 Rebecca S. Maynard
Rebecca S. Maynard Charlotte Hellmich
Charlotte Hellmich Kristian M. Bowles
Kristian M. Bowles Stuart A. Rushworth
Stuart A. Rushworth
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 29 June 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.924567
This article is part of the Research TopicDeciphering the Bone Marrow Microenvironment in Hematologic MalignanciesView all 5 articles
Acute myeloid leukaemia (AML) is a highly proliferative cancer characterised by infiltration of immature haematopoietic cells in the bone marrow (BM). AML predominantly affects older people and outcomes, particularly in this difficult to treat population remain poor, in part due to inadequate response to therapy, and treatment toxicity. Normal haematopoiesis is supported by numerous support cells within the BM microenvironment or niche, including adipocytes, stromal cells and endothelial cells. In steady state haematopoiesis, haematopoietic stem cells (HSCs) primarily acquire ATP through glycolysis. However, during stress-responses HSCs rapidly transition to oxidative phosphorylation, enabled by mitochondrial plasticity. Historically it was thought that cancer cells preferentially used glycolysis for ATP production, however recently it has become evident that many cancers, including AML primarily use the TCA cycle and oxidative phosphorylation for rapid proliferation. AML cells hijack the stress-response pathways of their non-malignant counterparts, utilising mitochondrial changes to drive expansion. In addition, amino acids are also utilised by leukaemic stem cells to aid their metabolic output. Together, these processes allow AML cells to maximise their ATP production, using multiple metabolites and fuelling rapid cell turnover which is a hallmark of the disease. This review of AML derived changes in the BM niche, which enable enhanced metabolism, will consider the important pathways and discuss future challenges with a view to understanding how AML cells are able to hijack metabolic pathways and how we may elucidate new targets for potential therapies.
AML is characterised by the malignant transformation of haematopoietic cells, which reside in the BM and ‘take over’ the environment of healthy haematopoietic stem and progenitor cells (1). AML is the most common acute leukaemia in adults and its incidence increases with age. Although treatment options have improved in the last decades (2), the prognosis for patients with AML remains poor, with high rates of relapse in patients who achieve initial remission, and many older patients unable to receive the most intensive treatments available (3, 4). It is therefore imperative that we further our understanding of the mechanisms of the disease, to elucidate more targets for treatment and drive the development of more effective and less toxic treatments.
One area of study is the metabolic changes that occur in the leukaemic BM niche. The BM is a structurally complex organ of blood vessels and a heterogeneous population of haematopoietic cells as well numerous support cells, including mesenchymal stromal cells (MSC), adipocytes, endothelial cells and osteoblasts (5). This creates the tissue specific niche to support the production, differentiation and maintenance of haematopoietic stem and their progeny (6) both in homeostasis and when required in response to systemic stresses. Of note the HSC niche is a hypoxic environment due to atrial blood being relatively deoxygenated when reaches the BM (7). This hypoxic state benefits long-term HSC health as it reduces oxidative stress and promotes HSC quiescence (8) and despite these relatively hypoxic conditions HSC are able to produce sufficient ATP for homeostasis (9).
A key function of HSCs is their ability to rapidly respond to external stress such as bleeding or infection and to increase their energy production in order to support rapid HSC expansion and production of mature blood cells. To achieve this, HSCs can transition rapidly to oxidative phosphorylation (OXPHOS) by acquiring mitochondria from BM stromal cells and increasing the uptake of other metabolites such as free fatty acids (FFA) and amino acids (10–12). We and others have shown that leukaemic cells are able to hijack these metabolic pathways in order to drive leukaemogenesis in preference to normal haematopoiesis (11, 13–19). This allows leukaemic cells to increase their metabolic output and ATP production and thus their proliferative potential. AML blasts also interact with the supporting BM cell populations and induce molecular changes to transform the normal haematopoietic niche into a ‘leukaemic niche’, which favours leukaemic growth over normal haematopoiesis (20). Here we will review the metabolism of the BM niche and how this changes in AML to promote leukaemogenesis. First, we will discuss the more permissible niche during normal metabolism of HSCs in the steady state and in response to stress stimuli. We will then contrast this with the leukaemic niche and how AML utilises the physiological pathways to its advantage. Finally, we will highlight new potential therapeutic targets based on these metabolic abnormalities for treating patients with leukaemia.
Haematopoiesis occurs within the BM niche, a supportive microenvironment that helps to regulate maintenance of HSCs and progenitor populations as well as blood cell production (6). Blood cell turnover is high, with 500 billion cells being produced daily (21), this process therefore not only needs to be regulated but also requires a constant and reliable energy supply. The supportive components of the BM niche provide a stable environment for this, as well as protecting HSCs from over-stimulation (22) and driving the required changes in cell expansion and differentiation in response to both local and systemic stress.
The complexity of the supportive BM niche is illustrated in Figure 1. The BM niche comprises many cell types including mesynchymal stem cells which are multipotent and differentiate into adipocytes, endothelial cells, MSC, osteoblasts, myocytes and chrondrocytes (23). Most of these cells directly interact with HSCs through cytokines and chemokines to regulate their maintenance and differentiation (6). Adipocytes are derived from MSCs and store high levels of fat as triglycerides in lipid droplets (24). They therefore provide an important source of energy, as well as regulating energy metabolism of HSCs and other cells residing in the BM niche (25). Endothelial cells form the lining of blood vessels and thus form the interface between circulating blood and the BM niche (5). They regulate trafficking and homing of HSCs and progenitor cells and secrete a number of cytokines, including granulocyte colony stimulating factor (G-CSF), granulocyte-macrophage colony stimulating factor (GM-CSF) and interleukin 6 (26). Finally, the osteolinage cells, including osteoblasts and osteoclasts, reside within the BM niche and are responsible for bone formation and resorption, which occurs alongside haematopoieisis within an overlapping microenvironment (27, 28). Whilst osteoblasts are thought to only be indirectly involved in the regulation of haematopoiesis (6), osteoclasts are not only derived from monocyte precursors (28) but have also been shown to be vital for HSC homing (29). All of the individual components of the BM niche work together to co-ordinate haematopoieisis, regulate HSC quiescence and drive differentiation.
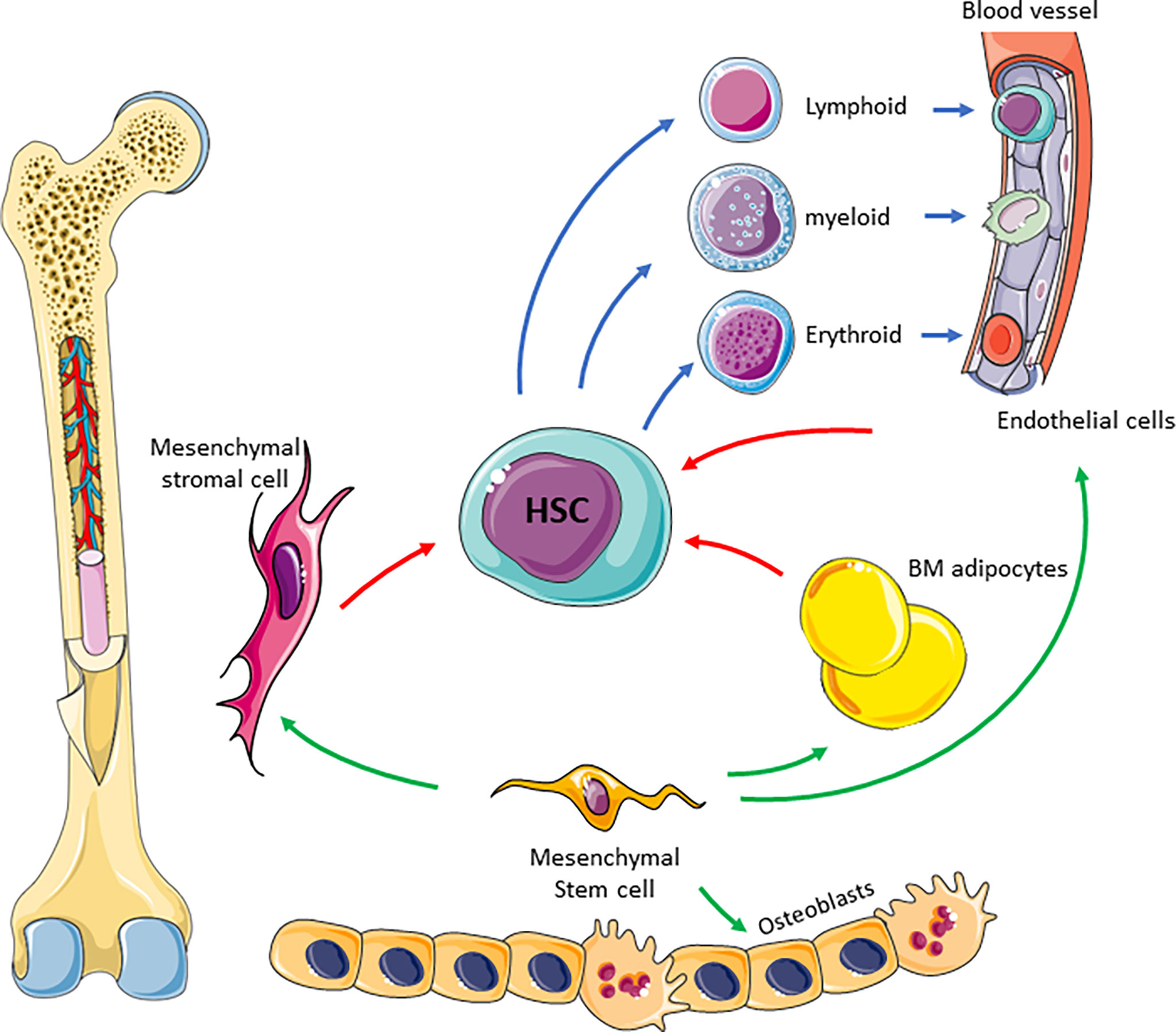
Figure 1 Schematic of BM niche. The complexity of the niche is highlighted showing many cell types and interaction between these cells to facilitate blood production. Mesenchymal stem cells differentiate into BM adipocytes, stromal cells, endothelial cells and osteoblasts (green arrow). Mesenchymal derived cells supports HSC differentiation (red arrows) in to blood progenitor cells (blue arrows).
Haematopoiesis is a dynamic process and cellular proliferation must respond to changes in the BM microenvironment as well as systemic changes and stress stimuli (5). Quiescent HSCs maintain a mitochondrial mass regulated by interplay between biogenesis and mitophagy (10) with low levels of reactive oxygen species (ROS) driving HSCs to favour anaerobic glycolysis. However, the metabolic conditions of HSCs change rapidly as they react to external systemic stressors such as infection or bleeding, amplifying blood production with minimal delay. In response to such stressors HSCs switch from glycolysis to OXPHOS to allow rapid HSC expansion and a several fold increase in production of mature haematopoietic cells (30) with an increase in ROS promoting HSC differentiation (10, 30). To support this switch to OXPHOS and supply the energy required, HSCs increase their mitochondrial mass initially via the transfer of mitochondria from BM stromal cells to HSCs via Connexin 43 Gap Junctions (10). Increased ROS levels have been shown to mediate PI3 kinase activation and subsequent gap junction formation (31). The influx of mitochondria from the environment supports the initial increase in energy demand and is only later in the response to stress that genes involved mitochondrial biogenesis are found to be upregulated (10). Thus, an increase in overall mitochondrial mass drives rapid HSC cycling and differentiation in response to pathogenic stress. Not all HSCs differentiate and a pool of HSCs remain metabolically quiescent and undifferentiated.
Furthermore, HSCs are able to utilise different metabolites within the BM niche to support their self-renewal and differentiation (32–35). BM adipocytes originate from mesenchymal stem cells (36) and with increasing age the proportion of BM mass occupied by adipose tissue gradually increases and can exceed 70% of the bone marrow compartment in older people (37, 38). The BM adipose tissue not only has a function in supplying energy but also as component of the endocrine system (39–41). The release of adipokines and inflammatory factors can alter the proliferative properties of the interacting haematopoietic cells (42). These factors form part of the signalling network that drives HSC differentiation and production of progenitor and mature haematopoietic cells during stress. At the same time lipolysis in BM adipose tissue provides a source of FFA for CPT1a mediated β-oxidation, which has been shown to play a role in regulating HSC asymmetric division and maintenance (32), and provide an additional source of energy to drive their expansion (43). A number of different proteins are involved in the transfer of FFA from the adipocytes, including CD206, CD36, fatty-acid-binding proteins (FABPs), and fatty-acid transport proteins (FATPs) (11, 44–46). In response to infection HSCs rely on the upregulation of the inducible fatty acid transporter, CD36, to facilitate the influx of FFA into HSCs. This promotes a further shift in the metabolic profile of the HSC from glycolysis at its steady state to β-oxidation in a stressed state (11). Together with the increase in OXPHOS this supports HSCs to maximise ATP production and facilitates rapid emergency haematopoiesis.
Amino acids are essential for HSC homeostasis and maintenance (34, 47). They are not only required for protein synthesis but also function as intermediate metabolites. They play a role in the interconnected metabolic pathways utilised by HSC, especially in response to stress and can directly enter the TCA cycle by generating acetyl-CoA (48). This can maintain the TCA cycle without the need for other substrates and thus amino acids can contribute in the same manner as glucose, FFA and lactate during the haematopoietic stress response. Furthermore, amino acids are involved in the regulation of the signalling pathways that determine the metabolic profile of HSCs. Branched chain amino acids have been shown to regulate levels of MEIS1 and p21, which are both involved in the maintenance of HSC quiescence and expansion (49). Amino acids, particularly glutamine have also been shown to play a role in HSC differentiation (33). Due to these many and somewhat contrasting effects, the roles of amino acids in HSC metabolism cannot at present be simply defined. It is likely amino acid function in HSC is dependent of the broader cellular and niche context. In AML, there is increasing evidence that the malignant cells utilise amino acids overall for growth advantage (16) and it seems likely that as with other pathways the tumour hijacks mechanisms inherent to HSCs in order to maximise growth potential. Thus, the increased understanding of amino acid metabolism in AML may help us to improve our knowledge of their role in HSCs both in the steady state and in response to stress.
Historically is was thought that cancer cells preferentially generate ATP through anaerobic glycolysis, as described by Warburg in 1956 (50). However, it has since become apparent that the metabolic processes of malignant cells are much more complex, and it has been suggested AML in fact has a bias towards mitochondrial OXPHOS (51). AML blasts have a very high proliferative potential and therefore equally high energy requirements. Thus it is not surprising that they utilise this most efficient form of ATP production, however as a result they have a need to increase their mitochondrial mass (52). It has been shown that they achieve this by hijacking the HSC response to stress to acquire mitochondria from BM stromal cells in their leukaemic niche (10, 17, 53). Studies have shown this can occur via multiple mechanisms in AML including transfer in extracellular vesicles or via leukaemia-derived tunnelling nanotubes, which requires cell-cell interactions (13, 17, 54, 55). As with HSCs and progenitor cells, increased ROS levels induce mitochondrial transfer. AML blasts can create oxidative stress in the BM stromal cells via NADPH oxidase-2, which generate superoxide (13) (Figure 2A). These conditions create a pro-tumoural environment in which AML blasts can acquire the mitochondria needed to generate the energy required for their increased proliferation. Understanding how AML blasts can maintain their high energy production required for their survival and proliferation can help to inform future treatment strategies. The transfer of mitochondria from BM stromal cells to AML blasts has been shown to rely on the transmembrane glycoprotein CD38 (56). This can be inhibited with the monoclonal antibody targetting CD38, already used widely in the treatment of myeloma (57) and preclinical data suggest it may be effective in treating AML (58, 59). The anti-CD38 antibody Daratumumab for example, has been shown to alter the metabolic profile of AML blasts and inhibit disease progression in vivo (56). This exemplifies how insight into the metabolic changes of malignant cells may be utilised to identify potential targets and inform future treatment strategies.
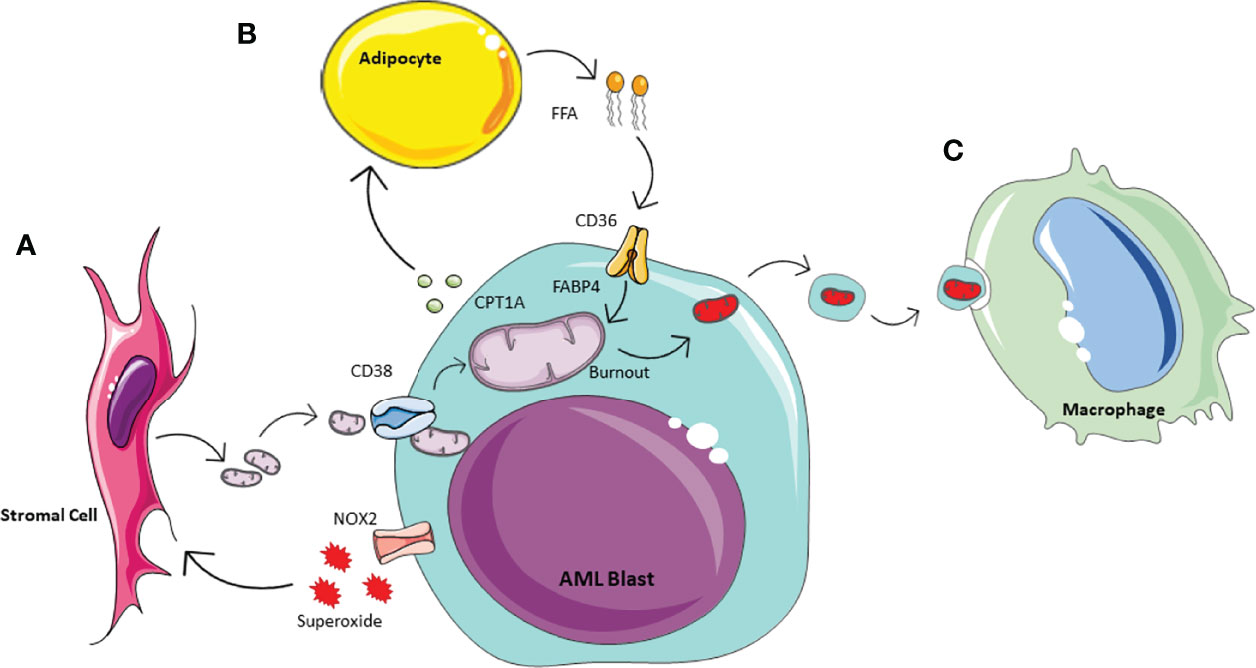
Figure 2 Schematic of AML metabolic of bone marrow microenvironment. Acute Myeliod Leukaemia (AML) is able to manipulate the surrounding environment to enhance its own metabolism. (A) In response to the increase energy demanding, AML produces NOX2 derived superoxide, whichacts on BM stormal cells to activate mitochondrial transfer. Mitochondrial uptake by AML is enable through tunneleing nanotubules by the transmembrane glycoprotien CD38. (B) AML manipulates adipocytes inducing phosphorylation of lipase, to activate lipolysis and te release of Free Fatty Acids (FFA). AML is able to icrease FFa uptake via increased expression of CD38 and fatty-acid binding protiens (FABPs). (C) To prevent the activation of apoptotic pathways, AML outsources its waste mitochondria. This process of outsourcing mitophagy utilises the macrophages in the microenvionment.
During normal haematopoiesis, long-term HSC health and quiescence is dependent on maintaining low levels of ROS (60, 61). By changing this fundamental aspect of the BM niche, AML not only impairs healthy haematopoiesis but it also has to adapt itself so that is can thrive in this environment (62). In fact some mutations seen in AML including the Fms-like tyrosine kinase 3 (FLT-3) receptor mutations, which occurs in 30-35% of cases (63) and is associated with particularly poor prognosis, are known to enhance ROS production by AML blasts further (64, 65) and high ROS levels are known to drive leukaemogenesis (65, 66). This creates a particular challenge in the treatment of AML, as many of the traditional chemotherapy agents, including daunorubicin and cytarabine, are known to increase ROS levels in order to promote apoptosis (67). Whilst even AML cells cannot withstand unlimited levels of ROS and these treatments are therefore effective to an extent, the increased levels of ROS also contribute to genomic instability, which further favors AML survival and promotes chemotherapy resistance (68, 69). Whether ROS production and its downstream effects on AML progression could be targeted in combination with other existing treatments remains to be explored (62). It is clear however that manipulation of ROS levels in the BM niche and the adaptability of AML blasts to tolerate this far better than HSCs provides another survival advantage to AML.
As the AML blasts acquire increasing numbers of mitochondria from the environment, they also accumulate dysfunctional mitochondria and other waste products. If not removed, dysfunctional mitochondria will initiate intra-cellular apoptotic pathways. Therefore, to limit this and minimise cell death, dysfunctional mitochondria are release in extracellular vesicles by AML blasts (70). These are then phagocytosed and removed from the microenvironment by macrophages (Figure 2C). The concept of outsourcing mitophagy has also been shown in mesychymal stem cells where this is mediated by arrestin domain-containing protein 1 (71). Thus, AML blasts are able to utilise another cell in their environment to help with the removal of ‘waste products’ which could otherwise be detrimental to their growth.
Although the AML blasts can acquire more mitochondria from their environment, they also need to maintain a constant supply of the substrates for OXPHOS and utilise as many metabolic pathways as possible to keep up with the energy supply demanded from rapid proliferation. As well as directly contributing to energy production substrates including glucose, lactate, amino acids and FFAs all feed into the TCA cycle to maximise energy output. As already described by Warburg, malignant cells including AML cells can increase their glucose uptake and upregulate their glucose metabolism (72, 73). Whilst we now know this is not solely to directly generate ATP through glycolysis but also to provide sufficient pyruvate for the TCA cycle and subsequent glycolysis, it nevertheless is an important component of the leukaemic metabolic profile. Another by-product of glycolysis is lactate which is known to promote a pro-tumoral microenvironment and has therefore been identified as a potential treatment target (74, 75). However, it is also evident that the conversion of pyruvate to lactate is reversible and therefore lactate can be converted back to pyruvate to then feed into the TCA cycle.
Amino acid metabolism presents a further source of fuel for ATP production in cancer cells (76–79). Cancer cells increase their uptake of amino acids which have a number of functions including protein biosynthesis, activation of signalling pathways as well as a substrate for the TCA cycle (80). Metabolism of amino acids, particularly glutamine, glutamate, and proline, has been shown to be increased in leukaemic stem cells isolated from AML patients (16). Glutamine is the most abundant amino acid in the human body and not only provides metabolites for the TCA cycle but is also required for numerous essential cell functions, including the production of antioxidants to regulate ROS, cell signalling and the synthesis of other amino acids, lipids and nucleic acids (81). Glutamine is a nitrogen donor and provides an amide group for de novo nucleotides and therefore can be a rate limiting factor for AML cell proliferation (81). The oncogenic protein c-Myc has a regulatory function on cell metabolism and can shift mitochondrial metabolism to increase its reliance on glutamine, promoting glutaminolysis, as well as increasing demethylation and inducing glutamine synthetase expression (82). The reliance of leukaemic stem cells on amino acid metabolism for production of ATP and the building of proteins for proliferation, makes them more susceptible to treatments targeting amino acid pathways (16). The combination treatment of venetoclax, a BCL-2 inhibitor and azacytidine, a hypomethylating agent, is now commonly used to treat older patients who are unable to tolerate more intensive treatments. Analysis has shown that treatment with venetoclax and azacytidine disrupts the TCA and OXPHOS in leukaemic stem cells (83) and that this results from a reduction in amino acid uptake (16). Thus, highlighting the potential therapeutic benefits of targeting the energy supply and metabolism of the leukaemic blast.
Similarly to HSCs in stressed conditions, AML blasts utilise β-oxidation of FFAs by accessing the abundant BM adipose tissue to further increase their energy supply to maintain their continuous and rapid self-renewal and differentiation capacity (11) (Figure 2B). It has been shown that a subset of leukaemic stem cells have upregulated CD36 expression to promote the use of FFA as a source of energy, and reside in an adipocytic niche (15). The proximity to adipocytes also increases the expression of fatty acid binding protein-4 (FABP4), another protein that aids the transfer of FFA, into AML blasts (11, 84). Furthermore, AML blasts can have a direct impact on adipocyte metabolism and induce phosphorylation of lipase to activate lipolysis and release FFA (84). This increase in FFA release combined with the increased expression of CD36 and FABP4 improves uptake of FFAs by AML blasts providing the necessary fuel for β-oxidation and therefore increased OXPHOS (84). The role of adipose tissue in AML survival and potential treatment resistance is of particular interest as the BM composition changes with age with an increase in BM adipose tissue (85) and AML is primarily a disease of the elderly (86). FFA metabolism has shown to play a particular role in relapsed AML, where they compensate for the reduced amino acid metabolism that is observed after some treatments which inhibit these pathways (16). Thus, the interaction between AML blasts and adipose tissue may be a useful target in the treatment of AML especially in the older population and in the prevention and treatment of relapsed disease.
AML is a highly proliferative malignancy, which for many is associated with poor clinical outcomes, even despite intensive chemotherapy regimens. While Warburg stated that all cancers preferentially undergo glycolysis for production of ATP (50), in recent years it has become evident that the metabolic profile of AML blasts is more complex and there is a bias towards utilising the most abundant metabolites in the BM niche (11, 13, 16). With this in mind it is therefore predictable that AML blasts would modulate the BM niche to create a pro-tumoral environment that supports their own growth over normal haematopoiesis. AML blasts utilise the pathways intended for the effective metabolic response of HSCs to stress in order to acquire the energy required for their survival, chemotherapy evasion and relentless high levels of proliferation (5, 87). Some of these pathways are already targeted by existing treatments, however improved understanding of these mechanisms will help to elucidate new targets and better tolerated treatments for this disease.
RM, CH, KB and SR conceptualised and wrote the paper. All authors contributed to the article and approved the submitted version.
CH is funded by Wellcome Trust Clinical Research Fellowship (220534/Z/20/Z) and was supported by the NNUH charitable fund. The work was supported from the MRC project grant SAR (MR/T02934X/1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Figure was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
1. Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med (2015) 373(12):1136–52. doi: 10.1056/NEJMra1406184
2. Liu H. Emerging Agents and Regimens for AML. J Hematol Oncol (2021) 14(1):49. doi: 10.1186/s13045-021-01062-w
3. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations From an International Expert Panel. Blood (2017) 129(4):424–47. doi: 10.1182/blood-2016-08-733196
4. Bertoli S, Tavitian S, Huynh A, Borel C, Guenounou S, Luquet I, et al. Improved Outcome for AML Patients Over the Years 2000-2014. Blood Cancer J (2017) 7(12):635. doi: 10.1038/s41408-017-0011-1
5. Mendelson A, Frenette PS. Hematopoietic Stem Cell Niche Maintenance During Homeostasis and Regeneration. Nat Med (2014) 20(8):833–46. doi: 10.1038/nm.3647
6. Morrison SJ, Scadden DT. The Bone Marrow Niche for Haematopoietic Stem Cells. Nature (2014) 505(7483):327–34. doi: 10.1038/nature12984
7. Cipolleschi MG, Dello Sbarba P, Olivotto M. The Role of Hypoxia in the Maintenance of Hematopoietic Stem Cells. Blood (1993) 82(7):2031–7. doi: 10.1182/blood.V82.7.2031.bloodjournal8272031
8. Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, et al. Regulation of the HIF-1alpha Level is Essential for Hematopoietic Stem Cells. Cell Stem Cell (2010) 7(3):391–402. doi: 10.1016/j.stem.2010.06.020
9. Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, et al. The Distinct Metabolic Profile of Hematopoietic Stem Cells Reflects Their Location in a Hypoxic Niche. Cell Stem Cell (2010) 7(3):380–90. doi: 10.1016/j.stem.2010.07.011
10. Mistry JJ, Marlein CR, Moore JA, Hellmich C, Wojtowicz EE, Smith JGW, et al. ROS-Mediated PI3K Activation Drives Mitochondrial Transfer From Stromal Cells to Hematopoietic Stem Cells in Response to Infection. Proc Natl Acad Sci U S A (2019) 116(49):24610–9. doi: 10.1073/pnas.1913278116
11. Mistry JJ, Hellmich C, Moore JA, Jibril A, Macaulay I, Moreno-Gonzalez M, et al. Free Fatty-Acid Transport via CD36 Drives β-Oxidation-Mediated Hematopoietic Stem Cell Response to Infection. Nat Commun (2021) 12(1):7130. doi: 10.1038/s41467-021-27460-9
12. Kreitz J, Schönfeld C, Seibert M, Stolp V, Alshamleh I, Oellerich T, et al. Metabolic Plasticity of Acute Myeloid Leukemia. Cells (2019) 8(8):805. doi: 10.3390/cells8080805
13. Marlein CR, Zaitseva L, Piddock RE, Robinson SD, Edwards DR, Shafat MS, et al. NADPH Oxidase-2 Derived Superoxide Drives Mitochondrial Transfer From Bone Marrow Stromal Cells to Leukemic Blasts. Blood (2017) 130(14):1649–60. doi: 10.1182/blood-2017-03-772939
14. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell (2013) 12(3):329–41. doi: 10.1016/j.stem.2012.12.013
15. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell (2016) 19(1):23–37. doi: 10.1016/j.stem.2016.06.001
16. Jones CL, Stevens BM, D'Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell (2018) 34(5):724–40.e4. doi: 10.1016/j.ccell.2018.10.005
17. Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, et al. Protective Mitochondrial Transfer From Bone Marrow Stromal Cells to Acute Myeloid Leukemic Cells During Chemotherapy. Blood (2016) 128(2):253–64. doi: 10.1182/blood-2015-07-655860
18. Jones CL, Stevens BM, D'Alessandro A, Culp-Hill R, Reisz JA, Pei S, et al. Cysteine Depletion Targets Leukemia Stem Cells Through Inhibition of Electron Transport Complex II. Blood (2019) 134(4):389–94. doi: 10.1182/blood.2019898114
19. Herst PM, Howman RA, Neeson PJ, Berridge MV, Ritchie DS. The Level of Glycolytic Metabolism in Acute Myeloid Leukemia Blasts at Diagnosis is Prognostic for Clinical Outcome. J Leukoc Biol (2011) 89(1):51–5. doi: 10.1189/jlb.0710417
20. Kumar B, Garcia M, Weng L, Jung X, Murakami JL, Hu X, et al. Acute Myeloid Leukemia Transforms the Bone Marrow Niche Into a Leukemia-Permissive Microenvironment Through Exosome Secretion. Leukemia (2018) 32(3):575–87. doi: 10.1038/leu.2017.259
21. Silva A, Anderson AR, Gatenby R. A Multiscale Model of the Bone Marrow and Hematopoiesis. Math Biosci Eng (2011) 8(2):643–58. doi: 10.3934/mbe.2011.8.643
22. Birbrair A, Frenette PS. Niche Heterogeneity in the Bone Marrow. Ann N Y Acad Sci (2016) 1370(1):82–96. doi: 10.1111/nyas.13016
23. Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ, et al. The Meaning, the Sense and the Significance: Translating the Science of Mesenchymal Stem Cells Into Medicine. Nat Med (2013) 19(1):35–42. doi: 10.1038/nm.3028
24. Martin S, Parton RG. Lipid Droplets: A Unified View of a Dynamic Organelle. Nat Rev Mol Cell Biol (2006) 7(5):373–8. doi: 10.1038/nrm1912
25. Lecka-Czernik B. Marrow Fat Metabolism is Linked to the Systemic Energy Metabolism. Bone (2012) 50(2):534–9. doi: 10.1016/j.bone.2011.06.032
26. Rafii S, Shapiro F, Pettengell R, Ferris B, Nachman RL, Moore MA, et al. Human Bone Marrow Microvascular Endothelial Cells Support Long-Term Proliferation and Differentiation of Myeloid and Megakaryocytic Progenitors. Blood (1995) 86(9):3353–63. doi: 10.1182/blood.V86.9.3353.bloodjournal8693353
27. Chen X, Wang Z, Duan N, Zhu G, Schwarz EM, Xie C. Osteoblast-Osteoclast Interactions. Connect Tissue Res (2018) 59(2):99–107. doi: 10.1080/03008207.2017.1290085
28. Calvi LM. Osteolineage Cells and Regulation of the Hematopoietic Stem Cell. Best Pract Res Clin Haematol (2013) 26(3):249–52. doi: 10.1016/j.beha.2013.10.004
29. Mansour A, Abou-Ezzi G, Sitnicka E, Jacobsen SE, Wakkach A, Blin-Wakkach C. Osteoclasts Promote the Formation of Hematopoietic Stem Cell Niches in the Bone Marrow. J Exp Med (2012) 209(3):537–49. doi: 10.1084/jem.20110994
30. Ludin A, Gur-Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C, et al. Reactive Oxygen Species Regulate Hematopoietic Stem Cell Self-Renewal, Migration and Development, as Well as Their Bone Marrow Microenvironment. Antioxid Redox Signal (2014) 21(11):1605–19. doi: 10.1089/ars.2014.5941
31. Taniguchi Ishikawa E, Gonzalez-Nieto D, Ghiaur G, Dunn SK, Ficker AM, Murali B, et al. Connexin-43 Prevents Hematopoietic Stem Cell Senescence Through Transfer of Reactive Oxygen Species to Bone Marrow Stromal Cells. Proc Natl Acad Sci U S A (2012) 109(23):9071–6. doi: 10.1073/pnas.1120358109
32. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML–PPAR-δ Pathway for Fatty Acid Oxidation Regulates Hematopoietic Stem Cell Maintenance. Nat Med (2012) 18(9):1350–8. doi: 10.1038/nm.2882
33. Oburoglu L, Tardito S, Fritz V, de Barros SC, Merida P, Craveiro M, et al. Glucose and Glutamine Metabolism Regulate Human Hematopoietic Stem Cell Lineage Specification. Cell Stem Cell (2014) 15(2):169–84. doi: 10.1016/j.stem.2014.06.002
34. Taya Y, Ota Y, Wilkinson AC, Kanazawa A, Watarai H, Kasai M, et al. Depleting Dietary Valine Permits Nonmyeloablative Mouse Hematopoietic Stem Cell Transplantation. Science (2016) 354(6316):1152–5. doi: 10.1126/science.aag3145
35. Kinder M, Wei C, Shelat SG, Kundu M, Zhao L, Blair IA, et al. Hematopoietic Stem Cell Function Requires 12/15-Lipoxygenase-Dependent Fatty Acid Metabolism. Blood (2010) 115(24):5012–22. doi: 10.1182/blood-2009-09-243139
36. Li Y, Meng Y, Yu X. The Unique Metabolic Characteristics of Bone Marrow Adipose Tissue. Front Endocrinol (Lausanne) (2019) 10:69. doi: 10.3389/fendo.2019.00069
37. Suchacki KJ, Tavares AAS, Mattiucci D, Scheller EL, Papanastasiou G, Gray C, et al. Bone Marrow Adipose Tissue is a Unique Adipose Subtype With Distinct Roles in Glucose Homeostasis. Nat Commun (2020) 11(1):3097. doi: 10.1038/s41467-020-16878-2
38. Prabhakar M, Ershler WB, Longo DL. Bone Marrow, Thymus and Blood: Changes Across the Lifespan. Aging Health (2009) 5(3):385–93. doi: 10.2217/ahe.09.31
39. Cawthorn WP, Scheller EL, Learman BS, Parlee SD, Simon BR, Mori H, et al. Bone Marrow Adipose Tissue is an Endocrine Organ That Contributes to Increased Circulating Adiponectin During Caloric Restriction. Cell Metab (2014) 20(2):368–75. doi: 10.1016/j.cmet.2014.06.003
40. Scheller EL, Doucette CR, Learman BS, Cawthorn WP, Khandaker S, Schell B, et al. Region-Specific Variation in the Properties of Skeletal Adipocytes Reveals Regulated and Constitutive Marrow Adipose Tissues. Nat Commun (2015) 6:7808. doi: 10.1038/ncomms8808
41. Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, et al. Endocrine Regulation of Energy Metabolism by the Skeleton. Cell (2007) 130(3):456–69. doi: 10.1016/j.cell.2007.05.047
42. Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O, et al. Bone Marrow Adipocytes Promote the Regeneration of Stem Cells and Haematopoiesis by Secreting SCF. Nat Cell Biol (2017) 19(8):891–903. doi: 10.1038/ncb3570
43. Han J, Koh YJ, Moon HR, Ryoo HG, Cho CH, Kim I, et al. Adipose Tissue is an Extramedullary Reservoir for Functional Hematopoietic Stem and Progenitor Cells. Blood (2010) 115(5):957–64. doi: 10.1182/blood-2009-05-219923
44. Coburn CT, Hajri T, Ibrahimi A, Abumrad NA. Role of CD36 in Membrane Transport and Utilization of Long-Chain Fatty Acids by Different Tissues. J Mol Neurosci (2001) 16(2-3):117–21. doi: 10.1385/JMN:16:2-3:117
45. Storch J, Veerkamp JH, Hsu KT. Similar Mechanisms of Fatty Acid Transfer From Human Anal Rodent Fatty Acid-Binding Proteins to Membranes: Liver, Intestine, Heart Muscle, and Adipose Tissue FABPs. Mol Cell Biochem (2002) 239(1-2):25–33. doi: 10.1007/978-1-4419-9270-3_4
46. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty Acid Transport Protein 2 Reprograms Neutrophils in Cancer. Nature (2019) 569(7754):73–8. doi: 10.1038/s41586-019-1118-2
47. Girotra M, Monnard C, Konz T, Sizzano F, Goulet L, Godin JP, et al. Mineral and Amino Acid Profiling of Different Hematopoietic Populations From the Mouse Bone Marrow. Int J Mol Sci (2020) 21(17):6444. doi: 10.3390/ijms21176444
48. Martínez-Reyes I, Chandel NS. Mitochondrial TCA Cycle Metabolites Control Physiology and Disease. Nat Commun (2020) 11(1):102. doi: 10.1038/s41467-019-13668-3
49. Liu X, Zhang F, Zhang Y, Li X, Chen C, Zhou M, et al. PPM1K Regulates Hematopoiesis and Leukemogenesis Through CDC20-Mediated Ubiquitination of MEIS1 and P21. Cell Rep (2018) 23(5):1461–75. doi: 10.1016/j.celrep.2018.03.140
50. Warburg O. On the Origin of Cancer Cells. Science (1956) 123(3191):309–14. doi: 10.1126/science.123.3191.309
51. Suganuma K, Miwa H, Imai N, Shikami M, Gotou M, Goto M, et al. Energy Metabolism of Leukemia Cells: Glycolysis Versus Oxidative Phosphorylation. Leuk Lymphoma (2010) 51(11):2112–9. doi: 10.3109/10428194.2010.512966
52. Boultwood J, Fidler C, Mills KI, Frodsham PM, Kusec R, Gaiger A, et al. Amplification of Mitochondrial DNA in Acute Myeloid Leukaemia. Br J Haematol (1996) 95(2):426–31. doi: 10.1046/j.1365-2141.1996.d01-1922.x
53. Burt R, Dey A, Aref S, Aguiar M, Akarca A, Bailey K, et al. Activated Stromal Cells Transfer Mitochondria to Rescue Acute Lymphoblastic Leukemia Cells From Oxidative Stress. Blood (2019) 134(17):1415–29. doi: 10.1182/blood.2019001398
54. Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, et al. Preferential Transfer of Mitochondria From Endothelial to Cancer Cells Through Tunneling Nanotubes Modulates Chemoresistance. J Transl Med (2013) 11:94. doi: 10.1186/1479-5876-11-94
55. Polak R, de Rooij B, Pieters R, den Boer ML. B-Cell Precursor Acute Lymphoblastic Leukemia Cells Use Tunneling Nanotubes to Orchestrate Their Microenvironment. Blood (2015) 126(21):2404–14. doi: 10.1182/blood-2015-03-634238
56. Mistry JJ, Moore JA, Kumar P, Marlein CR, Hellmich C, Pillinger G, et al. Daratumumab Inhibits Acute Myeloid Leukaemia Metabolic Capacity by Blocking Mitochondrial Transfer From Mesenchymal Stromal Cells. Haematologica (2021) 106(2):589–92. doi: 10.3324/haematol.2019.242974
57. Gandolfi S, Prada CP, Richardson PG. How I Treat the Young Patient With Multiple Myeloma. Blood (2018) 132(11):1114–24. doi: 10.1182/blood-2017-05-693606
58. Farber M, Chen Y, Arnold L, Möllmann M, Boog-Whiteside E, Lin YA, et al. Targeting CD38 in Acute Myeloid Leukemia Interferes With Leukemia Trafficking and Induces Phagocytosis. Sci Rep (2021) 11(1):22062. doi: 10.1038/s41598-021-01300-8
59. Naik J, Themeli M, de Jong-Korlaar R, Ruiter RWJ, Poddighe PJ, Yuan H, et al. CD38 as a Therapeutic Target for Adult Acute Myeloid Leukemia and T-Cell Acute Lymphoblastic Leukemia. Haematologica (2019) 104(3):e100–e3. doi: 10.3324/haematol.2018.192757
60. Eliasson P, Rehn M, Hammar P, Larsson P, Sirenko O, Flippin LA, et al. Hypoxia Mediates Low Cell-Cycle Activity and Increases the Proportion of Long-Term-Reconstituting Hematopoietic Stem Cells During In Vitro Culture. Exp Hematol (2010) 38(4):301–10.e2. doi: 10.1016/j.exphem.2010.01.005
61. Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, et al. Foxo3a is Essential for Maintenance of the Hematopoietic Stem Cell Pool. Cell Stem Cell (2007) 1(1):101–12. doi: 10.1016/j.stem.2007.02.001
62. Sillar JR, Germon ZP, DeIuliis GN, Dun MD. The Role of Reactive Oxygen Species in Acute Myeloid Leukaemia. Int J Mol Sci (2019) 20(23):6003. doi: 10.3390/ijms20236003
63. Staudt D, Murray HC, McLachlan T, Alvaro F, Enjeti AK, Verrills NM, et al. Targeting Oncogenic Signaling in Mutant FLT3 Acute Myeloid Leukemia: The Path to Least Resistance. Int J Mol Sci (2018) 19(10):3198. doi: 10.3390/ijms19103198
64. Sallmyr A, Fan J, Datta K, Kim KT, Grosu D, Shapiro P, et al. Internal Tandem Duplication of FLT3 (FLT3/ITD) Induces Increased ROS Production, DNA Damage, and Misrepair: Implications for Poor Prognosis in AML. Blood (2008) 111(6):3173–82. doi: 10.1182/blood-2007-05-092510
65. Stanicka J, Russell EG, Woolley JF, Cotter TG. NADPH Oxidase-Generated Hydrogen Peroxide Induces DNA Damage in Mutant FLT3-Expressing Leukemia Cells. J Biol Chem (2015) 290(15):9348–61. doi: 10.1074/jbc.M113.510495
66. Abdul-Aziz A, MacEwan DJ, Bowles KM, Rushworth SA. Oxidative Stress Responses and NRF2 in Human Leukaemia. Oxid Med Cell Longev (2015) 2015:454659. doi: 10.1155/2015/454659
67. Heasman SA, Zaitseva L, Bowles KM, Rushworth SA, Macewan DJ. Protection of Acute Myeloid Leukaemia Cells From Apoptosis Induced by Front-Line Chemotherapeutics is Mediated by Haem Oxygenase-1. Oncotarget (2011) 2(9):658–68. doi: 10.18632/oncotarget.321
68. Rassool FV, Gaymes TJ, Omidvar N, Brady N, Beurlet S, Pla M, et al. Reactive Oxygen Species, DNA Damage, and Error-Prone Repair: A Model for Genomic Instability With Progression in Myeloid Leukemia? Cancer Res (2007) 67(18):8762–71. doi: 10.1158/0008-5472.CAN-06-4807
69. Sallmyr A, Fan J, Rassool FV. Genomic Instability in Myeloid Malignancies: Increased Reactive Oxygen Species (ROS), DNA Double Strand Breaks (DSBs) and Error-Prone Repair. Cancer Lett (2008) 270(1):1–9. doi: 10.1016/j.canlet.2008.03.036
70. Moore JA, Mistry JJ, Hellmich C, Horton RH, Wojtowicz EE, Jibril A, et al. LC3-Associated Phagocytosis in Bone Marrow Macrophages Suppresses Acute Myeloid Leukemia Progression Through STING Activation. J Clin Invest (2022) 132(5):e153157. doi: 10.1172/JCI153157
71. Moore SG, Dawson KL. Red and Yellow Marrow in the Femur: Age-Related Changes in Appearance at MR Imaging. Radiology (1990) 175(1):219–23. doi: 10.1148/radiology.175.1.2315484
72. Pulikkottil AJ, Bamezai S, Ammer T, Mohr F, Feder K, Vegi NM, et al. TET3 Promotes AML Growth and Epigenetically Regulates Glucose Metabolism and Leukemic Stem Cell Associated Pathways. Leukemia (2022) 36(2):416–25. doi: 10.1038/s41375-021-01390-3
73. Åbacka H, Hansen JS, Huang P, Venskutonytė R, Hyrenius-Wittsten A, Poli G, et al. Targeting GLUT1 in Acute Myeloid Leukemia to Overcome Cytarabine Resistance. Haematologica (2021) 106(4):1163–6. doi: 10.3324/haematol.2020.246843
74. Chen Y, Feng Z, Kuang X, Zhao P, Chen B, Fang Q, et al. Increased Lactate in AML Blasts Upregulates TOX Expression, Leading to Exhaustion of CD8. Am J Cancer Res (2021) 11(11):5726–42.
75. Saulle E, Spinello I, Quaranta MT, Pasquini L, Pelosi E, Iorio E, et al. Targeting Lactate Metabolism by Inhibiting MCT1 or MCT4 Impairs Leukemic Cell Proliferation, Induces Two Different Related Death-Pathways and Increases Chemotherapeutic Sensitivity of Acute Myeloid Leukemia Cells. Front Oncol (2020) 10:621458. doi: 10.3389/fonc.2020.621458
76. Ni F, Yu WM, Li Z, Graham DK, Jin L, Kang S, et al. Critical Role of ASCT2-Mediated Amino Acid Metabolism in Promoting Leukaemia Development and Progression. Nat Metab (2019) 1(3):390–403. doi: 10.1038/s42255-019-0039-6
77. Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M, et al. Inhibiting Glutamine Uptake Represents an Attractive New Strategy for Treating Acute Myeloid Leukemia. Blood (2013) 122(20):3521–32. doi: 10.1182/blood-2013-03-493163
78. Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, et al. Tissue of Origin Dictates Branched-Chain Amino Acid Metabolism in Mutant Kras-Driven Cancers. Science (2016) 353(6304):1161–5. doi: 10.1126/science.aaf5171
79. Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, et al. Direct Evidence for Cancer-Cell-Autonomous Extracellular Protein Catabolism in Pancreatic Tumors. Nat Med (2017) 23(2):235–41. doi: 10.1038/nm.4256
80. Wei Z, Liu X, Cheng C, Yu W, Yi P. Metabolism of Amino Acids in Cancer. Front Cell Dev Biol (2020) 8:603837. doi: 10.3389/fcell.2020.603837
81. Yang L, Venneti S, Nagrath D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu Rev BioMed Eng (2017) 19:163–94. doi: 10.1146/annurev-bioeng-071516-044546
82. Bott AJ, Peng IC, Fan Y, Faubert B, Zhao L, Li J, et al. Oncogenic Myc Induces Expression of Glutamine Synthetase Through Promoter Demethylation. Cell Metab (2015) 22(6):1068–77. doi: 10.1016/j.cmet.2015.09.025
83. Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, et al. Venetoclax With Azacitidine Disrupts Energy Metabolism and Targets Leukemia Stem Cells in Patients With Acute Myeloid Leukemia. Nat Med (2018) 24(12):1859–66. doi: 10.1038/s41591-018-0233-1
84. Shafat MS, Oellerich T, Mohr S, Robinson SD, Edwards DR, Marlein CR, et al. Leukemic Blasts Program Bone Marrow Adipocytes to Generate a Protumoral Microenvironment. Blood (2017) 129(10):1320–32. doi: 10.1182/blood-2016-08-734798
85. Marlein CR. Bone Marrow. Chichester: John Wiley & Sons, Ltd (2018). doi: 10.1002/9780470015902.a0000505.pub2
86. Juliusson G, Antunovic P, Derolf A, Lehmann S, Möllgård L, Stockelberg D, et al. Age and Acute Myeloid Leukemia: Real World Data on Decision to Treat and Outcomes From the Swedish Acute Leukemia Registry. Blood (2009) 113(18):4179–87. doi: 10.1182/blood-2008-07-172007
87. Miraki-Moud F, Anjos-Afonso F, Hodby KA, Griessinger E, Rosignoli G, Lillington D, et al. Acute Myeloid Leukemia Does Not Deplete Normal Hematopoietic Stem Cells But Induces Cytopenias by Impeding Their Differentiation. Proc Natl Acad Sci U S A (2013) 110(33):13576–81. doi: 10.1073/pnas.1301891110
Keywords: acute myeloid leukaemia, bone marrow niche, metabolism, adipocytes, free fattty acids
Citation: Maynard RS, Hellmich C, Bowles KM and Rushworth SA (2022) Acute Myeloid Leukaemia Drives Metabolic Changes in the Bone Marrow Niche. Front. Oncol. 12:924567. doi: 10.3389/fonc.2022.924567
Received: 20 April 2022; Accepted: 03 June 2022;
Published: 29 June 2022.
Edited by:
Manja Wobus, Technical University Dresden, GermanyReviewed by:
Anju Kumari, National Cancer Institute (NIH), United StatesCopyright © 2022 Maynard, Hellmich, Bowles and Rushworth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stuart A. Rushworth, cy5ydXNod29ydGhAdWVhLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.