 Moon Ley Tung
Moon Ley Tung Bharatendu Chandra
Bharatendu Chandra Kyle Dillahunt
Kyle Dillahunt Matthew D. Gosse
Matthew D. Gosse T. Shawn Sato3
T. Shawn Sato3 Alpa Sidhu
Alpa Sidhu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 07 July 2022
Sec. Cancer Genetics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.925582
This article is part of the Research Topic Women in Cancer Genetics Vol II: 2022 View all 12 articles
Von Hippel Lindau(VHL)syndrome presents with cerebellar and spinal hemangioblastomas, renal cell cancer, neuroendocrine pancreatic tumor, and pheochromocytoma and it is caused by germline mutations in the VHL gene. Pathogenic germline variants in the succinate dehydrogenase A (SDHA) gene are associated with paraganglioma and pheochromocytoma. Here we report co-occurrence of germline pathogenic variants in both VHL and SDHA genes in a patient who presented with pancreatic neuroendocrine tumor. As these genes converge on the pseudo-hypoxia signaling pathway, further studies are warranted to determine the significance of co-occurrence of these variants in relation to tumor penetrance, disease severity, treatment response and clinical outcomes in this selected group of patients.
Von Hippel Lindau (VHL) syndrome caused by germline loss-of-function variants in the gene VHL, is an autosomal dominant cancer predisposition syndrome (1). Clinical features include cerebellar and spinal hemangioblastomas, renal cell cancer, neuroendocrine pancreatic tumor, pheochromocytoma, and paragangliomas. Germline pathogenic variants in the succinate dehydrogenase A (SDHA) gene are associated with familial paraganglioma and pheochromocytoma syndrome inherited in an autosomal dominant manner (2). At the cellular level, SDHA and VHL proteins interact and converge into a common molecular pathway via the hypoxia inducible factor alpha (HIF-α) (3). Here we report co-occurrence of germline pathogenic variants in both genes (VHL and SDHA) in a patient who presented with pancreatic neuroendocrine tumor. We describe the known molecular pathways involving VHL and SDHA and postulate that the disease prognosis may be dependent on the presence of co-occurring pathogenic variants in these genes through the involvement of HIF-α in the final common pathway. To our knowledge, this is the first case report of both VHL and SDHA pathogenic variants.
The proband, a 23-year-old female, was reviewed at our Genetics Cancer Predisposition clinic for evaluation and recommendations for concurrent germline pathogenic variants in the VHL and SDHA genes. She presented with recurrent episodes of abdominal pain, vomiting, and anorexia at 21-years of age. Initial radiological and endoscopic evaluations were unremarkable. Her symptoms continued to persist, and a review of her prior abdominal computed tomography (Figures 1A, B) and magnetic resonance imaging (MRI) study showed a mass in the pancreatic tail with hepatic metastases (Figures 1C–F). She underwent surgical resection of pancreatic tail mass as well as right liver lobectomy, splenectomy, and cholecystectomy for metastatic disease.
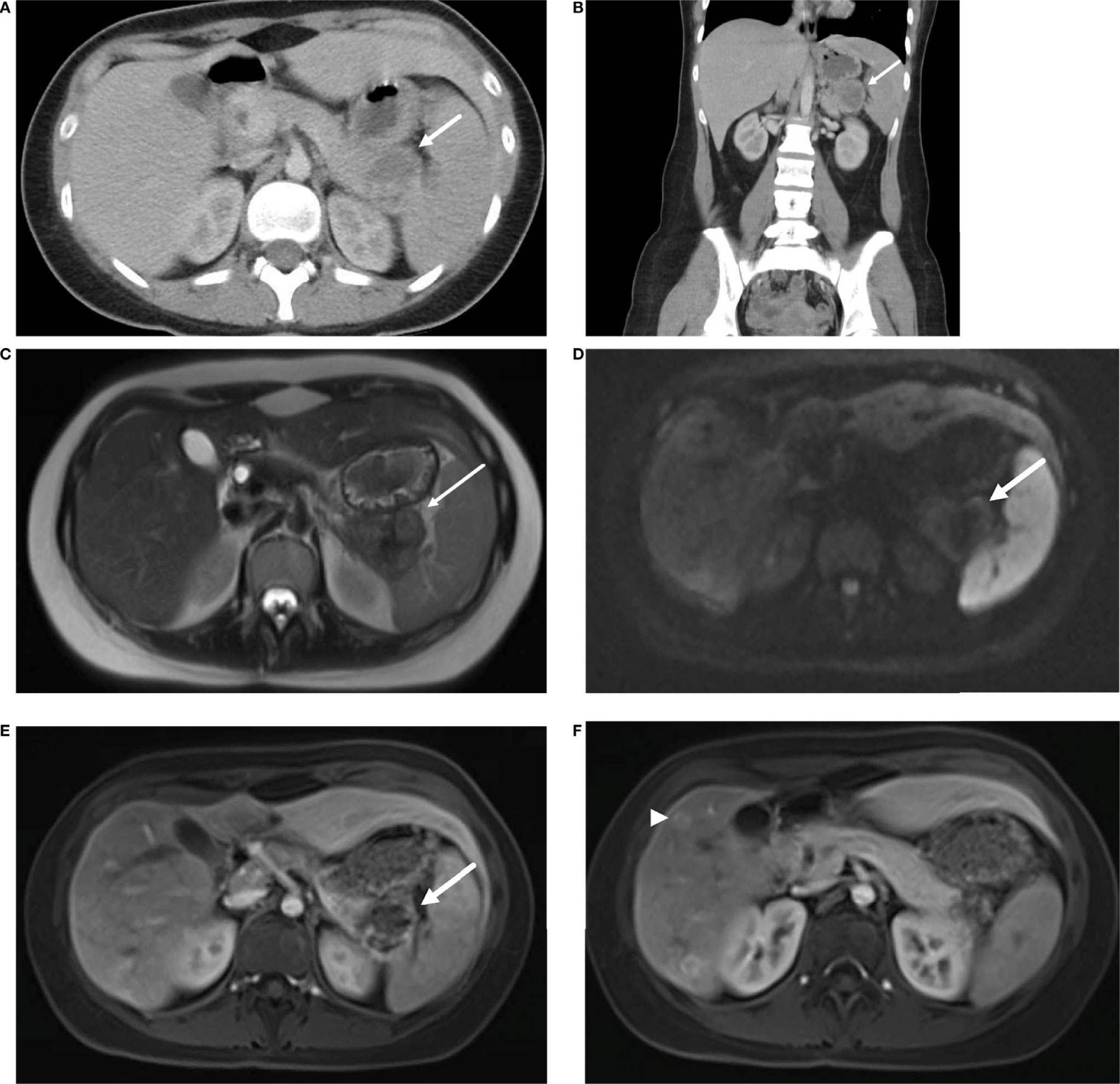
Figure 1 Axial (A) and coronal (B) contrast enhanced CT demonstrate a pancreatic tail mass with central necrosis (→) positioned between the stomach, spleen, and kidney. MRI of the abdomen reveals a mass in the tail of the pancreas (→) with intermediate T2 signal (C), diffusion restriction (D) and peripheral contrast enhancement (E). Additional enhancing lesions (▸) were seen in the liver consistent with metastatic disease (F).
Histopathologic examination of the pancreatic tail mass revealed a well-differentiated neuroendocrine tumor (Figure 2A). The primary tumor was centered in the distal pancreas, perineural invasion was present, and 4 mitoses per 2 mm2 were identified. Metastatic tumor deposits were identified in two of fifteen regional lymph nodes and multiple metastatic liver foci were identified (Figure 2B). A Ki-67 immunostain was performed on the primary tumor, local lymph node metastasis, and liver metastasis (Figure 2C) and the proliferation index was determined to be 14%, 8.5%, and 11%, respectively, using digital image analysis. This met criteria for a Grade 2 (intermediate) tumor by 2019 WHO classification using both mitotic count and Ki-67 proliferation index. Immunohistochemistry on the primary tumor was positive for synaptophysin, chromogranin, pan keratin, and CK7. and negative for CK20, CD10 and progesterone receptor (PR). Somatostatin receptor subtype 2A (SSTR2A) immunostaining was positive (3+, 90%) (Figure 2D) and ATRX immunostain was intact. Initial testing of tumor was performed at an outside institution and additional pathology samples were not available for SDHB immunohistochemistry.

Figure 2 (A) Hemotoxylin and eosin (H&E) staining of primary tumor. Cells are arranged in nests with finely stippled chromatin. The tumor cells expressed synaptophysin, chromogranin, and pankeratin (not shown) supporting the diagnosis of well-differentiated neuroendocrine tumor. (B) H&E stained section of metastatic tumor focus in the liver with similar morphology to the primary tumor. (C) Ki-67 immunostained section of the metastatic tumor focus in the liver. The proliferation index was found to be 11% in a similar hot spot by digital image analysis. (D) Somatostatin receptor subtype 2A (SSTR2A) immunostained section was positive (3+, 90%). (E) Tumor molecular profiling showing a variant in VHL (c.500G>A, p.R167Q) at a frequency of 66.3%). (F) Tumor molecular profiling showing a variant in SDHA (c.1054C>T, p.R352*) at a frequency of 50%). (G) Germline molecular profiling showing an identical variant in VHL (c.500G>A, p.R167Q). (H) Germline molecular profiling showing an identical variant in SDHA (c.1054C>T, p.R352*).
Molecular test performed on the tumor sample showed pathogenic variants in the VHL gene (c.500G>A, p.R167Q) (Figure 2E) involving 66% of tumor cells, and SDHA gene (c.1054C>T, p.R352*) (Figure 2F) involving 50% of tumor cells. Germline testing on peripheral blood sample showed identical pathogenic variants in VHL (c.500G>A, p.R167Q) (Figure 2G) and SDHA (c.1054C>T, p.R352*) genes (Figure 2H). Familial testing could not be performed due to patient’s adoptive status. A Positron Emission Tomography scan following the surgery was suggestive of multiple somatostatin receptor tracer uptake in the pancreatic head, duodenum, and the liver. She was initially treated with octreotide acetate and subsequently with Lantreotide.
The patient’s past medical history was significant for a right retinal hemangioma diagnosed at 18- years of age. Follow-up ophthalmological evaluation showed consistent exam without progression of disease. She was making good recovery from her initial diagnosis at the time of our review, and the pancreatic and hepatic lesions have remained stable. She was recommended studies per surveillance guidelines for her diagnosis of both VHL and SDHA-related paraganglioma and pheochromocytoma syndrome. She had normal brain, internal auditory canal, and spine magnetic resonance imaging studies without evidence of any VHL-associated lesions. There is limited prenatal, birth, and postnatal history available as she was adopted during infancy, and presented with early onset global developmental delay,undergoing interventional therapies during childhood. She was diagnosed with autism spectrum disorder and depression. To rule out underlying genetic etiology of global developmental delay, we performed chromosomal microarray and Fragile-X syndrome analysis. Her chromosomal microarray was normal whereas Fragile- X syndrome analysis incidentally revealed a premutation carrier status with 30 and 57 CGG repeats in the FMR1 gene. The patient and her family were provided comprehensive genetic counseling about these results.
Von Hippel-Lindau (VHL) is an autosomal dominant hereditary tumor predisposition syndrome caused due to a germline pathogenic variant in the VHL gene (4). VHL syndrome predisposes an individual to various tumors such as cerebellar and spinal hemangioblastomas, retinal angiomas, renal cell carcinoma, and pheochromocytoma (1, 5). Other tumors such as pancreatic neoplasms, pituitary hemangioblastomas, and duodenal carcinoid tumors have also been rarely reported. The disorder has a high disease penetrance that is estimated to be about 97% by the age of 60 years (6). Patients with VHL syndrome have a shortened life expectancy secondary to complications related to cerebellar hemangioblastoma, renal cell carcinoma, and pancreatic neoplasms (1). Although VHL-associated tumors usually manifest at a younger age compared to sporadic tumors (6), they appear to be more responsive to chemotherapeutics (7), and less aggressive with respect to their local recurrence and metastatic involvement (8). Present surveillance guidelines recommend age-based screening with dilated eye examination, plasma metanephrines, MRI of brain, spine, abdomen, and internal auditory canal.
Collectively, hereditary paraganglioma-pheochromocytoma (PGL/PCC) syndromes are rare neuroendocrine tumors with an estimated incidence of approximately 2-8 cases per million per year (9). Approximately 40% of all cases of PGL/PCC are associated with germline pathogenic variants (2, 10, 11) in the pseudo-hypoxic signaling pathway (cluster I), kinase signaling (cluster II), or Wingless and Int-1 (Wnt) signaling group (cluster III) (Figure 3) (12). Pathogenic variants in the SDHx genes (SDHA, SDHB, SDHC, and SDHD) are classified under the cluster I genes which results in dysfunction of succinate dehydrogenase (SDH) leading to competitive inhibition of the enzyme, prolyl hydroxylase, involved in the degradation of hypoxia-inducible factor 1- α (HIF1- α) (2, 13, 14). SDHx-related PGL/PCC are relatively new tumor predisposition syndromes that include PGL and PCC, and rarely renal cell carcinoma, pituitary adenoma, gastrointestinal stromal tumors, and pancreatic neuroendocrine tumor (PNET) (15). Patients with PGL/PCC can manifest tumor at any age, although a majority would present between the third and fifth decade of life (16) with symptoms of excess catecholamine production, including hypertension, headache, diaphoresis, palpitations, and tremors. SDHA pathogenic germline variants are present in up to 30% of wild-type gastrointestinal stromal tumors and 10% of patients with PGL and PCC (Boikos et al, 2016). A recent study identified only 10 patients harboring a pathogenic germline SDHA variant in 4,974 pediatric and adult patients with a variety of solid tumors (17). Out of these 10 patients with underlying SDHA pathogenic variant, 2 had gastrointestinal stromal tumor, and the remaining had melanoma, neuroblastoma, breast, colon, renal, prostate, endometrial, and bladder cancer.
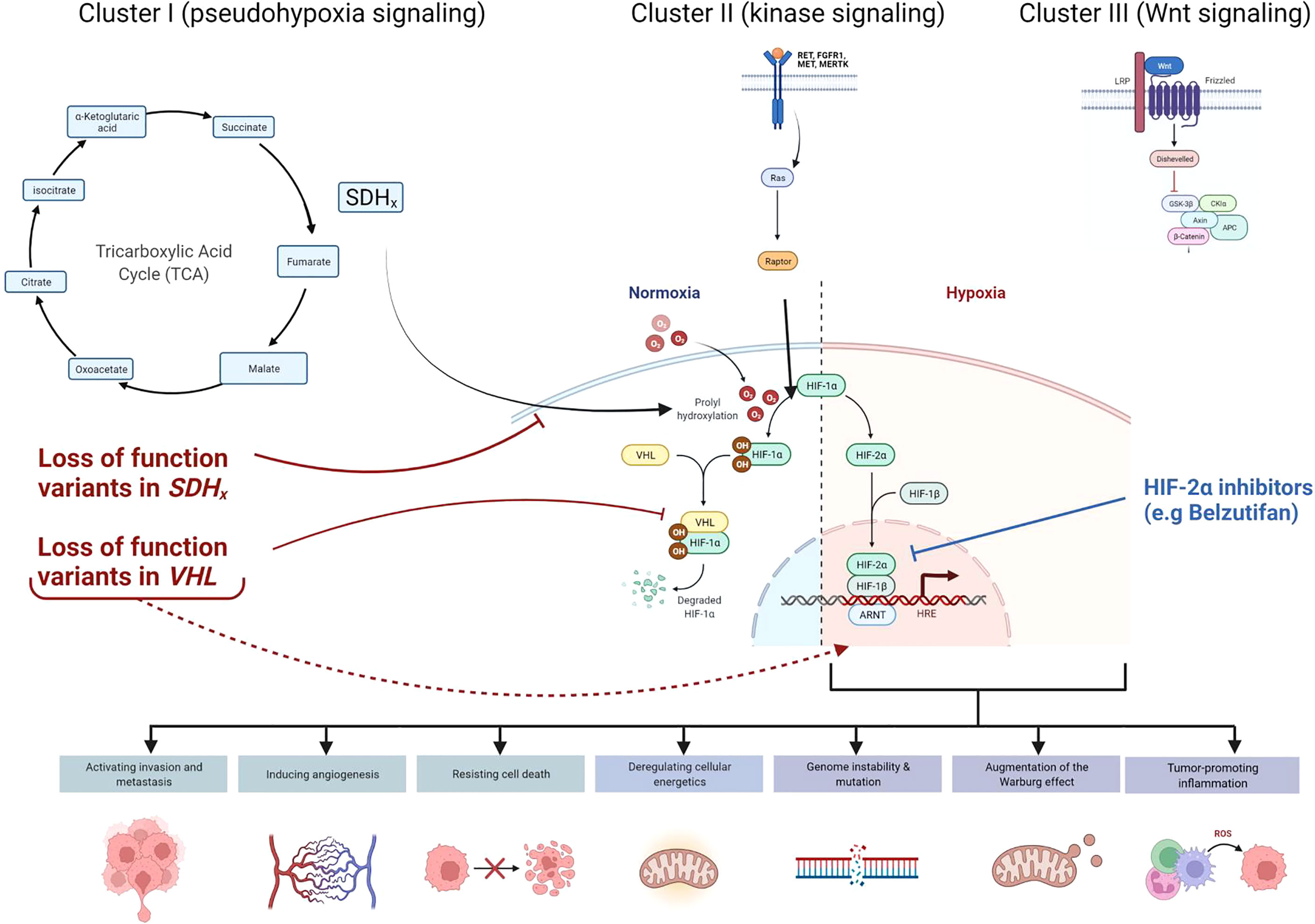
Figure 3 Cellular pathways and genes involved in hereditary paraganglioma-pheochromocytoma (PGL/PCC) syndrome and Von Hippel Lindau (VHL) disease. Approximately 40% of all cases of PGL/PCC cases are associated with germline pathogenic variants in the pseudo-hypoxic signaling pathway (Cluster I), kinase signaling (Cluster II), or Wingless and Int-1 (Wnt) signaling (Cluster III). Pathogenic variants in the SDHx genes (SDHA, SDHB, SDHC, and SDHD) and VHL gene are classified under the Cluster I genes. The SDH complex plays an important role in energy metabolism through the tricarboxylic acid (TCA) cycle. Both VHL and SDHx encode for proteins that target the protein HIF1-α for degradation. Pathogenic loss of function variants in VHL and SDHx genes leads to abnormal accumulation of HIF1-α and upregulation of downstream pathways, which results in increased expression of various proteins and augmentation of the Warburg effect. These negative effects may be mitigated by novel therapies such as HIF2-α inhibitors (e.g Belzutifan).
In contrast to SDHB which is a highly penetrant tumor predisposition gene, SDHA confers a much lower penetrance and severity, estimates of which are largely unknown (18, 19). Present surveillance guidelines for SDHx-associated PGL/PCC recommend plasma metanephrines, whole body and dedicated neck MRI, and complete blood count starting from age 6-8 years (19). Our patient represents the first case of a metastatic PNET in the setting of germline heterozygous pathogenic variants in the VHL and SDHA genes. PNETs are clinically heterogenous tumors originating from neuroendocrine cells of the pancreatic islets and usually follow a variable clinical course, with a low five-year survival rate in approximately 60% of patients (20). PNET occur in approximately 9 to 17% of patients with VHL disease (8) but have only been reported in one individual with a germline SDHD pathogenic variant (15). Since PNETs are seen in VHL and not observed so far in SDHA-related PGL/PCC syndrome, we postulate that the underlying genetic etiology would be the pathogenic variant with a second hit in the VHL gene. Although a second pathogenic variant was not reported by the tumor molecular testing, it is possible that the second hit could not be detected due to limitation of the tumor-based molecular analyses. These include possibility of somatic second hit being present in the promoter or deep enhancer region, or promoter methylation (21). Loss of heterozygosity (LOH) analysis could not be performedin the patient’s tumor type, as the laboratory only performs LOH analysis in ovarian tumors
Multi-locus inherited neoplasia allele syndrome (MINAS) is a relatively new entity that was initially reported in BRCA1/BRCA2-related cancers (21). With increasing adoption of next generation sequencing technologies for cancer susceptibility germline (CSG) testing, there has been a rise in reports of non-BRCA1/BRCA2 related MINAS. A recent study by McGuigan et al. reported that atypical tumor phenotypes comprised of about 15% of non-BRCA1/BRCA2 MINAS cases, which could be secondary to complex interactions between the relevant CSGs (21).
Interestingly, on a molecular level, both VHL and SDHA encode for proteins that target the protein HIF-α for degradation (22). Absence of these proteins would lead to abnormal accumulation and upregulation of HIF, resulting in increased expression of various proteins (e.g vascular endothelial growth factor, platelet-derived growth factor, matrix metalloproteinases and transforming growth factor-alpha), and augmentation of the Warburg effect by HIF-α. The Warburg effect relates to the phenomenon seen in tumor cells that effectively promotes growth and development. Even in normoxia, there is a metabolic shift in which tumor cells preferentially use glycolysis to generate adenosine triphosphate rather than the tricarboxylic acid (TCA) cycle (14). The SDH complex plays an important role in energy metabolism through the TCA cycle, and the reduction of ubiquinone to ubiquinol via the electron transport chain. Therefore, pathogenic variants in the genes involved in the SDH complex could result in accumulation of the ‘oncometabolite’ succinate, leading to disruption of the succinate to fumarate ratio that would inhibit the enzymatic degradation of HIF-α. Accumulation of succinate may also destabilize the redox state and cause mitochondrial dysfunction by increasing reactive oxygen species production and utilizing glutamine as an energy source (14). Epigenetic dysregulation has also been implicated in the malignant potential of SDHx-mutated PGL/PCC (3) and may be a contributing factor in the metastatic presentation of our patient’s PNET at diagnosis. As both these genes converge on the pseudo-hypoxia signaling pathway, we hypothesize that the co-occurrence of germline pathogenic variants in both the VHL and SDHA genes could have an impact on the long-term prognosis. Although our hypothesis is limited by the lack of functional studies and a single case report, further studies looking at this genotype-phenotype correlation in similar cases will be helpful. Co-occurrence of two germline variants in VHL and SDHA in our patient also creates an opportunity for the use of novel therapeutics based on the convergence of these two genes within the pseudo-hypoxia signaling pathway. Recently approved selective HIF inhibitors could be one of the therapeutic considerations in our patient as it has been shown to be effective in patients with renal cell carcinoma due to an underlying germline VHL pathogenic variant (23). HIF-2α overexpression is documented in VHL disease associated renal cell carcinoma (24). Preclinical studies indicated the potential efficacy of HIF-2α subunit inhibitors, which blocks the HIF pathway proximally and limits tumor growth in clear cell renal cell carcinoma (25, 26). Subsequent clinical trials documented the benefits and safety of HIF-2α inhibitors (e.g belzutifan) in both sporadic (27) and VHL-disease associated renal cell carcinoma (23). Belzutifan was also efficacious in reducing tumor size of non-renal cell carcinomas in VHL patients, including pancreatic neuroendocrine tumors, central nervous system hemangioblastomas and retinal hemangioblastomas (23). The U.S Food & Drug Administration (FDA) approved Belzutifan for renal cell carcinoma and non-renal cell neoplasms associated with VHL disease in August 2021 and remains a promising therapy for our patient who presented with both metastatic pancreatic neuroendocrine tumor as well as retinal hemangioma.
The co-occurrence of VHL and SDHA pathogenic variants implicated in PNET is described in this case report. It opens the way for additional exploratory studies to determine the significance of co-occurrence of these variants in terms of tumor penetrance, severity, and outcomes, as well as for the development of novel therapeutic approaches targeting the shared cellular pathway involved in VHL and SDHA-related etiopathogenesis.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study.
MT and BC drafted the original manuscript and created the figure, KD obtained consent and performed counseling for the testing coordinated in the patient. MG provided the pathology images and drafted the pathology description. TS provided radiology images and drafted the imaging description. AS provided patient information and performed manuscript revisions. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the patient and her family for consenting to participate in this publication. We would like to acknowledge Ambry Genetics for providing the IGV results of the germline sequencing data for our patient. We also acknowledge Foundation One®CDx for providing the IGV results of the tumor sequencing data for our patient. Figure 3 was created using BioRender.com.
1. Maddock IR, Moran A, Maher ER, Teare MD, Norman A, Payne SJ, et al. A Genetic Register for Von Hippel-Lindau Disease. J Med Genet (1996) 33(2):120–7. doi: 10.1136/jmg.33.2.120
2. Jhawar S, Arakawa Y, Kumar S, Varghese D, Kim YS, Roper N, et al. New Insights on the Genetics of Pheochromocytoma and Paraganglioma and Its Clinical Implications. Cancers (Basel) (2022) 14(3):594. doi: 10.3390/cancers14030594
3. Choudhry H, Harris AL. Advances in Hypoxia-Inducible Factor Biology. Cell Metab (2018) 27(2):281–98. doi: 10.1016/j.cmet.2017.10.005
4. Dahia PL. Pheochromocytomas and Paragangliomas, Genetically Diverse and Minimalist, All at Once! Cancer Cell (2017) 31(2):159–61. doi: 10.1016/j.ccell.2017.01.009
5. Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, et al. Von Hippel-Lindau Disease. Lancet (2003) 361(9374):2059–67. doi: 10.1016/S0140-6736(03)13643-4
6. Maher ER, Bentley E, Yates JR, Barton D, Jennings A, Fellows IW, et al. Mapping of Von Hippel-Lindau Disease to Chromosome 3p Confirmed by Genetic Linkage Analysis. J Neurol Sci (1990) 100(1-2):27–30. doi: 10.1016/0022-510X(90)90008-B
7. Roma A, Maruzzo M, Basso U, Brunello A, Zamarchi R, Bezzon E, et al. First-Line Sunitinib in Patients With Renal Cell Carcinoma (RCC) in Von Hippel-Lindau (VHL) Disease: Clinical Outcome and Patterns of Radiological Response. Fam Cancer (2015) 14(2):309–16. doi: 10.1007/s10689-014-9771-y
8. de Mestier L, Hammel P. Pancreatic Neuroendocrine Tumors in Von Hippel-Lindau Disease. Scand J Gastroenterol (2015) 50(8):1054–5. doi: 10.3109/00365521.2015.1022211
9. Chen H, Sippel RS, O’Dorisio MS, Vinik AI, Lloyd RV, Pacak K. The North American Neuroendocrine Tumor Society Consensus Guideline for the Diagnosis and Management of Neuroendocrine Tumors: Pheochromocytoma, Paraganglioma, and Medullary Thyroid Cancer. Pancreas. (2010) 39(6):775–83. doi: 10.1097/MPA.0b013e3181ebb4f0
10. Fishbein L, Nathanson KL. Pheochromocytoma and Paraganglioma Susceptibility Genes: Estimating the Associated Risk of Disease. JAMA Oncol (2017) 3(9):1212–3. doi: 10.1001/jamaoncol.2017.0222
11. Jochmanova I, Pacak K. Genomic Landscape of Pheochromocytoma and Paraganglioma. Trends Cancer. (2018) 4(1):6–9. doi: 10.1016/j.trecan.2017.11.001
12. Lam AK. Update on Paragangliomas and Pheochromocytomas. Turk Patoloji Derg (2015) 31 Suppl 1:105–12. doi: 10.5146/tjpath.2015.01318
13. Ilanchezhian M, Jha A, Pacak K, Del Rivero J. Emerging Treatments for Advanced/Metastatic Pheochromocytoma and Paraganglioma. Curr Treat Options Oncol (2020) 21(11):85. doi: 10.1007/s11864-020-00787-z
14. Vicha A, Taieb D, Pacak K. Current Views on Cell Metabolism in SDHx-Related Pheochromocytoma and Paraganglioma. Endocrine-related cancer. (2014) 21(3):R261–77. doi: 10.1530/ERC-13-0398
15. Niemeijer ND, Papathomas TG, Korpershoek E, de Krijger RR, Oudijk L, Morreau H, et al. Succinate Dehydrogenase (SDH)-Deficient Pancreatic Neuroendocrine Tumor Expands the SDH-Related Tumor Spectrum. J Clin Endocrinol Metab (2015) 100(10):E1386–93. doi: 10.1210/jc.2015-2689
16. Guerrero MA, Schreinemakers JM, Vriens MR, Suh I, Hwang J, Shen WT, et al. Clinical Spectrum of Pheochromocytoma. J Am Coll Surg (2009) 209(6):727–32. doi: 10.1016/j.jamcollsurg.2009.09.022
17. Dubard Gault M, Mandelker D, DeLair D, Stewart CR, Kemel Y, Sheehan MR, et al. Germline SDHA Mutations in Children and Adults With Cancer. Cold Spring Harb Mol Case Stud (2018) 4(4):a002584. doi: 10.1101/mcs.a002584
18. Maniam P, Zhou K, Lonergan M, Berg JN, Goudie DR, Newey PJ. Pathogenicity and Penetrance of Germline SDHA Variants in Pheochromocytoma and Paraganglioma (PPGL). J Endocr Soc (2018) 2(7):806–16. doi: 10.1210/js.2018-00120
19. Hanson H, Durkie M, Lalloo F, Izatt L, McVeigh TP, Cook JA, et al. UK Recommendations for SDHA Germline Genetic Testing and Surveillance in Clinical Practice. J Med Genet (2022). doi: 10.1136/jmedgenet-2021-108355
20. Lawrence B, Gustafsson BI, Chan A, Svejda B, Kidd M, Modlin IM. The Epidemiology of Gastroenteropancreatic Neuroendocrine Tumors. Endocrinol Metab Clinics North Am (2011) 40(1):1–18. doi: 10.1016/j.ecl.2010.12.005
21. McGuigan A, Whitworth J, Andreou A, Hearn T, Genomics England Research Consortium, Tischkowitz M, et al. Multilocus Inherited Neoplasia Allele Syndrome (MINAS): An Update. Eur J Hum Genet (2022) 30(3):265–70. doi: 10.1038/s41431-021-01013-6
22. Iliopoulos O. Von Hippel-Lindau Disease: Genetic and Clinical Observations. Front Horm Res (2001) 28. doi: 10.1159/000061052
23. Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, et al. Belzutifan for Renal Cell Carcinoma in Von Hippel–Lindau Disease. N Engl J Med (2021) 385(22):2036–46. doi: 10.1056/NEJMoa2103425
24. Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting Renal Cell Carcinoma With a HIF-2 Antagonist. Nature (2016) 539(7627):112–7. doi: 10.1038/nature19796
25. Acaba LA, Chenworth ML, Gold AS, Wildner AC, Ehlies FJ, Berrocal AM, et al. Terson’s Syndrome in a Patient With Von Hippel-Lindau Disease. Optom Vis Sci (2016) 93(9):1181–6. doi: 10.1097/OPX.0000000000000904
26. Cho H, Du X, Rizzi JP, Liberzon E, Chakraborty AA, Gao W, et al. On-Target Efficacy of a HIF-2α Antagonist in Preclinical Kidney Cancer Models. Nature (2016) 539(7627):107–11. doi: 10.1038/nature19795
Keywords: von Hippel-Lindau syndrome, SDHA-associated paraganglioma and pheochromocytoma syndrome, paraganglioma, genetics, cancer
Citation: Tung ML, Chandra B, Dillahunt K, Gosse MD, Sato TS and Sidhu A (2022) Co-occurrence of VHL and SDHA Pathogenic Variants: A Case Report. Front. Oncol. 12:925582. doi: 10.3389/fonc.2022.925582
Received: 21 April 2022; Accepted: 10 June 2022;
Published: 07 July 2022.
Edited by:
Israel Gomy, Oncoclinicas Group, BrazilReviewed by:
Raymond H Kim, University of Toronto, CanadaCopyright © 2022 Tung, Chandra, Dillahunt, Gosse, Sato and Sidhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alpa Sidhu, YWxwYS1zaWRodUB1aW93YS5lZHU=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.