 Shuyi Chen
Shuyi Chen Ling Li
Ling Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 18 August 2022
Sec. Molecular and Cellular Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.949688
This article is part of the Research TopicReviews in Molecular and Cellular OncologyView all 47 articles
Cyclin D1 has been reported to be upregulated in several solid and hematologic tumors, promoting cancer progression. Thus, decreasing cyclin D1 by degradation could be a promising target strategy for cancer therapy. This mini review summarizes the roles of cyclin D1 in tumorigenesis and progression and its degradation strategies. Besides, we proposed an exploration of the degradation of cyclin D1 by FBX4, an F box protein belonging to the E3 ligase SKP-CUL-F-box (SCF) complex, which mediates substrate ubiquitination, as well as a postulate about the concrete combination mode of FBX4 and cyclin D1. Furthermore, we proposed a possible photodynamic therapy strategythat is based on the above concrete combination mode for treating superficial cancer.
Loss of cell cycle control usually drives rapid and uncontrollable tumor growth, which has been considered an important feature for cancer cells (1). Cyclin D1, a key regulator of cell cycle progression, can integrate extracellular mitotic signals into DNA synthesis by binding to cyclin-dependent kinase 4/6 (CDK4/6), thus facilitating the cell cycle switch from G1 to S phase.
As its name suggests, cyclin D1 protein abundance fluctuates periodically throughout the cell cycle. In the late G1 phase, cyclin D1 levels increase and then activate CDK4/6, which helps trigger the progress of the cell cycle. In the S phase, however, the level of cyclin D1 falls. Cyclin D1 controls the G1-S phase transition in both CDK-dependent and CDK-independent manners.
In CDK-dependent manner, the cyclin D1/CDK4/6 complex phosphorylates SMAD3 and downregulates TGF-β family (2) (Figure 1A); the complex also phosphorylates RB1 and RBL1/2 and promotes E2F release, then upregulates cyclin E/CDK2 levels, further resulting in RB phosphorylation (3) (Figure 1B). Besides, the complex phosphorylates MEP50/PRMT5 and upregulates CDT1 activity via downregulating CUL4 activity (4) (Figure 1C). Additionally, the cyclin D1/CDK4/6 complex sequesters p21 and p27, indirectly upregulating cyclin E/CDK2 activity (5) (Figure 1D). All these molecular events mediate intracellular activities, including centrosome replication, mitochondrial metabolism, cell adhesion and motility, and cytoskeleton modeling (3) (Figure 1E).
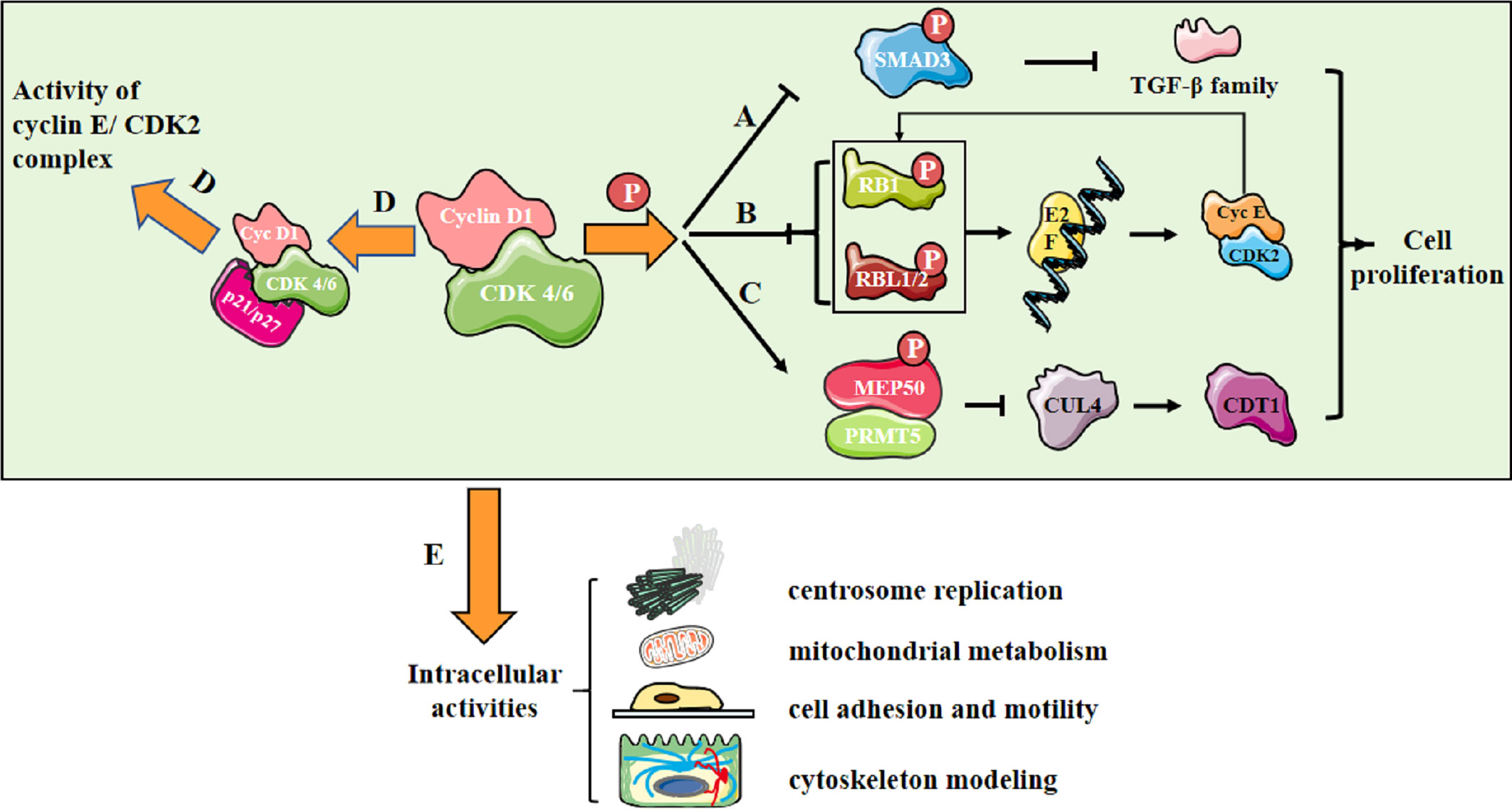
Figure 1 Cyclin D1/CDK4/6 promotes cell proliferation. (A) Cyclin D1/CDK 4/6 phosphorylates SMAD3 and downregulates the transcription of TGF-β family genes involving in growth inhibition; (B) Cyclin D1/CDK 4/6 phosphorylates RB1 and RBL1/2, leading to their inactivation and release of E2F family. E2F controls the transcription of genes essential for cyclin E synthesis, which will activate CDK2 and further promote RB phosphorylation, forming a positive feedback loop; (C) Cyclin D1/CDK4/6 triggers an increase in MEP50/PRMT5 activity and a decrease in the activity of CUL4, the E3 ligase of CDT1 and the replication licensing protein at the meantime; (D) Cyclin D1/CDK 4/6 sequesters the CDK inhibitors(CKIs) p21CIP1 and p27KIP1, indirectly upregulating cyclin E/CDK2 activity; (E) Molecular activities mediate intracellular activities, including centrosome replication, mitochondrial metabolism, cell adhesion and motility, and cytoskeleton modeling.
In addition to CDK-dependent cell cycle regulation activity, cyclin D1 itself has CDK-independent effects. Cyclin D1 is involved in the DNA damage response by interacting with RAD51 and BRCA2 (6). Besides, cyclin D1 regulates cell growth and differentiation by binding to several transcription factor families including nuclear hormone receptor family members [estrogen receptor-α (ERα), androgen receptor (AR), peroxisome proliferator-activated receptor-γ (PPAR γ)) and their co-activators (SRC1, GRIP1, and AIB1), BMYB, and the MYB-related transcription factor DMP1, the helix–loop–helix transcription factors neurogenic differentiation factor 1 (NEUROD1), as well as MYOD and C/EBPβ] (7–9). Cyclin D1 also promotes cell cycle progression by binding to signal transducer and activator of transcription 3 (STAT3) and TAFII250 (3).
Generally, all those activities are related to G1-S phase transition and cell proliferation. When dysregulated, upregulated cyclin D1 antagonizes the checkpoint induced cell cycle arrest, disturbs DNA replication, and allows damaged DNA to be replicated (4, 10, 11). Therefore, the overexpression of cyclin D1 or the failure of cyclin D1 degradation both accelerate G1-S transition, helping cancer cells to gain a survival advantage and an uncontrolled proliferation, which further promotes the invasiveness and malignance of cancer.
Cyclin D1 upregulation has been seen in at least 11 kinds of cancers, including head and neck squamous cell carcinoma (12), non-small-cell lung cancer (NSCLC) (13), endometrial cancer (14), melanoma (15), pancreatic cancer (16, 17), gastric cancer (18), breast cancer (19), colorectal cancer (20), mantle cell lymphoma (21), multiple myeloma (22), and prostate cancer (23). The prevailing view is that high levels of cyclin D1 expression have a positive correlation with poor prognosis in a wide variety of tumors such as nasopharyngeal carcinoma (NPC) (24), gastric cancer tissues (25), and squamous cell carcinoma of the head and neck (12, 26). Interestingly, a minority of reports suggest that a high level of cyclin D1 expression is associated with a favorable prognosis in breast cancer and clear renal cell carcinoma (27, 28).
Usually, a high level of cyclin D1 derives from enhanced mRNA expression of cyclin D1, splice variants, transcript aberrations, and downregulated cyclin D1 degradation (29–32), among which the dysregulated degradation of cyclin D1 could be a significant reason (3). In the early stage of cancer, failure to degrade cyclin D1 after double-strand DNA breaks (DSBs) may lead to the accumulation of DNA damage, thus promoting cancer development (33, 34). Furthermore, failure to degrade cyclin D1 compromises the intra-S-phase checkpoint (35) and results in high expression of cyclin D1, which is often witnessed in many kinds of malignant tumors, indicating poor prognosis (36–39).
Therefore, it becomes a necessity to seek methods for degrading cyclin D1. As a potential degradation complex of cyclin D1 (32), the binding between FBX4 and cyclin D1 is still being disputed by HURI1, NCBI2, and other official websites. Furthermore, it remains unclear about the specific binding motifs of FBX4 and cyclin D1. In this review, we will focus on the FBX4–cyclin D1 binding mode from the structural biology aspect to the application aspect.
Compared with cyclin D1 degradation, targeting CDK inhibition is more commonly observed clinically. The FDA has approved CDK inhibitors (CKIs) for patients with metastatic breast cancer (40). CKIs mainly act in the inhibition of RB phosphorylation and mediate cell cycle arrest. Besides, CKIs also play a role in modulating mitogenic kinase signaling and then inducing a senescence-like phenotype and enhancing cancel cell immunogenicity (41). However, CKIs usually lead to the avoidance of their conserved ATP-binding sites. Recently, researcher found that apart from CKIs that inhibiting cyclin D1, there also exist CDK degraders for degrading cyclin D1. Jiang et al. have developed Ikaros (IKZF1) and Aiolos (IKZF3) as imide-based CDK4/6 degraders in mantle cell lymphoma cell lines (42). The research and development of CDK degraders is still in the experimental stage, and patients usually are disturbed by the problems of decreased drug sensitivity in their later stages of CKI medication (43–45). Since both CDK degraders and CKIs have unavoidable defects, cyclin D1 targeted therapy is still a promising treatment. When cyclin D1 is degraded, G0/G1 arrest occurs, and then cells cease to proliferate over a period of time. The lower the amount of cyclin D1, the more likely it is to cause G0/G1 arrest.
Wu et al. (46) noted that acute cyclin D1 deficiency in the regenerating liver markedly inhibited hepatocyte proliferation after partial hepatectomy. Yang et al. (47) reported that myostatin, a transforming growth factor β-superfamily member, induces cell cycle arrest by degrading cyclin D1. Wei et al. (48) also observed that cyclin D1 degradation mediated by ERβ inhibited colon cell growth. From the opposite perspective, Li et al. (49) reported that USP5 stabilized cyclin D1 and downregulated its degradation, thus promoting glioblastoma multiforme (GBM) progression. Generally, methods for degrading cyclin D1 can be achieved through intracellular ubiquitin–proteasome degradation mechanisms that are exerted by extracellular factors.
1. Chelating agent: Smara et al. (50) applied iron chelator deferasirox (DFX) to mantle cell lymphoma (MCL) cells, and found that DFX lead to cyclin D1 proteolysis and degradation in a mechanism that requires its phosphorylation on T286 by glycogen synthase kinase-3β (GSK3β). Several iron chelators were proved to be effective anti-cancer agents and potential therapeutic potions, leading to the ubiquitin-proteasome degradation of cyclin D1.
2. Chemical osmosis: Casanovas et al. (51) illustrated that environmental stresses induced by chemical factors can also induce cyclin D1 degradation in vitro. Their further research showed that osmotic stresses activate p38SAPK2, thereby inducing cyclin D1 degradation. Herein, chemical osmotic stress can be induced by sodium chloride (NaCl), calcium chloride (CaCl2), magnesium chloride (MgCl2), hydrogen peroxide (H2O2) and sodium arsenite (NaAsO2). ODC-antizyme, a regulator of ornithine decarboxylase (ODC), has also been demonstrated to degrade cyclin D1 by altering chemical osmosis (52).
3. Organic compounds: Several kinds of natural and synthetic organic compounds have been demonstrated to degrade cyclin D1, such as all-trans retinoic acid (RA) (53), ML364 (54), and differentiation-inducing factors (DIFs) (55). RA suppresses the G1-S transition, thereby inhibiting human bronchial epithelial cell growth. ML364 is the inhibitor of the deubiquitinase USP2, which mediates the ubiquitination of cyclin D1. DIFs induce differentiation and restrict the cell cycle in the G0/G1 phase.
4. Drugs: There is proof from several pieces of evidence that some drugs can mediate cyclin D1 degradation. Nivelle et al. (56) proved that resveratrol, epsilon viniferin, and labruscol degraded cyclin D1 in melanoma cells. Zhu et al. (57) proved that arctigenin would mediate GSK3-dependent cyclin D1 degradation in ER-positive breast cancer cells. Additionally, there are other drugs targeting cyclin D1 degradation. Luzonicoside A inhibited the proliferation and migration of cancer cells significantly more than Luzonicoside D (58). Treatment with Gambogenic acid (GNA), benzofuroxan derivative-22 (BFD-22) and Hemsleyanol D induced G0/G1 arrest (59–61). Dehydroleucodine (DhL) degraded cyclin D1 in a concentration-dependent manner (62). Simvastatin enhanced the phosphorylation and protein degradation of cyclin D1 (63, 64). Rottlerin downregulated cyclin D1 in a p21-independent mechanism (65).
For the intracellular mechanism, namely, ubiquitin-proteasome degradation mechanism, two different E3 ubiquitin ligases were reported.
1. APC complex: The anaphase-promoting complex (APC) is a cell cycle regulated ubiquitin ligase. Peters et al. (66) stated in the review that cyclin D1 degradation was mediated by APC. Besides, Agami et al. reported that ionizing radiation (IR) induced cyclin D1 degradation requires an anaphase promoting complex (APC) (67).
2. AMBRA1: Activating molecule in beclin-1-regulated autophagy (AMBRA1) degrades cyclin D1 by interacting with cullin-RING family (CRL4-DDB1 complex) of E3 ubiquitin ligase, which activity depending on the phosphorylation of cyclin D1 at T286 (68, 69). Besides, CRL4AMBRA1 degrades three types of cyclin Ds (cyclin D1–3) through ubiquitin proteasome pathway (70).
3. SCF complex: Liu et al. (32) proposed that cyclin D1 undergoes ubiquitin-proteasome-dependent degradation in vivo, which is mediated by SCF complex subunit, FBX4. Besides, Okabe et al. (71) identified FBXW8 and FBXL12 as other cyclin D1 E3 ligases, which also belong to the SCF complex subunit.
Extracellular factors and intracellular ubiquitin-proteasome mechanisms mediated by APC have been thoroughly studied previously. However, the degradation of cyclin D1 specifically by the SCF complex, especially FBX4, has not been thoroughly studied. Moreover, the exact mechanism of interaction between FBX4 and cyclin D1 is still waiting to be explored. As the basis of structural biology reports on the binding of AMBRA1 and cyclin D1 is unclear yet, we would concentrate on the detailed binding mode between cyclin D1 and FBX4. Considering that CRL4AMBRA1 and the FBX complex have similar Cullin-RING, the analysis of the detailed binding mode between cyclin D1 and FBX4 may provide a basis for the structural biology research of AMBRA1.
The Skp–Cullin–F-box (SCF) complex is an E3 ubiquitin ligase that binds to protein substrates and mediates ubiquitin-proteasome degradation. The SCF complex consists of four parts: SKP1, Cullin1 (CUL1) or cullin7 (CUL7), RBX1, or ROC1, and F box protein (72) (Figure 2). In this structure, the amino acid sequence specificity of the F box determines the specificity of the SCF complex. Each F-box protein contains an SKP1 binding motif as well as a specific substrate-recognition motif (73). The F-box protein family can be divided into three classes3: FBWs, FBLs, and FBXs. FBWs are characterized by WD-40 domains. FBLs are characterized by leucine-rich repeats and by either different protein–protein interaction modules or no recognizable motifs. FBX4, which is also named FBXO4, belongs to the FBX class.
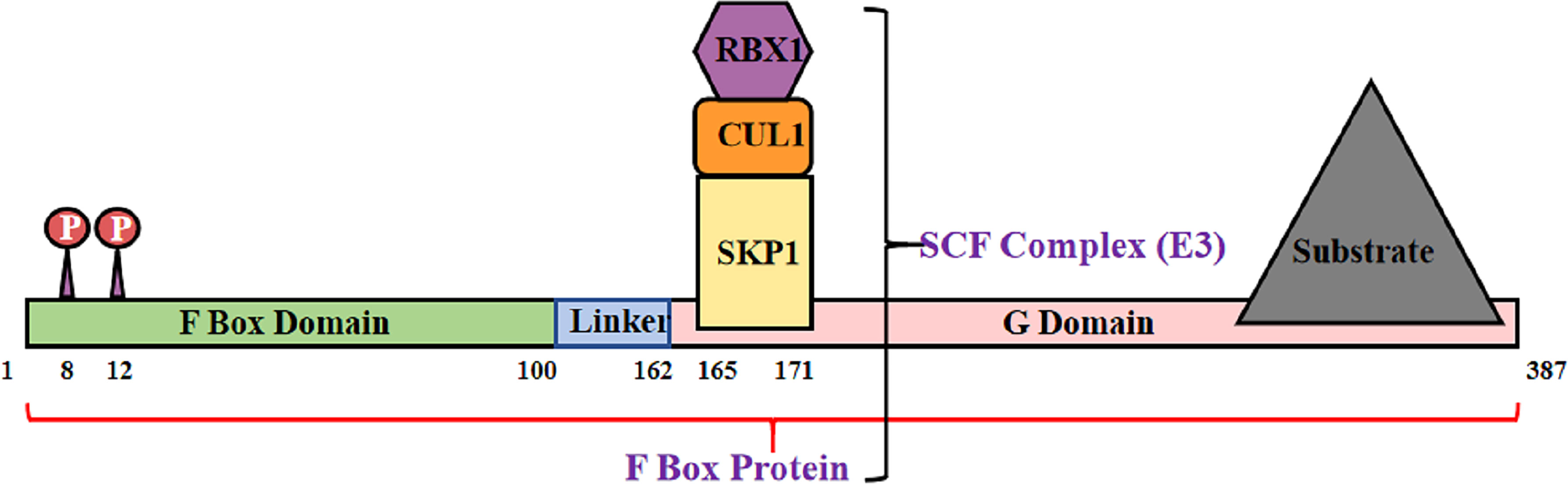
Figure 2 Structural diagram of FBX4. F box protein combine with Skp1, Cul1 and Rbx1, consisting SCF complex.
In the following section, we would summarize the conditions required for the combination of FBX4 and cyclin D1 and propose their possible combination mode.
Above all, the degradation of cyclin D1 relies on FBX4 dimerization in vivo, which process further depends on the phosphorylation of FBX4. Barbash et al. (74) found that GSK3β mediates the phosphorylation of FBX4 at Ser8 and Ser12, thereby promoting the dimerization of FBX4 and stimulating its activity as an E3 ligase (Figure 3A). Compared with GSK3β inhibited cells, cells with normal GSK3β expression showed a marked decrease in growth. Barbash et al. (74) have testified that Ser8 and Ser12 phosphorylation of enhances poly-ubiquitin linked cyclin D1, while FBX4 alone or FBX4, whereas was not phosphorylated showed low degradation activities.
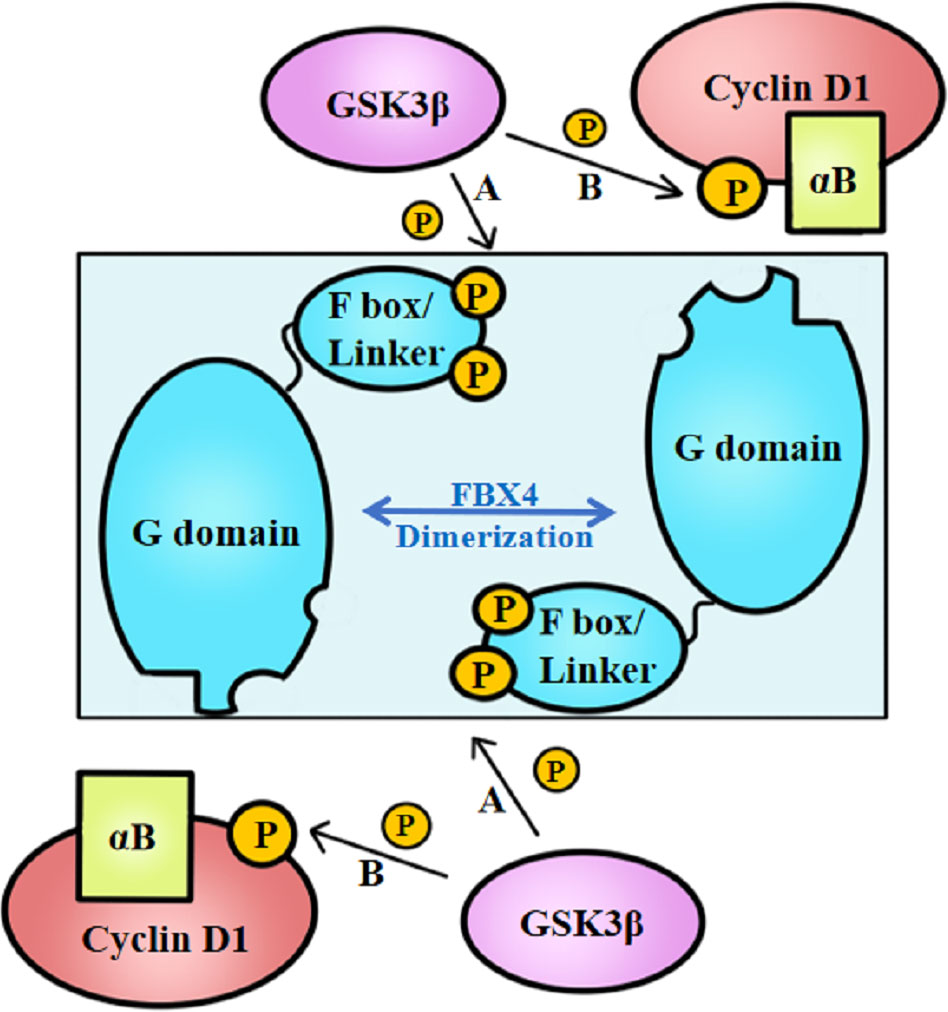
Figure 3 GSK3β-mediated FBX4 and cyclin D1 phosphorylation. (A) GSK3β phosphorylates FBX4 at Ser8 and Ser 12 of F box domain. (B) GSK3β phosphorylates cyclin D1 at Thr286. αB, alpha B-crystalline.
Besides, cyclin D1–FBX4 binding depends on alpha B-crystalline existence and cyclin D1 phosphorylation at Thr286 mediated by GSK3β (Figure 3B). Lin et al. (75) demonstrated that cyclin D1 binds to FBX4-alpha B-crystalline in a manner of cyclin D1 Thr286 phosphorylation in vitro since the cyclin D1 T286A mutant did not coprecipitate with alpha B-crystalline. What’s more, their experiments indicate that omitting alpha B-crystalline can inhibit the binding of FBX4 and cyclin D1. However, cyclin D1 precipitation mediated by FBX4 can also be detected in the absence of alpha B-crystalline, illustrating that alpha B-crystalline is not a necessity for cyclin D1–FBX4 binding.
Additionally, FBX4 associates with cyclin D1 in a relatively specific manner. Lin et al. (75) found that phosphorylated cyclin D1 peptides did not recruit other F box proteins such as FBW2 or SKP2. After IP-IB analysis of eight kinds of full-length F Box, Okabe et al. revealed that FBXW5, FBXW7, FBXW11, SKP2, and FBXL5 did not inhibit cyclin D1 expression (71).
As a supplement, the degradation of cyclin D by FBX4 may not be affected by the compensatory effect. According to the compensatory effect, if cyclin D1 alone was degraded, cyclin D2 and cyclin D3 would compensatively bind to CDK4/6 in place of cyclin D1 to promote cell cycle progression. Nevertheless, cyclin D1, cyclin D2, and cyclin D3 have similar phosphorylation sites for FBX4 binding (Thr286, Thr280, and Thr283) (3). Thus, cyclin D2 and D3 could also be degraded by FBX4 in the deduction. However, the expression of cyclin D2 and cyclin D3 is rare compared with the universal expression of cyclin D1 in most cancers (3), and that cyclin D3 plays a unique role mainly in T-cell leukemia (76). Considering the commonality of cyclin D degradation and the universality of cyclin D1, we focus on the cyclin D1–FBX4 combination.
At present, there are few reports about the binding mechanisms between FBX4 and cyclin D1. If the binding mode of FBX4 to cyclin D1 is determined, the binding motif itself may be used as a biological component for targeted degradation of cyclin D1.
Unfortunately, the binding of FBX4 and cyclin D1 is still being doubted. From NCBI Reference Sequence NM_012176.34, we found that (1): FBX4 is a protein with 387 amino acids (2); There exists two phosphorylation sites near the N-terminal of FBX4, which is coincident with the exploration of Barbash et al. (74) (3); Most importantly, the binding motif of FBX4 with another ascertained ligand—TRF1, is the peptides sequence near the C-terminal of amino acids from position 341 to 372. These information hints that the site where FBX4 binds to the substrate are supposed to be near the C-terminal.
Another article helped a lot. In this article, Li et al. (77) provided the overall structure of the SKP1-FBX4 core complex. Structurally, FBX4 has four essential domains essential for its function in the SCF complex: N-terminal dimerization domain (D domain), F box domain, linker domain, and C-terminal substrate-binding domain (G domain) (Figure 4) (77). According to the structural analyses by Li et al. (77), the interaction mode between the F box domain and G domain of FBX4 is head-to-tail dimerization, which is necessary for substrate binding and ubiquitin transfer.
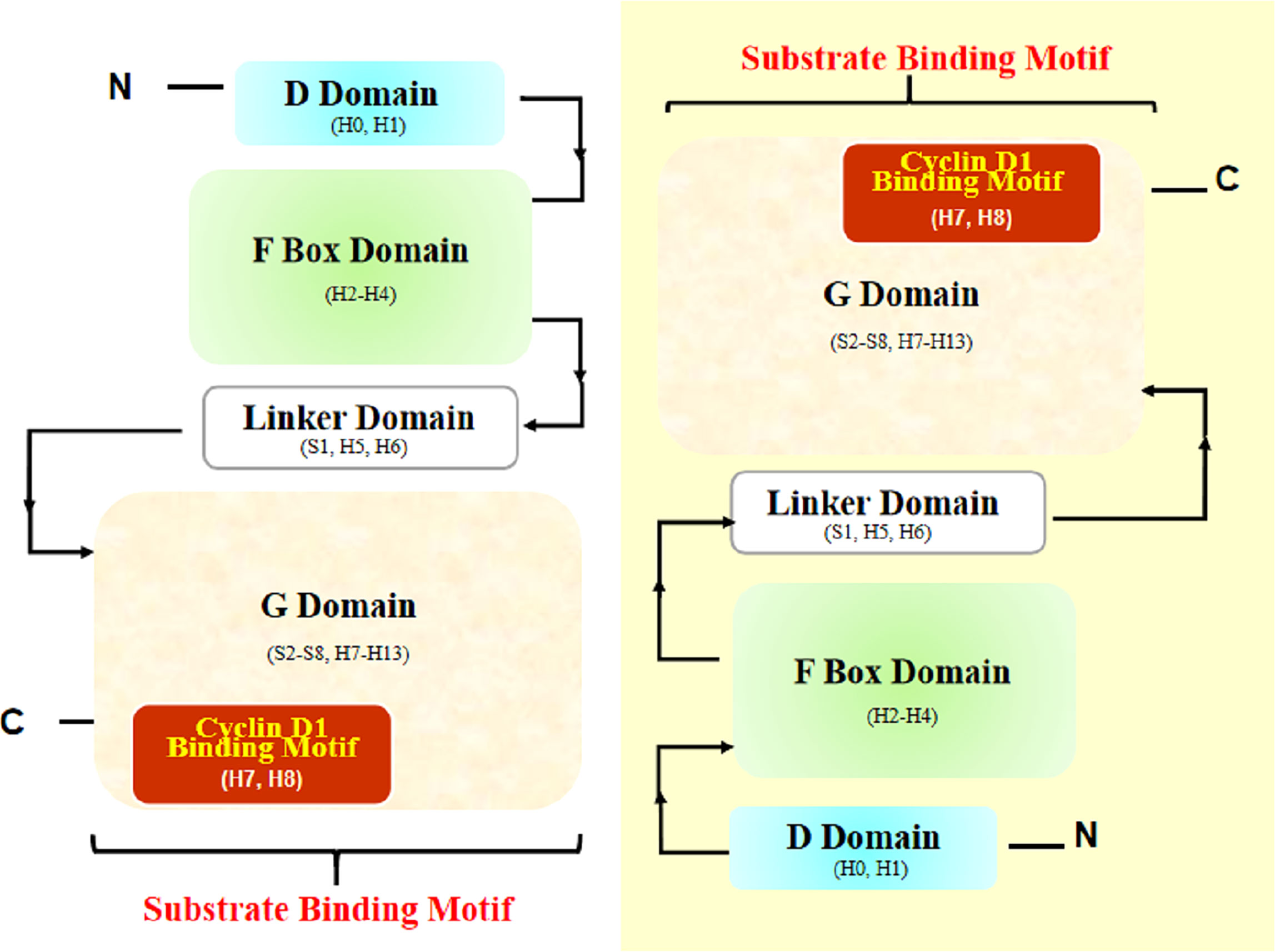
Figure 4 Structural diagram of FBX4 dimerization. Two FBX4 molecules dimerize in a head-to-tail manner and expose substrate binding motifs (with one FBX4 highlighted in yellow shadow). The presumed cyclin D1 binding motif is highlighted (in red shadow). H, helix; S, strand.
Thus, we can rationally deduce that the exposed end containing Helix7 (H7), Helix8 (H8), and Helix13 (H13) could be the substrate binding site. Considering the known facts that TRF1 combined with helix near the end H13 and cyclin D1 is a cyclin distinct-different from TRF1, the specifically binding motif of FBX4 and cyclin D1 could be at the other subdomain, that is, nearby H7 and H8. According to UniProt5, H7 and H8 are located at the positions corresponding to amino acids 193–212. However, whether the combination mode is exactly like this is waiting for experiments to testify.
To summarize, the specific binding manner of FBX4 and cyclin D1 is supposed to be as follows: cyclin D1 is phosphorylated at Thr286 and is accompanied by alpha B-crystalline binding. Cyclin D1 phosphorylation and alpha B-crystalline are two important conditions to ensure the degradation efficiency of cyclin D1. At the same time, FBX4 proceeds head-to-tail dimerization and recruits the rest of the SCF complex such as SKP1, CUL1, and RBX1. Then phosphorylated cyclin D1 binds to FBX4 at the G domain and is eventually ubiquitinated and degraded by the proteasome (Figure 5). As the specific binding motif between cyclin D1 and FBX4 has not been certified, the arrow from cyclin D1 to the G domain remains a question mark.
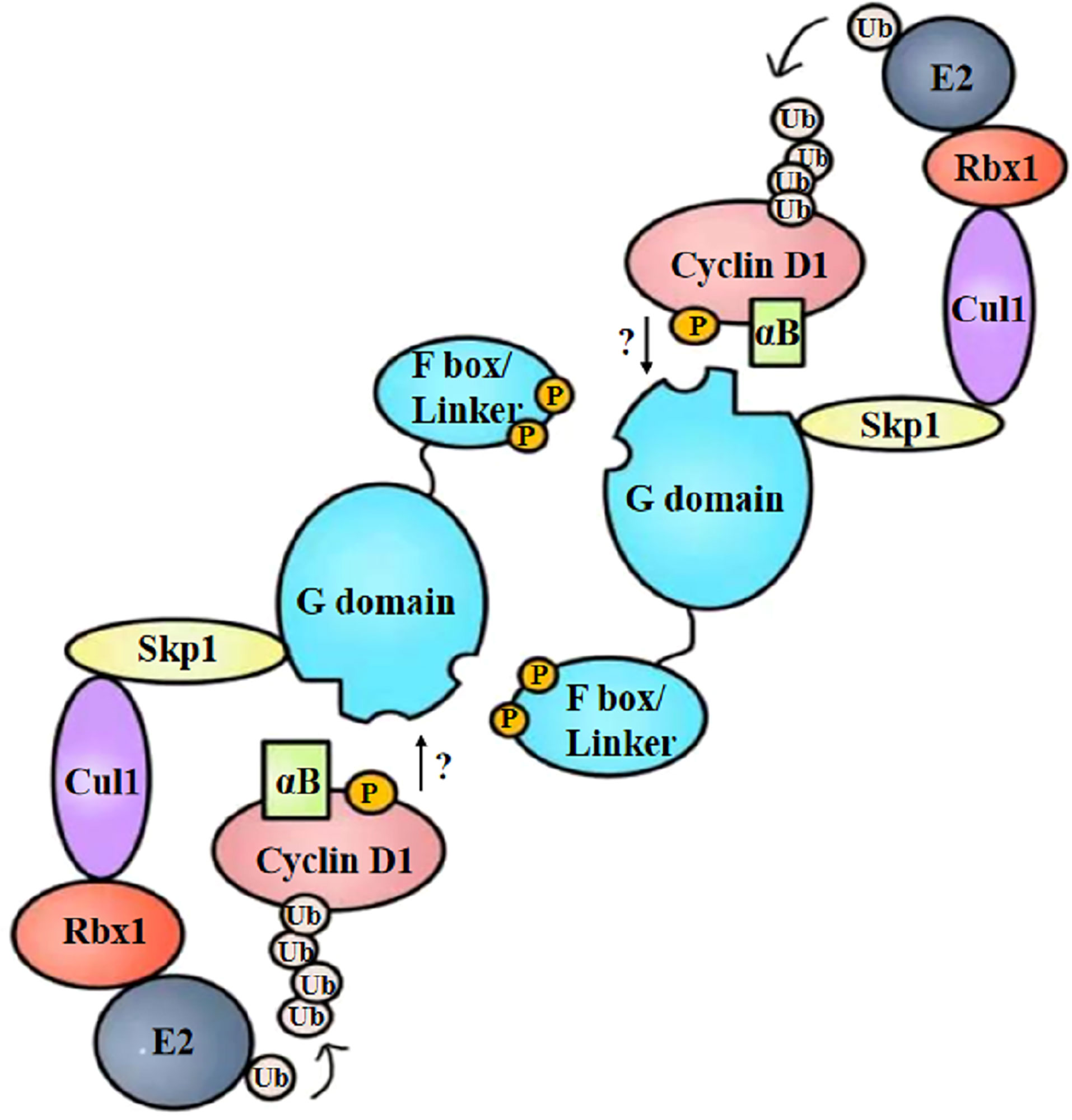
Figure 5 Structural diagram of FBX4-mediated cyclin D1 ubiquitination. After phosphorylation and alpha B-crystalline combination, cyclin D1 is recruited by dimerized FBX4. FBX4 also recruits the remainder of E3 ubiquitin ligase and E2 ubiquitin ligase, mediating cyclin D1 ubiquitination. αB, alpha B-crystalline; Ub, ubiquitin.
As for the application of ubiquitin mediated protein degradation, Luo et al. (78) reviewed major advances in PROteolysis TArgeting Chimeras (PROTAC), which mediates the transfer of poly-ubiquitin (Ub) chains from E3 ligase to the protein of interest (POI). At present, two powerful PROTAC candidates have entered phase 1/2 clinical trials developed by the Arvinas company, namely, ARV-110 targeting androgen receptors (AR) for treating prostate cancer (79), and ARV-471 targeting estrogen receptors (ER) for treating breast cancer (80). Although the traditional PROTAC has presented a great application value, there is still the problem of poor controllability and off-target effect. If the reversible optical switch can be added, these problems will be greatly solved. Among different controllable PROTACs, the photoswiching PROTAC strategy is reversible. By switching the light wavelength irradiating the controlling element of PROTAC, PROTAC can bind with POI and then mediate ubiquitin proteasome degradation. Otherwise, PROTAC would be separated from POI and then POI degradation would stop (81). The switch of a specific wavelength depends on the characteristics of the optical control element.
Photodynamic therapy is becoming a new hotspot in recent years. Ye et al. (82) constructed an optogenetic switch to regulate Fc-adiponectin expression in photosensitive cells by using far-red light and effectively reduced blood lipid and blood sugar, thus alleviating the symptoms of insulin resistance. Later, the team developed the REDMAP system (83), a red/far-red optogenetic switch used to regulate insulin expression and hypoglycemia in diabetic mice and rats. Their academic achievements indicate that the light-controlled system has a great application prospect. The NUDT IGEM team6 has made an achievement in integrating Trim21, a cytosolic ubiquitin ligase and antibody receptor with a blue light controlled model (CRY2 and its ligand CIB1) to target GFP degradation.
Based on the theoretical basis above, we then propose to modify it into a cyclin D1 degradation model by proposing the CIB1-FBX4 system. In this system, blue light mediates the combination of two photosensitive proteins of Arabidopsis, CIB1 and CRY2, by causing them to deform (84). As shown in Figure 6, CIB1 and Trim21 are designed by linking together, whereas CRY2 and the capture sequence of FBX4 are linked together with a segment of flexible peptide. If there were no light signal, the FBX4 binding motif could capture target cyclin D1 but does not cause its degradation. Upon blue light stimulation, CIB1 can bind to CRY2, which is then accompanied by the binding of Trim21 to cyclin D1, and subsequently the degradation of cyclin D1 (Figure 6). If the exact binding motif of FBX4 was integrated into their optical control model, rapid and efficient blue light-controlled cyclin D1 degradation would be achieved. Thus, we can synchronize the cycle of the tumor cells at the G0 phase. On the condition that the cycle of the tumor cells is synchronized before M phase, they stop proliferation and diffusion. This strategy is especially suitable for superficial cancers such as esophageal squamous carcinoma, because these lesions are easily irradiated by light. For the specific application, just take esophageal squamous carcinoma as an example. Specific implementation methods include modifying gastroscopy apparatus and transfecting the CIB1–FBX4 system into cancer cells. The proliferation of cancer cells will be slowed down by light therapy, which acts in a similar manner to gastroscopy.
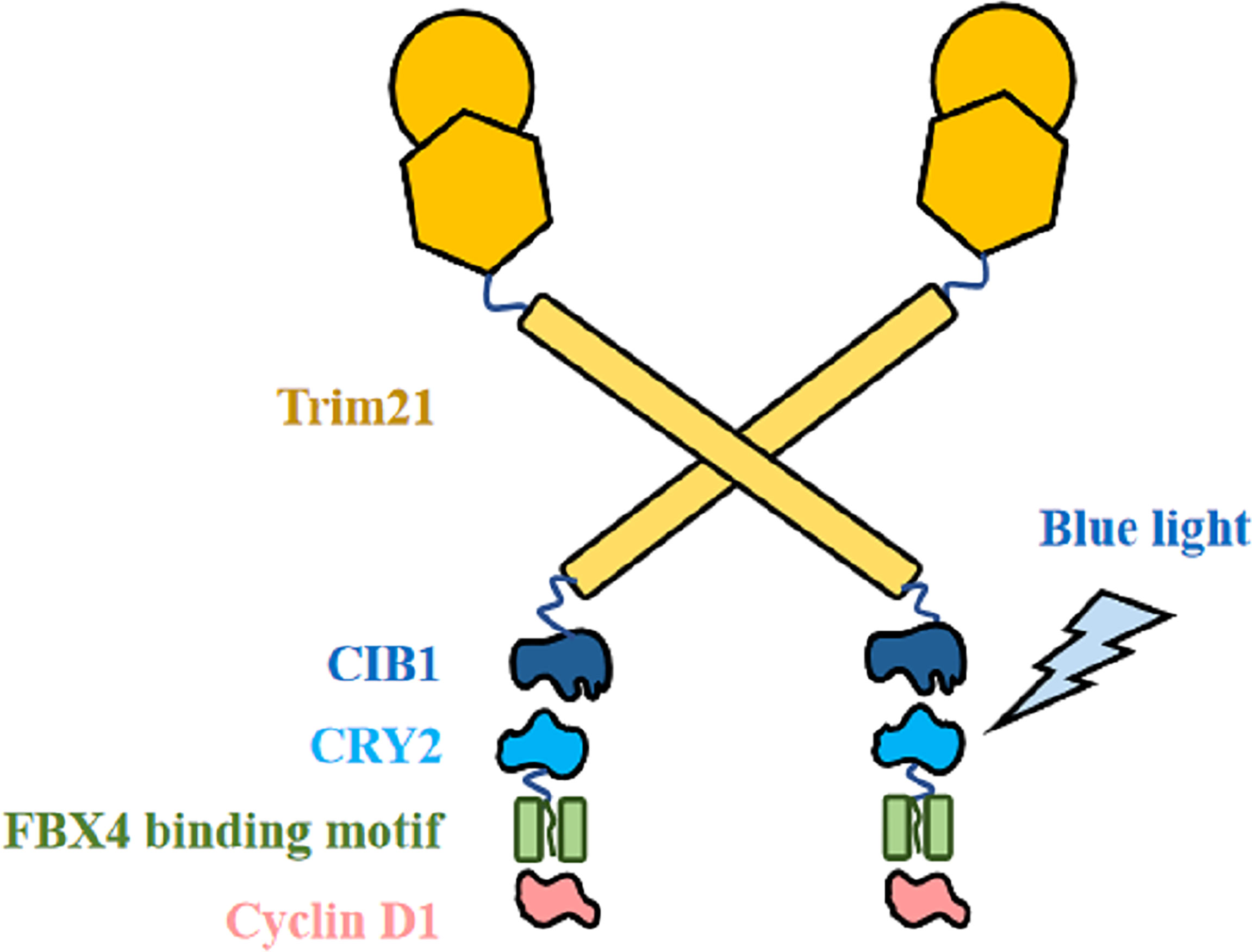
Figure 6 CIB1–FBX4 system. FBX4 binding motif captures cyclin D1. After blue light stimulation, CIB1 combines with CRY2 (84), leading to Trim21-mediated cyclin D1 degradation.
The outstanding features of the CIB1–FBX4 system include more reversible and sufficient degradation than the FBX4 whole sequence. It is inevitable that there are normal cells transfected with the CIB1–FBX4 system that experience G0/G1 arrest temporarily. However, the expression of cyclin D1 will be restored after light removal, and the normal cells will resume normal division after a while. To the greatest extent, it avoids “accidental injury.” In addition, it solves problems such as insufficient regulation ability of existing protein-targeted degradation tools on intracellular coding proteins, influence on normal physiological functions of cells, and limited application in vivo environment.
It is worth noting that there are drawbacks to this approach. As CIB1 and CRY2 come from Arabidopsis thaliana, a plant, it is unclear whether the system will cause immunological rejection in vivo. Therefore, the system is expected to be tested in organoids with an immune microenvironment. More light-controlled components are expected to be discovered and applied as well.
SC completed all gathering and arranging work under the guidance of LL. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discov (2022) 12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
2. Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of smads. Nature (2004) 430(6996):226–31. doi: 10.1038/nature02650
3. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin d as a therapeutic target in cancer. Nat Rev Cancer (2011) 11(8):558–72. doi: 10.1038/nrc3090
4. Aggarwal P, Lessie MD, Lin DI, Pontano L, Gladden AB, Nuskey B, et al. Nuclear accumulation of cyclin D1 during s phase inhibits Cul4-dependent Cdt1 proteolysis and triggers P53-dependent DNA rereplication. Genes Dev (2007) 21(22):2908–22. doi: 10.1101/gad.1586007
5. Tchakarska G, Sola B. The double dealing of cyclin D1. Cell Cycle (2020) 19(2):163–78. doi: 10.1080/15384101.2019.1706903
6. Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature (2011) 474(7350):230–4. doi: 10.1038/nature10155
7. Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, Marson A, et al. Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature (2010) 463(7279):374–8. doi: 10.1038/nature08684
8. Coqueret O. Linking cyclins to transcriptional control. Gene (2002) 299(1):35–55. doi: 10.1016/S0378-1119(02)01055-7
9. Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: Normal and abnormal functions. Endocrinology (2004) 145(12):5439–47. doi: 10.1210/en.2004-0959
10. Li Z, Jiao X, Wang C, Shirley LA, Elsaleh H, Dahl O, et al. Alternative cyclin D1 splice forms differentially regulate the DNA damage response. Cancer Res (2010) 70(21):8802–11. doi: 10.1158/0008-5472.CAN-10-0312
11. Shimura T, Ochiai Y, Noma N, Oikawa T, Sano Y, Fukumoto M. Cyclin D1 overexpression perturbs DNA replication and induces replication-associated DNA double-strand breaks in acquired radioresistant cells. Cell Cycle (2013) 12(5):773–82. doi: 10.4161/cc.23719
12. Michalides R, van Veelen N, Hart A, Loftus B, Wientjens E, Balm A. Overexpression of cyclin D1 correlates with recurrence in a group of forty-seven operable squamous cell carcinomas of the head and neck. Cancer Res (1995) 55(5):975–8.
13. Gautschi O, Ratschiller D, Gugger M, Betticher DC, Heighway J. Cyclin D1 in non-small cell lung cancer: A key driver of malignant transformation. Lung Cancer (2007) 55(1):1–14. doi: 10.1016/j.lungcan.2006.09.024
14. Yildirim HT, Nergiz D, Sadullahoglu C, Akgunduz Z, Yildirim S, Dogan S, et al. The extent of cyclin D1 expression in endometrial pathologies and relevance of cyclin D1 with the clinicopathological features of endometrioid endometrial carcinoma. Indian J Pathol Microbiol (2020) 63(3):412–7. doi: 10.4103/IJPM.IJPM_589_19
15. Donigan JM, De Luca J, Lum C. Cyclin D1 and P16 expression in blue nevi and malignant melanoma. Applied Immunohistochemistry & Molecular Morphology (2017) 25(2):91–4. doi: 10.1097/pai.0000000000000276
16. Zhang Y, Su Y, Zhao Y, Lv G, Luo Y. Microrna720 inhibits pancreatic cancer cell proliferation and invasion by directly targeting cyclin D1. Mol Med Rep (2017) 16(6):9256–62. doi: 10.3892/mmr.2017.7732
17. Kolsi LE, Leal AS, Yli-Kauhaluoma J, Liby KT, Moreira VM. Dehydroabietic oximes halt pancreatic cancer cell growth in the G1 phase through induction of P27 and downregulation of cyclin D1. Sci Rep (2018) 8(1):15923. doi: 10.1038/s41598-018-34131-1
18. Yuan C, Zhu X, Han Y, Song C, Liu C, Lu S, et al. Elevated Hoxa1 expression correlates with accelerated tumor cell proliferation and poor prognosis in gastric cancer partly Via cyclin D1. J Exp Clin Cancer Res (2016) 35:15. doi: 10.1186/s13046-016-0294-2
19. Abdalla AN, Qattan A, Malki WH, Shahid I, Hossain MA, Ahmed M. Significance of targeting vegfr-2 and cyclin D1 in luminal-a breast cancer. Molecules (2020) 25(20):4606. doi: 10.3390/molecules25204606
20. Albasri A, Yosef H, Hussainy AS, Sultan SA, Alhujaily A. Histopathological features of colorectal cancer in Al-madinah region of Saudi Arabia: 8 years experience. Asian Pac J Cancer Prev (2014) 15(7):3133–7. doi: 10.7314/apjcp.2014.15.7.3133
21. Navarro A, Bea S, Jares P, Campo E. Molecular pathogenesis of mantle cell lymphoma. Hematol Oncol Clin North Am (2020) 34(5):795–807. doi: 10.1016/j.hoc.2020.05.002
22. Sewify EM, Afifi OA, Mosad E, Zaki AH, El Gammal SA. Cyclin D1 amplification in multiple myeloma is associated with multidrug resistance expression. Clin Lymphoma Myeloma Leuk (2014) 14(3):215–22. doi: 10.1016/j.clml.2013.07.008
23. Casimiro MC, Di Sante G, Ju X, Li Z, Chen K, Crosariol M, et al. Cyclin D1 promotes androgen-dependent DNA damage repair in prostate cancer cells. Cancer Res (2016) 76(2):329–38. doi: 10.1158/0008-5472.CAN-15-0999
24. Zhou L, Liu R, Liang X, Zhang S, Bi W, Yang M, et al. Lncrna Rp11-624l4.1 is associated with unfavorable prognosis and promotes proliferation Via the Cdk4/6-cyclin D1-Rb-E2f1 pathway in npc. Mol Ther Nucleic Acids (2020) 22:1025–39. doi: 10.1016/j.omtn.2020.10.017
25. Shan YS, Hsu HP, Lai MD, Hung YH, Wang CY, Yen MC, et al. Cyclin D1 overexpression correlates with poor tumor differentiation and prognosis in gastric cancer. Oncol Lett (2017) 14(4):4517–26. doi: 10.3892/ol.2017.6736
26. Åkervall JA, Michalides RJAM, Mineta H, Balm A, Borg Å, Dictor MR, et al. Amplification of cyclin D1 in squamous cell carcinoma of the head and neck and the prognostic value of chromosomal abnormalities and cyclin D1 overexpression. Cancer (1997) 79(2):380–9. doi: 10.1002/(SICI)1097-0142(19970115)79:2<380::AID-CNCR22>3.0.CO;2-W
27. Siraj AK, Parvathareddy SK, Annaiyappanaidu P, Ahmed SO, Siraj N, Tulbah A, et al. High expression of cyclin D1 is an independent marker for favorable prognosis in middle Eastern breast cancer. OncoTargets Ther (2021) 14:3309–18. doi: 10.2147/ott.S309091
28. Wang QS, Li F, Liao ZQ, Li K, Yang XL, Lin YY, et al. Low level of cyclin-D1 correlates with worse prognosis of clear cell renal cell carcinoma patients. Cancer Med (2019) 8(9):4100–9. doi: 10.1002/cam4.2313
29. Wiestner A, Tehrani M, Chiorazzi M, Wright G, Gibellini F, Nakayama K, et al. Point mutations and genomic deletions in Ccnd1 create stable truncated cyclin D1 mrnas that are associated with increased proliferation rate and shorter survival. Blood (2007) 109(11):4599–606. doi: 10.1182/blood-2006-08-039859
30. Benzeno S, Lu F, Guo M, Barbash O, Zhang F, Herman JG, et al. Identification of mutations that disrupt phosphorylation-dependent nuclear export of cyclin D1. Oncogene (2006) 25(47):6291–303. doi: 10.1038/sj.onc.1209644
31. Moreno-Bueno G, Rodriguez-Perales S, Sanchez-Estevez C, Hardisson D, Sarrio D, Prat J, et al. Cyclin D1 gene (Ccnd1) mutations in endometrial cancer. Oncogene (2003) 22(38):6115–8. doi: 10.1038/sj.onc.1206868
32. Liu J, Yang H, Cheung PY, Tsao SW, Lv L, Cheung ALM. Fbx4 mediates rapid cyclin D1 proteolysis upon DNA damage in immortalized esophageal epithelial cells. Biochem Biophys Res Commun (2021) 554:76–82. doi: 10.1016/j.bbrc.2021.03.089
33. Sante GD, Rocco AD, Pupo C, Casimiro MC, Pestell RG. Hormone-induced DNA damage response and repair mediated by cyclin D1 in breast and prostate cancer. Oncotarget (2017) 8(47):1. doi: 10.18632/oncotarget.19413
34. Wu SY, Lan SH, Wu SR, Chiu YC, Lin XZ, Su IJ, et al. Hepatocellular carcinoma-related cyclin D1 is selectively regulated by autophagy degradation system. Hepatology (2018) 68(1):141–54. doi: 10.1002/hep.29781
35. Pontano LL, Aggarwal P, Barbash O, Brown EJ, Bassing CH, Diehl JA. Genotoxic stress-induced cyclin D1 phosphorylation and proteolysis are required for genomic stability. Mol Cell Biol (2008) 28(23):7245–58. doi: 10.1128/MCB.01085-08
36. Moradi Binabaj M, Bahrami A, Khazaei M, Ryzhikov M, Ferns GA, Avan A, et al. The prognostic value of cyclin D1 expression in the survival of cancer patients: A meta-analysis. Gene (2020) 728:144283. doi: 10.1016/j.gene.2019.144283
37. Villegas SL, Darb-Esfahani S, von Minckwitz G, Huober J, Weber K, Marmé F, et al. Expression of cyclin D1 protein in residual tumor after neoadjuvant chemotherapy for breast cancer. Breast Cancer Res Treat (2018) 168(1):179–87. doi: 10.1007/s10549-017-4581-1
38. Ortiz AB, Garcia D, Vicente Y, Palka M, Bellas C, Martin P. Prognostic significance of cyclin D1 protein expression and gene amplification in invasive breast carcinoma. PloS One (2017) 12(11):e0188068. doi: 10.1371/journal.pone.0188068
39. Mohammedi L, Doula FD, Mesli F, Senhadji R. Cyclin D1 overexpression in Algerian breast cancer women: Correlation with Ccnd1 amplification and clinicopathological parameters. Afr Health Sci (2019) 19(2):2140–6. doi: 10.4314/ahs.v19i2.38
40. Laderian B, Fojo T. Cdk4/6 inhibition as a therapeutic strategy in breast cancer: Palbociclib, ribociclib, and abemaciclib. Semin Oncol (2017) 44(6):395–403. doi: 10.1053/j.seminoncol.2018.03.006
41. Goel S, DeCristo MJ, McAllister SS, Zhao JJ. Cdk4/6 inhibition in cancer: Beyond cell cycle arrest. Trends Cell Biol (2018) 28(11):911–25. doi: 10.1016/j.tcb.2018.07.002
42. Jiang B, Wang ES, Donovan KA, Liang Y, Fischer ES, Zhang T, et al. Development of dual and selective degraders of cyclin-dependent kinases 4 and 6. Angewandte Chemie International Edition (2019) 131:6387–92. doi: 10.1002/anie.201901336
43. Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. Cdk4/6 inhibitors: The mechanism of action may not be as simple as once thought. Cancer Cell (2018) 34(1):9–20. doi: 10.1016/j.ccell.2018.03.023
44. Reinius MAV, Smyth E. Anti-cancer therapy with cyclin-dependent kinase inhibitors: Impact and challenges. Expert Rev Mol Med (2021) 23:e6. doi: 10.1017/erm.2021.3
45. Faluyi OO, Hull MA, Markham AF, Bonifer C, Coletta PL. Reduction in the resident intestinal myelomonocytic cell population occurs during Apc(Min/+) mouse intestinal tumorigenesis. Oncol Lett (2021) 21(4):263. doi: 10.3892/ol.2021.12524
46. Wu H, Reizel Y, Wang J, Lapiro J, Kren B, Schug J, et al. A negative reciprocal regulatory axis between cyclin D1 and Hnf4α modulates cell cycle progression and metabolism in the liver. Proc Natl Acad Sci (2020) 117:202002898. doi: 10.1073/pnas.2002898117
47. Yang W, Zhang Y, Li Y, Wu Z, Zhu D. Myostatin induces cyclin D1 degradation to cause cell cycle arrest through a phosphatidylinositol 3-Kinase/Akt/Gsk-3 beta pathway and is antagonized by insulin-like growth factor 1. J Biol Chem (2007) 282(6):3799–808. doi: 10.1074/jbc.M610185200
48. Wei Y, Huang C, Wu H, Huang J. Estrogen receptor beta (Erβ) mediated-Cyclind1 degradation Via autophagy plays an anti-proliferation role in colon cells. Int J Biol Sci (2019) 15(5):942–52. doi: 10.7150/ijbs.30930
49. Li G, Yang T, Chen Y, Bao J, Wu D, Hu X, et al. Usp5 sustains the proliferation of glioblastoma through stabilization of Cyclind1. Front Pharmacol (2021) 12:720307. doi: 10.3389/fphar.2021.720307
50. Samara A, Shapira S, Lubin I, Shpilberg O, Avigad S, Granot G, et al. Deferasirox induces cyclin D1 degradation and apoptosis in mantle cell lymphoma in a reactive oxygen species- and Gsk3beta-dependent mechanism. Br J Haematol (2021) 192(4):747–60. doi: 10.1111/bjh.17284
51. Casanovas O, Miro F, Estanyol JM, Itarte E, Agell N, Bachs O. Osmotic stress regulates the stability of cyclin D1 in a P38sapk2-dependent manner. J Biol Chem (2000) 275(45):35091–7. doi: 10.1074/jbc.M006324200
52. Newman RM, Mobascher A, Mangold U, Koike C, Diah S, Schmidt M, et al. Antizyme targets cyclin D1 for degradation. a novel mechanism for cell growth repression. J Biol Chem (2004) 279(40):41504–11. doi: 10.1074/jbc.M407349200
53. Langenfeld J, Kiyokawa H, Sekula D, Boyle J, Dmitrovsky E. Posttranslational regulation of cyclin D1 by retinoic acid: A chemoprevention mechanism. Proc Natl Acad Sci U.S.A. (1997) 94(22):12070–4. doi: 10.1073/pnas.94.22.12070
54. Davis MI, Pragani R, Fox JT, Shen M, Parmar K, Gaudiano EF, et al. Small molecule inhibition of the ubiquitin-specific protease Usp2 accelerates cyclin D1 degradation and leads to cell cycle arrest in colorectal cancer and mantle cell lymphoma models. Journal of Biological Chemistry (2016) 291:24628. doi: 10.1074/jbc.M116.738567
55. Mori J, Takahashi-Yanaga F, Miwa Y, Watanabe Y, Hirata M, Morimoto S, et al. Differentiation-inducing factor-1 induces cyclin D1 degradation through the phosphorylation of Thr286 in squamous cell carcinoma. Exp Cell Res (2005) 310(2):426–33. doi: 10.1016/j.yexcr.2005.07.024
56. Nivelle L, Aires V, Rioult D, Martiny L, Tarpin M, Delmas D. Molecular analysis of differential antiproliferative activity of resveratrol, epsilon viniferin and labruscol on melanoma cells and normal dermal cells. Food Chem Toxicol (2018) 116(Pt B):323–34. doi: 10.1016/j.fct.2018.04.043
57. Zhu L, Shen XB, Yuan PC, Shao TL, Liu XP. Arctigenin inhibits proliferation of er-positive breast cancer cells through cell cycle arrest mediated by Gsk3-dependent cyclin D1 degradation. Life Science (2020). 256:117983. doi: 10.1016/j.lfs.2020.117983
58. Malyarenko OS, Dyshlovoy SA, Kicha AA, Ivanchina NV, Malyarenko TV, Carsten B, et al. The inhibitory activity of luzonicosides from the starfish echinaster luzonicus against human melanoma cells. Marine Drugs (2017) 15(7):227. doi: 10.3390/md15070227
59. Li F, Wang Y, Yan Y. Gambogenic acid induces cell growth inhibition, cell cycle arrest and metastasis inhibition in choroidal melanoma in a dose-dependent manner. Exp Ther Med (2017) 13(5):2456–62. doi: 10.3892/etm.2017.4252
60. Ferreira AK, Pasqualoto KF, Kruyt FA, Palace-Berl F, Azevedo RA, Turra KM, et al. Bfd-22 a new potential inhibitor of braf inhibits the metastasis of B16f10 melanoma cells and simultaneously increased the tumor immunogenicity. Toxicol Appl Pharmacol (2016) 295:56–67. doi: 10.1016/j.taap.2016.02.008
61. Moriyama H, Moriyama M, Ninomiya K, Morikawa T, Hayakawa T. Inhibitory effects of oligostilbenoids from the bark of shorea roxburghii on malignant melanoma cell growth: Implications for novel topical anticancer candidates. Biol Pharm Bull (2016) 39(10):1675–82. doi: 10.1248/bpb.b16-00420
62. Costantino VV, Lobos-Gonzalez L, Ibañez J, Fernandez D, Cuello-Carrión FD, Valenzuela MA, et al. Dehydroleucodine inhibits tumor growth in a preclinical melanoma model by inducing cell cycle arrest, senescence and apoptosis. Cancer Lett (2016) 372(1):10–23. doi: 10.1016/j.canlet.2015.12.004
63. Tsubaki M, Takeda T, Kino T, Obata N, Itoh T, Imano M, et al. Statins improve survival by inhibiting spontaneous metastasis and tumor growth in a mouse melanoma model. Am J Cancer Res (2015) 5(10):3186–97.
64. Wang Y, Xu SL, Wu YZ, Zhao MS, Xu WJ, Yang HY, et al. Simvastatin induces caspase-dependent apoptosis and activates P53 in ocm-1 cells. Exp eye Res (2013) 113:128–34. doi: 10.1016/j.exer.2013.05.013
65. Daveri E, Valacchi G, Romagnoli R, Maellaro E, Maioli E. Antiproliferative effect of rottlerin on sk-Mel-28 melanoma cells. Evidence-Based Complementary Altern Med (2015) 2015:1. doi: 10.1155/2015/545838
66. Peters J-M. The anaphase-promoting complex: Proteolysis in mitosis and beyond. Mol Cell (2002) 9(5):931–43. doi: 10.1016/S1097-2765(02)00540-3
67. Agami R, Bernards R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell (2000) 102(1):55–66. doi: 10.1016/S0092-8674(00)00010-6
68. Maiani E, Milletti G, Nazio F, Holdgaard SG, Bartkova J, Rizza S, et al. Ambra1 regulates cyclin d to guard s-phase entry and genomic integrity. Nature (2021) 592(7856):799–803. doi: 10.1038/s41586-021-03422-5
69. Chaikovsky AC, Li C, Jeng EE, Loebell S, Lee MC, Murray CW, et al. The Ambra1 E3 ligase adaptor regulates the stability of cyclin d. Nature (2021) 592(7856):794–8. doi: 10.1038/s41586-021-03474-7
70. Simoneschi D, Rona G, Zhou N, Jeong YT, Jiang S, Milletti G, et al. Crl4(Ambra1) is a master regulator of d-type cyclins. Nature (2021) 592(7856):789–93. doi: 10.1038/s41586-021-03445-y
71. Okabe H, Lee SH, Phuchareon J, Albertson DG, McCormick F, Tetsu O. A critical role for Fbxw8 and mapk in cyclin D1 degradation and cancer cell proliferation. PloS One (2006) 1:e128. doi: 10.1371/journal.pone.0000128
72. Alao JP. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol Cancer (2007) 6:24. doi: 10.1186/1476-4598-6-24
73. Nakayama KI, Hatakeyama S, Nakayama K. Regulation of the cell cycle at the G1-s transition by proteolysis of cyclin e and P27kip1. Biochem Biophys Res Commun (2001) 282(4):853–60. doi: 10.1006/bbrc.2001.4627
74. Barbash O, Lee EK, Diehl JA. Phosphorylation-dependent regulation of Scf(Fbx4) dimerization and activity involves a novel component, 14-3-3varepsilon. Oncogene (2011) 30(17):1995–2002. doi: 10.1038/onc.2010.584
75. Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, et al. Phosphorylation-dependent ubiquitination of cyclin D1 by the Scf(Fbx4-alphab crystallin) complex. Mol Cell (2006) 24(3):355–66. doi: 10.1016/j.molcel.2006.09.007
76. Sawai CM, Freund J, Oh P, Ndiaye-Lobry D, Bretz JC, Strikoudis A, et al. Therapeutic targeting of the cyclin D3:Cdk4/6 complex in T cell leukemia. Cancer cell (2012) 22(4):452–65. doi: 10.1016/j.ccr.2012.09.016c
77. Li Y, Hao B. Structural basis of dimerization-dependent ubiquitination by the Scf(Fbx4) ubiquitin ligase. J Biol Chem (2010) 285(18):13896–906. doi: 10.1074/jbc.M110.111518
78. Luo H, Wu L, He Y, Qin C, Tang X. Major advances in emerging degrader technologies. Front Cell Dev Biol (2022) 10:921958. doi: 10.3389/fcell.2022.921958
79. Neklesa T, Snyder LB, Willard RR, Vitale N, Pizzano J, Gordon DA, et al. Arv-110: An oral androgen receptor protac degrader for prostate cancer. Am Soc Clin Oncol (ASCO) (2019) 1:1. doi: 10.1200/jco.2019.37.7_suppl.259
80. Jennifer P, Crew AP, Cadelina G, John H, John JF, Ian WT, et al. Abstract P5-04-18: Arv-471, an oral estrogen receptor protac degrader for breast cancer. Am Assoc Cancer Res (2019) 1:1. doi: 10.1158/1538-7445.SABCS18-P5-04-18
81. Welleman IM, Hoorens MWH, Feringa BL, Boersma HH, Szymański W eds. Photoresponsive molecular tools for emerging applications of light in medicine. Great Britain: THE ROYAL SOCIETY OF CHEMISTRY (2020).
82. Ye H, Xie M, Xue S, Charpin-El Hamri G, Yin J, Zulewski H, et al. Self-adjusting synthetic gene circuit for correcting insulin resistance. Nat Biomed Eng (2017) 1(1):5. doi: 10.1038/s41551-016-0005
83. Zhou Y, Kong D, Wang X, Yu G, Wu X, Guan N, et al. A small and highly sensitive Red/Far-red optogenetic switch for applications in mammals. Nat Biotechnol (2021) 40:262–70. doi: 10.1038/s41587-021-01036-w
Keywords: cyclin D1 degradation, cancer cells, FBX4, combination mode, photodynamic therapy
Citation: Chen S and Li L (2022) Degradation strategy of cyclin D1 in cancer cells and the potential clinical application. Front. Oncol. 12:949688. doi: 10.3389/fonc.2022.949688
Received: 23 May 2022; Accepted: 28 July 2022;
Published: 18 August 2022.
Edited by:
Sukhbir Kaur, National Institutes of Health (NIH), United StatesReviewed by:
Xuanbin Wang, Hubei University of Medicine, ChinaCopyright © 2022 Chen and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling Li, bGlsaW5nMjVAZm1tdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.