36); And other factors that regulate tissue structure and promote tumor cells migration, invasion and metastasis (37). TAMs lead to PDAC immunotherapy resistance, and ZEB1 is a key cytokine involved in this process in TME. Its main role is to maintain TAMs tumor-promoting function by promoting epithelial-mesenchymal transformation (EMT) of cancer cells. Clinical studies have shown that TAM infiltration in PDAC cells depends on the expression of CCR2 and ZEB1, of which CCL2 and CD74 determine poor prognosis (38). An important process in the therapeutic resistance of PDAC is the interaction between TAMs and cancer stem cells (CSCs), that is, TAMs can be recruited into TME to regulate the start-up state of pancreatic CSCs (39), which secrete IFN-β (40) and other factors to stimulate TAMs to maintain the active state, thus initiating the proliferation and differentiation effect of tumor cells. TAMs can express more Arg1 to interfere with the metabolism of effecting T cells, and TGF-β, IL-10, prostaglandin E2 (PGE2) and other factors released by TAMs can help Tregs recruit and inhibit CD8+T cells (41, 42). TAMs can also induce T cells apoptosis by expressing PD-L1 on its surface, which is similar to PDAC cells and MDSCs (43). TAMs play a dual role as “tumor promoter” and “immune suppressor”, helping to establish a pro-inflammatory microenvironment (44–46). TAMs also disrupts local immune surveillance, Because they indirectly inhibit T cell activity by expressing cell surface proteins or releasing soluble factors that demonstrate immunosuppressive function (e.g., argininase 1 (ARG1), IDO, IL-10, TGF-β) (37, 47) or by recruiting other immunosuppressive cells such as Tregs (48). The interaction between TAMs and immune checkpoint inhibitors and other components of TME leads to the inhibition of immune checkpoints such as PD-1/PD-L1 and CTLA-4, resulting in the elimination of inhibitory signals for T cell activation, thus enabling tumor reactive T cells to overcome regulatory mechanisms and produce effective antitumor responses, known as the expression pattern of checkpoint molecules (49, 50). Phosphatidylinositol 3-kinase γ (PI3Kγ), as a molecular switch, is closely related to the phenotype and classification of TAMs, and its involvement in immunosuppression mainly depends on turning off the “immune-stimulatory program” and turning on the “immunosuppression program” (51). Kaneda et al. showed that PI3Kγ determines the immunosuppressive properties of TAMs. Studies have shown that Tamsin lacks PI3Kγ activity, resulting in the expression of MHC-II and pro-inflammatory cytokines, while reducing immunosuppressive molecules including IL-10 and arginase. This significant change in TAMs also enhanced adaptive immunity of TME and significantly inhibited tumor progression (52). In summary, TAMs interact with a variety of cells in TME, using cytokines and intercellular signal transduction pathways as intermediate bridges to jointly promote PDAC progression and tumor immunity, resulting in treatment resistance and poor prognosis.
MDSCs
MDSCs represent another group of innate immune cells that suppress both innate and adaptive immunity (13). The marker of MDSCs is CD11b+, CD33+, HLA-DR- in humans. The levels of MDSC and pre-MDSC cytokines in peripheral blood of patients with PDAC are higher, and MDSCs in peripheral blood may be a predictive biomarker of chemotherapy failure in patients with PDAC (53). In addition, cultured bone marrow mesenchymal stem cells have been shown to induce Tregs development, whose function has been discussed previously, and targeted removal of bone marrow mesenchymal stem cell subpopulation GR-MDSC can lead to accumulation of activated CD8+T cells, tumor cell apoptosis, and tumor stromal remodeling (54). In PDAC, MDSCs are recruited to TME by tumor cells, mainly due to the production of granulocyte macrophage colony stimulating factor (GM-CSF) (55, 56). GM-CSF upregulation may be caused by KRASG12D mutation, which is detected in almost all cases of PDAC. Once inside the TME, MDSCs inhibit effector T cells, affecting several different pathways. For example, they can induce oxidative stress in T cells by producing reactive oxygen species (ROS), resulting in impaired T cell protein translation, leading to a lack of antigen-dependent proliferation (57). In addition, MDSCs inhibit T cell proliferation by depleting L-arginine from TME through a signal transductor and transcriptional activator3 (STAT3) dependent mechanism. Moreover, MDSCs also showed the ability to promote and maintain the development of Tregs to form tumor stroma (54). The process of this response is shown as follows. MDSCs produced by increased mobilization of granulocyte colony-stimulating factor (G-CSF)/IL-3/GM-CSF mediated progenitor cells express cysteine transporter Slc7A11, arginase and IDO are produced to isolate metabolites and ensure the initiation and maintenance of Tregs in tumors, thus directly inhibiting the proliferation of CD8+T cells (56, 58, 59). It can also directly produce carbon monoxide and reactive oxygen species through external stimulation and damage the effector T cells and their receptors to achieve immunosuppression effect (54). In simple terms, in PDAC, tumor cells produce GM-CSF, which promotes the accumulation of MDSCs in TME, thereby limiting T cell response.
Cancer stem cells
CSCs are subpopulations of small cells that are located between tumor cells and possess stem-like properties, cloning, long-term regeneration and self-renewal (60). CSCs in PDAC have a variety of specific markers, among which the most characteristic are CD44+, CD24+, CD133+ and ESA+ (61–63). It was found that CSCs positive for these specific markers increased tumorigenic potential several times, were more likely to mediate immunosuppression leading to therapeutic resistance and maintained their surface marker phenotypes after repeated passage as xenografts. Clinical data indicate that CD44+ is an important indicator of poor prognosis in PDAC patients (64). Some specific states in TME, such as acidosis and hypoxia, have great influence on the proliferation and differentiation of tumor stem cells, and thus directly affect tumor immunity. And the proliferation of PDAC stem cells and the immunosuppressive effect of PDAC stem cells are mainly dependent on the hypoxia state of TME. Hypoxic regions within tumors provide a favorable ecological niche for cancer cells to acquire and maintain stem cell properties, known as CSCs phenotypes, such as self-renewal, globular formation and metastasis capacity, and an undifferentiated state (60, 65). Pancreatic CSCs are highly heterogeneous in their surface and intracellular markers and in response to chemotherapy, radiotherapy, and hypoxia (66). During tumor progression, the expression of nestin, a pancreatic CSCs marker, is required to drive EMT and promote migration and invasion of PDAC cells. PDAC cells induced nestin expression through TGF-β1/Smad4 pathway under hypoxia. More importantly, elevated nestin expression promotes positive feedback constitutive activation of TGF-β1/Smad signaling by increasing the expression of TGF-β1, TGF-βR1 and TGF-βR2 in nestin-positive PDAC cells (67).
DCs
DCs from bone marrow are powerful antigen-presenting cells (APCs) with diverse and complex functions (68). After being activated in blood, dendritic cells migrate to lymph nodes, interact with effector T cells, and become the center of immune response, participating in innate and adaptive immunity. In addition, in this process, DCs recognizes endogenous and exogenous proteins associated with PDAC cells and degrades and integrates them, known as antigen internalization, which is presented on the cell surface and binds to native T cells, thereby initiating and regulating adaptive immunity and activating immunity against tumors (69). Traditional DC (cDC) is a branch of DCs lineage, which is closely related to the development of tumors. Studies have shown that some molecules in TME, as well as hypoxia environment and lactic acid inhibit the function of cDC, and specific receptor and ligand binding can target the activation and maturation of DC (70, 71). As mentioned above, the immune-related role of DCs is mainly to sense the microenvironment of tumor cells and form the corresponding adaptive immune response. DCs in PDAC are involved in the immunosuppressive effect of TME by differentiating the differentiation of different CD4+ T helper cells (Th), including Th1, Th2, Th17, through multiple phenotypic and functional heterogeneity subsets (72).
PSCs
PSCs are stellate stationary stromal cells that appear as myofibroblast-like cells located in the exocrine region of the pancreas (73, 74). PSCs are normally static in the body, and its main role is to participate in normal pancreas secretion, original immune response and maintenance of homeostasis of internal environment, as well as storage of vitamin A (75). Under the stimulation of special environmental factors, such as ROS, growth factors, cytokines, signal transduction factors, etc., PSCs transform into an activated state, which is called the activated state (76). This activation state shift is accompanied by loss of lipid droplets and expression of activation marker α -smooth muscle actin (αSMA) (77). Activated PSCs maintain their activation state through autocrine. Insulin-like growth factor 1 (IGF1), VEGF, PDGF, FGF, and CXCL12 can promote tumor angiogenesis, proliferation, migration, and immunosuppression through paracrine production (78). Studies have shown that activated PSCs can establish an interaction with PDAC cells by secreting cytokines such as TGF-β, IL-6, stromal cell derived factor-1 (SDF-1), hepatocyte growth factor (HGF) and galactose lectin-1, which contribute to promoting the immunosuppressive properties of TME and supporting the aggressibility of PDAC and the main mechanism is to promote EMT of tumor cells, which is mediated by IL-6 (79, 80). PSCs are closely related to the immunosuppression of pancreatic malignant tumors, one of the key points is to block the activation and inhibit the function of lymphocytes. For example, CXCL12 secreted by PSCs significantly reduces the migration of CD8+T cells to the peripheral stroma of pancreatic cancer, resulting in reduced anti-tumor activity of effector T cells (81). PSCs also achieve immunosuppression in PDAC microenvironment through galectin-1 mediated T cell apoptosis and Th2 cytokine secretion (82). Another factor is that PSCs play an immunosuppressive role in TME by combining with other immune-related cells and cytokines. The first pathway is IP-10/CXCL10 (interferon-inducible protein 10/CXC chemokine ligand 10), which is used by PSCs to recruit Tregs (83). In clinical studies, elevated IP-10 levels in patients with PDAC are often associated with poor prognosis and a larger invasive range of pancreatic tumors (84). The second pathway is IL-6/STAT3, through which PSCs induce the differentiation of peripheral blood monocytes into MDSCs, thereby inhibiting the proliferation of effector T cells (85).
CAFs
In the progression of pancreatic malignant tumors, fibroblasts mainly regulate the invasion of tumor cells, leading to extensive invasion and distant metastasis, but they are also closely related to tumor immunosuppression (86). Clinical studies have shown that PDAC FAP+ fibroblasts have at least two main states: periglandular αSMAhigh myofibroblastic CAFs (myCAFs) and diffusely distributed αSMAlow IL-6–positive inflammatory CAFs (87). Clinical results have shown that myCAFs have a synergistic effect on T cells and inhibit tumor growth, while inflammatory CAFs promote tumor growth and immunosuppressive response by secreting ECM proteins and cytokines such as IL-6, IL-11 and leukemia suppressors (88). The process of immunosuppression is reflected in that these cytokines can activate IL-6R+ malignant cells and myeloid cells, thereby activating STAT3 signaling and promoting tumor growth (89). As mentioned earlier, IL-6 is also strongly associated with cachexia and subsequent poor prognosis in PDAC (90). Targeting IL-6R and IL-11R in mouse models can significantly reduce STAT3 activation and enhance the effect of chemotherapy agents such as gemcitabine in tumor killing effect, suggesting that the interleukin pathway may be a good therapeutic target (91). Ber et al. Confirmed through RNA sequencing analysis that in PDAC, the increased expression of various cytokines produced by CAFs can affect the pathways of ECM remodeling and immune response to varying degrees, supporting their involvement in immunosuppression in PDAC, and ultimately leading to tumor invasion and metastasis (92). Recent studies have shown that Netrin G1 (NetG1) is the promoter of PDAC tumorigenesis in CAFs-related TME, and NetG1+CAFs stimulate immunosuppressive response and promote tumor progression through NetG1-mediated effects on glutamate/glutamine metabolism. In addition, the immunosuppressant effect of NetG1+CAFs are also reflected in the inhibition of natural killer cell-mediated killing of tumor cells, which is controlled by the downstream signaling pathway NetG1, which consists of AKT/4E-BP1, P38/FRA1, vesicular glutamate transporter and glutamine synthase (93). In the microenvironment of PDAC, CAFs can not only participate in the extensive fibrosis of the primary site, resulting in the proliferation of connective tissue, providing conditions for invasion and metastasis, but also participate in the immunosuppressive reaction by acting on a variety of immune cells such as effector T cells and NK cells, exacerbating progression.
Bregs
At the invasion stage of PDAC, a variety of immune cells participate in the tumor immune response of the body. Like Tregs, B cells also generate regulatory B cells (Bregs) through their own activation and release chemokine CXCL13 to infiltrate the microenvironment (94), which is both tumor-promoting and tumor-suppressive. One risk factor for PDAC is long-term chronic pancreatitis (95). An important microenvironmental change in chronic pancreatitis is extensive B lymphocyte infiltration. In human PDAC samples, PDAC patients with higher B cell content in TME have significantly shorter survival (96). When constructing mouse models, we found that during this process, extensive infiltration of Bregs increased the expression of IL-1β and decreased the activity of CD8+ T cells, promoting tumorigenesis (97), while reverse expression of PD-L1 and IL-35 in Bregs supported immune escape of tumor cells (98, 99). In addition, one of the characteristics of the occurrence and progression of PDAC is matrix reaction. In the study, it was found that Tregs produce PDGF-B, a pro-fibrosis molecule, and stimulate collagen generation through fibroblasts, resulting in massive production of CAFs to maintain the activation of tumor matrix and promote tumor metastasis (100). Bregs also secretes immunomodulatory cytokines such as IL-35 and IL-10, which induce Tregs production, stimulate tumor proliferation and promote local angiogenesis (101). Therefore, understanding the risk factors underlying inflammation in PDAC will have a profound impact on subsequent treatment regimen and prognostic monitoring.
Immune-related cellular biology in the progression of PDAC
Pre-metastatic niche
PDAC cells can facilitate the colonization of tumor cells by forming a supportive microenvironment at the site of metastasis through a series of cells and cytokines such as circulating tumor cells (CTCs), which is called the premetastatic niche. The formation and stable state of pre-metastatic niche is the key to the progression of PDAC and can also induce tumor dormancy at the metastatic site, and thus relapse at the metastatic site. Intercellular communication is the key to niche formation before metastases, the most common of which is liver metastases (102). The formation of an active environment is the basic mechanism that ensures the successful arrival and realization of tumor cells before they reach the secondary distal site. The primary tumor interacts with the environment of the metastatic organ to create an abundant microenvironment, while the spread of tumor cells requires many molecular and cellular changes in TME, thus providing fertile soil for the formation of secondary tumors in distant organs. The primary tumor secretes some essential soluble molecules such as TDSFs including tumor necrosis factor alpha (TNF-α, TGF-β, and VEGF along with extracellular vesicles (EVs) that are required for the preparation of distant receptive sites for pre-metastatic niche formation and organ-specific metastasis or organotropism (103). CAFs and tumor exosomes play important roles in regulating ECM remodeling for the preparation of the pre-metastatic niche. CAFs which are induced by hypoxic condition produce TGF-β2 that can preserve the stemness of the niche (104, 105). By secreting IL11, CAFs support distant metastasis through activating Glycoprotein 130 (GP130)/STAT3 signaling in PDAC cells (106). Also, increased STAT3 activity in CD11b+myeloid cells, motivates the local invasion of primary tumor cells into the blood or lymphatic vessels to move towards the metastatic sites and create pre-metastatic niche (107). Studies have shown that exosomes derived from PDAC can induce the formation of pre-metastatic niche in liver. The mechanism of pre-metastatic niche formation is that the uptake of PDAC exosomes by Kupffer cells leads to the secretion of TGF-β and up-regulation of fibronectin production by hepatic stellate cells, thus enhancing the recruitment of bone-derived macrophages. During this process, macrophage migration inhibitory factor (MIF) plays an important role. Studies have found that blocking MIF highly expressed in PDAC can effectively prevent the formation and metastasis of liver pre-metastatic niche (108). In the microenvironment of PDAC, a variety of immune-related cells are involved in the formation of liver pre-metastatic niche. TAMs secrete granular proteins to activate stellate cells to differentiate into myofibroblasts, forming a hepatic fibrosis microenvironment and supporting the growth of metastatic PDAC (109). CXCR2 is involved in the formation of pre-metastatic niche in the liver of PDAC through G-protein-coupled receptors that control neutrophil and MDSCs migration (110). The property of liver metastasis in PDAC is mainly reflected in hematopoietic stem cell (HSC) migration and homing, which is initiated by cancer-initiating cells (CIC) (111). CXCR4 and CXCL12 are key to tumor cell migration and directly affect the formation of pre-metastatic niche. Increased CXCL12 expression in hypoxic tissues is important for the homing of tumor stem cells in circulating tissues (112), CAFs secrete CXCL12, attract CXCR4-expressing tumor cells and endothelial progenitor cells, thereby promoting angiogenesis in the liver’s pre-metastatic niche (113, 114). Another important procedure is through the receptor tyrosine kinase Met (115), which shows that HGF and Met can drive the mobilization and migration of tumor stem cells in tissues (116, 117). CD44+CIC contributes to the activation of the Met signaling cascade (118), HGF-Met axis participates in angiogenesis through HGF expressing mesenchymal stem cells, and c-Met expression at high levels in CIC promotes liver implantation and metastasis of PDAC (119). In TME, pre-metastatic niche formation relies on EVs for extensive communication between tumor cells and others. The EV-capsuled factors not only enter circulation and reach distant organs to construct a pre-metastatic niche, but also facilitate tumor growth locally, include regulating angiogenesis, cellular metabolism, metastasis, cell survival, immune regulation and therapeutic resistance (120). At the level of gene expression, Rab27a GTPase is overexpressed in advanced tumors during the establishment of pre-metastatic niche and is used to regulate vesicle transport, which is related to the non-cellular autonomous control of tumor growth and metastasis. In addition, down-regulation of Rab27a can increase the expression of genes related to the EMT pathway and alter the characteristics of autonomous invasion of tumor cells. And these reveal that Rab27a can play divergent roles in regulating pro-metastatic propensity of PDAC cells: by generating pre-metastatic environment at the distant organ sites, and by suppressing invasive properties of the tumor cells (121). However, the specific mechanism of pre-metastatic niche formation remains unclear. Just as CTCs is the “seed” and the organ that is about to undergo distant metastasis is the “soil”, CTCs change the tissue microenvironment by secreting a variety of cytokines before planting to create conditions for tumor cells growth, such as hypoxia and acidosis (122).
Tumor dormancy
When PDAC cells spread to distant organs such as liver and lung, due to the limitation of growth microenvironment, tumor cells will be in non-proliferation or equilibrium state, that is, cell proliferation rate is equal to its death rate, which is called tumor dormancy. The mechanisms of PDAC cells dormancy can be divided into three categories: cell dormancy, angiogenic dormancy, and immune-mediated dormancy. The main processes include the cellular mechanisms that push a small number of DTCs (disseminated tumor cells) into a dormant state, the balance between tumor angiogenesis related cell proliferation and death, and the immune system maintaining a constant number of proliferating tumor cells (123–125). In addition, PDAC cells can sense changes in microenvironment and determine changes in dormancy state through expression of intracellular homologous ligands and receptors, thus promoting or inhibiting the growth of PDAC cells at distant metastasis sites (126). Studies have shown that the main significance of tumor dormancy is that it was demonstrated that they are a cellular reservoir for a swift relapse of pancreatic cancer following oncogene reactivation (127). DTCs and CTCs are the main mechanisms for tumor dormancy. The first two types of tumor cells also participate in the occurrence of immune escape. Among them, normal stem cell quiescence, extracellular and stromal microenvironment, autophagy, and epigenetics are the mechanisms that determine tumor cells dormancy (123). Clinical studies have found that PDAC cells in distant metastatic sites often remain clinically asymptomatic due to the dormancy of tumor cells, resulting in occult onset and ineffectiveness of conventional treatment (128–130). Under the pressure of high energy consumption of tumor cells and insufficient nutrition supply of local tissues, cancer cells secrete factors that inhibit the PI3K pathway, and this nutrient deficiency leads to the loss of tumor adhesion, further promoting short-term growth stagnation, and ultimately leading to stasis and autophagy induction. Therefore, reduced PI3K–AKT signaling has been linked to dormancy-like phenotypes (131–133). Related studies suggest that autophagy may be a survival mechanism during dormancy. Autophagy of tumor cells inhibits PI3K-Akt signaling while maintaining dormancy DTCs metabolic adaptability. The autophagy signaling mechanism can integrate stationery and survival signals to promote damage repair so that PDAC tumor cells can survive and develop therapeutic resistance during conventional chemotherapy as well as immunotherapy and targeted therapy (134–136). Although reduced mitogenic signaling can trigger quiescence, specific kinases such as dual specificity tyrosine-phosphorylation-regulated kinase 1B (DYRK1B) can actively induce this state (137). DYRK1B blocks the G0/G1/S conversion mechanism proteins, including cyclin D1, CDK4, and P27, in pancreatic malignant tumor cells (138, 139). DYRK1B also coordinates survival through antioxidant responses, inhibits and specifically kills resting pancreatic cancer cells through specific pathways, but does not kill normal resting cells (140). In conclusion, DYRK1A and DYRK1B may be markers of PDAC dormant cells. In addition, Lin et al. demonstrated that Kras mutation combined with c-MYC overexpression in PDAC tumor cells is closely associated with tumor dormancy in distant metastatic lesions (127). The dormancy of PDAC tumor cells does not mean the stagnation of cell cycle, but the existence of a dynamic equilibrium. Under the influence of TME and stem cells and corresponding cytokines, the cells were randomly transformed into activated states. Studies have shown that signals in normal stem cell niches seem to regulate dormancy, it also plays an important role in removing tumor dormancy. However, in dormant DTCs cell cycle arrest is coupled to a persistent but latent form of tumor-initiating or pluripotent capacity that provides an adaptive and survival advantage that eventually fuels tumor growth (114, 141). Tumor dormancy is closely related to immune cells and is involved in the immunosuppressive effect. T cell-induced tumor dormancy also involves crosstalk with endothelial cells and angiogenesis inhibition. Studies have shown that CD4+ T cell mediated antitumor effect is closely related to inducing tumor dormancy, and tumor progression is inhibited by its independent generation of tumor necrosis factor receptor 1 (TNFR1) and interferon -γ (IFN-γ) signals (142). IFN-γ signaling is a powerful inducer of growth stagnation and dormancy, acting mainly by removing immunosuppression of effector T cells. Meanwhile, IFN-γ can irreversibly induce tumor cell senescence and transform into a significant growth arrest phenotype in the T-antigen-induced PDAC model (143). Understanding tumor dormancy is important because dormant cells may be the source of tumor recurrence. Studies have shown that, overexpression of the Notch signaling ligand DLL4 (delta-like 4) in endothelial cells can promote T-ALL (T-cell acute lymphoblastic leukemia) cells to exit dormancy by binding to the Notch3 receptor on T-ALL cells, and this overexpression of DLL4 can be induced by VEGF (144). Regulating endothelial cell and dormancy DTCs interactions with VEGF inhibitors may be a new approach to prevent dormancy withdrawal. In addition to endothelial cells, other cell types may also facilitate the transition from dormancy, for example, recruitment of blood vessels through tissue factor signaling (CD105+) and myeloid cells (CD11b+ and F4/80+) can terminate the dormancy of tumor cells (145, 146). In this theory, inhibition of tumor cell activation in the dormant state can effectively improve the later survival rate of PDAC patients (126).
Signaling pathways important for PDAC immunosuppression
Immunosuppressive effect in microenvironment is an important factor in the progression of PDAC. In the process of immunosuppression, a variety of cells involved in immune function transmit information through cytokine pathways or intercellular transduction pathways, such as Notch, TGF-β, Wnt, Hippo, Fas/FasL, Hh, et. These signaling pathways are closely related to each other, with extensive mutual interference, and play a communication role in the immune-related microenvironment of tumors (Figure 2).
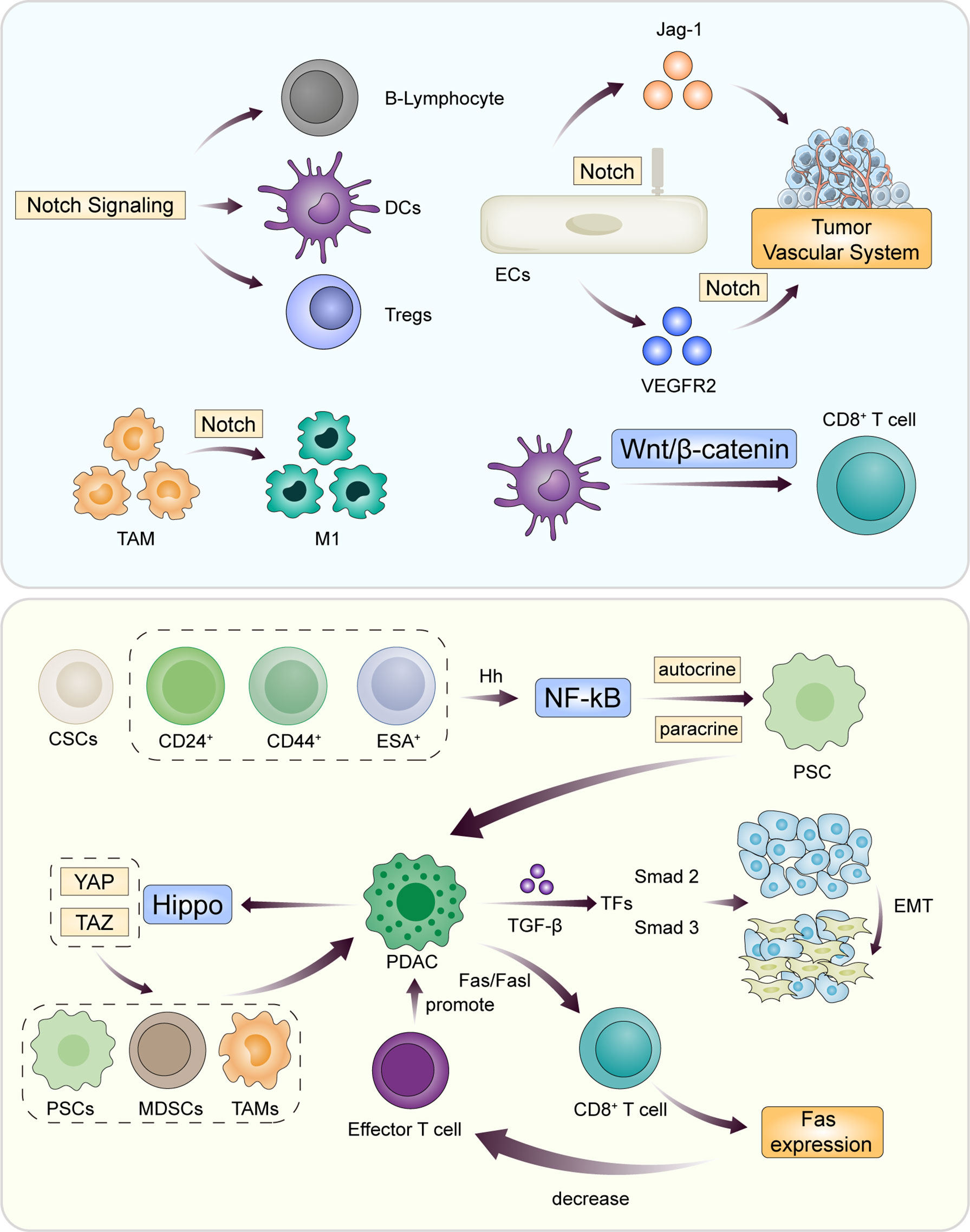
Figure 2 During the progression of PDAC, the innate immune system of the body is activated and antagonizes the immunosuppressive mechanism of tumor cells. Intercellular information transmission involves multiple complex signaling pathways through paracrine and autocrine. Classic signaling pathways include Notch, TGF-β, Wnt, Hippo, Fas/FasL, and Hh. The core of the pathway is to promote or inhibit the function of B cells and effector T cells by changing the cellular components of TME.
Notch
Notch signaling regulates many aspects of cancer biology in the PDAC microenvironment and plays an important role in immunosuppression. It also takes a significant part in regulating the crosstalk between the different compartments of TME (147). The mechanism of Notch signaling involved in tumor vascular remodeling is that the overexpression of Jag1 in endothelial cells (ECs) leads to the increase of tumor vascular system, while the loss of Jag1 function in ECs leads to the decrease of vascular system and tumor growth. Dll4/Notch controls the emergence of endothelial cells in TME. Notch-mediated VEGFR2 inhibition maintains the stem cell phenotype and avoids overexpression, thereby controlling tumor vascular structure (148). By regulating the tip/stalk ratio, Notch is also implicated in regulating the escape of metastasizing cancer cells from dormancy, as tip cells are associated with this process. Notch signaling is involved in cell lineage regulation of lymphocyte development, regulation of B-lymphocyte subsets in the marginal region, and differentiation and function of DCs, innate lymphocytes, and helper and regulatory T cells (149). Notch signaling has been found to play an important role in the regulation of CD8+ cytotoxic T cell activation (150). This also suggests that Notch signaling plays a positive role in modulating the antitumor activity of CD8+T cells, but it is also critical in modulating different components of immunosuppression. For example, Notch is important for the differentiation of TAMs, mainly because TAMs show transcriptional profiles associated with the Notch pathway, and Notch signaling is involved in the increased phenotype of M1 macrophages (151). Deletion of CBF-1/Su(H)/LAG1 (CSL) transcription factor in mononuclear cell lines can block TAMs differences and TAM-related immunosuppression functions (152). As mentioned above, CAFs express activating markers such as α-SMA, fibroblast activating protein, and many secretory factors that are involved in cell recruitment during extracellular matrix remodeling and immunosuppression. Notch1 is a major regulator of senescence secretion in fibroblasts, and Notch signaling in fibroblasts has a synergistic effect on the inhibition of tumor formation, possibly because the loss of Notch canonical signal is associated with CAFs differentiation (153, 154).
Fas/FasL
It was found that Fas/FasL pathway is mainly involved in mediating the apoptosis induced by cytotoxicity during T-cell development, while PDAC tumor cells antagonize the apoptosis of CD8+T cells in pancreatic cancer through Fas/FasL pathway, which leads to the down-regulation of Fas expression and the obstruction of the killing pathway of effector T cells to tumor cells, resulting in drug resistance (155). Kaplan-meier survival analysis showed that high levels of Fas cytoplasmic expression in PDAC cells were significantly associated with better prognosis (156).
Hh
Hedgehog (Hh) signaling mediates PDAC immune-related behavior, mainly through the regulation of CD44+CD24+epithelial-specific antigen (ESA)+ pancreatic cancer stem cells (157). The main mechanism is that overexpressed sonic Hh (Shh) in CSCs affects the transcription factor NF-κB through autocrine and paracrine pathways and secretes tumor growth factors against PSCs in the tumor-associated stroma to control inflammatory responses and immunosuppression in TME (158, 159). It has been reported that inhibition of Hh signaling can inhibit the self-renewal of pancreatic CSCs and reverse chemotherapy resistance (160).
TGF-β
Transforming growth factor-Beta (TGF-β) and it signaling pathway are key regulators of PDAC proliferation and differentiation, and their effects depend on TME. Changes in the microenvironment feedback to TGF-β signaling pathway may lead to tumor cells apoptosis or progression resulting in immunosuppression, they are generally viewed as separate fates for TGF-β-stimulated tumor cells, and opposite poles of the duality of TGF-β in cancer, or induce an EMT that promotes tumor invasion and metastasis and promote CSCs heterogeneity and drug resistance (161, 162). TGF-β is the major tumor suppressor signal of the pancreas, and its mechanism is that TGF-β activates Smad2 and Smad3 transcription factors (TFs) through membrane receptor kinases, which bind Smad4 to respond to the antitumor effects of immune cells (163). PDAC cells directly or indirectly induce EMT production by silencing TGF-β receptors or inactivation of Smad genes, promoting tumor aggressiveness and stem-like characteristics (164–166).
Wnt
Abnormal activation of Wnt/β-catenin signal transduction leads to the accumulation of β-catenin in the nucleus and promotes the transcription of many oncogenes, including c-Myc and CyclinD-1, and is involved in the initiation, progression, dormancy, immunity, and stem cell maintenance of pancreatic malignancies (167). In canonical Wnt pathway, β-catenin, and T-cell factor (TCF)/lymphoid enhancement factor (LEF) has been identified as signal transducers of the canonical Wnt pathway, in which β-catenin is a core molecule (168–171). β-catenin promotes the progression of tumors via suppressing the T-cell responses (172). When referring to tumor growth, Wnt/β-catenin signaling may play opposing roles in different tumor tissues. It has been reported that inhibition of β-catenin signaling could suppress pancreatic tumor growth by disrupting the nuclear β-catenin/TCF1 complex (173). As the main cause of tumor recurrence, Wnt signaling is often involved in the activation of tumor dormancy. β-catenin expression up-regulates urokinase plasminogen activator (uPA) expression and promotes invasion, metastasis, and dormancy of tumor cells (174). In addition, Wnt/β -catenin signaling is also involved in the regulation of tumor immunity. Activation of β -catenin in tumors mainly excludes T cells from infiltrating the TME. Thus, activation of beta-catenin may represent a mechanism for primary resistance to T cell immune tumor therapy (175). Cross-priming contributes to the production of anti-tumor CD8+T cells. β -catenin expression in DCs negatively regulates anti-tumor immunity by inhibiting its cross-priming ability, thereby inhibiting CD8+T cell response. Therefore, the Wnt/β -catenin signaling pathway may be a potential target for anti-tumor immunotherapy (176).
Hippo
Hippo signaling is a key regulator of organ size, tissue hemostasis and regeneration. Dysregulation of the Hippo pathway has been recognized in PDAC. YES-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) are the two major downstream effectors of the Hippo pathway. YAP and TAZ regulate the behavior of pancreatic stellate cells and the recruitment of TAMs and MDSCs. Among them, YAP plays a dominant role, whose activation is associated with immune dysfunction and induces the recruitment of myelogenous suppressor cells, while counter-inhibition supports the infiltration of antigen-presenting macrophages and the activation of effector T cells, leading to immunosuppression and progression (177, 178).
Clinical application and progress of immunotherapy
In today’s medical era, PDAC, as an incurable malignant tumor, escapes from chemotherapy or targeted therapy due to inherent genomic instability (179). Due to the instability of pancreatic cancer treatment, it is more likely to lead to resistance to surgical treatment, chemotherapy, and radiotherapy, at this time immunotherapy has become the fourth cornerstone of treatment. Immunotherapy is the best hope for preventing relapse and prolonging long-term survival by adapting the long-term memory function of the immune system (180, 181). Immunotherapy and its effect on treatment have been a hot topic in clinical research and may improve the prognosis of PDAC, mainly including targeted therapy and anti-tumor vaccination (182). It can also be divided into active or passive immunity according to the involvement of the host immune system (183). According to the different mechanisms of tumor activation, it can be divided into four categories: specific active immunotherapy, specific passive immunotherapy, non-specific adoptive immunotherapy and non-specific immunoregulation (184, 185). Immunotherapy for PDAC is a growing concern because conventional therapies such as chemotherapy are not effective in improving overall survival outcomes in patients with PDAC. However, it is still not a good solution for prolonging patient survival due to its unique tumor microenvironment and low tumor immunogenicity. Therefore, inducing more intratumor effector immune cells and reversing immunosuppression are the core of PDAC therapy (186). Immunotherapy has relatively mild side effects and has shifted the focus of treatment from the tumor itself to the immune system of the host. Immune cells in the TME have a key role in the development and progression. To overcome the deficiency of PDAC immune recognition, clinical researchers have explored several immunotherapy approaches in the form of vaccines to enhance antigen presentation and drive the expansion of tumor-specific T cell clones (187) to initiate new or enhance existing immune responses. Strategies for PDAC associated antigens (including telomerase (188), KRAS (189, 190), gastrin (191), CEA (192), MUC1 (193), and mesenchymal (194, 195)) include peptide-based vaccines (188–190), virus-based vaccines (196), listeria-based vaccines (195), DNA-based vaccines(neoantigens) (197, 198), and cell-based vaccines (199). Specific active immunotherapy mainly comprises therapeutic cancer vaccines, which aim to activate the immune system to eventually result in the expansion of tumor-specific T/B cells (200). Cancer vaccines can be generally divided into three major categories: cell-based vaccines, protein/peptide vaccines and genetic vaccines. Various genes or proteins have been targeted in recent I/II/III clinical trials of PDAC vaccines, such as telomerase and Wilms tumor gene (201, 202). Based on the results of many studies, vaccination methods have now been established to produce antigen-specific immune responses in PDAC patients (195, 203). Specific passive immunotherapy involves the direct infusion of tumor-specific immune effector cells or antibodies into a PDAC patient to mediate the immune response to the cancer. Monoclonal antibodies are currently the most widely adopted specific passive immunotherapy, including anti-EGFR and anti-VEGF antibodies, such as Erlotinib and Bevacizumab. A PD-1/PD-L1 blockade falls into this category of immunotherapy. A few studies have indicated that gemcitabine plus erlotinib shows favorable efficacy and safety and could significantly improve survival (204). Nonspecific adoptive immunotherapy involves the adoptive transfer of treated highly specific immune cells or cytokines into the patient to induce a passive immune response. In an early study, patients with advanced PDAC who received adoptive immunotherapy using intra-portal infusion of lymphoid-activated killer cells showed a lower rate of liver metastasis and a higher 3-year survival rate (205). Another study showed that adoptive immunotherapy of CTLs stimulated cells induced by co-culture with peripheral blood mononuclear cells and activated YPK-1 cells significantly inhibited postoperative liver recurrence of PDAC (206). Another immunotherapy direction for pancreatic tumors is to block the differentiation of macrophages induced by Galectin-9 (gal-9) (207), thereby enhancing the original immune response. Gal-9 is a member of the lectin β-galactoside-binding family and is highly expressed in PDAC. Binding of dectin-1 (208), an important natural immune receptor on the surface of macrophages, to Galectin-9 induces the transformation of macrophages to the M2 phenotype. Blocking galectin-9 can lead to reversal of immunosuppression, activation of CD4+ and CD8+ T cell effect and enhancement of anti-tumor effect, thus enhancing the killing effect of immune cells on PDAC cells and inhibiting growth and metastasis (207, 209). In addition, according to the immunosuppressive properties of NetG1+ CAF mentioned above, NetG1 is generally considered as a potential target for PDAC immunotherapy. In clinical application, inhibition of metabolic proteins in CAFs changes its immunosuppressive ability by blocking the NetG1 pathway with a neutralizing antibody (93). Therefore, immunotherapy is combined with other therapies such as surgery, chemotherapy, radiotherapy, targeted therapy, and other immunotherapies. Multiple therapies are used together to overcome resistance to immunotherapy (6, 210). Clinical trials have shown that a notable outcome in PDAC combination therapy is the combination of oxaliplatin (OXA) and gal-9 siRNA. OXA, part of folfirinox regimen for PDAC, triggers immunogenic cell death (ICD) effects in pancreatic tumor sites and kills tumor cells by inhibiting DNA synthesis and repair. OXA combined with gal-9 siRNA can block the galectin-9/Dectin-1 axis, reverse the immunosuppression induced by M2-TAMS, improve the efficiency of chemotherapy drug delivery and increased infiltration of antitumoral cytotoxic T lymphocytes, reversed immunosuppression, which has a significant effect on improving the quality of life of patients with advanced PDAC and controlling progression (207, 211). In terms of the current development of immunotherapy, immune checkpoint inhibition of T cells is a new technique in immunotherapy of PDAC, immune checkpoint inhibitors targeting cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein-1 (PD-1)/programmed cell death ligand-1 (PD-L1) pathways have shown significant potential in the treatment (210, 212, 213).
CTLA-4
CTLA-4 is an immune checkpoint receptor expressed on Tregs that activates conventional T cells (214). It’s also a negative regulator of T cell activation, also known as CD152. CTLA-4 is homologous to CD28 and has the same ligand. Both B7-1 (CD80) and B7-2 (CD86) ligands are expressed on antigen presenting cells (APCs) and can provide costimulatory signals to T cells. After these processes, the activated T cells expressed CTLA-4 on their surfaces. In addition, the binding of CTLA-4 to B7 inhibited T cell activation. CTLA-4 has a significantly higher affinity for B7 ligand than CD28. So, the CTLA-4 junction delivers inhibitory signals to T cells, while CD28 delivers stimulative signals (215, 216). In PDAC, anti-CTLA-4 antibody can block the interaction between CTLA-4 and B7, block the inhibitory signal, and down-regulate the immune system, to induce the host’s inhibitory effect on tumor and produce a lasting anti-tumor effect (217, 218). However, since T cell rejection is evident in PDAC, effector T cells are usually few in tumor tissue and confined to peripheral lymph nodes and lymphoid aggregates in PDAC (219). Therefore, the use of CTLA-4 antibody alone in the treatment of PDAC has little effect (220). This might be due to high tumor burden and the intrinsic nonimmunogenic nature of pancreatic cancer that cause immune quiescent, and the blockage of only one checkpoint is not enough for immunosuppressive reduction. And, with the development of clinical drug trials in recent years, the combination of CTLA-4 blocking drugs with surgical treatment or chemotherapy has gradually become a promising method in the treatment of PDAC.
PD-1/PDL-1
In the immunotherapy of pancreatic tumors, the inhibitory effect of PD-1 and PD-L1 on tumors has always been the expected result of clinical studies. PD-1 plays an important role in inhibiting immune response and promoting self-tolerance by regulating T Cell activity, activating antigen-specific T Cell apoptosis, and inhibiting regulatory T Cell apoptosis. PD-L1 is a transmembrane protein, which is regarded as an inhibitory factor of immune response. It can bind to PD-1, reduce the proliferation of PD-1 positive cells, inhibit the secretion of cytokines, and induce cell apoptosis. PD-L1 also plays an important role in various malignant tumors by weakening the host’s immune response to tumor cells. Based on these ideas, the PD-1/PD-L1 axis is responsible for cancer immune escape and has a huge impact on cancer treatment (221). These molecules can be blocked by monoclonal antibodies, such as ipilimumab, nivolumab and pembrolizumab. However, immune checkpoint suppression monotherapy may be ineffective in pancreatic cancer, possibly due to low PD-L1 expression, highly complex interactions between tumor and stroma, and connective tissue hyperplasia. PD-L1 was confirmed to increase T-cell apoptosis in vitro and in vivo and to protect tumor cells from being killed, which unlocked the door of T-cell-based cancer immunotherapy (222). The PD-1/PD-L1 pathway has become popular in immunotherapy for the following reasons. Multiple studies have shown that PD-1 can inhibit the overactivation of immune responses and help maintain immune tolerance to autoantigens (223–225). After antigen recognition, PD-1 was expressed on the surface of activated T cells. However, PD-L1 is expressed by a variety of cell types, including immune and tumor cells, after interacting with cytokines such as IFN-γ, which is produced by activated T cells (224, 226). PD-1 and PD-L1 (B7-H1) belong to CD28+ superfamily and B7 superfamily respectively (227). Preclinical studies and clinical trials in mouse models have recently evaluated anti-PD-1/PD-L1 as an immune checkpoint blocking therapy for overcoming fatal malignancies. Anti-PD-1/PD-L1 antibodies have been shown to have clinical efficacy in many cancer types (228, 229). In 2007, PD-L1 was first considered as a new prognostic factor for patients with pancreatic cancer, when PD-L1 was first demonstrated to be up-regulated in PDAC specimens (230, 231). It has been suggested that PD-L1 blockers can effectively inhibit preestablished pancreatic cancer in mouse models by increasing IFN-γ production and decreasing IL-10 production (232). In addition, the level of tumor infiltrating Tregs in PD-L1 positive tumors was higher than that in negative tumors (227). These results provide a theoretical basis for the treatment of PDAC by targeting PD-1/PD-L1 pathway. But in clinical practice, the efficacy of PD-1/PD-L1 blockers alone may be limited for two main reasons. First, immunosuppression due to high tumor load is the reason why PD-1/PD-L1 blockade alone does not cure PDAC. Second, PDAC is nonimmunogenic in nature (233). Immune resistance is also responsible for the failure of anti-PD-1/PD-L1 monotherapy in pancreatic cancer. Like chemotherapy resistance, immune resistance to immunotherapy (including PD-1/PD-L1 blockade) can be divided into two types: primary resistance and acquired resistance. Primary drug resistance is a clinical condition in which the cancer does not respond to initial immunotherapy. Acquired drug resistance is a clinical condition in which some initial responders’ relapse after a period of response (179). Adaptive resistance is a mechanism of resistance in which a cancer cell is recognized by the immune system, but it protects itself against immune attack by the immune system. PDAC is resistant to therapy, including immune checkpoint inhibitors. In addition, pancreatic tumors appear to evade the immune response by inducing development of immunosuppressive T cells. Current studies indicate that both internal and external factors of tumor cells are involved in the regulatory mechanism of drug resistance. The failure of T cell recognition due to tumor antigen deficiency has become the most direct factor to predict the lack of response to anti-PD-1/PD-L1 therapy (234). In the immunotherapy of PDAC, PD-L1 can be used as a biomarker to predict the anti-PD-1/PD-L1 immunotherapy response rate. Since tumor-associated PD-L1 has been shown to increase T cell apoptosis in vitro and in vivo, and is expressed in most cell types after IFN-γ treatment, it can represent effector T cell activity (235). PDAC patients with high PD-L1 levels may not respond to PD-L1 therapy, due to the surge in the number of tumor immunosuppressive cells, such as the high infiltration of Tregs/MDSCs/TAMs, where TGFβ stimulates dendritic cells to induce immunosuppression (236). And CSF-1R activates TAMs as a macrophage colony-stimulating growth factor receptor (179). Therefore, PD-L1 should not be the only predictive biomarker. Other biomarkers that affect TME and reflect effector T cell function, Tregs, MDSCs, and TAMs should be combined with PD-L1 to improve the accuracy of predicting anti-PD-1/PD-L1 immunotherapy responses. Therefore, accurate identification of tumor immune characteristics will help predict the response to anti-PD-1/PD-L1 therapy (210). The current studies indicate that a PD-1/PD-L1 blockade in combination with surgery, chemotherapy, radiotherapy, molecular targeted therapy, or other immunotherapies could modulate the immunoediting process, TME and the immune response in pancreatic cancer, which have been or will be validated in the latest clinical trials.
Drug combinations in immunotherapy
Drug combination in immunotherapy has always been a hot project in clinical practice (Table 1). Multiple drugs combined with each other can more effectively prevent PDAC from progressing due to treatment resistance. Among the drug combinations for PDAC, the most common is the combination of anti-PD-1/PD-L1 therapy with chemotherapy. Gemcitabine and 5-FU remain the main adjunctive treatment strategies for PDAC in clinical practice (237). Chemotherapeutic drugs can inhibit tumor growth by encouraging tumor cells to release antigens and reactivate anti-tumor immune responses (238). Immunotherapeutic agents are added to chemotherapy to enhance and synergistically enhance the anti-tumor effect of the two therapies (229). In addition, current studies have shown that specific molecules targeted against PD-1/PD-L1 therapy can simultaneously improve chemotherapy sensitivity and inhibit immunosuppression. Relevant data also clearly demonstrated that in the treatment environment of PDAC, anti-PD-1/PD-L1 therapy combined with chemotherapy (related clinical studies of various types of drugs) effectively extended the later survival rate of patients. In mouse models, the role of programmed cell death neutralizing antibody 1 (PD-1) and an OX40 agonist (which provides survival signals to activated T cells) in PDAC was evaluated. The combination of anti-PD-1 inhibitory and anti-OX40 agonist antibodies reduces the proportion of T-regulatory and exhausted T cells in PDAC and increases numbers of memory CD4+ and CD8+ T cells, eradicating all detectable tumor. This information can be used in development of immune-based combination therapies for PDAC (239). With the diversification of treatment methods, the combination of immunotherapy and targeted therapy has shown good results. Although targeting growth factor receptor inhibitors such as anti-EGFR antibodies (erlotinib) has significantly improved survival in pancreatic cancer, only a small population have benefitted (240). Various preclinical studies and clinical trials have shown that the frequency of BRCA1/2 mutations in pancreatic cancer is between 4% and 7% (241, 242). Poly (ADP-ribose) polymerase (PARP) inhibitors are a class of small-molecule drugs that inhibit the activity of PARP and may lead to cell death lacking homologous recombination repair (243). It is common in PDAC cells with BRCA1/2 mutations. A recent study confirmed the efficacy and safety of PARP inhibitors in PDAC (244). Therefore, anti-PD-1/PD-L1 combined with PARP inhibitors may be effective against PDAC with BRCA1/2 mutations. And it has been suggested that if agents could be developed to inactivate the WNT-β-catenin signaling pathway and prevent T-cell infiltration, it would be effective if combined with a PD-1/PD-L1 blockade (175). In immunotherapy, a specific method has been gradually used in clinical treatment in recent years. Based on the different mechanisms of various immunotherapies, anti-PD-1/PD-L1 therapy combined with other immunotherapies can often regulate TME and immune response and improve the anti-cancer efficacy. For example, combining cancer vaccines with other immune checkpoint inhibitors. Multiple clinical trials have shown that the combination of anti-PD-1/PD-L1 therapy with other types of immunotherapy is effective in treating PDAC, and the combination of GVAX (GM-CSF cell-based vaccines) vaccine and PD-1 blocker can significantly improve the survival of patients compared with anti-PD-1 or GVAX monotherapy (245). This is because GVAX in combination with PD-1 blockers increases CD8+TILs in PDAC and promotes the activation of CD8+T cells, resulting in the production of more tumor-specific IFN-γ in TME, which will help overcome Tregs, CTLA-4, and PD-1/PD-L1 immunosuppressive pathways (246).
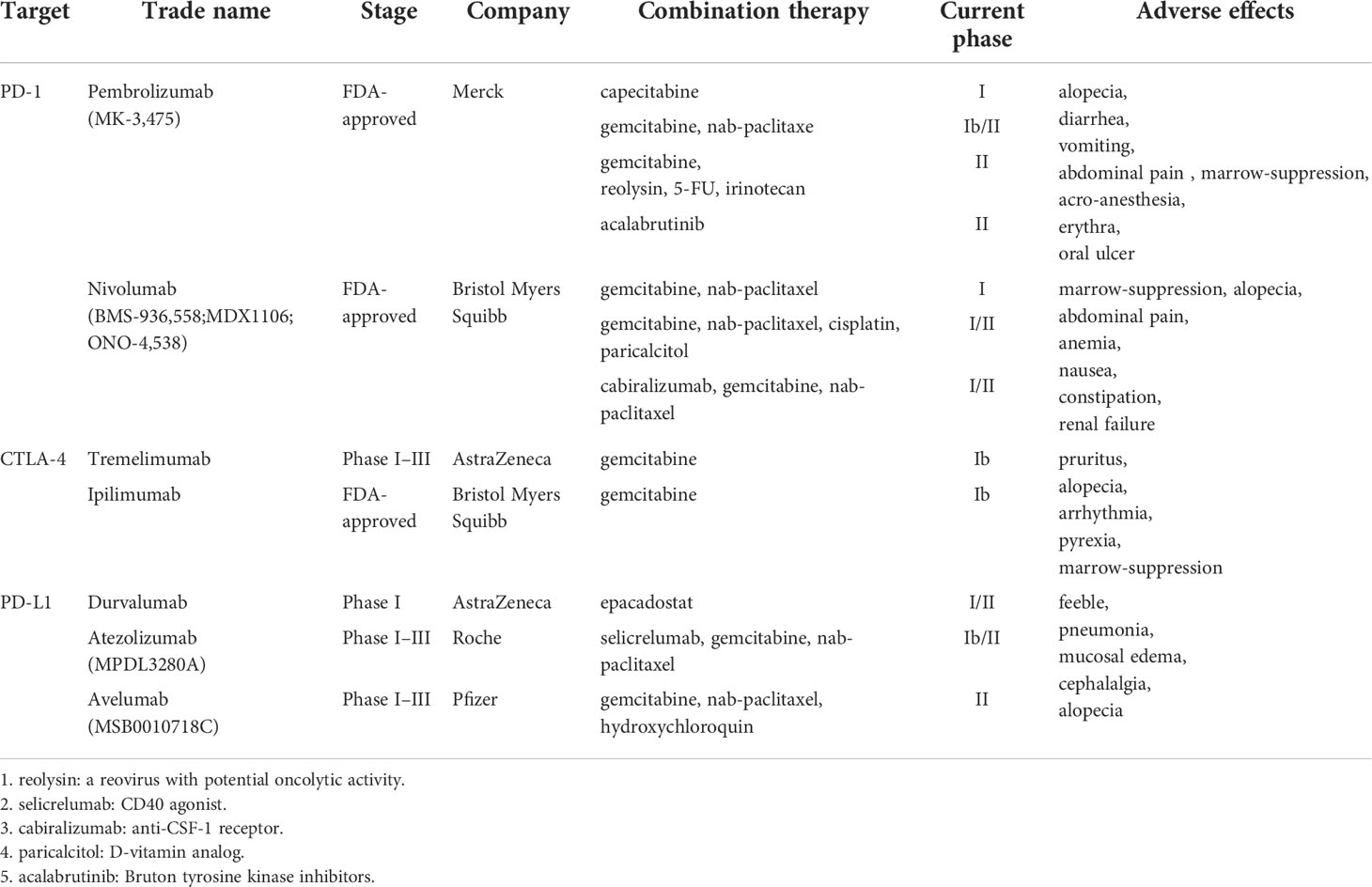
Table 1 Immune checkpoint inhibitors in combination with other therapies currently used in the clinical test medication.
In recent decades, encouraging results have been achieved in mouse models and clinical trials, and in various preclinical studies on the efficacy of immunotherapy and its potential therapeutic application in pancreatic cancer. The understanding of the biology and pathophysiology of TME in PDAC has improved significantly through in-depth research, and new immunotherapy approaches are emerging that may help improve the devastating prognosis of patients with PDAC.
Conclusions and perspectives
In PDAC, immune-related cells and cytokines in tumor microenvironment show tumor-promoting and tumor-suppressive activity to varying degrees. Tumor-promoting cells include MDSCs, CAFs, TAMs, etc., while tumor-suppressive immune cells include cytotoxic T cells (CTLs) and natural killer cells (NK cells). These cells recognize and to varying degrees alter tumor cells and their cellular environment with cytokines produced by paracrine and autocrine, which is also named tumor immune microenvironment (TIME). It is particularly important to accurately understand the immune components-related signal transduction pathways, in which the role of the signaling pathways involved in different parts of the TME is a prerequisite for better understanding of preclinical and clinical model data to design better treatments for signaling pathway targets in PDAC. In the treatment of PDAC, traditional chemotherapy has been regarded as an effective method of disease remission, but clinical data show that the effectiveness is only limited to a subset of patients. Therefore, immunotherapy has become a popular direction of clinical research, with its advantages of persistence, universal applicability, and relatively low toxicity. Checkpoint inhibitors have always been a hot spot in clinical research and application. In addition, the following clinical experiments show that PDAC immune checkpoint inhibitors combined with chemotherapy or radiotherapy can enhance the sensitivity of radiochemotherapy, reduce the toxic and side effects of radiochemotherapy, improve the efficacy of radiochemotherapy and reduce the occurrence of treatment resistance. Although immunotherapy is not an all-powerful treatment, it opens a new field in the treatment of PDAC with a very high degree of malignancy. With the progress of drug clinical research, immunotherapy will become one of the main treatment methods for tumors. Given the advances in our understanding of the PDAC microenvironment and emerging strategies, we have reason to be hopeful for the successful treatment of PDAC in the future.
Author contributions
JZ completed the main content of the article. SH and RL gave guidance and corrected the mistakes. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ansari D, Tingstedt B, Andersson B, Holmquist F, Sturesson C, Williamsson C, et al. Pancreatic cancer: Yesterday, today and tomorrow. Future Oncol (2016) 12(16):1929–46. doi: 10.2217/fon-2016-0010
2. Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet (2011) 378(9791):607–20. doi: 10.1016/S0140-6736(10)62307-0
3. Mostafa ME, Erbarut-Seven I, Pehlivanoglu B, Adsay V. Pathologic classification of "pancreatic cancers": Current concepts and challenges. Chin Clin Oncol (2017) 6(6):59. doi: 10.21037/cco.2017.12.01
4. Dhillon J, Betancourt M. Pancreatic ductal adenocarcinoma. Monogr Clin Cytol (2020) 26:74–91. doi: 10.1159/000455736
5. Panchal K, Sahoo RK, Gupta U, Chaurasiya A. Role of targeted immunotherapy for pancreatic ductal adenocarcinoma (PDAC) treatment: An overview. Int Immunopharmacol (2021) 95:107508. doi: 10.1016/j.intimp.2021.107508
6. Ren B, Cui M, Yang G, Wang H, Feng M, You L, et al. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol Cancer (2018) 17(1):108. doi: 10.1186/s12943-018-0858-1
7. Chronopoulos A, Robinson B, Sarper M, Cortes E, Auernheimer V, Lachowski D, et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun (2016) 7:12630. doi: 10.1038/ncomms12630
8. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov (2019) 18(3):175–96. doi: 10.1038/s41573-018-0006-z
9. Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol (2020) 17(8):807–21. doi: 10.1038/s41423-020-0488-6
10. Helmy KY, Patel SA, Nahas GR, Rameshwar P. Cancer immunotherapy: accomplishments to date and future promise. Ther Deliv (2013) 4(10):1307–20. doi: 10.4155/tde.13.88
11. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441(7090):235–8. doi: 10.1038/nature04753
12. Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep (2017) 20(3):558–71. doi: 10.1016/j.celrep.2017.06.062
13. Padoan A, Plebani M, Basso D. Inflammation and pancreatic cancer: Focus on metabolism, cytokines, and immunity. Int J Mol Sci (2019) 20(3):13. doi: 10.3390/ijms20030676
14. Grage-Griebenow E, Schäfer H, Sebens S. The fatal alliance of cancer and T cells: How pancreatic tumor cells gather immunosuppressive T cells. Oncoimmunology (2014) 3:e29382. doi: 10.4161/onci.29382
15. Pylayeva-Gupta Y, Lee KE, Bar-Sagi D. Microdissection and culture of murine pancreatic ductal epithelial cells. Methods Mol Biol (2013) 980:267–79. doi: 10.1007/978-1-62703-287-2_14
16. Wang X, Wang L, Mo Q, Dong Y, Wang G, Ji A. Changes of Th17/Treg cell and related cytokines in pancreatic cancer patients. Int J Clin Exp Pathol (2015) 8(5):5702–8.
17. Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin Cancer Res (2007) 13(15 Pt 1):4345–54. doi: 10.1158/1078-0432.CCR-07-0472
18. Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes a and b in human cytotoxic lymphocyte subsets and T regulatory cells. Blood (2004) 104(9):2840–8. doi: 10.1182/blood-2004-03-0859
19. Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme b-dependent, perforin-independent mechanism. J Immunol (2005) 174(4):1783–6. doi: 10.4049/jimmunol.174.4.1783
20. Ren X, Ye F, Jiang Z, Chu Y, Xiong S, Wang Y. Involvement of cellular death in TRAIL/DR5-dependent suppression induced by CD4(+)CD25(+) regulatory T cells. Cell Death Differ (2007) 14(12):2076–84. doi: 10.1038/sj.cdd.4402220
21. Garín MI, Chu CC, Golshayan D, Cernuda-Morollón E, Wait R, Lechler RI. Galectin-1: A key effector of regulation mediated by CD4+CD25+ T cells. Blood (2007) 109(5):2058–65. doi: 10.1182/blood-2006-04-016451
22. Beyer K, Normann L, Sendler M, Käding A, Heidecke CD, Partecke LI, et al. TRAIL promotes tumor growth in a syngeneic murine orthotopic pancreatic cancer model and affects the host immune response. Pancreas (2016) 45(3):401–8. doi: 10.1097/MPA.0000000000000469
23. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8(12):1353–62. doi: 10.1038/ni1536
24. Moon YW, Hajjar J, Hwu P, Naing A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J Immunother Cancer (2015) 3:51. doi: 10.1186/s40425-015-0094-9
25. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol (2003) 4(12):1206–12. doi: 10.1038/ni1003
26. Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T, et al. Treg cells promote the SREBP1-dependent metabolic fitness of tumor-promoting macrophages via repression of CD8(+) T cell-derived interferon-γ. Immunity (2019) 51(2):381–97.e6. doi: 10.1016/j.immuni.2019.06.017
27. Gordon S, Plüddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev (2014) 262(1):36–55. doi: 10.1111/imr.12223
28. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol (2004) 25(12):677–86. doi: 10.1016/j.it.2004.09.015
29. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep (2014) 6:13. doi: 10.12703/P6-13
30. Hu H, Hang JJ, Han T, Zhuo M, Jiao F, Wang LW. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumour Biol (2016) 37(7):8657–64. doi: 10.1007/s13277-015-4741-z
31. Yang M, Li J, Gu P, Fan X. The application of nanoparticles in cancer immunotherapy: Targeting tumor microenvironment. Bioact Mater (2021) 6(7):1973–87. doi: 10.1016/j.bioactmat.2020.12.010
32. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol (2017) 14(7):399–416. doi: 10.1038/nrclinonc.2016.217
33. Li X, Liu R, Su X, Pan Y, Han X, Shao C, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer (2019) 18(1):177. doi: 10.1186/s12943-019-1102-3
34. Lai Q, Vitale A, Manzia TM, Foschi FG, Levi Sandri GB, Gambato M, et al. Platelets and hepatocellular cancer: Bridging the bench to the clinics. Cancers (Basel) (2019) 11(10):7,8. doi: 10.3390/cancers11101568
35. Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers (2016) 2:16018. doi: 10.1038/nrdp.2016.18
36. Dhanasekaran R, Venkatesh SK, Torbenson MS, Roberts LR. Erratum to "Clinical implications of basic research in hepatocellular carcinoma" [J hepatol 2016;64:736-745]. J Hepatol (2016) 64(6):1462. doi: 10.1016/j.jhep.2016.02.036
37. Xiao P, Long X, Zhang L, Ye Y, Guo J, Liu P, et al. Neurotensin/IL-8 pathway orchestrates local inflammatory response and tumor invasion by inducing M2 polarization of tumor-associated macrophages and epithelial-mesenchymal transition of hepatocellular carcinoma cells. Oncoimmunology (2018) 7(7):e1440166. doi: 10.1080/2162402X.2018.1440166
38. Liu M, Zhang Y, Yang J, Zhan H, Zhou Z, Jiang Y, et al. Zinc-dependent regulation of ZEB1 and YAP1 coactivation promotes epithelial-mesenchymal transition plasticity and metastasis in pancreatic cancer. Gastroenterology (2021) 160(5):1771–83.e1. doi: 10.1053/j.gastro.2020.12.077
39. Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int J Oncol (2017) 51(5):1357–69. doi: 10.3892/ijo.2017.4129
40. Blaauboer A, Booy S, van Koetsveld PM, Karels B, Dogan F, van Zwienen S, et al. Interferon-beta enhances sensitivity to gemcitabine in pancreatic cancer. BMC Cancer (2020) 20(1):913. doi: 10.1186/s12885-020-07420-0
41. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi: 10.1158/0008-5472.CAN-04-0465
42. Ostuni R, Kratochvill F, Murray PJ, Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol (2015) 36(4):229–39. doi: 10.1016/j.it.2015.02.004
43. Anderson EM, Thomassian S, Gong J, Hendifar A, Osipov A. Advances in pancreatic ductal adenocarcinoma treatment. Cancers (Basel) (2021) 13(21)9, 10. doi: 10.3390/cancers13215510
44. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol (2009) 86(5):1065–73. doi: 10.1189/jlb.0609385
45. Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett (2008) 267(2):204–15. doi: 10.1016/j.canlet.2008.03.028
46. Szebeni GJ, Vizler C, Kitajka K, Puskas LG. Inflammation and cancer: Extra- and intracellular determinants of tumor-associated macrophages as tumor promoters. Mediators Inflamm (2017) 2017:9294018. doi: 10.1155/2017/9294018
47. Yang J, Zhang JX, Wang H, Wang GL, Hu QG, Zheng QC. Hepatocellular carcinoma and macrophage interaction induced tumor immunosuppression via treg requires TLR4 signaling. World J Gastroenterol (2012) 18(23):2938–47. doi: 10.3748/wjg.v18.i23.2938
48. Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, et al. Blocking triggering receptor expressed on myeloid cells-1-Positive tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-programmed cell death ligand 1 resistance in liver cancer. Hepatology (2019) 70(1):198–214. doi: 10.1002/hep.30593
49. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov (2018) 8(9):1069–86. doi: 10.1158/2159-8290.CD-18-0367
50. Sharma P, Allison JP. The future of immune checkpoint therapy. Science (2015) 348(6230):56–61. doi: 10.1126/science.aaa8172
51. Zheng W, Pollard JW. Inhibiting macrophage PI3Kγ to enhance immunotherapy. Cell Res (2016) 26(12):1267–8. doi: 10.1038/cr.2016.132
52. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature (2016) 539(7629):437–42. doi: 10.1038/nature19834
53. Markowitz J, Brooks TR, Duggan MC, Paul BK, Pan X, Wei L, et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol Immunother (2015) 64(2):149–59. doi: 10.1007/s00262-014-1618-8
54. Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut (2014) 63(11):1769–81. doi: 10.1136/gutjnl-2013-306271
55. Bulle A, Lim KH. Beyond just a tight fortress: contribution of stroma to epithelial-mesenchymal transition in pancreatic cancer. Signal Transduct Target Ther (2020) 5(1):249. doi: 10.1038/s41392-020-00341-1
56. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell (2012) 21(6):822–35. doi: 10.1016/j.ccr.2012.04.025
57. Otsuji M, Kimura Y, Aoe T, Okamoto Y, Saito T. Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses. Proc Natl Acad Sci U S A (1996) 93(23):13119–24. doi: 10.1073/pnas.93.23.13119
58. Gabrilovich DI. Myeloid-derived suppressor cells. Cancer Immunol Res (2017) 5(1):3–8. doi: 10.1158/2326-6066.CIR-16-0297
59. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell (2012) 21(6):836–47. doi: 10.1016/j.ccr.2012.04.024
60. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell (2007) 1(3):313–23. doi: 10.1016/j.stem.2007.06.002
61. Hong SP, Wen J, Bang S, Park S, Song SY. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int J Cancer (2009) 125(10):2323–31. doi: 10.1002/ijc.24573
62. Bao Q, Zhao Y, Renner A, Niess H, Seeliger H, Jauch KW, et al. Cancer stem cells in pancreatic cancer. Cancers (Basel) (2010) 2(3):1629–41. doi: 10.3390/cancers2031629
63. Rao CV, Mohammed A. New insights into pancreatic cancer stem cells. World J Stem Cells (2015) 7(3):547–55. doi: 10.4252/wjsc.v7.i3.547
64. Tan BT, Park CY, Ailles LE, Weissman IL. The cancer stem cell hypothesis: a work in progress. Lab Invest (2006) 86(12):1203–7. doi: 10.1038/labinvest.3700488
65. Hashimoto O, Shimizu K, Semba S, Chiba S, Ku Y, Yokozaki H, et al. Hypoxia induces tumor aggressiveness and the expansion of CD133-positive cells in a hypoxia-inducible factor-1α-dependent manner in pancreatic cancer cells. Pathobiology (2011) 78(4):181–92. doi: 10.1159/000325538
66. Jaiswal KR, Xin HW, Anderson A, Wiegand G, Kim B, Miller T, et al. Comparative testing of various pancreatic cancer stem cells results in a novel class of pancreatic-cancer-initiating cells. Stem Cell Res (2012) 9(3):249–60. doi: 10.1016/j.scr.2012.08.001
67. Su HT, Weng CC, Hsiao PJ, Chen LH, Kuo TL, Chen YW, et al. Stem cell marker nestin is critical for TGF-β1-mediated tumor progression in pancreatic cancer. Mol Cancer Res (2013) 11(7):768–79. doi: 10.1158/1541-7786.MCR-12-0511
68. Constantino J, Gomes C, Falcão A, Neves BM, Cruz MT. Dendritic cell-based immunotherapy: A basic review and recent advances. Immunol Res (2017) 65(4):798–810. doi: 10.1007/s12026-017-8931-1
69. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol (2016) 37(12):855–65. doi: 10.1016/j.it.2016.09.006
70. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res (2010) 70(19):7465–75. doi: 10.1158/0008-5472.CAN-10-1439
71. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
72. Yin X, Chen S, Eisenbarth SC. Dendritic cell regulation of T helper cells. Annu Rev Immunol (2021) 39:759–90. doi: 10.1146/annurev-immunol-101819-025146
73. Allam A, Thomsen AR, Gothwal M, Saha D, Maurer J, Brunner TB. Pancreatic stellate cells in pancreatic cancer: In focus. Pancreatology (2017) 17(4):514–22. doi: 10.1016/j.pan.2017.05.390
74. Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J Clin Invest (2007) 117(1):50–9. doi: 10.1172/JCI30082
75. Ferdek PE, Jakubowska MA. Biology of pancreatic stellate cells-more than just pancreatic cancer. Pflugers Arch (2017) 469(9):1039–50. doi: 10.1007/s00424-017-1968-0
76. Fu Y, Liu S, Zeng S, Shen H. The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol Cancer (2018) 17(1):62. doi: 10.1186/s12943-018-0815-z
77. Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, Kumar RK, et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas (2004) 29(3):179–87. doi: 10.1097/00006676-200410000-00002
78. Schnittert J, Bansal R, Prakash J. Targeting pancreatic stellate cells in cancer. Trends Cancer (2019) 5(2):128–42. doi: 10.1016/j.trecan.2019.01.001
79. Wu Q, Tian Y, Zhang J, Zhang H, Gu F, Lu Y, et al. Functions of pancreatic stellate cell-derived soluble factors in the microenvironment of pancreatic ductal carcinoma. Oncotarget (2017) 8(60):102721–38. doi: 10.18632/oncotarget.21970
80. Tang D, Wang D, Yuan Z, Xue X, Zhang Y, An Y, et al. Persistent activation of pancreatic stellate cells creates a microenvironment favorable for the malignant behavior of pancreatic ductal adenocarcinoma. Int J Cancer (2013) 132(5):993–1003. doi: 10.1002/ijc.27715
81. Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology (2013) 145(5):1121–32. doi: 10.1053/j.gastro.2013.07.025
82. Tang D, Yuan Z, Xue X, Lu Z, Zhang Y, Wang H, et al. High expression of galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int J Cancer (2012) 130(10):2337–48. doi: 10.1002/ijc.26290
83. Lunardi S, Lim SY, Muschel RJ, Brunner TB. IP-10/CXCL10 attracts regulatory T cells: Implication for pancreatic cancer. Oncoimmunology (2015) 4(9):e1027473. doi: 10.1080/2162402X.2015.1027473
84. Lunardi S, Jamieson NB, Lim SY, Griffiths KL, Carvalho-Gaspar M, Al-Assar O, et al. IP-10/CXCL10 induction in human pancreatic cancer stroma influences lymphocytes recruitment and correlates with poor survival. Oncotarget (2014) 5(22):11064–80. doi: 10.18632/oncotarget.2519
85. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res (2013) 73(10):3007–18. doi: 10.1158/0008-5472.CAN-12-4601
86. Dougan SK. The pancreatic cancer microenvironment. Cancer J (2017) 23(6):321–5. doi: 10.1097/PPO.0000000000000288
87. Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell (2015) 160(1-2):324–38. doi: 10.1016/j.cell.2014.12.021
88. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med (2017) 214(3):579–96. doi: 10.1084/jem.20162024
89. Panni RZ, Sanford DE, Belt BA, Mitchem JB, Worley LA, Goetz BD, et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother (2014) 63(5):513–28. doi: 10.1007/s00262-014-1527-x
90. Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, et al. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab (2016) 24(5):672–84. doi: 10.1016/j.cmet.2016.10.010
91. Long KB, Tooker G, Tooker E, Luque SL, Lee JW, Pan X, et al. IL6 receptor blockade enhances chemotherapy efficacy in pancreatic ductal adenocarcinoma. Mol Cancer Ther (2017) 16(9):1898–908. doi: 10.1158/1535-7163.MCT-16-0899
92. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
93. Francescone R, Barbosa Vendramini-Costa D, Franco-Barraza J, Wagner J, Muir A, Lau AN, et al. Netrin G1 promotes pancreatic tumorigenesis through cancer-associated fibroblast-driven nutritional support and immunosuppression. Cancer Discov (2021) 11(2):446–79. doi: 10.1158/2159-8290.CD-20-0775
94. Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-producing b cells promote the development of pancreatic neoplasia. Cancer Discov (2016) 6(3):247–55. doi: 10.1158/2159-8290.CD-15-0843
95. Marrache F, Tu SP, Bhagat G, Pendyala S, Osterreicher CH, Gordon S, et al. Overexpression of interleukin-1beta in the murine pancreas results in chronic pancreatitis. Gastroenterology (2008) 135(4):1277–87. doi: 10.1053/j.gastro.2008.06.078
96. Zhao Y, Shen M, Feng Y, He R, Xu X, Xie Y, et al. Regulatory b cells induced by pancreatic cancer cell-derived interleukin-18 promote immune tolerance via the PD-1/PD-L1 pathway. Oncotarget (2018) 9(19):14803–14. doi: 10.18632/oncotarget.22976
97. Takahashi R, Macchini M, Sunagawa M, Jiang Z, Tanaka T, Valenti G, et al. Interleukin-1β-induced pancreatitis promotes pancreatic ductal adenocarcinoma via b lymphocyte-mediated immune suppression. Gut (2021) 70(2):330–41. doi: 10.1136/gutjnl-2019-319912
98. Tong DN, Guan J, Sun JH, Zhao CY, Chen SG, Zhang ZY, et al. Characterization of b cell-mediated PD-1/PD-L1 interaction in pancreatic cancer patients. Clin Exp Pharmacol Physiol (2020) 47(8):1342–9. doi: 10.1111/1440-1681.13317
99. Jin P, Ren H, Sun W, Xin W, Zhang H, Hao J. Circulating IL-35 in pancreatic ductal adenocarcinoma patients. Hum Immunol (2014) 75(1):29–33. doi: 10.1016/j.humimm.2013.09.018
100. Minici C, Rigamonti E, Lanzillotta M, Monno A, Rovati L, Maehara T, et al. B lymphocytes contribute to stromal reaction in pancreatic ductal adenocarcinoma. Oncoimmunology (2020) 9(1):1794359. doi: 10.1080/2162402X.2020.1794359
101. Das S, Bar-Sagi D. BTK signaling drives CD1d(hi)CD5(+) regulatory b-cell differentiation to promote pancreatic carcinogenesis. Oncogene (2019) 38(17):3316–24. doi: 10.1038/s41388-018-0668-3
102. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature (2010) 467(7319):1114–7. doi: 10.1038/nature09515
103. Valcz G, Buzás EI, Szállási Z, Kalmár A, Krenács T, Tulassay Z, et al. Perspective: bidirectional exosomal transport between cancer stem cells and their fibroblast-rich microenvironment during metastasis formation. NPJ Breast Cancer (2018) 4:18. doi: 10.1038/s41523-018-0071-9
104. Dos Anjos Pultz B, da Luz FAC, Faria SS, de Souza LPF, Tavares PCB, Goulart VA, et al. The multifaceted role of extracellular vesicles in metastasis: Priming the soil for seeding. Int J Cancer (2017) 140(11):2397–407. doi: 10.1002/ijc.30595
105. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer (2009) 9(4):285–93. doi: 10.1038/nrc2621
106. Liu Y, Cao X. Characteristics and significance of the pre-metastatic niche. Cancer Cell (2016) 30(5):668–81. doi: 10.1016/j.ccell.2016.09.011
107. Batista S, Gregório AC, Hanada Otake A, Couto N, Costa-Silva B. The gastrointestinal tumor microenvironment: An updated biological and clinical perspective. J Oncol (2019) 2019:6240505. doi: 10.1155/2019/6240505
108. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol (2015) 17(6):816–26. doi: 10.1038/ncb3169
109. Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol (2016) 18(5):549–60. doi: 10.1038/ncb3340
110. Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, et al. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci Transl Med (2014) 6(237):237ra67. doi: 10.1126/scitranslmed.3007974
111. Brabletz T, Hlubek F, Spaderna S, Schmalhofer O, Hiendlmeyer E, Jung A, et al. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs (2005) 179(1-2):56–65. doi: 10.1159/000084509
112. Kucia M, Reca R, Miekus K, Wanzeck J, Wojakowski W, Janowska-Wieczorek A, et al. Trafficking of normal stem cells and metastasis of cancer stem cells involve similar mechanisms: pivotal role of the SDF-1-CXCR4 axis. Stem Cells (2005) 23(7):879–94. doi: 10.1634/stemcells.2004-0342
113. Begley L, Monteleon C, Shah RB, Macdonald JW, Macoska JA. CXCL12 overexpression and secretion by aging fibroblasts enhance human prostate epithelial proliferation in vitro. Aging Cell (2005) 4(6):291–8. doi: 10.1111/j.1474-9726.2005.00173.x
114. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature (2005) 438(7069):820–7. doi: 10.1038/nature04186
115. Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer (2006) 6(8):637–45. doi: 10.1038/nrc1912
116. Rosu-Myles M, Stewart E, Trowbridge J, Ito CY, Zandstra P, Bhatia M. A unique population of bone marrow cells migrates to skeletal muscle via hepatocyte growth factor/c-met axis. J Cell Sci (2005) 118(Pt 19):4343–52. doi: 10.1242/jcs.02555
117. Kucia M, Halasa M, Wysoczynski M, Baskiewicz-Masiuk M, Moldenhawer S, Zuba-Surma E, et al. Morphological and molecular characterization of novel population of CXCR4+ SSEA-4+ Oct-4+ very small embryonic-like cells purified from human cord blood: preliminary report. Leukemia (2007) 21(2):297–303. doi: 10.1038/sj.leu.2404470
118. Orian-Rousseau V, Morrison H, Matzke A, Kastilan T, Pace G, Herrlich P, et al. Hepatocyte growth factor-induced ras activation requires ERM proteins linked to both CD44v6 and f-actin. Mol Biol Cell (2007) 18(1):76–83. doi: 10.1091/mbc.e06-08-0674
119. Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer (2004) 4(11):839–49. doi: 10.1038/nrc1477
120. Chang CH, Pauklin S. Extracellular vesicles in pancreatic cancer progression and therapies. Cell Death Dis (2021) 12(11):973. doi: 10.1038/s41419-021-04258-7
121. Kren N, Michaud D, Bagchi S, Greene K, Pylayeva-Gupta Y. Rab27a plays a dual role in metastatic propensity of pancreatic cancer. Sci Rep (2020) 10(1):7390. doi: 10.1038/s41598-020-64248-1
122. High RA, Randtke EA, Jones KM, Lindeman LR, Ma JC, Zhang S, et al. Extracellular acidosis differentiates pancreatitis and pancreatic cancer in mouse models using acidoCEST MRI. Neoplasia (2019) 21(11):1085–90. doi: 10.1016/j.neo.2019.09.004
123. Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat Rev Cancer (2014) 14(9):611–22. doi: 10.1038/nrc3793
124. Pantel K, Alix-Panabières C, Riethdorf S. Cancer micrometastases. Nat Rev Clin Oncol (2009) 6(6):339–51. doi: 10.1038/nrclinonc.2009.44
125. Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer (2009) 9(4):302–12. doi: 10.1038/nrc2627
126. Omokehinde T, Johnson RW. Dormancy in the tumor microenvironment. Adv Exp Med Biol (2021) 1329:35–49. doi: 10.1007/978-3-030-73119-9_2
127. Lin WC, Rajbhandari N, Wagner KU. Cancer cell dormancy in novel mouse models for reversible pancreatic cancer: A lingering challenge in the development of targeted therapies. Cancer Res (2014) 74(8):2138–43. doi: 10.1158/0008-5472.CAN-13-3437
128. Klein CA. Framework models of tumor dormancy from patient-derived observations. Curr Opin Genet Dev (2011) 21(1):42–9. doi: 10.1016/j.gde.2010.10.011
129. Klein CA. Selection and adaptation during metastatic cancer progression. Nature (2013) 501(7467):365–72. doi: 10.1038/nature12628
130. Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer (2007) 7(11):834–46. doi: 10.1038/nrc2256
131. Jo H, Jia Y, Subramanian KK, Hattori H, Luo HR. Cancer cell-derived clusterin modulates the phosphatidylinositol 3'-kinase-Akt pathway through attenuation of insulin-like growth factor 1 during serum deprivation. Mol Cell Biol (2008) 28(13):4285–99. doi: 10.1128/MCB.01240-07
132. Humtsoe JO, Kramer RH. Differential epidermal growth factor receptor signaling regulates anchorage-independent growth by modulation of the PI3K/AKT pathway. Oncogene (2010) 29(8):1214–26. doi: 10.1038/onc.2009.419
133. Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci U S A (2008) 105(30):10519–24. doi: 10.1073/pnas.0800939105
134. Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest (2008) 118(12):3917–29. doi: 10.1172/JCI35512
135. Lee IH, Kawai Y, Fergusson MM, Rovira II, Bishop AJ, Motoyama N, et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science (2012) 336(6078):225–8. doi: 10.1126/science.1218395
136. Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med (2011) 208(3):455–67. doi: 10.1084/jem.20101145
137. Jin K, Ewton DZ, Park S, Hu J, Friedman E. Mirk regulates the exit of colon cancer cells from quiescence. J Biol Chem (2009) 284(34):22916–25. doi: 10.1074/jbc.M109.035519
138. Hu J, Nakhla H, Friedman E. Transient arrest in a quiescent state allows ovarian cancer cells to survive suboptimal growth conditions and is mediated by both Mirk/dyrk1b and p130/RB2. Int J Cancer (2011) 129(2):307–18. doi: 10.1002/ijc.25692
139. Jin K, Park S, Ewton DZ, Friedman E. The survival kinase Mirk/Dyrk1B is a downstream effector of oncogenic K-ras in pancreatic cancer. Cancer Res (2007) 67(15):7247–55. doi: 10.1158/0008-5472.CAN-06-4099
140. Deng X, Ewton DZ, Friedman E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res (2009) 69(8):3317–24. doi: 10.1158/0008-5472.CAN-08-2903
141. Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med (2011) 208(13):2641–55. doi: 10.1084/jem.20110840
142. Müller-Hermelink N, Braumüller H, Pichler B, Wieder T, Mailhammer R, Schaak K, et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell (2008) 13(6):507–18. doi: 10.1016/j.ccr.2008.04.001
143. Braumüller H, Wieder T, Brenner E, Aßmann S, Hahn M, Alkhaled M, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature (2013) 494(7437):361–5. doi: 10.1038/nature11824
144. Indraccolo S, Minuzzo S, Masiero M, Pusceddu I, Persano L, Moserle L, et al. Cross-talk between tumor and endothelial cells involving the Notch3-Dll4 interaction marks escape from tumor dormancy. Cancer Res (2009) 69(4):1314–23. doi: 10.1158/0008-5472.CAN-08-2791
145. Almog N, Ma L, Raychowdhury R, Schwager C, Erber R, Short S, et al. Transcriptional switch of dormant tumors to fast-growing angiogenic phenotype. Cancer Res (2009) 69(3):836–44. doi: 10.1158/0008-5472.CAN-08-2590
146. Magnus N, Garnier D, Meehan B, McGraw S, Lee TH, Caron M, et al. Tissue factor expression provokes escape from tumor dormancy and leads to genomic alterations. Proc Natl Acad Sci U S A (2014) 111(9):3544–9. doi: 10.1073/pnas.1314118111
147. Meurette O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer Cell (2018) 34(4):536–48. doi: 10.1016/j.ccell.2018.07.009
148. Charbonnier LM, Wang S, Georgiev P, Sefik E, Chatila TA. Control of peripheral tolerance by regulatory T cell-intrinsic notch signaling. Nat Immunol (2015) 16(11):1162–73. doi: 10.1038/ni.3288
149. Radtke F, MacDonald HR, Tacchini-Cottier F. Regulation of innate and adaptive immunity by notch. Nat Rev Immunol (2013) 13(6):427–37. doi: 10.1038/nri3445
150. Cho OH, Shin HM, Miele L, Golde TE, Fauq A, Minter LM, et al. Notch regulates cytolytic effector function in CD8+ T cells. J Immunol (2009) 182(6):3380–9. doi: 10.4049/jimmunol.0802598
151. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science (2014) 344(6186):921–5. doi: 10.1126/science.1252510
152. Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY, et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res (2010) 70(12):4840–9. doi: 10.1158/0008-5472.CAN-10-0269
153. Procopio MG, Laszlo C, Al Labban D, Kim DE, Bordignon P, Jo SH, et al. Combined CSL and p53 downregulation promotes cancer-associated fibroblast activation. Nat Cell Biol (2015) 17(9):1193–204. doi: 10.1038/ncb3228
154. Hoare M, Ito Y, Kang TW, Weekes MP, Matheson NJ, Patten DA, et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol (2016) 18(9):979–92. doi: 10.1038/ncb3397
155. von Bernstorff W, Spanjaard RA, Chan AK, Lockhart DC, Sadanaga N, Wood I, et al. Pancreatic cancer cells can evade immune surveillance via nonfunctional fas (APO-1/CD95) receptors and aberrant expression of functional fas ligand. Surgery (1999) 125(1):73–84. doi: 10.1016/S0039-6060(99)70291-6
156. He C, Jiang H, Geng S, Sheng H, Shen X, Zhang X, et al. Expression and prognostic value of c-myc and fas (CD95/APO1) in patients with pancreatic cancer. Int J Clin Exp Pathol (2014) 7(2):742–50.
157. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res (2007) 67(3):1030–7. doi: 10.1158/0008-5472.CAN-06-2030
158. Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A paracrine requirement for hedgehog signalling in cancer. Nature (2008) 455(7211):406–10. doi: 10.1038/nature07275
159. Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol (2005) 5(10):749–59. doi: 10.1038/nri1703
160. Han JB, Sang F, Chang JJ, Hua YQ, Shi WD, Tang LH, et al. Arsenic trioxide inhibits viability of pancreatic cancer stem cells in culture and in a xenograft model via binding to SHH-gli. Onco Targets Ther (2013) 6:1129–38. doi: 10.2147/OTT.S49148
161. Goggins M, Shekher M, Turnacioglu K, Yeo CJ, Hruban RH, Kern SE. Genetic alterations of the transforming growth factor beta receptor genes in pancreatic and biliary adenocarcinomas. Cancer Res (1998) 58(23):5329–32.
163. Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol (2012) 13(10):616–30. doi: 10.1038/nrm3434
164. Duda DG, Sunamura M, Lefter LP, Furukawa T, Yokoyama T, Yatsuoka T, et al. Restoration of SMAD4 by gene therapy reverses the invasive phenotype in pancreatic adenocarcinoma cells. Oncogene (2003) 22(44):6857–64. doi: 10.1038/sj.onc.1206751
165. Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFβ in cancer. FEBS Lett (2012) 586(14):1959–70. doi: 10.1016/j.febslet.2012.02.037
166. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell (2011) 147(2):275–92. doi: 10.1016/j.cell.2011.09.024
167. Shang S, Hua F, Hu ZW. The regulation of β-catenin activity and function in cancer: therapeutic opportunities. Oncotarget (2017) 8(20):33972–89. doi: 10.18632/oncotarget.15687
168. Behrens J, von Kries JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R, et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature (1996) 382(6592):638–42. doi: 10.1038/382638a0
169. Peifer M, Pai LM, Casey M. Phosphorylation of the drosophila adherens junction protein armadillo: roles for wingless signal and zeste-white 3 kinase. Dev Biol (1994) 166(2):543–56. doi: 10.1006/dbio.1994.1336
170. Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science (1996) 272(5264):1023–6. doi: 10.1126/science.272.5264.1023
171. Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in xenopus embryos by glycogen synthase kinase 3. Genes Dev (1996) 10(12):1443–54. doi: 10.1101/gad.10.12.1443
172. Hong Y, Manoharan I, Suryawanshi A, Majumdar T, Angus-Hill ML, Koni PA, et al. β-catenin promotes regulatory T-cell responses in tumors by inducing vitamin a metabolism in dendritic cells. Cancer Res (2015) 75(4):656–65. doi: 10.1158/0008-5472.CAN-14-2377
173. Pramanik KC, Fofaria NM, Gupta P, Ranjan A, Kim SH, Srivastava SK. Inhibition of β-catenin signaling suppresses pancreatic tumor growth by disrupting nuclear β-catenin/TCF-1 complex: Critical role of STAT-3. Oncotarget (2015) 6(13):11561–74. doi: 10.18632/oncotarget.3427
174. Hiendlmeyer E, Regus S, Wassermann S, Hlubek F, Haynl A, Dimmler A, et al. Beta-catenin up-regulates the expression of the urokinase plasminogen activator in human colorectal tumors. Cancer Res (2004) 64(4):1209–14. doi: 10.1158/0008-5472.CAN-3627-2
175. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature (2015) 523(7559):231–5. doi: 10.1038/nature14404
176. Fu C, Jiang A. β-catenin-mediated inhibition of cross-priming: A new mechanism for tumors to evade immunosurveillance. Oncoimmunology (2013) 2(12):e26920. doi: 10.4161/onci.26920
177. Ansari D, Ohlsson H, Althini C, Bauden M, Zhou Q, Hu D, et al. The hippo signaling pathway in pancreatic cancer. Anticancer Res (2019) 39(7):3317–21. doi: 10.21873/anticanres.13474
178. Murakami S, Shahbazian D, Surana R, Zhang W, Chen H, Graham GT, et al. Yes-associated protein mediates immune reprogramming in pancreatic ductal adenocarcinoma. Oncogene (2017) 36(9):1232–44. doi: 10.1038/onc.2016.288
179. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168(4):707–23. doi: 10.1016/j.cell.2017.01.017
180. Ibrahim AM, Wang YH. Viro-immune therapy: A new strategy for treatment of pancreatic cancer. World J Gastroenterol (2016) 22(2):748–63. doi: 10.3748/wjg.v22.i2.748
181. Finn OJ. Immuno-oncology: Understanding the function and dysfunction of the immune system in cancer. Ann Oncol (2012) 23 Suppl 8(Suppl 8):viii6–9. doi: 10.1093/annonc/mds256
182. Chiaravalli M, Reni M, O'Reilly EM. Pancreatic ductal adenocarcinoma: State-of-the-art 2017 and new therapeutic strategies. Cancer Treat Rev (2017) 60:32–43. doi: 10.1016/j.ctrv.2017.08.007
183. Paniccia A, Merkow J, Edil BH, Zhu Y. Immunotherapy for pancreatic ductal adenocarcinoma: An overview of clinical trials. Chin J Cancer Res (2015) 27(4):376–91. doi: 10.3978/j.issn.1000-9604.2015.05.01
184. Baxter D. Active and passive immunization for cancer. Hum Vaccin Immunother (2014) 10(7):2123–9. doi: 10.4161/hv.29604
185. Disis ML. Mechanism of action of immunotherapy. Semin Oncol (2014) 41 Suppl 5:S3–13. doi: 10.1053/j.seminoncol.2014.09.004
186. Zhou W, Zhou Y, Chen X, Ning T, Chen H, Guo Q, et al. Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials (2021) 268:120546. doi: 10.1016/j.biomaterials.2020.120546
187. Salman B, Zhou D, Jaffee EM, Edil BH, Zheng L. Vaccine therapy for pancreatic cancer. Oncoimmunology (2013) 2(12):e26662. doi: 10.4161/onci.26662
188. Bernhardt SL, Gjertsen MK, Trachsel S, Møller M, Eriksen JA, Meo M, et al. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: A dose escalating phase I/II study. Br J Cancer (2006) 95(11):1474–82. doi: 10.1038/sj.bjc.6603437
189. Gjertsen MK, Bakka A, Breivik J, Saeterdal I, Solheim BG, Søreide O, et al. Vaccination with mutant ras peptides and induction of T-cell responsiveness in pancreatic carcinoma patients carrying the corresponding RAS mutation. Lancet (1995) 346(8987):1399–400. doi: 10.1016/S0140-6736(95)92408-6
190. Gjertsen MK, Buanes T, Rosseland AR, Bakka A, Gladhaug I, Søreide O, et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int J Cancer (2001) 92(3):441–50. doi: 10.1002/ijc.1205
191. Gilliam AD, Broome P, Topuzov EG, Garin AM, Pulay I, Humphreys J, et al. An international multicenter randomized controlled trial of G17DT in patients with pancreatic cancer. Pancreas (2012) 41(3):374–9. doi: 10.1097/MPA.0b013e31822ade7e
192. Kaufman HL, Kim-Schulze S, Manson K, DeRaffele G, Mitcham J, Seo KS, et al. Poxvirus-based vaccine therapy for patients with advanced pancreatic cancer. J Transl Med (2007) 5:60. doi: 10.1186/1479-5876-5-60
193. Lepisto AJ, Moser AJ, Zeh H, Lee K, Bartlett D, McKolanis JR, et al. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther (2008) 6(B):955–64.
194. Laheru D, Lutz E, Burke J, Biedrzycki B, Solt S, Onners B, et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: A pilot study of safety, feasibility, and immune activation. Clin Cancer Res (2008) 14(5):1455–63. doi: 10.1158/1078-0432.CCR-07-0371
195. Le DT, Brockstedt DG, Nir-Paz R, Hampl J, Mathur S, Nemunaitis J, et al. A live-attenuated listeria vaccine (ANZ-100) and a live-attenuated listeria vaccine expressing mesothelin (CRS-207) for advanced cancers: Phase I studies of safety and immune induction. Clin Cancer Res (2012) 18(3):858–68. doi: 10.1158/1078-0432.CCR-11-2121
196. Gatti-Mays ME, Strauss J, Donahue RN, Palena C, Del Rivero J, Redman JM, et al. A phase I dose-escalation trial of BN-CV301, a recombinant poxviral vaccine targeting MUC1 and CEA with costimulatory molecules. Clin Cancer Res (2019) 25(16):4933–44. doi: 10.1158/1078-0432.CCR-19-0183
197. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol (2018) 18(3):168–82. doi: 10.1038/nri.2017.131
198. Kinkead HL, Hopkins A, Lutz E, Wu AA, Yarchoan M, Cruz K, et al. Combining STING-based neoantigen-targeted vaccine with checkpoint modulators enhances antitumor immunity in murine pancreatic cancer. JCI Insight (2018) 3(20):1. doi: 10.1172/jci.insight.122857
199. Jaffee EM, Hruban RH, Biedrzycki B, Laheru D, Schepers K, Sauter PR, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol (2001) 19(1):145–56. doi: 10.1200/JCO.2001.19.1.145
200. Lollini PL, Cavallo F, Nanni P, Forni G. Vaccines for tumour prevention. Nat Rev Cancer (2006) 6(3):204–16. doi: 10.1038/nrc1815
201. Günes C, Rudolph KL. The role of telomeres in stem cells and cancer. Cell (2013) 152(3):390–3. doi: 10.1016/j.cell.2013.01.010
202. Huff V. Wilms' tumours: about tumour suppressor genes, an oncogene and a chameleon gene. Nat Rev Cancer (2011) 11(2):111–21. doi: 10.1038/nrc3002
203. Lutz E, Yeo CJ, Lillemoe KD, Biedrzycki B, Kobrin B, Herman J, et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. a phase II trial of safety, efficacy, and immune activation. Ann Surg (2011) 253(2):328–35. doi: 10.1097/SLA.0b013e3181fd271c
204. Diaz Beveridge R, Alcolea V, Aparicio J, Segura Á, García J, Corbellas M, et al. Management of advanced pancreatic cancer with gemcitabine plus erlotinib: efficacy and safety results in clinical practice. Jop (2014) 15(1):19–24. doi: 10.6092/1590-8577/1570
205. Kobari M, Egawa S, Shibuya K, Sunamura M, Saitoh K, Matsuno S. Effect of intraportal adoptive immunotherapy on liver metastases after resection of pancreatic cancer. Br J Surg (2000) 87(1):43–8. doi: 10.1046/j.1365-2168.2000.01336.x
206. Kawaoka T, Oka M, Takashima M, Ueno T, Yamamoto K, Yahara N, et al. Adoptive immunotherapy for pancreatic cancer: cytotoxic T lymphocytes stimulated by the MUC1-expressing human pancreatic cancer cell line YPK-1. Oncol Rep (2008) 20(1):155–63. doi: 10.3892/or.20.1.155
207. Seifert AM, Reiche C, Heiduk M, Tannert A, Meinecke AC, Baier S, et al. Detection of pancreatic ductal adenocarcinoma with galectin-9 serum levels. Oncogene (2020) 39(15):3102–13. doi: 10.1038/s41388-020-1186-7
208. Daley D, Mani VR, Mohan N, Akkad N, Ochi A, Heindel DW, et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med (2017) 23(5):556–67. doi: 10.1038/nm.4314
209. Fan JQ, Wang MF, Chen HL, Shang D, Das JK, Song J. Current advances and outlooks in immunotherapy for pancreatic ductal adenocarcinoma. Mol Cancer (2020) 19(1):32. doi: 10.1186/s12943-020-01151-3
210. Feng M, Xiong G, Cao Z, Yang G, Zheng S, Song X, et al. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett (2017) 407:57–65. doi: 10.1016/j.canlet.2017.08.006
211. Feng B, Zhou F, Hou B, Wang D, Wang T, Fu Y, et al. Binary cooperative prodrug nanoparticles improve immunotherapy by synergistically modulating immune tumor microenvironment. Adv Mater (2018) 30(38):e1803001. doi: 10.1002/adma.201803001
212. Kimbara S, Kondo S. Immune checkpoint and inflammation as therapeutic targets in pancreatic carcinoma. World J Gastroenterol (2016) 22(33):7440–52. doi: 10.3748/wjg.v22.i33.7440
213. Henriksen A, Dyhl-Polk A, Chen I, Nielsen D. Checkpoint inhibitors in pancreatic cancer. Cancer Treat Rev (2019) 78:17–30. doi: 10.1016/j.ctrv.2019.06.005
214. McCoy KD, Le Gros G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol (1999) 77(1):1–10. doi: 10.1046/j.1440-1711.1999.00795.x
215. Huang RR, Jalil J, Economou JS, Chmielowski B, Koya RC, Mok S, et al. CTLA4 blockade induces frequent tumor infiltration by activated lymphocytes regardless of clinical responses in humans. Clin Cancer Res (2011) 17(12):4101–9. doi: 10.1158/1078-0432.CCR-11-0407
216. Ville S, Poirier N, Blancho G, Vanhove B. Co-Stimulatory blockade of the CD28/CD80-86/CTLA-4 balance in transplantation: Impact on memory T cells? Front Immunol (2015) 6:411. doi: 10.3389/fimmu.2015.00411
217. Callahan MK, Wolchok JD, Allison JP. Anti-CTLA-4 antibody therapy: immune monitoring during clinical development of a novel immunotherapy. Semin Oncol (2010) 37(5):473–84. doi: 10.1053/j.seminoncol.2010.09.001
218. Phan GQ, Weber JS, Sondak VK. CTLA-4 blockade with monoclonal antibodies in patients with metastatic cancer: surgical issues. Ann Surg Oncol (2008) 15(11):3014–21. doi: 10.1245/s10434-008-0104-y
219. Bengsch F, Knoblock DM, Liu A, McAllister F, Beatty GL. CTLA-4/CD80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol Immunother (2017) 66(12):1609–17. doi: 10.1007/s00262-017-2053-4
220. Walker LS, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol (2015) 36(2):63–70. doi: 10.1016/j.it.2014.12.001
221. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res (2020) 10(3):727–42.
222. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8(8):793–800. doi: 10.1038/nm730
223. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192(7):1027–34. doi: 10.1084/jem.192.7.1027
224. Homet Moreno B, Ribas A. Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. Br J Cancer (2015) 112(9):1421–7. doi: 10.1038/bjc.2015.124
225. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med (2006) 203(4):883–95. doi: 10.1084/jem.20051776
226. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature (2017) 541(7637):321–30. doi: 10.1038/nature21349
227. Loos M, Giese NA, Kleeff J, Giese T, Gaida MM, Bergmann F, et al. Clinical significance and regulation of the costimulatory molecule B7-H1 in pancreatic cancer. Cancer Lett (2008) 268(1):98–109. doi: 10.1016/j.canlet.2008.03.056
228. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell (2015) 161(2):205–14. doi: 10.1016/j.cell.2015.03.030
229. Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol (2016) 13(3):143–58. doi: 10.1038/nrclinonc.2015.209
230. Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res (2007) 13(7):2151–7. doi: 10.1158/1078-0432.CCR-06-2746
231. Geng L, Huang D, Liu J, Qian Y, Deng J, Li D, et al. B7-H1 up-regulated expression in human pancreatic carcinoma tissue associates with tumor progression. J Cancer Res Clin Oncol (2008) 134(9):1021–7. doi: 10.1007/s00432-008-0364-8
232. Okudaira K, Hokari R, Tsuzuki Y, Okada Y, Komoto S, Watanabe C, et al. Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse pancreatic cancer model. Int J Oncol (2009) 35(4):741–9. doi: 10.3892/ijo_00000387
233. Pico de Coaña Y, Choudhury A, Kiessling R. Checkpoint blockade for cancer therapy: revitalizing a suppressed immune system. Trends Mol Med (2015) 21(8):482–91. doi: 10.1016/j.molmed.2015.05.005
234. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515(7528):577–81. doi: 10.1038/nature13988
235. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
236. Lebrun JJ. The dual role of TGFβ in human cancer: From tumor suppression to cancer metastasis. ISRN Mol Biol (2012) 2012:381428. doi: 10.5402/2012/381428
237. Uesaka K, Boku N, Fukutomi A, Okamura Y, Konishi M, Matsumoto I, et al. Adjuvant chemotherapy of s-1 versus gemcitabine for resected pancreatic cancer: a phase 3, open-label, randomised, non-inferiority trial (JASPAC 01). Lancet (2016) 388(10041):248–57. doi: 10.1016/S0140-6736(16)30583-9
238. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity (2013) 39(1):74–88. doi: 10.1016/j.immuni.2013.06.014
239. Ma Y, Li J, Wang H, Chiu Y, Kingsley CV, Fry D, et al. Combination of PD-1 inhibitor and OX40 agonist induces tumor rejection and immune memory in mouse models of pancreatic cancer. Gastroenterology (2020) 159(1):306–19.e12. doi: 10.1053/j.gastro.2020.03.018
240. Ottaiano A, Capozzi M, De Divitiis C, De Stefano A, Botti G, Avallone A, et al. Gemcitabine mono-therapy versus gemcitabine plus targeted therapy in advanced pancreatic cancer: a meta-analysis of randomized phase III trials. Acta Oncol (2017) 56(3):377–83. doi: 10.1080/0284186X.2017.1288922
241. Liede A, Karlan BY, Narod SA. Cancer risks for male carriers of germline mutations in BRCA1 or BRCA2: a review of the literature. J Clin Oncol (2004) 22(4):735–42. doi: 10.1200/JCO.2004.05.055
242. Holter S, Borgida A, Dodd A, Grant R, Semotiuk K, Hedley D, et al. Germline BRCA mutations in a Large clinic-based cohort of patients with pancreatic adenocarcinoma. J Clin Oncol (2015) 33(28):3124–9. doi: 10.1200/JCO.2014.59.7401
243. Bao Z, Cao C, Geng X, Tian B, Wu Y, Zhang C, et al. Effectiveness and safety of poly (ADP-ribose) polymerase inhibitors in cancer therapy: A systematic review and meta-analysis. Oncotarget (2016) 7(7):7629–39. doi: 10.18632/oncotarget.5367
244. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol (2015) 33(3):244–50. doi: 10.1200/JCO.2014.56.2728
245. Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother (2013) 36(7):382–9. doi: 10.1097/CJI.0b013e31829fb7a2
Keywords: tumor microenvironment, immunotherapy, immunosuppression, PDAC, immune checkpoint inhibitor
Citation: Zhang J, Li R and Huang S (2022) The immunoregulation effect of tumor microenvironment in pancreatic ductal adenocarcinoma. Front. Oncol. 12:951019. doi: 10.3389/fonc.2022.951019
Received: 23 May 2022; Accepted: 04 July 2022;
Published: 28 July 2022.
Edited by:
Georg F. Weber, University of Erlangen Nuremberg, GermanyReviewed by:
Paul David, University Hospital Erlangen, GermanyDina Kouhestani, University Hospital Erlangen, Germany
Copyright © 2022 Zhang, Li and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renfeng Li, empjenpkeGR5ZnN5eTE5OTlAMTYzLmNvbQ==; Shuai Huang, ZmNjaHVhbmdzQHp6dS5lZHUuY24=