
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 01 August 2022
Sec. Pediatric Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.922928
This article is part of the Research TopicDevelopmental Therapeutics in Pediatric Neuro-oncologyView all 7 articles
 Kallen Schwark1
Kallen Schwark1 Dana Messinger1
Dana Messinger1 Jessica R. Cummings1Joshua Bradin1
Jessica R. Cummings1Joshua Bradin1 Abed Kawakibi1
Abed Kawakibi1 Clarissa M. Babila1Samantha Lyons1Sunjong Ji1Rodrigo T. Cartaxo1Seongbae Kong1Evan Cantor1
Clarissa M. Babila1Samantha Lyons1Sunjong Ji1Rodrigo T. Cartaxo1Seongbae Kong1Evan Cantor1 Carl Koschmann1
Carl Koschmann1 Viveka Nand Yadav1,2,3*
Viveka Nand Yadav1,2,3*Pediatric high-grade glioma (pHGG), including both diffuse midline glioma (DMG) and non-midline tumors, continues to be one of the deadliest oncologic diagnoses (both henceforth referred to as “pHGG”). Targeted therapy options aimed at key oncogenic receptor tyrosine kinase (RTK) drivers using small-molecule RTK inhibitors has been extensively studied, but the absence of proper in vivo modeling that recapitulate pHGG biology has historically been a research challenge. Thankfully, there have been many recent advances in animal modeling, including Cre-inducible transgenic models, as well as intra-uterine electroporation (IUE) models, which closely recapitulate the salient features of human pHGG tumors. Over 20% of pHGG have been found in sequencing studies to have alterations in platelet derived growth factor-alpha (PDGFRA), making growth factor modeling and inhibition via targeted tyrosine kinases a rich vein of interest. With commonly found alterations in other growth factors, including FGFR, EGFR, VEGFR as well as RET, MET, and ALK, it is necessary to model those receptors, as well. Here we review the recent advances in murine modeling and precision targeting of the most important RTKs in their clinical context. We additionally provide a review of current work in the field with several small molecule RTK inhibitors used in pre-clinical or clinical settings for treatment of pHGG.
Pediatric high-grade gliomas (pHGGs) are one of the leading causes of cancer-related death in children. pHGGs are further divided into multiple subgroups based on tumor location and mutation status. pHGGs encompass both hemispheric (non-midline) and diffuse midline gliomas (DMGs) and represent the most lethal pediatric brain tumors. Radiation remains the only proven therapy to extend survival to date and less than 20% of patients survive for five years past diagnosis (1). pHGG was identified as early as 1926; for several decades, its biological behavior was believed to be like adult high-grade gliomas and was treated with similar therapeutic regimens (2). These treatments failed to improve patient survival, which led researchers to investigate if pHGG has a biologically distinct phenotype from adult gliomas (3).
To combat the dismal prognosis for these patients, molecular drivers for this tumor subset are being investigated using sequencing and preclinical models. Several groups used high-throughput whole-genome, whole-exome, and transcriptome sequencing to characterize mutations in pediatric gliomas and differences from adult gliomas (4–6). Wu et al. identified that ~80% DMG contain a somatic point mutation in the H3K27M gene and 16% of cortical pHGG contain H3.3 G34R/V mutations (7). H3K27M mutations occur frequently in H3F3A and to a lesser extent in H3C2 encoding H3.1 (25% of cases), while H3.3G34R/V mutations occur exclusively in H3F3A. These onco-histone mutations in pHGG have been recognized as examples of key genetic drivers that play an integral role in tumor development (8–10). Pediatric gliomas have been found to have fewer drivers than their adult counterparts, which suggests that identifying the drivers that do exist via sequencing will lead to effective strategies for precision therapeutics (11).
Apart from histone mutations, the most common alterations in pHGG are overexpression of receptor tyrosine kinase (RTK) family members. Paugh et al. showed that most pHGGs demonstrated amplification of platelet-derived growth factor receptor A (PDGFRA)-driven signal, while adult HGG shows EGFR overexpression (6). Amplification of PDGFRA commonly leads to activation of the PI3K/mTOR or MAPK signaling pathways in pHGG (3, 12, 13). Alterations in PDGFRA are significant, as they are associated with worse prognoses (14). Many tyrosine kinase inhibitors (TKIs) have been developed as targeted therapies for cancers with similar RTK alterations, and research on their efficacy in pHGG is ongoing. A schematic of RTKs and TKIs that have been trialed in adult or pediatric brain tumors is shown in Figure 1.
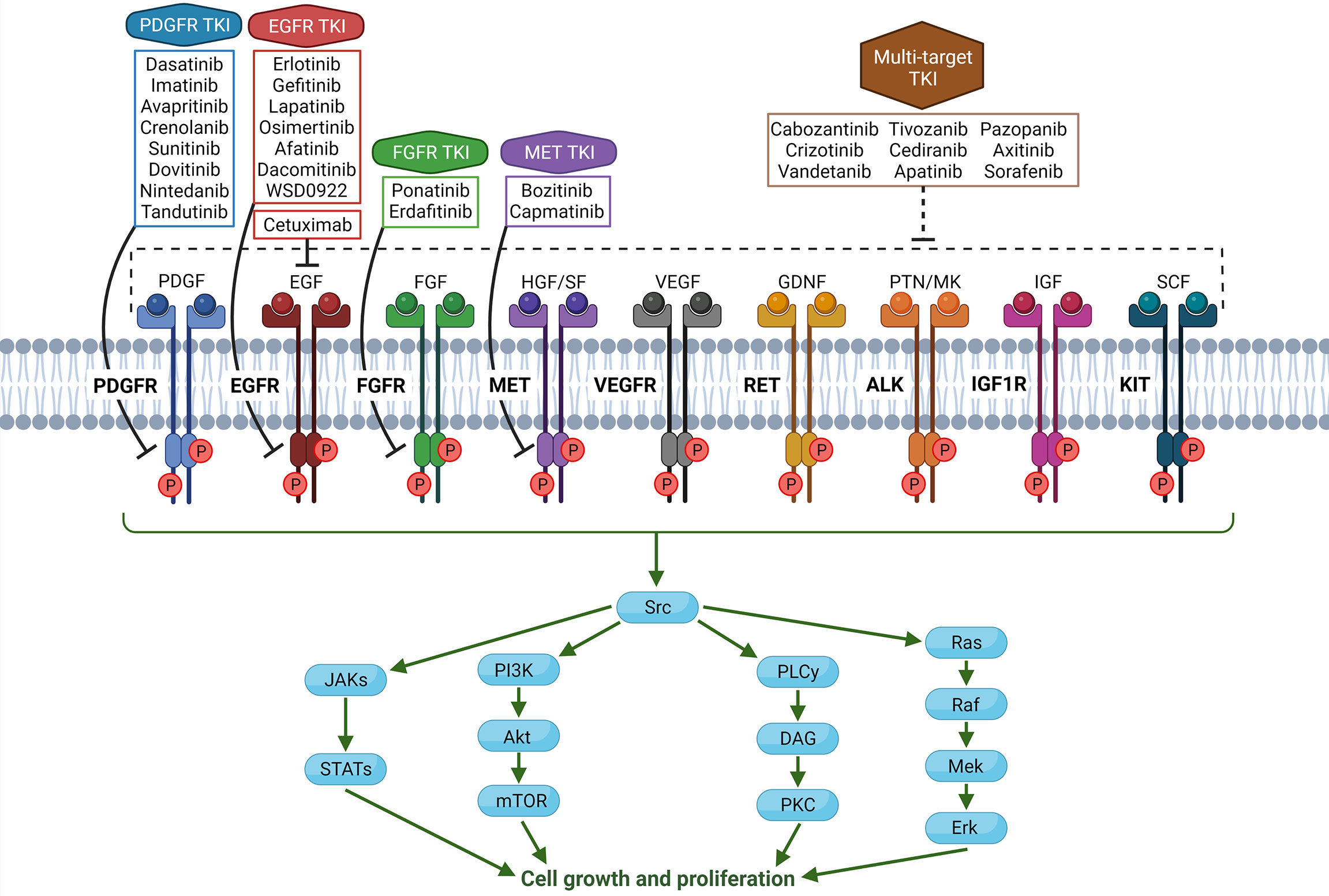
Figure 1 Schema of common and targetable receptor tyrosine kinases (RTKs) in the human body, along with associated tyrosine kinase inhibitors (TKIs). In glioma cells, overexpression, mutation, or amplification of RTKs can lead to tumorigenic phenotype. Note that all TKIs have varying degrees of off-target effects on other RTKs or kinases in the cell, and TKIs being specific to one receptor is an oversimplification. (Created with Biorender.com).
Understanding molecular mechanisms of specific tyrosine kinase alterations and their therapeutic impact on pHGG tumorigenesis warrants accurate genetic mouse models, which can recapitulate the salient features of pHGG for precise preclinical testing. Multiple models have been developed for the identification of potential therapeutics against pHGG, such as cell line xenografts and genetically engineered mouse models (15). Using these models, therapies that target these alterations can be developed and potentially lead to effective treatment strategies for pediatric gliomas. In this review, we outline our current understanding of RTK alterations in pediatric high-grade gliomas, as well as the status of their corresponding preclinical models and possible treatment methods. Illustrative description of all preclinical models of pHGG are depicted in Figure 2.
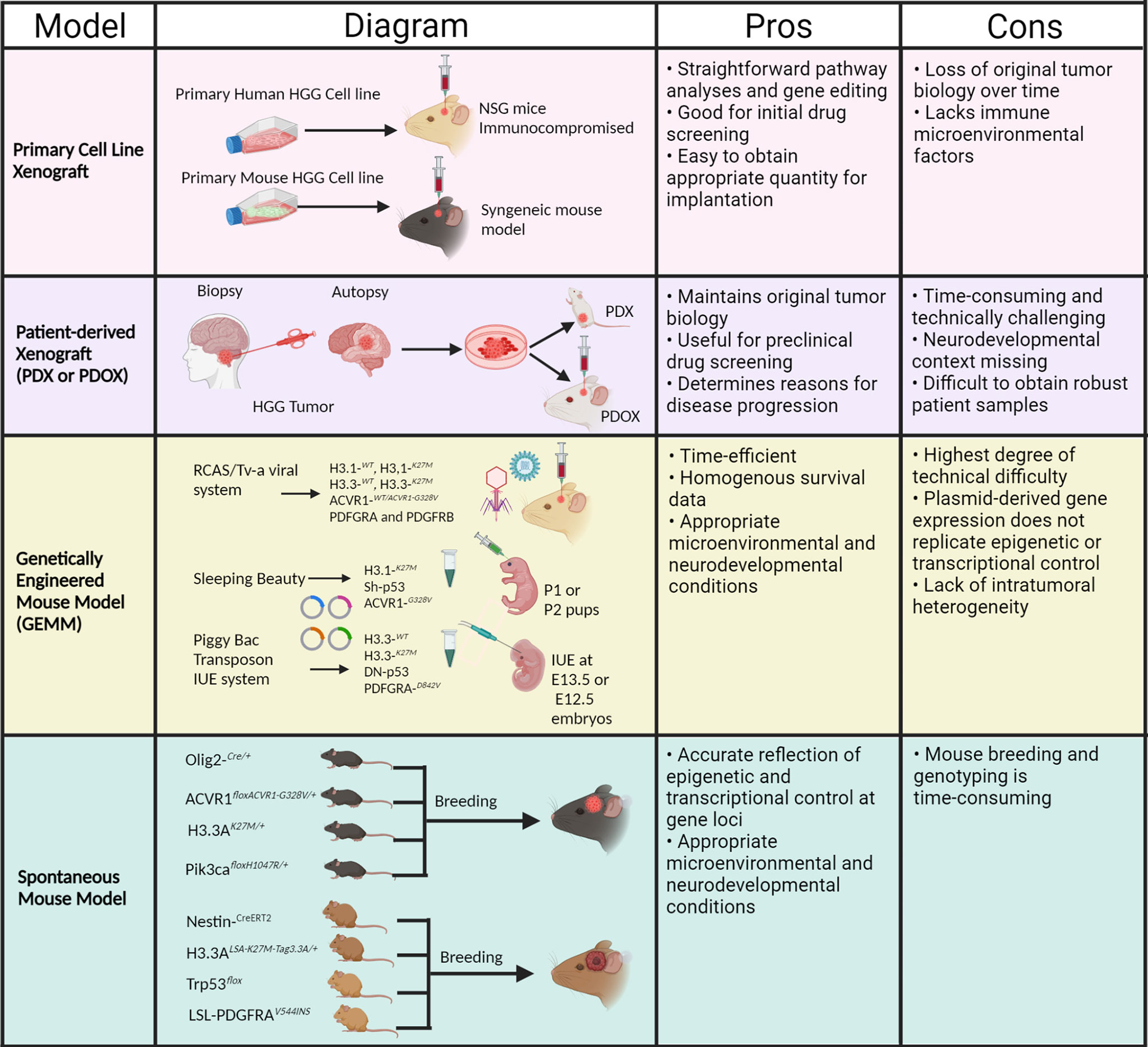
Figure 2 Schema of preclinical mouse models and associated pros and cons for pHGG. (Created with Biorender.com).
RTK pathway alterations are common in pediatric glioma. Wu et al. sequenced 127 pediatric HGGs and found RTK-RAS-PI3K signaling pathway alterations in 69% of diffuse intrinsic pontine gliomas (DIPGs) and 67% of non-brainstem-HGGs (NBS-HGGs) (7). PDGFRA alterations were found in 20% of DIPGs and 21% of NBS-HGGs, and EGFR alterations were found in 10% of NBS-HGGs (none in DIPGs). These alterations were mostly amplifications, with some indel mutations, missense mutations, and structural variants. Mackay et al. obtained copy number profiles from 834 cases of pHGGs and found 77 PDGFRA amplifications (9%), 32 EGFR amplifications (4%), and 19 MET amplifications (2%). They also analyzed sequencing data from 326 pHGG samples and found 19 PDGFRA mutations (6%), 11 EGFR mutations (3%), 12 FGFR1 or FGFR2 mutations (4%), 3 MET mutations (1%), and 1 IGF1R mutation (0.3%) (16). Guerriero Stucklin et al. performed a large-scale analysis of infantile hemispheric glioma and identified one subgroup of tumors that harbored alterations in ROS1 (21%), NTRK (21%), ALK (15%), and MET (6%) (17).
These alterations are significant because they change patient prognosis. In integrated sequencing datasets from 290 pediatric HGG patients, PDGFRA amplification significantly reduced overall survival in non-brainstem HGG when compared to patients without the amplification (14). Recently, Mondal et al. found pediatric bithalamic midline glial tumors that lacked the H3K27M mutation but harbored frequent insertion mutations in exon 20 or frameshift mutations in exon 7 of the EGFR oncogene (mutations - 11/13, 85%; amplifications – 1/13, 7.7%) that confer sensitivity to specific TKIs (osimertinib and afatinib) in vitro (13). Soon after, DNA methylation profiling was done to identify a group of pHGGs that were epigenetically similar to five of the bithalamic gliomas from Mondal et al.; this group was mostly composed of midline gliomas and frequently carried EGFR alterations (mutations – 20/30, 67%; amplifications – 16/58, 27%) (18). This contrasts with non-midline cortical pHGG, which do not commonly carry EGFR alterations (7); and is similar to adult glioblastoma (GBM), with as many as 60% of GBMs carrying EGFR amplification (19). With the optimization of patient-specific molecular profiling for high-grade brain tumors like pHGG, personalized therapeutics will be critical for effective treatments. To this end, developing personalized animal models with specific genetic lesions present in pHGG will be an important step in optimizing therapeutics in a preclinical setting.
Because PDGFRA is frequently amplified or mutated in pediatric gliomas, there is a need to replicate different PDGFRA variant types found in pHGG via modeling (6, 20). Paugh et al. introduced six PDGFRA activating mutants affecting different parts of the receptor, along with wildtype, into neural precursor cells (NPCs) cultured in vitro using retroviral constructs. They observed that PDGFRA mutants are constitutively active and have a proliferative advantage over empty vector controls in TP53-null primary mouse astrocytes (PMAs) (21). Further, Funato et al. showed that expression of PDGFRA-D842V synergizes with H3.3K27M and TP53 loss in NPCs, resulting in de-differentiation and neoplastic transformation (22).
In vitro PDGFRA-amplified models have also been generated through overexpressing wild-type PDGFRA. Pathania et al. found that in vitro cells from murine gliomas harboring H3.3K27M, TP53 knockdown, and PDGFRA-WT overexpression were more invasive than isogenic cells without induced PDGFRA amplification (21). Cells with PDGFRA-WT overexpression did not display receptor activation when in a ligand-free setting; however, when treated with PDGF-AA ligand, these cells had increased receptor activation to even higher levels than PDGFRA mutant cells. This suggested a possible increase in ligand dependence in PDGFRA-amplified variants compared to mutated variants (21).
Patient-derived xenograft (PDX) models provide a mechanism to study biologically relevant tumors in an in vivo setting. Coupled with anatomically relevant sites and tumors that maintain their original biology, PDX models can accurately recapitulate HGG and provide insight into the factors behind disease progression (23). These models are particularly valuable for preclinical screening of therapeutic agents and may be useful as a prognostic factor for many cancers (24).
Historically, because pHGG was typically diagnosed through features observed on imaging studies rather than surgical biopsies, there was a deficiency of the readily available tissue resources often used for preclinical therapeutic testing (25). In addition, growing tumors in the appropriate location in vivo has been challenging. Advances in xenograft technology have since allowed the establishment of anatomically accurate mouse models that have been used to examine patterns of growth and response to novel therapeutic agents in pHGG. These human derived glioma models retain the invasive patterns of growth and the morphological and pathological characteristics of the parental tumor (26). Furthermore, Brabetz et al. showed that PDX models show a differential response to therapy based on molecular drivers such as EGFR amplification (27).
Recently, He et al. established 21 PDX models for pHGG and verified their accurate recapitulation of tumor histopathology, mutations, and gene expression patterns. They tested a panel of 1134 FDA-approved drugs, including TKIs such as afatinib, ponatinib, and dasatinib, in pHGG cell lines derived directly from the xenograft models (28). Ultimately, they investigated dual therapy with PI3K/mTOR and MEK inhibitors, finding evidence of synergistic growth inhibition in vitro and significant survival extension in vivo. In summary, patient-derived xenografts allow for improved preclinical testing of new therapeutic targets for pHGG in a tumor- and organ-specific manner (28). However, PDX models do not recreate the brain developmental context and the impact of the immune microenvironment on tumor growth, leading the field to investigate genetically engineered models.
The replication-competent avian sarcoma-leucosis virus (RCAS)-TVA system is a genetically engineered mouse model that can be used to create tumors containing specific drivers of gliomagenesis, such as PDGF. The virus is modified to contain the genes to be inserted into the host genome, and TVA is the receptor for viral entry. Brain cells are specifically engineered to express the TVA receptor, allowing targeted gene transfer (29).
Using this model, Hambardzumyan et al. showed that Ntv-a mice overexpressing PDGFB can form tumors in multiple brain regions but form higher-grade tumors with increased efficiency when the mice also lack the Ink4a-ARF tumor suppressors (30). Barton et al. used the RCAS PDGF-B; Ink4a-ARF deficient mouse model to test the effects of the CDK4/6 inhibitor PD-0332991 (PD) on brainstem gliomas (31). More recently, Hoeman et al. introduced ACVR1 mutations into brainstem progenitor cells via RCAS and determined the R206H mutation works alongside H3.1K27M in promoting gliomagenesis (32). Finally, Halvorson et al. used cells derived from their RCAS mouse models for pHGG and identified the TKI BMS-754807 as a potent inhibitor of proliferation in tumor-derived cells (33). In summary, the RCAS-TVA system is a reliable method for creating pHGG models with specific tumor drivers and can be used for testing therapeutic efficacy of TKIs.
The Sleeping Beauty (SB) model is a DNA transposon system that has been used to induce HGGs in mice with specific genetic lesions. In this system, the SB transposase recognizes inverted repeat and direct repeat (IR/DR) sequences flanking the transposons and facilitates their stable integration into the host genome via a “cut and paste” mechanism. The SB method involves transfecting subventricular zone NPCs into 1-day-old mouse pups by injecting a combination of SB DNA transposon plasmids bearing mutations specific to pHGG. Different combinations of these plasmids have been used to generate highly penetrant gliomas using the SB model, including common mutations seen in HGG such as mutant H3.3K27M, ATRX, ACVR1-G328V, and IDH1, along with tumor drivers NRAS and shRNA KO TP53 (shp53) (34–36).
The SB model has been recently used to study the impact of the tumor microenvironment, specifically the role of myeloid cell immunosuppression on mutant IDH1 tumor growth (37). Recently, Mendez et al. used this model to develop a murine brainstem pHGG tumor using H3K27M and ACVR1-G328V genetic alterations, along with NRAS and shp53 as oncogenic drivers (38). SB has also been used to study RTK activity in gliomas; Qin et al. injected SB plasmids with HGF and MET cDNA as well as shRNA against p53 into lateral ventricles of neonatal mice and saw MET-driven glioma development (39). They tested two MET inhibitors, V-4084 and SGX523, and found sensitivity to both in isolated neurosphere cell lines. The SB model tumors arise de novo and recapitulate the human disease at both histological and molecular levels, providing a venue for testing targeted therapies in tumors with specific RTK drivers.
Studying the molecular role of growth factors (e.g., PDGFRA) in pHGG pathogenesis warrants accurate genetic and epigenetic models that retain features of pHGG. Recently, the in-utero electroporation (IUE) method has been used to generate pHGG models through transfecting NPCs of prenatal mice on embryonic day 12.5-13.5 (E12.5-13.5) using PiggyBac DNA transposon plasmids bearing characteristic mutations of pHGG (40, 41). This model reproduces the neurodevelopmental attributes of pHGGs in younger patients (i.e., tumor formation in a developing brain) and generates infiltrative de novo tumors within the developing mouse brain that are histologically consistent with human pHGG. Varying combinations of these plasmids have been used to generate highly penetrant gliomas throughout the evolution of this model, including mutations common in pHGG such as mutant H3.3K27M, ATRX, PDGFRA-D842V, and TP53.
Naturally, the IUE method requires significant precision in timing and gene specificity. Pathania et al. targeted NPCs in the lower rhombic lip of the developing hindbrain of E12.5 mice and found that H3.3K27M mutation and TP53 loss are sufficient to induce diffuse tumors characteristic of human pHGG in both hindbrain and forebrain regions after 6-8 months. In addition to these genes, they determined that including ATRX knockdown via shRNA resulted in more circumscribed tumors after 4 months, and further addition of PDGFRA to the IUE model shortened the period of tumor growth to 21 days (21). In contrast, H3.3K27M and TP53 loss did not result in tumorigenesis in a later study performed by Patel et al. at E13.5 in the hindbrain, suggesting an important developmental window between E12-14 for tumorigenesis with this specific combination. They also found that PDGFRA-D842V activating mutation and TP53 loss were able to cause malignant transformation without H3.3K27M mutation (40).
Combinations of PDGFRA-D842V, PDGFRA-WT, or PDGFB along with DNp53 and H3.3K27M have resulted in tumors with varying histological and molecular features seen in brainstem pHGG, suggesting that the IUE method can be used to learn about the pathological heterogeneity associated with oncogenic drivers of pHGG. Patel et al. found that PDGFB-induced tumors formed rapidly and displayed vascular remodeling and angiogenesis, while PDGFRA-D842V-induced tumors were highly invasive with minimal vascular abnormalities (40). Integration of PDGFRA-WT produced both high- and low-grade gliomas with extended latencies.
Recently, our group used IUE to generate murine pHGG models with plasmids expressing dnTP53, H3.3K27M and PDGFRA-D842V mutations, leading to highly invasive HGGs in the forebrains of mice (41). Total upregulation of PDGFRA was confirmed via IHC, and this pathway was targeted with co-administration of the TKI dasatinib and mTOR inhibitor everolimus. Co-treatment significantly extended the survival of tumor-bearing mice beyond that of dasatinib treatment, providing evidence for a potential improvement in targeted therapy for PDGFRA-altered tumors. As seen from these results, the IUE model can be instrumental in providing an effective model that closely mimics human tumor development for targeting RTKs in pHGG (41).
Genetically engineering a mouse model that endogenously expresses an activating mutation in PDGFRA helps define the role this mutation plays in spontaneous tumorigenesis. Specifically, use of a Cre-inducible system allows the effects of mutant PDGFRA to be studied solely in the brain (42). The LSL-PDGFRA-V544ins transgenic mouse model expresses a heterozygous duplication in a short segment of the transmembrane domain and is induced in mice shortly after birth to model pediatric disease. When combined with a TP53 knockout mutation (TP53 cKO), mice developed HGG brainstem tumors that closely mimicked traits of human DIPG (brain infiltration, variable astrocyte differentiation, nuclear staining of Olig2); in addition, the cells robustly expressed PDGFRA in the cytoplasm (42). These data indicate that PDGFRA activating mutations alone are not enough to drive tumorigenesis but will cooperate with knockout of TP53 to direct formation of HGG within the brainstem, significantly decreasing survival.
Similarly, Fortin et al. developed spontaneous midbrain and thalamic HGG models by knocking in H3.1K27M, ACVR1-G328V, and PIK3CA-H1047R. The respective endogenous loci were driven by Cre recombinase in Olig2-positive oligodendrocyte precursor cells (OPCs) (43). Zou et al. used Cre recombinase to knock in mutant PDGFRA combined with Ink4a/ARF -/- in OPCs of mice to cause development of brain tumors resembling anaplastic human gliomas, emphasizing the importance of PDGFRA as an early driver of malignant transformation of OPCs (44). This and similar models made with Cre recombinase would provide an accurate model for testing the therapeutic effects of TKIs in RTK-altered pHGG.
With the recent advances in surgical techniques, several groups have developed many pHGG cell lines using human tumor cells directly from biopsy and autopsy samples. These cell lines are very effective for in vitro studies, including drug screening, molecular pathway analysis, and specific gene editing. However, one major caveat is the fidelity of these cell lines compared to the actual tumor tissue. When cells are continuously passaged in artificial media, extensive clonal selection with high mutation frequency occurs. Recently, Filbin et al. compared the gene profiling of DIPG cell lines with the original corresponding tumor using single cell RNA sequencing analysis and found stark differences in expression (45). Of their models tested, tumor maintenance via PDX in mice was most accurate to the original tumor biology, followed by primary sphere cell lines, and the least accurate were adherent glioma cell line models.
Unlike xenografts from cell lines, PDXs have shown strong genetic and transcriptomic correlation with the original tumor when characterized, as well as retained histological features of the original tumor when implanted into immunodeficient mice (27, 46). However, PDX models are technically challenging and can take up to a year to develop tumors. Furthermore, both cell line and patient-derived xenografts can only be established in immunodeficient mice (e.g. athymic, nude, NSG models), which lack both innate and adaptive immune cells. This results in a lack of accuracy in modeling the human tumor microenvironment. Syngeneic mouse models are an alternative strategy, which use mouse tumor cell lines in immunocompetent mice (47); however, these are genetically distinct from human cancers. Though cell line and patient-derived xenografts are limited in reproducing pHGG biology, their high feasibility complements the sophistication of GEM models and allows efficient investigation of CNS penetration and pharmacodynamics of targeted therapies.
GEM models such as RCAS/TVA, SB, and IUE are very useful for recognizing the molecular events responsible for tumor initiation and growth. While they are technically more complex than PDX, mastery of GEM model generation can produce more time-efficient and more homogeneous survival data than xenografts. Within GEM models, spontaneous generation of tumors using the endogenous loci of the genes better reflects human epigenetic and transcriptional control when compared to overexpression of genes via plasmids as in IUE (42). GEM models can provide underlying molecular events during tumor growth in response to specific mutations, as well as allowing the role of the microenvironment to be deciphered (48). However, it is not clear whether the gene changes involved in these models truly mirror the tumor-associated events in human pHGG. GEM tumors are usually composed of cells with specific and homogeneous genetic changes and therefore cannot capture the intratumoral genomic and phenotypic heterogeneity of gliomas. These GEMMs require exogenous promoters to provide upregulated oncogenes, and they determine the geography of malignant transformation through deciding the specific location of plasmid injection/electroporation, both of which illustrate constraints of these models. However, these limitations have largely been addressed with the advent of new advances in germline knock-in mouse models. In neonatal nestin-positive cells throughout the developing murine brain, the mutation H3.3K27M has been knocked in to the endogenous H3F3A locus, in combination with TP53 loss and the constitutively active PDGFRAV544ins mutant, driven by a tamoxifen-inducible Cre recombinase. This model led to spontaneous malignant brain tumor formation, with H3.3K27M driving the hindbrain specificity of tumorigenesis, and PDGFRA signaling driving pHGG identity.
As previously stated, PDGFRA alteration has been found more frequently in pediatric HGG compared to adults and is associated with higher pro-tumorigenic potential and worse prognosis (14, 49). Chronic PDGF signaling was found to promote glioma formation in cell culture regardless of mutation profile (3, 50). Therefore, PDGFR pathway alterations in pediatric HGG provide a promising target for PDGFR inhibitors. In addition, Filbin et al. demonstrated that H3K27M-mutant OPC-like cells overexpress PDGFRA, potentially opening the door to targeting H3K27M with PDGFR inhibitors (45).
Treatment of thalamic HGG tumor cell cultures from a two-year-old patient with PDGFRA amplification with multiple tyrosine kinase inhibitors demonstrated that dasatinib is the most potent inhibitor of cell growth with nanomolar IC50 (14). Dasatinib was less effective in inhibiting proliferation in a pediatric cell culture that had no growth factor receptor amplifications (micromolar IC50). Although a phase II trial found insufficient activity in adult GBM at maximal tolerated dose (51), it is a promising agent in pediatric HGG with PDGF pathway alterations. Dasatinib is well tolerated orally, has moderate blood-brain penetration (52), and demonstrates improved inhibition of PDGFR signaling compared to previous generations of tyrosine kinase inhibitors.
Dasatinib therapy can also synergize with other compounds, such as the mTOR inhibitor everolimus. Although everolimus was being investigated for its blockade of P-gp, we found that this function required micromolar concentrations (too high for clinical use), whereas nanomolar levels of everolimus were able to synergize with dasatinib in vitro and prolong survival in vivo (41). Additionally, our group published data for four recurrent pHGG patients treated with dasatinib and everolimus that resulted in median progression-free survival (PFS) of 3 months and overall survival (OS) of 8.5 months; therapy was well-tolerated and did not require dose reduction for any patient. These factors indicate that further investigations of dasatinib’s clinical utility as a single agent or combination therapy in PDGFRA-altered tumors is warranted (14).
Avapritinib is a PDGFRA inhibitor that targets activation loop mutations (e.g. PDGFRA D842V) and is more specific to PDGFRA and KIT than dasatinib (53). Due to its high CNS penetration, avapritinib is a promising option for pHGG therapy and is being investigated in clinical trials (54). Crenolanib, a specific PDGFRA and PDGFRB inhibitor, demonstrates cytostatic effects in PDGFRA amplified and mutated cell lines with no significant effect on cell death (49). Therefore, combination therapy with PDGFR inhibitors may be beneficial for achieving cytotoxic response in pediatric HGG and triggering tumor regression.
There have been less exciting results using other PDGFR inhibitors in clinical trials for pediatric and adult glioma. A phase I trial using imatinib in 84 recurrent pHGG patients revealed significant risk of intratumoral hemorrhage; median PFS and OS were 7 and 11 months, respectively (55). A phase I trial using crenolanib enrolled 32 newly diagnosed and 23 recurrent pHGG patients. For new diagnoses, median PFS and OS were 7 and 12 months; for recurrent tumors, median PFS and OS were 2 and 18 months. No significant differences were seen between groups based on PDGFRA alteration, and all outcomes were similar to historical controls (56). Sunitinib was investigated in a phase II trial with 17 recurrent pHGG patients; the study was closed early due to lack of sustained response in all patients (57). The PDGFR inhibitors dovitinib, nintedanib, and tandutinib were tested in phase II adult GBM clinical trials; none showed changes in the survival (58–60).
Epidermal growth factor receptor (EGFR) is a member of the ErbB family of tyrosine kinase receptors, which are alpha-helix transmembrane proteins with cytoplasmic tyrosine kinase activity following homodimerization or heterodimerization (61). Many kinds of tumors have activating mutations in exons 18 to 22 of the EGFR gene, and the location of these mutations significantly change the effectiveness of EGFR inhibitors (62). EGFR amplifications have specifically been found at high frequency in bithalamic and other midline gliomas (13, 18). In cancer cells, inhibition of EGFR with TKIs or monoclonal antibodies can result in decreased cell movement, differentiation, and proliferation. The oral TKIs bind intracellularly and are most effective as monotherapy, while the intravenous monoclonal antibodies operate extracellularly with greater efficacy when used as adjunct therapy (63).
There are many FDA-approved TKIs and monoclonal antibodies for targeting EGFR in various types of cancers. Erlotinib and gefitinib are used for non-small cell lung cancer (NSCLC), and lapatinib is used for HER2-positive breast cancer. Cetuximab is used for colorectal, head and neck cancers, and NSCLC. Primary side effects of TKIs and monoclonal antibodies are diarrhea and acneiform rash, though interstitial lung disease has been reported. Resistance to these EGFR inhibitors arises from mutations in TK, other pathways of cellular proliferation that bypass EGFR, and variations in molecular activity downstream of EGFR (64). Osimertinib showed markedly increased blood-brain barrier penetration in a study with other EGFR inhibitors in brain metastases of NSCLC (65).
EGFR inhibitors have already been investigated in treating pHGG patients. A phase II trial studying gefitinib and radiation enrolled 44 patients and saw six patients with partial response, and three patients who remained progression-free after 36 months. No molecular data was collected, so it is unclear if the latter patients had EGFR alterations. Median PFS and OS were 7 and 12 months, similar to controls (66). A phase II study of erlotinib in newly diagnosed pHGG enrolled 41 patients and found median PFS and OS for anaplastic astrocytoma (AA) to be 11 and 15 months, and 6 and 12 months for GBM, respectively (67). These values were not improvements on historical controls. Lapatinib was tested in a phase II trial and enrolled 10 refractory pHGG patients; all cases showed progressive disease (68). Cetuximab was tested in a phase II trial with 25 newly diagnosed DIPG and 20 recurrent non-midline pHGG patients; median PFS was 7 months for DIPG patients and 9 months for non-midline patients, and median OS was 12 and 17 months (69). The PFS for non-midline patients did not meet the 1-year endpoint to continue the trial.
The EGFR inhibitors osimertinib and afatinib were trialed in three patients with bithalamic gliomas (1 grade II, 2 grade III) exhibiting EGFR exon 20 insertions (13). All patients exhibited slowly progressive disease on treatment (two alive at publication, 8-19 months after diagnosis; one passed 22 months after diagnosis). Given estimates of median OS for pediatric bithalamic gliomas being around 8 months (70), these results showed promise for moving forward to randomized clinical trials. In the context of adult GBM, afatinib was tested in combination with the alkylating agent temozolomide in a phase I trial; antitumor activity was seen in subsets, but many stopped treatment due to progression or adverse events (71). Dacomitinib was tested in a phase II trial that specifically recruited adult GBM patients with EGFR amplification and showed four patients who were progression-free at 6 months, demonstrating activity of the drug (72). Finally, WSD0922 has been investigated for its CNS penetration and is undergoing GBM clinical trials (73, 74).
Recent studies have implicated EGFR as an important modulator of cancer metabolism. In glioblastoma, the EGFRvIII mutation (deletion of exons 2-7) was found to regulate expression of hnRNPA1, which in turn spliced a Myc-interacting protein called Max. The splice product Delta Max was found to impart a Myc-dependent glycolytic gene expression pattern to glioblastoma that correlated with poor patient survival (75). Furthermore, kinase-independent EGFR signaling was found to rescue cancer cells from autophagic cell death by maintaining intracellular glucose levels using sodium/glucose cotransporter 1 (SGLT1) (76). As in glioblastoma, EGFR-driven metabolic phenotypes may be responsible for the resistance of pHGG to metabolic drugs such as the imipridones (e.g. ONC201) (77). Although the importance of EGFR in tumorigenesis is undisputed, these studies further highlight how combination therapy using EGFR inhibitors and metabolic modulators may be promising for pHGG.
Fibroblast growth factor receptors (FGFR) are a family (FGFR1-4) of tyrosine kinase receptors that have also been implicated in the development and progression of cancers. Aberrant expression of specific FGFR genes is frequently observed in tumors; FGFR1 is the most commonly altered gene of the four in pediatric gliomas, being altered in over a third of cases (78). FGFR2-4 amplification, mutation, and fusion have also been identified and linked to oncogenic activity. These findings indicate that the FGFRs may be a suitable biomarker and therapeutic target (79).
Multi-target TKIs, as well as selective inhibitors of the whole FGFR family, have demonstrated the potential to affect the FGFR pathways involved in tumor development and progression. A recent publication specifically addressing the multi-kinase inhibitor ponatinib CNS penetration by our group showed promising results (52); ponatinib has also demonstrated antiproliferative effects on glioma cells in vitro (80). Our group published off-trial use of ponatinib in a case of DIPG with FGFR3 activating mutation, with six months of stable disease before progressing (4-10 months from diagnosis) (78). Ponatinib did not show significant clinical activity in adult GBM (81).
In addition, several FGFR fusions have been discovered that may point to future targeted therapies (82, 83). However, since the FGFR family shares common intracellular signaling pathways with other TKRs, cancer cells may overcome these inhibition therapies by selecting TKR mutants or switching to a parallel signaling pathway (84). Thus, the efficacy of TKR inhibitors in personalized FGFR therapies will be reliant on multi-target TKIs or other drugs with different mechanisms of action.
The pan-FGFR inhibitor erdafitinib has been investigated in a phase I trial for solid tumors, with some glioblastoma patients showing partial responses (85). Both ponatinib and erdafitinib showed favorable CNS penetration based on pharmacokinetic analysis.
MET is an RTK that contributes to tumor growth and angiogenesis, and its alteration has been identified in recurrent pHGG. Capmatinib is a specific MET inhibitor that was studied in a phase II trial for MET-amplified adult GBM and showed no clear activity (86). The MET inhibitor bozitinib (PLB-1001) was tested in 18 recruited pHGG patients; two GBM patients showed partial response, with overall median PFS of 3 months (87).
Several VEGFR inhibitors have been investigated in the hope of combining the antiangiogenic effect with inhibition of common RTK pathways. Tivozanib failed to show antitumor activity in a phase II adult GBM trial, showing limitation of anti-VEGF monotherapy (88). The VEGFR inhibitor sorafenib was investigated in combination with the mTOR inhibitor temsirolimus and the EGFR inhibitor erlotinib in two different phase I/II adult GBM trials, and did not show efficacy in either trial (89, 90). Cediranib showed favorable responses and improved 6-month PFS (26%) in a phase II recurrent adult GBM trial of 31 patients, but did not meet phase III endpoints (91, 92). Apatinib was investigated in a phase II adult glioma trial combined with the topoisomerase inhibitor irinotecan; it enrolled 10 patients and found median PFS to be 8 months and objective response rate (ORR) of 55% (93). Apatinib was further investigated in combination with temozolomide in an adult GBM exploratory study, showing median PFS of 4 months and median OS of 8 months (94).
Cabozantinib, crizotinib, and vandetanib are all multi-tyrosine kinase inhibitors that have been trialed in preclinical pHGG models, targeting MET as well as RET, VEGFR, and ALK, among others. Cabozantinib has been studied preclinically in combination with dasatinib, with promising results (95). Cabozantinib was also investigated in a phase II recurrent adult GBM trial; among the 70 patients who had received prior antiangiogenic therapy, the ORR was 4.3%, described as modest clinical activity (96). Crizotinib was investigated in a phase I trial in combination with dasatinib in 25 recurrent pHGG patients; many dose-limiting toxicities occurred, and no objective radiologic responses were observed (97). A phase I trial using vandetanib in 21 newly diagnosed DIPG patients identified longer PFS in patients with higher VEGF levels before therapy (98). Vandetanib was later investigated in combination with dasatinib in another phase I trial, with median OS of 15 months (99). In addition, vandetanib and everolimus showed promising preclinical results and initial clinical case studies (100). Pazopanib targets several TKIs (among them VEGFR, KIT, PDGFR, and FGFR) to inhibit tumor angiogenesis; it was investigated in a phase II adult glioblastoma trial and did not prolong survival (101). The multi-kinase inhibitor axitinib was investigated in a phase II recurrent adult GBM study that enrolled 55 patients, with one arm adding the alkylating agent lomustine (102). Axitinib resulted in median PFS of 3 months and median OS of 7 months, which were improvements on historical controls. A summary of RTK inhibitors used in preclinical or clinical therapies for pediatric high-grade glioma is listed in Table 1.
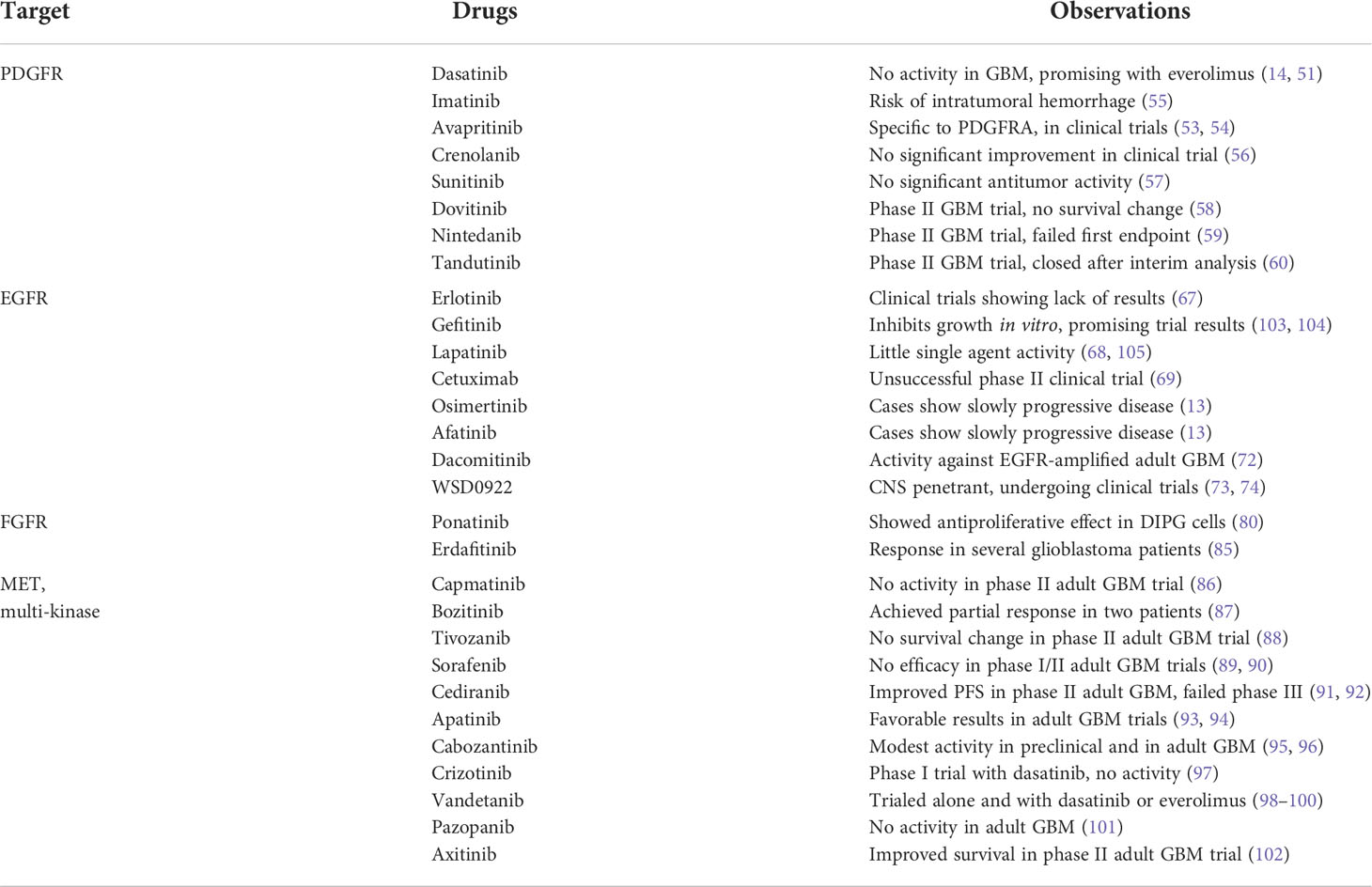
Table 1 Examples of RTK inhibitors used in preclinical or clinical therapies for pediatric high-grade glioma.
Due to the numerous RTK alterations in pediatric HGG, as well as potential therapies that already exist, the RTKs are a promising space for targeted therapy. In vitro and mouse models have helped discern the behavior of these variant tumors compared with controls, as well as their response to treatment. As modeling pediatric gliomas becomes more advanced, one emerging field is 3D organoid modeling. Recent developments in organoid cultures have harnessed the capacity of human embryonic stem cells (hESCs) or induced pluripotent stem cells (iPSCs) to form 3D aggregates called embryoid bodies. These embryoid bodies can self-organize into a structure that models the cytoarchitecture of organs in vitro, from the earliest developmental stages to more mature states (106). These 3D organoids can bridge limitations posed by the 2D cultures and the limitations of in vivo models and can serve to accommodate better precision in drug development for adult and pediatric brain tumors.
There are several different 3D organoid models that have been developed for adult brain tumors: glioblastoma organoids (GBOs), neoplastic cerebral organoids, cerebral organoid-glioblastoma cocultures, and bioprinted glioma organoids. GBOs were developed by incorporating minced pieces of GBM specimen with Matrigel in serum-free media (107). GBOs have been shown to have similar histological and transcriptional profiles to their respective patient tumor and recapitulate patient-specific responses and resistance to standard of care therapy (108, 109). A cryopreserved GBO repository has been developed and used to screen a library of 22 compounds that inhibit tumor invasion into surrounding tissue (110). GBOs have also been used to study response to chimeric antigen T (CAR-T) cell immunotherapy in the setting of the EGFRvIII variant (111), mTOR inhibitors in PTEN loss (112), and STAT inhibitors in Li-Fraumeni syndrome (113). These studies support that GBOs are a reliable tool for developing personalized therapies.
Neoplastic cerebral organoids (neoCORs) are iPSC-derived cerebral organoids that were genetically modified to develop GBM-like tumors by overexpression of oncogenic mutations or downregulation of tumor suppressor genes via CRISPR, with green fluorescent protein (GFP) commonly used as a reporter to visualize tumor cell growth and invasion (114, 115). The neoCOR model can be used to study the role of specific mutations in tumor formation and the identification of downstream pathways and specific drug responses. Recently, an ex vivo model of medulloblastoma was developed by overexpression of Otx2/c-MYC in cerebellar organoids which showed a disease-specific DNA methylation signature (116). Another recent study generated a malignant glioma model by overexpression of MEOX2, inhibition of p53, and loss of PTEN. They showed that MEOX2 increases proliferation in cerebral organoids by activation of the ERK signaling pathway (117).
Cerebral organoid-glioblastoma co-cultures (GLICO) have been developed by co-culturing patient-derived glioblastoma stem cells (GSCs) with cerebral organoids (118, 119). The GLICO model has been used to study the invasive behavior of GFP-marked GSC cell lines, showing stark differences between patients with respect to treatment response. Tumor cells derived from co-culturing also showed a high degree of invasiveness and tumorigenesis when implanted in mice (115). Finally, bioprinted glioma organoids are patient-derived GBM cells that were bioprinted with human umbilical vein endothelial cells and a defined brain extracellular matrix (ECM) (120). This model was shown to replicate patient-specific pathological features and responses to therapy. In summary, brain organoids offer a powerful platform to study the effect of disruptions of central pathways, for identification of novel drug targets, and their resistance mechanisms, and represent a reliable tool for developing personalized therapies.
Another potential strategy for modeling pHGG is the development of patient-specific models in real time. As modeling techniques become validated and efficient, they can be harnessed to generate xenografts that will behave and respond to treatment as the original tumor would. Sequencing can be incorporated to make GEM models for even more accurate and efficient tumor recapitulation. Currently, there is an ongoing clinical trial for medulloblastoma patients that uses biopsy tissue to develop individualized treatment plans (121). Currently, the strategy of characterizing PDXs for therapeutic validation would take weeks to months (27); however, as this technology becomes more efficient, further trials will be able to incorporate mouse models to refine targeted therapies for pHGG.
On the therapeutic side, one direction for RTK-altered pediatric gliomas is co-treatment of multiple TKIs, or one TKI with a synergistic drug to avoid or delay the development of drug resistance (100). As discussed, our group showed that the combination of PDGFRA inhibitor dasatinib with mTOR inhibitor everolimus significantly improved the survival of pediatric high-grade glioma than either treatment alone (41). A phase I/II clinical trial using bevacizumab, irinotecan, and erlotinib in recurrent DIPG enrolled nine patients and resulted in median OS of 13.8 months, compared to radiotherapy-only control median OS of 10 months (122). Due to the crossover between the RTK pathways, the resistance mechanisms that develop to one TKI may depend on another RTK. Akhavan et al. noted that EGFR resistance in GBM patients depends on PDGFRB deregulation and provided a strong rationale for combination therapy (123). Similarly, Day et al. found that glioblastoma cells treated with EGFR and MET inhibitors can develop resistance via FGFR signaling and that simultaneously inhibiting EGFR, FGFR, and MET overcomes the resistance by knocking out more downstream pathways (124). Experimentally, the combination of a PDGFR TKI with another inhibitor targeting either ERBB3 or IGF1R more potently suppressed the growth of GBM cells than either inhibitor alone. Therefore, identifying the RTKs responsible for resistance to one RTK inhibitor may synergistically enhance anti-glioma efficacy.
Another option would be to explore combinations of RTK-targeted therapy and TME-targeted therapy or immunotherapy, such as checkpoint inhibition or CAR-T cell therapy. Both adult and pediatric high-grade gliomas are considered immunologically “cold.” The TME plays an important role in the tumor immune invasion mechanism; tumor cells under TKI treatment may select to respond to the signals from the neighboring stromal cells leading to TME-mediated drug resistance (125). Combining a TKI inhibitor with TME growth factor inhibitors like FGF and SDF1 inhibition could help thwart these resistances. Myeloid-derived suppressor cells (MDSCs) are one of the significant components of the TME; a recent study showed that MDSCs suppress T-cell functions in the glioma TME (37). de Billy et al. combined IGF1R/IR inhibitors with GD2-CAR-T cells and saw increased antitumor activity from the combination in DMG cell lines (126). Exploring the combination of RTK therapy with MDSC inhibition, immune checkpoint inhibition, or CAR-T cell therapy could effectively increase the activation of CD8+ T-cells and change the TME to favor anti-tumor responses.
As we learn more about and experiment with different methods of modeling pediatric glioma, synergism studies between different treatment modalities will be essential in maximizing the efficacy of treatment. Currently, pediatric high-grade gliomas have very few proven effective treatment options. Some possible explanations for the lack of treatment options are the lack of intratumoral heterogeneity in current preclinical models, the limited capacity of small RTK inhibitors to cross the blood-brain barrier (BBB), and specificity/off-target effects of RTK inhibitors. Therefore, it is imperative to develop an individual treatment plan based on each patient’s tumor heterogeneity in terms of specific genetic alteration in combination with enhanced drug delivery approaches. Doing so may provide a much better outcome for pediatric high-grade gliomas, filling this dire need in the clinical setting.
KS wrote abstract and several sections, synthesized all sections together, and edited manuscript. DM wrote individual sections and helped review. JC, JB, AK, CB, SL, SJ, and RC wrote individual sections. SK helped review. EC initialized process of putting together manuscript. CK and VY oversaw synthesis of manuscript and provided feedback. All authors contributed to the article and approved the submitted version.
NIH Grant T32-GM007863 [KS]; NIH Grant T32-CA009676 [DM]; NIH/NINDS Grant R01-NS119231 and R01 NS124607 and Department of Defense Grant CA201129P1, University of Michigan Chad Carr Pediatric Brain Tumor Center, ChadTough Defeat DIPG Foundation, Catching Up With Jack, The Pediatric Brain Tumor Foundation, The Yuvaan Tiwari Foundation, The Morgan Behen Golf Classic, and Michael Miller Memorial Foundation [CK]; ChadTough Defeat DIPG Foundation and Joan and David Evans Prosper Road Foundation [VY].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Braunstein S, Raleigh D, Bindra R, Mueller S, Haas-Kogan D. Pediatric high-grade glioma: current molecular landscape and therapeutic approaches. J Neurooncol (2017) 134:541–9. doi: 10.1007/s11060-017-2393-0
2. Infinger LK, Stevenson CB. Re-examining the need for tissue diagnosis in pediatric diffuse intrinsic pontine gliomas: A review. Curr Neuropharmacol (2017) 15:129–33. doi: 10.2174/1570159X14666160425114024
3. Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol (2010) 28:3061–8. doi: 10.1200/JCO.2009.26.7252
4. Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature (2012) 482:226–31. doi: 10.1038/nature10833
5. Qu HQ, Jacob K, Fatet S, Ge B, Barnett D, Delattre O, et al. Genome-wide profiling using single-nucleotide polymorphism arrays identifies novel chromosomal imbalances in pediatric glioblastomas. Neuro Oncol (2010) 12:153–63. doi: 10.1093/neuonc/nop001
6. Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol (2011) 29:3999–4006. doi: 10.1200/JCO.2011.35.5677
7. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet (2014) 46:444–50. doi: 10.1038/ng.2938
8. Chen CCL, Deshmukh S, Jessa S, Hadjadj D, Lisi V, Andrade AF, et al. Histone H3.3G34-mutant interneuron progenitors Co-opt PDGFRA for gliomagenesis. Cell (2020) 183:1617–1633 e22. doi: 10.1016/j.cell.2020.11.012
9. Chung C, Sweha SR, Pratt D, Tamrazi B, Panwalkar P, Banda A, et al. Integrated metabolic and epigenomic reprograming by H3K27M mutations in diffuse intrinsic pontine gliomas. Cancer Cell (2020) 38:334–349 e9. doi: 10.1016/j.ccell.2020.07.008
10. Sweha SR, Chung C, Natarajan SK, Panwalkar P, Pun M, Ghali A, et al. Epigenetically defined therapeutic targeting in H3.3G34R/V high-grade gliomas. Sci Transl Med (2021) 13:eabf7860. doi: 10.1126/scitranslmed.abf7860
11. Miklja Z, Pasternak A, Stallard S, Nicolaides T, Kline-Nunnally C, Cole B, et al. Molecular profiling and targeted therapy in pediatric gliomas: review and consensus recommendations. Neuro Oncol (2019) 21:968–80. doi: 10.1093/neuonc/noz022
12. Bredel M, Pollack IF, Hamilton RL, James CD. Epidermal growth factor receptor expression and gene amplification in high-grade non-brainstem gliomas of childhood. Clin Cancer Res (1999) 5:1786–92.
13. Mondal G, Lee JC, Ravindranathan A, Villanueva-Meyer JE, Tran QT, Allen SJ, et al. Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol (2020) 139:1071–88. doi: 10.1007/s00401-020-02155-5
14. Koschmann C, Zamler D, MacKay A, Robinson D, Wu YM, Doherty R, et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget (2016) 7:65696–706. doi: 10.18632/oncotarget.11602
15. Hicks WH, Bird CE, Traylor JI, Shi DD, El Ahmadieh TY, Richardson TE, et al. Contemporary mouse models in glioma research. Cells (2021) 10:712. doi: 10.3390/cells10030712
16. Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell (2017) 32:520–537 e5. doi: 10.1016/j.ccell.2017.08.017
17. Guerreiro Stucklin AS, Ryall S, Fukuoka K, Zapotocky M, Lassaletta A, Li C, et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun (2019) 10:4343. doi: 10.1038/s41467-019-12187-5
18. Sievers P, Sill M, Schrimpf D, Stichel D, Reuss DE, Sturm D, et al. A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro Oncol (2021) 23:34–43. doi: 10.1093/neuonc/noaa251
19. Rhun E, Preusser M, Roth P, Reardon DA, van den Bent M, Wen P, et al. Molecular targeted therapy of glioblastoma. Cancer Treat Rev (2019) 80:101896. doi: 10.1016/j.ctrv.2019.101896
20. Alexandru O, Horescu C, Sevastre AS, Cioc CE, Baloi C, Oprita A, et al. Receptor tyrosine kinase targeting in glioblastoma: performance, limitations and future approaches. Contemp Oncol (Pozn) (2020) 24:55–66. doi: 10.5114/wo.2020.94726
21. Pathania M, De Jay N, Maestro N, Harutyunyan AS, Nitarska J, Pahlavan P, et al. H3.3(K27M) cooperates with Trp53 loss and PDGFRA gain in mouse embryonic neural progenitor cells to induce invasive high-grade gliomas. Cancer Cell (2017) 32:684–700 e9. doi: 10.1016/j.ccell.2017.09.014
22. Funato K, Major T, Lewis PW, Allis CD, Tabar V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science (2014) 346:1529–33. doi: 10.1126/science.1253799
23. Akter F, Simon B, de Boer NL, Redjal N, Wakimoto H, Shah K. Pre-clinical tumor models of primary brain tumors: Challenges and opportunities. Biochim Biophys Acta Rev Cancer (2021) 1875:188458. doi: 10.1016/j.bbcan.2020.188458
24. Joo KM, Kim J, Jin J, Kim M, Seol HJ, Muradov J, et al. Patient-specific orthotopic glioblastoma xenograft models recapitulate the histopathology and biology of human glioblastomas in situ. Cell Rep (2013) 3:260–73. doi: 10.1016/j.celrep.2012.12.013
25. Barkovich AJ, Krischer J, Kun LE, Packer R, Zimmerman RA, Freeman CR, et al. Brain stem gliomas: a classification system based on magnetic resonance imaging. Pediatr Neurosurg (1990) 16:73–83. doi: 10.1159/000120511
26. Hashizume R, Gupta N. Patient-derived tumor models for diffuse intrinsic pontine gliomas. Curr Neuropharmacol (2017) 15:98–103. doi: 10.2174/1570159X14666160523144117
27. Brabetz S, Leary SES, Grobner SN, Nakamoto MW, Seker-Cin H, Girard EJ, et al. A biobank of patient-derived pediatric brain tumor models. Nat Med (2018) 24:1752–61. doi: 10.1038/s41591-018-0207-3
28. He C, Xu K, Zhu X, Dunphy PS, Gudenas B, Lin W, et al. Patient-derived models recapitulate heterogeneity of molecular signatures and drug response in pediatric high-grade glioma. Nat Commun (2021) 12:4089. doi: 10.1038/s41467-021-24168-8
29. Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev (1998) 12:3675–85. doi: 10.1101/gad.12.23.3675
30. Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling adult gliomas using RCAS/t-va technology. Transl Oncol (2009) 2:89–95. doi: 10.1593/tlo.09100
31. Barton KL, Misuraca K, Cordero F, Dobrikova E, Min HD, Gromeier M, et al. PD-0332991, a CDK4/6 inhibitor, significantly prolongs survival in a genetically engineered mouse model of brainstem glioma. PloS One (2013) 8:e77639. doi: 10.1371/journal.pone.0077639
32. Hoeman CM, Cordero FJ, Hu G, Misuraca K, Romero MM, Cardona HJ, et al. ACVR1 R206H cooperates with H3.1K27M in promoting diffuse intrinsic pontine glioma pathogenesis. Nat Commun (2019) 10:1023. doi: 10.1038/s41467-019-08823-9
33. Halvorson KG, Barton KL, Schroeder K, Misuraca KL, Hoeman C, Chung A, et al. A high-throughput in vitro drug screen in a genetically engineered mouse model of diffuse intrinsic pontine glioma identifies BMS-754807 as a promising therapeutic agent. PloS One (2015) 10:e0118926. doi: 10.1371/journal.pone.0118926
34. Calinescu AA, Nunez FJ, Koschmann C, Kolb BL, Lowenstein PR, Castro MG. Transposon mediated integration of plasmid DNA into the subventricular zone of neonatal mice to generate novel models of glioblastoma. J Vis Exp (2015) 96:52443. doi: 10.3791/52443
35. Koschmann C, Calinescu AA, Nunez FJ, Mackay A, Fazal-Salom J, Thomas D, et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci Transl Med (2016) 8:328ra28. doi: 10.1126/scitranslmed.aac8228
36. Wiesner SM, Decker SA, Larson JD, Ericson K, Forster C, Gallardo JL, et al. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res (2009) 69:431–9. doi: 10.1158/0008-5472.CAN-08-1800
37. Alghamri MS, McClellan BL, Avvari RP, Thalla R, Carney S, Hartlage MS, et al. G-CSF secreted by mutant IDH1 glioma stem cells abolishes myeloid cell immunosuppression and enhances the efficacy of immunotherapy. Sci Adv (2021) 7:eabh3243. doi: 10.1126/sciadv.abh3243
38. Mendez F, Kadiyala P, Nunez FJ, Carney S, Nunez FM, Gauss JC, et al. Therapeutic efficacy of immune stimulatory thymidine kinase and fms-like tyrosine kinase 3 ligand (TK/Flt3L) gene therapy in a mouse model of high-grade brainstem glioma. Clin Cancer Res (2020) 26:4080–92. doi: 10.1158/1078-0432.CCR-19-3714
39. Qin Y, Musket A, Kou J, Preiszner J, Tschida BR, Qin A, et al. Overexpression of HGF/MET axis along with p53 inhibition induces de novo glioma formation in mice. Neurooncol Adv (2020) 2:vdaa067. doi: 10.1093/noajnl/vdaa067
40. Patel SK, Hartley RM, Wei X, Furnish R, Escobar-Riquelme F, Bear H, et al. Generation of diffuse intrinsic pontine glioma mouse models by brainstem-targeted in utero electroporation. Neuro Oncol (2020) 22:381–92. doi: 10.1093/neuonc/noz197
41. Miklja Z, Yadav VN, Cartaxo RT, Siada R, Thomas CC, Cummings JR, et al. Everolimus improves the efficacy of dasatinib in PDGFRalpha-driven glioma. J Clin Invest (2020) 130:5313–25. doi: 10.1172/JCI133310
42. Larson JD, Kasper LH, Paugh BS, Jin H, Wu G, Kwon CH, et al. Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell (2019) 35:140–155 e7. doi: 10.1016/j.ccell.2018.11.015
43. Fortin J, Tian R, Zarrabi I, Hill G, Williams E, Sanchez-Duffhues G, et al. Mutant ACVR1 arrests glial cell differentiation to drive tumorigenesis in pediatric gliomas. Cancer Cell (2020) 37:308–323 e12. doi: 10.1016/j.ccell.2020.02.002
44. Zou H, Feng R, Huang Y, Tripodi J, Najfeld V, Tsankova NM, et al. Double minute amplification of mutant PDGF receptor alpha in a mouse glioma model. Sci Rep (2015) 5:8468. doi: 10.1038/srep08468
45. Filbin MG, Tirosh I, Hovestadt V, Shaw ML, Escalante LE, Mathewson ND, et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science (2018) 360:331–5. doi: 10.1126/science.aao4750
46. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. Sellers, high-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med (2015) 21:1318–25. doi: 10.1038/nm.3954
47. Chatwin HV, Cruz Cruz J, Green AL. Pediatric high-grade glioma: moving toward subtype-specific multimodal therapy. FEBS J (2021) 288:6127–41. doi: 10.1111/febs.15739
48. Tian H, Lyu Y, Yang YG, Hu Z. Humanized rodent models for cancer research. Front Oncol (2020) 10:1696. doi: 10.3389/fonc.2020.01696
49. Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res (2013) 73:6219–29. doi: 10.1158/0008-5472.CAN-13-1491
50. Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev (2001) 15:1913–25. doi: 10.1101/gad.903001
51. Lassman AB, Pugh SL, Gilbert MR, Aldape KD, Geinoz S, Beumer JH, et al. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627). Neuro Oncol (2015) 17:992–8. doi: 10.1093/neuonc/nov011
52. Ravi K, Franson A, Homan MJ, Roberts H, Pai MP, Miklja Z, et al. Comparative pharmacokinetic analysis of the blood-brain barrier penetration of dasatinib and ponatinib in mice. Leukemia Lymphoma (2021) 62:1990–4. doi: 10.1080/10428194.2021.1894647
53. Evans EK, Gardino AK, Kim JL, Hodous BL, Shutes A, Davis A, et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci Transl Med (2017) 9:eaao1690. doi: 10.1126/scitranslmed.aao1690
54. A study of avapritinib in pediatric patients with solid tumors dependent on KIT or PDGFRA signaling. Available at: https://ClinicalTrials.gov/show/NCT04773782.
55. Pollack IF, Jakacki RI, Blaney SM, Hancock ML, Kieran MW, Phillips P, et al. Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: a pediatric brain tumor consortium report. Neuro Oncol (2007) 9:145–60. doi: 10.1215/15228517-2006-031
56. Tinkle CL, Broniscer A, Chiang J, Campagne O, Huang J, Orr BA, et al. Phase I study using crenolanib to target PDGFR kinase in children and young adults with newly diagnosed DIPG or recurrent high-grade glioma, including DIPG. Neurooncol Adv (2021) 3:vdab179. doi: 10.1093/noajnl/vdab179
57. Wetmore C, Daryani VM, Billups CA, Boyett JM, Leary S, Tanos R, et al. Phase II evaluation of sunitinib in the treatment of recurrent or refractory high-grade glioma or ependymoma in children: a children's oncology group study ACNS1021. Cancer Med (2016) 5:1416–24. doi: 10.1002/cam4.713
58. Sharma M, Schilero C, Peereboom DM, Hobbs BP, Elson P, Stevens GHJ, et al. Phase II study of dovitinib in recurrent glioblastoma. J Neurooncol (2019) 144:359–68. doi: 10.1007/s11060-019-03236-6
59. Muhic A, Poulsen HS, Sorensen M, Grunnet K, Lassen U. Phase II open-label study of nintedanib in patients with recurrent glioblastoma multiforme. J Neurooncol (2013) 111:205–12. doi: 10.1007/s11060-012-1009-y
60. Batchelor TT, Gerstner ER, Ye X, Desideri S, Duda DG, Peereboom D, et al. Feasibility, phase I, and phase II studies of tandutinib, an oral platelet-derived growth factor receptor-beta tyrosine kinase inhibitor, in patients with recurrent glioblastoma. Neuro Oncol (2017) 19:567–75. doi: 10.1093/neuonc/now185
61. Wells A. EGF receptor. Int J Biochem Cell Biol (1999) 31:637–43. doi: 10.1016/S1357-2725(99)00015-1
62. Kohsaka S, Nagano M, Ueno T, Suehara Y, Hayashi T, Shimada N, et al. A method of high-throughput functional evaluation of EGFR gene variants of unknown significance in cancer. Sci Transl Med (2017) 9:eaan6566. doi: 10.1126/scitranslmed.aan6566
63. da Cunha Santos G, Shepherd FA, Tsao MS. EGFR mutations and lung cancer. Annu Rev Pathol (2011) 6:49–69. doi: 10.1146/annurev-pathol-011110-130206
64. Gerber DE. EGFR inhibition in the treatment of non-small cell lung cancer. Drug Dev Res (2008) 69:359–72. doi: 10.1002/ddr.20268
65. Ballard P, Yates JW, Yang Z, Kim DW, Yang JC, Cantarini M, et al. Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin Cancer Res (2016) 22:5130–40. doi: 10.1158/1078-0432.CCR-16-0399
66. Pollack IF, Stewart CF, Kocak M, Poussaint TY, Broniscer A, Banerjee A, et al. A phase II study of gefitinib and irradiation in children with newly diagnosed brainstem gliomas: a report from the pediatric brain tumor consortium. Neuro Oncol (2011) 13:290–7. doi: 10.1093/neuonc/noq199
67. Qaddoumi I, Kocak M, Pai Panandiker AS, Armstrong GT, Wetmore C, Crawford JR, et al. Phase II trial of erlotinib during and after radiotherapy in children with newly diagnosed high-grade gliomas. Front Oncol (2014) 4:67. doi: 10.3389/fonc.2014.00067
68. Fouladi M, Stewart CF, Blaney SM, Onar-Thomas A, Schaiquevich P, Packer RJ, et al. A molecular biology and phase II trial of lapatinib in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Neurooncol (2013) 114:173–9. doi: 10.1007/s11060-013-1166-7
69. Macy ME, Kieran MW, Chi SN, Cohen KJ, MacDonald TJ, Smith AA, et al. A pediatric trial of radiation/cetuximab followed by irinotecan/cetuximab in newly diagnosed diffuse pontine gliomas and high-grade astrocytomas: A pediatric oncology experimental therapeutics investigators' consortium study. Pediatr Blood Cancer (2017) 64:e26621. doi: 10.1002/pbc.26621
70. Niu X, Wang T, Yang Y, Gan Y, Li J, Liu Y, et al. Prognostic factors for the survival outcome of bilateral thalamic glioma: An integrated survival analysis. World Neurosurg (2018) 110:e222–30. doi: 10.1016/j.wneu.2017.10.132
71. Saran F, Welsh L, James A, McBain C, Gattamaneni R, Jefferies S, et al. Afatinib and radiotherapy, with or without temozolomide, in patients with newly diagnosed glioblastoma: results of a phase I trial. J Neurooncol (2021) 155:307–17. doi: 10.1007/s11060-021-03877-6
72. Sepulveda-Sanchez JM, Vaz MA, Balana C, Gil-Gil M, Reynes G, Gallego O, et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro Oncol (2017) 19:1522–31. doi: 10.1093/neuonc/nox105
73. Zhong W, Zhang J, Mu Z, Sun C. DDIS-22. WSD0922: A BBB penetrable EGFR/EGFRVIII small molecule for the treatment of GBM and metastatic CNS tumor. Neuro Oncol (2018) 20:vi73–4. doi: 10.1093/neuonc/noy148.301
74. WSD0922-FU for the treatment of glioblastoma, anaplastic astrocytoma, or non-small cell lung cancer with central nervous system metastases . Available at: https://ClinicalTrials.gov/show/NCT04197934.
75. Babic I, Anderson ES, Tanaka K, Guo D, Masui K, Li B, et al. EGFR mutation-induced alternative splicing of max contributes to growth of glycolytic tumors in brain cancer. Cell Metab (2013) 17:1000–8. doi: 10.1016/j.cmet.2013.04.013
76. Weihua Z, Tsan R, Huang WC, Wu Q, Chiu CH, Fidler IJ, et al. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell (2008) 13:385–93. doi: 10.1016/j.ccr.2008.03.015
77. He Y, Li J, Koga T, Ma J, Dhawan S, Suzuki Y, et al. Epidermal growth factor receptor as a molecular determinant of glioblastoma response to dopamine receptor D2 inhibitors. Neuro Oncol (2021) 23:400–11. doi: 10.1093/neuonc/noaa188
78. Marini BL, Benitez LL, Zureick AH, Salloum R, Gauthier AC, Brown J, et al. Blood-brain barrier-adapted precision medicine therapy for pediatric brain tumors. Transl Res (2017) 188:27 e1–27 e14. doi: 10.1016/j.trsl.2017.08.001
79. Porta R, Borea R, Coelho A, Khan S, Araujo A, Reclusa P, et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit Rev Oncol Hematol (2017) 113:256–67. doi: 10.1016/j.critrevonc.2017.02.018
80. Schramm K, Iskar M, Statz B, Jager N, Haag D, Slabicki M, et al. DECIPHER pooled shRNA library screen identifies PP2A and FGFR signaling as potential therapeutic targets for diffuse intrinsic pontine gliomas. Neuro Oncol (2019) 21:867–77. doi: 10.1093/neuonc/noz057
81. Lee EQ, Muzikansky A, Duda DG, Gaffey S, Dietrich J, Nayak L, et al. Phase II trial of ponatinib in patients with bevacizumab-refractory glioblastoma. Cancer Med (2019) 8:5988–94. doi: 10.1002/cam4.2505
82. Parker BC, Engels M, Annala M, Zhang W. Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J Pathol (2014) 232:4–15. doi: 10.1002/path.4297
83. Chen L, Zhang Y, Yin L, Cai B, Huang P, Li X, et al. Fibroblast growth factor receptor fusions in cancer: opportunities and challenges. J Exp Clin Cancer Res (2021) 40:345. doi: 10.1186/s13046-021-02156-6
84. Jin H, Shi Y, Lv Y, Yuan S, Ramirez CFA, Lieftink C, et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature (2021) 595:730–4. doi: 10.1038/s41586-021-03741-7
85. Bahleda R, Italiano A, Hierro C, Mita A, Cervantes A, Chan N, et al. Multicenter phase I study of erdafitinib (JNJ-42756493), oral pan-fibroblast growth factor receptor inhibitor, in patients with advanced or refractory solid tumors. Clin Cancer Res (2019) 25:4888–97. doi: 10.1158/1078-0432.CCR-18-3334
86. van den Bent M, Azaro A, De Vos F, Sepulveda J, Yung WKA, Wen PY, et al. Open-label, multicenter study of INC280 (capmatinib) alone and in combination with buparlisib (BKM120) in adult patients with recurrent glioblastoma. J Neurooncol (2020) 146:79–89. doi: 10.1007/s11060-019-03337-2
87. Hu H, Mu Q, Bao Z, Chen Y, Liu Y, Chen J, et al. Mutational landscape of secondary glioblastoma guides MET-targeted trial in brain tumor. Cell (2018) 175:1665–1678 e18. doi: 10.1016/j.cell.2018.09.038
88. Kalpathy-Cramer J, Chandra V, Da X, Ou Y, Emblem KE, Muzikansky A, et al. Phase II study of tivozanib, an oral VEGFR inhibitor, in patients with recurrent glioblastoma. J Neurooncol (2017) 131:603–10. doi: 10.1007/s11060-016-2332-5
89. Schiff D, Jaeckle KA, Anderson SK, Galanis E, Giannini C, Buckner JC, et al. Phase 1/2 trial of temsirolimus and sorafenib in the treatment of patients with recurrent glioblastoma: North central cancer treatment group Study/Alliance N0572. Cancer (2018) 124:1455–63. doi: 10.1002/cncr.31219
90. Chen H, Kuhn J, Lamborn KR, Abrey LE, DeAngelis LM, Lieberman F, et al. Phase I/II study of sorafenib in combination with erlotinib for recurrent glioblastoma as part of a 3-arm sequential accrual clinical trial: NABTC 05-02. Neurooncol Adv (2020) 2:vdaa124. doi: 10.1093/noajnl/vdaa124
91. Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol (2010) 28:2817–23. doi: 10.1200/JCO.2009.26.3988
92. Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol (2013) 31:3212–8. doi: 10.1200/JCO.2012.47.2464
93. Wang L, Liang L, Yang T, Qiao Y, Xia Y, Liu L, et al. A pilot clinical study of apatinib plus irinotecan in patients with recurrent high-grade glioma: Clinical Trial/Experimental study. Medicine (Baltimore) (2017) 96:e9053. doi: 10.1097/MD.0000000000009053
94. Yao H, Liu J, Zhang C, Shao Y, Li X, Feng M, et al. Clinical study of apatinib plus temozolomide for the treatment of recurrent high-grade gliomas. J Clin Neurosci (2021) 90:82–8. doi: 10.1016/j.jocn.2021.05.032
95. Truffaux N, Philippe C, Paulsson J, Andreiuolo F, Guerrini-Rousseau L, Cornilleau G, et al. Preclinical evaluation of dasatinib alone and in combination with cabozantinib for the treatment of diffuse intrinsic pontine glioma. Neuro Oncol (2015) 17:953–64. doi: 10.1093/neuonc/nou330
96. Cloughesy TF, Drappatz J, de Groot J, Prados MD, Reardon DA, Schiff D, et al. Phase II study of cabozantinib in patients with progressive glioblastoma: subset analysis of patients with prior antiangiogenic therapy. Neuro Oncol (2018) 20:259–67. doi: 10.1093/neuonc/nox151
97. Broniscer A, Jia S, Mandrell B, Hamideh D, Huang J, Onar-Thomas A, et al. Phase 1 trial, pharmacokinetics, and pharmacodynamics of dasatinib combined with crizotinib in children with recurrent or progressive high-grade and diffuse intrinsic pontine glioma. Pediatr Blood Cancer (2018) 65:e27035. doi: 10.1002/pbc.27035
98. Broniscer A, Baker JN, Tagen M, Onar-Thomas A, Gilbertson RJ, Davidoff AM, et al. Phase I study of vandetanib during and after radiotherapy in children with diffuse intrinsic pontine glioma. J Clin Oncol (2010) 28:4762–8. doi: 10.1200/JCO.2010.30.3545
99. Broniscer A, Baker SD, Wetmore C, Pai Panandiker AS, Huang J, Davidoff AM, et al. Pharmacokinetics, and pharmacodynamics of vandetanib and dasatinib in children with newly diagnosed diffuse intrinsic pontine glioma. Clin Cancer Res (2013) 19:3050–8. doi: 10.1158/1078-0432.CCR-13-0306
100. Carvalho DM, Richardson PJ, Olaciregui N, Stankunaite R, Lavarino C, Molinari V, et al. Repurposing vandetanib plus everolimus for the treatment of ACVR1-mutant diffuse intrinsic pontine glioma. Cancer Discov (2021) 12:416–31. doi: 10.1158/2159-8290.CD-20-1201
101. Iwamoto FM, Lamborn KR, Robins HI, Mehta MP, Chang SM, Butowski NA, et al. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American brain tumor consortium study 06-02). Neuro Oncol (2010) 12:855–61. doi: 10.1093/neuonc/noq025
102. Duerinck J, Du Four S, Bouttens F, Andre C, Verschaeve V, Van Fraeyenhove F, et al. Randomized phase II trial comparing axitinib with the combination of axitinib and lomustine in patients with recurrent glioblastoma. J Neurooncol (2018) 136:115–25. doi: 10.1007/s11060-017-2629-z
103. Chang CY, Kuan YH, Ou YC, Li JR, Wu CC, Pan PH, et al. Autophagy contributes to gefitinib-induced glioma cell growth inhibition. Exp Cell Res (2014) 327:102–12. doi: 10.1016/j.yexcr.2014.05.011
104. Geyer JR, Stewart CF, Kocak M, Broniscer A, Phillips P, Douglas JG, et al. And biology study of gefitinib and radiation in children with newly diagnosed brain stem gliomas or supratentorial malignant gliomas. Eur J Cancer (2010) 46:3287–93. doi: 10.1016/j.ejca.2010.07.005
105. Fouladi M, Stewart CF, Blaney SM, Onar-Thomas A, Schaiquevich P, Packer RJ, et al. Phase I trial of lapatinib in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol (2010) 28:4221–7. doi: 10.1200/JCO.2010.28.4687
106. Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell (2016) 165:1238–54. doi: 10.1016/j.cell.2016.04.032
107. Hubert CG, Rivera M, Spangler LC, Wu Q, Mack SC, Prager BC, et al. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res (2016) 76:2465–77. doi: 10.1158/0008-5472.CAN-15-2402
108. Jacob F, Salinas RD, Zhang DY, Nguyen PTT, Schnoll JG, Wong SZH, et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell (2020) 180:188–204 e22. doi: 10.1016/j.cell.2019.11.036
109. Sundar SJ, Shakya S, Barnett A, Wallace LC, Jeon H, Sloan A, et al. Three-dimensional organoid culture unveils resistance to clinical therapies in adult and pediatric glioblastoma. Transl Oncol (2022) 15:101251. doi: 10.1016/j.tranon.2021.101251
110. Darrigues E, Zhao EH, De Loose A, Lee MP, Borrelli MJ, Eoff RL, et al. Biobanked glioblastoma patient-derived organoids as a precision medicine model to study inhibition of invasion. Int J Mol Sci (2021) 22:10720. doi: 10.3390/ijms221910720
111. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother (2019) 42:126–35. doi: 10.1097/CJI.0000000000000260
112. Loong HH, Wong AM, Chan DT, Cheung MS, Chow C, Ding X, et al. Patient-derived tumor organoid predicts drugs response in glioblastoma: A step forward in personalized cancer therapy? J Clin Neurosci (2020) 78:400–2. doi: 10.1016/j.jocn.2020.04.107
113. Reed MR, Lyle AG, De Loose A, Maddukuri L, Learned K, Beale HC, et al. A functional precision medicine pipeline combines comparative transcriptomics and tumor organoid modeling to identify bespoke treatment strategies for glioblastoma. Cells (2021) 10:3400. doi: 10.3390/cells10123400
114. Bian S, Repic M, Guo Z, Kavirayani A, Burkard T, Bagley JA, et al. Genetically engineered cerebral organoids model brain tumor formation. Nat Methods (2018) 15:631–9. doi: 10.1038/s41592-018-0070-7
115. Ogawa J, Pao GM, Shokhirev MN, Verma IM. Glioblastoma model using human cerebral organoids. Cell Rep (2018) 23:1220–9. doi: 10.1016/j.celrep.2018.03.105
116. Ballabio C, Anderle M, Gianesello M, Lago C, Miele E, Cardano M, et al. Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat Commun (2020) 11:583. doi: 10.1038/s41467-019-13989-3
117. Schonrock A, Heinzelmann E, Steffl B, Demirdizen E, Narayanan A, Krunic D, et al. MEOX2 homeobox gene promotes growth of malignant gliomas. Neuro Oncol (2022) noac110. doi: 10.1093/neuonc/noac110
118. da Silva B, Mathew RK, Polson ES, Williams J, Wurdak H. Spontaneous glioblastoma spheroid infiltration of early-stage cerebral organoids models brain tumor invasion. SLAS Discov (2018) 23:862–8. doi: 10.1177/2472555218764623
119. Linkous A, Balamatsias D, Snuderl M, Edwards L, Miyaguchi K, Milner T, et al. Modeling patient-derived glioblastoma with cerebral organoids. Cell Rep (2019) 26:3203–3211 e5. doi: 10.1016/j.celrep.2019.02.063
120. Yi HG, Jeong YH, Kim Y, Choi YJ, Moon HE, Park SH, et al. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat BioMed Eng (2019) 3:509–19. doi: 10.1038/s41551-019-0363-x
121. Individualized treatment plan in children and young adults with relapsed medulloblastoma . Available at: https://ClinicalTrials.gov/show/NCT05057702.
122. El-Khouly FE, Veldhuijzen van Zanten SEM, Jansen MHA, Bakker DP, Sanchez Aliaga E, Hendrikse NH, et al. A phase I/II study of bevacizumab, irinotecan and erlotinib in children with progressive diffuse intrinsic pontine glioma. J Neurooncol (2021) 153:263–71. doi: 10.1007/s11060-021-03763-1
123. Akhavan D, Pourzia AL, Nourian AA, Williams KJ, Nathanson D, Babic I, et al. De-repression of PDGFRbeta transcription promotes acquired resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients. Cancer Discov (2013) 3:534–47. doi: 10.1158/2159-8290.CD-12-0502
124. Day EK, Sosale NG, Xiao A, Zhong Q, Purow B, Lazzara MJ. Glioblastoma cell resistance to EGFR and MET inhibition can be overcome via blockade of FGFR-SPRY2 bypass signaling. Cell Rep (2020) 30:3383–3396 e7. doi: 10.1016/j.celrep.2020.02.014
125. Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer (2009) 9:665–74. doi: 10.1038/nrc2714
Keywords: glioma, TKI - tyrosine kinase inhibitor, RTK - receptor tyrosine kinase, pediatric, neuro-oncology - medical, high-grade glioma (HGG), preclinical (in vivo) studies, mouse models
Citation: Schwark K, Messinger D, Cummings JR, Bradin J, Kawakibi A, Babila CM, Lyons S, Ji S, Cartaxo RT, Kong S, Cantor E, Koschmann C and Yadav VN (2022) Receptor tyrosine kinase (RTK) targeting in pediatric high-grade glioma and diffuse midline glioma: Pre-clinical models and precision medicine. Front. Oncol. 12:922928. doi: 10.3389/fonc.2022.922928
Received: 18 April 2022; Accepted: 04 July 2022;
Published: 01 August 2022.
Edited by:
Yoshihiro Muragaki, Tokyo Women’s Medical University, JapanReviewed by:
Regina M. Graham, University of Miami Health System, United StatesCopyright © 2022 Schwark, Messinger, Cummings, Bradin, Kawakibi, Babila, Lyons, Ji, Cartaxo, Kong, Cantor, Koschmann and Yadav. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viveka Nand Yadav, dnlhZGF2QGNtaC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.