
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Oncol. , 10 November 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.947410
This article is part of the Research Topic Biological Aspects of Bone Marrow Failure View all 9 articles
 Eleni Gavriilaki1
Eleni Gavriilaki1 Athanasios Tragiannidis2
Athanasios Tragiannidis2 Maria Papathanasiou1
Maria Papathanasiou1 Sotiria Besikli1
Sotiria Besikli1 Paraskevi Karvouni3*Vassiliki Douka1Eleni Paphianou1
Paraskevi Karvouni3*Vassiliki Douka1Eleni Paphianou1 Emmanuel Hatzipantelis2Giorgos Papaioannou1Anastasia Athanasiadou1Anastasia Marvaki1
Emmanuel Hatzipantelis2Giorgos Papaioannou1Anastasia Athanasiadou1Anastasia Marvaki1 Alkistis-Kira Panteliadou1
Alkistis-Kira Panteliadou1 Anna Vardi1Ioannis Batsis1Antonia Syrigou1Despina Mallouri1
Anna Vardi1Ioannis Batsis1Antonia Syrigou1Despina Mallouri1 Chrysavgi Lalayanni1
Chrysavgi Lalayanni1 Ioanna Sakellari1
Ioanna Sakellari1Bone marrow failure (BMF) syndromes are a group of various hematological diseases with cytopenia as a main common characteristic. Given their rarity and continuous progress in the field, we aim to provide data considering the efficiency and safety of the therapeutic methods, focusing on the treatment of aplastic anemia(AA) and paroxysmal nocturnal hemoglobinuria (PNH). We enrolled consecutive patients diagnosed with BMF in two referral centers of Northern Greece from 2008 to 2020. We studied 43 patients with AA (37 adults and 6 children/adolescents) and 6 with classical PNH. Regarding classical PNH, 4 patients have received eculizumab treatment with 1/4 presenting extravascular hemolysis. Among 43 patients with aplastic anemia, PNH clones were detected in 11. Regarding patients that did not receive alloHCT (n=15), 14/15 were treated with ATG and cyclosporine as first line, with the addition of eltrombopag in patients treated after its approval (n=9). With a median follow-up of 16.7 (1.8-56.2) months from diagnosis, 12/14 (85.7%) are alive (4-year OS: 85.1%). AlloHCT was performed in 28 patients. Five patients developed TA-TMA which did not resolve in 3/5 (all with a pre-transplant PNH clone). With the follow-up among survivors reaching 86.3 (6.3-262.4) months, 10-year OS was 56.9%, independently associated with PNH clones after adjusting for age (p=0.024). In conclusion, our real-world experience confirms that novel treatments are changing the field of BMF syndromes. Nevertheless, there is still an unmet need to personalize algorithms in this field.
Bone marrow failure (BMF) syndromes are a group of various hematological diseases that have a main common characteristic: the cytopenia of one or more blood cell lines resulting in anemia, neutropenia and/or thrombopenia (1). Bone marrow failure syndromes can be divided in two main categories, acquired and congenital disorders (2). Acquired syndromes represent an abnormal immune response to an external factor such as infections mostly viral, drugs or chemicals and usually affect all three lines causing pancytopenia. On the other hand, inherited bone marrow failure syndromes (IBMF) occur due to mutations in the hematopoietic stem cell or other progenitor cells (3). The most common congenital disorders (1), eithercause pancytopenia such as Fanconi anemia (4) and dyskeratosis congenita (5) or primary affect one lineage such as Shwachman-Diamond syndrome (6), Diamond-Blackfan anemia (7), Kostmann syndrome and congenital amegakaryocytic thrombocytopenia (8). They also feature a predisposition for congenital malformations and progression to myelodysplasia, acute leukemias and solid tumors (9). Due to presentation variability increased awareness and continuous follow-up are always needed, even though acquired BMF syndromes are more frequent in both adults and children (10).
Aplastic anemia (AA) is the most common acquired BMF syndrome with an incidence of 2 per million in Western countries and up to 6 per million in Asia (11). AA is a diagnosis of exclusion with hypoplastic MDS and IBMF syndromes being the main conditions that need to be excluded especially in children aged <10 years. In favor of AA are the presence of peripheral pancytopenia, an ‘‘empty’’ bone marrow with a lack of dysplasia (12–14). In the context of acquired BMF syndromes, paroxysmal nocturnal hemoglobinuria (PNH) is a clonal disease caused by a somatic mutation in the PIGA-gene resulting in the deficiency of GPI-anchored proteins (such as CD55 and CD59) and leads to erythrocytes unable to control complement activation (15–18). This results in chronic intravascular hemolysis, thrombosis in unusual locations and BMF caused by cellular autoimmunity to HSCs (19–22). Apart from its classical form that is characterized by a cellular or even hypercellular marrow, a PNH clone can also be detected in up to 70% of patients with AA (23–25). In children, the percentage of PNH clone is much lower and varies between 21-53% according to different studies (26, 27). When the PNH clone increases, especially after immunosuppressive therapy, they may present with classic complications of PNH (28, 29).
There are two main upfront treatments for AA: the immunosuppressive treatment (IST), a choice suitable for patients older than 40 years, and allogeneic hematopoietic cell transplantation (HCT) for patients younger than 40 (30–32). The best cell source for HCT remains the bone marrow because it reduces the possibility of a Graft-versus-Host Disease (GVHD);the most suitable donor is a matched sibling donor (33–35). If a matched sibling donor isn’t available, a matched unrelated donor (UD) is searched, while other options are unrelated cord blood and haploidentical transplants (34, 36). HCT from UD can also be the frontline treatment for pediatric patients (younger than 20 years) (37). On the other hand, complement inhibitors are the main treatment for PNH. Eculizumab (a humanized monoclonal antibody against C5) has been the first-in-class inhibitor (38). Despite its benefits, new C5 inhibitors are being developed with the second-generation C5 inhibitors being approved, ravulizumaband crovalimab (a long acting anti-C5 monoclonal antibody) showing non-inferiority to eculizumab (39–42). The results from the development of upstream inhibitors with the C3 inhibitor pegcetacoplan receiving approval, and factors B and D being investigated within phase 3 registration trials are also encouraging (43–46).
Given the rarity of these entities and continuous progress in the field, we aim to provide data considering the efficiency and safety of the therapeutic methods, focusing on the treatment of AA and PNH.
We enrolled consecutive patients diagnosed with BMF in two referral centers of Northern Greece from 2008 to 2020: AHEPA Hospital for the pediatric and adolescent population and Papanikolaou Hospital for the adolescent and adult population. Patients diagnosed with hypoplastic myelodysplastic syndrome (MDS) were excluded from the present study, in order to avoid heterogeneity in the study population. All patients were tested for PNH clones using a standardized flow cytometry protocol based on FLAER (fluorescent aerolysin) detection (47). Disease was treated according to ongoing recommendations during each treatment period (33). In particular, patients younger than 40 years with a sibling donor proceeded to upfront alloHCT. Patients older than 40 years, or without a sibling donor, immunosuppression was the first-line therapy. Refractory or relapsed patients proceeded to alloHCT if eligible and with a suitable donor. HLA typing was performed at diagnosis for all patients. Standard of care was similar to both centers, according to current guidelines.
This study was a retrospective chart review, and it was approved by the institutional review board and ethics committee of G. Papanicolaou Hospital. All patients gave written informed consent. The study was conducted in compliance with the Helsinki Declaration.
BMF patients were admitted to neutropenic isolation rooms. According to ongoing protocols, patients received irradiated Red Blood Cell (RBC) and platelet transfusions only at a clinical indication (signs/symptoms of anemia or thrombocytopenia) or at Hemoglobin < 7 g/dl or platelet count < 10K/μL. GCSF (Granulocyte colony-stimulating factor) was administered in cases of persistent grade 4 neutropenia in patients with signs/symptoms of infection. Routine blood and urine cultures were performed once weekly in hospitalized patients. Wide spectrum antibiotics were administered according to ongoing protocols, with modification according to cultures. Prophylaxis for Pneumocystis jiroveci, herpes simplex, and Candida spp were also administered in hospitalized and neutropenic outpatient setting. Patients with PNH received prophylactic anticoagulation as standard practice. Prophylaxis was given to all patients, even those without thrombosis and was stopped after initiation of complement inhibition.
Conditioning regimens included Cyclophosphamide (50 mg/kg/day for 4 days) and Antithymocyte Globulin (rabbit ATG, Thymoglobulin 2.5 mg/kg/day for 3 days). In patients sensitized with multiple transfusions (48), modifications were performed accordingly: Cyclophosphamide (50 mg/kg/day for 4 days), Fludarabine (30 mg/m2/day for 4 days), ATG 7.5 mg/kg and 10 mg/kg for sibling and unrelated donors respectively, as previously described (49). GVHD prophylaxis consisted of Methotrexate and Cyclosporine. Cyclosporine was slowly tapered and stopped between 9-12 months post-transplant with a careful follow-up of blood counts. STR (short tandem repeat) fragment analysis was performed regularly (on day + 14, + 30, + 60, + 90) in unfractionated bone marrow for chimerism evaluation. Complete donor chimerism was defined as donor chimerism ≥99% (50). Regarding supportive care, patients were admitted to neutropenic isolation rooms with HEPA filters. Prophylaxis for Pneumocystisjiroveci, herpes simplex, and Candida spp was administered. Patients underwent Cytomegalovirus (CMV) and Epstein-Barr (EBV) surveillance using peripheral blood molecular assays (51).
Analysis was performed with SPSS 22.0 (IBM SPSS Statistics for Windows, Version 22.0. Armonk, NY: IBM Corp). The following demographic and laboratory/clinical factors were included: transfusions (red cells, platelets), pre-transplant lines of treatment, age, gender, hemoglobin, neutrophils, and platelets values at diagnosis, PNH clone. Additionally, transplant factors were registered: donor type (sibling/unrelated), graft source (bone marrow/peripheral), HLA matching, conditioning (Cyclophosphamide/ATG with/without fludarabine); post-transplant factors: neutrophil and platelet engraftment, severe acute (grade 2-4) and extensive chronic GVHD, TA-TMA (transplant-associated thrombotic microangiopathy), PTLD (Post-transplant lymphoproliferative disease), infections, late complications, relapse, and survival. Chi-square test, Student’s t-test or Mann-Whitney test were used to compare variables. Overall survival (OS) probability was calculated with Kaplan-Meier curves. Variables with p<0.1 in univariate analysis were included in multivariate analysis using Cox proportional hazards. Cumulative incidence of competing events analysis was calculated by the EZR software (52). Statistical significance was assessed by the Gray test and Fine and Gray regression modeling. Significance level was 0.05 and two-tailed.
In total, we examined43 patients with severe or very severe AA (37 adults and 6 children/adolescents, Table 1) and 6 adult patients with classical PNH. Regarding cytogenetics, bone marrow samples failed to yield metaphases in 12 patients and normal cytogenetics were detected in 26patients. Cytogenetic abnormalities were detected in 11patients: trisomy 6 in 6 patients, trisomy 8 in 4, andmonosomy 7 in 1 patient.

Table 1 Baseline patients’ characteristics for AA patients (n=43).
Regarding patients with classical PNH, 4 patients have received eculizumab treatment for a median of 5.1 years (range 2.1-8.2). Among them, 3 out of 4 have shown hemoglobin normalization and no transfusion requirements, while the fourth patient presented with extravascular hemolysis and regular transfusion requirements. No adverse event related to eculizumab was noted. Three patients remain under eculizumab treatment. Furthermore, 2 patients received crovalimab under the COMMODORE-1/2 open-label clinical trials: one switched from eculizumab (NCT04434092) and the other one started as a naïve patient (NCT04432584). One patient with classical PNH and history of thrombosis has never consented to receive complement inhibitors and remains with supportive treatment.
Among 43 patients with aplastic anemia, PNH clones were detected in 11 patients (median 4%, range 1-65%), of whom, 4 patients did not proceed to alloHCT. Only one patient was treated with eculizumab, two years after immunosuppressive therapy, due to prominent hemolytic anemia attributed to a PNH clone larger than 30% in neutrophils (65%). The patient achieved hemoglobin normalization. Table 1 summarizes baseline characteristics in patients that received alloHCT compared to those who did not.
Regarding patients that did not receive alloHCT (n=15), 14/15 were treated with rabbit ATG and cyclosporine as first line, with the addition of eltrombopag in patients treated after its approval (n=9). Only one adolescent patient, who was excluded from further analysis, did not receive ATG as first line due to comorbidities (schizophrenia) and poor performance status. The patient received only cyclosporine and steroids for a short period and succumbed due to septic shock.
The median follow-up was 32 (1.8-56.2) months from diagnosis. Grade II-III infections were detected in 3 patients (2/3 bacteremias). One patient died early due to Ps. Aeruginosa and Fusarium infection. Another patient was diagnosed with lung cancer 2 years after treatment and succumbed to lung cancer complications. Increased viral loads of CMV and EBV (quantitative PCR) were detected in 5 patients (CMV:2, EBV:5) and gradually decreased without treatment. Other toxicities rated as grade ≥II were: hepatotoxicity (7 patients), nephrotoxicity (3 patients), and polyneuropathy (1 patient). Twelve patients became transfusion independent for RBCs in a median period of 85 days (range 10-200) and for PLTs in a median period of 62 days (range 17-187), while the absolute neutrophil count reached >500 per cubic millimeter threshold in a median of 55 days (range 9-150).PNH clones were not associated with poor response in patients not receiving alloHCT. Complete response (CR) was achieved in 7/14 (50%) and partial response (PR) in 30% of patients, with an overall response rate (ORR) of 80%. There was no significant difference in patients with or without eltrombopag. Two adult patients with PR relapsed within 40 months from initial treatment, while one pediatric patient progressed to AML and received chemotherapy. In total, 12/14 (85.7%) are alive (4-year OS: 85.1%, Figure 1A). It should be highlighted that these results include patients that did not undergo alloHCT either because they had no indication or no suitable donor at any time-point.
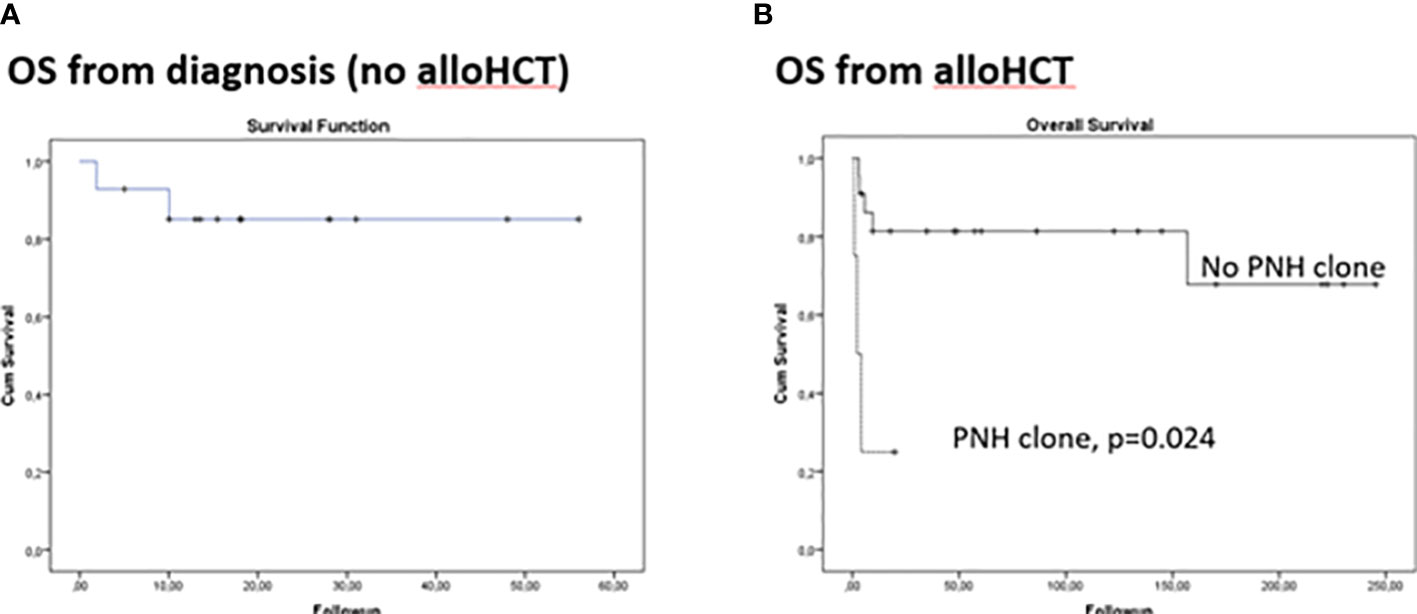
Figure 1 Kaplan Meier curves for overall survival in aplastic anemia patients. (A) From disease diagnosis in patients that did not receive allogeneic hematopoietic cell transplantation (alloHCT) (B) From alloHCT.
AlloHCT was performed in 28 patients, upfront in 12/28. Bone marrow grafts (25/28) and sibling donors (19/28) were preferred when available. Engraftment was evident at day 13 post-transplant (range 12-21) for neutrophils and 39 (16-121) for platelets. Complete donor chimerism was achieved in all patients. No graft rejection or failure was observed. Two patients presented with PTLD. The first patient had central nervous system involvement with a fatal outcome, whereas the second patients had a successful resolution following rituximab administration. Five patients (18%)were diagnosed with TA-TMA according to the International Working Group (IWG) criteria as an early complication, of whom 4/5 had detectable PNH clone pre-transplant that did not require treatment (<30%). PNH clones were no longer detectable post-transplant since they had achieved full donor chimerism. TA-TMA did not resolve in 3 out of 5 patients (all with a pre-transplant PNH clone) despite best available care: cyclosporine cessation (5/5), plasma exchange (3/5) and eculizumab (1/5). Patients were followed-up for a median of 47.5 months (0.9-262.4) post-transplant, with the follow-up among survivors reaching 86.3 (6.3-262.4) months. All survivors have a complete hematologic recovery. Cumulative incidence (CI) Grade 2-4 of acute GVHD was24.3%, while CI of moderate/severe chronic GVHD was 38.6%. TRM occurred only within the first-year post-transplant. 10-year CI of TRM was 14% was attributed to GVHD or TA-TMA. 10-year OS reached 56.9% and was independently associated with PNH clones after adjusting for age and donor type (p=0.024, Figure 1B, Table 2). Figure 1 presents comparison of OS from disease diagnosis in patients that received alloHCT (upfront or after immunosuppressive treatment) and not (p=0.877). No secondary malignancies or fatal long-term complications were documented.

Table 2 Cox regression analysis for Overall Survival.
Our study reflects the clinical spectrum of BMF presenting with several challenges in the real-world setting. Interestingly, AA was the most common diagnosis, with PNH clones being detected in many patients. Complement inhibition treatment has revolutionized the field providing safety and efficacy in treated patients, with or without AA. AlloHCT also showed safety and efficacy. Nevertheless, the presence of a PNH clone had an independent negative impact in survival post alloHCT.
Complement inhibition with eculizumab can indeed be efficient in patients with PNH clones, regardless of the existence or not of AA (53). As reflected by our rather small patient population, approximately 25% of PNH patients develop extravascular hemolysis. Since novel complement inhibitors are under advanced clinical development, these patients may benefit from upstream complement inhibition, such as pegcetacoplan that is currently FDA approved (54). Beyond novel complement inhibitors, the role of complement inhibitors in the transplant setting remains also to be clarified.
Despite that PNH clones are common in patients with aplastic anemia, only a few recent reports have considered the presence of PNH clones (55). Previous real-world reports have not taken this issue into consideration (56, 57). Recently, DeZern et al. reported successful outcomes with eculizumab bridging before alloHCT in 8 severe/very severe (SAA) patients (58). In addition, two recent studies have also explored outcomes of patients with PNH clones in the age of eculizumab (59, 60). Although both studies presented the potential benefits of eculizumab post alloHCT in 8 and 2 patients respectively, there was no clear comparison with a historical control group that did not receive eculizumab (59, 60). This comparison would clarify the role of complement inhibition, given that alloHCT mortality in SAA patients with PNH clones has been reported at approximately 30% (61).
PNH has been traditionally considered a negative predictor after alloHCT due to the heterogeneity of clinical presentations and severe signs of hemolysis and thrombocytopenia (62). Thrombosis is the major cause of death in PNH patients (63). Despite the multifactorial nature of thrombosis in PNH, complement inhibition seem to block this vicious cycle (63). However, early prediction of thrombotic or cardiovascular risk is not yet feasible, because little is known about patients post alloHCT (64). Interestingly, the incidence of TA-TMA reported in this cohort (18%) which is similar to the incidence of 16% previously reported in all patients receiving alloHCT in our center irrespective of indication (64).
In the group of SAA patients without a PNH clone, our results were comparable to those recently reported by the Aplastic Anemia Working Party of the European Group for Blood and Marrow Transplantation with the use of sibling or unrelated donors (34). The majority of recent previous reports has documented a risk of graft rejection/failure ranging from 3% to 33% (55). In our cohort, there was no graft rejection/failure. The use of fludarabine in the conditioning regimen along with ATG might have contributed to this result (65).
Our study is limited by its retrospective nature, the rather small number of participants and experience from two centers. In contrast, it reflects the local epidemiology from Northern Greece and reports data from the pediatric and adult hematology and BMT centers located in this area. In addition, our study was performed with both sibling and unrelated donors, since expansion of the donor pool using alternative donors remains currently under consideration as an alternative option for those patients (66, 67). It should be noted however that this study was conducted according to standard operating procedures with a long-term follow-up despite difficulties during the COVID-19 period (68).
In conclusion, our real-world experience confirms that novel treatments have revolutionized the field of BMF syndromes. Nevertheless, further studies are needed to personalize algorithms in the era of precision medicine.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
The studies involving human participants were reviewed and approved by Institutional Review Board of G Papanicolaou Hospital, Exochi, Thessaloniki, Greece. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
All authors have made contributions to the writing and design of the manuscript, collection or analysis of the data and drafting the article or revising it critically for important intellectual content. All authors have read and agreed to the published version of the manuscript.
The authors would like to acknowledge the valuable contribution of personnel involved in diagnosing and treating these patients. The authors would also like to thank Dr Maria Gavriilaki, an experienced writer, who revised the paper for typos and grammatical mistakes.
EG has received honoraria from Alexion and Omeros Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2. Bluteau O, Sebert M, Leblanc T, Peffault de Latour R, Quentin S, Lainey E, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood (2018) 131(7):717–32. doi: 10.1182/blood-2017-09-806489
3. Giudice V, Cardamone C, Triggiani M, Selleri C. Bone marrow failure syndromes, overlapping diseases with a common cytokine signature. Int J Mol Sci (2021) 22(2):705. doi: 10.3390/ijms22020705
4. Bagby GC, Alter BP. Fanconi anemia. Semin Hematol (2006) 43(3):147–56. doi: 10.1053/j.seminhematol.2006.04.005
5. Vulliamy T, Dokal I. Dyskeratosis congenita. Semin Hematol (2006) 43(3):157–66. doi: 10.1053/j.seminhematol.2006.04.001
6. Shimamura A. Shwachman-diamond syndrome. Semin Hematol (2006) 43(3):178–88. doi: 10.1053/j.seminhematol.2006.04.006
7. Lipton JM. Diamond blackfan anemia: New paradigms for a “not so pure” inherited red cell aplasia. Semin Hematol (2006) 43(3):167–77. doi: 10.1053/j.seminhematol.2006.04.002
8. Geddis AE. Inherited thrombocytopenia: Congenital amegakaryocytic thrombocytopenia and thrombocytopenia with absent radii. Semin Hematol (2006) 43(3):196–203.
9. Wilson DB, Link DC, Mason P. J, Bessler M. Inherited bone marrow failure syndromes in adolescents and young adults. Ann Med (2014) 46(6):353–63. doi: 10.3109/07853890.2014.915579
10. Sieff CA. Introduction to acquired and inherited bone marrow failure. Hematol Oncol Clin North Am (2018). 32(4):569–80. doi: 10.1016/j.hoc.2018.04.008
11. Ahmed P, Chaudhry QUN, Satti TM, Mahmood SK, Ghafoor T, Shahbaz N, et al. Epidemiology of aplastic anemia: a study of 1324 cases. Hematology (2020) 25(1):48–54. doi: 10.1080/16078454.2019.1711344
12. DeZern AE, Sekeres MA. The challenging world of cytopenias: distinguishing myelodysplastic syndromes from other disorders of marrow failure. Oncologist (2014) 19(7):735–45. doi: 10.1634/theoncologist.2014-0056
13. Gondek LP, DeZern AE. I Walk the line: how to tell MDS from other bone marrow failure conditions. Curr Hematol Malig Rep (2014) 9(4):389–99. doi: 10.1007/s11899-014-0224-3
14. Matsui WH, Brodsky RA, Smith BD, Borowitz M. J, Jones RJ. Quantitative analysis of bone marrow CD34 cells in aplastic anemia and hypoplastic myelodysplastic syndromes. Leukemia (2006) 20(3):458–62. doi: 10.1038/sj.leu.2404119
15. Miyata T, Takeda J, Iida Y, Yamada N, Inoue N, Takahashi M, et al. The cloning of PIG-a, a component in the early step of GPI-anchor biosynthesis. Science (1993) 259:1318–20.
16. Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-a gene in paroxysmal nocturnal hemoglobinuria. Cell (1993) 73:703–11.
17. Tomita M. Biochemical background of paroxysmal nocturnal hemoglobinuria. Biochim Biophys Acta (1999) 1455:269–86. doi: 10.1016/s0925-4439(99)00068-x
18. Walport MJ. Complement. first of two parts. N Engl J Med (2001) 344:1058–66. doi: 10.1056/NEJM200104053441406
19. Brodsky RA. Narrative review: paroxysmal nocturnal hemoglobinuria: the physiology of complement-related hemolytic anemia. Ann Intern Med (2008) 148(8):587–95. doi: 10.7326/0003-4819-148-8-200804150-00003
20. Nishimura J, Kanakura Y, Ware RE, Shichishima T, Nakakuma H, Ninomiya H, et al. Clinical course and flow cytometric analysis of paroxysmal nocturnal hemoglobinuria in the united states and Japan. Med (Baltimore) (2004) 83(3):193–207.
21. Risitano AM. Paroxysmal nocturnal hemoglobinuria and other complement-mediated hematological disorders. Immunobiology (2012) 217:1180–7. doi: 10.1016/j.imbio.2012.07.014
22. Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood (2006) 108:2509–19. doi: 10.1182/blood-2006-03-010777
23. Dezern AE, Borowitz MJ. ICCS/ESCCA consensus guidelines to detect GPI-deficient cells in paroxysmal nocturnal hemoglobinuria (PNH) and related disorders part 1 - clinical utility. Cytometry B Clin Cytom (2018) 94(1):16–22. doi: 10.1002/cyto.b.21608
24. DeZern AE, Symons HJ, Resar LS, Borowitz MJ, Armanios MY, Brodsky RA. Detection of paroxysmal nocturnal hemoglobinuria clones to exclude inherited bone marrow failure syndromes. Eur J Haematol (2014) 92(6):467–70. doi: 10.1111/ejh.12299
25. Hill A, DeZern AE, Kinoshita T, Brodsky RA. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers (2017) 3(1):17028. doi: 10.1038/nrdp.2017.28
26. Sutton KS, Shereck EB, Nemecek ER, Kurre P. Immune markers of disease severity and treatment response in pediatric acquired aplastic anemia. Pediatr Blood Cancer (2013) 60(3):455–60. doi: 10.1002/pbc.24247
27. Timeus F, Crescenzio N, Lorenzati A, Doria A, Foglia L, Pagliano S, et al. Paroxysmal nocturnal haemoglobinuria clones in children with acquired aplastic anaemia: a prospective single centre study. Br J Haematol (2010) 150(4):483–5. doi: 10.1111/j.1365-2141.2010.08219.x
28. Babushok DV, Perdigones N, Perin JC, Olson TS, Ye W, Roth JJ, et al. Emergence of clonal hematopoiesis in the majority of patients with acquired aplastic anemia. Cancer Genet (2015) 208(4):115–28.
29. Pu JJ, Mukhina G, Wang H, Savage WJ, Brodsky RA. Natural history of paroxysmal nocturnal hemoglobinuria clones in patients presenting as aplastic anemia. Eur J Haematol (2011) 87:37–45. doi: 10.1111/j.1600-0609.2011.01615.x
30. Bacigalupo A, Brand R, Oneto R, Bruno B, Socié G, Passweg J, et al. Treatment of acquired severe aplastic anemia: bone marrow transplantation compared with immunosuppressive therapy–the European group for blood and marrow transplantation experience. Semin Hematol (2000) 37(1):69–80.
31. Gupta V, Eapen M, Brazauskas R, Carreras J, Aljurf M, Gale RP, et al. Impact of age on outcomes after bone marrow transplantation for acquired aplastic anemia using HLA-matched sibling donors. Haematologica (2010) 95(12):2119–25.
32. Yoshida N, Kobayashi R, Yabe H, Kosaka Y, Yagasaki H, Watanabe K, et al. First-line treatment for severe aplastic anemia in children: bone marrow transplantation from a matched family donor versus immunosuppressive therapy. Haematologica (2014) 99(12):1784–91.
33. Bacigalupo A. How I treat acquired aplastic anemia. Blood (2017) 129(11):1428–36. doi: 10.1182/blood-2016-08-693481
34. Bacigalupo A, Socie G, Hamladji RM, Aljurf M, Maschan A, Kyrcz-Krzemien S, et al. Current outcome of HLA identical sibling versus unrelated donor transplants in severe aplastic anemia: An EBMT analysis. Haematologica (2015) 100(5):696–702. doi: 10.3324/haematol.2014.115345
35. Eapen M, Le Rademacher J, Antin JH, Champlin RE, Carreras J, Fay J, et al. Effect of stem cell source on outcomes after unrelated donor transplantation in severe aplastic anemia. Blood (2011) 118(9):2618–21.
36. Peffault de Latour R, Chevret S, Jubert C, Sirvent A, Galambrun C, Ruggeri A, et al. Unrelated cord blood transplantation in patients with idiopathic refractory severe aplastic anemia: a nationwide phase 2 study. Blood (2018) 132(7):750–4.
37. Dufour C, Veys P, Carraro E, Bhatnagar N, Pillon M, Wynn R, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. a study on behalf of the UK paediatric BMT working party, paediatric diseases working party and severe aplastic anaemia working party of EBMT. Br J Haematol (2015) 171(4):585–94.
38. Kelly RJ, Hill A, Arnold LM, Brooksbank GL, Richards SJ, Cullen M, et al. Long-term treatment with eculizumab in paroxysmal nocturnal hemoglobinuria: Sustained efficacy and improved survival. Blood (2011) 117:6786–92.
39. Kulasekararaj AG, Hill A, Rottinghaus ST, Langemeijer S, Wells R, Gonzalez-Fernandez FA, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: the 302 study. Blood (2019) 133(6):540–9.
40. Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, Lee L, Pessoa V, Gualandro S, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood (2018).
41. Risitano AM, Marotta S, Ricci P, Marano L, Frieri C, Cacace F, et al. Anti-complement treatment for paroxysmal nocturnal hemoglobinuria: Time for proximal complement inhibition? a position paper from the SAAWP of the EBMT. Front Immunol (2019) 10:1157.
42. Roth A, Nishimura JI, Nagy Z, Gaàl-Weisinger J, Panse J, Yoon SS, et al. The complement C5 inhibitor crovalimab in paroxysmal nocturnal hemoglobinuria. Blood (2020) 135(12):912–20.
43. Hillmen P, Szer J, Weitz I, Röth A, Höchsmann B, Panse J, et al. Pegcetacoplan versus eculiaumab in paroxysmal nocturnal hemoglobinuria. N Eng J Med (2021) 384(11):1028–37. doi: 10.1056/NEJMoa2029073
44. Kulasekararaj A, Risitano AM, Maciejewski JP, Notaro R, Browett P, Lee JW, et al. A phase 2 open-label study of danicopan (ACH0144471) in patients with paroxysmal nocturnal hemoglobinuria (PNH) who have an inadequate response to eculizumab monotherapy. Blood (2019) 138(20):1928–38. doi: 10.1182/blood.2021011388.
45. Risitano AM, Kulasekararaj AG, Lee JW, Maciejewski JP, Notaro R, Brodsky R, et al. Danicopan: An oral complement factor d inhibitor for paroxysmal nocturnal hemoglobinuria. Haematologica (2020) 106(12):3188–97. doi: 10.3324/haematol.2020.261826
46. Risitano AM, Roth A, Soret J, Frieri C, de Fontbrune FS, Marano L, et al. Addition of iptacopan, an oral factor b inhibitor, to eculizumab in patients with paroxysmal nocturnal haemoglobinuria and active haemolysis: an open-label, single-arm, phase 2, proof-of-concept trial. Lancet Haematol (2021) 8(5):e344–54.
47. Brodsky RA, Mukhina GL, Li S, Nelson KL, Chiurazzi PL, Buckley JT, et al. Improved detection and characterization of paroxysmal nocturnal hemoglobinuria using fluorescent aerolysin. Am J Clin Pathol (2000) 114(3):459–66. doi: 10.1093/ajcp/114.3.459
48. Shank B, Brochstein JA, Castro-Malaspina H, Yahalom J, Bonfiglio P, O'Reilly RJ. Immunosuppression prior to marrow transplantation for sensitized aplastic anemia patients: Comparison of TLI with TBI. Int J Radiat Oncol Biol Phys (1988) 14(6):1133–41. doi: 10.1016/0360-3016(88)90389-6
49. Young NS, Bacigalupo A, Marsh JC. Aplastic anemia: pathophysiology and treatment. Biol Blood Marrow Transpl (2010) 16(1 Suppl):S119–125. doi: 10.1016/j.bbmt.2009.09.013
50. Sakellari I, Mallouri D, Gavriilaki E, Batsis I, Kaliou M, Constantinou V, et al. Survival advantage and comparable toxicity in reduced-toxicity treosulfan-based versus reduced-intensity busulfan-based conditioning regimen in myelodysplastic syndrome and acute myeloid leukemia patients after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transpl (2017) 23(3):445–51. doi: 10.1016/j.bbmt.2016.11.023
51. Sakellari I, Papalexandri A, Mallouri D, Batsis I, Iskas M, Xochelli A, et al. Donor EBV at the time of hematopoietic cell transplantation: Is it time to adopt molecular assays? J Clin Virol (2018) 102:32–3. doi: 10.1016/j.jcv.2018.02.012
52. Kanda Y. Investigation of the freely available easy-to-use software 'EZR' for medical statistics. Bone Marrow Transpl (2013) 48(3):452–8. doi: 10.1038/bmt.2012.244
53. Lee JW, Peffault de Latour R, Brodsky RA, Jang JH, Hill A, Roth A, et al. Effectiveness of eculizumab in patients with paroxysmal nocturnal hemoglobinuria (PNH) with or without aplastic anemia in the international PNH registry. Am J Hematol (2019) 94(1):E37–41. doi: 10.1002/ajh.25334
54. Gavriilaki E, de Latour RP, Risitano AM. Advancing therapeutic complement inhibition in hematologic diseases: PNH and beyond. Blood (2022) 139(25):3571–82. doi: 10.1182/blood.2021012860
55. Georges GE, Doney K, Storb R. Severe aplastic anemia: allogeneic bone marrow transplantation as first-line treatment. Blood Adv (2018) 2(15):2020–8. doi: 10.1182/bloodadvances.2018021162
56. Dasgupta Y, Golovine K, Nieborowska-Skorska M, Luo L, Matlawska-Wasowska K, Mullighan CG, et al. Drugging DNA repair to target T-ALL cells. Leuk Lymphoma (2018) 59(7):1746–49. doi: 10.1080/10428194.2017.1397662
57. Vaht K, Göransson M, Carlson K, Isaksson C, Lenhoff S, Sandstedt A, et al. Incidence and outcome of acquired aplastic anemia: real-world data from patients diagnosed in Sweden from 2000-2011. Haematologica (2017) 102(10):1689–90. doi: 10.3324/haematol.2017.169862
58. DeZern AE, Jones RJ, Brodsky RA. Eculizumab bridging before bone marrow transplant for marrow failure disorders is safe and does not limit engraftment. Biol Blood Marrow Transplant (2018) 24(12):e26–30. doi: 10.1016/j.bbmt.2018.07.032
59. Cooper JP, Farah RJ, Stevenson PA, Gooley TA, Storb R, Scott BL. Hematopoietic cell transplantation for paroxysmal nocturnal hemoglobinuria in the age of eculizumab. Biol Blood Marrow Transpl (2019) 25(7): 1331–9. doi: 10.1016/j.bbmt.2019.01.033
60. Mei M, Gupta R, O'Donnell M, Al Malki MM, Aldoss I, Ali H, et al. Post-allogeneic hematopoietic stem cell transplantation eculizumab as prophylaxis against hemolysis and thrombosis for patients with hematologic disorders associated with paroxysmal nocturnal hemoglobinuria clones. Biol Blood Marrow Transpl (2019) 25(5):e183–5. doi: 10.1016/j.bbmt.2019.01.025
61. Vallet N, de Fontbrune FS, Loschi M, Desmier D, Villate A, Barraco F, et al. Hematopoietic stem cell transplantation for patients with paroxysmal nocturnal hemoglobinuria previously treated with eculizumab: a retrospective study of 21 patients from SFGM-TC centers. Haematologica (2018) 103(3):e103–5. doi: 10.3324/haematol.2017.182360
62. Kokoris SI, Gavriilaki E, Miari A, Travlou A, Kyriakou E, Anagnostopoulos A, et al. Renal involvement in paroxysmal nocturnal hemoglobinuria: an update on clinical features, pathophysiology and treatment. Hematology (2018) 23(8):558–66. doi: 10.1080/10245332.2018.1444563
63. Gavriilaki EB, Brodsky RA. Complementopathies and precision medicine. JCI (2020) 130(5): 2152–63. doi: 10.1172/JCI136094
64. Gavriilaki E, Gkaliagkousi E, Sakellari I, Anyfanti P, Douma S, Anagnostopoulos A. Early prediction of cardiovascular risk after hematopoietic cell transplantation: Are we there yet? Biol Blood Marrow Transpl (2019) 25(10):e310–6. doi: 10.1016/j.bbmt.2019.07.012
65. Kang HJ, Hong KT, Lee JW, Kim H, Park KD, Shin HY, et al. Improved outcome of a reduced toxicity-fludarabine, cyclophosphamide, plus antithymocyte globulin conditioning regimen for unrelated donor transplantation in severe aplastic anemia: Comparison of 2 multicenter prospective studies. Biol Blood Marrow Transpl (2016) 22(8):1455–9. doi: 10.1016/j.bbmt.2016.04.003
66. Bacigalupo A, Sica S. Alternative donor transplants for severe aplastic anemia: current experience. Semin Hematol (2016) 53(2):115–9. doi: 10.1053/j.seminhematol.2016.01.002
67. DeZern AE, Brodsky RA. Haploidentical donor bone marrow transplantation for severe aplastic anemia. Hematol Oncol Clin North Am (2018) 32(4):629–42. doi: 10.1016/j.hoc.2018.04.001
Keywords: bone marrow failure (BMF), bone marrow failure syndromes (BMFs), paroxysmal nocturnal hemoglobinuria, hematopoietic (stem) cell transplantation (HCST), fanconi anaemia
Citation: Gavriilaki E, Tragiannidis A, Papathanasiou M, Besikli S, Karvouni P, Douka V, Paphianou E, Hatzipantelis E, Papaioannou G, Athanasiadou A, Marvaki A, Panteliadou A-K, Vardi A, Batsis I, Syrigou A, Mallouri D, Lalayanni C and Sakellari I (2022) Aplastic anemia and paroxysmal nocturnal hemoglobinuria in children and adults in two centers of Northern Greece. Front. Oncol. 12:947410. doi: 10.3389/fonc.2022.947410
Received: 18 May 2022; Accepted: 17 October 2022;
Published: 10 November 2022.
Edited by:
Panagiotis Diamantopoulos, Laiko General Hospital of Athens, GreeceReviewed by:
Satheesh Chonat, School of Medicine, Emory University, United StatesCopyright © 2022 Gavriilaki, Tragiannidis, Papathanasiou, Besikli, Karvouni, Douka, Paphianou, Hatzipantelis, Papaioannou, Athanasiadou, Marvaki, Panteliadou, Vardi, Batsis, Syrigou, Mallouri, Lalayanni and Sakellari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paraskevi Karvouni, cGFyYXNrZXU5OEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.