 Zhiliang Zhang1,2†
Zhiliang Zhang1,2† Ruxia Cui
Ruxia Cui Roy Mridul
Roy Mridul Mingjun Xiang
Mingjun Xiang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 23 June 2022
Sec. Cancer Genetics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.922596
This article is part of the Research Topic Reviews in Cancer Genetics View all 20 articles
Inhibitor of apoptosis protein-related-like protein-2 (ILP-2), also known as BIRC-8, is a member of the inhibitor of apoptosis protein (IAPs) family, which mainly encodes the negative regulator of apoptosis. It is selectively overexpressed in a variety of human tumors and can help tumor cells evade apoptosis, promote tumor cell growth, increase tumor cell aggressiveness, and appears to be involved in tumor cell resistance to chemotherapeutic drugs. Several studies have shown that downregulation of ILP-2 expression increases apoptosis, inhibits metastasis, reduces cell growth potential, and sensitizes tumor cells to chemotherapeutic drugs. In addition, ILP-2 inhibits apoptosis in a unique manner; it does not directly inhibit the activity of caspases but induces apoptosis by cooperating with other apoptosis-related proteins. Here, we review the current understanding of the various roles of ILP-2 in the apoptotic cascade and explore the use of interfering ILP-2, and the combination of related anti-tumor agents, as a novel strategy for cancer therapy.
Cancer is the most important health problem that people in every country will face, and the most important obstacle to increasing human life expectancy (1). Tumor development is often a cytopathic process caused by a combination of high-risk factors. The characteristics of tumor cells are mainly anti-apoptosis, invasion and cell cycle disorders, and the control of this process is mainly caused by the abnormal expression of some key proteins (2). The inhibitor of apoptosis protein family (IAPs) are a class of cellular anti-apoptosis inhibitory proteins with high structural homology, highly conserved gene sequences and similar functional characteristics that have been found in yeast, invertebrates and vertebrates (3). In mammalian cells, the onset of apoptosis is tightly controlled by endogenous IAPs, and dysregulation of IAPs expression results in tumorigenesis and chemoresistance (4). The IAPs have widespread anti-apoptotic potential functions by inhibition caspases activity. Up to now, IAP-family members have been identified in humans cells: c IAP-1 (HIAP-2), c IAP-2 (HIAP-1), XIAP (ILP-1), ILP-2(BIRC-8), ML-IAP (Livin), Survivin and NAIP, and BRUCE (Apo1lon) (5, 6) (Figure 1). The members of the IAPs family are characterized by one or more repeats of a highly conserved 70 amino acids domain, termed the baculoviral IAP (BIR) and Ring structures (7). Notably, these structures have very important biological functions (8).
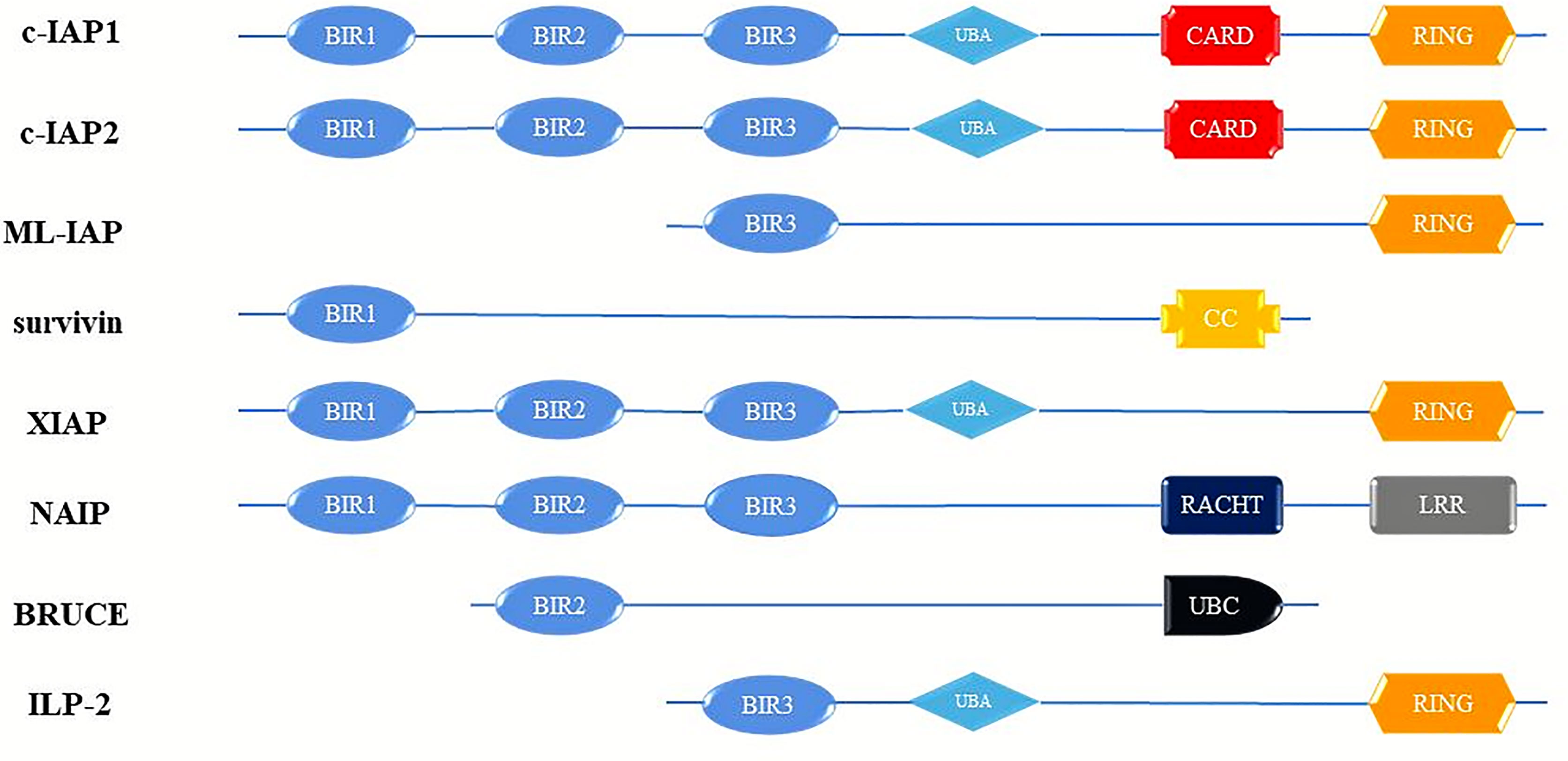
Figure 1 Schematic diagram of domain structure of human IAP proteins. BIR, baculovirus IAP repeats; CARD, caspase activating and recruitment domain; RING, ring zinc-finger. c-IAP1, cellular Inhibitor of apoptosis protein; c-IAP2, cellular Inhibitor of apoptosis protein; ML-IAP, livin/melanoma-Inhibitor of apoptosis protein; XIAP, Survivin, X-linked Inhibitor of apoptosis protein; NAIP, Neuronal apoptosis inhibitory protein; BRUCE, Baculoviral IAP repeat-containing ubiquitin-conjugating enzym; ILP-2, Inhibitor of apoptosis protein-like protein-2.
Inhibitor of apoptosis protein-related-like protein 2 (ILP-2), also known as BIRC-8, is a member of the inhibitor of apoptosis protein IAPs family, located on human chromosome 19 q13.3-13.4, and is highly conserved in genes (9). ILP-2 was first identified by RICHTER et al, while studied cDNA from RNA reverse transcription in peripheral blood cells and human genomic DNA for PCR amplification of XIPA gene was first identified as an apoptosis suppressor protein (10). Its coding sequence is very similar to that of XIAP (ILP-1 or BIRC4), with 80% identity and 95% homology at the amino acid level. In analysis the genomic organization of the BIRC4 locus by Lagacé, identified a cross-reactive band encodes a gene expresses a now 2-kb transcript homologous to ILP-1, hence the name ILP-2 (11). Initially, ILP-2 was found to be expressed only in human testicular tissue and later detected in the cytoplasm of lymphocytes. In subsequent studies, it was found that ILP-2 is highly expressed in various tumors such as nasopharyngeal carcinoma, breast cancer, liver cancer, hematological tumors, and neuroblastoma (12, 13). ILP-2 can protect cells from BAX-induced endogenous apoptosis, help tumor cells escape apoptosis, and antagonize clinical chemotherapy (14, 15). It is worth noting that ILP-2 may not operate independently and needs to cooperate with some proteins to jointly exert anti-apoptotic effects (16). At present, these key regulatory proteins are also under continuous exploration. Furthermore, since ILP-2 has multiple leukocyte Ig-like receptors, natural killer cells, ICAMs, and Fc receptors (FcRs), it appears to play a role in immune-related functions (17). This implies that the expression of ILP-2 may be closely related to the occurrence and development of tumors. In a follow-up study, it was found that by interfering with ILP-2 expression in tumor cells induced apoptosis, decreased cell migration and cell growth (18). It is confirmed that the abnormal expression of ILP-2 is closely related to the occurrence and development of tumors, indicating that ILP-2 may be a new target for human tumor therapy, and will be used as a potential new strategy for tumor clinical treatment by interfered with ILP-2 (19). This review would focus on the anti-apoptotic function of ILP-2 protein and its role in cancer development and progression, as well as ILP-2 as a potential target for cancer therapy.
The cDNA of ILP-2 contains 1 exon, 2022 bases, and the entire ILP-2 gene is about 2kb (Figure 2). Since the coding sequence of ILP-2 is very similar to that of XIAP (ILP-1), about 80% of the sequence is identical to XIAP and about 95% of the amino acids are in the same region as XIAP, so it is called ILP-2 (11). The ILP-2 gene produces a protein consisting of 236 amino acids with a molecular weight of 27 kDa and contains a BIR 3 domain, which is a baculovirus IAP repeat consisting of 70 amino acids (20). And also contains a UBA domain for ubiquitin binding and a RING domain with E3 ligase function (21), RING domain is a specific ring finger structure composed of 7 Cys and one His bound to 2 zinc ions (22).
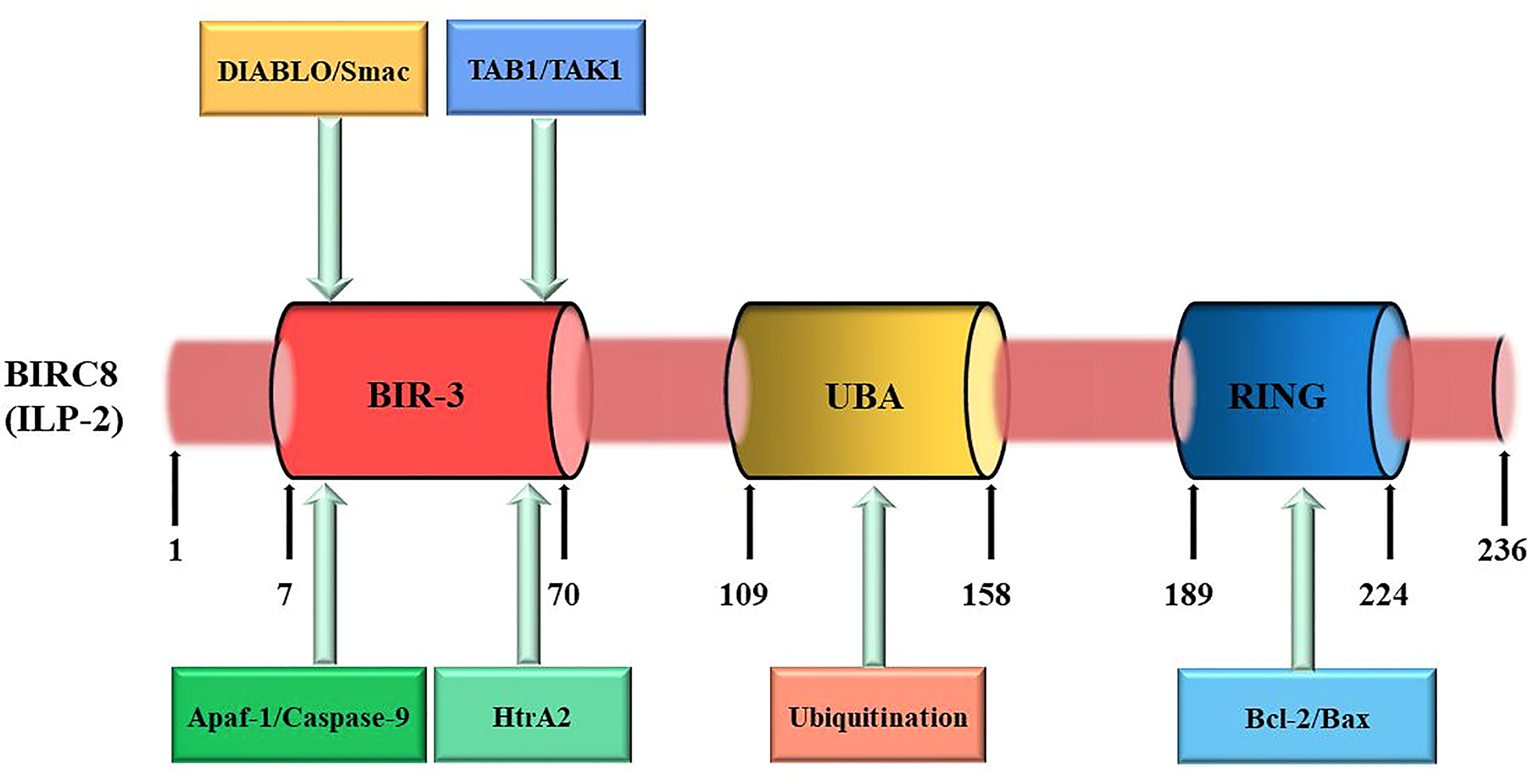
Figure 2 ILP-2 protein structure and function. ILP-2 (also kwon as BIRC8) protein structure presents 236 amino acids and is composed of a Baculovirus IAP Repeat (BIR) domain, an Ubiquitin-associated (UBA) domain, and a RING finger domain. Each structural domain of ILP-2 is capable of binding to the corresponding protein to perform the relevant function. The numbers refer to amino acids.
Both the N-terminal and C-terminal regions of ILP-2 protein have extremely important biological functions (23). The N-terminal domain of ILP-2, BIR 3 can specifically bind to caspase-9 to form a heterodimer, which inhibits the cleavage of the substrate by caspase-9, thereby preventing the occurrence of apoptosis; however, due to ILP Proline (Pro) 257/260 in the N-terminal domain of the -2 protein is easily mutated into Ala, and the lack of an amino acid sequence linked to BIR-3 leads to poor molecular conformational stability, so a small amount of ILP-2 The expression does not have obvious inhibitory effect on cell apoptosis (24). Only when it is overexpressed in cells or tissues and further inhibits the signaling pathway of Bax or apoptosis-promoting factor Apaf-1/caspase-9, it can exert its anti-apoptotic effect. The C-terminal domain of ILP-2 is a specific ring finger structure formed by 7 cysteines (Cys) and 1 histidine (His) combined with 2 zinc ions (25). It can induce the ubiquitination of itself and target proteins (caspase and Smac) by the activity of ligase E3, it can also cooperate with the caspase activating and recruitment (CARD) domain to mediate the ubiquitination of other proteins involved in cell death (26). ILP-2 specifically binds apoptosis-associated proteins Caspases and Smac (Second mitochondria-derived activator of caspase) through C-terminal ubiquitination, and then activates the ubiquitination pathway to degrade caspases and Smac in the cytoplasm to reduce their relative levels in the cytoplasm, resulting in their inability to express relevant activities and thus inhibit apoptosis (27). In addition, ILP-2 has multiple leukocyte Ig-like receptors, natural killer cells, ICAMs (Intercellular adhesion molecules), and Fc receptors (FcRs) in its protein structure, which appears to play a role in immune-related functions (28).
Apoptosis is a way of programmed cell death controlled by proteins, with a high degree of autonomy and order. For mammals, apoptosis is of great significance. In normal cells, the organism uses apoptosis to remove cells under adverse conditions, such as senescent damage or genetic mutations, and to prevent excessive cell proliferation. This is important for the proliferation and differentiation of the organism and for maintaining the homeostasis of the internal environment (29). When apoptotic signaling is inhibited and mutations occur, cells will not be able to recognize and remove damaged and mutated cells, resulting in abnormal cell proliferation and eventually tumorigenesis (30). To date, only two classical apoptotic pathways have been elucidated in detail in mammalian cells. One is the extrinsic pathway, primarily triggered by the Fas death receptor, a member of the TNF (Tumor necrosis factor) receptor superfamily (31, 32). The other, the intrinsic pathway, involves the involvement of mitochondria, which respond to various noxious stimuli and release caspase-activating proteins to trigger apoptosis (33). Although generally considered to be separate pathways and capable of acting independently, cross-talk between these pathways can occur, eventually converging on downstream effector caspases (34).
At present, it has been demonstrated in humans and Drosophila that some members of the IAPs family can directly bind and inhibit caspases through their BIR domains, thereby achieving the purpose of anti-apoptosis (35). For example, in the human X chromosome, XIAP proteins bind and inhibit caspase-9 activation via the BIR3 domain, and at the same time combine the intramolecular region between BIR1 and BIR2 to antagonize caspase-3 and caspase-7, thereby inhibiting apoptosis (36, 37). XIAP is the only mammalian IAP that has been shown to directly inhibit caspase activity. ILP-2 as a member of the IAPs family, shares great structural similarity with XIAP, which also contains a BIR3 domain and therefore considered to be an apoptosis inhibitory protein that directly binds and inhibits caspases (38). However, it has been reported that the inhibitory effect of ILP-2 on caspase-3 and caspase-9 is much weaker than that of XIAP. Currently, the mechanism of ILP-2 anti-apoptosis is quite controversial. It has been demonstrated that although ILP-2 contains all the surface elements required for caspase inhibition, its BIR domain is inherently unstable due to the natural truncation of the amino terminus and cannot act directly as a caspase inhibitor. ILP-2 requires some binding partners to stabilize its polypeptide structure, which in turn facilitates the caspase inhibitory effect of ILP-2 (39). One possibility is that ILP-2 binds to pro-apoptotic molecules, including caspases and Smac, through their IBM (Inhibitor of apoptosis protein binding motifs) interactions and degrades them via the ubiquitinated proteasome pathway (40). And these are achieved through their own RING structural domains or by forming complexes with other RING-containing IAPs (41). It is thus clear that ILP-2, as an inhibitor of apoptosis, has not a direct but an indirect anti-apoptotic effect on cells, requiring interaction with some proteins to exercise its anti-apoptotic effect. In summary, although ILP-2 contains a BIR domain, it is a poor inhibitor of caspases. ILP-2 has little direct effect on endogenous caspase activity, and the antiapoptotic effect of ILP-2 may be due to its antagonism of XIAP-Smac interaction rather than direct inhibition of caspases-9 (42). Recent studies suggest that ILP-2 may have a unique mechanism to inhibit caspases and requires some intermediate protein complex to facilitate the association between caspases and ILP-2 (Figure 3).
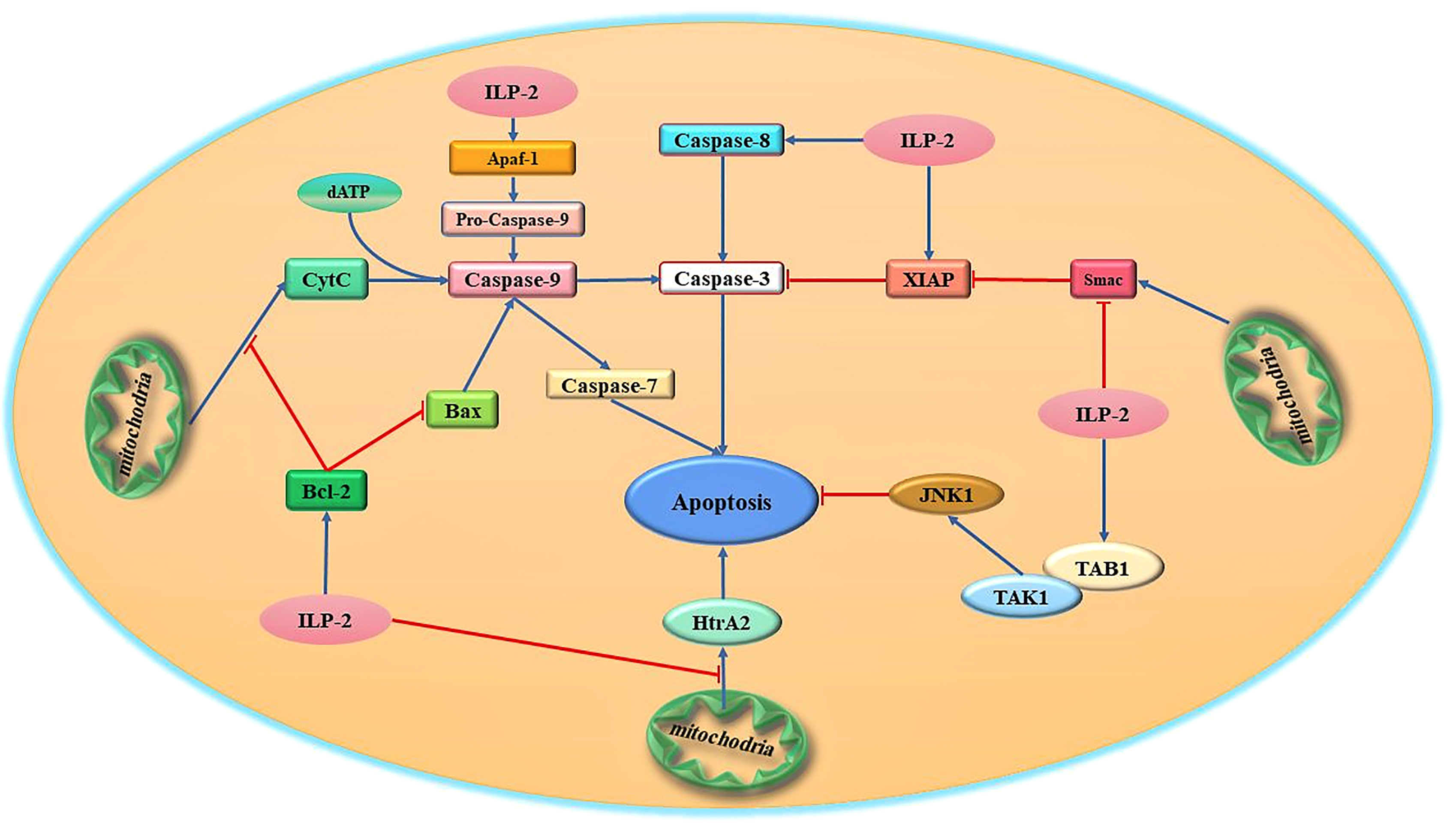
Figure 3 Schematic diagram of the major apoptotic signaling pathways related to ILP-2. ILP-2, which is highly expressed in tumor cells, is one of the high-risk factors for tumor discovery and development, and can activate multiple apoptosis-inhibiting signaling pathways and regulate the expression and activity of various apoptosis-related proteins to inhibit apoptosis.
Studies have shown that ILP-2 cannot inhibit apoptosis induced by Fas or TNF, but can effectively inhibit apoptosis induced by Bax (Bcl-2 associated X protein) pathway (43). In tumor cells, ILP-2 can inhibit apoptosis by regulating the endogenous pathway of apoptosis mediated by Bcl-2/Bax. ILP-2 can inhibit apoptosis by inducing the expression of Bcl-2 and inhibiting the production of Bax. The Bcl-2 protein family is composed of anti-apoptotic proteins (Bcl-2, Bcl-XL, etc.) and pro-apoptotic proteins (Bax, Bad, Bid, etc.) (44). This family of proteins usually functions as heterodimers or homodimers. When cells are stimulated by various intracellular and extracellular signals, the relative ratio between intracellular Bcl-2 and Bax determines whether apoptosis occurs or not. If the ratio of Bcl-2/Bax increases, the number of homodimers Bcl-2/Bcl-2 increases and apoptosis is inhibited. On the contrary, if the ratio of Bcl-2/Bax decreases, Bax/Bax homodimers are formed and apoptosis is promoted (45). It was found that ILP-2 could inhibit Bax production by promoting Bcl-2 expression, resulting in an increase in the Bcl-2/Bax ratio in cells, which in turn inhibited Bax pathway-induced apoptosis. More interestingly, ILP-2 can also inhibit the release of Cyt-c by inducing high expression of Bcl-2, preventing its entry into the cytoplasm, which in turn inhibits the binding of ATP/d ATP and inhibits the formation of apoptotic bodies (46, 47). This inhibits the activity and expression of caspase-9, resulting in the inability of caspase-9 to further cleave and activate the apoptosis effector caspase-3, thus achieving the goal of inhibiting apoptosis. Moreover, related studies have shown that ILP-2 can also interact with caspase-9 to form a heterodimer, which leads to the loss of its catalytic activity and the inability to cleave the effector caspase-3 zymogen to produce active caspase-3, thereby inhibiting apoptosis (48). However, it is still controversial whether ILP-2 can directly act on caspase-9 to inhibit apoptosis.
Bcl-2-like protein 1 (BCL2L1) is a member of the Bcl-2 family; it inhibits the activation of caspases and is a potent inhibitor of cell death (49). BCL2L1 can participate in calcium signaling regulation by binding to voltage-dependent anion channels (VDAC), reducing mitochondrial Ca2+ uptake and preventing the release of caspase activators from the mitochondrial membrane, thereby inhibiting cell death (50, 51). The apoptosis inhibitory protein ILP-2 is known to regulate apoptosis by controlling the expression of BCL2L1 and affecting the maintenance of mitochondrial morphology. Interestingly, BCL2L1 can also bind the tumor suppressor Beclin 1 to affect autophagy (52–54). Thus, ILP-2 regulates autophagy and affects apoptosis through a number of signaling pathways. Overexpression of BCL2L1 has been shown to protect endothelial cells from TNF-mediated apoptosis and inhibit NF-κB activation, thereby suppressing the upregulation of pro-inflammatory genes to participate in the inflammatory response (55, 56). In contrast, the protein of ILP-2 is closely linked to immune function as well as the development of inflammation, and thus its possible pathway is ILP-2 through promoting the expression of BCL2L1.
At present, studied have shown that ILP-2 protein can effectively inhibit apoptosis caused by (apoptotic protease activating factor-1) Apaf-1/caspase-9 pathway (10). Caspase-9 activation is achieved by the oligomerization of ILP-2 by the apoptotic proponent Apaf-1 in the presence of Cyt-c (cytochrome c). Cyt-c is a multifunctional protein produced by mitochondria and has the function of promoting apoptosis. In normal tissue cells, when cells receive apoptotic information, Cyt-c will be released from mitochondria into the cytoplasm if irreparable damage to DNA occurs, and binds to another apoptotic factor Apaf-1 to induce the onset of apoptosis (57). After cells receive apoptotic signals, Cyt-c is produced by mitochondria and released into the cytoplasm, where it interacts with Apaf-1 to enhance binding to ATP/d ATP, which in turn multimerizes the CARD domain of Apaf-1 protein to form a complex of protein and nucleotide containing 7-axis symmetry (apoptosome) (58). Apaf-1 is structurally altered in the apoptosome, recruits caspase-9 and causes caspase-9 to be cleaved into two segments for activation. The activated caspase-9 further cleaves and activates the activity of the apoptosis effector enzyme caspase-3, thereby promoting apoptosis. The Bcl-2 protein family plays an important role in maintaining the integrity of the mitochondrial membrane (59, 60). Studies have shown that overexpressed ILP-2 can effectively inhibit the Bax-induced apoptosis pathway and inhibit the release of Cyt-c from mitochondria. After ILP-2 binds to Apaf-1, Apaf-1 cannot recruit caspase-9, thereby inhibiting apoptosis induced by Apaf-1/caspase-9 pathway (61).
In the endogenous pathway of apoptosis, mitochondria are at the center (62). When cells are stimulated by internal apoptotic signals, they release relevant apoptotic factors to the cytoplasm by altering the molecular structure of the mitochondrial membrane and its permeability, thus affecting the occurrence of apoptosis (63). Among them, Cyt-c and Smac (second mitochondria derived activator of caspase) are represented. Smac, also known as DIABLO (direct IAP binding protein whit low pI), is a mitochondria-derived activator of caspase (64, 65). It is also a pro-apoptotic protein produced by mitochondria that contains IAP structural domains (IBMs) and is usually found in the membrane gap of mitochondria (66). It is released from mitochondria and binds to IAP, releasing caspases closed by IAP, relieving the inhibitory effect of IAP on caspases, activating the cascade response of caspases and promoting apoptosis (67, 68). Cyt-c and Smac are considered to be endogenous apoptosis activators (69). Smac is released from the mitochondria during apoptosis together with Cyt-c and Smac can further act on caspase-9 activity in the cytoplasm, resulting in the inability of caspase-9 to activate caspase-7/3 activity (70). The amount of Cyt-c and Smac in the cytoplasm is positively correlated with apoptosis is positively correlated. The dual requirement of both ensures that the caspases cascade is activated only when the signal is sufficient (71).
Studies have reported that Smac can bind to the BIR structural domain and deprive IAPs of their inhibitory effects through physical interactions (72). ILP-2 has a high affinity for Smac, but is significantly inferior to XIAP in inhibiting caspases (73). The highly active interaction of ILP-2 with Smac is similar to that of XIAP, and this particular interaction can be completely abolished by single amino acid mutation in the amino-terminal sequence of Smac or in the BIR domain of ILP-2 (74). The study reports that the high affinity of ILP-2 for Smac would effectively compete with the XIAP-Smac interaction and regulate apoptosis by sequestering Smac and preventing its resistance to XIAP-mediated inhibition of caspases (75). Thus, ILP-2 may become an inhibitor of apoptosis rather than a direct suppressor of caspases. This study further identifies the structural basis for this difference and shows that substitution of just three residues increases the caspase-9 inhibitory activity of ILP-2 to similar levels as observed in XIAP (76, 77). Therefore, ILP-2 mainly contributes to the inhibition of caspase by blocking the ability of Smac to disrupt the XIAP-caspase interaction. Recently, studies have reported that highly expressed ILP-2 can neutralize the pro-apoptotic effect of Smac in tumor cells (78). The highly expressed ILP-2 in the cytoplasm will produce a large amount of anti-apoptotic information, resulting in changes in the conformation of mitochondrial membrane molecules, and a large amount of ILP-2 specifically binds to Smac, so that Smac cannot release the blocking effect of caspase-IAP, thereby antagonizing Smac pro-apoptotic effect (79). Moreover, ILP-2 can also reduce the relative amount of free Smac in the cytoplasm by activating the ubiquitination pathway, and the low level of Smac in the cytoplasm cannot express its caspase protein kinase activity to activate related caspases, which leads to the inhibition of apoptotic signal generation and thus inhibits apoptosis (80, 81). In addition, ILP-2 is able to further bind activated caspases, which in turn inhibits the activity of caspases, thus achieving inhibition of apoptosis.
IAPs seem to have a feedback effect whereby a single protein can achieve the same purpose through different biochemical mechanisms (82). Although we know that most studies on ILP-2 have focused on its role as a caspase inhibitor, there is growing evidence that it also achieves this through other mechanisms (83). It is now accepted that the selective activation of the mitogen-activated protein kinase Jun NH2-terminal kinase 1 (JNK1) is the case for XIAP as well as ILP-2 (84). Studies have shown that ILP-2 can activate JNK1 by activating TAB1, and activated JNK1 is necessary to protect cells from apoptosis induced by TNF-α and interleukin converting enzyme (85). Although it is not clear what role the BIR domain of ILP-2 plays in the mitogen-activated protein kinase pathway, these results suggest that the mechanism of BIR domain function may also differ among IAP family members in response to different signaling molecules. In this way, it is likely that ILP-2 mediates the activation of JNK1 through the TAK1 (transforming growth factor protein kinase 1)/TAB1 (TAK1-binding protein) signaling cascade, thereby inhibiting apoptosis (86). Importantly, TAK1 and TAB1/2 act as important regulators in pyroptosis and necroptosis, which implies that ILP-2 may have a regulatory role in the immune inflammatory response induced by necroptosis and pyroptosis. Further studies of these phenomena may provide important information for disease treatment.
Mitochondrial serine protease (Omi/HtrA2), released from mitochondria into cytosol during apoptosis (87). In normal cells, HtrA2 can inhibit the activity of inhibitors of apoptosis IAPs and promote caspase-independent cell death by directly binding to the BIR domain of apoptosis protein inhibitors IAPs (88). Omi/HtrA2 can directly cleave various IAPs in vitro and the efficiency of cleavage is determined by its IAP-binding motif AVPS (89). Cleavage of IAPs such as ILP-2 greatly reduces their ability to inhibit and ubiquitinate caspases. Compared to the stoichiometric anti-IAP activity of Smac/DIABLO, cleavage of ILP-2 by Omi/HtrA2 was catalytic and irreversible, resulting in more efficient inactivation of IAPs and promotion of caspase activity (90, 91). In conclusion, these results suggest that, unlike Smac/DIABLO, catalytic cleavage of IAPs by Omi/HtrA2 is a key mechanism for its irreversible inactivation of IAPs and promotion of apoptosis (92). Importantly, when ILP-2 is mutated or dysregulated in expression, ILP-2 can inhibit the release of HtrA2 from mitochondria in multiple ways, antagonizing the inhibitory effect of HtrA2 on IAPs proteins and thus inhibiting apoptosis (93). However, this pathway needs to be further explored.
It is known that some HOX (homeobox) genes play an important role in a variety of tumor diseases (94). Human Hox genes can be divided into four gene clusters, namely HOXA, HOXB, HOXC and HOXD, which exist on different chromosomes. In addition, these genes can be divided into 13 paralogous families (paralogous families), represented by numbers, HOXD8 is a member of the HOXD family. Studies have shown that HOXD8 has an important regulatory role in tumorigenesis and progression (95). Studies have reported that HOXD8 can bind to the ILP-2 promoter and regulate the expression of ILP-2 (96). When the expression of ILP-2 increased, it inhibited the expression of HOXD8 through a negative feedback pathway. It has been reported that ILP-2, which is highly expressed in breast cancer, can inhibit the expression of HOXD8 in breast cancer tissues and cell lines through certain pathways. When the expression of HOXD8 is inhibited, it can further activate the PI3K/Akt signaling pathway to promote cancer cells. proliferation, invasion and migration (97). Furthermore, ILP-2 knockout could reverse the effects of HOXD8 knockdown on breast cancer cell proliferation, invasion, and migration. Thus, it was confirmed that the synergistic effect between the two has an important regulatory role for tumor cells in growth and invasion (Figure 4).
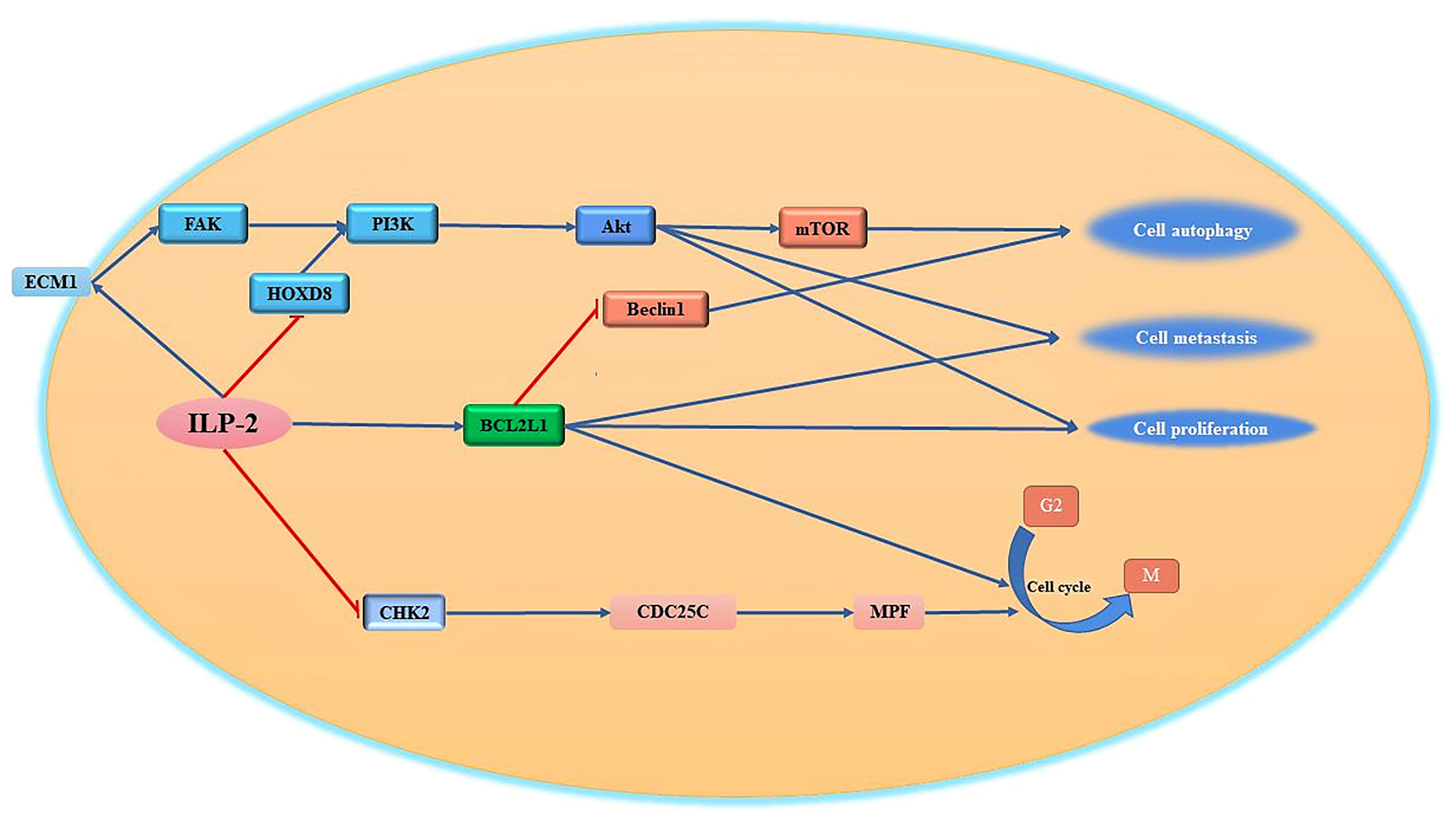
Figure 4 Regulation of cell proliferation, migration and cell cycle by ILP-2. ILP-2 activates Bcl-2, ECM1-Akt signaling pathway, inhibits CHK2 expression and promotes cell proliferation, autophagy, migration as well as cell cycle transition.
One of the most remarkable characteristics of tumor cells is their high invasiveness and their ability to escape the pro-apoptotic effects of various tumor drugs by metastasizing to other tissues through various pathways (98). The invasive ability of tumor cells is closely related to the tumor microenvironment composed of extracellular matrix (ECM) (99). The extracellular matrix can regulate cell adhesion, growth, proliferation and differentiation through various signaling pathways (100). At present, it has been reported that the apoptosis inhibitory protein ILP-2 interacts with extracellular matrix protein 1 (ECM1) to regulate cell growth and cell migration (101). ECM1 can bind to integrins on the cell surface and then bind to the N-terminal domain of focal adhesion kinase (FAK) to activate FAK, which can inhibit tumor cell apoptosis by mediating the Akt pathway, promote tumor cell surface adhesion, and activate phosphatidylinositol-3-kinase (PI3K) (102–104). It was reported that the high expression of ILP-2 in breast cancer cells further activates FAK activity by promoting cellular secretion of ECM1, and the activated FAK further activates downstream protein kinase PI3K activity through the signaling pathway, and the activated PI3K further transmits external signals to the cell interior through the second messenger to further transmit signals to the downstream pathway, activating Akt to regulate cell proliferation, differentiation and metastasis (105, 106) (Figure 4).
BCL2L1 is a regulator of the G2 checkpoint and cytokinesis during mitosis; it has an important regulatory role in cell proliferation (107). ILP-2 can promote the high expression of BCL2L1, activate the Bcl2L1 signaling pathway, and then promote cell proliferation. Importantly, ILP-2 has been shown to promote epithelial-mesenchymal transition, migration, invasion and proliferation in a variety of tumor cells. Cell cycle checkpoint protein kinase 2 (CHK2) is an important checkpoint in the cell cycle (108). It is a tumor suppressor gene or tumor-suppressor gene similar to P53 (109, 110). It is a negative regulator in the process of normal cell proliferation (111). At the checkpoint of the cycle, it can prevent the cycle transition or promote cell apoptosis (112). If the tumor suppressor gene is mutated or loses its function, it will cause the cell cycle to go out of control and over-proliferate, eventually causing the cell to become cancerous. It has been reported that increased expression of ILP-2 prevents the inhibitory activity of CHK2 on CDC25C (113), thereby promoting the transition of the tumor cell cycle from G2 to M phase and supporting tumor cell proliferation (114, 115).
In addition, ILP-2 may also regulate the tumor microenvironment by activating ECM1, which is a major component of the extracellular microenvironment, and ILP-2 may promote its expression in tumor cells by activating ECM1, thus helping tumor cells to detach from intercellular adhesion and promoting tumor cell invasion. mediated pathways, activating NF-κB and TNF signaling pathways, linking the tumor microenvironment with tumor cells, and playing an important regulatory role in the overall tumorigenesis and development. However, the effects of ILP-2 on the tumor microenvironment are still poorly understood and need to be explored more deeply.
Inflammation, one of the several features of tumors. Non-controllable inflammation is closely related to tumorigenesis and progression, as well as invasion and metastasis. In addition, with the help of certain factors, tumor cells are able to evade immune surveillance and thus maintain their survival. ILP-2 protein, in addition to its classical anti-apoptotic role, seems to be involved in the regulation of tumor immunity and inflammation and other related functions. ILP-2 belong to immunoglobin superfamily acts as a genetic home, having many leukocyte Ig-like receptors, killer cells, several ICAMs and PSG (pregnancy-specific glycoprotein) family and Fc receptors (FcRs) (17). This family of proteins can regulate cellular physiological activity by modulating T-cell receptor signaling as well as interleukin-mediated immune inflammatory pathways. ILP-2 can also promote ubiquitination and degradation pathways of the proteasome itself with several binding ligands through its E3 ubiquitination ligase function, including NF-κB, RIPK1 (Receptor-interacting The kinase activity of RIPK1 is essential for a variety of complexes that regulate inflammation (complex I), apoptosis (complex IIa), and necroptosis (complex IIb). phosphorylation at the Ser320 locus, which fails to inhibit the caspase-8-mediated pyroptosis and necroptosis pathways, thereby promoting tumor cell growth and migration (10, 21). Importantly, ILP-2 may also promote tumor cell development and progression by regulating immune and related inflammatory responses through regulation of Smac, BCL2L1 or TAB1-mediated pyroptosis or necroptosis pathways (32). However. The mechanisms by which ILP-2 regulates tumor immunity and inflammation are unclear and need to be further explored.
ILP-2 is undetectable in most normal tissues except testis, spinal cord and lymph nodes, but is present in transformed cells and some cancers including breast, liver, nasopharyngeal, neuroblastoma and hematologic tumors (116). Especially in breast cancer, nasopharyngeal cancer, hematological tumor and liver cancer cells showed overexpression of ILP-2 mRNA. In a comparison of serum samples from breast cancer patients with healthy women, women with mastopexy, women with other types of cancer or women after breast cancer surgery, ILP-2 expression was found to be increased in breast cancer patients (117). Furthermore, ILP-2 was found to be overexpressed in breast cancer tissues and breast cancer cells by immunohistochemistry and western blot (118). When the expression of ILP-2 was detected patients with chronic myeloid leukemia (CML), it was found that the expression of ILP-2 was significantly higher in CML patients than in normal hematopoietic cells. Moreover, when tyrosine kinase inhibitor resistance developed in CML patients, ILP-2 expression significantly decreased (15). Importantly, ILP-2 expression was significantly increased in bone marrow cells of patients with myelodysplastic syndrome (119). It was found that in hepatocellular carcinoma HepG2 cells were exposed to FumonisinB1 (FB1) and increased ILP-2 mRNA (5.7-fold) and protein (2.3-fold) expression was detected, indicating that ILP-2 can promote liver tumorigenesis (120). The guanine nucleotide exchange factor 3 gene ARHGEF3 of upregulation mediates ILP-2 expression contributing to nasopharyngeal carcinogenesis and progression (121). By inhibiting ILP-2 expression in a xenograft model of adult neuroblastoma, it is able to promote radio sensitization and lead to cell death (122). In recent studies, ILP-2 was found to be possibly involved in the progression of bladder cancer and was used as a marker for early recurrence. In summary, ILP-2 highly expressed in tumor cells, is closely associated with tumorigenesis and progression. It can affect cell proliferation, apoptosis and invasion by synergizing with several key signaling proteins, triggering cellular carcinogenesis and resulting in tumorigenesis (123). In most of the investigated ILP-2-expressing tumors, high levels of the protein are a predictive marker of tumor progression, which can provide prognosis-related information.
At present, the functional mechanism of IAPs regulating tumor cell growth is continuously deepened, and various strategies for innovative therapy of IAPs have begun to emerge (124). Based on its structural characteristics and prominent role in apoptosis, ILP-2 could be a new target for tumor therapy, suitable for molecular antagonists, vaccination strategies, small molecule inhibitors and gene therapy. The following is a brief discussion of a portfolio of novel cancer therapeutic strategies targeting ILP-2 at different biological levels (Figure 5).
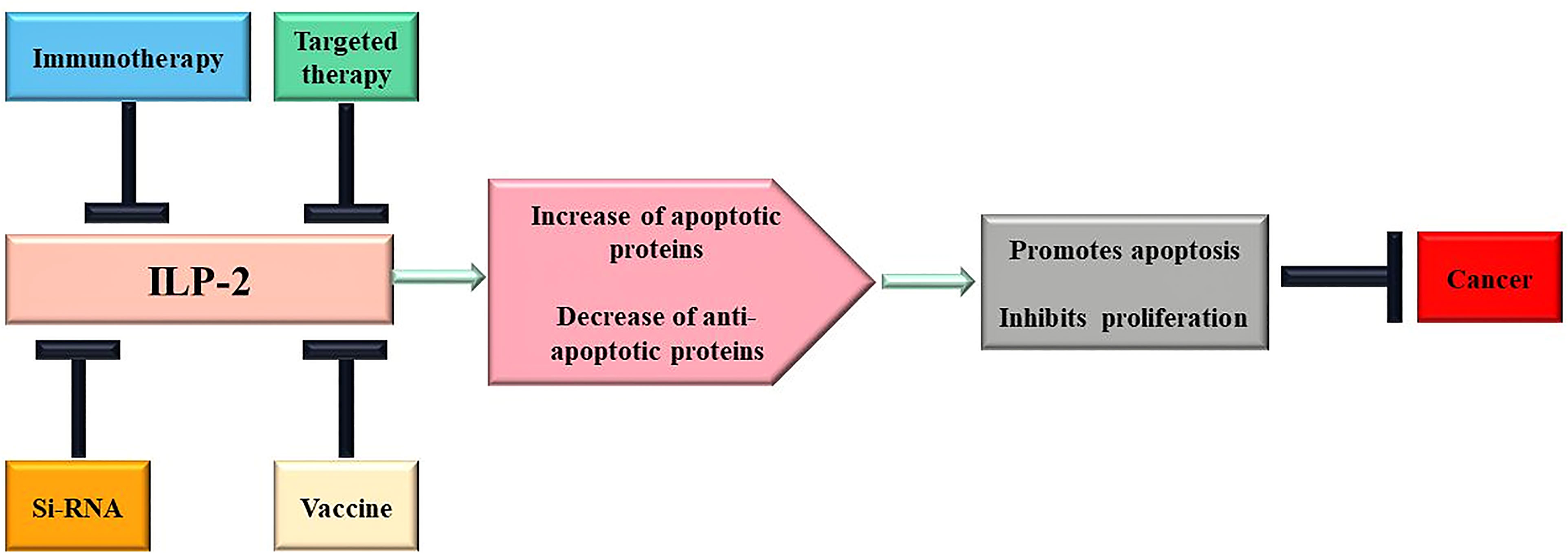
Figure 5 Scheme of therapeutic approaches potentially triggered by ILP-2. Multiple changes of apoptotic proteins contribute to cancer development and progression, and treatment targeting ILP-2 can be developed to restore normal sensitivity to apoptotic stimuli and, therefore, repress cancer.
Xiang et al. (117), analyzed 400 breast cancer serum samples and 40 non-cancer serum samples (i.e., healthy controls) using bidirectional gel electrophoresis (P<0.001). Western blot analysis (P<0.05) was then performed on 10 breast cancer serum samples and 10 non-cancer serum samples. Finally, 35 serum samples from healthy controls or subjects with breast cancer, other types of cancer, galactorrhea or postoperative breast cancer were analyzed by 2DE and enzyme-linked immunosorbent assay. The results showed that ILP-2 expression was significantly higher in serum from breast cancer patients than in serum samples from other groups of women.ILP-2 is a novel biomarker for breast cancer in peripheral blood.
Zhu et al (118), further found that ILP-2 was overexpressed in breast cancer tissues and breast cancer cell lines HCC-1937, MX-1 and MCF-7 by immunohistochemistry and western blot (P<0.01). Then, by knockdown the expression of ILP-2 via RNA interference technique, it was found that inhibition of ILP-2 could induce apoptosis and inhibit breast cancer cell migration, thus confirming the survival function of ILP-2 in breast cancer cells, involved in supports the migration ability of cells. In addition, proteomic studies have been showned that in breast cancer cells, high expression of ILP-2 can synergize with multiple signaling proteins to promote the growth and invasion of breast cancer cells and assist cells to escape apoptosis. These high-risk factors, together with ILP-2, are involved in the growth and proliferation of breast cancer cells, signal transduction and regulation of the immune system (125).
Glodkowska et al. (15), compared the expression of BIRC family genes in chronic myeloid leukemia (CML) patients cells and normal hematopoietic cells, found a significant decrease in ILP-2 expression after the development of tyrosine kinase inhibitor resistance in chronic phase CML patients. It was also hypothesized that ILP-2 does not act as a classical inhibitor of apoptosis in leukemic cells, but may actually act as a decoy for other anti-apoptotic proteins that are activated during the development of the disease. And raised the possibility of using ILP-2 expression level as a prognostic/predictive marker after further validation in clinical studies. This result suggests that ILP-2 does not display a classical apoptosis inhibitory function in leukemic cells. The potential role of ILP-2 in CML progression needs further investigation.
Abes et al. (119) studied myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) and found that ILP-2 expression was significantly increased in myeloid cells of patients with MDS (P<0.05). The experimental results show that these BIRC proteins are overexpressed in the early stage of leukemic transformation and can act as trigger factors to induce the expression of other IAPs family members to trigger the occurrence of the disease.
Sun Peng et al. (71) studied acute leukemia and found that the mRNA expression level of ILP-2 in the initial treatment group and the relapsed group was significantly higher than control group (P<0.05). Concluded that the development and progression of acute leukemia are closely related to the expression levels of ILP-2, and that high expression of ILP-2 may increase the insensitivity of leukemia to chemotherapeutic drugs, thereby reducing the therapeutic effect of the drugs.
Chuturgoon et al. (120) studied the inhibition of apoptosis in HepG2 cells by FumonisinB1 (FB 1), they found that the mycotoxin fumonisin B1 (FB1) produced by Fusarium sp can induce apoptosis resistance in HepG2 cell lines. In the process of increasing the dose of FB1, the results of qPCR showed that the expression of ILP-2 mRNA was significantly increased (P<0.001), while the expression of Caspase-9 and Caspase-3,7 was significantly down-regulated. Western blot results further showed that the expression of ILP-2 protein was increased, and the expression level of the pro-apoptotic protein Smac protein was significantly downregulated. The results show that the increased expression of ILP-2 can promote the growth of hepatoma cells and inhibit the apoptosis, which plays an important role in the process of hepatocarcinogenesis.
Liu et al. (121) found that upregulation of ARHGEF3 could promote ILP-2 expression and promote nasopharyngeal carcinogenesis and progression (P<0.01). In nasopharyngeal carcinoma cells, ARHGEF3 inhibits apoptosis by regulating the expression of ILP-2, which in turn inhibits the activation of caspases-3. The downregulation of ARHGEF3 by siRNA could inhibit the expression of ILP-2 and thus induce apoptosis. Thus, it is clear that the increased expression of ARHGEF3 prevents apoptosis through the upregulation of ILP-2 (P<0.01), which plays a key oncogenic role in the pathogenesis of nasopharyngeal carcinoma.
Veeraraghavan et al. (122) found that inhibiting the expression of ILP-2 (More than 8 times) in a neuroblastoma xenograft model promoted the expression of BAK1, BAX, Caspases, and CARD, which in turn promoted radio sensitization and resulted in cell death. This effect is caused by activation of pro-apoptotic signals and inhibition of anti-apoptotic genes, including IAPs family members NAIP and ILP-2 (P<0.01). Therefore, inhibiting the expression of ILP-2 may enhance the radiotherapy effect of neuroblastoma (NB).
Chen et al. (126) used immunohistochemical analysis in bladder transitional cell carcinoma and found that there were a large number of brown-yellow fine particles in the cytoplasm, indicating that the expression of ILP-2 was high in the cytoplasm. The positive expression level of ILP-2 in bladder transitional cell carcinoma was significantly higher than that in normal bladder tissue (P<0.05). The expression level has a certain effect on the growth and proliferation of tumor cells, mainly in the aspects of cell proliferation and infiltration ability.
In conclusion, it is undeniable that ILP-2 plays an extremely important role in tumorigenesis and development. Although the therapeutic use of ILP-2 remains to be determined, its potential utility in the early diagnosis of cancer is indisputable. Expression of ILP-2 in tumors is associated with a more aggressive phenotype, shorter survival time and reduced response to chemotherapy. Recent studies suggest that it can be used as a marker for early diagnosis, a prognostic indicator to aid patient management, or to monitor disease endpoints during and after treatment. Overall, the link between ILP-2 and tumor destruction outlined here should stimulate further work to target this protein for therapeutics.
At present, more and more evidences are pointing out that the abnormal expression of tumor suppressor gene ILP-2 is closely related to the occurrence of tumors, and it may be one of the main high-risk factors leading to cell carcinogenesis. In particular, abnormally high expression of ILP-2 in breast, blood and bladder cancers is closely associated with the development of these neoplastic diseases. The high expression of ILP-2 in tumor cells can help tumor cells escape apoptosis through various signaling pathways, and promote the growth, proliferation, migration and invasion of cancer cells (127). According to the related properties of ILP-2 in tumor cells, it is reasonable to speculate that ILP-2 can be used as a biomarker for early detection of tumors and is a new target for human tumor therapy. It can be seen that the in-depth study of the biological mechanism of ILP-2 in the process of tumor occurrence and development will provide new ideas for tumor treatment (128).
It is worth noting that the inhibitory effect of ILP-2 itself on apoptosis is weak. However, it is still highly expressed in most tumor cells, and cooperates with other key signaling proteins to further amplify its role to regulate the growth, proliferation, migration, apoptosis and other physiological functions of tumor cells. However, after knocking down the expression of ILP-2 by siRNA, the apoptosis rate of tumor cells was significantly increased, the migration ability was decreased, and tumor cell growth was significantly inhibited (129). In addition, many studies have also shown that the effect of radiotherapy and chemotherapy is significantly improved after combined with siRNA to knock down the expression of ILP-2. It can be seen that the use of siRNA to knock down the expression of ILP-2 has a very high potential in the clinical treatment of tumors. More interestingly, since ILP-2 protein has multiple leukocyte Ig-like receptors, natural killer cells, ICAMs and Fc receptors (FcRs), it seems to play an important role in immune-related functions (130). All these evidences directly or indirectly point to a possible close association of ILP-2 with tumorigenesis and development, which is of great research value.
Although the details of the multiple pathways from the ILP-2 network have not been fully elucidated, there is consensus that ILP-2 is an appropriate therapeutic target for the effective treatment of cancer. Today, targeted therapy for tumors is increasingly used in the clinical management of oncology patients. However, there are not many targets that can be used as targeted therapy for tumors, and there is also a certain degree of drug resistance. Therefore, it is urgent to find more effective targets for tumor therapy. We have reason to believe that with the continuous research on ILP-2, it will provide new ideas for the early clinical diagnosis and treatment of human tumors.
ZZ: Writing—original draft and review and editing, software, validation, and visualization. SX: Writing, data curation, and visualization. RC: Writing, formal analysis, and supervision. HP: Data curation, formal analysis, and supervision. RM: Data curation, formal analysis, and visualization. MX: Writing—review and editing, funding acquisition, investigation, and resources.
This study was supported by the National Natural Science Foundation of China (No.81360397), Hunan Provincial Natural Science Foundation of China (No.2020JJ4513), Scientific Research Foundation of Hunan Provincial Education Department of China (No.19A400).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We also thank the National Natural Science Foundation of China (No.81360397), Hunan Provincial Natural Science Foundation of China (No.2020JJ4513), Scientific Research Foundation of Hunan Provincial Education Department of China (No.19A400) for continued support of our research.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A, et al. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Boice A, Bouchier-Hayes L. Targeting Apoptotic Caspases in Cancer. Biochim Biophys Acta (BBA) - Mol Cell Res (2020) 1867(6):118688. doi: 10.1016/j.bbamcr.2020.118688
3. de Almagro MC, Vucic D. The Inhibitor of Apoptosis (IAP) Proteins are Critical Regulators of Signaling Pathways and Targets for Anti-Cancer Therapy. Exp Oncol (2012) 34(3):200–11. doi: 10.1007/s10549-012-2352-6
4. Cossu F, Milani M, Mastrangelo E, Lecis D. Targeting the BIR Domains of Inhibitor of Apoptosis (IAP) Proteins in Cancer Treatment. Comput Struct Biotechnol J (2019) 17:142–50. doi: 10.1016/j.csbj.2019.01.009
5. Nachmias B, Ashhab Y, Ben-Yehuda D. The Inhibitor of Apoptosis Protein Family (IAPs): An Emerging Therapeutic Target in Cancer. Semin Cancer Biol (2004) 14(4):231–43. doi: 10.1016/j.semcancer.2004.04.002
6. Srinivasula SM, Ashwell JD. IAPs: What's in a Name? Mol Cell (2008) 30(2):123–35. doi: 10.1016/j.molcel.2008.03.008
7. Hunter AM, LaCasse EC, Korneluk RG. The Inhibitors of Apoptosis (IAPs) as Cancer Targets. Apoptosis (2007) 12(9):1543–68. doi: 10.1007/s10495-007-0087-3
8. Smolewski P, Robak T. Inhibitors of Apoptosis Proteins (IAPs) as Potential Molecular Targets for Therapy of Hematological Malignancies. Curr Mol Med (2011) 11(8):633–49. doi: 10.2174/156652411797536723
9. Khalili S, Mohammadpour H, Shokrollahi Barough M, Kokhaei P. ILP-2 Modeling and Virtual Screening of an FDA-Approved Library:a Possible Anticancer Therapy. Turk J Med Sci (2016) 46(4):1135–43. doi: 10.3906/sag-1503-2
10. Richter BW, Mir SS, Eiben LJ. Molecular Cloning of ILP-2, a Novel Member of the Inhibitor of Apoptosis Protein Family. Mol Cell Biol (2001) 21(13):4292–301. doi: 10.1128/MCB.21.13.4292-4301.2001
11. Lagacé M, Xuan JY, Young SS. Genomic Organization of the X-Linked Inhibitor of Apoptosis and Identification of a Novel Testis-Specific Transcript. Genomics (2001) 77(3):181–8. doi: 10.1006/geno.2001.6635
12. LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG, et al. IAP-Targeted Therapies for Cancer. Oncogene (2008) 27(48):6252–75. doi: 10.1038/onc.2008.302
13. Hassan M, Watari H, AbuAlmaaty A, Ohba Y, Sakuragi N. Apoptosis and Molecular Targeting Therapy in Cancer. BioMed Res Int (2014) p 2014:150845. doi: 10.1155/2014/150845
14. Hu Y, Benedict MA, Wu D, Inohara N, Núñez G. Bcl-XL Interacts With Apaf-1 and Inhibits Apaf-1-Dependent Caspase-9 Activation. Proc Natl Acad Sci USA (1998) 95(8):4386–91. doi: 10.1073/pnas.95.8.4386
15. Glodkowska-Mrowka E, Solarska I, Mrowka P. Differential Expression of BIRC Family Genes in Chronic Myeloid Leukaemia–BIRC3 and BIRC8 as Potential New Candidates to Identify Disease Progression. Br J Haematol (2014) 164(5):740–2. doi: 10.1111/bjh.12663
16. Shin S, Sung BJ, Cho YS. An Anti-Apoptotic Protein Human Survivin is a Direct Inhibitor of Caspase-3 and -7. Biochemistry (2001) 40(4):1117–23. doi: 10.1021/bi001603q
17. Saleem M, Qadir MI, Perveen N. Inhibitors of Apoptotic Proteins: New Targets for Anticancer Therapy. Chem Biol Drug Des (2013) 82(3):243–51. doi: 10.1111/cbdd.12176
18. Oberoi-Khanuja TK, Murali A, Rajalingam K. IAPs on the Move: Role of Inhibitors of Apoptosis Proteins in Cell Migration. Cell Death Dis (2013) 4(9):e784. doi: 10.1038/cddis.2013.311
19. Dubrez L, Berthelet J, Glorian V. IAP Proteins as Targets for Drug Development in Oncology. Onco Targets Ther (2013) 9:1285–304. doi: 10.2147/OTT.S33375
20. Kim JY, Jeon YK, Jeon W, Nam MJ. Fisetin Induces Apoptosis in Huh-7 Cells via Downregulation of BIRC8 and Bcl2L2. Food Chem Toxicol (2010) 48(8-9):2259–64. doi: 10.1016/j.fct.2010.05.058
21. Bartke T, Pohl C, Pyrowolakis G, Jentsch S. Dual Role of BRUCE as an Antiapoptotic IAP and a Chimeric E2/E3 Ubiquitin Ligase. Mol Cell (2004) 14(6):801–11. doi: 10.1016/j.molcel.2004.05.018
22. Middleton AJ, Budhidarmo R, Day CL. Use of E2~ubiquitin Conjugates for the Characterization of Ubiquitin Transfer by RING E3 Ligases Such as the Inhibitor of Apoptosis Proteins. Methods Enzymol (2014) 545:243–63. doi: 10.1016/B978-0-12-801430-1.00010-X
23. Fulda S. Inhibitor of Apoptosis Proteins as Targets for Anticancer Therapy. Expert Rev Anticancer Ther (2007) 7(9):1255–64. doi: 10.1586/14737140.7.9.1255
24. Shin H, Renatus M, Eckelman Brendan P, Nunes Viviane A, Sampaio Claudio AM, Salvesen Guy S, et al. The BIR Domain of IAP-Like Protein 2 is Conformationally Unstable: Implications for Caspase Inhibition. Biochem J (2004) 385(1):1–10. doi: 10.1016/j.jss.2008.11.211
25. Feltham R, Khan N, Silke J. IAPS and Ubiquitylation. IUBMB Life (2012) 64(5):411–8. doi: 10.1002/iub.565
26. Gyrd-Hansen M, Darding M, Miasari M. IAPs Contain an Evolutionarily Conserved Ubiquitin-Binding Domain That Regulates NF-kappaB as Well as Cell Survival and Oncogenesis. Nat Cell Biol (2008) 10(11):1309–17. doi: 10.1038/ncb1789
27. Borden KL, Freemont PS. The RING Finger Domain: A Recent Example of a Sequence-Structure Family. Curr Opin Struct Biol (1996) 6(3):395–401. doi: 10.1016/S0959-440X(96)80060-1
28. Barathan M, Gopal K, Mohamed R. Chronic Hepatitis C Virus Infection Triggers Spontaneous Differential Expression of Biosignatures Associated With T Cell Exhaustion and Apoptosis Signaling in Peripheral Blood Mononucleocytes. Apoptosis (2015) 20(4):466–80. doi: 10.1007/s10495-014-1084-y
29. Hrdinka M, Yabal M. Inhibitor of Apoptosis Proteins in Human Health and Disease. Genes Immun (2019) 20(8):641–50. doi: 10.1038/s41435-019-0078-8
30. Finlay D, Teriete P, Vamos M, Cosford NDP, Vuori K. Inducing Death in Tumor Cells: Roles of the Inhibitor of Apoptosis Proteins. F1000Res (2017) 6:587. doi: 10.12688/f1000research.10625.1
31. Wu H, Tschopp J, Lin SC. Smac Mimetics and TNFalpha: A Dangerous Liaison? Cell (2007) 131(4):655–8. doi: 10.1016/j.cell.2007.10.042
32. Huerta S, Heinzerling JH, Anguiano-Hernandez YM. Modification of Gene Products Involved in Resistance to Apoptosis in Metastatic Colon Cancer Cells: Roles of Fas, Apaf-1, NFkappaB, IAPs, Smac/DIABLO, and AIF. J Surg Res (2007) 142(1):184–94. doi: 10.1016/j.jss.2006.12.551
33. Ilmarinen P, Moilanen E, Kankaanranta H. Mitochondria in the Center of Human Eosinophil Apoptosis and Survival. Int J Mol Sci (2014) 15(3):3952–69. doi: 10.3390/ijms15033952
34. Wen X, Lin ZQ, Liu B, Wei YQ. Caspase-Mediated Programmed Cell Death Pathways as Potential Therapeutic Targets in Cancer. Cell Prolif (2012) 45(3):217–24. doi: 10.1111/j.1365-2184.2012.00814.x
35. Fulda S. Targeting Inhibitor of Apoptosis Proteins (IAPs) for Cancer Therapy. Anticancer Agents Med Chem (2008) 8(5):533–9. doi: 10.2174/187152008784533107
36. Cullen SP, Martin SJ. Caspase Activation Pathways: Some Recent Progress. Cell Death Differ (2009) 16(7):935–8. doi: 10.1038/cdd.2009.59
37. Ng CP, Bonavida B. X-Linked Inhibitor of Apoptosis (XIAP) Blocks Apo2 Ligand/Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Mediated Apoptosis of Prostate Cancer Cells in the Presence of Mitochondrial Activation: Sensitization by Overexpression of Second Mitochondria-Derived Activator of Caspase/Direct IAP-Binding Protein With Low Pl (Smac/DIABLO). Mol Cancer Ther (2002) 1(12):1051–8.
38. Scott FL, Denault JB, Riedl SJ, Shin H, Renatus M, Salvesen GS. XIAP Inhibits Caspase-3 and -7 Using Two Binding Sites: Evolutionarily Conserved Mechanism of IAPs. EMBO J (2005) 24(3):645–55. doi: 10.1038/sj.emboj.7600544
39. Silke J, Vucic D. IAP Family of Cell Death and Signaling Regulators. Methods Enzymol (2014) 545:35–65. doi: 10.1016/B978-0-12-801430-1.00002-0
40. Duckett CS. IAP Proteins: Sticking it to Smac. Biochem J (2005) 385(Pt 1):e1–2. doi: 10.1042/BJ20041800
41. Gowda Saralamma VV, Lee HJ, Raha S. Inhibition of IAP's and Activation of P53 Leads to Caspase-Dependent Apoptosis in Gastric Cancer Cells Treated With Scutellarein. Oncotarget (2018) 9(5):5993–6006. doi: 10.18632/oncotarget.23202
42. Hao Y, Sekine K, Kawabata A. Apollon Ubiquitinates SMAC and Caspase-9, and has an Essential Cytoprotection Function. Nat Cell Biol (2004) 6(9):849–60. doi: 10.1038/ncb1159
43. Drouet A, Le Moigne F, Have L, Blondet R, Jacquin O, Chauvin F. Common Peroneal Nerve Palsy Following TNF-Based Isolated Limb Perfusion for Irresectable Extremity Desmoid Tumor. Orthop Traumatol Surg Res (2009) 95(8):639–44. doi: 10.1016/j.otsr.2009.09.009
44. Antonsson B, Martinou JC. The Bcl-2 Protein Family. Exp Cell Res (2000) 256(1):50–7. doi: 10.1006/excr.2000.4839
45. Cory S, Adams JM. Killing Cancer Cells by Flipping the Bcl-2/Bax Switch. Cancer Cell (2005) 8(1):5–6. doi: 10.1016/j.ccr.2005.06.012
46. Golestani Eimani B, Sanati MH, Houshmand M, Ataei M, Akbarian F, Shakhssalim N. Expression and Prognostic Significance of Bcl-2 and Bax in the Progression and Clinical Outcome of Transitional Bladder Cell Carcinoma. Cell J (2014) 15(4):356–63. doi: 10.1073/pnas.95.8.4386
47. Kuida K, Haydar TF, Kuan C-Y. Reduced Apoptosis and Cytochrome C–Mediated Caspase Activation in Mice Lacking Caspase 9. Cell (1998) 94(3):325–37. doi: 10.1016/S0092-8674(00)81476-2
48. Straub CS. Targeting IAPs as an Approach to Anti-Cancer Therapy. Curr Top Med Chem (2011) 11(3):291–316. doi: 10.2174/156802611794072623
49. Lee EF, Fairlie WD. The Structural Biology of Bcl-X(L). Int J Mol Sci (2019) 20(9):2234–18. doi: 10.3390/ijms20092234
50. Kurita T, Izumi H, Kagami S. Mitochondrial Transcription Factor A Regulates BCL2L1 Gene Expression and is a Prognostic Factor in Serous Ovarian Cancer. Cancer Sci (2012) 103(2):239–444. doi: 10.1111/j.1349-7006.2011.02156.x
51. Song S, Jacobson KN, McDermott KM. ATP Promotes Cell Survival via Regulation of Cytosolic [Ca2+] and Bcl-2/Bax Ratio in Lung Cancer Cells. Am J Physiol Cell Physiol (2016) 310(2):C99–114. doi: 10.1152/ajpcell.00092.2015
52. Lin F, Ghislat G, Luo S, Renna M, Siddiqi F, Rubinsztein DC. XIAP and Ciap1 Amplifications Induce Beclin 1-Dependent Autophagy Through Nfκb Activation. Hum Mol Genet (2015) 24(10):2899–913. doi: 10.1093/hmg/ddv052
53. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 Network Regulates Autophagy and Apoptosis. Cell Death Differ (2011) 18(4):571–80. doi: 10.1038/cdd.2010.191
54. White E. The Role for Autophagy in Cancer. J Clin Invest (2015) 125(1):42–6. doi: 10.1172/JCI73941
55. Schmidt N, Haydn T, Schneider I, Busch H, Boerries M, Fulda S. Smac Mimetic Induces an Early Wave of Gene Expression via NF-κb and AP-1 and a Second Wave via TNFR1 Signaling. Cancer Lett (2018) 421:170–85. doi: 10.1016/j.canlet.2018.01.082
56. Gozzelino R, Sole C, Llecha N. BCL-XL Regulates TNF-Alpha-Mediated Cell Death Independently of NF-Kappab, FLIP and IAPs. Cell Res (2008) 18(10):1020–36. doi: 10.1038/cr.2008.76
57. Zi M, Xu Y. Involvement of Cystatin C in Immunity and Apoptosis. Immunol Lett (2018) 196:80–90. doi: 10.1016/j.imlet.2018.01.006
58. Kim H, Jung YK, Kwon YK, Park SH. Assignment of Apoptotic Protease Activating Factor-1 Gene (APAF1) to Human Chromosome Band 12q23 by Fluorescence in Situ Hybridization. Cytogenet Cell Genet (1999) 87(3-4):252–3. doi: 10.1159/000015436
59. Warren CFA, Wong-Brown MW, Bowden NA. BCL-2 Family Isoforms in Apoptosis and Cancer. Cell Death Dis (2019) 10(3):177. doi: 10.1038/s41419-019-1407-6
60. Shi M, Yan S-G, Xie S-T, Wang H-N. Tip30-Induced Apoptosis Requires Translocation of Bax and Involves Mitochondrial Release of Cytochrome C and Smac/DIABLO in Hepatocellular Carcinoma Cells. Biochim Biophys Acta (BBA) - Mol Cell Res (2008) 1783(2):263–74. doi: 10.1016/j.bbamcr.2007.10.006
61. Kędzierska H, Piekiełko-Witkowska A. Splicing Factors of SR and hnRNP Families as Regulators of Apoptosis in Cancer. Cancer Lett (2017) 396:53–65. doi: 10.1016/j.canlet.2017.03.013
62. Martinez-Ruiz G, Maldonado V, Ceballos-Cancino G, Grajeda JP, Melendez-Zajgla J. Role of Smac/DIABLO in Cancer Progression. J Exp Clin Cancer Res (2008) 27(1):48. doi: 10.1186/1756-9966-27-48
63. Park SH, Shin I, Park SH, Kim ND, Shin I. An Inhibitor of the Interaction of Survivin With Smac in Mitochondria Promotes Apoptosis. Chem Asian J (2019) 14(22):4035–41. doi: 10.1002/asia.201900587
64. Qin S, Yang C, Li S, Xu C, Zhao Y, Ren H, et al. Smac: Its Role in Apoptosis Induction and Use in Lung Cancer Diagnosis and Treatment. Cancer Lett (2012) 318(1):9–13. doi: 10.1016/j.canlet.2011.12.024
65. Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y. Structural and Biochemical Basis of Apoptotic Activation by Smac/DIABLO. Nature (2000) 406(6798):855–62. doi: 10.1038/35022514
66. Paul A, Krelin Y, Arif T, Jeger R, Shoshan-Barmatz V. A New Role for the Mitochondrial Pro-Apoptotic Protein SMAC/Diablo in Phospholipid Synthesis Associated With Tumorigenesis. Mol Ther (2018) 26(3):680–94. doi: 10.1016/j.ymthe.2017.12.020
67. Allensworth JL, Sauer SJ, Lyerly HK, Morse MA, Devi GR. Smac Mimetic Birinapant Induces Apoptosis and Enhances TRAIL Potency in Inflammatory Breast Cancer Cells in an IAP-Dependent and TNF-α-Independent Mechanism. Breast Cancer Res Treat (2013) 137(2):359–71. doi: 10.1007/s10549-012-2352-6
68. Verhagen AM, Ekert PG, Pakusch M. Identification of DIABLO, a Mammalian Protein That Promotes Apoptosis by Binding to and Antagonizing IAP Proteins. Cell (2000) 102(1):43–53. doi: 10.1016/S0092-8674(00)00009-X
69. Mansour A, Nabil M, Ali-Labib R, Said H, Annos F. Reciprocal Expression of Survivin and SMAC/DIABLO in Primary Breast Cancer. Med Oncol (2012) 29(4):2535–42. doi: 10.1007/s12032-011-0129-0
70. Martinez-Velazquez M, Melendez-Zajgla J, Maldonado V. Apoptosis Induced by cAMP Requires Smac/DIABLO Transcriptional Upregulation. Cell Signal (2007) 19(6):1212–20. doi: 10.1016/j.cellsig.2007.01.001
71. Sun H, Liu L, Lu J. Cyclopeptide Smac Mimetics as Antagonists of IAP Proteins. Bioorg Med Chem Lett (2010) 20(10):3043–6. doi: 10.1016/j.bmcl.2010.03.114
72. Shiozaki EN, Shi Y. Caspases, IAPs and Smac/DIABLO: Mechanisms From Structural Biology. Trends Biochem Sci (2004) 29(9):486–94. doi: 10.1016/j.tibs.2004.07.003
73. Qiu XB, Goldberg AL. The Membrane-Associated Inhibitor of Apoptosis Protein, BRUCE/Apollon, Antagonizes Both the Precursor and Mature Forms of Smac and Caspase-9. J Biol Chem (2005) 280(1):174–82. doi: 10.1074/jbc.M411430200
74. Vucic D, Franklin MC, Wallweber HJ. Engineering ML-IAP to Produce an Extraordinarily Potent Caspase 9 Inhibitor: Implications for Smac-Dependent Anti-Apoptotic Activity of ML-IAP. Biochem J (2005) 385(Pt 1):11–20. doi: 10.1042/BJ20041108
75. Dineen S, Roland C, Greer R. Inhibiting XIAP With SMAC Mimetic JP-1201 Increases Sensitivity to Gemcitabine and Improves Survival in Murine Models of Pancreatic Cancer. J Surg Res (2009) 151(2):252–3. doi: 10.1016/j.jss.2008.11.211
76. De Oliveira Lima F, De Oliveira Costa H, Barrezueta LF. Immunoexpression of Inhibitors of Apoptosis Proteins and Their Antagonist SMAC/DIABLO in Colorectal Carcinoma: Correlation With Apoptotic Index, Cellular Proliferation and Prognosis. Oncol Rep (2009) 22(2):295–303. doi: 10.3892/or_00000437
77. Ogura A, Watanabe Y, Iizuka D. Radiation-Induced Apoptosis of Tumor Cells is Facilitated by Inhibition of the Interaction Between Survivin and Smac/DIABLO. Cancer Lett (2008) 259(1):71–81. doi: 10.1016/j.canlet.2007.09.017
78. Du C, Fang M, Li Y, Li L, Wang X. Smac, a Mitochondrial Protein That Promotes Cytochrome C-Dependent Caspase Activation by Eliminating IAP Inhibition. Cell (2000) 102(1):33–42. doi: 10.1016/S0092-8674(00)00008-8
79. McCormack D, McDonald D, McFadden D. Pterostilbene Induces Mitochondrially-Derived Apoptosis in Pancreatic Cancer Cells by Increasing MnSOD Activity and Release of Cytochrome C and Smac/DIABLO. Gastroenterology (2011), 140(5):1051–8. doi: 10.1016/S0016-5085(11)64263-7
80. Anguiano-Hernandez YM, Chartier A, Huerta S. Smac/DIABLO and Colon Cancer. Anticancer Agents Med Chem (2007) 7(4):467–73. doi: 10.2174/187152007781058631
81. Li W, Turaga RC, Li X. Overexpression of Smac by an Armed Vesicular Stomatitis Virus Overcomes Tumor Resistance. Mol Ther - Oncolytics (2019) 14:188–95. doi: 10.1016/j.omto.2019.05.006
82. Lv J, Qian Y, Ni X, Xu X, Dong X. Feedback Regulation of Methyl Methanesulfonate and Ultraviolet-Sensitive Gene Clone 81 via ATM/Chk2 Pathway Contributes to the Resistance of MCF-7 Breast Cancer Cells to Cisplatin. Tumour Biol (2017) 39(3):1010428317694307. doi: 10.1177/1010428317694307
83. Salvesen GS, Duckett CS. IAP Proteins: Blocking the Road to Death's Door. Nat Rev Mol Cell Biol (2002) 3(6):401–10. doi: 10.1038/nrm830
84. Cossu F, Camelliti S, Lecis D. Structure-Based Identification of a New IAP-Targeting Compound That Induces Cancer Cell Death Inducing NF-κb Pathway. Comput Struct Biotechnol J (2021) 19:6366–74. doi: 10.1016/j.csbj.2021.11.034
85. Dai Y, Lawrence TS, Xu L. Overcoming Cancer Therapy Resistance by Targeting Inhibitors of Apoptosis Proteins and Nuclear Factor-Kappa B. Am J Transl Res (2009) 1(1):1–15. doi: 10.3390/cancers13010010
86. Obexer P, Ausserlechner MJ. X-Linked Inhibitor of Apoptosis Protein - a Critical Death Resistance Regulator and Therapeutic Target for Personalized Cancer Therapy. Front Oncol (2014) 4:197. doi: 10.3389/fonc.2014.00197
87. Suzuki Y, Takahashi-Niki K, Akagi T, Hashikawa T, Takahashi R. Mitochondrial Protease Omi/HtrA2 Enhances Caspase Activation Through Multiple Pathways. Cell Death Differ (2004) 11(2):208–16. doi: 10.1038/sj.cdd.4401343
88. Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C. Omi/HtrA2 Catalytic Cleavage of Inhibitor of Apoptosis (IAP) Irreversibly Inactivates IAPs and Facilitates Caspase Activity in Apoptosis. Genes Dev (2003) 17(12):1487–96. doi: 10.1101/gad.1097903
89. Pruefer FG, Lizarraga F, Maldonado V, Melendez-Zajgla J. Participation of Omi Htra2 Serine-Protease Activity in the Apoptosis Induced by Cisplatin on SW480 Colon Cancer Cells. J Chemother (2008) 20(3):348–54. doi: 10.1179/joc.2008.20.3.348
90. Verhagen AM, Silke J, Ekert PG. HtrA2 Promotes Cell Death Through its Serine Protease Activity and its Ability to Antagonize Inhibitor of Apoptosis Proteins. J Biol Chem (2002) 277(1):445–54. doi: 10.1074/jbc.M109891200
91. Kaur I, Behl T, Sachdeva M, Bungau S, Venkatachalam T. Exploring the Mitochondrial Apoptotic Cell Death Landscape and Associated Components Serving as Molecular Targets, Primarily for Synthetic and Natural Drugs Targeting Oncology Therapeutics. Curr Mol Pharmacol (2021) 14(6):1066–82. doi: 10.2174/1874467214666210120145537
92. Bartling B, Lewensohn R, Zhivotovsky B. Endogenously Released Smac is Insufficient to Mediate Cell Death of Human Lung Carcinoma in Response to Etoposide. Exp Cell Res (2004) 298(1):83–95. doi: 10.1016/j.yexcr.2004.04.007
93. Mueller F, Friese A, Pathe C. Overlap of NatA and IAP Substrates Implicates N-Terminal Acetylation in Protein Stabilization. Sci Adv (2021) 7(3):8590–13. doi: 10.1126/sciadv.abc8590
94. Bhatlekar S, Fields JZ, Boman BM. HOX Genes and Their Role in the Development of Human Cancers. J Mol Med (Berl) (2014) 92(8):811–23. doi: 10.1007/s00109-014-1181-y
95. Paço A, Aparecida de Bessa Garcia S, Leitão Castro J, Costa-Pinto AR, Freitas R. Roles of the HOX Proteins in Cancer Invasion and Metastasis. Cancers (Basel) (2020) 13(1):10–19. doi: 10.3390/cancers13010010
96. Wen X, Chen Y, Fang X. Overexpression of HOXD8 Inhibits the Proliferation, Migration and Invasion of Breast Cancer Cells by Downregulating ILP2 Expression. Exp Ther Med (2021) 22(3):1006. doi: 10.3892/etm.2021.10439
97. Xu Z, Han X, Ou D. Targeting PI3K/AKT/mTOR-Mediated Autophagy for Tumor Therapy. Appl Microbiol Biotechnol (2020) 104(2):575–87. doi: 10.1007/s00253-019-10257-8
98. Pal S, Ganguly KK, Chatterjee A. Extracellular Matrix Protein Fibronectin Induces Matrix Metalloproteinases in Human Prostate Adenocarcinoma Cells PC-3. Cell Commun Adhes (2013) 20(5):105–14. doi: 10.3109/15419061.2013.833193
99. Sercu S, Zhang L, Merregaert J. The Extracellular Matrix Protein 1: Its Molecular Interaction and Implication in Tumor Progression. Cancer Invest (2008) 26(4):375–84. doi: 10.1080/07357900701788148
100. Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular Matrix Structure. Adv Drug Deliv Rev (2016) 97:4–27. doi: 10.1016/j.addr.2015.11.001
101. Han Z, Ni J, Smits P. Extracellular Matrix Protein 1 (ECM1) has Angiogenic Properties and is Expressed by Breast Tumor Cells. FASEB J (2001) 15(6):988–94. doi: 10.1096/fsb2fj990934com
102. Lal G, Hashimi S, Smith BJ. Extracellular Matrix 1 (ECM1) Expression Is a Novel Prognostic Marker for Poor Long-Term Survival in Breast Cancer: A Hospital-Based Cohort Study in Iowa. Ann Surg Oncol (2009) 16(8):2280–7. doi: 10.1245/s10434-009-0533-2
103. Gan L, Meng J, Xu M. Extracellular Matrix Protein 1 Promotes Cell Metastasis and Glucose Metabolism by Inducing Integrin β4/FAK/SOX2/HIF-1α Signaling Pathway in Gastric Cancer. Oncogene (2018) 37(6):744–55. doi: 10.1038/onc.2017.363
104. Hardy SA, Mabotuwana NS, Murtha LA. Novel Role of Extracellular Matrix Protein 1 (ECM1) in Cardiac Aging and Myocardial Infarction. PloS One (2019) 14(2):e0212230. doi: 10.1371/journal.pone.0212230
105. Abdel Salam R, El-Badry N, Rizk A, El-Sedfy A, Kamel N, El-Abd E. Expression of ECM1 and MMP-2 in Follicular Thyroid Lesions Among Egyptians. Cancer Biomark (2015) 15(4):441–58. doi: 10.3233/CBM-150481
106. Xu W, Yang Z, Lu N. A New Role for the PI3K/Akt Signaling Pathway in the Epithelial-Mesenchymal Transition. Cell Adh Migr (2015) 9(4):317–24. doi: 10.1080/19336918.2015.1016686
107. Sharifi S, Barar J, Hejazi MS, Samadi N. Doxorubicin Changes Bax /Bcl-xL Ratio, Caspase-8 and 9 in Breast Cancer Cells. Adv Pharm Bull (2015) 5(3):351–9. doi: 10.15171/apb.2015.049
108. Squatrito M, Brennan CW, Helmy K, Huse JT, Petrini JH, Holland EC. Loss of ATM/Chk2/p53 Pathway Components Accelerates Tumor Development and Contributes to Radiation Resistance in Gliomas. Cancer Cell (2010) 18(6):619–29. doi: 10.1016/j.ccr.2010.10.034
109. Lee HE, Han N, Kim MA. DNA Damage Response-Related Proteins in Gastric Cancer: ATM, Chk2 and P53 Expression and Their Prognostic Value. Pathobiology (2014) 81(1):25–35. doi: 10.1159/000351072
110. Bartkova J, Falck J, Rajpert-De Meyts E, Skakkebaek NE, Lukas J, Bartek J, et al. Chk2 Tumour Suppressor Protein in Human Spermatogenesis and Testicular Germ-Cell Tumours. Oncogene (2001) 20(41):5897–902. doi: 10.1038/sj.onc.1204746
111. Gogineni VR, Nalla AK, Gupta R, Dinh DH, Klopfenstein JD, Rao JS. Chk2-Mediated G2/M Cell Cycle Arrest Maintains Radiation Resistance in Malignant Meningioma Cells. Cancer Lett (2011) 313(1):64–75. doi: 10.1016/j.canlet.2011.08.022
112. Nie X, Guo E, Wu C. SALL4 Induces Radioresistance in Nasopharyngeal Carcinoma via the ATM/Chk2/p53 Pathway. Cancer Med (2019) 8(4):1779–92. doi: 10.1002/cam4.2056
113. Liu K, Zheng M, Lu R. The Role of CDC25C in Cell Cycle Regulation and Clinical Cancer Therapy: A Systematic Review. Cancer Cell Int (2020) 20:213. doi: 10.1186/s12935-020-01304-w
114. Ta HQ, Ivey ML, Frierson HF. Checkpoint Kinase 2 Negatively Regulates Androgen Sensitivity and Prostate Cancer Cell Growth. Cancer Res (2015) 75(23):5093–105. doi: 10.1158/0008-5472.CAN-15-0224
115. Neumann J, Yang Y, Köhler R. Mangrove Dolabrane-Type of Diterpenes Tagalsins Suppresses Tumor Growth via ROS-Mediated Apoptosis and ATM/ATR–Chk1/Chk2-Regulated Cell Cycle Arrest. Int J Cancer (2015) 137(11):2739–48. doi: 10.1002/ijc.29629
116. Liang J, Zhao W, Tong P. Comprehensive Molecular Characterization of Inhibitors of Apoptosis Proteins (IAPs) for Therapeutic Targeting in Cancer. BMC Med Genomics (2020) 13(1):7. doi: 10.1186/s12920-020-0661-x
117. Xiang M, Zhou W, Gao D, Fang X, Liu Q. Inhibitor of Apoptosis Protein-Like Protein-2 as a Novel Serological Biomarker for Breast Cancer. Int J Mol Sci (2012) 13(12):16737–50. doi: 10.3390/ijms131216737
118. Zhu L, Zhou W, Zhu X. Inhibitor of Apoptosis Protein−Like Protein−2: A Novel Growth Accelerator for Breast Cancer Cells. Oncol Rep (2018) 40(4):2047–55. doi: 10.1016/j.cellsig.2013.06.003
119. Abe S, Yamamoto K, Hasegawa M. Bone Marrow Cells of Myelodysplastic Syndromes Exhibit Significant Expression of Apollon, Livin and ILP-2 With Reduction After Transformation to Overt Leukemia. Leuk Res (2005) 29(9):1095–6. doi: 10.1016/j.leukres.2005.02.004
120. Chuturgoon AA, Phulukdaree A, Moodley D. Fumonisin B1 Inhibits Apoptosis in HepG2 Cells by Inducing Birc-8/ILP-2. Toxicol Lett (2015) 235(2):67–74. doi: 10.1016/j.toxlet.2015.03.006
121. Liu TH, Zheng F, Cai MY. The Putative Tumor Activator ARHGEF3 Promotes Nasopharyngeal Carcinoma Cell Pathogenesis by Inhibiting Cellular Apoptosis. Oncotarget (2016) 7(18):25836–48. doi: 10.18632/oncotarget.8283
122. Veeraraghavan J, Aravindan S, Natarajan M, Awasthi V, Herman TS, Aravindan N. Neem Leaf Extract Induces Radiosensitization in Human Neuroblastoma Xenograft Through Modulation of Apoptotic Pathway. Anticancer Res (2011) 31(1):161–70. doi: 10.1111/j.1365-2184.2012.00814
123. Li G, Chang H, Zhai YP, Xu W. Targeted Silencing of Inhibitors of Apoptosis Proteins With siRNAs: A Potential Anti-Cancer Strategy for Hepatocellular Carcinoma. Asian Pac J Cancer Prev (2013) 14(9):4943–52. doi: 10.7314/APJCP.2013.14.9.4943
124. Wang K, Lin B. Inhibitor of Apoptosis Proteins (IAPs) as Regulatory Factors of Hepatic Apoptosis. Cell Signal (2013) 25(10):1970–80. doi: 10.1016/j.cellsig.2013.06.003
125. Garg H, Suri P, Gupta JC, Talwar GP, Dubey S. Survivin: A Unique Target for Tumor Therapy. Cancer Cell Int (2016) 16:49. doi: 10.1186/s12935-016-0326-1
126. Chen Y, Li Z, Dong Z. 14-3-3ς Contributes to Radioresistance By Regulating DNA Repair and Cell Cycle via PARP1 and CHK2. Mol Cancer Res (2017) 15(4):418–28. doi: 10.1158/1541-7786.MCR-16-0366
127. Wang Z, Liu S, Ding K. Silencing Livin induces apoptotic and autophagic cell death, Increasing Chemotherapeutic Sensitivity to Cisplatin of Renal Carcinoma Cells. Tumor Biol (2016) 37(11):15133–43. doi: 10.1007/s13277-016-5395-1
128. Neophytou CM, Trougakos IP, Erin N, Papageorgis P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers (Basel) (2021) 13(17):4363–26. doi: 10.3390/cancers13174363
129. Olbryt M, Pigłowski W, Rajczykowski M, Pfeifer A, Student S, Fiszer-Kierzkowska A, et al. Genetic Profiling of Advanced Melanoma: Candidate Mutations for Predicting Sensitivity and Resistance to Targeted Therapy. Target Oncol (2020) 15(1):101–13. doi: 10.1007/s11523-020-00695-0
Keywords: ILP-2, IAPS, caspases, apoptosis, migration, tumor therapy
Citation: Zhang Z, Xiang S, Cui R, Peng H, Mridul R and Xiang M (2022) ILP-2: A New Bane and Therapeutic Target for Human Cancers. Front. Oncol. 12:922596. doi: 10.3389/fonc.2022.922596
Received: 18 April 2022; Accepted: 26 May 2022;
Published: 23 June 2022.
Edited by:
Patrícia Savio De Araujo-Souza, Federal University of Paraná, BrazilReviewed by:
Valéria Bumiller-Bini Hoch, Federal University of Paraná, BrazilCopyright © 2022 Zhang, Xiang, Cui, Peng, Mridul and Xiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingjun Xiang, eG1qNjg4QDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.