 Alessia Schirripa
Alessia Schirripa Veronika Sexl
Veronika Sexl Karoline Kollmann*
Karoline Kollmann*
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 11 August 2022
Sec. Molecular and Cellular Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.916682
This article is part of the Research Topic Women in Molecular and Cellular Oncology: 2021 View all 15 articles
The cell-cycle is a tightly orchestrated process where sequential steps guarantee cellular growth linked to a correct DNA replication. The entire cell division is controlled by cyclin-dependent kinases (CDKs). CDK activation is balanced by the activating cyclins and CDK inhibitors whose correct expression, accumulation and degradation schedule the time-flow through the cell cycle phases. Dysregulation of the cell cycle regulatory proteins causes the loss of a controlled cell division and is inevitably linked to neoplastic transformation. Due to their function as cell-cycle brakes, CDK inhibitors are considered as tumor suppressors. The CDK inhibitors p16INK4a and p15INK4b are among the most frequently altered genes in cancer, including hematopoietic malignancies. Aberrant cell cycle regulation in hematopoietic stem cells (HSCs) bears severe consequences on hematopoiesis and provokes hematological disorders with a broad array of symptoms. In this review, we focus on the importance and prevalence of deregulated CDK inhibitors in hematological malignancies.
Cell-cycle progression is a fundamental biological process which requires tight regulation to guarantee a correct cell division. Perturbations of cell cycle components may provoke an uncontrolled cell proliferation. Dysregulated G1-S transition is a common feature of tumor development and associated with genetic alterations of key regulators of the cell-cycle machinery (1). Based on their function as a cell cycle brake, CDK inhibitors (CKIs) mainly act as tumor suppressors and are frequently deactivated in human neoplasia (2–4).
Cyclin-dependent kinases (CDKs), their activating cyclins and CDK inhibitors guide cells through the cell cycle (Figure 1). Distinct cyclins are periodically produced and assemble to cyclin-CDK complexes that drive the specific cell-cycle steps, from G1 to M phase. Fine tuning is achieved by inhibitory phosphorylation or binding of CDK inhibitory subunits (CKls) (5–7).
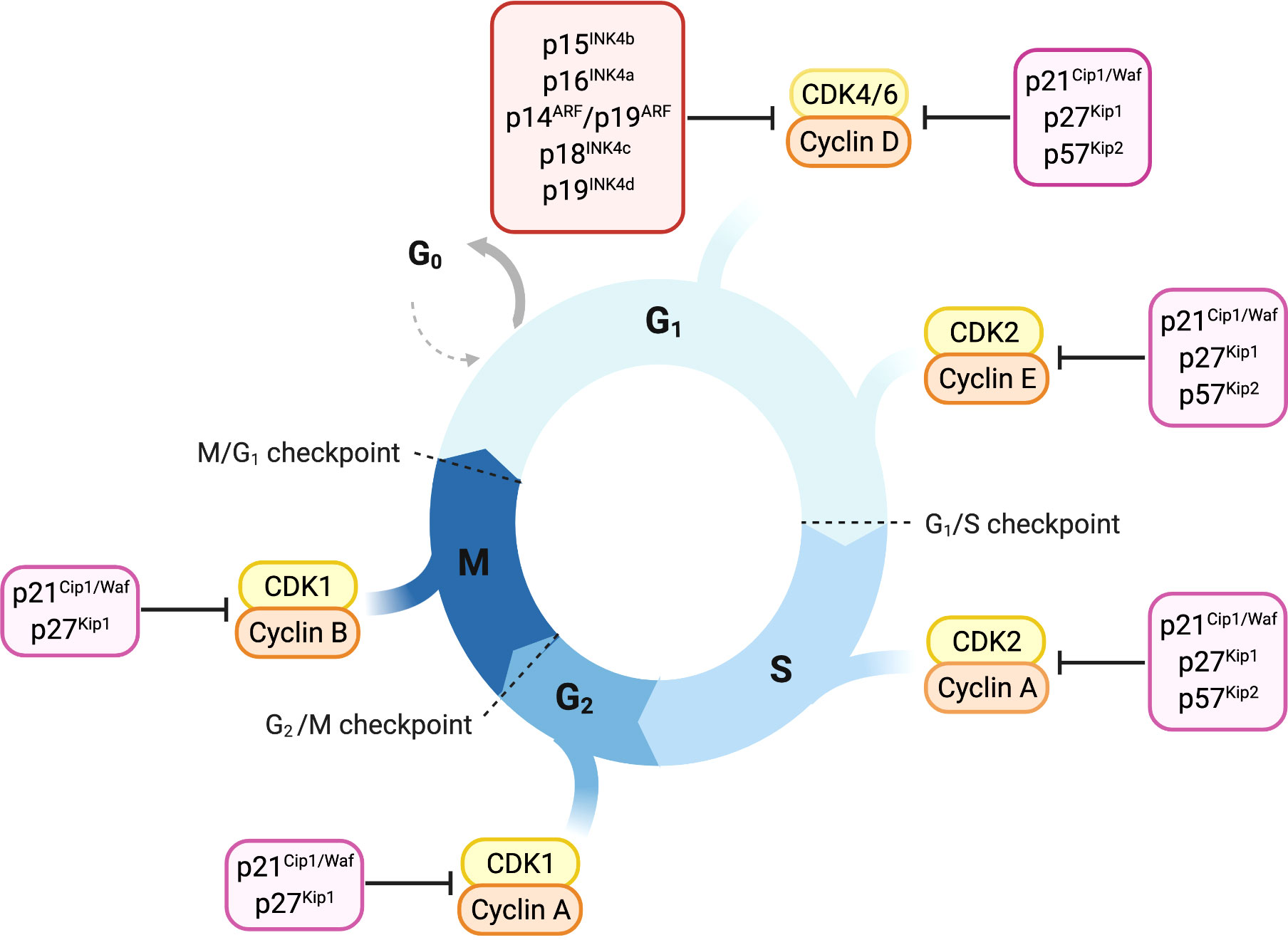
Figure 1 Overview of cell-cycle control and its main regulators. Progression through cell cycle phases is governed by different CDK-cyclin complexes and the respective cyclin-dependent kinase inhibitors. Members of the INK4 family, p16INK4a, p15INK4b, p18INK4c and p19INK4d, specifically bind and inhibit CDK4/6-cyclin D complexes promoting cell cycle arrest in the G1 phase. The Cip/Kip proteins including p21Cip1/Waf, p27kip1 and p57Kip2, play their role as cell-cycle inhibitors by counteracting a broader spectrum of CDK-cyclin complexes. p21Cip1/Waf, p27kip1 and p57Kip2 restrain cell-cycle both during early and late G1 phase by binding either CDK4/6-cyclin D or CDK2-cyclin E complexes. Later in the cell-cycle, they can bind and inhibit CDK2-cyclin A complex, thus imposing a brake during the S-phase. p21Cip1/Waf and p27kip1 are able to delay entry in the M phase by inhibiting CDK1-cyclin A complex and thereby prevent the progression through mitosis counteracting CDK1-cyclin B complex.
Cyclin-dependent kinase 4 (CDK4) and CDK6 are closely related serine/threonine kinases responsible for driving cells through the G1 phase. Mitogenic signals induce transcription of D-type cyclins (D1, D2 and D3). Their association with CDK4 and CDK6 leads to kinase activation and phosphorylation of the retinoblastoma protein (Rb) (8). CDK-dependent Rb phosphorylation releases Rb from E2F transcription factors and induces transcription of E2F target genes required for S-phase entry (9). G1-S transition is then initiated by CDK2-cyclin E/A complexes, which are active during the entire S-phase (10–12). CDK1 activity is low during G1/S transition but raises during G2-M phase, controlling the initiation of mitosis (13, 14).
CDK-cyclin activity is counterbalanced by members of the two CDK inhibitor families, the INK4 family and the Cip/Kip family (8). p16INK4a, p15INK4b, p18INK4c and p19INK4d are the members of the INK4 family and are specific for CDK4 and CDK6 (15). In response to anti-proliferative signals, INK4 proteins are transcribed and bind CDK4 and CDK6 causing a conformational change which reduces their affinity for D-type cyclins (16).
The Cip/Kip family consists of p21Cip1/Waf, p27Kip1 and p57Kip2. In contrast to INK4 proteins,
Cip/Kip proteins have the ability to bind CDK4/6-cyclin D and CDK-cyclin A/B/E complexes (8, 16–19). p21Cip1/Waf and p27Kip1 are described to have a dual function in cell cycle regulation. Whereas they mainly inhibit CDK-cyclin activity they have been reported to also enhance the assembly of CDK4/6-cyclin D complexes, resulting in a proliferative advantage for the cell (18, 20, 21).
When present at low levels, p21Cip1/Waf preferentially binds to CDK4/6-cyclin D complexes, facilitating complex formation, nuclear localization and cell-cycle progression. In response to DNA damage and p53 stimulation, p21Cip1/Waf accumulates at high levels in a cell and provokes a robust cell cycle arrest by inhibiting CDK2- cyclin E-A complexes (8, 22–25). The mechanism behind these observations is given by in vitro experiments showing that changes in p21Cip1/Waf stoichiometry reflect the conversion of active to inactive cyclin-CDK complexes. Active complexes contain a single p21Cip1/Waf molecule, while two molecules are required for complex inhibition (26, 27).
This double-faced role has been described also for p27Kip1. On the one hand, p27Kip1 binds to the conserved cyclin box residues thus promoting the subsequent complex formation between p27Kip1-cyclin A and CDK2. Upon complex formation, p27Kip1 induces a distortion on the CDK2 N-terminal lobe in proximity of CDK2 catalytic site, thereby preventing ATP binding. On the other hand, phosphorylated p27 Kip1 binds to CDK4 leading to a remodeling of the ATP site and results in increased RB phosphorylation. Data suggest a similar mechanism for p21Cip1/Waf activating CDK4 via phosphorylation sites (28).
p57Kip2 mainly functions during G1-S and G2-M transitions where it blocks any CDK-cyclin complexes. No cell cycle activating mechanisms have been described yet.
The Cip/Kip members, p57Kip2 and p21Cip1/Waf are major players in cellular stress responses, where they balance the induction of cell cycle arrest, apoptosis and senescence (29). p21Cip1/Waf has a unique role as it mediates cell cycle arrest downstream of the tumor suppressor p53 (22). A variety of cellular stresses, such as DNA damage and oncogene activation, stimulate p53 expression, which in turn transactivates its targets including the pro-apoptotic genes Bax, PUMA and Noxa as well as p21Cip1/Waf (30–32). Therefore, p21Cip1/Waf might be an exploitable candidate for therapeutic intervention in p53 mutated tumors.
Under homeostatic conditions, hematopoietic stem cells (HSCs) reside in the hypoxic bone marrow niche in a quiescent state (33–35). When needed, HSCs rapidly enter the cell cycle to replenish peripheral hematopoiesis. Self-renewal and differentiation are tightly balanced to maintain the stem cell pool while giving rise to hematopoietic progenitors, which ultimately differentiate into mature blood cells (35, 36). The delicate balance between quiescence and proliferation in HSCs requires a strictly controlled cell cycle progression.
Cyclin dependent kinase inhibitors (CKIs) represent a major break for cell cycle entry and the prevention of uncontrolled proliferation. Several studies started to unravel the impact of CKIs in HSCs (37–40).
p16INK4a is encoded by exons 1α, 2 and 3 of the INK4a locus (Figure 2). A different transcript derived from the same locus, encoded by the exons 1β, 2 and 3, encodes for the protein p19ARF (Figure 2) which has the capacity to block the cell cycle progression at the G1 and G2 phase (41–43). Thus, the INK4a locus represents a master growth regulator through its capacity to interface with both proliferation (Rb pathway via p16INK4a) and apoptosis (p53 pathway via p19ARF) (4, 44).
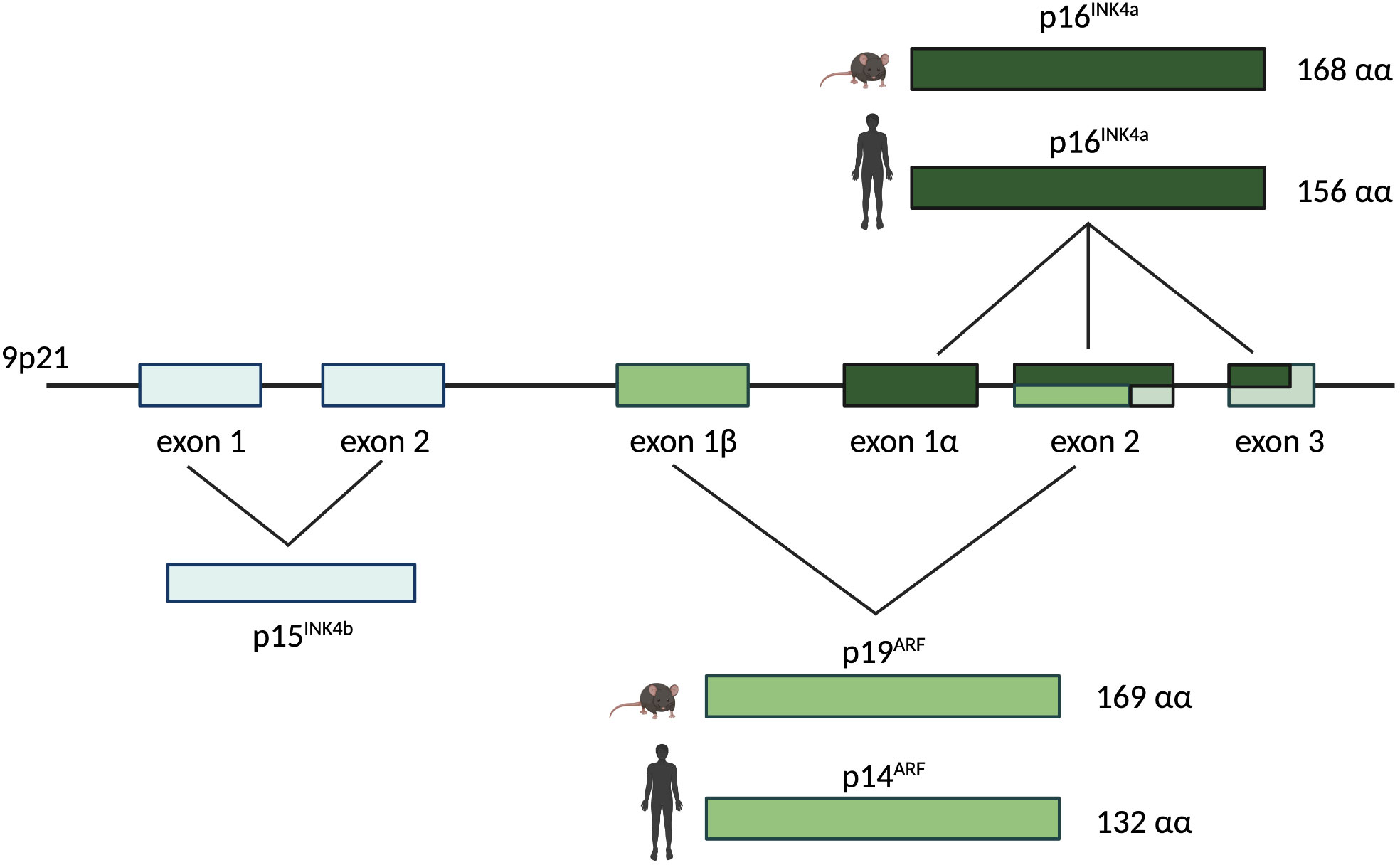
Figure 2 The human/murine INK4a/ARF locus. The INK4a/ARF locus resides on chromosome 9p21 and encodes for two different proteins in human and mouse: p16INK4a and p14ARF (named p19ARF in mouse). The INK4a gene is represented by exons 1α, 2, and 3 and it encodes for p16INK4a, a 168 amino acids protein in mouse and a 156 amino acids protein in human. The ARF gene is composed by exons 1β, 2, and 3. It encodes for p19ARF in mouse (169 amino acids) and for p14ARF in human (132 amino acids). Upstream of the INK4a and ARF genes on the same chromosome, exons 1 and 2 represent the INK4b gene encoding for p15INK4b.
The transcriptional repressor Bmi-1 is part of the Polycomb group and it is present at high levels in HSCs (45–47). Bmi-1 represses the INK4a locus, thus limiting p16INK4a and p19ARF expression (39, 48). Bmi-1 deficiency impairs HSCs self-renewal as it increases p16INK4a and p19ARF levels thereby leading to proliferative arrest and cell death (39). Mice lacking p16INK4a do not show any dramatic effect on hematopoiesis, which could be explained by the reported low p16INK4a expression in normal HSCs (49, 50).
p16INK4a expression increases in HSCs with aging and this is associated with lower HSC numbers. p16INK4a inhibition counteracts the reduced HSC maintenance associated with aging, improves their repopulation ability and mitigates apoptosis (51).
The role of p16INK4a and p19ARF for the regulation of hematopoietic progenitor cells becomes evident in mice harboring a targeted deletion of the INK4a locus that eliminates both proteins. Young p16INK4a-/-/p19ARF-/- mice show extramedullary hematopoiesis in the spleen with a high proportion of lymphoblasts and megakaryocytes in the red pulp and proliferative expansion of the white pulp. Aging aggravates this phenomenon and extends extramedullary hematopoiesis to nonlymphoid organs (49).
Among the CKIs, p18INK4c is the most powerful player and cell cycle inhibitor involved in murine HSC self-renewal (40, 52). p18INK4c deficient mice show HSCs with enhanced self-renewal ability which leads to the expansion of the HSC pool. This is also evident in serial transplantation experiments where p18INK4c deletion allows for an advanced HSC repopulation ability (40, 53).
Information on p15INK4b and p19INK4d in regulating HSC function is scarce. Characterization of the hematopoietic stem and progenitor cells of p15INK4b deficient mice revealed an increased frequency in common myeloid progenitors, but no alterations in the HSC compartment (54, 55).
The need to get first insights into the role of p19INK4d in HSCs leads to the characterization of the hematopoietic system of mice lacking p19INK4d. Knockout mice do not reveal any defect under homeostatic conditions (56). However, in vitro studies highlight the involvement of p19INK4d in megakaryopoiesis, where it regulates the endomitotic cell cycle arrest coupled to terminal differentiation (57).
Moreover, p19INK4d effects become evident when HSCs are exposed to genotoxic stress. In this context, p19INK4d is required to maintain HSCs in a quiescent state, protecting them from apoptosis as genotoxic substances act during the S-phase (58).
The p53 induced CKI p21Cip1/Waf also regulates effects upon stress. Bone marrow transplantation experiments, using cells derived from mice after 2 Gy irradiation show that p21Cip1/Waf deficiency leads to a significantly reduced repopulation ability (37, 59).
In contrast, p27Kip1 knock-out mice lack any perturbations in HSC number, self – renewal ability or cell-cycle state. The role of p27Kip1 is restricted to more committed progenitor cells where its deletion increases proliferation and the pool size of Sca1+Lin+ cells (38).
In quiescent HSCs p57Kip2 dominates as major CKI, where it is expressed at high levels. p57Kip2 deficiency reduces the HSC population, compromises the maintenance of quiescence and impairs repopulation capacity (60).
In summary this led us to conclude that CKIs have distinct essential roles in hematopoietic stem and progenitor cells that are only partially understood. Whereas Cip/Kip proteins are predominantly involved in stress responses, INK proteins dominate in the control of hemostatic conditions.
In human cancers the INK4a-ARF-INK4b locus at chromosome 9p21 is one of the most frequently mutated and epigenetically silenced sites (61–63). This locus encodes for the cyclin dependent kinase inhibitors p16INK4a and p15INK4b and for the tumor suppressor protein p14ARF (p19ARF in the mouse), which is induced upon p53 activation (Figure 2) (64, 65). Many solid tumors including melanoma, pancreatic adenocarcinomas, esophageal and non-small cell lung carcinoma, harbor mutations in the p16INK4a and p15INK4b genes. In hematological malignancies p16INK4a and p15INK4b are frequently deleted e.g. in chronic myeloid leukemia (CML) and acute lymphoblastic leukemia (ALL) (66–70).
p18INK4c and p19INK4d, mapped on chromosome 1p32 and 19p13.2 respectively (71, 72), are involved in the development of a more distinct set of tumors. Somatic mutations of p18INK4c are associated with medullary thyroid carcinoma, hepatocellular carcinoma and breast cancer (73–75). Only little information is available regarding the role of p19INK4d in human malignancies; frame shift mutations and rearrangements in the p19INK4d gene have been documented in osteosarcoma (76), while its loss or downregulation have been detected in hepatocellular carcinoma (77) and testicular germ cell tumors (78).
The deletion of the Cip/Kip proteins in mice leads to an increased development of malignancies (79–81), underlining their main role as tumor suppressors. Contradictorily, in some tumor types Cip/Kip proteins also display an oncogenic activity when relocated to the cytoplasm (82–84).
Low p27Kip1 levels are associated with more aggressiveness and poor prognosis in several human cancers (85–87). Control of p27Kip1 levels involves a nuclear to cytoplasmic redistribution which is regulated by phosphorylation sites on distinct residues. Mitogenic signals induce p27Kip1 phosphorylation on Ser10, inducing nuclear export (88, 89), while phosphorylation on Thr198, mediated by PKB/Akt, promotes p27Kip1 association with 14-3-3 proteins and its transport to the cytoplasm (90).
Whereas nuclear p27Kip1 inhibits cell proliferation and suppresses tumor formation, cytoplasmatic p27Kip1 is involved in cytoskeleton rearrangement and contributes to cell migration (82, 89) and may promote metastasis. In some hematologic malignancies (91–93) and carcinomas (such as breast, esophagus, cervix and uterus tumors) (94–98), a positive association of cytoplasmic p27Kip1 levels with a poor clinical outcome has been reported.
p21Cip1/Waf acts as a tumor suppressor in breast, colorectal, gastric, ovarian and oral cancers. Similar to p27Kip1 it may display oncogenic activities when retained in the cytoplasm. p21Cip1/Waf cytoplasmic accumulation is caused by phosphorylation at Thr145 by activated AKT1 (99). Through the association with proteins involved in the apoptotic process, cytoplasmatic p21Cip1/Waf mediates their inhibition, thus exhibiting anti-apoptotic effects. As such, cytoplasmic p21Cip1/Waf is indicative for aggressiveness and poor survival in prostate, cervical, breast and squamous cell carcinomas (100).
In contrast, the role of p57Kip2 is limited at being a tumor suppressor, as there is so far no evidence of an oncogenic role so far (101–104).
Given the extensive knowledge regarding the role of CDK inhibitors in tumor biology there is increasing interest in exploiting them as potential target for cancer treatments. Here we review and discuss the importance they play in hematopoietic malignancies.
Hematologic malignancies consist of a spectrum of malignant neoplasms that affect bone marrow, blood and lymph nodes and originate from the uncontrolled proliferation of hematopoietic cells. They are driven by genetic and epigenetic aberrations, which can be exploited for diagnosis and therapeutic decisions. The dominant alterations of CKIs are reviewed below and illustrated in Figures 3, 4.
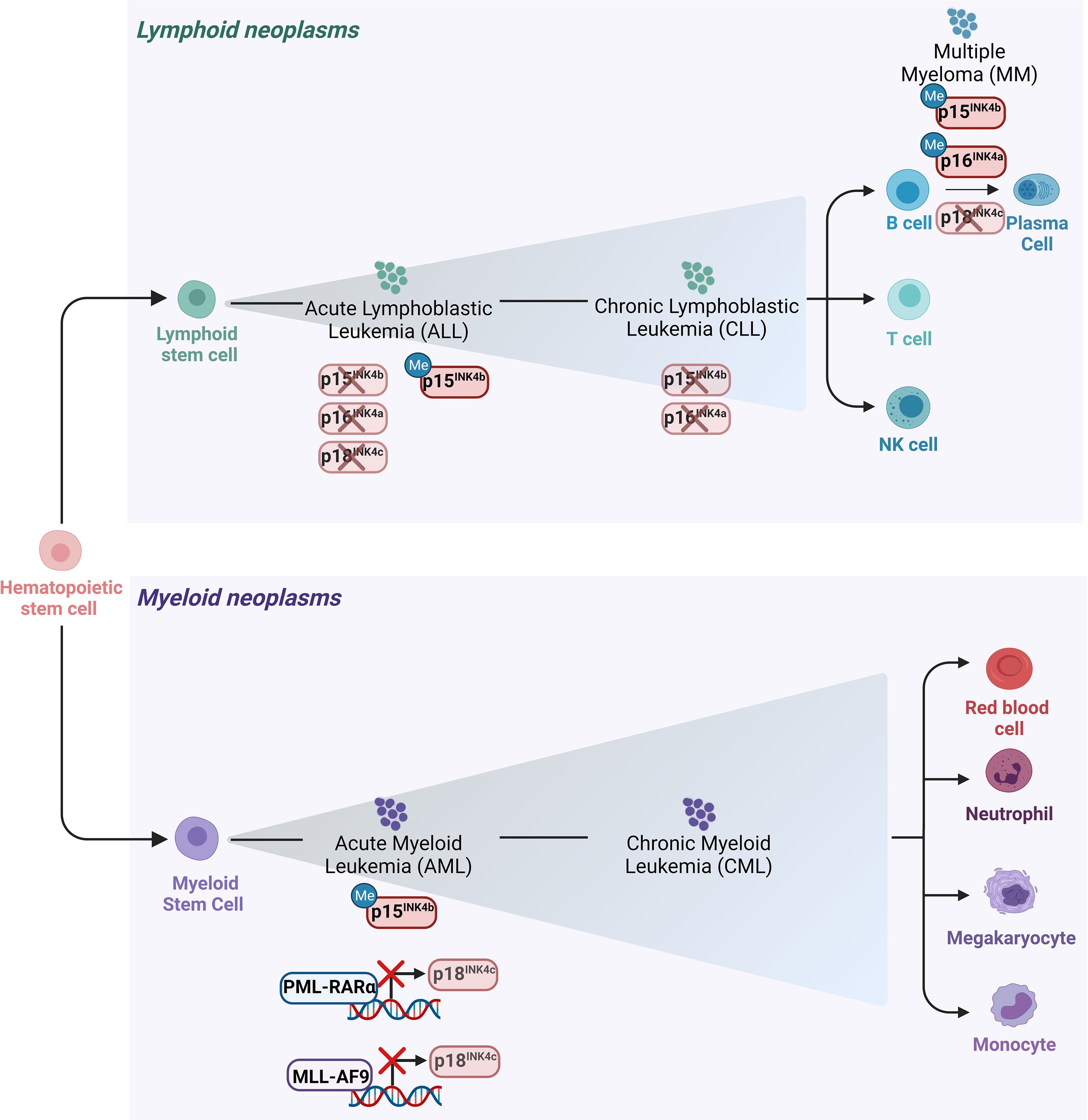
Figure 3 Main alterations of the INK4 proteins in leukemia and lymphomas. Schematic representation of the hematopoietic tree and main alterations affecting the INK4 proteins in different hematopoietic malignancies. Deletion of p15INK4b and p16INK4a together with their 5’ CpG islands hypermethylation in their promoter regions are the most frequent modes of p15INK4b and of p16INK4a inactivation in various subtypes of hematopoietic neoplasms including ALL and CLL. Deletion of p18INK4c has been rarely observed in ALL, whereas it is frequently deleted in MM. p18INK4c is subjected to a transcriptional repression imposed by the oncofusion protein PML-RARα in APL blasts and it is similarly downregulated by MLL-AF9 in cell lines derived from AML patients.
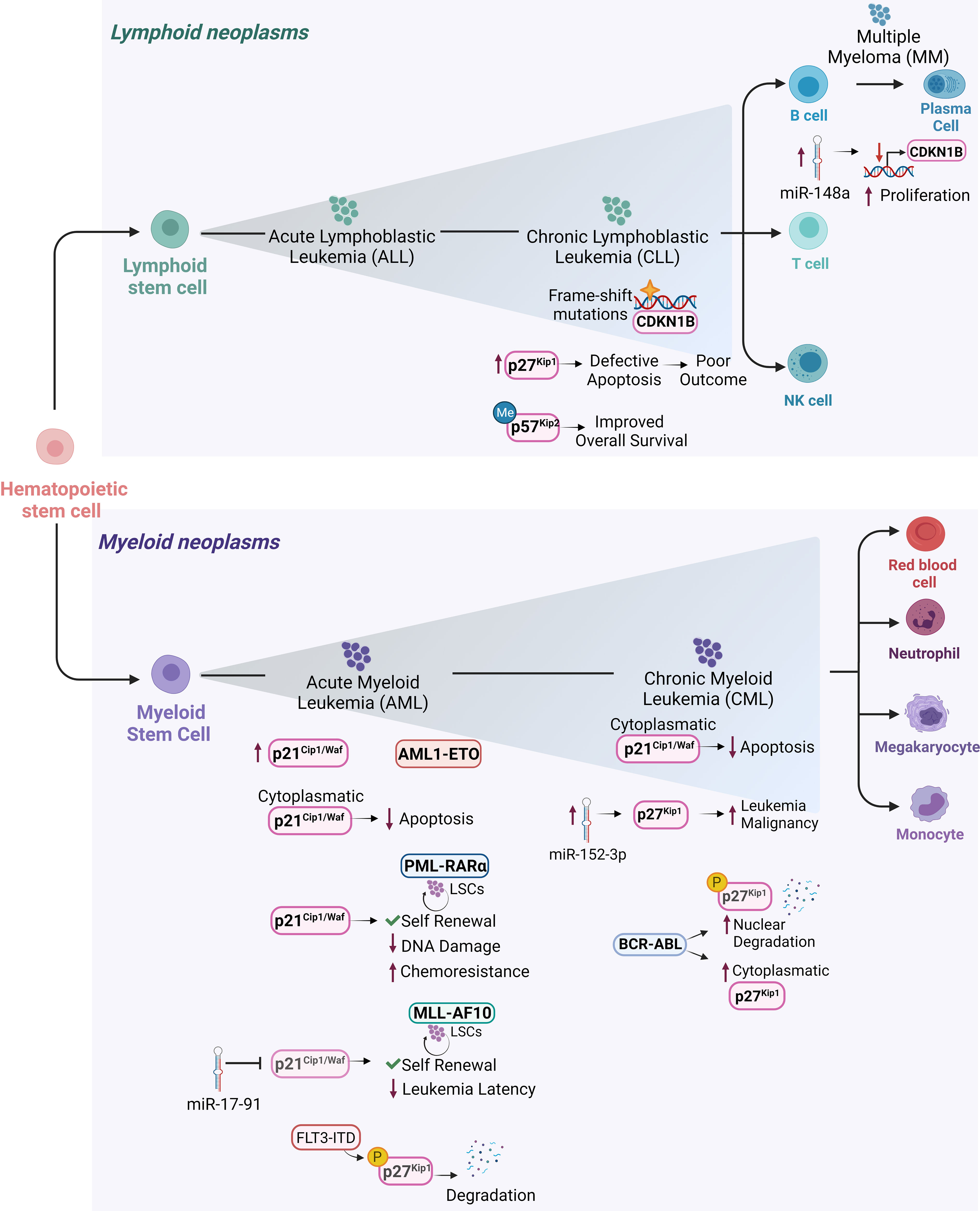
Figure 4 Cip/Kip proteins main deregulations and functions in different hematopoietic malignancies. Schematic representation of the hematopoietic tree and main functions exerted by Cip/Kip proteins in different hematopoietic malignancies. Increased p21Cip1/Waf levels have been reported in AML1-ETO positive AML patients, where it is believed to support LSCs maintenance and self-renewal ability. p21Cip1/Waf anti-apoptotic functions associated with its cytoplasmatic localization have been observed in AML blasts and in cell lines derived from human CML in blast crisis. In PML-RARα LSCs, p21Cip1/Waf expression maintains self-renewal of LSCs and limits DNA damage, thus protecting them from functional exhaustion and conferring chemoresistance. In MLL-AF10 induced AML, p21Cip1/Waf suppression mediated by miR-17-91 leads to decreased leukemia latency. Elevated p27Kip1 levels in B-CLL where they confer protection against apoptosis, are associated with poor outcome. In hairy cell leukemia, a form of B-CLL, CDKN1B gene encoding for p27Kip1 is the second most common altered gene by frame shift mutations. In MM, higher miR-148a levels correlate with decreased CDKN1B expression leading to sustained proliferation. In CML, overexpression of miR-152-3p targets p27Kip1 and promotes leukemia malignancy. In AML, p27Kip1 is subjected to FLT3-ITD phosphorylation (pY88- p27Kip1) which mediates p27Kip1 degradation. BCR-ABL1+ CML can promote degradation of nuclear p27Kip1 and to increased cytoplasmatic p27Kip1, thus compromising p27Kip1 tumor suppressor activity and promoting leukemic cell survival. p57Kip2 gene has been frequently found methylated in diffuse large B-cell lymphoma patients, where the low-risk group it is associated with a more favorable overall survival.
The CDKN2A/B locus encodes for p16INK4a, p14ARF (p19ARF in mice) and p15INK4b. This locus is affected by deletion, mutation or promoter hyper-methylation (62, 63) and frequently altered in patients with hematologic malignancies (4, 105, 106). The design of mouse strains with single or multiple targeted disruptions of the p16INK4a, p19ARF and p15INK4b loci shed light on their distinct roles.
p19ARF-/- mice spontaneously develop a variety of tumors already by the age of 2 months. Analysis of diseased mice shows that T cell lymphoma is the second most common tumor type (107, 108). In line, p19ARF-/- newborn mice exposed either to X-ray or to γ-irradiation develop anaplastic T cell lymphoma (107, 108). In an acute lymphoblastic leukemia (ALL) model, the loss of p19ARF initiates a more aggressive disease BCR-ABL1+ transformation. In this model, p19ARF deletion also confers resistance to the kinase inhibitor imatinib (109). These data suggest a specific role for p19ARF in the lymphoid lineage. Therefore, it would be interesting to analyze if p19ARF could serve as a marker for prognosis and therapeutic outcome.
Homozygous deletion of p16INK4a is not associated with an increased spontaneous cancer development. Of note, the concomitant heterozygous loss of p19ARF in p16INK4a-/- animals increases tumorigenesis and provokes the development of a wide spectrum of malignancies, including lymphoma (110). Importantly, the spontaneous tumors originating from mice harboring the heterozygous loss of p19ARF and p16INK4a homozygous deletion, retain the second p19ARF allele. However, the observed increased tumorigenesis in p16INK4a-/- mice upon heterozygous p19ARF loss underlines the cooperation of the two tumor suppressors.
Young mice show spontaneous tumorigenesis and a higher sensitivity to carcinogenic treatments, especially B cell lymphoma (49).
p15INK4b-/- mice show lymphoproliferative disorders including lymphoid hyperplasia in the spleen and formation of secondary follicles in lymph nodes but rarely develop lymphoma. This suggests that p15INK4b controls homeostasis of the hematopoietic compartment, rather than acting as a tumor suppressor (111).
Although p15INK4b and p16INK4a function as repressors of the cell cycle, in view of the phenotypes shown by the mouse models described above, they seem to have roles in different contexts. p15INK4b is mainly responsible for homeostasis and p16INK4a, together with p19ARF, is more involved in regulating the response to oncogenic stress. This suggests that p16INK4a might function as a sensor of oncogenic signals thus representing a safeguard against neoplasia.
CDK4R24C/CDK6R31C double knock-in mice have been used to address the importance of INK4 inhibitors in regulating CDK4 and CDK6. INK4 binding is prevented by introducing point mutations in CDK4 (R24C) and CDK6 (R31C). The CDK4R24C mutation has been initially identified in hereditary melanoma and shows elevated CDK4 kinase activity (112). So far the CDK6R31C mutation has not been found in patients but is used to investigate CDK6-INK4 interactions. CDK4R24C/CDK6R31C mice show a shortened survival caused by the onset of primary endocrine epithelial or hematopoietic malignancies. Mice injected with CDK4R24C/CDK6R31C BCR-ABL1 transformed cell lines display accelerated tumor growth and reduced disease latency (113). This analysis highlights the crucial importance of INK4 binding to control CDK4/CDK6 activity in hematopoiesis. Therefore, it is attractive to conclude that CDK4/6 inhibitors are effective in patients that lack appropriate INK4-mediated control.
First evidence indicated that the CDKN2 locus in human tumor cell lines derived from solid tumors is predominantly homozygously deleted and thereby p16INK4a becomes inactivated. This was later verified also for leukemia and lymphoma; only a low frequency of point mutations has so far been documented (114–118).
Studies in primary leukemia also identified alterations in p15INK4b. The highest frequency of homozygous deletions of p16INK4a or p15INK4b occurs in ALL, while they are heterozygously deleted in chronic lymphocytic leukemia (CLL) (114, 119–121). T-ALL is most frequently associated with p16INK4a loss, while p15INK4b deletions are more often observed in pediatric ALL (70, 106, 119, 122–127). Initial studies focused their attention on the frequency of p16INK4a and p15INK4b mutations in adult and childhood ALL (70, 114, 120, 122, 128). Only at later stages the potential of these genes as prognostic factors was taken into account.
The overall incidence of p16INK4a deletion is higher than p15INK4b. Patients with p15INK4b deletions harbor p16INK4a co-deletions, which is not consistently observed vice versa. Cases with homozygous p16INK4a deletion either maintain an unmutated p15INK4b gene or show a hemizygous p15INK4b deletion. These findings point at p16INK4a as the central target of deletions which play the central role for ALL leukemogenesis (70, 119, 120, 123).
The prognostic significance of p16INK4a and p15INK4b deletions remains a matter of debate with contradictory reports: some studies showed an adverse prognostic effect (122, 123, 127, 129–133), which was not confirmed by others (70, 134–136).
Analysis of mixed leukemia types, small patient cohorts or insensitive molecular techniques, like polymerase chain reaction (PCR), immunocytochemistry and fluorescence in situ hybridization (FISH) may have complicated the interpretation. The conclusion of some studies still leaves the potential implication of p16INK4a and p15INK4b deletions in patient prognosis elusive.
Point mutations in the CDKN2A/CDKN2B genes, encoding for p16INK4a and p15INK4b respectively, are sporadically found in human hematopoietic disorders. A comprehensive analysis of 264 T-ALL cases, searching for mutations in cell cycle genes, found CDKN2A/CDKN2B as the most mutated ones (137). Inactivation of p15INK4b and p16INK4a genes can also be based on hypermethylation of the 5’ CpG islands in their promoter regions which induces transcriptional silencing (138). This mode of p16INK4a inactivation is commonly found in breast and colon cancer (139) but also in leukemia and lymphoma. Normal hematopoietic cells lack p15INK4b and p16INK4a promoter hypermethylation, which only occurs de novo upon malignant transformation (140). Interestingly, p15INK4b or p16INK4a seem unaffected at any stage of CML (140), whereas hypermethylation of p15INK4b and p16INK4a is a common event in multiple myeloma (MM) (141). Selective p15INK4b promoter hypermethylation, without p16INK4a alterations, is observed in acute myeloid leukemia (AML), myelodysplastic syndrome and ALL (140, 142–146), whereas Burkitt’s lymphoma and Hodgkin’s lymphoma present p16INK4a hypermethylation (140, 141, 147–150).
Overall, the current available data show that inactivation of p15INK4b and p16INK4a in human hematopoietic malignancies is caused by genetic deletion or promoter hypermethylation. Linking these alterations in a well-evaluated cohort of patients would be extremely precious to finally define their role for disease progression and their prognostic relevance. The frequency of their alterations in leukemia and lymphoma is indicative of a central role and renders them promising candidates for novel therapeutic approaches.
Being the functionally most relevant INK in HSC regulation under stress conditions, it is not surprising that the absence of p18INK4c provokes hematopoietic abnormalities and extramedullary hematopoiesis (111). Mice lacking p18INK4cexperience the consequences of the absence of its tumor suppressor function and its role in controlling lymphocyte homeostasis (111, 151). p18INK4c-/- mice spontaneously develop neoplasia including angiosarcoma, testicular tumors, pituitary tumors and lymphoma.
p18INK4c mutations in human hematopoietic malignancies are surprisingly rare in acute leukemias, as they have not been identified in AML and deletions have been reported in just some cases of adult ALL (70, 152, 153). p18INK4c maps on the chromosomal region 1p32. In line with data showing no involvement of p18INK4c in childhood AML (70), no alterations of the 1p region in childhood ALL have been found so far (154). Similarly, no evidence for p18INK4c promoter hypermethylation in acute leukemia has been reported (155).
In MM, p18INK4c is frequently deleted, whereas no point mutations have been detected (156, 157).
In normal B-cells, p18INK4c controls the cell cycle and is involved in the terminal differentiation of B-cells into plasma cells through the inhibition of CDK6 (158, 159). Despite that role, p18INK4c expression is preserved in most lymphoid malignancies (68, 118). The hemizygous loss of p18INK4c has been reported in mantle cell lymphoma, but not in Hodgkin’s lymphoma, where p18INK4c is frequently repressed due to promoter hypermethylation (160–162).
The oncofusion protein PML-RARα which drives acute promyelocytic leukemia (APL) directly suppresses p18INK4c expression which is downregulated in APL blasts compared to normal promyelocytes (163).
ChIP-seq experiments of MLL and AF9 in THP-1 cells reveal the CDKN2C locus, encoding for p18INK4c, as a binding region. This indicates that p18INK4c expression is subject to MLL-AF9 mediated regulation (164).
A detailed map of p18INK4c regulation in different leukemic subtypes is still missing and would help clarifying the role of p18INK4c in hematopoietic malignancies and leukemic stem cells (LSCs). The data currently available are indicative for sporadic alterations of p18INK4c in hematologic malignancies.
The analysis of p19INK4d knock-out mice failed to detect any tumor suppressing effects of p19INK4d. Mice lacking p19INK4d do not spontaneously develop tumors and no abnormalities of the hematopoietic system are evident (56). In line, alterations of p19INK4d are not general hallmarks of hematopoietic neoplasms (76, 165) albeit the data available are scarce. The absence of a mouse phenotype in terms of enhanced cell proliferation and tumor development upon p19INK4d loss suggests a functional compensation exerted by the other INK4 or Cip/Kip proteins.
p21Cip1/Waf is a key mediator of p53-dependent tumor suppressor functions (22) and acts as a negative regulator of cell cycle progression. p21Cip1/Waf and its role in cellular proliferation have been described in a vast body of literature. Its negative function on cell cycle progression indicates that p21Cip1/Waf may exert tumor suppressive roles and participates in leukemia development even under wild type p53 conditions.
p21Cip1/Waf deficient mice are viable and fertile (166, 167). In those mice, harboring wild type p53, spontaneous tumor development occurs late in life at an average age of 16 months. The variety of malignancies includes tumors of hematopoietic, vascular and epithelial origin. For instance, 14% of all tumors are B-cell lymphoma (168).
The tumor spectrum developed by p21Cip1/Waf deficient mice is remarkably similar to the one observed in p53 deficient mice, which is not surprising keeping in mind the p21Cip1/Waf activation by p53. However, p53 deficient mice are characterized by longer latency. However, p21Cip1/Waf deficient mice do not develop T-cell lymphoma, one of the most frequent tumors arising in p53 deficient mice.
The clinical relevance and potential as a prognostic marker of aberrant p21Cip1/Waf expression has been assessed in various types of human cancers.
Loss of p21Cip1/Waf protein levels correlates with a more advanced tumor stage and worse prognosis in pancreatic cancer (169), while its overexpression has been shown to be associated with poor prognosis in non-small cell lung cancer (170) and in esophageal squamous cell carcinoma patients (171).
Interestingly, other studies report low p21Cip1/Waf expression being associated with reduced survival in patients affected by esophageal carcinoma (172, 173).
The relationship between p21Cip1/Waf expression and gastric cancer remains controversial as well. Some authors reported a positive correlation between p21Cip1/Waf expression and favorable prognosis (174, 175), whereas others observed that p21Cip1/Waf expression is associated with poor survival (176).
Analysis of deletions and mutations of p21Cip1/Waf has been carried out in few human hematological malignancies and could be mapped in few subtypes. p21Cip1/Waf alterations are rare in typical mantle cell lymphoma (MCL), but loss of p21Cip1/Waf expression is present in aggressive MCLs harboring wild-type p53 gene (177).
In a large cohort of AML patient blasts, high p21Cip1/Waf expression was found in AML1-ETO positive leukemia (178) with unknown significance. Given its role in maintaining the HSC-pool during normal hematopoiesis (37), one may speculate that it plays a role for LSCs by supporting their self-renewal capacity.
p21Cip1/Waf mutations appear to be not involved in childhood T-ALL pathogenesis, despite extensive studies no mutations were detected (179).
p21Cip1/Waf methylation status in leukemia still remains a debated topic. p21Cip1/Waf hypermethylation was observed in bone marrow cells derived from ALL patients, where it is indicative of a poor prognosis (180). Other studies failed to find any evidence for p21Cip1/Waf methylation in ALL and AML (155, 181, 182).
For instance, p21Cip1/Waf expression appears independent of its promoter methylation status in AML cell lines but correlates with demethylation of p73, a homologue of p53 and a known upstream transcriptional activator of p21Cip1/Waf (183). Treatment of AML cell lines with the methylation inhibitor 5-Aza-2′-deoxycytidine (5-Aza-CdR) results in the induced p21Cip1/Waf expression by p73 demethylation, provoking a cell cycle arrest in the G1 phase (184, 185). Decreased p21Cip1/Waf expression, without any signs of methylation, has been linked to higher disease aggressiveness in myelodysplastic syndrome (MDS). In line with the data from AML patients, reduced p21Cip1/Waf expression was commonly correlated to p73 methylation (186).
More studies are required to precisely understand how the p21Cip1/Waf methylation status interferes with disease progression and if p73 methylation can be used as a marker for the p21Cip1/Waf status.
In addition to growth arrest, p21Cip1/Waf is involved in apoptosis, DNA repair and senescence. For instance, one of the most extensively studied functions of p21Cip1/Waf is the protection of cells against apoptosis.
An example is given by the usage of histone deacetylase inhibitors (HDACI) to induce apoptosis (187–189). p21Cip1/Waf expression is upregulated by an increased histone acetylation of H3K4 at the p21Cip1/Waf promoter region, which is mediated by the HDACI SAHA (suberoylanilide hydroxamic acid) (190). p21Cip1/Waf overexpression confers resistance to SAHA-induced apoptosis which was shown in human AML cells. SAHA treatment promotes apoptotic cell death in leukemic cells by inducing pro-apoptotic genes such as TRAIL (TNF-related apoptosis-inducing ligand) and its downstream effector caspase-8. One mechanism through which p21Cip1/Waf exerts anti-apoptotic effects in AML cell lines is the inhibition of caspase-8 cleavage to suppress TRAIL-mediated apoptosis (191).
A second anti-apoptotic function of p21Cip1/Waf was also reported for AML blasts. There, high cytoplasmatic p21Cip1/Waf protein levels provide protection against cytotoxic agents. Blasts with cytoplasmatic p21Cip1/Waf levels show reduced etoposide (VP-16) mediated apoptosis (192). Similarly, the enforced expression of p21Cip1/Waf in CML blast cells confers resistance to Imatinib induced apoptosis (193). These studies suggest that p21Cip1/Waf expression should be investigated to act as a marker for therapeutic outcome.
p21Cip1/Waf expression is essential for the initiation and maintenance of leukemogenesis induced by PML/RAR-transformed HSCs. Under this condition p21Cip1/Waf is required to maintain the self-renewal capacity of LSCs and to limit DNA-damage. p21Cip1/Waf protects from functional exhaustion (194). In line p21Cip1/Waf is crucial for the maintenance of self-renewal and chemoresistance of LSCs in a murine model of T-ALL (195).
In MLL-AF10-induced AML p21Cip1/Waf suppression is achieved by the oncomir miR-17-91, that is associated with enhanced LSC self-renewal and decreased leukemia latency (196). Functional studies for the role of p21Cip1/Waf have been mainly carried out in cell lines from different leukemia subtypes. The literature on primary patient samples is scarce. It appears that the involvement of p21Cip1/Waf is highly context dependent and relies on the differentiation status of the cells and on the driver oncogenes.
The fact that p21Cip1/Waf is important to maintain stem cell self-renewal might provide a basis for novel attempts to target p21Cip1/Waf to induce exhaustion.
p27Kip1 regulates cell proliferation by inhibiting CDK complexes and arresting cell proliferation in response to anti-mitogenic signals (Figure 1) (8, 197–199).
Analysis of p27Kip1 knock-out mice highlighted the importance of p27Kip1 as cell cycle regulator: p27Kip1 deficient mice have an overall augmented cell proliferation which is reflected in increased body size and hyperplastic organs. Tumor formation becomes manifested spontaneously; pituitary and parathyroid tumors evolve and the mice show an increased susceptibility to tumorigenesis upon γ-irradiation or treatment by the chemical carcinogen N-ethyl-N-nitrosourea (ENU) (79, 80, 200). These studies defined p27Kip1 as tumor suppressor.
Mutations in the p27Kip1 gene and its homozygous inactivation are generally rare in human cancers. In people CDKN1B, encoding for p27Kip1, has been identified as the second most common altered gene by frame-shift mutations in heterozygosity in hairy cell leukemia (HCL), a form of B-cell CLL. In most patients the CDKN1B mutation is clonal, thereby suggesting an early role in the pathogenesis of HCL (201, 202).
The subcellular location of p27Kip1 and its concentration determine the impact on malignant transformation. On the one hand, p27Kip1 acts as a tumor suppressor by inhibiting CDK-cyclin complexes and cell cycle progression when present in the nucleus. On the other hand, a localization shift of p27Kip1 from the nucleus to the cytoplasm, may promote tumor formation by regulating cytoskeletal structure and cell migration (89).
Augmented levels of p27Kip1 and its cytoplasmic localization have been correlated with poor prognosis and increased metastasis in diverse solid tumors including breast (94), cervix (97) and esophagus (95) carcinomas, as well as in some lymphoma and leukemia (91–93).
Despite a rare mutation rate, p27Kip1 deregulation is one of the key events promoting leukemogenesis. Several mechanisms altering p27Kip1 expression and localization have been described. miRNAs play a prominent role and abundance of p27Kip1 subjected to miRNA-mediated regulation: oncogenic expression of miRNA targeting p27Kip1 translation can cause p27Kip1 loss (203). In CML patients, increased miR-152-3p promotes aggressive behavior of CML cells by targeting p27Kip1 (204). Similarly, miR-148a correlates with low p27Kip1 expression and increased proliferation in MM cells (205).
In lymphoma, low p27Kip1 levels correlate with a poor prognosis (206). Vice versa, high p27Kip1 levels are associated with enhanced disease-free survival in AML, indicative for disease progression (207).
In contrast, AML patients with low p27Kip1 due to deletion of the chromosomal region 12p13, have a better overall survival. Although together with CDKN1B, nine other genes are located in the 12p13 chromosomal region, the reported improved clinical outcome can be ascribed to reduced CDKN1B expression levels which might lead to higher cell proliferation which makes leukemic cells more susceptible to cytotoxic agents (208).
Besides the genomic alterations, also the phosphorylation sites play an important role for p27Kip1 levels. p27Kip1 is a substrate of FLT3 and FLT3-ITD in AML patient samples, where they phosphorylate p27Kip1 at the residue Y88 which is required for subsequent p27Kip1 phosphorylation at T187 by the CDK2-cyclin complex marking p27Kip1 for SCFSkp2-mediated degradation. FLT3 inhibition reduces pY88-p27Kip1 and increases p27Kip1 levels leading to cell cycle arrest (209).
High p27Kip1 levels are associated with a poor outcome in B-cell chronic lymphocytic leukemia (B-CLL). In B-CLL disease progression does not result from uncontrolled cell proliferation but is the result of defective apoptosis and enhanced cell survival. High p27Kip1 expression is discussed to contribute to the protection against apoptotic stimuli like p21Cip1/Waf (93).
The presence of high p27Kip1 levels in CLL was confirmed by others who also found an inverse correlation with c-Myc protein levels. C-Myc deregulation is a frequent event in leukemia and lymphoma (210, 211). Low Myc levels are associated with low expression of its target gene Skp2, a component of the SCFSkp2 ubiquitin ligase complex that degrades p27Kip1. The reduced Skp2-mediated degradation leads to the p27Kip1 accumulation which confers resistance to apoptosis (210).
In untransformed CD34+ progenitor cells, β1-integrin engagement increases p27Kip1 nuclear levels, which in turn decrease CDK2 activity thus restraining G1/S-phase progression. BCR-ABL expression in CML CD34+ cells induces elevated cytoplasmatic p27Kip1 levels. In this context, such high p27Kip1 levels do not restrain CML cell proliferation due to its cytoplasmatic relocation, thereby contributing to the loss of integrin-mediated proliferation inhibition observed in normal CD34+ cells (212).
More recent studies demonstrate that BCR-ABL1 promotes leukemia by subverting nuclear p27Kip1 tumor-suppressor function via two independent mechanisms. In a kinase-dependent manner, BCR-ABL1 induces SCFSkp2 expression through the PI3K pathway (213), promoting the degradation of nuclear p27Kip1, thus compromising its tumor-suppressor activity. In a kinase-independent fashion it increases cytoplasmatic p27Kip1 abundance, preventing apoptosis and thereby promoting leukemic cell survival (214, 215).
The overexpression of a stable p27Kip1 harboring two point mutations which prevent its phosphorylation on sites responsible for its SCFSkp2-mediated nuclear degradation (T187A) and for its PI3K-directed cytoplasmatic sequestration (T157A) causes a G1/S arrest, markedly inhibiting proliferation of BCR-ABL+ cells (216).
The complexity of the regulation mechanism regulation location and degradation require further investigations to define disease entities where p27Kip1 may serve as clinical marker.
Based on its ability to inhibit G1-S phase cyclin-CDK complexes, p57Kip2 is considered a tumor suppressor. As mentioned above for p21Cip1/Waf and p27Kip1, p57Kip2 is involved in many cellular processes including apoptosis, and cellular migration.
The fact that p57Kip2 has a crucial role during embryogenesis and is required for normal embryonic development makes it unique under der CKI family. p57Kip2 knock-out mice show severe developmental defects and display increased embryonic and perinatal lethality (217, 218) which complicated further studies on tumorigenesis in mice and most studies rely on human patient samples.
Reduced p57Kip2 expression is associated with high tumor aggressiveness and poor prognosis in several types of tumors, such as gastric, colorectal, pancreatic, breast and lung carcinoma as well as leukemia (103, 104, 219–221). p57Kip2 expression is decreased in MDS, in particular in patients with a poor karyotype. Low expression results from an impaired response to the SDF-1/CXCR4 signal which induces p57Kip2 expression (222). p57Kip2 knock-out mice show hyperproliferation and differentiation delay in several tissues (218), which are features associated with the pathogenesis of MDS (223).
Another described mechanism how p57Kip2 expression is altered is promoter methylation. Hypermethylation of the CDKN1C gene, encoding for p57Kip2, occurs in diffuse large B-cell lymphoma (DLBCL), follicular lymphoma, ALL (224, 225) and nodal DLBCL (226). In the low-risk group of DLBCL, CDKN1C methylation is associated with a more favorable overall survival. The authors proposed aberrant CDKN1C promoter methylation as a biological marker in patients with DLBCL (226). Another study in DLBCL patients suggested that the analysis of CDKN1C methylation status may serve as a biomarker for the detection of minimal residual disease, underlining the importance of p57Kip2 for determining leukemia relapse risk (227).
Analysis of the p57Kip2 methylation status in adult and childhood ALL found a rate of 50% CDKN1C hypermethylation in adult ALL but only 7% hypermethylation in childhood leukemia (226). Interestingly, in 53% of the childhood ALL samples p57Kip2 was absent without methylation and overall p57Kip2 levels were 8-fold lower compared to normal lymphocytes. The low expression points at additional ways to regulate p57Kip2 in this particular disease class (228). In line, p57Kip2 methylation and protein expression in adult ALL patients does not show any correlation as 10 out of 15 patients with CDKN1C hypermethylation expressed p57Kip2 (229).
Overall, methylation status of p57Kip2 does not seem to be a reliable marker for p57Kip2 levels. Conditional knockout mice would be a useful tool to study the role of p57Kip2 in hematopoietic diseases in more detail.
CDK kinase inhibitors are under extensive investigation in numerous preclinical and clinical studies in a variety of solid tumors and they are currently tested in hematological neoplasms (230, 231).
Pan-CDK inhibitors represented the very first generation of CDK inhibitors with the function to restrain cell proliferation via the inhibition of the CDK enzymatic activity. Flavopiridol was the first CDK inhibitor used in clinical trials and tested for the treatment of ALL, AML and CLL (232–234). Due to their low selectivity causing severe cytotoxic effects in healthy cells and a wide range of side effects, pan-CDK inhibitors have been discontinued in clinical trials (113, 235).
Considering the key role of CDK6 in malignant hematopoiesis it represents an effective therapeutic target (236–238). This is underlined by the high frequency of p15INK4b and p16INK4a inactivation in leukemia and lymphoma. The development of more specific CDK inhibitors, including CDK4/6-kinase inhibitors, represented an exciting turn over in the field (239).
Palbociclib is a CDK4/6 kinase inhibitor that acts by blocking enzymatic functions by mimicking INK4 binding. Palbociclib has been FDA approved to treat breast cancer patients and clinical trials exploring its effects in hematological malignancies are ongoing. Richter et al. present in their recent work (231) an extensive and detailed collection of preclinical and clinical studies conducted with several CDK4/6 inhibitors in hematological diseases.
Palbociclib resistance is a common phenomenon in breast cancer patients (240, 241). In breast cancer and AML high levels of p16INK4a and p18INK4c are associated with resistance to Palbociclib and to a CDK6 protein degrader that is based on the structure of Palbociclib. Despite this correlation, low p16INK4a levels are not predictive for Palbociclib sensitivity (242). All INK4 proteins are in principle capable to prevent Palbociclib binding to CDK6 and thereby capable to induce resistance. Whether this fact is also true for other CDK inhibitors needs to be investigated. The cell-type specific expression of INK4 proteins needs also to be taken into consideration when studying CDK-inhibitors resistance.
The challenge in the development of novel inhibitors is in the design of molecules able to reduce the side effects and to overcome drug resistance. An innovative approach of CDK inhibition would consider the possibility to mimic the functions of INK4 proteins for a selective inactivation of CDKs. However, intensive research is needed to fill the need of X-ray crystal structures of most of the CDKs and CDKs/INK4/Cip/Kip complexes and to make this creative approach possible.
INK4 and Cip/Kip proteins were initially identified as CDK inhibitors and negative regulators of cell cycle progression. Only recently, the involvement in other cellular processes including apoptosis and cell migration was uncovered. Thereby CKIs bridge cell cycle regulation to other cellular functions. Under certain circumstances CKIs may even promote cancer progression.
Tumor cells frequently display mutations in CKIs which underscores the significance of these proteins for tumorigenesis. We here summarize the dominant alterations of CKIs in hematopoietic malignancies and discuss their consequences for disease development, maintenance, and diagnosis.
Within the INK4 family, p15INK4b and p16INK4a are most frequently inactivated in leukemia and lymphoma either by deletion or hypermethylation of 5’ CpG islands in their promoter regions (114–116, 118, 140–150). The prognostic importance of these alterations in distinct disease entities remains unclear. Considering the unique functions of each INK4 proteins, especially their role under stress conditions, one could speculate that distinct expression patterns lead to different disease subtypes and dictates therapeutic outcomes.
CDK4/6 specific inhibitors represent a promising valuable choice for the treatment of hematological malignancies. However, resistance to CDK inhibitor therapy has been frequently observed. INK4 proteins are capable of inducing resistance by binding to CDK6. Studies are needed to evaluate whether this holds true for other CDK inhibitors.
As proliferation and cell cycle control are essential features of a cell, the components of the cell cycle machinery are present in multiple variants, which can substitute for each other. INK4 proteins share common tasks and, in a similar manner, CDKs may substitute for each other. This complexity makes it exceedingly difficult to generalize any consequence upon loss or mutations of a single player. Effects will also be context and cell type dependent.
This enormous plasticity of the cell cycle machinery to adapt ensures cell proliferation and presents a major challenge when it comes to predict therapeutic outcomes of drugs interfering with CDKs or INKs. The removal or inhibition of a single player may be rapidly compensated by a rearrangement of CDK complexes.
Another layer of complexity is induced by the emerging CDK6 kinase-independent functions that regulate transcriptional processes relevant for leukemia. The involvement of CDK6 in LSCs biology makes it an attractive target for leukemia therapy (238, 243). It is unclear how CKIs binding to CDK6 interferes with the transcriptional role of CDK6. It is also unknown whether INK4 or Cip/Kip binding to CDK6 alters the composition of CDK6 containing transcriptional complexes and/or chromatin location. We need to understand how CDK-CKIs complexes interfere with cell cycle-independent functions to reliable predict treatment outcomes. Moreover, effects of kinase inhibitor treatment on the kinase-independent functions of CDK6 are still enigmatic. The frequent upregulation of CDK6 (237, 235) in hematopoietic tumors (243, 244) and the fact that alterations of INK4 proteins are commonly found in hematopoietic tumors demands for the understanding of any CDK6-INK4 correlation in leukemia/lymphoma to exploit CDK4/6 inhibitors in hematopoietic malignancies.
Despite the importance of p18INK4d for HSC self-renewal under homeostatic and stress conditions (40, 52,53), p18INK4d mutations are not a hallmark of hematopoietic malignancies. p18INK4d deregulation is rarely observed in hematopoietic neoplasms. Alterations on the transcriptional/translational level cannot be entirely excluded. As such the oncogene MLL-AF9 regulates p18INK4d. In line, the comparison of AML subtypes identified distinct INK4 expression patterns for different AML entities. The global analysis of the protein levels of individual CIKs in respect to their hematopoietic disease type is required to design tailored treatment strategies.
We are only starting to understand and appreciate functions of the Cip/Kip proteins in regulating apoptosis and cell migration. The involvement of Cip/Kip in tumorigenesis is an attractive emerging field of research and will open novel innovative therapeutic avenues.
p21Cip1/Waf has a dual context-dependent role in leukemogenesis and acts as tumor suppressor and promoter. In cell lines, the anti-apoptotic effect of cytoplasmatic p21Cip1/Waf confers a survival advantage and mediates chemoresistance. Inhibition of p21Cip1/Waf under these conditions bears the potential to sensitize leukemic cells to chemotherapy. Similarly, cytoplasmatic p27Kip1 prevents apoptosis and may be exploited as potential therapeutic target. Most studies rely on cell lines and this only partially reflects the in vivo situation. The reality-check in patients is still missing to judge the clinical relevance of these observations. Therapeutic strategies that simultaneously target oncogenic Cip/Kip functions while preserving tumor suppressive functions would represent an innovative optimal approach.
All authors made substantial, direct, and intellectual contributions to the work. KK was the principal investigator and takes primary responsibility for the paper. AS, VS and KK wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Open Access Funding by the Austrian Science Fund (FWF), project grant P 31773 (KK), and the European Research Council under the European Union’s Horizon 2020 research and innovation programme, grant agreement no. 694354 (VS).
Graphics were created with BioRender.com (24 March 2022). Open Access Funding by the University of Veterinary Medicine Vienna.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
3. Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer (2001) 1:222–31. doi: 10.1038/35106065
4. Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta (1998) 1378:F115–177. doi: 10.1016/s0304-419x(98)00017-1
6. Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol (1997) 13:261–91. doi: 10.1146/annurev.cellbio.13.1.261
7. Hartwell LH, Weinert TA. Checkpoints: Controls that ensure the order of cell cycle events. Science (1989) 246:629–34. doi: 10.1126/science.2683079
8. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev (1999) 13:1501–12. doi: 10.1101/gad.13.12.1501
9. Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer (2002) 2:910–7. doi: 10.1038/nrc950
10. Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin a is required at two points in the human cell cycle. EMBO J (1992) 11:961–71. doi: 10.1002/j.1460-2075.1992.tb05135.x
11. Pagano M, Pepperkok R, Lukas J, Baldin V, Ansorge W, Bartek J, et al. Regulation of the cell cycle by the cdk2 protein kinase in cultured human fibroblasts. J Cell Biol (1993) 121:101–11. doi: 10.1083/jcb.121.1.101
12. Tsai LH, Lees E, Faha B, Harlow E, Riabowol K. The cdk2 kinase is required for the G1-to-S transition in mammalian cells. Oncogene (1993) 8:1593–602.
13. Liao H, Ji F, Ying S. CDK1: beyond cell cycle regulation. Aging (Albany NY) (2017) 9:2465–6. doi: 10.18632/aging.101348
14. Bashir T, Pagano M. Cdk1: the dominant sibling of Cdk2. Nat Cell Biol (2005) 7:779–81. doi: 10.1038/ncb0805-779
15. Ekholm SV, Reed SI. Regulation of G1 cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol (2000) 12:676–84. doi: 10.1016/S0955-0674(00)00151-4
16. Russo AA, Tong L, Lee JO, Jeffrey PD, Pavletich NP. Structural basis for inhibition of the cyclin-dependent kinase Cdk6 by the tumour suppressor p16INK4a. Nature (1998) 395:237–43. doi: 10.1038/26155
17. Blain SW, Montalvo E, Massagué J. Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 with cyclin a-Cdk2 and cyclin D2-Cdk4. J Biol Chem (1997) 272:25863–72. doi: 10.1074/jbc.272.41.25863
18. LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, et al. New functional activities for the p21 family of CDK inhibitors. Genes Dev (1997) 11:847–62. doi: 10.1101/gad.11.7.847
19. James MK, Ray A, Leznova D, Blain SW. Differential modification of p27Kip1 controls its cyclin d-cdk4 inhibitory activity. Mol Cell Biol (2008) 28:498–510. doi: 10.1128/MCB.02171-06
20. Sugimoto M, Martin N, Wilks DP, Tamai K, Huot TJG, Pantoja C, et al. Activation of cyclin D1-kinase in murine fibroblasts lacking both p21(Cip1) and p27(Kip1). Oncogene (2002) 21:8067–74. doi: 10.1038/sj.onc.1206019
21. Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, et al. The p21(Cip1) and p27(Kip1) CDK “inhibitors” are essential activators of cyclin d-dependent kinases in murine fibroblasts. EMBO J (1999) 18:1571–83. doi: 10.1093/emboj/18.6.1571
22. El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell (1993) 75:817–25. doi: 10.1016/0092-8674(93)90500-P
23. Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell (1993) 75:805–16. doi: 10.1016/0092-8674(93)90499-g
24. Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev (1995) 9:1149–63. doi: 10.1101/gad.9.10.1149
25. Leonardo AD, Linke SP, Clarkin K, Wahl GM. DNA Damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev (1994) 8:2540–51. doi: 10.1101/gad.8.21.2540
26. Zhang H, Hannon GJ, Beach D. p21-containing cyclin kinases exist in both active and inactive states. Genes Dev (1994) 8:1750–8. doi: 10.1101/gad.8.15.1750
27. Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai LH, Zhang P, et al. Inhibition of cyclin-dependent kinases by p21. MBoC (1995) 6:387–400. doi: 10.1091/mbc.6.4.387
28. Guiley KZ, Stevenson JW, Lou K, Barkovich KJ, Kumarasamy V, Wijeratne TU, et al. p27 allosterically activates cyclin-dependent kinase 4 and antagonizes palbociclib inhibition. Science (2019) 366:eaaw2106. doi: 10.1126/science.aaw2106
29. Rossi MN, Antonangeli F. Cellular response upon stress: p57 contribution to the final outcome. Mediators Inflammation (2015) 2015:259325. doi: 10.1155/2015/259325
30. El-Deiry WS. The role of p53 in chemosensitivity and radiosensitivity. Oncogene (2003) 22:7486–95. doi: 10.1038/sj.onc.1206949
31. Mandinova A, Lee SW. The p53 pathway as a target in cancer therapeutics: Obstacles and promise. Sci Trans Med (2011) 3:64rv1–1. doi: 10.1126/scitranslmed.3001366
32. Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell (2009) 137:413–31. doi: 10.1016/j.cell.2009.04.037
33. Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells (1978) 4:7–25.
34. Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol (2006) 6:93–106. doi: 10.1038/nri1779
35. Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med (2010) 2:640–53. doi: 10.1002/wsbm.86
36. Giebel B, Bruns I. Self-renewal versus differentiation in hematopoietic stem and progenitor cells: a focus on asymmetric cell divisions. Curr Stem Cell Res Ther (2008) 3:9–16. doi: 10.2174/157488808783489444
37. Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science (2000) 287:1804–8. doi: 10.1126/science.287.5459.1804
38. Cheng T, Rodrigues N, Dombkowski D, Stier S, Scadden DT. Stem cell repopulation efficiency but not pool size is governed by p27(kip1). Nat Med (2000) 6:1235–40. doi: 10.1038/81335
39. Park I, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature (2003) 423:302–5. doi: 10.1038/nature01587
40. Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat Cell Biol (2004) 6:436–42. doi: 10.1038/ncb1126
41. Stone S, Jiang P, Dayananth P, Tavtigian SV, Katcher H, Parry D, et al. Complex structure and regulation of the P16 (MTS1) locus. Cancer Res (1995) 55:2988–94.
42. Mao L, Merlo A, Bedi G, Shapiro GI, Edwards CD, Rollins BJ, et al. A novel p16INK4A transcript. Cancer Res (1995) 55:2995–7.
43. Ouelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell (1995) 83:993–1000. doi: 10.1016/0092-8674(95)90214-7
44. Sharpless NE, DePinho RA. The INK4A/ARF locus and its two gene products. Curr Opin Genet Dev (1999) 9:22–30. doi: 10.1016/s0959-437x(99)80004-5
45. Lessard J, Baban S, Sauvageau G. Stage-specific expression of polycomb group genes in human bone marrow cells. Blood (1998) 91:1216–24. doi: 10.1182/blood.V91.4.1216
46. Lessard J, Schumacher A, Thorsteinsdottir U, van Lohuizen M, Magnuson T, Sauvageau G. Functional antagonism of the polycomb-group genes eed and Bmi1 in hemopoietic cell proliferation. Genes Dev (1999) 13:2691–703. doi: 10.1101/gad.13.20.2691
47. Park I-K, He Y, Lin F, Laerum OD, Tian Q, Bumgarner R, et al. Differential gene expression profiling of adult murine hematopoietic stem cells. Blood (2002) 99:488–98. doi: 10.1182/blood.v99.2.488
48. Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature (2003) 423:255–60. doi: 10.1038/nature01572
49. Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell (1996) 85:27–37. doi: 10.1016/s0092-8674(00)81079-x
50. Passegué E, Wagers AJ, Giuriato S, Anderson WC, Weissman IL. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med (2005) 202:1599–611. doi: 10.1084/jem.20050967
51. Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature (2006) 443:421–6. doi: 10.1038/nature05159
52. Gao Y, Yang P, Shen H, Yu H, Song X, Zhang L, et al. Small-molecule inhibitors targeting INK4 protein p18(INK4C) enhance ex vivo expansion of haematopoietic stem cells. Nat Commun (2015) 6:6328. doi: 10.1038/ncomms7328
53. Yu H, Yuan Y, Shen H, Cheng T. Hematopoietic stem cell exhaustion impacted by p18 INK4C and p21 Cip1/Waf1 in opposite manners. Blood (2006) 107:1200–6. doi: 10.1182/blood-2005-02-0685
54. Rosu-Myles M, Wolff L. p15Ink4b: dual function in myelopoiesis and inactivation in myeloid disease. Blood Cells Mol Dis (2008) 40:406–9. doi: 10.1016/j.bcmd.2007.09.005
55. Rosu-Myles M, Taylor BJ, Wolff L. Loss of the tumor suppressor p15Ink4b enhances myeloid progenitor formation from common myeloid progenitors. Exp Hematol (2007) 35:394–406. doi: 10.1016/j.exphem.2006.11.005
56. Zindy F, van Deursen J, Grosveld G, Sherr CJ, Roussel MF. INK4d-deficient mice are fertile despite testicular atrophy. Mol Cell Biol (2000) 20:372–8. doi: 10.1128/MCB.20.1.372-378.2000
57. Gilles L, Guièze R, Bluteau D, Cordette-Lagarde V, Lacout C, Favier R, et al. P19INK4D links endomitotic arrest and megakaryocyte maturation and is regulated by AML-1. Blood (2008) 111:4081–91. doi: 10.1182/blood-2007-09-113266
58. Hilpert M, Legrand C, Bluteau D, Balayn N, Betems A, Bluteau O, et al. p19INK4d controls hematopoietic stem cells in a cell-autonomous manner during genotoxic stress and through the microenvironment during aging. Stem Cell Rep (2014) 3:1085–102. doi: 10.1016/j.stemcr.2014.10.005
59. van Os R, Kamminga LM, Ausema A, Bystrykh LV, Draijer DP, van Pelt K, et al. A limited role for p21Cip1/Waf1 in maintaining normal hematopoietic stem cell functioning. Stem Cells (2007) 25:836–43. doi: 10.1634/stemcells.2006-0631
60. Matsumoto A, Takeishi S, Kanie T, Susaki E, Onoyama I, Tateishi Y, et al. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell (2011) 9:262–71. doi: 10.1016/j.stem.2011.06.014
61. Ortega S, Malumbres M, Barbacid M. Cyclin d-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta (2002) 1602:73–87. doi: 10.1016/s0304-419x(02)00037-9
62. Gil J, Peters G. Regulation of the INK4b–ARF–INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol (2006) 7:667–77. doi: 10.1038/nrm1987
63. Gu F, Pfeiffer RM, Bhattacharjee S, Han SS, Taylor PR, Berndt S, et al. Common genetic variants in the 9p21 region and their associations with multiple tumours. Br J Cancer (2013) 108:1378–86. doi: 10.1038/bjc.2013.7
64. Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar arf sequesters Mdm2 and activates p53. Nat Cell Biol (1999) 1:20–6. doi: 10.1038/8991
65. Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol (2001) 2:731–7. doi: 10.1038/35096061
66. Liggett WH, Sidransky D. Role of the p16 tumor suppressor gene in cancer. J Clin Oncol (1998) 16:1197–206. doi: 10.1200/JCO.1998.16.3.1197
67. Okamoto A, Demetrick DJ, Spillare EA, Hagiwara K, Hussain SP, Bennett WP, et al. Mutations and altered expression of p16INK4 in human cancer. Proc Natl Acad Sci U.S.A. (1994) 91:11045–9. doi: 10.1073/pnas.91.23.11045
68. Otsuki T, Clark HM, Wellmann A, Jaffe ES, Raffeld M. Involvement of CDKN2 (p16INK4A/MTS1) and p15INK4B/MTS2 in human leukemias and lymphomas. Cancer Res (1995) 55:1436–40.
69. Sill H, Goldman JM, Cross NCP. Homozygous deletions of the p16 tumor-suppressor gene are associated with lymphoid transformation of chronic myeloid leukemia. Blood (1995) 85:2013–6. doi: 10.1182/blood.V85.8.2013.bloodjournal8582013
70. Takeuchi S, Bartram CR, Seriu T, Miller CW, Tobler A, Janssen JW, et al. Analysis of a family of cyclin-dependent kinase inhibitors: p15/MTS2/INK4B, p16/MTS1/INK4A, and p18 genes in acute lymphoblastic leukemia of childhood. Blood (1995) 86:755–60. doi: 10.1182/blood.V86.2.755.bloodjournal862755
71. Guan KL, Jenkins CW, Li Y, Nichols MA, Wu X, O’Keefe CL, et al. Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev (1994) 8:2939–52. doi: 10.1101/gad.8.24.2939
72. Guan KL, Jenkins CW, Li Y, O’Keefe CL, Noh S, Wu X, et al. Isolation and characterization of p19INK4d, a p16-related inhibitor specific to CDK6 and CDK4. Mol Biol Cell (1996) 7:57–70. doi: 10.1091/mbc.7.1.57
73. van Veelen W, Klompmaker R, Gloerich M, van Gasteren CJR, Kalkhoven E, Berger R, et al. P18 is a tumor suppressor gene involved in human medullary thyroid carcinoma and pheochromocytoma development. Int J Cancer (2009) 124:339–45. doi: 10.1002/ijc.23977
74. Morishita A, Masaki T, Yoshiji H, Nakai S, Ogi T, Miyauchi Y, et al. Reduced expression of cell cycle regulator p18(INK4C) in human hepatocellular carcinoma. Hepatology (2004) 40:677–86. doi: 10.1002/hep.20337
75. Lapointe J, Lachance Y, Labrie Y, Labrie C. A p18 mutant defective in CDK6 binding in human breast cancer cells. Cancer Res (1996) 56:4586–9.
76. Miller CW, Yeon C, Aslo A, Mendoza S, Aytac U, Koeffler HP. The p19INK4D cyclin dependent kinase inhibitor gene is altered in osteosarcoma. Oncogene (1997) 15:231–5. doi: 10.1038/sj.onc.1201185
77. Morishita A, Gong J, Deguchi A, Tani J, Miyoshi H, Yoshida H, et al. Frequent loss of p19INK4D expression in hepatocellular carcinoma: relationship to tumor differentiation and patient survival. Oncol Rep (2011) 26:1363–8. doi: 10.3892/or.2011.1452
78. Bartkova J, Thullberg M, Rajpert-De Meyts E, Skakkebaek NE, Bartek J. Lack of p19INK4d in human testicular germ-cell tumours contrasts with high expression during normal spermatogenesis. Oncogene (2000) 19:4146–50. doi: 10.1038/sj.onc.1203769
79. Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, et al. Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell (1996) 85:707–20. doi: 10.1016/S0092-8674(00)81237-4
80. Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27Kip1-deficient mice. Cell (1996) 85:733–44. doi: 10.1016/S0092-8674(00)81239-8
81. Denicourt C, Dowdy SF. Cip/Kip proteins: more than just CDKs inhibitors. Genes Dev (2004) 18:851–5. doi: 10.1101/gad.1205304
82. Besson A, Assoian RK, Roberts JM. Regulation of the cytoskeleton: an oncogenic function for cdk inhibitors? Nat Rev Cancer (2004) 4:948–55. doi: 10.1038/nrc1501
83. Roninson IB. Oncogenic functions of tumour suppressor p21(Waf1/Cip1/Sdi1): association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett (2002) 179:1–14. doi: 10.1016/s0304-3835(01)00847-3
84. Gartel AL. Is p21 an oncogene? Mol Cancer Ther (2006) 5:1385–6. doi: 10.1158/1535-7163.MCT-06-0163
85. Slingerland J, Pagano M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol (2000) 183:10–7. doi: 10.1002/(SICI)1097-4652(200004)183:1<10::AID-JCP2>3.0.CO;2-I
86. Philipp-Staheli J, Payne SR, Kemp CJ. p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res (2001) 264:148–68. doi: 10.1006/excr.2000.5143
87. Bloom J, Pagano M. Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin Cancer Biol (2003) 13:41–7. doi: 10.1016/s1044-579x(02)00098-6
88. Boehm M, Yoshimoto T, Crook MF, Nallamshetty S, True A, Nabel GJ, et al. A growth factor-dependent nuclear kinase phosphorylates p27(Kip1) and regulates cell cycle progression. EMBO J (2002) 21:3390–401. doi: 10.1093/emboj/cdf343
89. McAllister SS, Becker-Hapak M, Pintucci G, Pagano M, Dowdy SF. Novel p27(kip1) c-terminal scatter domain mediates rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol (2003) 23:216–28. doi: 10.1128/MCB.23.1.216-228.2003
90. Fujita N, Sato S, Katayama K, Tsuruo T. Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J Biol Chem (2002) 277:28706–13. doi: 10.1074/jbc.M203668200
91. Sáez A, Sánchez E, Sánchez-Beato M, Cruz MA, Chacón I, Muñoz E, et al. p27KIP1 is abnormally expressed in diffuse Large b-cell lymphomas and is associated with an adverse clinical outcome. Br J Cancer (1999) 80:1427–34. doi: 10.1038/sj.bjc.6690539
92. Sánchez-Beato M, Camacho FI, Martínez-Montero JC, Sáez AI, Villuendas R, Sánchez-Verde L, et al. Anomalous high p27/KIP1 expression in a subset of aggressive b-cell lymphomas is associated with cyclin D3 overexpression. p27/KIP1-cyclin D3 colocalization in tumor cells. Blood (1999) 94:765–72. doi: 10.1182/blood.V94.2.765
93. Vrhovac R, Delmer A, Tang R, Marie JP, Zittoun R, Ajchenbaum-Cymbalista F. Prognostic significance of the cell cycle inhibitor p27Kip1 in chronic b-cell lymphocytic leukemia. Blood (1998) 91:4694–700. doi: 10.1182/blood.V91.12.4694
94. Kouvaraki M, Gorgoulis VG, Rassidakis GZ, Liodis P, Markopoulos C, Gogas J, et al. High expression levels of p27 correlate with lymph node status in a subset of advanced invasive breast carcinomas: relation to e-cadherin alterations, proliferative activity, and ploidy of the tumors. Cancer (2002) 94:2454–65. doi: 10.1002/cncr.10505
95. Anayama T, Furihata M, Ishikawa T, Ohtsuki Y, Ogoshi S. Positive correlation between p27Kip1 expression and progression of human esophageal squamous cell carcinoma. Int J Cancer (1998) 79:439–43. doi: 10.1002/(sici)1097-0215(19980821)79:4<439::aid-ijc22>3.0.co;2-z
96. Watanabe J, Sato H, Kanai T, Kamata Y, Jobo T, Hata H, et al. Paradoxical expression of cell cycle inhibitor p27 in endometrioid adenocarcinoma of the uterine corpus - correlation with proliferation and clinicopathological parameters. Br J Cancer (2002) 87:81–5. doi: 10.1038/sj.bjc.6600434
97. Shiozawa T, Shiohara S, Kanai M, Konishi I, Fujii S, Nikaido T. Expression of the cell cycle regulator p27(Kip1) in normal squamous epithelium, cervical intraepithelial neoplasia, and invasive squamous cell carcinoma of the uterine cervix. immunohistochemistry and functional aspects of p27(Kip1). Cancer (2001) 92:3005–11. doi: 10.1002/1097-0142(20011215)92:12<3005::aid-cncr10153>3.0.co;2-6
98. Dellas A, Schultheiss E, Leivas MR, Moch H, Torhorst J. Association of p27Kip1, cyclin e and c-myc expression with progression and prognosis in HPV-positive cervical neoplasms. Anticancer Res (1998) 18:3991–8.
99. Winters ZE, Leek RD, Bradburn MJ, Norbury CJ, Harris AL. Cytoplasmic p21WAF1/CIP1 expression is correlated with HER-2/ neu in breast cancer and is an independent predictor of prognosis. Breast Cancer Res (2003) 5:R242–9. doi: 10.1186/bcr654
100. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer (2009) 9:400–14. doi: 10.1038/nrc2657
101. Guo H, Lv Y, Tian T, Hu TH, Wang WJ, Sui X, et al. Downregulation of p57 accelerates the growth and invasion of hepatocellular carcinoma. Carcinogenesis (2011) 32:1897–904. doi: 10.1093/carcin/bgr220
102. Guo S-X, Taki T, Ohnishi H, Piao H-Y, Tabuchi K, Bessho F, et al. Hypermethylation of p16 and p15 genes and RB protein expression in acute leukemia. Leukemia Res (2000) 24:39–46. doi: 10.1016/S0145-2126(99)00158-7
103. Borriello A, Caldarelli I, Bencivenga D, Criscuolo M, Cucciolla V, Tramontano A, et al. p57(Kip2) and cancer: time for a critical appraisal. Mol Cancer Res (2011) 9:1269–84. doi: 10.1158/1541-7786.MCR-11-0220
104. Kavanagh E, Joseph B. The hallmarks of CDKN1C (p57, KIP2) in cancer. Biochim Biophys Acta (2011) 1816:50–6. doi: 10.1016/j.bbcan.2011.03.002
105. Krug U, Ganser A, Koeffler HP. Tumor suppressor genes in normal and malignant hematopoiesis. Oncogene (2002) 21:3475–95. doi: 10.1038/sj.onc.1205322
106. Sulong S, Moorman AV, Irving JAE, Strefford JC, Konn ZJ, Case MC, et al. A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups. Blood (2009) 113:100–7. doi: 10.1182/blood-2008-07-166801
107. Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19 ARF. Cell (1997) 91:649–59. doi: 10.1016/S0092-8674(00)80452-3
108. Kamijo T, Bodner S, van de KE, DH R, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res (1999) 59:2217–22.
109. Williams RT, Roussel MF, Sherr CJ. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of bcr-abl-induced acute lymphoblastic leukemia. Proc Natl Acad Sci U.S.A. (2006) 103:6688–93. doi: 10.1073/pnas.0602030103
110. Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature (2001) 413:83–6. doi: 10.1038/35092584
111. Latres E, Malumbres M, Sotillo R, Martín J, Ortega S, Martín-Caballero J, et al. Limited overlapping roles of P15INK4b and P18INK4c cell cycle inhibitors in proliferation and tumorigenesis. EMBO J (2000) 19:3496–506. doi: 10.1093/emboj/19.13.3496
112. Wölfel T, Hauer M, Schneider J, Serrano M, Wölfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science (1995) 269:1281–4. doi: 10.1126/science.7652577
113. Rodríguez-Díez E, Quereda V, Bellutti F, Prchal-Murphy M, Partida D, Eguren M, et al. Cdk4 and Cdk6 cooperate in counteracting the INK4 family of inhibitors during murine leukemogenesis. Blood (2014) 124:2380–90. doi: 10.1182/blood-2014-02-555292
114. Ogawa S, Hirano N, Sato N, Takahashi T, Hangaishi A, Tanaka K, et al. Homozygous loss of the cyclin-dependent kinase 4-inhibitor (p16) gene in human leukemias. Blood (1994) 84:2431–5. doi: 10.1182/blood.V84.8.2431.2431
115. Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science (1994) 264:436–40. doi: 10.1126/science.8153634
116. Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature (1994) 368:753–6. doi: 10.1038/368753a0
117. Hirama T, Koeffler H. Role of the cyclin-dependent kinase inhibitors in the development of cancer. Blood (1995) 86:841–54. doi: 10.1182/blood.V86.3.841.841
118. Siebert R, Willers CP, Opalka B. Role of the cyclin-dependent kinase 4 and 6 inhibitor gene family p15, p16, p18 and p19 in leukemia and lymphoma. Leuk Lymphoma (1996) 23:505–20. doi: 10.3109/10428199609054859
119. Haidar MA, Cao XB, Manshouri T, Chan LL, Glassman A, Kantarjian HM, et al. p16INK4A and p15INK4B gene deletions in primary leukemias. Blood (1995) 86:311–5. doi: 10.1182/blood.V86.1.311.bloodjournal861311
120. Hebert J, Cayuela JM, Berkeley J, Sigaux F. Candidate tumor-suppressor genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous deletions in primary cells from T- but not from b-cell lineage acute lymphoblastic leukemias. Blood (1994) 84:4038–44. doi: 10.1182/blood.V84.12.4038.bloodjournal84124038
121. Duro D, Flexor MA, Bernard O, d’Agay MF, Berger R, Larsen CJ. Alterations of the putative tumor suppressor gene p16/MTS1 in human hematological malignancies. C R Acad Sci III (1994) 317:913–9.
122. Okuda T, Shurtleff SA, Valentine MB, Raimondi SC, Head DR, Behm F, et al. Frequent deletion of p16INK4a/MTS1 and p15INK4b/MTS2 in pediatric acute lymphoblastic leukemia. Blood (1995) 85:2321–30. doi: 10.1182/blood.V85.9.2321.bloodjournal8592321
123. Diccianni MB, Batova A, Yu J, Vu T, Pullen J, Amylon M, et al. Shortened survival after relapse in T-cell acute lymphoblastic leukemia patients with p16/p15 deletions. Leuk Res (1997) 21:549–58. doi: 10.1016/s0145-2126(97)00007-6
124. Genescà E, Lazarenkov A, Morgades M, Berbis G, Ruíz-Xivillé N, Gómez-Marzo P, et al. Frequency and clinical impact of CDKN2A/ARF/CDKN2B gene deletions as assessed by in-depth genetic analyses in adult T cell acute lymphoblastic leukemia. J Hematol Oncol (2018) 11:96. doi: 10.1186/s13045-018-0639-8
125. Zhang W, Kuang P, Liu T. Prognostic significance of CDKN2A/B deletions in acute lymphoblastic leukaemia: a meta-analysis. Ann Med (2019) 51:28–40. doi: 10.1080/07853890.2018.1564359
126. Carrasco Salas P, Fernández L, Vela M, Bueno D, González B, Valentín J, et al. The role of CDKN2A/B deletions in pediatric acute lymphoblastic leukemia. Pediatr Hematol Oncol (2016) 33:415–22. doi: 10.1080/08880018.2016.1251518
127. Wang H-P, Zhou Y-L, Huang X, Zhang Y, Qian J-J, Li J-H, et al. CDKN2A deletions are associated with poor outcomes in 101 adults with T-cell acute lymphoblastic leukemia. Am J Hematol (2021) 96:312–9. doi: 10.1002/ajh.26069
128. Quesnel B, Preudhomme C, Philippe N, Vanrumbeke M, Dervite I, Lai JL, et al. pl6 gene homozygous deletions in acute lymphoblastic leukemia. Blood (1995) 85:657–63. doi: 10.1182/blood.V85.3.657.bloodjournal853657
129. Zhou M, Gu L, Yeager AM, Findley HW. Incidence and clinical significance of CDKN2/MTS1/P16ink4A and MTS2/P15ink4B gene deletions in childhood acute lymphoblastic leukemia. Pediatr Hematol Oncol (1997) 14:141–50. doi: 10.3109/08880019709030900
130. Kees UR, Burton PR, Lü C, Baker DL. Homozygous deletion of the p16/MTS1 gene in pediatric acute lymphoblastic leukemia is associated with unfavorable clinical outcome. Blood (1997) 89:4161–6. doi: 10.1182/blood.V89.11.4161
131. Carter TL, Watt PM, Kumar R, Burton PR, Reaman GH, Sather HN, et al. Hemizygous p16(INK4A) deletion in pediatric acute lymphoblastic leukemia predicts independent risk of relapse. Blood (2001) 97:572–4. doi: 10.1182/blood.v97.2.572
132. Dalle JH, Fournier M, Nelken B, Mazingue F, Laıü J-L, Bauters F, et al. p16INK4a immunocytochemical analysis is an independent prognostic factor in childhood acute lymphoblastic leukemia. Blood (2002) 99:2620–3. doi: 10.1182/blood.V99.7.2620
133. Xiao X, Xu N, Zhang J, Cao R, Huang Y, Xiao Y, et al. [Comparison of clinical implications of p16 deletion in childhood and adult b-lineage acute lymphoblastic leukemia]. Zhonghua Xue Ye Xue Za Zhi (2013) 34:389–94. doi: 10.3760/cma.j.issn.0253-2727.2013.05.003
134. Ohnishi H, Guo SX, Ida K, Taki T, Naritaka S, Bessho F, et al. Alterations of p16 and p15 genes in acute leukemia with MLL gene rearrangements and their correlation with clinical features. Leukemia (1997) 11:2120–4. doi: 10.1038/sj.leu.2400872
135. van Zutven LJCM, van Drunen E, de Bont JM, Wattel MM, Den Boer ML, Pieters R, et al. CDKN2 deletions have no prognostic value in childhood precursor-b acute lymphoblastic leukaemia. Leukemia (2005) 19:1281–4. doi: 10.1038/sj.leu.2403769
136. Mirebeau D, Acquaviva C, Suciu S, Bertin R, Dastugue N, Robert A, et al. The prognostic significance of CDKN2A, CDKN2B and MTAP inactivation in b-lineage acute lymphoblastic leukemia of childhood. results of the EORTC studies 58881 and 58951. Haematologica (2006) 91:881–5.
137. Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet (2017) 49:1211–8. doi: 10.1038/ng.3909
138. Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, et al. 5’ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med (1995) 1:686–92. doi: 10.1038/nm0795-686
139. Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res (1995) 55:4525–30.
140. Herman JG, Civin CI, Issa JP, Collector MI, Sharkis SJ, Baylin SB. Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Res (1997) 57:837–41.
141. Ng MHL, Chung YF, Lo KW, Wickham NWR, Lee JCK, Huang DP. Frequent hypermethylation of p16 and p15 genes in multiple myeloma. Blood (1997) 89:2500–6. doi: 10.1182/blood.V89.7.2500
142. Iravani M, Dhat R, Price CM. Methylation of the multi tumor suppressor gene-2 (MTS2, CDKN1, p15INK4B) in childhood acute lymphoblastic leukemia. Oncogene (1997) 15:2609–14. doi: 10.1038/sj.onc.1201428
143. Batova A, Diccianni MB, Yu JC, Nobori T, Link MP, Pullen J, et al. Frequent and selective methylation of p15 and deletion of both p15 and p16 in T-cell acute lymphoblastic leukemia. Cancer Res (1997) 57:832–6.
144. Uchida T, Kinoshita T, Nagai H, Nakahara Y, Saito H, Hotta T, et al. Hypermethylation of the p15INK4B gene in myelodysplastic syndromes. Blood (1997) 90:1403–9. doi: 10.1182/blood.V90.4.1403
145. Quesnel B, Guillerm G, Vereecque R, Wattel E, Preudhomme C, Bauters F, et al. Methylation of the p15(INK4b) gene in myelodysplastic syndromes is frequent and acquired during disease progression. Blood (1998) 91:2985–90. doi: 10.1182/blood.V91.8.2985.2985_2985_2990
146. Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Methylation of p15INK4B is common, is associated with deletion of genes on chromosome arm 7q and predicts a poor prognosis in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia (2003) 17:1813–9. doi: 10.1038/sj.leu.2403054
147. Klangby U, Okan I, Magnusson KP, Wendland M, Lind P, Wiman KG. p16/INK4a and p15/INK4b gene methylation and absence of p16/INK4a mRNA and protein expression in burkitt’s lymphoma. Blood (1998) 91:1680–7. doi: 10.1182/blood.V91.5.1680
148. García MJ, Martínez-Delgado B, Cebrian A, Martínez Á, Benítez J, Rivas C. Different incidence and pattern of p15INK4b and p16INK4a promoter region hypermethylation in hodgkin’s and CD30-positive non-hodgkin’s lymphomas. Am J Pathol (2002) 161:1007–13. doi: 10.1016/S0002-9440(10)64261-7
149. Lo YM, Wong IH, Zhang J, Tein MS, Ng MH, Hjelm NM. Quantitative analysis of aberrant p16 methylation using real-time quantitative methylation-specific polymerase chain reaction. Cancer Res (1999) 59:3899–903.
150. González M, Mateos MV, García-Sanz R, Balanzategui A, López-Pérez R, Chillón MC, et al. De novo methylation of tumor suppressor gene p16/INK4a is a frequent finding in multiple myeloma patients at diagnosis. Leukemia (2000) 14:183–7. doi: 10.1038/sj.leu.2401617
151. Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, et al. CDK inhibitors p18INK4c and p27Kip1 mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev (1998) 12:2899–911. doi: 10.1101/gad.12.18.2899
152. Nakamaki T, Kawamata N, Schwaller J, Tobler A, Fey M, Pakkala S, et al. Structural integrity of the cyclin-dependent kinase inhibitor genes, p15, p16 and p18 in myeloid leukaemias. Br J Haematol (1995) 91:139–49. doi: 10.1111/j.1365-2141.1995.tb05259.x
153. Drexler HG. Review of alterations of the cyclin-dependent kinase inhibitor INK4 family genes p15, p16, p18 and p19 in human leukemia-lymphoma cells. Leukemia (1998) 12:845–59. doi: 10.1038/sj.leu.2401043
154. Johansson B, Mertens F, Mitelman F. Cytogenetic deletion maps of hematologic neoplasms: Circumstantial evidence for tumor suppressor loci. Genes Chromosomes Cancer (1993) 8:205–18. doi: 10.1002/gcc.2870080402
155. Chim CS, Wong ASY, Kwong YL. Epigenetic inactivation of INK4/CDK/RB cell cycle pathway in acute leukemias. Ann Hematol (2003) 82:738–42. doi: 10.1007/s00277-003-0744-8
156. Tasaka T, Berenson J, Vescio R, Hirama T, Miller CW, Nagai M, et al. Analysis of the p16INK4A, p15INK4B and p18INK4C genes in multiple myeloma. Br J Haematol (1997) 96:98–102. doi: 10.1046/j.1365-2141.1997.8552482.x
157. Kulkarni MS, Daggett JL, Bender TP, Kuehl WM, Bergsagel PL, Williams ME. Frequent inactivation of the cyclin-dependent kinase inhibitor p18 by homozygous deletion in multiple myeloma cell lines: ectopic p18 expression inhibits growth and induces apoptosis. Leukemia (2002) 16:127–34. doi: 10.1038/sj.leu.2402328
158. Morse L, Chen D, Franklin D, Xiong Y, Chen-Kiang S. Induction of cell cycle arrest and b cell terminal differentiation by CDK inhibitor p18 INK4c and IL-6. Immunity (1997) 6:47–56. doi: 10.1016/S1074-7613(00)80241-1
159. Tourigny MR, Ursini-Siegel J, Lee H, Toellner K-M, Cunningham AF, Franklin DS, et al. CDK inhibitor p18(INK4c) is required for the generation of functional plasma cells. Immunity (2002) 17:179–89. doi: 10.1016/s1074-7613(02)00364-3
160. Koduru PR, Zariwala M, Soni M, Gong JZ, Xiong Y, Broome JD. Deletion of cyclin-dependent kinase 4 inhibitor genes P15 and P16 in non-hodgkin’s lymphoma. Blood (1995) 86:2900–5. doi: 10.1182/blood.V86.8.2900.2900
161. Williams ME, Whitefield M, Swerdlow SH. Analysis of the cyclin-dependent kinase inhibitors p18 and p19 in mantle-cell lymphoma and chronic lymphocytic leukemia. Ann Oncol (1997) 8 Suppl 2:71–3. doi: 10.1093/annonc/8.suppl_2.S71
162. Shiohara M, Gombart AF, Morosetti R, Said JW, Spirin K, Koeffler HP. Mutational analysis of CDKIs in a large series of non-hodgkins lymphomas. Blood (1995) 86(10 Suppl 1):823A.
163. Wang X, Tan Y, Li Y, Li J, Jin W, Wang K. Repression of CDKN2C caused by PML/RARα binding promotes the proliferation and differentiation block in acute promyelocytic leukemia. Front Med (2016) 10:420–9. doi: 10.1007/s11684-016-0478-3
164. Prange KHM, Mandoli A, Kuznetsova T, Wang S-Y, Sotoca AM, Marneth AE, et al. MLL-AF9 and MLL-AF4 oncofusion proteins bind a distinct enhancer repertoire and target the RUNX1 program in 11q23 acute myeloid leukemia. Oncogene (2017) 36:3346–56. doi: 10.1038/onc.2016.488
165. Shiohara M, Spirin K, Said JW, Gombart AF, Nakamaki T, Takeuchi S, et al. Alterations of the cyclin-dependent kinase inhibitor p19 (INK4D) is rare in hematopoietic malignancies. Leukemia (1996) 10:1897–900.
166. Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature (1995) 377:552–7. doi: 10.1038/377552a0
167. Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell (1995) 82:675–84. doi: 10.1016/0092-8674(95)90039-x
168. Martín-Caballero J, Flores JM, García-Palencia P, Serrano M. Tumor susceptibility of p21Waf1/Cip1-deficient mice. Cancer Res (2001) 61:6234–8.
169. Sun Y, Yang S, Sun N, Chen J. Differential expression of STAT1 and p21 proteins predicts pancreatic cancer progression and prognosis. Pancreas (2014) 43:619–23. doi: 10.1097/MPA.0000000000000074
170. Xie D, Lan L, Huang K, Chen L, Xu C, Wang R, et al. Association of p53/p21 expression and cigarette smoking with tumor progression and poor prognosis in non-small cell lung cancer patients. Oncol Rep (2014) 32:2517–26. doi: 10.3892/or.2014.3538
171. Goan Y-G, Hsu H-K, Chang H-C, Chou Y-P, Chiang K-H, Cheng J-T. Deregulated p21WAF1 overexpression impacts survival of surgically resected esophageal squamous cell carcinoma patients. Ann Thorac Surg (2005) 80:1007–16. doi: 10.1016/j.athoracsur.2005.03.050
172. Natsugoe S, Nakashima S, Matsumoto M, Xiangming C, Okumura H, Kijima F, et al. Expression of p21WAF1/Cip1 in the p53-dependent pathway is related to prognosis in patients with advanced esophageal carcinoma. Clin Cancer Res (1999) 5:2445–9.
173. Lin Y, Shen L-Y, Fu H, Dong B, Yang H-L, Yan W-P, et al. P21, COX-2, and e-cadherin are potential prognostic factors for esophageal squamous cell carcinoma. Dis Esophagus (2017) 30:1–10. doi: 10.1111/dote.12522
174. Gamboa-Dominguez A, Seidl S, Reyes-Gutierrez E, Hermannstädter C, Quintanilla-Martinez L, Busch R, et al. Prognostic significance of p21WAF1/CIP1, p27Kip1, p53 and e-cadherin expression in gastric cancer. J Clin Pathol (2007) 60:756–61. doi: 10.1136/jcp.2006.038976
175. Kouraklis G, Katsoulis IE, Theocharis S, Tsourouflis G, Xipolitas N, Glinavou A, et al. Does the expression of cyclin e, pRb, and p21 correlate with prognosis in gastric adenocarcinoma? Dig Dis Sci (2009) 54:1015–20. doi: 10.1007/s10620-008-0464-y
176. Liu X, Yu H, Cai H, Wang Y. Expression of CD24, p21, p53, and c-myc in alpha-fetoprotein-producing gastric cancer: Correlation with clinicopathologic characteristics and survival. J Surg Oncol (2014) 109:859–64. doi: 10.1002/jso.23599
177. Pinyol M, Hernandez L, Cazorla M, Balbıín M, Jares P, Fernandez PL, et al. Deletions and loss of expression of P16INK4a and P21Waf1 genes are associated with aggressive variants of mantle cell lymphomas. Blood (1997) 89:272–80. doi: 10.1182/blood.V89.1.272
178. Berg T, Fliegauf M, Burger J, Staege MS, Liu S, Martinez N, et al. Transcriptional upregulation of p21/WAF/Cip1 in myeloid leukemic blasts expressing AML1-ETO. Haematologica (2008) 93:1728–33. doi: 10.3324/haematol.13044
179. Kawamura M, Ohnishi H, Guo S-X, Sheng XM, Minegishi M, Hanada R, et al. Alterations of the p53, p21, p16, p15 and RAS genes in childhood T-cell acute lymphoblastic leukemia. Leukemia Res (1999) 23:115–26. doi: 10.1016/S0145-2126(98)00146-5
180. Roman-Gomez J, Castillejo JA, Jimenez A, Gonzalez MG, Moreno F, Rodriguez M del C, et al. 5′ CpG island hypermethylation is associated with transcriptional silencing of the p21CIP1/WAF1/SDI1 gene and confers poor prognosis in acute lymphoblastic leukemia. Blood (2002) 99:2291–6. doi: 10.1182/blood.V99.7.2291
181. Shen L, Kondo Y, Issa J-P, Garcia-Manero G. Lack of p21CIP1 DNA methylation in acute lymphocytic leukemia. Blood (2002) 100:3432–3. doi: 10.1182/blood-2002-07-1990
182. De Cave F, Petrucci MT, Gregorj C, Ricciardi MR, Decandia S, Bergamo P, et al. Protein expression of p15 and p21 plays an unfavorable prognostic role in adult acute lymphoblastic leukemia (ALL) patients independently of their gene promoter methylation status. Blood (2007) 110:2802. doi: 10.1182/blood.V110.11.2802.2802
183. Melino G, De Laurenzi V, Vousden KH. p73: Friend or foe in tumorigenesis. Nat Rev Cancer (2002) 2:605–15. doi: 10.1038/nrc861
184. Schmelz K, Wagner M, Dörken B, Tamm I. 5-Aza-2′-deoxycytidine induces p21WAF expression by demethylation of p73 leading to p53-independent apoptosis in myeloid leukemia. Int J Cancer (2005) 114:683–95. doi: 10.1002/ijc.20797
185. Schmelz K, Sattler N, Wagner M, Lübbert M, Dörken B, Tamm I. Induction of gene expression by 5-Aza-2’-deoxycytidine in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) but not epithelial cells by DNA-methylation-dependent and -independent mechanisms. Leukemia (2005) 19:103–11. doi: 10.1038/sj.leu.2403552
186. Zhao Y, Guo J, Zhang X, Zhang Z, Gu S, Fei C, et al. Downregulation of p21 in myelodysplastic syndrome is associated with p73 promoter hypermethylation and indicates poor prognosis. Am J Clin Pathol (2013) 140:819–27. doi: 10.1309/AJCPZ5E6IWPWSZXE
187. Vrana JA, Decker RH, Johnson CR, Wang Z, Jarvis WD, Richon VM, et al. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by bcl-2/Bcl-XL, c-jun, and p21CIP1, but independent of p53. Oncogene (1999) 18:7016–25. doi: 10.1038/sj.onc.1203176
188. Burgess AJ, Pavey S, Warrener R, Hunter LJ, Piva TJ, Musgrove EA, et al. Up-regulation of p21(WAF1/CIP1) by histone deacetylase inhibitors reduces their cytotoxicity. Mol Pharmacol (2001) 60:828–37.
189. Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, et al. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci U.S.A. (1999) 96:4592–7. doi: 10.1073/pnas.96.8.4592
190. Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci U.S.A. (2000) 97:10014–9. doi: 10.1073/pnas.180316197
191. Wu X, Yang N, Zhou W, Xu J, Chen J, Zheng F, et al. Up-regulation of P21 inhibits TRAIL-mediated extrinsic apoptosis, contributing resistance to SAHA in acute myeloid leukemia cells. CPB (2014) 34:506–18. doi: 10.1159/000363018
192. Schepers H, Geugien M, Eggen BJL, Vellenga E. Constitutive cytoplasmic localization of p21Waf1/Cip1 affects the apoptotic process in monocytic leukaemia. Leukemia (2003) 17:2113–21. doi: 10.1038/sj.leu.2403106
193. Ferrandiz N, Caraballo JM, Albajar M, Gomez-Casares MT, Lopez-Jorge CE, Blanco R, et al. p21(Cip1) confers resistance to imatinib in human chronic myeloid leukemia cells. Cancer Lett (2010) 292:133–9. doi: 10.1016/j.canlet.2009.11.017
194. Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, Bossi D, et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature (2009) 457:51–6. doi: 10.1038/nature07618
195. Tremblay CS, Saw J, Chiu SK, Wong NC, Tsyganov K, Ghotb S, et al. Restricted cell cycle is essential for clonal evolution and therapeutic resistance of pre-leukemic stem cells. Nat Commun (2018) 9:3535. doi: 10.1038/s41467-018-06021-7
196. Wong P, Iwasaki M, Somervaille TCP, Ficara F, Carico C, Arnold C, et al. The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res (2010) 70:3833–42. doi: 10.1158/0008-5472.CAN-09-3268
197. Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, et al. p27Kip1, a cyclin-cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev (1994) 8:9–22. doi: 10.1101/gad.8.1.9
198. Polyak K, Lee MH, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell (1994) 78:59–66. doi: 10.1016/0092-8674(94)90572-x
199. Coats S, Flanagan WM, Nourse J, Roberts JM. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science (1996) 272:877–80. doi: 10.1126/science.272.5263.877
200. Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27Kip1. Cell (1996) 85:721–32. doi: 10.1016/S0092-8674(00)81238-6
201. Dietrich S, Hüllein J, Lee SC-W, Hutter B, Gonzalez D, Jayne S, et al. Recurrent CDKN1B (p27) mutations in hairy cell leukemia. Blood (2015) 126:1005–8. doi: 10.1182/blood-2015-04-643361
202. Robak T, Smolewski P. New mutation in hairy cell leukemia. Blood (2015) 126:930–1. doi: 10.1182/blood-2015-06-652065
203. le Sage C, Nagel R, Agami R. Diverse ways to control p27Kip1 function: miRNAs come into play. Cell Cycle (2007) 6:2742–9. doi: 10.4161/cc.6.22.4900
204. Wang L, Wang Y, Lin J. MiR-152-3p promotes the development of chronic myeloid leukemia by inhibiting p27. Eur Rev Med Pharmacol Sci (2018) 22:8789–96. doi: 10.26355/eurrev_201812_16646
205. Lang T, Nie Y. MiR-148a participates in the growth of RPMI8226 multiple myeloma cells by regulating CDKN1B. Biomed Pharmacother (2016) 84:1967–71. doi: 10.1016/j.biopha.2016.11.002
206. Erlanson M, Portin C, Linderholm B, Lindh J, Roos G, Landberg G. Expression of cyclin e and the cyclin-dependent kinase inhibitor p27 in malignant lymphomas-prognostic implications. Blood (1998) 92:770–7. doi: 10.1182/blood.V92.3.770
207. Yokozawa T, Towatari M, Iida H, Takeya K, Tanimoto M, Kiyoi H, et al. Prognostic significance of the cell cycle inhibitor p27Kip1 in acute myeloid leukemia. Leukemia (2000) 14:28–33. doi: 10.1038/sj.leu.2401640
208. Haferlach C, Bacher U, Kohlmann A, Schindela S, Alpermann T, Kern W, et al. CDKN1B, encoding the cyclin-dependent kinase inhibitor 1B (p27), is located in the minimally deleted region of 12p abnormalities in myeloid malignancies and its low expression is a favorable prognostic marker in acute myeloid leukemia. Haematologica (2011) 96:829–36. doi: 10.3324/haematol.2010.035584
209. Peschel I, Podmirseg SR, Taschler M, Duyster J, Götze KS, Sill H, et al. FLT3 and FLT3-ITD phosphorylate and inactivate the cyclin-dependent kinase inhibitor p27Kip1 in acute myeloid leukemia. Haematologica (2017) 102:1378–89. doi: 10.3324/haematol.2016.160101
210. Caraballo JM, Acosta JC, Cortés MA, Albajar M, Gómez-Casares TM, Batlle-López A, et al. High p27 protein levels in chronic lymphocytic leukemia are associated to low myc and Skp2 expression, confer resistance to apoptosis and antagonize myc effects on cell cycle. Oncotarget (2014) 5:4694–708. doi: 10.18632/oncotarget.2100
211. Delgado MD, León J. Myc roles in hematopoiesis and leukemia. Genes Cancer (2010) 1:605–16. doi: 10.1177/1947601910377495
212. Jiang Y, Zhao RCH, Verfaillie CM. Abnormal integrin-mediated regulation of chronic myelogenous leukemia CD34+ cell proliferation: BCR/ABL up-regulates the cyclin-dependent kinase inhibitor, p27Kip, which is relocated to the cell cytoplasm and incapable of regulating cdk2 activity. PNAS (2000) 97:10538–43. doi: 10.1073/pnas.190104497
213. Andreu EJ, Lledó E, Poch E, Ivorra C, Albero MP, Martínez-Climent JA, et al. BCR-ABL induces the expression of Skp2 through the PI3K pathway to promote p27Kip1 degradation and proliferation of chronic myelogenous leukemia cells. Cancer Res (2005) 65:3264–72. doi: 10.1158/0008-5472.CAN-04-1357
214. Grimmler M, Wang Y, Mund T, Cilenšek Z, Keidel E-M, Waddell MB, et al. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell (2007) 128:269–80. doi: 10.1016/j.cell.2006.11.047
215. Agarwal A, Mackenzie RJ, Besson A, Jeng S, Carey A, LaTocha DH, et al. BCR-ABL1 promotes leukemia by converting p27 into a cytoplasmic oncoprotein. Blood (2014) 124:3260–73. doi: 10.1182/blood-2013-04-497040
216. Sengupta A, Banerjee D, Chandra S, Banerjee S. Gene therapy for BCR-ABL+ human CML with dual phosphorylation resistant p27Kip1 and stable RNA interference using an EBV vector. J Gene Med (2006) 8:1251–61. doi: 10.1002/jgm.959
217. Yan Y, Frisén J, Lee MH, Massagué J, Barbacid M. Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev (1997) 11:973–83. doi: 10.1101/gad.11.8.973
218. Zhang P, Liégeois NJ, Wong C, Finegold M, Hou H, Thompson JC, et al. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in beckwith-wiedemann syndrome. Nature (1997) 387:151–8. doi: 10.1038/387151a0
219. Li J-Q, Wu F, Usuki H, Kubo A, Masaki T, Fujita J, et al. Loss of p57KIP2 is associated with colorectal carcinogenesis. Int J Oncol (2003) 23:1537–43. doi: 10.3892/ijo.23.6.1537
220. Pateras IS, Apostolopoulou K, Koutsami M, Evangelou K, Tsantoulis P, Liloglou T, et al. Downregulation of the KIP family members p27KIP1 and p57KIP2 by SKP2 and the role of methylation in p57KIP2 inactivation in nonsmall cell lung cancer. Int J Cancer (2006) 119:2546–56. doi: 10.1002/ijc.22214
221. Qiu Z, Li Y, Zeng B, Guan X, Li H. Downregulated CDKN1C/p57kip2 drives tumorigenesis and associates with poor overall survival in breast cancer. Biochem Biophys Res Commun (2018) 497:187–93. doi: 10.1016/j.bbrc.2018.02.052
222. Zhao Y, Guo J, Gu S, Zhang X, Li X, Chang C. SDF-1/CXCR4 signal is involved in decreased expression of p57kip2 in de novo MDS patients. Hematology (2012) 17:220–8. doi: 10.1179/1607845412Y.0000000005
223. Bejar R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of myelodysplastic syndromes. J Clin Oncol (2011) 29:504–15. doi: 10.1200/JCO.2010.31.1175
224. Li Y, Nagai H, Ohno T, Yuge M, Hatano S, Ito E, et al. Aberrant DNA methylation ofp57KIP2 gene in the promoter region in lymphoid malignancies of b-cell phenotype. Blood (2002) 100:2572–7. doi: 10.1182/blood-2001-11-0026
225. Shen L, Toyota M, Kondo Y, Obata T, Daniel S, Pierce S, et al. Aberrant DNA methylation of p57KIP2 identifies a cell-cycle regulatory pathway with prognostic impact in adult acute lymphocytic leukemia. Blood (2003) 101(10):4131–6. doi: 10.1182/BLOOD-2002-08-2466
226. Lee SM, Lee EJ, Ko Y-H, Lee SH, Maeng L, Kim K-M. Prognostic significance of O6-methylguanine DNA methyltransferase and p57 methylation in patients with diffuse large b-cell lymphomas. APMIS (2009) 117:87–94. doi: 10.1111/j.1600-0463.2008.00017.x
227. Hagiwara K, Li Y, Kinoshita T, Kunishma S, Ohashi H, Hotta T, et al. Aberrant DNA methylation of the p57KIP2 gene is a sensitive biomarker for detecting minimal residual disease in diffuse large b cell lymphoma. Leuk Res (2010) 34:50–4. doi: 10.1016/j.leukres.2009.06.028
228. Gutiérrez MI, Siraj AK, Ibrahim MM, Hussain A, Bhatia K. Childhood and adult ALL: Differences in epigenetic lesions associated with cell cycle genes. Am J Hematol (2005) 80:158–60. doi: 10.1002/ajh.20458
229. Bueso-Ramos C, Xu Y, McDonnell TJ, Brisbay S, Pierce S, Kantarjian H, et al. Protein expression of a triad of frequently methylated genes, p73, p57Kip2, and p15, has prognostic value in adult acute lymphocytic leukemia independently of its methylation status. JCO (2005) 23:3932–9. doi: 10.1200/JCO.2005.02.998
230. Zhang M, Zhang L, Hei R, Li X, Cai H, Wu X, et al. CDK inhibitors in cancer therapy, an overview of recent development. Am J Cancer Res (2021) 11:1913–35.
231. Richter A, Schoenwaelder N, Sender S, Junghanss C, Maletzki C. Cyclin-dependent kinase inhibitors in hematological malignancies-current understanding, (Pre-)Clinical application and promising approaches. Cancers (Basel) (2021) 13:2497. doi: 10.3390/cancers13102497
232. Zeidner JF, Karp JE. Clinical activity of alvocidib (flavopiridol) in acute myeloid leukemia. Leuk Res (2015) 39:1312–8. doi: 10.1016/j.leukres.2015.10.010
233. Phelps MA, Lin TS, Johnson AJ, Hurh E, Rozewski DM, Farley KL, et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood (2009) 113:2637–45. doi: 10.1182/blood-2008-07-168583
234. Parker BW, Kaur G, Nieves-Neira W, Taimi M, Kohlhagen G, Shimizu T, et al. Early induction of apoptosis in hematopoietic cell lines after exposure to flavopiridol. Blood (1998) 91:458–65. doi: 10.1182/blood.V91.2.458
235. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discovery (2015) 14:130–46. doi: 10.1038/nrd4504
236. Malumbres M, Sotillo R, Santamarıía D, Galán J, Cerezo A, Ortega S, et al. Mammalian cells cycle without the d-type cyclin-dependent kinases Cdk4 and Cdk6. Cell (2004) 118:493–504. doi: 10.1016/j.cell.2004.08.002
237. Scheicher R, Hoelbl-Kovacic A, Bellutti F, Tigan A-S, Prchal-Murphy M, Heller G, et al. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood (2015) 125:90–101. doi: 10.1182/blood-2014-06-584417
238. Uras IZ, Sexl V, Kollmann K. CDK6 inhibition: A novel approach in AML management. Int J Mol Sci (2020) 21:2528. doi: 10.3390/ijms21072528
239. Yuan K, Wang X, Dong H, Min W, Hao H, Yang P. Selective inhibition of CDK4/6: A safe and effective strategy for developing anticancer drugs. Acta Pharm Sin B (2021) 11:30–54. doi: 10.1016/j.apsb.2020.05.001
240. Green JL, Okerberg ES, Sejd J, Palafox M, Monserrat L, Alemayehu S, et al. Direct CDKN2 modulation of CDK4 alters target engagement of CDK4 inhibitor drugs. Mol Cancer Ther (2019) 18:771–9. doi: 10.1158/1535-7163.MCT-18-0755
241. McCartney A, Migliaccio I, Bonechi M, Biagioni C, Romagnoli D, De Luca F, et al. Mechanisms of resistance to CDK4/6 inhibitors: Potential implications and biomarkers for clinical practice. Front Oncol (2019) 9:666. doi: 10.3389/fonc.2019.00666
242. Schmalzbauer BS, Thondanpallil T, Heller G, Schirripa A, Sperl C-M, Mayer IM, et al. CDK6 degradation is counteracted by p16INK4A and p18INK4C in AML. Cancers (Basel) (2022) 14:1554. doi: 10.3390/cancers14061554
243. Kollmann K, Heller G, Schneckenleithner C, Warsch W, Scheicher R, Ott RG, et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell (2013) 24:167–81. doi: 10.1016/j.ccr.2013.07.012
Keywords: cyclin-dependent kinase inhibitors, hematopoiesis, hematopoietic diseases, INK4 family, Cip/Kip family
Citation: Schirripa A, Sexl V and Kollmann K (2022) Cyclin-dependent kinase inhibitors in malignant hematopoiesis. Front. Oncol. 12:916682. doi: 10.3389/fonc.2022.916682
Received: 09 April 2022; Accepted: 18 July 2022;
Published: 11 August 2022.
Edited by:
Laura Rosanò, National Research Council (CNR), ItalyReviewed by:
Florian Perner, Universitätsmedizin Greifswald, GermanyCopyright © 2022 Schirripa, Sexl and Kollmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karoline Kollmann, a2Fyb2xpbmUua29sbG1hbm5AdmV0bWVkdW5pLmFjLmF0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.