
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 27 September 2022
Sec. Genitourinary Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.911466
This article is part of the Research TopicMechanism of Prostate Cancer Initiation, Progression and Drug ResistanceView all 9 articles
 Changze Song1,2
Changze Song1,2 Jianong Zhang2Xiao Liu3,4
Jianong Zhang2Xiao Liu3,4 Meilu Li5
Meilu Li5 Dejie Wang2Zhijian Kang3,4Jiaao Yu3,4Jiuwei Chen3,4Hongxin Pan3,4,6Honglei Wang3,4Guangbin Li3,4
Dejie Wang2Zhijian Kang3,4Jiaao Yu3,4Jiuwei Chen3,4Hongxin Pan3,4,6Honglei Wang3,4Guangbin Li3,4 Haojie Huang2,7,8*
Haojie Huang2,7,8*Rationale: Fructose-1,6-bisphosphatase (FBP1) is a tumor suppressor and a key enzyme negatively regulating Warburg effect in cancer. However, regulation of FBP1 protein expression and its exact role in prostate cancer (PCa) is largely unclear. Phosphatase and tensin homolog (PTEN) is one of the most frequently deleted tumor suppressor genes in human PCa. However, the role of PTEN loss in aberrant Warburg effect in cancer remains poorly understood.
Methods: Expression of PTEN and FBP1 was analyzed in several PCa cell lines and prostate tumor tissues in mice. Western blot (WB) and RT-PCR approaches were used to examine how PTEN regulates FBP1 expression. Co-immunoprecipitation (co-IP) and in vivo ubiquitination assays were used to define the regulatory mechanisms. A PCa xenograft model was employed to determine the impact of PTEN regulation of FBP1 on PCa growth in vivo.
Result: We demonstrated that in a manner dependent of PI3K/AKT signal pathway PTEN regulated FBP1 expression in various PCa cell lines and tumors in mice. We confirmed that this regulation took place at the protein level and was mediated by SKP2 E3 ubiquitin ligase. Mechanistically, we showed that serine 271 phosphorylation of FBP1 by cyclin-dependent kinases (CDKs) was essential for SKP2-mediated degradation of FBP1 protein induced by PTEN loss. Most importantly, we further showed that loss of PTEN expression enhanced Warburg effect and PCa growth in mice in a manner dependent, at least partially on FBP1 protein degradation.
Conclusions: Our results reveal a novel tumor-suppressive feature of PTEN in restraining FBP1 degradation and the Warburg effect. These results also suggest that prohibiting FBP1 protein degradation could be a viable therapeutic strategy for PTEN-deficient PCa.
Non-malignant cells primarily rely on mitochondrial oxidative phosphorylation to produce the energy needed in the processes of life. In contrast, one of the common characteristics of tumor cells is that they metabolize glucose into lactic acid even if there is sufficient oxygen, which is called aerobic glycolysis or “Warburg effect” (1). Indeed, mounting evidence suggests that this effect is closely associated with tumor carcinogenesis and progression (2). Furthermore, current research has suggested that this metabolic change can be accomplished by a variety of mechanisms (3), including the abnormal regulation of related enzymes in glucose metabolism (4).
Gluconeogenesis plays important roles in glucose homeostasis in normal cells. It is also essential in regulating aerobic glycolysis in cancerous cells (5, 6). Fructose-1,6-bisphosphatase (FBP1) is the rate-limiting enzyme in gluconeogenesis (7) and plays a key function in Warburg effect (4, 8–10). The loss of FBP1 may be an essential tumorigenic event that promotes the development of basal-like breast cancer cells in epithelial mesenchymal transformation (4). In gastric cancer, colon cancer and hepatocellular carcinoma, the expression of FBP1 is downregulated (8, 9, 11), and its deletion is related to the poor prognosis of clear cell renal cell carcinoma and hepatocellular carcinoma (10, 11). These studies have shown that FBP1 exerts a necessary function in regulating tumor glucose metabolism and cancer progression.
Prostate cancer (PCa) is the most commonly diagnosed malignancy and the second leading causes of death in American men (12). Despite the high morbidity and mortality of PCa, the understanding of molecular mechanisms related to the occurrence and evolution of this malignant disease is incomplete.
PTEN functions as a lipid phosphatase to dephosphorylate phosphatidylinositol (3–5) trisphosphate (PtdIns (3–5) P3 or PIP3) (13). It is a multi-functional tumor suppressor and is often lost in human cancer, including PCa (14–17). Elevated activation of the signaling networks through the phosphoinositide 3-kinase (PI3K) group of lipid kinases by loss of PTEN is a characteristic of most cancers such as PCa (18–21). Additionally, PTEN, as the main negative regulator of the PI3K/AKT pathway, is a key regulator controlling lipid and glucose metabolism and mitochondrial functions in cell (22). Previous studies have shown that the serine/threonine kinase AKT, as a downstream target of PI3K/PTEN, can phosphorylate a variety of metabolic related proteins, such as AS160, GSK3, FOXO and PGC-1a (23–26).
In this study, we found that PTEN inhibited the degradation of FBP1 protein, which was mediated via phosphorylation-dependent ubiquitination. Our results also suggest that the deregulation of FBP1 may be the key mechanism of tumor progression driven by Warburg effect in PTEN-deficient PCa.
22Rv1, DU145 and 293T cell lines were purchased from ATCC (Manassas, VA). PTEN WT and -null MEFs were originally generated from Pten knockout mice and kindly provided by Dr. Zhenbang Chen (Meharry Medical College, Nashville, TN). 22Rv1 and DU145 cells were cultured in RPMI 1640 medium (Corning Cellgro) with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific). 293T cells and MEFs were cultured in Dulbecco’s modified Eagle’s medium (Corning Cellgro) supplied with 10% FBS. All cell lines were routinely cultured in 37°C, 5% CO2 incubator.
The expression plasmids for Flag-FBP1, HA-FBP1, HA-Ub and HA-SKP2 had been generated in our lab. The Flag-FBP1 mutant S271A was generated by using the KOD-Plus Mutagenesis Kit (Toyobo). The antibodies used in this study include: FBP1 (Abcam); PTEN, AKT and pS473 AKT (Cell Signaling Technology); SKP2, ERK2 and p27Kip1 (Santa Cruz Biotechnology); Ser/Thr-p (BD Scientific); HA tag (Covance); β-Tubulin and Flag tag (Sigma). The chemicals used include: LY294002 (Invitrogen), MG132 (Millipore), CHX (Sigma) and Roscovitine (MedChemExpress).
The cells were harvested and lysed in RIPA buffer for more than 15 min on ice, and the lysate was centrifuged at 15,600 xg at 4°C for 10 min. After the supernatant was quantified using the BCA protein quantitative kit (Thermo Fisher Scientific), 4x DTT containing loading buffer (Thermo Fisher Scientific) was added to protein samples and the mixed samples were heated in 100°C for 5 min. The samples were separated on SDS-PAGE and transferred to nitrocellulose membrane (Thermo Fisher Scientific). The membrane was pre-blocked with 5% skim milk for 1 h at room temperature and incubated with primary antibody at 4°C overnight. Membranes were washed in 1x TBST for 3 times, 5 min each and then incubated with horseradish peroxidase-conjugated secondary antibody at room temperature for 1 h. Blots were finally visualized using SuperSignal West Pico Stable Peroxide Solution (Thermo Fisher Scientific).
Total RNA was isolated from cells by directly adding Trizol reagent (Thermo Fisher Scientific) into cultured cells. The first strand cDNA was synthesized from 1 µg of total RNA with GoScript kit (Promega). Real-time polymerase chain reaction (PCR) was carried by using SYBR green mix (Bio-Rad), C1000 Touch Thermal Cycler and CFX96 real-time system (Bio-Rad). All PCR signals were normalized to the internal control GAPDH cDNA, and the fold change was calculated using the 2-△Ct method. The DNA sequence information of primers used for RT-qPCR is provided in Table S1.
Co-IP was performed using the method reported previously (27). Cells transfected with expression plasmids were harvested and cells were lysed in IP buffer (Sigma-Aldrich, St. Louis, MO) on ice for 15 min or longer, and the cell lysate was centrifuged at 15,600 xg for 10 min at 4°C. The supernatant was transferred to a fresh tube. The supernatant was then incubated with protein A/G agarose beads (Thermo Fisher Scientific) and primary antibodies at 4°C overnight. The IP beads were washed extensively (six times) on ice with IP buffer, and then re-suspended in 1 × SDS loading buffer followed by SDS-PAGE and Western blot analysis.
293T cells were transfected with plasmids for HA–ubiquitin and related genes. After 36 h of transfection, the cells were treated with 30 μM of MG132 for 6 h, and then lysed in 1% SDS buffer and boiled for 10 min. The cell lysate was incubated with anti-FLAG M2 agarose beads (Sigma) at 4°C for 4 h. The beads was washed for 4 times using BC100 buffer containing 0.2% of Triton X-100. The pulldown proteins were eluted with 3X FLAG polypeptide at 4°C. Ubiquitinated proteins were analyzed by SDS-PAGE and Western blot.
The procedure for colony formation assay was carried out as previously described (28, 29). In short, 1×103 cells were plated into each well in 6-well plates. The next day, the cells were treated with vehicle or Roscovitine (10 μM, Cat# HY-30237, MedChemExpress). After about 16 days of treatment, the cells were fixed with 4% paraformaldehyde for 15 min, stained with (0.5% w/v) crystal violet for 1 h, and then gently rinsed with running water. The cell colonies with more than 50 cells were counted, and the survival curve was generated using linear regression.
Cell viability was measured using an MTS kit (Promega) in accordance with the instruction from the manufacturer. Briefly, the cells were seeded into 96-well plates with about 1,000 cells per well. After the cells attached to the bottom of the well, the designated drugs were added to each well at the indicated concentration. At the specified time points, CellTiter 96R Aqueous One Solution Reagent with a final concentration of 10% (V/V) was added into the wells to measure cell viability. Absorbance was measured after incubating the cell culture plates in the cell incubator at 37°C for 60 min. The absorbance of 490 nm was read in a microplate reader.
Lentivirus-based control and gene-specific shRNAs were purchased from Sigma-Aldrich. 293T cells were transfected using Lipofectamine 2000 with shRNA plasmids and virus package plasmids (PMD2.G and pSPAX2). Twelve hours after transfection, the spent medium was replaced with fresh DMEM containing 10% fetal bovine serum (FBS) and 1 mM sodium pyruvate. After 36 hours of transfection, the culture medium containing packaged virus was filtered using 0.45 μm filters and added to target cells by supplying 8 µg/mL polybrene. shRNA sequences are provided in Table S2.
Cells (4.0 × 105/well) were seeded into each well of 6-well plates and cultured in phenol red-free RPMI 1640 medium (Thermo Fisher Scientific) for 24 h. The spent medium was collected and the glucose concentration of the spent medium was measured using the Glucose (GO) Assay Kit in accordance with the manufacturer’s instructions (Sigma-Aldrich). Glucose consumption was calculated based on the difference of glucose concentration between the spent and fresh medium. Lactate concentration of the spent medium was quantitatively measured using a L-Lactate Assay kit according to the manufacturer’s instructions (Eton Bioscience). Lactate production was calculated according to the difference of lactate concentration between the spent and fresh medium. The optical densities were measured at 570 nm wave-length in a plate reader.
All animal studies were approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC). Mice were housed with a 12 h light/12 h dark cycle and free to eat and drink. Probasin (Pb)-driven Cre4 recombinase transgenic mice (C57BL/DBA2) were originally generated in the laboratory of Dr. Pradip Roy-Burnam at the University of Southern California, Los Angeles, CA (17) and acquired from the National Cancer Institute (NCI) Mouse Repository. PtenLoxp/Loxp conditioned knockout mice (129/Balb/c) were originally generated in the laboratory of Dr. Hong Wu at the University of California, Los Angeles, CA (18), and purchased from Jackson Laboratory (004597). Littermate mice were used for control and target mice to ensure comparable genetic backgrounds. The sequence information of all the PCR primers used is listed in Table S1.
SCID mice, at 6 weeks of age, were bred in house and randomly divided into different groups. The animal study was approved by the IACUC at Mayo Clinic. All mice were housed in standard conditions with a 12 h light/dark cycle and access to food and water ad libitum. DU145 cells (5×106 in 100 μl 1×PBS) infected with lentivirus expressing empty vector (EV) or PTEN-specific shRNAs in combination with Flag-tagged FBP1-WT or S271A mutant were first mixed with Matrigel (BD Biosciences, 100 μl) on ice and then the mixed cells were injected s.c. into the right flank of mice. Xenograft growth was measured in a blinded fashion using a digital caliper and the volume of xenografts was estimated using the formula (L×W2)/2, where L is the length of tumor and W is the width. After the completion of measurement, mice were euthanized with CO2 and xenograft tumors were isolated and weighed. Tumor tissues were divided, and a portion was formalin-fixed and paraffin-embedded (FFPE) and the rest was frozen for protein and RNA extraction.
Unless otherwise specified, all experiments were repeated three or more times. One-sided or two-sided paired Student’s t test was used for single comparison, and multi comparisons were carried out using one-way ANOVA test. The P < 0.05 is considered statistically significant.
Previous studies have shown that the loss of PTEN tumor suppressor leads to the abnormal activation of PI3K signaling pathway, which is considered to be one of the most common oncogenic events in the pathogenesis of PCa (21). FBP1 is a key rate-limiting enzyme in gluconeogenesis (30). Increasing evidence shows that FBP1 is a key enzyme in regulating tumor glucose metabolism, and the deletion of FBP1 gene is related to cancer progression (4, 8–10). We were interested in investigating whether the expression of PTEN affected cancer metabolism by regulating the level of FBP1 in PCa cells. Therefore, we knocked down endogenous PTEN by the usage of two independent gene-specific small hairpin RNAs (shRNAs) in 22Rv1 and DU145 PCa cell lines. WB and RT-PCR showed that PTEN knockdown (KD) reduced the expression of FBP1 at the protein level, but had little impact on the level of FBP1 mRNA (Figures 1A, B). Restoring PTEN in PTEN-negative PC-3 and C4-2 PCa cell lines induced FBP1 protein expression, but exerted little impact on FBP1 mRNA expression (Figures 1C, D). Furthermore, we showed that deletion of Pten gene in MEFs induced downregulation of Fbp1 protein, but not Fbp1 mRNA expression (Figures 1E, F). IHC analyses further showed that knockout of Pten gene in the murine prostate downregulated Fbp1 protein level in mice (Figure 1G). These results suggest that PTEN positively regulates FBP1 expression in different human PCa cell lines and mouse prostate tumors.
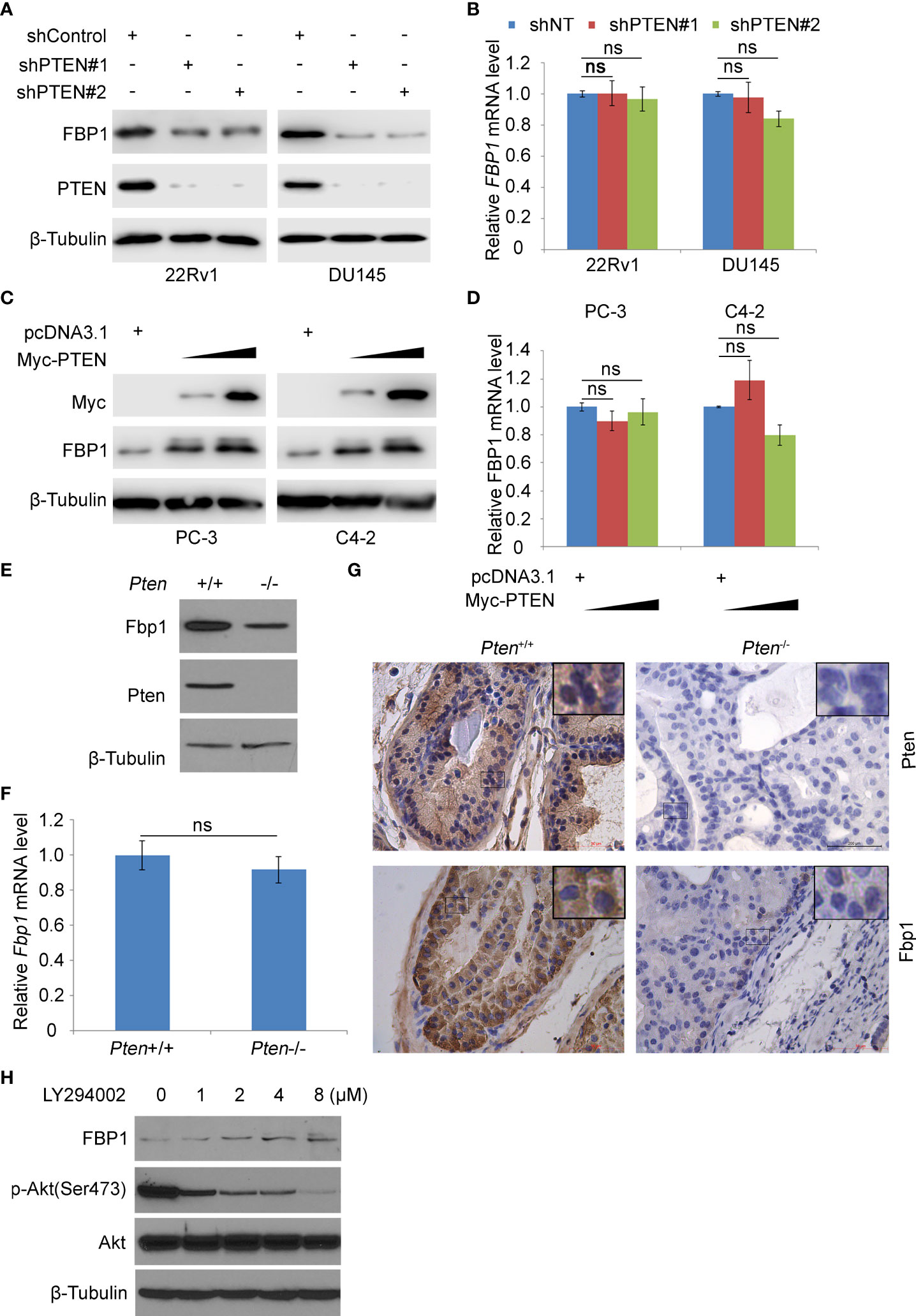
Figure 1 PTEN positively regulates FBP1 in human PCa cell lines and murine prostate tumors. (A, B) WB (A) and RT-PCR (B) were performed in 22Rv1 and DU145 cells infected with lentivirus expressing control or PTEN shRNAs for 48 h. β-Tubulin was used as a WB loading control. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. n.s., not significant. (C, D) WB (C) and RT-PCR (D) were performed in PC-3 and C4-2 cells transfected with pcDNA3.1 or FBP1 expression plasmid for 24 h. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. n.s., not significant. (E, F) WB (E) and RT-PCR (F) were performed in MEFs generated from Ptenp/p conditional mice infected with or without infected with lentivirus expressing CMV-driven Cre. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. n.s., not significant. (G) Photos of IHC of Fbp1 and Pten protein in the FFPE prostate tissues of Cre-negative mice (left column) and prostate tumors in Cre-positive mice (right column) at 5 months of age. (H) LNCaP cells were treated with LY294002 at the indicated concentrations for 24 h and then analyzed by WB.
PTEN is an established negative regulator of PI3K/AKT signaling pathway (31). To determine how inhibition of this pathway affected FBP1 expression, PTEN-negative LNCaP PCa cells were treated with different doses of the PI3K inhibitor LY294002. We demonstrated that inhibition of AKT phosphorylation by LY294002 correlated with upregulation of FBP1 protein dose-dependently (Figure 1H). Collectively, these data suggest that PTEN regulates FBP1 protein expression via the PI3K/AKT signaling pathway.
It has been reported that FBP1 can be degraded by ubiquitination (32). We sought to determine whether loss of PTEN regulated this process. To this end, we treated control and PTEN knockdown cells with the protein de novo synthesis inhibitor cycloheximide (CHX) and examined FBP1 protein expression level at different time points. We demonstrated that PTEN depletion largely decreased the half-life of FBP1 protein in 22Rv1 and DU145 cells (Figures 2A, B), suggesting that PTEN might negatively influence FBP1 protein degradation via proteasome pathway. To test this hypothesis, PTEN small hairpin RNAs (shPTENs) were transferred to 22Rv1 and DU145 cells and then treated with MG132. We found that PTEN loss-induced downregulation of FBP1 protein was blocked by MG132 treatment in both 22Rv1 and DU145 cells, but little or no effect was observed at the mRNA level (Figures 2C–F). In agreement with these results, the ubiquitin assays showed that PTEN depletion largely augmented FBP1 protein polyubiquitination (Figure 2G). Thus, these data suggest that loss of PTEN promotes the ubiquitination and proteasomal degradation of FBP1 protein in PCa cells.
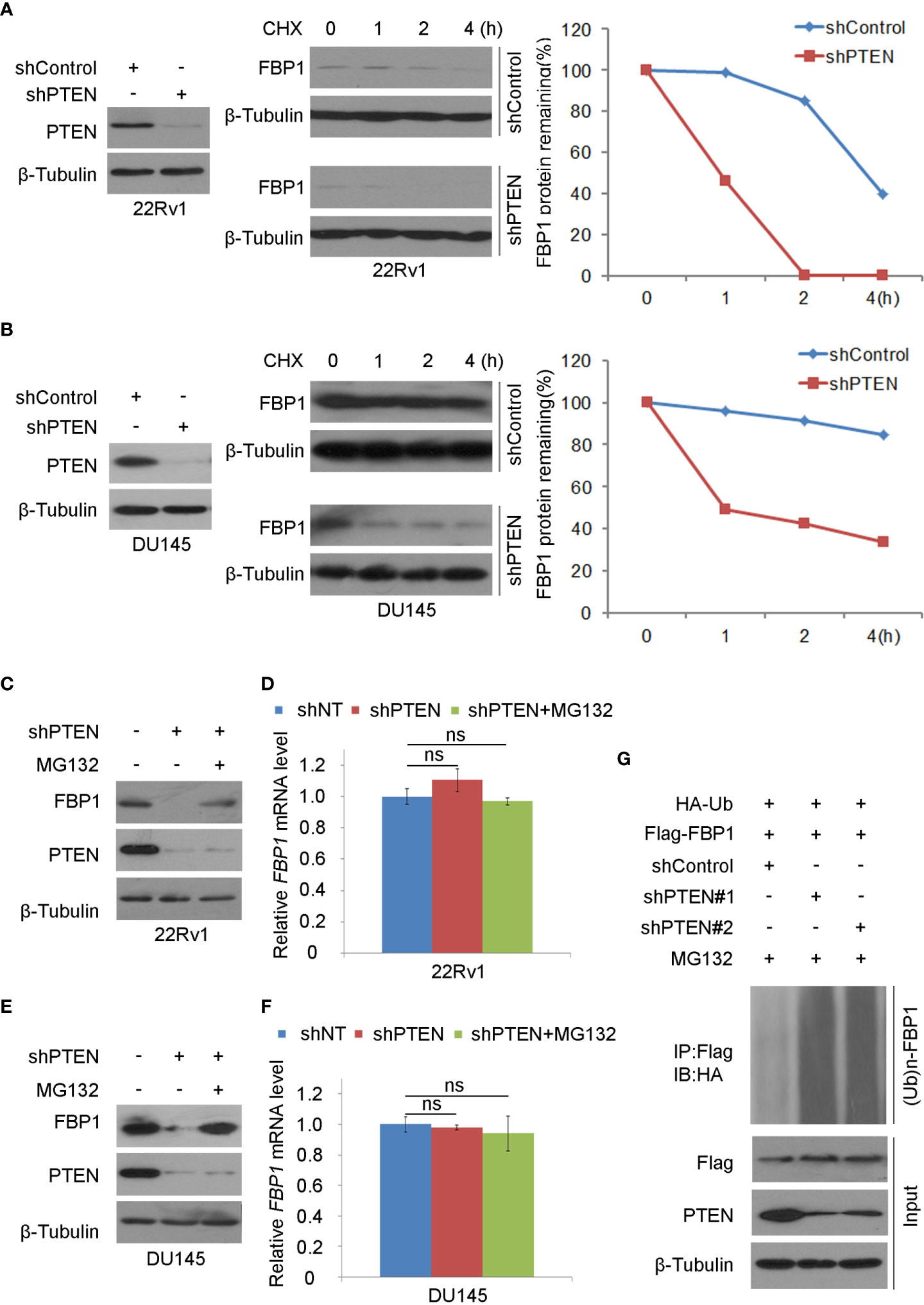
Figure 2 Loss of PTEN promotes FBP1 protein ubiquitination and degradation (A, B) WB and quantification of WB bands were carried out in 22Rv1 (A) and DU145 cells (B) infected with shControl or PTEN shRNAs for 48 h and followed by treatment with 50 μg/ml cycloheximide (CHX) for specific intervals of time. At every time point, the level of FBP1 protein was normalized to the level of β-Tubulin (a WB loading control) first and then to the value at the 0-h time point. (C, D) WB (C) and RT-PCR (D) were performed in 22Rv1 cells infected with lentivirus expressing control or PTEN shRNAs for 48 h and further treated with MG132 at 20 μM for 12 hours. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. n.s., not significant. (E, F) WB (E) and RT-PCR (F) were conducted in DU145 cells infected with lentivirus expressing control or PTEN shRNAs for 48 h and exposed with MG132 at 20 μM for another 12 hours. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. n.s., not significant. (G) WB evaluation was carried out using whole-cell lysate and co-IP samples obtained in 293T cells transfected with the indicated constructs and further treatment with 20 μM of MG132 for 12 more hours.
Consistent with the previous reports that inhibition of the PI3K/AKT signaling decreases expression of SKP2, an adaptor protein of the SKIP1-CULLIN1-F-Box protein (SCF) E3 ligase complex (33, 34), we found that depletion of PTEN increased mRNA and protein expression of SKP2 in 22Rv1 and DU145, which co-occurred with downregulation of FBP1 protein (Figures 3A, B). We, therefore, sought to determine whether SKP2 mediates FBP1 degradation induced by PTEN loss in PCa cells. To this end, shSKP2 was transfected into 22Rv1 and DU145 cells and protein expression was determined by WB. We showed that knockdown of SKP2 increased FBP1 expression at protein, but not mRNA level (Figures 3C, D). Moreover, we found that ectopic expression of SKP2 decreased FBP1 expression in a dose-dependent manner at the protein level, but had no drastic impact on FBP1 mRNA expression (Figures 3E, F). Furthermore, we showed that knockdown of endogenous SKP2 caused stabilization of FBP1 protein in both 22Rv1 and DU145 cell lines (Figures 3G, H). Next, we sought to determine whether SKP2 mediates FBP1 degradation induced by PTEN loss. To this end, we knocked down PTEN and/or SKP2 in 22Rv1 and DU145 cells and we found that PTEN loss-induced downregulation of FBP1 protein was completed blocked by knocking down SKP2 in these two cell lines (Figures 3I, J). These data indicate that SKP2 mediates FBP1 degradation induced by PTEN loss in PCa cells.
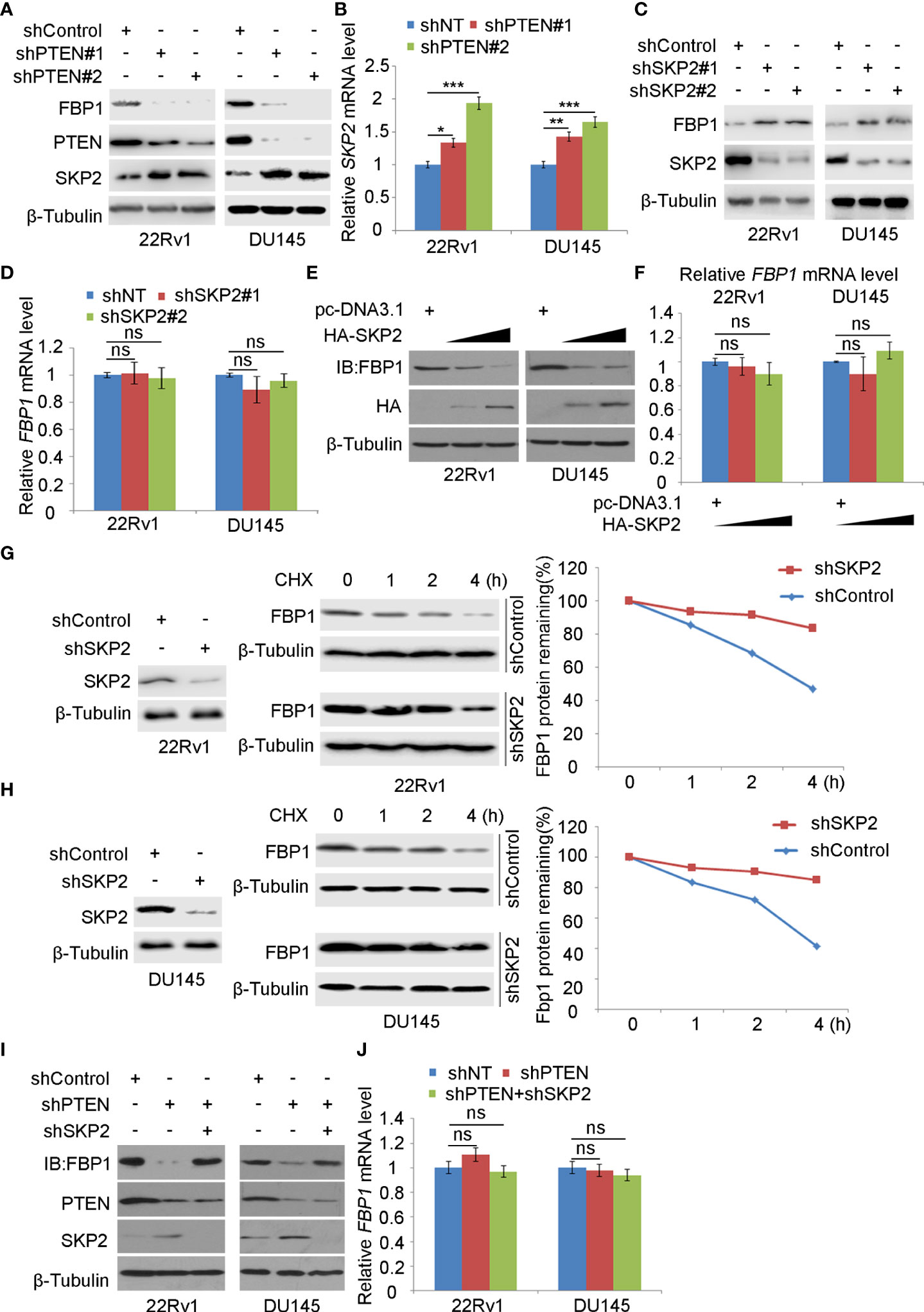
Figure 3 SKP2 mediates FBP1 protein ubiquitination and degradation induced by PTEN loss (A, B) WB (A) and quantitative RT-PCR (B) analyses were carried out in 22Rv1 and DU145 cells infected with lentivirus expressing control or PTEN-specific shRNAs for 48 hours. β-Tubulin was used as a WB loading control. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. *P < 0.05, **P<0.01, ***P<0.001. (C, D) WB (C) and quantitative RT-PCR (D) analyses were performed in 22Rv1 and DU145 cells infected with lentivirus expressing control or SKP2-specific shRNAs for 48 hours. β-Tubulin was used as a WB loading control. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. n.s., not significant. (E, F) WB (E) and quantitative RT-PCR (F) were conducted using samples obtained from 22Rv1 and DU145 cells transfected with pcDNA3.1 or HA-SKP2 expression plasmid for 24 hours. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. n.s., not significant. (G, H) WB and quantification of WB bands were carried out in 22Rv1 (G) and DU145 cells (H) infected with shControl or SKP2-specific shRNAs for 48 hours and treated with 50 μg/ml cycloheximide (CHX) for different periods of time. At each time point, the intensity of FBP1 WB band was normalized to the intensity of β-Tubulin (a WB loading control) first and then to the value at the 0-h time point. (I, J) Western blot (I) and quantitative RT-PCR (J) analyses were conducted in 22Rv1 and DU145 cells infected with lentivirus expressing the indicated shRNAs for 48 hours. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. n.s., not significant.
It has been known that SKP2 induces K48-linked polyubiquitination and proteasomal degradation of p27Kip1 protein in a manner dependent on cyclin-dependent kinase-2 (CDK2)-mediated phosphorylation of p27Kip1 (35–40). Also, a study has shown that the deletion of PTEN leads to the activation of CDK2 by down-regulating p27Kip1 (41). As expected, we found that PTEN knockdown decreased p27Kip1 expression but most importantly, PTEN loss increased FBP1 phosphorylation (Figure 4A). To determine whether CDKs play any role in PTEN loss-induced downregulation of FBP1 protein, we knocked down PTEN in 22Rv1 and DU145 cells and treated cells with or without Roscovitine, a broad inhibitor of CDKs including CDK2 (Figure 4B). We demonstrated that Roscovitine treatment blocked the down-regulation of FBP1 protein caused by PTEN deletion in both cell lines, but without obvious impact on mRNA expression (Figures 4B, C). Similar results were obtained in wild-type (WT) and Pten knockout MEF cells (Figures 4D, E). We further showed that CDK inhibition diminished FBP1 protein decay in 22Rv1 and DU145 cells (Figures 4F, G). Together, these data indicate that CDKs mediate FBP1 degradation induced by PTEN loss and this process is blocked by CDK inhibitor.
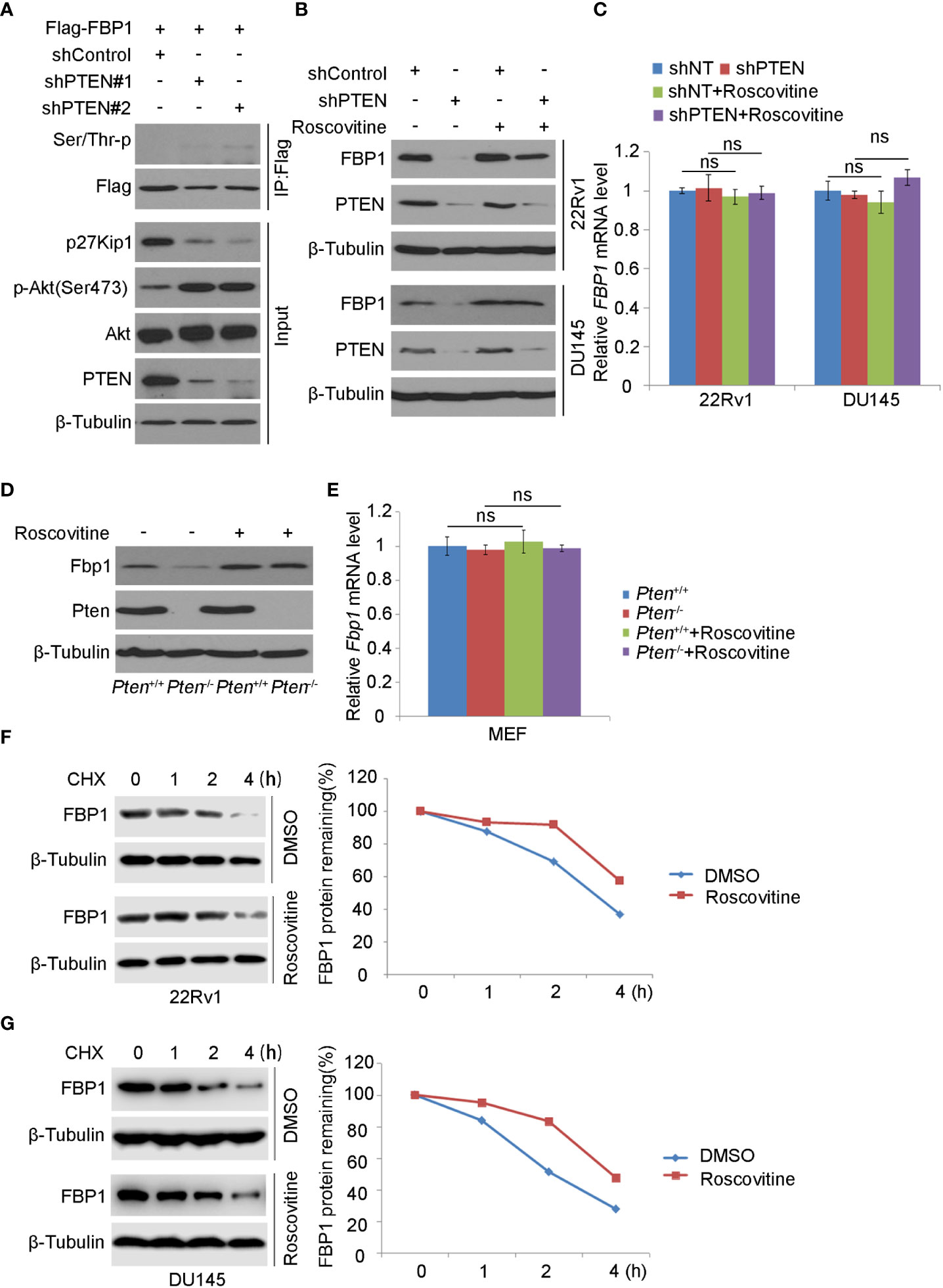
Figure 4 CDK inhibitor blocks FBP1 degradation induced by PTEN loss (A) WB was carried out using the whole-cell lysate and co-IP samples obtained from 293T cells transfected with the indicated constructs. β-Tubulin was used as a WB loading control. (B, C) Western blot (B) and quantitative RT-PCR (C) analysis in 22Rv1 and DU145 cells infected with lentivirus expressing control or PTEN-specific shRNAs for 48 hours and treated with Roscovitine for 24 hours. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. n.s., not significant. (D, E) WB (D) and quantitative RT-PCR (E) were carried out in MEFs generated from Ptenp/p conditional mice infected with or without lentivirus expression CMV-driven Cre and treated with Roscovitine for 24 hours. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. n.s., not significant. (F, G) WB and quantification of WB bands were performed in 22Rv1 (F) and DU145 cells (G) treated with Roscovitine for 48 hours followed by treatment with 50 μg/ml cycloheximide (CHX) for different periods of time. At each time point, the intensity of FBP1 was normalized to the intensity of β-Tubulin (a WB loading control) first and then to the value at the 0-h time point.
It is known that CDKs regulate the protein level and function of their downstream substrates through protein phosphorylation (42). We noticed that akin to known CDK substrates such as RB, H1B, FOXO1, EZH2, ATM and p27Kip1, there is a putative CDK phosphorylation site (270KSPNG274) which is similar to the CDK phosphorylation consensus motif K/R-T/S-P-X-K/R (X, any amino acid) (Figure 5A). To determine whether the serine 271 (S271) residue plays any role in PTEN loss-induced FBP1 degradation, we generated an FBP1 S271A phosphorylation-resistant mutant. We demonstrated that knockdown of PTEN largely increased phosphorylation of Flag-FBP1-WT, but not the Flag-FBP1-S271A mutant in 293T cells (Figure 5B), suggesting that PTEN loss results in S271 phosphorylation on FBP1. We further showed that PTEN knockdown-induced downregulation of FBP1 was blocked by S271A mutation, but this mutant had no obvious effect on the mRNA expression of FBP1 (Figures 5C, D). In agreement with these observations, in vivo ubiquitination assays showed that loss of PTEN robustly enhanced polyubiquitination of FBP1-WT, but not the FBP1-S271A phosphorylation-resistant mutant (Figure 5E). Thus, these data indicate that the serine 271 phosphorylation on FBP1 is necessary for FBP1 protein degradation induced by PTEN loss.
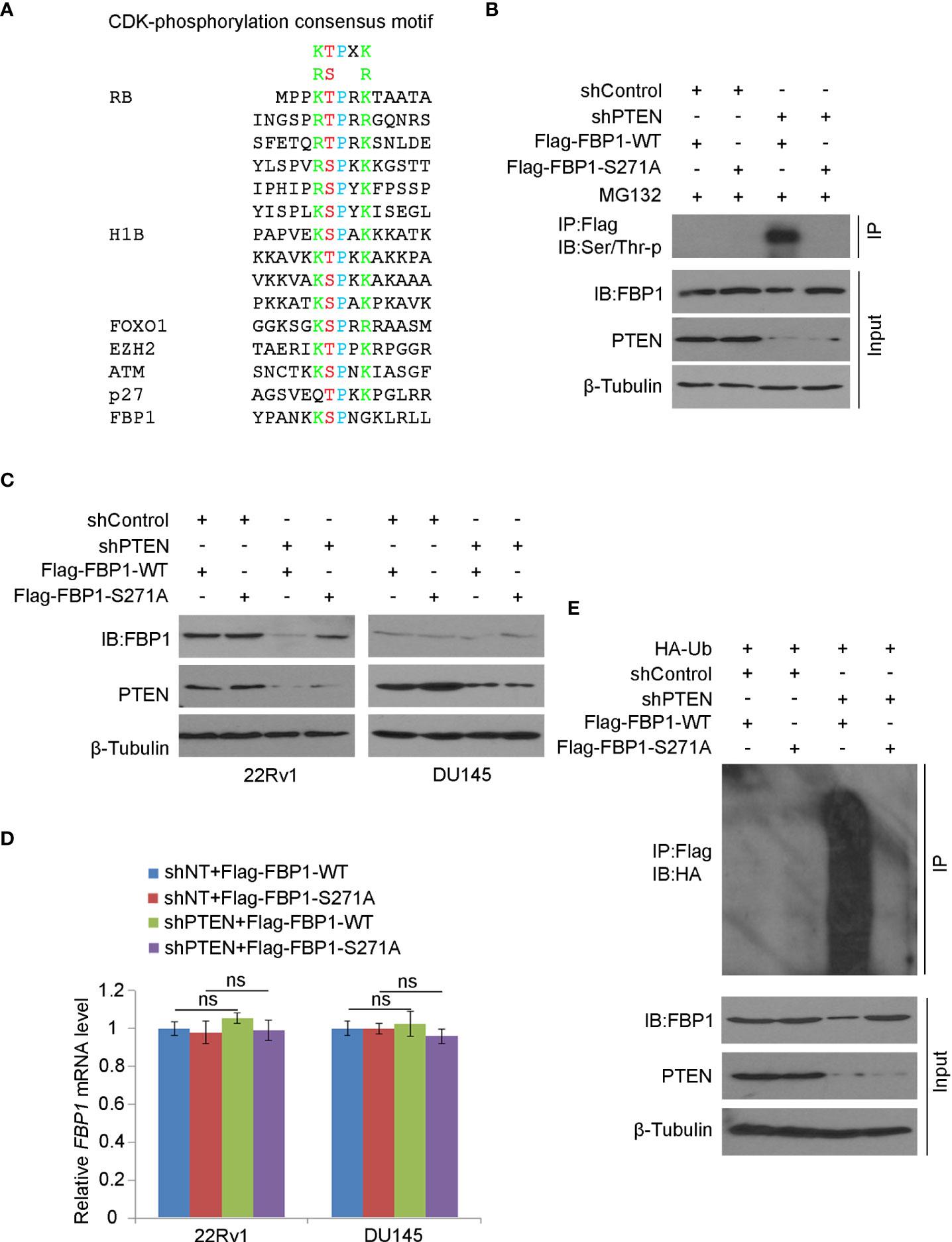
Figure 5 Serine 271 phosphorylation is important for FBP1 degradation induced by PTEN loss (A) Amino acid sequence alignment among CDK phosphorylation consensus motif, those identified in known CDK substrates, and the putative CDK phosphorylation site (serine 271) on FBP1. (B) WB analysis in the whole cell lysate and co-IP samples in 293T cells transfected with indicated plasmids or infected with indicated lentivirus for 48 hours. (C, D) WB (C) and quantitative RT-PCR (D) analyses were performed in 22Rv1 and DU145 cells infected with lentivirus expressing control or PTEN-specific shRNAs and/or transfected with plasmid for Flag-tagged FBP1 WT or S271A for 48 hours. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. n.s., not significant. (E) WB analysis was carried out using the whole cell lysate and co-IP samples in 293T cells transfected with indicated plasmids or infected with indicated lentivirus for 48 hours.
Given that FBP1 is a major negative regulator of glycolysis, and cancer cells rely heavily on aerobic glycolysis (Warburg effect) for growth, we were very interested to determine whether restored expression of FBP1 inhibited the growth of PCa cells by blocking Warburg effect. As expected, we found that knockdown of PTEN increased glucose metabolism and lactate production in both 22Rv1 and DU145 cells (Figures 6A, B). This impact was largely inhibited by ectopic expression of the phosphorylation/degradation-resistant mutant Flag-tagged FBP1-S271A, but only modestly diminished by ectopic expression of Flag-tagged degradable FBP1-WT (Figures 6A, B). Similar results were seen in the cell proliferation assays in the two cell lines (Figures 6C, D). Next, we evaluated the impact of restored expression of FBP1 on tumor growth in mice. We infected DU145 cells with lentivirus expressing shControl or shPTEN in combination with or without Flag-tagged FBP1-WT or FBP1-S271A and inoculated these groups of cells into SCID mice. We discovered that FBP1-S271A expression, but not FBP1-WT inhibited PTEN knockdown-enhanced tumor growth in vivo (Figures 6E, F). Therefore, our data suggest that degradation of FBP1 plays a significant role in contributing to PTEN deletion-induced PCa growth in vitro and in vivo.
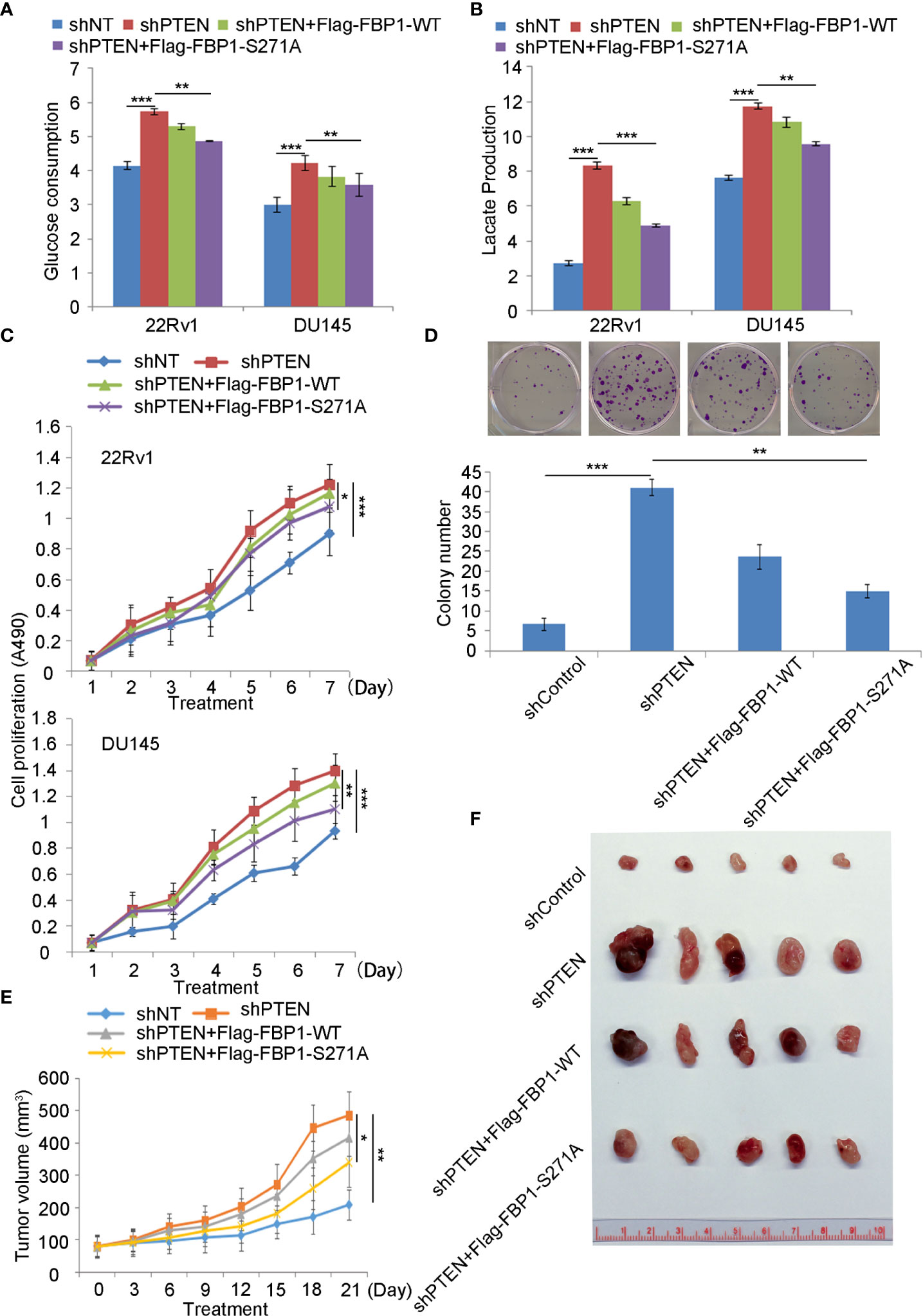
Figure 6 Serine 271 phosphorylation of FBP1 is important in regulating the Warburg effect and tumor growth in PCa (A, B) Glucose consumption and lactate production were measured in the spent medium of 22Rv1 and DU145 cells 48 hours after transfected with indicated constructs. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. **P<0.01, ***P<0.001. (C) MTS assay was performed in 22Rv1 and DU145 cells 48 hours after transfected with indicated constructs for different periods of time. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. *P<0.05, ***P<0.001. (D) Representative images were taken and quantification of the results of colony formation assays was performed in DU145 cells transfected with indicated constructs for 13 days. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. **P<0.01, ***P<0.001. (E, F) Tumor growth curve over the time (E) and photograph (F) of tumors at the endpoint of inoculation of DU145 cells infected with lentivirus expressing indicated shRNAs or expression vectors. All quantitative data are shown as Mean ± SD (n=3). Student’s t test. **P<0.01.
The major finding of the current study is the identification of a previously unrecognized molecular mechanism that promotes Warburg effect and cancer progression. FBP1, a recognized tumor suppressor, is known to inhibit cancer development via inhibition of aerobic glycolysis and suppression of the Warburg effect in different cancer types (4, 10, 43). In this study, we have revealed that PTEN plays a key function in regulating FBP1 expression and tumor progression through FBP1 phosphorylation-dependent ubiquitin degradation in PCa, thus defining a new role of PTEN in metabolism and tumor progression. We have found that the loss of PTEN promotes the degradation of FBP1 protein by activating the PI3K/AKT pathway via two mechanisms. On the one hand, activated AKT down-regulates p27, thus increasing the activity of its substrate proteins such as CDKs and promoting the CDK-dependent phosphorylation of FBP1. On the other hand, activated AKT upregulates SKP2 mRNA and then increases the expression of SKP2 protein, which can ubiquitinate CDK-phosphorylated FBP1, and lead to CDK phosphorylation-dependent degradation of FBP1 (Figure 7). Our observation further support the notion that mutation in the CDK-phosphorylation site on FBP1 can hinder the degradation of FBP1 and increases FBP1 expression, thereby restraining Warburg effect and PCa growth. These findings not only provide an mechanistic explanation for the observed PTEN loss-induced downregulation of FBP1 protein in PCa, but also provide mechanistic insight into the tumor growth augmented by the Warburg effect.
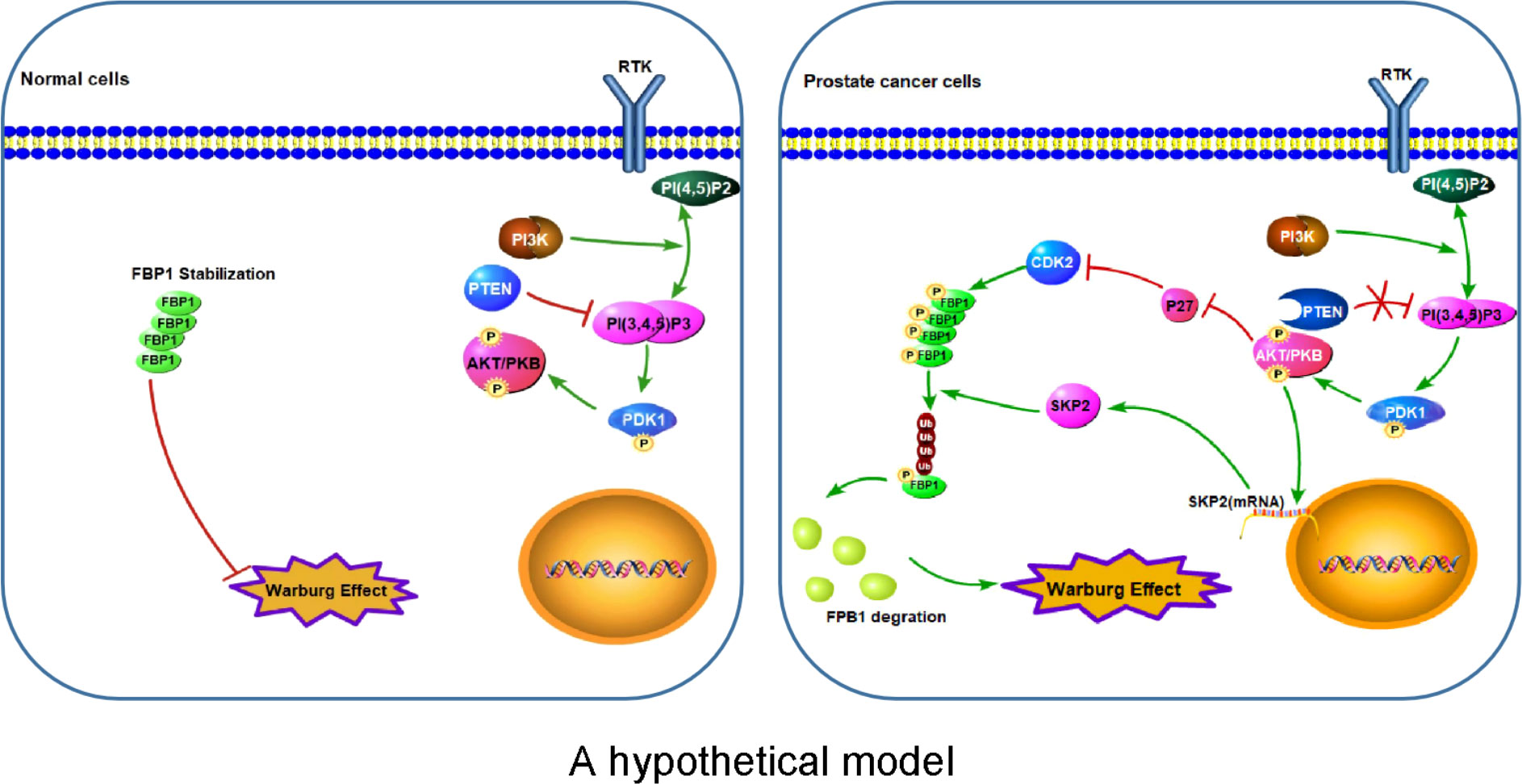
Figure 7 A hypothetical model Left: Stabilization of FBP1 protein prevents over-activation of the Warburg effect in normal cells. Right: Aberrant activation of the PI3K/AKT pathway due to deregulation of signaling such as loss of PTEN leads to CDK-dependent phosphorylation and ubiquitination and degradation of FBP1 mediated by SKP2 E3 ubiquitin ligase, thereby leading to abnormal activation of the Warburg effect and cell growth of cancer such as PCa. P letter with circle represents protein phosphorylation.
FBP1 is the rate-limiting enzyme in the process of gluconeogenesis, which converts fructose-1,6-biphosphate to fructose-6-phophate and inorganic phosphate (44). Previous studies have shown that FBP1 is frequently downregulated in a variety of solid tumors (4, 10, 45). Its down-regulation mechanism may be related to DNA hypermethylation of gene promoter or loss of the copy number of the gene (4, 46), histone deacetylation caused by deregulation of HDACs (11) or degradation of FBP1 in tumor cells caused by the E3 ligase TRIM28. It is important that FBP1 downregulation is tightly related to the Warburg effect, which is a key metabolic feature of many cancers. In this study, we have demonstrated that FBP1 protein is destructed after phosphorylation in PTEN-deficient PCa. Consistent with the finding that FBP1 is often downregulated in human cancers, we provide evidence that loss of FBP1 contributes to tumor growth through the Warburg effect in PTEN-deficient PCa. Thus, our findings define a new role of FBP1 in regulating cancer progression.
PTEN plays an important role in cancer development and progression. Previous studies have reported that loss of PTEN plays a role in tumor metabolism. In our study, we have demonstrated that PTEN loss leads to phosphorylation of FBP1 at the CDK site and promotes its degradation. This effect is attenuated by mutation of CDK phosphorylation site on FBP1. Our findings reveal PTEN as an upstream protector of FBP1 and uncover a new mechanism by which PTEN regulates metabolism in cancer. The significance of these findings is further accentuated by our observations that expression of PTEN and FBP1 is positively corrected in multiple PCa cell lines and a PCa mouse model.
In summary, we have discovered a new role of PTEN in antagonizing Warburg effect by regulating expression of FBP1 and its effect on cell metabolism and tumor growth. Mechanistically, we show that PTEN inhibits phosphorylation-dependent ubiquitination-mediated degradation of FBP1. This finding highlights the importance of FBP1 protein destruction in augmented Warburg effect and growth of PTEN-deficient PCa. Our findings also suggest that targeting PTEN loss-induced FBP1 protein degradation could be a viable strategy for effective treatment of cancers such as PCa with aberrant activation of PI3K/AKT.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
All animal study was approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC).
HH conceived the study. CS, JZ, XL, ML, ZK, JY, and JC performed experiments and collected and analyzed the data. HH and DW supervised the study. CS, JZ, ML and HH wrote the manuscript. All authors contributed to the article and approved the submitted version. CS, JZ and XL have contributed equally to this work.
We thank Lan Shen for the technical assistance. The work was supported by funding from the Mayo Clinic Foundation (to HH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.911466/full#supplementary-material
1. Aguilar M, Ali Cavasonza L, Alpat B, Ambrosi G, Arruda L, Attig N, et al. Towards understanding the origin of cosmic-ray electrons. Phys Rev Lett (2019) 122(10):101101. doi: 10.1103/PhysRevLett.122.101101
2. Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell (2008) 134(5):703–7. doi: 10.1016/j.cell.2008.08.021
3. Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res (2011) 71(22):6921–5. doi: 10.1158/0008-5472.Can-11-1457
4. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, et al. Loss of FBP1 by snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell (2013) 23(3):316–31. doi: 10.1016/j.ccr.2013.01.022
5. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
6. DeBerardinis RJ, Thompson CB. Cellular metabolism and disease: what do metabolic outliers teach us? Cell (2012) 148(6):1132–44. doi: 10.1016/j.cell.2012.02.032
8. Liu X, Wang X, Zhang J, Lam EK, Shin VY, Cheng AS, et al. Warburg effect revisited: an epigenetic link between glycolysis and gastric carcinogenesis. Oncogene (2010) 29(3):442–50. doi: 10.1038/onc.2009.332
9. Chen M, Zhang J, Li N, Qian Z, Zhu M, Li Q, et al. Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PloS One (2011) 6(10):e25564. doi: 10.1371/journal.pone.0025564
10. Li B, Qiu B, Lee DS, Walton ZE, Ochocki JD, Mathew LK, et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature (2014) 513(7517):251–5. doi: 10.1038/nature13557
11. Yang J, Jin X, Yan Y, Shao Y, Pan Y, Roberts LR, et al. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci Rep (2017) 7:43864. doi: 10.1038/srep43864
12. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin (2019) 69(1):7–34. doi: 10.3322/caac.21551
13. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem (1998) 273(22):13375–8. doi: 10.1074/jbc.273.22.13375
14. Álvarez-Garcia V, Tawil Y, Wise HM, Leslie NR. Mechanisms of PTEN loss in cancer: It's all about diversity. Semin Cancer Biol (2019) 59:66–79. doi: 10.1016/j.semcancer.2019.02.001
15. Wang SI, Parsons R, Ittmann M. Homozygous deletion of the PTEN tumor suppressor gene in a subset of prostate adenocarcinomas. Clin Cancer Res (1998) 4(3):811–5.
16. Suzuki H, Freije D, Nusskern DR, Okami K, Cairns P, Sidransky D, et al. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res (1998) 58(2):204–9.
17. Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, et al. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res (1997) 57(22):4997–5000.
18. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene (2008) 27(41):5497–510. doi: 10.1038/onc.2008.245
19. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell (2010) 18(1):11–22. doi: 10.1016/j.ccr.2010.05.026
20. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol (2012) 13(5):283–96. doi: 10.1038/nrm3330
21. Wise HM, Hermida MA, Leslie NR. Prostate cancer, PI3K, PTEN and prognosis. Clin Sci (Lond) (2017) 131(3):197–210. doi: 10.1042/cs20160026
22. Chen CY, Chen J, He L, Stiles BL. PTEN: Tumor suppressor and metabolic regulator. Front Endocrinol (Lausanne) (2018) 9:338. doi: 10.3389/fendo.2018.00338
23. Sano H, Kane S, Sano E, Mîinea CP, Asara JM, Lane WS, et al. Insulin-stimulated phosphorylation of a rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem (2003) 278(17):14599–602. doi: 10.1074/jbc.C300063200
24. Stiles BL. Phosphatase and tensin homologue deleted on chromosome 10: extending its PTENtacles. Int J Biochem Cell Biol (2009) 41(4):757–61. doi: 10.1016/j.biocel.2008.09.022
25. Gross DN, Wan M, Birnbaum MJ. The role of FOXO in the regulation of metabolism. Curr Diabetes Rep (2009) 9(3):208–14. doi: 10.1007/s11892-009-0034-5
26. Xiong S, Salazar G, San Martin A, Ahmad M, Patrushev N, Hilenski L, et al. PGC-1 alpha serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J Biol Chem (2010) 285(4):2474–87. doi: 10.1074/jbc.M109.065235
27. Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science (2006) 314(5797):294–7. doi: 10.1126/science.1130512
28. Franken NAP, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc (2006) 1(5):2315–9. doi: 10.1038/nprot.2006.339
29. Yan Y, An J, Yang Y, Wu D, Bai Y, Cao W, et al. Dual inhibition of AKT-mTOR and AR signaling by targeting HDAC3 in PTEN- or SPOP-mutated prostate cancer. EMBO Mol Med (2018) 10(4):e8478. doi: 10.15252/emmm.201708478
30. Timson DJ. Fructose 1,6-bisphosphatase: getting the message across. Biosci Rep (2019) 39(3):BSR20190124. doi: 10.1042/bsr20190124
31. Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer (2002) 2(7):489–501. doi: 10.1038/nrc839
32. Jin X, Pan Y, Wang L, Zhang L, Ravichandran R, Potts PR, et al. MAGE-TRIM28 complex promotes the warburg effect and hepatocellular carcinoma progression by targeting FBP1 for degradation. Oncogenesis (2017) 6(4):e312. doi: 10.1038/oncsis.2017.21
33. Mamillapalli R, Gavrilova N, Mihaylova VT, Tsvetkov LM, Wu H, Zhang H, et al. PTEN regulates the ubiquitin-dependent degradation of the CDK inhibitor p27(KIP1) through the ubiquitin E3 ligase SCF(SKP2). Curr Biol (2001) 11(4):263–7. doi: 10.1016/s0960-9822(01)00065-3
34. Auld CA, Caccia CD, Morrison RF. Hormonal induction of adipogenesis induces Skp2 expression through PI3K and MAPK pathways. J Cell Biochem (2007) 100(1):204–16. doi: 10.1002/jcb.21063
35. Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, et al. Targeted disruption of Skp2 results in accumulation of cyclin e and p27(Kip1), polyploidy and centrosome overduplication. EMBO J (2000) 19(9):2069–81. doi: 10.1093/emboj/19.9.2069
36. Sutterlüty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Müller U, et al. p45SKP2 promotes p27Kip1 degradation and induces s phase in quiescent cells. Nat Cell Biol (1999) 1(4):207–14. doi: 10.1038/12027
37. Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, et al. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell (2004) 6(5):661–72. doi: 10.1016/s1534-5807(04)00131-5
38. Russo AA, Jeffrey PD, Patten AK, Massagué J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin a-Cdk2 complex. Nature (1996) 382(6589):325–31. doi: 10.1038/382325a0
39. Ray A, James MK, Larochelle S, Fisher RP, Blain SW. p27Kip1 inhibits cyclin d-cyclin-dependent kinase 4 by two independent modes. Mol Cell Biol (2009) 29(4):986–99. doi: 10.1128/mcb.00898-08
40. Aleem E, Kiyokawa H, Kaldis P. Cdc2-cyclin e complexes regulate the G1/S phase transition. Nat Cell Biol (2005) 7(8):831–6. doi: 10.1038/ncb1284
41. Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, et al. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and akt/protein kinase b signaling pathway. Proc Natl Acad Sci USA (1999) 96(11):6199–204. doi: 10.1073/pnas.96.11.6199
42. Swaffer MP, Jones AW, Flynn HR, Snijders AP, Nurse P. CDK substrate phosphorylation and ordering the cell cycle. Cell (2016) 167(7):1750–61.e16. doi: 10.1016/j.cell.2016.11.034
43. Chen J, Lee HJ, Wu X, Huo L, Kim SJ, Xu L, et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res (2015) 75(3):554–65. doi: 10.1158/0008-5472.Can-14-2268
44. Jin X, Pan Y, Wang L, Ma T, Zhang L, Tang AH, et al. Fructose-1,6-bisphosphatase inhibits ERK activation and bypasses gemcitabine resistance in pancreatic cancer by blocking IQGAP1-MAPK interaction. Cancer Res (2017) 77(16):4328–41. doi: 10.1158/0008-5472.Can-16-3143
45. Phillips R. Kidney cancer: FBP1 depletion feeds ccRCC. Nat Rev Urol (2014) 11(9):482. doi: 10.1038/nrurol.2014.200
Keywords: PTEN, SKP2, ubiquitination, PCa, FBP1
Citation: Song C, Zhang J, Liu X, Li M, Wang D, Kang Z, Yu J, Chen J, Pan H, Wang H, Li G and Huang H (2022) PTEN loss promotes Warburg effect and prostate cancer cell growth by inducing FBP1 degradation. Front. Oncol. 12:911466. doi: 10.3389/fonc.2022.911466
Received: 02 April 2022; Accepted: 29 July 2022;
Published: 27 September 2022.
Edited by:
Eneda Toska, Johns Hopkins Medicine, United StatesReviewed by:
Italia Falcone, Regina Elena National Cancer Institute (IRCCS), ItalyCopyright © 2022 Song, Zhang, Liu, Li, Wang, Kang, Yu, Chen, Pan, Wang, Li and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haojie Huang, aHVhbmcuaGFvamllQG1heW8uZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.