 Luise Victoria Claaß1†
Luise Victoria Claaß1† Christoph Schultheiß1†
Christoph Schultheiß1† Rebekka Scholz1†
Rebekka Scholz1† Lisa Paschold1
Lisa Paschold1 Donjete Simnica1
Donjete Simnica1 Volker Heinemann2
Volker Heinemann2 Sebastian Stintzing3Mascha Binder1*
Sebastian Stintzing3Mascha Binder1*- 1Internal Medicine IV, University Hospital Halle, Martin-Luther University Halle-Wittenberg, Halle (Saale), Germany
- 2Department of Hematology/Oncology, Ludwig-Maximilians-University Munich (LMU) Klinikum, University of Munich and Comprehensive Cancer Center Munich, Munich, Germany
- 3Department of Hematology, Oncology, and Cancer Immunology (CCM), Charité - Universitätsmedizin Berlin, Berlin, Germany
The two most common antibody targeting principles in oncology are the induction of direct antitumor effects and the release of antitumor T cell immunity by immune checkpoint blockade. These two principles, however, may be overlapping if the targeted checkpoint molecule is not located on the immune cell but on the tumor cell itself. Secondary resistance by epitope escape may therefore remain a challenge in both settings. We previously reported epitope escape through L88S and truncating programmed cell death ligand 1 (PD-L1) gene mutations in colorectal cancer patients on selective pressure with avelumab, a PD-L1-directed checkpoint blocker that—in addition to T cell disinhibition—allows direct tumor cell killing via its unmodified Fc portion. Here, we confirmed this principle by liquid biopsy monitoring in a colorectal cancer patient from an independent clinical trial. In this patient, both PD-L1 L88E and L88fs mutations emerged under selective pressure with avelumab. By ectopically expressing PD-L1 L88E, we show that this mutation leads to a reduction of full-length glycosylated PD-L1 and greatly reduced avelumab surface binding. Further experiments indicated that PD-L1 L88E represents a phosphomimetic variant of PD-L1 L88S leading to loss of protein stability and increased proteasomal degradation. The association of this PD-L1 mutation with the high-affinity FCGR3A single nucleotide polymorphism rs396991 confirms prior evidence that patients harboring this polymorphism experience the strongest selective pressure by avelumab. Together, position 88 of PD-L1 is a hotspot residue critically regulating PD-L1 cell surface expression with clinical significance in the context of immune checkpoint blockade.
Introduction
Programmed cell death ligand 1 (PD-L1) is a membrane protein expressed on some cells of the tumor microenvironment such as macrophages and dendritic cells as well as on the surface of cancer cells (1, 2). It consists of IgV- and IgC-like domains, a trans-membrane, and a short intracellular domain. The interaction of the IgV domain of PD-L1 with its binding partner programmed cell death 1 (PD-1) on activated T cells triggers the formation of clusters with the T cell receptor and CD28, resulting eventually in abrogation of CD28 signaling by recruitment of Src homology 2 domain-containing tyrosine phosphatase 2 (SHP2) (3). As a result, T cells are inactivated and antitumor immune reactions dampened. To prevent tumor immune escape, a number of PD-1 and PD-L1 blocking antibodies have been developed and commercialized over the past decade resulting in significant therapeutic improvements for patients with a broad spectrum of tumors (4). Since PD-1 is expressed on the T cell itself, targeting antibodies like nivolumab or pembrolizumab were equipped with IgG4 constant regions showing reduced ability to bind complement or Fc gamma receptors on effector cells (5) to prevent the elimination of T cells by complement-dependent (CDC), antibody-dependent cellular cytotoxicity (ADCC), or antibody-dependent cell phagocytosis (ADCP) (6, 7). PD-L1 antibodies like durvalumab or atezolizumab harbor IgG1 instead of IgG4, but these regions have been genetically engineered to significantly reduce CDC or ADCC potentially directed not only at the tumor cell but also at PD-L1-positive cells within the tumor microenvironment (8, 9). In contrast, avelumab is a PD-L1 antibody with a functional IgG1 region that blocks the PD-L1/PD-1 axis, but may also directly promote ADCC against tumor cells (10). In a phase II trial where avelumab was given in addition to chemotherapy and EGFR-targeted therapy in a cohort of patients with predominantly microsatellite-stable metastatic colorectal cancer, we identified three patients with emerging tumor subclones on avelumab treatment that harbored PD-L1 sequence variants (11). In two patients, we found a PD-L1 L88S point mutation located outside the avelumab epitope resulting in lower membrane expression. A third patient developed a PD-L1 K162fs variant inducing a premature stop codon, thereby resulting in complete loss of PD-L1 membrane expression due to a lack of the transmembrane domain. Cells transduced to express these variants triggered significantly less natural killer (NK) cell degranulation than cells expressing the PD-L1 wild-type, but T cell-mediated killing was increased. Interestingly, these PD-L1 mutations were significantly associated with the homo- and heterozygous Fc gamma receptor IIIa (FCGR3A) single nucleotide polymorphism (SNP) rs396991 (V/V and V/F) that is known to mediate high-affinity binding of NK cells to IgG1 (12). This suggested that patients with microsatellite-stable metastatic colorectal cancer with the high-affinity FCGR3A SNP may be a subset with the highest selective pressure exerted by avelumab.
In the data presented here, we confirm the selection of two PD-L1 mutants at position 88 on avelumab treatment in a patient with microsatellite-stable metastatic colorectal cancer. This patient—treated on an independent clinical trial—again showed homozygosity for the high-affinity FCGR3A SNP. We go on to show that PD-L1 L88E represents a phosphomimetic mutation to PD-L1 L88S leading to decreased protein stability and degradation. Together, our data suggest that position 88 may represent a clinically relevant PD-L1 hotspot position regulating protein stability.
Methods
Patient and Biomaterial
Patient 05-001 was a patient enrolled on the FIRE-6 (AIO KRK-0118) trial with histologically confirmed, treatment-naive, RAS/RAF wild-type metastatic colorectal cancer (MSS) that agreed to take part in a biomarker substudy after informed consent [Ethical Commission of the Ludwig-Maximilians-University Munich (LMU): 18-0933]. The patient received 4 cycles of treatment with FOLFIRI and cetuximab, followed by 4 cycles of FOLFIRI/cetuximab with additional avelumab. These 8 cycles were followed by avelumab maintenance treatment for another 10 weeks until the end of treatment (EOT) due to progression in week 19. Plasma samples were collected at regular intervals in cell-free DNA BCT tubes (Streck, USA) and processed according to the manufacturer’s instructions. Plasma was immediately frozen at −80°C until extraction of cell-free DNA, and the leukocyte pellet was stored in 1 ml of heat-inactivated fetal bovine serum (Life Technologies, USA) with 10% of dimethyl sulfoxide (Sigma, Germany). Additionally, the paraffin-embedded tumor biopsy obtained before treatment initiation was used for genetic analyses as described below.
Next-Generation Sequencing and Data Analysis
For mutational profiling, 100 ng of cell-free DNA (cfDNA) was used. Genes were selected through a review of published literature and by using the cBio Cancer Genomics Portal (13). Either the entire coding region or the hotspot regions containing known pathogenic or resistance variants were targeted, and sequencing libraries were constructed using QIAseq Targeted DNA Custom Panel (Qiagen, Germany). Quantification and quality control of the libraries were conducted using the Qubit high-sensitivity double-strand DNA assay kit (Thermo Fisher Scientific Inc., USA) and Agilent 2100 Bioanalyzer (Agilent Technologies, Germany). Sequencing was performed on the Illumina NextSeq or HiSeq platform (San Diego, USA) with 2 × 151 cycles at an average coverage of 13,500. Variant calling of unique molecular identifier (UMI)-based sequencing data was performed using the CLC workbench (Qiagen, Hilden, Germany). Variants were filtered for a minimum coverage of 10 UMIs, minimum count of 2 UMIs, minimal average base quality of 35, and minimum frequency of 1% for cfDNA or 10% for DNA from formalin-fixed paraffin-embedded tissue. Analyses were carried out and data plotting was performed using R (version 3.4.4) (14) as well as GraphPad Prism 7 (San Diego, CA). A p-value of <0.05 was considered statistically significant.
Digital Droplet PCR Workflow
Digital droplet PCR (ddPCR) custom assays were designed by and ordered from IDT (Iowa, USA). To increase specificity, locked nucleic acid bases were incorporated into the probes. Probes were HPLC-purified and contained either a 5′-HEX (wild-type probe) or a 5′-FAM (mutant probe) reporter dye and a 3′Iowa Black® Fluorescent quencher. The probe and primer sequences are listed in Supplementary Table S1. Each reaction (22 µl) contained 11 µl of 2×ddPCR SuperMix for probes (no UTP) (Bio-Rad, Feldkirchen, Germany), template DNA, and primers and probes at a final concentration of 1.8 µM and 500 nM, respectively. After droplet generation [AutoDG, Bio-Rad (Feldkirchen, Germany)], the plate was sealed with a pierceable foil heat seal [PX1 PCR plate sealer, Bio-Rad (Feldkirchen, Germany)], and the PCR was performed on a C1000 Touch thermal cycler (Bio-Rad, Feldkirchen, Germany). The cycling conditions were as follows: 95°C, 10 min; (94°C, 30 s; 56°C, 2 min] × 50; 98°C, 10 min; and 12°C hold for at least 4 h. Prior to the readout on the QX200 droplet reader (Bio-Rad, Feldkirchen, Germany), the plate was left at room temperature for 10 min. ddPCR data were analyzed using the QX Manager 1.0 Standard Edition (Bio-Rad, Feldkirchen, Germany). Assay specificity was tested on gBlocks (IDT, Iowa, USA). Thresholds for positive/negative droplets were set manually according to the positive control (gBlocks, IDT, Supplementary Figure S1).
Generation and Evaluation of Cell Lines Stably Expressing PD-L1 Variants
For functional validation of PD-L1 L88E, we used the UT-SCC-14 (tongue squamous cell carcinoma) and HT-29 (colorectal adenocarcinoma, KRAS wild-type) cell lines that were depleted of endogenous PD-L1 by CRISPR/Cas9 as described in Stein et al. (11). The PD-L1 L88E (c.262_263delinsGA) variant and L88M (c.262T>A) as control variants were generated by site-directed mutagenesis of the PD-L1 wt cDNA cloned into the Lentiviral Gene Ontology (LeGO) vector LeGO-iC2-Puro+ via AsiSI/EcoRI. Mutagenesis was performed using the QuikChange® II XL Site-Directed Mutagenesis Kit according to the manufacturer’s instructions. For ectopic re-expression, the PD-L1 constructs were lentivirally transduced and selected as described elsewhere (15). Cell lysates were generated using RIPA buffer supplemented with protease and phosphatase inhibitors (both from Roche, Basel, Switzerland) and quantified using the Pierce™ Detergent Compatible Bradford Assay Kit according to the manufacturer’s standard. For semidry immunoblotting, 20 µg of the extract was separated using the NuPAGE™ Bis-Tris gel system (Thermo Fisher, Hennigsdorf, Germany). The following antibodies were used for detection: anti-PD-L1, goat polyclonal IgG (R&D Systems, Minneapolis, United States); anti-GAPDH, mouse monoclonal IgG (Santa Cruz, Heidelberg, Germany); anti-goat-IgG-HRP, produced in mouse (Santa Cruz, Heidelberg, Germany); and anti-mouse-IgG-HRP, produced in goat (R&D Systems, Minneapolis, United States).
qRT-PCR
Total RNA was extracted with the Quick-RNA kit (Zymo Research, Freiburg, Germany) and reverse-transcribed with SuperScript III (Thermo Fisher, Hennigsdorf, Germany). Target amplification was performed on the CFX96 System (Bio-Rad, Feldkirchen, Germany) using the SYBR Select Master Mix CFX as suggested by the manufacturer and using the following primer pairs: PD-L1 (forward: TGCTGAACGCATTTACTGTCAC; reverse: TCTGTCTGTAGCTA CTATGCTG), mCherry (forward: AGGAGGATAACATGGCCATCAT; reverse: ACCC TTGGTCACCTTCAGCT), and HPRT1 (forward: TGACACTGGCAAAACAATGCA; reverse: GGTCCTTTTCACCAGCAAGCT). The relative gene expression levels were normalized to HPRT1 and calculated according to the comparative Ct (ΔΔCt) method.
Flow Cytometry
To quantify PD-L1 surface expression, 100,000 cells per well were seeded on a 96-well plate and incubated with 100 ng of avelumab or a human IgG pool [Intratec® (Biotest Pharma, Dreieich, Germany)] as isotype controls for 45 min at 4°C. After washing, avelumab was detected using a FITC-coupled α-human-IgG antibody (Sigma-Aldrich, Taufkirchen, Germany). Readout was performed on a BD FACSCelesta flow cytometer (Becton Dickinson, Kelberg, Germany) and analyzed using FlowJo™ (Becton Dickinson, Kelberg, Germany).
PD-L1 Degradation Assays
To assess the turnover of PD-L1 variants, compound C/dorsomorphin and MG132 were obtained from Selleck Chem (Planegg, Germany). To analyze the degradation kinetics of PD-L1 variants, 2 million cells were seeded in a six-well format and either incubated with 10 µM of compound C for 2 and 4 h or with 20 µM of MG132 for 4 h. Proteins were isolated as described above and subjected to immunoblotting.
Results
Colorectal Cancer Patient With Acquired PD-L1 Mutations on Avelumab Treatment
The FIRE-6 trial combined chemotherapy with PD-L1 inhibition and EGFR inhibition in patients with metastatic colorectal cancer. The PD-L1 inhibitor avelumab was used which represents an antibody with preserved effector functions that have been previously shown to induce direct tumor cell killing, especially in subjects expressing the FCGR3A high-affinity SNP rs396991 (Figure 1A). We initially subjected 29 patients enrolled in the FIRE-6 trial to liquid biopsy testing at EOT using a gene panel covering the most frequent driver and resistance mutations in mCRC including the full coding region of PD-L1. We identified one patient with study ID 05-001 who showed a PD-L1 L88E mutation at the end of treatment and profiled the patient consecutively with liquid biopsy at additional timepoints. While there was no PD-L1 L88 mutation at baseline, we found a PD-L1 L88fs mutation at week 9 of the treatment protocol (Figures 1B, C, Table 1). Recapitulation of nucleotide exchanges suggested that the alteration had developed in several steps: at first, a TT to GA mutation (PD-L1 L88E) followed by a GA to G deletion (PD-L1 L88fs) leading to a premature stop codon at position 104 (Figures 1C, D). Interestingly, patient 05-001 showed homozygosity for the FCGR3A high-affinity SNP rs396991 that was found associated with PD-L1 alterations in the AVETUX trial (11). To confirm the mutations detected by next-generation sequencing (NGS) with a more sensitive and specific method, we designed ddPCR primers for both genetic lesions and subjected all cfDNAs collected in patient 05-001 to serial ddPCR testing. This analysis confirmed the mutations identified via NGS showing comparable variant allele frequencies at week 9 and EOT (Table 1, Supplementary Figure S1), while baseline tumor tissue as well as germline leukocyte DNA was not mutated in this position. Notably, we did not detect acquired EGFR mutations mediating secondary resistance toward cetuximab.
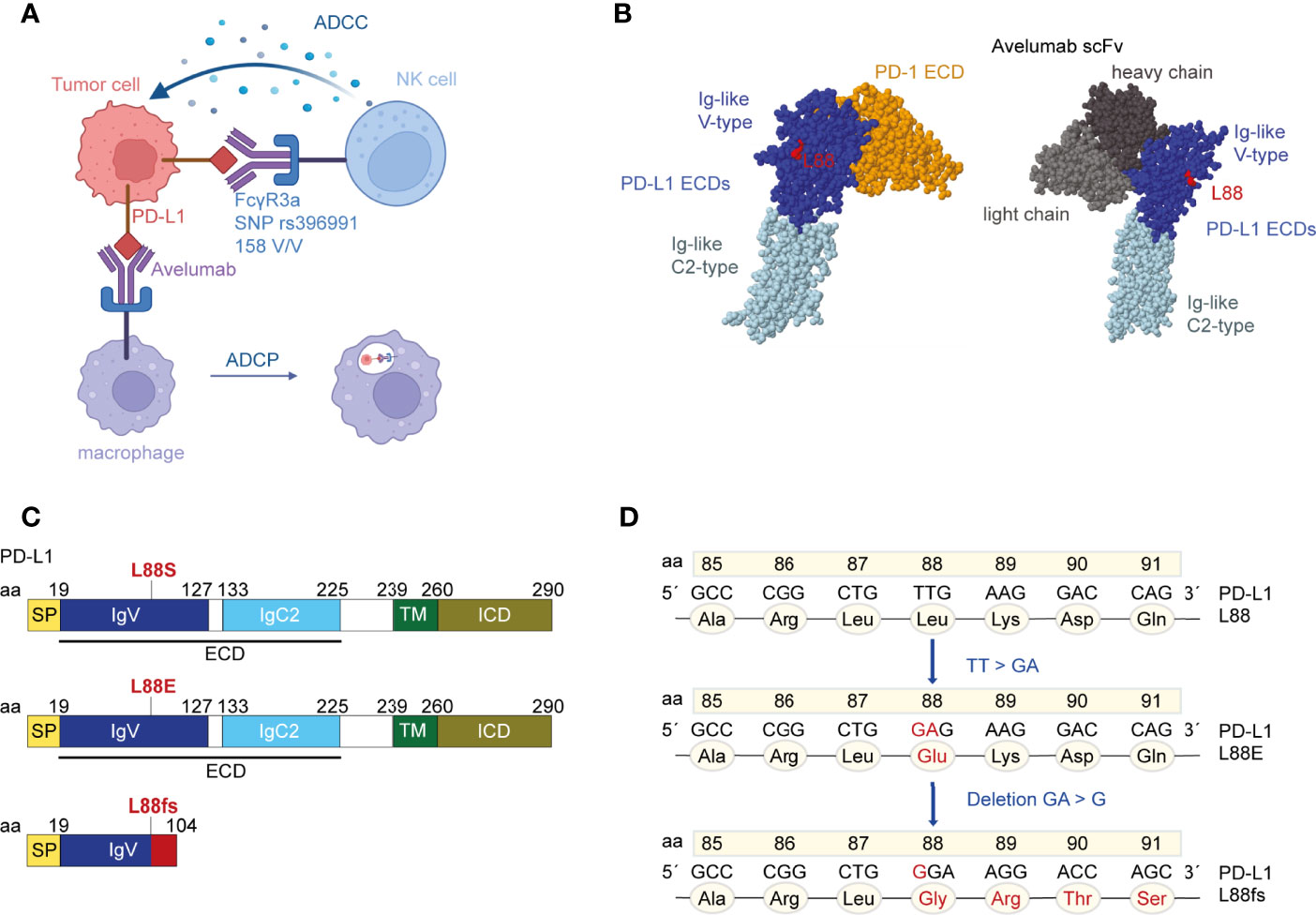
Figure 1 The localization of residue leucine 88 in the extracellular domains (ECDs) of programmed cell death ligand 1 (PD-L1). (A) Binding of avelumab to PD-L1 promotes antibody-dependent cell-mediated cytotoxicity (ADCC) or antibody-dependent cell phagocytosis (ADCP) against tumor cells via the Fcγ receptor. Patient 05-001 showed homozygosity for the FCGR3A SNP rs396991 (amino acid 158V/V, also known as 176V/V) that is known to mediate high-affinity binding of NK cells to IgG1 (12, 16). The figure was created with BioRender.com. (B) Crystal structure of PD-L1 ECDs in complex with PD-1 ECD [PDB ID: 3BIK (17)] and crystal structure of PD-L1 ECDs in complex with an avelumab single-chain variable fragment (scFv) [PDB ID: 5GRJ (18)]. Leucine 88 is highlighted in red. Space-filling models were created with RasMol (19). (C) Schematic structure of the human PD-L1 peptide with indications where the novel mutations theoretically introduce changes. PD-L1 L88fs leads to a premature termination codon and consecutively to loss of the transmembrane domain. (D) Nucleotide and amino acid exchanges found in patient 05-001 at week 9 and EOT in the PD-L1 L88 position.

Table 1 PD-L1 mutation frequencies in patient 05-001 across different timepoints.
PD-L1 L88E Leads to Reduced Cellular PD-L1 Levels
As part of the extracellular domain, the PD-L1 L88fs mutation leads to full abrogation of PD-L1 cell surface expression due to loss of the transmembrane domain (Figure 1C). This has been previously shown for a different frameshift mutation (PD-L1 K162fs) that was equally localized in the extracellular domain (11). In contrast, the functional consequences of PD-L1 L88E are unknown. We recently detected expanding subclones with PD-L1 L88S mutations in two colorectal cancer patients from the AVETUX trial (11). The introduction of a new serine phosphorylation site diminished protein stability and fostered proteasomal degradation of PD-L1 (11). Since glutamic acid (E) or aspartic acid (D) can mimic the functional effects of a phosphorylated serine residue when replacing it in the phosphorylated target site (phosphomimetic mutations) (20), we hypothesized that this mechanism also applies to PD-L1 L88E. To test this hypothesis, we lentivirally transduced a PD-L1 L88E expression construct into the UT-SSC-14 and HT-29 cell lines that were depleted of endogenous PD-L1 expression by CRISPR/Cas9 (11). PD-L1 depletion of these cell lines did not affect their viability (11). As a control, the PD-L1 wild-type as well as PD-L1 L88A and L88M—two mutants without suspected phosphomimetic properties—was used. In addition, UT-SSC-14 and HT-29 cells overexpressing PD-L1 L88S served as control for enhanced degradation. While all cells expressing PD-L1 variants showed equal or slightly elevated transcript levels as compared to the wild-type (Figure 2A), protein abundancy varied depending on the expressed variant but with comparable patterns in flow cytometry and Western blotting: while the expression of the non-phosphomimetic L88A and L88M control mutants was equal (UT-SSC-14) or only minimally reduced (HT-29) as compared to the wild-type, both PD-L1 L88S and PD-L1 L88E showed substantial reduction of surface PD-L1 (Figure 2B) as well as total PD-L1 levels in the cell (Figure 2C).
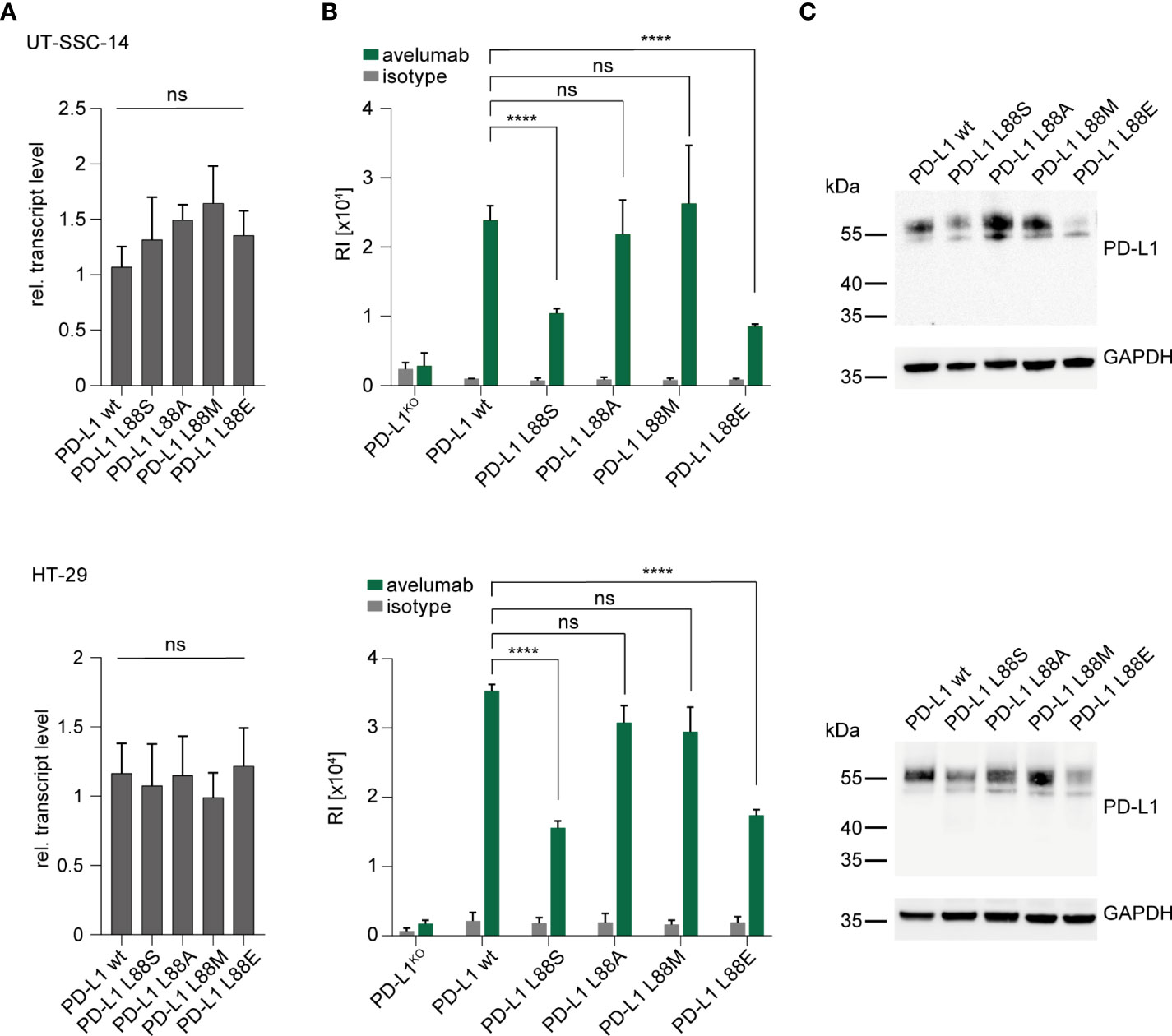
Figure 2 Introduction of new PD-L1 variants into PD-L1-deficient tumor cells. (A–C) UT-SSC-14 and HT-29 cells ectopically expressing PD-L1 L88S, L88A, L88M, and L88E were analyzed by (A) qRT-PCR (replicates n = 4), (B) flow cytometry (replicates n = 4-6), and (C) Western blotting in comparison with PD-L1 wt. Data are presented as mean ± SD. RI, relative mean fluorescence intensity. Statistics: Welch’s ANOVA over all groups (A) with post-hoc Dunnett’s test (B). Asterisks indicate p-value range (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns > 0.05).
PD-L1 L88E Induces Increased Proteasomal Degradation
The interplay of phosphorylation-induced degradation and glycosylation of PD-L1 is a key feature of homeostatic PD-L1 turnover and stability (21–23). To test the phosphomimetic potential of PD-L1 L88E, we first assessed whether L88E is reactive to inhibition of the AMP-activated protein kinase (AMPK) which was shown to phosphorylate serine on codon 88 of PD-L1, thus enhancing degradation (11). For this purpose, we treated the cell models with the AMPK inhibitor compound C (dorsomorphin) and quantified protein levels after 2 and 4 h using immunoblotting. As shown in Figures 3A, B, the protein levels of the PD-L1 wild-type as well as the L88A, L88M, and L88E variants were not affected by AMPK inhibition, while the amounts of PD-L1 L88S were enriched over time. This suggested that PD-L1 E88 was not phosphorylated by this kinase (Figure 3).
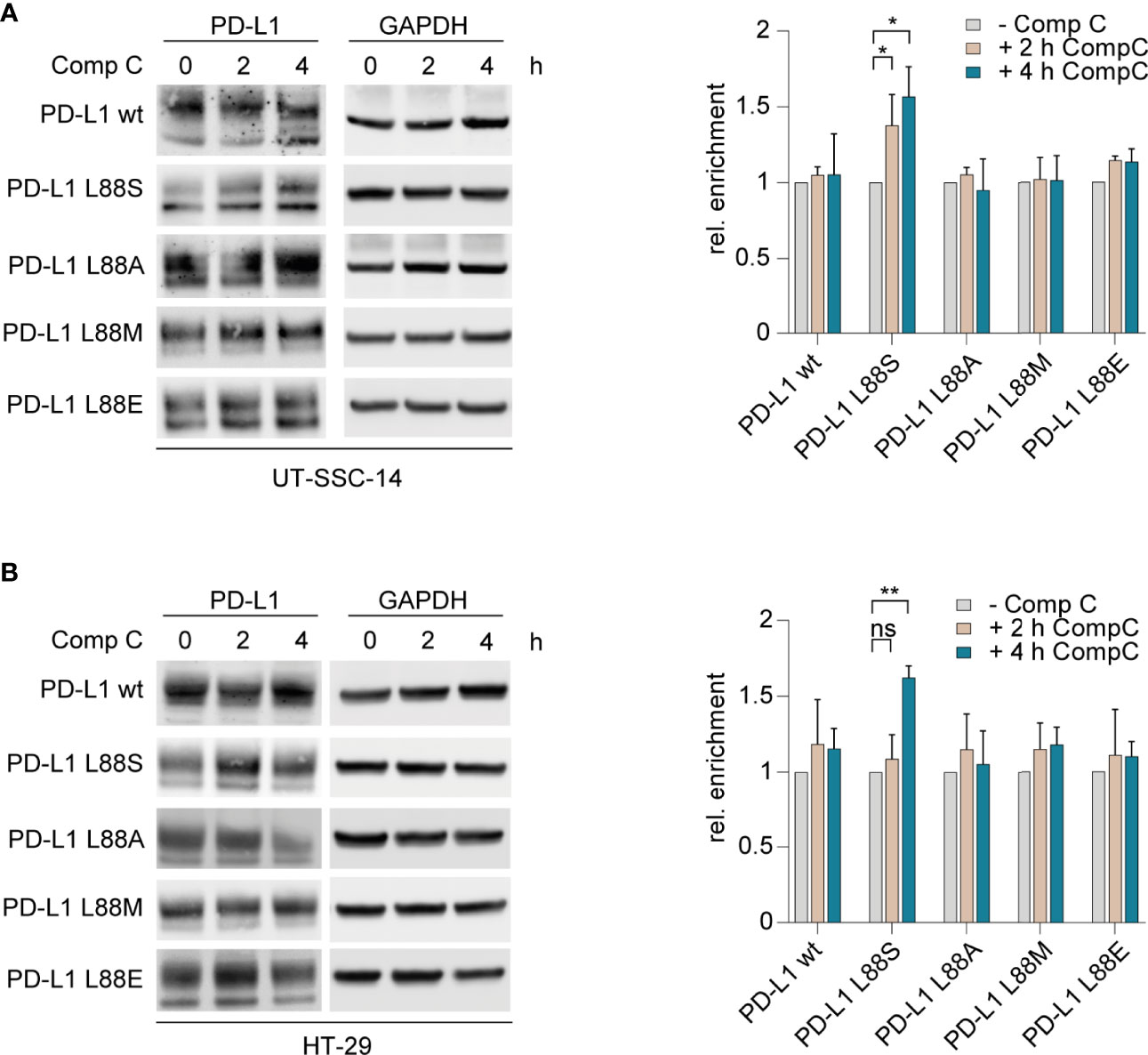
Figure 3 Inhibition of AMPK and examination of its effect on different PD-L1 variants. (A) UT-SCC-14 or (B) HT-29 cells overexpressing five different PD-L1 variants were treated with compound C (CompC) to inhibit AMPK, and cell lysates were analyzed by Western blotting (n = 3–4) before and after 2 and 4 h of treatment. Band intensities of at least two independent experiments were quantified using ImageJ, and enrichment of PD-L1 was calculated relative to untreated controls after normalization to GAPDH (24). Data are presented as mean ± SD. Statistics: two-sided unpaired t-test. Asterisks indicate p-value range (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns > 0.05).
To confirm the phosphomimetic properties of the L88E mutation, we next investigated the degradation of the PD-L1 variants as readout. For this purpose, we treated the cell models with MG132, an inhibitor of the 26S proteasome, for 4 h. Immunoblotting revealed that PD-L1 L88S and L88E were enriched faster than the wild-type and the L88A and L88M variants (Figures 4A, B).
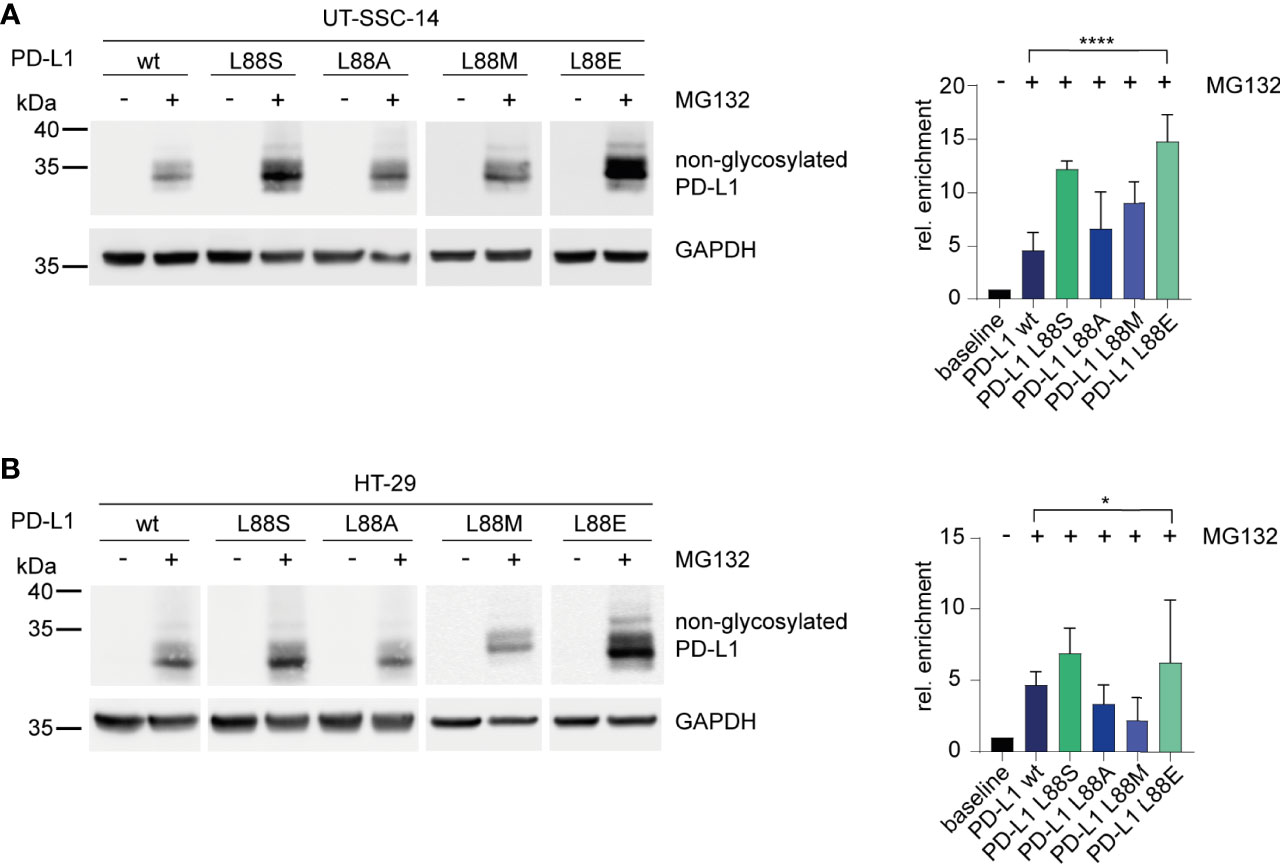
Figure 4 Inhibition of the 26S proteasome and examination of its effect on different PD-L1 variants. (A) UT-SCC-14 or (B) HT-29 cells overexpressing five different PD-L1 variants were treated with MG132 to block the 26S proteasome, and cell lysates were analyzed by Western blotting before and after treatment. Band intensities of non-glycosylated PD-L1 derived from at least three independent experiments were quantified using ImageJ, and enrichment of PD-L1 was calculated relative to untreated samples (=baseline) after normalization to GAPDH concentrations (24). Data are presented as mean ± SD. Statistics: one-way ANOVA over all groups. Asterisks indicate p-value range (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns > 0.05).
Discussion
We previously showed that direct tumor cell killing by ADCC can be considered a clinically relevant mechanism of action of avelumab, a PD-L1-directed immune checkpoint inhibiting antibody with preserved effector function (11). Secondary resistance by epitope escape—as seen in many cancer therapies targeted at a single antigen at the tumor cell surface [e.g (25–27).,]—is therefore not unexpected.
In the work presented here, we dissect the functional consequences of mutations at amino acid position 88 of PD-L1 that have been acquired in colorectal cancer patients across two clinical trials including the PD-L1 antibody avelumab as part of their experimental protocol. In the AVETUX and FIRE-6 studies, 72 patients were sequenced for the germline FCGR3A SNP as well as for PD-L1 mutations at baseline and end of treatment. None of the patients showed a PD-L1 mutation at baseline. In total, 3 of 7 patients that were homozygous for the FCGR3A SNP and 1 of 22 patients heterozygous for the FCGR3A SNP acquired a PD-L1 mutation on treatment. None of the 43 FCGR3A wild-type patients acquired a PD-L1 mutation in the course of avelumab treatment. The emergence of such mutations exclusively in patients expressing the FCGR3A V allele indicates that avelumab’s therapeutic pressure may largely be mediated by tumor-infiltrating innate immune effector cells, especially NK cells. NK cells from individuals with the SNP rs396991 (V/V and V/F) have been shown to have an especially high affinity to IgG (12, 28), thus enhancing their ability to mediate effective ADCC toward tumor cells coated with avelumab.
Two different alterations at position 88 of PD-L1 have been identified in human patient samples by our group: an exchange from leucine to serine introducing a novel phosphorylation site that facilitates PD-L1 degradation and the exchange from leucine to glutamic acid. The experiments presented here demonstrate that E88 represents a phosphomimetic exchange with comparable consequences as S88 in terms of reduced PD-L1 protein stability and membrane expression resulting in significantly reduced avelumab binding. Notably, our data are in agreement with the post-translational regulation of PD-L1 stability which represents a key mechanism mediating its biological functions (21, 22, 29, 30). The stability of PD-L1 is conferred by its glycosylation (asparagine residues 192, 200, and 219) which masks targeted phosphosites (threonine 180, serine 184, serine 195) and/or prevents the binding of several kinases like GSK3β (21), CDK4/6 (22), or AMPK (29) which phosphorylate PD-L1 and thus induce its degradation via the proteasome or the ER-associated protein degradation (ERAD). The acquisition of another phosphorylated serine outside this region allows glycosylation-independent phosphorylation-induced proteasomal degradation. In contrast, the PD-L1 L88fs leads to full PD-L1 loss.
The detection of PD-L1 L88fs at an earlier timepoint (week 9) than PD-L1 L88E (detected at EOT) appeared counterintuitive since our nucleotide sequence analysis suggested that the L88fs variant emerged from the L88E variant. Interestingly, the “precursor” variant L88E was undetectable both by NGS and ddPCR at week 9. It appears likely that this is rather a matter of sensitivity of detection, but it shows that this variant—at this point in treatment—may have shown a lower biological fitness than the full PD-L1 loss variant L88fs. The strong selective advantage of the L88fs variant in the early treatment phase was likely due to its potent escape from direct antitumor pressure exerted by avelumab. In later phases, this selective advantage might have been counterbalanced by a higher T cell pressure suppressing this PD-L1-negative clone. In the long run, the reduced but not completely absent PD-L1 expression of clone PD-L1 L88E may have offered protection from avelumab’s direct antitumor effects while at the same time counteracting T cell pressure. Notably, other therapeutic anti-PD-L1 antibodies like atezolizumab and durvalumab have IgG regions specifically designed to avoid effector functions (8, 9). Since they only function as blockers of the PD-1/PD-L1 interaction, it is very likely that patients treated with these antibodies will not select PD-L1 escape mutations as observed under avelumab treatment.
From a tumor biological perspective, these data are interesting since they show the complexity of subclonal selection principles with drugs that have different mechanisms of action. Under such conditions, only tumor subclones with a multidimensional escape to different selective forces will establish themselves successfully in the long run. On the other hand, our data show the importance to target surface molecules and epitopes that are of biological importance for the tumor. If this is not the case, the tumor will abrogate surface expression or express an epitope-disrupted but still functional receptor as has been previously shown for EGFR antibodies and CD19-targeting cell therapies (25–27, 31, 32). The finding of a close association between FCGR3A V allele expression and PD-L1 escape mutation suggests that the selective direct antitumor pressure needs to be extremely high to allow for the selection of such mutants that—on the other hand—show compromised biological fitness due to reduced inactivation of T cells.
Taken together, our mutational data from cancer patients and the subsequent dissection of functional consequences of these mutations show that amino acid position 88 of PD-L1 represents a hotspot that critically influences PD-L1 membrane expression. These data have relevance for immune checkpoint blockade in solid tumors using PD-L1 antibodies with preserved effector functions.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/ena, PRJEB51424.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethical Commission of the Ludwig-Maximilians-University Munich (LMU): 18-0933. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
Conception and design: LC, CS, and MB. Collection and assembly of data: VH, SS, LC, CS, RS, LP, and DS. Data analysis and interpretation: LC, CS, RS, LP, and DS. Manuscript writing and final approval of the manuscript: all authors.
Funding
This work was funded by Deutsche Krebshilfe (70114663 to MB).
Conflict of Interest
SS received honoraria for talks and advisory board role from Amgen, Astra-Zeneca, Bayer, BMS, ESAI, LEO Pharma, Lilly, Merck KGaA, MSD, Pierre-Fabre, Roche, Sanofi, Servier, Taiho, and Takeda and research funding from Merck KGaA, Pierre-Fabre, Servier, and Roche. VH received honoraria for talks and advisory board role from Merck, Amgen, Roche, Sanofi, Sirtex, Servier, Pfizer, Pierre-Fabre, AstraZeneca, BMS, MSD, Novartis, Boehringer Ingelheim, Pierre-Fabre, Celgene, Terumo, Oncosil, and Seagen and research funding from Merck, Amgen, Roche, Sanofi, Pfizer, Boehringer-Ingelheim, Sirtex, and Servier.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank all patients and families as well as all participating study centers, especially the University Hospital Munich (LMU) and Charité – Universaetsmedizin Berlin. We thank Aline Patzschke, Christoph Wosiek, and Jenny Wehde for excellent technical assistance. We would also like to thank Alexander Navarrete-Santos and the cell sorting core facility at the University Hospital Halle (Saale).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.941666/full#supplementary-material
References
1. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-Associated B7-H1 Promotes T-Cell Apoptosis: A Potential Mechanism of Immune Evasion. Nat Med (2002) 8(8):793–800. doi: 10.1038/nm730
2. Lau J, Cheung J, Navarro A, Lianoglou S, Haley B, Totpal K, et al. Tumour and Host Cell PD-L1 Is Required to Mediate Suppression of Anti-Tumour Immunity in Mice. Nat Commun (2017) 8:14572. doi: 10.1038/ncomms14572
3. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T Cell Costimulatory Receptor CD28 Is a Primary Target for PD-1-Mediated Inhibition. Science (2017) 355(6332):1428–33. doi: 10.1126/science.aaf1292
4. Ribas A, Wolchok JD. Cancer Immunotherapy Using Checkpoint Blockade. Science (2018) 359(6382):1350–5. doi: 10.1126/science.aar4060
5. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and Affinity of Human Fcgamma Receptors and Their Polymorphic Variants for Human IgG Subclasses. Blood (2009) 113(16):3716–25. doi: 10.1182/blood-2008-09-179754
6. Wang C, Thudium KB, Han M, Wang XT, Huang H, Feingersh D, et al. In Vitro Characterization of the Anti-PD-1 Antibody Nivolumab, BMS-936558, and In Vivo Toxicology in non-Human Primates. Cancer Immunol Res (2014) 2(9):846–56. doi: 10.1158/2326-6066.CIR-14-0040
7. Zhang T, Song X, Xu L, Ma J, Zhang Y, Gong W, et al. The Binding of an Anti-PD-1 Antibody to FcgammaRIota has a Profound Impact on its Biological Functions. Cancer Immunol Immunother (2018) 67(7):1079–90. doi: 10.1007/s00262-018-2160-x
8. Stewart R, Morrow M, Hammond SA, Mulgrew K, Marcus D, Poon E, et al. Identification and Characterization of MEDI4736, an Antagonistic Anti-PD-L1 Monoclonal Antibody. Cancer Immunol Res (2015) 3(9):1052–62. doi: 10.1158/2326-6066.CIR-14-0191
9. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (Anti-PD-L1) Treatment Leads to Clinical Activity in Metastatic Bladder Cancer. Nature (2014) 515(7528):558–62. doi: 10.1038/nature13904
10. Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, et al. Antibody-Dependent Cellular Cytotoxicity Activity of a Novel Anti-PD-L1 Antibody Avelumab (MSB0010718C) on Human Tumor Cells. Cancer Immunol Res (2015) 3(10):1148–57. doi: 10.1158/2326-6066.CIR-15-0059
11. Stein A, Simnica D, Schultheiss C, Scholz R, Tintelnot J, Gokkurt E, et al. PD-L1 Targeting and Subclonal Immune Escape Mediated by PD-L1 Mutations in Metastatic Colorectal Cancer. J Immunother Cancer (2021) 9(7):e002844. doi: 10.1136/jitc-2021-002844
12. Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Fcγriiia-158V/F Polymorphism Influences the Binding of IgG by Natural Killer Cell Fcγriiia, Independently of the Fcγriiia-48L/R/H Phenotype. Blood (1997) 90(3):1109–14. doi: 10.1182/blood.V90.3.1109
13. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The Cbio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov (2012) 2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095
14. Team RC. R: A Language and Environment for Statistical Computing Vienna, Austria: R Foundation for Statistical Computing (2018). Available at: https://www.R-project.org.
15. Tintelnot J, Baum N, Schultheiss C, Braig F, Trentmann M, Finter J, et al. Nanobody Targeting of Epidermal Growth Factor Receptor (EGFR) Ectodomain Variants Overcomes Resistance to Therapeutic EGFR Antibodies. Mol Cancer Ther (2019) 18(4):823–33. doi: 10.1158/1535-7163.MCT-18-0849
16. Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, et al. A Novel Polymorphism of FcgammaRIIIa (CD16) Alters Receptor Function and Predisposes to Autoimmune Disease. J Clin Invest (1997) 100(5):1059–70. doi: 10.1172/JCI119616
17. Lin DY, Tanaka Y, Iwasaki M, Gittis AG, Su HP, Mikami B, et al. The PD-1/PD-L1 Complex Resembles the Antigen-Binding Fv Domains of Antibodies and T Cell Receptors. receptors. Proc Natl Acad Sci USA (2008) 105(8):3011–6. doi: 10.1073/pnas.0712278105
18. Liu K, Tan S, Chai Y, Chen D, Song H, Zhang CW-H, et al. Structural Basis of Anti-PD-L1 Nonoclonal Antibody Avelumab for Tumor Therapy. Cell Res (2016) 27(1):151–3. doi: 10.1038/cr.2016.102
19. Sayle RA, Milner-White EJ. RASMOL: Biomolecular Graphics for All. Trends Biochem Sci (1995) 20(9):374. doi: 10.1016/S0968-0004(00)89080-5
20. Pearlman SM, Serber Z, Ferrell JE Jr. A Mechanism for the Evolution of Phosphorylation Sites. Cell (2011) 147(4):934–46. doi: 10.1016/j.cell.2011.08.052
21. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and Stabilization of Programmed Death Ligand-1 Suppresses T-Cell Activity. Nat Commun (2016) 7:12632. doi: 10.1038/ncomms12632
22. Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 Kinase Destabilizes PD-L1 via Cullin 3-SPOP to Control Cancer Immune Surveillance. Nature (2018) 553(7686):91–5. doi: 10.1038/nature25015
23. Chan LC, Li CW, Xia W, Hsu JM, Lee HH, Cha JH, et al. IL-6/JAK1 Pathway Drives PD-L1 Y112 Phosphorylation to Promote Cancer Immune Evasion. J Clin Invest (2019) 129(8):3324–38. doi: 10.1172/JCI126022
24. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 Years of Image Analysis. Nat Methods (2012) 9(7):671–5. doi: 10.1038/nmeth.2089
25. Braig F, März M, Schieferdecker A, Schulte A, Voigt M, Stein A, et al. Epidermal Growth Factor Receptor Mutation Mediates Cross-Resistance to Panitumumab and Cetuximab in Gastrointestinal Cancer. Oncotarget (2015) 6(14):12035–47. doi: 10.18632/oncotarget.3574
26. Braig F, Brandt A, Goebeler M, Tony HP, Kurze AK, Nollau P, et al. Resistance to Anti-CD19/CD3 BiTE in Acute Lymphoblastic Leukemia May Be Mediated by Disrupted CD19 Membrane Trafficking. Blood (2017) 129(1):100–4. doi: 10.1182/blood-2016-05-718395
27. Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal Evolution and Resistance to EGFR Blockade in the Blood of Colorectal Cancer Patients. Nat Med (2015) 21(7):795–801. doi: 10.1038/nm.3870
28. Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High Resolution Mapping of the Binding Site on Human IgG1 for Fc Gamma RI, Fc Gamma RII, Fc Gamma RIII, and FcRn and Design of IgG1 Variants With Improved Binding to the Fc Gamma R. J Biol Chem (2001) 276(9):6591–604. doi: 10.1074/jbc.M009483200
29. Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol Cell (2018) 71(4):606–20 e7. doi: 10.1016/j.molcel.2018.07.030
30. Wang Y, Wang H, Yao H, Li C, Fang JY, Xu J. Regulation of PD-L1: Emerging Routes for Targeting Tumor Immune Evasion. Front Pharmacol (2018) 9:536. doi: 10.3389/fphar.2018.00536
31. Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, et al. Identification of a Mutation in the Extracellular Domain of the Epidermal Growth Factor Receptor Conferring Cetuximab Resistance in Colorectal Cancer. Nat Med (2012) 18(2):221–3. doi: 10.1038/nm.2609
Keywords: immune checkpoint blockade, PD-L1, avelumab, resistance, ddPCR
Citation: Claaß LV, Schultheiß C, Scholz R, Paschold L, Simnica D, Heinemann V, Stintzing S and Binder M (2022) PD-L1 Amino Acid Position 88 Represents a Hotspot for PD-L1 Stability With Relevance for PD-L1 Inhibition. Front. Oncol. 12:941666. doi: 10.3389/fonc.2022.941666
Received: 11 May 2022; Accepted: 14 June 2022;
Published: 22 July 2022.
Edited by:
Lekh N. Dahal, University of Liverpool, United KingdomReviewed by:
Ali Roghanian, University of Southampton, United KingdomPingli Mo, Xiamen University, China
Copyright © 2022 Claaß, Schultheiß, Scholz, Paschold, Simnica, Heinemann, Stintzing and Binder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mascha Binder, TWFzY2hhLkJpbmRlckB1ay1oYWxsZS5kZQ==
†These authors have contributed equally to this work