 Meng-Ying Cui
Meng-Ying Cui Xing Yi
Xing Yi Dan-Xia Zhu*
Dan-Xia Zhu*- Department of Oncology, The Third Affiliated Hospital of Soochow University, Changzhou, China
Gastric cancer has been one of the most common cancers worldwide with extensive metastasis and high mortality. Chemotherapy has been found as a main treatment for metastatic gastric cancer, whereas drug resistance limits the effectiveness of chemotherapy and leads to treatment failure. Chemotherapy resistance in gastric cancer has a complex and multifactorial mechanism, among which lipid metabolism plays a vital role. Increased synthesis of new lipids or uptake of exogenous lipids can facilitate the rapid growth of cancer cells and tumor formation. Lipids form the structural basis of biofilms while serving as signal molecules and energy sources. It is noteworthy that lipid metabolism is capable of inducing drug resistance in gastric cancer cells by reshaping the tumor micro-environment. In this study, new mechanisms of lipid metabolism in gastric cancer and the metabolic pathways correlated with chemotherapy resistance are reviewed. In particular, we discuss the effects of lipid metabolism on autophagy, biomarkers treatment and drug resistance in gastric cancer from the perspective of lipid metabolism. In brief, new insights can be gained into the development of promising therapies through an in-depth investigation of the mechanism of lipid metabolism reprogramming and resensitization to chemotherapy in gastric cancer cells, and scientific treatment can be provided by applying lipid-key enzyme inhibitors as cancer chemical sensitizers in clinical settings.
Background
Despite significant advances in clinical practice over the past few years, gastric cancer (GC) remains recognized as the leading cause of cancer-related deaths worldwide. Chemotherapy takes on a critical significance in the treatment of local and metastatic gastric cancer, whereas chemotherapy resistance has limited the efficacy of chemotherapy. Chemotherapeutic drug resistance was found as a complex multi-factor phenomenon, whether inherent or acquired, which was linked to tumor cells and tumor microenvironment (1). Abnormal lipid metabolism has been found as one of the critical metabolic features of tumor cells. In addition, cancer cells obtain fatty acid through lipid decomposition, thereby promoting fatty acid (FA) synthesis and increasing lipid uptake. FAs were found to play a certain role in various aspects of tumorigenesis and tumor progression (2). However, the specific process of lipid reprogramming in gastric cancer remains unclear, and the biological functions and regulatory mechanisms of lipid reprogramming have been rarely investigated. Under normal physiological conditions, gastric epithelial cells are constantly renewed. When damages to the stomach due to various factors occur, the epithelial cells tend to renew more rapidly. During the occurrence of gastric cancer, gastric epithelial cells proliferated rapidly, leading to the transformation from normal gastric mucosa to atrophic gastritis, intestinal metaplasia, dysplasia, as well as gastric cancer (3). Abnormal lipid metabolism can affect all aspects of gastric cancer growth. An abnormal expression of genes correlated with fatty acid synthesis or oxidation was found to be significantly correlated with malignant phenotypes (e.g., tumor metastasis, drug resistance, and recurrence) (4). Low serum high-density lipoprotein levels predicted a higher risk of gastric cancer, higher rates of lymphatic and vascular infiltration, advanced lymph node metastasis, as well as poor prognosis (5–7). Adipocytes and fatty acids were found to promote gastric cancer metastasis, which indicated a poor prognosis of gastric cancer (8–10). In a mouse gastric cancer model, it was found that tumor tissues were often characterized by the up-regulated expression of fatty acid metabolism-related proteins (11, 12). Accordingly, targeting lipid metabolism has been recognized as a promising strategy for the treatment of gastric cancer patients. However, specific strategies targeting the vital regulatory factors for lipid metabolism reprogramming to treat gastric cancer remain challenging in existing medical research. In this study, the mechanisms of lipid metabolism in gastric cancer cells are briefly reviewed, with an emphasis on the pathways of lipid origin, utilization and storage, as well as the vital enzymes of lipid metabolism used as biomarkers for monitoring gastric cancer metastasis, therapeutic effect and prognosis. Targeting and blocking pathways in gastric cancer can enhance chemotherapeutic resistance and increase possible therapeutic benefits. This study aims to identify novel prognostic biomarkers to reduce risk, stratify patients, and guide subsequent research into potential new therapeutic targets.
Reprogramming of Lipid Metabolism in Gastric Cancer
Exogenous FAs Absorption
CD36 accounts for free fatty acid and cholesterol uptake and intracellular signaling, playing a critical role in cancer-related antigen presentation, inflammation and angiogenesis (13). CD36 was found to increase the uptake of exogenous palmitic acid in gastric cancer and induce metastasis via the AKT/GSK-3β/β-catenin signaling pathway (14). Furthermore, fatty acid-induced up-regulation of CD36 led to an increase in the fat uptake by GC cells, thus leading to a formation of a vicious cycle that could promote GC metastasis (10). The results of this study suggest that CD36 may serve as a potential therapeutic target for metastatic gastric cancer. Absorption of fatty acids is also aided by fatty acid-binding proteins (FABPs), a family of proteins involved in the regulation of FAs storage and distribution (15). FABP5, members of the FABPs family, was correlated with tumorigenesis. Zhao et al. found that FABP-5 was involved in gastric cancer cell cycle regulation and apoptosis, and its expression level was significantly correlated with the invasiveness of gastric cancer cells (16).
De Novo Synthesis of Lipids
Acetyl-CoA is produced by ATP citrate lyase (ACLY) catalyzing the conversion of citric acid into oxaloacetic acid in the cytoplasm, which has been found as a significant component of endogenous FAs and cholesterol biosynthesis. ACLY is often highly expressed in gastric adenocarcinoma patients (17). Acetyl-CoA synthase 2 (ACSS2) is an enzyme catalyzing the conversion of acetate to acetyl-CoA. It is overexpressed under hypoxia and lipid reduction conditions. ACSS2 has been found as the only enzyme that recycles acetate from cytoplasmic and nuclear deacetylation (e.g., deacetylation of proteins and metabolites) (18). Surprisingly, loss of ACSS2 expression was identified in a high proportion of gastric cancers (62.6%), whereas this was more commonly observed in poorly differentiated gastric adenocarcinoma or signet ring cell carcinoma (19). Cancer cells form a capable nascent FAS mechanism with the increased activity of key adipogenic enzymes (e.g., acetyl-CoA carboxylase (ACC) and fatty acid synthase (FASN)). He et al. reported a significant negative correlation between ACC expression and the immune characteristics in GC, which revealed that the inhibition of ACC could enhance anti-tumor immunity in GC (20). Besides, as revealed by the result of the experimental study conducted by Fang et al., with the advancement of disease stage and lymph node metastasis, the expression of the phosphorylated form of ACC decreased, which further supported the key role of ACC in the occurrence and development of GC (21). De novo lipogenesis can produce saturated fatty acids, which are converted to polyunsaturated fatty acids (MUFAs) via stearoyl-CoA desaturases (SCDs), the preferred substrates for triacylglycerols (TAGs) formation. Tumor cell proliferation is significantly dependent on MUFAs; in the absence of exogenous sources of MUFAs, they are completely dependent on SCD1 activity (22). By conducting gene expression manipulation and bioinformatics analysis, Wang et al. suggested that SCD1 could facilitate the migration of gastric cancer cells and anti-iron cell apoptosis and growth, illustrating the potential of SCD1 as a therapeutic target for gastric cancer (23). However, based on the needs of tumor cells, synthetic monounsaturated fatty acids are preferentially converted to triglycerides for adipose tissue energy storage, or phospholipids for membrane formation and signaling functions (24). The expression levels of the aforementioned enzymes are regulated by sterol regulatory element binding protein 1 (SREBP-1), which is a major transcription factor that controls lipid metabolism. In GC, activation of sterol regulatory element-binding protein 1 c (SREBP-1c) leads to changes in lipogenic enzymes, including SCD1 and FASN up-regulation and fatty acid elongase 6 (ELOVL6) down-regulation. The combined action of these enzymes was found to lead to a reduction in phosphatidic acid (PA), which was treated in gastric cancer cells at high concentrations (25).
Formation and lipolysis of lipid droplets
Cancer cells are characterized by the up-regulation of fatty acid synthesis (FAS) and FAs uptake, which lead to an increased LDs accumulation, and in gastric cancer, this is not an exception. As reported by Enjoji et al., fat production and lipid storage generally increased in the process of gastric carcinogenesis, and SREBP1c and DGAT2 genes were also significantly up-regulated. In contrast, genes promoting intracellular lipid consumption/removal (β-oxidation and lipolysis) were consistently down-regulated in tumor mucosa (26). Excessive fatty acids and cholesterol in cells can be converted to triglycerides (TGs) and cholesteryl esters (CEs) by diacylglycerol O-acyltransferase 1/2 (DGAT1/2) and sterol O-acyltransferase 1 (SOAT1)/Acetyl-CoA acyltransferase 1 (ACAT1) to form LDs. He et al. suggested that a high expression of DGAT1 led to lower overall survival in poorly differentiated gastric cancer patients. Inhibition of DGAT1 may serve as a promising strategy for gastric cancer therapy (27). As revealed by another experimental study, adipocytes could provide FAs to GC cells, thereby facilitating NADPH synthesis and anoikis resistance. The above adipocyte-derived FAs are transported to GC cells to form lipid droplets, followed by the induction of re-esterification of adipocyte FAs by DGAT2. During peritoneal metastasis, the up-regulation of DGAT2 increased intracellular lipid metabolism and provided NADPH for scavenging reactive oxygen species (ROS) (28).
It was found that excessive lipids in cells did not exist as non-esterified free fatty acids (FFAs) since high concentrations of FFAs are potentially cytotoxic (29). As a result, cells stored excessive fatty acids and cholesterol in neutral, inert biomolecules (e.g., sterols and TG) in cellular structures, termed lipid droplets or LDs (30, 31). LDs were deposited inside the cell by lipids surrounded by a layer of phospholipids; lastly, structural proteins termed perilipins (PLINs) were separated from the hydrophilic cytoplasm (32–34). Perilipin2 (PLIN2), also known as adipose differentiation-related protein (ADRP), is a member of the tail-interacting protein (PAT) family of PLIN, which involved in lipid droplet formation (35). Sun et al. confirmed that periilipin2 could facilitate the regulation of gastric cancer cell proliferation and apoptosis by inhibiting ferroptosis (36).
Lipid droplets are dynamic organelles that can also be hydrolyzed by intracellular lipase to release FAs when required (e.g., under nutritional stress). Adipose triglyceride lipase (ATGL) initiated degradation of intracellular TAG and hydrolyze TAG to generate diacylglycerol (DAG). Al-zoughbi et al. found that low ATGL mRNA levels were correlated with significantly reduced survival in patients with gastric cancer (37). DAG is hydrolyzed by hormone-sensitive lipase (HSL) to generate monoacylglycerol (MAG). Monoacylglycerol lipase (MAGL) was found to hydrolyze MAG, release the glycerol backbone, and release FAs (38). In gastrointestinal stromal tumors (GISTs), overexpression of monoglyceride lipase (MGLL) is correlated with adverse clinicopathological factors, suggesting a pathogenic role in the aggressive phenotype of primary localized gastrointestinal stromal tumors. The study by Li et al. confirmed the disturbance of cellular lipid metabolism in gastrointestinal stromal tumors and identified MGLL as the top candidate gene playing a crucial role in the progression of GIST (39). Besides, lipolysis stimulated lipoprotein receptor (LSR) was found as a lipoprotein receptor binding to triglyceride-rich lipoproteins with increased affinity when activated by FFAs (40). It was found that inhibition of LSR reduced lipid droplet storage, which revealed that a high expression of LSR could up-regulate lipid metabolism (41). Consistently, Sugase et al. reported that the Janus kinase/signal transduction and transcription activator (JAK/STAT) and phosphatidylinositol-3-kinase (PI3K) signaling pathways were enhanced after administration of low-density lipoprotein (VLDL) but inhibited in GC cells. LSR was inhibited by monoclonal antibody (#1-25) against human LSR (42). Lipid metabolism plays a role in GC cell proliferation via LSR. Notably, lipid droplets can serve as a tumor-promoting source of FAs. FA lipolysis mediated by MAGL has been reported to facilitate tumor migration, invasion, survival, and growth. Therefore, targeting FAs esterification and lipolysis are considered potential therapeutic strategies requiring further investigation.
Cholesterol Synthesis
In mammalian cells, cholesterol is synthesized from acetyl-CoA via the mevalonate acid pathway. First, 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) is synthesized from three acetyl-CoA molecules. HMG-CoA is reduced to mevalonate acid under the action of HMG-CoA reductase (HMGCR). As reported by Li et al., HMGCR activates Hedgehog/Gli1 gene expression that promotes Gli1, thereby facilitating the growth and migration of gastric cancer cells (43). In an enzymatic reaction, mevalonate acid is converted to Farnesyl diphosphate (FPP). Two FPP molecules condense to squalene. Subsequently, squalene is oxidized by squalene epoxidase (SQLE) to 2,3-oxidosqualene, which is cycled to lanosterol. Lanosterol is ultimately converted to cholesterol (44). Primary and metastatic gastric cancer cells exhibit different sensitivities to drugs affecting isoprenoid synthesis, metabolism, and cholesterol uptake. Isoprenoids play a vital role in the growth and survival of the above two types of cells, whereas the effects of free cholesterol and esterified cholesterol on the survival of metastatic gastric cancer cells are not as significant as those of primary gastric cancer cells. Differences in low-density lipoprotein receptor (LDLR) expression due to mevalonate acid pathway inhibition suggest different cholesterol uptake regulation between primary and metastatic cancer cells (45). Besides cholesterol biosynthesis, most cells acquire extracellular cholesterol through LDLR (46). LDLR refers to a transmembrane protein that mediates cellular cholesterol uptake. Plasma oxidized low-density lipoprotein (oxLDL) is considered a risk factor for tumor development in patients with abnormal lipid metabolism. LDL binds to LDL, transporting cholesterol to cancer cells. Ox-LDL up-regulates the expression of vascular endothelial growth factor C (VEGF-C) by binding to lectin-like oxidized low -density lipoprotein receptor-1 (LOX-1) -mediated nuclear factor κB (NF-κB) signaling pathway, it also interacts with a cluster of differentiated cells (CD36). Lastly, proliferation and distant metastasis of gastric cancer cells were induced (47).
Excessive cholesterol is exported from cells by ATP-binding box transporters, including ATP-binding box transporter A1 (ABCA1) and ATP-binding box G transporters (ABCGs), or by sterol O-acyltransferase 1/acyl-CoA: cholesterol acyltransferase (SOAT1/ACATs) transformed into more harmful cholesterol (48). As reported by Zhu et al., SOAT1 up-regulated cholesterol metabolism gene sterol regulatory element-binding protein 1 (SREBP1) in the development of gastric cancer. SREBP1 and sterol regulatory element-binding protein 2 (SREBP2) induce lymphatic angiogenesis by increasing VEGF-C expression. Overexpression of SOAT1 enhanced the proliferation, migration, and invasion of GC cells (49). The above CEs are stored in LDs or secreted into lipoproteins. Chang et al. found that lipoprotein-mediated cholesterol entry and steroid production are biological signals that promote gastric cancer progression (50).
Cholesterol concentrations are regulated by SREBP-2. The oxysterol receptor liver X receptor α and β (LXRα and LXRβ) are nuclear receptors having a significant effect on the transcriptional control of lipid metabolism. Transcriptional activity of Liver X receptors (LXRs) is induced in response to the increase in cellular cholesterol levels. LXRs are capable of binding to and regulating the expression of genes encoding cholesterol absorption, transportation, outflow, excretion, and conversion to bile acids. Furthermore, LXRs were found to regulate fatty acid metabolism by adjusting the adipose formation transcription factor sterol regulatory element-binding protein 1c and genes encoding fatty acid elongation and desaturation proteins (51). LDLR expression is regulated at the transcriptional and post-translational levels. SREBP2 serves as a major transcription factor for LDLR. In response to low cholesterol levels, SREBP2 drove the transcription of genes encoding proteins participating in cholesterol biosynthesis and the uptake of LDL cholesterol (52). LXRβ agonists inhibit GC cell proliferation by inhibiting Wnt signaling through LXRβ relocalization (53). Treatment with activated LXRβ and paclitaxel could inhibit the proliferation and induce the apoptosis of gastric cancer cells and up-regulate the expression of activated transcription factor 4 (ATF4) in a concentration-dependent manner (54).
Synthesis of Long-Chain Acyl-CoA and Fatty Acids Oxidation (FAO)
The Acyl-CoA synthetase long-chain family members (ACSL) play a part in essential lipid metabolism pathways (e.g., the peroxisome adipocytokine signaling pathway and fatty acid metabolism). It is therefore revealed that the ACSL family is a vital determinant of lipid metabolism. Acyl-CoA synthetase long-chain family member (ACSL4) is one of the isoforms of long-chain fatty acid acyl-CoA synthase. Ye et al. proved that the increase or decrease of ACSL4 expression in GC cell lines significantly affected cell proliferation and migration, which revealed that ACSL4 could play a vital role in inhibiting GC growth and metastasis (55). Furthermore, arachidonate 15-lipoxygenase (ALOX15) refers to a polyunsaturated fatty acid metabolism enzyme that, similar to other lipoxygenases, has various physiological and pathological central products. Kalamkari et al. reported that the ALOX15 gene product played a role in anti-inflammation, membrane remodeling, as well as cancer development and metastasis. Thyroid peptide receptors (FPR1, 2 and 3) are the pattern recognition receptor (PRR) family of G-protein-coupled receptors (GPCR), which are capable of recognizing exogenous and endogenous “danger” signals and triggering inflammation and immune response (56). Chronic inflammation can result from inadequate engagement of resolution mechanisms. This phenomenon is primarily due to the metabolic activity of lipoxygenase (ALOX5/15) on ω-6 or ω-3 essential polyunsaturated fatty acids (PUFAs) (57). Prevete et al. reported that genetic modulation of formylpeptide receptor-1 (FPR1) in gastric cancer cells can regulate ALOX5/15 expression and increase the potential for angiogenesis and tumorigenesis (58). Liu et al. suggested that Honokiol inhibited gastric carcinogenesis by activating 15-lipoxygenase-1 (15-LOX-1) and subsequently inhibiting peroxisome proliferator-activated receptor-g (PPAR-g) and cox-2-dependent signal transduction. 15-LOX-1-regulated PPAR-g and targeting the cyclooxygenase-2 (COX-2) signaling pathways may serve as a promising therapeutic approach (59).
Fatty acids resulting from adipose decomposition are decomposed via the mitochondrial fatty acid β -oxidation pathway to comply with the energy requirements of rapidly proliferating cells. FAO involves a series of cyclic reactions leading to the oxidation of fatty acid beta-carbon. The respective cycle of FAO was found to result in two carbon-shortened fatty acids and the production of reduced nicotinamide adenine dinucleotide (NADH), reduced flavin adenine dinucleotide 2 (FADH2) and acetyl-CoA. NADH and FADH2 produced by FAO entered the electron transport chain (ETC) to produce ATP (60). FAO plays a vital role in satisfying the energy needs of cancer cells, which can ensure their survival and proliferation in the acidic and low-oxygen tumor environment. Ezzeddini et al. found that the up-regulation of Hypoxia-inducible factor-1α(HIF-1α) and the down-regulation of lipid catabolic genes may be parallel to the down-regulation of FAO mediated by PPAR γ in patients with gastric adenocarcinoma (GA) and primary gastric cancer. This metabolic adaptation to hypoxic conditions may contribute to the pathogenesis of GA and be of clinical and therapeutic significance in patients with GA (61).
In gastric cancer, the rate of lipogenesis increases significantly, and the level of mitochondrial fatty acid β-oxidation increases, which is largely involved in lipid rafts and lipid-modified signaling molecules, thus promoting a wide variety of signal transduction in cancer cells. Fatty acids are employed to meet the needs of cell membrane synthesis, and the degradation of fatty acids is promoted by β -oxidation, leading to the accumulation of fatty acids. Fatty acids (e.g., hexadecenoic acid, docosahexaenoic acid, valeric acid, and β-hydroxybutyric acid) are significantly more prevalent in gastric cancer than in benign tissues (e.g., chronic superficial gastritis) (62). Elevated levels of octadecanoic acid have also been detected in blood samples from patients with gastric cancer. To be specific, β-hydroxybutyric acid was found as a typical product of fatty acid degradation by β-oxidation, which indicated that fatty acid decomposition could be more intense in the microenvironment (63). As revealed by Currie’s study, fatty acid degradation pathways are activated during the development of gastric cancer. Activation of fatty acid degradation was reported as a common feature of all cancers, as cell proliferation requires fatty acids to synthesize cell membranes and label molecules (64).
Carnitine acyltransferase 1 (CPT1) is a mitochondrial outer membrane enzyme catalyzing the rate-limiting step of FAO by transporting FAs across the mitochondrial membrane. Its activity is dependent on tissue-specific needs in FA metabolism and energy consumption. Tumor cells could acquire FAs through lipolysis, thus facilitating tumor proliferation, survival, drug resistance, as well as stem cell formation (65–67). The experimental study of Wang et al. revealed that overexpression of carnitine acyltransferase 1 A (CPT1A) was correlated with pathological stage, lymph node metastasis and poor prognosis in gastric cancer patients. Overexpression of CPT1A also facilitated the proliferation, invasion and EMT processes of GC cells (68). Chen et al. found that hypoxia-induced high expression of carnitine acyltransferase 1 C(CPT1C) is closely correlated with poor prognosis and can promote GC cell proliferation (69).
Furthermore, acyl-CoA thioesterase (ACOT) is involved in FAO. ACOT is capable of catalyzing the hydrolysis of acyl-CoA to the corresponding non-esterified fatty acids and coenzyme A (CoA-SH). The above enzymes play their role by maintaining cellular levels and appropriate proportions of free and activated fatty acids and CoA-SH. Wang et al. recently found that ACOT1 was abnormally overexpressed in gastric cancer tissues, which significantly correlated with a poor prognosis of gastric cancer patients (70). Li et al. reported that the overexpression of ACOT4 in cancer-associated fibroblasts (CAFs) was correlated with the survival rate of GC patients. Individualized therapy targeting ACOT4 might also be a promising modality for the treatment of gastric cancer patients (71). (The above process is shown in Figure 1).
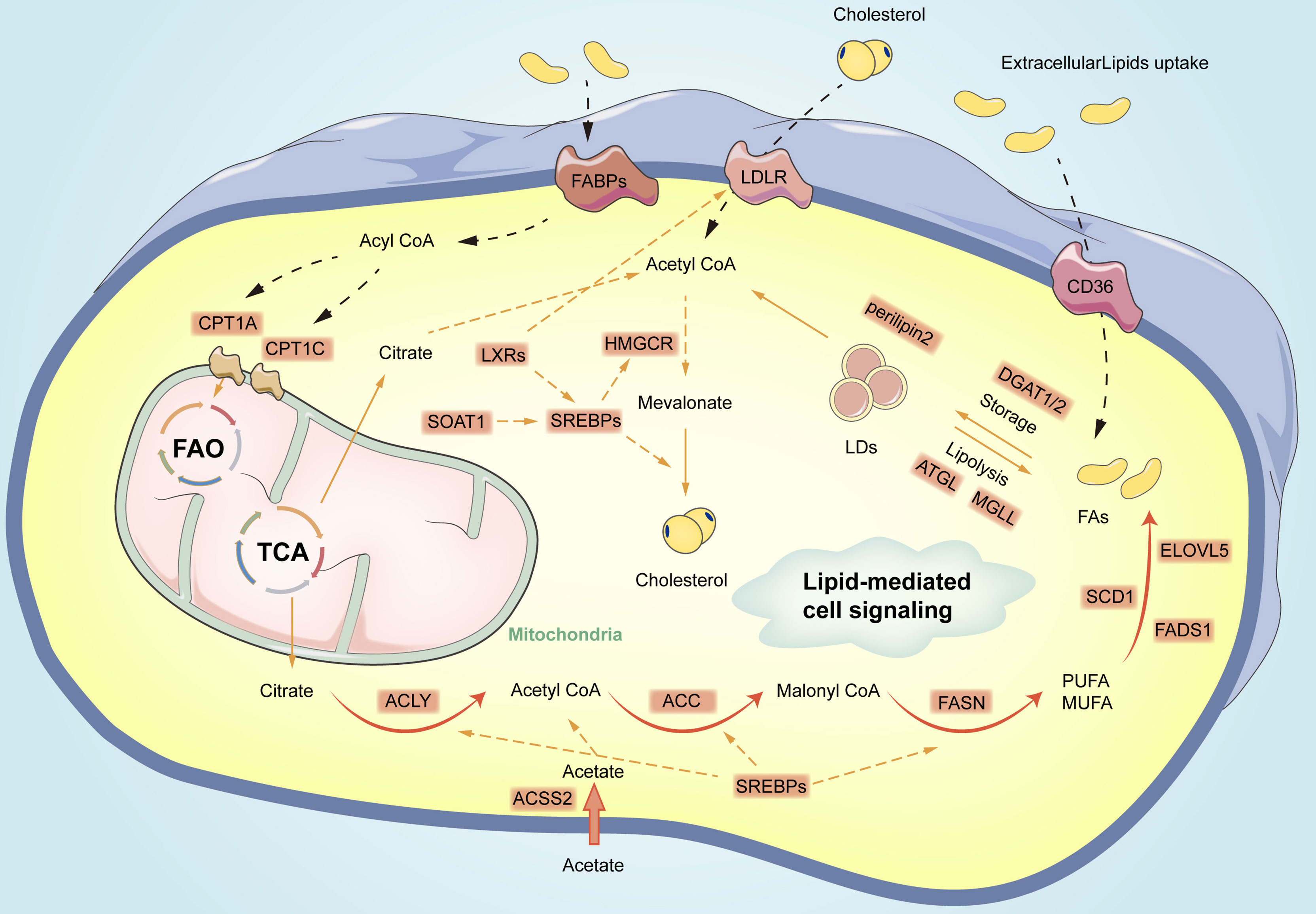
Figure 1 Overview of fatty acid metabolism reprogramming in gastric cancer cells. The process primarily includes de novo lipid synthesis, fatty acid oxidation, cholesterol synthesis, formation, as well as lipolysis of lipid droplets. Orange circles represent vital enzymes in lipid metabolism pathways. CPT1, carnitine palmitoyl transferase 1; FAs, fatty acids; FAO, fatty acid oxidation; TCA, Tricarboxylic Acid; LDs, lipid droplets; MUFA, Mono-unsaturated fatty acids; PUFA, polyunsaturated fatty acids; DGAT1/2, diacylglycerol O-acyltransferases 2; ATGL, Adipose triglyceride lipase; MGLL, monoglyceride lipase; ELOVL5, elongation of very long-chain fatty acid protein 5; FADS1, fatty acid desaturase 1; SOAT1, Sterol O-acyltransferase 1; LXRs, Liver X receptors; CD36, Cluster of differentiation 36; LDLR, low-density lipoprotein receptor; FABPs, fatty acid-binding proteins; ACSS2, acetyl-CoA synthetase 2;CPT1A, carnitine acyltransferase 1 A; CPT1C, carnitine acyltransferase 1 C; ATGL, adipose triglyceride lipase; HSL, hormone-sensitive lipase; MGLL, monoglyceride lipase.
Different Gastric Cancer Types Are Heterogenous in Terms of Lipid Metabolism
Gastric cancers are heterogeneous, and their lipid metabolism is not identical. Gastric mucosal tumors fall into two distinct main entities, the diffuse and intestinal subtypes, in accordance with Lauren’s histological criteria. Ortiz et al. suggested that LDL receptor levels were lower in primary gastric tumor tissue, but only in the diffuse type (72). As revealed by Karthik et al., the intestinal subtype of gastric cancer was primarily dependent on glucose and glutamine metabolism, and the diffuse subtype of gastric cancer relied heavily on dysregulated fatty acid metabolism. Most fatty acid metabolism genes were enriched in diffuse subtype gastric tumors. Notably, fatty acid biosynthesis, fatty acid oxidation, fatty acid transport, and fat differentiation (fatty acid storage) genes were enriched in the diffuse subtype of gastric tumors. Gastric tumors of the diffuse subtype also achieve a high expression of fatty acid metabolism genes (e.g., FASN, ACC, and ACLY). Two distinct metabolic alterations have been identified to play different roles in the intestinal and diffuse subtypes of gastric tumors. Moreover, possible metabolic differences between the intestinal and diffuse subtypes of epithelial and mesenchymal gastric cancers have been proposed (73). The regulation of cholesterol uptake also differs between primary and metastatic cancer cells. Several advanced and metastatic cancer cells may tend to synthesize rather than absorb new cholesterol to provide supply. Primary and metastatic gastric cancer cells have shown different sensitivities to drugs affecting isoprenoid synthesis and cholesterol metabolic uptake. This phenomenon suggests that isoprenoids contribute to the growth and viability of the above two types of cells. However, the effects of free cholesterol and esterified cholesterol on the survival of metastatic gastric cancer cells are not as significant as those on primary gastric cancer cells (45). Zhu et al. identified four specific gastric cancer subgroups, including cholesterol-derived, glycolytic, mixed, and resting, based on cholesterol production and glycolysis pathways. The cholesterol subtypes have been found to have the worst prognosis in gastric cancer. The abnormal expression of TP53 and MYC may facilitate the malignant progression of gastric cancer by facilitating cholesterol synthesis and affecting the use of cholesterol. From the genotyping of the metabolic genes of gastric cancer, differences may exist in patients subjected to different metabolic subtypes of gastric cancer in clinical characteristics (e.g., survival), and a clinically feasible gastric cancer grading scheme can be developed to guide the design of gastric cancer targeted therapy (74).
The Signaling Pathways Correlated With Lipid Metabolism in Gastric Cancer
PI3K/Akt/mTOR Signal Pathway
It has been widely recognized that the accumulation of genetic alterations leads to the development of gastric cancer (75). Most gene mutations in gastric cancer were correlated with changes in biological signals (e.g., the mammalian target of PI3K/Akt/mTOR pathway) (76). The PI3K signaling pathway was reported as a vital regulator of numerous critical cellular processes (e.g., cell growth, metabolism, survival, metastasis, and chemotherapy resistance) (77). SREBP1 preferentially regulates the genes related to fatty acid biosynthesis, while SREBP2 mainly regulates cholesterol pathway genes. Both fatty acid synthesis and uptake are stimulated by mTOR signals. Thus, both mTORC1 and mTORC2 stimulate the expression and proteolysis of SREBP1, a major transcription factor that induces de novo lipid synthesis (78). Furthermore, mTORC1 was found to promote SREBP1 activity by preventing the nuclear entry of LIPIN1, the negative regulator of SREBP1, while mTORC2 promoted the expression of AKT, SREBP1’s downstream effector (79, 80), while preventing SREBP1 degradation in cancer cells (81). In gastric cancer, the activation of SREBP-1c could lead to changes in lipogenic enzymes (e.g., up-regulation of SCD1 and FASN) (25).
Hippo Signal Pathway
YAP and TAZ, two key mediators of the Hippo pathway, play critical roles in carcinogenic signal transduction and cell attachment and their expression levels were significantly correlated with lipid metabolism. Abnormal regulation of the Hippo pathway was found to facilitate the proliferation and metastasis of gastric cancer, and the inhibition of the Hippo pathway effector proteins YAP and TAZ have shown important application prospects in treating gastric cancer (38). YAP/TAZ has been reported to interact with mature SREBPs in the nucleus, resulting in enhanced transcriptional activity and up-regulated expression of downstream targets (e.g., HMGCR and fatty acid synthase (FAS)) (82). In brief, the activity of mevalonate pathway-associated oncogenic proteins (YAP/TAZ) was activated by SREBP2 (83), which in turn stabilize lipid-producing enzymes and localize to the nucleus (84). Besides the typical hippo signaling pathway, the atypical Hippo signaling pathway also contributes to the reprogramming of lipid metabolism. LATS2 could inhibit the maturation of SREBPs by interacting with SREBPs precursors located in the endoplasmic reticulum to regulate lipid metabolism in a YAP/TAZ-independent manner (85).
Wnt/β-Catenin Signal Pathway
The Wnt/β-catenin pathway has been found to play a vital role in the pathogenesis of gastric cancer, and its complex functions are significantly dependent on the transcriptional output of β-catenin (86). Activation of β-catenin correlates with the metastasis of gastric cancer. A positive feedback loop between YAP and β-catenin has been reported, and YAP/TAZ has been found to inhibit the Wnt/β-catenin signaling pathway in vivo and in vitro (87, 88). In addition, SCD1 can facilitate the synthesis of unsaturated fatty acids. Unsaturated fatty acids activate Wnt ligands. The activated Wnt ligand could bind to FZD4 receptor, destroy the complex, stabilize the activity of β-catenin and YAP/TAZ proteins, promote the accumulation of β-catenin and YAP/TAZ proteins in the nucleus, and play a role of transcriptional regulation (89). The canonical Wnt/β-catenin pathway was found to regulate de novo lipogenesis (DNL) and fatty acid monounsaturation in adipocytes. Moreover, β-catenin was found to mediate the effects arising from Wnt signaling on lipid metabolism in part by the transcriptional regulation of SREBF1 (90). Simvastatin is capable of inhibiting HMGCR and down-regulating GER anylgerany L pyrophosphate (GGPP), which is a significant substrate for the post-translational modification of RhoA. Subsequently, the activity of RhoA is reduced, and reaches the inhibition level of YAP and β-catenin activity. The reduced activity level of YAP and β-catenin was found to eventually promote the apoptosis of gastric cancer cells and inhibit the proliferation and metastasis of cancer cells (91).
Hedgehog Signal Pathway
Sterols play a vital role in Hh protein production, secretion, transport, and Hh signal transduction. Mature Hh proteins require a double lipid modification, cholesterol necessary for Hh gradient, as well as the palmitate portion required for interactions with PTCH. When PTCH bound to Hh, the inhibition of SMO was released and activated by sterols interacting with CRD (92). The role played by cholesterol in the HH pathway has long been found since active HH proteins are covalently modified with cholesterol, which makes this post-translational modification unique among all known proteins (93). Hedgehog/Gli1 signaling pathway has been found to be frequently activated in gastric cancer. Binding hedgehog ligands to the receptor patch could activate smoothing and initiate a signaling cascade promoting the nuclear localization of Gli1 and activating the expression of downstream genomes (43). The Sonic hedgehog (SHH) pathway interacts with the genes of gastric cancer cells. Statins target HMGCR, hydroxysteroids, a rate-limiting enzyme synthesizing cholesterol, and 7-hydroxycholesterol (a precursor of vitamin D). Some metabolites of the mevalonate acid pathway have been found as active regulators of the Hh pathway, while vitamin D3 is inhibitory. Accordingly, statins were found to significantly regulate HH signaling (93). 93).(The above process is shown in Figure 2).
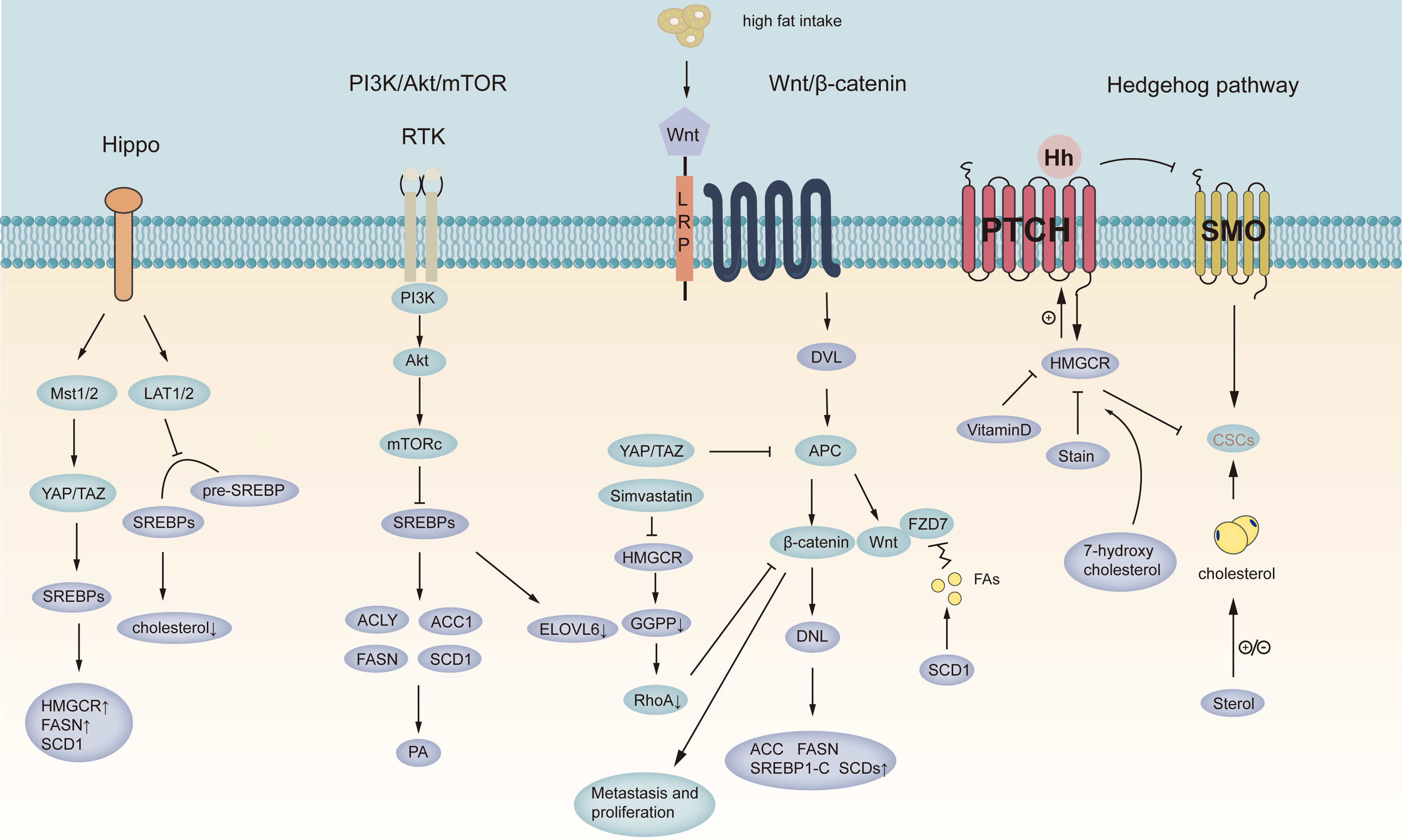
Figure 2 Targeting signaling pathways in gastric cancer. Schematic representation of the Wnt, hedgehog, PI-3K, and Hippo pathways in GC. The purple circles represent lipid metabolism-related pathways in gastric cancer, and the blue circles represent signaling pathways in gastric cancer. YAT/PAZ, Yes-associated protein/transcriptional coactivator with PDZ-binding motif; LAT1/2 represents Large tumor suppressor 2; MST1/2, Macrophage stimulating 1/2; SREBPs, sterol regulatory element-binding protein; Pre-SREBPs, Pre-sterol regulatory element-binding protein; HMGCR, 3-hydroxy-3-methyl-glutaryl-CoA reductase; FASN, fatty acid synthase; SCD1, stearoyl-CoA desaturase 1; ACLY, ATP-citrate lyase; ACC1, acetyl-CoA carboxylase; ELOVL6, fatty acid elongate 6; PA, phosphatidic acid; PI-3K/AKT/MTORC1, phosphoinositide 3-kinase (PI3K)-protein kinase B (AKT)-mechanistic target of rapamycin (mTOR) signaling; DVL, Doppler Velocity Log; APC, Adenomatous polyposis coli; GGPP, geranylgeranyl pyrophosphate; CSCs, cancer stem cell; FZD7, Wnt receptor frizzled7.
The Function of Lipid Metabolism Related Enzymes
Disease stage and lymph node metastasis status remain the most critical determinants for designing treatment strategies and predicting prognosis in gastric cancer patients (94–97). In gastric cancer, the enzymes catalyzing the de novo synthesis of FAs (e.g., ACLY, ACC, FASN, and SCDs) were significantly up-regulated. The roles of the above proteins are described, and their potential as biomarkers in the diagnosis and treatment of gastric cancer is discussed.
Lipid Fatty and Cholesterol Transporters
CD36
Lipid uptake from the exogenous environment has been found as another vital method for cells to acquire fatty acids besides de novo synthesis. CD36 was found to transport fatty acids into cells and play a vital role in cancer cell growth, metastasis, as well as epithelial-mesenchymal transition (98–100). The stomachs of CD36 mice were characterized by altered gland organization and secretion, more fibronectin, and inflammation. Other symptoms could be detected (e.g., reduced tissue respiration and mitochondrial efficiency, phospholipids increasing, as well as triglycerides decreasing). CD36 and its absence have been found to act in chronic diseases predisposing to malignancy (101).
FABPs
FABPs are a superfamily of lipid-binding proteins that play essential roles in fatty acid uptake, transport, as well as oxidation. They are capable of adjusting the concentration of intracellular free fatty acids by regulating the uptake and intracellular transport, maintaining a stable internal environment of lipid metabolism, and participating in the development and progression of various malignant tumors by affecting the proliferation, transformation, and migration of tumors (102–105). A high expression of FABP3 was found in the gastrointestinal stroma, which revealed that FABP3 might play a growth-promoting role in the gastrointestinal stroma. A possible mechanism for FABP3 to facilitate growth is that the proliferation of such tumors may require fatty acids formed by lipid metabolism. High expression of FABP3 was significantly correlated with poor 5-year overall survival, which indicates its prognostic value in patients with gastrointestinal stromal tumors (GISTs) (106).
The main function of FABP4 is lipid metabolism control, it plays a role in inflammatory responses, cell growth and differentiation, as well as apoptosis regulation. The expression of FABP4 is regulated by fatty acids, PPARγ, and PPARγ agonists. Lipids have essential regulatory and energy supply functions in vivo. Patients with elevated FABP4 expression had a poorer prognosis, and the 5-year survival rate was significantly reduced. Thus, FABP4 expression was confirmed as a novel and valuable marker for determining the prognosis of GISTs, as well as an independent indicator of GIST risk (106).
LDLR
Cholesterol was reported as a vital structural component of cell membranes (107). Cholesterol can be de novo synthesized by cells or by internalizing low-density lipoprotein (LDL). LDL was found to bind to the LDLR and was internalized before entering lysosomes, where free cholesterol was released (107). LDLR-related protein 1B (LRP1B) also belongs to the LDLR family (108). LRP1B binds extracellular ligands, apoE-loaded lipoproteins, and urokinase plasminogen activator (uPAR); on that basis, it mediates the internalization of the above ligands. Impaired endocytosis of the cytoplasmic domain of LRP1B results in insufficient ligand internalization, which is correlated with poor prognosis in patients with diffuse-type gastric cancer. Cytoplasmic LRP1B immune response significantly correlated with low clinicopathological stage and good prognosis in patients with diffuse-type gastric cancer but not intestinal-type gastric cancer. Cytoplasmic LRP1B immune response is of independent prognostic value in diffuse gastric cancer, whereas it was not found to be significantly correlated with patient prognosis in intestinal-type gastric cancer (109). Chang et al. investigated cholesterol importing via lipoprotein/receptors (L/R route), steroidogenic enzymes, and steroid receptors in patients’ prognosis with gastric cancer. The results revealed that lipoprotein-mediated cholesterol entry and steroidogenesis are biosignatures of GC progression (50).
De Novo Lipid Synthesis
ACSS2
The conversion of acetate to ACSSs makes acetate a vital molecule for lipid synthesis and histone acetylation. In mammalian cells, ACSS isoforms 1 and 3 are localized in mitochondria, while isoform 2 is present in the cytoplasm and nucleus (110). SREBPs regulate the expression of isoform 2. Mammalian ACSS2 was first cloned as a target of the SREBP transcription factor regulating lipid homeostasis (111). By exposing mice to a high-fat diet (HFD) or prolonged fasting, it was found that this simple metabolic enzyme could promote the proper storage or utilization of fat depending on the fed or fasted state. Thus, ACSS2 was found to play an unexpected role in controlling systemic lipid metabolism (112). Only ACSS2 down-regulation significantly inhibited acetate-mediated lipid synthesis and histone modification. In clinical practice, the deficiency of ACSS2 expression could serve as an independent prognostic factor predicting poorer prognosis in patients with gastric cancer, which was found to be especially correlated with Microsatellite Instability (MSI) (19).
ACLY
ACLY is a recognized vital enzyme in the de novo synthesis of fatty acids, and ACLY catalyzes the conversion of citrate to acetyl-CoA. It is a vital source of energy for the growth and metabolism of cancer cells. The ACLY-mediated conversion of citrate to acetyl-CoA is the basis for fatty acid synthesis (113). The protein released by HP results in the up-regulation of ACLY gene expression, promotes the synthesis of acetyl-CoA, and enhances the interaction between H. pylori (HP) and gastric epithelial cells, eventually leading to gastritis and epithelial damage (114). The study by Cheng et al. found that the overexpression of miR-133b could reduce the proliferation and invasion ability of gastric cancer cells by inhibiting the expression and activation of ACLY (114). The study by Qian et al. found that high expression of ACLY was correlated with overall survival, stage, and lymph node metastasis. ACLY can serve as a biomarker for predicting disease progression and prognosis in patients with gastric adenocarcinoma (17).
ACC
ACC are enzymes that catalyze the carboxylation of acetyl-CoA to form malonyl-CoA. In mammals, ACC1 and ACC2 are the two members of ACCs. ACC1 is localized in the cytoplasm and is the first rate-limiting enzyme in the de novo fatty acid synthesis pathway. ACC2 localizes to the mitochondrial outer membrane, produces malonyl-CoA, and regulates the activity of CPT1 involved in fatty acid β-oxidation (115). Increased activation of ACC is ordinary in human gastric cancer. pACC down-regulation is a significant prognostic factor, and pACC deficiency may play a vital role in developing GC. High pACC expression strongly correlated with better survival in all gastric cancer patients. Low/absent expression of pACC was significantly correlated with (1) advanced tumor stage and lymph node metastasis (2); poor GC cells differentiation status (3); shorter survival than those with high pACC expression, especially at an early stage. In addition, metformin inactivated ACC, which was characterized by ACC phosphorylation and significantly inhibited cell proliferation and growth (21). ACC expression in GC was significantly negatively correlated with immune signals. Specifically, the expression of ACC was significantly negatively correlated with the infiltration level of CD8+ T cells and immune cytolytic activity in GC, which suggested that inhibiting ACC could enhance antitumor immunity in gastric cancer. ACC might be a valuable biomarker to differentiate the response to immunotherapy in gastric cancer patients or a promising intervention target to enhance anti-tumor immunity and immunotherapy response in gastric cancer (20).
FASN
FASN refers to the major enzyme involved in the anabolic conversion of dietary carbohydrates to fatty acids. By using acetyl-CoA as a primer, malonyl-CoA as a two-carbon donor, and NADPH as an intermediate reducing agent, FASN catalyzes the synthesis of long-chain lipids fatty acids, which largely covers synthetic palmitate (80%), myristate (10%), as well as stearate (10%). FAS is highly expressed in gastric cancer, adenoma, regenerating epithelium, as well as intestinal metaplasia. FAS expression occurs at an early stage of tumorigenesis, it plays a vital role in forming precancerous lesions in gastric cancer (116). FASN is overexpressed in GC tissues, correlated with worse survival outcomes, and largely contributes to GC carcinogenesis. FASN expression was closely correlated with the immune infiltration levels of CD8+ T, CD4+ T, neutrophils, macrophages, and dendritic cells. FASN is an oncogene in GC and can serve as a potential biomarker. Existing studies have found that FASN is significantly overexpressed in GC tissues, and its high expression leads to poor survival outcomes in GC patients. Furthermore, FASN expression is significantly correlated with immune infiltration and may play a vital role in GC-related immunology (117). GC cells are highly resistant to Anoikis, and Anoikis resistance promotes the proliferation, migration, and invasion of GC cells while inhibiting GC cell apoptosis. Down-regulation of FASN has an inhibitory effect on Anoikis resistance (AR) in GC cells and is correlated with the inhibition of the p-ERK1/2/Bcl-xL signaling pathway. In brief, as revealed by the results and data of this study, FASN may be a novel target for preventing tumor metastasis, which provides a new reference for anticancer therapy (118).
SCD1
SCD1 refers to a novel endoplasmic reticulum-related enzyme synthesizing fatty acid synthase (FAS), which catalyzes the desaturation of saturated fatty acids (SFAs) to Δ9monounsaturated counterparts (MUFAs) (e.g., Stearic acid (18:0) and palmitic acid (16:0) yield oleic acid (18:1) and palmitoleic acid (16:1) (119)). Both SFAs and MUFAs are significant components of human cellular lipids, essential components of biological membranes, and sources of energy and signaling molecules (e.g., cholesteryl esters) (120). SCD1 enhances GC stemness via the Hippo/YAP pathway. SCD1 inhibition reduced the migratory/invasive ability and wound healing ability of GC cells. SCD1 overexpression is correlated with mesenchymal markers in GC tissues. After SCD1 down-regulation, GC cells show a mesenchymal-to-epithelial transition phenotype. YAP and TEAD1, the levels of essential proteins of the Hippo signaling pathway are correlated with SCD1 expression, and SCD1 inhibition leads to YAP inhibition (121). In addition, SCD1 down-regulation reduced p-YAP expression, leading to YAP disassembly in the GC nucleus, which also attenuated the activation of the Hippo pathway. Furthermore, SCD1 plays a crucial role in regulating lipid metabolism and ferroptosis in gastric cancer stem cells (GCSCs) (122). SCD1 is capable of accelerating the migration of gastric cancer cells, inhibiting cell death and growth, as well as predicting a poorer prognosis for gastric cancer patients. Thus, the potential of SCD1 as a biomarker for early diagnosis and a therapeutic target for gastric cancer has been demonstrated (23).
Elongation of Very Long-Chain Fatty Acid Protein 5 (ELOVL5) and Fatty Acid Desaturase 1 (FADS1)
The elongation and desaturation reactions of PUFAs in cells are dependent on the expression of elongates and desaturases. Intracellular PUFAs can be elongated by the extension of the elongates of very long-chain fatty acids (ELOVLs). Desaturation reactions are catalyzed by FADS1 and FADS2 gene products (123). Lee et al. found that ELOVL5 and FADS1 were up-regulated in mesenchymal gastric cancer cells, leading to ferroptosis sensitization. In contrast, DNA methylation silences the above enzymes in intestinal-type GCs, rendering the cells resistant to ferroptosis (124).
SREBPs
SREBPs refer to a family of basic-helix-loop-helix leucine zipper transcription factors that regulate de novo synthesis of fatty acids and cholesterol and cholesterol uptake. Mammalian cells express three SREBP proteins, SREBP-1a, -1c, and -2, which are encoded by two genes, sterol regulatory element-binding transcription factor 1(SREBF1), as well as sterol regulatory element-binding transcription factor 2(SREBF2). SREBF1 encodes SREBP-1a and -1c proteins through selective transcriptional start sites. SREBP-1a protein nh2 terminal is approximately 24 amino acids longer than -1c and is highly transcriptional active. SREBP-1a regulates the synthesis of fatty acid and cholesterol as well as cholesterol uptake, while SREBP-1c mainly controls fatty acid synthesis. SREBF2 encodes the SREBP-2 protein and plays a vital role in regulating cholesterol synthesis and uptake (125). The study by Zhao et al. confirmed that SREBP-1a could bind to the GPX4 promoter region to promote the transcription of glutathione peroxidase 4(GPX4) in gastric cancer cells. Apatinib inhibited the transcription of GPX4 by regulating the srebp1a-mediated pathway and culminating in lipid peroxidation-induced ferroptosis in gastric cancer (126). Sun et al. found that SREBP-1c was activated in human gastric cancer tissues. Furthermore, the srebp-1c-dependent adipogenic pathway can be stimulated by human gastric cancer, promoting the expression of a series of fatty acid synthesis-related genes, such as SCD1 and FASN. Knockdown of the SREBP1c gene can significantly inhibit the proliferation, invasion, and migration of gastric cancer cells, thus providing novel evidence supporting SREBP-1c as a potential therapeutic biomarker target for gastric cancer. In addition, low expression of the SREBP-1c gene was correlated with better 5-year survival rates in gastric cancer patients, and most patients with stage III gastric cancer achieved higher 5-year survival rates (25).
Lipid Storage/Lipid Droplets
DGAT1
Cellular lipids in excess were converted to triglycerides and cholesteryl esters in the ER, leading to the formation of lipid droplets observed in gastric cancer (32). Diglyceride acyltransferase 1/2 (DGAT1/2) can synthesize triglycerides from diglycerides and acyl-CoA (127). This enzyme is highly expressed in gastric glands, and its expression level is negatively correlated with prognosis and patient survival (27).
Sterol o-Acyltransferase 1 (SOAT1)/Acyl-CoA Acyltransferase 1 (ACAT1)
SOAT1, i.e., ACAT1, can convert cholesterol into cholesterol esters and store them in lipid droplets. SOAT1 regulates the expression of cholesterol metabolism genes SREBP1 and SREBP2, which induce lymph angiogenesis by increasing the expression of vascular endothelial growth factor-C (VEGF-C). SOAT1 is highly expressed in cancerous tissues and correlated with advanced tumor stage and lymph node metastasis, leading to a poor prognosis of GC (49).
Cholesterol Synthesis
HMGCR
Cholesterol synthesized from mevalonate plays a crucial role in forming and maintaining cell membrane structure and function. It is also a precursor to steroid hormones, vitamin D, and bile acids (128). The mevalonate pathway itself is regulated by transcriptional and translational machinery (129). The rate-limiting step in cholesterol production is mediated by HMGCR, making it the most controlled part of the pathway (130). Li et al. found that HMGCR positively regulates the oncogenic role of the Hedgehog/Gli1 signaling pathway in gastric cancer. Overexpression of HMGCR promoted the growth and migration of gastric cancer cells, whereas the down-regulation of HMGCR expression had an inhibitory effect. In addition, HMGCR also activated the transcriptional activity of Gli1. The above results suggest that HMGCR may play a vital role in the development of gastric cancer, and targeting it in therapy will have excellent effects (43).
Fatty Acid Oxidation
CPT1A and CPT1C
Carnitine palmitoyltransferase (CPT), including CPT1 and CPT2, plays a critical role in FAO. CPT1, located in the outer layer of the mitochondrial membrane, is considered a vital enzyme in FAO that converts carnitine to fatty acylcarnitine (131, 132). CPT1 consists of three isozymes (e.g., CPT1a, CPT1b, and CPT1c). CPT1c is inactive, while CPT2 is located in the mitochondrial membrane, where it promotes the β-oxidation of fatty acids (FAs) by facilitating the conversion of acetyl coenzyme A (CoA) to fatty acid coenzyme A. CPT takes on a critical significance in the oxidation of long-chain FAs (133). CPT1A protein expression is correlated with the grade, pathological stage, lymph node metastasis, as well as poor prognosis in patients with GC (68). High expression of CPT1C induced by hypoxia is closely correlated with poor prognosis (69).
Lipolysis
ATGL and MGLL
ATGL initiates the lipolytic catabolism of TGs by converting TGs to DGs and FAs. The subsequent steps of lipolysis are catalyzed by hormone-sensitive lipase (HSL), which hydrolyzes DGs into MG and FAs. Lastly, mg is hydrolyzed to FAs and glycerol by monoacylglycerol lipase (MGL) (134). The study by Al-Zoughbi et al. found that low levels of ATGL mRNA were correlated with significantly reduced survival in gastric cancer patients. MGLL mRNA levels were significantly increased from adjacent normal tissues to non-high-risk groups, and gastrointestinal stromal tumors were significantly elevated and correlated with immune expression levels from non-high-risk to high-risk groups (37). MGLL overexpression strongly predicted poorer disease-free survival and overall survival. In brief, MGLL is a lipid-metabolizing enzyme correlated with the progression of gastrointestinal stromal tumors with adverse clinicopathologic factors and independent adverse prognostic effects (39).
The Limitations of the Quality of Lipid Biomarkers
Clinically applicable biomarkers for gastric cancer are required for early detection of the disease, as well as for accurate diagnosis, prognostic stratification, and post-treatment monitoring of gastric cancer. We hope that vital enzymes of lipid metabolism can serve as biomarkers to assist clinicians when predicting the survival and prognosis of GC patients and facilitate personalized treatment. However, current evidence supporting the association of lipid biomarkers is experimental and usually retrospective, leading to significant limitations on their application. It is generally considered that an effective biomarker of malignancy should exhibit specific characteristics. To be specific, it should be detectable at high levels in cancer-affected patients and undetectable or present at low levels otherwise. Besides, it should be easily quantifiable in clinical samples and demonstrate functional correlation with disease progression to provide prognostic or diagnostic information (135, 136). However, little is known about the lipid metabolism-related pathways and the dynamic regulation of critical enzymes in the regulation of gastric carcinogenesis. As a result, the development of lipid biomarkers remains at an early stage, safety, efficacy, and adverse reactions have not been explored. The above will hinder the generalization of the biomarker-related results in clinical trials to the entire gastric cancer patient population. Furthermore, existing well-established biomarkers (e.g., Human epidermal growth factor receptor 2 (HER2) and programmed cell death-ligand 1(PD-L1)) are capable of detecting tumors early and guide specific molecule-based targeted therapy to adjust the characteristics of gastric cancer cells. The combination of lipids and conventional biomarkers will produce more accurate prediction.
Targeting Lipid Metabolism for GC Therapy
Chemical inhibitors targeting critical enzymes of lipid metabolism in GC are currently under preclinical and clinical investigations. Representative inhibitors are listed in Table 1 and Figure 3. A considerable number of natural and synthetic inhibitors of ACLY have been tested for anticancer effects in preclinical and clinical studies (146). Other drugs (e.g., proton pump inhibitors) have been employed to suppress the FASN gene in cancer, and clinical trials are being performed on one of the above drugs, omeprazole, for several cancer types. As revealed by the study of Chen et al., treating gastric cancer epithelial cells with omeprazole significantly inhibited the expression of FASN and ACLY, while inhibiting the synthesis of new lipids, which would result in lower lipid content (147). Orlistat was found as an over-the-counter anti-obesity drug reducing dietary fat absorption by covalently modifying an enzyme that inhibits gastric and pancreatic lipase (148). It was also reported as an excellent inhibitor of FAS (149). Inhibition of FAS by orlistat exposure resulted in rapid cellular damage followed by a direct but transient autophagic response. Long-term survival studies in rats and other mouse strains should be conducted to further assess orlistat’s potential to inhibit GI cancers (142). Significant efficacy of C75/imatinib combination therapy was observed on imatinib-resistant GIST cells and xenotransplantation. Exposure to C75 alone or in combination with C75/imatinib combination attenuated AKT activity (141). SCD1 and its catalytically active products are key drivers of tumor transformation and cancer progression. Accordingly, it is reasonable to consider the use of SCD1 inhibitors as possible anticancer drugs. The inhibition of SCD by a small molecule inhibitor (A939572) can impair the proliferation of gastric cancer cells (150, 151). Besides targeting de novo synthesis, blocking FA uptake may be another effective strategy for cancer therapy. CD36 expression is up-regulated in gastric cancer (10). Combination therapy targeting lipogenesis and lipid absorption would be a promising approach. JC61.3, an antibody targeting CD36, inhibits the uptake of FA and LDL proteins only and is potent and specific for lymph node metastases (99). JC61.3 has been tested in preclinical models of gastric cancer and found to inhibit in vitro cell migration and in vivo metastasis of gastric cancer (14). Inhibiting the ability of cancer cells to utilize FA as an energy source represents another potential therapeutic opportunity. The inhibition of lipid uptake within mitochondria to limit energy production has been an attractive therapeutic target for years. In preclinical studies, etomoxir, an irreversible inhibitor of CPT1 acting on AGS/BGC823 cells that overexpress CPT1A, has been one of the most used inhibitors of mitochondrial FAO. Inhibition of FAO with etomoxir was found to inhibit the growth, migration, and EMT process of CPT1A-overexpressing GC cells (68). Besides, the combination of oxaliplatin and perhexiline significantly inhibited the progression of gastrointestinal tumors in a patient-derived xenograft model (PDX) (137). Lipid storage has been found as a novel and vital mechanism affecting the survival and metastasis of cancer cells. Over the past few years, targeting the TAG-producing DGAT enzyme and storing it in LD has become an exciting target for obesity and type 2 diabetes research. The inhibitory effect of PF06424439 on DGAT2 has been tested in preclinical models, and PF06424439 reduced LD formation and metastasis of gastric cancer in vitro and vivo (28). Several inhibitors of the mevalonate acid pathway have been tested in gastric cancer, and statin cholesterol-lowering drugs have been tested in clinical trials. Simvastatin combined with Capecitable/cisplatin (CDDP) can limit the survival and growth of primary gastric cancer cells (NCT01099085) (152). Lovastatin plus docetaxel has been recognized as a promising anticancer strategy for sensitive or resistant tumors (153).Other drugs interfering with the mevalonate acid pathway (e.g., zoledronic bisphosphonate acids and aureoyl and geranyl transferase inhibitors affecting protein prenylation) have also been employed and tested in clinical trials. Avasimibe is a prominent anticancer drug that significantly reduces cholesteryl ester storage by inhibiting vesicule transport, integrin, and transforming growth factor beta (154). We found for the first time that avasimibe significantly inhibited the proliferation, migration, and invasion of GC cells in a dose-dependent manner. The vital enzymes of different steps of mevalonate acid have different therapeutic effects on primary and metastatic gastric cancer (distant liver metastasis). Statins and avasimibe have been considered as promising new antitumor drugs in primary gastric cancer but not in gastric cancer cells, which are highly resistant. Moreover, terbinafine (a clinically used enzyme inhibitor), which is much less enzymatic in nature than HMGCR, has been proven to be a very promising chemo-preventive natural compound, and it does inhibit monooxygenase, lower serum cholesterol. Accordingly, it can be a fascinating target for cancer prevention (45). Furthermore, exemestane (a type II aromatase inhibitor) also significantly inhibited the growth of gastric cancer cells at a pharmacologically tolerable dose in vitro (50).
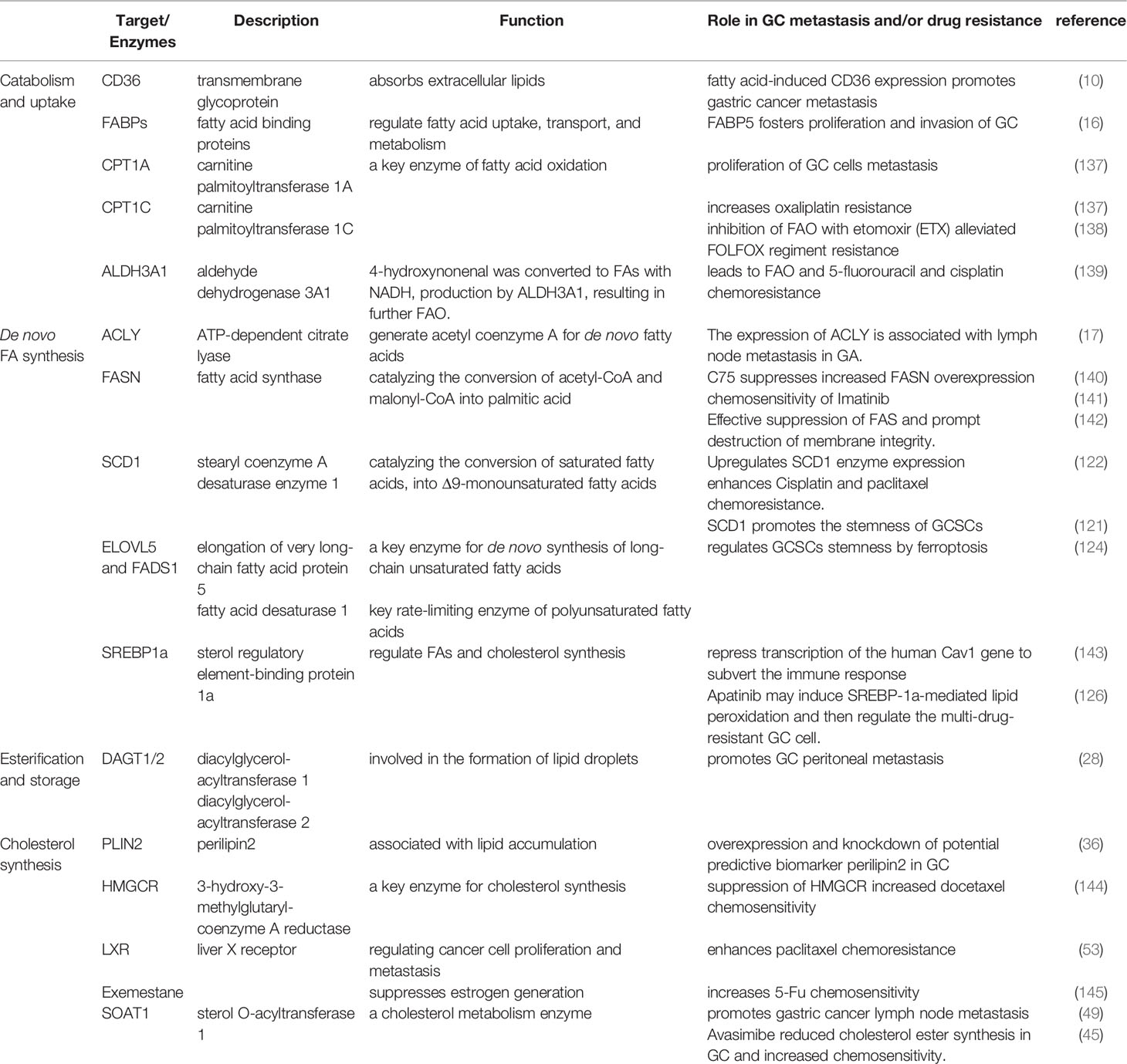
Table 1 Summary of fatty acids related targets/enzymes and their links to GC metastasis and drug resistance.
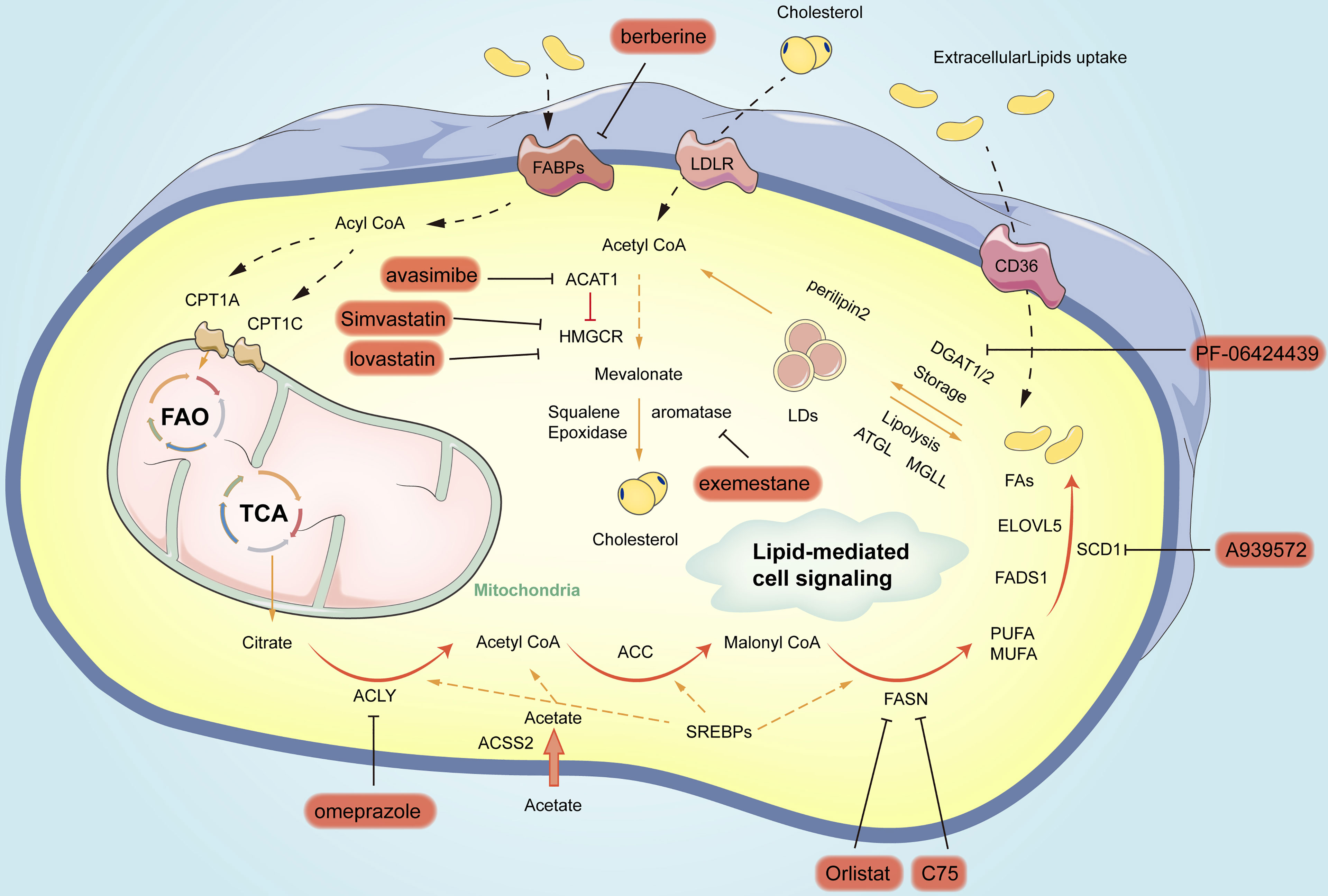
Figure 3 Summary of small-molecule inhibitors of lipogenic enzymes in gastric cancer. In orange circles are the names of inhibitors for different vital enzymes for blocking the related pathways.
Further Studies Are Required to Investigate the Role of Lipid Metabolism in the Drug Treatment of Gastric Cancer
Targeting lipid metabolism is a promising therapeutic approach, but its application in the treatment of gastric cancer patients still requires extensive research and exploration.
First, the specific metabolic regulation in gastric cancer remains unclear. Accordingly, further studies on the specific mechanisms involved in the reprogramming of lipid metabolism in gastric cancer cells and their dual roles in multiple tumor-related signaling pathways are required to allow more effective identification of therapeutic targets. Further clarification of the roles of different lipid signaling molecules will provide new therapeutic opportunities for the development of anticancer drugs.
Second, numerous lipids and lipid analogs serve as critical regulators of gastric carcinogenesis. Currently, most of the information originates from studies conducted in vitro tumor cells or experimental animals, while there have been rare clinical studies in patients. As revealed by the results of preclinical studies, a combination therapy consisting of cytostatic, targeted therapy drugs, and lipid metabolism inhibitors would be more effective in cancer treatment (155). Whether we can use a combination of drugs to counteract the adaptation of gastric cancer cells to metabolic reprogramming or improve the effectiveness of current immunotherapy remains unclear. The synergy, safety, efficacy, side effects, and other aspects of lipid metabolism therapy and traditional anticancer drugs should be further assessed in clinical trials.
Again, relatively few molecules make it through the drug development process due to the chemical instability of enzymes, rapid metabolism in the body, and side effects in some cases. Currently, the above approaches are in their infancy, and there are few examples of the application of the above strategies. Further experimental studies are currently required to optimize the above potential lead molecules in cells and experimental animals for clinical application (155).There are no standard drugs as inhibitors of certain vital enzymes of lipid metabolism. Despite the powerful effects of natural compounds, their side effects are still concerning. To maintain the efficacy of certain biologically active alkaloids, high-frequency dosing is required, resulting in low patient compliance and even cumulative toxicity in patients. Thus, additional clinical trials and efficacy evaluations are needed to identify strategies to mitigate adverse effects when using the above natural compounds as treatments (155).
Lastly, the lipid profiles of different gastric cancer subtypes vary significantly with study designs, study subjects, methods, and analyses. Therefore, clinical studies are needed to elucidate the characteristics and patterns of lipid profiles in patients with different gastric cancer subtypes, the underlying mechanisms of lipid-related enzymes and proteins, as well as the standardized methods of targeting lipid metabolism (156).
The Body Lipid Metabolism Influences the Inter-Cellular Lipid Metabolism in Gastric Cancer
In gastric cancer, the body’s lipid metabolism is capable of regulating the internal processes of cells and playing a certain role in cell-to-cell lipid metabolism. Interactions between cancer cells and neighboring immune cells support tumor growth and development by altering lipid metabolism (157). Yuan et al. confirmed that in the gastric cancer microenvironment, tumors could up-regulate the expression of Forkhead transcription factor 3 (Foxp3) in tumor-infiltrating Treg cells and inhibit the proliferation of autologous CD4(+) CD25(−) T cells by producing COX-2/PGE (2) (158). Lin et al. reported that gastric adenocarcinoma cells outperformed tissue-resident memory T (Trm) cells in lipid uptake and induced Trm cell death in a gastric adenocarcinoma cell-T cell co-culture system. When the expression of FABP4 and FABP5 in gastric adenocarcinoma tumor cells was down-regulated, the expression of Fabp4/5 in Trm cells was inhibited, and the uptake of lipids by Trm cells increased, thus increasing the survival rate of Trm cells in vitro and vivo (159). When macrophage receptor 1(Msr1) was up-regulated in tumor exposed dendritic cells (DCs), they continued to take up surrounding cholesterol and FAs, which accumulated in the cytoplasm. The results suggest that DCs have a reduced ability to cross-express and present Ag, ultimately leading to insufficient antigen-specific activation of CD8+ T cells in tumor cells (160, 161).The study by Luo et al. confirmed that lipid accumulation in tumor-associated macrophages (TAMs) could primarily arise from the increased uptake of extracellular lipids by gastric cancer cells, thus leading to the up-regulated expression of the gamma isoform of phosphoinositide 3-kinase (PI3K-γ). As a result, TAMs could be M2-like spectral polarization, which is manifested as decreased phagocytic ability and enhanced anti-tumor immunity (162).
Lipid Metabolism Helps Maintain Drug Resistance in Gastric Cancer
Chemotherapy is the preferred treatment for patients with advanced gastric cancer. 5-Fluorouracil (5-FU) -based chemotherapy regimens, such as 5-FU combined with cisplatin or oxaliplatin, are generally considered first-line treatment options for advanced gastric cancer (163). However, drug resistance reduces the effectiveness of chemotherapy and triggers treatment failure. Based on the role of gastric cancer cells in chemotherapy resistance, tumor recurrence, and metastasis, novel strategies in cancer therapy should be urgently developed to eradicate this aggressive cell population.
Currently, changes in lipid metabolism are critical mediators of antitumor drug resistance. Reprogramming of lipid metabolism in drug-resistant cancer cells involves altered pathways of new fat production and lipid breakdown. Gastric cancer drug resistance appears to be correlated with the up-regulation of endogenous lipogenesis or lipolytic enzyme expression. This study reviews the correlation between the changes of lipid metabolism and drug resistance in gastric cancer and its significance, as well as methods to reduce drug resistance by abnormal lipid metabolism.(Tables 1–5 lists the evidence from studies demonstrating the correlation between lipid metabolism and drug resistance in cancers).

Table 2 Summary of targeting phosphatidylcholine metabolism for drug resistance.
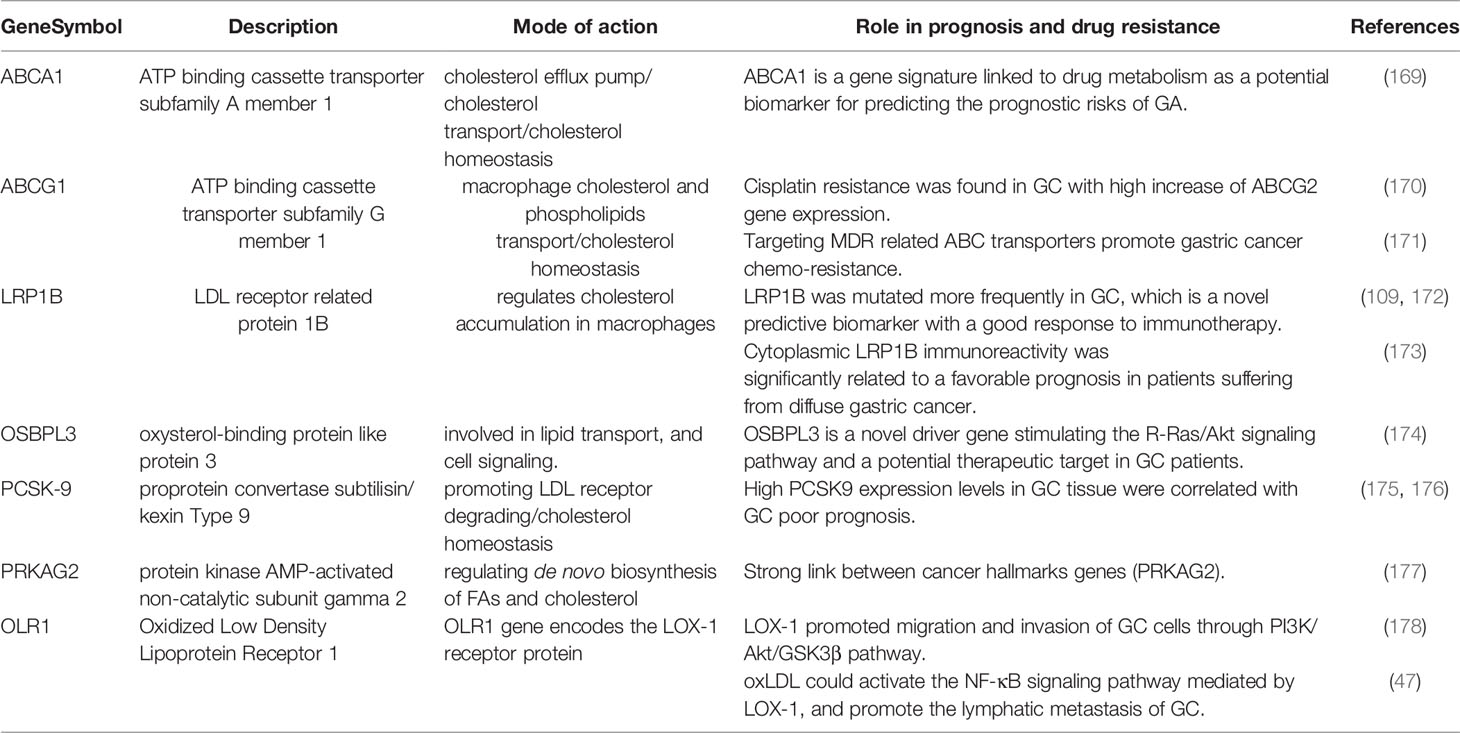
Table 3 Summary of cholesterol related genes and their link to GC metastasis, EMT and/or drug resistance.
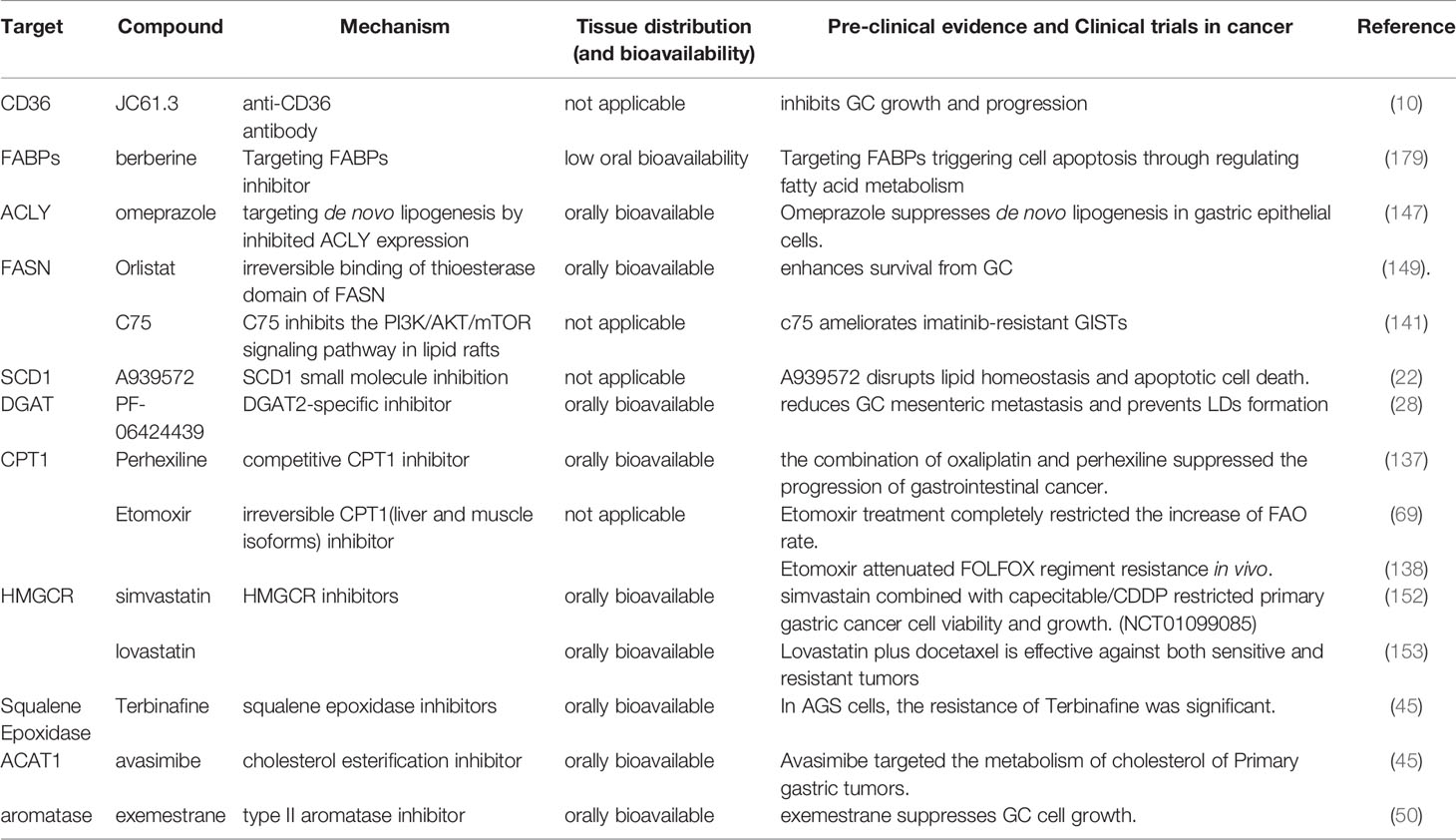
Table 4 Summary of small-molecule inhibitors of lipogenic enzymes.
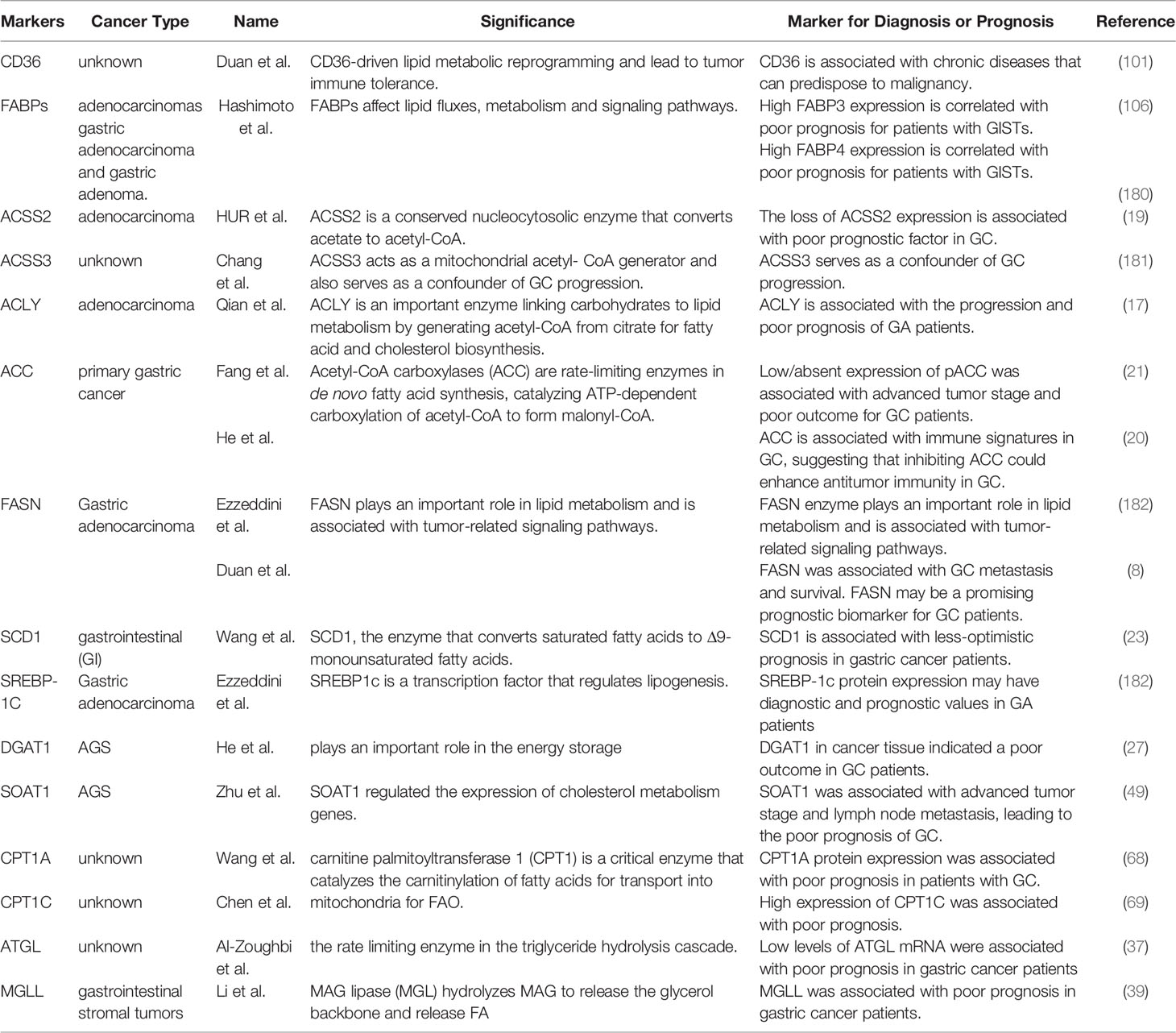
Table 5 Lipid key enzymes as biomarkers for gastric cancer prognosis.
The Effect of De Novo Synthesis and Decomposition of Fatty Acids on Chemotherapy Resistance of Gastric Cancer
Abnormal de novo fatty acids biosynthesis constantly supports membrane synthesis, signaling molecules, and the energy matrix of tumor cells that grow rapidly and continuously adapt to a wide variety of adverse microenvironmental conditions (e.g., limited nutrients and oxygen) (183). Lipid anabolic reorganization is capable of supporting disease recurrence and metastatic spread and changing the response to anticancer therapy, ultimately leading to drug resistance. resistance.(The above process is shown in Figure 4).
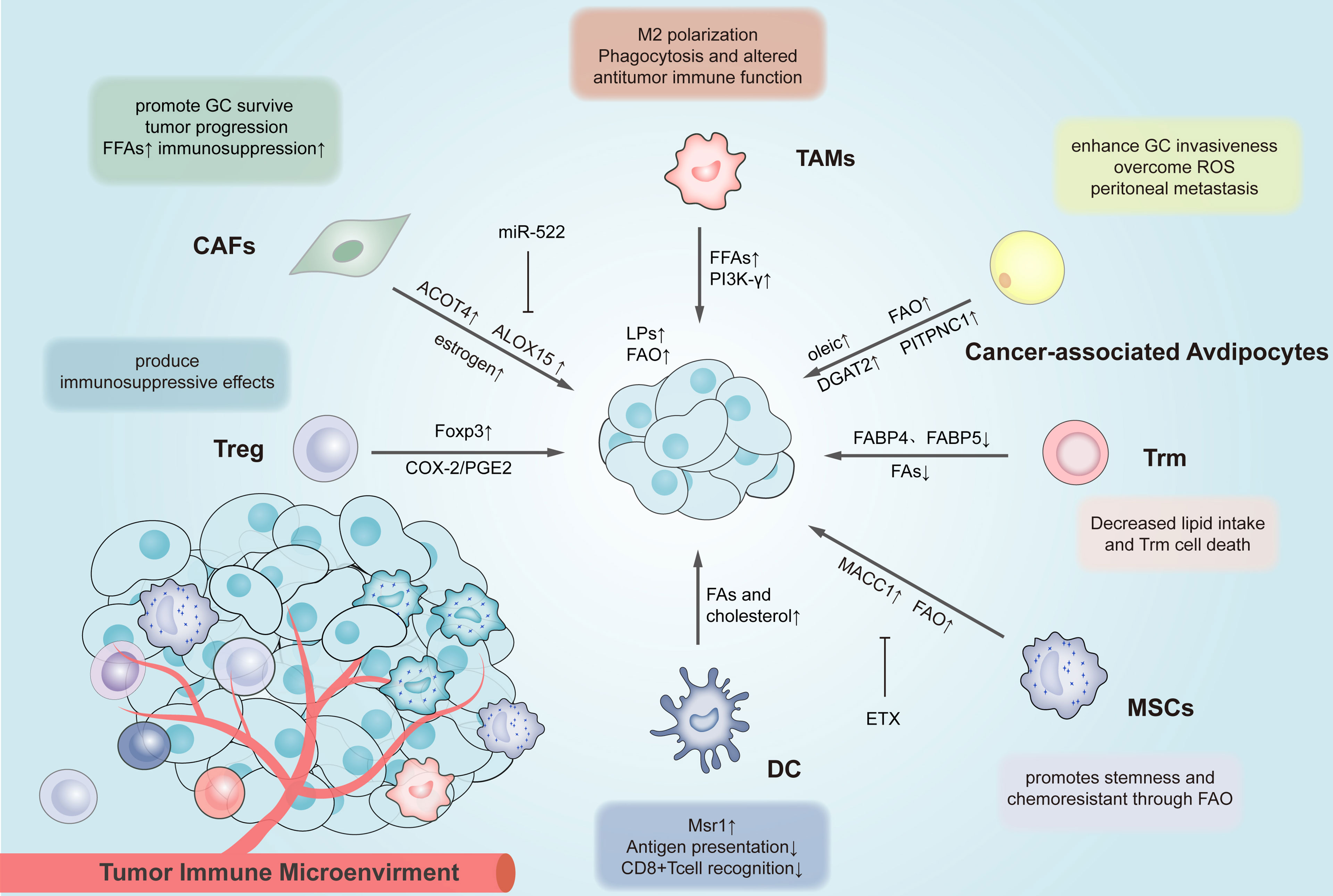
Figure 4 Lipid reprogramming in the tumor microenvironment affects the inter-cellular lipid metabolism and drug resistance of immune cells. Different immune cells in the TME of gastric cancer have different lipid metabolism changes, thus affecting their functions. The above metabolically reprogrammed immune cells exert different effects on GC. MSCs, Mesenchymal stem cell; CAFs, Cancer-associated fibroblasts; FFAs, free fatty acids; MACC1, Metastasis Associated in Colon Cancer 1; PITPNC1, Phosphatidylinositol transfer protein represents cytoplasmic 1; ACOT4, The acyl-CoA thioesterase 4; ALOX15, arachidonate lipoxygenase 15; Treg cells, regulatory T cells; TAMs, tumor-associated macrophages; DCs, dendritic cells; TAMs, tumor-associated macrophages; Trm cell, tissue-resident memory T cell.
Cancer cells rely on increased adipogenesis to maintain their rapid proliferation, which requires high levels of anabolic lipid biogenesis for membrane synthesis. FAS is a key enzyme in the adipogenesis pathway, and its overexpression triggers cancer resistance to genotoxic drugs by increasing DNA repair. Shawn et al. reported that fatty acid synthesis was almost restricted by limiting the availability of exogenous lipids and inhibiting FAS with orlistat. Since membrane phospholipids are highly dynamic, blocking the supply of fatty acids may rapidly reduce membrane integrity, thus activating autophagy and leading to death in a short period (142). Based on the positive association of metastasis-related 1 (MACC1) in colon cancer, Duan et al. drove GC’s chemotherapy resistance to oxaliplatin by up-regulating the central enzyme formed by fat. Small interfering RNA and C75 inhibited FASN when applied experimentally, significantly reversing the down-regulation of platinum-induced chemosensitivity MACC1 overexpression. The results reveal that FASN MACC1-induced platinum resistance is a crucial link, and the inhibition of chem-resistance FASN may also serve as a therapeutic strategy (140). As the evidence demonstrating the link between the above membrane changes and drug resistance emerge, drug intervention has been focusing on critical pathways and enzymes that drive changes in the lipid profile of tumor cells. Thus, pharmacologically targeting fatty acid synthase can sensitize multiple types of cancer cells to chemotherapy in vitro (184, 185) and in vivo (186, 187). Zhang et al. found that IncFERO up-regulated the expression of SCD1 enzyme to adjust lipid metabolism, and iron supplementation enhanced chemotherapeutic resistance of cisplatin and paclitaxel (122). Targeting FAO as a chemosensitization strategy is also of great significance since it increases tumor cell survival by generating energy and maintaining redox balance. A recent study found that penicillin combined with oxaliplatin effectively inhibited gastric cancer progression in an in vivo preclinical model. In the above model, hexidine resensitized cells to oxaliplatin by inhibiting FAO and facilitating intracellular ROS accumulation (137).
Recent studies have demonstrated that cholesterol and its oxygenated derivatives could be also correlated with drug resistance in gastric cancer. Drug resistance may be due to the effect of cholesterol on cellular signaling pathways or direct effects on the expression and activity of multidrug transporters.
Targeting Cholesterol Affecting the Activity of Drug Transporters
The acquisition of chemotherapy resistance after platinum-based therapy was regulated under the cholesterol-mediated effects on ABCG2 expression. Tuvshinjargal et al., found that patients with gastric cancer acquired cisplatin resistance, and ABCG2 gene expression was significantly up-regulated (188). Wu et al. reported that MiR-129-5p was hyper-methylated, and the overexpression led to the resistance of gastric cancer cells to the chemotherapeutic drugs, including 5-FU, vincristine (VCR), and CDDP. Through bioinformatics analysis and reporter gene analysis, three members of ABC transporters (ABC subfamily B member 1 (ABCB1), ATP-binding box C member 5 (ABCC5), and ATP-Binding box G transporter 1(ABCG1)), were identified to correlate with multidrug-resistance (MDR) and as direct targets of miR-129-5p (171). Yin et al. found that the drug metabolism-related gene ABCA1 could predict the prognosis of gastric adenocarcinoma (169). Notably, ABCB1-mediated drug outflow can be changed by drug regulation of membrane fluidity (e.g., by polyunsaturated fatty acid supplementation), suggesting that clinical lipid modifiers or dietary interventions may be promising chemo-sensitization strategies. With their relatively low proportion of polyunsaturated fat acyl groups in cells, chemo-resistant tumor cells are more likely to undergo toxic lipid peroxidation (capable of inducing apoptosis and ferroptosis) in response to oxidative stress induction chemotherapy agents.
Targeting Cholesterol-Related Signaling Pathways
Cholesterol Affects Drug-Resistant Cell Signaling Pathways, Thus Enhancing Chemotherapeutic Drug Retention
In gastric cancer, simvastatin can enhance capecitabine by inhibiting markers of proliferation, invasion, angiogenesis and metastasis regulated by NF-κ B (144). Oxysterol-binding protein-like protein three (OSBPL3) is involved in lipid transport, lipid metabolism, and cell signaling transduction. OSBPL3 is a novel driver gene stimulating the R- Ras/Akt signaling pathway and a potential therapeutic target in GC patients (174).
Cholesterol and Its Oxygenated Derivatives Linked to GC Chemoresistance
Follet et al. confirmed that effective suppression of HMGCR enhanced the chemosensitivity of docetaxel (153). As reported by Yang et al., targeted Ar inhibitors (ARIs) could inhibit the production of estrogen and enhance the chemotherapeutic sensitivity of 5-FU (145). Avasimibe is capable of inhibiting the synthesis of cholesterol esters and lipids in GC cells and increasing the sensitivity to chemotherapy (45).
Phosphatidylcholine and Its Metabolites Contribute to Drug Resistance in Gastric Cancer
Lipid mediators from phosphatidylcholine (PtdCho) metabolism are conducive to the treatment of drug resistance. PtdCho is hydrolyzed by Phospholipases A2 (PLA2) to produce lysophosphatidylcholine (lyso-PC) and arachidonic acid (AA). Liao et al. reported that when cytosolic phospholipase A2alpha (cPLA2α) is targeted, Ras/MEK/ERK and Akt/β-catenin signaling pathways are inhibited, ultimately enhancing the effect of chemotherapy (168).COX-2 catalyzes the conversion of AA to prostaglandin E2 (PGE2). Choi et al. reported that celecoxib could overcome the chemoresistance of 5-FU in gastric cancer by modulating the activity of COX-2 (189). The study by Qiu et al. reported that the expression level of COX-2 is significantly correlated with resistance to various chemotherapeutic drugs. (e.g., vincristine, 5-fluorouracil, oxaliplatin, mitomycin, as well as epirubicin) (190). Yu et al. found that pseudoplastic acid B circumvented MDR and increased chemotherapeutic drug sensitivity by down-regulating COX-2 expression (191). LPCAT1 converts lysophosphatidylcholine (LPC) into phosphatidylcholine (PC) in the presence of acyl-CoA in the Lands’ cycle. In gastric cancer, the expression of LPCAT1 was significantly up-regulated and facilitated the proliferation, increased the survival and enhanced the migration of cancer cells (167).
PLA2G2A hydrolyzes glycerophospholipids at the sn-2 position, releasing fatty acids and lysophospholipids (192). Chen et al. proved that artificial inhibition of PLA2G2A increased the chemosensitivity of gastric cancer cells to 5-Fu (193). In addition, PtdCho is also hydrolyzed by phosphatidylcholine-specific phospholipase C (PC-PLC) and phosphatidylcholine-specific phospholipase D(PC-PLD) to glycerol diacyl (DAG) and phosphatidic acid. Cho et al. found that the overexpression of phospholipase D (PLD) could inhibit taxotere-induced gastric cancer cell death and enhance chemotherapy resistance (165).Lin et al. demonstrated that VEGFR2 has an antiproliferative effect and chemosensitivity to Apatinib in gastric cancer cells via the phospholipase C (PLC)- extracellular regulated protein kinases 1/2(ERK1/2)-dependent pathway (166).DAG activates the protein kinase C (PKC) pathway. PA is the key to the activity of mTOR and promotes the proliferation and survival of cancer cells. All the above decomposition products of PtdCho are significantly correlated with chemical resistance.
Lipid Metabolism Affects Chemoresistance by Altering the Immune Microenvironment
Mesenchymal Stromal Cells (MSCs)
Interstitial cells in the tumor microenvironment have been recognized as a vital factor in promoting the development of chemotherapeutic resistance. As a vital part of the tumor environment, MSCs can promote chemotherapeutic resistance and help cancer cells overcome the anticancer effects of chemotherapeutic drugs by secreting protective cytokines, even generating gene mutations and changing transcriptional expression (194, 195). To be specific, MSCs promoted self-renewal and chemotherapeutic resistance of GC cells through FAO. FAO inhibitor Etomoxir (ETX) significantly reversed stem cell characteristics and resistance to 5-FU and oxaliplatin, suggesting metabolic reprogramming of GC cells when interacting with MSCs (138). Interestingly, as revealed by an existing study, MSC co-culture activated FAO in GC cells, which led to an enhancement of chemotherapeutic resistance (196).
Cancer-Associated Adipocyte
Adipocytes refer to the extracellular source of lipids in cancer cells (197). Co-cultured adipocytes are capable of increasing the proliferation of gastric cancer cells through lipolysis and provision of FAs. The above adipocyte-derived FAs are transported to GC cells to form lipid droplets and provide NADPH for ROS elimination during peritoneal metastasis. When DGAT2 was inhibited in vivo, it synergized with chemotherapeutic drugs to significantly inhibit peritoneal metastasis of gastric cancer (198). Tan et al. found that phosphatidylinositol transfer protein, cytoplasmic 1 (PITPNC1), interacts with adipocytes and GC omental metastasis to promote anoikis resistance by enhancing FAO (9).
CAFs
As a significant part of tumor stroma, CAFs are activated during carcinogenesis. CAFs interactions with tumor cells may contribute to the development of invasive phenotypes of cancer cells, including metastatic potential and chemotherapy resistance (199). Zhang et al. suggested that arachidonic acid lipoxygenase 15 (ALOX15), the primary mediator of lipid ROS production in GC cells, was significantly down-regulated, and the sensitivity of gastric cancer cells to cisplatin and paclitaxel decreased significantly. ACOT4, a subtype of the ACOTs family, can catalyze the hydrolysis of fatty acid acyl-CoA to CoA-SH and free fatty acids. The above enzymes have been found to play crucial roles in lipid metabolism (200). CAFs are capable of inhibiting ferroptosis by targeting ALOX15 and blocking lipid-ROS accumulation and secreting exosome Mir-522 (201). As reported by another existing study, estrogen could stimulate gastric CAFs to secrete IL-6 and activate the STAT3 signaling pathway in GC cells (202). As a result, the proliferation and invasion ability of gastric cancer cells could be enhanced, and the chemotherapy resistance of GC cells was also confirmed to be crucial (203).
Targeting Lipid Raft Formation in Gastric Cancer Cells
We recently reported that the anti-epidermal growth factor receptor (EGFR) -targeted drug cetuximab facilitated the formation of DISC in gastric cancer cell lipid rafts, which promoted TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis (204). It was found that β-element increased the anticancer activity of doxorubicin in multidrug-resistant gastric cancer cells by down-regulating Akt phosphorylation and P-GP expression (205). As revealed by recent studies, oxaliplatin, cisplatin, epirubicin, 5-fluorouracil and other chemotherapy drugs could increase the sensitivity of gastric cancer cells to TRAIL (206–208). Furthermore, keratin 6 regulated lipid raft formation, thereby enhancing the chemotherapy resistance to cisplatin (209).
Lipid Metabolism Alters Chemotherapy Resistance by Affecting Autophagy in Gastric Cancer
Autophagy is a significant mechanism leading to chemotherapy resistance in gastric cancer. Autophagy could protect gastric cancer cells from the cytotoxicity of chemotherapeutic drugs and promote the formation of chemotherapy resistance (210).Lipid metabolism plays different roles in gastric cancer chemotherapy resistance by regulating autophagy. If properly applied, targeted autophagy may be a significant strategy for the prevention and treatment of chemotherapeutic resistance (211). COX-2 refers to a rate-limiting enzyme of PGE2 biosynthesis and is often overexpressed in helicobacter pylori infection, precancerous lesions and gastric cancer patients (212, 213). COX-2 promotes carcinogenesis by supporting the production of PGE2. PGE2, an active lipid compound derived from arachidonic acid, regulates different stages of the immune response such as chronic infections or cancer. Pro-inflammatory eicosanes were found to play a vital role in tumorigenesis by transducing their signals through EP1-4 and PGE2 and facilitating cell proliferation, angiogenesis, invasion and metastasis by inducing a sustained inflammatory response (212, 214, 215). Bacterial infection and PGE2 signaling are required for gastric tumorigenesis in mice; they jointly up-regulate CC-chemokine ligand 2 (CCL2), in which macrophages are recruited to gastric tumors. The expression of CCL2 is correlated with invasion, metastasis, and drug resistance of tumors. It was found that DDP-resistant gastric cancer cell lines (e.g., BGC823/DDP cells and SGC7901/DDP cells) secreted more CCL2 to maintain DDP resistance (216, 217). Moreover, CCL2 overexpression up-regulated the expression of p62 by activating the PI3K/AKT/mTOR signaling pathway. The increase in p62 expression activated the transcription of CCL2, inhibited autophagy, and formed a positive feedback loop to maintain drug resistance (218). Chi et al. initially demonstrated the methylation of MGMT promoter in gastric tissue through COX-2/PGE2 axis in COX-2 transgenic mice (219).
As reported by Lei et al., a high expression of promoter methylation of the DNA repair gene O-methylguanine DNA methyltransferase (MGMT) was significantly correlated with a low expression of autophagy-related gene 4B (ATG4B), which revealed a reasonable prognosis of gastric cancer. DDP was found to down-regulate MGMT expression in a dose- and time-dependent manner, and a low MGMT expression could induce autophagy and cisplatin resistance. Overexpression of MGMT could inhibit autophagy and reverse DDP resistance in vivo and in vitro (220). As revealed by the results, the COX-2 specific inhibitor celecoxib or the non-selective COX-2 inhibitor sulindac could synergistically inhibit GC with decitabine in vitro and in vivo (219). Indomethacin, a non-steroidal anti-inflammatory drug, was reported as an adjunct to anticancer drugs with satisfactory efficacy (221, 222). Vallecillo-Hernandez et al. suggested that indomethacin induces accumulation of p62 and BRCA1 (NBR1) neighbors in AGS cells, impairs lysosomal function, inhibits autophagic degradation, and increases oxa-induced cell death (211). It was reported that oral administration of LPA lipids in Chinese herbal medicines could increase the production of prostaglandin E2, thus leading to a high expression of LPA2 in gastric cancer cells. The above could prevent indomethacin-induced cell death and stimulate the proliferation of gastric cancer cells (223).
Lipid Metabolism Contributes to Chemoresistance by Causing Oxidative Stress in GC
Lipid droplets were found to decrease ROS toxicity, thus increasing cancer cell survival (224). In numerous types of cancer, LDs facilitated tumor growth, while enhancing metastasis, chemotherapy resistance, as well as disease recurrence (225–227). Lipid droplets can act as additional lipid sources for fatty acid oxidation under nutritional stress or as “precipitates” for isolating hydrophobic drugs, leading directly to chemotherapy resistance (228). For instance, tumor cell lines with high LDs content have increased chemotherapy resistance to 5-FU and oxaliplatin. Stimulated by lysophosphatidylcholine acyltransferase 2 (LPCAT2), the above cells further accumulated LPCAT2 during the chemotherapy response, and the increased expression of LPCAT2 prevented the endoplasmic reticulum stress induced by chemotherapy, further highlighting the influence of LDs on the cell chemotherapy response (226, 229). Enjoji et al. reported a significant accumulation of lipid droplets in gastric cancer cells, primarily in the formation of new lipid and lipid storage (26). As reported by Choi et al., ROS levels in gastric cancer cells increased dose-dependently after 5-FU treatment, while ROS levels in GC were maintained at a relatively low level. GC was found with increased OXPHOS and paradoxically maintained a low ROS level through FAO-mediated NADPH regeneration (230). On that basis, the sensitivity to chemotherapeutic drugs (5-FU) decreased. N-3 docosahexaenoic acid (DHA) was found to induce ROS-dependent apoptosis in cisplatin-resistant gastric cancer (231).
The Degree of Lipid Saturation Affects the Physicochemical Properties of Cell Membranes
Increased levels of saturated fatty acids make cell membranes more resistant to ROS-dependent peroxidation and cell death attributed to ferroptosis. For instance, orlistat was found to reduce the absorption of exogenous dietary fat by inhibiting gastric and pancreatic lipase, and it was also indicated as an excellent FAS inhibitor (142). Chen et al. found that LDLR-reduced LPC abundance perturbed the phospholipid homeostasis of the Lands cycle in LDs, thus reducing intracellular platinum transport during DNA adduct formation and then increasing cisplatin sensitivity in gastric cancer (232).
Conclusions
Abnormal lipid metabolism represents a crucial component of tumor metabolic adaptation, which is conducive to enhancing treatment resistance and facilitating metastasis. In this study, the specific process of lipid metabolism reprogramming in gastric cancer is described. Moreover, the roles of lipid metabolism in gastric cancer treatment, novel biomarkers, and chemotherapy resistance of gastric cancer cells are summarized. Numerous drugs targeting lipid metabolism pathways have been developed for cancer treatment. However, the role of these drugs in gastric cancer remains unclear. Accordingly, further research is needed to gain a comprehensive and in-depth understanding of the lipid metabolism of GC.
Author Contributions
M-YC, D-XZ, and JW contributed to the study conception and design. Material preparation, data collection and analysis were performed by M-YC and XY. The first draft of the manuscript was written by M-YC and all authors commented on existing versions of the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by grants from National Natural Science Foundation of China (nos. 81770212, to D-XZ) and Changzhou City Health Commission major scientific and technological projects (nos. ZD201901, to JW).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Morin PJ. Drug Resistance and the Microenvironment: Nature and Nurture. Drug Resist Update (2003) 6:169–72. doi: 10.1016/S1368-7646(03)00059-1
2. Liu Q, Luo Q, Halim A, Song G. Targeting Lipid Metabolism of Cancer Cells: A Promising Therapeutic Strategy for Cancer. Cancer Lett (2017) 401:39–45. doi: 10.1016/j.canlet.2017.05.002
3. Li K, Dan Z, Nie YQ. Gastric Cancer Stem Cells in Gastric Carcinogenesis, Progression, Prevention and Treatment. World J Gastroenterol (2014) 20:5420–6. doi: 10.3748/wjg.v20.i18.5420
4. Kuo CY, Ann DK. When Fats Commit Crimes: Fatty Acid Metabolism, Cancer Stemness and Therapeutic Resistance. Cancer Commun (Lond) (2018) 38:47. doi: 10.1186/s40880-018-0317-9
5. Guo E, Chen L, Xie Q, Chen J, Tang Z, Wu Y. Serum HDL-C as a Potential Biomarker for Nodal Stages in Gastric Cancer. Ann Surg Oncol (2007) 14:2528–34. doi: 10.1245/s10434-007-9401-0
6. Tamura T, Inagawa S, Hisakura K, Enomoto T, Ohkohchi N. Evaluation of Serum High-Density Lipoprotein Cholesterol Levels as a Prognostic Factor in Gastric Cancer Patients. J Gastroenterol Hepatol (2012) 27:1635–40. doi: 10.1111/j.1440-1746.2012.07189.x
7. Nam SY, Park BJ, Nam JH, Kook MC. Effect of Helicobacter Pylori Eradication and High-Density Lipoprotein on the Risk of De Novo Gastric Cancer Development. Gastrointest Endosc (2019) 90:448–56.e1. doi: 10.1016/j.gie.2019.04.232
8. Duan J, Sun L, Huang H, Wu Z, Wang L, Liao W. Overexpression of Fatty Acid Synthase Predicts a Poor Prognosis for Human Gastric Cancer. Mol Med Rep (2016) 13:3027–35. doi: 10.3892/mmr.2016.4902
9. Tan Y, Lin K, Zhao Y, Wu Q, Chen D, Wang J, et al. Adipocytes Fuel Gastric Cancer Omental Metastasis via PITPNC1-Mediated Fatty Acid Metabolic Reprogramming. Theranostics (2018) 8:5452–68. doi: 10.7150/thno.28219
10. Jiang M, Wu N, Xu B, Chu Y, Li X, Su S, et al. Fatty Acid-Induced CD36 Expression via O-GlcNAcylation Drives Gastric Cancer Metastasis. Theranostics (2019) 9:5359–73. doi: 10.7150/thno.34024
11. Bénarouche A, Sams L, Bourlieu C, Vié V, Point V, Cavalier JF, et al. Studying Gastric Lipase Adsorption Onto Phospholipid Monolayers by Surface Tensiometry, Ellipsometry, and Atomic Force Microscopy. Methods Enzymol (2017) 583:255–78. doi: 10.1016/bs.mie.2016.09.039
12. Hirata Y, Sezaki T, Tamura-Nakano M, Oyama C, Hagiwara T, Ishikawa T, et al. Fatty Acids in a High-Fat Diet Potentially Induce Gastric Parietal-Cell Damage and Metaplasia in Mice. J Gastroenterol (2017) 52:889–903. doi: 10.1007/s00535-016-1291-0
13. DeFilippis RA, Chang H, Dumont N, Rabban JT, Chen YY, Fontenay GV, et al. CD36 Repression Activates a Multicellular Stromal Program Shared by High Mammographic Density and Tumor Tissues. Cancer Discovery (2012) 2:826–39. doi: 10.1158/2159-8290.CD-12-0107
14. Pan J, Fan Z, Wang Z, Dai Q, Xiang Z, Yuan F, et al. CD36 Mediates Palmitate Acid-Induced Metastasis of Gastric Cancer via AKT/GSK-3β/β-Catenin Pathway. J Exp Clin Cancer Res (2019) 38:52. doi: 10.1186/s13046-019-1049-7
15. Furuhashi M, Hotamisligil GS. Fatty Acid-Binding Proteins: Role in Metabolic Diseases and Potential as Drug Targets. Nat Rev Drug Discovery (2008) 7:489–503. doi: 10.1038/nrd2589
16. Zhao G, Wu M, Wang X, Du Z, Zhang G. Effect of FABP5 Gene Silencing on the Proliferation, Apoptosis and Invasion of Human Gastric SGC-7901 Cancer Cells. Oncol Lett (2017) 14:4772–8. doi: 10.3892/ol.2017.6748
17. Qian X, Hu J, Zhao J, Chen H. ATP Citrate Lyase Expression is Associated With Advanced Stage and Prognosis in Gastric Adenocarcinoma. Int J Clin Exp Med (2015) 8:7855–60.
18. Moffett JR, Puthillathu N, Vengilote R, Jaworski DM, Namboodiri AM. Acetate Revisited: A Key Biomolecule at the Nexus of Metabolism, Epigenetics, and Oncogenesis - Part 2: Acetate and ACSS2 in Health and Disease. Front Physiol (2020) 11:580171. doi: 10.3389/fphys.2020.580171
19. Hur H, Kim YB, Ham IH, Lee D. Loss of ACSS2 Expression Predicts Poor Prognosis in Patients With Gastric Cancer. J Surg Oncol (2015) 112:585–91. doi: 10.1002/jso.24043
20. He Y, Wang X. Identification of Molecular Features Correlating With Tumor Immunity in Gastric Cancer by Multi-Omics Data Analysis. Ann Transl Med (2020) 8:1050. doi: 10.21037/atm-20-922
21. Fang W, Cui H, Yu D, Chen Y, Wang J, Yu G. Increased Expression of Phospho-Acetyl-CoA Carboxylase Protein is an Independent Prognostic Factor for Human Gastric Cancer Without Lymph Node Metastasis. Med Oncol (2014) 31:15. doi: 10.1007/s12032-014-0015-7
22. Roongta UV, Pabalan JG, Wang X, Ryseck RP, Fargnoli J, Henley BJ, et al. Cancer Cell Dependence on Unsaturated Fatty Acids Implicates Stearoyl-CoA Desaturase as a Target for Cancer Therapy. Mol Cancer Res (2011) 9:1551–61. doi: 10.1158/1541-7786.MCR-11-0126
23. Wang C, Shi M, Ji J, Cai Q, Zhao Q, Jiang J, et al. Stearoyl-CoA Desaturase 1 (SCD1) Facilitates the Growth and Anti-Ferroptosis of Gastric Cancer Cells and Predicts Poor Prognosis of Gastric Cancer. Aging (Albany NY) (2020) 12:15374–91. doi: 10.18632/aging.103598
24. Peck B, Schulze A. Lipid Desaturation - The Next Step in Targeting Lipogenesis in Cancer? FEBS J (2016) 283:2767–78. doi: 10.1111/febs.13681
25. Sun Q, Yu X, Peng C, Liu N, Chen W, Xu H, et al. Activation of SREBP-1c Alters Lipogenesis and Promotes Tumor Growth and Metastasis in Gastric Cancer. BioMed Pharmacother (2020) 128:110274. doi: 10.1016/j.biopha.2020.110274
26. Enjoji M, Kohjima M, Ohtsu K, Matsunaga K, Murata Y, Nakamuta , et al. Intracellular Mechanisms Underlying Lipid Accumulation (White Opaque Substance) in Gastric Epithelial Neoplasms: A Pilot Study of Expression Profiles of Lipid-Metabolism-Associated Genes. J Gastroenterol Hepatol (2016) 31:776–81. doi: 10.1111/jgh.13216
27. He P, Cheng S, Hu F, Ma Z, Xia Y. Up-Regulation of DGAT1 in Cancer Tissues and Tumor-Infiltrating Macrophages Influenced Survival of Patients With Gastric Cancer. BMC Cancer (2021) 21:252. doi: 10.1186/s12885-021-07976-5
28. Li S, Wu T, Lu YX, Wang JX, Yu FH, Yang MZ, et al. Obesity Promotes Gastric Cancer Metastasis via Diacylglycerol Acyltransferase 2-Dependent Lipid Droplets Accumulation and Redox Homeostasis. Redox Biol (2020) 36:101596. doi: 10.1016/j.redox.2020.101596
29. Unger RH. Lipotoxic Diseases. Annu Rev Med (2002) 53:319–36. doi: 10.1146/annurev.med.53.082901.104057
30. Thiele C, Spandl J. Cell Biology of Lipid Droplets. Curr Opin Cell Biol (2008) 20:378–85. doi: 10.1016/j.ceb.2008.05.009
31. Martin S, Parton RG. Lipid Droplets: A Unified View of a Dynamic Organelle. Nat Rev Mol Cell Biol (2006) 7:373–8. doi: 10.1038/nrm1912
32. Walther TC, Farese RV Jr. Lipid Droplets and Cellular Lipid Metabolism. Annu Rev Biochem (2012) 81:687–714. doi: 10.1146/annurev-biochem-061009-102430
33. Brasaemle DL. Thematic Review Series: Adipocyte Biology. The Perilipin Family of Structural Lipid Droplet Proteins: Stabilization of Lipid Droplets and Control of Lipolysis. J Lipid Res (2007) 48:2547–59. doi: 10.1194/jlr.R700014-JLR200
34. Pol A, Gross SP, Parton RG. Review: Biogenesis of the Multifunctional Lipid Droplet: Lipids, Proteins, and Sites. J Cell Biol (2014) 204:635–46. doi: 10.1083/jcb.201311051
35. Orlicky DJ, Degala G, Greenwood C, Bales ES, Russell TD, McManaman JL, et al. Multiple Functions Encoded by the N-Terminal PAT Domain of Adipophilin. J Cell Sci (2008) 121:2921–9. doi: 10.1242/jcs.026153
36. Sun X, Yang S, Feng X, Zheng Y, Zhou J, Wang H, et al. The Modification of Ferroptosis and Abnormal Lipometabolism Through Overexpression and Knockdown of Potential Prognostic Biomarker Perilipin2 in Gastric Carcinoma. Gastric Cancer (2020) 23:241–59. doi: 10.1007/s10120-019-01004-z
37. Al-Zoughbi W, Pichler M, Gorkiewicz G, Guertl-Lackner B, Haybaeck J, Jahn SW, et al. Loss of Adipose Triglyceride Lipase is Associated With Human Cancer and Induces Mouse Pulmonary Neoplasia. Oncotarget (2016) 7:33832–40. doi: 10.18632/oncotarget.9418
38. Broadfield LA, Pane AA, Talebi A, Swinnen JV, Fendt SM. Lipid Metabolism in Cancer: New Perspectives and Emerging Mechanisms. Dev Cell (2021) 56:1363–93. doi: 10.1016/j.devcel.2021.04.013
39. Li CF, Chuang IC, Liu TT, Chen KC, Chen YY, Fang FM, et al. Transcriptomic Reappraisal Identifies MGLL Overexpression as an Unfavorable Prognosticator in Primary Gastrointestinal Stromal Tumors. Oncotarget (2016) 7:49986–97. doi: 10.18632/oncotarget.10304
40. Bihain BE, Yen FT. The Lipolysis Stimulated Receptor: A Gene at Last. Curr Opin Lipidol (1998) 9:221–4. doi: 10.1097/00041433-199806000-00006
41. Hiramatsu K, Serada S, Enomoto T, Takahashi Y, Nakagawa S, Nojima S, et al. LSR Antibody Therapy Inhibits Ovarian Epithelial Tumor Growth by Inhibiting Lipid Uptake. Cancer Res (2018) 78:516–27. doi: 10.1158/0008-5472.CAN-17-0910
42. Sugase T, Takahashi T, Serada S, Fujimoto M, Ohkawara T, Hiramatsu K, et al. Lipolysis-Stimulated Lipoprotein Receptor Overexpression is a Novel Predictor of Poor Clinical Prognosis and a Potential Therapeutic Target in Gastric Cancer. Oncotarget (2018) 9:32917–28. doi: 10.18632/oncotarget.25952
43. Chushi L, Wei W, Kangkang X, Yongzeng F, Ning X, Xiaolei C, et al. HMGCR is Up-Regulated in Gastric Cancer and Promotes the Growth and Migration of the Cancer Cells. Gene (2016) 587:42–7. doi: 10.1016/j.gene.2016.04.029
44. Huang B, Song BL, Xu C. Cholesterol Metabolism in Cancer: Mechanisms and Therapeutic Opportunities. Nat Metab (2020) 2:132–41. doi: 10.1038/s42255-020-0174-0
45. Ortiz N, Delgado-Carazo JC, Díaz C. Importance of Mevalonate Pathway Lipids on the Growth and Survival of Primary and Metastatic Gastric Carcinoma Cells. Clin Exp Gastroenterol (2021) 14:217–28. doi: 10.2147/CEG.S310235
46. Goldstein JL, Brown MS. The LDL Receptor. Arterioscler Thromb Vasc Biol (2009) 29:431–8. doi: 10.1161/ATVBAHA.108.179564
47. Ma C, Xie J, Luo C, Yin H, Li R, Wang X, et al. OxLDL Promotes Lymphangiogenesis and Lymphatic Metastasis in Gastric Cancer by Upregulating VEGF−C Expression and Secretion. Int J Oncol (2019) 54:572–84. doi: 10.3892/ijo.2018.4648
48. Wang YJ, Bian Y, Luo J, Lu M, Xiong Y, Guo SY, et al. Cholesterol and Fatty Acids Regulate Cysteine Ubiquitylation of ACAT2 Through Competitive Oxidation. Nat Cell Biol (2017) 19:808–19. doi: 10.1038/ncb3551
49. Zhu T, Wang Z, Zou T, Xu L, Zhang S, Chen Y, et al. SOAT1 Promotes Gastric Cancer Lymph Node Metastasis Through Lipid Synthesis. Front Pharmacol (2021) 12:769647. doi: 10.3389/fphar.2021.769647
50. Chang WC, Huang SF, Lee YM, Lai HC, Cheng BH, Cheng WC, et al. Cholesterol Import and Steroidogenesis are Biosignatures for Gastric Cancer Patient Survival. Oncotarget (2017) 8:692–704. doi: 10.18632/oncotarget.13524
51. Wang B, Tontonoz P. Liver X Receptors in Lipid Signalling and Membrane Homeostasis. Nat Rev Endocrinol (2018) 14:452–63. doi: 10.1038/s41574-018-0037-x
52. Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, et al. SREBP-2, A Second Basic-Helix-Loop-Helix-Leucine Zipper Protein That Stimulates Transcription by Binding to a Sterol Regulatory Element. Proc Natl Acad Sci U.S.A. (1993) 90:11603–7. doi: 10.1073/pnas.90.24.11603
53. Wang Q, Feng F, Wang J, Ren M, Shi Z, Mao X, et al. Liver X Receptor Activation Reduces Gastric Cancer Cell Proliferation by Suppressing Wnt Signalling via Lxrβ Relocalization. J Cell Mol Med (2019) 23:789–97. doi: 10.1111/jcmm.13974
54. Liu J, Qi YB. Activation of Lxrβ Inhibits Proliferation, Promotes Apoptosis, and Increases Chemosensitivity of Gastric Cancer Cells by Upregulating the Expression of ATF4. J Cell Biochem (2019) 120:14336–47. doi: 10.1002/jcb.28558
55. Ye X, Zhang Y, Wang X, Li Y, Gao Y. Tumor-Suppressive Functions of Long-Chain Acyl-CoA Synthetase 4 in Gastric Cancer. IUBMB Life (2016) 68:320–7. doi: 10.1002/iub.1486
56. Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, et al. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the Formyl Peptide Receptor (FPR) Family. Pharmacol Rev (2009) 61:119–61. doi: 10.1124/pr.109.001578
57. Serhan CN, Chiang N, Van Dyke TE. Resolving Inflammation: Dual Anti-Inflammatory and Pro-Resolution Lipid Mediators. Nat Rev Immunol (2008) 8:349–61. doi: 10.1038/nri2294
58. Prevete N, Liotti F, Illiano A, Amoresano A, Pucci P, de Paulis A, et al. Formyl Peptide Receptor 1 Suppresses Gastric Cancer Angiogenesis and Growth by Exploiting Inflammation Resolution Pathways. Oncoimmunology (2017) 6:e1293213. doi: 10.1080/2162402X.2017.1293213
59. Liu SH, Shen CC, Yi YC, Tsai JJ, Wang CC, Chueh JT, et al. Honokiol Inhibits Gastric Tumourigenesis by Activation of 15-Lipoxygenase-1 and Consequent Inhibition of Peroxisome Proliferator-Activated Receptor-Gamma and COX-2-Dependent Signals. Br J Pharmacol (2010) 160:1963–72. doi: 10.1111/j.1476-5381.2010.00804.x
60. Houten SM, Violante S, Ventura FV, Wanders RJ. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu Rev Physiol (2016) 78:23–44. doi: 10.1146/annurev-physiol-021115-105045
61. Ezzeddini R, Taghikhani M, Salek Farrokhi A, Somi MH, Samadi N, Esfahani A, et al. Downregulation of Fatty Acid Oxidation by Involvement of HIF-1α and Pparγ in Human Gastric Adenocarcinoma and Related Clinical Significance. J Physiol Biochem (2021) 77:249–60. doi: 10.1007/s13105-021-00791-3
62. Aa JY, Yu LZ, Sun M, Liu LS, Li MJ, Cao B, et al. Metabolic Features of the Tumor Microenvironment of Gastric Cancer and the Link to the Systemic Macroenvironment. Metabolomics (2012) 8:164–73. doi: 10.1007/s11306-011-0297-0
63. Song H, Wang L, Liu HL, Wu XB, Wang HS, Liu ZH, et al. Tissue Metabolomic Fingerprinting Reveals Metabolic Disorders Associated With Human Gastric Cancer Morbidity. Oncol Rep (2011) 26:431–8. doi: 10.3892/or.2011.1302
64. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr.. Cellular Fatty Acid Metabolism and Cancer. Cell Metab (2013) 18:153–61. doi: 10.1016/j.cmet.2013.05.017
65. Carracedo A, Cantley LC, Pandolfi PP. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat Rev Cancer (2013) 13:227–32. doi: 10.1038/nrc3483
66. Park JH, Vithayathil S, Kumar S, Sung PL, Dobrolecki LE, Putluri V, et al. Fatty Acid Oxidation-Driven Src Links Mitochondrial Energy Reprogramming and Oncogenic Properties in Triple-Negative Breast Cancer. Cell Rep (2016) 14:2154–65. doi: 10.1016/j.celrep.2016.02.004
67. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML–PPAR-δ Pathway for Fatty Acid Oxidation Regulates Hematopoietic Stem Cell Maintenance. Nat Med (2012) 18:1350–8. doi: 10.1038/nm.2882
68. Wang L, Li C, Song Y, Yan Z. Inhibition of Carnitine Palmitoyl Transferase 1A-Induced Fatty Acid Oxidation Suppresses Cell Progression in Gastric Cancer. Arch Biochem Biophys (2020) 696:108664. doi: 10.1016/j.abb.2020.108664
69. Chen T, Wu G, Hu H, Wu C. Enhanced Fatty Acid Oxidation Mediated by CPT1C Promotes Gastric Cancer Progression. J Gastrointest Oncol (2020) 11:695–707. doi: 10.21037/jgo-20-157
70. Wang F, Wu J, Qiu Z, Ge X, Liu X, Zhang C, et al. ACOT1 Expression is Associated With Poor Prognosis in Gastric Adenocarcinoma. Hum Pathol (2018) 77:35–44. doi: 10.1016/j.humpath.2018.03.013
71. Li Q, Yang Y, Jiang X, Jin Y, Wu J, Qin Y, et al. The Combined Expressions of B7H4 and ACOT4 in Cancer-Associated Fibroblasts are Related to Poor Prognosis in Patients With Gastric Carcinoma. Int J Clin Exp Pathol (2019) 12:2672–81.
72. Ortiz N, Díaz C. Mevalonate Pathway as a Novel Target for the Treatment of Metastatic Gastric Cancer. Oncol Lett (2020) 20:320. doi: 10.3892/ol.2020.12183
73. Balakrishnan K, Ganesan K. Occurrence of Differing Metabolic Dysregulations, A Glucose Driven and Another Fatty Acid Centric in Gastric Cancer Subtypes. Funct Integr Genomics (2020) 20:813–24. doi: 10.1007/s10142-020-00753-w
74. Zhu Z, Qin J, Dong C, Yang J, Yang M, Tia J, et al. Identification of Four Gastric Cancer Subtypes Based on Genetic Analysis of Cholesterogenic and Glycolytic Pathways. Bioengineered (2021) 12:4780–93. doi: 10.1080/21655979.2021.1956247
75. Deng N, Goh LK, Wang H, Das K, Tao J, Tan IB, et al. A Comprehensive Survey of Genomic Alterations in Gastric Cancer Reveals Systematic Patterns of Molecular Exclusivity and Co-Occurrence Among Distinct Therapeutic Targets. Gut (2012) 61:673–84. doi: 10.1136/gutjnl-2011-301839
76. Wadhwa R, Song S, Lee JS, Yao Y, Wei Q, Ajani JA. Gastric Cancer-Molecular and Clinical Dimensions. Nat Rev Clin Oncol (2013) 10:643–55. doi: 10.1038/nrclinonc.2013.170
77. Willems L, Tamburini J, Chapuis N, Lacombe C, Mayeux P, Bouscary D, et al. PI3K and mTOR Signaling Pathways in Cancer: New Data on Targeted Therapies. Curr Oncol Rep (2012) 14:129–38. doi: 10.1007/s11912-012-0227-y
78. Mossmann D, Park S, Hall MN. mTOR Signalling and Cellular Metabolism are Mutual Determinants in Cancer. Nat Rev Cancer (2018) 18:744–57. doi: 10.1038/s41568-018-0074-8
79. Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, et al. Hepatic Mtorc2 Activates Glycolysis and Lipogenesis Through Akt, Glucokinase, and SREBP1c. Cell Metab (2012) 15:725–38. doi: 10.1016/j.cmet.2012.03.015
80. Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, et al. Mtorc2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell (2017) 32:807–23.e12. doi: 10.1016/j.ccell.2017.11.011
81. Li S, Oh YT, Yue P, Khuri FR, Sun SY. Inhibition of mTOR Complex 2 Induces GSK3/FBXW7-Dependent Degradation of Sterol Regulatory Element-Binding Protein 1 (SREBP1) and Suppresses Lipogenesis in Cancer Cells. Oncogene (2016) 35:642–50. doi: 10.1038/onc.2015.123
82. Shu Z, Gao Y, Zhang G, Zhou Y, Cao J, Wan D, et al. A Functional Interaction Between Hippo-YAP Signalling and SREBPs Mediates Hepatic Steatosis in Diabetic Mice. J Cell Mol Med (2019) 23:3616–28. doi: 10.1111/jcmm.14262
83. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, et al. Metabolic Control of YAP and TAZ by the Mevalonate Pathway. Nat Cell Biol (2014) 16:357–66. doi: 10.1038/ncb2936
84. Noto A, De Vitis C, Pisanu ME, Roscilli G, Ricci G, Catizone A, et al. Stearoyl-CoA-Desaturase 1 Regulates Lung Cancer Stemness via Stabilization and Nuclear Localization of YAP/TAZ. Oncogene (2017) 36:4573–84. doi: 10.1038/onc.2017.75
85. Aylon Y, Gershoni A, Rotkopf R, Biton IE, Porat Z, Koh AP, et al. The LATS2 Tumor Suppressor Inhibits SREBP and Suppresses Hepatic Cholesterol Accumulation. Genes Dev (2016) 30:786–97. doi: 10.1101/gad.274167.115
86. Niehrs C. The Complex World of WNT Receptor Signalling. Nat Rev Mol Cell Biol (2012) 13:767–79. doi: 10.1038/nrm3470
87. Imajo M, Miyatake K, Iimura A, Miyamoto A, Nishida E. A Molecular Mechanism That Links Hippo Signalling to the Inhibition of Wnt/β-Catenin Signalling. EMBO J (2012) 31:1109–22. doi: 10.1038/emboj.2011.487
88. Barry ER, Morikawa T, Butler BL, Shrestha K, de laRosa R, Yan KS, et al. Restriction of Intestinal Stem Cell Expansion and the Regenerative Response by YAP. Nature (2013) 493:106–10. doi: 10.1038/nature11693
89. Zhang X, Zhao H, Li Y, Xia D, Yang L, Ma Y, et al. The Role of YAP/TAZ Activity in Cancer Metabolic Reprogramming. Mol Cancer (2018) 17:134. doi: 10.1186/s12943-018-0882-1
90. Bagchi DP, Nishii A, Li Z, DelProposto JB, Corsa CA, Mori H, et al. Wnt/β-Catenin Signaling Regulates Adipose Tissue Lipogenesis and Adipocyte-Specific Loss Is Rigorously Defended by Neighboring Stromal-Vascular Cells. Mol Metab (2020) 42:101078. doi: 10.1016/j.molmet.2020.101078
91. Liu Q, Xia H, Zhou S, Tang Q, Zhou J, Ren M, et al. Simvastatin Inhibits the Malignant Behaviors of Gastric Cancer Cells by Simultaneously Suppressing YAP and β-Catenin Signaling. Onco Targets Ther (2020) 13:2057–66. doi: 10.2147/OTT.S237693
92. Gu Y, Liu X, Liao L, Gao Y, Shi Y, Ni J, et al. Relationship Between Lipid Metabolism and Hedgehog Signaling Pathway. J Steroid Biochem Mol Biol (2021) 209:105825. doi: 10.1016/j.jsbmb.2021.105825
93. Riobo NA. Cholesterol and its Derivatives in Sonic Hedgehog Signaling and Cancer. Curr Opin Pharmacol (2012) 12:736–41. doi: 10.1016/j.coph.2012.07.002
94. Nagini S. Carcinoma of the Stomach: A Review of Epidemiology, Pathogenesis, Molecular Genetics and Chemoprevention. World J Gastrointest Oncol (2012) 4:156–69. doi: 10.4251/wjgo.v4.i7.156
95. Wang J, Yu JC, Kang WM, Ma ZQ. Treatment Strategy for Early Gastric Cancer. Surg Oncol (2012) 21:119–23. doi: 10.1016/j.suronc.2010.12.004
96. Meyer HJ, Wilke H. [Gastric Cancer: Current Status of Multimodality Treatment]. Zentralbl Chir (2011) 136:317–24. doi: 10.1055/s-0031-1271580
97. Mackenzie M, Spithoff K, Jonker D. Systemic Therapy for Advanced Gastric Cancer: A Clinical Practice Guideline. Curr Oncol (2011) 18:e202–9. doi: 10.3747/co.v18i4.737
98. Zhao J, Zhi Z, Wang C, Xing H, Song G, Yu X, et al. Exogenous Lipids Promote the Growth of Breast Cancer Cells via CD36. Oncol Rep (2017) 38:2105–15. doi: 10.3892/or.2017.5864
99. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS, et al. Targeting Metastasis-Initiating Cells Through the Fatty Acid Receptor CD36. Nature (2017) 541:41–5. doi: 10.1038/nature20791
100. Nath A, Li I, Roberts LR, Chan C. Elevated Free Fatty Acid Uptake via CD36 Promotes Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma. Sci Rep (2015) 5:14752. doi: 10.1038/srep14752
101. Jacome-Sosa M, Miao ZF, Peche VS, Morris EF, Narendran R, Pietka KM, et al. CD36 Maintains the Gastric Mucosa and Associates With Gastric Disease. Commun Biol (2021) 4:1247. doi: 10.1038/s42003-021-02765-z
102. Ma X, Ren X, Han P, Hu S, Wang J, Yin J. SiRNA Against Fabp5 Induces 3T3-L1 Cells Apoptosis During Adipocytic Induction. Mol Biol Rep (2010) 37:4003–11. doi: 10.1007/s11033-010-0059-5
103. Scaglia N, Tyekucheva S, Zadra G, Photopoulos C, Loda M, et al. De Novo Fatty Acid Synthesis at the Mitotic Exit is Required to Complete Cellular Division. Cell Cycle (2014) 13:859–68. doi: 10.4161/cc.27767
104. Yarla NS, Bishayee A, Sethi G, Reddanna P, Kalle AM, Dhananjaya BL, et al. Targeting Arachidonic Acid Pathway by Natural Products for Cancer Prevention and Therapy. Semin Cancer Biol (2016), 40–41:48-81. doi: 10.1016/j.semcancer.2016.02.001
105. Levi L, Wang Z, Doud MK, Hazen SL, Noy N. Saturated Fatty Acids Regulate Retinoic Acid Signalling and Suppress Tumorigenesis by Targeting Fatty Acid-Binding Protein 5. Nat Commun (2015) 6:8794. doi: 10.1038/ncomms9794
106. Chen X, Hu SL, Feng Y, Li P, Mao QS, Xue WJ, et al. Expression of Fatty Acid-Binding Protein-3 in Gastrointestinal Stromal Tumors and Its Significance for Prognosis. J Surg Res (2021) 260:462–6. doi: 10.1016/j.jss.2020.11.003
107. Maxfield FR, Tabas I. Role of Cholesterol and Lipid Organization in Disease. Nature (2005) 438:612–21. doi: 10.1038/nature04399
108. Liu CX, Li Y, Obermoeller-McCormick LM, Schwartz AL, Bu G. The Putative Tumor Suppressor LRP1B, a Novel Member of the Low Density Lipoprotein (LDL) Receptor Family, Exhibits Both Overlapping and Distinct Properties With the LDL Receptor-Related Protein. J Biol Chem (2001) 276:28889–96. doi: 10.1074/jbc.M102727200
109. Yasufuku I, Saigo C, Kito Y, Yoshida K, Takeuchi T. Prognostic Significance of LDL Receptor-Related Protein 1B in Patients With Gastric Cancer. J Mol Histol (2021) 52:165–72. doi: 10.1007/s10735-020-09932-2
110. Watkins PA, Maiguel D, Jia Z, Pevsner J. Evidence for 26 Distinct Acyl-Coenzyme A Synthetase Genes in the Human Genome. J Lipid Res (2007) 48:2736–50. doi: 10.1194/jlr.M700378-JLR200
111. Xu H, Luo J, Ma G, Zhang X, Yao D, Li M, et al. Acyl-CoA Synthetase Short-Chain Family Member 2 (ACSS2) is Regulated by SREBP-1 and Plays a Role in Fatty Acid Synthesis in Caprine Mammary Epithelial Cells. J Cell Physiol (2018) 233:1005–16. doi: 10.1002/jcp.25954
112. Huang Z, Zhang M, Plec AA, Estill SJ, Cai L, Repa JJ, et al. ACSS2 Promotes Systemic Fat Storage and Utilization Through Selective Regulation of Genes Involved in Lipid Metabolism. Proc Natl Acad Sci U.S.A. (2018) 115:E9499–e506. doi: 10.1073/pnas.1806635115
113. Jha V, Galati S, Volpi V, Ciccone L, Minutolo F, Rizzolio F, et al. Discovery of a New ATP-Citrate Lyase (ACLY) Inhibitor Identified by a Pharmacophore-Based Virtual Screening Study. J Biomol Struct Dyn (2021) 39:3996–4004. doi: 10.1080/07391102.2020.1773314
114. Kim N, Park WY, Kim JM, Park JH, Kim JS, Jung HC, et al. Gene Expression of AGS Cells Stimulated With Released Proteins by Helicobacter Pylori. J Gastroenterol Hepatol (2008) 23:643–51. doi: 10.1111/j.1440-1746.2007.05241.x
115. Wang Y, Yu W, Li S, Guo D, He J, Wang Y. Acetyl-CoA Carboxylases and Diseases. Front Oncol (2022) 12:836058. doi: 10.3389/fonc.2022.836058
116. Kusakabe T, Nashimoto A, Honma K, Suzuki T. Fatty Acid Synthase is Highly Expressed in Carcinoma, Adenoma and in Regenerative Epithelium and Intestinal Metaplasia of the Stomach. Histopathology (2002) 40:71–9. doi: 10.1046/j.1365-2559.2002.01289.x
117. Zhou Y, Su W, Liu H, Chen T, Höti N, Pei H, et al. Fatty Acid Synthase Is a Prognostic Marker and Associated With Immune Infiltrating in Gastric Cancers Precision Medicine. biomark Med (2020) 14:185–99. doi: 10.2217/bmm-2019-0476
118. Yu L, Wang X, Du Y, Zhang X, Ling Y. FASN Knockdown Inhibited Anoikis Resistance of Gastric Cancer Cells via P-ERK1/2/Bcl-xL Pathway. Gastroenterol Res Pract (2021) 2021:6674204. doi: 10.1155/2021/6674204
119. Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, et al. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells From Ferroptotic Cell Death. Cancer Res (2019) 79:5355–66. doi: 10.1158/0008-5472.CAN-19-0369
120. Tracz-Gaszewska Z, Dobrzyn P. Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers (Basel) (2019) 11:948. doi: 10.3390/cancers11070948
121. Gao Y, Li J, Xi H, Cui J, Zhang K, Zhang J, et al. Stearoyl-CoA-Desaturase-1 Regulates Gastric Cancer Stem-Like Properties and Promotes Tumour Metastasis via Hippo/YAP Pathway. Br J Cancer (2020) 122:1837–47. doi: 10.1038/s41416-020-0827-5
122. Zhang H, Wang M, He Y, Deng T, Liu R, Wang W, et al. Chemotoxicity-Induced Exosomal lncFERO Regulates Ferroptosis and Stemness in Gastric Cancer Stem Cells. Cell Death Dis (2021) 12:1116. doi: 10.1038/s41419-021-04406-z
123. Robichaud PP, Munganyiki JE, Boilard E, Surette ME. Polyunsaturated Fatty Acid Elongation and Desaturation in Activated Human T-Cells: ELOVL5 is the Key Elongase. J Lipid Res (2018) 59:2383–96. doi: 10.1194/jlr.M090050
124. Lee JY, Nam M, Son HY, Hyun K, Jang SY, Kim JW, et al. Polyunsaturated Fatty Acid Biosynthesis Pathway Determines Ferroptosis Sensitivity in Gastric Cancer. Proc Natl Acad Sci U.S.A. (2020) 117:32433–42. doi: 10.1073/pnas.2006828117
125. Cheng C, Geng F, Cheng X, Guo D. Lipid Metabolism Reprogramming and its Potential Targets in Cancer. Cancer Commun (Lond) (2018) 38:27. doi: 10.1186/s40880-018-0301-4
126. Zhao L, Peng Y, He S, Li R, Wang Z, Huang J, et al. Apatinib Induced Ferroptosis by Lipid Peroxidation in Gastric Cancer. Gastric Cancer (2021) 24:642–54. doi: 10.1007/s10120-021-01159-8
127. Harris CA, Haas JT, Streeper RS, Stone SJ, Kumari M, Yang K, et al. DGAT Enzymes are Required for Triacylglycerol Synthesis and Lipid Droplets in Adipocytes. J Lipid Res (2011) 52:657–67. doi: 10.1194/jlr.M013003
128. Göbel A, Rauner M, Hofbauer LC, Rachner TD. Cholesterol and Beyond - The Role of the Mevalonate Pathway in Cancer Biology. Biochim Biophys Acta Rev Cancer (2020) 1873:188351. doi: 10.1016/j.bbcan.2020.188351
129. Sharpe LJ, Brown AJ. Controlling Cholesterol Synthesis Beyond 3-Hydroxy-3-Methylglutaryl-CoA Reductase (HMGCR). J Biol Chem (2013) 288:18707–15. doi: 10.1074/jbc.R113.479808
130. DeBose-Boyd RA. Feedback Regulation of Cholesterol Synthesis: Sterol-Accelerated Ubiquitination and Degradation of HMG CoA Reductase. Cell Res (2008) 18:609–21. doi: 10.1038/cr.2008.61
131. Foster DW. The Role of the Carnitine System in Human Metabolism. Ann N Y Acad Sci (2004) 1033:1–16. doi: 10.1196/annals.1320.001
132. van der Leij FR, Kram AM, Bartelds B, Roelofsen H, Smid GB, Takens J, et al. Cytological Evidence That the C-Terminus of Carnitine Palmitoyltransferase I is on the Cytosolic Face of the Mitochondrial Outer Membrane. Biochem J (1999) 341(Pt 3):777–84. doi: 10.1042/bj3410777
133. Wang J, Xiang H, Lu Y, Wu T, Ji G, et al. The Role and Therapeutic Implication of CPTs in Fatty Acid Oxidation and Cancers Progression. Am J Cancer Res (2021) 11:2477–94.
134. Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, et al. FAT SIGNALS–lipases and Lipolysis in Lipid Metabolism and Signaling. Cell Metab (2012) 15:279–91. doi: 10.1016/j.cmet.2011.12.018
135. Fenn JB, Mann M, Meng CK, Wong SF. Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science (1989) 246:64–71. doi: 10.1126/science.2675315
136. Karas M, Hillenkamp F. Laser Desorption Ionization of Proteins With Molecular Masses Exceeding 10,000 Daltons. Anal Chem (1988) 60:2299–301. doi: 10.1021/ac00171a028
137. Wang Y, Lu JH, Wang F, Wang YN, He MM, Wu QN, et al. Inhibition of Fatty Acid Catabolism Augments the Efficacy of Oxaliplatin-Based Chemotherapy in Gastrointestinal Cancers. Cancer Lett (2020) 473:74–89. doi: 10.1016/j.canlet.2019.12.036
138. He W, Liang B, Wang C, Li S, Zhao Y, Huang Q, et al. MSC-Regulated lncRNA MACC1-AS1 Promotes Stemness and Chemoresistance Through Fatty Acid Oxidation in Gastric Cancer. Oncogene (2019) 38:4637–54. doi: 10.1038/s41388-019-0747-0
139. Lee JS, Kim SH, Lee S, Kang JH, Lee SH, Cheong JH, et al. Gastric Cancer Depends on Aldehyde Dehydrogenase 3A1 for Fatty Acid Oxidation. Sci Rep (2019) 9:16313. doi: 10.1038/s41598-019-52814-1
140. Duan J, Chen L, Zhou M, Zhang J, Sun L, Huang N, et al. MACC1 Decreases the Chemosensitivity of Gastric Cancer Cells to Oxaliplatin by Regulating FASN Expression. Oncol Rep (2017) 37:2583–92. doi: 10.3892/or.2017.5519
141. Li CF, Fang FM, Chen YY, Liu TT, Chan TC, Yu SC, et al. Overexpressed Fatty Acid Synthase in Gastrointestinal Stromal Tumors: Targeting a Progression-Associated Metabolic Driver Enhances the Antitumor Effect of Imatinib. Clin Cancer Res (2017) 23:4908–18. doi: 10.1158/1078-0432.CCR-16-2770
142. Dowling S, Cox J, Cenedella RJ. Inhibition of Fatty Acid Synthase by Orlistat Accelerates Gastric Tumor Cell Apoptosis in Culture and Increases Survival Rates in Gastric Tumor Bearing Mice In Vivo. Lipids (2009) 44:489–98. doi: 10.1007/s11745-009-3298-2
143. Hitkova I, Yuan G, Anderl F, Gerhard M, Kirchner T, Reu S, et al. Caveolin-1 Protects B6129 Mice Against Helicobacter Pylori Gastritis. PloS Pathog (2013) 9:e1003251. doi: 10.1371/journal.ppat.1003251
144. Manu KA, Shanmugam MK, Li F, Chen L, Siveen KS, Ahn KS, et al. Simvastatin Sensitizes Human Gastric Cancer Xenograft in Nude Mice to Capecitabine by Suppressing Nuclear Factor-Kappa B-Regulated Gene Products. J Mol Med (Berl) (2014) 92:267–76. doi: 10.1007/s00109-013-1095-0
145. Yang JC, Chang N, Wu DC, Cheng WC, Chung WM, Chang WC, et al. Preclinical Evaluation of Exemestane as a Novel Chemotherapy for Gastric Cancer. J Cell Mol Med (2019) 23:7417–26. doi: 10.1111/jcmm.14605
146. Granchi C. ATP Citrate Lyase (ACLY) Inhibitors: An Anti-Cancer Strategy at the Crossroads of Glucose and Lipid Metabolism. Eur J Med Chem (2018) 157:1276–91. doi: 10.1016/j.ejmech.2018.09.001
147. Chen P, Li L, Wang H, Zhao J, Cheng Y, Xie J, et al. Omeprazole, an Inhibitor of Proton Pump, Suppresses De Novo Lipogenesis in Gastric Epithelial Cells. BioMed Pharmacother (2020) 130:110472. doi: 10.1016/j.biopha.2020.110472
148. McEwen DR. PDR Guide to Drug Interactions, Side Effects, Indications. AORN J (1994) 59:731–. doi: 10.1016/S0001-2092(07)69995-5
149. Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. Orlistat is a Novel Inhibitor of Fatty Acid Synthase With Antitumor Activity. Cancer Res (2004) 64:2070–5. doi: 10.1158/0008-5472.CAN-03-3645
150. Piao C, Cui X, Zhan B, Li J, Li Z, Li Z, et al. Inhibition of Stearoyl CoA Desaturase-1 Activity Suppresses Tumour Progression and Improves Prognosis in Human Bladder Cancer. J Cell Mol Med (2019) 23:2064–76. doi: 10.1111/jcmm.14114
151. von Roemeling CA, Marlow LA, Wei JJ, Cooper SJ, Caulfield TR, Wu K, et al. Stearoyl-CoA Desaturase 1 Is a Novel Molecular Therapeutic Target for Clear Cell Renal Cell Carcinoma. Clin Cancer Res (2013) 19:2368–80. doi: 10.1158/1078-0432.CCR-12-3249
152. Kim ST, Kang JH, Lee J, Park SH, Park JO, Park YS, et al. Simvastatin Plus Capecitabine-Cisplatin Versus Placebo Plus Capecitabine-Cisplatin in Patients With Previously Untreated Advanced Gastric Cancer: A Double-Blind Randomised Phase 3 Study. Eur J Cancer (2014) 50:2822–30. doi: 10.1016/j.ejca.2014.08.005
153. Follet J, Corcos L, Baffet G, Ezan F, Morel F, Simon B, et al. The Association of Statins and Taxanes: An Efficient Combination Trigger of Cancer Cell Apoptosis. Br J Cancer (2012) 106:685–92. doi: 10.1038/bjc.2012.6
154. Jiang Y, Sun A, Zhao Y, Ying W, Sun H, Yang X, et al. Proteomics Identifies New Therapeutic Targets of Early-Stage Hepatocellular Carcinoma. Nature (2019) 567:257–61. doi: 10.1038/s41586-019-0987-8
155. Murray M, Hraiki A, Bebawy M, Pazderka C, Rawling T, et al. Anti-Tumor Activities of Lipids and Lipid Analogues and Their Development as Potential Anticancer Drugs. Pharmacol Ther (2015) 150:109–28. doi: 10.1016/j.pharmthera.2015.01.008
156. Zhang L, Zhu B, Zeng Y, Shen H, Zhang J, Wang X, et al. Clinical Lipidomics in Understanding of Lung Cancer: Opportunity and Challenge. Cancer Lett (2020) 470:75–83. doi: 10.1016/j.canlet.2019.08.014
157. Corn KC, Windham MA, Rafat M. Lipids in the Tumor Microenvironment: From Cancer Progression to Treatment. Prog Lipid Res (2020) 80:101055. doi: 10.1016/j.plipres.2020.101055
158. Yuan XL, Chen L, Li MX, Dong P, Xue J, Wang J, et al. Elevated Expression of Foxp3 in Tumor-Infiltrating Treg Cells Suppresses T-Cell Proliferation and Contributes to Gastric Cancer Progression in a COX-2-Dependent Manner. Clin Immunol (2010) 134:277–88. doi: 10.1016/j.clim.2009.10.005
159. Lin R, Zhang H, Yuan Y, He Q, Zhou J, Li S, et al. Fatty Acid Oxidation Controls CD8(+) Tissue-Resident Memory T-Cell Survival in Gastric Adenocarcinoma. Cancer Immunol Res (2020) 8:479–92. doi: 10.1158/2326-6066.CIR-19-0702
160. Gabrilovich D. Mechanisms and Functional Significance of Tumour-Induced Dendritic-Cell Defects. Nat Rev Immunol (2004) 4:941–52. doi: 10.1038/nri1498
161. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid Accumulation and Dendritic Cell Dysfunction in Cancer. Nat Med (2010) 16:880–6. doi: 10.1038/nm.2172
162. Luo Q, Zheng N, Jiang L, Wang T, Zhang P, Liu Y, et al. Lipid Accumulation in Macrophages Confers Protumorigenic Polarization and Immunity in Gastric Cancer. Cancer Sci (2020) 111:4000–11. doi: 10.1111/cas.14616
163. Catalano V, Bisonni R, Graziano F, Giordani P, Alessandroni P, Baldelli AM, et al. A Phase II Study of Modified FOLFOX as First-Line Chemotherapy for Metastatic Gastric Cancer in Elderly Patients With Associated Diseases. Gastric Cancer (2013) 16:411–9. doi: 10.1007/s10120-012-0204-z
164. Mao XM, Fu QR, Li HL, Zheng YH, Chen SM, Hu XY, et al. Crocodile Choline From Crocodylus Siamensis Induces Apoptosis of Human Gastric Cancer. Tumour Biol (2017) 39:1010428317694320. doi: 10.1177/1010428317694320
165. Cho JH, Hong SK, Kim EY, Park SY, Park CH, Kim JM, et al. Overexpression of Phospholipase D Suppresses Taxotere-Induced Cell Death in Stomach Cancer Cells. Biochim Biophys Acta (2008) 1783:912–23. doi: 10.1016/j.bbamcr.2007.11.019
166. Lin Y, Zhai E, Liao B, Xu L, Zhang X, Peng S, et al. Autocrine VEGF Signaling Promotes Cell Proliferation Through a PLC-Dependent Pathway and Modulates Apatinib Treatment Efficacy in Gastric Cancer. Oncotarget (2017) 8:11990–2002. doi: 10.18632/oncotarget.14467
167. Uehara T, Kikuchi H, Miyazaki S, Iino I, Setoguchi T, Hiramatsu Y, et al. Overexpression of Lysophosphatidylcholine Acyltransferase 1 and Concomitant Lipid Alterations in Gastric Cancer. Ann Surg Oncol (2016) 23 Suppl 2:S206–13. doi: 10.1245/s10434-015-4459-6
168. Liao Y, Chen W, Shi W, Zha H. Targeting Cpla2α Inhibits Gastric Cancer and Augments Chemotherapy Efficacy via Suppressing Ras/MEK/ERK and Akt/β-Catenin Pathways. Cancer Chemother Pharmacol (2021) 88:689–97. doi: 10.1007/s00280-021-04322-1
169. Yin HM, He Q, Chen J, Li Z, Yang W, Hu X, et al. Drug Metabolism-Related Eight-Gene Signature can Predict the Prognosis of Gastric Adenocarcinoma. J Clin Lab Anal (2021) 35:e24085. doi: 10.1002/jcla.24085
170. Gotovdorj T, Lee E, Lim Y, Cha EJ, Kwon D, Hong E, et al. 2,3,7,8-Tetrachlorodibenzo-P-Dioxin Induced Cell-Specific Drug Transporters With Acquired Cisplatin Resistance in Cisplatin Sensitive Cancer Cells. J Korean Med Sci (2014) 29:1188–98. doi: 10.3346/jkms.2014.29.9.1188
171. Wu Q, Yang Z, Xia L, Nie Y, Wu K, Shi Y, et al. Methylation of miR-129-5p CpG Island Modulates Multi-Drug Resistance in Gastric Cancer by Targeting ABC Transporters. Oncotarget (2014) 5:11552–63. doi: 10.18632/oncotarget.2594
172. Hu S, Zhao X, Qian F, Jin C, Hou K. Correlation Between LRP1B Mutations and Tumor Mutation Burden in Gastric Cancer. Comput Math Methods Med (2021) 2021:1522250. doi: 10.1155/2021/1522250
173. Thorsen K, Schepeler T, Øster B, Rasmussen MH, Vang S, Wang K, et al. Tumor-Specific Usage of Alternative Transcription Start Sites in Colorectal Cancer Identified by Genome-Wide Exon Array Analysis. BMC Genomics (2011) 12:505. doi: 10.1186/1471-2164-12-505
174. Hu Q, Masuda T, Koike K, Sato K, Tobo T, Kuramitsu S, et al. Oxysterol Binding Protein-Like 3 (OSBPL3) is a Novel Driver Gene That Promotes Tumor Growth in Part Through R-Ras/Akt Signaling in Gastric Cancer. Sci Rep (2021) 11:19178. doi: 10.1038/s41598-021-98485-9
175. Xu B, Li S, Fang Y, Zou Y, Song D, Zhang S, et al. Proprotein Convertase Subtilisin/Kexin Type 9 Promotes Gastric Cancer Metastasis and Suppresses Apoptosis by Facilitating MAPK Signaling Pathway Through HSP70 Up-Regulation. Front Oncol (2020) 10:609663. doi: 10.3389/fonc.2020.609663
176. Remes SM, Leijon H, Vesterinen T, Louhimo J, Pulkkinen V, Ezer S, et al. PCSK2 Expression in Neuroendocrine Tumors Points to a Midgut, Pulmonary, or Pheochromocytoma-Paraganglioma Origin. Apmis (2020) 128:563–72. doi: 10.1111/apm.13071
177. Kim J, Yum S, Kang C, Kang SJ. Gene-Gene Interactions in Gastrointestinal Cancer Susceptibility. Oncotarget (2016) 7:67612–25. doi: 10.18632/oncotarget.11701
178. Li C, Zhang J, Wu H, Li L, Yang C, Song S, et al. Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1 Facilitates Metastasis of Gastric Cancer Through Driving Epithelial-Mesenchymal Transition and PI3K/Akt/Gsk3β Activation. Sci Rep (2017) 7:45275. doi: 10.1038/srep45275
179. Li L, Peng Z, Hu Q, Xu L, Zou X, Yu Y, et al. Berberine Suppressed Tumor Growth Through Regulating Fatty Acid Metabolism and Triggering Cell Apoptosis via Targeting FABPs. Evid Based Complement Alternat Med (2020) 2020:6195050. doi: 10.1155/2020/6195050
180. Zang WJ, Wang ZN, Hu YL, Huang H, Ma P. Expression of Fatty Acid-Binding Protein-4 in Gastrointestinal Stromal Tumors and its Significance for Prognosis. J Clin Lab Anal (2021) 35:e24017. doi: 10.1002/jcla.24017
181. Chang WC, Cheng WC, Cheng BH, Chen L, Ju LJ, Ou YJ, et al. Mitochondrial Acetyl-CoA Synthetase 3 Is Biosignature of Gastric Cancer Progression. Cancer Med (2018) 7:1240–52. doi: 10.1002/cam4.1295
182. Ezzeddini R, Taghikhani M, Somi MH, Samadi N, Rasaee MJ. Clinical Importance of FASN in Relation to HIF-1α and SREBP-1c in Gastric Adenocarcinoma. Life Sci (2019) 224:169–76. doi: 10.1016/j.lfs.2019.03.056
183. Bacci M, Lorito N, Smiriglia A, Morandi A. Fat and Furious: Lipid Metabolism in Antitumoral Therapy Response and Resistance. Trends Cancer (2021) 7:198–213. doi: 10.1016/j.trecan.2020.10.004
184. Bauerschlag DO, Maass N, Leonhardt P, Verburg FA, Pecks U, Zeppernick F, et al. Fatty Acid Synthase Overexpression: Target for Therapy and Reversal of Chemoresistance in Ovarian Cancer. J Transl Med (2015) 13:146. doi: 10.1186/s12967-015-0511-3
185. Wu X, Dong Z, Wang CJ, Barlow LJ, Fako V, Serrano MA, et al. FASN Regulates Cellular Response to Genotoxic Treatments by Increasing PARP-1 Expression and DNA Repair Activity via NF-κb and SP1. Proc Natl Acad Sci U.S.A. (2016) 113:E6965–e73. doi: 10.1073/pnas.1609934113
186. Kant S, Kumar A, Singh SM. Tumor Growth Retardation and Chemosensitizing Action of Fatty Acid Synthase Inhibitor Orlistat on T Cell Lymphoma: Implication of Reconstituted Tumor Microenvironment and Multidrug Resistance Phenotype. Biochim Biophys Acta (2014) 1840:294–302. doi: 10.1016/j.bbagen.2013.09.020
187. Papaevangelou E, Almeida GS, Box C, deSouza NM, Chung YL. The Effect of FASN Inhibition on the Growth and Metabolism of a Cisplatin-Resistant Ovarian Carcinoma Model. Int J Cancer (2018) 143:992–1002. doi: 10.1002/ijc.31392
188. Wu Y, Si R, Tang H, He Z, Zhu H, Wang L, et al. Cholesterol Reduces the Sensitivity to Platinum-Based Chemotherapy via Upregulating ABCG2 in Lung Adenocarcinoma. Biochem Biophys Res Commun (2015) 457:614–20. doi: 10.1016/j.bbrc.2015.01.035
189. Choi SM, Cho YS, Park G, Lee SK, Chun KS. Celecoxib Induces Apoptosis Through Akt Inhibition in 5-Fluorouracil-Resistant Gastric Cancer Cells. Toxicol Res (2021) 37:25–33. doi: 10.1007/s43188-020-00044-3
190. Qiu ZQ, Qiu ZR. Sensitivity of Gastric Cancer Cells to Chemotherapy Drugs in Elderly Patients and its Correlation With Cyclooxygenase-2 Expression. Asian Pac J Cancer Prev (2015) 16:3447–50. doi: 10.7314/APJCP.2015.16.8.3447
191. Yu F, Li K, Chen S, Liu Y, Li Y. Pseudolaric Acid B Circumvents Multidrug Resistance Phenotype in Human Gastric Cancer SGC7901/ADR Cells by Downregulating Cox-2 and P-Gp Expression. Cell Biochem Biophys (2015) 71:119–26. doi: 10.1007/s12013-014-0170-7
192. Kuefner MS, Stephenson E, Savikj M, Smallwood HS, Dong Q, Payré C, et al. Group IIA Secreted Phospholipase A2 (PLA2G2A) Augments Adipose Tissue Thermogenesis. FASEB J (2021) 35:e21881. doi: 10.1096/fj.202002481RR
193. Chen D, Jiao XL, Liu ZK, Zhang MS, Niu M. Knockdown of PLA2G2A Sensitizes Gastric Cancer Cells to 5-FU In Vitro. Eur Rev Med Pharmacol Sci (2013) 17:1703–8.
194. Houthuijzen JM, Daenen LG, Roodhart JM, Voest EE. The Role of Mesenchymal Stem Cells in Anti-Cancer Drug Resistance and Tumour Progression. Br J Cancer (2012) 106:1901–6. doi: 10.1038/bjc.2012.201
195. Meads MB, Hazlehurst LA, Dalton WS. The Bone Marrow Microenvironment as a Tumor Sanctuary and Contributor to Drug Resistance. Clin Cancer Res (2008) 14:2519–26. doi: 10.1158/1078-0432.CCR-07-2223
196. Wu H, Liu B, Chen Z, Li G, Zhang Z, et al. MSC-Induced lncRNA HCP5 Drove Fatty Acid Oxidation Through miR-3619-5p/AMPK/Pgc1α/CEBPB Axis to Promote Stemness and Chemo-Resistance of Gastric Cancer. Cell Death Dis (2020) 11:233. doi: 10.1038/s41419-020-2426-z
197. Li F, Simon MC. Cancer Cells Don't Live Alone: Metabolic Communication Within Tumor Microenvironments. Dev Cell (2020) 54:183–95. doi: 10.1016/j.devcel.2020.06.018
198. Kopecka J, Trouillas P, Gašparović A, Gazzano E, Assaraf YG, Riganti C. Phospholipids and Cholesterol: Inducers of Cancer Multidrug Resistance and Therapeutic Targets. Drug Resist Update (2020) 49:100670. doi: 10.1016/j.drup.2019.100670
199. Yamaguchi H, Sakai R. Direct Interaction Between Carcinoma Cells and Cancer Associated Fibroblasts for the Regulation of Cancer Invasion. Cancers (Basel) (2015) 7:2054–62. doi: 10.3390/cancers7040876
200. Brocker C, Carpenter C, Nebert DW, Vasiliou V. Evolutionary Divergence and Functions of the Human Acyl-CoA Thioesterase Gene ( ACOT ) Family. Hum Genomics (2010) 4:411–20. doi: 10.1186/1479-7364-4-6-411
201. Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. CAF Secreted miR-522 Suppresses Ferroptosis and Promotes Acquired Chemo-Resistance in Gastric Cancer. Mol Cancer (2020) 19:43. doi: 10.1186/s12943-020-01168-8
202. Zhang Y, Cong X, Li Z, Xue Y. Estrogen Facilitates Gastric Cancer Cell Proliferation and Invasion Through Promoting the Secretion of Interleukin-6 by Cancer-Associated Fibroblasts. Int Immunopharmacol (2020) 78:105937. doi: 10.1016/j.intimp.2019.105937
203. Ham IH, Oh HJ, Jin H, Bae CA, Jeon SM, Choi KS, et al. Targeting Interleukin-6 as a Strategy to Overcome Stroma-Induced Resistance to Chemotherapy in Gastric Cancer. Mol Cancer (2019) 18:68. doi: 10.1186/s12943-019-0972-8
204. Xu L, Hu X, Qu X, Hou K, Zheng H, Liu Y, et al. Cetuximab Enhances TRAIL-Induced Gastric Cancer Cell Apoptosis by Promoting DISC Formation in Lipid Rafts. Biochem Biophys Res Commun (2013) 439:285–90. doi: 10.1016/j.bbrc.2013.08.040
205. Zhang X, Zhang Y, Li Y. β-Elemene Decreases Cell Invasion by Upregulating E-Cadherin Expression in MCF-7 Human Breast Cancer Cells. Oncol Rep (2013) 30:745–50. doi: 10.3892/or.2013.2519
206. Xu L, Qu X, Zhang Y, Hu X, Yang X, Hou K, et al. Oxaliplatin Enhances TRAIL-Induced Apoptosis in Gastric Cancer Cells by CBL-Regulated Death Receptor Redistribution in Lipid Rafts. FEBS Lett (2009) 583:943–8. doi: 10.1016/j.febslet.2009.02.014
207. Xu L, Qu X, Luo Y, Zhang Y, Liu J, Qu J, et al. Epirubicin Enhances TRAIL-Induced Apoptosis in Gastric Cancer Cells by Promoting Death Receptor Clustering in Lipid Rafts. Mol Med Rep (2011) 4:407–11. doi: 10.3892/mmr.2011.439
208. Hu JK, Yang K, Li CM, Zhang B, Chen ZX, Chen XZ, et al. The Expression of TRAIL and its Receptors in Gastric Cancer and the Apoptotic Effect of Rh-TRAIL on SGC7901 Cells. Oncol Rep (2009) 21:681–8. doi: 10.3892/or_00000271
209. Lim SC, Parajuli KR, Han SI. Keratin 6, Induced by Chronic Cisplatin Exposure, Confers Chemoresistance in Human Gastric Carcinoma Cells. Oncol Rep (2019) 42:797–804. doi: 10.3892/or.2019.7201
210. Russi S, Verma HK, Laurino S, Mazzone P, Storto G, Nardelli A, et al. Adapting and Surviving: Intra and Extra-Cellular Remodeling in Drug-Resistant Gastric Cancer Cells. Int J Mol Sci (2019) 20:3736. doi: 10.3390/ijms20153736
211. Xu JL, Yuan L, Tang YC, Xu ZY, Xu HD, Cheng XD, et al. The Role of Autophagy in Gastric Cancer Chemoresistance: Friend or Foe? Front Cell Dev Biol (2020) 8:621428. doi: 10.3389/fcell.2020.621428
212. Mrena J, Wiksten JP, Kokkola A, Nordling S, Ristimäki A, Haglund C. COX-2 Is Associated With Proliferation and Apoptosis Markers and Serves as an Independent Prognostic Factor in Gastric Cancer. Tumour Biol (2010) 31:1–7. doi: 10.1007/s13277-009-0001-4
213. Lim HY, Joo HJ, Choi JH, Yi JW, Yang MS, Cho DY, et al. Increased Expression of Cyclooxygenase-2 Protein in Human Gastric Carcinoma. Clin Cancer Res (2000) 6:519–25.
214. Uefuji K, Ichikura T, Mochizuki H. Cyclooxygenase-2 Expression is Related to Prostaglandin Biosynthesis and Angiogenesis in Human Gastric Cancer. Clin Cancer Res (2000) 6:135–8.
215. Murata H, Kawano S, Tsuji S, Tsuji M, Sawaoka H, Kimura Y, et al. Cyclooxygenase-2 Overexpression Enhances Lymphatic Invasion and Metastasis in Human Gastric Carcinoma. Am J Gastroenterol (1999) 94:451–5. doi: 10.1111/j.1572-0241.1999.876_e.x
216. Zhong B, Li Y, Liu X, Wang D. Association of Mast Cell Infiltration With Gastric Cancer Progression. Oncol Lett (2018) 15:755–64. doi: 10.3892/ol.2017.7380
217. Hao Q, Vadgama JV, Wang P. CCL2/CCR2 Signaling in Cancer Pathogenesis. Cell Commun Signal (2020) 18:82. doi: 10.1186/s12964-020-00589-8
218. Xu W, Wei Q, Han M, Zhou B, Wang H, Zhang J, et al. CCL2-SQSTM1 Positive Feedback Loop Suppresses Autophagy to Promote Chemoresistance in Gastric Cancer. Int J Biol Sci (2018) 14:1054–66. doi: 10.7150/ijbs.25349
219. Wong CC, Kang W, Xu J, Qian Y, Luk STY, Chen H, et al. Prostaglandin E(2) Induces DNA Hypermethylation in Gastric Cancer In Vitro and In Vivo. Theranostics (2019) 9:6256–68. doi: 10.7150/thno.35766
220. Lei Y, Tang L, Hu J, Wang S, Liu Y, Yang M, et al. Inhibition of MGMT-Mediated Autophagy Suppression Decreases Cisplatin Chemosensitivity in Gastric Cancer. BioMed Pharmacother (2020) 125:109896. doi: 10.1016/j.biopha.2020.109896
221. López-Contreras F, Muñoz-Uribe M, Pérez-Laines J, Ascencio-Leal L, Rivera-Dictter A, Martin-Martin A, et al. Searching for Drug Synergy Against Cancer Through Polyamine Metabolism Impairment: Insight Into the Metabolic Effect of Indomethacin on Lung Cancer Cells. Front Pharmacol (2019) 10:1670. doi: 10.3389/fphar.2019.01670
222. Seetha A, Devaraj H, Sudhandiran G. Indomethacin and Juglone Inhibit Inflammatory Molecules to Induce Apoptosis in Colon Cancer Cells. J Biochem Mol Toxicol (2020) 34:e22433. doi: 10.1002/jbt.22433
223. Afroz S, Yagi A, Fujikawa K, Rahman MM, Morito K, Fukuta T, et al. Lysophosphatidic Acid in Medicinal Herbs Enhances Prostaglandin E(2) and Protects Against Indomethacin-Induced Gastric Cell Damage In Vivo and In Vitro. Prostaglandins Other Lipid Mediat (2018) 135:36–44. doi: 10.1016/j.prostaglandins.2018.01.003
224. Sunami Y, Rebelo A, Kleeff J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers (Basel) (2017) 10:3. doi: 10.3390/cancers10010003
225. Butler LM, Perone Y, Dehairs J, Lupien LE, de Laat V, Talebi A, et al. Lipids and Cancer: Emerging Roles in Pathogenesis, Diagnosis and Therapeutic Intervention. Adv Drug Delivery Rev (2020) 159:245–93. doi: 10.1016/j.addr.2020.07.013
226. Shyu P Jr., Wong XFA, Crasta K, Thibault G. Dropping in on Lipid Droplets: Insights Into Cellular Stress and Cancer. Biosci Rep (2018) 38:BSR20180764. doi: 10.1042/BSR20180764
227. Rak S, De Zan T, Stefulj J, Kosović M, Gamulin O, Osmak M. FTIR Spectroscopy Reveals Lipid Droplets in Drug Resistant Laryngeal Carcinoma Cells Through Detection of Increased Ester Vibrational Bands Intensity. Analyst (2014) 139:3407–15. doi: 10.1039/C4AN00412D
228. Dubey R, Stivala CE, Nguyen HQ, Goo YH, Paul A, Carette JE, et al. Lipid Droplets can Promote Drug Accumulation and Activation. Nat Chem Biol (2020) 16:206–13. doi: 10.1038/s41589-019-0447-7
229. Cotte AK, Aires V, Fredon M, Limagne E, Derangère V, Thibaudin M, et al. Lysophosphatidylcholine Acyltransferase 2-Mediated Lipid Droplet Production Supports Colorectal Cancer Chemoresistance. Nat Commun (2018) 9:322. doi: 10.1038/s41467-017-02732-5
230. Choi HJ, Jhe YL, Kim J, Lim JY, Lee JE, Shin MK, et al. FoxM1-Dependent and Fatty Acid Oxidation-Mediated ROS Modulation is a Cell-Intrinsic Drug Resistance Mechanism in Cancer Stem-Like Cells. Redox Biol (2020) 36:101589. doi: 10.1016/j.redox.2020.101589
231. Shin JI, Jeon YJ, Lee S, Lee YG, Kim JB, Lee K, et al. G-Protein-Coupled Receptor 120 Mediates DHA-Induced Apoptosis by Regulating IP3R, ROS and, ER Stress Levels in Cisplatin-Resistant Cancer Cells. Mol Cells (2019) 42:252–61.
Keywords: gastric cancer, lipid metabolism, biomarkers, treatment, chemoresistance
Citation: Cui M-Y, Yi X, Zhu D-X and Wu J (2022) The Role of Lipid Metabolism in Gastric Cancer. Front. Oncol. 12:916661. doi: 10.3389/fonc.2022.916661
Received: 09 April 2022; Accepted: 23 May 2022;
Published: 15 June 2022.
Edited by:
Wenkai Ni, Fudan University, ChinaReviewed by:
Renfang Mao, Nantong University, ChinaZeYu Huang, Sichuan University, China
Wenjie Zheng, Affiliated Hospital of Nantong University, China
Copyright © 2022 Cui, Yi, Zhu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Wu, d3VqdW4xMzc4QDE2My5jb20=; Dan-Xia Zhu, emh1ZGFueGlhNjZAMTYzLmNvbQ==
†These authors have contributed equally to this work