
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 04 January 2023
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.910199
This article is part of the Research Topic Meningioma: From Basic Research to Clinical Translational Study, Volume II View all 32 articles
 Elaina J. Wang1
Elaina J. Wang1 Alexander F. Haddad2
Alexander F. Haddad2 Jacob S. Young2
Jacob S. Young2 Ramin A. Morshed2
Ramin A. Morshed2 Joshua P. H. Wu1
Joshua P. H. Wu1 Diana M. Salha2
Diana M. Salha2 Nicholas Butowski2
Nicholas Butowski2 Manish K. Aghi2*
Manish K. Aghi2*Meningiomas are the most common primary intracranial neoplasm. While traditionally viewed as benign, meningiomas are associated with significant patient morbidity, and certain meningioma subgroups display more aggressive and malignant behavior with higher rates of recurrence. Historically, the risk stratification of meningioma recurrence has been primarily associated with the World Health Organization histopathological grade and surgical extent of resection. However, a growing body of literature has highlighted the value of utilizing molecular characteristics to assess meningioma aggressiveness and recurrence risk. In this review, we discuss preclinical and clinical evidence surrounding the use of molecular classification schemes for meningioma prognostication. We also highlight how molecular data may inform meningioma treatment strategies and future directions.
Meningiomas account for up to 40% of all primary central nervous system (CNS) tumors, making them the most common primary intracranial tumor (1). While they are thought to be derived from arachnoid cap cells due to cytological similarities (2, 3), the actual cell of origin for meningiomas remains unknown. It is possible they are derived from arachnoid barrier cells, a meningeal cell layer between pia and dura mater separating cerebrospinal fluid from underlying blood vessels, since meningiomas and arachnoid barrier cells have shared expression of prostaglandin D synthase (PGDS) (4, 5). Understanding the origin and natural history of meningiomas is important, since the incidence of meningiomas has been steadily rising, secondary to improvements in imaging resolution and more frequent use of various imaging modalities by providers (6, 7).
The 2021 World Health Organization (WHO) classification system describes 15 meningioma variants categorized into 3 grades based off of histopathological features and molecular biomarkers, with atypical and anaplastic criteria now applied to each of the subtypes (8). Eighty percent of meningiomas fall under the grade 1 category and can be treated with maximally safe resection with the goal of gross total resection (9). However, there remains a 5% 5-year recurrence rate for grade 1 meningiomas that increases to 40% for grade 2 meningiomas (3), and there is no established standard of care (SOC) for grade 2 and 3 meningiomas (10). While meningiomas have been traditionally risk stratified using the World Health Organization (WHO) histopathological grade and extent of surgical resection (8, 11), advances in molecular profiling have highlighted the benefits of utilizing genetic and epigenetic changes to further characterize meningioma aggressiveness and recurrence risk. The inter-observer variability inherent to histopathological diagnoses (12), coupled with advances in genetic and epigenetic technologies that have changed our understanding of meningioma tumor biology, lend support to the need for new molecular classifications for diagnosis and treatment. This review summarizes the use of genetic biomarkers and other forms of molecular data to inform meningioma prognostication and treatment strategies.
Sporadic mutations in the NF2 gene on chromosome 22 are implicated in 40 to 60% of meningioma patients (3), while 50 to 75% of patients with germline mutations develop meningiomas (13) (Table 1). NF2 encodes for the tumor suppressor protein merlin (14), the loss of which is a well-studied driver mutation commonly implicated in high-grade meningiomas (15). NF2 loss-of-function mutations occur through a double-hit mechanism in meningiomas, either through a germline mutation and a second hit with a somatic mutation in syndromic cases, or with a somatic single nucleotide variation or insertion/deletion mutation and an overlapping chromosome 22 deletion event as commonly seen in sporadic cases (15). While 95% of NF2-associated meningiomas remain grade 1 (13), the presence of an NF2 mutation has been associated with increased tumor size and cell proliferation, and it has been suggested that NF2 loss may be the primary and sole initiator of meningioma tumorigenesis in both cranial and spinal meningiomas (16, 17). Two phase II clinical trials (Table 2) are currently underway to test FAK inhibitor GSK2256098 and AZD2014 in patients with NF2-mutated meningiomas and NF2 patients with symptomatic meningiomas respectively (19, 20).
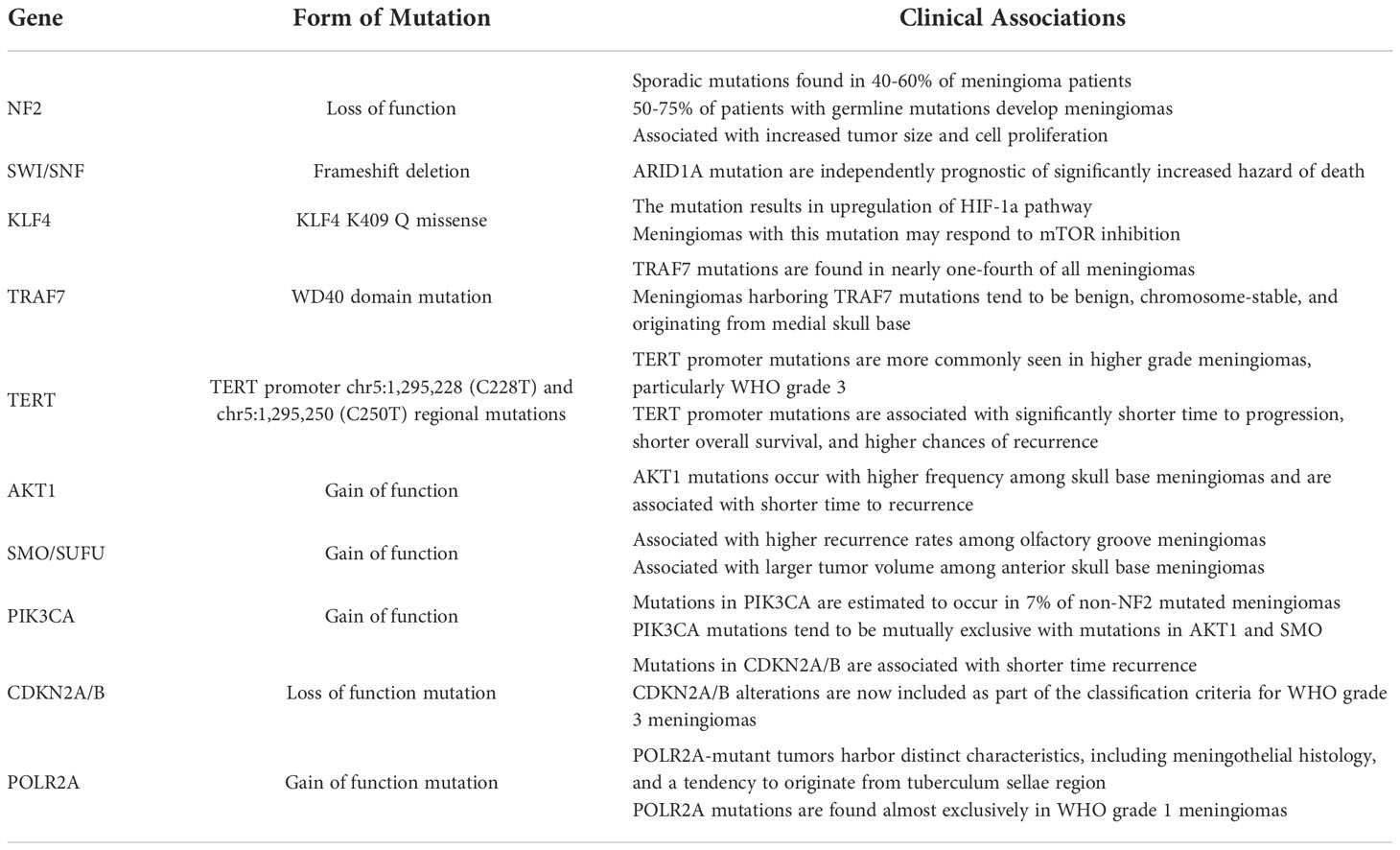
Table 1 Commonly identified germline and somatic mutations in meningiomas.

Table 2 Phase 2 clinical trials targeting genetic mutations in meningiomas.
Germline mutations in two SWI/SNF chromatin remodeling complex subunits have also been implicated in meningioma tumorigenesis: SMARCB1 and SMARCE1 (21). Mutations in SMARCB1 have been linked to the development of multiple meningiomas, while SMARCE1 loss of function mutations have been implicated in patients with familial multiple spinal meningiomas with clear cell histology (22, 23).
Germline mutations in known tumor suppressor SUFU on chromosome 10 have long been associated with childhood medulloblastoma, with loss of SUFU leading to disruptions in the sonic hedgehog signaling pathway (24, 25). Germline disruptions in SUFU are also thought to predispose to development of additional cancers such as basal cell carcinoma, gonadal tumors, and meningiomas (26). Mutations in SUFU have been linked to development of isolated familial meningiomas and development of multiple meningiomas (27). In a case series of four related family members with three that had a history of meningiomas, a frameshift mutation in SUFU leading to a premature stop codon was isolated and is posited to be related to development of meningiomas (28).
KLF4 is a transcriptional regulator known to maintain stemness and found to perform both oncogenic and tumor suppressor functions in a variety of cancers, including but not limited to bladder, esophageal, and gastric cancers (29–32). KLF loss of function has been implicated in colon cancer, while its overexpression has been shown to lead to decreased tumorigenicity of colon cancer cells in vivo (33, 34). In meningiomas, it is one of two genes found to be mutated in whole-exome sequencing of sixteen secretory meningiomas (35). KLF4 overexpression in anaplastic meningiomas has been associated with increased expression of tumor suppressor proteins such as p21, p53, and BAX, demonstrating a potential anti-tumor role in higher grade meningiomas (36). Recent in vitro data has also suggested that skull-based meningiomas with KLF K409Q mutations have a unique tumor phenotype that may respond to mammalian target of rapamycin (mTOR) inhibition with temsirolimus (37).
TRAF7 encodes for a ubiquitin E3 ligase and is the second most commonly mutated gene in meningiomas (38). It catalyzes a variety of ubiquitination reactions, including that of tumor suppressor p53, which has been shown to promote tumor progression in hepatocellular cancer while stabilizing p53’s anti-tumoral effects in breast cancer (39, 40). TRAF7 and KLF4 mutations often co-occur in secretory meningiomas (35), with 40% of TRAF7-mutated meningiomas harboring a KLF4 mutation as well (41). TRAF7 is also one of four genes including KLF4, AKT1, and SMO, likely to be mutated in non-NF2 mutated meningiomas found at the skull base (38). TRAF7 mutations are also closely associated with hyperostosis and often found in spheno-orbital meningiomas (42).
TERT encodes for telomerase reverse transcriptase, a catalytic subunit of telomerase that promotes cell immortalization via telomere elongation (43). Mutations in the chr5:1,295,228 (C228T) and chr5:1,295,250 (C250T) regions of the TERT promoter have been associated with uncontrolled proliferation in several cancers (44–46) and recently in meningiomas that demonstrate histological malignant transformation (47). TERT promoter mutations are more commonly seen in higher grade meningiomas, with mutations found in 1.7%, 5.7% and 20% of 2007 WHO classification grade 1, 2, and 3 meningiomas respectively (18). Clinically, mutations in the TERT promoter region have been associated with significantly shorter time to progression, shorter overall survival, and higher chances of recurrence (18, 48, 49). TERT promoter mutations are now included in the 2021 WHO classification of grade 3 meningiomas (8).
AKT1 encodes for AKT1 kinase, which regulates cell growth and survival through a variety of pathways (50). AKT1 mutations have been shown to lead to PI3K/AKT pathway activation (51). In a study that applied exome sequencing to 300 meningiomas, mutations in AKT1 were found in 13% of tumors (38). Among skull base meningiomas, AKT1 mutations were found at a higher frequency of 30% and was shown to be associated with shorter time to recurrence (52). In the same study, mutations in AKT1 were found to activate mTOR and ERK1/2 signaling pathways (52). AKT inhibitors have been shown to downregulate the expression of osteoglycin (OGN), an oncogene implicated in meningioma growth, in vitro and to stabilize meningotheliomatous meningioma growth in the lung of a patient with multiple intra- and extra-cranial tumors (53, 54).
SMO is a G-protein coupled receptor involved in the Hedgehog (Hh) signaling pathway (55). Mutations in SMO have been detected in 3 to 6% of all meningiomas, 28% of olfactory groove meningiomas, and 11% of anterior skull base meningiomas (56–59). Compared to meningiomas with AKT1 mutations, SMO-mutated olfactory groove meningiomas had higher recurrence rates, and when compared to AKT1-mutated or wild type meningiomas, SMO-mutated anterior skull base meningiomas had significantly larger tumor volume (58, 59). Given the targetable nature of SMO mutations, a clinical trial is currently underway to test the SMO inhibitor vismodegib in SMO-mutant meningiomas (19).
PIK3CA encodes for a catalytic subunit of phosphatidylinositol 3-kinase (PI3K) that has been implicated in several human cancers (60). Mutations in PIK3CA are estimated to occur in 7% of non-NF2 mutated meningiomas and tend to be mutually exclusive with aforementioned mutations in AKT1 and SMO (57). In a study assessing 55 meningioma patient samples, PIK3CA mutations were found in two patients who had atypical and anaplastic meningiomas respectively (61). PI3K alterations have also been seen to co-occur with TRAF7 mutations, with these tumors demonstrating lower levels of chromosomal instability and clinical tendencies to arise in the skull base (57). Targeting PIK3CA has long been an area of therapeutic interest given the role that increased protein expression in the PI3K/AKT pathway plays in more aggressive malignant meningiomas (62). Two phase II clinical trials are currently underway targeting the PI3K/AKT pathway with vistusertib and capivasertib respectively (19, 63).
The cyclin-dependent kinase inhibitor A and B (CDKN2A/B) gene encodes for three tumor suppressor proteins, the loss of which has been demonstrated to contribute to spontaneous development of melanomas in CDKN2A/B knockout mice (64). In addition to NF2 inactivation, loss of CDKN2A/B contributes to meningioma progression and has been associated with shorter time to recurrence in mice (65). Among a series of 17 recurrent and 13 non-recurrent meningiomas, CDKN2A/B alterations were found only in recurrent meningiomas, and a novel SNV (p.Ala148Thr) was identified in 5 recurrent meningiomas, further supporting the association between CDKN2A/B alterations and meningioma recurrence (66). Along with TERT promoter mutations, CDKN2AB alterations are now included in categorizing grade III meningiomas (8).
Changes in SUFU lead to dysregulation of the hedgehog (Hh) signaling pathway, the activation of which has been shown to play a critical role in meningioma growth and development, with 72% of Hh signaling pathway genes being differentially expressed in meningiomas compared to normal tissue (67). Across 850 meningiomas that underwent genomic analyses, SUFU mutations were identified in 23 patients and seen to co-occur with PTEN and ARID1A mutations (68).
POLR2A, the catalytic subunit of RNA polymerase II, has been shown to harbor mutations that characterize a distinct subset of meningiomas that lack the aforementioned mutations commonly seen in other meningiomas (69). Meningiomas with mutations in POLR2A were exclusively benign with distinct meningothelial histology and were more likely to arise from the tuberculum sellae (69).
There are several copy number variations (CNVs) associated with meningioma pathology (Table 3). The initial loss of chromosome 22q in meningioma tumorigenesis has long been established as an early chromosomal event linked to both NF2 mutated and non-NF2 mutated tumors (3, 70). It is estimated to be found in 60 to 70% of all meningiomas, with significantly increased odds among older patients (71, 72). While the loss of 22q occurs in many patients with established neurofibromatosis type 2 syndrome, somatic mutations have also been discovered in around 47% of sporadic meningiomas (73, 74).

Table 3 Commonly identified cytogenetic alterations in meningiomas.
In the 40% of meningiomas that do not have NF2 inactivation, alternative explanations are sought to explain meningioma tumorigenesis (75). Losses in chromosomal locations 1p and 14q have been identified in higher grade meningiomas (71). Specifically, loss of 1p is the second most common chromosomal event after loss of 22q, and it has been linked to higher rates of tumor recurrence and progression (76). The frequency of mutations in 1p increases roughly from 30% in grade 1 (2000 WHO classification) tumors to 80% and 100% of grade 2 and 3 tumors respectively (76). Losses in 14q are the third most common cytogenetic change detected among meningiomas, with similar frequencies among WHO grade 1 through 3 tumors as 1p losses (76). Similar to 1p losses, a loss at chromosomal arm 14q has also been correlated to increased risk of tumor recurrence (77).
The concurrent loss of 1p and 3p has also been detected in a small subset of meningiomas without detectable losses in chromosome 22. These losses are hypothesized to contribute to meningioma growth, as changes in chromosome 3 have been linked to other cancers such as breast and small cell lung cancer (78, 79). Other chromosomes such as 6 have been found to harbor genes such as HIST1H1C and CTGF, changes in which are associated with meningioma recurrence (80). Loss of heterozygosity at certain sites of chromosome 10 have also been predictive of recurrence and worse prognosis in meningioma patients (81).
Greater number of chromosomal anomalies within a tumor has also been associated with higher tumor grade. For instance, in Pfisterer et al., the distribution of chromosomal 1, 14, and 22 anomalies was examined among 77 meningioma cases (82). The loss of 1p, 14q, or 22q alone was only found in grade I meningiomas, while 23% of meningiomas with both 1p and 14q deletions were grade II meningiomas, and 80% of meningiomas with losses in all three chromosomes were grade III (82). In the literature, losses of 1p were found in 75% of high-grade versus 23% of low-grade tumors, 14q losses in 67% of high-grade versus 31% of benign tumors, and chromosomal 22 deletions in 47% of grade II versus 19% of grade I tumors (77, 83). Bi et al. examined the genomes of 39 high-grade meningiomas and found that high-grade tumors were more likely to have loss of chromosome 22 and 1p than low-grade meningiomas, with high-grade meningiomas also commonly exhibiting losses of 1p, 6q, 10q, 18q and gains of 17q and 20q (84).
Meningiomas are one of the most common tumors to arise after radiotherapy, particularly in the pediatric population (85, 86). Radiation-induced meningiomas have a tendency to behave more aggressively than sporadic meningiomas and often arise two decades after radiation treatment (87, 88). Unlike sporadic meningiomas that commonly harbor NF2 mutations, the same mutations are not seen in radiation-induced meningiomas (87). Instead, loss of chromosomal segment 1p was found to play a larger role in development of radiation-induced meningiomas, followed by changes in chromosomal locations 9p, 19q, and 22q (87). Among 16 patients with radiation-induced meningiomas, cytogenetic analyses revealed changes in chromosome 1p in 89% of cases and changes in chromosome 6 in 67% of cases (89). Further work exploring the genetics underlying the aggressive behavior of radiation-induced meningiomas may shed light on how to best distinguish benign from malignant sporadic meningiomas.
Epigenetic changes have been found to be useful biomarkers of cancer diagnosis and predictors of recurrence and treatment response. For instance, alterations in hypermethylation of O6-methylguanine DNA methyltransferase (MGMT) in glioblastoma can be useful indicators of chemotherapy response (90). In meningiomas, the absence of trimethylation of H3K27 (H3K27me3) has been shown to be associated with more aggressive growth of tumor, as well as NF2 and SUFU mutations, allowing us to further stratify grade 1 and 2 tumors according to the 2016 WHO classification system (91). In another study examining 1268 meningiomas, the loss of H3K27me3 was found to be a significant predictor of negative prognosis and recurrence, further underlying the importance of methylation profiling in categorizing meningiomas (92).
Methylation profiling has taken on a similar role in identifying and distinguishing meningiomas. When compared to bulk RNA-sequencing, DNA methylation profiling was found to more accurately identify meningioma metastases to the liver in one case report (93). While both bulk RNA-sequencing and DNA methylation profiling could separate the liver metastasis from the primary intracranial meningioma, DNA methylation could better establish the diagnosis of the liver metastasis as a meningioma while hepatocyte-specific gene expression confounded similar findings using bulk RNA-sequencing (93). However, it is important to note that these findings were in the context of one case report, and the improved accuracy of DNA methylation profiling in this instance cannot be generalized. In another study, application of DNA methylation profiling to more than 3000 meningiomas identified an epigenetically distinct cluster of 31 tumors, the majority of which were histopathologically diagnosed as clear cell meningiomas (94). Several studies that have integrated methylation studies with other techniques to classify meningiomas will be discussed in the upcoming section.
Advances in computational molecular profiling techniques have allowed for new classifications of meningiomas that account for findings at the DNA level rather than histopathological analysis (95). A variety of techniques including sequencing, methylation profiles, and copy number variation analysis have also been used to generate scores that may better predict prognosis in meningioma patients when compared to standard WHO grading systems.
Genomic sequencing analyses have also been utilized to further classify meningiomas. Patel et al. applied bulk RNA-sequencing and whole-exome sequencing to 160 tumors from 140 meningioma patients to identify 3 classes of meningiomas that were found to predict recurrence more accurately than the standard 2016 WHO grading system (96). Among the three groups (designated type A, B, and C), type C had the highest MIB1 proliferative index, the highest proportion of men, and the shortest recurrence-free survival despite containing primarily WHO grade 1 tumors (96). Type C tumors were also found to have increased expression of FOXM1, leading to loss of the repressive DREAM complex (96). Work by Vasudevan et al. similarly revealed two distinct groups of 280 meningiomas, with aggressive tumors marked by increased expression of FOXM1 (97). FOXM1 has also been implicated as one of three genes upregulated in primary atypical meningiomas, which have also been found to demonstrate NF2 loss, genomic instability, mutations in SMARCB1, and a hypermethylated phenotype (98).
Genomic analyses applied to aggressive meningiomas have also identified 3 distinct groups of meningiomas organized by NF2 status: NF2-mutant, NF2-agnostic, and NF2-wild type (68). NF2-mutant meningiomas were more often associated with men and mutations in CDKN2A/B while NF2-agnostic meningiomas were often associated with TERT and TP53 mutations (68). The third group of NF2-wild type tumors predominantly lacked NF2 mutations and were split further into 3 subgroups: those containing chromatin regulator mutations in BAP1 or PBRM1, skull-based meningiomas with AKT1, PIK3CA, and SMO mutations, and meningiomas with a mix of mutations that shared no discernable pattern (68). Genomic analysis of 300 meningiomas by Clark et al. also revealed benign tumors at the skull base express mutations in TRAF7, KLF4, AKT1, and SMO while higher grade tumors often contained NF2 mutations and were found at the cerebral and cerebellar hemispheres (38).
Genome-wide DNA methylation profiles on 497 meningiomas revealed 6 distinct methylation classes that were found to predict meningioma progression more accurately when compared to 2016 WHO grade 1 tumors and meningioma recurrence more accurately when compared to 2016 WHO grade 2 tumors (99). Similarly, Nassiri et al. applied copy number variation analysis, somatic point mutations, methylation profiles, and messenger RNA abundance to 121 meningioma patient samples to create four molecular groups of meningiomas: immunogenic, benign NF2 wild-type, hypermetabolic, and proliferative (100). The four groups were able to predict patient outcomes more accurately when compared to the existing 2016 WHO classification system and to classification systems based off DNA methylation (100). Methylation analysis by Olar et al. revealed two distinct groups with favorable and unfavorable prognoses respectively (101). Tumors in the unfavorable prognosis group were found to have a higher proportion of copy number aberrations than those in the favorable prognosis group, including losses of 1p and 14q (101).
The integration of histologic findings with genetic profiling including methylation profiling and copy number analysis has further helped improve the precision of meningioma stratification. Using data across 3031 meningiomas, Maas et al. developed an integrated score that predicted risk of meningioma progression more accurately than existing 2016 WHO grading by classifying tumors into low, intermediate, and high-risk groups based off of histology, methylation classes, and CNV analysis (102). While CNV- and methylation-based subtypes independently demonstrated increased prediction accuracy compared to the existing 2016 WHO grading system, the integrated score resulted in further improved accuracy, emphasizing the value of both histology and molecular risk stratification in meningiomas (102).
Integrated use of DNA methylation, RNA sequencing, and cytogenetic profiling by Patel et al. on 110 grade 1 and II meningiomas according to the 2016 WHO classification system revealed two benign and one malignant tumor group closely resembling the previously established type A, B, and C classifications (103). Methylation analysis further distinguished these groups as tumors with balanced methylation (Meth 1), hypomethylation (Meth 2), and hypermethylation (Meth 3) (103). When comparing these groups to those established by Olar et al., Meth 2 and 3 tumors were conferred a favorable and unfavorable prognosis respectively, corresponding with clinical outcomes (101, 103). Further analysis revealed 75% of tumors classified via methylation analysis corresponded to a tumor group established by either transcriptional classification (type A, B, C) or cytogenetic classification (no loss, 22q loss, 1p/22q loss) (103).
In a similar vein, Driver et al. created an integrated scoring system by combining chromosomal losses, CDKN2A losses, and mitotic count to separate meningiomas into three separate groups (104). The subsequent grading system reclassified 32% of meningiomas to a higher or low grade when compared to their original WHO grade and was able to more reliably predict risk of recurrence compared to the existing 2007 and 2016 WHO grading system (104). Key molecular alterations associated with high grade included higher mitotic count, 1p del, 3p del, 4p/q del, 6 p/q del, 10 p/q del, 14q del, 18 p/q del, 19 p/q del, and del CDKN2A/B (104). While both Driver and Maas et al. emphasized 22q, 1p, 6q, and 14q loss as the most frequent deletions encountered, Driver et al. also included 3p, 4p/q, 10p/q, 18p/q, 19p/q alterations and mitotic count instead of methylation families in their scoring system (102, 104).
Most recently, DNA methylation profiling on 565 meningiomas was performed and integrated with single cell, proteomic, and other genetic, transcriptomic, biochemical approaches to categorize meningiomas into three distinct clinical groups: merlin-intact meningiomas, immune-enriched meningiomas, and hypermitotic meningiomas (105). Merlin-intact meningiomas were characterized by gain of function in chromosome 5, loss of function in chromosome 6p, and intact NF2 expression, with the best overall survival among the three groups (105). Immune-enriched meningiomas exhibited gain of function in 6p and loss of function in 22q, and notably, lymphocytes in these tumor microenvironments were prone to exhibiting exhaustion markers, potentially explaining why immune checkpoint blockade has not had the same effect on survival in meningiomas as it has in other cancers (106). Hypermitotic meningiomas had the worst overall survival of all three groups and were distinguished by upregulation of FOXM1 expression, which was found to regulate DNA damage response, potentially increasing hypermitotic meningioma resistance to cytotoxic therapy (105).
While the information afforded by integrated scoring systems is an essential first step to guiding meningioma management, many questions remain to be answered particularly regarding adjuvant therapies and post-operative imaging follow-up. For instance, while most studies do not include Simpson grade or gross versus subtotal resection as part of their grading criteria, these are essential factors that guide decisions regarding post-operative management and consideration of radiotherapy. It is important to determine whether there are specific genetic changes that make a tumor amenable to systemic therapies such as upfront radiation versus observation. Since new classification schemes including the recent changes to the WHO grading system may alter how aggressively a tumor is treated, it is also important to determine whether new classification systems are changing outcomes such as recurrence rates.
In addition, with several novel integrated systems of meningioma classification, it will be crucial to compare the accuracy of different systems through further investigation, including cross study group comparisons. Another potential factor limiting the widespread use of novel integrated classification systems is the high cost and limited availability of some of the technologies utilized, including DNA methylation and next generation sequencing. When applying these technologies to categorize meningiomas, it is also important to take note of intratumoral heterogeneity among meningiomas, with spatially distinct areas of the same tumor exhibiting distinct gene expression patterns (107). Moving forward, it is important to determine how the new groupings afforded by integrated scoring systems may change future management of meningiomas.
Meningiomas are the most common primary intracranial neoplasm and among the most genetically well-studied intracranial tumors. While pre-existing classification schemes by the WHO have traditionally been used to predict meningioma prognosis and risk of recurrence, advances in molecular profiling have allowed for development of several new classification schemes utilizing DNA-level rather than histopathological findings. It is critical to continue applying new sequencing and computational technologies to better predict meningioma behavior in the clinical setting.
Conceptualization: MA, NB, RM, JY, AH, EW; Writing - Original draft: EW; Writing - revisions: EW, JW; Visualization: AH, EW, DS; Review & editing: MA, NB, RM, JY, AH, JW; Supervision: MA, NB. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Buerki RA, Horbinski CM, Kruser T, Horowitz PM, James CD, Lukas RV. An overview of meningiomas. Future Oncol (2018) 14(21):2161–77. doi: 10.2217/fon-2018-0006
2. SHIBUYA M. Pathology and molecular genetics of meningioma: Recent advances. Neurol Med Chir (Tokyo). (2015) 55(1):14–27. doi: 10.2176/nmc.ra.2014-0233
3. Riemenschneider MJ, Perry A, Reifenberger G. Histological classification and molecular genetics of meningiomas. Lancet Neurol (2006) 5(12):1045–54. doi: 10.1016/S1474-4422(06)70625-1
4. Yamashima T, Sakuda K, Tohma Y, Yamashita J, Oda H, Irikura D, et al. Prostaglandin d synthase (β-trace) in human arachnoid and meningioma cells: Roles as a cell marker or in cerebrospinal fluid absorption, tumorigenesis, and calcification process. J Neurosci (1997) 17(7):2376–82. doi: 10.1523/JNEUROSCI.17-07-02376.1997
5. Yasuda K, Cline C, Vogel P, Onciu M, Fatima S, Sorrentino BP, et al. Drug transporters on arachnoid barrier cells contribute to the blood–cerebrospinal fluid barrier. Drug Metab Dispos (2013) 41(4):923–31. doi: 10.1124/dmd.112.050344
6. Hoffman S, Propp JM, McCarthy BJ. Temporal trends in incidence of primary brain tumors in the united states, 1985-1999. Neuro-Oncol. (2006) 8(1):27–37. doi: 10.1215/S1522851705000323
7. Campbell BA, Jhamb A, Maguire JA, Toyota B, Ma R. Meningiomas in 2009: Controversies and future challenges. Am J Clin Oncol (2009) 32(1):73–85. doi: 10.1097/COC.0b013e31816fc920
8. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncol. (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
9. Brastianos PK, Horowitz PM, Santagata S, Jones RT, McKenna A, Getz G, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet (2013) 45(3):285–9. doi: 10.1038/ng.2526
10. Wang YC, Chuang CC, Wei KC, Chang CN, Lee ST, Wu CT, et al. Long term surgical outcome and prognostic factors of atypical and malignant meningiomas. Sci Rep (2016) 6:35743. doi: 10.1038/srep35743
11. Simpson D. THE RECURRENCE OF INTRACRANIAL MENINGIOMAS AFTER SURGICAL TREATMENT. J Neurol Neurosurg Psychiatry (1957) 20(1):22–39. doi: 10.1136/jnnp.20.1.22
12. van den Bent MJ. Interobserver variation of the histopathological diagnosis in clinical trials on glioma: a clinician’s perspective. Acta Neuropathol (Berl). (2010) 120(3):297–304. doi: 10.1007/s00401-010-0725-7
13. Goutagny S, Bah AB, Henin D, Parfait B, Grayeli AB, Sterkers O, et al. Long-term follow-up of 287 meningiomas in neurofibromatosis type 2 patients: clinical, radiological, and molecular features. Neuro-Oncol (2012) 14(8):1090–6. doi: 10.1093/neuonc/nos129
14. Xu Hm, Gutmann DH. Merlin differentially associates with the microtubule and actin cytoskeleton. J Neurosci Res (1998) 51(3):403–15. doi: 10.1002/(SICI)1097-4547(19980201)51:3<403::AID-JNR13>3.0.CO;2-7
15. Lee S, Karas PJ, Hadley CC, Bayley V JC, Khan AB, Jalali A, et al. The role of Merlin/NF2 loss in meningioma biology. Cancers (2019) 11(11):1633. doi: 10.3390/cancers11111633
16. Loewenstern J, Rutland J, Gill C, Arib H, Pain M, Umphlett M, et al. Comparative genomic analysis of driver mutations in matched primary and recurrent meningiomas. Oncotarget. (2019) 10(37):3506–17. doi: 10.18632/oncotarget.26941
17. Pemov A, Dewan R, Hansen NF, Chandrasekharappa SC, Ray-Chaudhury A, Jones K, et al. Comparative clinical and genomic analysis of neurofibromatosis type 2-associated cranial and spinal meningiomas. Sci Rep (2020) 10(1):12563. doi: 10.1038/s41598-020-69074-z
18. Sahm F, Schrimpf D, Olar A, Koelsche C, Reuss D, Bissel J, et al. TERT promoter mutations and risk of recurrence in meningioma. JNCI J Natl Cancer Inst (2015) 108(5):djv377. doi: 10.1093/jnci/djv377
19. Alliance for Clinical Trials in Oncology. Phase II trial of SMO/ AKT/ NF2/CDK inhibitors in progressive meningiomas with SMO/ AKT/ NF2/CDK pathway mutations (2021). Available at: https://clinicaltrials.gov/ct2/show/NCT02523014.
20. PhD SRP MD. A single arm phase 2 study of the dual mTORC1/mTORC2 inhibitor AZD2014 provided on an intermittent schedule for neurofibromatosis 2 patients with progressive or symptomatic meningiomas (2020). Available at: https://clinicaltrials.gov/ct2/show/NCT02831257.
21. Smith MJ. Germline and somatic mutations in meningiomas. Cancer Genet (2015) 208(4):107–14. doi: 10.1016/j.cancergen.2015.02.003
22. Christiaans I, Kenter SB, Brink HC, Os TAMv, Baas F, Munckhof Pvd, et al. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J Med Genet (2011) 48(2):93–7. doi: 10.1136/jmg.2010.082420
23. Smith MJ, O’Sullivan J, Bhaskar SS, Hadfield KD, Poke G, Caird J, et al. Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat Genet (2013) 45(3):295–8. doi: 10.1038/ng.2552
24. Guerrini-Rousseau L, Dufour C, Varlet P, Masliah-Planchon J, Bourdeaut F, Guillaud-Bataille M, et al. Germline SUFU mutation carriers and medulloblastoma: clinical characteristics, cancer risk, and prognosis. Neuro-Oncol (2018) 20(8):1122–32. doi: 10.1093/neuonc/nox228
25. Taylor MD, Liu L, Raffel C, Hui Cc, Mainprize TG, Zhang X, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet (2002) 31(3):306–10. doi: 10.1038/ng916
26. Guerrini-Rousseau L, Masliah-Planchon J, Waszak SM, Alhopuro P, Benusiglio PR, Bourdeaut F, et al. Cancer risk and tumour spectrum in 172 patients with a germline SUFU pathogenic variation: a collaborative study of the SIOPE host genome working group. J Med Genet (2022) 59(11):1123–32. doi: 10.1136/jmedgenet-2021-108385
27. Aavikko M, Li SP, Saarinen S, Alhopuro P, Kaasinen E, Morgunova E, et al. Loss of SUFU function in familial multiple meningioma. Am J Hum Genet (2012) 91(3):520–6. doi: 10.1016/j.ajhg.2012.07.015
28. Askaner G, Lei U, Bertelsen B, Venzo A, Wadt K. Novel SUFU frameshift variant leading to meningioma in three generations in a family with gorlin syndrome. Case Rep Genet (2019) 2019:e9650184. doi: 10.1155/2019/9650184
29. Ghaleb AM, Yang VW. Krüppel-like factor 4 (KLF4): What we currently know. Gene. (2017) 611:27–37. doi: 10.1016/j.gene.2017.02.025
30. Ohnishi S, Ohnami S, Laub F, Aoki K, Suzuki K, Kanai Y, et al. Downregulation and growth inhibitory effect of epithelial-type krüppel-like transcription factor KLF4, but not KLF5, in bladder cancer. Biochem Biophys Res Commun (2003) 308(2):251–6. doi: 10.1016/S0006-291X(03)01356-1
31. Wei D, Gong W, Kanai M, Schlunk C, Wang L, Yao JC, et al. Drastic down-regulation of krüppel-like factor 4 expression is critical in human gastric cancer development and progression. Cancer Res (2005) 65(7):2746–54. doi: 10.1158/0008-5472.CAN-04-3619
32. Luo A, Kong J, Hu G, Liew CC, Xiong M, Wang X, et al. Discovery of Ca2+-relevant and differentiation-associated genes downregulated in esophageal squamous cell carcinoma using cDNA microarray. Oncogene. (2004) 23(6):1291–9. doi: 10.1038/sj.onc.1207218
33. Dang DT, Chen X, Feng J, Torbenson M, Dang LH, Yang VW. Overexpression of krüppel-like factor 4 in the human colon cancer cell line RKO leads to reduced tumorigenecity. Oncogene (2003) 22(22):3424–30. doi: 10.1038/sj.onc.1206413
34. Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW. Identification of krüppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene (2004) 23(2):395–402. doi: 10.1038/sj.onc.1207067
35. Reuss DE, Piro RM, Jones DTW, Simon M, Ketter R, Kool M, et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol (Berl) (2013) 125(3):351–8. doi: 10.1007/s00401-013-1093-x
36. Tang H, Zhu H, Wang X, Hua L, Li J, Xie Q, et al. KLF4 is a tumor suppressor in anaplastic meningioma stem-like cells and human meningiomas. J Mol Cell Biol (2017) 9(4):315–24. doi: 10.1093/jmcb/mjx023
37. von Spreckelsen N, Waldt N, Poetschke R, Kesseler C, Dohmen H, Jiao HK, et al. KLF4K409Q–mutated meningiomas show enhanced hypoxia signaling and respond to mTORC1 inhibitor treatment. Acta Neuropathol Commun (2020) 8(1):41. doi: 10.1186/s40478-020-00912-x
38. Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Özduman K, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Sci [Internet] (2013) 7(1):1–8. doi: 10.1126/science.1233009
39. Zhang Q, Zhang X, Dong W. TRAF7 contributes to tumor progression by promoting ubiquitin-proteasome mediated degradation of P53 in hepatocellular carcinoma. Cell Death Discov (2021) 7(1):1–8. doi: 10.1038/s41420-021-00749-w
40. Wang L, Wang L, Zhang S, Qu G, Zhang D, Li S, et al. Downregulation of ubiquitin E3 ligase TNF receptor-associated factor 7 leads to stabilization of p53 in breast cancer. Oncol Rep (2013) 29(1):283–7. doi: 10.3892/or.2012.2121
41. Najm P, Zhao P, Steklov M, Sewduth RN, Baietti MF, Pandolfi S, et al. Loss-of-Function mutations in TRAF7 and KLF4 cooperatively activate RAS-like GTPase signaling and promote meningioma development. Cancer Res (2021) 81(16):4218–29. doi: 10.1158/0008-5472.CAN-20-3669
42. Jin L, Youngblood MW, Gupte TP, Vetsa S, Nadar A, Barak T, et al. Type of bony involvement predicts genomic subgroup in sphenoid wing meningiomas. J Neurooncol. (2021) 154(2):237–46. doi: 10.1007/s11060-021-03819-2
43. Dratwa M, Wysoczańska B, Łacina P, Kubik T, Bogunia-Kubik K. TERT–regulation and roles in cancer formation. Front Immunol (2020) 11:2930. doi: 10.3389/fimmu.2020.589929
44. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. (2013) 339(6122):957–9. doi: 10.1126/science.1229259
45. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT promoter mutations in familial and sporadic melanoma. Sci [Internet] (2013) 5(6):1515–25. doi: 10.1126/science.1230062
46. Killela PJ, Pirozzi CJ, Healy P, Reitman ZJ, Lipp E, Rasheed BA, et al. Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget (2014) 5(6):1515–25. doi: 10.18632/oncotarget.1765
47. Goutagny StéChecktae, Nault J, Mallet M, Henin D, Rossi J, Kalamarides M. High incidence of activating TERT promoter mutations in meningiomas undergoing malignant progression. Brain Pathol (2014) 108(5):184–9. doi: 10.1111/bpa.12110
48. Spiegl-Kreinecker S, Lötsch D, Neumayer K, Kastler L, Gojo J, Pirker C, et al. TERT promoter mutations are associated with poor prognosis and cell immortalization in meningioma. Neuro-Oncol (2018) 20(12):1584–93. doi: 10.1093/neuonc/noy104
49. Mirian C, Duun-Henriksen AK, Juratli T, Sahm F, Spiegl-Kreinecker S, Peyre M, et al. Poor prognosis associated with TERT gene alterations in meningioma is independent of the WHO classification: an individual patient data meta-analysis. J Neurol Neurosurg Psychiatry (2020) 91(4):378–87. doi: 10.1136/jnnp-2019-322257
50. Duggal S, Jailkhani N, Midha MK, Agrawal N, Rao KVS, Kumar A. Defining the Akt1 interactome and its role in regulating the cell cycle. Sci Rep (2018) 8(1):1303. doi: 10.1038/s41598-018-19689-0
51. Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. (2007) 448(7152):439–44. doi: 10.1038/nature05933
52. Yesilöz Ü, Kirches E, Hartmann C, Scholz J, Kropf S, Sahm F, et al. Frequent AKT1E17K mutations in skull base meningiomas are associated with mTOR and ERK1/2 activation and reduced time to tumor recurrence. Neuro-Oncol. (2017) 19(8):1088–96. doi: 10.1093/neuonc/nox018
53. Weller M, Roth P, Sahm F, Burghardt I, Schuknecht B, Rushing EJ, et al. Durable control of metastatic AKT1-mutant WHO grade 1 meningothelial meningioma by the AKT inhibitor, AZD5363. JNCI J Natl Cancer Inst (2017) 109(3):djw320. doi: 10.1093/jnci/djw320
54. Mei Y, Du Z, Hu C, Greenwald NF, Abedalthagafi M, Agar NYR, et al. Osteoglycin promotes meningioma development through downregulation of NF2 and activation of mTOR signaling. Cell Commun Signal (2017) 15(1):34. doi: 10.1186/s12964-017-0189-7
55. Ruiz-Gómez A, Molnar C, Holguín H, Mayor F, de Celis JF. The cell biology of smo signalling and its relationships with GPCRs. Biochim Biophys Acta BBA - Biomembr. (2007) 1768(4):901–12. doi: 10.1016/j.bbamem.2006.09.020
56. Yuzawa S, Nishihara H, Tanaka S. Genetic landscape of meningioma. Brain Tumor Pathol (2016) 33(4):237–47. doi: 10.1007/s10014-016-0271-7
57. Abedalthagafi M, Bi WL, Aizer AA, Merrill PH, Brewster R, Agarwalla PK, et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro-Oncol (2016) 18(5):649–55. doi: 10.1093/neuonc/nov316
58. Boetto J, Bielle F, Sanson M, Peyre M, Kalamarides M. SMO mutation status defines a distinct and frequent molecular subgroup in olfactory groove meningiomas. Neuro-Oncol (2017) 19(3):345–51. doi: 10.1093/neuonc/now276
59. Strickland MR, Gill CM, Nayyar N, D’Andrea MR, Thiede C, Juratli TA, et al. Targeted sequencing of SMO and AKT1 in anterior skull base meningiomas. J Neurosurg (2017) 127(2):438–44. doi: 10.3171/2016.8.JNS161076
60. Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer. (2006) 94(4):455–9. doi: 10.1038/sj.bjc.6602970
61. Bujko M, Kober P, Tysarowski A, Matyja E, Mandat T, Bonicki W, et al. EGFR, PIK3CA, KRAS and BRAF mutations in meningiomas. Oncol Lett (2014) 7(6):2019–22. doi: 10.3892/ol.2014.2042
62. Mawrin C, Sasse T, Kirches E, Kropf S, Schneider T, Grimm C, et al. Different activation of mitogen-activated protein kinase and akt signaling is associated with aggressive phenotype of human meningiomas. Clin Cancer Res (2005) 11(11):4074–82. doi: 10.1158/1078-0432.CCR-04-2550
63. PhD SRP MD. A single arm phase II study of the dual mTORC1/mTORC2 inhibitor vistusertib (AZD2014) provided on an intermittent schedule for sporadic patients with grade II-III meningiomas that recur or progress after surgery and radiation [Internet] (2021). Available at: https://clinicaltrials.gov/ct2/show/NCT03071874.
64. Krimpenfort P, Snoek M, Lambooij JP, Song JY, van der Weide R, Bhaskaran R, et al. A natural WNT signaling variant potently synergizes with Cdkn2ab loss in skin carcinogenesis. Nat Commun (2019) 10(1):1425. doi: 10.1038/s41467-019-09321-8
65. Peyre M, Stemmer-Rachamimov A, Clermont-Taranchon E, Quentin S, El-Taraya N, Walczak C, et al. Meningioma progression in mice triggered by Nf2 and Cdkn2ab inactivation. Oncogene (2013) 32(36):4264–72. doi: 10.1038/onc.2012.436
66. Guyot A, Duchesne M, Robert S, Lia AS, Derouault P, Scaon E, et al. Analysis of CDKN2A gene alterations in recurrent and non-recurrent meningioma. J Neurooncol (2019) 145(3):449–59. doi: 10.1007/s11060-019-03333-6
67. Laurendeau I, Ferrer M, Garrido D, D’Haene N, Ciavarelli P, Basso A, et al. Gene expression profiling of the hedgehog signaling pathway in human meningiomas. Mol Med (2010) 16(7):262–70. doi: 10.2119/molmed.2010.00005
68. Williams EA, Santagata S, Wakimoto H, Shankar GM, Barker FG, Sharaf R, et al. Distinct genomic subclasses of high-grade/progressive meningiomas: NF2-associated, NF2-exclusive, and NF2-agnostic. Acta Neuropathol Commun (2020) 8(1):171. doi: 10.1186/s40478-020-01040-2
69. Clark VE, Harmancı AS, Bai H, Youngblood MW, Lee TI, Baranoski JF, et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat Genet (2016) 48(10):1253–9. doi: 10.1038/ng.3651
70. Choy W, Kim W, Nagasawa D, Stramotas S, Yew A, Gopen Q, et al. The molecular genetics and tumor pathogenesis of meningiomas and the future directions of meningioma treatments. Neurosurg Focus (2011) 30(5):E6. doi: 10.3171/2011.2.FOCUS1116
71. Mawrin C, Perry A. Pathological classification and molecular genetics of meningiomas. J Neurooncol (2010) 99(3):379–91. doi: 10.1007/s11060-010-0342-2
72. Baser ME, Poussaint TY. Age associated increase in the prevalence of chromosome 22q loss of heterozygosity in histological subsets of benign meningioma. J Med Genet (2006) 43(3):285–7. doi: 10.1136/jmg.2005.035162
73. Hansson CM, Buckley PG, Grigelioniene G, Piotrowski A, Hellström AR, Mantripragada K, et al. Comprehensive genetic and epigenetic analysis of sporadic meningioma for macro-mutations on 22q and micro-mutations within the NF2 locus. BMC Genomics (2007) 8(1):16. doi: 10.1186/1471-2164-8-16
74. Heinrich B, Hartmann C, Stemmer-Rachamimov AO, Louis DN, MacCollin M. Multiple meningiomas: Investigating the molecular basis of sporadic and familial forms. Int J Cancer (2003) 103(4):483–8. doi: 10.1002/ijc.10840
75. Carlson KM, Bruder C, Nordenskjöld M, Dumanski JP. 1p and 3p deletions in meningiomas without detectable aberrations of chromosome 22 identified by comparative genomic hybridization. Genes Chromosomes Cancer. (1997) 20(4):419–24. doi: 10.1002/(SICI)1098-2264(199712)20:4<419::AID-GCC15>3.0.CO;2-H
76. Lamszus K. Meningioma pathology, genetics, and biology. J Neuropathol Exp Neurol (2004) 63(4):275–86. doi: 10.1093/jnen/63.4.275
77. Cai DX, Banerjee R, Scheithauer BW, Lohse CM, Kleinschmidt-Demasters BK, Perry A. Chromosome 1p and 14q FISH analysis in clinicopathologic subsets of meningioma: Diagnostic and prognostic implications. J Neuropathol Exp Neurol (2001) 60(6):628–36. doi: 10.1093/jnen/60.6.628
78. Martinez A, Walker RA, Shaw JA, Dearing SJ, Maher ER, Latif F. Chromosome 3p allele loss in early invasive breast cancer: detailed mapping and association with clinicopathological features. Mol Pathol (2001) 54(5):300–6. doi: 10.1136/mp.54.5.300
79. Daly MC, Xiang RH, Buchhagen D, Hensel CH, Garcia DK, Killary AM, et al. A homozygous deletion on chromosome 3 in a small cell lung cancer cell line correlates with a region of tumor suppressor activity. Oncogene. (1993) 8(7):1721–9.
80. He S, Pham MH, Pease M, Zada G, Giannotta SL, Wang K, et al. A review of epigenetic and gene expression alterations associated with intracranial meningiomas. Neurosurg Focus. (2013) 35(6):E5. doi: 10.3171/2013.10.FOCUS13360
81. Mihaila D, Jankowski M, Gutiérrez JA, Rosenblum ML, Newsham IF, Bögler O, et al. Meningiomas: loss of heterozygosity on chromosome 10 and marker-specific correlations with grade, recurrence, and survival. Clin Cancer Res Off J Am Assoc Cancer Res (2003) 9(12):4443–51.
82. Pfisterer WK, Hank NC, Preul MC, Hendricks WP, Pueschel J, Coons SW, et al. Diagnostic and prognostic significance of genetic regional heterogeneity in meningiomas. Neuro-Oncol. (2004) 6(4):290–9. doi: 10.1215/S1152851704000158
83. Zattara-Cannoni H, Gambarelli D, Dufour H, Figarella D, Vollot F, Grisoli F, et al. Contribution of cytogenetics and FISH in the diagnosis of meningiomas. a study of 189 tumors. Ann Genet (1998) 41(3):164–75.
84. Bi WL, Greenwald NF, Abedalthagafi M, Wala J, Gibson WJ, Agarwalla PK, et al. Genomic landscape of high-grade meningiomas. NPJ Genomic Med (2017) 2(1):1–14. doi: 10.1038/s41525-017-0014-7
85. Neglia JP, Robison LL, Stovall M, Liu Y, Packer RJ, Hammond S, et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the childhood cancer survivor study. JNCI J Natl Cancer Inst (2006) 98(21):1528–37. doi: 10.1093/jnci/djj411
86. Broniscer A, Ke W, Fuller CE, Wu J, Gajjar A, Kun LE. Second neoplasms in pediatric patients with primary central nervous system tumors. Cancer. (2004) 100(10):2246–52. doi: 10.1002/cncr.20253
87. Shoshan Y, Chernova O, Juen SS, Somerville RP, Israel Z, Barnett GH, et al. Radiation-induced meningioma: a distinct molecular genetic pattern? J Neuropathol Exp Neurol (2000) 59(7):614–20. doi: 10.1093/jnen/59.7.614
88. Yamanaka R, Hayano A, Kanayama T. Radiation-induced meningiomas: An exhaustive review of the literature. World Neurosurg (2017) 97:635–644.e8. doi: 10.1016/j.wneu.2016.09.094
89. Al-Mefty O, Topsakal C, Pravdenkova S, Sawyer JR, Harrison MJ. Radiation-induced meningiomas: clinical, pathological, cytokinetic, and cytogenetic characteristics. J Neurosurg (2004) 100(6):1002–13. doi: 10.3171/jns.2004.100.6.1002
90. Heyn H, Esteller M. DNA Methylation profiling in the clinic: applications and challenges. Nat Rev Genet (2012) 13(10):679–92. doi: 10.1038/nrg3270
91. Katz LM, Hielscher T, Liechty B, Silverman J, Zagzag D, Sen R, et al. Loss of histone H3K27me3 identifies a subset of meningiomas with increased risk of recurrence. Acta Neuropathol (Berl). (2018) 135(6):955–63. doi: 10.1007/s00401-018-1844-9
92. Behling F, Fodi C, Gepfner-Tuma I, Kaltenbach K, Renovanz M, Paulsen F, et al. H3K27me3 loss indicates an increased risk of recurrence in the tübingen meningioma cohort. Neuro-Oncol (2021) 23(8):1273–81. doi: 10.1093/neuonc/noaa303
93. Vasudevan HN, Castro MRH, Lee JC, Villanueva-Meyer JE, Bush NAO, McDermott MW, et al. DNA Methylation profiling demonstrates superior diagnostic classification to RNA-sequencing in a case of metastatic meningioma. Acta Neuropathol Commun (2020) 8:82. doi: 10.1186/s40478-020-00952-3
94. Sievers P, Sill M, Blume C, Tauziede-Espariat A, Schrimpf D, Stichel D, et al. Clear cell meningiomas are defined by a highly distinct DNA methylation profile and mutations in SMARCE1. Acta Neuropathol (Berl) (2021) 141(2):281–90. doi: 10.1007/s00401-020-02247-2
95. Bi WL, Abedalthagafi M, Horowitz P, Agarwalla PK, Mei Y, Aizer AA, et al. Genomic landscape of intracranial meningiomas. J Neurosurg (2016) 125(3):525–35. doi: 10.3171/2015.6.JNS15591
96. Patel AJ, Wan YW, Al-Ouran R, Revelli JP, Cardenas MF, Oneissi M, et al. Molecular profiling predicts meningioma recurrence and reveals loss of DREAM complex repression in aggressive tumors. Proc Natl Acad Sci (2019) 116(43):21715–26. doi: 10.1073/pnas.1912858116
97. Vasudevan HN, Braunstein SE, Phillips JJ, Pekmezci M, Tomlin BA, Wu A, et al. Comprehensive molecular profiling identifies FOXM1 as a key transcription factor for meningioma proliferation. Cell Rep (2018) 22(13):3672–83. doi: 10.1016/j.celrep.2018.03.013
98. Harmancı AS, Youngblood MW, Clark VE, Coşkun S, Henegariu O, Duran D, et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat Commun (2017) 8(1):14433. doi: 10.1038/ncomms14433
99. Sahm F, Schrimpf D, Stichel D, Jones DTW, Hielscher T, Schefzyk S, et al. DNA Methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol (2017) 18(5):682–94. doi: 10.1016/S1470-2045(17)30155-9
100. Nassiri F, Liu J, Patil V, Mamatjan Y, Wang JZ, Hugh-White R, et al. A clinically applicable integrative molecular classification of meningiomas. Nature. (2021) 597(7874):119–25. doi: 10.1038/s41586-021-03850-3
101. Olar A, Wani KM, Wilson CD, Zadeh G, DeMonte F, Jones DTW, et al. Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol (Berl). (2017) 133(3):431–44. doi: 10.1007/s00401-017-1678-x
102. Maas SLN, Stichel D, Hielscher T, Sievers P, Berghoff AS, Schrimpf D, et al. Integrated molecular-morphologic meningioma classification: A multicenter retrospective analysis, retrospectively and prospectively validated. J Clin Oncol (2021) 39(34):3839–52. doi: 10.1200/JCO.21.00784
103. Bayley JC, Hadley CC, Harmanci AO, Harmanci AS, Klisch TJ, Patel AJ. Multiple approaches converge on three biological subtypes of meningioma and extract new insights from published studies. Sci Adv (2022) 8(5):eabm6247. doi: 10.1126/sciadv.abm6247
104. Driver J, Hoffman SE, Tavakol S, Woodward E, Maury EA, Bhave V, et al. A molecularly integrated grade for meningioma. Neuro-Oncol. (2021) 11:noab213. doi: 10.14791/btrt.2022.10.F-2747
105. Choudhury A, Magill ST, Eaton CD, Prager BC, Chen WC, Cady MA, et al. Meningioma DNA methylation groups identify biological drivers and therapeutic vulnerabilities. Nat Genet (2022) 54(5):649–59. doi: 10.1038/s41588-022-01061-8
106. Bi WL, Nayak L, Meredith DM, Driver J, Du Z, Hoffman S, et al. Activity of PD-1 blockade with nivolumab among patients with recurrent atypical/anaplastic meningioma: phase II trial results. Neuro-Oncol. (2022) 24(1):101–13. doi: 10.1093/neuonc/noab118
Keywords: meningioma, cytogenetics, genomics, epigenetics, methylation, atypical, anaplastic
Citation: Wang EJ, Haddad AF, Young JS, Morshed RA, Wu JPH, Salha DM, Butowski N and Aghi MK (2023) Recent advances in the molecular prognostication of meningiomas. Front. Oncol. 12:910199. doi: 10.3389/fonc.2022.910199
Received: 01 April 2022; Accepted: 17 November 2022;
Published: 04 January 2023.
Edited by:
Hailiang Tang, Fudan University, ChinaReviewed by:
Carlos Eduardo Da Silva, Federal University of Health Sciences of Porto Alegre, BrazilCopyright © 2023 Wang, Haddad, Young, Morshed, Wu, Salha, Butowski and Aghi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manish K. Aghi, bWFuaXNoLmFnaGlAdWNzZi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.