 Thomas Yul Avery
Thomas Yul Avery Natalie Köhler
Natalie Köhler Robert Zeiser
Robert Zeiser Tilman Brummer
Tilman Brummer Dietrich Alexander Ruess
Dietrich Alexander Ruess
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 27 July 2022
Sec. Molecular and Cellular Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.931774
This article is part of the Research Topic Kinase Inhibitors in Cancer Therapy View all 9 articles
Hyperactivation of the RAS-RAF-MEK-ERK cascade - a mitogen-activated protein kinase pathway – has a well-known association with oncogenesis of leading tumor entities, including non-small cell lung cancer, colorectal carcinoma, pancreatic ductal adenocarcinoma, and malignant melanoma. Increasing evidence shows that genetic alterations leading to RAS-RAF-MEK-ERK pathway hyperactivation mediate contact- and soluble-dependent crosstalk between tumor, tumor microenvironment (TME) and the immune system resulting in immune escape mechanisms and establishment of a tumor-sustaining environment. Consequently, pharmacological interruption of this pathway not only leads to tumor-cell intrinsic disruptive effects but also modification of the TME and anti-tumor immunomodulation. At the same time, the importance of ERK signaling in immune cell physiology and potentiation of anti-tumor immune responses through ERK signaling inhibition within immune cell subsets has received growing appreciation. Specifically, a strong case was made for targeted MEK inhibition due to promising associated immune cell intrinsic modulatory effects. However, the successful transition of therapeutic agents interrupting RAS-RAF-MEK-ERK hyperactivation is still being hampered by significant limitations regarding durable efficacy, therapy resistance and toxicity. We here collate and summarize the multifaceted role of RAS-RAF-MEK-ERK signaling in physiology and oncoimmunology and outline the rationale and concepts for exploitation of immunomodulatory properties of RAS-RAF-MEK-ERK inhibition while accentuating the role of MEK inhibition in combinatorial and intermittent anticancer therapy. Furthermore, we point out the extensive scientific efforts dedicated to overcoming the challenges encountered during the clinical transition of various therapeutic agents in the search for the most effective and safe patient- and tumor-tailored treatment approach.
As stated in the World Health Organization’s (WHO) Global Cancer Observatory (GLOBOCAN) report of 2021, cancer incidence and mortality are rapidly growing on a global scale, forming an increasing health burden and important barrier to increasing life expectancy (1). As reported in the WHO report on Global Health Estimates 2020, cancer has become the first or second leading cause of death before the age of 70 years in 112 of 183 countries, surpassing mortality rates of stroke and coronary heart disease in many countries (2). Estimating future development based on growth and aging of the population, worldwide annual new cancer cases are projected to grow 47% in the year 2040 to an estimated 28.4 million cases per year compared to 19.3 million cases in 2020 (1). This projection will very likely be an underestimation of the true development in the light of increasing prevalence of cancer-associated risk factors in many countries of the world (1). Therefore, it comes as no surprise that a tremendous amount of scientific effort is being devoted to cancer research in order to increase our knowledge in the areas of prevention, screening, diagnosis, effective interventions, and surveillance and to aid in their tailored integration into national health care plans to reduce the future burden and suffering from cancer.
In an attempt to understand the mechanistic background of cancer on a molecular basis, scientific investigations in the recent decades have led to the discovery of the causal relationship between the pathologically hyperactivated RAS-RAF-MEK-ERK signaling cascade – a mitogen-activated protein kinase (MAPK) pathway – and development and progression of various leading tumor entities, including non-small cell lung cancer (NSCLC), colorectal carcinoma (CRC), pancreatic ductal adenocarcinoma (PDAC), and malignant melanoma (3). Elements of this signaling cascade have been identified as potential oncogenes, ultimately leading to pathway hyperactivation and promotion of proliferative and tumorigenic signals (3). Of the associated oncogenes, mutant Kirsten rat sarcoma viral oncogene homolog (KRAS) and BRAF have been identified as the major driving forces behind the RAS-RAF-MEK-ERK pathway hyperactivation. Mutant KRAS appears to be involved in over 90% of PDAC, about 50% of CRC, about 30% of NSCLC and to a lesser extent in other tumors, while mutant BRAF has primarily been observed in melanomas (3–5). Consequently, the RAS-RAF-MEK-ERK pathway and its associated regulatory feedback loops as well as upstream activators and downstream effector proteins have been brought into the focus of the search for potential therapeutic targets in the attempt to slow down, put a halt to, or even reverse the oncogene-driven process (3). These efforts have led to the identification of promising targets for pharmacological inhibition and interruption of RAS-RAF-MEK-ERK hyperactivation. Furthermore, an increasing body of evidence shows that oncogenic mutations are capable of mediating autocrine and paracrine crosstalk between tumor cells, the tumor microenvironment (TME) and various subsets of immune cells in order to establish and maintain a pro-tumorigenic environment and employ effective immune evasion mechanisms at different stages of the so-called cancer-immunity cycle (6, 7).
Selective pharmacologic targeting of elements of the RAS-RAF-MEK-ERK signaling cascade has led to encouraging results in both preclinical and clinical studies through direct inhibition of oncogenic signaling (8). Importantly, a closer look at the role of the immune system in tumor growth and its influence on the establishment and maintenance of the TME revealed that the systemically administered targeted therapies are capable of achieving multifaceted anti-tumorigenic immunomodulation (9–15). Within this context, especially MEK inhibitors (MEKi) have received a substantial amount of scientific attention due to their preclinically observed potential to significantly alter the tumor-associated pro-tumorigenic immune response towards an anti-tumorigenic inflammatory response (10, 13). However, preclinical studies have not uniformly shown consistency in the aforementioned beneficial immunomodulatory effects of MEK inhibition. Furthermore, transition to the clinical setting has been hampered due to significant toxicity, occurrence of resistance mechanisms and unequivocal clinical results (Table 1) (10, 23–26). Nevertheless, pharmacological interruption of ERK signaling is being investigated extensively in both the preclinical and clinical setting, especially as part of various combinatorial and/or intermittently administered therapy regimens (Table 2) (8). Furthermore, the combination of immune checkpoint blockade (ICB) and RAS-RAF-MEK-ERK signaling inhibition has been identified as a promising approach in tumors with high immunogenicity (9, 10, 27). However, further research elucidating the immunomodulatory anti-tumor effects and associated consequences for the complex interactions in the TME is still necessary.
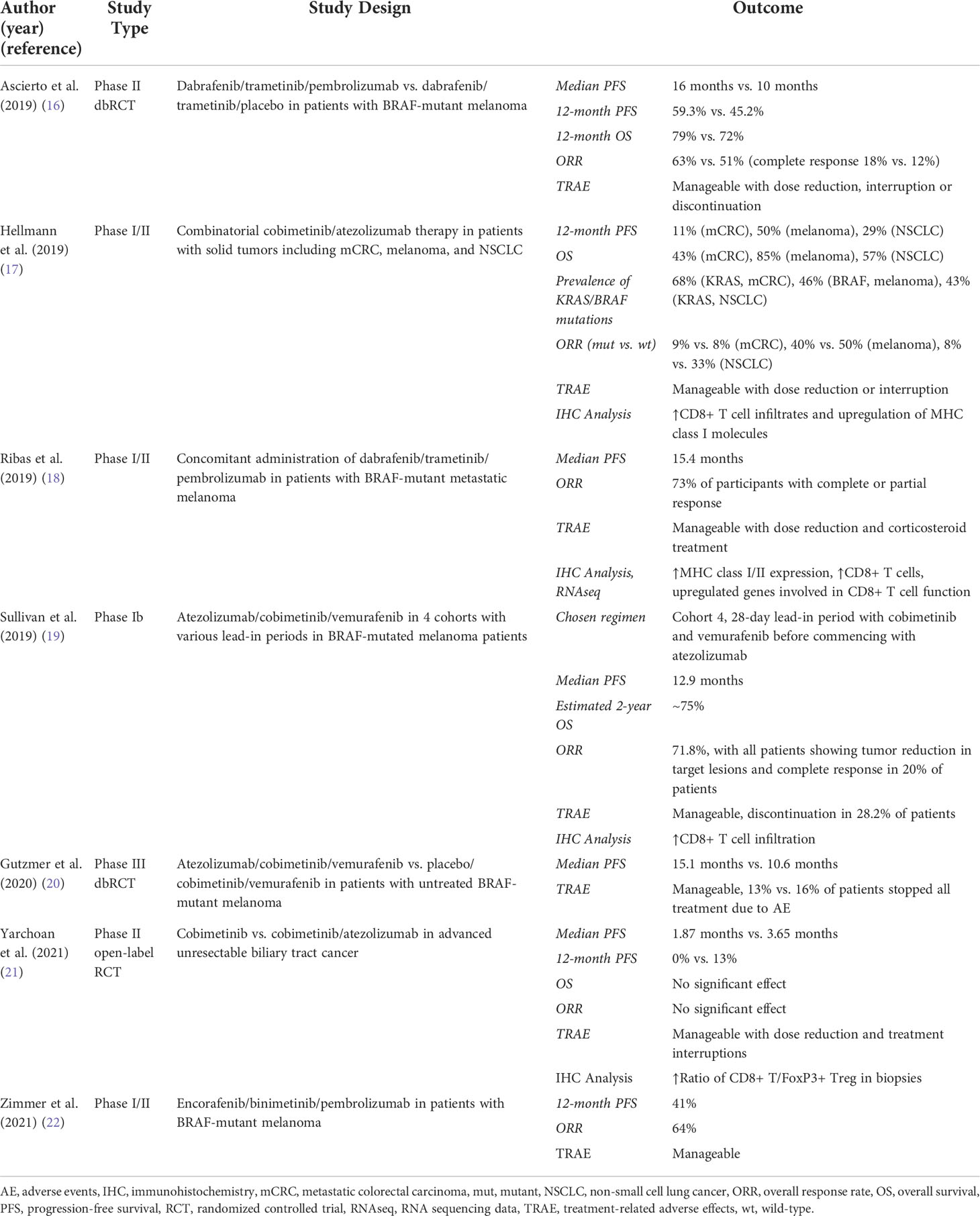
Table 1 Results from selected completed clinical trials investigating MEK inhibition as part of dual and triple therapy regimens in patients with KRAS-/BRAF-mutant solid tumors and melanoma.
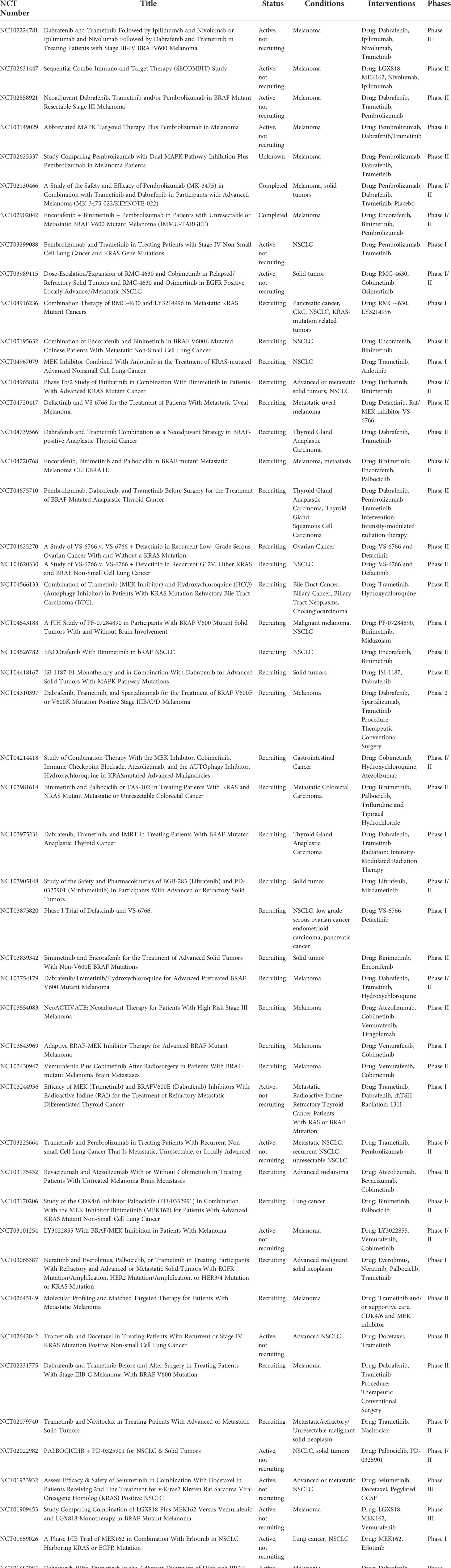
Table 2 Overview of currently active clinical trials investigating various combinations of MEK inhibition, other small molecule inhibitors and immunotherapy in mutant KRAS- and BRAF-driven malignancies.
We here collate and summarize the properties of the RAS-RAF-MEK-ERK signaling pathway from physiology to oncoimmunology, highlighting its function in physiologic cell signaling, its implications in tumor development, tumor maintenance and immune-evasion, but also its importance in homeostasis of immune cells. We further outline the rationale and concepts for exploitation of immunomodulatory properties of RAS-RAF-MEK-ERK inhibition with a focus on novel therapeutic agents and therapy regimens. In this update, we discuss relevant results from selected recent (pre-)clinical studies and in particular the anti-tumor and immunomodulatory effects associated with MEK inhibition to point out its potential in the context of combinatorial and intermittent anticancer therapy.
The intracellular RAS-RAF-MEK-ERK signaling cascade is classified as a MAPK pathway, a group of signaling pathways each consisting of three distinct cytosolic protein kinase components, that form a functional signaling module serving the purpose of relaying extracellular signals to the cell nucleus in order to alter the expression pattern of genes promoting proliferation and/or differentiation. In an upstream-to-downstream fashion, the three protein kinase groups are collectively called MAP kinase kinase kinases (MAPKKK), MAP kinase kinases (MAPKK), and MAP kinases (MAPK), also known as the classical three-tiered MAPKKK-MAPKK-MAP kinase cascade. There are several kinases that belong to each of the kinase groups and form a functional unit or signaling pathway with each of their upstream and downstream counterparts (28). The MAP kinases can be classified into conventional and atypical enzymes dependent on the ability of MAPKK members to phosphorylate and activate them, as reviewed extensively elsewhere (29, 30). Conventional MAP kinases are substrates of the MAPKK family and as such are regulated by the classical three-tiered MAPKKK-MAPKK-MAP kinase cascade, whereas the regulation and physiological function of atypical MAP kinases is much more complex and still unclear (29). There are four important members belonging to the group of conventional MAP kinases, i.e. the extracellular signal-regulated protein kinases (ERK1/2), ERK5, the p38 MAP kinases, and the c-Jun NH2-terminal kinases (JNK) (28). Atypical MAP kinases are ERK3/4, the Nemo-like kinase (NLK), and ERK7/8 (29). Despite the similarities in structure and regulation, in particular at the level of MAPKK and MAPKs, the various typical MAPK pathways fulfil partially overlapping but also unique functions, such as promoting proliferation and differentiation in case of ERK, while relaying stress and pro-apoptotic signals in case of the p38 kinases. In vitro many MAPK share several substrates, but to what extent the various MAPK can complement each other in these phosphorylation events in living cells remains an area of investigation. Lastly, it should be kept in mind that in mammalian cells almost every MAPK pathway-signaling element is represented by various isoforms as a result of gene duplication and splice variants, thereby contributing to the fine tuning of signaling intensities but also complicating pharmacological intervention.
Focusing on the RAS-RAF-MEK-ERK pathway, the ERK1/2 MAP kinases are effector proteins of the MAPKKs MEK1/2 (28). The MAPKKs themselves are activated by different MAPKKKs, most importantly the group of RAF kinases, which are in turn activated by various upstream signals (31). In order to activate the MAPK pathways, an extracellular signal has to be first recognized by the cell through the means of various receptors on its membrane surface, including transmembrane receptor tyrosine kinases (RTKs), cytokine receptors, integrin receptors, G-protein coupled receptors (GPCRs) and TGFβ-receptors. These receptors then transmit the signal to the intracellular space by either phosphorylation or binding and therefore activation of specific kinases. This step leads to the formation of an intracellular signaling complex that can relay the signal onward. Major effector proteins of RTKs and activators of the RAF-MEK-ERK pathway are proteins belonging to the RAS family, which consists of monomeric guanosine triphosphate (GTP)-loaded GTPases capable of activating various different signaling pathways besides the RAF-MEK-ERK pathway, e.g. the PI3K-AKT pathway and the Ral Guanine Nucleotide Dissociation Stimulator (RalGDS) pathway (32, 33). After RAF-MEK-ERK activation, pERK1/2 phosphorylates numerous cytoplasmic substrates, but also translocates into the cell nucleus where a large spectrum of substrates and transcription factors are phosphorylated and activated leading to increased transcriptional activity of genes associated with cell metabolism, cell cycle progression, survival, and differentiation (34). Additionally, the seemingly linear cascade of the MAPK pathway is regulated by complex crosstalk and regulatory feedback loops at every level, which together lead to a finely tuned homeostasis as comprehensively reviewed elsewhere (3, 35). Regarding functional consequences of the activation of the different members of the RAS-RAF-MEK-ERK pathway, studies using genetically engineered mouse models have revealed that the three isoforms of the RAF protein, i.e., RAF1 (c-RAF), ARAF, and BRAF, are all capable of activating MEK1/2 by phosphorylation (3, 36). Yet, differences in the functional consequence dependent on the active RAF protein isoform exist for embryonic development. In mice, deletion of Raf1 led to increased levels of apoptosis in several tissues and defects in vascularization and placental development (37, 38). Deletion of Braf on the other hand led to general growth retardation and vascular defects due to endothelial cell apoptosis (39). Araf-deficient mice showed a wide range of phenotypic expression ranging from minor neurological abnormalities with long-term survival to more severe neurological and intestinal defects and postnatal death (3). It is noteworthy, that systemic ablation of Raf1 or Braf in adult mice, alone or in combination, did not lead to significant toxicities (40). However, in the case of ablation of all three RAF isoforms, cell proliferation ceased and death followed (41). Lastly, it needs to be kept in mind that the MAPKKK and proto-oncogene product TPL2/COT confer RAF independent activation downstream of Toll-like receptors in inflammatory settings (42). Regarding the function of MEK1/2, deletion of Mek2 in mice showed dispensability for embryonic development and adult homeostasis (3). However, Mek1-deficient mice died during embryonic development due to placental defects. Deletion of both genes also led to embryonic death (43). Systemic ablation of Mek1 and Mek2 in adult mice caused rapid death due to severe intestinal defects (40). Similar results were found in studies regarding ERK-deficient mice, showing dispensability of Erk1 and embryonic lethality in case of Erk2 disruption (44). Taken together, the mentioned studies revealed significant dependency of embryonic development and adult homeostasis on all three main nodes of the RAF-MEK-ERK pathway (3).
ERK signaling regulates many physiologic cellular processes and responses in a wide variety of immune cells (45). Here, we focus specifically on T and B lymphocytes, NK cells, NKT cells, macrophages, DCs, and neutrophils, since these immune cell types have been demonstrated to take on important roles in the interaction with tumor cells and the associated TME tilting the balance either towards a pro- or anti-tumor setting. Since selective pharmacologic targeting of elements of the MAPK pathway has shown promise in the battle against cancer, it is of great interest to identify possible implications of MAPK and especially RAS-RAF-MEK-ERK signaling interference in specific immune cell types.
In T lymphocytes, distinctive mechanisms activate the three classical three-tiered MAPK pathways described earlier. For example, the activation of the T cell antigen receptor (TCR) can lead to ERK activation, whereas co-stimulation by accessory molecules of the TCR, e.g. CD28, can lead to JNK activation and therefore transcription of different subsets of genes (28). The MAPK pathways are already involved in early thymocyte development when thymocytes go through the stages of double negative (CD4-CD8-) (DN) thymocytes, double positive (CD4+CD8+) (DP) thymocytes and finally maturation into either CD4+CD8- Thelper cells or CD4-CD8+ Tcytotoxic cells (Figure 1). While the p38 MAPK pathway is involved in the progression from DN to DP thymocytes, the ERK pathway seems to be crucial for differentiation and proliferation of immature DP thymocytes after activation of the pre-TCR. Also, the ERK pathway was shown to be involved in the positive selection and lineage commitment of future Thelper or Tcytotoxic cells (Figure 1) (28). Interestingly, various studies have shown that pharmacological inhibition of the ERK pathway seems to skew thymocyte lineage commitment towards CD8+ Tcytotoxic cells, mediated by the kinase ERK2 (46, 47). The JNK pathway on the other hand was suggested to be involved in the negative selection (apoptosis) of DP thymocytes with insufficient affinity of the pre-TCR to the corresponding major histocompatibility complex (MHC) molecule or autoreactivity to self-antigens (28). After positive selection and lineage commitment, differentiation of CD4+ Thelper cells into Thelper 1 (Th1) or Thelper 2 (Th2) cells does not seem to be solely dependent on the presence of specific transcription factors involved in cytokine gene regulation, e.g., T-bet or GATA3, but seems to be regulated by activation of MAPK pathways. It was shown, that the ERK1/2 signaling pathway is required for the differentiation of CD4+ T cells into Th2 cells through stabilization of GATA3, whereas intact JNK and p38 signaling appeared to be a requirement for interferon (IFN)γ production and subsequent differentiation of CD4+ T cells into Th1 cells (28, 48). How the MAPK signaling cascade determines developmental and functional aspects of other Thelper subsets, including Th9, Th17, Th22 or T follicular helper (Tfh) cells has not yet been completely clarified and requires more research. In effector CD8+ Tcytotoxic cells, the cytotoxic activity seems to be regulated by the ERK pathway, as studies on pharmacologic ERK inhibition or in Erk deficient mouse models have suggested (28, 47). The role of the JNK pathway in CD8+ T cells is unclear, whereas p38 signaling was demonstrated to lead to the induction of apoptosis in CD8+ T cells but not in CD4+ T cells by decreased expression of the anti-apoptotic protein Bcl-2 (49). Furthermore, intact ERK signaling was shown to be required for the regulation of the expression and release of cytokines such as interleukin-2 (IL-2), a crucial cytokine for the proliferation and differentiation of T cells and priming with subsequent proliferation and survival of naïve T cells in response to antigen presentation (Figure 1) (11, 50, 51). Interestingly, it was discovered that specific loss or inhibition of Erk2 leads to a severe defect in IL-2 production accompanied by decreased expansion and survival of Erk2T-/- CD8+ T cells in vitro and in vivo of a murine model (47). Exogenous IL-2 could only partially rescue the Erk2-deficient CD8+ T cells. IL-2 was also shown to be critical for regulatory T cell function, which promote an immunosuppressive tumor microenvironment (52). Activation and functional capacity remained intact in Erk2-deficient CD8+ T cells, as well as priming and early expansion in response to antigen presentation (47). In contrast, absence of Erk1 was found to be largely dispensable for CD8+ T cell proliferation and survival (47). Further investigation into the regulation of survival and apoptosis in Erk2T-/- CD8+ T cells showed that upregulation of pro-survival regulators and downregulation of pro-apoptotic regulators are Erk2 dependent (Figure 1) (47). The Erk2-dependency of survival and apoptosis seems particularly relevant in T cell response contraction after activation. In this context, Programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) signaling have been identified to exert immunoregulatory functions, i.e., suppression of T cell activation, reduction of proinflammatory cytokine expression and induction of apoptosis, in response to TCR-mediated activation and proliferation (10).
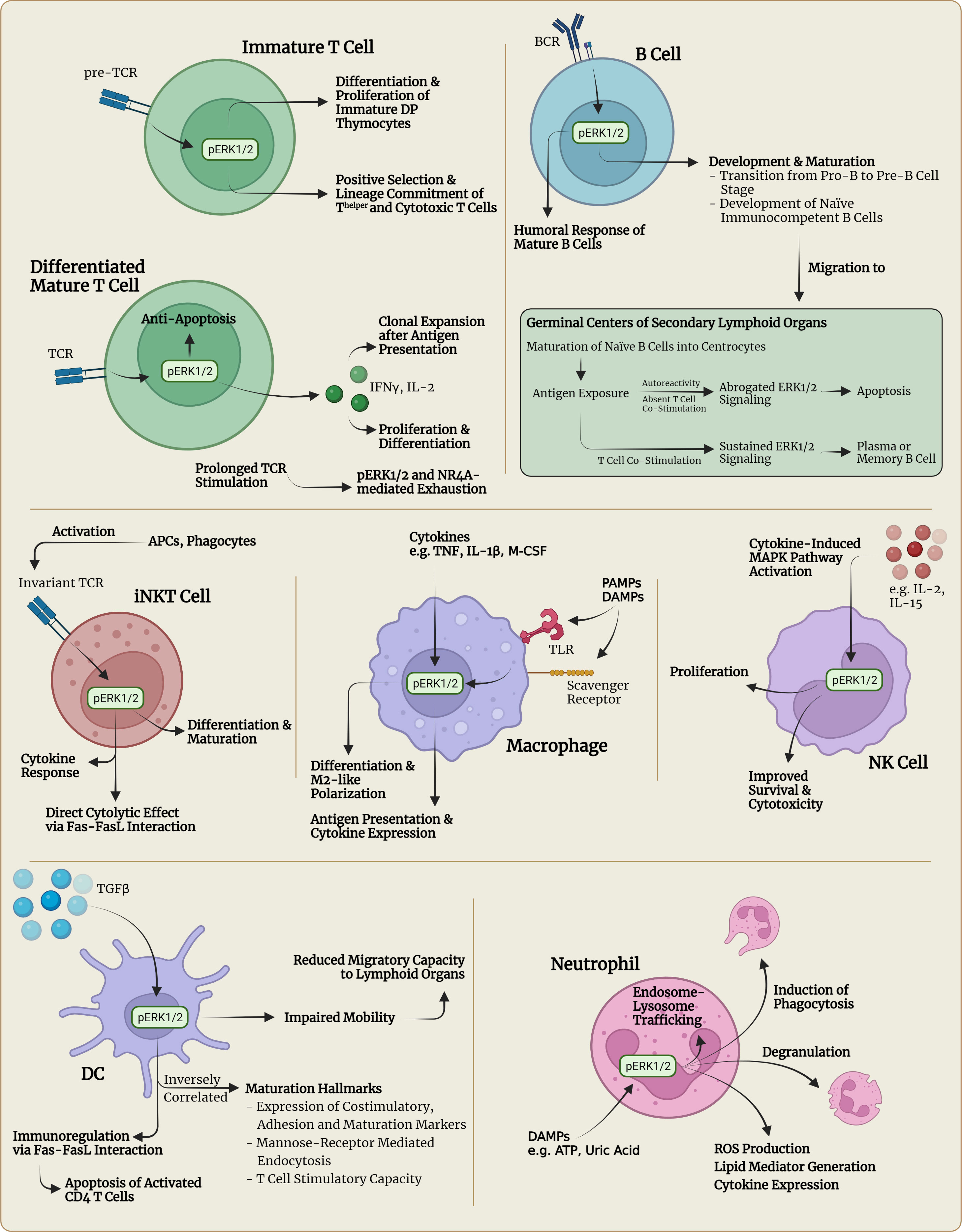
Figure 1 Visualization of selected immunophysiological consequences of pERK1/2 signaling in different immune cell types. APC, antigen-presenting cell; ATP, adenosine triphosphate; BCR, B cell receptor, DAMPs, damage-associated molecular patterns; DC, dendritic cell; DP thymocytes, double positive (CD4+CD8+) thymocytes; IL, interleukin; IFN, interferon; M-CSF, macrophage colony-stimulating factor; NK, natural killer; NKT, natural killer T; PAMPs, pathogen-associated molecular patterns; ROS, reactive oxygen species; TCR, T cell receptor; TLR, Toll-like receptor; TNF, tumor-necrosis factor.
In B lymphocytes, intact RAS-RAF-MEK-ERK signaling has been implicated in several developmental stages such as maturation, selection, expansion, differentiation and survival (53). For instance, the RAS-RAF-MEK-ERK pathway has been found to be crucial in successful cell cycle progression in the process of B cell development, particularly regarding the pre-B cell receptor driven transition from the pro-B to the pre-B cell stage (Figure 1) (54, 55). Pre-BCR but also B cell antigen receptor (BCR) signaling has been shown to activate three distinct protein tyrosine kinases, LYN, SYK and BTK, which are capable of activating various signaling pathways including the RAS-RAF-MEK-ERK pathway (56, 57). Once pre-B cells continue development and reach the stage of naïve immunocompetent B cells, they undergo a complex priming and maturation process within microanatomic structures located in secondary lymphoid organs called germinal centers (GC) (Figure 1). In the event of BCR stimulation in combination with costimulatory signals from Thelper cells, naïve B cells transform into centroblasts that undergo clonal expansion and somatic hypermutation (58, 59). Subsequently, centroblasts differentiate into centrocytes and migrate towards the light zone of a GC where the centrocytes are confronted with follicular dendritic cells (FDCs) that display unprocessed antigen on their surface (59). In case the centrocytes express BCRs with high affinity to the presented antigen, they take up the antigen from FDCs, internalize and process it so that it can be presented to T cells. If these T cells recognize the presented antigen, they provide costimulatory signaling including CD40-ligation to induce survival and further differentiation of centrocytes into antibody secreting plasma cells or memory B cells. Autoreactive centrocytes on the other hand do not receive costimulatory T cell signaling and therefore go into BCR-triggered apoptosis (Figure 1) (59). Investigations into the mechanisms of selection have revealed that the outcome of the underlying BCR signaling has a biphasic course of action (60). In the first 12h after antigen presentation, BCR signaling activates ERK1/2 (early signaling) with subsequent phosphorylation of prosurvival Bcl-2 family proteins and proapoptotic Bim. The phosphorylation of Bcl-2 proteins was shown to lead to a delay in BCR-induced apoptosis, whereas phosphorylation of Bim inhibited its proapoptotic activity by preventing its association with Bax, a proapoptotic protein (60–62). The resulting anti-apoptotic signaling then gives GC B cells time to process and present antigenic protein structures to Thelper cells. Once the GC B cells receive Thelper-mediated CD40L-CD40 costimulatory signals, the RAF-MEK-ERK-mediated anti-apoptotic signaling is sustained and the selected B cells can differentiate into memory B cells or plasma cells (60). Indeed, ablation of ERK1/2 in GC B cells was shown to significantly compromise plasma cell differentiation (63). However, if no costimulatory signal is received after antigenic presentation to Thelper cells, ERK1/2 activity undergoes BCR-induced inhibition (late signaling) and as a result the B cells will die within the next 12 h through apoptosis induced by downregulation of Bcl-2 proteins and accumulation of Bim (Figure 1) (60). Interestingly, intact KRAS-signaling was shown to play a significant role in mediating BCR-induced RAF-MEK-ERK signaling and consequent cell proliferation and survival in the development and differentiation of mature B cells in a murine model, as KRAS deficiency led to a marked reduction of mature B cells and impairment of cell proliferation and survival (64). However, the in vivo B cell humoral response was only minimally affected by KRAS deficiency, suggesting that the remaining ERK1/2 activity in the absence of KRAS is sufficient for mounting a B-cell immune response with comparable antibody production levels as seen in control conditions (64). Concluding, it has become clear that RAF-MEK-ERK signaling is crucial in key developmental stages in the production of immunocompetent, functional B lymphocytes but not in mounting a humoral response. Also, RAF-MEK-ERK signaling is not just an all-or-nothing phenomenon regarding cell survival or apoptosis, but can show contextual ambiguity. To which degree the RAF-MEK-ERK signaling cascade is involved in other areas of B lymphocyte physiology is still being actively researched today. A very recent study investigated the role of BRAF and RAF1 in murine B cell development. Although both BRAF and RAF1 are expressed in B cells and are both activated by BCR stimulation, their requirement for B cell development and function appears context dependent as the conditional knock-out of both isoforms mainly affected the transition of pro-B cells into pre-B cells and the differentiation of activated B cells into plasma cells (65). This study, however, did not functionally address to which extent ARAF and the enigmatic KSR pseudokinases could potentially compensate for the loss of BRAF and RAF in circumstances in which these two isoforms were found to be less relevant. Nevertheless, BRAF plays an important role in some B cell neoplasms as virtually 100% of typical hairy cell leukemia and about 1 to 5% of chronic lymphocytic leukemia and myeloma contain BRAF mutations (66). As recently reviewed elsewhere, the impact of the neoantigen-driven humoral response of B lymphocytes on tumorigenesis is increasingly being acknowledged and valued in understanding the complex interactions of the immune system with the TME (67).
As comprehensively reviewed elsewhere, natural killer (NK) cells are a subset of the heterogeneous group of innate lymphoid cells (ILC) that develop from common lymphoid progenitor cells but do not carry a genetically rearranged antigen receptor (68). Besides their role in the protection against pathogens, NK cells are known to be capable of carrying out receptor-mediated anti-tumor cell cytotoxicity and specifically modulating the DC and T lymphocyte immune response in a contact- and cytokine/chemokine-dependent way (68). Similar to T lymphocytes, the RAS-RAF-MEK-ERK signaling cascade has been implicated to be a crucial signaling node in the process of proliferation, survival and cytotoxicity of NK cells (Figure 1) (69–71). In this context, especially IL-15, the most important cytokine for NK cell development, and IL-2 were shown to be capable of activating downstream signaling pathways including the RAS-MEK-ERK, JAK-STAT5 and PI3K/AKT pathways to sustain cell expansion and cytolytic function (72, 73).
Natural killer T (NKT) cells are developmentally related to conventional T lymphocytes and carry surface markers characteristic of both NK cells and memory T lymphocytes – hence the label NKT cells (74, 75). Importantly, type 1 NKT (iNKT) cells express an invariant TCR capable of recognizing lipid-presenting CD1 molecules – mainly expressed by professional APCs and phagocytes - and carry important functions in both innate and adaptive immunity regulation by rapidly secreting large amounts of cytokines upon TCR stimulation and bridging both types of immune responses, reviewed comprehensively elsewhere (Figure 1) (74, 76). Furthermore, NKT cells were implied to play an important role in tumor regression through cytokine-mediated NK or cytolytic T cell activation, stimulation of pro-inflammatory IL-12 production by DCs and subsequent enhanced T cell proliferation, and direct cytolytic effects through Fas-FasL interaction (Figure 1) (74). Regarding the impact of the RAF-MEK-ERK signaling cascade on NKT cells, previous studies discovered a large dependency of NKT cell differentiation and maturation on the RAS-MEK-ERK pathway, similar to the development and differentiation of conventional T lymphocytes as described above (77). However, intact RAS-MEK-ERK signaling did not appear to be important in NKT cell survival, but was rather dependent on the mammalian target of rapamycin (mTOR) signaling pathway (78). In inflammatory responses, previous studies identified a crucial role of the RAS-RAF-MEK-ERK and JNK pathways in mounting the pivotal cytokine response of NKT cells responsible for their multifaceted effector functions (78).
Macrophages are phagocytic cells that act on various pathogen- and/or damage-associated molecular patterns (PAMPs, DAMPs) detected by specific pattern recognition receptors such as Toll-like receptors (TLRs), scavenger receptors and integrins (79–81). The recognition of such PAMPs and DAMPs, the presence of proinflammatory cytokines, e.g. tumor necrosis factor (TNF) and IL-1β, and physical-chemical changes in the extracellular milieu caused by environmental stress all lead to the initiation of signaling pathways cumulating in the activation of NF-κB and MAP kinases (Figure 1). The resulting production of proinflammatory and regulatory cytokines and internalization and degradation of recognized PAMPs and/or DAMPs leads to enhanced antigen presentation to CD4+ Thelper and invariant NKT cells (81, 82). The development of macrophages is dependent on the presence of the cytokine M-CSF, which has been shown to activate the ERK1/2 pathway, amongst others, driving macrophage growth and development (81). Further investigations into the importance of intact ERK1/2 signaling have revealed a critical dependence specifically in the terminal stages of macrophage development from monocytes, as ablation of ERK1/2 was shown to lead to a defective cell proliferation and differentiation in said late stage development of macrophages but not in myeloid precursor cells or monocytes (81). The physiological role of BRAF in the monocytic-macrophage/dendritic cell lineage remains ill-defined, but somatic BRAF and MEK mutations are increasingly found in histiocytic neoplasms and represent a target for inhibitors blocking these kinases (83).
DCs act as the sentinels of the immune system, continuously scanning their environment for pathogens and foreign antigens. Once a pathogen or foreign antigen is encountered, the DCs are activated and migrate to secondary lymphoid tissues in order to execute their function as antigen-presenting cells (APCs). DCs are potent APCs responsible for the initiation of primary immune responses by interacting with naïve CD4+ Thelper cells and leading to CD8+ T cell activation via cross-presentation. Furthermore, they contribute to the induction and maintenance of immunological tolerance through various mechanisms such as immunosuppressive action as well as induction of apoptosis of activated effector T cells (84, 85). It has been proposed that different subsets of DCs at different developmental and functional stages could explain the large variety of DC functions, as described elsewhere (86). Interestingly, recent research efforts have revealed the existence of DCs with immunoregulatory capacities that express TGF β-induced and ERK1/2-mediated high levels of Fas ligand (FasL) and are capable of inducing apoptosis of activated CD4+ T cells via Fas-FasL interaction in order to negatively regulate T cell responses and maintain immune homeostasis (Figure 1) (86). In regard to the maturation process of DCs, activity of the RAF-MEK-ERK signaling pathway was found to be inversely correlated with maturation hallmarks including expression of costimulatory, adhesion and maturation surface proteins, loss of mannose-receptor-mediated endocytosis, and IL-12 and T-lymphocyte stimulatory capacity (Figure 1). Activity levels of the p38 MAPK pathway on the other hand were shown to be positively correlated with the overall maturation process (87). Also, ERK1/2 activity seems to be correlated with impaired mobility of DCs, inhibiting migratory capacity to egress from tissues and migrate to lymphoid tissues (88).
As comprehensively reviewed elsewhere (89, 90), neutrophils play a central role in the inflammatory response of innate immunity as professional phagocytes and source of various cytokines and chemokines influencing initiation, orchestration and maintenance of adaptive immune responses. Regarding the importance of the RAF-MEK-ERK pathway and other MAPK pathways for neutrophil physiology, previous studies have indicated that the ERK1/2 and p38 MAPK signaling pathways are involved in key physiologic cellular processes such as reactive oxygen species (ROS) production, lipid mediator generation, cytokine production, the process of degranulation, and regulation of chemotaxis (Figure 1) (91–93). Furthermore, it was shown that traumatic injury and tissue necrosis initiate a systemic inflammatory response syndrome (SIRS) through the release of damage associated molecular patterns (DAMPs), such as ATP (94) or uric acid (Figure 1) (95), which are readily recognized by circulating neutrophils and activate the ERK1/2 and p38 pathways leading to the mounting of a pro-inflammatory response (96). Also, neutrophil extracellular traps (NETs) - decondensed nuclear chromatin associated with proteins and released by neutrophils during an inflammatory response in order to prevent pathogen dissemination and exert immunomodulatory effects – were shown to activate effector functions of neutrophils through the activation of ERK1/2, p38 and AKT signaling cascades (97). In contrast, the JNK pathway was shown to take on a less pivotal role in neutrophil function than the ERK1/2 and p38 pathways (98). Intact JNK signaling was implicated in controlling delayed apoptosis in neutrophils (98). Furthermore, RAF-MEK-ERK signaling in concert with PI3K/AKT signaling was shown to regulate endosome-lysosome trafficking and the induction of phagocytosis, which is critical for inflammatory resolution (99). Surprisingly, it was shown in a neutrophil-specific fashion, that MEK and ERK can be activated independently of each other dependent on the extracellular stimulatory signal, resulting in either complementary or redundant effects, e.g., cytokine generation and delayed apoptosis, respectively (93). Under discrete stimulatory conditions, ERK1/2 is activated by a TGFβ-activated kinase 1 (TAK1)-controlled signaling cascade rather than the MAPK pathway and appears to be a pivotal signaling molecule regulating cytokine transcription (93). This phenomenon should be taken into account in inflammatory responses consisting of a strong neutrophilic component and might warrant the utilization of concurrent ERK inhibition, especially when investigating therapeutic strategies involving MEK inhibition (93).
Taken together, MAPK- in general and RAS-RAF-MEK-ERK-signaling in particular bears important implications in immune cell physiology from development to activation and homeostasis. The resulting consequences for targeted inhibition in an oncologic context will be discussed later on.
Hyperactivation of various elements of the RAS-RAF-MEK-ERK pathway is known to play a crucial role in a large variety of tumors. The respective pathologically altered genes have been recognized as oncogenes (3). The most frequently observed mutations occur in KRAS, an isoform of the RAS family that code for various families of monomeric GTPases. Once activated by binding to GTP, the GTPases transmit intracellular signals and catalyze the inactivation of the bound GTP molecule to GDP. Most of these mutations are missense mutations of the codon 12, corresponding to glycine (G), which lead to changes from glycine to another amino acid, most frequently aspartic acid (D), valin (V), or cysteine (C) (33, 100). Oncogenic mutant RAS proteins can have varying degrees of intrinsic catalytic ability dependent on specific mutations and consequently remain GTP-bound and activated for varying durations (32, 100, 101). Furthermore, it has also been shown that mutant RAS is capable of increased self-activation and therefore decreased dependence on its associated regulatory proteins by spontaneously exchanging GDP for GTP (101). Also, mutant RAS isoforms can be partially or even completely insensitive to associated regulatory GTPase activating proteins (GAPs), such as neurofibromin (NF1) or p120GAP, and guanine nucleotide exchange factors (GEFs), such as Son of Sevenless homolog 1/2 (SOS1/2) and Ras guanyl nucleotide-releasing protein (RasGRP) (100). The occurrence of these phenomena and the associated frequency auf auto-activation are highly dependent on the specific mutation of the RAS protein, as extensively discussed elsewhere (101). Other mutated isoforms of RAS, namely the H- and N-isoforms, have also been implicated in human cancer, e.g. melanoma, bladder cancer, and acute myeloid leukemia, however to a lower extent in comparison to KRAS (5).
Other mutations frequently observed in human cancers regard the RAF isoform BRAF. In >90% of cases, the substitution of valine 600 by a glutamic acid residue (V600E) leads to a constitutively active kinase independent of upstream RAS signaling, while other BRAF mutations still require post-translational dimerization but also function independent of upstream signaling (102). In mechanistic murine studies on RAS-driven tumors, Braf and Raf1 have been discovered to be either crucial or completely dispensable in the initiation of tumor development dependent on disease entity (3, 40, 103–106). Ablation of Raf1 induced significant tumor regression, including a complete disappearance in 10% of tumors, likely as a consequence of increased rates of apoptosis (3). Various studies have shown that Raf1 is capable of phosphorylating proteins outside of the canonical MAPK pathway, e.g., RB1, MST2, ASK IκB, and Bad, implying the ability of Raf1 to influence pro- and anti-apoptotic processes and to control cellular migration and adhesion via ROKα (107–111).
Less frequent mutations within the RAF-MEK-ERK cascade occur in the genes ARAF, MEK1, MEK2 and ERK1/2. Although these genes have been reported to function as oncogenes, they are either rarely found in human tumors or their implication in tumorigenesis has yet to be defined more clearly (112). Mutations regarding regulatory proteins of RAS have also been implicated in enhancing tumorigenesis as well as development of therapy resistance and include NF1. NF1 is frequently co-mutated with codon 13 mutations of KRAS and PTPN11. PTPN11 encodes the Src homology region 2 domain-containing phosphatase-2 (SHP2) and can lead to so-called RASopathies, i.e., clinically defined genetic syndromes caused by germline mutations of regulators or components of the MAPK pathway (100).
In a physiologic state, the activation of RAS triggered by activated RTKs is usually short-lived. Tyrosine-specific protein phosphatases reverse the phosphorylation which contributed to the activation of RAS. The nature of an extracellular signaling molecule, e.g., EGF or NGF, influences the subsequent duration and extent of the MAPK pathway activation by modulating the intracellular signaling complex and influencing the response of positive and negative feedback loops (35). Depending on this modulation, the cellular response can vary greatly from induced proliferation to cessation of proliferation and a switch to differentiation. The presence of built-in negative feedback loops on many different levels however makes sure that, once triggered, the MAP kinase module is shut off at a given time to restore homeostasis. In oncogene-driven tumorigenesis, the regulatory feedback mechanisms are rendered ineffective by perpetuated hyperactivation of the MAPK pathway (3). Different pathologically altered mechanisms can lead to distorted intracellular signaling culminating in the aforementioned hyperactivation including continuous expression of various oncogenes such as RTK mutations and amplification of wild-type genes (3, 113).
Since the most frequently observed mutation leading to RAS-RAF-MEK-ERK pathway hyperactivation regards mutant KRAS, the KRAS oncoproteins have been the focus of a tremendous amount of research efforts in the search of therapeutic interventions targeting KRAS. However, efforts to develop selective inhibitors of KRAS oncoproteins have historically been hampered due to its small molecule size, relatively smooth and shallow surface lacking apparent binding sites for small molecules, interaction with multiple associated regulatory proteins and its picomolar affinity for GTP/GDP. Thus, potential candidates targeting RAS oncoproteins – alone or in combination regimens - proved to be either clinically insignificant or too toxic in early-phase clinical trials. The successful development of KRASG12C specific small molecule inhibitors covalently binding to the codon 12 cysteine residue therefore implicates nothing less than a paradigm shift (33). Promising results in phase I/II clinical trials in patients suffering from KRAS-driven NSCLC evaluating sotorasib, a highly selective and irreversible inhibitor leading to trapping of KRAS in an inactive GDP-bound state, have led to a recent approval by the Food and Drug Administration (FDA) (114–119). In the phase II trial by Skoulidis et al., the objective response rate to sotorasib was 37.1%, the median progression-free survival 6.8 months and the median overall survival 12.5 months with comparably low toxicity, providing supportive evidence of the importance and potential of sotorasib in the treatment of patients with pretreated advanced KRASG12C-mutated NSCLC (119). A recent trial evaluating another KRASG12C inhibitor adagrasib by Jänne et al. uncovered similar results in patients with pretreated advanced NSCLC, paving the way for another impending medical approval (120). However, the most commonly observed KRAS mutations in PDAC and CRC, KRASG12D and KRASG12V, are not targetable with cysteine-directed molecules like sotorasib (33). Interestingly, a noncovalent, potent KRASG12D inhibitor was recently discovered through extensive structure-based drug design with promising and robust anti-tumor efficacy in a murine model awaiting further substantiation (121). However, even though KRASG12C inhibitors have shown promising activity in patients harboring the KRASG12C mutation, progression of the disease nevertheless occurs due to mechanisms of acquired resistance as a consequence of secondary KRAS mutations or acquired bypass mechanisms on multiple levels, e.g. activating mutations in NRAS or BRAF, oncogenic fusions involving ALK or RAF1, or epithelial-mesenchymal-transition (EMT) and dedifferentiation (33). In continuation of drug discovery, several preclinical studies on selective, direct-acting covalent inhibitors of KRASG12C are currently active with no efficacy results reported yet (33). Furthermore, KRAS-directed therapeutic approaches based on novel mechanisms such as vaccines, adoptive T cell therapy, proteolysis-targeting chimeras (PROTACs), and CRISPR/Cas9 technology are emerging and are being currently investigated (33).
The first important drug capable of inhibiting RAF-signaling was sorafenib, a multi-kinase inhibitor, which was ultimately approved for treatment of kidney and liver tumors (122). In the following years, inhibitors with higher specificity toward specific mutant RAF isoforms were developed, e.g. vemurafenib, dabrafenib, or encorafenib, which target and inhibit mutant BRAFV600E/V600K proteins in melanoma (123). However, these RAF inhibitors were found to induce paradoxical ERK activation through a mechanism in which active (not necessarily mutant) RAS promotes the formation of BRAF homo- or heterodimers in which the drug-bound protomer induces allosteric transactivation of the drug-free partner and subsequent MEK/ERK activation (102, 124). This paradoxical ERK activation promoted the development of squamous skin cancer in patients carrying latent RAS mutations in skin cells. Furthermore, these RAF inhibitors were shown to promote the development of secondary neoplasms such as various types of leukemia and solid tumors including melanoma and PDAC as reported in vemurafenib-treated patients (125, 126). In an effort to prevent such paradoxical inhibitor action, RAF inhibitors known as paradox breakers were developed, e.g. PLX4032, which are capable of disrupting BRAF-containing dimers (127, 128). Other efforts to develop specific RAF inhibitors, in particular RAF dimer inhibitors, have not yet yielded significant results or are limited by toxicity and are still in early phase clinical trials (3). In KRAS-driven lung cancer, targeting of RAF1 was suggested to be a tumor-selective strategy since RAF1 was found to be essential for tumor initiation (40, 129, 130). In a murine model, systemic ablation of Raf1 did not seem to cause significant toxicities, which is a major advantage compared to MEK or ERK inhibitors (131). However, RAF1 inhibition requires high specificity since interference with other RAF isoforms could lead to significant toxicity, as studies on panRAF kinase inhibitors have shown (132). Attempts to target RAF1 more specifically have led to the discovery of therapeutics interfering with the interaction of RAF1 and its selective pro-apoptotic effectors, e.g. ROK α, ASK1, and MST2 (133). Furthermore, pharmacological, proteasome-mediated selective degradation of RAF1 by means of proteolysis-targeting chimeras (PROTAC) could lead to another strategy in specifically targeting RAF1 (134). To date, however, RAF1 degradation by PROTACs has noy yet been successfully tested (135, 136).
Important druggable targets in the RAS-RAF-MEK-ERK pathway were found to be MEK1/2, which can be considered central components of this signaling pathway. Of the MEKi that have been developed so far, some have already been approved for the treatment of melanoma, e.g. trametinib, in combination with selective BRAFV600E inhibitors, e.g. dabrafenib (137). In other cancer entities, therapy regimens with MEKi as monotherapy or in combination with chemotherapy have failed to demonstrate significant survival benefits (138–140). Possibly, administration of adequate dosages for a sufficient anti-tumor response was limited by the occurrence of significant toxicities at higher dosages, e.g. anemia, thrombocytopenia, febrile neutropenia, transaminase elevation, and uveitis besides more common side effects including skin toxicity, diarrhea and fatigue (3, 32). Also, inhibition of one effector pathway might result in compensatory activation of parallel survival and proliferation pathways, e.g., the PI3K-AKT pathway and STAT3 activation (32, 140, 141). Other concerning observations were either (i) the presence of intrinsic resistance of tumor cells mediated by for example a feedback loop that activates EGFR signaling and induces cell survival/proliferation, (ii) the rapid development of acquired resistance to the administered MEKi through various mechanisms, including insufficient inhibition of ERK activity, loss of feedback-inhibition, (re-)activation or upregulation of diverse RTKs such as MET, IGF-1R, EGFR or HER2 leading to cell growth and proliferation, newly acquired mutations in MEK1/2 or the emergence of other oncoproteins, such as RAS/RAF amplification or mutation, that eventually lead to ERK1/2 reactivation, and (iii) the evolution of cross-resistance between targeted MAPK pathway inhibition and immunotherapy in sequential combinatorial therapeutic approaches (123, 141–147).
Particularly fueled by reactivation of ERK1/2 after previous treatment with MEKi, ERK kinase inhibitors (ERKi) were developed, e.g. ulixertinib and ONC201, with the intention of overcoming MEKi-associated limitations. ERKi are currently being evaluated in various phase I/II clinical trials either as monotherapy or in combination with chemotherapy, i.e., paclitaxel and gemcitabine (32, 123, 132). Similar to MEKi, ERKi have been shown to cause significant toxicities when administered in high dosages. Studies are currently investigating the possibility of selectively inhibiting key substrates further downstream of ERK1/2 with the hope of causing less toxicities (3).
Nevertheless, targeted therapies and immunotherapies have revolutionized the treatment of patients with metastatic cancer with durable tumor control in specific patient subsets warranting further investigation and refinement of these therapeutic modalities, also to increase understanding of resistance mechanisms and how to avoid or circumvent these mechanisms (3, 8).
In recent years, regulatory proteins of RAS activity have been identified as eligible drug targets including the SHP2 and Son of SOS1/2. SHP2 is a non-receptor protein tyrosine phosphatase (PTP) encoded by PTPN11 that is involved in relaying signals downstream of several growth factor, cytokine and integrin RTKs (148). The activity of SHP2 has been shown to be required for full activation of the RAS-MAPK pathway. Furthermore, SHP2 has a role as a negative regulator of the JAK-STAT signaling cascade (148–150). The involvement and requirement of SHP2 in these signaling pathways has uncovered its essential role in oncogenic signaling, for example linked to PDAC and NSCLC (151). Extensive research efforts have demonstrated that SHP2 seems to be required for proper function of mutant KRAS, prominently during carcinogenesis (151). Loss or inhibition of SHP2 in established tumors was shown to slow down tumor progression. Furthermore, once downstream RAS effector proteins are pharmacologically inhibited, intact SHP2 signaling appeared be required for the reestablishment of RAS signaling through a multiple RTK-dependent feedback reactivation mechanism, e.g., EGFR, HER and FGFR signaling (152). Therefore, one could argue that intact SHP2 signaling plays an important role in keeping up intrinsic and acquired resistance mechanisms to circumvent pharmacologic inhibition of the RAF-MEK-ERK signaling cascade. Indeed, Fedele et al. and Ryan et al. found indications supporting this notion as the development of adaptive resistance to prolonged MEK inhibition or KRASG12C inhibition in a murine model could be prevented by co-administration of a SHP2 inhibitor (152, 153). Taken together, these findings warrant further investigation in the human setting. Currently, multiple clinical trials are actively recruiting participants to this end, combining targeted inhibition of MEK or ERK with SHP2 inhibition (e.g., NCT03989115, NCT04916236) (Table 2).
In the recent years, fragment-based and high-throughput screening approaches have led to the discovery of small molecules capable of disrupting the KRAS-SOS1 interaction (154). Interestingly, some of these small molecules were shown to activate rather than inhibit the SOS1-mediated nucleotide exchange leading to a biphasic modulation of RAS signaling through negative feedback on SOS1 (154, 155). As an example, a recent study demonstrated that selective inhibition of SOS1 with a nanomolar inhibitor effectively downregulated levels of active RAS in tumor cells (154). Importantly, in wild-type KRAS cells a complete inhibition of the RAS-RAF-MEK-ERK pathway was observed, whereas in cells with mutated KRASG12C SOS1 inhibition led to a reduction in ERK phosphorylation by approx. 50% (154). This finding raised the question whether combinatorial inhibition of additional targets could enhance the observed partial inhibitory effect on the RAS-RAF-MEK-ERK signaling cascade. Indeed, vertical dual inhibition with a covalent inhibitor of KRASG12C resulted in synergistic anti-tumor activity (154). Therefore, SOS1 inhibition has been identified as a viable option and promising tool in therapeutic regimens consisting of various combinatorial therapeutic agents targeting mutant RAS-driven tumors and other tumors with EGFR and NF1 alterations (154, 156). First clinical trials have been initiated investigating the potential of SOS1 inhibition in patients with solid tumor malignancies with KRASG12C mutation, however with no results reported yet (NCT04111458, NCT04975256).
Increasing evidence has shown that oncogenic mutations such as KRAS mutations are capable of mediating crosstalk with the immune system via oncogenic signaling and can lead to immune escape mechanisms at different stages of the cancer-immunity cycle (Figure 2) (6, 7). During tumorigenesis, the degree of inflammation and inflammatory responses play a key role and concurrently influence the efficacy of various therapeutic approaches. A large variety of tumor-infiltrating immune cells, including CD4+ and CD8+ T lymphocytes, regulatory T cells (Tregs), B cells, Th17 cells, NK cells, DCs, MDSCs and neutrophils, interact with tumor cells in a complex way, mediated by the TME (Figure 2). The TME can be seen as a functional entity made up of the infiltrating immune cell types and stromal cells consisting of fibroblasts, adipocytes, endothelial cells, and the extracellular matrix (ECM) (7, 157, 158). These different cell types interact with each other in a contact- or soluble molecule-dependent manner (autocrine and/or paracrine) and ultimately tilt the balance towards either a pro- or anti-tumorigenic state. Regarding the interaction with the immune system, the two following characteristics of cancer cells have been branded hallmarks of cancer: (i) the ability to evade immunological destruction and (ii) maintenance of tumor-promoting inflammation (159). The advancement of the understanding of KRAS-driven carcinogenesis and its underlying mechanisms has shown that KRAS mutations are not only capable of leading to sustained proliferation and reduced apoptosis in cancer cells but can take direct influence on the TME through autocrine and paracrine effects. Subsequently, the surrounding stromal cells are remodeled by a cytokine-, chemokine- and/or growth-factor driven process in order to establish tumor-promoting inflammation and evasion from the immune system. The resulting immunosuppressive microenvironment prevents tumor antigen (neoantigen) presentation by antigen-presenting cells (APCs) for T cell priming and promotes polarization of macrophages towards the tumor-supportive M2-like phenotype (160–163).
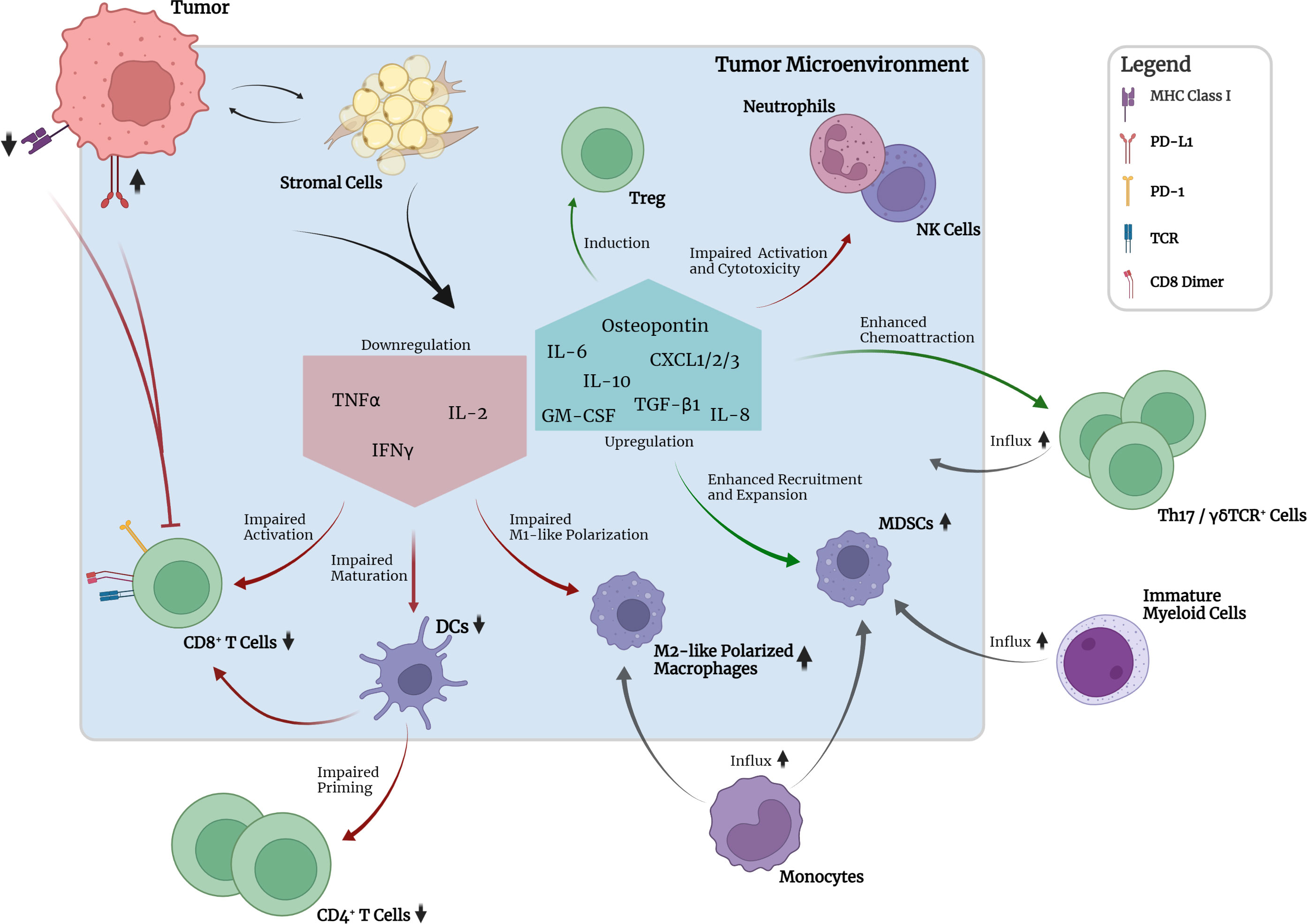
Figure 2 Crosstalk between tumor, TME and selected immune cell types in the absence of pharmacological interruption. Mutant KRAS-associated downregulation of MHC class I molecules and increased expression of PD-L1 on tumor cells leads to reduced detectability and pronounced inhibition of CD8+ T cells. Crosstalk between tumor and stromal cells leads to downregulation of pro-inflammatory cytokine expression, e.g., TNFα, IL-2, and IFNγ, by stromal cells. Consequently, activation of CD8+ T cells, maturation of DCs and M1 polarization of macrophages are impaired. Subsequent priming of CD4+ T cells is impaired. Upregulation of IL-10 and TGF-β1 leads to induction of suppressive peripheral Treg. GM-CSF in the TME together with tumor-secreted osteopontin leads to recruitment and expansion of immunosuppressive myeloid- and monocyte-derived MDSCs. Upregulated expression of chemokines leads to chemoattraction of Th17 and γδTCR+ cells which have been implicated in promotion of tumor cell proliferation and angiogenesis. Neutrophil and NK cell function seem to be impaired regarding activation and cytotoxicity. Upregulation of IL-6 production promotes cell survival and establishment of an inflammatory microenvironment required for optimal tumor growth.
Regarding the establishment of tumor-promoting inflammation, KRAS was shown to be capable of inducing cytokine secretion such as IL-6 and reduced secretion of IFNγ, TNF, and IL-2 by different stromal cell types, e.g., fibroblasts, myoblasts and epithelial cells. This reduced secretion of IFNγ, TNF, and IL-2 was shown to lead to impaired maturation of DCs, CD8+ T cell activation and expansion and polarization of M1-like (proinflammatory) macrophages (161). Increased IL-6 secretion was ultimately shown to lead to IL-6-mediated Janus activated kinase (JAK) activation in tumor cells with subsequent downstream phosphorylation and activation of the transcription factor signal transducer and activator of transcription 3 (STAT3) (164). In various studies, this so-called IL-6/JAK/STAT3 axis has been implicated to be the main contributor to several tumorigenic cellular processes, especially in lung and pancreatic cancer, by promoting cell survival and upregulation of the reactive oxygen species (ROS) detoxification program via activated MAPK and PI3K/AKT pathways (165–168). Furthermore, IL-6 was shown to be necessary to maintain the activation status of stromal fibroblasts and the inflammatory microenvironment required for optimal tumor growth (168). Besides IL-6, scientific evidence has also pointed towards IL-8, the ligand of CXCR2, to be a critical player in the maintenance of inflammation, tumor growth and also angiogenesis (169). Various studies have uncovered a KRAS/IL-8 link mediated by the MAPK or PI3K/AKT signaling pathways in different human cancer cell lines and tumor specimens (170). Similar to IL-6, IL-8 and CXCR2 signaling was shown to affect cancer-associated fibroblasts and cause increased secretion of pro-tumorigenic cytokines mediated by the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) transcription factor (170). NF-κB itself was also discovered to be a key mediator and inducer of tumor-promoting inflammatory responses (171).
Other more recent scientific efforts have indicated that activating KRAS mutations could lead to the chemoattraction of macrophages and Th17 cells, which have both been implicated in inflammation-induced tumorigenesis by secretion of proinflammatory cytokines, e.g., TNF, and proteases, e.g., matrix metalloprotease-9 (MMP-9) (172–176). Moreover, activation of KRAS was also reported to be associated with increased numbers of γδTCR+ inflammatory cells, which are a non-MHC-restricted lymphocyte subset closely associated with innate immunity and implicated in accelerated tumor formation (174). γδTCR+ cells were shown to produce high levels of IL-17, leading to promotion of tumor cell proliferation, angiogenesis, production of proinflammatory cytokines and chemokines, metalloproteases, and MDSC attraction (173). Furthermore, γδTCR+ cells were found to suppress tumor-infiltrating and anti-tumorigenic αβTCR+ cells through checkpoint receptor ligation (177). In line with these findings, genetic or pharmacologic ablation of γδTCR+ cells led to protective effects regarding tumorigenesis in a murine model (177). Furthermore, recent studies have identified the inflammasome, a danger-sensing multimeric protein complex part of the innate immune response, to play a crucial role in KRAS-driven leukemia (178). Oncogenic KRAS in leukemia was shown to cause the activation of the NLRP3 inflammasome, which appeared to take on a key role in the development of cytopenia, splenomegaly and myeloproliferation (178). Whether this finding is also relevant in KRAS-driven solid tumors requires further research.
Besides the establishment of a tumor-promoting inflammation in the TME through a wide variety of mechanisms, KRAS itself has also been associated with immunomodulatory and immune escape effects (Figure 2). As such, oncogenic KRAS mutations have been linked to programmed cell death 1 ligand 1 (PD-L1) expression in cancer cells of lung or pancreatic origin (179). Tumor cells in for instance lung and pancreatic cancer have been found to be capable of acquiring the ability to (over)express PD-L1 which facilitates immune evasion by reduction of the tumor-specific T cell function and is correlated with poor OS in several types of solid tumors (179, 180). In KRAS-mutant tumors, upregulation of PD-L1 expression was shown to be regulated by MAPK-dependent transcriptional activity of ERK1/2, AP-1 and partially STAT3 (13, 179, 181). However, one study showed that PD-L1 expression was instead regulated by the activation of PI3K/AKT downstream of KRAS (182). Besides upregulating the transcriptional activity of the PD-L1 gene, KRAS-mutant tumor cells were also shown to be able to increase PD-L1 mRNA stability via MAPK-dependent signaling (183). Importantly, the associations between KRAS activation and the expression of PD-L1 should always be interpreted in the context of the respective tumor origin, as studies have revealed that KRAS mutations in CRC can result in inverted effects on PD-L1 expression levels compared to lung or pancreatic cancer (184, 185). Turning to other immunomodulating mechanisms, KRAS-mutant tumors also appeared to enhance local immunosuppression by peripheral induction of regulatory T cells (Tregs) in the TME through increased IL-10 and TGF-β1 secretion mediated by the MEK-ERK and AP-1 pathway (186). Another immunomodulatory effect of KRAS-mutant tumors was found to be the downregulation of MHC class I molecules on the cell surface of tumor cells through MAPK-driven internalization and intracellular sequestration. As a result, the recognition of tumor-associated antigens or neoantigens by effector CD8+ T cells through MHC class I molecule and TCR interaction was hindered, rendering the tumor cells less recognizable and less sensitive to lysis (7, 10, 161, 187). Furthermore, KRAS mutations were shown to upregulate GM-CSF expression in the TME, enhancing the infiltration of MDSCs which are known to be potent suppressors of effector T cell responses and therefore contribute to the evasion of anti-tumor immunity (187–190). Another mechanism involved in the chemoattraction of MDSCs to the TME was shown to be oncogenic KRAS-induced repression of interferon regulatory factor 2 (IRF2) resulting in higher expression of CXCL3 which binds to CXCR2 on MDSCs and promotes their migration to the TME (191).
Taken together, research efforts of the past have clarified that oncogenic KRAS-signaling is capable of inhibiting or modulating immune responses on various levels in order to dampen or even completely silence anti-tumor immune responses at different stages of the cancer-immunity cycle and enable unchecked tumor growth. This increased understanding has led to the discovery of many new opportunities regarding therapeutic approaches in the ongoing fight against cancer.
Since immunomodulatory effects of KRAS-driven oncogenic signaling have been identified to play a major role in tumorigenesis and maintenance. Moreover, MEKi have been shown to be promising and feasible in particularly combinatorial therapy regimens not only regarding direct anti-cancer effects but also anti-tumor immunomodulatory effects. Although MEKi have primarily been developed for inhibition of oncogenic signaling, recent scientific interest in their immunomodulatory effects when administered systemically has substantially risen due to promising results after targeted inhibition of MEK in combination with immunotherapeutic agents in certain tumor types with high immunogenicity (10). As various studies have shown, the involvement of the MAPK pathway in tumorigenesis and immune function, e.g. initiation of innate immunity, activation of adaptive immunity or establishment of the TME, is complex and context-dependent and requires further elaboration regarding MEKi-associated alterations to immune regulation of specific immune cell subsets and the TME (10).
MEKi have been shown to influence T lymphocyte physiology and function on various levels. In vitro priming of T cells revealed that MEK inhibition blocks priming and expansion of naïve CD8+ T cells in response to anti-CD3 and anti-CD28 stimulation (11). In order to identify whether MEK inhibition would also influence priming in vivo, tumor-draining lymph nodes were evaluated for conversion of naïve CD8+ T cells into fully differentiated cytotoxic T-bethiEomeslo T cells (11). In line with the in vitro findings, MEKi treatment led to reduced numbers of T-bethiEomeslo T cells in tumor-draining lymph nodes. Also, the number of tumor-antigen-specific T cells in the lymph nodes was reduced. Withdrawal from MEKi treatment restored the MEKi-associated effects, indicating that MEKi treatment does not deplete naïve precursor cells and that its effect on tumor-antigen-specific T cells is reversible (11). In a different murine study, DC function, antigen-induced T cell priming and proliferation upon antigen presentation showed slight reduction at MEKi dosages sufficient for suppression of tumor growth only when co-administered with anti-CD40 agonistic Ab, supporting the notion that CD40 signaling could overcome the impaired priming (192). Other in vitro studies have shown that MEKi are capable of suppressing proliferation and cytokine production, e.g., IL-2 through TCR signaling blockade (11, 13, 193, 194). Cell culturing in the presence of trametinib showed partial inhibition of CD4+ T cell proliferation after 3 days of treatment. Interestingly, the inhibitory effect vanished after 7 days of cell culturing (161). The negative effects found in vitro could however not fully translate to in vivo models (10). In a BRAFV600E-driven melanoma mouse model, the T cell inhibitory effects seen in vitro could not be observed in vivo (195). T cell lytic activity, infiltration, cytotoxicity and their response to stimulation with subsequent cytokine release (IFNγ) appeared to be normal (195). However, in another study on trametinib in an ovarian cancer mouse model, effector CD8+ T cells were shown to proliferate to a lesser extent after treatment with the MEKi. Also, IFNγ production by MEKi-treated CD8+ and CD4+ T cells isolated from the mouse tumors appeared to be significantly reduced (24). The inconsistent results found by the afore mentioned studies could be explained by differences in treatment schedules and the co-administration of exogenous IL-2 in the utilized functional assays in ref. 153. Interestingly, in a different study, decreased T cell proliferation and IFNγ production in the presence of MEKi was shown to be the result of decreased IL-2 secretion. This effect was shown to be reversible upon exogenous IL-2 administration (10, 23).
When looking closer at tumor-infiltrating CD8+ T cells after MEK inhibition, notable differences in the expression of the transcription factors T-bet and Eomes were observed (11). In control tumors without MEK inhibition, the infiltrating CD8+ T cell population primarily consisted of the T-betloEomeslo phenotype with virtually no T-bethiEomeshi cells (11). In MEKi-treated tumors, the majority of tumor-infiltrating CD8+ T cells (approx. 70%) expressed T-bet. 20 – 25% of the CD8+ T cells were double positive for T-bet and Eomes. A closer look at antigen-specificity of these T cells revealed that the MEKi-induced appearance of T-bethiEomeslo and T-bethiEomeshi CD8+ T cells included the accumulation of tumor-antigen-specific effector T cells that could directly target the tumor cells (11). Upon in vitro restimulation, CD8+ T cells from the MEKi-treated tumors exhibited more pronounced effector characteristics than T cells from control tumors measured by IFNγ production, supporting the notion of MEK inhibition leading to the presence of a more potent tumor-infiltrating effector T cell population (11). These observations were further substantiated in a recent study on MEKi-induced metabolic reprogramming of effector CD8+ T cells (196). MEK inhibition led to the generation of antigen-experienced T memory stem cells (TSCM) with a very strong cellular fitness giving rise to highly activated and less exhausted CD8+ T cells with high antitumor activity (196). In chronic virus infections and cancer, the occurrence of T cell exhaustion ultimately leading to apoptosis as a consequence to chronic exposure to antigens and inflammatory signals has been frequently observed (11). Typically, inhibitory surface molecules including PD-1, CTLA-4, T cell immunoglobulin and mucin-domain containing 3 (TIM-3), and Lymphocyte activation gene 3 (LAG-3) are significantly upregulated, limiting response to antigen-mediated TCR stimulation, proliferation, and secretion of effector cytokines (197). In exhaustion, the NR4A transcription factor family has been implicated as a master regulator of especially CD8+ T cells (197). Interestingly, maximal NR4A activity in response to TCR stimulation has been shown to be dependent on intact ERK signaling, offering a rationale for the reported MEKi-associated effects on T cell exhaustion (196, 198). Although negative feedback cycles, such as activation of the PD-1 receptor, could also slow down or prevent exhaustion in T cells, maintenance of highly active cytotoxic T lymphocyte effectors is critical for cell-mediated anti-tumor responses (11). Prolonged blockade of TCR signaling by means of MEK inhibition was shown to interfere with effector function and proliferation of T cells at the tumor site (24). It was also shown that temporary MEK inhibition would transiently inhibit cell-cycle progression after TCR-mediated cell activation in naïve CD8+ T cells, creating a window of opportunity for the generation of functionally and metabolically enhanced CD8+ TSCM (196). Later on, after withdrawal of the MEKi and upon restimulation, these CD8+ TSCM would then reinstate their proliferative capacities to generate highly potent and robust CD8+ T cells, as described above (196).
Taken together, the potential effects of MEK inhibition on T lymphocytes have been investigated extensively (Figure 3). Nevertheless, it has become apparent that an unambiguous conclusion cannot be drawn yet due to inconsistent results on priming, proliferation and effector function in various studies. Importantly, there is a need for a standardized approach with regard to experimental design in order to further clarify the true MEKi-associated effects on T lymphocytes.
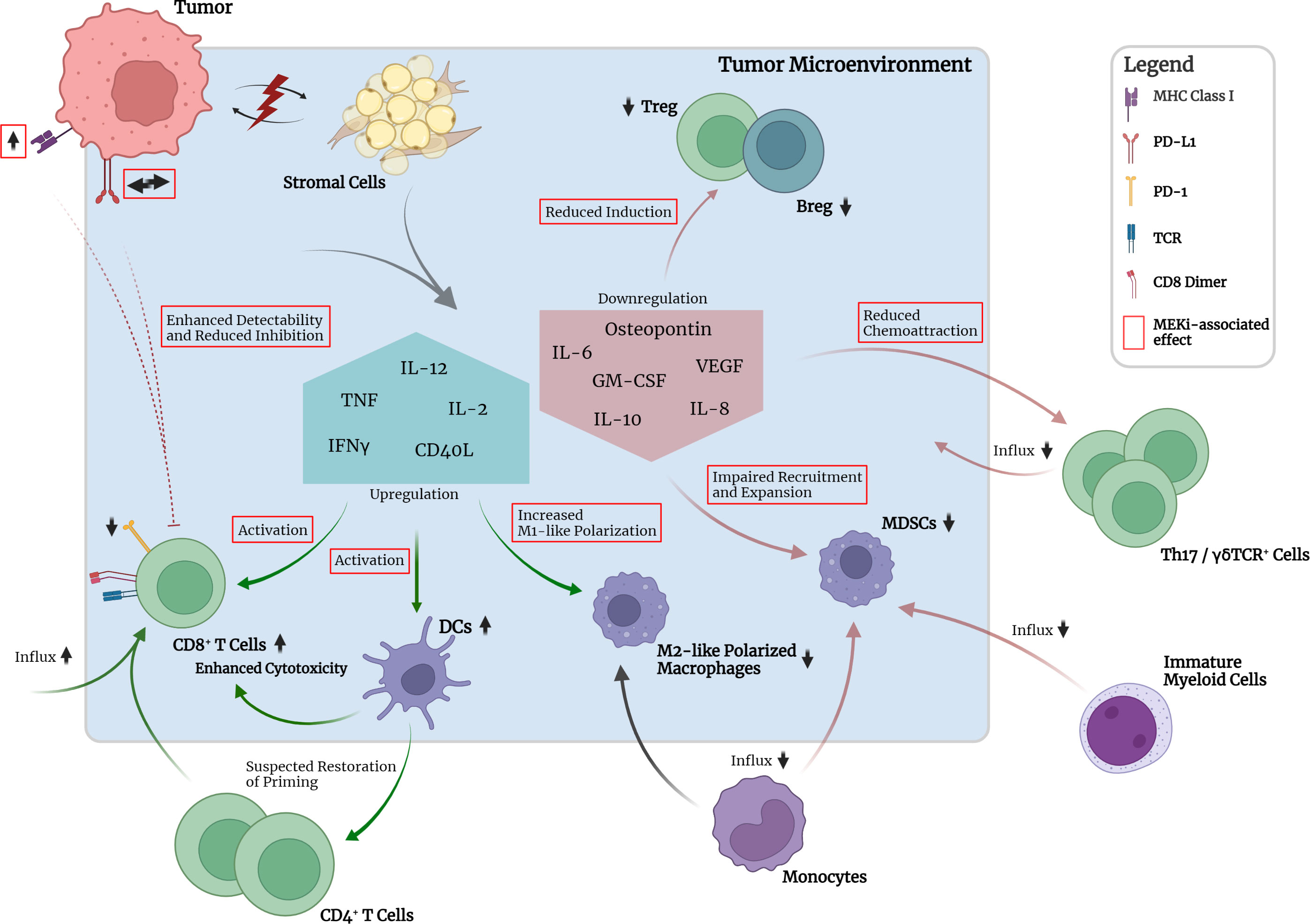
Figure 3 MEKi-associated cell intrinsic effects in selected immune cell types. MEKi leads to transient suppressed proliferation and cytokine production, e.g., IFNγ and IL-2, in CD4+ and CD8+ T cells through prolonged TCR signaling blockade. Infiltration, cytotoxicity and cytokine release upon (re-)stimulation however appear normal. Tumor-infiltrating CD8+ T cells showed MEKi-induced increased expression of the transcription factors T-bet and Eomes leading to accumulation of more potent tumor-antigen-specific CD8+ T effector cells. Transient MEKi-associated cell-cycle progression halt leads to generation of metabolically enhanced T memory stem cells (TSCM), which give rise to potent and robust CD8+ T effector cells upon restimulation. Lower expression of PD-1 was observed in tumor-infiltrating CD8+ T cells, suggesting these cells being less prone to exhaustion and inhibition. MEK inhibition disrupts chronic BCR signaling in Breg resulting in diminished expression of suppressive surface molecules and reduction of Breg numbers removing inhibitory action on CD4+ and CD8+ T cells. MEKi treatment of NK cells does not impair viability, even at high MEKi concentrations. However, proliferation, expression of activation markers and cytolytic capacities are significantly reduced. Cell cycle progression and cellular survival mediated through activated eukaryotic translation initiation factor 4E (eIF4E) is regulated by the MAPK pathway. MEKi disrupts RAS-RAF-MEK-ERK dependent M2 polarization and shifts macrophages towards p38 MAPK dependent M1 polarization. MEKi can restore production of IL-12 and TNF in DCs reversing tumor-induced downregulation of costimulatory molecules and activation status. MEKi-associated reduction of IL-6 expression by tumor and stromal cells restores expression of costimulatory molecules, activation markers, antigen presenting receptor CD1a and functionality. In MDSCs, MEK inhibition with subsequent prevention of ERK phosphorylation led to strongly reduced accumulation in the tumor microenvironment by inhibiting cell expansion and promoting apoptosis. Furthermore, MEKi can abrogate cytokine-induced MDSC expansion.
Although the precise role of B lymphocytes in the anti-tumor immune response has received comparably little scientific attention, recent research efforts have been gathering an emerging body of evidence recognizing a role for B cells in modulating the immune response in cancer. Regulatory B cells (Bregs) have been identified to be a heterogeneous cell population capable of suppressing effector T cell function and promoting immune tolerance (10, 199). The BCR is known to activate the downstream positioned RAS-RAF-MEK-ERK pathway in order to induce B cell responses to the recognized antigen, as discussed above (200). MEK inhibition was found to reduce Bregs in vitro and in vivo in a model of CRC. Although the precise mechanism behind this finding remains unclear, it is proposed that MEK inhibition disrupts chronic BCR signaling resulting in diminished expression of specific suppressive surface molecules and leading to the reduction of Breg numbers (Figure 3). In line with the decreased Breg population upon MEK inhibition, numbers of tumor-infiltrating CD8+ and CD4+ T cells were found to be increased (199).
Natural killer (NK) cells together with cytolytic T lymphocytes (CTL) are known to play an important role in cancer immune-surveillance through their potent natural cytotoxic activity. Two distinct mechanisms of cytotoxicity have been identified, i.e. perforin and granzyme-dependent and Fas ligand-dependent (201). Interestingly, it has been proposed in the past that healthy individuals with low overall cytotoxic activity are at a significantly higher risk of developing cancer compared to individuals with medium or high cytotoxic activity (202). Moreover, increased tumor-infiltration by NK cells correlates with a better prognosis in different human tumors (203–205). In vitro MEKi treatment of NK cells does not impair viability, even at high MEKi concentrations. However, proliferation, expression of activation markers and cytolytic capacities are significantly reduced (Figure 3). IL-15/IL-18 substitution could rescue the cells from the observed detrimental MEKi-associated effects (201). In line with these findings, another study revealed cell cycle progression and cellular survival of NK cells mediated through activated eukaryotic translation initiation factor 4E (eIF4E) to be regulated by the MAPK pathway. Furthermore, it was shown that IL-2 and IL-15 were both capable of activating the MAPK pathway leading to increased eIF4E activity, which could explain the rescue of NK cell functionality in the presence of IL-15 (201, 206). It is of great importance to investigate whether these effects seen in in vitro experiments can be reproduced in vivo and how these potentially negative effects of MEKi on NK cell expansion and function could be countered.
Recent studies investigating the effect of MEKi on macrophage polarization in the TME revealed a shift in the macrophage subset balance upon MEK inhibition from tumor-nurturing M2 macrophages towards the proinflammatory M1 macrophage type. M2 macrophages were shown to critically depend on the intact signaling cascade of the RAS-RAF-MEK-ERK pathway, whereas M1 macrophages rely more heavily on the p38 MAPK signaling pathway rescuing them from cell death under MEK inhibition (Figure 3) (9). This finding indicates that MEK inhibition can not only mediate direct antiproliferative and cytotoxic effects on tumor cells but could also shift the balance of the immune setting towards an anti-tumorigenic immune setting by depletion of immunosuppressive macrophages (9, 192).
It has been proposed that differentiation and function of DCs in cancer patients are impaired due to interaction with tumor cells or tumor-derived cytokines, in particular IL-6. Interestingly, it was shown that the tumor-derived cytokines are able to activate the p38 MAPK pathway and STAT3 signaling in DCs, leading to lower expression levels of costimulatory molecules (i.e., CD40, CD80), activation markers (e.g., HLA-DR) and CD1a, and functionally abnormal DCs (Figure 3) (15). These findings are in contrast to the earlier reported maturation-enhancing influence of p38 pathway activity in the maturation process of DCs, possibly pointing at opposite outcomes of p38 activity dependent on maturation status (87). Indeed, in vitro neutralization of IL-6 and inhibition of p38 restored the observed abnormalities and function of the DCs (15). In another study, DCs were exposed to in vivo MEK inhibition in a mouse model and did not seem to be impaired in their function regarding antigen uptake, processing and presentation to lymphocytes, opening the door to DC-based vaccination as a combinatorial approach in anticancer therapy (14, 192). In particular, Fischetti et al. reported cytokine release in response to TLR ligation to be predominantly dependent on intact p38 and JNK signaling but not ERK signaling (207). Also, MEKi treatment was shown to partially or completely restore the suppressed production of IL-12 and TNF in DCs mediated by melanoma cells and reverse the melanoma-induced downregulation of costimulatory molecules and activation markers of DCs (14).
Chronic inflammatory responses and expression of specific chemoattractants such as CXCL1/2/3/8 by tumor cells during tumorigenesis promote the pathologic recruitment and expansion of MDSCs in the TME which contribute to the occurrence of immune evasion from adaptive and non-specific immune responses (12, 208). Normal myeloid cell differentiation is diverted from its intrinsic pathway of terminal differentiation into mature myeloid cells, i.e., DCs, macrophages and granulocytes, towards the generation of pathologically activated MDSCs. Tumor-derived factors have been demonstrated to regulate myeloid cell responses and initiate immunosuppressive pathways committing immature myeloid cells to become MDSCs (12). MEK and ERK signaling have been identified to play an important role in lineage commitment in myeloid cells from hematopoietic stem cells and multipotent progenitor cells (209, 210). In tumor-driven MDSCs, the requirement for RAS-RAF-MEK-ERK signaling in cell expansion has not been studied extensively (211). However, MEK inhibition with subsequent prevention of ERK phosphorylation led to strongly reduced accumulation of MDSCs in mouse tumor models by inhibiting the inflammation-induced cell expansion and promotion of apoptosis (12, 192, 212). Additionally, the secretion of tumor-derived osteopontin, a chemoattractant for MDSCs, was reduced after MEKi treatment correlating with a decrease in MDSCs (12). Furthermore, in vitro studies showed that MEK inhibition is capable of abrogating cytokine-induced MDSC expansion (12). It has been proposed that specifically IL-6 is capable of promoting MDSC expansion and also significantly improves the immunosuppressive capacity of MDSCs (208). Subsequent RNA sequencing and proteomic analyses in MDSCs treated with IL-6 revealed that especially the RAS-RAF-MEK-ERK signaling cascade was upregulated, suggesting a relationship between IL-6, cell expansion and enhanced immunosuppression (Figure 3) (208). These findings suggest that MEK inhibition does not only contribute to creating MDSC-hostile conditions in the TME by attenuating the expression of immunosuppressive factors by tumor and stromal cells but might also directly influence intracellular, RAS-RAF-MEK-ERK-dependent proliferative and prosurvival processes in MDSCs, leading to an overall increased protective anti-tumor immune response (12, 208).
Regarding the capability of KRAS and BRAF mutant tumors to create an immunosuppressive microenvironment by interfering with the cancer-immunity cycle, various studies have shown that MEKi can restore an immune stimulatory and anti-tumorigenic microenvironment. This effect was achieved by increasing the expression and/or release of proinflammatory molecules, e.g. CD40L or IFNγ, while reducing the presence of immunosuppressive cytokines, e.g. IL-6 and IL-10, and suppressive immune cells such as Tregs, MDSCs, and tumor-associated macrophages (TAMs) (Figure 4) (161). KRAS activation in non-hematopoietic microenvironment caused malignant transformation of hematopoietic cells (213). Gene expression analysis of sorted nonhematopoietic BM niche cells from KrasG12D mice revealed upregulation of multiple inflammation-related genes including IL1-superfamily members (Il1α, Il1β, Il1f9) and the NLPR3 inflammasome (213). The immunosuppressive cytokines were shown to promote the accumulation of immunosuppressive cells leading to inhibition of anti-tumor immunity through various mechanisms, e.g. depletion of arginine necessary for T cell growth, recruitment of Tregs and increase in (ROS) (13). Utilization of MEKi was shown to result in decreased mRNA and protein levels of such immunosuppressive cytokines and subsequent restoration of a proinflammatory environment with increased activity of anti-tumor immunity (13). Moreover, MEKi were shown to hinder the recruitment of monocytic MDSCs to the TME and inhibit monocyte differentiation into TAM. MDSCs and TAM are known to be heterogeneous and plastic cell populations capable of hindering anti-tumor immunity by blocking effector T cell functioning through iNOS and arginase production, which would respectively lead to increased ROS production and arginine depletion (12). Furthermore, these cell populations have been implicated in the direct promotion of tumor growth through stimulation of angiogenesis and metastasis formation (12). MEKi treatment could therefore slow these processes from occurring and support the restoration of an adequate anti-tumor immune response (10).
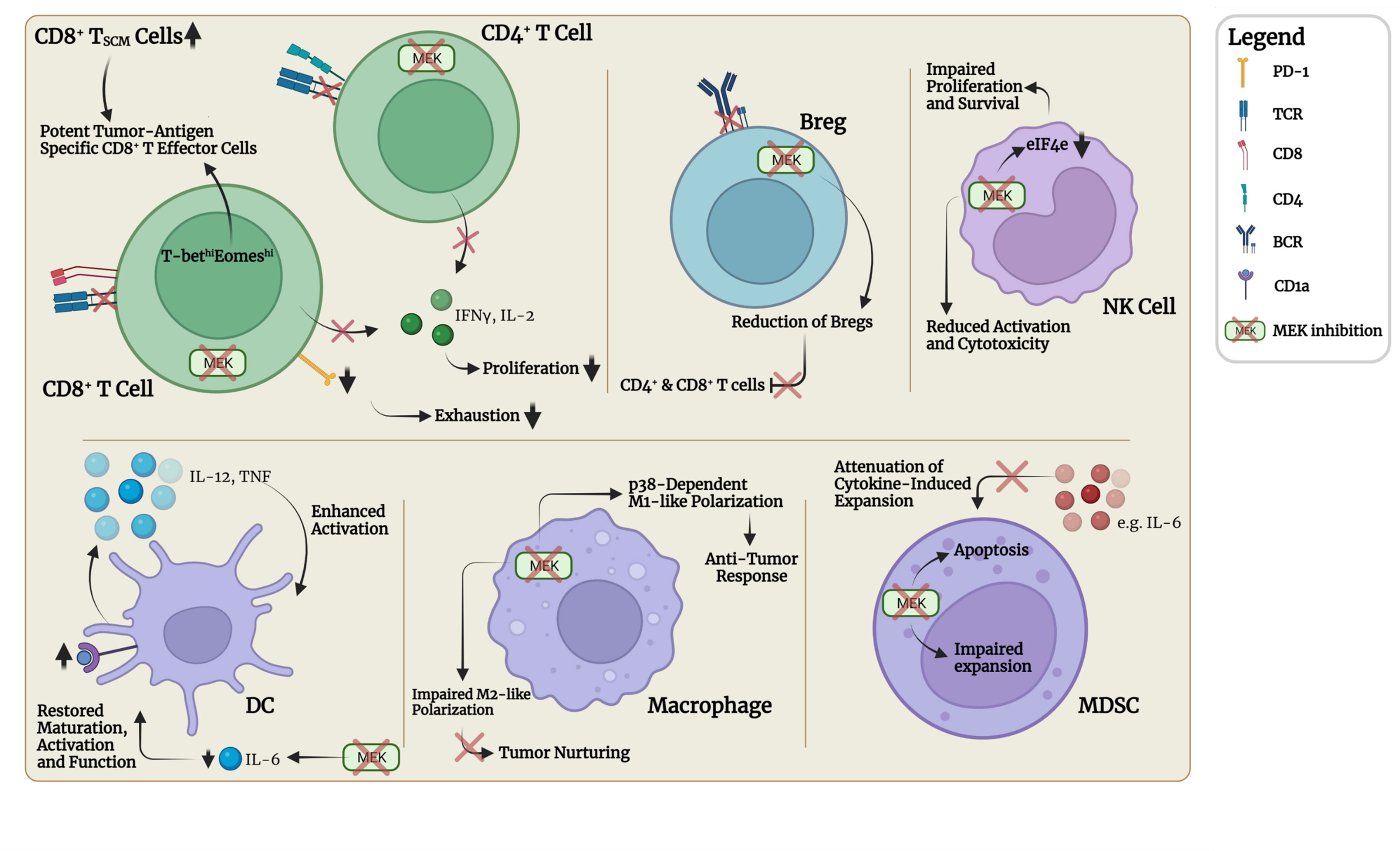
Figure 4 MEK inhibition and associated immunomodulatory effects in the TME (marked with red boxes). MEKi interrupts immune evasion mechanisms and tumor crosstalk with stromal cells and restores an anti-tumorigenic microenvironment by increasing the expression and/or release of proinflammatory molecules, e.g., CD40L, IFNγ, TNF, and CD40L, while simultaneously reducing the presence of immunosuppressive cytokines, e.g., IL-6, IL-10, GM-CSF, and IL-6, and suppressive cell populations such as Tregs, Bregs, MDSCs and M2-polarized macrophages. MEKi also induces upregulation of MHC class I molecules, thereby increasing detectability of tumor cells by CD8+ T effector cells and leading to enhanced infiltration. PD-L1 expression by tumor cells remains either unchanged or variable. Loss of VEGF expression by tumor cells could potentially improve CD8+ T cell infiltration through reduction of abnormal formations of tumor vessels. Furthermore, DC activation and expression of costimulatory molecules is restored leading to enhanced priming and activation of CD4+ and CD8+ T cells.
Next, application of different MEKi induced the upregulation of MHC class I molecules in various tumor models, suggesting increased detectability of tumor cells by effector CD8+ T cells (Figure 4) (161). In line with this finding, a pooled human kinome shRNA interference-based approach led to the identification of MEK to be a negative regulator of MHC class I expression (214). Furthermore, several studies linked MEKi-associated CD8+ T cell infiltration of tumor tissue with the loss of vascular endothelial growth factor (VEGF) expression by tumor cells (161, 215). Abnormal formations of tumor vessels due to hyperexpression of VEGF have been hypothesized to impede the influx and subsequent penetration of tumor tissue by immune cells (216).
Regarding the important role of PD-L1 expression by tumor cells, as described above, several studies have seen no or variable changes in PD-L1 expression in various cancer cell lines upon administration of MEKi, dependent on specific cell line, experimental context or duration of MEK inhibition (13, 183). Interestingly, analysis of PD-1 expression on tumor-infiltrating CD8+ T cells revealed that in MEKi-treated tumors almost 40% of the infiltrating CD8+ T cells were PD-1lo, whereas in control tumors 85% of infiltrating CD8+ T cells expressed high levels of PD-1 (11). A precise understanding of these unexpected and in some cases contradictory observations is still lacking, although escape mechanisms through acquisition of molecular resistance pathways have been proposed. A better understanding of the genetic and proteomic background on tumor PD-L1 regulation is required (13). Besides investigating the effect of PD-L1/PD-1 interruption, inhibition of other immune checkpoints including cytotoxic CTLA-4 and TIM-3 in the presence of MEK inhibition has also come into the focus of recent research efforts (27, 217, 218).
RAS, MEK or ERK kinase inhibitors have the potential to cause significant toxicities at higher dosages. Furthermore, various resistance mechanisms against these kinase inhibitors have been shown to lead to transient anti-tumor effects. Therefore, therapy regimens utilizing combinations of different selective inhibitors targeting both upstream and downstream proteins or parallel pathways have received continuously growing scientific attention in an attempt to overcome these hurdles (32). For example, the combination of MEK or KRASG12C inhibitors and SHP2 or SOS1 inhibitors has led to promising results in preclinical studies for the treatment of KRAS-driven tumors (151, 154, 219). Various clinical trials are currently active, investigating the clinical effectiveness of these therapeutic strategies (Table 2) (33). Also, combinatorial approaches utilizing RAF, MEK and/or ERK inhibitors and autophagy inhibitors, which are thought to increase antigen presentation for enhanced immune responses, are being investigated in clinical trials (e.g., NCT04214418, Table 2) (220–222). Particularly simultaneous inhibition of two elements of the RAF-MEK-ERK cascade, also known as vertical inhibition, was proven to result in highly synergistic and apoptotic activity in murine tumor cells that could not be replicated with the administration of only one single inhibitor, even at high dosage regimens (223). Concurrent inhibition of RAF and ERK was shown to be most effective with induction of tumor regression even at low doses without causing significant toxicity (223). In line with this finding, another research group identified the so-called multiple low dose (MLD) therapy with RAF (RAFi) and ERKi as part of either a triple or quadruple vertical combination therapy to be effective in a lung cancer mouse model leading to significant tumor regression without causing major toxicities or acquired resistance in specific patient subsets (224).
In addition to finding the most effective approach in RAS-RAF-MEK-ERK interference, the increased understanding of the impact of tumor-driven immune modulation and crosstalk with the TME has led to extensive investigational work on therapy regimens combining the immunomodulatory effects of MEKi with another selective inhibitor and an immunotherapeutic agent targeting immune checkpoint molecules (ICB), e.g., CTLA-4, PD-1 or PD-L1, or addressing immunostimulatory receptors, e.g., CD40, CD134 or CD137. These efforts have led to the identification of treatment modalities with significant clinical efficacy in specific patient subsets (9, 10, 27). For instance, the interruption of the PD-L1/PD-1 signaling axis in combination with MEKi and BRAFi was shown to have the potential to increase overall response rates and reduce the rate of MEKi/BRAFi-associated resistance in preclinical models (225). In this regimen, comparably lower but longer response rates to PD-L1/PD-1 blockade and higher but shorter response rates to MEKi/BRAFi treatment complement each other for the most optimal therapy efficacy (26, 226–228). Multiple other advantages linked to anti-PD-L1/PD-1 treatment have been reported, including (i) previous immune checkpoint inhibition and lead-in therapy with two doses of anti-PD-L1/PD-1 enhancing subsequent MAPK pathway inhibition and anti-tumor efficacy and (ii) increased tumor-infiltration by iNOS+ M1-like tumor-associated macrophages (TAMs), Th1-like Tbethi CD4+ T cells and granzyme Bhi CD8+ cytotoxic T cells after sequential-combinatorial therapy with improved durability of tumor regression (225). Another interesting approach has been identified to be the concurrent administration of MEKi and ICB together with chemotherapy, which was very recently shown to result in synergistic effects regarding the TME-associated cytokine profile, CD8+ T cell recruitment and sensitization to ICB treatment (229).
With immunostimulatory targeting, the administration of for instance an agonistic anti-CD40 Ab alongside a MEKi in a murine model led to the discovery of a potent synergistic impact on MEKi-induced immunomodulation through a dual CD40-mitigated mechanism of action, i.e., enhancement of T cell immunity and modulation of the macrophage TME infiltrate towards a higher M1/M2 ratio by activating the p38 MAPK signaling cascade (9). In line with these findings, a previous murine study by Baumann et al. also demonstrated potent synergy between the investigated MEKi and anti-CD40 treatment with significant tumor growth suppression (192). Mechanistically, the authors showed the observed effects to be T cell-dependent and noticed an increased CD8+ T/CD4+ Treg ratio after MEKi and anti-CD40 Ab treatment. Furthermore, the authors demonstrated a similar effect of MEKi/CD40 Ab treatment on the M1/M2 ratio in the TME tilting the balance towards the anti-tumorigenic M1 subtype (9, 192). Other promising immunostimulatory targets have been identified to be the two members of the tumor necrosis factor receptor (TNFR) superfamily CD134 (also known as OX40) and CD137. CD137 signaling has been implicated in the activation of CD8+ T cells, while CD134 activity was shown to be a driver of T cell-mediated anti-tumor immunity, both having the potential to significantly improve the anti-tumorigenic effect of combinatorial targeted therapy (162). However, an important caveat in combinatorial approaches utilizing sequential ERK pathway inhibition and immunotherapy has been identified to be the risk of development of cross-resistance to immunotherapeutics. After development of BRAFi/MEKi/ERKi resistance, the development of cross-resistance led to the acquisition of a strongly immune-evasive state determined by an remarkably immunosuppressive TME (147). Within the TME associated with cross-resistance, a pivotal role was attributed to CD103+ DCs which were identified to be reduced and functionally impaired in cross-resistant tumors (147). Importantly, restoration of DC functionality was shown to be sufficient to restore immunotherapy responsiveness (147). These findings underpin the importance of understanding the process of cancer cell evolution and TME modification during and after targeted therapy and/or immunotherapy. Uniting this improved understanding with data and reports from most recent clinical trials investigating various dual and triple therapy regimens combining MEK inhibition with other inhibitory agents (Table 1) will profoundly aid in the ongoing search for effective combinatorial therapeutic strategies.
Besides improving combinatorial therapy regimens, the precise timing and duration of drug administration has also come into focus due to reports of improved efficacy, reduced odds of resistance induction and reduced toxicity, especially with intermittent administration of MEKi either as monotherapy or in combination with other therapeutic agents (215, 230). Furthermore, the continuous administration of MEKi has been reported to potentially interfere with intracellular cell signaling and physiology in immune cells, as described above. The potential benefits of intermittent administration of cobimetinib in combination with the PI3K inhibitor pictilisib were investigated in murine KRAS- and BRAF-driven tumors (106). This approach led to synergistic cell growth inhibition and increased apoptosis. The intermittent dosing of both compounds led to transient pathway knockdown with a positive correlation between dosage and tumor inhibition/regression (106). These findings were further substantiated by another murine study investigating intermittent administration of selumetinib or trametinib (215). However, contrary to expectations based on preclinical studies, a phase I/II clinical trial investigating continuous as well as intermittent combinatorial administration of cobimetinib and pictilisib in patients diagnosed with CRC, NSCLC or PDAC, reported low tolerance and insignificant anti-tumor activity (231). Similarly, a phase II clinical trial in patients with advanced melanoma showed that intermittent administration of cobimetinib with vemurafenib did not result in statistically significant OS benefit compared to continuous administration. Toxicity was only slightly reduced in the intermittent dosage group (232). Two other study groups investigated intermittent dosing trametinib in combination with either afuresertib or lapatinib in patients with various solid tumors and melanoma in phase I/II clinical trials (233, 234). The dosage tolerability of trametinib and afuresertib was well below the recommended dosage due to significant toxicities. A clinically meaningful dosing schedule could not be achieved (234). Huijberts et al. investigated the combination of trametinib with lapatinib and found that toxicities were manageable at approx. 50% of their single agent dosages. The intermittent dosing regimen seemed more tolerable regarding toxicities. Interestingly, histologically confirmed suppression of MAPK pathway activity did not correlate with clinical activity in CRC, suggesting underlying resistance or escape mechanisms dependent on tumor entity (233). A similar observation was made by Van Brummelen et al. who conducted a phase I trial investigating intermittent administration of selumetinib in combination with afatinib in patients with CRC, NSCLC and PDAC. The majority of enrolled patients were diagnosed with CRC (73%). Although confirmed target engagement was observed, a clinical correlation could not be made, possibly due to disease-associated factors of CRC rendering the therapeutic agents less effective (235). Also in patients with BRAF-mutant malignant melanoma, no advantage of the intermittent administration of trametinib with dabrafenib could be found (236). Based on the findings of clinical trials so far, the potential benefit of intermittent MEKi administration remains unclear. Nevertheless, a currently active phase I/II clinical trial (NCT03581487) is seeking to clarify whether intermittent MEKi treatment combined with ICB therapy could lead to significant benefits (27).
Besides investigating therapeutic agents with single target engagement, a treatment regimen with the novel MEK-pan-RAF inhibitor R05126766 has also come into focus regarding intermittent administration. Guo et al. investigated intermittent dosing schedules and anti-tumor activity in a phase I clinical trial in patients with RAS/RAF-mutant solid tumors and multiple myeloma (237). The investigated inhibitor appeared to be tolerable in intermittent dosing schedules with anti-tumor activity in a large fraction of the patients across various tumor types. Therefore, the authors concluded that the inhibitor R05126766 could be considered in a single-agent regimen (237). This finding is in line with another study on R05126766 with similar findings, warranting further evaluation (238).
The transition from the promising preclinical setting towards the human setting with regard to intermittent MEKi either as monotherapy or in combination with various other agents has proven to be challenging with so far discouraging results. More studies are required to further evaluate the combination of multiple selective inhibitors with or without immunotherapeutic agents and further elucidate the precise role of the immune system in MEKi-associated tumor reduction. Moreover, more studies are required regarding different MEKi agents, as their potency, target specificity and half-life (T1/2) can vary and lead to different effects on the immune system or tumor response (215). Furthermore, tumor entity-associated factors potentially dictating treatment response need to be further evaluated in order to improve prediction of target response and identify individualized therapeutic approaches as the shortcomings of the “one-size fits all” conviction have been demonstrated more than once.
Urged by the global need for improved therapeutic strategies in the treatment of cancer, scientific efforts have accumulated an increasing body of knowledge on tumor-associated molecular biology, the tumor microenvironment and the involvement of the immune system in various tumor entities. In this context, the precise role of the oncogenic RAS-RAF-MEK-ERK signaling cascade in the establishment and maintenance of a tumor-sustaining and immune evasive microenvironment has been and is still being elucidated as its complexity has not yet been fully comprehended. The increasing knowledge has led to the discovery of various selective and immunomodulatory therapeutic approaches in an attempt to disrupt pathologically hyperactivated RAS-RAF-MEK-ERK signaling and tilt the balance of the tumor-TME-immune system crosstalk towards an anti-tumorigenic setting. Tremendous efforts have been and are being undertaken to achieve a successful transition of selective anti-cancer treatment modalities, particularly MEK inhibition and disruption of the PD-L1/PD-1 axis, from the promising preclinical stage to clinical trials and approval by medical authorities. However, important limitations hampering the clinical transition have been identified concerning limited effectiveness, significant treatment-related toxicities and occurrence of various resistance mechanisms – possibly dependent on disease entities – rendering therapeutic agents less effective than aspired. The challenge of the most recent scientific efforts has therefore been overcoming these limitations while identifying the most effective therapy regimens combining selective inhibitory targeting, immunomodulatory effects of particularly MEKi and ICB or immunostimulatory agents and also chemotherapy. Besides combining different agents in treatment strategies, a more recent step towards treatment optimization has been the development of intermittent dosing schedules or sequential drug administration with pre-specified lead-in periods. Multiple clinical trials investigating these approaches are currently active and incoming results are highly anticipated. Fortunately, the entirety of the ongoing scientific efforts has led to a slow but steady progress and has continued to successfully bring forward new and promising therapeutic agents, e.g., the most recently approved agent Sotorasib, despite significant challenges. Therefore, it is our conviction based on the recent developments that interventions interfering with the hyperactivation of the RAS-RAF-MEK-ERK pathway still carry great potential in the treatment of associated tumor entities, giving cause for new hope in affected patients.
TA developed the first draft of the manuscript. TA and DAR contributed to the planning, organization, and writing of the manuscript. All authors reviewed the manuscript and provided critical edits. The final version of the manuscript was approved by all authors.
This study was supported by the Deutsche Forschungsgemeinschaft, Germany, SFB-1479 – Project ID: 441891347 (to NK, RZ, TB, and DR). TB is further supported by the DFG (Heisenberg professorship, BR 3662/5–1 and BR3662/4–1), the German Cancer Consortium DKTK (projects SORATRAM, NextGen LOGGIC and MTB TAILOR) and the Ministry for Science, Research and Arts of the State of Baden-Wuerttemberg, project BW-VAPO. DR is supported by the German Cancer Aid (grant number 70113697).
We would like to thankfully acknowledge support by the Open Access Publication Fund of the University of Freiburg, Germany.
All figures were created with BioRender.com. The illustrations in the figures do not claim to be exhaustive. The involved cell types, cytokines, and interactions depicted in the figures are exemplary.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. World Health Organization (WHO). WHO. global health estimates 2020: Deaths by cause, age, sex, by country and by region, 2000-2019. Switzerland: WHO (2020).
3. Drosten M, Barbacid M. Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell (2020) 37(4):543–50. doi: 10.1016/j.ccell.2020.03.013
4. Ryan MB, Der CJ, Wang-Gillam A, Cox AD. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer (2015) 1(3):183–98. doi: 10.1016/j.trecan.2015.10.001
5. Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci (2016) 129(7):1287–92. doi: 10.1242/jcs.182873
6. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
7. Hamarsheh S, Groß O, Brummer T, Zeiser R. Immune modulatory effects of oncogenic KRAS in cancer. Nat Commun (2020) 11(1):1–11. doi: 10.1038/s41467-020-19288-6
8. Luke JJ, Flaherty KT, Ribas A, Long GV. Targeted agents and immunotherapies: optimizing outcomes in melanoma. Nat Rev Clin Oncol (2017) 14(8):463–82. doi: 10.1038/nrclinonc.2017.43
9. Baumann D, Drebant J, Hägele T, Burger L, Serger C, Lauenstein C, et al. p38 MAPK signaling in M1 macrophages results in selective elimination of M2 macrophages by MEK inhibition. J ImmunoTherapy Cancer (2021) 9(7):e002319. doi: 10.1136/jitc-2020-002319
10. Dennison L, Mohan AA, Yarchoan M. Tumor and systemic immunomodulatory effects of MEK inhibition. Curr Oncol Rep (2021) 23(2):1–14. doi: 10.1007/s11912-020-01008-4
11. Ebert PJ, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity (2016) 44(3):609–21. doi: 10.1016/j.immuni.2016.01.024
12. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12(4):253–68. doi: 10.1038/nri3175
13. Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res (2015) 21(7):1639–51. doi: 10.1158/1078-0432.CCR-14-2339
14. Ott PA, Henry T, Baranda SJ, Frleta D, Manches O, Bogunovic D, et al. Inhibition of both BRAF and MEK in BRAFV600E mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol Immunother (2013) 62(4):811–22. doi: 10.1007/s00262-012-1389-z
15. Wang S, Hong S, Yang J, Qian J, Zhang X, Shpall E, et al. Optimizing immunotherapy in multiple myeloma: Restoring the function of patients' monocyte-derived dendritic cells by inhibiting p38 or activating MEK/ERK MAPK and neutralizing interleukin-6 in progenitor cells. Blood (2006) 108(13):4071–7. doi: 10.1182/blood-2006-04-016980
16. Ascierto PA, Ferrucci PF, Fisher R, Del Vecchio M, Atkinson V, Schmidt H, et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat Med (2019) 25(6):941–6. doi: 10.1038/s41591-019-0448-9
17. Hellmann M, Kim T-W, Lee C, Goh B-C, Miller W, Oh D-Y, et al. Phase ib study of atezolizumab combined with cobimetinib in patients with solid tumors. Ann Oncol (2019) 30(7):1134–42. doi: 10.1093/annonc/mdz113
18. Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH, Carlino MS, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat Med (2019) 25(6):936–40. doi: 10.1038/s41591-019-0476-5
19. Sullivan RJ, Hamid O, Gonzalez R, Infante JR, Patel MR, Hodi FS, et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat Med (2019) 25(6):929–35. doi: 10.1038/s41591-019-0474-7
20. Gutzmer R, Stroyakovskiy D, Gogas H, Robert C, Lewis K, Protsenko S, et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAFV600 mutation-positive melanoma (IMspire150): primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (2020) 395(10240):1835–44. doi: 10.1016/S0140-6736(20)30934-X
21. Yarchoan M, Cope L, Ruggieri AN, Anders RA, Noonan AM, Goff LW, et al. Multicenter randomized phase II trial of atezolizumab with or without cobimetinib in biliary tract cancers. J Clin Invest (2021) 131(24):e152670. doi: 10.1172/JCI152670
22. Zimmer L, Livingstone E, Krackhardt A, Schultz ES, Göppner D, Assaf C, et al. Encorafenib, binimetinib plus pembrolizumab triplet therapy in patients with advanced BRAFV600 mutant melanoma: safety and tolerability results from the phase I IMMU-TARGET trial. Eur J Cancer (2021) 158:72–84. doi: 10.1016/j.ejca.2021.09.011
23. Dumont FJ, Staruch MJ, Fischer P, DaSilva C, Camacho R. Inhibition of T cell activation by pharmacologic disruption of the MEK1/ERK MAP kinase or calcineurin signaling pathways results in differential modulation of cytokine production. J Immunol (1998) 160(6):2579–89.
24. Dushyanthen S, Teo ZL, Caramia F, Savas P, Mintoff CP, Virassamy B, et al. Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer. Nat Commun (2017) 8(1):1–18. doi: 10.1038/s41467-017-00728-9
25. Eng C, Kim TW, Bendell J, Argilés G, Tebbutt NC, Di Bartolomeo M, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol (2019) 20(6):849–61. doi: 10.1016/S1470-2045(19)30027-0
26. Hu-Lieskovan S, Robert L, Moreno BH, Ribas A. Combining targeted therapy with immunotherapy in BRAF-mutant melanoma: promise and challenges. J Clin Oncol (2014) 32(21):2248. doi: 10.1200/JCO.2013.52.1377
27. Gaudreau P-O, Lee JJ, Heymach JV, Gibbons DL. Phase I/II trial of immunotherapy with durvalumab and tremelimumab with continuous or intermittent MEK inhibitor selumetinib in NSCLC: early trial report. Clin Lung Cancer (2020) 21(4):384–8. doi: 10.1016/j.cllc.2020.02.019
28. Rincon M, Flavell RA, Davis RJ. Signal transduction by MAP kinases in T lymphocytes. Oncogene (2001) 20(19):2490–7. doi: 10.1038/sj.onc.1204382
29. Coulombe P, Meloche S. Atypical mitogen-activated protein kinases: structure, regulation and functions. Biochim Biophys Acta Mol Cell Res (2007) 1773(8):1376–87. doi: 10.1016/j.bbamcr.2006.11.001
30. Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev (2011) 75(1):50–83. doi: 10.1128/MMBR.00031-10
31. Lavoie H, Gagnon J, Therrien M. ERK signalling: a master regulator of cell behaviour, life and fate. Nat Rev Mol Cell Biol (2020) 21(10):607–32. doi: 10.1038/s41580-020-0255-7
32. Van Cutsem E, Van De Velde H, Karasek P, Oettle H, Vervenne W, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol (2004) 22(8):1430–8. doi: 10.1200/JCO.2004.10.112
33. Nagasaka M, Potugari B, Nguyen A, Sukari A, Azmi AS, Ou S-HI. KRAS inhibitors–yes but what next? direct targeting of KRAS–vaccines, adoptive T cell therapy and beyond. Cancer Treat Rev (2021) 101:102309. doi: 10.1016/j.ctrv.2021.102309
34. Ünal EB, Uhlitz F, Blüthgen N. A compendium of ERK targets. FEBS Lett (2017) 591(17):2607–15. doi: 10.1002/1873-3468.12740
35. Reth M, Brummer TJNRI. Feedback regulation of lymphocyte signalling. Nat Rev Immunol (2004) 4(4):269–78. doi: 10.1038/nri1335
36. Dorard C, Vucak G, Baccarini M. Deciphering the RAS/ERK pathway in vivo. Biochem Soc Trans (2017) 45(1):27–36. doi: 10.1042/BST20160135
37. Hüser M, Luckett J, Chiloeches A, Mercer K, Iwobi M, Giblett S, et al. MEK kinase activity is not necessary for raf-1 function. EMBO J (2001) 20(8):1940–51. doi: 10.1093/emboj/20.8.1940
38. Mikula M, Schreiber M, Husak Z, Kucerova L, Rüth J, Wieser R, et al. Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. EMBO J (2001) 20(8):1952–62. doi: 10.1093/emboj/20.8.1952
39. Wojnowski L, Zimmer AM, Beck TW, Hahn H, Bernal R, Rapp UR, et al. Endothelial apoptosis in braf-deficient mice. Nat Genet (1997) 16(3):293–7. doi: 10.1038/ng0797-293
40. Blasco RB, Francoz S, Santamaría D, Cañamero M, Dubus P, Charron J, et al. C-raf, but not b-raf, is essential for development of K-ras oncogene-driven non-small cell lung carcinoma. Cancer Cell (2011) 19(5):652–63. doi: 10.1016/j.ccr.2011.04.002
41. Drosten M, Sum EY, Lechuga CG, Simón-Carrasco L, Jacob HK, García-Medina R, et al. Loss of p53 induces cell proliferation via ras-independent activation of the Raf/Mek/Erk signaling pathway. Proc Natl Acad Sci (2014) 111(42):15155–60. doi: 10.1073/pnas.1417549111
42. Banerjee A, Gugasyan R, McMahon M, Gerondakis S. Diverse toll-like receptors utilize Tpl2 to activate extracellular signal-regulated kinase (ERK) in hemopoietic cells. Proc Natl Acad Sci (2006) 103(9):3274–9. doi: 10.1073/pnas.0511113103
43. Scholl FA, Dumesic PA, Barragan DI, Harada K, Bissonauth V, Charron J, et al. Mek1/2 MAPK kinases are essential for mammalian development, homeostasis, and raf-induced hyperplasia. Dev Cell (2007) 12(4):615–29. doi: 10.1016/j.devcel.2007.03.009
44. Frémin C, Saba-El-Leil MK, Lévesque K, Ang S-L, Meloche S. Functional redundancy of ERK1 and ERK2 MAP kinases during development. Cell Rep (2015) 12(6):913–21. doi: 10.1016/j.celrep.2015.07.011
45. Liu Y, Shepherd EG, Nelin LD. MAPK phosphatases–regulating the immune response. Nat Rev Immunol (2007) 7(3):202–12. doi: 10.1038/nri2035
46. Sharp LL, Hedrick SM. Commitment to the CD4 lineage mediated by extracellular signal-related kinase mitogen-activated protein kinase and lck signaling. J Immunol (1999) 163(12):6598–605.
47. D'Souza WN, Chang C-F, Fischer AM, Li M, Hedrick SM. The Erk2 MAPK regulates CD8 T cell proliferation and survival. J Immunol (2008) 181(11):7617–29. doi: 10.4049/jimmunol.181.11.7617
48. Yamashita M, Shinnakasu R, Asou H, Kimura M, Hasegawa A, Hashimoto K, et al. Ras-ERK MAPK cascade regulates GATA3 stability and Th2 differentiation through ubiquitin-proteasome pathway. J Biol Chem (2005) 280(33):29409–19. doi: 10.1074/jbc.M502333200
49. Merritt C, Enslen H, Diehl N, Conze D, Davis RJ, Rincón M. Activation of p38 mitogen-activated protein kinase in vivo selectively induces apoptosis of CD8+ but not CD4+ T cells. Mol Cell Biol (2000) 20(3):936–46. doi: 10.1128/MCB.20.3.936-946.2000
50. Pandit M, Acharya S, Gu Y, Seo SU, Kweon MN, Kang B, et al. Geranylgeranyl pyrophosphate amplifies treg differentiation via increased IL-2 expression to ameliorate DSS-induced colitis. Eur J Immunol (2021) 51(6):1461–72. doi: 10.1002/eji.202048991
51. Papoutsopoulou S, Symons A, Tharmalingham T, Belich MP, Kaiser F, Kioussis D, et al. ABIN-2 is required for optimal activation of erk MAP kinase in innate immune responses. Nat Immunol (2006) 7(6):606–15. doi: 10.1038/ni1334
52. Zeiser R, Nguyen VH, Beilhack A, Buess M, Schulz S, Baker J, et al. Inhibition of CD4+ CD25+ regulatory T-cell function by calcineurin-dependent interleukin-2 production. Blood (2006) 108(1):390–9. doi: 10.1182/blood-2006-01-0329
53. Greaves SA, Peterson JN, Torres RM, Pelanda R. Activation of the MEK-ERK pathway is necessary but not sufficient for breaking central b cell tolerance. Front Immunol (2018) 9:707. doi: 10.3389/fimmu.2018.00707
54. Yasuda T, Kurosaki T. Regulation of lymphocyte fate by Ras/ERK signals. Cell Cycle (2008) 7(23):3634–40. doi: 10.4161/cc.7.23.7103
55. Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B-cell anergy: from transgenic models to naturally occurring anergic b cells? Nat Rev Immunol (2007) 7(8):633–43. doi: 10.1038/nri2133
56. Kurosaki T, Shinohara H, Baba Y. B cell signaling and fate decision. Annu Rev Immunol (2009) 28:21–55. doi: 10.1146/annurev.immunol.021908.132541
57. Oh-Hora M, Johmura S, Hashimoto A, Hikida M. Requirement for ras guanine nucleotide releasing protein 3 in coupling phospholipase c-γ2 to ras in b cell receptor signaling. J Exp Med (2003) 198(12):1841–51. doi: 10.1084/jem.20031547
58. Li Z, Woo CJ, Iglesias-Ussel MD, Ronai D, Scharff MD. The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes Dev (2004) 18(1):1–11. doi: 10.1101/gad.1161904
59. Klein U, Tu Y, Stolovitzky GA, Keller JL, Haddad J, Miljkovic V, et al. Transcriptional analysis of the b cell germinal center reaction. Proc Natl Acad Sci (2003) 100(5):2639–44. doi: 10.1073/pnas.0437996100
60. Adem J, Hämäläinen A, Ropponen A, Eeva J, Eray M, Nuutinen U, et al. ERK1/2 has an essential role in b cell receptor-and CD40-induced signaling in an in vitro model of germinal center b cell selection. Mol Immunol (2015) 67(2):240–7. doi: 10.1016/j.molimm.2015.05.017
61. Harada H, Quearry B, Ruiz-Vela A, Korsmeyer SJ. Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc Natl Acad Sci (2004) 101(43):15313–7. doi: 10.1073/pnas.0406837101
62. Craxton A, Draves KE, Gruppi A, Clark EA. BAFF regulates b cell survival by downregulating the BH3-only family member bim via the ERK pathway. J Exp Med (2005) 202(10):1363–74. doi: 10.1084/jem.20051283
63. Yasuda T, Kometani K, Takahashi N, Imai Y, Aiba Y, Kurosaki T. ERKs induce expression of the transcriptional repressor blimp-1 and subsequent plasma cell differentiation. Sci Signaling (2011) 4(169):ra25–ra. doi: 10.1126/scisignal.2001592
64. Zheng Y, Chen Y, You X, Yu M, Fu G, Su X, et al. Kras is critical for b cell lymphopoiesis. J Immunol (2015) 126(23):3588. doi: 10.1182/blood.V126.23.3588.3588
65. Scheffler L, Feicht S, Babushku T, Kuhn LB, Ehrenberg S, Frankenberger S, et al. ERK phosphorylation is RAF independent in naïve and activated b cells but RAF dependent in plasma cell differentiation. Sci Signaling (2021) 14(682):eabc1648. doi: 10.1126/scisignal.abc1648
66. Machnicki MM, Stoklosa T. BRAF–a new player in hematological neoplasms. Blood Cells Mol Dis (2014) 53(1-2):77–83. doi: 10.1016/j.bcmd.2014.01.001
67. Fridman WH, Petitprez F, Meylan M, Chen TW-W, Sun C-M, Roumenina LT, et al. B cells and cancer: To b or not to b? J Exp Med (2021) 126(23):3588. doi: 10.1084/jem.20200851
68. Chiossone L, Dumas P-Y, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol (2018) 18(11):671–88. doi: 10.1038/s41577-018-0061-z
69. May RM, Okumura M, Hsu C-J, Bassiri H, Yang E, Rak G, et al. Murine natural killer immunoreceptors use distinct proximal signaling complexes to direct cell function. Blood J Am Soc Hematol (2013) 121(16):3135–46. doi: 10.1182/blood-2012-12-474361
70. Briercheck EL, Trotta R, Chen L, Hartlage AS, Cole JP, Cole TD, et al. PTEN is a negative regulator of NK cell cytolytic function. J Immunol (2015) 194(4):1832–40. doi: 10.4049/jimmunol.1401224
71. Liang S, Zhang J, Wei H, Sun R, Tian Z. Differential roles of constitutively activated ERK1/2 and NF-κB in cytotoxicity and proliferation by human NK cell lines. Int Immunopharmacol (2005) 5(5):839–48. doi: 10.1016/j.intimp.2004.12.016
72. Wang X, Zhao X-Y. Transcription factors associated with IL-15 cytokine signaling during NK cell development. Front Immunol (2021) 12:785. doi: 10.3389/fimmu.2021.610789
73. Benson DM Jr., Yu J, Becknell B, Wei M, Freud AG, Ferketich AK, et al. Stem cell factor and interleukin-2/15 combine to enhance MAPK-mediated proliferation of human natural killer cells. Blood J Am Soc Hematol (2009) 113(12):2706–14. doi: 10.1182/blood-2008-05-159285
74. Vincent MS, Gumperz JE, Brenner MB. Understanding the function of CD1-restricted T cells. Nat Immunol (2003) 4(6):517–23. doi: 10.1038/ni0603-517
75. Engel I, Kronenberg M. Making memory at birth: understanding the differentiation of natural killer T cells. Curr Opin Immunol (2012) 24(2):184–90. doi: 10.1016/j.coi.2012.01.012
76. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol (2007) 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711
77. Hu T, Gimferrer I, Simmons A, Wiest D, Alberola-Ila J. The Ras/MAPK pathway is required for generation of iNKT cells. PloS One (2011) 6(5):e19890. doi: 10.1371/journal.pone.0019890
78. Tian J, Liu L, Wang X, Sun X, Mu S, Wu C, et al. The differential roles of mTOR, ERK, and JNK pathways in invariant natural killer T-cell function and survival. Inflammation (2014) 37(6):2013–9. doi: 10.1007/s10753-014-9933-y
79. Wooten RM, Ma Y, Yoder RA, Brown JP, Weis JH, Zachary JF, et al. Toll-like receptor 2 is required for innate, but not acquired, host defense to borrelia burgdorferi. J Immunol (2002) 168(1):348–55. doi: 10.4049/jimmunol.168.1.348
80. Behera AK, Hildebrand E, Uematsu S, Akira S, Coburn J, Hu LT. Identification of a TLR-independent pathway for borrelia burgdorferi-induced expression of matrix metalloproteinases and inflammatory mediators through binding to integrin α3β1. J Immunol (2006) 177(1):657–64. doi: 10.4049/jimmunol.177.1.657
81. Richardson ET, Shukla S, Nagy N, Boom WH, Beck RC, Zhou L, et al. ERK signaling is essential for macrophage development. PloS One (2015) 10(10):e0140064. doi: 10.1371/journal.pone.0140064
82. Hawley K, Navasa N, Olson CM Jr., Bates TC, Garg R, Hedrick MN, et al. Macrophage p38 mitogen-activated protein kinase activity regulates invariant natural killer T-cell responses during borrelia burgdorferi infection. J Infect Dis (2012) 206(2):283–91. doi: 10.1093/infdis/jis332
83. Chakraborty R, Abdel-Wahab O, Durham BH. MAP-kinase-driven hematopoietic neoplasms: a decade of progress in the molecular age. Cold Spring Harbor Perspect Med (2021) 11(5):a034892. doi: 10.1101/cshperspect.a034892
84. Shortman K, Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol (2007) 7(1):19–30. doi: 10.1038/nri1996
85. Heath WR, Carbone FR. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat Immunol (2009) 10(12):1237–44. doi: 10.1038/ni.1822
86. Xu X, Yi H, Guo Z, Qian C, Xia S, Yao Y, et al. Splenic stroma-educated regulatory dendritic cells induce apoptosis of activated CD4 T cells via fas ligand-enhanced IFN-γ and nitric oxide. J Immunol (2012) 188(3):1168–77. doi: 10.4049/jimmunol.1101696
87. Puig-Kroger A, Relloso M, Fernández-Capetillo O, Zubiaga A, Silva A, Bernabeu C, et al. Extracellular signal–regulated protein kinase signaling pathway negatively regulates the phenotypic and functional maturation of monocyte-derived human dendritic cells. Blood J Am Soc Hematol (2001) 98(7):2175–82. doi: 10.1182/blood.V98.7.2175
88. Hogstad B, Berres M-L, Chakraborty R, Tang J, Bigenwald C, Serasinghe M, et al. RAF/MEK/extracellular signal–related kinase pathway suppresses dendritic cell migration and traps dendritic cells in langerhans cell histiocytosis lesions. J Exp Med (2018) 215(1):319–36. doi: 10.1084/jem.20161881
89. Luster AD. Chemokines–chemotactic cytokines that mediate inflammation. New Engl J Med (1998) 338(7):436–45. doi: 10.1056/NEJM199802123380706
90. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol (2006) 6(3):173–82. doi: 10.1038/nri1785
91. Ran T, Zhang Y, Diao N, Geng S, Chen K, Lee C, et al. Enhanced neutrophil immune homeostasis due to deletion of PHLPP. Front Immunol (2019) 2127. doi: 10.3389/fimmu.2019.02127
92. Heit B, Tavener S, Raharjo E, Kubes P. An intracellular signaling hierarchy determines direction of migration in opposing chemotactic gradients. J Cell Biol (2002) 159(1):91–102. doi: 10.1083/jcb.200202114
93. Simard FA, Cloutier A, Ear T, Vardhan H, McDonald PP. MEK-independent ERK activation in human neutrophils and its impact on functional responses. J Leukocyte Biol (2015) 98(4):565–73. doi: 10.1189/jlb.2MA1214-599R
94. Wilhelm K, Ganesan J, Müller T, Dürr C, Grimm M, Beilhack A, et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat Med (2010) 16(12):1434–8. doi: 10.1038/nm.2242
95. Jankovic D, Ganesan J, Bscheider M, Stickel N, Weber FC, Guarda G, et al. The Nlrp3 inflammasome regulates acute graft-versus-host disease. J Exp Med (2013) 210(10):1899–910. doi: 10.1084/jem.20130084
96. Hazeldine J, Hampson P, Opoku FA, Foster M, Lord JM. N-formyl peptides drive mitochondrial damage associated molecular pattern induced neutrophil activation through ERK1/2 and P38 MAP kinase signalling pathways. Injury (2015) 46(6):975–84. doi: 10.1016/j.injury.2015.03.028
97. Dömer D, Walther T, Möller S, Behnen M, Laskay T. Neutrophil extracellular traps activate proinflammatory functions of human neutrophils. Front Immunol (2021) 12:1190. doi: 10.3389/fimmu.2021.636954
98. Avdi NJ, Nick JA, Whitlock BB, Billstrom MA, Henson PM, Johnson GL, et al. Tumor necrosis factor-α activation of the c-jun n-terminal kinase pathway in human neutrophils: integrin involvement in a pathway leading from cytoplasmic tyrosine kinases to apoptosis. J Biol Chem (2001) 276(3):2189–99. doi: 10.1074/jbc.M007527200
99. Hawkins P, Stephens L. PI3K signalling in inflammation. Biochim Biophys Acta Mol Cell Biol Lipids (2015) 1851(6):882–97. doi: 10.1016/j.bbalip.2014.12.006
100. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discovery (2020) 19(8):533–52. doi: 10.1038/s41573-020-0068-6
101. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res (2015) 13(9):1325–35. doi: 10.1158/1541-7786.MCR-15-0203
102. Brummer T, McInnes C. RAF Kinase dimerization: Implications for drug discovery and clinical outcomes. Oncogene (2020) 39(21):4155–69. doi: 10.1038/s41388-020-1263-y
103. Kern F, Doma E, Rupp C, Niault T, Baccarini M. Essential, non-redundant roles of b-raf and raf-1 in ras-driven skin tumorigenesis. Oncogene (2013) 32(19):2483–92. doi: 10.1038/onc.2012.254
104. Ehrenreiter K, Kern F, Velamoor V, Meissl K, Galabova-Kovacs G, Sibilia M, et al. Raf-1 addiction in ras-induced skin carcinogenesis. Cancer Cell (2009) 16(2):149–60. doi: 10.1016/j.ccr.2009.06.008
105. Blasco MT, Navas C, Martín-Serrano G, Graña-Castro O, Lechuga CG, Martín-Díaz L, et al. Complete regression of advanced pancreatic ductal adenocarcinomas upon combined inhibition of EGFR and c-RAF. Cancer Cell (2019) 35(4):573–87. e6. doi: 10.1016/j.ccell.2019.03.002
106. Hoeflich KP, Merchant M, Orr C, Chan J, Den Otter D, Berry L, et al. Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res (2012) 72(1):210–9. doi: 10.1158/0008-5472.CAN-11-1515
107. Kinkade R, Dasgupta P, Carie A, Pernazza D, Carless M, Pillai S, et al. A small molecule disruptor of Rb/Raf-1 interaction inhibits cell proliferation, angiogenesis, and growth of human tumor xenografts in nude mice. Cancer Res (2008) 68(10):3810–8. doi: 10.1158/0008-5472.CAN-07-6672
108. Baumann B, Weber CK, Troppmair J, Whiteside S, Israel A, Rapp UR, et al. Raf induces NF-κB by membrane shuttle kinase MEKK1, a signaling pathway critical for transformation. Proc Natl Acad Sci (2000) 97(9):4615–20. doi: 10.1073/pnas.080583397
109. Wang H-G, Rapp UR, Reed JC. Bcl-2 targets the protein kinase raf-1 to mitochondria. Cell (1996) 87(4):629–38. doi: 10.1016/S0092-8674(00)81383-5
110. Piazzolla D, Meissl K, Kucerova L, Rubiolo C, Baccarini M. Raf-1 sets the threshold of fas sensitivity by modulating rok-α signaling. J Cell Biol (2005) 171(6):1013–22. doi: 10.1083/jcb.200504137
111. Varga A, Ehrenreiter K, Aschenbrenner B, Kocieniewski P, Kochanczyk M, Lipniacki T, et al. RAF1/BRAF dimerization integrates the signal from RAS to ERK and ROKα. Sci Signaling (2017) 10(469):eaai8482. doi: 10.1126/scisignal.aai8482
112. Gao Y, Chang MT, McKay D, Na N, Zhou B, Yaeger R, et al. Allele-specific mechanisms of activation of MEK1 mutants determine their properties. Cancer Discov (2018) 8(5):648–61. doi: 10.1158/2159-8290.CD-17-1452
113. Wong GS, Zhou J, Liu JB, Wu Z, Xu X, Li T, et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med (2018) 24(7):968–77. doi: 10.1038/s41591-018-0022-x
114. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov (2020) 10(1):54–71. doi: 10.1158/2159-8290.CD-19-1167
115. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. (G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature (2019) 575(7781):217–23. doi: 10.1038/s41586-019-1694-1
116. Nakajima EC, Drezner N, Li X, Mishra-Kalyani PS, Liu Y, Zhao H, et al. FDA approval summary: sotorasib for KRAS G12C-mutated metastatic NSCLC. Clin Cancer Res (2022) 28(8):1482–86.
117. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature (2013) 503(7477):548–51. doi: 10.1038/nature12796
118. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRASG12C inhibition with sotorasib in advanced solid tumors. N Engl J Med (2020) 383(13):1207–17. doi: 10.1056/NEJMoa1917239
119. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p. G12C mutation. N Engl J Med (2021) 384(25):2371–81.
120. Jänne PA, Riely GJ, Gadgeel SM, Heist RS, Ou S-HI, Pacheco JM, et al. Adagrasib in non–Small-Cell lung cancer harboring a KRASG12C mutation. N Engl J Med (online ahead of print) (online ahead of print) (2022) 387:120–131. doi: 10.1056/NEJMoa2204619
121. Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12D inhibitor. J Med Chem (2021) 65(4):3123–33. doi: 10.1021/acs.jmedchem.1c01688
122. Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov (2006) 5(10):835–44. doi: 10.1038/nrd2130
123. Savoia P, Fava P, Casoni F, Cremona O. Targeting the ERK signaling pathway in melanoma. Int J Mol Sci (2019) 20(6):1483. doi: 10.3390/ijms20061483
124. Cook FA, Cook SJ. Inhibition of RAF dimers: it takes two to tango. Biochem Soc Trans (2021) 49(1):237–51. doi: 10.1042/BST20200485
125. Yaktapour N, Meiss F, Mastroianni J, Zenz T, Andrlova H, Mathew NR, et al. BRAF inhibitor–associated ERK activation drives development of chronic lymphocytic leukemia. J Clin Invest (2014) 124(11):5074–84. doi: 10.1172/JCI76539
126. Dietrich S, Pircher A, Endris V, Peyrade F, Wendtner C-M, Follows GA, et al. BRAF inhibition in hairy cell leukemia with low-dose vemurafenib. Blood J Am Soc Hematol (2016) 127(23):2847–55. doi: 10.1182/blood-2015-11-680074
127. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF Inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature (2010) 464(7287):427–30. doi: 10.1038/nature08902
128. Yao Z, Gao Y, Su W, Yaeger R, Tao J, Na N, et al. RAF Inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat Med (2019) 25(2):284–91. doi: 10.1038/s41591-018-0274-5
129. Karreth FA, Frese KK, DeNicola GM, Baccarini M, Tuveson DA. C-raf is required for the initiation of lung cancer by K-RasG12D. Cancer Discov (2011) 1(2):128–36. doi: 10.1158/2159-8290.CD-10-0044
130. Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, et al. Selective requirement of PI3K/PDK1 signaling for kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell (2013) 23(3):406–20. doi: 10.1016/j.ccr.2013.01.023
131. Sanclemente M, Francoz S, Esteban-Burgos L, Bousquet-Mur E, Djurec M, Lopez-Casas PP, et al. C-RAF ablation induces regression of advanced Kras/Trp53 mutant lung adenocarcinomas by a mechanism independent of MAPK signaling. Cancer Cell (2018) 33(2):217–28. e4. doi: 10.1016/j.ccell.2017.12.014
132. Ryan MB, Corcoran RB. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol (2018) 15(11):709–20. doi: 10.1038/s41571-018-0105-0
133. McCormick F. C-raf in KRas mutant cancers: A moving target. Cancer Cell (2018) 33(2):158–9. doi: 10.1016/j.ccell.2018.01.017
134. Gu S, Cui D, Chen X, Xiong X, Zhao Y. PROTACs: an emerging targeting technique for protein degradation in drug discovery. BioEssays (2018) 40(4):1700247. doi: 10.1002/bies.201700247
135. Sanclemente M, Nieto P, Garcia-Alonso S, Fernández-García F, Esteban-Burgos L, Guerra C, et al. RAF1 kinase activity is dispensable for KRAS/p53 mutant lung tumor progression. Cancer Cell (2021) 39(3):294–6. doi: 10.1016/j.ccell.2021.01.008
136. Li Y, Meng Q, Wang P, Liu X, Fu Q, Xie Y, et al. Identification of PDE6D as a potential target of sorafenib via PROTAC technology. bioRxiv (2020). doi: 10.1101/2020.05.06.079947
137. Yaeger R, Corcoran RB. Targeting alterations in the RAF–MEK pathway. Cancer Discov (2019) 9(3):329–41. doi: 10.1158/2159-8290.CD-18-1321
138. Jänne PA, Van Den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crinò L, et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with kras-mutant advanced non–small cell lung cancer: The select-1 randomized clinical trial. Jama (2017) 317(18):1844–53. doi: 10.1001/jama.2017.3438
139. Infante JR, Somer BG, Park JO, Li C-P, Scheulen ME, Kasubhai SM, et al. A randomized, double-blind, placebo-controlled trial of trametinib, a MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Am Soc Clin Oncol (2013) 31(4_suppl):291. doi: 10.1200/jco.2013.31.4_suppl.291
140. Oddo D, Sennott EM, Barault L, Valtorta E, Arena S, Cassingena A, et al. Molecular landscape of acquired resistance to targeted therapy combinations in BRAF-mutant colorectal cancer. Cancer Res (2016) 76(15):4504–15. doi: 10.1158/0008-5472.CAN-16-0396
141. Samatar AA, Poulikakos PI. Targeting RAS–ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov (2014) 13(12):928–42. doi: 10.1038/nrd4281
142. Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep (2014) 7(1):86–93. doi: 10.1016/j.celrep.2014.02.045
143. Ebi H. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition. Ann Oncol (2012) 23:xi41. doi: 10.1016/S0923-7534(20)32033-0
144. Misale S, Di Nicolantonio F, Sartore-Bianchi A, Siena S, Bardelli A. Resistance to anti-EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov (2014) 4(11):1269–80. doi: 10.1158/2159-8290.CD-14-0462
145. Kun E, Tsang Y, Ng C, Gershenson D, Wong K. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat Rev (2021) 92:102137. doi: 10.1016/j.ctrv.2020.102137
146. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired resistance to KRASG12C inhibition in cancer. New Engl J Med (2021) 384(25):2382–93. doi: 10.1056/NEJMoa2105281
147. Haas L, Elewaut A, Gerard CL, Umkehrer C, Leiendecker L, Pedersen M, et al. Acquired resistance to anti-MAPK targeted therapy confers an immune-evasive tumor microenvironment and cross-resistance to immunotherapy in melanoma. Nat Cancer (2021) 2(7):693–708. doi: 10.1038/s43018-021-00221-9
148. Neel BG, Gu H, Pao L. The ‘Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci (2003) 28(6):284–93. doi: 10.1016/S0968-0004(03)00091-4
149. Chan G, Kalaitzidis D, Neel BG. The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev (2008) 27(2):179–92. doi: 10.1007/s10555-008-9126-y
150. Xu D, Qu C-K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci: J Virtual Library (2008) 13:4925. doi: 10.2741/3051
151. Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med (2018) 24(7):954–60. doi: 10.1038/s41591-018-0024-8
152. Ryan MB, de la Cruz FF, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical pathway inhibition overcomes adaptive feedback resistance to KRASG12C inhibition. Clin Cancer Res (2020) 26(7):1633–43. doi: 10.1158/1078-0432.CCR-19-3523
153. Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, et al. SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov (2018) 8(10):1237–49. doi: 10.1158/2159-8290.CD-18-0444
154. Hillig RC, Sautier B, Schroeder J, Moosmayer D, Hilpmann A, Stegmann CM, et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc Natl Acad Sci (2019) 116(7):2551–60. doi: 10.1073/pnas.1812963116
155. Howes JE, Akan DT, Burns MC, Rossanese OW, Waterson AG, Fesik SW. Small molecule–mediated acti34 8vation of RAS elicits biphasic modulation of phospho-ERK levels that are regulated through negative feedback on SOS1. Mol Cancer Ther (2018) 17(5):1051–60. doi: 10.1158/1535-7163.MCT-17-0666
156. Kessler D, Gerlach D, Kraut N, McConnell DB. Targeting son of sevenless 1: the pacemaker of KRAS. Curr Opin Chem Biol (2021) 62:109–18. doi: 10.1016/j.cbpa.2021.02.014
157. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell (2012) 21(3):309–22. doi: 10.1016/j.ccr.2012.02.022
158. Carvalho PD, Guimaraes CF, Cardoso AP, Mendonça S, Costa ÂM, Oliveira MJ, et al. KRAS oncogenic signaling extends beyond cancer cells to orchestrate the microenvironment. Cancer Res (2018) 78(1):7–14. doi: 10.1158/0008-5472.CAN-17-2084
159. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
160. Liu P, Wang Y, Li X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm Sin B (2019) 9(5):871–9. doi: 10.1016/j.apsb.2019.03.002
161. Kuske M, Westphal D, Wehner R, Schmitz M, Beissert S, Praetorius C, et al. Immunomodulatory effects of BRAF and MEK inhibitors: Implications for melanoma therapy. Pharmacol Res (2018) 136:151–9. doi: 10.1016/j.phrs.2018.08.019
162. Homet Moreno B, Mok S, Comin-Anduix B, Hu-Lieskovan S, Ribas A. Combined treatment with dabrafenib and trametinib with immune-stimulating antibodies for BRAF mutant melanoma. Oncoimmunology (2016) 5(7):e1052212. doi: 10.1080/2162402X.2015.1052212
163. Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
164. Ancrile B, Lim K-H, Counter CM. Oncogenic ras-induced secretion of IL6 is required for tumorigenesis. Genes Dev (2007) 21(14):1714–9. doi: 10.1101/gad.1549407
165. Brooks GD, McLeod L, Alhayyani S, Miller A, Russell PA, Ferlin W, et al. IL6 trans-signaling promotes KRAS-driven lung carcinogenesis. Cancer Res (2016) 76(4):866–76. doi: 10.1158/0008-5472.CAN-15-2388
166. Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Klöppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell (2011) 19(4):456–69. doi: 10.1016/j.ccr.2011.03.009
167. Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res (2011) 71(14):5020–9. doi: 10.1158/0008-5472.CAN-11-0908
168. Zhang Y, Yan W, Collins MA, Bednar F, Rakshit S, Zetter BR, et al. Interleukin-6 is required for pancreatic cancer progression by promoting MAPK signaling activation and oxidative stress resistance. Cancer Res (2013) 73(20):6359–74. doi: 10.1158/0008-5472.CAN-13-1558-T
169. Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell (2004) 6(5):447–58. doi: 10.1016/j.ccr.2004.09.028
170. Awaji M, Saxena S, Wu L, Prajapati DR, Purohit A, Varney ML, et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J (2020) 34(7):9405–18. doi: 10.1096/fj.201902990R
171. Ling J, Kang Y, Zhao R, Xia Q, Lee D-F, Chang Z, et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell (2012) 21(1):105–20. doi: 10.1016/j.ccr.2011.12.006
172. Liou G-Y, Döppler H, Necela B, Edenfield B, Zhang L, Dawson DW, et al. Mutant KRAS–induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov (2015) 5(1):52–63. doi: 10.1158/2159-8290.CD-14-0474
173. Chang SH, Mirabolfathinejad SG, Katta H, Cumpian AM, Gong L, Caetano MS, et al. T Helper 17 cells play a critical pathogenic role in lung cancer. Proc Natl Acad Sci (2014) 111(15):5664–9. doi: 10.1073/pnas.1319051111
174. McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, et al. Oncogenic kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell (2014) 25(5):621–37. doi: 10.1016/j.ccr.2014.03.014
175. Liou G-Y, Storz P. Inflammatory macrophages in pancreatic acinar cell metaplasia and initiation of pancreatic cancer. Oncoscience (2015) 2(3):247. doi: 10.18632/oncoscience.151
176. Storz P. The crosstalk between acinar cells with kras mutations and M1-polarized macrophages leads to initiation of pancreatic precancerous lesions. Oncoimmunology (2015) 4(6):e1008794. doi: 10.1080/2162402X.2015.1008794
177. Daley D, Zambirinis CP, Seifert L, Akkad N, Mohan N, Werba G, et al. γδ T cells support pancreatic oncogenesis by restraining αβ T cell activation. Cell (2016) 166(6):1485–99. e15. doi: 10.1016/j.cell.2016.07.046
178. Hamarsheh S, Osswald L, Saller BS, Unger S, De Feo D, Vinnakota JM, et al. Oncogenic KrasG12D causes myeloproliferation via NLRP3 inflammasome activation. Nat Commun (2020) 11(1):1–17. doi: 10.1038/s41467-020-15497-1
179. Sumimoto H, Takano A, Teramoto K, Daigo Y. RAS–mitogen-activated protein kinase signal is required for enhanced PD-L1 expression in human lung cancers. PloS One (2016) 11(11):e0166626. doi: 10.1371/journal.pone.0166626
180. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
181. Chen N, Fang W, Lin Z, Peng P, Wang J, Zhan J, et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol Immunother (2017) 66(9):1175–87. doi: 10.1007/s00262-017-2005-z
182. Lastwika KJ, Wilson W, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT–mTOR pathway in non–small cell lung cancer. Cancer Res (2016) 76(2):227–38. doi: 10.1158/0008-5472.CAN-14-3362
183. Coelho MA, de Carné Trécesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity (2017) 47(6):1083–99. e6. doi: 10.1016/j.immuni.2017.11.016
184. Rosenbaum MW, Bledsoe JR, Morales-Oyarvide V, Huynh TG, Mino-Kenudson M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Modern Pathol (2016) 29(9):1104–12. doi: 10.1038/modpathol.2016.95
185. Albitar M, Sudarsanam S, Ma W, Jiang S, Chen W, Funari VA, et al. Expression of PD-L1 in colorectal cancer that lack mutations in RAS or TP53 genes. Am Soc Clin Oncol (2017) 35(15_suppl):e14500. doi: 10.1200/JCO.2017.35.15_suppl.e14500
186. Zdanov S, Mandapathil M, Eid RA, Adamson-Fadeyi S, Wilson W, Qian J, et al. Mutant KRAS conversion of conventional T cells into regulatory T cells. Cancer Immunol Res (2016) 4(4):354–65. doi: 10.1158/2326-6066.CIR-15-0241
187. Bradley SD, Chen Z, Melendez B, Talukder A, Khalili JS, Rodriguez-Cruz T, et al. BRAFV600E co-opts a conserved MHC class I internalization pathway to diminish antigen presentation and CD8+ T-cell recognition of melanoma. Cancer Immunol Res (2015) 3(6):602–9. doi: 10.1158/2326-6066.CIR-15-0030
188. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell (2012) 21(6):836–47. doi: 10.1016/j.ccr.2012.04.024
189. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell (2012) 21(6):822–35. doi: 10.1016/j.ccr.2012.04.025
190. Petanidis S, Anestakis D, Argyraki M, Hadzopoulou-Cladaras M, Salifoglou A. Differential expression of IL-17, 22 and 23 in the progression of colorectal cancer in patients with K-ras mutation: Ras signal inhibition and crosstalk with GM-CSF and IFN-γ. PloS One (2013) 8(9):e73616. doi: 10.1371/journal.pone.0073616
191. Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell (2019) 35(4):559–72. e7. doi: 10.1016/j.ccell.2019.02.008
192. Baumann D, Hägele T, Mochayedi J, Drebant J, Vent C, Blobner S, et al. Proimmunogenic impact of MEK inhibition synergizes with agonist anti-CD40 immunostimulatory antibodies in tumor therapy. Nat Commun (2020) 11(1):1–18. doi: 10.1038/s41467-020-15979-2
193. Kane LP, Lin J, Weiss A. Signal transduction by the TCR for antigen. Curr Opin Immunol (2000) 12(3):242–9. doi: 10.1016/S0952-7915(00)00083-2
194. Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res (2014) 2(1):70–9. doi: 10.1158/2326-6066.CIR-13-0160
195. Hu-Lieskovan S, Mok S, Moreno BH, Tsoi J, Robert L, Goedert L, et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci Trans Med (2015) 7(279):279ra41–ra41. doi: 10.1126/scitranslmed.aaa4691
196. Verma V, Jafarzadeh N, Boi S, Kundu S, Jiang Z, Fan Y, et al. MEK inhibition reprograms CD8+ T lymphocytes into memory stem cells with potent antitumor effects. Nat Immunol (2021) 22(1):53–66. doi: 10.1038/s41590-020-00818-9
197. Ando M, Ito M, Srirat T, Kondo T, Yoshimura A. Memory T cell, exhaustion, and tumor immunity. J Immunol (2020) 43(1):1–9. doi: 10.1080/25785826.2019.1698261
198. Jennings E, Elliot TA, Thawait N, Kanabar S, Yam-Puc JC, Ono M, et al. Nr4a1 and Nr4a3 reporter mice are differentially sensitive to T cell receptor signal strength and duration. Cell Reports (2020) 33(5):108328. doi: 10.1016/j.celrep.2020.108328
199. Yarchoan M, Mohan AA, Dennison L, Vithayathil T, Ruggieri A, Lesinski GB, et al. MEK inhibition suppresses b regulatory cells and augments anti-tumor immunity. PloS One (2019) 14(10):e0224600. doi: 10.1371/journal.pone.0224600
200. Richards JD, Davé SH, Chou C-HG, Mamchak AA, DeFranco AL. Inhibition of the MEK/ERK signaling pathway blocks a subset of b cell responses to antigen. J Immunol (2001) 166(6):3855–64. doi: 10.4049/jimmunol.166.6.3855
201. Manzini C, Venè R, Cossu I, Gualco M, Zupo S, Dono M, et al. Cytokines can counteract the inhibitory effect of MEK-i on NK-cell function. Oncotarget (2016) 7(38):60858. doi: 10.18632/oncotarget.11504
202. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet (2000) 356(9244):1795–9. doi: 10.1016/S0140-6736(00)03231-1
203. Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer: Interdiscip Int J Am Cancer Soc (1997) 79(12):2320–8. doi: 10.1002/(SICI)1097-0142(19970615)79:12<2320::AID-CNCR5>3.0.CO;2-P
204. Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, et al. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer (2000) 88(3):577–83. doi: 10.1002/(SICI)1097-0142(20000201)88:3<577::AID-CNCR13>3.0.CO;2-V
205. Villegas FR, Coca S, Villarrubia VG, Jiménez R, Chillón M, Jareño J, et al. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer (2002) 35(1):23–8. doi: 10.1016/S0169-5002(01)00292-6
206. Grund EM, Spyropoulos DD, Watson DK, Muise-Helmericks RC. Interleukins 2 and 15 regulate Ets1 expression via ERK1/2 and MNK1 in human natural killer cells. J Biol Chem (2005) 280(6):4772–8. doi: 10.1074/jbc.M408356200
207. Fischetti L, Zhong Z, Pinder CL, Tregoning JS, Shattock RJ. The synergistic effects of combining TLR ligand based adjuvants on the cytokine response are dependent upon p38/JNK signalling. Cytokine (2017) 99:287–96. doi: 10.1016/j.cyto.2017.08.009
208. Franklin DA, Sharick JT, Ericsson-Gonzalez PI, Sanchez V, Dean PT, Opalenik SR, et al. MEK activation modulates glycolysis and supports suppressive myeloid cells in TNBC. JCI Insight (2020) 5(15):e134290. doi: 10.1172/jci.insight.134290
209. Hsu C-L, Kikuchi K, Kondo M. Activation of mitogen-activated protein kinase kinase (MEK)/extracellular signal–regulated kinase (ERK) signaling pathway is involved in myeloid lineage commitment. Blood J Am Soc Hematol (2007) 110(5):1420–8. doi: 10.1182/blood-2007-02-071761
210. Staser K, Park S-J, Rhodes SD, Zeng Y, He YZ, Shew MA, et al. Normal hematopoiesis and neurofibromin-deficient myeloproliferative disease require erk. J Clin Invest (2012) 123(1):329–34. doi: 10.1172/JCI66167
211. Allegrezza MJ, Rutkowski MR, Stephen TL, Svoronos N, Perales-Puchalt A, Nguyen JM, et al. Trametinib drives t-cell–dependent control of KRAS-mutated tumors by inhibiting pathological myelopoiesis. Cancer Res (2016) 76(21):6253–65. doi: 10.1158/0008-5472.CAN-16-1308
212. Yu J, Li H, Zhang Z, Lin W, Wei X, Shao B. Targeting the MDSCs of tumors In situ with inhibitors of the MAPK signaling pathway to promote tumor regression. Front Oncol (2021) 11:796. doi: 10.3389/fonc.2021.647312
213. Osswald L, Uhl FM, Andrieux G, Klein C, Dierks C, Duquesne S, et al. Oncogenic KrasG12D activation in the nonhematopoietic bone marrow microenvironment causes myelodysplastic syndrome in mice. Mol Cancer Res (2021) 19(9):1596–608. doi: 10.1158/1541-7786.MCR-20-0275
214. Brea EJ, Oh CY, Manchado E, Budhu S, Gejman RS, Mo G, et al. Kinase regulation of human MHC class I molecule expression on cancer cells. Cancer Immunol Res (2016) 4(11):936–47. doi: 10.1158/2326-6066.CIR-16-0177
215. Choi H, Deng J, Li S, Silk T, Dong L, Brea EJ, et al. Pulsatile MEK inhibition improves anti-tumor immunity and T cell function in murine kras mutant lung cancer. Cell Rep (2019) 27(3):806–19. e5. doi: 10.1016/j.celrep.2019.03.066
216. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov (2011) 10(6):417–27. doi: 10.1038/nrd3455
217. Liu Y, Cai P, Wang N, Zhang Q, Chen F, Shi L, et al. Combined blockade of Tim-3 and MEK inhibitor enhances the efficacy against melanoma. Biochem Biophys Res Commun (2017) 484(2):378–84. doi: 10.1016/j.bbrc.2017.01.128
218. Poon E, Mullins S, Watkins A, Williams GS, Koopmann J-O, Di Genova G, et al. The MEK inhibitor selumetinib complements CTLA-4 blockade by reprogramming the tumor immune microenvironment. J Immunother Cancer (2017) 5(1):1–14. doi: 10.1186/s40425-017-0268-8
219. Mainardi S, Mulero-Sánchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, et al. SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med (2018) 24(7):961–7. doi: 10.1038/s41591-018-0023-9
220. Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med (2019) 25(4):628–40. doi: 10.1038/s41591-019-0368-8
221. Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF→ MEK→ ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med (2019) 25(4):620–7. doi: 10.1038/s41591-019-0367-9
222. Raufi A, Wong W, Lee SM, Manji GA. MEKiAUTO: A phase I/II open-label study of combination therapy with the MEK inhibitor cobimetinib, immune-checkpoint blockade with atezolizumab, and the AUTOphagy inhibitor hydroxychloroquine in KRAS-mutated advanced malignancies. Am Soc Clin Oncol (2021) 39(3_suppl):TPS450. doi: 10.1200/JCO.2021.39.3_suppl.TPS450
223. Ozkan-Dagliyan I, Diehl JN, George SD, Schaefer A, Papke B, Klotz-Noack K, et al. Low-dose vertical inhibition of the RAF-MEK-ERK cascade causes apoptotic death of KRAS mutant cancers. Cell Rep (2020) 31(11):107764. doi: 10.1016/j.celrep.2020.107764
224. Fernandes Neto JM, Nadal E, Bosdriesz E, Ooft SN, Farre L, McLean C, et al. Multiple low dose therapy as an effective strategy to treat EGFR inhibitor-resistant NSCLC tumours. Nat Commun (2020) 11(1):1–9. doi: 10.1038/s41467-020-16952-9
225. Wang Y, Liu S, Yang Z, Algazi AP, Lomeli SH, Wang Y, et al. Anti-PD-1/L1 lead-in before MAPK inhibitor combination maximizes antitumor immunity and efficacy. Cancer Cell (2021) 39(10):1375–87. e6. doi: 10.1016/j.ccell.2021.07.023
226. Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. New Engl J Med (2014) 371(20):1867–76. doi: 10.1056/NEJMoa1408868
227. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. New Engl J Med (2014) 371(20):1877–88. doi: 10.1056/NEJMoa1406037
228. Deken MA, Gadiot J, Jordanova ES, Lacroix R, van Gool M, Kroon P, et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology (2016) 5(12):e1238557. doi: 10.1080/2162402X.2016.1238557
229. Limagne E, Nuttin L, Thibaudin M, Jacquin E, Aucagne R, Bon M, et al. MEK inhibition overcomes chemoimmunotherapy resistance by inducing CXCL10 in cancer cells. Cancer Cell (2022) 40(2):136–152.e12. doi: 10.1016/j.ccell.2021.12.009
230. Xue Y, Martelotto L, Baslan T, Vides A, Solomon M, Mai TT, et al. An approach to suppress the evolution of resistance in BRAF V600E-mutant cancer. Nat Med (2017) 23(8):929–37. doi: 10.1038/nm.4369
231. Shapiro GI, LoRusso P, Kwak E, Pandya S, Rudin CM, Kurkjian C, et al. Phase ib study of the MEK inhibitor cobimetinib (GDC-0973) in combination with the PI3K inhibitor pictilisib (GDC-0941) in patients with advanced solid tumors. Investigational New Drugs (2020) 38(2):419–32. doi: 10.1007/s10637-019-00776-6
232. Gonzalez-Cao M, Mayo de las Casas C, Oramas J, Berciano-Guerrero MA, de la Cruz L, Cerezuela P, et al. Intermittent BRAF inhibition in advanced BRAF mutated melanoma results of a phase II randomized trial. Nat Commun (2021) 12(1):1–6. doi: 10.1038/s41467-021-26572-6
233. Huijberts SC, van Geel RM, van Brummelen EM, Opdam FL, Marchetti S, Steeghs N, et al. Phase I study of lapatinib plus trametinib in patients with KRAS-mutant colorectal, non-small cell lung, and pancreatic cancer. Cancer Chemother Pharmacol (2020) 85(5):917–30. doi: 10.1007/s00280-020-04066-4
234. Tolcher AW, Patnaik A, Papadopoulos KP, Rasco DW, Becerra CR, Allred AJ, et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother Pharmacol (2015) 75(1):183–9. doi: 10.1007/s00280-014-2615-5
235. Brummelen EM, Huijberts S, Herpen C, Desar I, Opdam F, Geel R, et al. Phase I study of afatinib and selumetinib in patients with KRAS-mutated colorectal, non-small cell lung, and pancreatic cancer. Oncol (2021) 26(4):290–e545. doi: 10.1002/onco.13631
236. Algazi AP, Othus M, Daud AI, Lo RS, Mehnert JM, Truong T-G, et al. Continuous versus intermittent BRAF and MEK inhibition in patients with BRAF-mutated melanoma: a randomized phase 2 trial. Nat Med (2020) 26(10):1564–8. doi: 10.1038/s41591-020-1060-8
237. Guo C, Chénard-Poirier M, Roda D, de Miguel M, Harris SJ, Candilejo IM, et al. Intermittent schedules of the oral RAF–MEK inhibitor CH5126766/VS-6766 in patients with RAS/RAF-mutant solid tumours and multiple myeloma: A single-centre, open-label, phase 1 dose-escalation and basket dose-expansion study. Lancet Oncol (2020) 21(11):1478–88. doi: 10.1016/S1470-2045(20)30464-2
238. Martinez-Garcia M, Banerji U, Albanell J, Bahleda R, Dolly S, Kraeber-Bodéré F, et al. First-in-human, phase I dose-escalation study of the safety, pharmacokinetics, and pharmacodynamics of RO5126766, a first-in-class dual MEK/RAF inhibitor in patients with solid tumors. Clin Cancer Res (2012) 18(17):4806–19. doi: 10.1158/1078-0432.CCR-12-0742
Keywords: MAPK signaling, KRAS/BRAF mutations, targeted inhibition, tumor immunity, immune escape, immunomodulation, immunotherapy
Citation: Avery TY, Köhler N, Zeiser R, Brummer T and Ruess DA (2022) Onco-immunomodulatory properties of pharmacological interference with RAS-RAF-MEK-ERK pathway hyperactivation. Front. Oncol. 12:931774. doi: 10.3389/fonc.2022.931774
Received: 29 April 2022; Accepted: 30 June 2022;
Published: 27 July 2022.
Edited by:
Dharmendra Kumar Yadav, aa University, South KoreaReviewed by:
Italia Falcone, Regina Elena National Cancer Institute (IRCCS), ItalyCopyright © 2022 Avery, Köhler, Zeiser, Brummer and Ruess. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Yul Avery, dGhvbWFzLnl1bC5hdmVyeUB1bmlrbGluaWstZnJlaWJ1cmcuZGU=; Dietrich Alexander Ruess, ZGlldHJpY2gucnVlc3NAdW5pa2xpbmlrLWZyZWlidXJnLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.