 Lin Gao1,2
Lin Gao1,2 Bo Han
Bo Han Xuesen Dong
Xuesen Dong- 1Department of Urologic Sciences, Faculty of Medicine, University of British Columbia, Vancouver, BC, Canada
- 2The Key Laboratory of Experimental Teratology, Ministry of Education and Department of Pathology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, China
While the androgen receptor (AR) signalling is the mainstay therapeutic target for metastatic prostate cancers, these tumours will inevitably develop therapy resistance to AR pathway inhibitors suggesting that prostate tumour cells possess the capability to develop mechanisms to bypass their dependency on androgens and/or AR to survive and progress. In many studies, protein kinases such as Src are reported to promote prostate tumour progression. Specifically, the pro-oncogene tyrosine Src kinase regulates prostate cancer cell proliferation, adhesion, invasion, and metastasis. Not only can Src be activated under androgen depletion, low androgen, and supraphysiological androgen conditions, but also through crosstalk with other oncogenic pathways. Reciprocal activations between Src and AR proteins had also been reported. These findings rationalize Src inhibitors to be used to treat castrate-resistant prostate tumours. Although several Src inhibitors had advanced to clinical trials, the failure to observe patient benefits from these studies suggests that further evaluation of the roles of Src in prostate tumours is required. Here, we summarize the interplay between Src and AR signalling during castrate-resistant prostate cancer progression to provide insights on possible approaches to treat prostate cancer patients.
Introduction
Androgens and the androgen receptor (AR) play key roles during the development of prostate cancer (PCa). They are also tightly coupled with PCa progression to the castrate-resistant prostate cancer (CRPC) state. Targeting AR signalling has been a mainstay therapeutic option to manage locally advanced and metastatic PCa (1, 2). These therapies include inhibitors of androgen synthesis through the hypothalamus-pituitary-adrenal axis (e.g., leuprorelin) (3, 4), AR antagonist that prevent androgens from binding to AR (e.g., bicalutamide) (5), and more potent new generation AR inhibitors (e.g., abiraterone acetate and enzalutamide) (6, 7). Despite the maximal androgen ablation therapies that could be possibly applied to patients, therapy-resistant tumours are inevitably developed (6, 7). While the majority of CRPC remains AR-positive and presents an adenocarcinoma phenotype, more potent AR pathway inhibitor (ARPI) treatment often induced AR negative neuroendocrine prostate cancer (NEPC) or double-negative prostate tumours (DNPC) accounting for ~20% of CRPC (8–11). These findings indicate that the phenotypes of CRPC tumour cells are heterogeneous and are associated with the potency and duration of ARPI treatments. AR target therapies trigger various cellular mechanisms that promote tumour evolution to bypass the dependency of androgens and/or AR. Supporting this notion, PCa patients can benefit from intermittent androgen deprivation therapy (1) and bipolar androgen therapy (12–14) that either recover endogenous androgens or supplement exogenous androgens to delay disease progression. Therefore, CRPC is not an initial clinical presentation of prostate cancer, but the consequence of anti-AR therapies.
Castrate-Resistant Prostate Cancer Progression Is Coupled With AR Signalling
Except for small cell carcinoma and DNPC, most CRPC tumours express AR proteins (8–11). The questions remain whether an AR-positive CRPC tumour is driven by the AR signalling, or it is AR indifferent and other oncogenic pathways replace AR to promote tumour progression. If the AR drives the progression of the CRPC tumour, does the AR rely on the castrate levels of androgens or act in a ligand-independent manner? It is important to differentiate tumours by various modes of action of AR so that effective ARPI treatments could be used.
Studies using in vitro cell models indicated that the ligand-dependent and ligand-independent AR signalling, and AR bypass mechanisms can all be possibly adopted by cancer cells. The LNCaP cell model, androgen-dependent tumour, can form xenografts only in non-castrated mice (15). These tumours are initially responsive to castration surgery but will progress into castrate-resistant xenografts (16). The CRPC LNCaP tumours are still responsive to ARPIs such as enzalutamide, emphasizing that these tumours still rely on ligand-dependent AR signalling (17). In contrast, the LNCaP95 cell model was derived from LNCaP cells but cultured under prolonged androgen deprivation conditions (18). These cells are naturally resistant to enzalutamide in vitro and in vivo, and can only be inoculated in castrated mice to form xenografts (19, 20). LNCaP95 xenografts are androgen-independent tumours, expressing high levels of AR, AR splice variants (AR-Vs), and AR target genes such as PSA. AR gene disruption by CRISPR demolishes the expression of AR and AR-V7 and reduces LNCaP95 cell growth, supporting that AR-mediated signalling is still functional to promote tumour progression (20, 21). However, the establishment of LNCaP95 cell clones with complete disruption of AR and AR-V protein expression indicates that the AR and AR-Vs do not necessitate the viability of LNCaP95 cells, highlighting the existence of AR bypass mechanisms. It had been shown that signalling mediated by glucocorticoid receptors may replace AR to promote CRPC progression (22). In PTEN deficient cancer cells, there exists a reciprocal activation between the AR and PI3K/Akt signalling. Inhibition of one will activate the other to maintain tumour cell survival (23, 24). Since genomic alterations of the PTEN gene and genes associated with the PI3K/Akt signalling are common in CRPC, this AR bypass mechanism is frequently used by PCa to counteract AR target therapy. These model systems indicate that PCa cells possess phenotypical plasticity to adapt to ARPI treatments by using various intracellular mechanisms to switch off the ligand-dependent AR signalling and switch on the ligand-independent AR signalling, and bypass AR through other oncogenic kinase pathways (Figure 1).
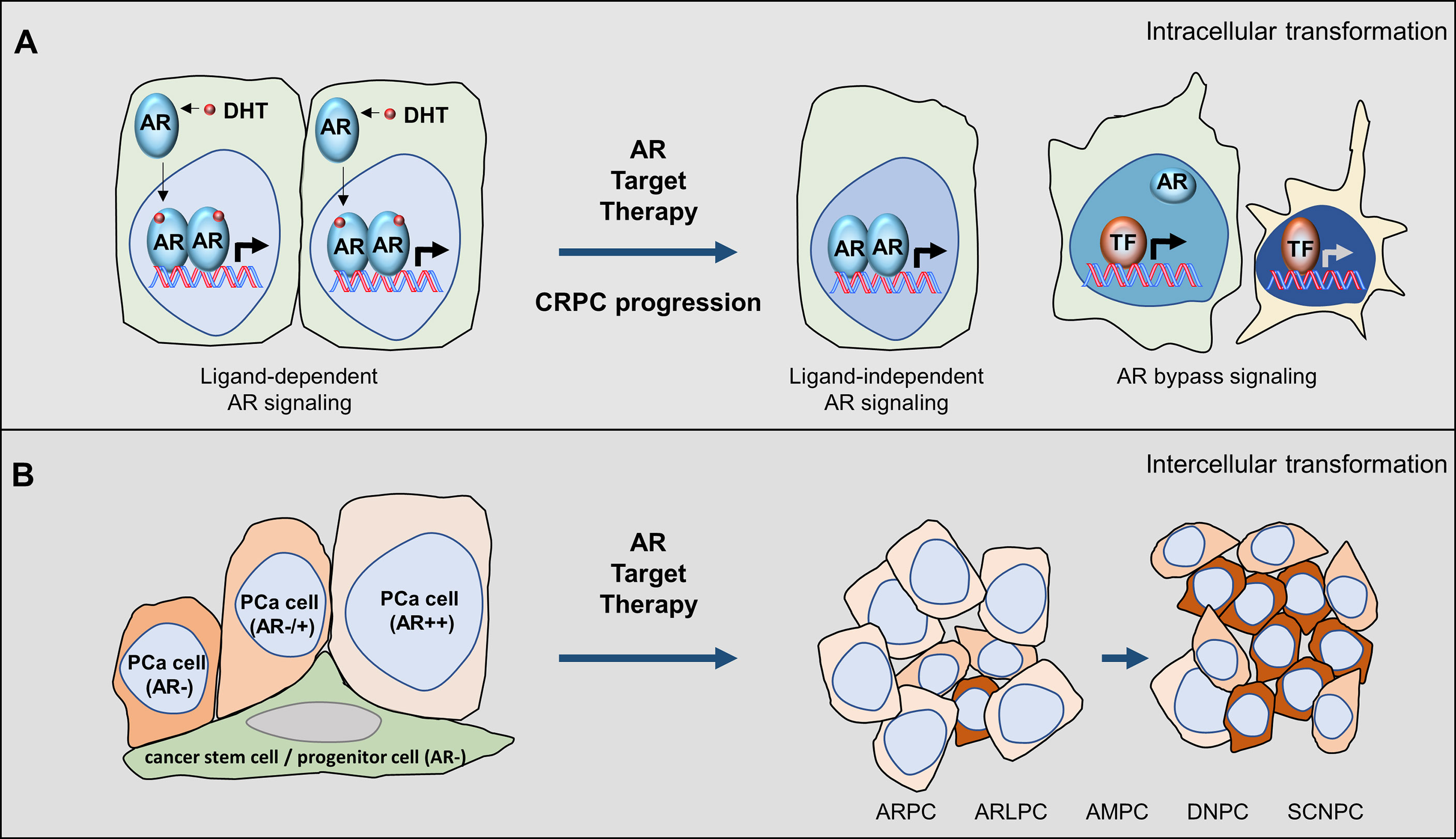
Figure 1 Castrate-resistant prostate cancer progression is coupled with AR pathway inhibitions. (A) Intracellular mechanisms can be adopted by PCa cells under ARPI treatments that shift the ligand-dependent AR signalling to the ligand-independent AR signalling and AR bypass oncogenic pathways. (B) Intercellular mechanisms can be adopted by tumours under ARPI treatments to select tumours that are less dependent on androgens to survive and populate. During this process, tumours exhibit heterogeneous phenotypes that can be classified by luminal epithelial markers such as AR and PSA and neuroendocrine markers such as chromogranin.
Based on these findings from cell models, we can speculate that AR target therapy will only provide short-term tumour suppression. Prolonged ARPI treatment will eventually transform the LNCaP type of tumours into the LNCaP95 type of tumours. Even though drugs can be successfully developed to degrade AR and AR splice variant proteins, LNCaP95 cells with complete AR gene destruction by CRISPR sets up an example that AR is not essential for PCa cell viability after prolonged AR target therapy. This hypothesis is supported by that small cell carcinoma, DNPC, and even AR indifferent tumours become more prevalent in patients treated with new generation ARPIs. Nevertheless, there still exists a therapeutic window for AR target therapy during the early stage of tumour progression.
Heterogeneity of Castrate-Resistant Prostate Cancer
Tumour heterogeneity also complicates CRPC progression. The epithelium in the adult prostate contains luminal epithelial cells, neuroendocrine (NE) cells, and basal cells that are believed to be sustained in a homeostasis state by stem/progenitor cells within the basal layer through differentiation (25). The AR is expressed in luminal epithelial cells when they are terminally differentiated from progenitor cells (26), and acts to maintain the differentiation but does not regulate cell proliferation (27). While PCa is predominantly derived from luminal epithelial cells, the AR signal in PCa cells can also promote PCa cell proliferation and metastasis (28, 29). ARPIs would thereby not only inhibit cell growth but also trigger PCa cells to de-differentiate and gain lineage plasticity (30). Because heterogeneous cell populations co-exist in PCa tissues, ARPIs can block the androgen-sensitive cells from growing and simultaneously select androgen-insensitive cells to populate. These surviving cells may also contain cells that are derived from prostate progenitor cells but have not differentiated to the luminal epithelial lineage. There are at least five different CRPC tumours reported in patients that can be classified based on AR signature and NE biomarkers: 1) AR-high tumours (ARPC) containing cancer cells expressing uniform AR and AR target genes and no NE markers; 2) AR-low tumours (ARLPC) contains cells with weak or heterogenous AR and PSA expression with no NE markers; 3) amphicrine PCa (AMPC) containing cells co-expressing AR, PSA, and NE markers; 4) small cell neuroendocrine PCa (SCNPC) containing cells with classic small cell carcinoma histology and NE marker positive and no AR and PSA expression; and 5) DNPC containing cells with neither NE marker and AR expression (8, 9). These observations are recapitulated in patient-derived xenografts (8, 9, 31–34). Since treatment naïve PCa is predominantly AR and PSA positive tumours, the presentation of ARLPC, SCNPC, and DNPC at the late stage of tumour progression suggests that AR bypass mechanisms are adopted by these tumours.
The mechanisms by which the transition of AR high PCa to AR negative SCNPC or DNPC are not fully defined. Two possible mechanisms have been proposed (Figure 1A). The co-expression of AR signalling and NE biomarkers in the same cancer cells supports a “trans-differentiation” mechanism by which existing AR-positive PCa cells undergo de-differentiation by ARPIs and gain an NE lineage. It had been shown that androgen deprivation, chemotherapy reagents, UV light exposure, cytokines as well as hypoxia can all transiently induce NE differentiation of androgen-sensitive PCa cells (35–40). The other possibility is the “clonal selection” mechanism by which the prostate stem-like/progenitor cells gain oncogenic capacities and differentiate into basal, stemness, luminal, and NE lineages (Figure 1B). These progenitor cells are AR negative and have a low proliferation index, thereby, are resistant to ARPIs and/or systematic radiation and chemotherapy. The androgen-sensitive LNCaP model was also reported to gain stem cell and neural phenotypes and tumour-initiating potential, in an androgen-free neural/neural crest (N/NC) stem medium (41). These findings indicate that PCa cells have an intrinsic capability to switch on differentiation to a different lineage under ARPI treatments.
In summary, the heterogeneity of prostate tumours is a natural challenge to AR target therapy. CRPC tumour cells have different genetic and epigenetic backgrounds, cell lineages, or evolutional stages (from being androgen-dependent to AR indifferent) within a tumour. Targeting AR signalling in the long-term is predictably ineffective since it blocks one cancer cell population and permits others to expand. Combinational therapies would be a future therapeutic option.
Ligand-Independent AR Signalling in Prostate Cancer Cells
AR can exert its transcriptional activity in the absence of androgens through its N-terminal domain (NTD) (42). Androgen binding to AR triggers intramolecular interaction between NTD and the ligand-binding domain (LBD) that is required for ligand-dependent AR activity (43, 44). However, deletion of the LBD renders AR constitutively active (45). Endogenously expressed AR-Vs with truncated LBD have been demonstrated to mediate ligand-independent AR signalling to drive CRPC progression (46–48). AR-Vs are generated by the RNA splicing process, which is tightly coupled with AR gene transcription initiation and elongation rates (49). ARPIs increase the transcription rate of the AR gene (50) resulting in more AR pre-mRNA copies processed into mRNAs of either AR or AR-Vs by the RNA splicing machinery (49, 51). Therefore, when the LBD is truncated or blocked pharmacologically by ARPIs, it provides an opportunity for NTD to enable AR activation ligand independently.
The NTD is a domain with a flexible conformation that can interact with various coregulators to activate AR. It possesses the Tau5 domain (aa360-485) that mediates the ligand-independent activation of AR (52). Interestingly, the Src kinase uses its Src homology domain 3 (SH3) domain to recognize a polyproline motif (aa371-381) within AR and phosphorylates the Tyr534, which is required for AR activation by growth factors under androgen-depleted conditions (53). AR Tyr534 phosphorylation by Src also enhances AR protein stability through a mechanism that prevents AR from being recognized by the chaperone-associated ubiquitin ligase COOH terminus of Hsp70-interacting protein (CHIP) for proteasomal degradation (54).
Blocking the LBD by ARPIs creates opportunities for NTD to interact with growth factors, cytokines, and intracellular kinases to activate AR signalling. Epidermal growth factor (EGF) acts through its membrane receptor to stimulate Src activation, which in turn activates AR Tyr534 phosphorylation (53). AR phosphorylation at Tyr267 and Tyr363 can also be stimulated through Cdc42-associated kinase Ack by HER2 activation (55). IL-6 enhances the transcriptional activity of the AR NTD through STAT3, which action is associated with IL-6 stimulated MAPK/ERK pathways (56). The activation of the MAPK/ERK kinase pathways can enhance ligand-independent AR actions through modulating coactivators such as steroid receptor coactivator-1 (57). Furthermore, IL-8 can also stimulate ligand-independent activation of AR through the FAK and Src pathways (58). Given these growth factors and tumour-promoting cytokines are enriched in the tumour microenvironment, it is reasonable to believe that AR target therapy provides an opportunity for NTD to mediate ligand-independent AR signalling through crosstalk with intracellular kinases to confer tumours therapy resistance. Furthermore, activated kinases (e.g. Src) can trigger its downstream signal pathways to promote PCa cell survival, proliferation, and metastasis that may eventually bypass the requirement of the AR for tumour progression as we discuss later.
The Src Tyrosine Kinases
Intracellular kinases play critical roles in regulating ligand-independent AR signalling, among which the proto-oncogene tyrosine kinase Src had been demonstrated to contribute to CRPC progression (59, 60). It is the founding member of the Src family of structurally-related protein tyrosine kinases that contains at least eleven members: Src, Yes, Fyn, Fgr, Blk, Hck, Lck, Lyn, Frk, Srm, and Brk (61). Src, Yes, and Fyn are widely expressed in many types of tissues, while Src is the most well-studied member. It has four functional domains (Figure 2): 1) the amino-terminal SH4 domain has myristoylation and palmitoylation post-translational modifications that are important for Src anchorage to the cell membrane, tyrosine kinase activity, and protein stability (62, 63); 2) the SH3 domain can be folded in a way that it interacts with the linker between the SH2 domain and the amino-terminal lobe of the SH1 domain to keep Src in an inactive conformation. The SH3 domain also recognizes the consensus sequence of PxxP as a minimal consensus target site within Src substrates (64). These PxxP motifs could be positioned in two opposite orientations defined by a positively charged residue (+xxPxxP or xPxxPx+) that interacts with a negatively charged SH3 peptide-binding surface (65, 66). However, there are several non-consensus SH3 targeting peptides have been reported (67), indicating that the SH3 domain could recognize atypical motifs that permit Src to interact with broader ranges of protein substrates to regulate complex intracellular signalling; 3) the SH2 domain has a conserved arginine residue that is required for the high affinity of the SH2 domain to interact with phospho-peptide (e.g., pYEEI) (68, 69). This domain forms intradomain interactions with the SH1 domain through the phosphor-Tyr530 to inactivate Src kinase activity (70); and 4) the SH1 tyrosine kinase domain, which contains the amino-terminal lobe that plays regulatory roles for Src kinase activity and the carboxyl-terminal lobe that has the intrinsic kinase activity (71). It contains a catalytic kinase domain which is activated by Tyr419 autophosphorylation and inactivated when Tyr530 is phosphorylated by C-terminal Src kinase (CSK).
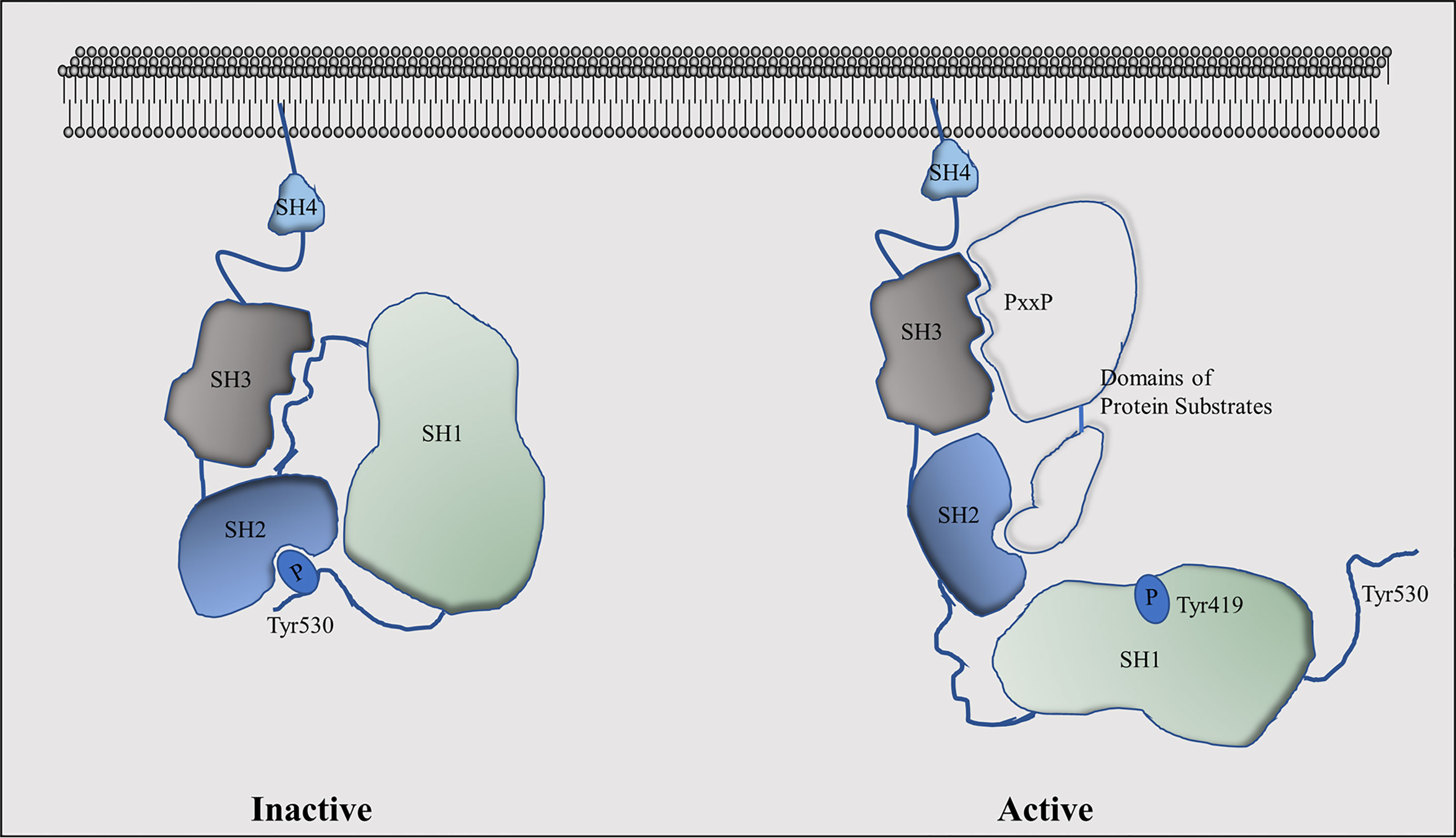
Figure 2 Functional domains of the Src kinase and their conformational changes in active and inactive stages.
Src activation is mainly controlled by its intramolecular phosphorylation modifications and by Src interacting proteins. Src is inactivated when Tyr530 is phosphorylated by CSK and its homolog CHK, and recognized by the SH2 domain resulting in a closed inactive Src conformation (72). In contrast, several protein phosphatases such as protein tyrosine phosphatase-α (PTPα), PTP1, SH2-containing phosphatase 1 (SHP1), and SHP2 can serve as activators of Src by dephosphorylating the Tyr530 (73–75). X-ray crystallography studies had revealed that the phospho-Tyr530 forms an intramolecular interaction with the SH2 domain. In conjunction with other intramolecular interactions between SH3 and SH1 domains, Src kinase activity is silenced. In contrast, phosphorylation of Tyr419 by either auto-phosphorylation or other kinases (76, 77) is critical for Src activation. This phosphorylation site is located in the “activation loop” that is conserved among other Src family members (78). When phosphorylated, it permits the substrate-binding pocket of the SH1 kinase domain to be exposed to allow the kinase to access its substrates (71), while mutation of this tyrosine will dramatically reduce Src activity. Another key factor to determine Src activity is the Src interacting proteins. While the SH2 and SH3 domains mediate intramolecular interactions with the SH1 kinase domain to keep Src in an inactivated state, peptides from Src interacting proteins could disrupt these intramolecular interactions to activate Src. It is now known that several tyrosine kinase receptors such as focal adhesion kinase (FAK) (79–81), epidermal growth factor receptor (EGFR) (82), HER2 (83), platelet-derived growth factor receptor (PDGFR) (84), and fibroblast growth factor receptor (85) can activate Src activity. Therefore, the activation of Src involves the selective recognization of SH2 and SH3 domains to their targeted ligand peptides in addition to SH1 recognization of its substrates, which model had been termed as ‘‘turned on by touch’’ mode (86).
Src was reported to play an important role in regulating cancer cell adhesion, migration, and invasion in addition to its established roles in cell proliferation. Src is required for the transition of the G2 to M phase of mammalian fibroblasts (87), as it forms a protein complex with p27 (88) and phosphorylates its Tyr88 and Tyr74 resulting in reduced p27 protein expression and subsequent cyclin-dependent kinases (CDKs) activation (89). Blocking Src by the PP2 inhibitor in lymphoma cells caused G2/M cell cycle arrest by suppressing the WEE1-CDK1 axis (90). Src inhibition by PD173955 induces mitosis arrest post chromosome condensation in the prophase phase but before the spindle assembly (91), which is consistent with the report showing that Src controls the G2 phase DNA damage checkpoint through activating ataxia telangiectasia mutated ATM, ATR, and Chk1 kinases (92). However, Src was shown to have more potent effects on cell adhesion and mobility in association with tumour metastasis. The cell-cell adhesion is formed by adherens junctions, desmosomes, tight junctions, and gap junctions et al. (93). Activated Src by protein tyrosine phosphatase 1B (PTP1B) can form a protein complex with the adherens junctions, which in turn promotes ubiquitination and subsequent endocytosis of E-cadherin for degradation (94). Activated Src can also stimulate the JNK signal pathway to regulate the expression of matrix metalloproteinases MMP2 and MMP9, and several tissue inhibitors of metalloproteinases (TIMPs) to break down ECM (95, 96). These Src functions together allow cancer cells to disassociate from adjacent cells and ECM and invade through ECM for metastatic dissemination.
The Interplay Between Src and AR Signalling
The AR and Src can form a protein complex in PCa cells indicating that there exists crosstalk between both factors (97). Genetically engineered mouse models carrying overexpression of both AR and Src, but not alone, in normal prostate epithelial cells, can induce poorly or non-differentiated adenocarcinoma (98), supporting co-activation of AR and Src is required for prostate tumourigenesis. While AR gene amplification and overexpression are frequently observed in CRPC, upregulation of phospho-Src (Tyr419) and downregulation of CSK, an Src inhibitor, had also been reported (99, 100), indicating that active AR and Src signalling also co-exist during CRPC progression (Figure 3).
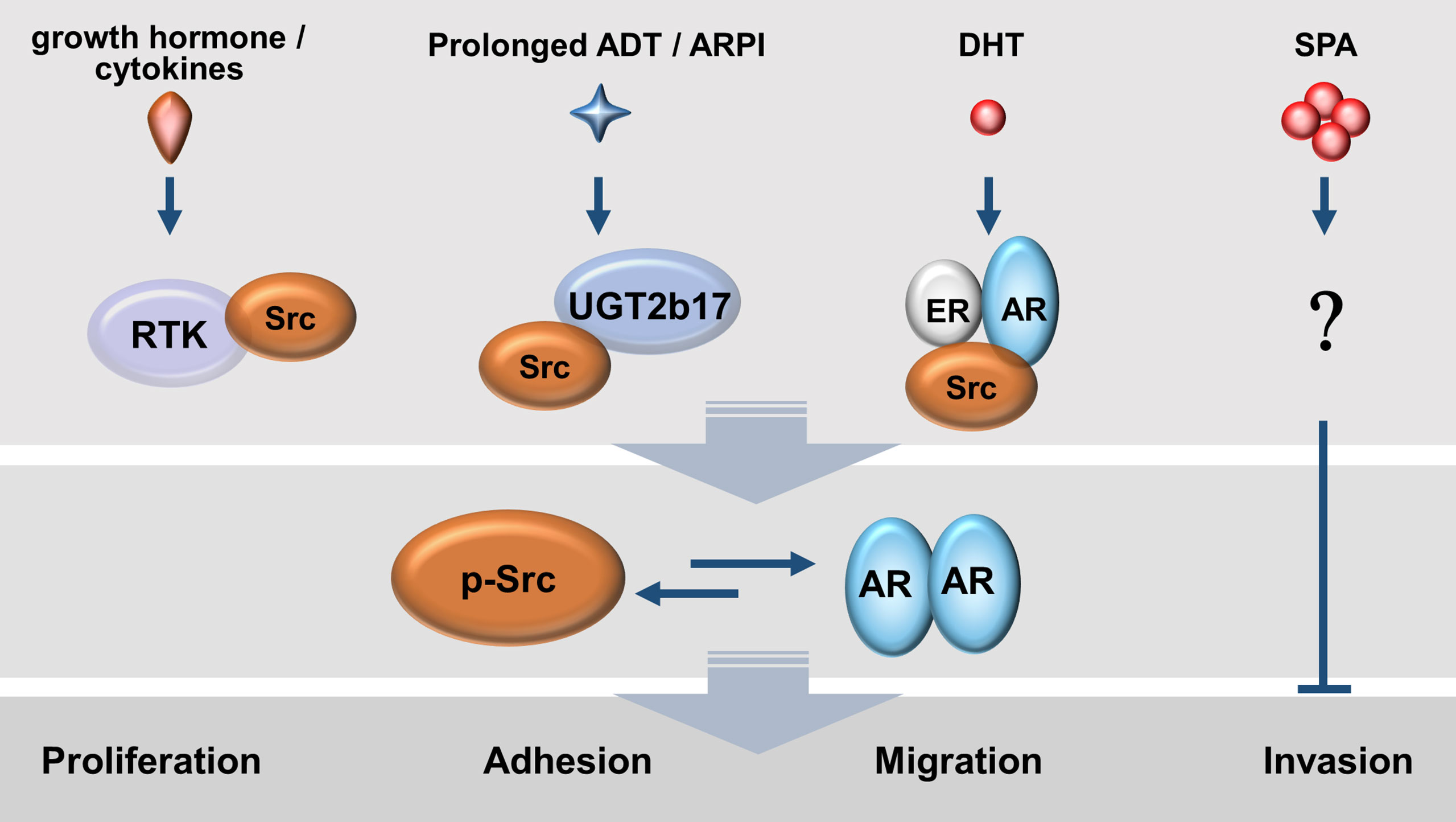
Figure 3 Mutual activations between AR and Src kinase exist in prostate cancer cells under various androgen conditions. Enriched growth factors and tumour-promoting cytokines as well as de novo androgen synthesis by tumour cells allow a persistent activation of the AR-Src axis.
Mutual activation between AR and Src can exist not only under androgen depletion conditions but also in the presence of androgens. Under androgen deprivation conditions, growth factors (e.g., IGF-1 and EGF) and cytokines (e.g., IL-6 and IL-8) can mediate signalling to induce tyrosine phosphorylation of AR, and activate the transcriptional activity of AR (53, 58, 101). One key mediator of all these signal pathways is the Src (53, 54, 98). The mutant Src with inactive kinase activity downregulates AR transcriptional activity, while the constitutively active Src mutant (Y527F) induces AR nuclear translocation and AR activity in the absence of androgens (102). Blocking Src activities by small molecules inhibited ligand-independent, but not ligand-dependent AR activity and AR target gene expression (102, 103). Furthermore, prolonged androgen deprivation enhances UGT2b17 expression which in turn accelerates androgen clearance inside CRPC cells and at the same time promotes the formation of a UGT2b17-Src protein complex and subsequent Src activation (100). These findings indicate that growth factors, cytokines, and androgen catabolism enzymes can activate AR through Src in the absence of androgens.
In the presence of androgens, androgen-sensitive LNCaP cells were reported to stimulate Src autophosphorylation and its kinase activity by ligand-activated AR or by ligand-activated estrogen receptor beta (ERβ) (97, 104). The same results were replicated in breast cancer cells co-expressing AR and ER alpha (ERα) in the presence of estradiol. DHT or estradiol promotes a protein complex of AR/ER/Src. Src uses its SH2 domain to recognize the Tyr537 in ER, while the SH3 domain recognizes the PxxP motif in AR. In contrast, androgen deprivation abolishes Src autophosphorylation and kinase activity in LNCaP cells (97). This AR-activated Src activity, in turn, promotes PCa cell proliferation through accelerating G1-S phase transition (105), which effects can be blocked by Src inhibitors (104).
However, when androgen-sensitive PCa cell lines, as well as ex vivo PCa tissue, were challenged with supraphysiological doses of androgens (SPA), it induces cell cycle arrest at the G1 phase and cell senescence through cell cycling regulators such as CDK inhibitor p16, Rb1, and E2F1 (106). The SPA-induced cell cycle arrest can be partially alleviated by small molecule inhibitors against Src and Akt (106). These results suggest that Src has biphasic impacts on cell cycling depending on the levels of androgens exposed to PCa cells. It remains to be clarified how SPA-induced cell arrest is associated with Src activation.
Furthermore, Src can act independently of AR to stimulate PCa cell proliferation and migration. It stimulates AR negative PC3 and DU145 cell proliferation by accelerating the G1 phase of cell cycling (107). Inhibition of the Src activity by AZD0530 reduced the binding of β-catenin to cyclin D1 and c-Myc to block cell growth in vitro, and orthotopic DU145 xenograft growth in vivo. In addition, DU145 and PC3 cells have higher mobility activity than AR-positive LNCaP, which is correlated with high FAK/Src kinase activities. Treatment of PP2, the Src inhibitor, to both cell lines dramatically suppressed cell migration rates (108). The transgenic adenocarcinoma of mouse prostate (TRAMP) model is commonly used to study neuroendocrine prostate tumours since adenocarcinoma is initially developed in TRAMP mice and will commonly progress to neuroendocrine tumours (109). However, TRAMP mice with Src knockout have reduced adenocarcinoma establishment and metastasis, but have no impact on neuroendocrine tumour formation (110).
These studies indicate that Src can be activated by various mechanisms under different androgen regimes and exert biphasic actions on AR-positive PCa cell proliferation. Src also acts independently of AR signalling to regulate tumour cell proliferation and mobility. While multiple oncogenic pathways (e.g., aberrant AR, growth hormone, and tumour-promoting cytokines) all serve as the upstream regulator of Src activation, and enhanced Src activation is associated with CRPC progression. However, whether blocking Src activity will suppress CRPC remains to be defined. Src knockout mice were viable at birth but died due to impaired osteoclast function (111), suggesting that Src is not essential for benign cell viability. Transgenic mouse overexpressing Src alone in normal prostate epithelial cells did not induce malignancy (98), suggesting that Src itself is not critical for PCa development. Gain of overactive Src activity may be an adaptive response of PCa cells to survival under ARPI treatment. There may exist subpopulations of Src-dependent CRPC cells or a therapeutic window to co-target AR and Src during the early stage of tumour progression. In this case, the identification of biomarkers to stratify Src-dependent tumours will be critical to approving Src inhibitors to be used to treat patients.
Src Inhibitors Tested in Prostate Cancer Patients
Results from preclinical studies using multiple PCa cells and xenograft models had built a reasonable rationale that targeting Src kinases would effectively suppress prostate tumour growth and progression in patients. Several phase I and II clinical trials in various tumour settings had also shown that dasatinib and saracatinib are well tolerated by patients with acceptable side effects, and the optimal dosing had been determined (112–115). Dasatinib had been approved by FDA to treat chronic myelogenous leukemia and acutely lymphoblastic leukemia (116). These results have encouraged the efforts to evaluate dasatinib in PCa patients (Table 1). Dasatinib had been tested in phase II trials as a monotherapy in chemotherapy naïve patients (114, 115), in which studies dasatinib was shown to be well tolerated with moderate treatment-related adverse effects. The most common treatment-related adverse events are fatigue, nausea and diarrhea (114, 115, 117–119). A further phase III READY clinical trial on 1522 PCa patients treated with docetaxel plus prednisolone with either placebo or dasatinib were performed (117). However, the results were disappointing since dasatinib did not show improved overall survival for chemotherapy naïve CRPC patients. Since Src had been demonstrated to contribute to tumours developing resistance to AR target therapy, a phase II trial was also designed to test 26 metastatic CRPC patients with no prior chemotherapy who received abiraterone plus prednisone together with placebo or dasatinib (118). Dasatinib did not show a significant improvement in progression-free survival. Another phase II trial has tested on abiraterone-resistant CRPC tumours treated with either dasatinib or sunitinib, an inhibitor targeting multiple receptor tyrosine kinases including VEGFR, Kit, and FLT-3 et al. (120). No difference in overall survival and time to treatment failure was observed between the dasatinib and sunitinib arms. These results raised doubts about the oncogenic property of Src during CRPC progression and urged more extensive characterization of the Src signalling under the context of heterogeneity of CRPC.
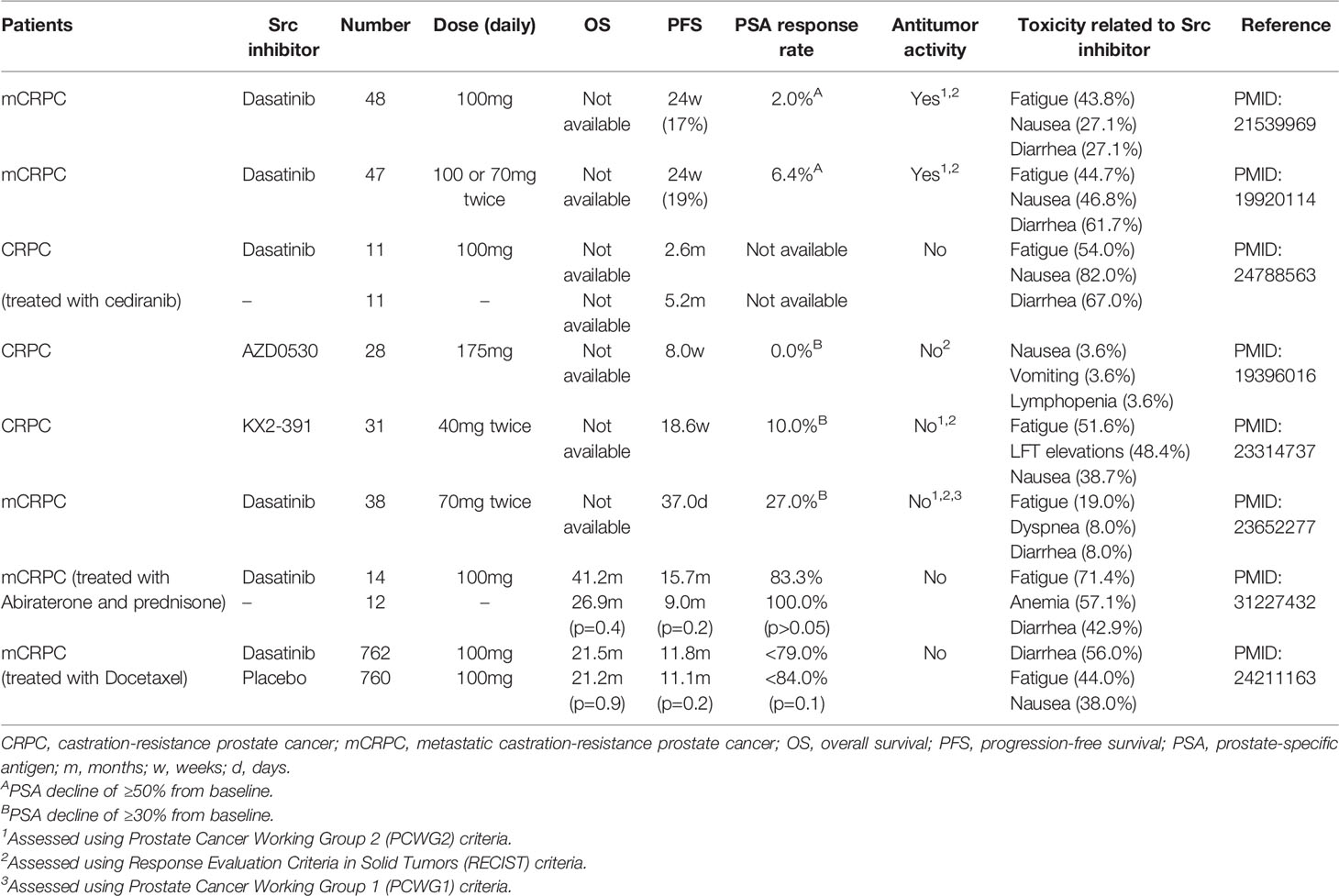
Table 1 Findings for clinical trials using Src inhibitor.
The inconsistent results from pre-clinical and clinical studies raised several questions. Is the Src activity critical for CRPC progression? Genetically engineered mouse model studies did not support that Src is essential for cell viability (98). The association of overactive Src with CRPC progression suggests that increased Src activity is an adaptive response of PCa cells for survival. However, it does not warrant that blocking Src will reduce PCa growth. Several other signallings such as PI3K/Akt or reprogrammed AR signalling may also be adopted by cancer cells. Src inhibition can force cancer cells to detour and use alternative survival mechanisms. In addition, pre-clinical studies using the PCa cell models and xenografts had shown that Src inhibitors induce cell cycle arrest and suppress tumour growth and cancer cell mobility. However, Src inhibitors may have limited capability to kill cancer cells to reduce tumour volumes. Whether they can cause PCa cell death and decrease xenograft volume needs further investigation.
Do all tumours/tumour cells respond to Src inhibitors? There exist inter-patient and intra-tumour heterogeneity of prostate tumours. Tumour cells carrying heterogeneous genetic and epigenetic backgrounds may respond to Src inhibitors differently. Particularly, prostate tumours in these clinical trials had progressed to the metastatic stage post castration therapies (117), previously receive radiation and chemotherapies (120), or had developed abiraterone resistance (120). The heterogeneity of these tumours is greater than that in treatment-naive tumours. Pre-clinical studies using xenografts derived from cell models may not capitulate the complete landscape of cancer cells in patients. Developing biomarkers that can be used to stratify Src-dependent tumours may help identify tumours sensitive to Src inhibition.
Are Src activities sufficiently blocked within patient tumours? Some clinical trials had used phosphor-Src protein (121) or mRNAs of CSF2, CD40L, GZMB, and IL-2 from peripheral blood cells (122) as surrogate biomarkers to confirm Src inhibition within tumours. However, the concentration of dasatinib that is sufficient to block Src in blood cells may not be sufficient for prostate tumour cells. As we discussed in previous sections, AR and AR-Vs could use their PxxP motifs to stimulate Src activity. Under prolonged androgen deprivation conditions, UGT2b17 is upregulated and forms a protein complex with Src, and stimulates its kinase activity. FAK is an upstream regulator of Src. FAK gene amplification is frequent in CRPC (123) and global phosphoproteomic profiling had found that overactivation of FAK kinase is a common mechanism by which PCa develops therapy resistance (124). These factors (e.g., AR, UGT2b17, and FAK) in PCa cells may be constitutively activating Src to counteract Src inhibitors. Neither did the phase II trials (114, 125) nor the phase III READY trial (117) successfully retrieve data confirming that Src is sufficiently inhibited by dasatinib inside the prostate tumours as discussed by the investigators. Together, there are several possibilities that Src inhibitors failed to show clinical benefits to PCa patients, and further investigations are needed.
Conclusion
The Src signalling plays an important role to promote PCa development and CRPC progression. The interplay between Src and AR regulates PCa cell proliferation and metastasis under various androgen regimes, implying potential Src target therapy for CRPC. The negative results of Src inhibitors in clinical trials on PCa urge further mechanistic investigations at the molecular level on the roles of Src in PCa.
Author Contributions
Writing-original draft preparation, LG and XD; writing-review and editing, BH and XD; All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by Canadian Institute of Health Research operating grants (MOP-137007 & PJT-156150) to XD.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gleave M, Goldenberg SL, Bruchovsky N, Rennie P. Intermittent Androgen Suppression for Prostate Cancer: Rationale and Clinical Experience. Prostate Cancer Prostatic Dis (1998) 1(6):289–96. doi: 10.1038/sj.pcan.4500260
2. Albertsen PC, Hanley JA, Fine J. 20-Year Outcomes Following Conservative Management of Clinically Localized Prostate Cancer. JAMA (2005) 293(17):2095–101. doi: 10.1001/jama.293.17.2095
3. Crawford ED, Moul JW, Sartor O, Shore ND. Extended Release, 6-Month Formulations of Leuprolide Acetate for the Treatment of Advanced Prostate Cancer: Achieving Testosterone Levels Below 20 Ng/Dl. Expert Opin Drug Metab Toxicol (2015) 11(9):1465–74. doi: 10.1517/17425255.2015.1073711
4. Klotz L, Gleave M, Goldenberg SL. Neoadjuvant Hormone Therapy: The Canadian Trials. Mol Urol (2000) 4(3):233–7;discussion 239.
5. Culig Z, Bartsch G, Hobisch A. Antiandrogens in Prostate Cancer Endocrine Therapy. Curr Cancer Drug Targets (2004) 4(5):455–61. doi: 10.2174/1568009043332925
6. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and Increased Survival in Metastatic Prostate Cancer. New Engl J Med (2011) 364(21):1995–2005. doi: 10.1056/NEJMoa1014618
7. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased Survival With Enzalutamide in Prostate Cancer After Chemotherapy. N Engl J Med (2012) 367(13):1187–97. doi: 10.1056/NEJMoa1207506
8. Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained Through FGF Signaling. Cancer Cell (2017) 32(4):474–89.e6. doi: 10.1016/j.ccell.2017.09.003
9. Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B, et al. Molecular Profiling Stratifies Diverse Phenotypes of Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer. J Clin Invest (2019) 129(10):4492–505. doi: 10.1172/JCI128212
10. Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular Characterization of Neuroendocrine Prostate Cancer and Identification of New Drug Targets. Cancer Discov (2011) 1(6):487–95. doi: 10.1158/2159-8290.CD-11-0130
11. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent Clonal Evolution of Castration-Resistant Neuroendocrine Prostate Cancer. Nat Med (2016) 22(3):298–305. doi: 10.1038/nm.4045
12. Schweizer MT, Wang H, Luber B, Nadal R, Spitz A, Rosen DM, et al. Bipolar Androgen Therapy for Men With Androgen Ablation Naive Prostate Cancer: Results From the Phase II BATMAN Study. Prostate (2016) 76(13):1218–26. doi: 10.1002/pros.23209
13. Schweizer MT, Antonarakis ES, Wang H, Ajiboye AS, Spitz A, Cao H, et al. Effect of Bipolar Androgen Therapy for Asymptomatic Men With Castration-Resistant Prostate Cancer: Results From a Pilot Clinical Study. Sci Transl Med (2015) 7(269):269ra2. doi: 10.1126/scitranslmed.3010563
14. Sena LA, Wang H, Lim Sc MS, Rifkind I, Ngomba N, Isaacs JT, et al. Bipolar Androgen Therapy Sensitizes Castration-Resistant Prostate Cancer to Subsequent Androgen Receptor Ablative Therapy. Eur J Cancer (2021) 144:302–9. doi: 10.1016/j.ejca.2020.11.043
15. Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, et al. LNCaP Model of Human Prostatic Carcinoma. Cancer Res (1983) 43(4):1809–18. doi: 10.1016/0304-3835(83)90174-X
16. Lim DJ, Liu XL, Sutkowski DM, Braun EJ, Lee C, Kozlowski JM. Growth of an Androgen-Sensitive Human Prostate Cancer Cell Line, LNCaP, in Nude Mice. Prostate (1993) 22(2):109–18. doi: 10.1002/pros.2990220203
17. Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, et al. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science (2009) 324(5928):787–90. doi: 10.1126/science.1168175
18. Pflug BR, Reiter RE, Nelson JB. Caveolin Expression is Decreased Following Androgen Deprivation in Human Prostate Cancer Cell Lines. Prostate (1999) 40(4):269–73. doi: 10.1002/(SICI)1097-0045(19990901)40:4<269:AID-PROS9>3.0.CO;2-6
19. Leung JK, Tam T, Wang J, Sadar MD. Isolation and Characterization of Castration-Resistant Prostate Cancer LNCaP95 Clones. Hum Cell (2021) 34(1):211–8. doi: 10.1007/s13577-020-00435-6
20. Zhu Y, Dalrymple SL, Coleman I, Zheng SL, Xu J, Hooper JE, et al. Role of Androgen Receptor Splice Variant-7 (AR-V7) in Prostate Cancer Resistance to 2nd-Generation Androgen Receptor Signaling Inhibitors. Oncogene (2020) 39(45):6935–49. doi: 10.1038/s41388-020-01479-6
21. Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct Transcriptional Programs Mediated by the Ligand-Dependent Full-Length Androgen Receptor and Its Splice Variants in Castration-Resistant Prostate Cancer. Cancer Res (2012) 72(14):3457–62. doi: 10.1158/0008-5472.CAN-11-3892
22. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. Glucocorticoid Receptor Confers Resistance to Antiandrogens by Bypassing Androgen Receptor Blockade. Cell (2013) 155(6):1309–22. doi: 10.1016/j.cell.2013.11.012
23. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell (2011) 19(5):575–86. doi: 10.1016/j.ccr.2011.04.008
24. Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell Autonomous Role of PTEN in Regulating Castration-Resistant Prostate Cancer Growth. Cancer Cell (2011) 19(6):792–804. doi: 10.1016/j.ccr.2011.05.006
25. Isaacs JT, Coffey DS. Etiology and Disease Process of Benign Prostatic Hyperplasia. Prostate Suppl (1989) 2:33–50. doi: 10.1002/pros.2990150506
26. Cooke PS, Young P, Cunha GR. Androgen Receptor Expression in Developing Male Reproductive Organs. Endocrinology (1991) 128(6):2867–73. doi: 10.1210/endo-128-6-2867
27. Vander Griend DJ, Litvinov IV, Isaacs JT. Conversion of Androgen Receptor Signaling From a Growth Suppressor in Normal Prostate Epithelial Cells to an Oncogene in Prostate Cancer Cells Involves a Gain of Function in C-Myc Regulation. Int J Biol Sci (2014) 10(6):627–42. doi: 10.7150/ijbs.8756
28. Gao S, Gao Y, He HH, Han D, Han W, Avery A, et al. Androgen Receptor Tumor Suppressor Function Is Mediated by Recruitment of Retinoblastoma Protein. Cell Rep (2016) 17(4):966–76. doi: 10.1016/j.celrep.2016.09.064
29. Heinlein CA, Chang C. Androgen Receptor in Prostate Cancer. Endocr Rev (2004) 25(2):276–308. doi: 10.1210/er.2002-0032
30. Formaggio N, Rubin MA, Theurillat JP. Loss and Revival of Androgen Receptor Signaling in Advanced Prostate Cancer. Oncogene (2021) 40(7):1205–16. doi: 10.1038/s41388-020-01598-0
31. Brennen WN, Zhu Y, Coleman IM, Dalrymple SL, Antony L, Patel RA, et al. Resistance to Androgen Receptor Signaling Inhibition Does Not Necessitate Development of Neuroendocrine Prostate Cancer. JCI Insight (2021) 6(8):146827–46. doi: 10.1172/jci.insight.146827
32. Lin D, Wyatt AW, Xue H, Wang Y, Dong X, Haegert A, et al. High Fidelity Patient-Derived Xenografts for Accelerating Prostate Cancer Discovery and Drug Development. Cancer Res (2014) 74(4):1272–83. doi: 10.1158/0008-5472.CAN-13-2921-T
33. Lam HM, McMullin R, Nguyen HM, Coleman I, Gormley M, Gulati R, et al. Characterization of an Abiraterone Ultraresponsive Phenotype in Castration-Resistant Prostate Cancer Patient-Derived Xenografts. Clin Cancer Res (2017) 23(9):2301–12. doi: 10.1158/1078-0432.CCR-16-2054
34. Zhang X, Coleman IM, Brown LG, True LD, Kollath L, Lucas JM, et al. SRRM4 Expression and the Loss of REST Activity May Promote the Emergence of the Neuroendocrine Phenotype in Castration-Resistant Prostate Cancer. Clin Cancer Res (2015) 21(20):4698–708. doi: 10.1158/1078-0432.CCR-15-0157
35. Deeble PD, Murphy DJ, Parsons SJ, Cox ME. Interleukin-6- and Cyclic AMP-Mediated Signaling Potentiates Neuroendocrine Differentiation of LNCaP Prostate Tumor Cells. Mol Cell Biol (2001) 21(24):8471–82. doi: 10.1128/MCB.21.24.8471-8482.2001
36. Bang YJ, Pirnia F, Fang WG, Kang WK, Sartor O, Whitesell L, et al. Terminal Neuroendocrine Differentiation of Human Prostate Carcinoma Cells in Response to Increased Intracellular Cyclic AMP. Proc Natl Acad Sci USA (1994) 91(12):5330–4. doi: 10.1073/pnas.91.12.5330
37. Cox ME, Deeble PD, Lakhani S, Parsons SJ. Acquisition of Neuroendocrine Characteristics by Prostate Tumor Cells is Reversible: Implications for Prostate Cancer Progression. Cancer Res (1999) 59(15):3821–30.
38. Zhu Y, Liu C, Cui Y, Nadiminty N, Lou W, Gao AC. Interleukin-6 Induces Neuroendocrine Differentiation (NED) Through Suppression of RE-1 Silencing Transcription Factor (REST). Prostate (2014) 74(11):1086–94. doi: 10.1002/pros.22819
39. Lin TP, Chang YT, Lee SY, Campbell M, Wang TC, Shen SH, et al. REST Reduction is Essential for Hypoxia-Induced Neuroendocrine Differentiation of Prostate Cancer Cells by Activating Autophagy Signaling. Oncotarget (2016) 7(18):26137–51. doi: 10.18632/oncotarget.8433
40. Deng X, Elzey BD, Poulson JM, Morrison WB, Ko SC, Hahn NM, et al. Ionizing Radiation Induces Neuroendocrine Differentiation of Prostate Cancer Cells In Vitro, In Vivo and in Prostate Cancer Patients. Am J Cancer Res (2011) 1(7):834–44.
41. Nouri M, Caradec J, Lubik AA, Li N, Hollier BG, Takhar M, et al. Therapy-Induced Developmental Reprogramming of Prostate Cancer Cells and Acquired Therapy Resistance. Oncotarget (2017) 8(12):18949–67. doi: 10.18632/oncotarget.14850
42. Dehm SM, Tindall DJ. Androgen Receptor Structural and Functional Elements: Role and Regulation in Prostate Cancer. Mol Endocrinol (2007) 21(12):2855–63. doi: 10.1210/me.2007-0223
43. He B, Kemppainen JA, Wilson EM. FXXLF and WXXLF Sequences Mediate the NH2-Terminal Interaction With the Ligand Binding Domain of the Androgen Receptor. J Biol Chem (2000) 275(30):22986–94. doi: 10.1074/jbc.M002807200
44. He B, Bowen NT, Minges JT, Wilson EM. Androgen-Induced NH2- and COOH-Terminal Interaction Inhibits P160 Coactivator Recruitment by Activation Function 2. J Biol Chem (2001) 276(45):42293–301. doi: 10.1074/jbc.M107492200
45. Jenster G, van der Korput HA, van Vroonhoven C, van der Kwast TH, Trapman J, Brinkmann AO. Domains of the Human Androgen Receptor Involved in Steroid Binding, Transcriptional Activation, and Subcellular Localization. Mol Endocrinol (1991) 5(10):1396–404. doi: 10.1210/mend-5-10-1396
46. Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A Novel Androgen Receptor Splice Variant is Up-Regulated During Prostate Cancer Progression and Promotes Androgen Depletion-Resistant Growth. Cancer Res (2009) 69(6):2305–13. doi: 10.1158/0008-5472.CAN-08-3795
47. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-Independent Androgen Receptor Variants Derived From Splicing of Cryptic Exons Signify Hormone-Refractory Prostate Cancer. Cancer Res (2009) 69(1):16–22. doi: 10.1158/0008-5472.CAN-08-2764
48. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen Receptor Splice Variants Mediate Enzalutamide Resistance in Castration-Resistant Prostate Cancer Cell Lines. Cancer Res (2013) 73(2):483–9. doi: 10.1158/0008-5472.CAN-12-3630
49. Liu LL, Xie N, Sun S, Plymate S, Mostaghel E, Dong X. Mechanisms of the Androgen Receptor Splicing in Prostate Cancer Cells. Oncogene (2014) 33(24):3140–50. doi: 10.1038/onc.2013.284
50. Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen Receptor Gene Expression in Prostate Cancer is Directly Suppressed by the Androgen Receptor Through Recruitment of Lysine-Specific Demethylase 1. Cancer Cell (2011) 20(4):457–71. doi: 10.1016/j.ccr.2011.09.001
51. Yu Z, Chen S, Sowalsky AG, Voznesensky OS, Mostaghel EA, Nelson PS, et al. Rapid Induction of Androgen Receptor Splice Variants by Androgen Deprivation in Prostate Cancer. Clin Cancer Res (2014) 20(6):1590–600. doi: 10.1158/1078-0432.CCR-13-1863
52. Jenster G, van der Korput HA, Trapman J, Brinkmann AO. Identification of Two Transcription Activation Units in the N-Terminal Domain of the Human Androgen Receptor. J Biol Chem (1995) 270(13):7341–6. doi: 10.1074/jbc.270.13.7341
53. Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, et al. Regulation of Androgen Receptor Activity by Tyrosine Phosphorylation. Cancer Cell (2006) 10(4):309–19. doi: 10.1016/j.ccr.2006.08.021
54. DaSilva J, Gioeli D, Weber MJ, Parsons SJ. The Neuroendocrine-Derived Peptide Parathyroid Hormone-Related Protein Promotes Prostate Cancer Cell Growth by Stabilizing the Androgen Receptor. Cancer Res (2009) 69(18):7402–11. doi: 10.1158/0008-5472.CAN-08-4687
55. Mahajan NP, Liu Y, Majumder S, Warren MR, Parker CE, Mohler JL, et al. Activated Cdc42-Associated Kinase Ack1 Promotes Prostate Cancer Progression via Androgen Receptor Tyrosine Phosphorylation. Proc Natl Acad Sci USA (2007) 104(20):8438–43. doi: 10.1073/pnas.0700420104
56. Ueda T, Bruchovsky N, Sadar MD. Activation of the Androgen Receptor N-Terminal Domain by Interleukin-6 via MAPK and STAT3 Signal Transduction Pathways. J Biol Chem (2002) 277(9):7076–85. doi: 10.1074/jbc.M108255200
57. Ueda T, Mawji NR, Bruchovsky N, Sadar MD. Ligand-Independent Activation of the Androgen Receptor by Interleukin-6 and the Role of Steroid Receptor Coactivator-1 in Prostate Cancer Cells. J Biol Chem (2002) 277(41):38087–94. doi: 10.1074/jbc.M203313200
58. Lee LF, Louie MC, Desai SJ, Yang J, Chen HW, Evans CP, et al. Interleukin-8 Confers Androgen-Independent Growth and Migration of LNCaP: Differential Effects of Tyrosine Kinases Src and FAK. Oncogene (2004) 23(12):2197–205. doi: 10.1038/sj.onc.1207344
59. Varkaris A, Katsiampoura AD, Araujo JC, Gallick GE, Corn PG. Src Signaling Pathways in Prostate Cancer. Cancer Metastasis Rev (2014) 33(2-3):595–606. doi: 10.1007/s10555-013-9481-1
60. Drake JM, Graham NA, Lee JK, Stoyanova T, Faltermeier CM, Sud S, et al. Metastatic Castration-Resistant Prostate Cancer Reveals Intrapatient Similarity and Interpatient Heterogeneity of Therapeutic Kinase Targets. Proc Natl Acad Sci USA (2013) 110(49):E4762–9. doi: 10.1073/pnas.1319948110
61. Quintaje SB, Orchard S. The Annotation of Both Human and Mouse Kinomes in UniProtKB/Swiss-Prot: One Small Step in Manual Annotation, One Giant Leap for Full Comprehension of Genomes. Mol Cell Proteomics (2008) 7(8):1409–19. doi: 10.1074/mcp.R700001-MCP200
62. Patwardhan P, Resh MD. Myristoylation and Membrane Binding Regulate C-Src Stability and Kinase Activity. Mol Cell Biol (2010) 30(17):4094–107. doi: 10.1128/MCB.00246-10
63. Alland L, Peseckis SM, Atherton RE, Berthiaume L, Resh MD. Dual Myristylation and Palmitylation of Src Family Member P59fyn Affects Subcellular Localization. J Biol Chem (1994) 269(24):16701–5. doi: 10.1016/S0021-9258(19)89447-4
64. Ren R, Mayer BJ, Cicchetti P, Baltimore D. Identification of a Ten-Amino Acid Proline-Rich SH3 Binding Site. Science (1993) 259(5098):1157–61. doi: 10.1126/science.8438166
65. Feng S, Chen JK, Yu H, Simon JA, Schreiber SL. Two Binding Orientations for Peptides to the Src SH3 Domain: Development of a General Model for SH3-Ligand Interactions. Science (1994) 266(5188):1241–7. doi: 10.1126/science.7526465
66. Lim WA, Richards FM, Fox RO. Structural Determinants of Peptide-Binding Orientation and of Sequence Specificity in SH3 Domains. Nature (1994) 372(6504):375–9. doi: 10.1038/372375a0
67. Saksela K, Permi P. SH3 Domain Ligand Binding: What's the Consensus and Where's the Specificity? FEBS Lett (2012) 586(17):2609–14. doi: 10.1016/j.febslet.2012.04.042
68. Waksman G, Kominos D, Robertson SC, Pant N, Baltimore D, Birge RB, et al. Crystal Structure of the Phosphotyrosine Recognition Domain SH2 of V-Src Complexed With Tyrosine-Phosphorylated Peptides. Nature (1992) 358(6388):646–53. doi: 10.1038/358646a0
69. Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, et al. SH2 Domains Recognize Specific Phosphopeptide Sequences. Cell (1993) 72(5):767–78. doi: 10.1016/0092-8674(93)90404-E
70. Waksman G, Kuriyan J. Structure and Specificity of the SH2 Domain. Cell (2004) 116(2 Suppl):S45–8, 3 p following S8. doi: 10.1016/S0092-8674(04)00043-1
71. Xu W, Harrison SC, Eck MJ. Three-Dimensional Structure of the Tyrosine Kinase C-Src. Nature (1997) 385(6617):595–602. doi: 10.1038/385595a0
72. Cooper JA, Gould KL, Cartwright CA, Hunter T. Tyr527 is Phosphorylated in Pp60c-Src: Implications for Regulation. Science (1986) 231(4744):1431–4. doi: 10.1126/science.2420005
73. Zheng XM, Wang Y, Pallen CJ. Cell Transformation and Activation of Pp60c-Src by Overexpression of a Protein Tyrosine Phosphatase. Nature (1992) 359(6393):336–9. doi: 10.1038/359336a0
74. Jung EJ, Kim CW. Interaction Between Chicken Protein Tyrosine Phosphatase 1 (CPTP1)-Like Rat Protein Phosphatase 1 (PTP1) and P60(V-Src) in V-Src-Transformed Rat-1 Fibroblasts. Exp Mol Med (2002) 34(6):476–80. doi: 10.1038/emm.2002.66
75. Bjorge JD, Pang A, Fujita DJ. Identification of Protein-Tyrosine Phosphatase 1B as the Major Tyrosine Phosphatase Activity Capable of Dephosphorylating and Activating C-Src in Several Human Breast Cancer Cell Lines. J Biol Chem (2000) 275(52):41439–46. doi: 10.1074/jbc.M004852200
76. Smart JE, Oppermann H, Czernilofsky AP, Purchio AF, Erikson RL, Bishop JM. Characterization of Sites for Tyrosine Phosphorylation in the Transforming Protein of Rous Sarcoma Virus (Pp60v-Src) and its Normal Cellular Homologue (Pp60c-Src). Proc Natl Acad Sci USA (1981) 78(10):6013–7. doi: 10.1073/pnas.78.10.6013
77. Chiang GG, Sefton BM. Phosphorylation of a Src Kinase at the Autophosphorylation Site in the Absence of Src Kinase Activity. J Biol Chem (2000) 275(9):6055–8. doi: 10.1074/jbc.275.9.6055
78. Johnson LN, Noble ME, Owen DJ. Active and Inactive Protein Kinases: Structural Basis for Regulation. Cell (1996) 85(2):149–58. doi: 10.1016/S0092-8674(00)81092-2
79. Cobb BS, Schaller MD, Leu TH, Parsons JT. Stable Association of Pp60src and Pp59fyn With the Focal Adhesion-Associated Protein Tyrosine Kinase, Pp125fak. Mol Cell Biol (1994) 14(1):147–55. doi: 10.1128/mcb.14.1.147-155.1994
80. Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the Focal Adhesion Kinase, Pp125fak, Directs SH2-Dependent Binding of Pp60src. Mol Cell Biol (1994) 14(3):1680–8. doi: 10.1128/mcb.14.3.1680-1688.1994
81. Thomas JW, Ellis B, Boerner RJ, Knight WB, White GC2, Schaller MD. SH2- and SH3-Mediated Interactions Between Focal Adhesion Kinase and Src. J Biol Chem (1998) 273(1):577–83. doi: 10.1074/jbc.273.1.577
82. Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of Biological Synergy Between Cellular Src and Epidermal Growth Factor Receptor. Proc Natl Acad Sci U S A (1999) 96(4):1415–20. doi: 10.1073/pnas.96.4.1415
83. Muthuswamy SK, Siegel PM, Dankort DL, Webster MA, Muller WJ. Mammary Tumors Expressing the Neu Proto-Oncogene Possess Elevated C-Src Tyrosine Kinase Activity. Mol Cell Biol (1994) 14(1):735–43. doi: 10.1128/mcb.14.1.735-743.1994
84. Kypta RM, Goldberg Y, Ulug ET, Courtneidge SA. Association Between the PDGF Receptor and Members of the Src Family of Tyrosine Kinases. Cell (1990) 62(3):481–92. doi: 10.1016/0092-8674(90)90013-5
85. Landgren E, Blume-Jensen P, Courtneidge SA, Claesson-Welsh L. Fibroblast Growth Factor Receptor-1 Regulation of Src Family Kinases. Oncogene (1995) 10(10):2027–35. doi: 10.1016/0092-8674(95)90085-3
86. Boggon TJ, Eck MJ. Structure and Regulation of Src Family Kinases. Oncogene (2004) 23(48):7918–27. doi: 10.1038/sj.onc.1208081
87. Roche S, Fumagalli S, Courtneidge SA. Requirement for Src Family Protein Tyrosine Kinases in G2 for Fibroblast Cell Division. Science (1995) 269(5230):1567–9. doi: 10.1126/science.7545311
88. Chu I, Sun J, Arnaout A, Kahn H, Hanna W, Narod S, et al. P27 Phosphorylation by Src Regulates Inhibition of Cyclin E-Cdk2. Cell (2007) 128(2):281–94. doi: 10.1016/j.cell.2006.11.049
89. Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, et al. Cdk-Inhibitory Activity and Stability of p27Kip1 are Directly Regulated by Oncogenic Tyrosine Kinases. Cell (2007) 128(2):269–80. doi: 10.1016/j.cell.2006.11.047
90. Cozzi M, Giorgi F, Marcelli E, Pentimalli F, Forte IM, Schenone S, et al. Antitumor Activity of New Pyrazolo[3,4-D]Pyrimidine SRC Kinase Inhibitors in Burkitt Lymphoma Cell Lines and its Enhancement by WEE1 Inhibition. Cell Cycle (2012) 11(5):1029–39. doi: 10.4161/cc.11.5.19519
91. Moasser MM, Srethapakdi M, Sachar KS, Kraker AJ, Rosen N. Inhibition of Src Kinases by a Selective Tyrosine Kinase Inhibitor Causes Mitotic Arrest. Cancer Res (1999) 59(24):6145–52.
92. Fukumoto Y, Morii M, Miura T, Kubota S, Ishibashi K, Honda T, et al. Src Family Kinases Promote Silencing of ATR-Chk1 Signaling in Termination of DNA Damage Checkpoint. J Biol Chem (2014) 289(18):12313–29. doi: 10.1074/jbc.M113.533752
93. Janiszewska M, Primi MC, Izard T. Cell Adhesion in Cancer: Beyond the Migration of Single Cells. J Biol Chem (2020) 295(8):2495–505. doi: 10.1074/jbc.REV119.007759
94. Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, et al. Hakai, a C-Cbl-Like Protein, Ubiquitinates and Induces Endocytosis of the E-Cadherin Complex. Nat Cell Biol (2002) 4(3):222–31. doi: 10.1038/ncb758
95. Kuo L, Chang HC, Leu TH, Maa MC, Hung WC. Src Oncogene Activates MMP-2 Expression via the ERK/Sp1 Pathway. J Cell Physiol (2006) 207(3):729–34. doi: 10.1002/jcp.20616
96. Kim H, Lee HS, Kim TH, Lee JS, Lee ST, Lee SJ. Growth-Stimulatory Activity of TIMP-2 is Mediated Through C-Src Activation Followed by Activation of FAK, PI3-Kinase/AKT, and ERK1/2 Independent of MMP Inhibition in Lung Adenocarcinoma Cells. Oncotarget (2015) 6(40):42905–22. doi: 10.18632/oncotarget.5466
97. Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, et al. Steroid-Induced Androgen Receptor-Oestradiol Receptor Beta-Src Complex Triggers Prostate Cancer Cell Proliferation. EMBO J (2000) 19(20):5406–17. doi: 10.1093/emboj/19.20.5406
98. Cai H, Babic I, Wei X, Huang J, Witte ON. Invasive Prostate Carcinoma Driven by C-Src and Androgen Receptor Synergy. Cancer Res (2011) 71(3):862–72. doi: 10.1158/0008-5472.CAN-10-1605
99. Yang CC, Fazli L, Loguercio S, Zharkikh I, Aza-Blanc P, Gleave ME, et al. Downregulation of C-SRC Kinase CSK Promotes Castration Resistant Prostate Cancer and Pinpoints a Novel Disease Subclass. Oncotarget (2015) 6(26):22060–71. doi: 10.18632/oncotarget.4279
100. Li H, Xie N, Chen R, Verreault M, Fazli L, Gleave ME, et al. UGT2B17 Expedites Progression of Castration-Resistant Prostate Cancers by Promoting Ligand-Independent AR Signaling. Cancer Res (2016) 76(22):6701–11. doi: 10.1158/0008-5472.CAN-16-1518
101. Migliaccio A, Domenico MD, Castoria G, Nanayakkara M, Lombardi M, de Falco A, et al. Steroid Receptor Regulation of Epidermal Growth Factor Signaling Through Src in Breast and Prostate Cancer Cells: Steroid Antagonist Action. Cancer Res (2005) 65(22):10585–93. doi: 10.1158/0008-5472.CAN-05-0912
102. Asim M, Siddiqui IA, Hafeez BB, Baniahmad A, Mukhtar H. Src Kinase Potentiates Androgen Receptor Transactivation Function and Invasion of Androgen-Independent Prostate Cancer C4-2 Cells. Oncogene (2008) 27(25):3596–604. doi: 10.1038/sj.onc.1211016
103. Liu Y, Karaca M, Zhang Z, Gioeli D, Earp HS, Whang YE. Dasatinib Inhibits Site-Specific Tyrosine Phosphorylation of Androgen Receptor by Ack1 and Src Kinases. Oncogene (2010) 29(22):3208–16. doi: 10.1038/onc.2010.103
104. Zhoul J, Hernandez G, Tu SW, Huang CL, Tseng CP, Hsieh JT. The Role of DOC-2/DAB2 in Modulating Androgen Receptor-Mediated Cell Growth via the Nongenomic C-Src-Mediated Pathway in Normal Prostatic Epithelium and Cancer. Cancer Res (2005) 65(21):9906–13. doi: 10.1158/0008-5472.CAN-05-1481
105. Migliaccio A, Castoria G, Domenico MD, de Falco A, Bilancio A, Lombardi M, et al. Sex Steroid Hormones Act as Growth Factors. J Steroid Biochem Mol Biol (2002) 83(1-5):31–5. doi: 10.1016/S0960-0760(02)00264-9
106. Roediger J, Hessenkemper W, Bartsch S, Manvelyan M, Huettner SS, Liehr T, et al. Supraphysiological Androgen Levels Induce Cellular Senescence in Human Prostate Cancer Cells Through the Src-Akt Pathway. Mol Cancer (2014) 13:214. doi: 10.1186/1476-4598-13-214
107. Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src Family Kinase Oncogenic Potential and Pathways in Prostate Cancer as Revealed by AZD0530. Oncogene (2008) 27(49):6365–75. doi: 10.1038/onc.2008.250
108. Slack JK, Adams RB, Rovin JD, Bissonette EA, Stoker CE, Parsons JT. Alterations in the Focal Adhesion Kinase/Src Signal Transduction Pathway Correlate With Increased Migratory Capacity of Prostate Carcinoma Cells. Oncogene (2001) 20(10):1152–63. doi: 10.1038/sj.onc.1204208
109. Kaplan-Lefko PJ, Chen TM, Ittmann MM, Barrios RJ, Ayala GE, Huss WJ, et al. Pathobiology of Autochthonous Prostate Cancer in a Pre-Clinical Transgenic Mouse Model. Prostate (2003) 55(3):219–37. doi: 10.1002/pros.10215
110. Gelman IH, Peresie J, Eng KH, Foster BA. Differential Requirement for Src Family Tyrosine Kinases in the Initiation, Progression, and Metastasis of Prostate Cancer. Mol Cancer Res (2014) 12(10):1470–9. doi: 10.1158/1541-7786.MCR-13-0490-T
111. Soriano P, Montgomery C, Geske R, Bradley A. Targeted Disruption of the C-Src Proto-Oncogene Leads to Osteopetrosis in Mice. Cell (1991) 64(4):693–702. doi: 10.1016/0092-8674(91)90499-O
112. Naqvi K, Jabbour E, Skinner J, Yilmaz M, Ferrajoli A, Bose P, et al. Early Results of Lower Dose Dasatinib (50 Mg Daily) as Frontline Therapy for Newly Diagnosed Chronic-Phase Chronic Myeloid Leukemia. Cancer (2018) 124(13):2740–7. doi: 10.1002/cncr.31357
113. Robison NJ, Yeo KK, Berliner AP, Malvar J, Sheard MA, Margol AS, et al. Phase I Trial of Dasatinib, Lenalidomide, and Temozolomide in Children With Relapsed or Refractory Central Nervous System Tumors. J Neurooncol (2018) 138(1):199–207. doi: 10.1007/s11060-018-2791-y
114. Yu EY, Wilding G, Posadas E, Gross M, Culine S, Massard C, et al. Phase II Study of Dasatinib in Patients With Metastatic Castration-Resistant Prostate Cancer. Clin Cancer Res (2009) 15(23):7421–8. doi: 10.1158/1078-0432.CCR-09-1691
115. Yu EY, Massard C, Gross ME, Carducci MA, Culine S, Hudes G, et al. Once-Daily Dasatinib: Expansion of Phase II Study Evaluating Safety and Efficacy of Dasatinib in Patients With Metastatic Castration-Resistant Prostate Cancer. Urology (2011) 77(5):1166–71. doi: 10.1016/j.urology.2011.01.006
116. Keating GM. Dasatinib: A Review in Chronic Myeloid Leukaemia and Ph+ Acute Lymphoblastic Leukaemia. Drugs (2017) 77(1):85–96. doi: 10.1007/s40265-016-0677-x
117. Araujo JC, Trudel GC, Saad F, Armstrong AJ, Yu EY, Bellmunt J, et al. Docetaxel and Dasatinib or Placebo in Men With Metastatic Castration-Resistant Prostate Cancer (READY): A Randomised, Double-Blind Phase 3 Trial. Lancet Oncol (2013) 14(13):1307–16. doi: 10.1016/S1470-2045(13)70479-0
118. Dorff TB, Quinn DI, Pinski JK, Goldkorn A, Sadeghi S, Tsao-Wei D, et al. Randomized Phase II Trial of Abiraterone Alone or With Dasatinib in Men With Metastatic Castration-Resistant Prostate Cancer (mCRPC). Clin Genitourin Cancer (2019) 17(4):241–7.e1. doi: 10.1016/j.clgc.2019.02.010
119. Spreafico A, Chi KN, Sridhar SS, Smith DC, Carducci MA, Kavsak P, et al. A Randomized Phase II Study of Cediranib Alone Versus Cediranib in Combination With Dasatinib in Docetaxel Resistant, Castration Resistant Prostate Cancer Patients. Invest New Drugs (2014) 32(5):1005–16. doi: 10.1007/s10637-014-0106-5
120. Spetsieris N, Boukovala M, Weldon JA, Tsikkinis A, Hoang A, Aparicio A, et al. A Phase 2 Trial of Abiraterone Followed by Randomization to Addition of Dasatinib or Sunitinib in Men With Metastatic Castration-Resistant Prostate Cancer. Clin Genitourin Cancer (2021) 19(1):22–31.e5. doi: 10.1016/j.clgc.2020.05.013
121. Luo FR, Luo FR, Barrett Y, Ji P, Holly P, McCann E, et al. Dasatinib (BMS-354825) Pharmacokinetics Correlate With pSRC Pharmacodynamics in Phase I Studies of Patients With Cancer (CA180002, Ca180003). J Clin Oncol (2006) 24(18_suppl):3046. doi: 10.1200/jco.2006.24.18_suppl.3046
122. Twardowski PW, Beumer JH, Chen CS, Kraft AS, Chatta GS, Mitsuhashi M, et al. A Phase II Trial of Dasatinib in Patients With Metastatic Castration-Resistant Prostate Cancer Treated Previously With Chemotherapy. Anticancer Drugs (2013) 24(7):743–53. doi: 10.1097/CAD.0b013e328361feb0
123. Goto Y, Ando T, Izumi H, Feng X, Arang N, Gilardi M, et al. Muscarinic Receptors Promote Castration-Resistant Growth of Prostate Cancer Through a FAK-YAP Signaling Axis. Oncogene (2020) 39(20):4014–27. doi: 10.1038/s41388-020-1272-x
124. Alfano RW, Leppla SH, Liu S, Bugge TH, Ortiz JM, Lairmore TC, et al. Inhibition of Tumor Angiogenesis by the Matrix Metalloproteinase-Activated Anthrax Lethal Toxin in an Orthotopic Model of Anaplastic Thyroid Carcinoma. Mol Cancer Ther (2010) 9(1):190–201. doi: 10.1158/1535-7163.MCT-09-0694
Keywords: prostate cancer, Src kinase, androgen receptor, castrate-resistant prostate cancer, UGT2B17
Citation: Gao L, Han B and Dong X (2022) The Androgen Receptor and Its Crosstalk With the Src Kinase During Castrate-Resistant Prostate Cancer Progression. Front. Oncol. 12:905398. doi: 10.3389/fonc.2022.905398
Received: 27 March 2022; Accepted: 26 May 2022;
Published: 27 June 2022.
Edited by:
Maximilian Marhold, Medical University of Vienna, AustriaReviewed by:
Xiuping Yu, Louisiana State University Health Shreveport, United StatesXiaohong Li, University of Toledo, United States
Kalpit Shah, Genentech, Inc., United States
Copyright © 2022 Gao, Han and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuesen Dong, eGRvbmdAcHJvc3RhdGVjZW50cmUuY29t