 Mauro Di Vito Nolfi†
Mauro Di Vito Nolfi† Davide Vecchiotti†Irene Flati
Davide Vecchiotti†Irene Flati Daniela VerzellaMonica Di Padova
Daniela VerzellaMonica Di Padova Edoardo Alesse
Edoardo Alesse Daria Capece*
Daria Capece* Francesca Zazzeroni*
Francesca Zazzeroni*- Department of Biotechnological and Applied Clinical Sciences (DISCAB), University of L’Aquila, L’Aquila, Italy
Drug resistance is a major impediment to patient survival and remains the primary cause of unsuccessful cancer therapy. Drug resistance occurs in many tumors and is frequently induced by chemotherapy which triggers a defensive response both in cancerous and cancer-associated cells that constitute the tumor microenvironment (TME). Cell to cell communication within the TME is often mediated by extracellular vesicles (EVs) which carry specific tumor-promoting factors able to activate survival pathways and immune escape mechanisms, thus sustaining tumor progression and therapy resistance. NF-κB has been recognized as a crucial player in this context. NF-κB activation is involved in EVs release and EVs, in turn, can trigger NF-κB pathway activation in specific contexts, based on secreting cytotype and their specific delivered cargo. In this review, we discuss the role of NF-κB/EVs interplay that sustain chemoresistance in the TME by focusing on the molecular mechanisms that underlie inflammation, EVs release, and acquired drug resistance.
Introduction
Cancer chemotherapy resistance is the innate and/or acquired ability of cancer cells to escape the effects of chemotherapeutics and represents a great challenge in cancer therapy to improve clinical outcomes. The development of resistance occurs in many tumors and depends partially on genetic instability, heterogeneity, speedy mutation in the tumor cell, cytogenetic changes, and intra-neoplastic diversity (1, 2). The tumor microenvironment (TME) is also considered to be a factor for resistance development in many cancers, as chemotherapy frequently triggers a defensive response not only in cancerous cells but also in cancer-associated cells within the TME (3). Activation of survival pathways and immune escape mechanisms are often mediate by extracellular vesicles (EVs), which release their cargo in recipient cells within TME. EVs are lipid-contained vesicle, which are classified based on their size, biogenesis, and release mechanism. EVs include microvesicles (MVs), exosomes (EXs) and apoptotic bodies (ABs) (4). MVs are 100 to 1000 nm in size, EXs are smaller and range in size from 30 to 150 nm, while ABs show a size ranging from 50 to 5000 nm. MVs and ABs directly bud from cytoplasmic membrane, while EXs are produced by inward budding of plasma membrane and formation of endosomes, which mature into multivesicular bodies (MVBs) and subsequently were secreted into extracellular space (5, 6). Among EVs, exosomes are the best characterized. After formation, early endosome is processed in MVBs by the endosomal sorting complexes required for transport (ESCRT), directing by the invagination of MVBs outer membrane and packaging of biomolecules (7). EVs support intracellular communications within the TME by carrying specific tumor-promoting factors that positively regulate several pro-survival pathways including NF-κB, which plays a pivotal role in this context.
NF-κB transcription factor family consists of a group of inducible effectors which regulate a plethora of genes involved in several physiological processes such as inflammation (8), differentiation (9), survival (10), proliferation (11), and immunity (12). However, dysregulated NF-κB signaling is often found in many pathological conditions including inflammatory disorders (13), autoimmunity (14), and cancer (15). NF-κB family comprises five structurally related members, specifically, p65 (RelA), RelB, c-Rel, p50 (NF-κB1), and p52 (NF-κB2) that form homodimers and heterodimers able to specifically bind the consensus κB site 5’-GGGPuNNPyPyCC-3 (16) with different transcriptional activity (17). p50 and p52 are active forms of precursors proteins p105 and p100 respectively that, in turn, operate both as NF-κB precursors and inhibitors of NF-κB dimers (18). Activation of NF-κB pathway occurs through two major mechanisms, namely canonical (or classical) and non-canonical (or alternative) pathways (19). In absence of stimuli, NF-κB dimers are retained within the cytosol in inactive forms through their interaction with IκB (inhibitor of kappa B) proteins (20). In the canonical pathway, upon stimulation [i.e., lipopolysaccharide (LPS), tumor necrosis factor α (TNF-α)], IKK (IκB kinase) (21) proteins (IKKα, IKKβ, IKKγ) assemble into multiproteic complexes and trigger NF-κB activation. Specifically, in presence of stimuli, activated IKK complex phosphorylates IκB proteins, thus inducing its ubiquitination (22) and proteasomal degradation and allowing, in turn, NF-κB dimers to translocate into the nucleus and activate its target genes. In the non-canonical pathway, induced by a subset of tumor necrosis factor receptor (TNFR) superfamily members upon stimulation by several factors such as lymphotoxin B and BAFF, the activation of the heterodimers p100/RelB is triggered by NF-κB inducing kinase (NIK) that activates IKKα complex and in turn promote the processing of p100 in p52 and the nuclear translocation of the active p52/RelB dimer.
While the role of NF-κB in promoting cancer progression and drug resistance is well-known, emerging evidence are pointing out for a crucial interplay between NF-κB and EVs that sustain TME remodeling, tumor inflammation and therapy resistance. Here, we discuss this crosstalk by focusing on the molecular mechanisms that underlie inflammation, EVs release, and acquired drug resistance.
NF-κB: A Master Regulator of Inflammation and Therapy Resistance in Cancer
NF-κB Signaling in Inflammation and Cancer
During carcinogenesis, parenchymal cells continuously interact with the surrounding environment establishing a plethora of interactions with stromal cells (i.e., fibroblasts, endothelial cells, adipose cells, mesenchymal stem cells), immune cells, and extracellular matrix that constitute the TME (23). In this context, inflammation plays a key role in contributing to carcinogenesis and promoting the metastatic phenotype (24, 25). NF-κB constitutive activation is widely recognized as a hallmark of many types of tumors including hepatocellular carcinoma (26), breast cancer (27), lymphoid malignancies (28), colorectal (29) and prostate cancer (30). In addition to promoting tumor cell survival, oncogenic NF-κB signaling operates in the TME, thereby linking inflammation and cancer (15, 31). The inflammatory response is mainly induced by TNF-α, Interleukin-1β (IL-1β), and Interleukin-6 (IL-6). These cytokines usually are not overexpressed in healthy tissues but are significant upregulated in response to several pathological stimuli. Although released to protect the host, these cytokines often trigger a positive feedback mechanism promoting a chronic inflammation that, in turn, sustains, carcinogenesis and tumor progression (32). Accordingly, NF-κB activation in non-malignant tumor-associated cells has been shown to amplify the production of cytokines and other specialized effectors that promote tumor-cell proliferation, invasion and therapy resistance, while suppressing anti-tumor immune responses (33). Damage-associated molecular patterns (DAMPs), are endogenous molecules produced by dying cancer cells in response to stress and cell injury (34). After production, DAMPs are secreted through several mechanisms including extracellular vesicles (EVs) (35); these are recognized by specific pattern recognition receptors (PRRs) expressed on several cells, such as monocytes and macrophages, which activate different inflammatory pathways including NF-κB pathway. In turn, NF-κB activation causes the release of proinflammatory cytokines, such as IL-1β (36). As part of a positive feedback loop, active IL-1β binds to IL-1 receptors (IL-1R) on cancer cells and further stimulate NF-κB signaling, thus inducing the expression of pro-inflammatory cytokines TNF-α and IL-6 and sustaining NF-κB-mediated chronic inflammation (37–39). Moreover, enhanced or deregulated NF-κB activity in fibroblasts and macrophages promotes their switching to cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs), respectively, thus supporting tumor progression, vascularization and tumor growth, as observed in several cancers (40–45). NF-κB signaling also plays an important role in macrophages polarization (46); M1-type macrophages have a pro-inflammatory activity and tissue damaging properties, while M2 macrophages, with their anti-inflammatory phenotype, promote cell proliferation and tissue repair. Interestingly, although NF-κB represents a key transcription factor in M1 macrophage during the early stage of tumorigenesis, it also plays a pivotal role in advanced stages where it polarizes TAMs toward the immunosuppressive and tumor-promoting M2 phenotype. Indeed, we have demonstrated that NF-κB activation, through its target gene GADD45B, prevents TAM polarization to M1, thus inhibiting their antitumor activity (47, 48). Furthermore, it was observed that NF-κB p50 protein suppresses M1-type polarization and supports M2 immunosuppressive phenotype (49). The role of NF-κB in CAFs and TAMs polarization represents a hallmark of inflammatory TME and has been well explained in many excellent reviews (50, 51). In summary, as discussed below, NF-κB-mediated inflammation constitutes an important link between EVs activity and acquired drug resistance.
NF-κB-Driven Drug Resistance
In addition to promote initiation and tumor progression, NF-κB signaling fosters cancer resistance to chemotherapy. Although chemotherapy is the gold standard option for many types of cancers, multi-drug resistance (MDR) occurring in late/advanced stages significantly limits its long-term efficacy. Several authors reported that various anti-cancer drugs can activate NF-κB pathway by different mechanisms (Figure 1). The microtubule stabilizer paclitaxel, triggers NF-κB cascade by binding the toll-like receptor 4 (TLR4) (52). Other microtubule polymerization inhibitors, such as vinblastine and vincristine, can activate NF-κB, specifically by inducing protein kinase C-mediated phosphorylation and subsequent degradation of IκBα (53, 54). Another class of drugs able to induce NF-κB are the topoisomerases inhibitors, such as doxorubicin and SN38, that act by directly activating IKK complex (55). Once activated, NF-κB promotes chemoresistance in different ways, including the induction of anti-apoptotic genes, thus increasing resistance to drug-induced damage, and the overexpression of efflux pumps to prevent xenobiotic accumulation. In A549 human lung adenocarcinoma cells, chemotherapy-induced NF-κB activation leads to the expression of anti-apoptotic proteins like BCL-xL and BFL1 that in turn promote cancer cell survival (56). Furthermore, NF-κB induces resistance to apoptosis by upregulating the expression of inhibitors of apoptosis proteins (IAPs) (57) and suppressing the TNF-related apoptosis-inducing ligand (TRAIL) pathway (58). In addition, activation of NF-κB signaling can lead to chemoresistance by directly inducing the expression of efflux pumps proteins, such as human multidrug resistance protein 1 (MDR1), also known as P-glycoprotein 1 (P-gp), and ATP-binding cassette sub-family B member 1 (ABCB1). P-gp is an ATP-dependent transporter with a broad spectrum of activity, and it is able to efflux nonionic and amphipathic xenobiotics like anthracyclines, vinca-alkaloids and taxanes. It has been observed that NF-κB can transactivate the promoter of MDR1 (59) and that the inhibition of this signaling results in the downregulation of MDR1 in different types of cancers (60–62). NF-κB is also a transcriptional regulator of cyclooxygenase-2 (COX-2) (63), whose activity showed a strong correlation with P-gp expression in hepatocellular carcinoma (64) and colorectal cancer (65), and with multidrug resistance protein 4 (MRP4) in lung cancer (66). Another important mechanism in NF-κB-mediated drug resistance involves microRNAs (miRNAs). In this regard TRAIL cascade activation induces NF-κB with subsequent upregulation of miR-21 and miR-100, which in turn activate TNF Receptor Associated Factor 7 (TRAF7) and sustain NF-κB signaling, thus generating a positive feedback loop that suppresses TRAIL-induced apoptosis (67). NF-κB is also involved in radiotherapy resistance through the transcription of prosurvival genes that mediate resistance to ionizing radiation such as DNA-damage sensor ataxia-telangiectasia mutated (ATM) and cyclin B1 (68). In addition, overexpression of several miRNAs, such as miR-125b and miR-668, leads to NF-κB activation by targeting TNF alpha induced protein 3 (TNFAIP3/A20) and IκBα, respectively, thus promoting radioresistance in nasopharyngeal carcinoma (69) and in breast cancer (70). These findings show that NF-κB activation induces drug resistance at multiple levels and that its inhibition could represent an efficient approach to improve clinical outcome.
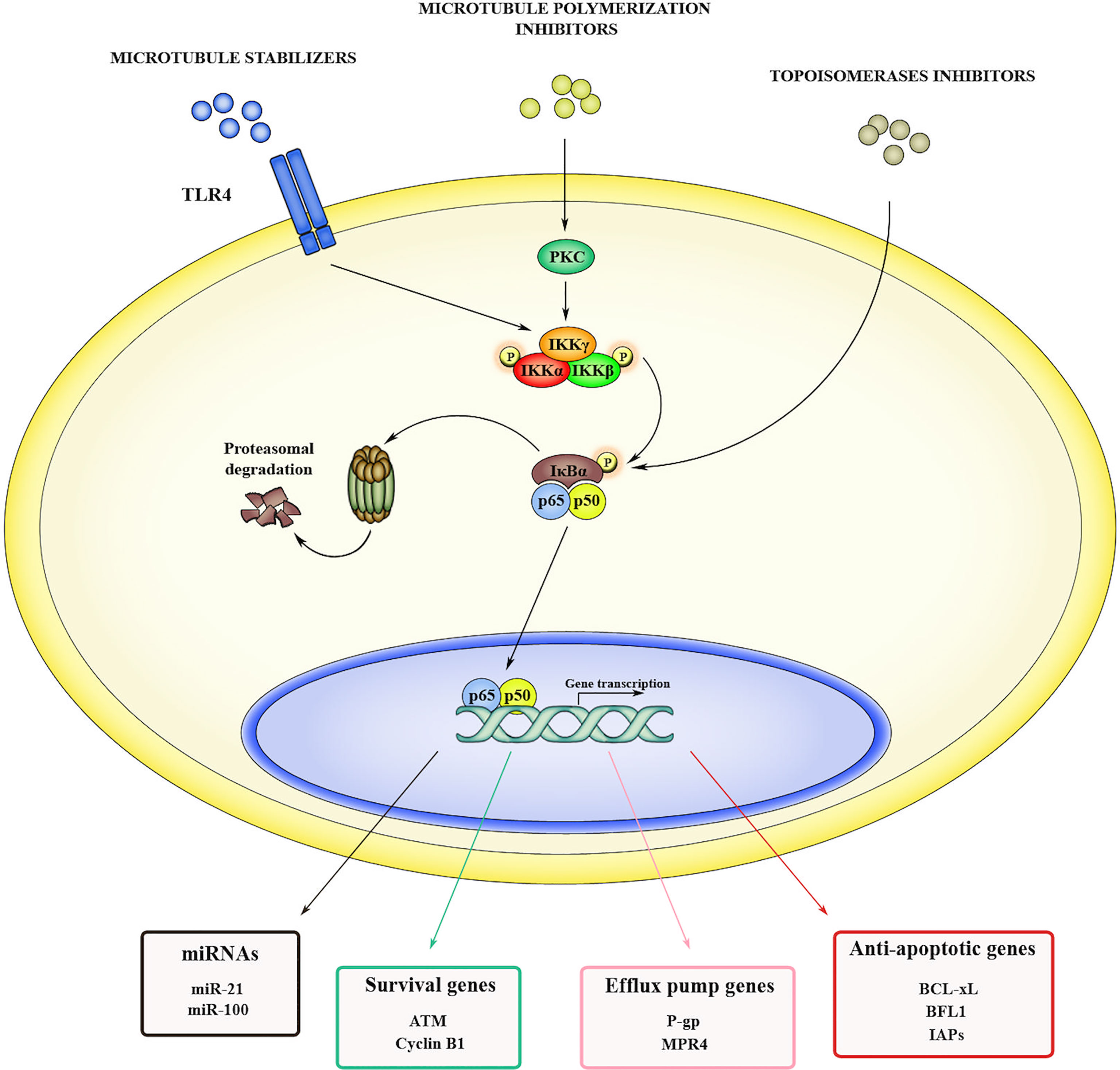
Figure 1 Mechanisms of NF-κB-induced drug resistance. NF-κB can be activated in cancer cells in response to several anticancer drugs, including microtubule stabilizers (i.e., paclitaxel), microtubule polymerization inhibitors (i.e., vinblastine and vincristine) and topoisomerases inhibitors (i.e., doxorubicin and SN38). When activated, NF-κB promotes drug resistance by inducing the transcription of genes such as miRNAs, as well as genes codifying for prosurvival factors, anti-apoptotic effectors, and efflux pumps.
EVs in TME-Cancer Cell Crosstalk and Drug Resistance
EVs Roles in the Bidirectional Cell to Cell Communication Within the TME
The crosstalk between cancerous and surrounding cells in TME through EVs is essential to sustain tumor growth, change cell phenotype and induce metastatic switch (71) (Figure 2). The key role of EVs in TME is due to their capacity to transfer proteins, lipids, nucleic acids, and membrane receptors, thus providing different information based on the specific composition of their cargo. Exosome secretion and its intracellular trafficking involve a subset of small GTPase named Rabs which are demonstrated to be expressed in a context-dependent manner (72–74). In some cancers, Rabs overexpression is linked with tumor progression and worse clinical outcome (75). Once released, exosomes interact with target cells through specific receptors or molecules such as intercellular adhesion molecule 1 (ICAM1) and heat shock protein 70 (Hsp70), expressed on dendritic cell-derived and on mast cell-derived exosomes, respectively. While ICAM 1 is recognized by lymphocyte function-associated antigen 1 (LFA-1) (76, 77), Hsp70 interacts with low density lipoprotein receptor-related protein 1 (LRP1/CD91) on antigen presenting cells (78). Due to their crucial role in cell-cell communications, exosomes activity in TME has been associated with tumor progression and induction of metastatic phenotype (79, 80). Importantly, the bidirectional cell to cell communication between tumor and stromal cells sustains cancer progression toward advanced stages (81). Within TME, the communication between CAFs and TAMs with tumor cells is mediated by CAFs/TAMs derived exosomes. It was observed that CAF-derived exosomes (CDEs) containing intact metabolites are able to promote oncogenic transformation, by inhibiting oxidative phosphorylation and increasing glycolytic metabolism (82, 83). Moreover, CAFs induce tumor growth and metastatic phenotype switching by producing exosomes with high concentration of TGF-1β (84). Lan et al. demonstrated that TAMs-derived exosomes also mediate cell migration and invasion in colon cancer via miRNAs (miR-21-5p and miR-155-5p)-mediated downregulation of BRG1 gene (85), whose decreased expression is responsible for Wnt/β-catenin signaling activation (86). On the other hand, tumor cells modulate and “re-educate” surrounding cells by secreting tumor-derived exosomes (TEXs). Xiao et al. showed that melanoma-derived EVs promote CAFs transformation by inducing VCAM-1 expression via extracellular signal-regulated kinase 1/2 (ERK1/2)-activation (87). Moreover, miR-155-5p in melanoma-derived exosomes triggers the proangiogenic switch of CAFs by targeting suppressor of cytokine signaling 1 (SOCS1), thus activating the janus kinase (JAK)2/signal transducer and activator of transcription (STAT)3 (JAK2/STAT3) pathway (88). As to TAMs transformation, Hsu and collaborators reported that in hypoxic lung cancer miR-103a-loaded EVs promoted M2 macrophage polarization by inhibiting phosphatase and tensin homolog (PTEN) and enhancing protein kinase B (PKB/Akt) and STAT3 activity (89). Moreover, ovarian cancer-derived exosomes induce macrophages polarization by delivering miR-222-3p which target SOCS3, thus sustaining STAT3 signaling (90). Furthermore, cancer stem cell (CSCs)-derived EVs (CSCDEs) play a significant role in TME remodeling. Sun et al. showed that glioblastoma stem cell (GSCs)-derived exosomes are able to confer stemness traits and enhance tumorigenicity in non-GSC glioma cells by delivering notch receptor 1 (notch1) protein and activating Notch signaling in recipient cells (91). Collectively, these studies underline the central roles of EVs in sustaining TME remodeling and fostering cancer progression.
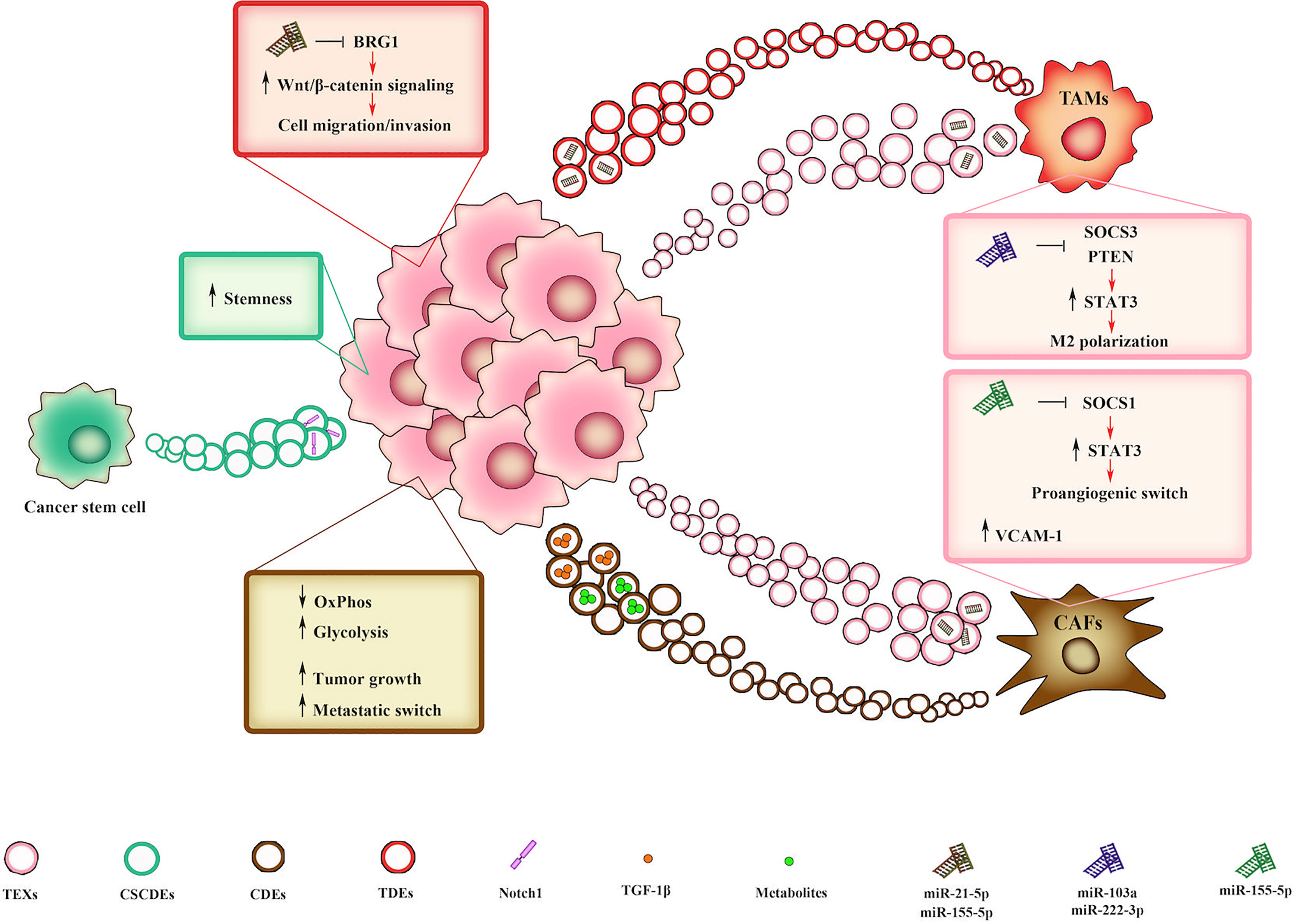
Figure 2 EV-mediated interplay within the TME mediates tumor progression. CAF-derived exosomes (CDEs) promote either metabolism deregulation or metastatic switch in tumor cells by delivering intact metabolites or TGF-β, respectively. TAMs-derived exosomes (TDEs) loaded with miR-21-5p and miR-155-5p can promote cell migration and invasion by downregulating BRG1 gene, thus sustaining Wnt/β-catenin signaling. CSCs-derived EVs (CSCDEs) confer stemness traits to recipient cells by delivering notch receptor 1 (notch1) protein. In turn, tumor-derived exosomes (TEXs) induce CAF transformation and macrophage M2 polarization by delivering miR-155-5p, miR-103a and miR-222-3p, that in turn affect STAT3 signaling.
EVs-Mediated Chemotherapy Resistance
In addition to promote tumor growth and progression, EVs also play a pivotal role in cancer drug resistance by taking part in several processes (i.e., direct removal of drugs, incorporation of efflux pumps and miRNAs/long non-coding RNAs (lncRNAs) delivery) (Figure 3). Direct removal of drugs from intracellular space has been reported by Shedden et colleagues; they observed the release of doxorubicin-containing vesicles in doxorubicin-treated MCF-7 breast cancer cells and demonstrated that vesicles accumulated the drug passively. Moreover, they also founded a correlation between vesicle-shedding-associated gene expression and doxorubicin resistance in several cancer cell lines (92). Furthermore, Federici and collaborators identified cisplatin-enriched exosomes in human metastatic melanoma cells and founded that EVs-mediated drug elimination is enhanced by tumor acidic microenvironment (93). EVs are also able to incorporate efflux pumps which can actively transport drugs into intraluminal space; drug resistant cancer cells can transfer efflux pumps via EVs to the sensitive surrounding ones, thus conferring them resistance traits (94–97). In addition, EVs can incorporate factors that induce the expression of efflux pumps. In this regard, Ma et al. demonstrated that adriamycin-resistant breast cancer cell line MCF-7/ADM showed elevated levels of Ca2+-permeable channel TRPC5, and that TRPC5 expression is essential for P-gp induction (98). The suppression of TRPC5 activity as well as of P-gp expression reduced drug resistance and tumor growth both in vitro and in vivo suggesting that inhibiting either P-gp or TRPC5 could be an attractive strategy to overcome drug resistance (98). They also founded that TRPC5-carrying EVs released from MCF-7/ADM cells can transfer chemoresistance properties to drug sensitive recipient cells (99). Accordingly, Wang et al. showed that exosomes-carrying Ca2+-permeable channel TRPC5 acts as a noninvasive chemoresistance marker that could predict chemoresistance in metastatic breast cancer patients (100). EVs can also transport drug-metabolizing enzymes, such as glutathione S-transferase P1 (GSTP1), that contribute to the intracellular detoxification as observed in chemotherapy-resistant breast cancer (101). Another crucial mechanism in drug resistance involves miRNA and lncRNA. As reported by Lunavat and collaborators, miR-211–5p, which is overexpressed in response to treatment with BRAF inhibitors (Vemurafenib and Dabrafenib), induced drug resistance in melanoma cells (102). Furthermore, Mikamori et al. evidenced that long exposure to gemcitabine increased miR-155-loaded EVs secretion in pancreatic ductal adenocarcinoma, thus conferring drug resistance through inhibition of pro-apoptotic stress-induced p53 target gene tumor protein p53-inducible nuclear protein 1 (TP53INP1) (103). Again, Shen et al. showed that breast cancer cells treated with sublethal dose of chemotherapeutic drugs released miR-9-5p, miR-195-5p, and miR-203a-3p-enriched EVs, which simultaneously targeted transcription factor One Cut Homeobox 2 (ONECUT2), thus conferring stemness and resistance traits in recipient cells (104). Qu et al. founded that, in renal cell carcinoma (RCC), lncARSR (lncRNA Activated in RCC with Sunitinib Resistance)-loaded exosomes competitively bind miR-34/miR-449 and enhance AXL and c-MET receptors signaling that are responsible for Sunitinib resistance through activation of STAT3, AKT, and ERK signaling (105). In light of this finding, lncARSR represent a potential therapeutic target to overcome sunitinib resistance in RCC (105). Other two lncRNAs involved in drug resistance are lncRNA urothelial carcinoma-associated 1 (UCA1) and lncRNA prostate androgen-regulated transcript 1 (PART1); Yang et al. founded that exosomal UCA1 is associated with Cetuximab resistance in colorectal cancer and could predict clinical outcome of Cetuximab therapy (106), while Kang and collaborators demonstrated that lncRNA PART1 is able to confer resistance to Gefitinib in esophageal squamous cell carcinoma by inducing B-cell lymphoma 2 (Bcl-2) expression through inhibition of miR-129 (107).
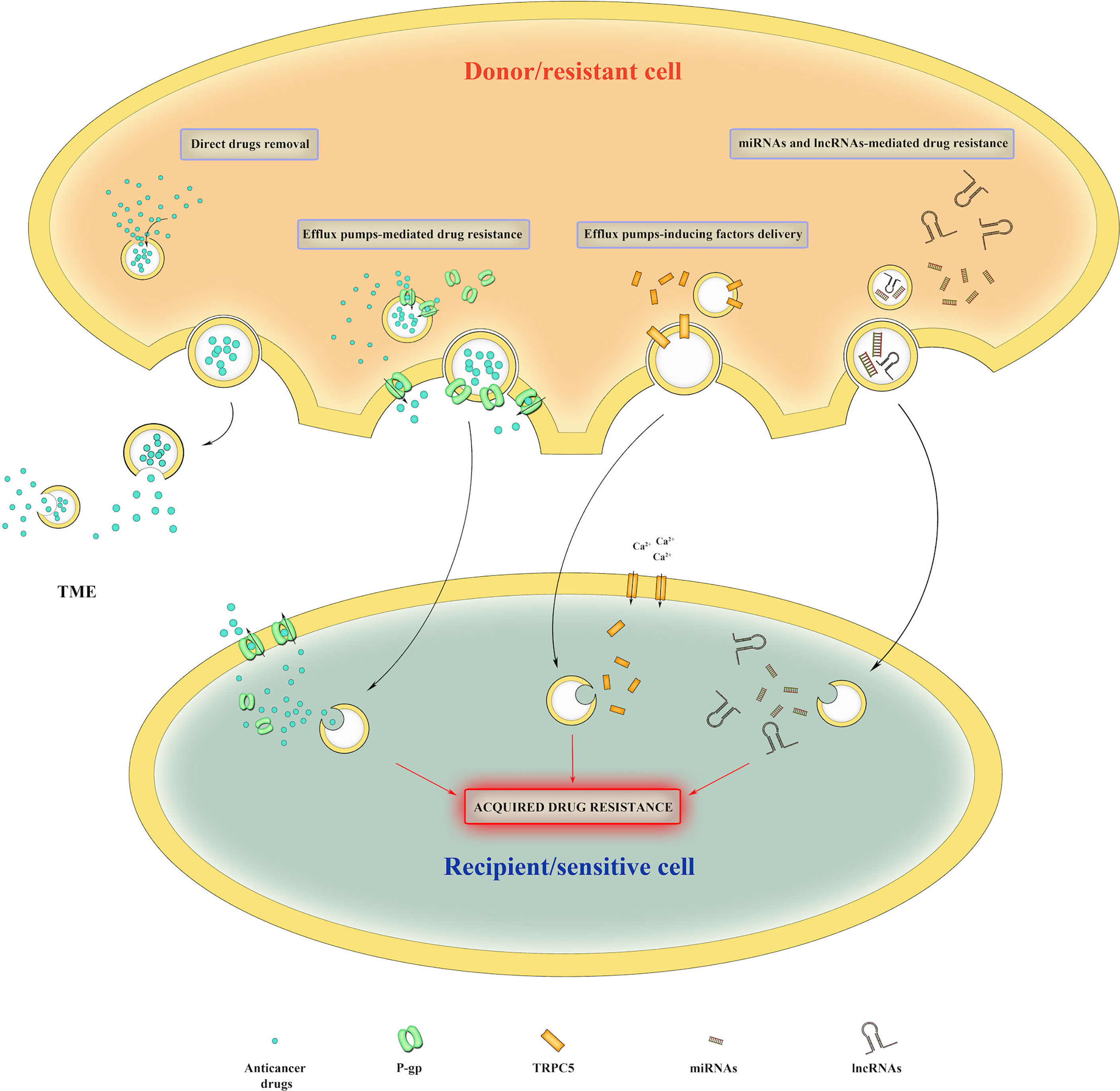
Figure 3 Mechanisms of EV-mediated drug resistance. EVs can directly remove intracellular drugs from cancer cells. They can also deliver efflux pumps, efflux pumps-inducing proteins, miRNAs and lncRNAs to recipient cells, thus conferring them drug resistance traits.
In summary, these findings show how EVs can exploit different mechanisms to sustain the acquisition of drug resistance by cancer cells, making them a central player in tumor evolution.
Linking NF-κB and EVs Activity in Cancer Progression and Therapy Resistance
Reciprocal Regulation Between EVs and NF-κB Signaling in the TME
NF-κB is directly involved in EVs trafficking and EVs-mediated chemoresistance, and, at the same time, EVs are responsible for NF-κB activation (Figure 4).
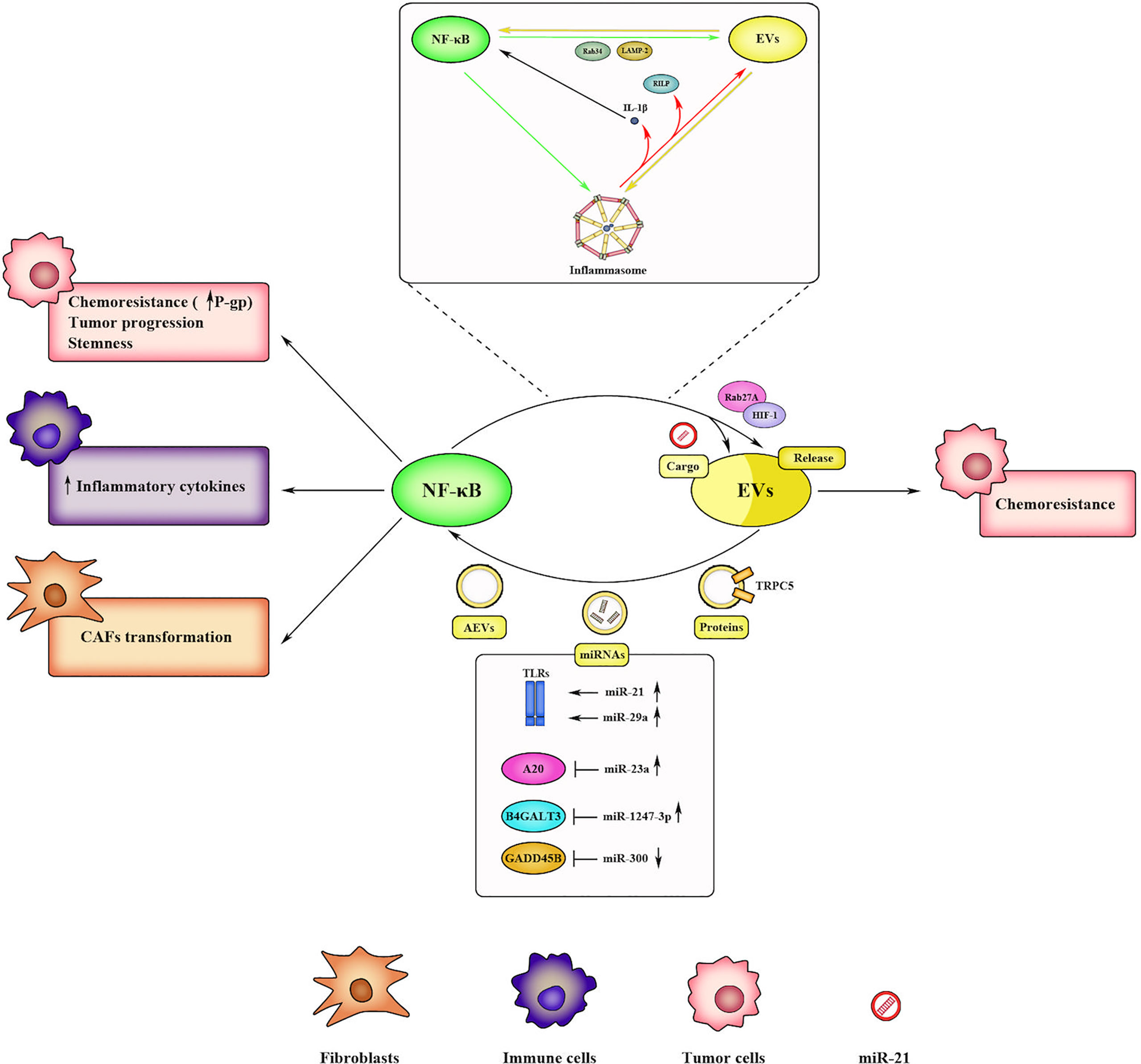
Figure 4 Bidirectional communication between NF-κB and EVs within the TME. NF-κB promotes EV-mediated chemoresistance in cancer cells by regulating the expression of EV-releasing factors (i.e., Rab27A, Rab34, LAMP-2, HIF-1). NF-κB also affects EV cargo by inducing miRNA transcription (i.e., miR-21). Reciprocally, EVs activate NF-κB through several mechanisms, such as by acting as DAMPs or delivering specific proteins and miRNAs. Upon activation, NF-κB induces chemoresistance, progression, and stemness in tumor cells, inflammatory cytokines release in immune cells, and CAFs transformation in fibroblasts. Inflammasome often mediates the crosstalk between NF-κB and EVs. Activated inflammasome promotes EV release through specific factors (i.e., RILP) and sustains NF-κB signaling through IL-1β production. In turn, NF-κB regulates inflammasome activation by promoting priming step, and reciprocally, EVs induce inflammasome signaling, thus generating a positive feedback loop.
Hypoxia is recognized as a hallmark of cancer (108) and promotes the immunosuppressive phenotype within the TME (109). In addition, hypoxia promotes EVs biogenesis and release in order to support intercellular communication and to compensate nutrient’s deficiency and oxygen starvation in cancer cells (110–112). In response to oxygen starvation, cancer cells activate hypoxia-inducible factors (HIFs) (113), a family of transcription factors involved in adaptation to hypoxic condition through modulation of angiogenesis, metastasis, and drug resistance (114), as well as in the regulation of small Rab GTPases (i.e., Rab22A) which in turn control intra- and intercellular trafficking of EVs (112). It is well-known that NF-κB, in response to inflammatory stimuli, upregulates HIF-1α and HIF-1β expression (115–117). Notably, hypoxia, per sé, can induce NF-κB, leading to an inflammatory response (118). Indeed, it has been observed that inhibition of oxygen sensors prolyl hydroxylases (PHDs) by low oxygen tension, stabilizes IKKβ with subsequent p65 nuclear accumulation and signaling transduction (119). Importantly, NF-κB is also able to directly regulate Rab proteins, as observed by Feng et al. in colon cancer stem cells. They identified a functional NF-κB binding site in the Rab27A promoter and observed that increased p65 levels induced Rab27A expression and enhanced EVs secretion in HT29 cells (120). In turn, Rab27A can promote tumor proliferation and chemoresistance via NF-κB, in bladder cancer (121). Indeed, Rab27A overexpression was associated with increased phosphorylation of p65 and increased expression of the antiapoptotic gene Bcl-2, and conversely, pharmacological inhibition of NF-κB by BAY 11-7082 abrogated cisplatin resistance and cancer cell survival (121). NF-κB activation also influence EVs cargo. Yang et al. reported an altered exosomal protein profile in NF-κB knockout mice following ischemia-reperfusion (I/R) injury in skeletal muscles, suggesting that NF-κB contributes to EVs production. Specifically, in the exosomes of NF-κB knockout mice, they observed upregulation of several proteins such as protease serine 1 and glyceraldehyde-3-phosphate dehydrogenase-like isoform 1, and downregulation of other factors including apolipoprotein B, complement component C3 prepropeptide, and immunoglobulin kappa light chain variable region (122). It has been reported that NF-κB can also drive transcription of specific miRNAs including miR-21; indeed, it was observed that miR-21-carrying exosomes released by TAMs induced cisplatin resistance in gastric cancer cells modulating PTEN/PI3K/AKT signaling pathway (123).
In accordance with the reciprocal interplay between EVs and NF-κB signaling, EVs can modulate NF-κB activity in recipient cells (Figure 4). Bretz et al. showed that exosomes from various body fluids stimulate the NF-κB-dependent production of pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-6 in monocytic cell line THP-1 via TLR2 and TLR4 activation (124). Gastric (125) and breast cancer (126) derived TEXs induce the expression of pro-inflammatory cytokines by macrophages via NF-κB, and the NF-κB activation in myeloid cells could involve TLR2 signaling (126). NF-κB pathway can be also regulated by specific EVs-delivered factors (i.e., proteins, miRNAs, lncRNAs), which can directly or indirectly activate/inhibit this pathway depending on the context. As previously mentioned, exosomes-carrying Ca2+-permeable channel TRPC5, responsible of P-gp-induced chemoresistance, are associated with IL-6 expression and increased phosphorylation of p65 in nasal polyps (127), suggesting a possible role of NF-κB in TRPC5-mediated drug resistance. Delivered miR-21 and miR-29a, can directly trigger NF-κB-mediated inflammatory response by binding murine TLR7 and human TLR8 (128). Commonly, miRNAs modulate NF-κB pathway in an indirect manner. Fang et al. showed that miR-1247-3p-carrying TEXs promote liver cancer stemness and chemoresistance via inhibition of beta-1,4-galactosyltransferase 3 (B4GALT3) and activation of β1-integrin–NF-κB pathway in fibroblasts (129). Activated CAFs further induce tumor progression by secreting pro-inflammatory cytokines, including IL-6 and IL-8 (129). Furthermore, Li and colleagues observed that miR-23a-enriched exosomes activate NF-κB pathway in macrophages by targeting its negative regulator A20 (130) while Chen and collaborators founded that exosomal miR-300 controls melanoma cell progression targeting GADD45B expression, a NF-κB-induced pro-survival factor (131). MiRNAs can also be downregulated to sustain NF-κB pathway. Wang et al. demonstrated that when the usually low expressed miR-192-5p is overexpressed in TAMs-derived exosomes, it suppresses endometrial cancer progression through NF-κB inhibition (132). With respect to lncRNAs, Li et al. founded that exosomal lncRNA FMR1-AS1 (FMR1 antisense RNA 1) is associated with CSC-like phenotype by binding TLR7 in female esophageal carcinoma (133). Moreover, expression of lncRNA BORG (BMP/OP-responsive gene) in triple-negative breast cancer (TNBC) (134) and lncRNA HOTAIR (HOX antisense intergenic RNA) (135) in colorectal cancer promotes chemoresistance via NF-κB. NF-κB activation can be triggered also by so-called “apoptotic exosome-like vesicles” (AEVs). Recently, Park and collaborators demonstrated that AEVs, during apoptotic process, act as DAMPs and activate NF-κB pathway, thus promoting an inflammatory response (136).
EVs and NF-κB Interplay: The Crucial Role of Inflammasome
Inflammasomes represent an important link between NF-κB and EVs activity in the TME (Figure 4). Inflammasomes are multimeric protein complexes that are assembled upon recognition of specific pathogen-associated molecular patterns (PAMPs) and DAMPs by PRRs (137) during innate immune response. Inflammasome consists of cytoplasmic PRRs ‘sensors’ such as nucleotide-binding domain and leucine-rich repeat receptors (NLRs), the ‘adapter’ apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain (ASC), and an ‘effector’ pro-caspase 1 (138). In this context, NF-κB plays a leading role; indeed, DAMPs are recognized by toll-like receptors (TLRs), a subset of membrane-bound PRRs which trigger NF-κB cascade and subsequent transcription of IL-1β (139). IL-1β is released as precursor protein (pro-IL-1β) and is processed in its active form by caspase-1 (140) through the formation of a multi-protein complex termed inflammasome. Although the existence of several NLRs has been reported, nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family pyrin domain containing 3 (NLRP3) is the best characterized inflammasome. Activated NF-κB upregulates NLRP3 (141, 142) that is necessary for the inflammasome assembly and for the pro-IL-1β processing. The process that lead to the inflammasome activation consist of two phases, priming and activation (143). In the priming step, NF-κB induce transcriptional upregulation of NLRP3 inflammasome components while the activation step led to full induction and inflammasome complex formation. In detail, NLRs act as DAMPs sensors leading to recruitment of ASC and subsequent activation of caspase-1 thus promoting processing of pro-IL-1β in active IL-1β. Importantly, it is known that inflammasome activation induce EVs secretion (144) by several mechanisms. One of those could be represented by de novo synthesis and production of IL-1β, which, in turn, activate NF-κB signaling and induce the expression of factors involved in EVs secretion. Gutierrez et al. demonstrated that activation of NF-κB can induce several membrane-trafficking regulators such as lysosome-associated membrane protein 2 (LAMP-2) and ras-related protein Rab34 (145). EVs secretion can also be promoted by inflammasome itself through caspase-1-dependent cleavage of the trafficking adaptor protein rab interacting lysosomal protein (RILP) (146). Furthermore, inflammasome-derived exosomes can activate NF-κB in macrophages leading to their pyroptosis via up-regulation of NLRP3 and pro-IL1β (147). While these findings show how inflammasome affects EVs secretion, it is worthy to note that EVs can also influence inflammasome activity, thus revealing a bidirectional crosstalk. Hence, EVs can either positively or negatively affect inflammasome activity depending on the nature of the EVs releasing cells (148). Exosomes from LPS-treated macrophages are able to activate NLRP3 inflammasome in AML-12 hepatocytes (149). Again, EVs from palmitate-treated Huh7 hepatocytes induce production of IL-1β in mouse bone marrow-derived macrophages (150) and exosomes from ARPE-19 exposed to photooxidative blue-light activate NLRP3 inflammasome in vitro (151). However, inflammasome activation is repressed in THP1 cells treated in vitro with human amniotic fluid derived EVs, as well as in cardiomyocytes isolated from a mouse model of doxorubicin-induced cardiotoxicity following treatment with embryonic stem cell-derived EVs (152, 153). Although inflammasome involvement in cancer is still debated and sometimes controversial, several findings underline its role in tumorigenesis, cancer progression and drug resistance (154). In this context, NLRP3 inflammasome activity is associated with carcinogenesis in head and neck squamous cell carcinoma (155) and with proliferation and migration in A549 lung cancer cells (156).
Further, TEX-mediated inflammasome activation in non-cancer cells within the TME was shown to exacerbate inflammatory response and sustain tumor progression. In prostate cancer, EVs released from advanced-stage tumor cells were found to activate inflammasome in non-cancerous prostate cells and induce M2 polarization in THP1 (157). Moreover, Liang et al. showed that tripartite motive containing 59 (TRIM59)-loaded EVs released from lung cancer cells promoted tumor progression in vitro and in vivo. Mechanistically, TRIM59 promoted abhydrolase domain containing 5 (ABHD5) degradation and activation of NLRP3 inflammasome in macrophages, which in turn released proinflammatory cytokines, thus sustaining cancer cell proliferation and invasion (158). Hwang et al. also demonstrated that colorectal CSCs-derived exosomes induced IL-1β expression in neutrophils via NF-κB, thus promoting tumorigenesis in colorectal cancer cells (159). Moreover, inhibition or deletion of inflammasome components NLRP3, ASC, or caspase-1 is protective against pancreatic ductal adenocarcinoma as it is associated with the reprogramming of innate and adaptive immunity in TME (160). Inhibition of NLRP3 inflammasome also suppresses metastatic potential of melanoma cancer cells (161). Inflammasome is also involved in drug resistance and can be activated following chemotherapeutic treatments, likely as result of NF-κB stimulation. Zhai et al. founded that NLRP1 inflammasome activation induces drug resistance in melanoma cells through release of IL-1β (162). Again, Theivanthiran et al. observed activation of NLRP3 inflammasome following treatment with anti-PD-1 checkpoint inhibitor with subsequent infiltration of granulocytic myeloid-derived suppressor cells and reduction of antitumor response (163). As reported by Feng et al. inflammasome activation is also involved in in 5-fluorouracil resistance of oral squamous cell carcinoma (164).
All together, these findings highlight an intricate interplay between NF-κB and EVs activity which influence tumor growth and drug resistance. Although further studies are needed to fully elucidate the molecular mechanisms underlying this reciprocal regulation between NF-κB and EVs, it could represent yet another way through which NF-κB orchestrate tumor behavior within the TME.
Conclusions
The bulk of evidence summarized herein shows that EVs play a key role in drug resistance through several mechanisms, including direct load and expulsion of chemotherapeutics, as well as delivery of pro-survival, anti-apoptotic, and stemness-associated factor cargo. The pharmacological target of EV biogenesis, uptake, and transfer to suppress chemoresistance has shown promise, but most of the data obtained were generated in vitro (165), and further in vivo investigation are needed. Another potential issue is represented by the heterogeneity of EVs, as not all EV communication is pro-tumorigenic. Thus, a better understanding of the molecular mechanisms underlying EV processes will significantly contribute to develop more specific approaches for overcoming drug resistance. In this review, we discuss the complex crosstalk between NF-κB pathway and EVs, as NF-κB activation affects EVs formation and release, and EVs, in turn, can trigger NF-κB activation. Since NF-κB/EVs interplay profoundly contribute to fuel aggressive disease and desensitize cancer cells to drugs, this axis could represent a promising target. However, as seen for EVs, targeting NF-κB core pathway produces off target effects due to the lack of anti-cancer specificity, as this factor is a master regulator of several biological processes. Therefore, the identification of NF-κB targets involved in this interplay in specific cancer models could be a useful strategy to overcome NF-κB/EV-mediated resistance.
Author Contributions
MDVN and DVec wrote the manuscript. IF, DVer and MP made the figures. EA DC and FZ revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The work was supported by the MIUR PRIN grant n°2017WLKYAM_1 to FZ. IF is supported by Experimental Medicine PhD programme of University of L’Aquila.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Qin S, Jiang J, Lu Y, Nice EC, Huang C, Zhang J, et al. Emerging Role of Tumor Cell Plasticity in Modifying Therapeutic Response. Signal Transduction Targeting Ther (2020) 5(1):1–36. doi: 10.1038/s41392-020-00313-5
2. Roberts CM, Cardenas C, Tedja R. The Role of Intra-Tumoral Heterogeneity and Its Clinical Relevance in Epithelial Ovarian Cancer Recurrence and Metastasis. Cancers (2019) 11:1083. doi: 10.3390/cancers11081083
3. Wang X, Zhang H, Chen X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist (2019) 2:141–60. doi: 10.20517/cdr.2019.10
4. Doyle LM, Wang MZ. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells (2019) 8:727. doi: 10.3390/cells8070727
5. Heijnen HFG, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated Platelets Release Two Types of Membrane Vesicles: Microvesicles by Surface Shedding and Exosomes Derived From Exocytosis of Multivesicular Bodies and α-Granules. Blood (1999) 94:3791–9. doi: 10.1182/blood.V94.11.3791
6. Mathivanan S, Ji H, Simpson RJ. Exosomes: Extracellular Organelles Important in Intercellular Communication. J Proteomics (2010) 73:1907–20. doi: 10.1016/j.jprot.2010.06.006
7. Wollert T, Hurley JH. Molecular Mechanism of Multivesicular Body Biogenesis by ESCRT Complexes. Nature (2010) 464:864. doi: 10.1038/nature08849
8. Liu T, Zhang L, Joo D, Sun SC. NF-κb Signaling in Inflammation. Signal Transduction Targeting Ther (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
9. Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr.. NF-κb Controls Cell Growth and Differentiation Through Transcriptional Regulation of Cyclin D1. Mol Cell Biol (1999) 19:5785. doi: 10.1128/MCB.19.8.5785
10. Piva R, Belardo G, Santoro MG. NF-Kappab: A Stress-Regulated Switch for Cell Survival. Antioxid Redox Signal (2006) 8:478–86. doi: 10.1089/ars.2006.8.478
11. Brantley DM, Chen CL, Muraoka RS, Bushdid PB, Bradberry JL, Kittrell F, et al. Nuclear Factor-κb (NF-κb) Regulates Proliferation and Branching in Mouse Mammary Epithelium. Mol Biol Cell (2001) 12:1445–55. doi: 10.1091/mbc.12.5.1445
13. Tak PP, Firestein GS. NF-κb: A Key Role in Inflammatory Diseases. J Clin Invest (2001) 107:7–11. doi: 10.1172/JCI11830
14. Sun SC, Chang JH, Jin J. Regulation of Nuclear Factor-κb in Autoimmunity. Trends Immunol (2013) 34:282–9. doi: 10.1016/j.it.2013.01.004
15. Xia Y, Shen S, Verma IM. NF-κb, an Active Player in Human Cancers. Cancer Immunol Res (2014) 2:823–30. doi: 10.1158/2326-6066.CIR-14-0112
16. Chen FE, Ghosh G. Regulation of DNA Binding by Rel/NF-κb Transcription Factors: Structural Views. Oncogene (1999) 18:6845–52. doi: 10.1038/sj.onc.1203224
17. Hoffmann A, Natoli G, Ghosh G. Transcriptional Regulation via the NF-kappaB Signaling Module. Oncogene (2006) 25:6706–16. doi: 10.1038/sj.onc.1209933
18. Savinova OV, Hoffmann A, Ghosh G. The Nfkb1 and Nfkb2 Proteins P105 and P100 Function as the Core of High-Molecular-Weight Heterogeneous Complexes. Mol Cell (2009) 34:591–602. doi: 10.1016/j.molcel.2009.04.033
19. Bonizzi G, Karin M. The Two NF-kappaB Activation Pathways and Their Role in Innate and Adaptive Immunity. Trends Immunol (2004) 25:280–8. doi: 10.1016/j.it.2004.03.008
20. Baeuerle PA, Baltimore D. I Kappa B: A Specific Inhibitor of the NF-Kappa B Transcription Factor. Science (1988) 242:540–6 March 19, 2016. doi: 10.1126/science.3140380
21. Hans H, Michael K. Regulation and Function of IKK and IKK-Related Kinases. Sci STKE (2006) 2006:re13–re13. doi: 10.1126/stke.3572006re13
22. Karin M, Ben-Neriah Y. Phosphorylation Meets Ubiquitination: The Control of NF-[Kappa]B Activity. Annu Rev Immunol (2000) 18:621–63. doi: 10.1146/annurev.immunol.18.1.621
23. Anderson NM, Simon MC. Tumor Microenvironment. Curr Biol (2020) 30:R921. doi: 10.1016/j.cub.2020.06.081
24. Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The Inflammatory Micro-Environment in Tumor Progression: The Role of Tumor-Associated Macrophages. Crit Rev Oncol Hematol (2008) 66:1–9. doi: 10.1016/j.critrevonc.2007.07.004
25. de Visser KE, Coussens LM. The Inflammatory Tumor Microenvironment and its Impact on Cancer Development. Contrib Microbiol (2006) 13:118–37. doi: 10.1159/000092969
26. Li W, Tan D, Zenali MJ, Brown RE. Constitutive Activation of Nuclear Factor-Kappa B (NF-κb) Signaling Pathway in Fibrolamellar Hepatocellular Carcinoma. Int J Clin Exp Pathol (2010) 3:238–43.
27. Yamaguchi N, Ito T, Azuma S, Ito E, Honma R, Yanagisawa Y, et al. Constitutive Activation of Nuclear Factor-κb Is Preferentially Involved in the Proliferation of Basal-Like Subtype Breast Cancer Cell Lines. Cancer Sci (2009) 100:1668–74. doi: 10.1111/j.1349-7006.2009.01228.x
28. Nagel D, Vincendeau M, Eitelhuber AC, Krappmann D. Mechanisms and Consequences of Constitutive NF-κb Activation in B-Cell Lymphoid Malignancies. Oncogene (2014) 33:5655–65. doi: 10.1038/onc.2013.565
29. Sakamoto K, Maeda S, Hikiba Y, Nakagawa H, Hayakawa Y, Shibata W, et al. Constitutive NF-κb Activation in Colorectal Carcinoma Plays a Key Role in Angiogenesis, Promoting Tumor Growth. Clin Cancer Res (2009) 15:2248–58. doi: 10.1158/1078-0432.CCR-08-1383
30. Verzella D, Fischietti M, Capece D, Vecchiotti D, Del Vecchio F, Cicciarelli G, et al. Targeting the NF-κb Pathway in Prostate Cancer: A Promising Therapeutic Approach? Curr Drug Targets (2016) 17:311–20. doi: 10.2174/1389450116666150907100715
31. Didonato JA, Mercurio F, Karin M. NF-κb and the Link Between Inflammation and Cancer. Immunol Rev (2012) 246:379–400. doi: 10.1111/j.1600-065X.2012.01099.x
32. Dinarello CA. The Paradox of Pro-Inflammatory Cytokines in Cancer. Cancer Metastasis Rev (2006) 25:307–13. doi: 10.1007/s10555-006-9000-8
33. Bao B, Thakur A, Li Y, Ahmad A, Azmi AS, Banerjee S, et al. The Immunological Contribution of NF-κb Within the Tumor Microenvironment: A Potential Protective Role of Zinc as an Anti-Tumor Agent. Biochim Biophys Acta (2012) 1825:160–72. doi: 10.1016/J.BBCAN.2011.11.002
34. Vénéreau E, Ceriotti C, Bianchi ME. DAMPs From Cell Death to New Life. Front Immunol (2015) 6:1. doi: 10.3389/FIMMU.2015.00422
35. Murao A, Aziz M, Wang H, Brenner M, Wang P. Release Mechanisms of Major DAMPs. Apoptosis (2021) 26:152–62. doi: 10.1007/s10495-021-01663-3/TABLES/1
36. Lawrence T. The Nuclear Factor NF-κb Pathway in Inflammation. Cold Spring Harb Perspect Biol (2009) 1:a001651. doi: 10.1101/cshperspect.a001651
37. Gabay C. Interleukin-6 and Chronic Inflammation. Arthritis Res Ther (2006) 8:S3. doi: 10.1186/ar1917
38. Terzić J, Grivennikov S, Karin E, Karin M. Inflammation and Colon Cancer. Gastroenterology (2010) 138:2101–2114.e5. doi: 10.1053/j.gastro.2010.01.058
39. Oh K, Lee OY, Park Y, Seo MW, Lee DS. IL-1β Induces IL-6 Production and Increases Invasiveness and Estrogen-Independent Growth in a TG2-Dependent Manner in Human Breast Cancer Cells. BMC Cancer (2016) 16:1–11. doi: 10.1186/s12885-016-2746-7/FIGURES/6
40. Raudenská M, Svobodová M, Gumulec J, Falk M, Masařík M. The Importance of Cancer-Associated Fibroblasts in the Pathogenesis of Head and Neck Cancers. Klin Onkol (2020) 33:39–48. doi: 10.14735/amko202039
41. Wu HJ, Hao M, Yeo SK, Guan JL. FAK Signaling in Cancer-Associated Fibroblasts Promotes Breast Cancer Cell Migration and Metastasis by Exosomal miRNAs-Mediated Intercellular Communication. Oncogene (2020) 39:2539–49. doi: 10.1038/s41388-020-1162-2
42. Vosseler S, Lederle W, Airola K, Obermueller E, Fusenig NE, Mueller MM. Distinct Progression-Associated Expression of Tumor and Stromal MMPs in HaCaT Skin SCCs Correlates With Onset of Invasion. Int J Cancer (2009) 125:2296–306. doi: 10.1002/ijc.24589
43. Guo C, Buranych A, Sarkar D, Fisher PB, Wang XY. The Role of Tumor-Associated Macrophages in Tumor Vascularization. Vasc Cell (2013) 5:20. doi: 10.1186/2045-824X-5-20
44. Luput L, Licarete E, Sesarman A, Patras L, Alupei MC, Banciu M. Tumor-Associated Macrophages Favor C26 Murine Colon Carcinoma Cell Proliferation in an Oxidative Stress-Dependent Manner. Oncol Rep (2017) 37:2472–80. doi: 10.3892/or.2017.5466
45. Cui X, Morales RTT, Qian W, Wang H, Gagner JP, Dolgalev I, et al. Hacking Macrophage-Associated Immunosuppression for Regulating Glioblastoma Angiogenesis. Biomaterials (2018) 161:164–78. doi: 10.1016/j.biomaterials.2018.01.053
46. Mills CD. M1 and M2 Macrophages: Oracles of Health and Disease. Crit Rev Immunol (2012) 32:463–88. doi: 10.1615/CritRevImmunol.v32.i6.10
47. Verzella D, Bennett J, Fischietti M, Thotakura AK, Recordati C, Pasqualini F, et al. Gadd45β Loss Ablates Innate Immunosuppression in Cancer. Cancer Res (2018) 78:1275–92. doi: 10.1158/0008-5472.CAN-17-1833
48. Capece D, D’Andrea D, Verzella D, Tornatore L, Begalli F, Bennett J, et al. Turning an Old GADDget Into a Troublemaker. Cell Death Differ (2018) 25(4):642–4. doi: 10.1038/s41418-018-0087-6
49. Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. Tolerance and M2 (Alternative) Macrophage Polarization are Related Processes Orchestrated by P50 Nuclear Factor Kappab. Proc Natl Acad Sci USA (2009) 106:14978–83. doi: 10.1073/pnas.0809784106
50. Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. ‘Re-Educating’ Tumor-Associated Macrophages by Targeting NF-Kappab. J Exp Med (2008) 205:1261–8. doi: 10.1084/jem.20080108
51. Erez N, Truitt M, Olson P, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell (2010) 17:135–47. doi: 10.1016/j.ccr.2009.12.041
52. Kelly MG, Alvero AB, Chen R, Silasi DA, Abrahams VM, Chan S, et al. TLR-4 Signaling Promotes Tumor Growth and Paclitaxel Chemoresistance in Ovarian Cancer. Cancer Res (2006) 66:3859–68. doi: 10.1158/0008-5472.CAN-05-3948
53. Rosette C, Karin M. Cytoskeletal Control of Gene Expression: Depolymerization of Microtubules Activates NF-Kappa B. J Cell Biol (1995) 128:1111–9. doi: 10.1083/jcb.128.6.1111
54. Das KC, White CW. Activation of NF-κb by Antineoplastic Agents: ROLE OF PROTEIN KINASE C *. J Biol Chem (1997) 272:14914–20. doi: 10.1074/jbc.272.23.14914
55. Bottero V, Busuttil V, Loubat A, Magné N, Fischel JL, Milano G, et al. Activation of Nuclear Factor kappaB Through the IKK Complex by the Topoisomerase Poisons SN38 and Doxorubicin: A Brake to Apoptosis in HeLa Human Carcinoma Cells. Cancer Res (2001) 61:7785–91.
56. Cheng Q, Lee HH, Li Y, Parks TP, Cheng G. Upregulation of Bcl-X and Bfl-1 as a Potential Mechanism of Chemoresistance, Which can be Overcome by NF-κb Inhibition. Oncogene (2000) 19(42):4936–40. doi: 10.1038/sj.onc.1203861
57. Shi Y. Mechanisms of Caspase Activation and Inhibition During Apoptosis. Mol Cell (2002) 9:459–70. doi: 10.1016/S1097-2765(02)00482-3
58. Chen X, Kandasamy K, Srivastava RK. Differential Roles of RelA (P65) and C-Rel Subunits of Nuclear Factor κb in Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Signaling. Cancer Res (2003) 63:1059–1066.
59. Bentires-Alj M, Barbu V, Fillet M, Chariot A, Relic B, Jacobs N, et al. NF-κb Transcription Factor Induces Drug Resistance Through MDR1 Expression in Cancer Cells. Oncogene (2003) 22(1):90–7. doi: 10.1038/sj.onc.1206056
60. Seubwai W, Vaeteewoottacharn K, Kraiklang R, Umezawa K, Okada S, Wongkham S. Inhibition of NF-κb Activity Enhances Sensitivity to Anticancer Drugs in Cholangiocarcinoma Cells. Oncol Res (2016) 23:21. doi: 10.3727/096504015X14424348426071
61. Liu T, Wei R, Zhang Y, Chen W, Liu H. Association Between NF-κb Expression and Drug Resistance of Liver Cancer. Oncol Lett (2019) 17:1030–4. doi: 10.3892/OL.2018.9640/HTML
62. Abdin SM, Tolba MF, Zaher DM, Omar HA. Nuclear Factor-κb Signaling Inhibitors Revert Multidrug-Resistance in Breast Cancer Cells. Chem Biol Interact (2021) 340:109450. doi: 10.1016/j.cbi.2021.109450
63. Lim JW, Kim H, Kim KH. Nuclear factor-kappaB Regulates Cyclooxygenase-2 Expression and Cell Proliferation in Human Gastric Cancer Cells. Lab Invest (2001) 81:349–60. doi: 10.1038/labinvest.3780243
64. Fantappiè O, Masini E, Sardi I, Raimondi L, Bani D, Solazzo M, et al. The MDR Phenotype is Associated With the Expression of COX-2 and iNOS in a Human Hepatocellular Carcinoma Cell Line. Hepatology (2002) 35:843–52. doi: 10.1053/jhep.2002.32469
65. Sui H, Zhou S, Wang Y, Liu X, Zhou L, Yin P, et al. COX-2 Contributes to P-Glycoprotein-Mediated Multidrug Resistance via Phosphorylation of C-Jun at Ser63/73 in Colorectal Cancer. Carcinogenesis (2011) 32:667–75. doi: 10.1093/carcin/bgr016
66. Maeng HJ, Lee WJ, Jin QR, Chang JE, Shim WS. Upregulation of COX-2 in the Lung Cancer Promotes Overexpression of Multidrug Resistance Protein 4 (MRP4) via PGE2-Dependent Pathway. Eur J Pharm Sci (2014) 62:189–96. doi: 10.1016/j.ejps.2014.05.023
67. Jeon YJ, Middleton J, Kim T, Laganà A, Piovan C, Secchiero P, et al. A Set of NF-κb - Regulated microRNAs Induces Acquired TRAIL Resistance in Lung Cancer. Proc Natl Acad Sci USA (2015) 112:E3355–64. doi: 10.1073/PNAS.1504630112/-/DCSUPPLEMENTAL
68. Mokim Ahmed K, Li JJ. NF-κb-Mediated Adaptive Resistance to Ionizing Radiation. Free Radic Biol Med (2008) 44:1. doi: 10.1016/j.freeradbiomed.2007.09.022
69. Li LN, Xiao T, Yi HM, Zheng Z, Qu JQ, Huang W, et al. MiR-125b Increases Nasopharyngeal Carcinoma Radioresistance by Targeting A20/NF-κb Signaling Pathway. Mol Cancer Ther (2017) 16:2094–106. doi: 10.1158/1535-7163.MCT-17-0385
70. Luo M, Ding L, Li Q, Yao H. miR-668 Enhances the Radioresistance of Human Breast Cancer Cell by Targeting Iκbα. Breast Cancer (2017) 24:673–82. doi: 10.1007/s12282-017-0756-1
71. Tao SC, Guo SC. Role of Extracellular Vesicles in Tumour Microenvironment. Cell Commun Signal (2020) 18(1):1–24. doi: 10.1186/s12964-020-00643-5
72. Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, et al. Rab27a and Rab27b Control Different Steps of the Exosome Secretion Pathway. Nat Cell Biol (2010) 12:19–30. doi: 10.1038/ncb2000
73. Savina A, Fader CM, Damiani MT, Colombo MI. Rab11 Promotes Docking and Fusion of Multivesicular Bodies in a Calcium-Dependent Manner. Traffic (2005) 6:131–43. doi: 10.1111/j.1600-0854.2004.00257.x
74. Hsu C, Morohashi Y, Yoshimura SI, Manrique-Hoyos N, Jung SY, Lauterbach MA, et al. Regulation of Exosome Secretion by Rab35 and Its GTPase-Activating Proteins TBC1D10A-C. J Cell Biol (2010) 189:223–32. doi: 10.1083/jcb.200911018
75. Dong WW, Mou Q, Chen J, Cui JT, Li WM, Xiao WH. Differential Expression of Rab27A/B Correlates With Clinical Outcome in Hepatocellular Carcinoma. World J Gastroenterol (2012) 18:1806–13. doi: 10.3748/wjg.v18.i15.1806
76. Morelli AE, Larregina AT, Shufesky WJ, Sullivan MLG, Stolz DB, Papworth GD, et al. Endocytosis, Intracellular Sorting, and Processing of Exosomes by Dendritic Cells. Blood (2004) 104:3257–66. doi: 10.1182/blood-2004-03-0824
77. Segura E, Guérin C, Hogg N, Amigorena S, Théry C. CD8+ Dendritic Cells Use LFA-1 to Capture MHC-Peptide Complexes From Exosomes In Vivo. J Immunol (2007) 179:1489–96. doi: 10.4049/jimmunol.179.3.1489
78. Skokos D, Botros HG, Demeure C, Morin J, Peronet R, Birkenmeier G, et al. Mast Cell-Derived Exosomes Induce Phenotypic and Functional Maturation of Dendritic Cells and Elicit Specific Immune Responses In Vivo. J Immunol (2003) 170:3037–45. doi: 10.4049/jimmunol.170.6.3037
79. Sullivan R, Maresh G, Zhang X, Salomon C, Hooper J, Margolin D, et al. The Emerging Roles of Extracellular Vesicles as Communication Vehicles Within the Tumor Microenvironment and Beyond. Front Endocrinol (Lausanne) (2017) 8:194. doi: 10.3389/fendo.2017.00194/BIBTEX
80. Tian W, Liu S, Li B. Potential Role of Exosomes in Cancer Metastasis. BioMed Res Int (2019) 2019. doi: 10.1155/2019/4649705
81. Valcz G, Buzás EI, Szállási Z, Kalmár A, Krenács T, Tulassay Z, et al. Perspective: Bidirectional Exosomal Transport Between Cancer Stem Cells and Their Fibroblast-Rich Microenvironment During Metastasis Formation. NPJ Breast Cancer (2018) 4(1):1–7. doi: 10.1038/s41523-018-0071-9
82. Whiteside TL. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv Clin Chem (2016) 74:103–41. doi: 10.1016/bs.acc.2015.12.005
83. Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, et al. Tumor Microenvironment Derived Exosomes Pleiotropically Modulate Cancer Cell Metabolism. Elife (2016) 5:e10250. doi: 10.7554/eLife.10250
84. Calon A, Tauriello DVF, Batlle E. TGF-Beta in CAF-Mediated Tumor Growth and Metastasis. Semin Cancer Biol (2014) 25:15–22. doi: 10.1016/j.semcancer.2013.12.008
85. Lan J, Sun L, Xu F, Liu L, Hu F, Song D, et al. M2 Macrophage-Derived Exosomes Promote Cell Migration and Invasion in Colon Cancer. Cancer Res (2019) 79:146–58. doi: 10.1158/0008-5472.CAN-18-0014
86. Wang G, Fu Y, Yang X, Luo X, Wang J, Gong J, et al. Brg-1 Targeting of Novel Mir550a-5p/RNF43/Wnt Signaling Axis Regulates Colorectal Cancer Metastasis. Oncogene (2015) 35(5):651–61. doi: 10.1038/onc.2015.124
87. Zhao XP, Wang M, Song Y, Song K, Yan lin T, Wang L, et al. Membrane Microvesicles as Mediators for Melanoma-Fibroblasts Communication: Roles of the VCAM-1/VLA-4 Axis and the ERK1/2 Signal Pathway. Cancer Lett (2015) 360:125–33. doi: 10.1016/j.canlet.2015.01.032
88. Zhou X, Yan T, Huang C, Xu Z, Wang L, Jiang E, et al. Melanoma Cell-Secreted Exosomal miR-155-5p Induce Proangiogenic Switch of Cancer-Associated Fibroblasts via SOCS1/JAK2/STAT3 Signaling Pathway. J Exp Clin Cancer Res (2018) 37:1–15. doi: 10.1186/s13046-018-0911-3/FIGURES/7
89. Hsu Y-L, Hung J-Y, Chang W-A, Jian S-F, Lin Y-S, Pan Y-C, et al. Hypoxic Lung-Cancer-Derived Extracellular Vesicle MicroRNA-103a Increases the Oncogenic Effects of Macrophages by Targeting PTEN. Mol Ther (2018) 26:568. doi: 10.1016/j.ymthe.2017.11.016
90. Ying X, Wu Q, Wu X, Zhu Q, Wang X, Jiang L, et al. Epithelial Ovarian Cancer-Secreted Exosomal miR-222-3p Induces Polarization of Tumor-Associated Macrophages. Oncotarget (2016) 7:43076–87. doi: 10.18632/oncotarget.9246
91. Sun Z, Wang L, Zhou Y, Dong L, Ma W, Lv L, et al. Glioblastoma Stem Cell-Derived Exosomes Enhance Stemness and Tumorigenicity of Glioma Cells by Transferring Notch1 Protein. Cell Mol Neurobiol (2020) 40:767–84. doi: 10.1007/s10571-019-00771-8
92. Shedden K, Xie T, Chandaroy P, Chang YT, Rosania GR. Expulsion of Small Molecules in Vesicles Shed by Cancer Cells: Association With Gene Expression and Chemosensitivity Profiles 1,2. Cancer Res (2003) 63:4331–7 February 8, 2022.
93. Federici C, Petrucci F, Caimi S, Cesolini A, Logozzi M, Borghi M, et al. Exosome Release and Low pH Belong to a Framework of Resistance of Human Melanoma Cells to Cisplatin. PloS One (2014) 9:e88193. doi: 10.1371/journal.pone.0088193
94. Bebawy M, Combes V, Lee E, Jaiswal R, Gong J, Bonhoure A, et al. Membrane Microparticles Mediate Transfer of P-Glycoprotein to Drug Sensitive Cancer Cells. Leukemia (2009) 23:1643–9. doi: 10.1038/leu.2009.76
95. Sousa D, Lima RT, Vasconcelos MH. Intercellular Transfer of Cancer Drug Resistance Traits by Extracellular Vesicles. Trends Mol Med (2015) 21:595–608. doi: 10.1016/j.molmed.2015.08.002
96. Levchenko A, Mehta BM, Niu X, Kang G, Villafania L, Way D, et al. Intercellular Transfer of P-Glycoprotein Mediates Acquired Multidrug Resistance in Tumor Cells. Proc Natl Acad Sci U S A (2005) 102:1933–8. doi: 10.1073/pnas.0401851102
97. Ifergan I, Scheffer GL, Assaraf YG. Novel Extracellular Vesicles Mediate an ABCG2-Dependent Anticancer Drug Sequestration and Resistance. Cancer Res (2005) 65:10952–8. doi: 10.1158/0008-5472.CAN-05-2021
98. Ma X, Cai Y, He D, Zou C, Zhang P, Lo CY, et al. Transient Receptor Potential Channel TRPC5 Is Essential for P-Glycoprotein Induction in Drug-Resistant Cancer Cells. Proc Natl Acad Sci USA (2012) 109:16282–7. doi: 10.1073/pnas.1202989109/-/DCSUPPLEMENTAL
99. Ma X, Chen Z, Hua D, He D, Wang L, Zhang P, et al. Essential Role for TrpC5-Containing Extracellular Vesicles in Breast Cancer With Chemotherapeutic Resistance. Proc Natl Acad Sci USA (2014) 111:6389–94. doi: 10.1073/pnas.1400272111/-/DCSUPPLEMENTAL
100. Wang T, Ning K, Lu TX, Sun X, Jin L, Qi X, et al. Increasing Circulating Exosomes-Carrying TRPC5 Predicts Chemoresistance in Metastatic Breast Cancer Patients. Cancer Sci (2017) 108:448–54. doi: 10.1111/cas.13150
101. Yang SJ, Wang DD, Li J, Xu HZ, Shen HY, Chen X, et al. Predictive Role of GSTP1-Containing Exosomes in Chemotherapy-Resistant Breast Cancer. Gene (2017) 623:5–14. doi: 10.1016/j.gene.2017.04.031
102. Lunavat TR, Cheng L, Einarsdottir BO, Bagge RO, Muralidharan SV, Sharples RA, et al. BRAFV600 Inhibition Alters the microRNA Cargo in the Vesicular Secretome of Malignant Melanoma Cells. Proc Natl Acad Sci USA (2017) 114:E5930–9. doi: 10.1073/PNAS.1705206114/-/DCSUPPLEMENTAL
103. Mikamori M, Yamada D, Eguchi H, Hasegawa S, Kishimoto T, Tomimaru Y, et al. MicroRNA-155 Controls Exosome Synthesis and Promotes Gemcitabine Resistance in Pancreatic Ductal Adenocarcinoma. Sci Rep (2017) 71:1–14. doi: 10.1038/srep42339
104. Shen M, Dong C, Ruan X, Yan W, Cao M, Pizzo D, et al. Chemotherapy-Induced Extracellular Vesicle miRNAs Promote Breast Cancer Stemness by Targeting Onecut2. Cancer Res (2019) 79:3608–21. doi: 10.1158/0008-5472.CAN-18-4055
105. Qu L, Ding J, Chen C, Wu ZJ, Liu B, Gao Y, et al. Exosome-Transmitted lncARSR Promotes Sunitinib Resistance in Renal Cancer by Acting as a Competing Endogenous RNA. Cancer Cell (2016) 29:653–68. doi: 10.1016/j.ccell.2016.03.004
106. Yang YN, Zhang R, Du JW, Yuan HH, Li YJ, Wei XL, et al. Predictive Role of UCA1-Containing Exosomes in Cetuximab-Resistant Colorectal Cancer. Cancer Cell Int (2018) 18:164. doi: 10.1186/s12935-018-0660-6
107. Kang M, Ren M, Li Y, Fu Y, Deng M, Li C. Exosome-Mediated Transfer of lncRNA PART1 Induces Gefitinib Resistance in Esophageal Squamous Cell Carcinoma via Functioning as a Competing Endogenous RNA. J Exp Clin Cancer Res (2018) 37:171. doi: 10.1186/s13046-018-0845-9
108. Vaupel P, Kallinowski F, Okunieff P. Blood Flow, Oxygen Consumption and Tissue Oxygenation of Human Tumors. Adv Exp Med Biol (1990) 277:895–905. doi: 10.1007/978-1-4684-8181-5_103
109. Lee CT, MacE T, Repasky EA. Hypoxia-Driven Immunosuppression: A New Reason to Use Thermal Therapy in the Treatment of Cancer? Int J Hyperthermia (2010) 26:232. doi: 10.3109/02656731003601745
110. Park JE, Tan Sen H, Datta A, Lai RC, Zhang H, Meng W, et al. Hypoxic Tumor Cell Modulates Its Microenvironment to Enhance Angiogenic and Metastatic Potential by Secretion of Proteins and Exosomes. Mol Cell Proteomics (2010) 9:1085–99. doi: 10.1074/mcp.M900381-MCP200
111. King HW, Michael MZ, Gleadle JM. Hypoxic Enhancement of Exosome Release by Breast Cancer Cells. BMC Cancer (2012) 12:421. doi: 10.1186/1471-2407-12-421
112. Wang T, Gilkes DM, Takano N, Xiang L, Luo W, Bishop CJ, et al. Hypoxia-Inducible Factors and RAB22A Mediate Formation of Microvesicles That Stimulate Breast Cancer Invasion and Metastasis. Proc Natl Acad Sci USA (2014) 111:E3234–42. doi: 10.1073/pnas.1410041111/-/DCSUPPLEMENTAL
113. Semenza GL. Hypoxia-Inducible Factors in Physiology and Medicine. Cell (2012) 148:399–408. doi: 10.1016/j.cell.2012.01.021
114. Semenza GL. Targeting HIF-1 for Cancer Therapy. Nat Rev Cancer (2003) 3:721–32. doi: 10.1038/nrc1187
115. van Uden P, Kenneth NS, Webster R, Müller HA, Mudie S, Rocha S. Evolutionary Conserved Regulation of HIF-1β by NF-κb. PloS Genet (2011) 7:1001285. doi: 10.1371/journal.pgen.1001285
116. Van Uden P, Kenneth NS, Rocha S. Regulation of Hypoxia-Inducible Factor-1α by NF-κb. Biochem J (2008) 412:477. doi: 10.1042/BJ20080476
117. Jung YJ, Isaacs JS, Lee S, Trepel J, Liu ZG, Neckers L. Hypoxia-Inducible Factor Induction by Tumour Necrosis Factor in Normoxic Cells Requires Receptor-Interacting Protein-Dependent Nuclear Factor Kappa B Activation. Biochem J (2003) 370:1011–7. doi: 10.1042/bj20021279
118. Fitzpatrick SF, Tambuwala MM, Bruning U, Schaible B, Scholz CC, Byrne A, et al. An Intact Canonical NF-κb Pathway is Required for Inflammatory Gene Expression in Response to Hypoxia. J Immunol (2011) 186:1091–6. doi: 10.4049/jimmunol.1002256
119. Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, et al. Prolyl Hydroxylase-1 Negatively Regulates Iκb Kinase-β, Giving Insight Into Hypoxia-Induced Nfκb Activity. Proc Natl Acad Sci USA (2006) 103:18154. doi: 10.1073/pnas.0602235103
120. Feng F, Jiang Y, Lu H, Lu X, Wang S, Wang L, et al. Rab27A Mediated by NF-κb Promotes the Stemness of Colon Cancer Cells via Up-Regulation of Cytokine Secretion. Oncotarget (2016) 7:63342. doi: 10.18632/oncotarget.11454
121. Liu J, Gong X, Zhu X, Xue D, Liu Y, Wang P. Rab27A Overexpression Promotes Bladder Cancer Proliferation and Chemoresistance Through Regulation of NF-κb Signaling. Oncotarget (2017) 8:75272. doi: 10.18632/oncotarget.20775
122. Yang JCS, Lin MW, Rau CS, Jeng SF, Lu TH, Wu YC, et al. Altered Exosomal Protein Expression in the Serum of NF-κb Knockout Mice Following Skeletal Muscle Ischemia-Reperfusion Injury. J Biomed Sci (2015) 22:40. doi: 10.1186/S12929-015-0147-X
123. Zheng P, Chen L, Yuan X, Luo Q, Liu Y, Xie G, et al. Exosomal Transfer of Tumor-Associated Macrophage-Derived miR-21 Confers Cisplatin Resistance in Gastric Cancer Cells. J Exp Clin Cancer Res (2017) 36:1–13. doi: 10.1186/s13046-017-0528-y/FIGURES/6
124. Bretz NP, Ridinger J, Rupp AK, Rimbach K, Keller S, Rupp C, et al. Body Fluid Exosomes Promote Secretion of Inflammatory Cytokines in Monocytic Cells via Toll-Like Receptor Signaling. J Biol Chem (2013) 288:36691–702. doi: 10.1074/jbc.M113.512806
125. Wu L, Zhang X, Zhang B, Shi H, Yuan X, Sun Y, et al. Exosomes Derived From Gastric Cancer Cells Activate NF-κb Pathway in Macrophages to Promote Cancer Progression. Tumour Biol (2016) 37:12169–80. doi: 10.1007/s13277-016-5071-5
126. Chow A, Zhou W, Liu L, Fong MY, Champer J, Van Haute D, et al. Macrophage Immunomodulation by Breast Cancer-Derived Exosomes Requires Toll-Like Receptor 2-Mediated Activation of NF-κb. Sci Rep (2014) 4:5750. doi: 10.1038/srep05750
127. Fu Z, Gu L, Li N, Ma Z, Ling M, Wang Y. Upregulated TRPC5 Plays an Important Role in Development of Nasal Polyps by Activating Eosinophilic Inflammation and NF-κb Signaling Pathways. Int J Clin Exp Pathol (2018) 11:1935. February 11, 2022
128. Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, et al. MicroRNAs Bind to Toll-Like Receptors to Induce Prometastatic Inflammatory Response. Proc Natl Acad Sci USA (2012) 109:E2110. doi: 10.1073/pnas.1209414109/-/DCSUPPLEMENTAL
129. Fang T, Lv H, Lv G, Li T, Wang C, Han Q, et al. Tumor-Derived Exosomal miR-1247-3p Induces Cancer-Associated Fibroblast Activation to Foster Lung Metastasis of Liver Cancer. Nat Commun (2018) 9:191. doi: 10.1038/s41467-017-02583-0
130. Li ZL, Lv LL, Tang TT, Wang B, Feng Y, Zhou LT, et al. HIF-1α Inducing Exosomal microRNA-23a Expression Mediates the Cross-Talk Between Tubular Epithelial Cells and Macrophages in Tubulointerstitial Inflammation. Kidney Int (2019) 95:388–404. doi: 10.1016/j.kint.2018.09.013
131. Chen L, Karisma VW, Liu H, Zhong L. MicroRNA-300: A Transcellular Mediator in Exosome Regulates Melanoma Progression. Front Oncol (2019) 9:1005. doi: 10.3389/fonc.2019.01005/BIBTEX
132. Wang Y, Ma H, Li Y, Su R. MiR-192-5p-Modified Tumor-Associated Macrophages-Derived Exosome Suppressed Endometrial Cancer Progression Through Targeting IRAK1/NF-κb Signaling. Reprod Sci (2022) 29(2):436–47. doi: 10.1007/s43032-021-00789-8
133. Li W, Zhang L, Guo B, Deng J, Wu S, Li F, et al. Exosomal FMR1-AS1 Facilitates Maintaining Cancer Stem-Like Cell Dynamic Equilibrium via TLR7/Nfκb/C-Myc Signaling in Female Esophageal Carcinoma. Mol Cancer (2019) 18:22. doi: 10.1186/s12943-019-0949-7
134. Gooding AJ, Zhang B, Gunawardane L, Beard A, Valadkhan S, Schiemann WP. The lncRNA BORG Facilitates the Survival and Chemoresistance of Triple-Negative Breast Cancers. Oncogene (2018) 38(12):2020–41. doi: 10.1038/s41388-018-0586-4
135. Li P, Zhang X, Wang L, Du L, Yang Y, Liu T, et al. lncRNA HOTAIR Contributes to 5FU Resistance Through Suppressing miR-218 and Activating NF-κb/TS Signaling in Colorectal Cancer. Mol Ther Nucleic Acids (2017) 8:356–69. doi: 10.1016/j.omtn.2017.07.007/ATTACHMENT/E36387CD-4CD8-49F8-94E1-F5244014DF21/MMC1.PDF
136. Park SJ, Kim JM, Kim J, Hur J, Park S, Kim K, et al. Molecular Mechanisms of Biogenesis of Apoptotic Exosome-Like Vesicles and Their Roles as Damage-Associated Molecular Patterns. Proc Natl Acad Sci USA (2018) 115:E11721–30. doi: 10.1073/pnas.1811432115/-/DCSUPPLEMENTAL
137. Broz P, Dixit VM. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat Rev Immunol (2016) 16:407–20. doi: 10.1038/nri.2016.58
138. Schroder K, Tschopp J. The Inflammasomes. Cell (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
139. Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, Magez S, et al. Stimulation of Toll-Like Receptor 3 and 4 Induces Interleukin-1β Maturation by Caspase-8. J Exp Med (2008) 205:1967. doi: 10.1084/jem.20071632
140. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A Novel Heterodimeric Cysteine Protease is Required for Interleukin-1 Beta Processing in Monocytes. Nature (1992) 356:768–74. doi: 10.1038/356768A0
141. Boaru SG, Borkham-Kamphorst E, Van DeLeur E, Lehnen E, Liedtke C, Weiskirchen R.. NLRP3 Inflammasome Expression Is Driven by NF-κb in Cultured Hepatocytes. Biochem Biophys Res Commun (2015) 458:700–6. doi: 10.1016/j.bbrc.2015.02.029
142. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. NF-kB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J Immunol (2009) 183:787. doi: 10.4049/jimmunol.0901363
143. Swanson KV, Deng M, Ting JPY. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat Rev Immunol (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
144. Cypryk W, Nyman TA, Matikainen S. From Inflammasome to Exosome—Does Extracellular Vesicle Secretion Constitute an Inflammasome-Dependent Immune Response? Front Immunol (2018) 9:2188. doi: 10.3389/fimmu.2018.02188
145. Gutierrez MG, Mishra BB, Jordao L, Elliott E, Anes E, Griffiths G. NF-κb Activation Controls Phagolysosome Fusion-Mediated Killing of Mycobacteria by Macrophages. J Immunol (2008) 181:2651–63. doi: 10.4049/jimmunol.181.4.2651
146. Wozniak AL, Adams A, King KE, Dunn W, Christenson LK, Hung WT, et al. The RNA Binding Protein FMR1 Controls Selective Exosomal miRNA Cargo Loading During Inflammation. J Cell Biol (2020) 219:e201912074. doi: 10.1083/jcb.201912074
147. Zhang Y, Liu F, Yuan Y, Jin C, Chang C, Zhu Y, et al. Inflammasome-Derived Exosomes Activate NF-κb Signaling in Macrophages. J Proteome Res (2017) 16:170–8. doi: 10.1021/acs.jproteome.6b00599
148. Mezzasoma L, Bellezza I, Romani R, Talesa VN. Extracellular Vesicles and the Inflammasome: An Intricate Network Sustaining Chemoresistance. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.888135
149. Wang G, Jin S, Ling X, Li Y, Hu Y, Zhang Y, et al. Proteomic Profiling of LPS-Induced Macrophage-Derived Exosomes Indicates Their Involvement in Acute Liver Injury. Proteomics (2019) 19:e1800274. doi: 10.1002/pmic.201800274
150. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology (2016) 150:956. doi: 10.1053/j.gastro.2015.12.037
151. Zhang W, Ma Y, Zhang Y, Yang J, He G, Chen S. Photo-Oxidative Blue-Light Stimulation in Retinal Pigment Epithelium Cells Promotes Exosome Secretion and Increases the Activity of the NLRP3 Inflammasome. Curr Eye Res (2019) 44:67–75. doi: 10.1080/02713683.2018.1518458
152. Mezzasoma L, Bellezza I, Orvietani P, Manni G, Gargaro M, Sagini K, et al. Amniotic Fluid Stem Cell-Derived Extracellular Vesicles are Independent Metabolic Units Capable of Modulating Inflammasome Activation in THP-1 Cells. FASEB J (2022) 36:e22218. doi: 10.1096/fj.202101657R
153. Singla DK, Johnson TA, Dargani ZT. Exosome Treatment Enhances Anti-Inflammatory M2 Macrophages and Reduces Inflammation-Induced Pyroptosis in Doxorubicin-Induced Cardiomyopathy. Cells (2019) 8:1224. doi: 10.3390/cells8101224
154. Kolb R, Liu GH, Janowski AM, Sutterwala FS, Zhang W. Inflammasomes in Cancer: A Double-Edged Sword. Protein Cell (2014) 5:12. doi: 10.1007/s13238-013-0001-4
155. Huang CF, Chen L, Li YC, Wu L, Yu GT, Zhang WF, et al. NLRP3 Inflammasome Activation Promotes Inflammation-Induced Carcinogenesis in Head and Neck Squamous Cell Carcinoma. J Exp Clin Cancer Res (2017) 36:1–13. doi: 10.1186/s13046-017-0589-y/FIGURES/5
156. Wang Y, Kong H, Zeng X, Liu W, Wang Z, Yan X, et al. Activation of NLRP3 Inflammasome Enhances the Proliferation and Migration of A549 Lung Cancer Cells. Oncol Rep (2016) 35:2053–64. doi: 10.3892/or.2016.4569/HTML
157. Mezzasoma L, Costanzi E, Scarpelli P, Talesa VN, Bellezza I. Extracellular Vesicles From Human Advanced-Stage Prostate Cancer Cells Modify the Inflammatory Response of Microenvironment-Residing Cells. Cancers (Basel) (2019) 11. doi: 10.3390/cancers11091276
158. Liang M, Chen X, Wang L, Qin L, Wang H, Sun Z, et al. Cancer-Derived Exosomal TRIM59 Regulates Macrophage NLRP3 Inflammasome Activation to Promote Lung Cancer Progression. J Exp Clin Cancer Res (2020) 39:176. doi: 10.1186/s13046-020-01688-7
159. Hwang WL, Lan HY, Cheng WC, Huang SC, Yang MH. Tumor Stem-Like Cell-Derived Exosomal RNAs Prime Neutrophils for Facilitating Tumorigenesis of Colon Cancer. J Hematol Oncol (2019) 12:1–17. doi: 10.1186/s13045-019-0699-4/FIGURES/6
160. Daley D, Mani VR, Mohan N, Akkad N, Balasubramania Pandian GSD, Savadkar S, et al. NLRP3 Signaling Drives Macrophage-Induced Adaptive Immune Suppression in Pancreatic Carcinoma. J Exp Med (2017) 214:1711–24. doi: 10.1084/jem.20161707
161. Lee HE, Lee JY, Yang G, Kang HC, Cho YY, Lee HS, et al. Inhibition of NLRP3 Inflammasome in Tumor Microenvironment Leads to Suppression of Metastatic Potential of Cancer Cells. Sci Rep (2019) 9:12277. doi: 10.1038/s41598-019-48794-x
162. Zhai Z, Samson JM, Yamauchi T, Vaddi PK, Matsumoto Y, Dinarello CA, et al. Inflammasome Sensor NLRP1 Confers Acquired Drug Resistance to Temozolomide in Human Melanoma. Cancers (Basel) (2020) 12:1–22. doi: 10.3390/cancers12092518
163. Theivanthiran B, Evans KS, de Vito NC, Plebanek M, Sturdivant M, Wachsmuth LP, et al. A Tumor-Intrinsic PD-L1/NLRP3 Inflammasome Signaling Pathway Drives Resistance to Anti–PD-1 Immunotherapy. J Clin Invest (2020) 130:2570–86. doi: 10.1172/JCI133055
164. Feng X, Luo Q, Zhang H, Wang H, Chen W, Meng G, et al. The Role of NLRP3 Inflammasome in 5-Fluorouracil Resistance of Oral Squamous Cell Carcinoma. J Exp Clin Cancer Res (2017) 36:81. doi: 10.1186/s13046-017-0553-x
Keywords: extracellular vesicles, NF-κB, drug resistance, tumor microenvironment, inflammasome
Citation: Di Vito Nolfi M, Vecchiotti D, Flati I, Verzella D, Di Padova M, Alesse E, Capece D and Zazzeroni F (2022) EV-Mediated Chemoresistance in the Tumor Microenvironment: Is NF-κB a Player? Front. Oncol. 12:933922. doi: 10.3389/fonc.2022.933922
Received: 01 May 2022; Accepted: 23 May 2022;
Published: 22 June 2022.
Edited by:
Antonio Giordano, Temple University, United StatesReviewed by:
Letizia Mezzasoma, University of Perugia, ItalyTommaso Neri, University of Pisa, Italy
Cinzia Antognelli, University of Perugia, Italy
Copyright © 2022 Di Vito Nolfi, Vecchiotti, Flati, Verzella, Di Padova, Alesse, Capece and Zazzeroni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Zazzeroni, ZnJhbmNlc2NhLnphenplcm9uaUB1bml2YXEuaXQ=; Daria Capece, ZGFyaWEuY2FwZWNlQHVuaXZhcS5pdA==
†These authors share first authorship