 Hua Liu
Hua Liu Biao Chen
Biao Chen Lian-Lin Liu1
Lian-Lin Liu1 Yong Cheng
Yong Cheng- 1Key Laboratory of Ethnomedicine for Ministry of Education, Center on Translational Neuroscience, School of Pharmacy, Minzu University of China, Beijing, China
- 2Department of General Surgery, People’s Hospital of Tibet Autonomous Region, Lhasa, China
- 3Department of General Surgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
Colorectal cancer (CRC) is the third most common cancer with a high global incidence and mortality. Mutated genes or dysregulated pathways responsible for CRC progression have been identified and employed as biomarkers for diagnosis and prognosis. In this study, a ubiquitination regulator, MARCH9, was shown to accelerate CRC progression both in vitro and in vivo. CRC samples from The Cancer Genome Atlas (TCGA) showed significantly upregulated MARCH9 expression by individual cancer stage, histological subtype, and nodal metastasis status. Knockdown of MARCH9 inhibited, while MARCH9 overexpression promoted, CRC cell proliferation and migration. Knockdown of MARCH9 also induced CRC cell apoptosis and caused cell cycle arrest. Further investigation showed that MARCH9 promoted CRC progression by downregulating the expression of a deubiquitinase cylindromatosis (CYLD) gene and activating p65, a member of the nuclear factor-κB (NF-κB) protein family. Finally, in vivo xenograft studies confirmed that MARCH9 knockdown suppressed tumor growth in nude mice. Thus, this study demonstrated that MARCH9 may be a novel and effective therapeutic target for CRC therapy.
Introduction
According to the International Agency for Research on Cancer (IARC)’s GLOBOCAN 2020 database, there were more than 1.9 million new cases of colorectal cancer (CRC, including anus), with 935,000 deaths and 10% of all new cancer cases and deaths in 2020, respectively (1). In China, CRC is the fifth and fourth leading cause of cancer morbidity among men and women, respectively, the fifth leading cause of cancer mortality for both men and women (2). Current CRC treatment includes surgical therapy, postoperative chemotherapy, neoadjuvant radiotherapy and chemotherapy, maintenance therapy, immunotherapy, or targeted therapy according to each patient’s clinical situation and the known epidemiology in particular regions (3). CRC develops through a series of genetic, histological, and morphological changes that accumulate over time (4). Thus, it is necessary to define the potential mechanism of CRC tumorigenesis and progression.
Several studies have suggested that dysregulation of ubiquitin-mediated degradation may correlate with the occurrence and development of cancers (5). Membrane-associated RING-CH (MARCH) proteins were confirmed as ubiquitination regulators of major histocompatibility complex (MHC) proteins firstly (6). They include 11 family members, MARCH1 to MARCH 11, that play a role in immune regulation, dendritic cell (DC) maturation, antigen presentation, T cell development, thyroid hormone activity regulation, bile transport, endosomal trafficking, cell polarity, spermiogenesis, mitochondrial dynamics, protein quality control, and Toll-like receptor (TLR) signaling (7). Recent studies have found that MARCH protein expression correlates with cancer progression. MARCH1, MARCH2, MARCH3, and MARCH8 are associated with CRC (8–11), MARCH1, MARCH3, and MARCH6 are correlated with hepatocellular carcinoma (12–14), and MARCH1, MARCH5, MARCH7, and MARCH10 are associated with ovarian cancer (15–18).
MARCH9 is found in lysosomes and the trans-Golgi network (TGN) and is highly expressed in lymph tissue, lung tissue, DCs, T cells, and B cells (19–21). MARCH9 may participate in cellular immune regulation by downregulating MHC1, CD4, and ICAM-1 expression (22, 23). Luoto et al. used computational analysis to show that the CDK4-MARCH9 locus was expressed in the glioblastoma immune microenvironment and could potentially be used to facilitate patient stratification and improve personalized immunotherapy (24). Shen et al. reported that MARCH9 suppressed the progression of lung adenocarcinoma, and downregulation correlated with poor clinical outcomes (23). This manuscript explores a potential function and possible mechanism for MARCH9 in CRC.
Materials and methods
Clinical samples from the genome cancer atlas (TGCA)
CRC samples (n=471) and control samples (n=41) were downloaded from the TCGA-Colon Adenocarcinoma (COAD) database and MARCH9 expression was assessed by individual cancer stage (stage 1-stage 4), histological subtype (adenocarcinoma and mucinous adenocarcinoma), and nodal metastasis status (N0-N2).
Cell culture and transfection
For cell lines, the human colonic cancer cell lines, HCT116 and RKO, were chosen and purchased from ATCC. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Hyclone) that was supplemented with 10% fetal bovine serum (FBS, Hyclone) and 1% penicillin-streptomycin solution (Solarbio), and incubated in 5% CO2 (PHcbi, MCO-18AC). Lipofectamine RNA-iMax reagent (Invitrogen) was used for cell transfection. The sequences of siMARCH9#1 (5’-GCAGTGGAAGGTCCTAAATTA-3’), siMARCH9#2 (5’-GTCCAGATTGCTGCCATAGTT-3’) and siCtrl(5’-UUCUCCGAACGUGUCACGU-3’) were synthesized from GenePharma (Shanghai). The VigoFect repeat (Vigorous Biotechnology) was used for MARCH9 overexpression.
Quantitative real-time polymerase chain reaction (RT-qPCR)
The total RNAs of the HCT116 and RKO cells were isolated using Trizol Reagent. (Ambion, Life Technologies). ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo) was used for reverse transcription and 2* SYBR Green qPCR Master Mix (Low ROX) (Servicebio) was used for real-time PCR. The following primers were utilized: MARCH9-F: 5’-CTCCTCTGTCTACCGCATCTT-3’; MARCH9-R: 5’-TCTCCTCCTATGTCCTTGGTCT-3’; GAPDH-F: 5’-GACTCATGACCACAGTCCATGC-3’; and GAPDH-R: 5’-AGAGGCAGGGATGATGTTCTG-3’.
Western blotting
Whole-cell lysates were prepared in RIPA lysis buffer (Beyotime) containing a protease cocktail (Beyotime). BCA Protein Assay Kit (Thermo) was used to evaluate total protein concentrations. Proteins were separated with 10% SDS-PAGE and transferred to a nitrocellulose membrane, and the membrane was blocked with 0.5% bovine serum albumin (BSA) for 1 h at 25°C. Anti-MARCH9 (proteintech, 1:1000), anti-CYLD (Abcam 1:1000), anti-p-p65 (1:1000), and anti-vinculin (ABclonal, 1:1000) antibodies were added to the membranes and incubated at 4°C overnight, separately. After washing three times with TBST, secondary antibodies were added and the membrane was incubated for 1 h at 25°C. IRDye@800CW reagent (LI-COR) and Odyssey CLx imager (LI-COR) were used to visualize the protein bands.
Colony formation assay
Two CRC cell lines containing about 300 cells each were planted into 6 cm tissue culture plates and 4 mL DMEM was added. The cells were cultured for 3 weeks to form colonies. The cells were then fixed in 100% methanol for 20 min and stained with 0.1% crystal violet solution (Solarbio) for 10 min. The positive colonies were photographed and quantitated using a digital camera.
Transwell migration assay
Transfected HCT116 and RKO cells were seeded in the upper chamber of a Transwell plate in DMEM containing 10% FBS. DMEM containing 20% FBS was added to the lower chamber. An 8.0 μm pore membrane was inserted to allow the cells to migrate, and the plate was incubated at 37°C for 24 h. The migrated cells were fixed, stained with crystal violet, and ten random fields of each well were counted.
Cell apoptosis and cell cycle assays
Cell apoptosis in transfected cells was analyzed using a FITC-Annexin V apoptosis kit (Yeasen, Shanghai). In brief, cultured cells were suspended, centrifuged, washed with PBS, and resuspended in cold Binding Buffer. FITC-Annexin V reagent (1.25 μL) was added to the cells and apoptotic cells were assessed using a A00-1-1122 flow cytometer (Beckman Kurt Biotechnology (Suzhou) Co., Ltd).
For cell cycle analysis, the transfected cells were fixed with 70% cold ethanol (700 μL) at 4°C for 4 h, washed three times with PBS, and stained with the fluorescent dye propidium iodide (PI; Sigma) at 37°C for 30 min in the dark. The ratio of cells in various cell cycle phases was assessed using a Beckman flow cytometer.
Measurement of caspase 3/7 activity
In brief, a total of 104 siCtrl, siMARCH9#1 and siMARCH9#2 HCT116 and RKO cells were seeded into 96-well plates. 1.5 hours later, the activity of caspase 3/7 was measured by using Caspase-Glo reagent (Promega), according to the manufacturers’ protocols.
Xenograft model assay
Female BALB/c nude mice that were 6 weeks old (Beijing vital-river Co.td) were raised in a pathogen-free facility and divided into a shMARCH9 group (n=5) and a shCtrl group (n=5). The mice were subcutaneously injected with shMARCH9#1 or shCtrl transfected HCT116 cells (4 × 106 cells) in the right axilla. After 45 days, the mice were anesthetized and sacrificed, and the tumors were excised, weighed, and photographed. Tumor tissues were used to detect the expression of MARCH9 by western blotting.
Statistical analysis
All the experimental results was used to analyze in GraphPad Prism 7 software and data were expressed as the mean ± SD. The Student’s t-test was used to compare differences between the groups. Statistical significance was considered at p < 0.05.
Results
MARCH9 was highly expressed in CRC tissue
We firstly explored the clinical significance of MARCH9 in CRC. Based on TCGA data, MARCH9 expression was significantly higher in CRC than adjacent normal tissue samples (Figure 1A). MARCH9 expression was also much higher in CRC stages 1-4 (Figure 1B), adenocarcinoma and mucinous adenocarcinoma (Figure 1C), and N0-N2 samples (Figure 1D).
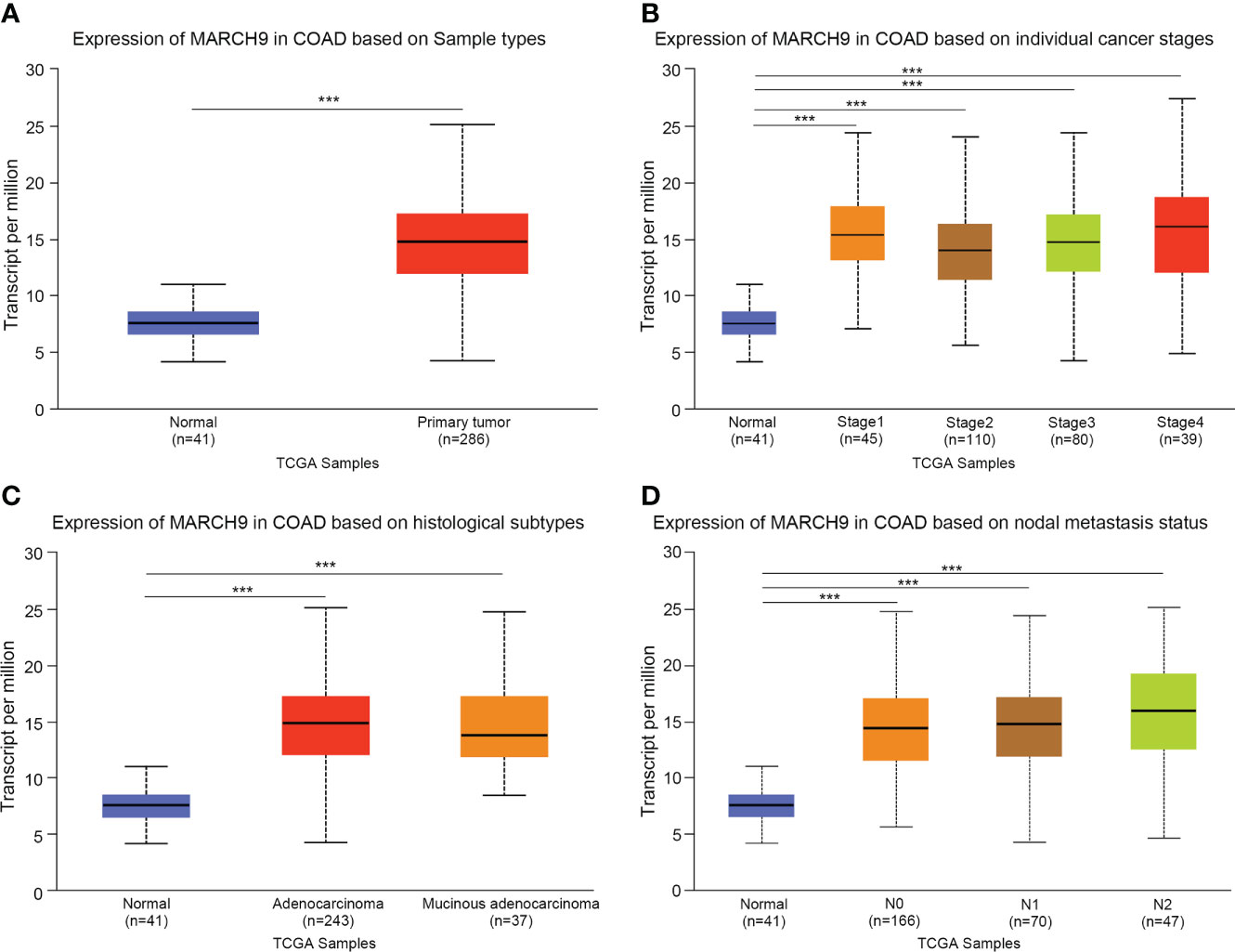
Figure 1 MARCH9 was highly expressed in CRC tissue. (A) Expression of MARCH9 in tumors vs normal controls. (B) Expression of MARCH9 in different cancer stages. (C) Expression of MARCH9 in different histological subtypes. (D) Expression of MARCH9 in nodal metastasis ***p<0.001.
Knockdown of MARCH9 inhibited CRC cell proliferation and migration
Next, we investigated the role of MARCH9 in CRC by using loss-of-function assay. As shown by RT-qPCR and Western blot analysis, both siMARCH9#1 and siMARCH9#2 could effectively knock down MARCH9 expression in two CRC cell lines (Figures 2A, B). The CCK-8 assay was used to investigate whether MARCH9 knockdown influenced CRC cell proliferation. CRC cell lines transfected with siMARCH9#1 and siMARCH9#2 had significantly lower viability than the siCtrl group (Figure 2C). Knockdown of MARCH9 also inhibited the colony formation and migration of CRC cells (Figures 2D, E). We also checked the effect of MARCH9 on the proliferation of colorectal normal cells. The results showed that knockdown of MARCH9 suppressed the proliferation of FHC cells (Supplementary Figure 1). Therefore, MARCH9 expression is critical for the proliferation and growth of colorectal normal and CRC cells.
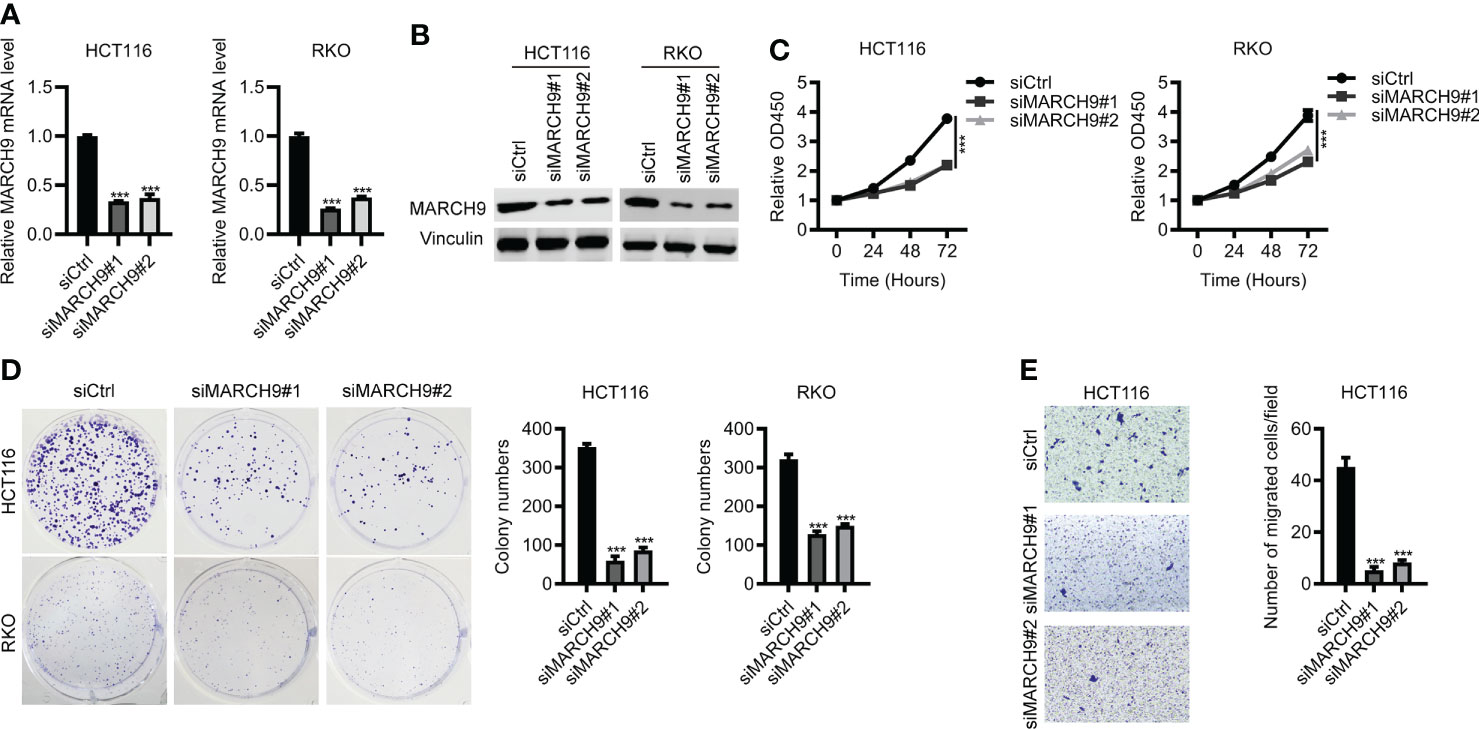
Figure 2 Knockdown of MARCH9 inhibited CRC cell proliferation and migration. (A, B) RT-qPCR and Western blot results of MARCH9 mRNA and protein levels in two CRC cell lines transfected with two MARCH9 siRNAs for 48 (h) (C) CCK-8 assay results of cell viability in two CRC cell lines transfected with two MARCH9 siRNAs at 24, 48, and 72h. (D) Cell colony formation results of two CRC cell lines transfected with two MARCH9 siRNAs at 14 days. (E) Transwell assay results of cell migration ability in two CRC cell lines transfected with two MARCH9 siRNAs at 48 (h) ***P < 0.001.
Overexpression of MARCH9 promoted CRC cell proliferation and migration
To confirm the function of MARC9 in CRC, we then performed gain-of-function experiments. As shown by RT-qPCR and Western blot analysis, endogenous MARCH9 expression was greatly upregulated after MARCH9 overexpression in two CRC cell lines (Figures 3A, B). This resulted in an increase in cell viability and proliferation (Figures 3C, D). MARCH9 overexpression also enhanced CRC cell migration, supporting an oncogenic role for this protein in CRC. These results confirmed the oncogenic function of MARCH9 in CRC (Figure 3E).
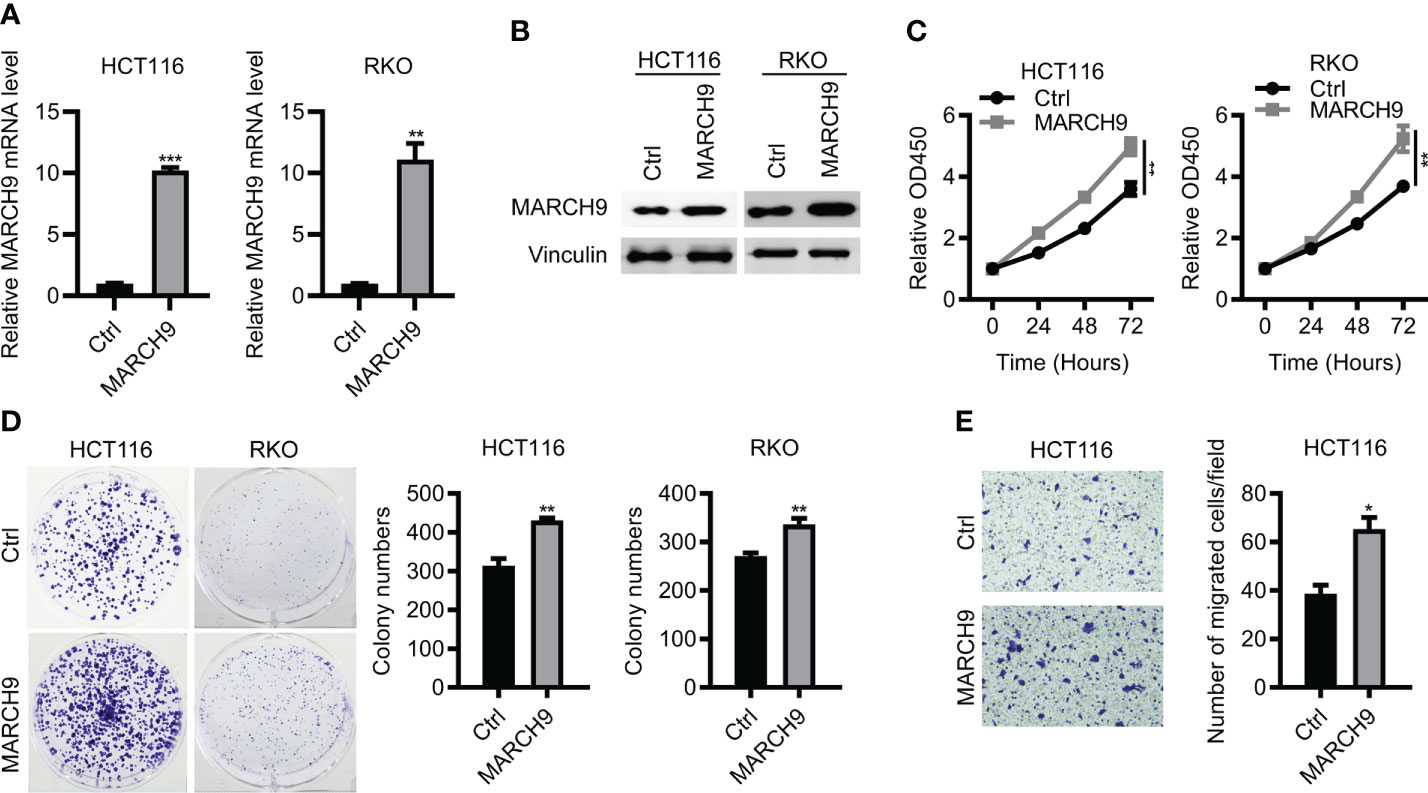
Figure 3 Overexpression of MARCH9 promoted CRC cell proliferation and migration. (A, B) RT-qPCR and western blot results of MARCH9 mRNA and protein levels in CRC cell lines transfected with MARCH9 or Ctrl plasmids at 48 (h) (C) CCK-8 assay results of cell viability in two CRC cell lines transfected with MARCH9 plasmids and Ctrl at 24, 48, and 72h. (D) Cell colony formation results of two CRC cell lines transfected with MARCH9 plasmids and Ctrl at 14 days. (E) The migration ability of two CRC cell lines transfected with MARCH9 plasmids and Ctrl at 48 (h) *P < 0.05; **P < 0.01; ***P < 0.001.
MARCH9 regulated CRC cell apoptosis and cell cycle
We then explore the role of MARCH9 in CRC cell apoptosis and cell cycle progression. MARCH9 knockdown significantly promoted apoptosis in HCT116 and RKO cells as shown by flow cytometry (Figures 4A, B). Furthermore, downregulation of MARCH9 enhanced the caspase 3/7 activity in both cell lines (Figure S2), suggesting the underlying mechanisms how MARCH9 regulates apoptosis. There was a higher number of cells in the G0/G1 phase of the cell cycle and a lower number in the S and G2/M phases in two CRC cell lines (Figures 4C, D). These results indicated that MARCH9 downregulation induced cell apoptosis and blocked the cell cycle in the G0/G1 phase.
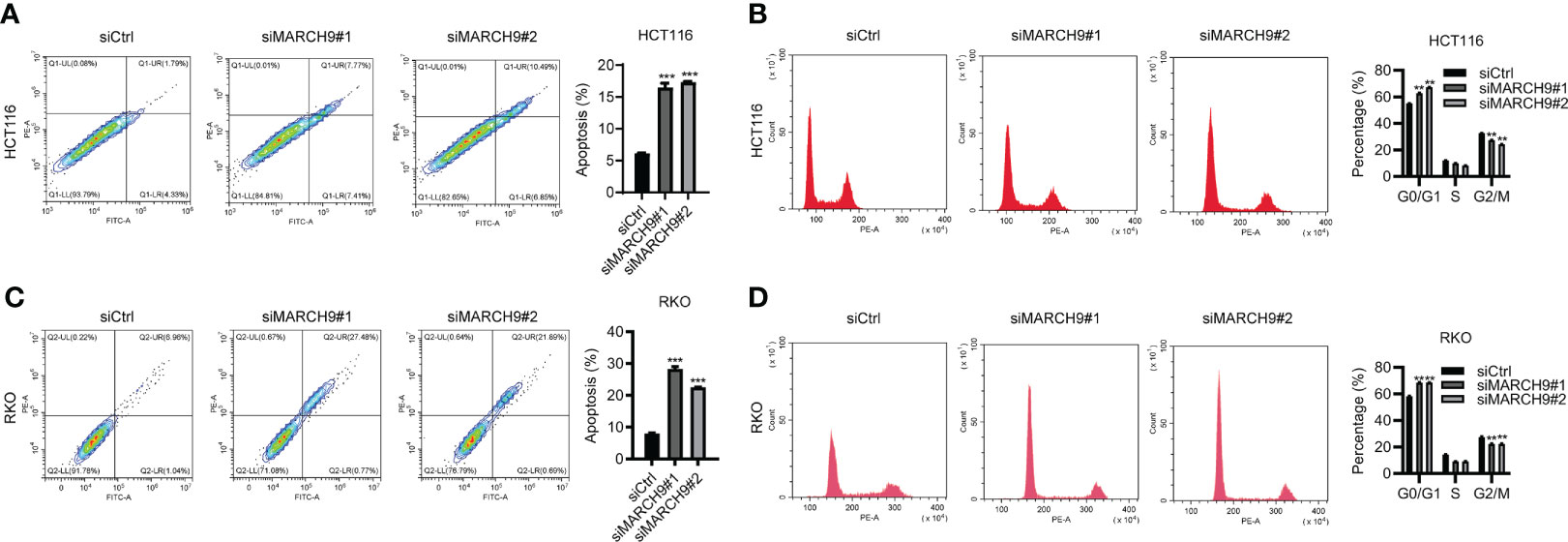
Figure 4 MARCH9 regulated CRC cell apoptosis and cell cycle. (A, B) The cell apoptosis rate of HCT116 and RKO cells after MARCH9 knockdown at 48 (h) (C, D) The cell cycle distribution of HCT116 and RKO cells after MARCH9 knockdown at 48 (h) **P < 0.01; ***P < 0.001.
MARCH9 downregulated CYLD and activated P65
Cylindromatosis (CYLD) is an important tumor suppressor gene that inhibits the development of various cancers through inactivation of NFκB (p65) (25, 26). A wide range of studies have shown that the expression of CYLD could be suppressed at transcription level by different non-coding RNAs (26–28), whereas whether CYLD abundance was repressed at post-transcription manner in colorectal cancer should be determined. Based on western blot results, we showed that knockdown of MARCH9 induced CYLD protein expression and reduced p65 phosphorylation in two CRC cell lines (Figure 5A), while overexpression of MARCH9 had the opposite effect on CYLD expression and p65 phosphorylation (Figure 5B). Total protein expression of p65 did not change after knocking down or overexpressing MARCH9 (Figure 5), indicating that MARCH9 regulates p65 phosphorylation but not expression levels. These results suggest that MARCH9 suppresses the expression of CYLD at post-transcription manner, leading to activation of p65.
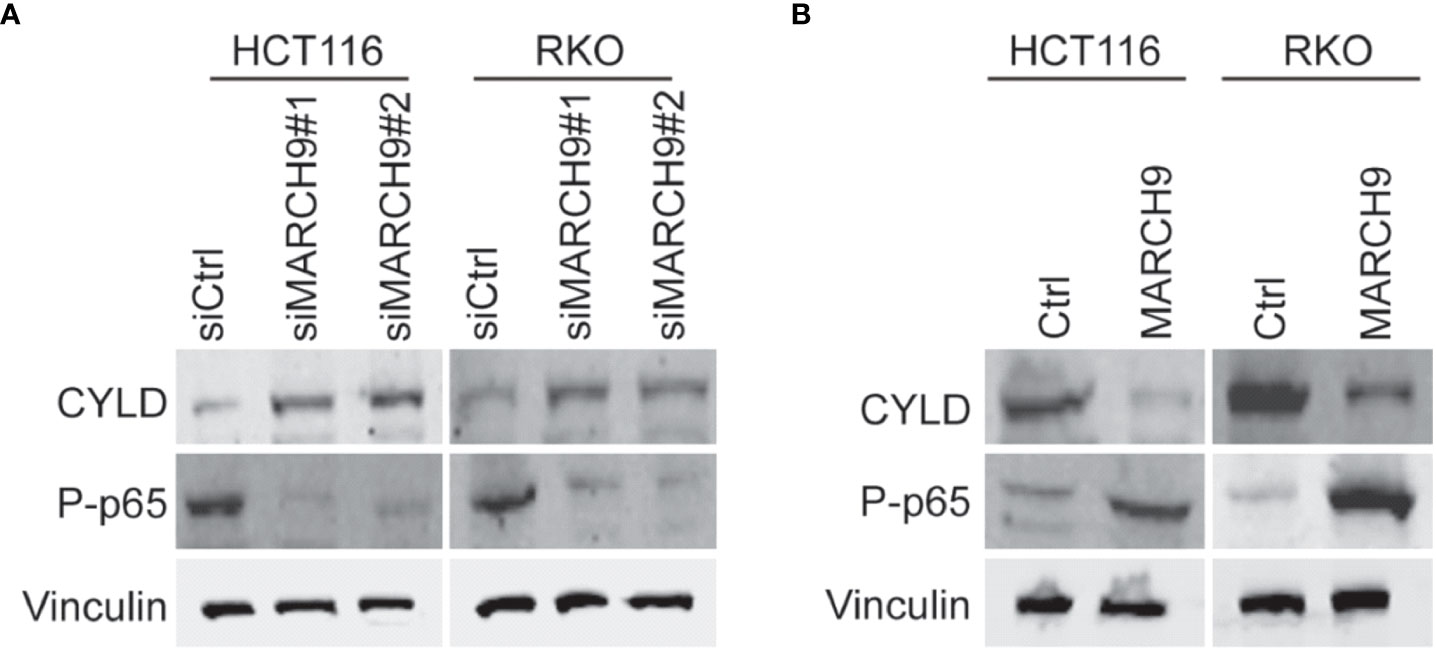
Figure 5 MARCH9 downregulated CYLD and activates P65. (A) Expression of CYLD, p65 and P-p65 in two CRC cell lines after MARCH9 knockdown by western blotting. (B) The expression of CYLD, p65 and P-p65 in two CRC cell lines following MARCH9 overexpression by western blotting.
MARCH9 accelerated CRC progression by promoting p65
To explore the role of p65 activity regulated by MARCH9 in CRC, we treated Ctrl and MARCH9 CRC cells with a specific p65 inhibitor, named Caffeic Acid Phenethyl Ester (CAFE). While MARCH9 overexpression promoted CRC cell viability (Figure 6A), increased the number of cell colonies (Figure 6B), and increased cell migration (Figure 6C), co-expression of CAFE weakened these tumor-promoting effects. By contrast, the inhibitory effect of CAFÉ on the growth and migration of Ctrl cells was smaller than that of MARCH9 overexpressing cells (Figures 6A-C). These results suggest that MARCH9 may accelerate CRC progression by regulating p65.
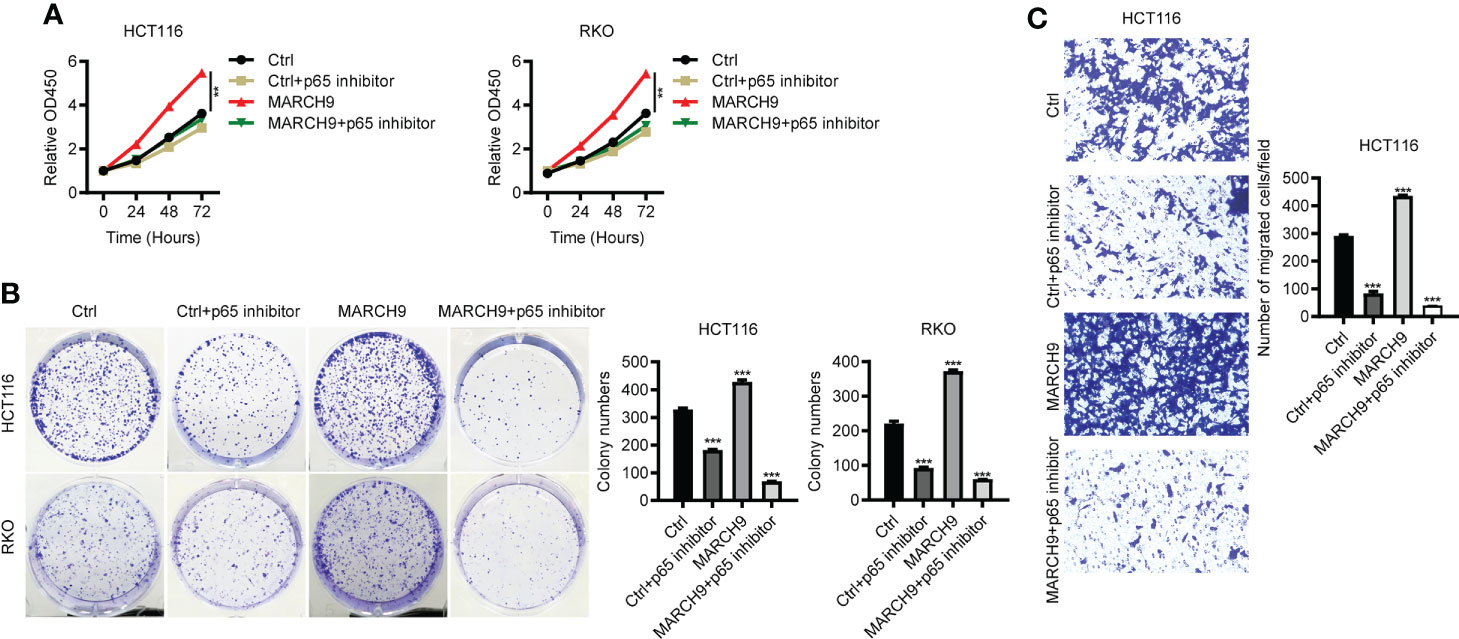
Figure 6 MARCH9 accelerated CRC progression by promoting p65. (A) The cell viability of two CRC cell lines transfected with Ctrl plasmids, Ctrl plasmids+p65 inhibitor, MARCH9 plasmids and MARCH9 plasmids+p65 inhibitor at 24, 48, and 72h using a CCK-8 assay. (B) Cell colony formation results of two CRC cell lines transfected with Ctrl plasmids, Ctrl plasmids+p65 inhibitor, MARCH9 plasmids and MARCH9 plasmids+p65 inhibitor at 14 days. (C) The migration ability of HCT116 and RKO cells transfected with Ctrl plasmids, Ctrl plasmids+p65 inhibitor, MARCH9 plasmids and MARCH9 plasmids+p65 inhibitor at 48 (h) **P < 0.01; ***P < 0.001.
MARCH9 knockdown suppressed tumor growth in vivo
Lastly, we studied the in vivo function of MARCH9 by implanting shCtrl and shMARCH9 CRC into the BALB/C nude mice. The shMARCH9#1 group of BALB/C nude mice had smaller tumors with significantly lower weight than the shCtrl group (Figures 7A, B). Western blot analysis revealed lower expression of MARCH9 and higher expression of CYLD in the xenograft tumor tissues from the shMARCH9#1 group than tissues from the siCtrl group (Figure 7C). In addition, MARCH9 knockdown suppressed the phosphorylation of p65, while had no effect on the total protein expression of p65 in the tumor tissues (Figure 7C). Collectively, these data suggest that MARCH9 knockdown suppressed HCT116 cell tumorigenicity.
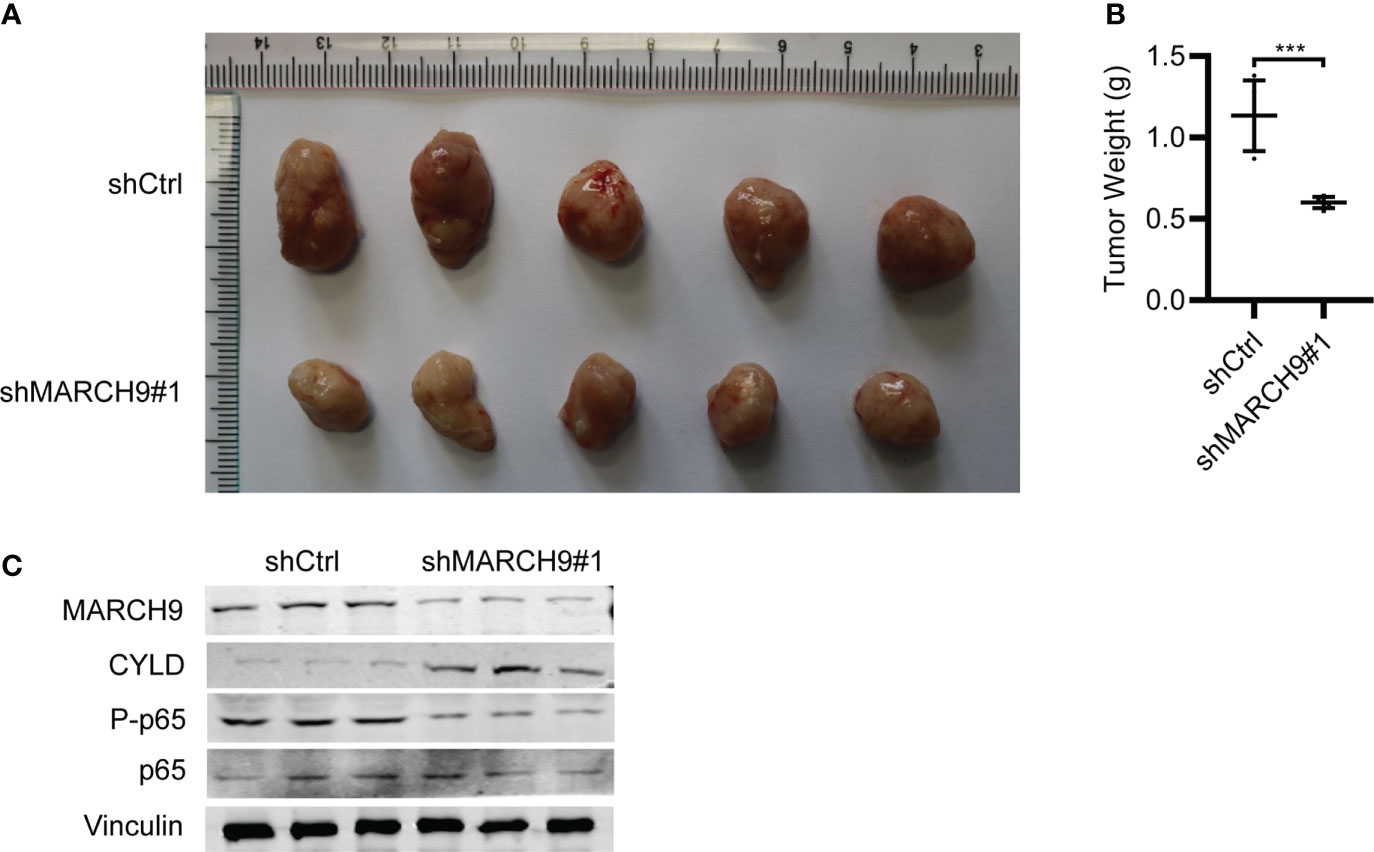
Figure 7 MARCH9 knockdown suppressed tumor growth in vivo. (A) Xenograft tumors were excised from BALB/c nude mice on day 45 and compared between shMARCH9#1 and shCtrl groups. (B) Tumor weight of BALB/c nude mice between shMARCH9#1 and shCtrl groups in statistics. (C) MARCH9, CYLD, P-p65 and p65 expression in xenograft tumor tissues between shMARCH9#1and shCtrl groups by western blotting. ***P < 0.001.
Discussion
Ubiquitination and deubiquitination are reversible post-translational modification processes that regulate both tumor-suppressing and -promoting pathways (29). MARCH9 is a member of the MARCH ubiquitin ligase family that targets cell surface receptors for degradation (22). MARCH9 plays a pivotal role in antigen-presenting cells by regulating the protein stability of human leukocyte antigen (HLA)-DM (30). In addition, MARCH9 modulates the export of major histocompatibility complex class I (MHC I) based on the regulation of MARCH9 on MHC I’s ubiquitination (31). These results indicate the significance of MARCH9 in immune system. Despite these studies, the clinical significance and the precise role of MARCH9 during the development of malignant tumors, including CRC remain to be determined. In this study, we explored the role of MARCH9 on CRC progression both in vitro and in vivo. MARCH9 expression was significantly upregulated in CRC samples from the TCGA database by individual cancer stage, histological subtype, and nodal metastasis status. While knockdown of MARCH9 inhibited CRC cell growth and migration, overexpression of MARCH9 had the opposite effect. In vivo xenograft studies confirmed that MARCH9 knockdown suppressed tumor growth, resulting in smaller and lower weight tumors than observed in the control group. Knockdown of MARCH9 also promoted CRC cell apoptosis and caused cell cycle arrest, indicating that MARCH9 functions as an oncogene during this disease. Taken together, MARCH9 is a promising diagnosis biomarker and therapeutic target for CRC.
Previous studies have shown that MARCH9 targets a wide range of downstream substrates involved in immune system, whereas the substrates of MARCH9 in cancer development are less clear. Identifying the molecular mechanisms contributing to the MARCH9-mediated CRC progression will help us develop effective drugs to treat the CRC patients with highly expressed MARCH9. Based on Western blots, we showed that MARCH9 could downregulate CYLD expression and activate p65 expression. CYLD is a K63-specific deubiquitinase that functions as a tumor suppressor in many cancers (32). Yang et al. reported that CYLD could inhibit growth and promote apoptosis of CRC cells (26). This is consistent with the results of the current study. In addition, CYLD disrupts key protein-protein interactions that are important for the activation of the Nuclear Factor-κB (NF-κB) pathway (33). NF-κB plays an important role in cancer-related processes and regulates cell proliferation, apoptosis, angiogenesis, and metastasis during CRC (34). p65 is one of the five known members of the NF-κB protein family, which form hetero or homodimers and bind to inhibitory proteins (35). Findings from the current study showed that CYLD expression was upregulated and p65 was downregulated in HCT116 and RKO cells after MARCH9 knockdown. Conversely, CYLD expression was downregulated and p65 was upregulated following MARCH9 overexpression. These results indicate that MARCH9 regulated CYLD and p65 expression in CRC cells. Importantly, cell viability, colony formation, and migration assays confirmed that MARCH9 overexpression conferred higher sensitivity of CRC cells to the treatment of p65 inhibitors, suggesting that MARCH9 promoted CRC progression by activating p65. These results also implied that p65 inhibitors might be a therapeutic strategy for the treatment of CRC patients with overexpression of MARCH9.
In summary, our results confirmed that MARCH9 was overexpressed in CRC patients and its overexpression contributed to the proliferation, migration and tumorigenesis of CRC cells, suggesting that it may function as an oncogene during this disease. The findings also illustrated that MARCH9 could serve as a very promising biomarker for CRC therapy. Importantly, MARCH9 activated p65 signaling and inhibition of p65 completely reversed the oncogenic function of MARCH9, implying that p65 could be the therapeutic target for MARCH9 overexpressed CRC patients. However, further clinical trials are needed to confirm these results.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal study was reviewed and approved by Animal Care and Use Committee of the Minzu University of China (ECMUC2019001AO).
Author contributions
YC and LC conceived and designed this study. BC and HL carried out the experiment and analyzed the experiment data. HL, BC, and L-LL drafted the manuscript with critical revisions from YC and LC. All authors critically reviewed the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.906897/full#supplementary-material
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, et al. Cancer statistics in China, 2015. CA Cancer J Clin (2016) 66(2):115–32. doi: 10.3322/caac.21338
3. Zhang Y, Chen Z, Li J. The current status of treatment for colorectal cancer in China: A systematic review. Med (Baltimore) (2017) 96(40):e8242. doi: 10.1097/MD.0000000000008242
4. Dariya B, Aliya S, Merchant N, Alam A, Nagaraju GP, et al. Colorectal cancer biology, diagnosis, and therapeutic approaches. Crit Rev Oncog (2020) 25(2):71–94. doi: 10.1615/CritRevOncog.2020035067
5. Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer (2011) 11(11):792–804. doi: 10.1038/nrc3139
6. De Gassart A, Camosseto V, Thibodeau J, Ceppi M, Catalan N, Pierre P, et al. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc Natl Acad Sci U.S.A. (2008) 105(9):3491–6. doi: 10.1073/pnas.0708874105
7. Nakamura N. The role of the transmembrane RING finger proteins in cellular and organelle function. Membranes (Basel) (2011) 1(4):354–93. doi: 10.3390/membranes1040354
8. Wang N, Yang L, Dai J, Wu Y, Zhang R, Jia X, et al. 5-FU inhibits migration and invasion of CRC cells through PI3K/AKT pathway regulated by MARCH1. Cell Biol Int (2021) 45(2):368–81. doi: 10.1002/cbin.11493
9. Xia D, Ji W, Xu C, Lin X, Wang X, Xia Y, et al. Knockout of MARCH2 inhibits the growth of HCT116 colon cancer cells by inducing endoplasmic reticulum stress. Cell Death Dis (2017) 8(7):e2957. doi: 10.1038/cddis.2017.347
10. Lin H, Feng L, Cui KS, Zeng LW, Gao D, Zhang LX, et al. The membrane-associated E3 ubiquitin ligase MARCH3 downregulates the IL-6 receptor and suppresses colitis-associated carcinogenesis. Cell Mol Immunol (2021) 18(12):2648–59. doi: 10.1038/s41423-021-00799-1
11. Zhang X, Feng B, Zhu F, Yu C, Lu J, Pan M, et al. N-myc downstream-regulated gene 1 promotes apoptosis in colorectal cancer via up-regulating death receptor 4. Oncotarget (2017) 8(47):82593–608. doi: 10.18632/oncotarget.19658
12. Xie L, Dai H, Li M, Yang W, Yu G, Wang X, et al. MARCH1 encourages tumour progression of hepatocellular carcinoma via regulation of PI3K-AKT-beta-catenin pathways. J Cell Mol Med (2019) 23(5):3386–401. doi: 10.1111/jcmm.14235
13. Wu Y, Wang L, Wang X, Zhao Y, Mao A, Zhang N, et al. RNA Sequencing analysis reveals the competing endogenous RNAs interplay in resected hepatocellular carcinoma patients who received interferon-alpha therapy. Cancer Cell Int (2021) 21(1):464. doi: 10.1186/s12935-021-02170-w
14. Sun J, Dong Z, Chang Z, Liu H, Jiang Q, Zhang D, et al. MARCH6 promotes hepatocellular carcinoma development through up-regulation of ATF2. BMC Cancer (2021) 21(1):827. doi: 10.1186/s12885-021-08540-x
15. Meng Y, Hu J, Chen Y, Yu T, Hu L. Silencing MARCH1 suppresses proliferation, migration and invasion of ovarian cancer SKOV3 cells via downregulation of NF-kappaB and wnt/beta-catenin pathways. Oncol Rep (2016) 36(5):2463–70. doi: 10.3892/or.2016.5076
16. Hu J, Meng Y, Zhang Z, Yan Q, Jiang X, Lv Z, et al. MARCH5 RNA promotes autophagy, migration, and invasion of ovarian cancer cells. Autophagy (2017) 13(2):333–44. doi: 10.1080/15548627.2016.1256520
17. Hu J, Zhang L, Mei Z, Jiang Y, Yi Y, Liu L, et al. Interaction of E3 ubiquitin ligase MARCH7 with long noncoding RNA MALAT1 and autophagy-related protein ATG7 promotes autophagy and invasion in ovarian cancer. Cell Physiol Biochem (2018) 47(2):654–66. doi: 10.1159/000490020
18. Coan M, Rampioni Vinciguerra GL, Cesaratto L, Gardenal E, Bianchet R, Dassi E, et al. Exploring the role of fallopian ciliated cells in the pathogenesis of high-grade serous ovarian cancer. Int J Mol Sci (2018) 19(9):2512–32. doi: 10.3390/ijms19092512
19. Hoer S, Smith L, Lehner PJ. MARCH-IX mediates ubiquitination and downregulation of ICAM-1. FEBS Lett (2007) 581(1):45–51. doi: 10.1016/j.febslet.2006.11.075
20. Bartee E, Mansouri M, Hovey Nerenberg BT, Gouveia K, Früh K. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J Virol (2004) 78(3):1109–20. doi: 10.1128/JVI.78.3.1109-1120.2004
21. Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U.S.A. (2002) 99(7):4465–70. doi: 10.1073/pnas.012025199
22. Hor S, Ziv T, Admon A, Lehner PJ. Stable isotope labeling by amino acids in cell culture and differential plasma membrane proteome quantitation identify new substrates for the MARCH9 transmembrane E3 ligase. Mol Cell Proteomics (2009) 8(8):1959–71. doi: 10.1074/mcp.M900174-MCP200
23. Shen QM, Wang HY, Xu S. MARCH9 suppresses lung adenocarcinoma progression by downregulating ICAM-1. Cell Physiol Biochem (2018) 50(1):92–107. doi: 10.1159/000493961
24. Luoto S, Hermelo I, Vuorinen EM, Hannus P, Kesseli J, Nykter M, et al. Computational characterization of suppressive immune microenvironments in glioblastoma. Cancer Res (2018) 78(19):5574–85. doi: 10.1158/0008-5472.CAN-17-3714
25. Nikolaou K, Tsagaratou A, Eftychi C, Kollias G, Mosialos G, Talianidis I, et al. Inactivation of the deubiquitinase CYLD in hepatocytes causes apoptosis, inflammation, fibrosis, and cancer. Cancer Cell (2012) 21(6):738–50. doi: 10.1016/j.ccr.2012.04.026
26. Yang X, Sun Y, Zhang Y, Han S. Downregulation of miR−181b inhibits human colon cancer cell proliferation by targeting CYLD and inhibiting the NF−κB signaling pathway. Int J Mol Med (2020) 46(5):1755–64. doi: 10.3892/ijmm.2020.4720
27. Pan S, Bao D, Li Y, Liu D, Quan S, Wang R, et al. SOX4 induces drug resistance of colorectal cancer cells by downregulating CYLD through transcriptional activation of microRNA-17. J Biochem Mol Toxicol (2022) 36(1):e22910. doi: 10.1002/jbt.22910
28. Liang H, Hu AP, Li SL, Xie JP, Ma QZ, Liu JY, et al. MiR-454 prompts cell proliferation of human colorectal cancer cells by repressing CYLD expression. Asian Pacific J Cancer Prevent: APJCP (2015) 16(6):2397–402. doi: 10.7314/APJCP.2015.16.6.2397
29. Mansour MA. Ubiquitination: Friend and foe in cancer. Int J Biochem Cell Biol (2018) 101:80–93. doi: 10.1016/j.biocel.2018.06.001
30. Jahnke M, Trowsdale J, Kelly AP. Ubiquitination of human leukocyte antigen (HLA)-DM by different membrane-associated RING-CH (MARCH) protein family E3 ligases targets different endocytic pathways. J Biol Chem (2012) 287(10):7256–64. doi: 10.1074/jbc.M111.305961
31. De Angelis Rigotti F, De Gassart A, Pforr C, Cano F, N'Guessan P, Combes A, et al. MARCH9-mediated ubiquitination regulates MHC I export from the TGN. Immunol Cell Biol (2017) 95(9):753–64. doi: 10.1038/icb.2017.44
32. Cui Z, Kang H, Grandis JR, Johnson DE. CYLD alterations in the tumorigenesis and progression of human papillomavirus-associated head and neck cancers. Mol Cancer Res (2021) 19(1):14–24. doi: 10.1158/1541-7786.MCR-20-0565
33. Sun SC. CYLD: A tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ (2010) 17(1):25–34. doi: 10.1038/cdd.2009.43
34. Patel M, Horgan PG, McMillan DC, Edwards J. NF-kappaB pathways in the development and progression of colorectal cancer. Transl Res (2018) 197:43–56. doi: 10.1016/j.trsl.2018.02.002
Keywords: MARCH9, CYLD, p65, colorectal cancer, progression
Citation: Liu H, Chen B, Liu L-L, Cong L and Cheng Y (2022) The role of MARCH9 in colorectal cancer progression. Front. Oncol. 12:906897. doi: 10.3389/fonc.2022.906897
Received: 29 March 2022; Accepted: 26 August 2022;
Published: 16 September 2022.
Edited by:
Yohei Shirakami, Gifu University, JapanReviewed by:
Paul Dent, Virginia Commonwealth University, United StatesAndrea Morrione, Temple University, United States
Copyright © 2022 Liu, Chen, Liu, Cong and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Cong, Y29uZ2xpbjIwMDgyMDA4QGFsaXl1bi5jb20=; Yong Cheng, eW9uZ2NoZW5nQG11Yy5lZHUuY24=
†These authors have contributed equally to this work and share first authorship