 Linlin Lv
Linlin Lv Shilei Yang
Shilei Yang Yanna Zhu
Yanna Zhu Xiaohan Zhai
Xiaohan Zhai Shuai Li
Shuai Li Xufeng Tao
Xufeng Tao Deshi Dong
Deshi Dong
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 18 August 2022
Sec. Cancer Metabolism
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.942064
This article is part of the Research Topic Metabolic Reprogramming in Breast Cancer View all 7 articles
Breast cancer is the leading cause of cancer death in women. At present, chemotherapy is the main method to treat breast cancer in addition to surgery and radiotherapy, but the process of chemotherapy is often accompanied by the development of drug resistance, which leads to a reduction in drug efficacy. Furthermore, mounting evidence indicates that drug resistance is caused by dysregulated cellular metabolism, and metabolic reprogramming, including enhanced glucose metabolism, fatty acid synthesis and glutamine metabolic rates, is one of the hallmarks of cancer. Changes in metabolism have been considered one of the most important causes of resistance to treatment, and knowledge of the mechanisms involved will help in identifying potential treatment deficiencies. To improve women’s survival outcomes, it is vital to elucidate the relationship between metabolic reprogramming and drug resistance in breast cancer. This review analyzes and investigates the reprogramming of metabolism and resistance to breast cancer therapy, and the results offer promise for novel targeted and cell-based therapies.
Breast cancer is the primary cause of cancer-related death in women. The WHO reported approximately 2.26 million newly diagnosed cases of female breast cancer worldwide in 2020, which is equivalent to 1 in 8 cancer patients being breast cancer patients, rendering it the most common cancer in the world (1). According to GLOBOCAN data from 2020, China has the highest ASIR (age‐standardized rates of cancer incidence) of breast cancer, approximately 39.10 per 100,000 people (2). In 2019, GBD (Global Burden of Disease) data estimated breast cancer to be the main cause of the DALY (disability‐adjusted life year) burden in young and middle-aged women in the US and UK (3).
Currently, treatments for breast cancer are based on three broad classes: estrogen receptor α-positive or progesterone receptor-positive breast cancer, human epidermal growth factor receptor 2-enriched breast cancer, and triple-negative breast cancer that expresses none of these three receptors (4). Endocrine therapy can be effective for treating cancers in which either or both of the ER and PR proteins are overexpressed. Selective estrogen receptor modifiers (SERMs), aromatase inhibitors (AIs), and/or selective estrogen receptor degraders (SERDs) are among the endocrine therapies available. The subtype overexpressing HER2 was identified using HER2-targeted therapy. HER-2 targeted therapy can be achieved by monoclonal antibodies that are humanized, including trastuzumab and epratuzumab (5–7). Alternatively, HER2-positive patients may be treated with tyrosine kinase inhibitors, such as lapatinib (8) and neratinib (9–11). As a result, a lack of appropriate targeted therapies for breast cancers are classified as triple negative, which are therapies with cytotoxic chemotherapeutic agents, including taxane-based, platin-based, and other DNA damage-causing drugs (12–15).
However, the development of drug resistance reduces treatment effectiveness in breast cancer patients and is an important cause of cancer-related death. Endocrine and HER-2 resistance can result in disappointing outcomes, similar to chemotherapy resistance. Therefore, elucidating the mechanism of drug resistance in breast cancer is crucial to improving rates of survival. It is known that cancer cells possess distinct metabolic properties; among the metabolic properties of cancer cells are increased aerobic glycolysis, fatty acid synthesis, and glutaminolysis (10, 16). (Figure 1) Much more attention has recently been paid to targeting metabolic enzymes in cancer therapies and overcoming drug resistance (17, 18). The purpose of this review is to discuss metabolic reprogramming and progress in targeting metabolic pathways to treat breast cancer.
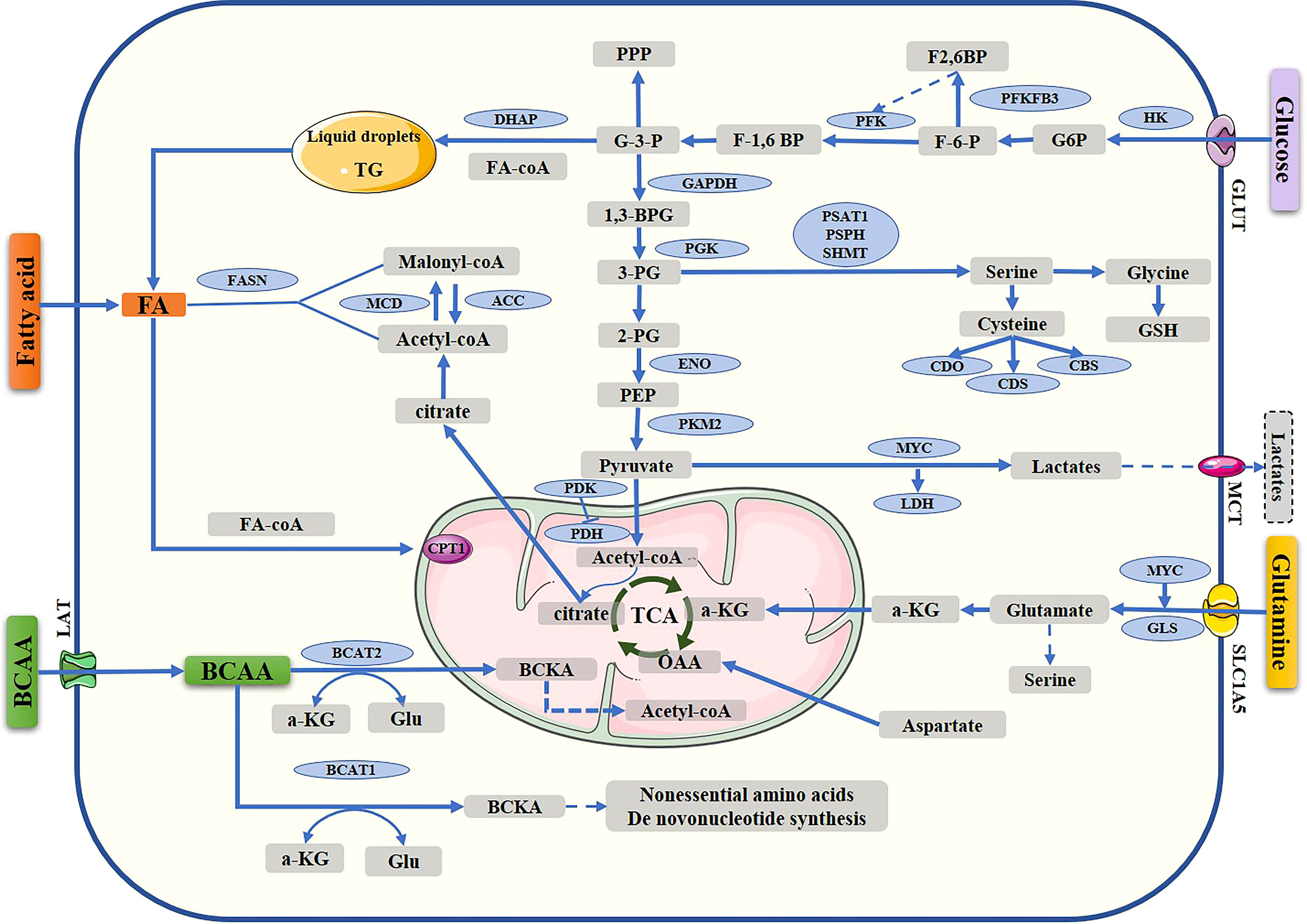
Figure 1 Metabolic pathway in breast cancer cells. Reprogramming of the metabolism, including glucose metabolism, fatty acid synthesis, and amino acid metabolism. TCA, tricarboxylic acid cycle; G-6-P, glucose-6-phosphate; F6P, fructose-6-phosphate; F1,6P, fructose-1,6-bisphosphate; G-3-P, glyceraldehyde 3 phosphate; DHAP, dihydroxyacetone phosphate; 1,3-BPG, 1,3-bisphosphoglycerate, 3-PG, 3-phosphoglycerate; 2-PG, 2-phosphoglycerate; PEP, phosphoenolpyruvate; OAA, oxaloacetate; α-KG, α-ketoglutarate; GLUT, glucose transporter; HK, hexokinase; PKM2, pyruvate kinase isozyme type 2; LDHA, lactate dehydrogenase A; MCT1, monocarboxylate transporter 1; PDK, pyruvate dehydrogenase kinase; PDH, pyruvate dehydrogenase; acetyl-CoA carboxylase; FASN, fatty acid synthase; CPT1, carnitine palmitoyl transferase 1; 3PG, 3-phospho-glycerate; GSH, reduced glutathione; GLU, glutamate; GLUT, glucose transporter; PHGDH, phosphoglycerate dehydrogenase; PSAT1, phosphoserine aminotransferase 1; SLC1A5, solute carrier family 1 member 5; GLS, glutaminase; PSPH, 1-3-phosphoserine phosphatase; BCAAs, branched-chain amino acids; BCAT1, branched-chain amino acid transaminase 1; BCAT2, branched-chain amino acid transaminase 2; BCKA, branched-chain a-keto acid; MCD, malonyl-CoA decarboxylase; ACC, Acetyl-CoA carboxylase; MCT Monocarboxylate transporter.
A recent focus in various cancer studies has been the dysregulated metabolism of cancer cells, identifying intratumoral heterogeneity and metabolic abnormalities in cancer cells as likely causes of chemotherapeutic resistance. The various clinical challenges in cancer therapy, such as immunosuppression, inevitable recurrence, anticancer drug resistance, cancer progression and metastasis, also contribute to metabolic abnormalities (19). It has been suggested that the specific metabolic characteristics of tumor cells can overcome the toxic effects of anticancer drugs, possibly leading to drug resistance in tumor cells (20) or promoting lipid synthesis and inducing mutations in such harsh environments (21). Tumor cells may be endowed with a highly adaptive metabolic capacity or benefit from metabolism in the microenvironment and are more likely to evade drug toxicity (22). Overall, metabolic reprogramming is now recognized as a hallmark of cancer. Increasing evidence indicates that metabolic reprogramming is associated with drug resistance in cancer therapy (23).
Metabolism of glucose differs significantly between normal and tumor cells. In normally differentiated cells, energy for growth is cells mainly provided by mitochondrial oxidative phosphorylation; in many tumor cells, even when oxygen is sufficient, cells still mainly rely on glycolysis for productivity, which is known as the “Warburg effect”. This phenomenon has been regarded as a phenotype of all cancers (23, 24). The Warburg effect produces substrates that become available for other metabolic pathways, including fat, nucleotide, and amino acid syntheses, that are crucial for oncogenesis (25). In cancer cells, glycolysis is the major metabolic process that produces ATP; the pyruvate formed from glucose must be converted to lactate for it to exert its effects and not be incorporated into the TCA cycle (26). The “reverse Warburg effect” states that cancer-associated fibroblasts can generate lactic acid through aerobic glycolysis, which is then provided via a paracrine route to adjacent cells, activating mitochondria, increasing oxidative phosphorylation in adjacent cells and promoting the growth of tumors (27). In general, the “Warburg effect” and “reverse Warburg effect” are both crucial to the development of cancer. Emerging studies suggest that various cancer cell subsets depend on different energy-producing pathways (28).
The reprogramming of glucose metabolism that occurs in many cancers is to meet the energy requirements of growing rapidly cancer cells (29, 30). Many enzymes play roles in metabolism of glucose, which provides cancer cells with energy. As glycolysis regulators, abnormal expression of glycolytic-related enzymes results in glycolysis dysregulation, which gives rise to oncogenesis, tumor growth, and treatment resistance (31). Researchers have demonstrated the effectiveness of treatments that target metabolism in improving anticancer therapies or reversing drug resistance in breast cancer cells, such as resistance to chemotherapy, endocrine therapy, and HER-2 targeted treatment.
Hexokinases contribute significantly to the initiation and maintenance of tumors and catalyze the first reaction of glycolysis. This step is a rate-limiting reaction in glycolysis, transforming glucose into glucose 6-phosphate (32, 33). The human hexokinase family consists of three members: HK1, HK2, and HK3 (34). HK2 is highly expressed in many tumors. Some studies indicate that breast cancer cells exhibit a high level of HK2 expression (32). Chemotherapy resistance can also be induced by upregulating HK2 expression, which is an enzyme of crucial importance that is involved in resistance to breast cancer and its prognosis through tumor glycolysis (35).
One study reports that curcumin increases sensitivity to TAM in breast cancer cells by regulating the HK2 pathway. SLUG may also regulate HK2 expression through activation of transcription. Hence, HK2 and TAM resistance may be closely related (36). The mechanism by which HK2 causes TAM resistance was described in depth in another study. When comparing TAMR and MCF-7 cells, TAMR cells exhibit higher HK2 expression and higher glycolysis rates. Both HK2 and mTORC1 are primary sensors of glucose ingestion and metabolism (35). HK2 binds to voltage-dependent anion channels (VDACs) and inhibits apoptosis (37). Additionally, HK2 can be phosphorylated at Thr473, which can cause resistance to paclitaxel (38). Furthermore, dephosphorylation of HK2 at Thr473, SMI 4a resensitizes paclitaxel-resistant cell lines. In preclinical studies, two-deoxyglucose (2-DG), three-bromopyruvate (3-BrPA), and lonidamine (LND) acted as HK2 inhibitors. Trastuzumab resistance correlates with increased glycolysis. It has been demonstrated that trastuzumab combined with 2-DG inhibits glycolysis in breast cancer cells in vitro and in vivo (39). HK2 knockdown inhibits the proliferation of MDA-MB-231 breast cancer cells and enhances the ability of 5-FU to kill them. When HK2 is downregulated in breast cancer cells, lactate secretion and glycolysis baseline are significantly reduced (40). DZNep, as an indirect inhibitor of histone methyltransferases, potently induces degradation of NSD2 protein and inhibits expression of NSD2 target genes (HK2, G6PD, GLUT1 and TIGAR) involved in the pentose phosphate pathway (PPP). These findings suggest that DZNep-such as agents can be developed to target NSD2 histone methyltransferase for effective treatment of tamoxifen-resistant breast cancer (41). Zhu et al. reported that ETV4, as a pivotal transcription factor, regulates gene expression associated with glycolysis. In the presence of loss of ETV4, glycolytic enzymes, such as HK2 and LDHA, and glucose uptake are inhibited (42). Liu et al. (35) demonstrated that by suppressing the mTOR-S6K signaling pathway, upregulation of HK2 promotes autophagy, subsequently conferring tamoxifen resistance to MCF 7 breast cancer cells.
Phosphofructokinase-1 (PFK1) catalyzes conversion of fructose-6 phosphate to fructose-1,6 bisphosphate in the third step of glycolysis. Fructose 2,6-biphosphate, which is produced by the enzyme 6-phosphofructo-2-kinase/fructose2,6-bisphosphatase 3 (PFKFB3) from fructose-6 phosphate, is thought to allosterically activate PFK1. PFKFB3 is expressed at high levels in many cancers (43). PFKFB3 is important for sustaining glycolysis in the tumorigenic environment, even under unfavorable conditions, promoting metabolic reprogramming, cell proliferation, DNA repair, and drug resistance (44). Tamoxifen-resistant LCC9 cells express twofold higher levels of PFKFB3 mRNA and protein than MCF-7 cells. Combining an inhibitor of PFKFB3 with TAM suppresses the growth of both TAMR LCC9 and MCF-7 cells, demonstrating the role of PFKB3 in TAM resistance (45). A combination of PFKFB inhibitors and ER-targeted therapies block tumorsphere formation in several models of advanced breast cancer, such as tamoxifen (TamR)- and paclitaxel (TaxR)-resistant cell models, ER+ patient-derived organoids (PDxO) and murine tumor cells (46).
The glycolysis regulator PFKFB3 is key during BC progression and drug resistance. PIM2 has been identified as a novel binding protein for PFKFB3. PIM2 can directly bind and change the phosphorylation status of PFKFB3 at Ser478 to enhance stability through the ubiquitin−proteasome pathway and to promote glycolysis, BC cell growth, and paclitaxel resistance together with PIM2 in vitro and in vivo (47). Studies have shown that PFKFB3 stimulation of lactic acid production may mediate activation of the TLR4 signaling pathway to some extent, leading to drug resistance to paclitaxel (48). PFKFB3 is a hub for coordinating the cell cycle and glucose metabolism. PFKFB3 binding results in accumulation of the CDK4 protein by inhibiting ubiquitin proteasome degradation mediated by the heat shock protein 90-Cdc37–CDK4 complex. Proteasome-dependent degradation of CDK4 is accelerated via disruption of the interaction of PFKFB3 with CDK4 through lysine 147 to alanine mutation. Blocking the PFKFB3–CDK4 interaction improves the therapeutic effect of the FDA-approved CDK4 inhibitor palbociclib against breast cancer (49). PO is also an inhibitor of PFKFB3, and a study showed that it increases the effectiveness of resistance combined with other anticancer agents (50).
Pyruvate kinase is a key enzyme that catalyzes the last step in glycolysis. It participates in the process of transferring a phosphate from PEP to pyruvate and converting ADP to ATP. M1 and M2 are the two isoforms of pyruvate kinase, but cancer cells express only the latter. The results of recent studies indicate that many tumors express PKM2, which is a growth factor and an inhibitor of apoptosis. It has a major impact on tumor growth and metabolism (51, 52). Researchers have investigated expression of PKM2 in breast cancer cells, both nuclear and cytoplasmic (53). As a transcription coactivator, PKM2 translocates to the nucleus and increases chemotherapy resistance. In advanced breast cancer, PKM2 expression correlates with cisplatin resistance (54). In MCF-7 breast cancer cells, PKM2 cooperates with sterile 20-like kinase 1 and prevents caspase-3, resulting in inactivation of TAM-induced apoptosis. PKM2 is important in regulating breast cancer cell viability (55). Chemotherapy resistance is also promoted by PKM2 in ER+ breast cancer via increased aerobic glycolysis. Accordingly, 2-deoxy-D-glucose (2-DG) is a PKM2 inhibitor that can suppress glycolysis and reverse adriamycin sensitivity in MCF-7 and T47D cells (56).
Enolase (EN) is a critical enzyme involved in the Warburg effect. During the phosphorylation reaction, ENO catalyzes conversion of 2-phosphoenolpyruvate (2-PG) to phosphoenolpyruvate (PEP) and ATP. Three ENO isoforms exist: ENO-1, ENO-2, and ENO-3. ENO1 regulates transcription, apoptosis, and cell differentiation. It is also is essential for glycolysis (57). Moreover, analysis of 244 samples of breast cancer tissue revealed strong expression of ENO-1 in ER breast cancer, showing that it is also an important marker for BC (58). In breast cancer, elevated expression of ENO1 has been reported to be closely related to tamoxifen resistance and adriamycin resistance (59), and silencing of ENO expression significantly increases the cytotoxicity of 100 nM tamoxifen in tamoxifen-resistant breast cancer cells. Upregulated ENO-1 suppresses expression of c-Myc, resulting in the survival of resistant cells (60). In MCF7 cells, TAM induces mRNA expression of ENO-1 by activating ERα and NF-κB. As a result, drug-induced apoptosis is inhibited (61, 62).
The correlation between ENO1 and MDR in breast cancer may be regulated by activating the ERK1/2 pathway, and it is likely to be regulated by c-Myc. Therefore, ENO1 alters the concentration of extracellular ATP and further influences tumor cell proliferation (63, 64). Doxorubicin-resistant MCF-7R cells lacks E-cadherin expression and show upregulated Vimentin expression and higher EGFR and ENO-1 levels (65). Furthermore, proteomics profiling studies have indicated that knockdown of ENO-1 expression restores oxidative phosphorylation (66). Above all, an innovative strategy for treating drug-resistant breast cancer is to target EN-1.
Lactate dehydrogenase A (LDHA) is one version of the enzyme LDH. In aerobic glycolysis, LDHA catalyzes the last step of the process, converting pyruvate to lactate, which causes the formation of NAD from NADH. Breast tissue expresses high levels of LDHA, which is one of the most prominent isoforms of LDH (67). According to a recent study, LDHA also plays a significant role in acquired tamoxifen resistance in breast cancer by facilitating autophagy (68). In addition, tamoxifen resistance is associated with changes in LDHA and LDHB gene expression and increased lactate concentrations (69). Thus, LDHA is a great target for controlling TAM resistance in breast cancer. ErbB2 signaling enhances glycolysis via LDHA-dependent upregulation of HSF1. Taking a targeted approach to glucose metabolism may help overcome Herceptin resistance in breast cancer. Glycolytic inhibitors combined with chemotherapy overcome resistance and lead to more potent inhibition of glycolysis in ErbB2-positive breast cancer (39).
LDHA correlates with trastuzumab-based therapy resistance (70). LDHA inhibitors suppress proliferation of HER-2-overexpressing cells in breast cancer and increase sensitivity to drug therapy (71). When MDA MB231 cells are subjected to sustained exposure to NAMPT inhibitors, such as FK866, drug resistance is induced based on glycolytic metabolism shifts and LDHA activity (72). Compared to cisplatin alone, electrical pulses (EP) + cisplatin (CsP) cause a switch in metabolism with LDHA downregulation, which impacts TNBC growth, proliferation, invasiveness, chemotherapeutic resistance and poor therapeutic response (73). We observed that LDHA and MCT1 are upregulated in Taxol-resistant breast cancer cells (74). Oxamate, an LDHA inhibitor, combined with paclitaxel induces apoptosis in paclitaxel-resistant breast cells by inhibiting cellular glycolysis. Thus, LDHA may serve as a therapeutic target for breast cancer resistance (75).
The pyruvate dehydrogenase complex (PDC) contains three types of enzymes that perform catalytic functions, known as E1, E2 and E3. Cellular metabolic flexibility is provided by the PDC, which integrates glycolysis, fatty acid metabolism, and the TCA cycle (76). PDH is an E1 enzyme that is a component of the PDC that converts pyruvate to acetyl-CoA (77). PDH activity is mainly controlled by pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase. By phosphorylating PDH, PDK inhibits PDH activity, whereas pyruvate dehydrogenase phosphatase activates it by reversing phosphorylation. Four different types of PDKs participate in glycolysis. They exert their effects on chemoresistance in tumor therapy and include PDK1-4 (78).
The role of overexpressed PDK in aerobic glycolysis, chemotherapeutic resistance, and metastasis in cancer has been widely studied (79). Researchers have discovered that inhibiting PDK reduces neoplasm development by controlling aerobic glycolysis (80). When PDK1 inhibitors such as triciribine or tetrandrine are combined with tamoxifen, breast cancer becomes more sensitive to the drug (81).
Researchers have shown that hypoxia-inducible factor (HIF)-1α regulates expression of pyruvate dehydrogenase kinase 3 (PDK3), thereby inducing resistance to chemotherapy under hypoxic conditions (82). Additionally, PDK4 alters regulation of PDH and is associated with antiestrogen resistance in breast cancer (83). The pyruvate dehydrogenase kinase (PDK) inhibitor dichloroacetate (DCA) PDK regulates pyruvate dehydrogenase, which aids in the conversion of pyruvate to acetyl-CoA, illustrating the proliferation-inhibiting properties of DCA in highly metastatic diseases (84). By decreasing expression of EGFR, MCF7 cells can be sensitized to tamoxifen-induced apoptosis by DCA (85). In trastuzumab-resistant HER2+ cancers, neuromedin U (NmU) is upregulated, and ectopic expression of NmU increases glycolysis, likely via PDK activity, suggesting a possible treatment strategy (86). OSU-03012, which is based on celecoxib as a scaffold to develop a COX-2-inactive PDK-1 inhibitor, potentiates trastuzumab’s antiproliferative effect in HER2-positive cells, especially in SKBR3/IGF-IR cells, through downregulation of PDK-1/Akt signaling (87). By blocking PDK-1/Akt signaling, tamoxifen can be used to sensitize ER-negative breast cancer cells to its antitumor effects (88).
Although many previous studies have focused on catabolic glycolysis, recent studies work reveals that Fructose-1,6-bisphosphatase (FBP), as a rate-limiting enzyme that regulates conversion of fructose 1,6-diphosphate to fructose 6-phosphate, is essential for the genesis and development of cancers. Furthermore, the function of FBP in chemoresistance has attracted attention (59). Two types of FBP exist in mammals, FBP1 and FBP2. FBP1 plays a regulatory role in gluconeogenesis, though the physiological role of FBP2 remains unclear. Fructose-bisphosphatase 1 (FBP1) is a target gene of CELF6, and CUG-BP Elav-like family member 6 (CELF6) was identified as an RNA-binding protein. Stable CELF6-overexpressing BT549 and MDA-MB-231 cell lines have been established, and CELF6 overexpression-mediated inhibition of TNBC growth relies on FBP1. CELF6 acts as a tumor suppressor by upregulating FBP1 expression through mRNA stabilization to inhibit TNBC progression and increase sensitivity to PTX treatment (60).
GLUTs are glucose transporters in mammalian cells. The GLUT family comprises 14 members. GLUTs increase uptake of glucose by the cytomembrane and play a critical role in glycolysis (89). Due to oncogenic signaling, it is possible that increased glucose utilization and activated glycolysis, resulting in lactate accumulation, can occur even in cancer cells with oxygen present. Glucose transporter-1 (GLUT-1) expression is higher in TNBC than in non-TNBC (90). Upregulation of GLUT1, GLUT3, and GLUT4 has been related to cancer resistance in several studies. Inhibition of GLUT enhances the anticancer effects of chemotherapy compounds (91). Researchers have recently discovered that GLUT1 plays a role in enhancing autophagy and resistance to TAM in MCF-7 breast cancer cells (92).
Increased GLUT1 transcription and membrane translocation leads to increased glucose uptake and glycolysis through the Akt signaling cascade. Targeting glycolysis via Skp2 increases HER2+ tumor sensitivity to trastuzumab treatment (93). Therefore, GLUT inhibitors have been used in a variety of combinations with chemotherapeutics, such as doxorubicin, paclitaxel, and cytarabine, and they exhibit synergistic or additive cancer-fighting effects with reduced chemo-, radio-, and immuno-resistance. Using glucose transporter (GLUT) inhibitors in combination with chemotherapeutic agents reduces chemotherapeutic toxicity compared to monotherapy due to reduced therapeutic doses required to achieve desired effects (94).
In addition, palbociclib in combination with paclitaxel inhibits proliferation of cells and induces apoptosis. By pretreating cells with palbociclib and then removing it before paclitaxel treatment, cell cycle reentry from G1 to S phase can be synchronized. Moreover, palbociclib inhibits glucose transport by reducing GLUT-1 glucose uptake through the Rb/E2F/c-Myc signaling pathway. Furthermore, expression of HIF-1α, a key factor in tumorigenesis, is inhibited. Researchers have shown a high level of GLUT1 in breast and primary colon cancers (95). Phloretin is a GLUT1 inhibitor that inhibits glucose transport and glycolysis. Additionally, phloretin increases the sensitivity of tumor cells to daunorubicin under hypoxic conditions (96). Compound WZB117 has been shown to inhibit GLUT1 in MCF-7 breast cancer cells. There is also evidence of synergistic anticancer effects when WZB117 is combined with cisplatin and paclitaxel. WZB117 inhibits cell proliferation more effectively in combination with a mitochondrial inhibitor, which indicates that it might be more effective against aggressive cancer cells, which are invariably mitochondrial deficient (96). However, combined use of other targeted therapies along with GLUT inhibitors may also be a key strategy for overcoming drug resistance; nevertheless, GLUTs are present in a variety of organs and cells, which makes them difficult to target (97). Consequently, improvement of GLUT inhibitor selectivity and affinity is a major area of study in anticancer research.
Contrary to previous studies, it has been proposed that ablation of GLUT1 attenuates apoptosis and increases drug resistance via upregulation of p-Akt/p-GSK-3β (Ser9)/β-catenin/survivin. These results indicate that the potential of Glut1 as a therapeutic target should be carefully re-evaluated (98).
Proteins are composed of the amino acids and have structural and functional roles in organisms. Among the various requirements of biosynthesis, amino acid metabolism is vital to maintaining cellular homeostasis, energy production, and redox equilibrium. Furthermore, tumor-specific metabolites, such as polyamines, that play an important role in tumor progression and growth are produced by amino acids (99). Cells resistant to hormonal treatment regulate amino acid anabolism and catabolism to ensure survival and growth. Breast cancer therapeutic resistance is thought to be associated with amino acid metabolites.
In addition to glucose, glutamine is the most abundant circulating amino acid and functions as a key carbon and energy source for cancer cells. It is known that glutamine is important in cancer because it contributes nitrogen and carbon for a variety of reactions that result in proliferation, invasion, and metastatic spread of cancer cells (100–102). First, by generating α-ketoglutarate (αKG), glutamine serves to provide carbon sources for entry into the TCA cycle. Second, glutamine is an important source of nitrogen for the synthesis of nucleotides and other nonessential amino acids. Last, glutaminolysis-generated glutamate is a precursor of glutathione and helps maintain redox balance (103).
Tamoxifen-induced apoptosis is inhibited by glutamine, and the cooperates between glutamine and stromal cells results in chemoresistance (104). Furthermore, the interaction between the stroma and the epithelium is critical to cancer progression and metastasis. Cancer-associated fibroblast cells produce glutamine, which is then secreted into the tumor via autophagy. Glutamine is taken up from the tumor microenvironment and converted to glutamate and ammonia. Upon conversion to α-ketoglutarate, glutamate is used in the TCA cycle, increasing mitochondrial activity. By inhibiting the p53-induced protein TIGAR, glutamine decreases glycolysis, apoptosis, and autophagy (105). Additionally, tumor epithelial cells release ammonia into the microenvironment, where it enters stromal cells, activates autophagy and inhibits Cav-1 expression. Autophagy is proposed to be a common survival mechanism during resistance to TAM (106). Myc is activated in breast cancer cells in the presence of acquired endocrine resistance. In addition to regulating various cell processes, Myc, a proto-oncogene, is involved in glutamine and glucose metabolism (107). Myc inhibition in TAMR cells decreases cell viability, growth, and glucose uptake. Thus, appropriate regulation of the glutaminase-glutamine synthase system (GLS/GAC-GLUL) by Myc is crucial for maintenance of antiestrogen-resistant phenotypes (108). In TAMR cells, endoplasmic reticulum stress is associated with marked upregulation of the unfolded protein response. When glucose is depleted, glutamine induces apoptosis and inhibits autophagy through a pathway mediated by the unfolded protein response (UPR) (109). In breast cancer, c-Myc overexpression may be sufficient to cause antiestrogen resistance (110), and MYC expression is upregulated by crosstalk between ER and HER2 in aromatase inhibitor-resistant breast cancer cells. MYC-mediated glutamine metabolism is associated with AI resistance in breast cancer (111). Re-expression of ERRα in resistant cells triggers metabolic adaptations favoring mitochondrial energy metabolism through increased glutamine metabolism, as well as ROS detoxification required for cell survival under therapeutic stress conditions. Pharmacological inhibition of ERRa activity represents a viable mechanism to counteract lapatinib resistance in breast cancer and to impact metabolic adaptations occurring in resistant tumors (112). It is also notable that the master regulator of mitochondrial metabolism PGC-1a regulates a significant number of pathways implicated in therapy resistance, including OXPHOS (113), oxidative stress response (114), glutamine metabolism (115), and glutathione metabolism (116). The context-dependent roles of PGC-1a may therefore underpin specific metabolic vulnerabilities in both doxorubicin and epirubicin resistance in breast cancer. Targeting global regulators of metabolic plasticity, such as PGC-1a, is promising as a broad strategy for treating therapeutic-resistant cancers (117).
Resistance is common in breast cancer cells, and glutamine addiction is a way to escape drug treatment. As a potential pharmacological target to reverse cancer cell resistance to chemotherapy, glutamine transporters or glutaminolysis have emerged as promising candidates. The amino acid transporter SLC6A14, also called ATB0,+, is upregulated in ER-positive breast cancer in women. The features of SLC6A14 include concentrative transport of leucine, glutamine, and arginine. It is possible to inhibit mTOR activity, activate autophagy, and cause cell death by blocking SLC6A14 (118). AI-resistant breast cancer cells show significant upregulation of the glutamine transporters SLC1A5 and GLS, and inhibition of MYC, SLC1A5, and GLS decrease cell proliferation in AI-resistant cells (119). EphA2 is highly expressed in HER2+ tumors, with increased dependence on glutamine metabolism through enhanced transcription of SLC1A5 and GLS, which is recognized as a new target of therapy in HER2+ tumors (120). The glutamine transporter SNAT2 is the AA transporter most frequently induced by hypoxia in breast cancer and is regulated by hypoxia both in vitro and in vivo in xenografts. SNAT2 induction in MCF7 cells is also regulated by ERα, but it is predominantly a hypoxia-inducible factor 1α (HIF-1α)-dependent gene under hypoxia. A switch in regulation of SNAT2 between ERα and HIF-1α leads to endocrine resistance in hypoxia. The development of drugs targeting SNAT2 may be of value for a subset of hormone-resistant breast cancers (121).
Recent studies have drawn attention to glutaminase, an enzyme that catalyzes glutamine to glutamate and has become a potential target for cancer therapy. A pair of novel glutaminase inhibitors has been found: CB-839 (122, 123) and 968 (124). CB-839 exerts the strongest inhibition of proliferation in TNBC cells but not in ER-positive cells. CB-839 shows significant antitumor activity in xenograft models, whether used alone or in combination with paclitaxel. Compound 968 has the strongest cytotoxic effect against MDA-MB-231 breast cancer cells. Genome analysis indicates that Compound 968 can inhibit apoptosis or promote metastasis gene expression and modify histones. Hence, MDA-MB-231 cells are more likely to be apoptotic and less invasive. When combined with doxorubicin, Compound 968 also increases the chemosensitivity of breast cancer cells.
The branched-chain amino acids leucine, isoleucine, and valine play a pivotal function in tumorigenesis (125). It has been found that breast cancer patients’ plasma and tissues contain higher levels of BCAAs (126). BCAA metabolism enhances the proliferation and growth of breast cancer cells through modulation of mitochondrial biogenesis and function. Catabolism of BCAAs is triggered by the enzyme branched-chain amino acid transaminase 1 (BCAT1). In addition to enhancing citrate synthase activity, BCAT1 increases the quantity of ATP and reduces ROS generation. AMPK, SIRT1, and mTOR are nutritional sensors involved in mitochondrial activity, and experiments have demonstrated that mitochondrial biogenesis is promoted by BCAT1 through its selective mTOR signaling activation. Rapamycin may inhibit BCAT1 by repressing mTOR (126). In TAM-resistant cells, mTORC1 phosphorylates (activates) p70S6 kinase by activating ER signaling pathways independent of estrogen (127). According to one study, leucine uptake is crucial for tamoxifen-resistant cells to grow under nutrient stress conditions. Leucine enters a cell via the SLC7A5 transporter (128). The protein LLGL scrawl cell polarity complex component 2 (LLGL2) regulates expression of the SLC7A5 transporter on the cell surface. LLGL2 interacts with SLCA5 and then binds to the YKT6 protein to form a trimeric complex, which results in an increase in transporters on the surface of the cell. Estrogen regulates LLGL2 expression. Overall, expression of SLC7A5 is elevated in TAM-resistant MCF-7 cells (129).
L-type amino acid transporter-1 (LAT1) is involved in chemotherapeutic resistance and may represent a new treatment target in breast cancer. Metabolites of cancer and branched-chain amino acids are also important in energy production and drug resistance in MCF-7 cells treated with chemotherapy, despite reduced glucose metabolism (129).
Serine is considered a key factor in glucose metabolism. Indeed, many cancers are associated with upregulation of the serine biosynthesis pathway (130, 131). Glycine is produced by catabolism of serine. Together, serine and glycine provide the primary one-carbon units needed for synthesis of nucleic acids, lipids, proteins, and cofactors (132). Various enzymes are involved in serine metabolism, including phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase 1 (PSAT1), and 1-3-phosphoserine phosphatase (PSPH), which are highly expressed in TNBC. Additionally, serine and glycine depletion in culture media reduces proliferation of TNBC cells (133).
A variety of cancers highly express 3-phosphoglycerate dehydrogenase (PHGDH), the enzyme responsible for de novo serine biosynthesis. In addition to contributing to tumorigenicity, PHGDH may contribute to innate or acquired resistance to current chemotherapies in cancer (134). In vitro and in vivo, small molecules inhibit the serine synthesis pathway of PHGDH, resulting in a lower proliferation rate of breast cancer cells expressing PHGDH (135, 136). CBR-5884 is a PHGDH inhibitor that suppresses proliferation of PHGDH-dependent TNBC tumor cells (137).
The phosphoserine aminotransferase (PSAT1) gene encodes a key aminotransferase that contributes to serine biosynthesis; 3-phosphohydroxypyruvate is converted to phosphoserine by this enzyme during the oxidation reaction. Transcriptional and immunohistochemical analyses have revealed that ER-positive breast cancer patients who receive TAM are more likely to have a poor prognosis if PSAT1 is overexpressed (138). PSAT1 knockdown sensitizes tamoxifen-resistant MCF7 breast cancer cells to tamoxifen, suggesting that PSAT1 contributes to tamoxifen resistance in MCF7 breast cancer cells. Additionally, combination treatment with YAP/TAZ or PSAT1 siRNA and tamoxifen significantly reduces mTORC1 activity and survivin expression in tamoxifen-resistant MCF7 breast cancer cells. These data suggest that targeting the YAP/TAZ-PSAT1 axis might sensitize tamoxifen-resistant MCF7 breast cancer cells by modulating the mTORC1 survivin pathway (139). In tamoxifen-sensitive MCF-7 cells, overexpression of PSAT1 decreases the inhibition of cell proliferation by 4-OHT. In contrast, silencing either PSAT1 or PHGDH results in a higher response to 4-OHT treatment in tamoxifen-resistant LCC9 cells. Combining a PHGDH inhibitor with 4-OHT also reduces proliferation of LCC9 cells. Overall, these findings suggest that ER+ BC is more likely to develop tamoxifen resistance due to overexpression of serine synthase enzymes. It is capable of being targeted as a novel combinatorial treatment option (140). Studies indicate that kinase inhibitors (KIs) and biguanide agents target various types of cancers in a synergistic and selective manner. The ability of KI/biguanides to effectively treat disease is determined by synthesis of nonessential amino acids (NEAAs). Aspartate, asparagine, and serine synthesis are controlled by the mTORC1/4E-BP axis in response to mRNA translation, and eliminating 4E-BP1/2 significantly reduces breast cancer sensitivity (141).
TAMR MCF-7 cells have a significantly higher level of cystine metabolism than MCF-7 cells, leading to increased glutathione and taurine synthesis. A higher amount of enzymes related to cysteine consumption is found in the TAMR MCF-7 cells, including methionine adenosyl transferase (MAT), cystathionine b-synthase (CbS), cysteine dioxygenase (CDO), and cysteine sulfinate decarboxylase (CSD). TAMR cells grown in medium lacking sulfur amino acids (SAAD) results in a decrease in cell viability (142). In breast cancer cells, CDO1 restoration leads to increased ROS levels, resulting in reduced viability and growth, as well as anthracycline sensitization. This demonstrates the importance of CDO1 inactivation in breast cancer and its potential as a biomarker and treatment target for overcoming anthracycline resistance (143).
Autophagy is activated due to depletion of the amino acid pool within drug-resistant cells. To cope with increasing amino acid requirements, TAMR cells promote import of aspartate and glutamate by expressing the SLC1A2 transporter on the cell surface (144). In comparison to normal cells, cancer cells require different amounts of metabolites when they proliferate. Researchers have studied the role of aspartate in cancer. In general, aspartate is necessary for biosynthesis of purine and pyrimidine nucleotides to generate AMP from inosine 5’-monophosphate via aspartate (145). The oxidative phosphorylation process provides electron acceptors for aspartate biosynthesis. A lack of electron acceptors prevents proliferative activity in cells. Exogenous aspartate supplementation can increase cell proliferation in cells with insufficient oxidative phosphorylation (146). One study found that ursolic acid increases nuclear accumulation of doxorubicin (Dox) by increasing the amount entering cells and decreasing levels of intracellular alanine, lactate, pyruvate, glucose, α-ketoglutarate, glutamate and various amino acids in the body to reverse MDR. According to the study, UA has potential as an adjuvant antitumor herbal medicine to resensitize cells with chemotherapeutic resistance (143). Consequently, studies have shown that levels of different amino acids and their metabolizing genes determine when treatment ends. Several amino acids are believed to contribute to acquired drug resistance, including serine, cysteine, aspartate, glutamate, and glutamine. Thus, we need to better understand the action of amino acids themselves and their precursors as oncogenic metabolites.
Targeting lipid metabolism is an emerging strategy to enhance the efficacy of anti-HER2 therapies in HER2-positive breast cancer (147). A large amount of lipid and cholesterol is required by cancer cells, which is met by either taking up more exogenous lipids and lipoproteins or promoting de novo lipogenesis and cholesterol biosynthesis. Lipid synthesis is crucial to satisfying the anabolic needs of cancer cells (148). As the key enzyme in the fatty acid synthesis pathway, acetyl-CoA carboxylase converts malonyl-CoA to the long-chain fatty acids palmitate and stearic acid (149). Acetyl-CoA carboxylase carboxylates acetyl-CoA to malonyl-CoA. FASN has been found to be upregulated in premalignant lesions as well as in most human cancers. TNBC tumor cells overexpress fatty acid synthase (FASN) (150), and the combination of FASN inhibitors and anti-EGFR signaling agents has significant antitumor effects in preclinical models of TNBC tumors. Overall, FASN activity may play an important role in doxorubicin resistance in TNBC (151).
Blocking FASN inhibits transcription of HER2 by upregulating PEA3, a transcriptional repressor of HER2. Trastuzumab inhibits the HER-2-induced upregulation of FASN expression and fatty synthesis triggered by HER-2 overexpression. In combination with a FASN inhibitor, trastuzumab resensitizes trastuzumab-resistant breast cancers by downregulating HER-2 expression (152, 153). Researchers have demonstrated that FASN regulates HER2 bidirectionally, which should increase sensitivity to trastuzumab (154). Additionally, the FASN inhibitor cerulenin exhibits synergistic effects with docetaxel in HER-2-overexpressing and docetaxel-resistant SK-BR-3 cells, suggesting involvement of FASN in HER-2-induced breast cancer (155). In addition, FASN blockade may promote synergistic chemosensitization of breast cancer cells to other treatments, such as paclitaxel, adriamycin, 5-FU, and vinorelbine (156–159). One study revealed that crosstalk between AKT and AMPK influences autophagy and metabolism (FAO). In turn, AKT activation, autophagy, and FAO are among the mechanisms promoting endoxifen resistance through AMPK (160).
During the cancer process, cancer cells experience oxidative stress, which enhances HIF-1α expression and stimulates TGF-α and Caveolin (Cav1) protein loss, downregulating TGF-α; moreover, stromal cells undergo autophagy and become CAFs (161). By undergoing glycolytic metabolism or the Warburg effect, these CAFs provide energy to nearby cancer cells (162). CAFs in the mammary gland are a major component of the tumor microenvironment, greatly contributing to progression of breast cancer. There is a link between drug resistance and enhanced growth, anti-apoptosis, and cell survival processes. However, recent evidence suggests that multidrug resistance in breast cancer cells may also be caused by autophagy (163, 164). (Figure 2)
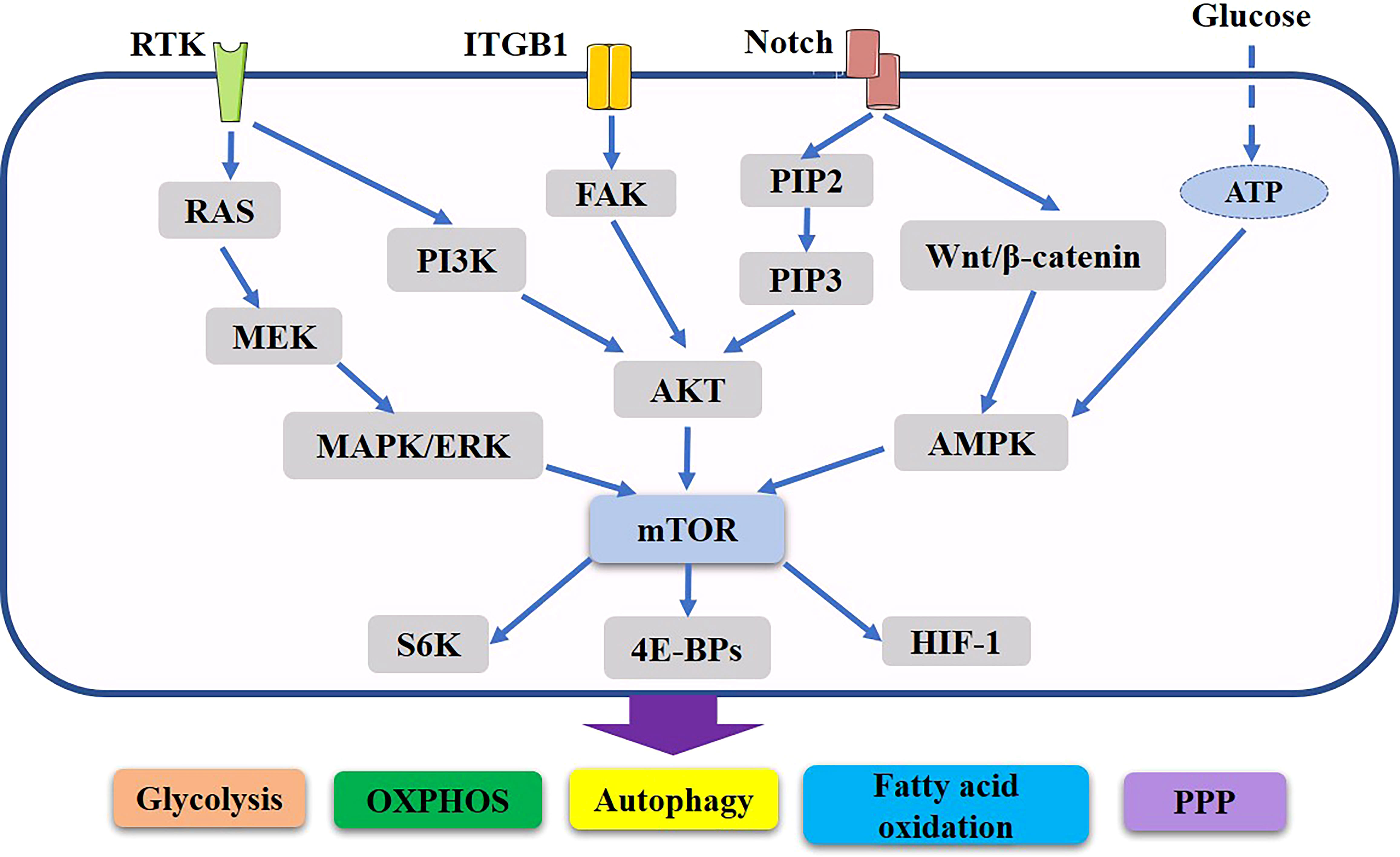
Figure 2 Important role of mTOR related pathway in metabolic reorganization of breast cancer. PI3K and Ras regulate Akt and ERK, which in turn induce changes in intermediate metabolism to promote anabolic processes. Potential Notch signaling crosstalk with other pathways in breast cancer. In addition, they also induce the activation of mTORC1, thus further supporting the rewiring of cellular metabolism and anabolic metabolism progress. Through various mechanisms Akt, ERK and mTORC1 stimulate aerobic glycolysis, lipid synthesis, the pentose phosphate, oxidative phosphorylation, autophagy, thus producing the major components necessary for cell growth and proliferation. These networks of signaling cascades, their interconnection and regulation allow the cells to maintain energetic balance and allow for the physiological adaptation to the ever-changing environment.
In autophagy, broken organelles such as mitochondria and unfolded proteins are scavenged by autophagy-related proteins, and autophagy-related proteins can also modulate key metabolic enzymes to regulate metabolic reprogramming. Cancer cells can survive by increasing glycolysis when autophagy activity is impaired (165). Researchers have found that autophagy promotes resistance to lapatinib, a HER2/EGFR tyrosine kinase inhibitor, in HER2-positive breast cancer (166) as well as the anti-HER2 monoclonal antibody trastuzumab (167). Enhanced autophagy activity has been demonstrated in doxorubicin- and 5-fluorouracil-resistant TNBC cells (168).
According to a previous study, GPR30-mediated autophagy can reduce apoptosis, thereby conferring resistance to TAMs in breast cancer cells (169). Additionally, CAFs may contribute to TAM-acquired resistance in breast cancer cells via the paracrine action of HMGB1, and it has been demonstrated that CAF-expressed GPR30 initiates this interaction. This interaction depends on transcriptional regulation through the GPR30/PI3K/AKT pathway in CAFs and MEK/ERK signaling-induced autophagy in ERα+ breast cancer cells, contributing to TAM resistance (170).
There is evidence that Beclin-1 (BECN1) acts as a suppressor of cancer and is involved in improving autophagy with lysosomal degradation; its expression levels are reduced in mammary carcinomas, particularly TNBC (171, 172). Autophagy-related BECN1 may therefore promote mammary carcinogenesis by negatively regulating metabolic rewiring. As a consequence, loss of BECN1 and autophagy may be linked to metabolic reprogramming and carcinogenesis in TNBC (173).
The proliferation-inducing ligand TNFSF13 (tumor necrosis factor superfamily member 13), which is the ligand for TNFRSF17/BCMA, was identified as an essential gene for B-cell development, autoimmunity, and cancer (174–176). By suppressing the Akt-mTOR pathway, TNFSF13 induces autophagy and therefore desensitizes TNBC cells to chemotherapy drugs such as paclitaxel, doxorubicin, and anthracyclines. Furthermore, TNFSF13-induced autophagy is a useful biomarker for predicting chemotherapeutic efficacy and a potential therapeutic target for reversing chemoresistance in TNBC (177).
In a recent study, it was found that acetylation of lysine 254 (K254) increases activity of GAPDH in response to glucose, which promotes the proliferation of tumor cells (178). The acetyltransferase PCAF and the deacetylase HDAC5 are also involved in reversibly regulating GAPDH acetylation (K254). In addition to increasing glycolysis, GAPDH promotes autophagy of damaged mitochondria, helping to protect cells against caspase-independent cell death (179).
3PO, a PFKFB3 inhibitor, reduces the size of tumors in HER2+ mice with breast cancer (50). One interesting finding (180) is that 3PO treatment-related induction of autophagy provides a mechanism that promotes survival. Consequently, the combination of an autophagy inhibitor and 3PO is recommended to enhance antitumor efficacy. Inhibiting LDHA causes apoptosis and suppresses autophagy in tamoxifen-resistant BC cells, reversing resistance to tamoxifen in MCF-7 and T47D cells (68).
Paclitaxel induction of ER stress in breast cancer cells leads to RNF5 association with and ubiquitination and degradation of SLC1A5/38A2. As a result, Gln uptake decreases, TCA cycle components are reduced, mTOR signaling decreases, and autophagy and cell death are increased (181).
By regulating chaperone-mediated autophagy (CMA), PKM2 K305 acetylation decreases enzyme activity and promotes lysosomal degradation. After acetylation of PKM2, it interacts with HSC70, a chaperone for CMA, and associates with lysosomes. Glycolytic intermediates accumulate in cells expressing the acetylation mimetic mutant K305Q, causing cell proliferation and tumor development. It appears that pyruvate kinase is regulated by lysine acetylation, and the link between lysine acetylation and CMA has been revealed (182).
In tumor cells, increased glucose consumption creates a hypoglycemic microenvironment, and these nutritional deficiencies are regarded by tumor cells as stress signals, which activate the stress signaling pathway to induce autophagy and escape apoptosis. Upstream of the metabolic pathway, several molecules activate the proliferation signaling pathway, promote tumor metabolism, increase glycolysis activity, and inhibit glycolytic enzyme activity while causing drug resistance as a result (25). Several pathways, such as the PI3K/Akt signaling and the Ras/ERK signaling, play a role in anabolic reprogramming (183) (Figure 2).
Additionally, MCF-7 cells resistant to tamoxifen exhibit enhanced HK2 and mTOR expression. A mechanism of resistance to tamoxifen occurs by increasing autophagy through inactivation of mTOR-S6K via HK2 (35). It was found that drugs with lower mTOR activity were more resistant. Cancer cells maintain aerobic glycolysis and HIF-1α stability despite the absence of hypoxia by the AKT/mTOR pathway or AMPK signaling pathway (184). When inhibition of HK2 suppresses the AKT/mTOR/HIF-1α axis, MCF-7 cells become resensitized to tamoxifen. Through downregulation of EGFR signaling, tamoxifen and dichloroacetate inhibits tamoxifen-resistant MCF-7-cell growth (85). Based on these studies, tamoxifen resistance in breast cancer may be related to AKT/mTOR/AMPK signaling (184, 185).
The PI3K/mTOR, Ras, MAPK and Src pathways are constitutively activated by oncogenic mutations in both normoxia and hypoxia, which increases the level of HIF-1α expression (186). Breast cancer is associated with upregulation of HER-2 levels and activation of PI3K/AKT, which leads to increased stability of HIF-1 via mTOR. Blocking the PI3K/Akt/mTOR pathway enhances the radiation response of breast cancer models in vitro (187), and phase II clinical trials have shown that CCI-779, an mTOR inhibitor, is an effective treatment for breast cancer (188). Several novel PI3K/Akt inhibitors have been developed in recent years, including SF1126, PI-103, and P529, increasing the effectiveness of radiation therapy and chemotherapy.
It is also believed that FASN is modulated by the PI3k-Akt and MAPK pathways (149, 189). FASN gene expression is increased under hypoxic conditions via Akt activation and subsequent SREBP-1 induction (190). In MCF7 cells, MAP kinase inhibition decreases transcription from FASN promoters as well as FASN expression (191). FASN may also be inhibited by rapamycin, an inhibitor of mTOR (192). Recent research indicates that regulation of FASN and HER2 occurs in a bidirectional manner through the HER2-FASN axis (193).
It is known that integrins play multiple functions, including adhesion, migration, and proliferation. They are controlled by the mTOR, HIF-1 and AMPK signaling pathways. In turn, signaling via β1-integrin/FAK/PI3K/AKT/mTOR also controls other glucose metabolic pathways (194). Blocking β1-integrin with an antibody before doxorubicin treatment enhances its cytotoxic activity (195). Furthermore, enhanced signaling between fibroblast growth factor (FGF) and fibroblast growth factor receptor (FGFR) is observed in BC cells that are resistant to doxorubicin. Downstream signaling is involved in a variety of oncogenic processes, including angiogenesis, resistance to therapy, and metastasis. FGFR plays a role in increased glycolysis and doxorubicin resistance, according to gene expression microarrays. Furthermore, blocking FGF-FGFR-ERK1/2 signaling with drug inhibitors targeting FGFR4 and ERK1/2 can resensitize drug-resistant phenotypes to adriamycin therapy (196). Although metformin is a hypoglycemic drug, its antiproliferative effects have been demonstrated in various breast cancer cell lines, and it was able to sensitize the multidrug resistance phenotype (197). When metformin is combined with doxorubicin, metformin acts via the IFN-α signaling pathway and induces cellular oxidative stress in resistant breast cancer cells, showing higher cytotoxicity than doxorubicin alone (198).
Notch signaling is an evolutionarily conserved pathway. Dysregulation of Notch signaling, for instance, by activating Notch receptor mutations, overexpressing Notch ligands and/or receptors, or overexpressing its target genes, contributes to increased proliferation, cell transformation, and drug resistance in various cancers, including breast cancer, multiple myeloma, prostate cancer, and T-cell acute lymphoblastic leukemia (199). It is known that HER2-driven cancers are aggressive; furthermore, 70% of patients are resistant to targeted treatment (200). Studies have shown that resistance may be caused by direct control of the ERBB2 gene by Notch1 (201), in turn, increased HER2 may activate Notch (202), possibly creating a positive feedback loop. Specifically, trastuzumab-resistant cells express higher levels of NOTCH1, JAG1, and their targets, including HEY1, DTX1, and HES5. However, a decrease in Notch1 expression using siRNA sensitizes these cells to trastuzumab (203).
Great progress in the metabolic reprogramming of tumor cells has occurred in recent decades. A number of molecules act synergistically upstream of the metabolic pathway to regulate the signaling pathway of cell proliferation and increase glycolysis activity and glycolytic enzyme generation and activation, ultimately leading to chemotherapeutic resistance. Hence, drug-resistant cells are endowed with adaptive, proliferative, and survival advantages because of altered metabolism (25). Nevertheless, the notion that resistant tumor cells rely more on mitochondrial OXPHOS and respiration and less on glycolysis challenges the idea that tumors primarily invoke glycolytic metabolism and possess defective mitochondria, as originally proposed by Warburg (204). Metabolic plasticity has been observed in some tumor cells, suggesting a transformation from glycolysis to mitochondrial OXPHOS to produce vast amounts of energy (205).
One study revealed that miRNA-211 controls transcription of PDK4 and that inhibiting PDK4 by miRNA-211 causes BC MDA cells to shift from glycolytic to OXPHOS dominance (77). RNA sequencing has also been applied to analyze differentially expressed genes in tamoxifen-resistant cells. Gene expression patterns suggest dysfunctional mitochondria and translate to OXPHOS (206). Metastatic cancers resistant to hormonal therapies express high levels of CD133 and IL6 and low levels of ER. CD133hi/ERlo also reduces mitochondrial OXPHOS (207). Additionally, two other studies have demonstrate the importance of OXPHOS and highlight the metabolic plasticity of TNBC through enhanced susceptibility to fatty acid oxidation inhibitors (208, 209).
There is growing evidence that OXPHOS participates in tumorigenesis and chemotherapeutic resistance (210). Breast cancer research has shown that OXPHOS supplies most of the ATP required (211). Moreover, OXPHOS influences tumor treatment in a number of ways. Due to the large amount of ATP produced, it stimulates the activity of some transporters, including drug transporters. ABC transporters in breast cancer cells use the ATP produced from OXPHOS to promote efflux of DOX and onset of chemotherapy resistance (205). OXPHOS-induced drug resistance is also associated with tumor stem cells. Mitochondria OXPHOS can cause tumor stem cells to spread and may lead to tumor cell resistance (212). Increasing STAT3 enhances mitochondrial complex I and II activity and thus OXPHOS in mitochondria. Activation of OXPHOS is a mechanism for resistance to TKI treatment (213). In TNBC stem cells, MYC and MCL1 are often overexpressed together, acting as enhancers of mitochondria. They enhance mitochondrial OXPHOS and upregulate HIF-1α expression in synergy; this enhanced mitochondrial OXPHOS promotes BCSC enrichment in TNBC, leading to an increase in chemoresistance. HIF-1α inhibition decreases BCSC enrichment, enhancing chemosensitivity in TNBC cells (212).
Rather than an overly glycolysis-dependent phenotype, recent research suggests that cancer cells can achieve mixed phenotypes of glycolysis and OXPHOS in which ATP production is a result of both glycolysis and oxidative phosphorylation and is critical to supporting the physiological activity of individual cells and thus influencing aggressiveness and therapy resistance (214, 215).
Furthermore, stromal cells interact with cancer cells, promoting tumor metabolism. Stromal cells are induced by cancer cells to invoke aerobic glycolysis, and metabolites accumulated by stromal cells are utilized by cancer cells for the mitochondrial OXPHOS pathway. To more effectively fight cancer, both aerobic glycolysis and mitochondrial metabolism should be targeted (216).
Lactate production and secretion increase as a result of aerobic glycolysis, which eventually results in acidification of the cancer microenvironment. Cancer progression is enhanced by release of lactate into the tumor microenvironment (35).
Monocarboxylate transporter (MCT) is a lactate efflux transporter that is necessary for maintaining pH and regulating glycolysis. MCTs belong to the solute carrier (SLC) family of 14 members (217). MCT-1 is the key element facilitating lactate import, and MCT-4 is a lactate exporter (218). These proteins are present almost ubiquitously in the body, and they are particularly upregulated in cancer cells and CAFs, where lactate is generated and transported. As a result, their overexpression can be used as a biomarker for various types and subtypes of cancer (219). Indeed, there is an association between drug resistance and abnormal expression of the MCT family. For example, MCT1 expression correlates with aggressiveness, recurrence, decreased survival, and tumorigenicity in breast cancer (220). It has been reported that high MCT1 expression causes increases in intratumoral lactic acid, which is associated with poor prognosis (221). MCT1 is a major transporter that assists 3-bromopyruvate (3-BrPA) (222), and MCT1 overexpression in cancer cells increases tumor xenograft sensitivity to 3-BrPA. The study by Morais-Santos et al. found that various subtypes of breast cancer are sensitive to MCT1 inhibitors in different ways (223). A high level of MCT1 expression is observed in TNBC (224). As a direct target of miR-342-3p, MCT1 is increased when miR-342-3p is silenced, enhancing the glycolytic profile of TNBC cells and rendering them more aggressive (225).
In breast cancer lesions, MCT4 is associated with immune cell infiltration, PKM2 and HK3 expression, and glycolytic rate-limiting enzymes. Additionally, MCT4 may play an important role in maintaining the tumor immune microenvironment through metabolic reprogramming. Therefore, these enzymes of the glycolysis pathway (MCT4, PKM2, and HK3) may serve as new targets for modulating the tumor immune microenvironment and enhancing immunotherapy effectiveness (226). MCT4 downregulation overcomes resistance to antiangiogenic therapy (227). Based on studies using xenograft models, MCT4, as a transporter of monocarboxylate across cell membranes, appears to be responsible for secretion of lactate by breast tumor cells. After being secreted, lactate is transported into endothelial cells expressing MCT-1, which triggers the autocrine NF-κB/IL-8 pathway. As a result, lactate signaling induces cell migration and tube formation in endothelial cells, promoting tumor artery morphogenesis and perfusion.
Cancer research has recently concentrated on the dysregulation of metabolism within cancer cells; metabolic reprogramming is now considered one of the hallmarks of cancer. Increasing evidence suggests that dysregulated cellular metabolism may contribute to drug resistance in cancer patients. According to the Warburg effect, cancer cells invoke glycolysis irrespective of whether they are aerobic or anaerobic, meaning that mitochondrial dysfunction is present (228). Metabolic reprogramming, includes glucose metabolism, fatty acid synthesis, and amino acid metabolism. The fact that metabolic reprogramming occurs in resistant cells and may occur in the majority of tumors has important therapeutic implications and shows that metabolic vulnerabilities might be exploited therapeutically.
In addition, the emergence of the “reverse Warburg effect” indicates that lactic acid serves as a material that provides energy; it can be converted into pyruvate, resulting in stimulated mitochondria and OXPHOS in neighboring cells, and mitochondria are important in many aspects of cellular metabolism (229). Recently, several studies have demonstrated that tumor cells also display metabolic plasticity. When tumor cells are surrounded by ample oxygen or when the external environment changes, glycolysis can moderately transform into OXPHOS. This review, by unveiling key regulatory events, further contributes to our knowledge of the relationship between breast cancer metabolism and drug resistance. To target cancer metabolism in the context of treatment, it is vital to alter the metabolic characteristics of tumorigenesis and the plasticity of cancer cells to switch between different metabolic pathways, survival, and apoptosis inhibition. There are several agents that target specific enzymes in the metabolic pathways of breast cancer, including HK, PK, PDC, GLUTs and lactate, in addition to that targeting metabolism-related molecular pathways and genes in the tumor microenvironment. And potential molecular mechanisms and new methods of treatment have been studied or hypothesized. Several of these agents have been shown to improve the efficacy of current treatments and resensitize resistant cancer cells and have now entered clinical trials. Combining strategies that modulate glycolytic and mitochondrial pathways may be an effective way to eliminate drug-resistant cells.
Overall, proteomic and metabolomic analyses of tumor metabolism provide physicians with insight into therapeutic targets, leading to successful clinical translation. Our hope is that targeting tumor metabolic pathways will play an important role in treating breast cancer in the near future.
LL reviewed the literature, collected data and wrote the manuscript. SY and YZ revised the manuscript. XZ and SL rechecked the manuscript. XT and DD designed and revised themanuscript. All authors contributed to the article and approved the submitted version.
This work was financially supported by grants from the National Natural Science Foundation of China (No. 62072070).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Cao W, Chen HD, Yu YW, Li N, Chen WQ. Changing profiles of cancer burden worldwide and in China: A secondary analysis of the global cancer statistics 2020. Chin Med J (2021) 134:783–91. doi: 10.1097/Cm9.0000000000001474
2. Global cancer observatory: Cancer today. Lyon, France: International Agency for Research on Cancer (2020). Available at: http://gco.iarc.fr/2020.
3. The Institute for Health Metrics and Evaluation. Global burden of disease. (2020). Available at: http://www.healthdata.org/gbd/20192020.
4. Dai XF, Xiang LJ, Li T, Bai ZH. Cancer hallmarks, biomarkers and breast cancer molecular subtypes. J Cancer (2016) 7:1281–94. doi: 10.7150/jca.13141
5. Gianni L, Pienkowski T, Im YH, Tseng LM, Liu MC, Lluch A, et al. 5-year analysis of neoadjuvant pertuzumab and trastuzumab in patients with locally advanced, inflammatory, or early-stage Her2-positive breast cancer (Neosphere): A multicentre, open-label, phase 2 randomised trial. Lancet Oncol (2016) 17:791–800. doi: 10.1016/S1470-2045(16)00163-7
6. Howie LJ, Scher NS, Amiri-Kordestani L, Zhang LJ, King-Kallimanis BL, Choudhry Y, et al. Fda approval summary: Pertuzumab for adjuvant treatment of Her2-positive early breast cancer. Clin Cancer Res (2019) 25:2949–55. doi: 10.1158/1078-0432.Ccr-18-3003
7. Hudis CA. Drug therapy: Trastuzumab - mechanism of action and use in clinical practice. New Engl J Med (2007) 357:39–51. doi: 10.1056/NEJMra043186
8. Jones J, Takeda A, Picot J, Von Keyserlingk C, Clegg A. Lapatinib for the treatment of Her2-overexpressing breast cancer. Health Technol Assess (2009) 13:1–6. doi: 10.3310/hta13suppl3/01
9. Echavarria I, López-Tarruella S, Márquez-Rodas I, Jerez Y, Martin M. Neratinib for the treatment of Her2-positive early stage breast cancer. Expert Rev Anticancer Ther (2017) 17:669–79. doi: 10.1080/14737140.2017.1338954
10. Singh H, Walker AJ, Amiri-Kordestani L, Cheng J, Tang SH, Balcazar P, et al. Us food and drug administration approval: Neratinib for the extended adjuvant treatment of early-stage Her2-positive breast cancer. Clin Cancer Res (2018) 24:3486–91. doi: 10.1158/1078-0432.Ccr-17-3628
11. Martin M, Holmes FA, Ejlertsen B, Delaloge S, Moy B, Iwata H, et al. Neratinib after trastuzumab-based adjuvant therapy in Her2-positive breast cancer (Extenet): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol (2017) 18:1688–700. doi: 10.1016/S1470-2045(17)30717-9
12. Hatzis C, Symmans WF, Zhang Y, Gould RE, Moulder SL, Hunt KK, et al. Relationship between complete pathologic response to neoadjuvant chemotherapy and survival in triple-negative breast cancer. Clin Cancer Res (2015) 22:26–33. doi: 10.1158/1078-0432.CCR-14-3304
13. Ishikawa T, Shimizu D, Yamada A, Sasaki T, Morita S, Tanabe M, et al. Impacts and predictors of cytotoxic anticancer agents in different breast cancer subtypes. Oncol Res (2012) 20:71–9. doi: 10.3727/096504012X13473664562565
14. Pal SK, Bh C, Pegram M. Triple negative breast cancer: Unmet medical needs. BMC Med (2015) 13:303. doi: 10.1007/s10549-010-1293-1
15. Prat A, Fan C, Fernández A, Hoadley KA, Martinello R, Vidal M, et al. Response and survival of breast cancer intrinsic subtypes following multi-agent neoadjuvant chemotherapy. BMC Med (2015) 13:303 doi: 10.1186/s12916-015-0540-z
16. Cheng TL, Sudderth J, Yang CD, Mullen AR, Jin ES, Mates JM, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci USA (2011) 108:8674–9. doi: 10.1073/pnas.1016627108
17. Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature (2012) 491:364–73. doi: 10.1038/nature11706
18. Tennant DA, Duran RV, Boulahbel H, Gottlieb E. Metabolic transformation in cancer. Carcinogenesis (2009) 30:1269–80. doi: 10.1093/carcin/bgp070
19. Fu Y, Liu S, Yin S, Niu W, Xiong W, Tan M, et al. The reverse warburg effect is likely to be an achilles' heel of cancer that can be exploited for cancer therapy. Oncotarget. (2017) 8:57813–25. doi: 10.18632/oncotarget.18175
20. Catanzaro D, Gaude E, Orso G, Giordano C, Guzzo G, Rasola A, et al. Inhibition of glucose-6-Phosphate dehydrogenase sensitizes cisplatin-resistant cells to death. Oncotarget. (2015) 6:30102–14. doi: 10.18632/oncotarget.4945
21. Havas KM, Milchevskaya V, Radic K, Alladin A, Kafkia E, Garcia M, et al. Metabolic shifts in residual breast cancer drive tumor recurrence. J Clin Invest. (2017) 127:2091–105. doi: 10.1172/JCI89914
22. Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, et al. Environment dictates dependence on mitochondrial complex I for nad+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab (2016) 24:716–27. doi: 10.1016/j.cmet.2016.09.006
23. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
24. Cairns RA, Is H, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11:85–95. doi: 10.1038/nrc2981
25. Ma L, Zong X. Metabolic symbiosis in chemoresistance: Refocusing the role of aerobic glycolysis. Front Oncol (2020) 10:5. doi: 10.3389/fonc.2020.00005
26. Vander Heiden MG, Cantley Lc, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi: 10.1126/science.1160809
27. Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, et al. The reverse warburg effect: Glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle (2010) 9:1960–71. doi: 10.4161/cc.9.10.11601
28. Nakajima EC, Van Houten B. Metabolic symbiosis in cancer: Refocusing the warburg lens. Mol Carcinog (2013) 52:329–37. doi: 10.1002/mc.21863
29. Bose S, Le A. Glucose metabolism in cancer. Trans Res Breast Cancer (2018) 1063:3–12. doi: 10.1007/978-3-319-77736-8_1
30. Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol (2006) 18:598–608. doi: 10.1016/j.ceb.2006.10.005
31. Varghese E, Samuel S, Líšková A, Samec M, Kubatka P, Büsselberg D. Targeting glucose metabolism to overcome resistance to anticancer chemotherapy in breast cancer. - Cancers (Basel) (2020) 12:2252. doi: 10.3390/Cancers12082252[Doi]Lid-2252
32. Patra KC, Wang Q, Bhaskar PT, Miller L, Wang ZB, Wheaton W, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer (Vol 24, pg 213, 2013). Cancer Cell (2013) 24:399–9. doi: 10.1016/j.ccr.2013.08.029
33. Shinohara Y, Yamamoto K, Kogure K, Ichihara J, Terada H. Steady state transcript levels of the type ii hexokinase and type 1 glucose transporter in human tumor cell lines. Cancer Lett (1994) 82:27–32. doi: 10.1016/0304-3835(94)90142-2
34. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci (2014) 39:347–54. doi: 10.1016/j.tibs.2014.06.005
35. Liu XC, Miao WL, Huang M, Li L, Dai XX, Wang YS. Elevated hexokinase ii expression confers acquired resistance to 4-hydroxytamoxifen in breast cancer cells. Mol Cell Proteomics. (2019) 18:2273–84. doi: 10.1074/mcp.RA119.001576
36. Geng C, Li J, Ding F, Wu G, Yang Q, Sun Y, et al. Curcumin suppresses 4-hydroxytamoxifen resistance in breast cancer cells by targeting Slug/Hexokinase 2 pathway. Biochem Biophys Res Commun (2016) 473:147–53. doi: 10.1016/j.bbrc.2016.03.067
37. Krasnov GS, Dmitriev AA, Lakunina VA, Kirpiy AA, Kudryavtseva AV. Targeting vdac-bound hexokinase ii: A promising approach for concomitant anti-cancer therapy. Expert Opin Ther Targets (2013) 17:1221–33. doi: 10.1517/14728222.2013.833607
38. Yang TT, Ren CN, Qiao PY, Han X, Wang L, Lv SJ, et al. Pim2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer (Vol 37, pg 5997, 2019). Oncogene. (2020) 39:720–1. doi: 10.1038/s41388-019-0982-4
39. Zhao YH, Liu H, Liu ZX, Ding Y, Ledoux SP, Wilson GL, et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res (2011) 71:4585–97. doi: 10.1158/0008-5472.Can-11-0127
40. Wang J, Tao M, Wang T, Wang Z, Xiao J, Ding S, et al. [Knockdown of hexokinase 2 (Hk2) inhibits breast cancer cell proliferation and reduces their resistance to fluorouracil]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. (2021) 37:722–7.
41. Wang Q, Zheng J, Zou JX, Xu J, Han F, Xiang S, et al. S-adenosylhomocysteine (Adohcy)-dependent methyltransferase inhibitor dznep overcomes breast cancer tamoxifen resistance Via induction of Nsd2 degradation and suppression of Nsd2-driven redox homeostasis. Chem Biol Interact (2020) 317:108965. doi: 10.1016/j.cbi.2020.108965
42. Zhu T, Zheng J, Zhuo W, Pan P, Li M, Zhang W, et al. Etv4 promotes breast cancer cell stemness by activating glycolysis and Cxcr4-mediated sonic hedgehog signaling. Cell Death Discovery (2021) 7:126. doi: 10.1038/s41420-021-00508-x
43. Shi L, Pan H, Liu Z, Xie J, Han W. Roles of Pfkfb3 in cancer. Signal Transduct Target Ther (2017) 2:17044. doi: 10.1038/sigtrans.2017.44
44. Galindo CM, Oliveira Ganzella FA, Klassen G, Souza Ramos EA, Acco A. Nuances of Pfkfb3 signaling in breast cancer. Clin Breast Cancer (2022) 22:e604–e614. doi: 10.1016/j.clbc.2022.01.002
45. Sengupta S, Sevigny CM, Liu X, Lu J, Clarke R. Abstract 907: Targeting glycolysis enzyme, Pfkfb3, in endocrine therapy resistant breast cancers. Cancer Res (2018) 78:907–7. doi: 10.1158/1538-7445.AM2018-907
46. Truong TH, Benner EA, Hagen KM, Temiz NA, Kerkvliet CP, Wang Y, et al. Pelp1/Src-3-Dependent regulation of metabolic pfkfb kinases drives therapy resistant er(+) breast cancer. Oncogene. (2021) 40:4384–97. doi: 10.1038/s41388-021-01871-w
47. Lu C, Qiao P, Sun Y, Ren C, Yu Z. Positive regulation of Pfkfb3 by Pim2 promotes glycolysis and paclitaxel resistance in breast cancer. Clin Transl Med (2021) 11:e400. doi: 10.1002/ctm2.400
48. Ge X, Cao Z, Gu Y, Wang F, Li J, Han M, et al. Pfkfb3 potentially contributes to paclitaxel resistance in breast cancer cells through Tlr4 activation by stimulating lactate production. Cell Mol Biol (2016) 62:119–25. doi: 10.14715/cmb/2016.62.6.22
49. Jia WZ, Zhao XP, Zhao L, Yan H, Li JJ, Yang H, et al. Non-canonical roles of Pfkfb3 in regulation of cell cycle through binding to Cdk4. Oncogene. (2018) 37:1685–98. doi: 10.1038/s41388-017-0072-4
50. O'neal J, Clem A, Reynolds L, Dougherty S, Imbert-Fernandez Y, Telang S, et al. Inhibition of 6-Phosphofructo-2-Kinase (Pfkfb3) suppresses glucose metabolism and the growth of Her2+ breast cancer. Breast Cancer Res Treat (2016) 160:29–40. doi: 10.1007/s10549-016-3968-8
51. Christofk HR, Heiden M, Harris MH, Ramanathan A, Gerszten RE, Ru W, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. (2008) 452:230–3. doi: 10.1038/nature06734
52. Dong G, Mao Q, Xia W, Xu Y, Wang J, Xu L, et al. Pkm2 and cancer: The function of Pkm2 beyond glycolysis. Oncol Lett (2016) 11:1980–6. doi: 10.3892/ol.2016.4168
53. Ge X, Zhao Y, Dong LL, Seng JJ, Zhang XY, Dou DW. Nampt regulates Pkm2 nuclear location through 14-3-3 zeta: Conferring resistance to tamoxifen in breast cancer. J Cell Physiol (2019) 234:23409–20. doi: 10.1002/jcp.28910
54. Su QL, Luo SP, Tan QH, Deng J, Zhou SC, Peng M, et al. The role of pyruvate kinase M2 in anticancer therapeutic treatments. Oncol Letters. (2019) 18:5663–72. doi: 10.3892/ol.2019.10948
55. Ji FH, Guo BQ, Wang NA, Zhong CL, Huang LY, Huang YX, et al. Pyruvate kinase M2 interacts with mammalian sterile 20-like kinase 1 and inhibits tamoxifen-induced apoptosis in human breast cancer cells. Tumor Biol (2017) 39:1–11. doi: 10.1177/1010428317692251
56. Qian YJ, Bi LN, Yang YX, Wang D. Effect of pyruvate kinase M2-regulating aerobic glycolysis on chemotherapy resistance of estrogen receptor-positive breast cancer. Anti-Cancer Drugs (2018) 29:616–27. doi: 10.1097/Cad.0000000000000624
57. Lebioda L, Stec B. Crystal structure of enolase indicates that enolase and pyruvate kinase evolved from a common ancestor. Nature (1988) 333:683–6. doi: 10.1038/333683a0
58. Cancemi P, Buttacavoli M, Roz E, Feo S. Expression of alpha-enolase (Eno1), myc promoter-binding protein-1 (Mbp-1) and matrix metalloproteinases (Mmp-2 and mmp-9) reflect the nature and aggressiveness of breast tumors. Int J Mol Sci (2019) 20:3952. doi: 10.3390/ijms20163952
59. Chuthapisith S, Layfield R, Kerr ID, Hughes C, Eremin O. Proteomic profiling of mcf-7 breast cancer cells with chemoresistance to different types of anti-cancer drugs. Int J Oncol (2007) 30:1545–51. doi: 10.3892/ijo.30.6.1545
60. Tu SH, Chang CC, Chen CS, Tam KW, Wang YJ, Lee CH, et al. Increased expression of enolase alpha in human breast cancer confers tamoxifen resistance in human breast cancer cells. Breast Cancer Res Treat (2010) 121:539–53. doi: 10.1007/s10549-009-0492-0
61. Subramanian A, Miller DM. Structural analysis of alpha-enolase. mapping the functional domains involved in down-regulation of the c-myc protooncogene. J Biol Chem (2000) 275:5958–65. doi: 10.1074/jbc.275.8.5958
62. Ray RB. Induction of cell death in murine fibroblasts by a c-myc promoter binding protein. Cell Growth Differ (1995) 6:1089–96.
63. Mizukami Y, Iwamatsu A, Aki T, Kimura M, Nakamura K, Nao T, et al. Erk1/2 regulates intracellular atp levels through alpha-enolase expression in cardiomyocytes exposed to ischemic hypoxia and reoxygenation. J Biol Chem (2004) 279:50120–31. doi: 10.1074/jbc.M402299200
64. Abreu RD, Penalva LO, Marcotte EM, Vogel C. Global signatures of protein and mrna expression levels. Mol Biosystems. (2009) 5:1512–26. doi: 10.1039/b908315d
65. Perconti G, Maranto C, Romancino DP, Rubino P, Feo S, Bongiovanni A, et al. Pro-invasive stimuli and the interacting protein Hsp70 favour the route of alpha-enolase to the cell surface. Sci Rep (2017) 7:3841. doi: 10.1038/s41598-017-04185-8
66. Capello M, Ferri-Borgogno S, Riganti C, Chattaragada MS, Principe M, Roux C, et al. Targeting the warburg effect in cancer cells through Eno1 knockdown rescues oxidative phosphorylation and induces growth arrest. Oncotarget. (2016) 7:5598–612. doi: 10.18632/oncotarget.6798
67. Fantin VR, St-Pierre J, Leder P. Attenuation of ldh-a expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell (2006) 9:425–34. doi: 10.1016/j.ccr.2006.04.023
68. Das CK, Parekh A, Parida PK, Bhutia SK, Mandal M. Lactate dehydrogenase a regulates autophagy and tamoxifen resistance in breast cancer. Biochim Et Biophys Acta-Molecular Cell Res (2019) 1866:1004–18. doi: 10.1016/j.bbamcr.2019.03.004
69. Hamadneh L, Al-Lakkis L, Alhusban AA, Tarawneh S, Abu-Irmaileh B, Albustanji S, et al. Changes in lactate production, lactate dehydrogenase genes expression and DNA methylation in response to tamoxifen resistance development in mcf-7 cell line. Genes. (2021) 12:777. doi: 10.3390/genes12050777
70. Yang T, Fu Z, Zhang Y, Wang M, Mao C, Ge W. Serum proteomics analysis of candidate predictive biomarker panel for the diagnosis of trastuzumab-based therapy resistant breast cancer. BioMed Pharmacother (2020) 129:110465. doi: 10.1016/j.biopha.2020.110465
71. Zhao YH, Zhou M, Liu H, Ding Y, Khong HT, Yu D, et al. Upregulation of lactate dehydrogenase a by Erbb2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. (2009) 28:3689–701. doi: 10.1038/onc.2009.229
72. Thongon N, Zucal C, D'agostino VG, Tebaldi T, Ravera S, Zamporlini F, et al. Cancer cell metabolic plasticity allows resistance to nampt inhibition but invariably induces dependence on ldha. Cancer Metab (2018) 6:1. doi: 10.1186/s40170-018-0174-7
73. Mittal L, Aryal UK, Camarillo IG, Ferreira RM, Sundararajan R. Quantitative proteomic analysis of enhanced cellular effects of electrochemotherapy with cisplatin in triple-negative breast cancer cells. Sci Rep (2019) 9:13916. doi: 10.1038/s41598-019-50048-9
74. Hou L, Zhao Y, Song GQ, Ma YH, Jin XH, Jin SL, et al. Interfering cellular lactate homeostasis overcomes taxol resistance of breast cancer cells through the microrna-124-Mediated lactate transporter (Mct1) inhibition. Cancer Cell Int (2019) 19:193. doi: 10.1186/s12935-019-0904-0
75. Zhou M, Zhao YH, Ding Y, Liu H, Liu ZX, Fodstad O, et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-a re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer. (2010) 9:33. doi: 10.1186/1476-4598-9-33
76. Sugden MC, Holness MJ. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem (2006) 112:139–49. doi: 10.1080/13813450600935263
77. Guda MR, Asuthkar S, Labak CM, Tsung AJ, Alexandrov I, Mackenzie MJ, et al. Targeting Pdk4 inhibits breast cancer metabolism. Am J Cancer Res (2018) 8:1725–38.
78. Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem (2008) 283:28106–14. doi: 10.1074/jbc.M803508200
79. Sradhanjali S, Reddy MM. Inhibition of pyruvate dehydrogenase kinase as a therapeutic strategy against cancer. Curr Topics Medicinal Chem (2018) 18:444–53. doi: 10.2174/1568026618666180523105756
80. Saunier E, Benelli C, Bortoli S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int J Cancer (2016) 138:809–17. doi: 10.1002/ijc.29564
81. Iorns E, Lord CJ, Ashworth A. Parallel rnai and compound screens identify the Pdk1 pathway as a target for tamoxifen sensitization. Biochem J (2009) 417:361–70. doi: 10.1042/Bj20081682
82. Gordan JD, Thompson CB, Simon MC. Hif and c-myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell (2007) 12:108–13. doi: 10.1016/j.ccr.2007.07.006
83. Walter W, Thomalla J, Bruhn J, Fagan DH, Zehowski C, Yee D, et al. Altered regulation of Pdk4 expression promotes antiestrogen resistance in human breast cancer cells. Springerplus. (2015) 4:689. doi: 10.1186/s40064-015-1444-2
84. Gudi R, Bowker-Kinley MM, Kedishvili NY, Zhao Y, Popov KM. Diversity of the pyruvate dehydrogenase kinase gene family in humans. J Biol Chem (1995) 270:28989–94. doi: 10.1074/jbc.270.48.28989
85. Woo SH, Seo SK, Park Y, Kim EK, Seong MK, Kim HA, et al. Dichloroacetate potentiates tamoxifen-induced cell death in breast cancer cells Via downregulation of the epidermal growth factor receptor. Oncotarget. (2016) 7:59809–19. doi: 10.18632/oncotarget.10999
86. Martinez VG, Crown J, Porter RK, O'driscoll L. Neuromedin U alters bioenergetics and expands the cancer stem cell phenotype in Her2-positive breast cancer. Int J Cancer (2017) 140:2771–84. doi: 10.1002/ijc.30705
87. Tseng PH, Wang YC, Weng SC, Weng JR, Chen CS, Brueggemeier RW, et al. Overcoming trastuzumab resistance in Her2-overexpressing breast cancer cells by using a novel celecoxib-derived phosphoinositide-dependent kinase-1 inhibitor. Mol Pharmacol (2006) 70:1534–41. doi: 10.1124/mol.106.023911
88. Weng SC, Kashida Y, Kulp SK, Wang D, Brueggemeier RW, Shapiro CL, et al. Sensitizing estrogen receptor-negative breast cancer cells to tamoxifen with osu-03012, a novel celecoxib-derived phosphoinositide-dependent protein kinase-1/Akt signaling inhibitor. Mol Cancer Ther (2008) 7:800–8. doi: 10.1158/1535-7163.Mct-07-0434
89. Barron CC, Bilan PJ, Tsakiridis T, Tsiani E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism-Clinical Exp (2016) 65:124–39. doi: 10.1016/j.metabol.2015.10.007
90. Sun XY, Wang MZ, Wang MS, Yu XT, Guo JY, Sun T, et al. Metabolic reprogramming in triple-negative breast cancer. Front Oncol (2020) 10:428. doi: 10.3389/fonc.2020.00428
91. Jiang T, Zhou ML, Fan J. Inhibition of glut-1 expression and the Pi3k/Akt pathway to enhance the chemosensitivity of laryngeal carcinoma cells in vitro. Onco Targets Ther (2018) 11:7865–72. doi: 10.2147/OTT.S176818
92. Sun MQ, Zhao S, Duan YC, Ma YM, Wang YC, Ji HF, et al. Glut1 participates in tamoxifen resistance in breast cancer cells through autophagy regulation. Naunyn-Schmiedebergs Arch Pharmacol (2021) 394:205–16. doi: 10.1007/s00210-020-01893-3
93. Chan CH, Li CF, Yang WL, Gao Y, Lee SW, Feng ZZ, et al. The Skp2-scf E3 ligase regulates akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. (2012) 149:1098–111. doi: 10.1016/j.cell.2012.02.065
94. Tilekar K, Upadhyay N, Iancu CV, Pokrovsky V, Choe JY, Ramaa CS. Power of two: combination of therapeutic approaches involving glucose transporter (GLUT) inhibitors to combat cancer. Biochim Biophys Acta Rev Cancer (2020) 1874:188457. doi: 10.1016/j.bbcan.2020.188457
95. Cretella D, Fumarola C, Bonelli M, Alfieri R, La Monica S, Digiacomo G, et al. Pre-treatment with the Cdk4/6 inhibitor palbociclib improves the efficacy of paclitaxel in tnbc cells. Sci Rep (2019) 9:13014. doi: 10.1038/s41598-019-49484-4
96. Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H, et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Ther (2012) 11:1672–82. doi: 10.1158/1535-7163.MCT-12-0131
97. Shi YL, Liu SN, Ahmad S, Gao QZ. Targeting key transporters in tumor glycolysis as a novel anticancer strate. Curr Topics Medicinal Chem (2018) 18:454–66. doi: 10.2174/1568026618666180523105234
98. Oh S, Kim H, Nam K, Shin I. Silencing of Glut1 induces chemoresistance Via modulation of Akt/Gsk-3beta/Beta-Catenin/Survivin signaling pathway in breast cancer cells. Arch Biochem Biophys (2017) 636:110–22. doi: 10.1016/j.abb.2017.08.009
99. Gerner EW, Meyskens FL. Polyamines and cancer: Old molecules, new understanding. Nat Rev Cancer (2004) 4:781–92. doi: 10.1038/nrc1454
100. Wise DR, Thompson CB. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem Sci (2010) 35:427–33. doi: 10.1016/j.tibs.2010.05.003
101. Yang LF, Venneti S, Nagrath D. Glutaminolysis: A hallmark of cancer metabolism. Annu Rev Biomed Engineering Vol 19 (2017) 19:163–94. doi: 10.1146/annurev-bioeng-071516-044546
102. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16:773–3. doi: 10.1038/nrc.2016.131
103. Lampa M, Arlt H, He T, Ospina B, Reeves J, Zhang BL, et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mtor inhibition. PLoS One (2017) 12:e0185092. doi: 10.1371/journal.pone.0185092
104. Ko YH, Lin Z, Flomenberg N, Pestell RG, Howell A, Sotgia F, et al. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells implications for preventing chemotherapy resistance. Cancer Biol Ther (2011) 12:1085–97. doi: 10.4161/cbt.12.12.18671
105. Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, et al. Tigar, a P53-inducible regulator of glycolysis and apoptosis. Cell. (2006) 126:107–20. doi: 10.1016/j.cell.2006.05.036
106. Liu JX, Yue W, Chen HY. The correlation between autophagy and tamoxifen resistance in breast cancer. Int J Clin Exp Pathol (2019) 12:2066–74.
107. Wasylishen AR, Penn LZ. Myc: The beauty and the beast. Genes Cancer. (2010) 1:532–41. doi: 10.1177/1947601910378024
108. Miller TW, Balko JM, Ghazoui Z, Dunbier A, Anderson H, Dowsett M, et al. A gene expression signature from human breast cancer cells with acquired hormone independence identifies myc as a mediator of antiestrogen resistance. Clin Cancer Res (2011) 17:2024–34. doi: 10.1158/1078-0432.Ccr-10-2567
109. Shajahan-Haq AN, Cook KL, Schwartz-Roberts JL, Eltayeb AE, Demas DM, Warri AM, et al. Myc regulates the unfolded protein response and glucose and glutamine uptake in endocrine resistant breast cancer. Mol Cancer. (2014) 13:239. doi: 10.1186/1476-4598-13-239
110. Venditti M, Iwasiow B, Orr FW, Shiu R. C-myc gene expression alone is sufficient to confer resistance to antiestrogen in human breast cancer cells. Int J Cancer (2002) 99:35–42. doi: 10.1002/ijc.10269
111. Chen ZK, Wang YZ, Warden C, Chen SA. Cross-talk between er and Her2 regulates c-Myc-Mediated glutamine metabolism in aromatase inhibitor resistant breast cancer cells. J Steroid Biochem Mol Biol (2015) 149:118–27. doi: 10.1016/j.jsbmb.2015.02.004
112. Deblois G, Smith HW, Tam IS, Gravel SP, Caron M, Savage P, et al. Erralpha mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nat Commun (2016) 7:12156. doi: 10.1038/ncomms12156
113. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. Pgc-1alpha-Responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet (2003) 34:267–73. doi: 10.1038/ng1180
114. St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, et al. Suppression of reactive oxygen species and neurodegeneration by the pgc-1 transcriptional coactivators. Cell. (2006) 127:397–408. doi: 10.1016/j.cell.2006.09.024
115. Mcguirk S, Audet-Delage Y, St-Pierre J. Metabolic fitness and plasticity in cancer progression. Trends Cancer (2020) 6:49–61. doi: 10.1016/j.trecan.2019.11.009
116. Guo X, Jiang Q, Tuccitto A, Chan D, Alqawlaq S, Won GJ, et al. The ampk-Pgc-1α signaling axis regulates the astrocyte glutathione system to protect against oxidative and metabolic injury. Neurobiol Disease. (2018) 113:59–69. doi: 10.1016/j.nbd.2018.02.004
117. Mcguirk S, Audet-Delage Y, Annis MG, Xue Y, Vernier M, Zhao K, et al. Resistance to different anthracycline chemotherapeutics elicits distinct and actionable primary metabolic dependencies in breast cancer. Elife. (2021) 10:e65150. doi: 10.7554/eLife.65150
118. Karunakaran S, Ramachandran S, Coothankandaswamy V, Elangovan S, Babu E, Periyasamy-Thandavan S, et al. Slc6a14 (Atb(0,+)) protein, a highly concentrative and broad specific amino acid transporter, is a novel and effective drug target for treatment of estrogen receptor-positive breast cancer. J Biol Chem (2011) 286:31830–8. doi: 10.1074/jbc.M111.229518
119. Andrade-Vieira R, Goguen D, Bentley HA, Bowen CV, Marignani PA. Pre-clinical study of drug combinations that reduce breast cancer burden due to aberrant mtor and metabolism promoted by Lkb1 loss. Oncotarget (2014) 5:12738–52. doi: 10.18632/oncotarget.2818
120. Edwards DN, Ngwa VM, Wang S, Shiuan E, Brantley-Sieders DM, Kim LC, et al. The receptor tyrosine kinase Epha2 promotes glutamine metabolism in tumors by activating the transcriptional coactivators yap and taz. Sci Signaling (2017) 10:eaan4667. doi: 10.1126/scisignal.aan4667
121. Morotti MA-O, Bridges E, Valli A, Choudhry H, Sheldon H, Wigfield S, et al. Hypoxia-induced switch in Snat2/Slc38a2 regulation generates endocrine resistance in breast cancer. Proc Natl Acad Sci USA (2019) 116:12452–61. doi: 10.1073/pnas.1818521116
122. Gross MI, Sd D, Dennison JB, Chen L, - Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor cb-839 in triple-negative breast cancer. Mol Cancer Ther (2014) 13:890–901. doi: 10.1158/1535-7163
123. Demas DM, Demo S, Fallah Y, Clarke R, Nephew KP, Althouse S, et al. Glutamine metabolism drives growth in advanced hormone receptor positive breast cancer. Front Oncol (2019) 9:686. doi: 10.3389/fonc.2019.00686
124. Simpson NE, Tryndyak VP, Pogribna M, Beland FA, Pogribny IP. Modifying metabolically sensitive histone marks by inhibiting glutamine metabolism affects gene expression and alters cancer cell phenotype. Epigenetics (2012) 7:1413–20. doi: 10.4161/epi.22713
125. Ananieva EA, Wilkinson AC. Branched-chain amino acid metabolism in cancer. Curr Opin Clin Nutr Metab Care (2018) 21:64–70. doi: 10.1097/MCO.0000000000000430
126. Zhang L, Han JQ. Branched-chain amino acid transaminase 1 (Bcat1) promotes the growth of breast cancer cells through improving mtor-mediated mitochondrial biogenesis and function. Biochem Biophys Res Commun (2017) 486:224–31. doi: 10.1016/j.bbrc.2017.02.101
127. Yamnik RL, Holz MK. Mtor/S6k1 and Mapk/Rsk signaling pathways coordinately regulate estrogen receptor alpha serine 167 phosphorylation. FEBS Lett (2010) 584:124–8. doi: 10.1016/j.febslet.2009.11.041
128. Scalise M, Galluccio M, Console L, Pochini L, Indiveri C. The human Slc7a5 (Lat1): The intriguing Histidine/Large neutral amino acid transporter and its relevance to human health. Front Chem (2018) 6:243. doi: 10.3389/fchem.2018.00243
129. Saito Y, Li L, Coyaud E, Luna A, Sander C, Raught B, et al. Llgl2 rescues nutrient stress by promoting leucine uptake in er(+) breast cancer. Nature. (2019) 569:275–9. doi: 10.1038/s41586-019-1126-2
130. Snell K. Enzymes of serine metabolism in normal, developing and neoplastic rat tissues. Adv Enzyme Regul (1984) 22:325–400. doi: 10.1016/0065-2571(84)90021-9
131. Mattaini KR, Sullivan MR, Vander Heiden MG. The importance of serine metabolism in cancer. J Cell Biol (2016) 214:248–57. doi: 10.1083/jcb.201604085
132. Kim SK, Jung WH, Koo JS. Differential expression of enzymes associated with Serine/Glycine metabolism in different breast cancer subtypes. PLoS One (2014) 9:e101004. doi: 10.1371/journal.pone.0101004
133. Labuschagne CF, van den Broek NJF, Mackay GM, Vousden KH, Maddocks ODK. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep (2014) 7:1248–58. doi: 10.1016/j.celrep.2014.04.045
134. Rathore R, Schutt CR, Tine B. Phgdh as a mechanism for resistance in metabolically-driven cancers. Cancer Drug Resist (2020) 3:762–74. doi: 10.20517/cdr.2020.46
135. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature (2011) 476:346–U119. doi: 10.1038/nature10350
136. Samanta D, Park Y, Andrabi SA, Shelton LM, Gilkes DM, Semenza GL. Phgdh expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res (2016) 76:4430–42. doi: 10.1158/0008-5472.Can-16-0530
137. Mullarky E, Lucki NC, Zavareh RB, Anglin JL, Gomes AP, Nicolay BN, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers (Vol 113, pg 1778, 2016). Proc Natl Acad Sci USA (2016) 113:E1585–5. doi: 10.1073/pnas.1602228113
138. De Marchi T, Timmermans MA, Sieuwerts AM, Smid M, Look MP, Grebenchtchikov N, et al. Phosphoserine aminotransferase 1 is associated to poor outcome on tamoxifen therapy in recurrent breast cancer. Sci Rep (2017) 7:2099. doi: 10.1038/s41598-017-02296-w
139. Kim YJ, Jang SK, Hong SE, Park CS, Seong MK, Kim HA, et al. Knockdown of Yap/Taz sensitizes tamoxifen-resistant Mcf7 breast cancer cells. Biochem Biophys Res Commun (2022) 601:73–8. doi: 10.1016/j.bbrc.2022.02.083
140. Metcalf S, Petri BJ, Kruer T, Green B, Dougherty S, Wittliff JL, et al. Serine synthesis influences tamoxifen response in er plus human breast carcinoma. Endocrine-Related Cancer. (2021) 28:27–37. doi: 10.1530/Erc-19-0510
141. Hulea L, Gravel SP, Morita M, Cargnello M, Uchenunu O, Im YK, et al. Translational and hif-1α-Dependent metabolic reprogramming underpin metabolic plasticity and responses to kinase inhibitors and biguanides. Cell Metab (2018) 28:817–832.e8. doi: 10.1016/j.cmet.2018.09.001
142. Ryu CS, Kwak HC, Lee JY, Oh SJ, Phuong NT, Kang KW, et al. Elevation of cysteine consumption in tamoxifen-resistant mcf-7 cells. Biochem Pharmacol (2013) 85:197–206. doi: 10.1016/j.bcp.2012.10.021
143. Zong L, Cheng G, Liu S, Pi Z, Liu Z, Song F. Reversal of multidrug resistance in breast cancer cells by a combination of ursolic acid with doxorubicin. J Pharm BioMed Anal (2019) 165:268–75. doi: 10.1016/j.jpba.2018.11.057
144. Bacci M, Lorito N, Ippolito L, Ramazzotti M, Luti S, Romagnoli S, et al. Reprogramming of amino acid transporters to support aspartate and glutamate dependency sustains endocrine resistance in breast cancer. Cell Rep (2019) 28:104–118 e108. doi: 10.1016/j.celrep.2019.06.010
145. Lunt SY, Vander Heiden MG. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
146. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell (2015) 162:552–63. doi: 10.1016/j.cell.2015.07.017
147. Ligorio F, Pellegrini I, Castagnoli L, Vingiani A, Lobefaro R, Zattarin E, et al. Targeting lipid metabolism is an emerging strategy to enhance the efficacy of anti-Her2 therapies in Her2-positive breast cancer. Cancer Letters. (2021) 511:77–87. doi: 10.1016/j.canlet.2021.04.023
148. Wang YP, Lei QY. Perspectives of reprogramming breast cancer metabolism. Trans Res Breast Cancer (2017) 1026:217–32. doi: 10.1007/978-981-10-6020-5_10
149. Kuhajda FP. Amp-activated protein kinase and human cancer: Cancer metabolism revisited. Int J Obes (Lond). (2008) 32 Suppl 4:S36–41. doi: 10.1038/ijo.2008.121
150. Giro-Perafita A, Sarrats A, Perez-Bueno F, Oliveras G, Buxo M, Brunet J, et al. Fatty acid synthase expression and its association with clinico-histopathological features in triple-negative breast cancer. Oncotarget. (2017) 8:74391–405. doi: 10.18632/oncotarget.20152
151. Giro-Perafita A, Palomeras S, Lum DH, Blancafort A, Vinas G, Oliveras G, et al. Preclinical evaluation of fatty acid synthase and egfr inhibition in triple-negative breast cancer. Clin Cancer Res (2016) 22:4687–97. doi: 10.1158/1078-0432.CCR-15-3133
152. Menendez JA, Vellon L, Mehmi I, Oza BP, Ropero S, Colomer R, et al. Inhibition of fatty acid synthase (Fas) suppresses Her2/Neu (Erbb-2) oncogene overexpression in cancer cells. Proc Natl Acad Sci USA (2004) 101:10715–20. doi: 10.1073/pnas.0403390101
153. Vazquez-Martin A, Colomer R, Brunet J, Menendez JA. Pharmacological blockade of fatty acid synthase (Fasn) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting 'Her2 super-expression' occurring in high-dose trastuzumab-conditioned Skbr3/Tzb100 breast cancer cells. Int J Oncol (2007) 31:769–76. doi: 10.3892/ijo.31.4.769
154. Menendez JA, Vellon L, Lupu R. Targeting fatty acid synthase-driven lipid rafts: A novel strategy to overcome trastuzumab resistance in breast cancer cells. Med Hypotheses (2005) 64:997–1001. doi: 10.1016/j.mehy.2004.09.027
155. Menendez JA, Lupu R, Colomer R. Inhibition of tumor-associated fatty acid synthase hyperactivity induces synergistic chemosensitization of her-2/Neu-Overexpressing human breast cancer cells to docetaxel (Taxotere). Breast Cancer Res Treat (2004) 84:183–95. doi: 10.1023/B:BREA.0000018409.59448.60
156. Menendez JA, Vellon L, Colomer R, Lupu R. Pharmacological and small interference rna-mediated inhibition of breast cancer-associated fatty acid synthase (Oncogenic antigen-519) synergistically enhances taxol (Paclitaxel)-induced cytotoxicity. Int J Cancer (2005) 115:19–35. doi: 10.1002/ijc.20754
157. Liu H, Liu Y, Zhang JT. A new mechanism of drug resistance in breast cancer cells: Fatty acid synthase overexpression-mediated palmitate overproduction. Mol Cancer Ther (2008) 7:263–70. doi: 10.1158/1535-7163.MCT-07-0445
158. Vazquez-Martin A, Ropero S, Brunet J, Colomer R, Menendez JA. Inhibition of fatty acid synthase (Fasn) synergistically enhances the efficacy of 5-fluorouracil in breast carcinoma cells. Oncol Rep (2007) 18:973–80. doi: 10.3892/or.18.4.973
159. Menendez JA, Colomer R, Lupu R. Inhibition of tumor-associated fatty acid synthase activity enhances vinorelbine (Navelbine)-induced cytotoxicity and apoptotic cell death in human breast cancer cells. Oncol Rep (2004) 12:411–22. doi: 10.3892/or.12.2.411
160. Duan L, Calhoun S, Shim D, Perez RE, Blatter LA, Maki CG. Fatty acid oxidation and autophagy promote endoxifen resistance and counter the effect of akt inhibition in er-positive breast cancer cells. J Mol Cell Biol (2021) 13:433–44. doi: 10.1093/jmcb/mjab018
161. Shan M, Dai D, Vudem A,, Varner JD, Stroock AD. Multi-scale computational study of the warburg effect, reverse warburg effect and glutamine addiction in solid tumors. PLoS Comput Biol (2018) 14:e1006584. doi: 10.1371/journal.pcbi.1006584
162. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Lisanti MP. The reverse warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle (2009) 8:3984–4001. doi: 10.4161/cc.8.23.10238
163. Gomes LR, Vessoni AT, Menck CFM. Microenvironment and autophagy cross-talk: Implications in cancer therapy. Pharmacol Res Off J Ital Pharmacol Society. (2016) 107:300–7. doi: 10.1016/j.phrs.2016.03.031
164. Viedma-Rodríguez R, Baiza-Gutman L, Salamanca-Gómez F, Diaz-Zaragoza M, Martínez-Hernández G, Ruiz Esparza-Garrido R, et al. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer (Review). Oncol Rep (2014) 32:3–15. doi: 10.3892/or.2014.3190
165. Jiao L, Zhang HL, Li DD, Yang KL, Tang J, Li X, et al. Regulation of glycolytic metabolism by autophagy in liver cancer involves selective autophagic degradation of Hk2 (Hexokinase 2). Taylor Francis (2018) 14:671–84. doi: 10.1080/15548627.2017.1381804
166. Chen SN, Zhu XM, Qiao HY, Ye MX, Lai XF, Yu ST, et al. Protective autophagy promotes the resistance of Her2-positive breast cancer cells to lapatinib. Tumor Biol (2015) 37:2321–31. doi: 10.1007/s13277-015-3800-9
167. Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Autophagy facilitates the development of breast cancer resistance to the anti-Her2 monoclonal antibody trastuzumab. PLoS One (2009) 4:e6251. doi: 10.1371/journal.pone.0006251
168. Zhang LH, Yang AJ, Wang M, Wei L, Min L. Enhanced autophagy reveals vulnerability of p-gp mediated epirubicin resistance in triple negative breast cancer cells. APOPTOSIS (2016) 21:473–88. doi: 10.1007/s10495-016-1214-9
169. Pan B, Chen D, Huang J, Rui W, Chen L. Hmgb1-mediated autophagy promotes docetaxel resistance in human lung adenocarcinoma. Mol Cancer (2014) 13:165. doi: 10.1186/1476-4598-13-165
170. Liu L, Liu S, Luo H, Chen C, Zhang X, He L, et al. Gpr30-mediated Hmgb1 upregulation in cafs induces autophagy and tamoxifen resistance in erα-positive breast cancer cells. Aging (Albany NY). (2021) 13:16178–97. doi: 10.18632/aging.203145
171. . Funderburk SF, Wang QJ, Yue Z. The beclin 1-Vps34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol (2010) 20:355–62. doi: 10.1016/j.tcb.2010.03.002
172. Cicchini M, Chakrabarti R, Kongara S, Price S, Nahar R, Lozy F, et al. Autophagy regulator Becn1 suppresses mammary tumorigenesis driven by Wnt1 activation and following parity. Autophagy. (2014) 10:2036–52. doi: 10.4161/auto.34398
173. Koboldt DC, Fulton RS, Mclellan MD, Schmidt H, Kalicki-Veizer J, Mcmichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. (2012) 490:61. doi: 10.1038/nature11412
174. Stein JV, Lópezfraga M, Elustondo FA, Carvalhopinto CE, Rodríguez D, Gómezcaro R, et al. April Modulates b and T cell immunity. J Clin Invest (2002) 109:1587–98. doi: 10.1172/JCI0215034
175. Dillon SR, Gross JA, Ansell SM, Novak AJ. An April to remember: Novel tnf ligands as therapeutic targets. Nat Rev Drug Discovery (2006) 5:235. doi: 10.1038/nrd1982
176. Xu S, Lam KP. B-cell maturation protein, which binds the tumor necrosis factor family members baff and April, is dispensable for humoral immune responses. Mol Cell Biol (2001) 21:4067–74. doi: 10.1128/MCB.21.12.4067-4074.2001
177. Lin HY, Kuei CH, Lee HH, Lin CH, Lin YF. Tnfsf13 upregulation confers chemotherapeutic resistance Via triggering autophagy initiation in triple-negative breast cancer. J Mol Med (2020) 98:1255–67. doi: 10.1007/s00109-020-01952-5
178. Li T, Liu M, Feng X, Wang Z, Das I, Xu Y, et al. Glyceraldehyde-3-Phosphate dehydrogenase is activated by lysine 254 acetylation in response to glucose signal. J Biol Chem (2014) 289:3775–85. doi: 10.1074/jbc.M113.531640
179. Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, et al. Gapdh and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. (2007) 129:983–97. doi: 10.1016/j.cell.2007.03.045
180. Klarer AC, O’neal J, Imbert-Fernandez Y, Clem A, Ellis SR, Clark J, et al. Inhibition of 6-Phosphofructo-2-Kinase (Pfkfb3) induces autophagy as a survival mechanism. Cancer Metab (2014) 2:2–2. doi: 10.1186/2049-3002-2-2
181. Jeon Y, Khelifa S, Ratnikov B, Scott D, Feng Y, Parisi F, et al. Regulation of glutamine carrier proteins by Rnf5 determines breast cancer response to er stress-inducing chemotherapies. Cancer Cell (2015) 27:354–69. doi: 10.1016/j.ccell.2015.02.006
182. Lv L, Li D, Zhao D, Lin R, Lei QY. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell (2011) 42:719–30. doi: 10.1016/j.molcel.2011.04.025
183. Gomes AP, Blenis J. A nexus for cellular homeostasis: The interplay between metabolic and signal transduction pathways. Curr Opin Biotechnol (2015) 34:110–7. doi: 10.1016/j.copbio.2014.12.007
184. Mi WY, Yubin S, Ji LE, Sunyoung L, Hun JS, Kyung KH, et al. Inhibition of aerobic glycolysis represses Akt/Mtor/Hif-1α axis and restores tamoxifen sensitivity in antiestrogen-resistant breast cancer cells. PLoS One (2015) 10:e0132285. doi: 10.1371/journal.pone.0132285
185. Mayer I. Role of mTOR inhibition in preventing resistance and restoring sensitivity to hormone-targeted and Her2-targeted therapies in breast cancer. Clin Adv Hematol oncology: H&O. (2013) 11:217–24.
186. Semenza GL. Targeting hif-1 for cancer therapy. Nat Rev Cancer. (2003) 3:721–32. doi: 10.1038/nrc1187
187. Albert Jm KK, Cao C, Lu B. Targeting the Akt/Mammalian target of rapamycin pathway for radiosensitization of breast cancer. Mol Cancer Ther (2006) 5:1183. doi: 10.1158/1535-7163.MCT-05-0400
188. Chan S. Phase ii study of temsirolimus (Cci-779), a novel inhibitor of mtor, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol Off J Am Soc Clin Oncol (2005) 23:5314. doi: 10.1200/JCO.2005.66.130
189. Menendez J. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. (2007) 7:763–77. doi: 10.1038/nrc2222
190. Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo YY, et al. Fatty acid synthase gene is up-regulated by hypoxia Via activation of akt and sterol regulatory element binding protein-1. Cancer Res (2008) 68:1003–11. doi: 10.1158/0008-5472.CAN-07-2489
191. Yang YA, Wan FH, Morin PJ, Chrest FJ, Pizer ES. Activation of fatty acid synthesis during neoplastic transformation: Role of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Exp Cell Res (2002) 279:80–90. doi: 10.1006/excr.2002.5600
192. Yan C, Wei H, Zheng M, Yan X, Yang J, Liu W, et al. The mtor inhibitor rapamycin synergizes with a fatty acid synthase inhibitor to induce cytotoxicity in Er/Her2-positive breast cancer cells. PLoS One (2014) 9:e97697. doi: 10.1371/journal.pone.0097697
193. Corominas-Faja B, Vellon L, Cuyàs E, Buxó M, Lupu R. Clinical and therapeutic relevance of the metabolic oncogene fatty acid synthase in Her2+ breast cancer. Histol histopathols (2016) 32:11830. doi: 10.14670/HH-11-830
194. Moldogazieva NT, Mokhosoev IM, Terentiev AA. Metabolic heterogeneity of cancer cells: An interplay between hif-1, gluts, and ampk. Cancers (2020) 12:862. doi: 10.3390/cancers12040862
195. Lovitt CJ, Shelper TB, Avery VM. Doxorubicin resistance in breast cancer cells is mediated by extracellular matrix proteins. BMC Cancer (2018) 18:41. doi: 10.1186/s12885-017-3953-6
196. Xu M, Chen S, Yang W, Cheng X, Ye Y, Mao J, et al. Fgfr4 links glucose metabolism and chemotherapy resistance in breast cancer. Cell Physiol Biochem (2018) 47:151–60. doi: 10.1159/000489759
197. Qu C, Zhang W, Zheng G, Zhang Z, Yin J, He Z. Metformin reverses multidrug resistance and epithelial-mesenchymal transition (Emt) Via activating amp-activated protein kinase (Ampk) in human breast cancer cells. Mol Cell Biochem (2014) 386:63–71. doi: 10.1007/s11010-013-1845-x
198. Marinello PC, Panis C, Silva TNX, Binato R, Abdelhay E, Rodrigues JA, et al. Metformin prevention of doxorubicin resistance in mcf-7 and mda-Mb-231 involves oxidative stress generation and modulation of cell adaptation genes. Sci Rep (2019) 9:5864. doi: 10.1038/s41598-019-42357-w
199. Bolós V, Grego-Bessa J, Luis D. Notch signaling in development and cancer. Endocrine Rev (2007) 28:339–63. doi: 10.1210/er.2006-0046
200. Wong A, Lee S-C. Mechanisms of resistance to trastuzumab and novel therapeutic strategies in Her2-positive breast cancer. Int J Breast Cancer (2012) 2012:415170. doi: 10.1155/2012/415170
201. Guo S, Liu M, Gonzalez-Perez RR. Role of notch and its oncogenic signaling crosstalk in breast cancer. Biochim Et Biophys Acta (2011) 1815:197–213. doi: 10.1016/j.bbcan.2010.12.002
202. Lindsay J, Jiao X, Sakamaki T, Casimiro MC, Shirley LA, Tran TH, et al. Erbb2 induces Notch1 activity and function in breast cancer cells. doi: 10.1111/j.1752-8062.2008.00041.x
203. Osipo C, Patel P, Rizzo P, Clementz AG, Hao L, Golde TE, et al. Erbb-2 inhibition activates notch-1 and sensitizes breast cancer cells to a Γ-secretase inhibitor. Oncogene Basingstoke (2008) 27:5019–32. doi: 10.1038/onc.2008.149
204. Bosc C, Selak MA, Sarry JE. Resistance is futile: Targeting mitochondrial energetics and metabolism to overcome drug resistance in cancer treatment. Cell Metab (2017) 26:705–7. doi: 10.1016/j.cmet.2017.10.013
205. Alexa-Stratulat T, Pešić M, Gašparović A, Trougakos IP, Riganti C. What sustains the multidrug resistance phenotype beyond abc efflux transporters? looking beyond the tip of the iceberg. Drug Resist Updat. (2019) 46:100643. doi: 10.1016/j.drup.2019.100643
206. Huber-Keener KJ, Liu X, Wang Z, Wang Y, Freeman W, Wu S, et al. Differential gene expression in tamoxifen-resistant breast cancer cells revealed by a new analytical model of rna-seq data. PLoS One (2012) 7:e41333. doi: 10.1371/journal.pone.0041333
207. Sansone P, Ceccarelli C, Berishaj M, Chang Q, Rajasekhar VK, Perna F, et al. Self-renewal of Cd133(Hi) cells by Il6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat Commun (2016) 7:10442. doi: 10.1038/ncomms10442
208. Camarda R, Zhou AY, Kohnz RA-O, Balakrishnan S, Mahieu C, Anderton B, et al. Inhibition of fatty acid oxidation as a therapy for myc-overexpressing triple-negative breast cancer. Nat Med (2016) 22:427–32. doi: 10.1038/nm.4055
209. Park JH, Vithayathil S, Kumar S, Sung PL, Dobrolecki LE, Putluri V, et al. Fatty acid oxidation-driven src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep (2016) 14:2154–65. doi: 10.1016/j.celrep.2016.02.004
210. Guerra F, Arbini AA, Moro L. Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg. (2017) 1858:686–99. doi: 10.1016/j.bbabio.2017.01.012
211. Deus CM, Coelho AR, Serafim TL, Oliveira PJ. Targeting mitochondrial function for the treatment of breast cancer. Future Med Chem (2014) 6:1499–513. doi: 10.4155/fmc.14.100
212. Lee KM, Giltnane JM, Balko JM, Schwarz LJ, Guerrero-Zotano AL, Hutchinson KE, et al. Myc and Mcl1 cooperatively promote chemotherapy-resistant breast cancer stem cells Via regulation of mitochondrial oxidative phosphorylation. Cell Metab (2017) 26:633–647.e637. doi: 10.1016/j.cmet.2017.09.009
213. Lee M, Hirpara JL, Eu JQ, Sethi G, Wang L, Goh BC, et al. Targeting Stat3 and oxidative phosphorylation in oncogene-addicted tumors. Redox Biol (2019) 25:101073. doi: 10.1016/j.redox.2018.101073
214. Jia D, Park JH, Jung KH, Levine H, Kaipparettu BA. Elucidating the metabolic plasticity of cancer: Mitochondrial reprogramming and hybrid metabolic states. Cells (2018) 7:21. doi: 10.3390/cells7030021
215. Roth KG, Mambetsariev I, Kulkarni P, Salgia R. The mitochondrion as an emerging therapeutic target in cancer. Trends Mol Med (2020) 26:119–34. doi: 10.1016/j.molmed.2019.06.009
216. Lee M, Yoon JH. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse warburg effect and its therapeutic implication. World J Biol Chem (2015) 6:148–61. doi: 10.4331/wjbc.v6.i3.148
217. Boasquevisque PH, Schoeneberger V, Caporiccio L, Vellanki RN, Wouters BG. Targeting lactate transporters mct-1 and mct-4 to inhibit the growth of hypoxic hnscc cells in vitro. Int J Radiat OncologyBiologyPhysics (2017) 99:E579. doi: 10.1016/j.ijrobp.2017.06.1994
218. Gotanda Y, Akagi Y, Kawahara A, Kinugasa T, Shirouzu K. Expression of monocarboxylate transporter (Mct)-4 in colorectal cancer and its role: Mct4 contributes to the growth of colorectal cancer with vascular endothelial growth factor. Anticancer Res (2013) 33:2941–7.
219. Javaeed A, Ghauri SK. Mct4 has a potential to be used as a prognostic biomarker - a systematic review and meta-analysis. Oncol Rev (2019) 13:403. doi: 10.4081/oncol.2019.403
220. Johnson JM, Paolo C, Roberto F, Lekha M, Chen J, Daniele C, et al. Mct1 in invasive ductal carcinoma: Monocarboxylate metabolism and aggressive breast cancer. Front Cell Dev Biol (2017) 5. doi: 10.3389/fcell.2017.00027
221. Apicella M, Giannoni E, Fiore S, Ferrari KJ, Fernández-Pérez D, Isella C, et al. Increased lactate secretion by cancer cells sustains non-Cell-Autonomous adaptive resistance to met and egfr targeted therapies. Cell Metab (2018) 28:848–65.e6. doi: 10.1016/j.cmet.2018.08.006
222. Qixiang LI, Pei Z, Fang L, Xianzhi W, Lu LI, Zhongkun W, et al. Monocarboxylate transporter 1 enhances the sensitivity of breast cancer cells to 3-bromopyruvate in vitro. J South Med Univ (2017) 37:588–93. doi: 10.3969/j.issn.1673-4254.2017.05.04
223. Morais-Santos F, Miranda-Goncalves V, Pinheiro S, Vieira AF, Paredes J, Schmitt FC, et al. Differential sensitivities to lactate transport inhibitors of breast cancer cell lines. Endocrine-Related Cancer. (2013) 21:27–38. doi: 10.1530/ERC-13-0132
224. Pinheiro C, Albergaria A, Paredes J, Sousa B, Dufloth R, Vieira D, et al. Monocarboxylate transporter 1 is up-regulated in basal-like breast carcinoma. Histopathology. (2010) 56:860–7. doi: 10.1111/j.1365-2559.2010.03560.x
225. Romero-Cordoba SL, Sergio RC, Veronica BP, Antonio MA, Elvira D, Giulia C, et al. Loss of function of mir-342-3p results in Mct1 over-expression and contributes to oncogenic metabolic reprogramming in triple negative breast cancer. entific Rep (2018) 8:12252–. doi: 10.1038/s41598-018-29708-9
226. Yuan C, Zhang J, Lou J, Wang S, Wang S. Comprehensive analysis of monocarboxylate transporter 4 (Mct4) expression in breast cancer prognosis and immune infiltration Via integrated bioinformatics analysis. Bioengineered (2021) 12:3850–63. doi: 10.1080/21655979.2021.1951928
227. Pisarsky L, Bill R, Fagiani E, Dimeloe S, Goosen RW, Hagmann JR, et al. Targeting metabolic symbiosis to overcome resistance to anti-angiogenic therapy. Cell Rep (2016) 15:1161–74. doi: 10.1016/j.celrep.2016.04.028
228. Warburg O. On the origin of cancer cells. Science (1956) 123:309–14. doi: 10.1126/science.123.3191.309
Keywords: Breast cancer, drug resistance, metabolic reprogramming, glucose metabolism, fatty acid synthesis
Citation: Lv L, Yang S, Zhu Y, Zhai X, Li S, Tao X and Dong D (2022) Relationship between metabolic reprogramming and drug resistance in breast cancer. Front. Oncol. 12:942064. doi: 10.3389/fonc.2022.942064
Received: 12 May 2022; Accepted: 01 August 2022;
Published: 18 August 2022.
Edited by:
Cristian Scatena, University of Pisa, ItalyReviewed by:
Gary Xiao, Dalian University of Technology, ChinaCopyright © 2022 Lv, Yang, Zhu, Zhai, Li, Tao and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xufeng Tao, dGFveHVmZW5nLjIwMDhAMTYzLmNvbQ==; Deshi Dong, ZGVzaGlkb25nQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.