 Dipta Dey1†
Dipta Dey1† Mohammad Mehedi Hasan2
Mohammad Mehedi Hasan2 Partha Biswas3,4†
Partha Biswas3,4† Stavros P. Papadakos5
Stavros P. Papadakos5 Rehab A. Rayan6
Rehab A. Rayan6 Sabiha Tasnim7Muhammad Bilal8Mohammod Johirul Islam2Farzana Alam Arshe9
Sabiha Tasnim7Muhammad Bilal8Mohammod Johirul Islam2Farzana Alam Arshe9 Efat Muhammad Arshad9Maisha Farzana10
Efat Muhammad Arshad9Maisha Farzana10 Tanjim Ishraq Rahaman11
Tanjim Ishraq Rahaman11 Sumit Kumar Baral12Priyanka Paul1
Sumit Kumar Baral12Priyanka Paul1 Shabana Bibi13,14
Shabana Bibi13,14 Md. Ataur Rahman15,16,17*
Md. Ataur Rahman15,16,17* Bonglee Kim16,17*
Bonglee Kim16,17*- 1Biochemistry and Molecular Biology department, Life Science Faculty, Bangabandhu Sheikh Mujibur Rahman Science and Technology University, Gopalgonj, Bangladesh
- 2Department of Biochemistry and Molecular Biology, Faculty of Life Science, Mawlana Bhashani Science and Technology University, Tangail, Bangladesh
- 3Department of Genetic Engineering and Biotechnology, Faculty of Biological Science and Technology, Jashore University of Science and Technology (JUST), Jashore, Bangladesh
- 4ABEx Bio-Research Center, East Azampur, Dhaka, Bangladesh
- 5First Department of Pathology, School of Medicine, National and Kapodistrian University of Athens (NKUA), Athens, Greece
- 6Department of Epidemiology, High Institute of Public Health, Alexandria University, Alexandria, Egypt
- 7Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Dhaka, Dhaka, Bangladesh
- 8College of Pharmacy, Liaquat University of Medical and Health Sciences, Jamshoro, Pakistan
- 9Department of Biochemistry and Microbiology, North South University, Dhaka, Bangladesh
- 10College of Medical, Veterinary and Life Sciences, University of Glasgow, University Avenue, Glasgow, United Kingdom
- 11Department of Biotechnology and Genetic Engineering, Faculty of Life Science, Bangabandhu Sheikh Mujibur Rahman Science and Technology University, Gopalganj, Bangladesh
- 12Microbiology department, Jagannath University, Dhaka, Bangladesh
- 13Yunnan Herbal Laboratory, College of Ecology and Environmental Sciences, Yunnan University, Kunming, China
- 14Department of Biological Sciences, International Islamic University, Islamabad, Pakistan
- 15Global Biotechnology & Biomedical Research Network (GBBRN), Department of Biotechnology and Genetic Engineering, Faculty of Biological Sciences, Islamic University, Kushtia, Bangladesh
- 16Department of Pathology, College of Korean Medicine, Kyung Hee University, Seoul, South Korea
- 17Korean Medicine-Based Drug Repositioning Cancer Research Center, College of Korean Medicine, Kyung Hee University, Seoul, South Korea
Salvicine is a new diterpenoid quinone substance from a natural source, specifically in a Chinese herb. It has powerful growth-controlling abilities against a broad range of human cancer cells in both in vitro and in vivo environments. A significant inhibitory effect of salvicine on multidrug-resistant (MDR) cells has also been discovered. Several research studies have examined the activities of salvicine on topoisomerase II (Topo II) by inducing reactive oxygen species (ROS) signaling. As opposed to the well-known Topo II toxin etoposide, salvicine mostly decreases the catalytic activity with a negligible DNA breakage effect, as revealed by several enzymatic experiments. Interestingly, salvicine dramatically reduces lung metastatic formation in the MDA-MB-435 orthotopic lung cancer cell line. Recent investigations have established that salvicine is a new non-intercalative Topo II toxin by interacting with the ATPase domains, increasing DNA–Topo II interaction, and suppressing DNA relegation and ATP hydrolysis. In addition, investigations have revealed that salvicine-induced ROS play a critical role in the anticancer-mediated signaling pathway, involving Topo II suppression, DNA damage, overcoming multidrug resistance, and tumor cell adhesion suppression, among other things. In the current study, we demonstrate the role of salvicine in regulating the ROS signaling pathway and the DNA damage response (DDR) in suppressing the progression of cancer cells. We depict the mechanism of action of salvicine in suppressing the DNA–Topo II complex through ROS induction along with a brief discussion of the anticancer perspective of salvicine.
Introduction
Cancer is a menace to humankind that causes millions of deaths worldwide every year (1). Cancer occurs when there is an uncontrollable cell division in any tissues of a living body, and it usually proliferates, causing the invasion of other healthy tissues in an individual. It is still one of the most challenging issues in the medical and public health sectors (2–4). The American Cancer Society evaluates cancer cases annually in the USA, and its most recent estimate in 2021 revealed 1,898,160 cases with 6,08,570 mortalities (5–8). Furthermore, a lot of conditions are associated with the most concerning risk factors for cancers, notably age, sex, genetic and/or epigenetic factors, coexisting complications, and so forth (9, 10). Smoking, pollution, and obesity affect the rate of lung cancer and cancers related to reproductive organs, such as prostate, ovarian, cervical, and breast cancers (11–14). Likewise, other risk factors such as solar exposure, lousy diet, and radiation exposure affect cancers of the stomach, colon, liver, pancreas, kidney, and bladder, as well as glioblastoma and skin cancers. Additionally, polluted air with benzene can trigger some effects on leukemia, lymphoma, and myeloma (15). As maintained by the National Cancer Institute, breast cancer in women is the most common cancer type (approximately 2.3 million new cases occur annually), prostate the second, and the third is lung cancer. Colorectal cancer takes the fourth position in the survey; however, liver and intrahepatic bile duct cancers have the least number of new cases (16, 17). As a matter of fact, mutations in the BRCA1/BRCA2 genes stimulate early-onset breast cancer even if patients do not have any family history of cancer (18, 19). The World Cancer Research Fund claimed that the consumption of healthy foods could significantly reduce cancer risks, and some diets like the Mediterranean diet can reduce the severity of gastric, glioblastoma, colon, rectal, bladder, liver, and lung cancers. Notably, a high intake of roughage is known to minimize the risk of colorectal cancers (20). Multiple phytocompounds, without any doubt, have been applied in an anticancer perspective and have shown positive outcomes in clinical trials, along with acting as potential anticancer agents (21–24).
Salvicine is a potential bioactive phytomolecule belonging to the diterpenoquinone derivatives extracted from Salvia prionitis Hance, a Chinese herbal medicinal plant. It has a chemically active quinone moiety and imparts antitumor activity by modulating the generation of reactive oxygen species (ROS) (25). However, salvicine directly acts toward topoisomerase II (Topo II), or simply Topo II enzyme, as Topo II is especially crucial in regulating the DNA topology, notably DNA replication, transcription, recombination, and cell cycle (26, 27). Type II topoisomerases include two main subdivisions—notably Topo IIα and Topo IIβ—and hence human cells express such enzymes from Topo IIα and Topo IIβ genes, respectively (28). However, Topo IIα plays a key role in the cell division stage of the S and M phases of the cancer cell cycle at the same time as its role in both DNA multiplication and chromosome separation for mitotic cell division. Conversely, Topo IIα is not related to cell sustainability, sometimes trapped by transcriptional factors of multiple cancerous cells; therefore, it is not related to cell division (29, 30). Importantly, Topo II is the principal drug target site for cancer treatment via following its anti-proliferative activity, DNA double-strand breaks (DSBs), and inducing apoptosis-related pathways (31, 32). Accordingly, salvicine effectively elevates the level of intracellular ROS, which potentially initiates the DSBs through the N-acetylation of cysteine (NAC) residues (33). Multiple scientific reports have demonstrated that salvicine upregulates the level of ROS in the cellular system, which subsequently blocks the enzyme activity of Topo II and mediates DNA DSBs, apoptosis, and the cytotoxicity mechanism (34–36).
Additionally, salvicine mediates the activation of the following pathways: ataxia telangiectasia mutated (ATM) kinase, ataxia telangiectasia and Rad3-related (ATR) kinase, and histone H2AX; therefore, the complete experimental trail occurs in lung carcinoma A549 cells (37). It is noteworthy that it directly shuts off the activity of the telomerase enzyme and subsequently inhibits lung carcinoma, as well as the initiation of apoptotic pathways, as shown in a research work on the A549 and HL-60 cell lines. (33, 38). Salvicine causes genomic DNA DSBs in HL-60 human promyelocytic leukemia cells and MCF-7 breast cancer cells (39). It shows activity toward not only several cancer cases but also in different cell lines including K562, HL-60, U-937, K-562, HL-60, A02, SGC-7901, MKN-28, A549, SPC-A4, NCI-H23, NCI-H522, S-180, MCF-7, and HeLa. All these are shown in detail in Table 1. In leukemia, this novel compound follows diverse pathways, among them are enhancement of cytotoxic activity, induction of the apoptotic pathway, initiation of promoters mediating DNA damage, reduction of the telomerase enzyme activity, decrease of mdr-1 via blocking Topo II, repression of the activation of the transcription factor c-Jun, and production of the ROS moiety involved in cell death with P-glycoprotein (P-gp) activity reduction (58–64). At the same time, its cytotoxic activity has been proven in several cell lines, such as in K562, HL-60, U-937, K-562, A02, SGC-7901, and MKN-28, with such activity being more dose-dependent. In contrast, salvicine is involved in anti-stomach cancer activity by following cytotoxicity, apoptotic, and antitumor mechanisms. It is important to note that the experiment was not only performed on human patients but also involved the different cell lines mentioned, i.e., SGC-7901 and MKN-28 (64, 65). Several scientific reports have demonstrated that the A549, SPC-A4, NCI-H23, and NCI-H522 cell lines also showed more positive results in lung carcinoma after the administration of salvicine at specific doses via following mechanisms such as telomerase inhibition, decreased p53 gene levels, telomeric protein TRF2 damage, and anti-angiogenic activity (66–69). Additionally, the MCF-7, ADR, and MDA-MB-435 cell lines were used for the analysis of the anti-breast cancer activity of salvicine. Moreover, this compound maintains diverse mechanisms including DNA double-strand damage, apoptosis, derangement of cell adhesion to the extracellular matrix (ECM), and cytotoxic activity (61, 70–72). Other more authentic scientific reports have stated that salvicine has more anticancer activity in sarcoma, pancreatic cancer, cervical cancer, and oral carcinoma (61, 65, 73, 74). In tumor metastasis, salvicine influences the cell adhesion genes and decreases the expressions of different integrin proteins, i.e., integrin a3/a6/aE/b3/b5/b8, paxillin, and focal adhesion kinase (FAK), in human breast cancer; its anti-metastatic efficiency has also been suggested (75). On the other hand, it elicits the activation of extracellular signal-regulated kinase (ERK) and p38 mitogen-activated protein kinase (p38 MAPK). It has also shown effects on integrin-mediated cell adhesion in U0126 and SB203580, as well as on the inhibition of both p38 MAPK and MAPK/ERK (55). Salvicine remarkably reduces the expression level of the Rho protein, mainly RhoC in primary tumors (without affecting RhoA), and it significantly disrupts the Rho-dependent stress fiber and subsequently restricts the cell adhesion and motility, also downregulating the gene expression along with control of functional proteins such as fibronectin, integrins, FAK, RhoC, and paxillin, which are associated with these pathways (75). Another research group showed that salvicine decreases the level of RhoA (GTP-mediated) and damages actin stress fiber networks crucial to cell adhesion via ROS-related inhibition of the activity of RhoA (55). Salvicine also possesses cytotoxic properties against a variety of multidrug-resistant (MDR) tumor cells via downregulating P-gp expression. It suppresses the expression of the mdr-1 gene, and hence P-gp expression, by stimulating the gene expression of mainly c-Jun in MDR K562/A02 cells.

Table 1 Potential activities of salvicine in numerous cancer types and cancer cell lines with their significant mechanisms of actions.
The studies included in the current review specifically emphasize that salvicine potentially blocks the DNA Topo II enzyme, which is an interesting target for cancer treatment, along with diterpenoquinone compounds that impart the ROS molecule and are directly involved in DNA double-strand damage, DNA DSBs, apoptosis, and cytotoxicity mechanism. Furthermore, we included here multiple anticancer perspectives of salvicine from published scientific reports.
Overview of Salvicine
Salvicine is a new diterpenoid quinone compound that is produced by modifying the composition of a natural product lead found in the Chinese herb S. prionitis Hance (Labiatae) (76). It was chemically synthesized in 1999 by Sheng et al. and has been demonstrated to have substantial inhibitory effects against a broad spectrum of human tumor cells both in vitro and in mice harboring human tumor xenografts (77, 78). Specifically, salvicine and its analogs are non-intercalative Topo II inhibitors that have demonstrated potent anticancer efficacy both in vitro and in vivo, as well as a broad spectrum of anti-MDR activities in animal models (79). It has been reported that salvicine causes the breakage of two strands of DNA by promoting Topo II activity and blocking re-ligation, which is correlated with the suppression of tumor growth. Salvicine also promotes apoptosis in human cancer cells via inducing DNA damage to specific genes. It has also been shown that salvicine possesses anticancer activity manifested in a variety of ways, including downregulation of Topo II, DNA damage, bypassing multidrug resistance, and prevention of tumor cell adhesion (78). It has been revealed that a new family of salvicine compounds exhibited significant cytotoxicity against tumor cell lines (80). Salvicine and its derivatives showed significant activity against solid tumor cells, particularly lung and gastric cancer cells, in comparison to its modest cytotoxicity on leukemia cell lines (46). In vivo experiments revealed that salvicine exhibited strong anticancer efficacy against murine S-180 sarcoma and Lewis’s lung cancer, as well as in human lung adenocarcinoma xenograft models, in addition to other cancer types (48). Furthermore, a study found that the anticancer activity salvicine was correlated with its ability to cause tumor cell apoptosis with comparable potency against both human leukemia cells and gastric carcinoma cells, showing its particular impact on solid tumor cells (47). Three cell lines (K-562/A02, MCF-7/ADM, and MKN28/VCR) were found to be resistant to a lot of different drugs, but the cytotoxicity of salvicine was not affected by P-gp. These research findings indicate that salvicine is a promising anticancer drug. It is currently undergoing clinical trials in several countries (33).
ROS Signaling Pathway, DNA Damage Response, and Cancer
ROS are a collection of transient elements initially delineated as free radicals in skeletal muscle, such as O2, OH, and H2O2 (81). They were first thought of as dangerous derivatives of respiration by the mitochondria, but recent breakthroughs have shed light on the cellular operational functions of ROS from meliorating immunity, e.g., oxidative changes in scavenger cells, to getting rid of microorganisms and to acting as signaling elements, such as H2O2 modulating the MAPK and NF-κB pathways (82, 83). ROS are produced internally via the mitochondria, with O2 acting as the ultimate electron recipient for electron transmission series (84); NADPH oxidase, a cellular membrane-linked enzyme (85); peroxisomes, which contain enzymes for the production of H2O2, such as polyamine oxidase (86); and the endoplasmic reticulum, which induces H2O2 as a derivative in folding peptides or during vulnerability to exogenic tension such as chemotherapy, ionizing radiation (IR), or ecological breaches that impact the previously mentioned cell organs and enzymes (87). The production of ROS has connections with the outcomes of radiation therapy or chemotherapy through its impacts on backward cellular viability or fatality rate ranges (88, 89). Therefore, ROS modulators could be employed in malignant neoplasm primary prevention or to intensify treatment outcomes (90). There have been trivial advancements in the understanding of ROS from the research laboratory to the medical institution (40). For instance, despite the auspicious in vitro data, some antioxidant trials in the prevention of malignant neoplasm have shown contradictory findings, highlighting the demand for further understanding of such procedures in cells (91).
DNA impairment denotes chemical or physical alterations of cellular DNA, which could impact the reading and sending of inherited data. DNA can be altered by a variety of endogenic and exogenic injuries involving radiation, chemicals, free radicals, and topologic alterations, all of which stimulate well-defined types of impairment (92). Cells have developed composite procedures for managing impairments to the genome. Regarding the reason for injury to the DNA, certain pathway/s are triggered for ease of determining the altered parts and their repair (93). Indeed, DNA DSB is a serious mutagenic injury because of the arrangements inside the chromosomes or failure of hereditary data for the wrong DNA repair. As a reaction to DNA impairment, a system of events jointly called DNA damage response (DDR) is triggered (Figure 1). Such an effect involves identifying the DNA impairment, stimulating checkpoints, halting the cellular processes, and, finally, the processes of repair, apoptosis, and immune clearance (94–96). The building blocks of DSB-generated DDR have been analyzed and sorted into three main units: sensors to determine the impairment, transducers to organize signaling, and effectors to coordinate final events (97). Different DNA impairment outcome/repair pathways involve mismatch repair for incompatible bases, nucleotide excision repair (NER) for the cross-linking of intra-strand thymidine dimers, base excision repair (BER) for base alterations, single-strand annealing for single-strand DNA (ssDNA) damage, and transcription-coupled repair (TCR) for transcription-mediated impairments. However, the DDR motivated by DSBs triggers a system of similar pathways involving homologous recombination (HR), non-homologous end-joining (NHEJ), microhomology-mediated end-joining (MMEJ), and Fanconi anemia (FA) repair (93). The reaction to DSBs especially applies to carcinogenesis and chemotherapy, as many of the factors of the pathway are changed in malignant neoplasm, and so much of today’s radiotherapy and chemotherapy overwork such flaws (98).
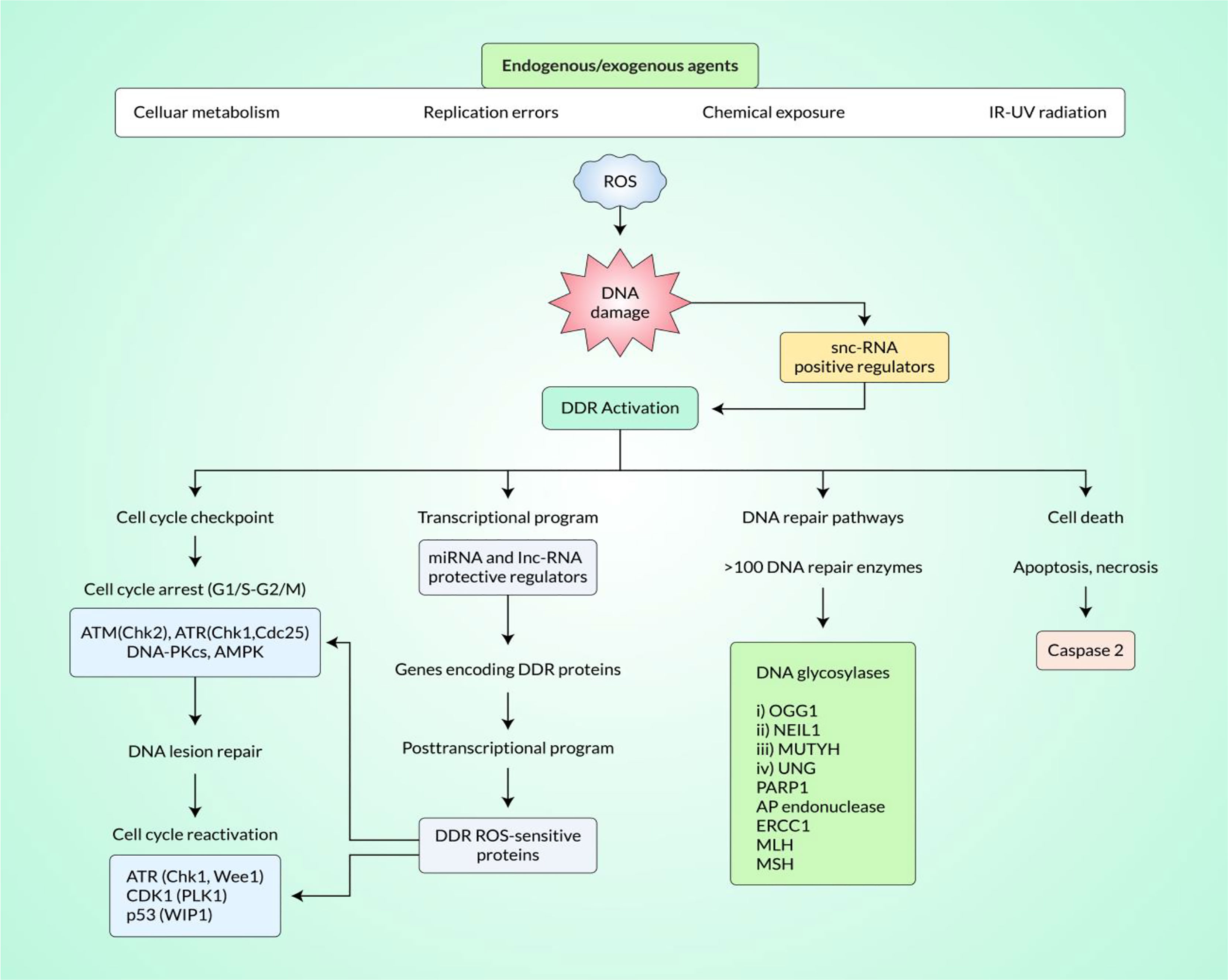
Figure 1 Induction of DNA damage response (DDR) by reactive oxygen species (ROS) signaling in cancer tissue. ROS signaling pathways can effectively induce the DDR in cancer tissue. Small non-coding RNA (snc-RNA) positive regulators play important roles in DDR activation. DDR activation effectively modulates the cell cycle checkpoint, transcriptional program, DNA repair pathways, and programmed cell death.
ROS in DNA Damage by Genotoxin
ROS have been recognized to mediate DNA impairment. For instance, IR stimulates DSBs via immediate great power impairment of the sugar pillar of DNA, besides via cellular free radicals, mainly the water’s OH. Chemotherapeutics such as cisplatin and doxorubicin increase the level of ROS, adding to their genotoxicity (99). ROS have likewise been denoted to instantly stimulate different types of DNA impairments via oxidizing nucleoside bases, for example, by producing 8-oxoguanine that could result in G–A or G–T transversions when unrestored (100). The BER pathway identifies and fixes oxidized bases; however, if they appear at the same time on opposing strands, BER could directly initiate DSBs (101). ROS generation likewise involves injuries, strand cracks, and decay of mitochondrial DNA (102).
ROS-Mediated Oncogenic Stress in DNA Damage
The replication stress triggered by oncogenes is an influential origin of endogenous DNA damage and the production of DSBs in malignant neoplasms (103). Proto-oncogenes assist in cell proliferation and growth; however, overexpression or mutations could transform them into oncogenes, which causes continuous cellular development and carcinogenicity. Oncogenic cellular processes are linked to replication stress, which has been delineated as an abnormal replication fork series and formation of DNA (104). Replication stress finally leads to genetic imbalance and drives neoplasm evolution via the aggregation of additional pro-carcinogenic alterations (103). The DDR acts as a roadblock that confines the elaboration of unusual cellular replication; hence, it controls an exclusive force for carcinogenic-related DDR defects (105). Replication stress has diverse origins including abnormal source blasting, the action of physical obstacles, and disconnected DNA polymerase helicase from the replicating fork (104). The stimulation of oncogenes gives rise to ROS that affect replication stress events (106). dNTPs are oxidized by ROS to impact polymerase activity and, hence, decrease the speed of the replication forks in vitro (107). ROS can likewise impact replication fork advancement via the separation of peroxiredoxin 2 oligomers (PRDX2), where it creates a replisome-related ROS sensor that binds to the catalyst of the fork (TIMELESS). Increased ROS results in the separation of TIMELESS and PRDX2, therefore decreasing the replication fork velocity (108). Oxidized bases resulting from the activity of ROS likewise show physical barriers to replication forks (42), leading to the collapse of the replication forks at breakable positions along the genome, further resulting in DSBs and, finally, to over- or under-replicated DNA, with accompanying genomic imbalance in the neoplasm. The modulation of replication stress through ROS has medical indications, with various improved factors, such as the WEE1 and ATR inhibitors illustrated in Figure 1, focusing on replication stress in neoplasms (103).
ROS in Detecting Double-Strand Breaks
Generally, the DNA is continually harmed by both internal and external agents, such as chemicals and radiation. A DNA DSB is arguably the most important stage of damage because it can cause cell death if left unrepaired and chromosomal translocations if misrepaired, all of which are early steps in the etiology of carcinogenesis (109). DNA DSBs are unlikely to be directly caused to any significant degree by endogenously produced ROS, which mainly initiate base damage and single-strand breaks that are close to or during the reconstruction of other lesions (110, 111). In particular, DSB is the final lesion of a wide variety of DNA-damaging agents where ROS are not restricted to radical superoxide (O2·), hydrogen peroxide (H2O2), or hydroxyl radical (OH−) as endogenous DNA injury, including DSB (H2O2) (112). Importantly, base alteration is a typical ROS DNA injury, illustrated in Figure 2. Karanjawala et al. showed that ROS cause the most lethal form of DNA damage, DSBs (113). ROS can contribute to carcinogenic treatment via promotion of DNA damage, inactivation of the functions of key proteins, and insufficient regulation of specific cell growth regulators (114). A research study has revealed that, in comparison to healthy cells, the level of protein kinase CK-2 is higher in tumor cells. Researchers have also discovered that DMAT (2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole), unlike TBB (4,5,6,7-tetrabromo-1H-benzotriazole), induced ROS and DNA DSBs. Consequently, inhibition of the CK-2 enzyme, ROS, and DNA DSBs ultimately initiates the apoptotic pathway by DMAT (115). Many research studies have demonstrated that the activation of the STAT/MAP kinase/RAS-MAP kinase/PI3K pathways regulates the generation of ROS complexes. Some myeloid leukemias involve aggressive mutations in members of the RAS family in receptor tyrosine kinases (e.g., FMS-like tyrosine kinase receptor 3, FLT3) (NF-1) (116–118). These mutations collectively account for up to 50% of all acute myeloid leukemia (AML) cases and present leukemic cell proliferation and survival benefits by activating the genes that signal the RAS-MAP kinase, PI3K, and STAT pathways. FLT3 and internal tandem duplications (ITDs) enhance the production of ROS complexes (119, 120). In addition, approximately 30% of chronic carcinomas activate NRAS and KRAS mutations, and several laboratory studies have shown that they can modulate ROS and genomic instability (121, 122). Moreover, BCR–ABL tyrosine kinases are generated by the fusion of portions of BCR and ABL genes that initiate ROS development and a genomic instability cycle that can trigger DNA damage, especially DSBs in human carcinoma (41).
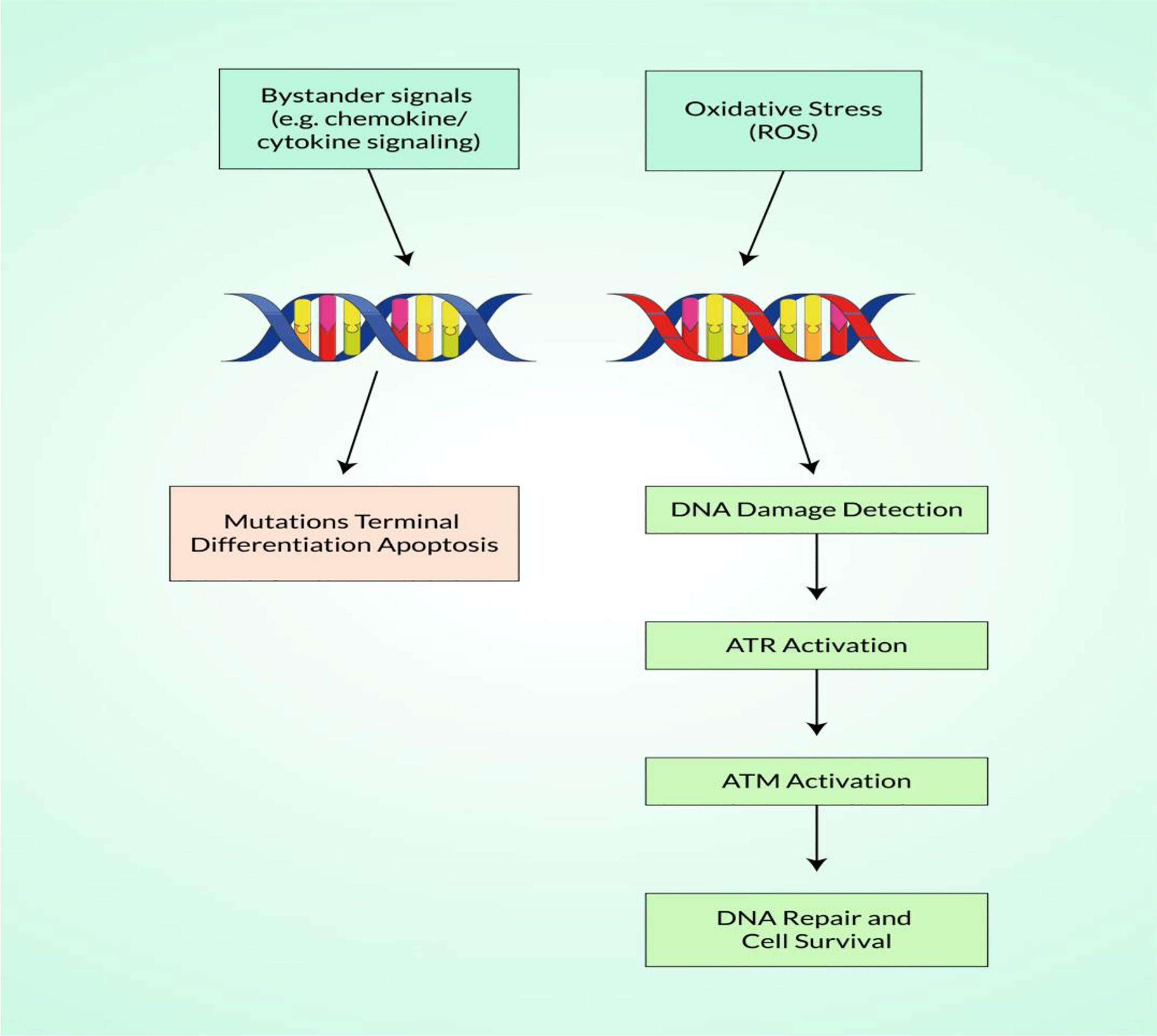
Figure 2 Schematic illustration of the detection of reactive oxygen species (ROS)-mediated DNA damage with DNA repair.
ROS-Mediated Signal Transduction in DNA Damage Response and Potential Implications
Overexpression of ROS is closely related to the enhancement of the DDR response, which may be significant and positively related to genetic instability. DNA damage induces DNA oxidation, leading to a wide range of DNA changes, likely “indels,” i.e., insertions/deletions, base pair mutations, and DSBs, among which deletions and translocations are the most damaging DNA lesions (41, 123). Inadequate remediation of the DNA damage and DSBs by NHEJ and HR also contributes to genomic instability (114). Damages to the DNA are strictly regulated, and irregular function can significantly affect cells, leading to improvements in how cells can cope with genotoxic stress (95, 124, 125). The MRE11/RAD50/NBS1 (MRN), ATR-interacting protein (ATRIP), or Ku complexes, which activate ATM, ATR, and the DNA-dependent protein kinase catalytic subunit (DNA-PKcs), respectively, detect DNA DSBs, and several downstream proteins, including p53, are then targeted (126, 127). These damage sensors either provide the signals of the DNA repair pathways to repair the damage depending on the severity of DNA damage or induce cells to undergo apoptosis if the damage is excessive. At least two DSB repair processes, HR and NHEJ, are available. HR is an error-free repair route that involves RAD52, the end-binding DNA protein, and RAD51, which forms filaments across the unwound DNA strand to support the invasion. The resected 30 end is present in a homologous duplex of DNA and is expanded by DNA polymerase (124). NHEJ involves split DNA ends and minimal homology; this mechanism is not always accurate and can lead to small areas of non-model nucleotides around the DNA break site (microhomologies). NHEJ has two phases: a fast process that mixes basic DSB with compatible ends and a slower step that repairs DSB with complex non-compatible ends that require intervention (128).
Furthermore, new evidence has emerged for alternative end-to-end pathways that play significant roles in DSB repair but are less well defined. These routes lead to translocations and deletions coordinated by repair using significant microhomological DNA sequence levels and appear to be involved while the main NHEJ route remains missing or downregulated (129). In the absence of core NHEJ proteins, such as DNA ligase IV and Ku, Wang et al. reported that important end-joining in response to irradiation, and in the absence of Ku or DNA ligase IV, invalidates the end-joining results in translocations (130). In addition, although NHEJ is assumed to join the breakpoints of chromosomal translocations, alternative NHEJ will be the only primary mechanism of DNA end-joining in NHEJ-mutant mice and is very rare in humans with an NHEJ mutation (130). A research study revealed that the oxidative DNA damage in leukemia cells could lead to a wide range of DNA changes, including base pair mutations, insertions, and deletions (123).
Increases in NHEJ activity and downregulation in the faithful NHEJ pathway have been observed in combination with oncogenic FLT3/ITD and BCR–ABL signaling (Figure 3) (131, 132). Therefore, further research has recently been done to explore particular mechanisms leading to the development of ROS and ROS-mediated damage in FLT3/ITD-positive AML cells to DNA and genome stability (133).
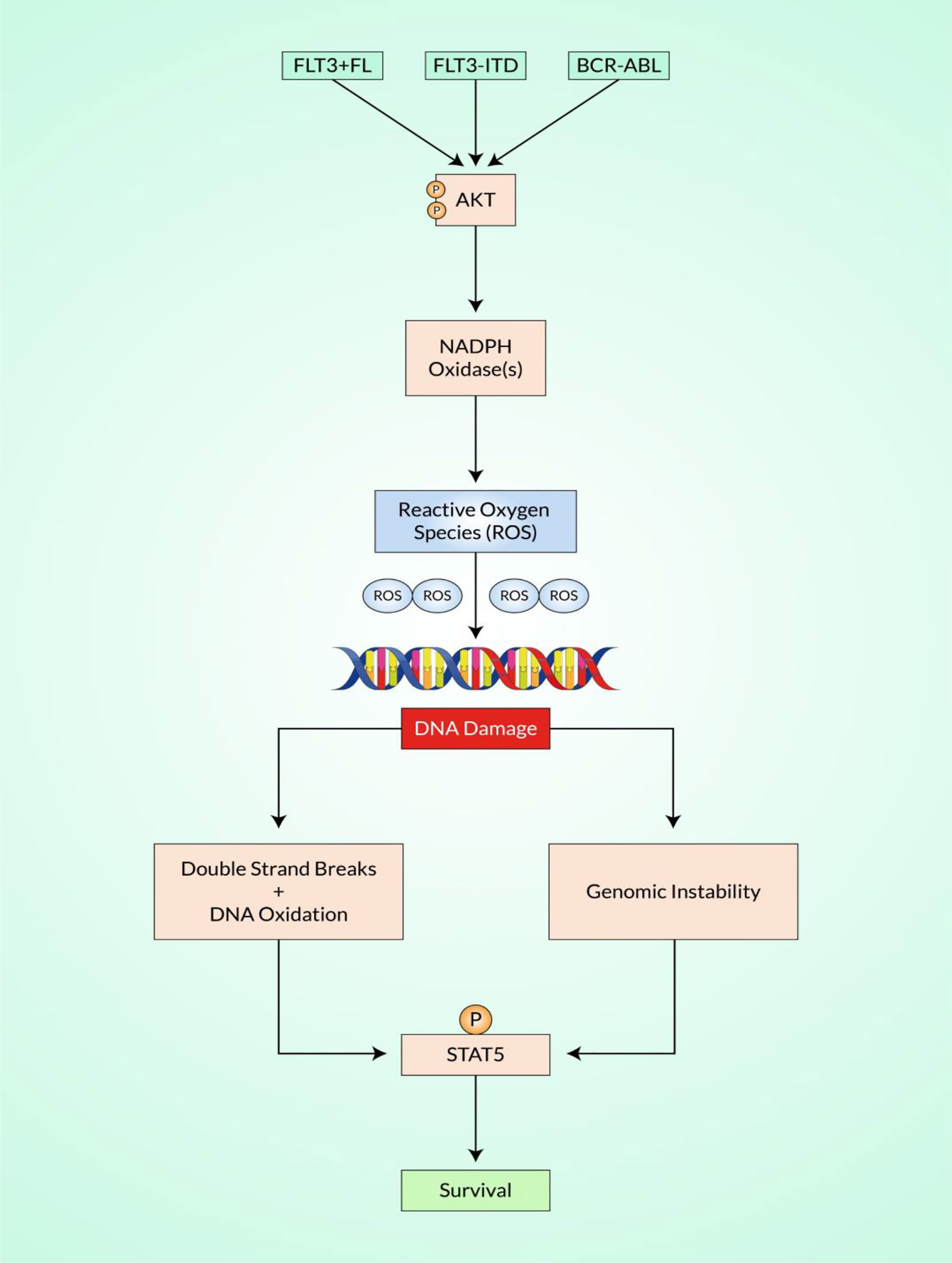
Figure 3 Schematic representation of reactive oxygen species (ROS)-mediated DNA damage response (DDR). Shown is a significant strategy in which the altered development of ROS increases the sources of endogenous DNA damage in various cancers, such as myeloid malignancies. In chronic myeloid leukemia (CML), the fusion gene BCR–ABL produces ROS, as do FLT3/ITD mutations in acute myeloid leukemia (AML) and RAS mutations in myelodysplastic syndromes (MDS)/myeloproliferative diseases (MPDs). Increased ROS can cause a sequence of genomic instabilities by Akt and NADPH oxidases, resulting in DNA double-strand breaks (DSBs) and altered repair, further leading to the acquisition of genomic modifications. There is accumulating evidence that defects in the primary signaling pathways for DSBs, non-homologous end-joining (NHEJ), and activation of the RAS/PI3K/STAT signaling pathways result in the increased expression of complementary or “backup” recovery, which can result in chromosomal deletions and translocations.
Mechanism of Action of Salvicine in Inhibiting Topoisomerase II
Topo II is vital for the metabolism of DNA since it plays a role in the regulation of topology during the processes of DNA replication, recombination, transcription, and in cell cycle (134). This enzyme has been recommended as a clinically crucial key route for various medical treatments such as chemotherapy against tumors, and molecules that inhibit this enzyme are the main constituents of several medical regimens (135). These Topo II inhibitors can be broadly classified based on their mode of action: Topo II poisons and its catalytic inhibitors (Figure 4). Topo II poisons have the capacity to sustain the reversible covalent Topo II–DNA aggregates generally known as cleavage complex, while the catalytic inhibitors follow different routes in the catalytic stages without trapping the covalent aggregates. The former are more often recommended clinically for different treatments of human cancers, but they still have some drawbacks including partial toxicities and resistance to drugs, which result in failure of treatment after the initial therapeutic promise. Furthermore, drugs synthesized from various chemical moieties, which have uniform cellular targets, show various levels of antitumor efficacy. Therefore, there is still an keen need for investigations and the design of novel anticancer molecules focusing on human Topo II (136).
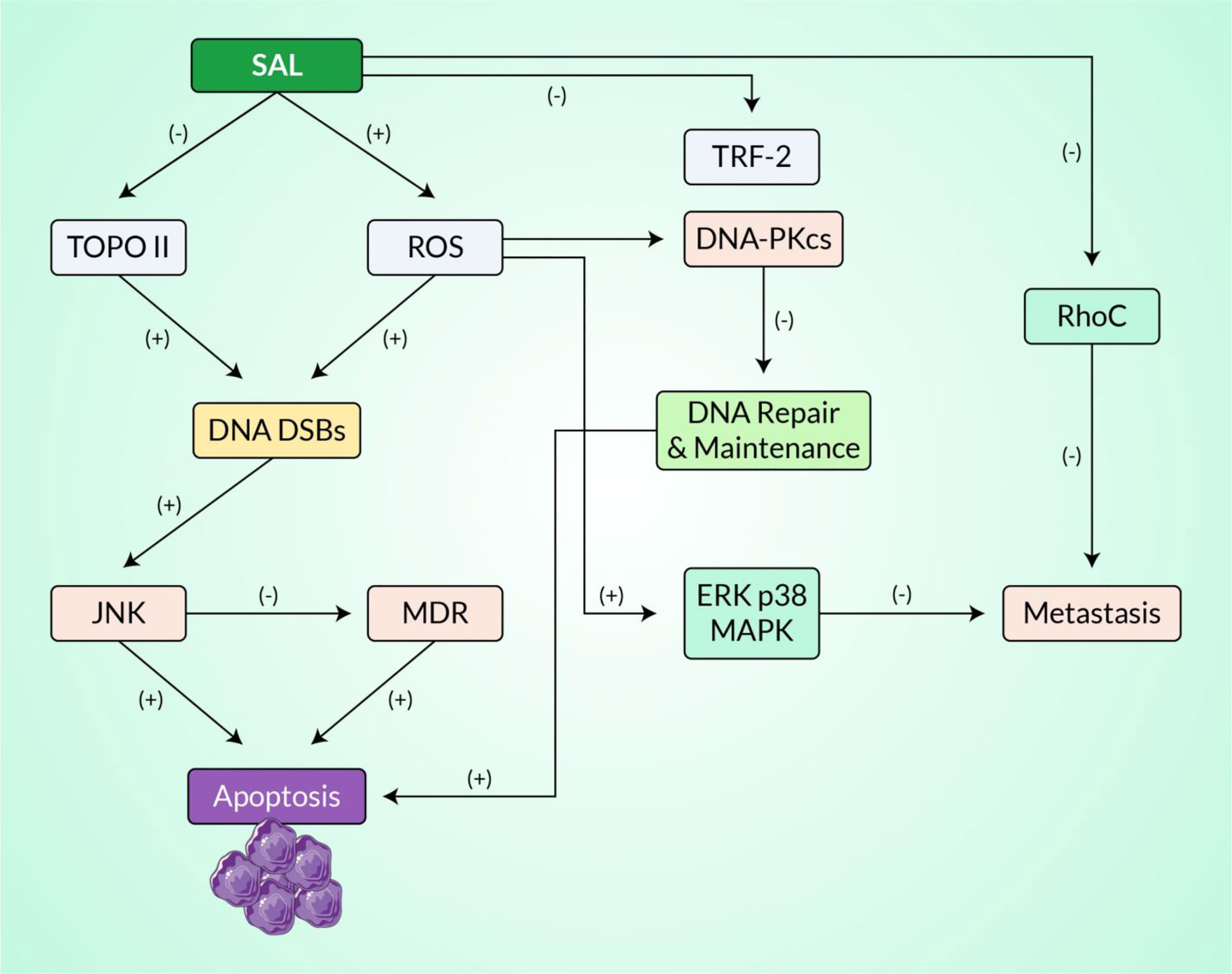
Figure 4 An estimated signaling pathway exhibiting the mechanisms of action of salvicine in inhibiting topoisomerase II and inducing DNA damage. All of these activities are regulated by ROS generation. Here, the positive sign represents stimulation or enhancement and the negative sign represents inhibition.
Salvicine is a diterpenoid quinone molecule extracted from a Chinese herbal plant that showed therapeutic potential against a broad range of human cancerous cells when tested in vitro and in vivo in mice transfected with human tumor xenografts (46, 137). This compound showed prominent cytotoxic efficacy on MDR cells. Furthermore, it drastically regulated the lung metastatic foci of the MDA-MB-435 orthotopic xenograft without observable resistance in the initial tumor cells. Drugs that mainly act on Topo II are also recommended for hematological and solid tumors (138). They catalyze enzymatic activities that disintegrate and re-ligate DNA DSBs in order to reduce the superhelical phase of genomic DNA or unscramble linked chromosomes (139). Topo IIα and Topo IIβ genes are expressed in human cells, with Topo IIα being essential in the cancer cell cycle because it is expressed during the S and M phases of cell division and is required for DNA multiplication and chromosome separation during mitosis (99, 140). On the other hand, Topo IIβ is superfluous for cell sustainability, and its expression is not related to cell division (141). Furthermore, it is frequently entrapped by malignant transcriptional factors that provide a proliferative transcriptome to cancer cells (142). Topo II poisons are responsible for the DNA disruption to kill the growth of cancerous cells. The limitation of this mechanism of action (MOA) is that Topo II-induced DNA disruption is found in non-cancerous or normal cells. Although the NHEJ pathway can overcome DNA breakage and rescue cells, it also allows for gene deletions and recombination, both of which are malignant. Molecules such as etoposide and teniposide, which are used in therapy for secondary leukemias, have been associated with alterations in the MLL gene on chromosome 11q23 (143). In addition to this, if these cancerous cells overcome the Topo II poison dose, their treatment becomes quite tedious as they possess heterogeneous mutations. T60, on the other hand, uses a different MOA to block Topo II activity and cell growth, resulting in fewer DNA breaks and less cytotoxicity. It may also reduce the ability of other anticancer medications to maintain maximum cell growth inhibition. As a result, T60 and its analogs may have clinical benefit when used separately or in combination with other anticancer agents (144). Overall, salvicine was discovered to be a unique Topo II inhibitor with a specific spectrum of activity involving the production of ROS, increase of Topo II–DNA interaction, and blockage of Topo II-mediated DNA disruption. Therefore, elucidating the pharmacokinetic and pharmacodynamic behavior of salvicine demands more research investigations in both preclinical and clinical settings.
Furthermore, poison inhibitors block the catalytic phases of Topo II post-DNA disruption, thus enhancing the limitations of Topo II–DNA cleavage complexes, which play vital functions as genotoxic entities among cancerous cells (145). Moreover, according to the Drug Bank database, there are approx. 19 clinically accepted molecules that target Topo II, and 15 drugs are still under investigation. However, the toxicity of these drugs is widely known, with symptoms including myelosuppression and initiation of secondary cancers (146). A unique morphological feature of these drugs is the occurrence of a planar moiety that intercalates DNA. The connecting pathways of such ligands show unique bipartite association when the planar section interacts with the DNA, while the other actively functional moieties of these drugs aggregate with proteins (147). The first inhibitor co-crystallized with the Topo II protein was etoposide (PDB ID: 3QX3); therefore, most molecular docking studies were based on this structure to emphasize on model protein−ligand interactions.
ROS-Induced and Topo II-Dependent DNA Damage Response and Apoptosis Mediated by Salvicine
Topo II poisons are well known to possess anti-proliferative activity by inducing DNA DSBs, which can trigger DNA reactivity and ultimately apoptosis. It is noteworthy that salvicine effectively increases the intracellular ROS production and thereby induces DSBs through NAC (33). NAC also prevents the generation of salvicine-induced Topo II–DNA cleavable complexes and the growth suppression of salvicine-treated JN394top2-4 yeast cells, suggesting that Topo II is a target of salvicine-induced ROS (54). Heat treatment that reversed the salvicine-trapped DNA–Topo II cleavage complex also reversed the accumulation of DNA DSB, revealing that salvicine-induced DNA damage was caused by ROS and was mediated by Topo II. DNA intercalators (e.g., doxorubicin), enzyme binders (e.g., VP16), DNA lesions (e.g., abasic sites), or oxidative stress can all disrupt the DNA–Topo II breakage and/or reunion reaction, resulting in Topo II-mediated DNA damage (35, 148). Salvicine contains a chemically active quinone moiety, and the majority of anticancer drugs containing quinones are believed to stimulate ROS as part of their anticancer potential. Salvicine contributes to the entire biological implications of salvicine therapy, such as DNA DSBs, apoptosis, and tumor cell cytotoxicity, by generating ROS to control Topo II-mediated DNA damage (35, 36).
Salvicine-induced DNA DSBs activate ATM and ATR kinases, as well as histone H2AX phosphorylation, in A549 lung cancer cells, which has been widely described in DSB-induced cellular responses (149, 150). Salvicine inhibits the catalytic subunit of DNA-dependent protein kinase (DNA-PK), but not the Ku70 or Ku86 subunits, which is surprising. In MCF-7 cells, salvicine treatment decreases DNA-PK activity, which could be due to a decrease in the (DNA-PK) protein. DNA-PK is made up of around 450 kDa DNA-PKcs and two smaller Ku subunits (Ku70 and Ku86), and it is an important element of the NHEJ pathway, which is the most common mechanism of DSB repair (including Topo II-medicated mammalian DNA repair) (151, 152). Salvicine damages the DNA at the same time, disrupting the DNA repair route that could improve its therapeutic effectiveness and overcome DNA repair resistance. NAC pretreatment abrogates the effects of salvicine on the protein level and the DNA-PK activity, which suggests that salvicine-induced DNA damage and repair involves ROS generation. The mechanism of salvicine-induced ROS on Topo II and DNA-PK provides new insights into the broad range of biological functions of ROS (34).
Salvicine causes DNA DSBs in human promyelocytic leukemia HL-60 cells and MCF-7 breast cancer cells (39). The findings of a study using a genetic yeast system demonstrated that DNA damage is strongly associated with cell growth suppression, indicating that Topo II is the principal cellular target of salvicine (33). Short-term salvicine treatment generated DNA damage that could be partially repaired, although early DNA breaks resulted in the induction of apoptosis in the cells. The c-myc oncogene region of the P2 promoter has detected preference damages; in both HL-60 and MCF-7 cells, no clear damage to DNA was detected in the 3′ region of the same gene. Salvicine reduces c-myc gene transcription in a dose-dependent manner while simultaneously increasing c-Jun expression (39, 53). It is possible that DNA damage can develop early in these areas, leading to growth inhibition through changes in the expression of specific genes governing gene proliferation, including c-myc and c-Jun, and eventually cell death. Notably, salvicine shut down the oncogene expression along with the induced DSB pathway. It is also worth noting that blocking of the c-myc gene by conducting salvicine at the MCF-7 breast cancer cell line has been clinically proven. Salvicine, in addition to inducing genomic DNA damage, has been demonstrated to cause telomere erosion and to decrease the functioning of the telomerase enzyme (39, 49). The trf2 gene encodes the TRF2 (telomere repeat binding factor 2) protein, which is a major telomerase and plays a critical role in telomere maintenance with DDR (51, 153). Such noble compound is directly involved in blocking the overexpressed TRF2 protein via transcription and proteasome degradation, eventually downregulating cellular proliferation and halting cancer progression. Recent reports have proven that salvicine directly deters the function of the telomerase enzyme, at the same time inhibiting lung carcinoma on the A549 cell line. However, it also followed the disruption of the telomerase enzyme along with the initiation of apoptosis in the HL-60 cell line (human leukemia) (33, 38). The work of Zhang et al. (52) demonstrated that salvicine blocked the function of integrin β1, which plays a crucial role in cell–ECM adhesion and maintains the signaling cascade reaction, along with regulation of cell proliferation, cellular activation, and homeostasis. Blocking the integrin protein directly halts neoangiogenesis and protects the human body from a metastatic cancer state, with both in vivo and in vitro reports also showing proof of the significant anti-angiogenesis effect of salvicine on A549 cells (represented in Figure 5). Overall, salvicine produces ROS, which alters DNA damage (Topo II-mediated), leading to its broad physiological effects such as cytotoxicity, DSBs, and apoptosis in cancer cells. More clinical studies should be conducted to validate the mechanisms of ROS-induced and Topo II-dependent DDR and apoptosis of tumor cells mediated by salvicine.
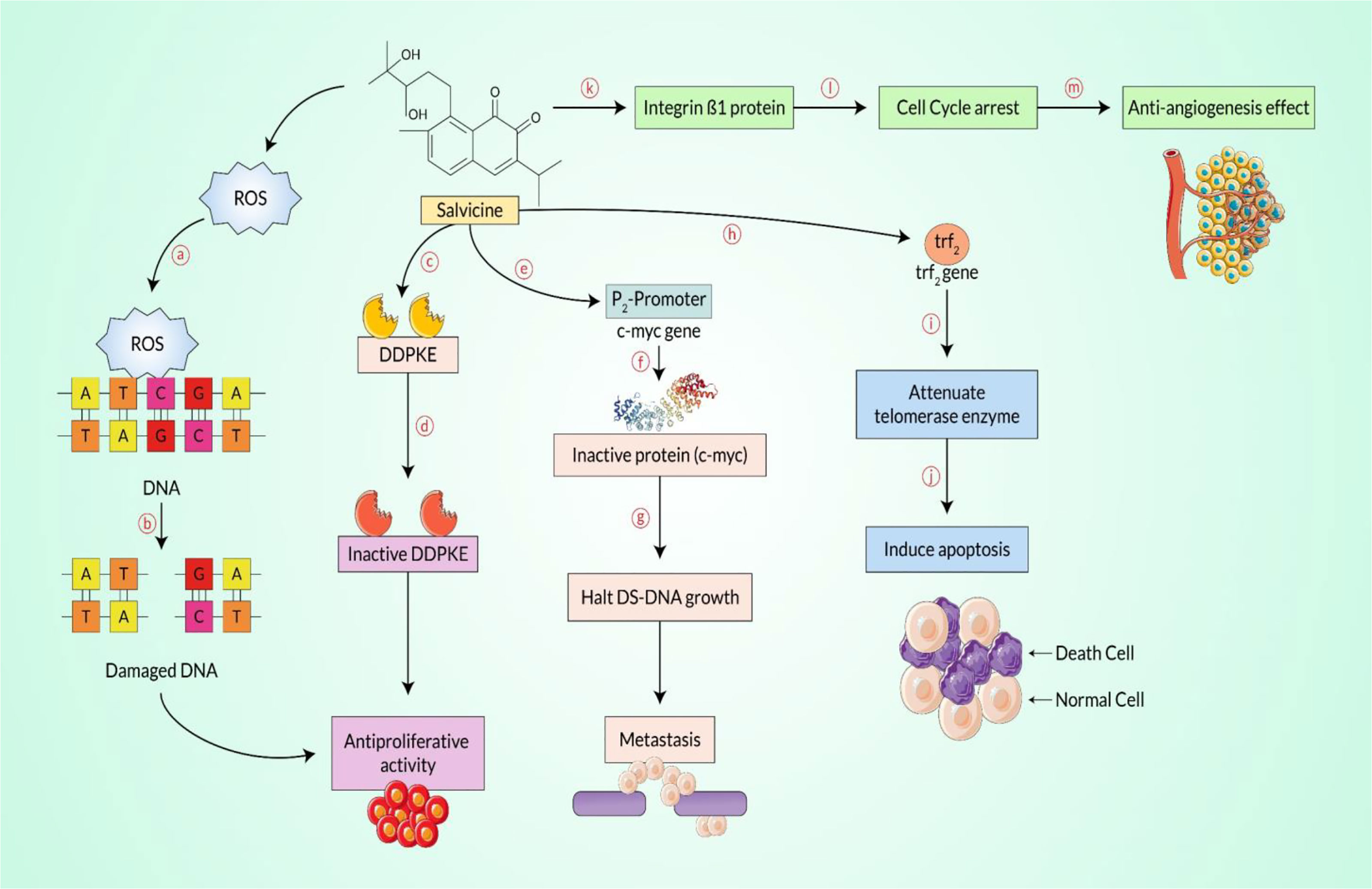
Figure 5 Salvicine-mediated anti-proliferative and/or anticancer effect pathway. As an initial step, salvicine produces reactive oxygen species (ROS) and binds to the double-strand (DS)-DNA moiety, the cysteine residue (a). At the same time, it enables disruption of the double-strand DNA (b), additionally inhibiting the rapidly proliferating cells, including cancerous cells. Salvicine directly interacts with the DNA-dependent protein kinase enzyme (DDPKE) (c), which is why the inactive enzyme functions with the inhibition of cellular growth (d), halting the rapidly propagating cancerous cells. On the contrary, the c-myc protein plays a key regulatory role during DS-DNA growth, but it directly binds with the promoter region of the c-myc gene (e) and interestingly yields a malfunction enzyme (f), which damages the DS-DNA (g) and subsequently downregulates metastasis. However, telomere repeat binding factor-2 is synthesized by the trf2 gene that plays a fundamental role in normal cells, and salvicine binds to the trf2 gene (h) and shuts down the expression of the telomerase protein (i) that induces apoptosis (j), which mediates the anticancer effect. Finally, salvicine disrupts the cell–extracellular matrix adhesion protein, mainly integrin β1 (k), which is involved in cell cycle arrest (l) and hinders neoangiogenesis (m) and even cancer metastasis.
Anticancer Perspective
Salvicine inhibited the growth of a variety of human tumor cells in vitro and in mice with human tumor xenografts (154). In particular, it exhibited superior anti-carcinogenic activity against stomach and lung cancer cells. The potential of salvicine to cause apoptosis in K-562 and SGC-7901 cells was also discovered to be connected with its anticancer therapeutic activity (64). Salvicine exhibited substantial anticancer effects against S-180 sarcoma and Lewis lung cancer in mice, as well as in human lung cancer xenografts A-549 and LAX-83 (65). In this section, we exhibit both the clinical and experimental outcomes of salvicine as a potential anticancer agent in several types of cancer cell lines and in in vivo research models.
Leukemia
Qing et al. investigated in vitro the cytotoxic effects of salvicine in various cell lines isolated from hematopoietic and solid malignancies. Apart from salvicine in K562 cells, the HL-60 and U-937 cell lines were grown in the presence of etoposide (VP16) and vincristine (VCR). Salvicine and VP16 showed comparable efficacy (IC50), with VCR being the most cytotoxic compound. K562 cells were grown in higher salvicine concentrations and in a steady drug concentration at increasing time intervals, establishing the drug concentration and time dependence of cell inhibition. After prolonged exposure (more than 24 h) to salvicine, cells tend to arrest in the G1 phase, expressing an inability to duplicate their genetic material (46). Qing et al. reconfirmed these effects of salvicine on K562 cells and further documented the induction of apoptosis. Specifically, they highlighted the structural characteristics of apoptosis such as DNA fragmentation in multiples of 180–185 bases and condensed nuclei in a concentration- and time-dependent manner (155). Unpublished data also revealed that DNA–Topo II was inhibited by salvicine. Meng et al. took a step further in order to shed light on the molecular events regarding salvicine-mediated cytotoxicity and apoptosis induction (74). HL-60 is a cell line in which amplified c-myc drives carcinogenesis, implying that chromatin in the specific loci is relaxed. This renders the c-myc gene more accessible to topoisomerase II and prone to salvicine-mediated DSBs. At least the P2 promoter is predisposed to salvicine-induced DSB triggering apoptosis (39). The lack of correlation between growth retardation and the damage on the P2 promoter suggests that there are other crucial loci that need to be investigated. Contrary to c-myc, the transcription factors c-Fos and c-Jun were upregulated. c-Fos and c-Jun are implicated in signaling pathways regulating cellular proliferation, differentiation, and apoptosis (118, 156). Salvicine-induced apoptosis in HL-60 cells was studied by Liu et al., who found that telomerase played a role. They documented an inhibition of telomerase activity within the first 2 h, while the expression of hTR (hemolytic transfusion reaction) remained stable and those of TP1 (transition protein 1) and hTERT (telomerase reverse transcriptase) decreased at 6 h, assuming that a posttranslational mechanism is responsible. Protein phosphatase 2A (PP2A) was found to be upregulated, while okadaic acid, a protein phosphatase inhibitor, in co-culture with salvicine profoundly inhibited apoptosis. Conclusively, these data suggest that the stimulation of PP2A has negative effects on the activity of telomerase. There are no convincing data to implicate any caspase in the activation of PP2A despite the fact that caspase-3, in contrast with caspase-1, which remained stable, was found to be time- and dose-dependently upregulated (38). Qing et al. reported on the effects of actinomycin D (AcD) as an add-on to salvicine in K562 cells. The rationale was the documented cytotoxic and apoptotic effects of AcD in leukemic cell lines (157). Interestingly, despite the synergistic inhibition of cell growth, AcD totally suppressed the salvicine-induced apoptosis (43). By definition, the chemotherapeutic regimen should induce apoptosis since the other mechanisms of cell death elicited unwanted immune responses. Miao et al. investigated the effect of salvicine on MDR cell lines. It appears that, in the K562/A02 cell line, salvicine outweighed the effects of VCR, doxorubicin, and VP16. It also potently induced apoptosis, increasing caspase-1 and caspase-3 and decreasing bcl-2. Additionally, the encoding gene of P-gp was downregulated, in contrast to the MRP (multiple resistance proteins) and LRP (lung resistance proteins) genes (44). With regard to the downregulation of the mdr-1 gene, Miao et al. documented a pathogenic signaling pathway. Specifically, they noticed that, at the gene and protein levels, the expression of c-Jun precedes that of P-gp. Antisense oligodeoxynucleotides interfere with the translation of hybridizing messenger RNA (mRNA) in order to prevent ribosome binding. When c-Jun AcD applied, the expression levels of c-Jun protein, mdr-1 mRNA, and P-gp were found to be decreased. In addition, c-Jun AcD diminished apoptosis and cellular growth. Collectively, Miao et al. provided sufficient evidence that the transcription factor c-Jun regulates the expression of the mdr-1 gene in salvicine-treated K562/A02 cells (158). In conclusion, vitamin C might add a synergistic effect to the effects of salvicine. Cai et al. demonstrated a shift toward the production of ROS in the K562 and MDR cell lines, in parallel with the exhaustion of glutathione (GSH). Growth inhibition was responsive to the administration of antioxidants, e.g., NAC, GSH, and catalase, and the H2O2 scavenger. Furthermore, NAC inhibited the salvicine-mediated DNA damage, the subsequent trigger of apoptosis, and the activation of c-Jun and its aforementioned downstream effects. Conversely, H2O2 and vitamin C enhanced the effects of salvicine regarding cell growth inhibition and apoptosis induction in both K562/A02 and the parental cell lines (45).
Gastric Carcinoma
Zhang et al. highlighted the signs of growth inhibition in SGC-7901, a gastric carcinoma cell line extracted from the pathologic lymph node of a patient with stage 4 metastatic disease (48, 159). Qing et al. exposed the SGC-7901 and MKN-28 cell lines to salvicine, VCR, and VP16. The IC50 values for VCR and VP16 in SGC-7901 cells exceeded 100 μmol/L, while that of salvicine was near to 7.8 μmol/L. The results of the MKN-28 cell line showed the same trend, signifying a nearly 10-fold greater potency over VCR and VP16 (46). SGC-7901 cells were exposed to increasing doses of salvicine for 24 h, establishing a dose-dependent inhibition of cellular growth, as revealed by Qing et al. Morphological signs of programmed cell death were evident and became more apparent when cells were exposed to higher drug concentrations (47).
Lung Adenocarcinoma
Indications of cytotoxicity against lung cancer cell lines were documented by Qing et al., specifically treatment of the A549, SPCA-4, NCI-H23, and NCI-H522 cell lines with salvicine, VCR, and VP16. It was demonstrated that salvicine caused more damage in cancerous cell lines than did the classical chemotherapeutic agents (46). Miao et al. delineated the characteristics of the A549 cell line. This cell line exhibited significant resistance to salvicine and antimetabolites, while alkylating and platinum agents showed nearly the same sensitivity as that in the parental cell line. The authors reported a dose-dependent induction of apoptosis in the higher dose spectrum than in the parental A549 cells, which is another indication of resistance to salvicine. In cultures, A549/salvicine cells showed a more diffuse growth pattern, with longer doubling time than that in the parental cells. At the molecular level, p53 and Bax were downregulated in A549 cells, without enhancement of the genes regulating drug resistance (50). Liu et al., after elucidating the implication of telomerase in HL-60 leukemic cells, investigated the role of telomerase in A549 lung adenocarcinoma cells. At a low dose for longer time intervals, salvicine was the most potent telomerase inhibitor among those most commonly used and showed the unique characteristic of inhibiting telomerase when administered at a high dose for a short duration. In the range from 25 to 50 μM, salvicine dose-dependently inhibited telomerase, and increasing the treatment duration with a salvicine dose of 50 μM further reduced the enzyme’s activity. Additionally, salvicine did not appear either to interfere with telomerase directly or with the transcriptional expressions of hTERT, hTP1, and hTR, the subunits of telomerase. For example, in the HL-60 cell line, okadaic acid reversed the salvicine-induced telomerase downregulation, implying the occurrence of posttranslational telomerase modifications (49). Another research group investigated the effects of TRF2 on DNA damage repair. In addition to the salvicine-induced DSBs and telomere damage, the authors documented alterations in TRF2 function. Experimenting with trf2 gene enhancement and transcriptional silencing, they also recorded the fundamental effects of TRF2 on the protection of genomic material from salvicine damage. They also reported that these effects were mediated by ATR, even though both ATM and ATR were overexpressed in salvicine-mediated damage. Finally, Zhang and his research collaborators, taking into consideration the anti-integrin effects of salvicine that emerged in the literature, explored the potential anti-metastatic activity of salvicine. They documented a dose-dependent inhibition of both A549 cells and human microvascular endothelial cells (HMECs). In HMECs, salvicine altered the mobility of the cells and prevented vessel tube formation. Furthermore, salvicine downregulated the expression of basic fibroblast growth factor, while the expression of vascular endothelial growth factor remained unaffected (52).
Breast Cancer
The potency of salvicine in the MDR cell line MCF-7/ADR was discovered by Miao et al. Interestingly, they documented salvicine as being more potent than doxorubicin, VCR, and VP16. The IC50 values were 1.4, 4.48, 13.85, and 58.05, respectively, and the resistance factors, the IC50 ratios between the parental and the MDR cell lines, were 1.42, 233.19, 344.35, and 71.22, respectively (44). Lu et al. investigated the interplay between DNA damage and cellular development. They documented that 2.5 μM of salvicine was sufficient to cause DNA damage and 7.69% of growth inhibition. Higher doses of the drug were responsible for greater growth inhibition. The main type of damage was DSB, and similarly to what has been reported by Meng et al., for HL-60 cells, salvicine caused preferential damage to the P2 promoter of the c-myc gene, a critical transcription factor for the development of the disease (39). The c-myc gene was not the only gene affected. The expression of c-Jun was downregulated, and it is noteworthy that its downregulation preceded that of c-myc. It is not clear whether there is a causative relationship between the two events. Researchers have also documented that p53 and its downstream molecules were not implicated in the repair of salvicine-mediated DNA injury, at least in the dosage spectrum tested. As observed in other cell lines, salvicine caused a concentration-dependent induction of apoptosis. Lu et al., in order to elucidate the antineoplastic effects of salvicine, investigated whether salvicine induces the generation of ROS, with notable results. Additionally, they reported that salvicine contributed to the production of ROS. The NAC-mediated reversal of oxidative stress decreased the burden of DSBs, implying an underlying causative relationship. Salvicine-induced ROS generation affected the DNA repair pathways, interacting with DNA-PK kinase. Remarkably, pretreatment with NAC in the MCF-7 cell line reduced programmed cell death and growth inhibition, which strongly suggests that salvicine-induced ROS mediated a significant part of the effect of salvicine (54). Zhou et al., furthermore, documented the implications of ROS generation on the metastatic potential of the MCF-7 cell line. Utilizing NAC and U0126 and SB203580, which respectively inhibit ERK and p38 MAPK, the authors reported that salvicine generating ROS modified the activity of integrin β1, decreasing its migration and metastatic cell profile (55).
Sarcoma
Salvicine has shown significant antineoplastic activity against murine S-180 sarcoma (33). In vivo antitumor activity studies were conducted against murine S-180 sarcoma animal models, where the results revealed remarkable activity against the models, indicating salvicine as a promising drug against sarcoma. The results are displayed in Table 1 (48). A research study using the sarcoma S-180 murine model has demonstrated that salvicine accomplished tumor killing effects comparable to those of VP16 (48).
Pancreatic Cancer
An experiment conducted by Yao et al. on gemcitabine-resistant SW1990 cells documented the downregulation of the deoxycytidine kinase (dCK) gene and the upregulation of ribonucleotide reductase (RR) and mdr-1 genes. Treatment with a therapeutic dose of 4 nmol/L of salvicine did not alter the expression of mdr-1. At toxic concentrations, salvicine displayed a concentration-dependent decrease of mdr-1 expression. Therefore, the research group concluded that salvicine is only able to reverse the drug resistance in pancreatic cancer cell lines when it is used at toxic concentrations (56). According to Boreddy and Srivastava, despite advancements in traditional cancer therapies such as chemotherapy, surgery, and radiation, pancreatic cancer is still the fourth deadliest cancer in the USA. As a result, phytochemicals from natural sources such as salvicine have received attention for the treatment of pancreatic cancer (160). Miao et al. found that salvicine initiates caspase-1- and caspase-3-dependent apoptosis. Although it has been assumed that caspase-1 does not cause apoptosis, recent reports have suggested that, in human pancreatic cancer cells, caspase-1 plays an important role in cell death by inducing interferon gamma (44).
Cervical Cancer
Salvicine has anticancer properties that have been shown to positively contribute toward its efficacy to inhibit the multiplication and growth of solid tumors, such as in lung, ovary, colon, and cervical cancers (158). It has been proven to be 4.2 and 5.4 times more potent than the positive controls used, VCR and VP16, respectively. With its novel chemical properties and mode of action, salvicine, which is under phase II clinical trials, is a promising anticancer candidate that also has the benefit of low toxic side effects when used for the treatment of various cancers (161).
Cai et al. explored the role of GSH as an oxidative stress inducer in HeLa, a cervical cancer cell line. It was discovered that salvicine caused the exhaustion of intracellular GSH, modifying it directly. This led to the excessive production of H2O2, causing oxidative stress. In contrast to superoxide dismutase (SOD), NADPH oxidase inhibitors, and Trolox, NAC and GSH reversed the cell toxicity caused by salvicine. Furthermore, GSH resisted the DNA injuries and the Topo IIA inhibition induced by salvicine (57).
Oral Carcinoma
Various chemotherapeutic drugs including cisplatin (cis-[PtCl2(NH3)2]) (CDDP), 5-fluorouracil (5-FU), and teniposide (Vm-26) have been used for the treatment of head and neck cancers, which function by inducing specific apoptotic pathways (162). The studies of Tong et al. and others have found that the apoptosis induced by 5-FU in oral squamous cell carcinoma (OSCC) is caspase-dependent, similar to that of salvicine (163, 164). Due to their effectiveness in targeting topoisomerases and causing DNA damage or tumor cell apoptosis, plant-derived compounds such as teniposide and camptothecin (CPT) have been successfully used as anticancer drugs (165). Salvicine is also a Topo II inhibitor with greater potential for use as an anticancer drug as it possesses caspase-3-dependent apoptosis-inducing properties. A study by Miao et al. (44) on the effects of salvicine on three different caspases showed that it activated caspase-1 and caspase-3 proportionally to the dose administered and the duration of treatment. Miao et al. (44) also exposed KB/VCR, an MDR oral carcinoma cell line, to salvicine, doxorubicin, VP16, and VCR. The IC50 values in the parental cell line were 2.26, 0.08, 1.88, 0.004, respectively, and the resistant factors were 1.93, 106.2, 28.9, and 926.86, respectively. These results indicate that salvicine is a potent inhibitor for the KB/VCR cell line (44).
Bladder Cancer
Heyder et al. found that the invasion ability of the T24 human bladder carcinoma cell line can be reduced by inhibiting the integrin β1 subunit, which results in the reduced adhesion and mobility of T24 cells (166). Zhou et al. demonstrated that salvicine is able to reduce the function of integrin β1 by inhibiting the binding of the cell to the ECM. As a result, salvicine showed antimetastatic efficacy that can be useful for inhibiting the integrin β1 of bladder cancer and decreasing the adhesion and mobility of T24 cells (55).
Prostate Cancer
Miao and Ding found that salvicine was able to increase the expression of c-Jun and decrease the expression of P-gp by inhibiting the expression of the mdr-1 gene. It was also clear from the study that an increase in the c-Jun level is required for the downregulation of P-gp by salvicine (158). In another study, Wartenberg et al. stated that ROS were able to downregulate the expression of P-gp and activate c-Jun N-terminal kinase (JNK) in prostate tumors (167). From these two studies, it can be said that, as salvicine can also decrease P-gp expression via the increased c-Jun level, it might be effective against prostate cancer as well (158, 167). Another study demonstrated that extracts from several Salvia species were able to show growth inhibitory activity against the prostate cancer cell line MDA Pca2b. Although the authors did not use S. prionitis Hance, the ability of the other species certainly showed the potential of salvicine to work against prostate cancer cells (168). Zhou et al. revealed that salvicine was able to activate ERK1/2 by triggering ROS generation (55). Chen et al. found that shikonin can reduce the mobility of prostate cancer cells by using the ROS–ERK1/2 pathway, similarly to salvicine. Therefore, salvicine might have the potential to work similarly against prostate cancer (169).
Anti-Proliferation Activity via the ROS-Dependent p38 MAPK Pathway
Salvicine-induced ROS are involved in several cellular functions including DNA damage, mitigating multidrug resistance, Topo II inhibition, cell adhesion inhibition, and anti-metastatic activity (75, 77). Without affecting the growth of the primary tumor, salvicine effectively suppressed the lung metastatic foci of MDA-MB-435 orthotopic xenograft (75). It is widely recognized that cell–cell interactions between cancer cells and the endothelium of distant tissues aid in the progression of cancer metastasis (170). Cell adhesion molecules such as selectins and integrins are the key players in the cell–cell interaction of tumor cells that results in tumor progression and metastasis (171, 172). Transmembrane proteins known as integrins bind to the ECM, assemble at the binding site, and initiate focal adhesion by attracting cytoplasmic proteins such as FAK, c-Src, and paxillin to the binding site (173, 174). Since cell–cell interaction is a fundamental step in the metastatic cascade, disruption of this interaction by the downregulation of the cell adhesion molecules could be a potential therapeutic approach to halting metastasis. Salvicine was found to significantly influence the cell adhesion genes involved in tumor metastasis. Human breast cancer cells treated with salvicine showed lower levels of genes for integrin a3, integrin a6, integration E, integrin b3, integrin b5, integrin b8, paxillin, and FAK.
The in vitro anti-metastatic efficacy of Salvicine on MDA-MB-435 orthotopic xenograft is intimately associated with Rho-dependent pathway. (75). The effect of salvicine on integrin-mediated cell adhesion has been investigated to determine its underlying anti-metastatic mechanism. Salvicine inhibited cell adherence to fibronectin and collagen in human breast cancer MDA-MB-435 cells, reduced the fibronectin-dependent establishment of focal adhesion, and disrupted the actin stress fiber networks, resulting in a rounded cell shape in the test subjects. Moreover, salvicine dephosphorylated FAK and paxillin, resulting in a downregulated integrin β1 ligand affinity, clustering, and signaling. Conversely, the anti-inflammatory drug salvicine induced an increase in the activity of ERK and p38 MAPK. Notably, U0126 and SB203580, which are inhibitors of MAPK–ERK1/2 and p38 MAPK, respectively, abolished the effect of salvicine on integrin-mediated cell adhesion (Figure 6) (55). According to evidence, the p38 MAPK and ERK pathways were activated in response to ROS, which raises the question whether salvicine induced ROS, which in turn activated the ERK and p38 MAPK pathways (175, 176). It has been demonstrated that the addition of NAC, a ROS scavenger, to MDA-MB-435 cells reversed the salvicine-induced activation of the p38 MAPK and ERK pathways, which facilitated the formation of integrin-mediated cell adhesion and the promotion of metastasis in cancer cells (55). Together, the results exhibited that the anti-metastatic action of salvicine on cancer cells was mediated by the ROS-triggered p38 MAPK and ERK pathways.
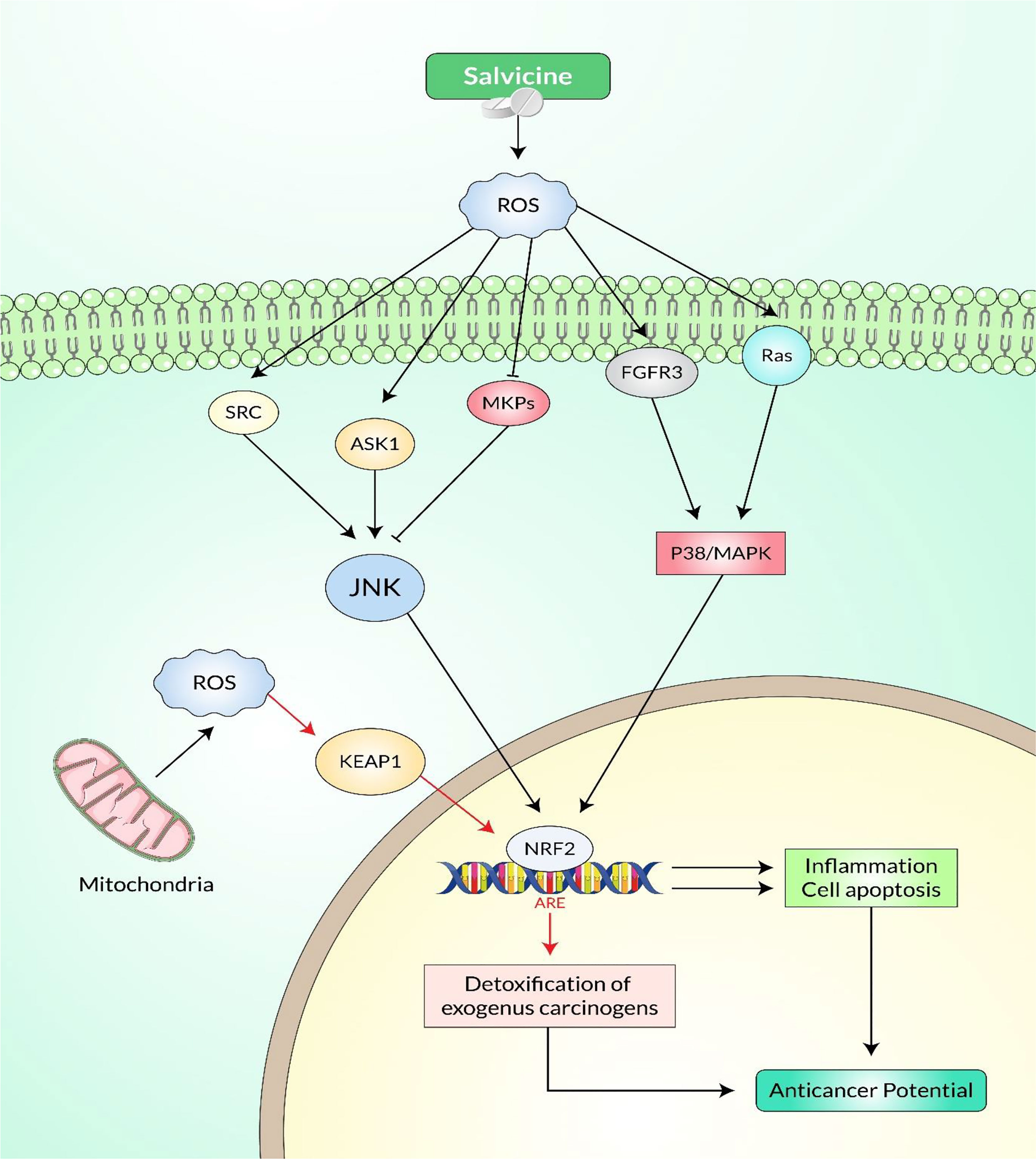
Figure 6 Cellular signaling pathways involved in salvicine-induced anticancer potential via the p38 MAPK and c-Jun N-terminal kinase (JNK) cascades.
Anti-Metastatic Activity via the Rho-Dependent Pathway
Metastasis is the process by which cancer spreads and eventually colonizes from the primary mass to distant organs. The key factors in metastasis include the microenvironment of the host tissue, i.e., angiogenesis, signals from the autocrine, paracrine, or the endocrine pathways, and the interconversion capability of cellular identities, i.e., epithelial-to-mesenchymal (EMT) and mesenchymal-to-epithelial transitions (MET) (177). EMT is in charge of local invasion, intravasation, and extravasation in metastasis, whereas MET is in charge of the recapitulation of the initial mass at distant locations in the disease progression process (178).
Migration, invasion, and adhesion are the primary stages of cancer metastatic progression. It is essential for cancer metastasis because it stimulates cell proliferation and integrin interactions, both of which are necessary for motile function and the survival of cancer cells. At the cytosolic and extracellular sites, cell detachment depends on both mechanical forces and protease-mediated cleavage. Migration occurs via two different mechanisms: a protease-dependent mesenchymal cell migration and a protease-independent amoeboid cell migration, which is faster than mesenchymal cell migration (179).
According to Lang et al., lung metastasis due to human breast cancer is inhibited by the anti-metastatic action of salvicine via the Rho-dependent pathway. Rho proteins are essential regulators of cell motility, and their overexpression is intimately correlated with metastasis. When administered in a dose- and time-dependent manner, salvicine decreased the expression of RhoC without changing the levels of RhoA, and it further decreased the level of RhoC protein in primary tumors. There are two types of G-protein-coupled receptors (G12/G13) that control how RhoA and RhoC move from the cytosol to the membrane. Lysophosphatidic acid (LPA) encourages the movement of both of these receptors. Salvicine effectively antagonizes LPA in a time- and dose-dependent manner, but independent of G12/13. Salvicine also reduces the expression of active RhoC without affecting RhoA due to its different regulatory mechanism. Additionally, salvicine significantly impairs Rho-induced stress fiber formation, impairing cell adhesion and motility and downregulating the expression of genes involved in this pathway, including integrins, fibronectin, FAK, paxillin, and RhoC (75). It was later discovered that the RhoA protein is a downstream effector of ROS-induced cytoskeleton disruption in a separate study by Zhou et al. The production of ROS caused the hydrolysis of GTP-bound RhoA, which promoted cell adhesion. Because NAC pretreatment reverses salvicine-induced damage and negatively regulates cell adhesion, salvicine drastically lowered the level of GTP-bound RhoA and destroyed the actin stress fiber networks that are critical for cell adhesion (55). Salvicine stimulated p38 MAPK and ERK by causing ROS production, which allowed inactivation of the activity of integrin β1 and retardation of cell adhesion. Overall, the anti-metastatic action of salvicine is linked to the ROS-activated p38 MAPK and Rho-dependent signaling cascade. The connection between these two signaling cascades warrants additional investigation.
Mitigating the P-Glycoprotein-Mediated Multidrug Resistance of Chemotherapeutic Agents
Multidrug resistance is described as the ability of a tumor cell to survive and develop resistance to a wide variety of anticancer treatments. It is one of the vital reasons for the failure of cancer therapies that affect patients suffering from a variety of cancers (180, 181). When tumor cells resist anticancer drugs, the mdr-1 gene and its product, P-gp, are overexpressed. Drugs can be expelled from cells by P-gp, an ATP-binding cassette (ABC) transporter with the potential to do so (182, 183). P-gp can attach to various anticancer drugs such as vinblastine, taxol, and doxorubicin, which causes the efflux of drug molecules into the extracellular space. Consequently, there is decreased accumulation of anticancer drugs in cells and increased resistance of cancer cells to the therapy (184, 185). The damaging role of P-gp in anticancer regimens points out a constant need for effective cytotoxic P-gp inhibitors to mitigate P-gp-associated multidrug resistance in order to improve patient outcomes.
Studies have shown that salvicine possesses cytotoxic properties against a variety of MDR tumor cells via the downregulation of P-gp expression. Salvicine also exhibited cytotoxicity against the MDR sub-lines K562/A02, KB/VCR, and MCF-7/ADM, with IC50 values (1.55, 4.50, and 1.40 µm, respectively) quite close to those (0.87, 2.26, and 2.61 µm) of the corresponding parental cell lines: K562, KB, and MCF-7, respectively. In MDR K562/A02 cells, salvicine downregulated the expressions of mdr-1 and P-gp in a dose-dependent manner (44). It was found that salvicine suppressed the expressions of mdr-1 and P-gp by stimulating the expression of the c-Jun gene in MDR K562/A02 cells. Salvicine stimulated JNK phosphorylation, which enhanced the expression of c-Jun; in turn, the activated c-Jun bound to the activator protein-1 (AP-1) target element in the mdr-1 gene promoter region, which resulted in the decreased expressions of mdr-1 mRNA and P-gp in MDR K562/A02 cells (158). Further study was carried out to elucidate the role of salvicine-induced ROS on P-gp expression to circumvent multidrug resistance. Salvicine produced ROS equally in both MDR K562/A02 and parental K562 cell lines (45). Pretreatment of MDR K562/A02 cells with NAC significantly reversed the salvicine-induced P-gp downregulation and JNK phosphorylation. Collectively, salvicine-induced oxidative stress increased the phosphorylation of JNK, resulting in the activation of c-Jun. The activated c-Jun enhanced the expression of c-Jun, suppressed the transcription of mdr-1, and activated pro-apoptotic pathways, leading to a reduced mdr-1 expression and apoptosis. Overall, these results suggest the potential of salvicine to overcome multidrug resistance and promote P-gp downregulation via ROS generation.
Conclusion and Future Perspectives
Specifically, salvicine is a novel Topo II toxin that binds to the ATPase domain of Topo II, increasing DNA–Topo II binding while simultaneously decreasing DNA relegation and ATP hydrolysis. Salvicine showed more potency than VCR and VP-16 at killing 12 types of solid tumor cells than they are at killing healthy cells. The potential of salvicine to cause apoptosis in K-562 and SGC-7901 cells was discovered to be connected to its anticancer activity. Additionally, salvicine induces DNA damage, bypassed by P-glycoprotein of multidrug resistance mechanism, and prevents tumor cells from sticking to each other. In MCF-7 cells, it increased intracellular ROS generation and caused significant DNA DSBs. NAC reduced the salvicine-induced ROS increase and reversed the subsequent DNA breakpoints (DSBs). It mostly inhibited pre-strand Topo II-mediated DNA re-ligation and showed little effect on the catalytic activities of the cleavage complex. Interestingly, salvicine had no effect on pBR322 relaxation mediated by Topo I, which suggests that its Topo II activity is quite selective. The Topo II catalytic cycle may be broken down into six distinct phases, each with its own mode of action. In the intended concentration range, salvicine failed to intercalate into DNA. Topo II is attacked by the antineoplastic-induced ROS, resulting in DNA DSBs. As the first experimental evidence for the role of ROS in DNA-PKcs modulation, salvicine and NAC prevented salvicine-induced cell death and cytotoxicity in MCF-7 cells. Salvicine inhibited the activity of DNA-PK, a key component of the NHEJ repair process. The downregulation of a catalytic subunit of DNA-PK might be due to one mechanism. IBC, BEL-7402, HO8910, and HCT116 nude xenografts showed no growth inhibitory effects when exposed to salvicine. All of these activities are reliant on ROS in certain ways, and the salvicine-elicited ROS appear to play a significant role in ROS generation and participation in the anticancer activity of salvicine. Salvicine can thus be employed as an anticancer medication candidate and a tool for exploring the intricate roles of ROS in the physiological functioning of tumor cells in diverse disorders, which will provide useful information for future clinical trials.
Our findings in this work suggest that salvicine-induced ROS disrupted Topo II and DNA-PK simultaneously in cultured tumor cells, affecting two components of DNA damage and repair and accounting for at least some of its anticancer effects. This is due to Topo II being able to attach to ellipticine and VP16 in the absence of DNA, according to previous research. The drug binding sites have yet to be identified. We also attempted to focus out the multidrug resistance against the cancer effects of salvicine, which are used in cancer treatments. These findings suggest that targeting certain DNA repair proteins might help enhance the existing DNA-damaging anticancer medicines and lead to the development of novel agents or treatments for cancer based on salvicine.
In this review study, we have broadly demonstrated the complicated activities of ROS in various physiological processes in tumor cells to aid in the creation of crucial DNA repair proteins or anticancer drugs by permitting the logical design of salvicine-like anti-Topo II compounds. We have exhibited that salvicine possessed potent anticancer effectivity against several cancer research models. However, further research studies are needed to evaluate its anticancer potential and its synergistic effects in combination with several conventional cancer drugs. Therefore, it can be concluded that, after a number of in vitro and in vivo clinical studies in cancer research models, salvicine can be developed as an alternative therapeutic option for cancer treatment.
Author Contributions
DD, PB, and MMH: Research idea development and conceptualization. DD, MMH, PB, SP, RR, ST, MB, MJI, FAA, EMA, MF, SKB, and PP: Writing and main draft preparation. DD, PB, MMH: Writing, reviewing, and editing. TIR: Drawing of figures. SB, MAR, and BK: Visualization and supervision. BK: Project fund. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2020R1I1A2066868), the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT; no. 2020R1A5A2019413), a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant no. HF20C0116), and a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant no. HF20C0038).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Morounke SG, Ayorinde JB, Benedict AO, Adedayo FF, et al. Epidemiology and Incidence of Common Cancers in Nigeria. J Cancer Biol Res (2017) 5(3):1105.
2. Al Saber M, Biswas P, Dey D, Kaium M, Islam M, Tripty MIA, et al. Comprehensive Review of Recent Advancements in Cancer Immunotherapy and Generation of CAR T Cell by CRISPR-Cas9. Processes (2022) 10:16. doi: 10.3390/pr10010016
3. Dey D, Quispe C, Hossain R, Jain D, Ahmed Khan R, Janmeda P, et al. Ethnomedicinal Use, Phytochemistry, and Pharmacology of Xylocarpus Granatum J. Koenig. Evid Based Complement Altern Med (2021) 2021:8922196. doi: 10.1155/2021/8922196
4. Gupta GP, Massagué J. Cancer Metastasis: Building a Framework. Cell (2006) 127:679–95. doi: 10.1016/j.cell.2006.11.001
5. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA: Cancer J Clin (2021) 71:7–33. doi: 10.3322/caac.21654
6. Biswas P, Hasan MM, Dey D, dos Santos Costa AC, Polash SA, Bibi S, et al. Candidate Antiviral Drugs for COVID-19 and Their Environmental Implications: A Comprehensive Analysis. Environ Sci Pollut Res (2021) 28:59570–93. doi: 10.1007/s11356-021-16096-3
7. Dipta D, Tanzila Ismail E, Partha BISWAS SA, Shoeba ISLAM URR, FIROZ M, AHMED SZ, et al. Antiviral Effects of Bacteriocin Against Animal-to-Human Transmittable Mutated Sars-Cov-2: A Systematic Review. Front Agric Sci Eng (2021) 8:603–22. doi: 10.15302/J-FASE-2021397
8. Hasan A, Biswas P, Bondhon TA, Jannat K, Paul TK, Paul AK, et al. Can Artemisia Herba-Alba Be Useful for Managing COVID-19 and Comorbidities? Molecules (2022) 27:492. doi: 10.3390/molecules27020492
9. Assaad S, Avrillon V, Fournier M-L, Mastroianni B, Russias B, Swalduz A, et al. High Mortality Rate in Cancer Patients With Symptoms of COVID-19 With or Without Detectable SARS-COV-2 on RT-PCR. Eur J Cancer (2020) 135:251–9. doi: 10.1016/j.ejca.2020.05.028
10. Rahman M, Rahman M, Hossain M, Biswas P, Islam R, Uddin MJ, et al. Molecular Insights Into the Multifunctional Role of Natural Compounds: Autophagy Modulation and Cancer Prevention. Biomedicines (2020) 8:517. doi: 10.3390/biomedicines8110517
11. Yim SH, Huang T, Ho JM, Lam AS, Yau ST, Yuen TW, et al. Rise and Fall of Lung Cancers in Relation to Tobacco Smoking and Air Pollution: A Global Trend Analysis From 1990 to 2012. Atmos Environ (2022) 269:118835. doi: 10.1016/j.atmosenv.2021.118835
12. Shahadin MS, Mutalib NSA, Latif MT, Greene CM, Hassan T. Challenges and Future Direction of Molecular Research in Air Pollution-Related Lung Cancers. Lung Cancer (2018) 118:69–75. doi: 10.1016/j.lungcan.2018.01.016
13. Feng RM, Zong YN, Cao SM, Xu RH. Current Cancer Situation in China: Good or Bad News From the 2018 Global Cancer Statistics? Cancer Commun (2019) 39:1–12. doi: 10.1186/s40880-019-0368-6
14. Biswas P, Dey D, Rahman A, Islam M, Susmi TF, Kaium M, et al. Analysis of SYK Gene as a Prognostic Biomarker and Suggested Potential Bioactive Phytochemicals as an Alternative Therapeutic Option for Colorectal Cancer: An In-Silico Pharmaco-Informatics Investigation. J Pers Med (2021) 11:888. doi: 10.3390/jpm11090888
15. Soerjomataram I, Shield K, Marant-Micallef C, Vignat J, Hill C, Rogel A, et al. Cancers Related to Lifestyle and Environmental Factors in France in 2015. Eur J Cancer (Oxf Engl 1990) (2018) 105:103–13. doi: 10.1016/j.ejca.2018.09.009
16. Noone A, Howlader N, Krapcho MA, Miller D, Brest A, Yu M, et al. SEER Cancer Statistics Review, 1975-2015 Vol. 4. Bethesda: MD: National Cancer Institute (2018).
17. Figueroa JD, Gray E, Pashayan N, Deandrea S, Karch A, Vale DB, et al. The Impact of the Covid-19 Pandemic on Breast Cancer Early Detection and Screening. Prev Med (2021) 151:106585. doi: 10.1016/j.ypmed.2021.106585
18. Okano M, Nomizu T, Tachibana K, Nagatsuka M, Matsuzaki M, Katagata N, et al. The Relationship Between BRCA-Associated Breast Cancer and Age Factors: An Analysis of the Japanese HBOC Consortium Database. J Hum Genet (2021) 66:307–14. doi: 10.1038/s10038-020-00849-y
19. Sohel M, Biswas P, Al Amin M, Hossain M, Sultana H, Dey D, et al. Genistein, a Potential Phytochemical Against Breast Cancer Treatment-Insight Into the Molecular Mechanisms. Processes (2022) 10:415. doi: 10.3390/pr10020415
20. Morze J, Danielewicz A, Przybyłowicz K, Zeng H, Hoffmann G, Schwingshackl L. An Updated Systematic Review and Meta-Analysis on Adherence to Mediterranean Diet and Risk of Cancer. Eur J Nutr (2021) 60:1561–86. doi: 10.1007/s00394-020-02346-6
21. Kaiser AE, Baniasadi M, Giansiracusa D, Giansiracusa M, Garcia M, Fryda Z, et al. Sulforaphane: A Broccoli Bioactive Phytocompound With Cancer Preventive Potential. Cancers (2021) 13:4796. doi: 10.3390/cancers13194796
22. Choudhari AS, Mandave PC, Deshpande M, Ranjekar P, Prakash O. Phytochemicals in Cancer Treatment: From Preclinical Studies to Clinical Practice. Front Pharmacol (2020) 10:1614. doi: 10.3389/fphar.2020.00175
23. Paul P, Biswas P, Dey D, Saikat ASM, Islam M, Sohel M, et al. Exhaustive Plant Profile of “Dimocarpus Longan Lour” With Significant Phytomedicinal Properties: A Literature Based-Review. Processes (2021) 9:1803. doi: 10.3390/pr9101803
24. Hussain H, Hamdan N, Sim EU-H. Anticancer and Antimicrobial Peptides From Medicinal Plants of Borneo Island in Sarawak. Adv Tradit Med (2021) 21:189–97. doi: 10.1007/s13596-020-00504-z
25. Lichota A, Gwozdzinski K. Anticancer Activity of Natural Compounds From Plant and Marine Environment. Int J Mol Sci (2018) 19:3533. doi: 10.3390/ijms19113533
26. Meng L-h, Zhang J-s, Ding J. Salvicine, a Novel DNA Topoisomerase II Inhibitor, Exerting Its Effects by Trapping Enzyme-DNA Cleavage Complexes. Biochem Pharmacol (2001) 62:733–41. doi: 10.1016/S0006-2952(01)00732-8
27. Branzei D, Foiani M. Regulation of DNA Repair Throughout the Cell Cycle. Nat Rev Mol Cell Biol (2008) 9:297–308. doi: 10.1038/nrm2351
28. Skok ZI, Zidar N, Kikelj D, Ilaš J. Dual Inhibitors of Human DNA Topoisomerase II and Other Cancer-Related Targets. J Med Chem (2019) 63:884–904. doi: 10.1021/acs.jmedchem.9b00726
29. Depowski PL, Rosenthal SI, Brien TP, Stylos S, Johnson RL, Ross JS. Topoisomerase Iiα Expression in Breast Cancer: Correlation With Outcome Variables. Modern Pathol (2000) 13:542–7. doi: 10.1038/modpathol.3880094
30. Lee JH, Berger JM. Cell Cycle-Dependent Control and Roles of DNA Topoisomerase II. Genes (2019) 10:859. doi: 10.3390/genes10110859
31. Wu F, Zhou L, Jin W, Yang W, Wang Y, Yan B, et al. Anti-Proliferative and Apoptosis-Inducing Effect of Theabrownin Against Non-Small Cell Lung Adenocarcinoma A549 Cells. Front Pharmacol (2016) 7:465. doi: 10.3389/fphar.2016.00465
32. Brel V, Annereau J-P, Vispé S, Kruczynski A, Bailly C, Guilbaud N. Cytotoxicity and Cell Death Mechanisms Induced by the Polyamine-Vectorized Anti-Cancer Drug F14512 Targeting Topoisomerase II. Biochem Pharmacol (2011) 82:1843–52. doi: 10.1016/j.bcp.2011.08.028
33. Meng LH, Ding J. Salvicine, A Novel Topoisomerase II Inhibitor, Exerts its Potent Anticancer Activity by ROS Generation. Acta Pharmacol Sin (2007) 28:1460–5. doi: 10.1111/j.1745-7254.2007.00698.x
34. Ding J, Miao ZH, Meng LH, Geng MY. Emerging Cancer Therapeutic Opportunities Target DNA-Repair Systems. Trends Pharmacol Sci (2006) 27:338–44. doi: 10.1016/j.tips.2006.04.007
35. Wang H, Mao Y, Chen AY, Zhou N, LaVoie EJ, Liu LF. Stimulation of Topoisomerase II-Mediated DNA Damage Via Mech Involving Protein Thiolation. Biochemistry (2001) 40:3316–23. doi: 10.1021/bi002786j
36. Shiah SG, Chuang SE, Chau YP, Shen SC, Kuo ML. Activation of C-Jun NH2-Terminal Kinase and Subsequent CPP32/Yama During Topoisomerase Inhibitor Beta-Lapachone-Induced Apoptosis Through an Oxidation-Dependent Pathway. Cancer Res (1999) 59:391–8.
37. Furuta T, Takemura H, Liao Z-Y, Aune GJ, Redon C, Sedelnikova OA, et al. Phosphorylation of Histone H2AX and Activation of Mre11, Rad50, and Nbs1 in Response to Replication-Dependent DNA Double-Strand Breaks Induced by Mammalian DNA Topoisomerase I Cleavage Complexes. J Biol Chem (2003) 278:20303–12. doi: 10.1074/jbc.M300198200
38. Liu WJ, Jiang JF, Xiao D, Ding J. Down-Regulation of Telomerase Activity via Protein Phosphatase 2A Activation in Salvicine-Induced Human Leukemia HL-60 Cell Apoptosis. Biochem Pharmacol (2002) 64:1677–87. doi: 10.1016/s0006-2952(02)01424-7
39. Meng L, Ding J. Induction of Bulk and C-Myc P2 Promoter-Specific DNA Damage by an Anti-Topoisomerase II Agent Salvicine Is an Early Event Leading to Apoptosis in HL-60 Cells. FEBS Lett (2001) 501:59–64. doi: 10.1016/s0014-5793(01)02633-3
40. Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA Damage Response in Cancer. Redox Biol (2019) 25:101084. doi: 10.1016/j.redox.2018.101084
41. Osarogiagbon UR, McGlave PB. Chronic Myelogenous Leukemia. Curr Opin Hematol (1999) 6:241–6. doi: 10.1097/00062752-199907000-00008
42. Sedletska Y, Radicella JP, Sage E. Replication Fork Collapse Is a Major Cause of the High Mutation Frequency at Three-Base Lesion Clusters. Nucleic Acids Res (2013) 41:9339–48. doi: 10.1093/nar/gkt731
43. Qing C, Miao ZH, Tong LJ, Zhang JS, Ding J. Actinomycin D Inhibiting K562 Cell Apoptosis Elicited by Salvicine But Not Decreasing its Cytotoxicity. Acta Pharmacol Sin (2003) 24:415–21.
44. Miao ZH, Tang T, Zhang YX, Zhang JS, Ding J. Cytotoxicity, Apoptosis Induction and Downregulation of MDR-1 Expression by the Anti-Topoisomerase II Agent, Salvicine, in Multidrug-Resistant Tumor Cells. Int J Cancer (2003) 106:108–15. doi: 10.1002/ijc.11174
45. Cai Y, Lu J, Miao Z, Lin L, Ding J. Reactive Oxygen Species Contribute to Cell Killing and P-Glycoprotein Downregulation by Salvicine in Multidrug Resistant K562/A02 Cells. Cancer Biol Ther (2007) 6:1794–9. doi: 10.4161/cbt.6.11.4860
46. Qing C, Zhang JS, Ding J. In Vitro Cytotoxicity of Salvicine, a Novel Diterpenoid Quinone. Zhongguo Yao Li Xue Bao Acta Pharmacol Sin (1999) 20:297–302.
47. Qing C, Jiang C, Zhang JS, Ding J. Induction of Apoptosis in Human Leukemia K-562 and Gastric Carcinoma SGC-7901 Cells by Salvicine, a Novel Anticancer Compound. Anti-Cancer Drugs (2001) 12:51–6. doi: 10.1097/00001813-200101000-00007
48. Zhang JS, Ding J, Tang QM, Li M, Zhao M, Lu LJ, et al. Synthesis and Antitumour Activity of Novel Diterpenequinone Salvicine and the Analogs. Bioorg Med Chem Lett (1999) 9:2731–6. doi: 10.1016/s0960-894x(99)00472-2
49. Liu WJ, Zhang YW, Shen Y, Jiang JF, Miao ZH, Ding J. Telomerase Inhibition Is a Specific Early Event in Salvicine-Treated Human Lung Adenocarcinoma A549 Cells. Biochem Biophys Res Commun (2004) 323:660–7. doi: 10.1016/j.bbrc.2004.08.135
50. Miao ZH, Tong LJ, Zhang JS, Han JX, Ding J. Characterization of Salvicine-Resistant Lung Adenocarcinoma A549/SAL Cell Line. Int J Cancer (2004) 110:627–32. doi: 10.1002/ijc.20026
51. Zhang YW, Zhang ZX, Miao ZH, Ding J. The Telomeric Protein TRF2 is Critical for the Protection of A549 Cells From Both Telomere Erosion and DNA Double-Strand Breaks Driven by Salvicine. Mol Pharmacol (2008) 73:824–32. doi: 10.1124/mol.107.039081
52. Zhang Y, Wang L, Chen Y, Qing C. Anti-Angiogenic Activity of Salvicine. Pharm Biol (2013) 51:1061–5. doi: 10.3109/13880209.2013.776612
53. Lu HR, Meng LH, Huang M, Zhu H, Miao ZH, Ding J. DNA Damage, C-Myc Suppression and Apoptosis Induced by the Novel Topoisomerase II Inhibitor, Salvicine, in Human Breast Cancer MCF-7 Cells. Cancer Chemother Pharmacol (2005) 55:286–94. doi: 10.1007/s00280-004-0877-z
54. Lu HR, Zhu H, Huang M, Chen Y, Cai YJ, Miao ZH, et al. Reactive Oxygen Species Elicit Apoptosis by Concurrently Disrupting Topoisomerase II and DNA-Dependent Protein Kinase. Mol Pharmacol (2005) 68:983–94. doi: 10.1124/mol.105.011544
55. Zhou J, Chen Y, Lang JY, Lu JJ, Ding J. Salvicine Inactivates Beta 1 Integrin and Inhibits Adhesion of MDA-MB-435 Cells to Fibronectin via Reactive Oxygen Species Signaling. Mol Cancer Res MCR (2008) 6:194–204. doi: 10.1158/1541-7786.Mcr-07-0197
56. Yao J, Feng FY, Lin C, Zhang XY, Fu M, Liang X, et al. [The Mechanism of Resistance to 2', 2'-Difluorodeoxycytidine (Gemcitabine) in a Pancreatic Cancer Cell Line]. Zhonghua Zhong Liu Za Zhi [Chin J Oncol] (2005) 27:721–6.
57. Cai YJ, Lu JJ, Zhu H, Xie H, Huang M, Lin LP, et al. Salvicine Triggers DNA Double-Strand Breaks and Apoptosis by GSH-Depletion-Driven H2O2 Generation and Topoisomerase II Inhibition. Free Radical Biol Med (2008) 45:627–35. doi: 10.1016/j.freeradbiomed.2008.05.017
58. Pendleton M, Lindsey RH Jr., Felix CA, Grimwade D, Osheroff N. Topoisomerase II and Leukemia. Ann N Y Acad Sci (2014) 1310:98–110. doi: 10.1111/nyas.12358
59. Meng L-h, Ding J. Induction of Bulk and C-Myc P2 Promoter-Specific DNA Damage by an Anti-Topoisomerase II Agent Salvicine Is an Early Event Leading to Apoptosis in HL-60 Cells. FEBS Lett (2001) 501:59–64. doi: 10.1016/S0014-5793(01)02633-3
60. Qing C, Miao Z-H, Tong L-J, Zhang J-S, Ding J. Actinomycin D Inhibiting K562 Cell Apoptosis Elicited by Salvicine But Not Decreasing Its Cytotoxicity. Acta Pharmacol Sin (2003) 24:415–21.
61. Miao ZH, Tang T, Zhang YX, Zhang JS, Ding J. Cytotoxicity, Apoptosis Induction and Downregulation of MDR-1 Expression by the Anti-Topoisomerase II Agent, Salvicine, in Multidrug-Resistant Tumor Cells. Int J Cancer (2003) 106:108–15. doi: 10.1002/ijc.11174
62. Shiah S-G, Chuang S-E, Chau Y-P, Shen S-C, Kuo M-L. Activation of C-Jun NH2-Terminal Kinase and Subsequent CPP32/Yama During Topoisomerase Inhibitor β-Lapachone-Induced Apoptosis Through an Oxidation-Dependent Pathway. Cancer Res (1999) 59:391–8.
63. Cai Y, Lu J, Miao Z, Lin L, Ding J. Reactive Oxygen Species Contribute to Cell Killing and P-Glycoprotein Downregulation by Salvicine in Multidrug Resistant K562/A02 Cells. Cancer Biol Ther (2007) 6:1794–9. doi: 10.4161/cbt.6.11.4860
64. Qing C, Jiang C, Zhang J-S, Ding J. Induction of Apoptosis in Human Leukemia K-562 and Gastric Carcinoma SGC-7901 Cells by Salvicine, A Novel Anticancer Compound. Anti-Cancer Drugs (2001) 12:51–6. doi: 10.1097/00001813-200101000-00007
65. Zhang J-S, Ding J, Tang Q-M, Li M, Zhao M, Lu L-J, et al. Synthesis and Antitumour Activity of Novel Diterpenequinone Salvicine and the Analogs. Bioorg Med Chem Lett (1999) 9:2731–6. doi: 10.1016/S0960-894X(99)00472-2
66. Liu W-J, Zhang Y-W, Shen Y, Jiang J-F, Miao Z-H, Ding J. Telomerase Inhibition Is a Specific Early Event in Salvicine-Treated Human Lung Adenocarcinoma A549 Cells. Biochem Biophys Res Commun (2004) 323:660–7. doi: 10.1016/j.bbrc.2004.08.135
67. Zhang Y-W, Zhang Z-X, Miao Z-H, Ding J. The Telomeric Protein TRF2 is Critical for the Protection of A549 Cells From Both Telomere Erosion and DNA Double-Strand Breaks Driven by Salvicine. Mol Pharmacol (2008) 73:824–32. doi: 10.1124/mol.107.039081
68. Zhang Y, Wang L, Chen Y, Qing C. Anti-Angiogenic Activity of Salvicine. Pharm Biol (2013) 51:1061–5. doi: 10.3109/13880209.2013.776612
69. Miao ZH, Tong LJ, Zhang JS, Han JX, Ding J. Characterization of Salvicine-Resistant Lung Adenocarcinoma A549/SAL Cell Line. Int J Cancer (2004) 110:627–32. doi: 10.1002/ijc.20026
70. Lu H-R, Zhu H, Huang M, Chen Y, Cai Y-J, Miao Z-H, et al. Reactive Oxygen Species Elicit Apoptosis by Concurrently Disrupting Topoisomerase II and DNA-Dependent Protein Kinase. Mol Pharmacol (2005) 68:983–94. doi: 10.1124/mol.105.011544
71. Lu H-R, Meng L-H, Huang M, Zhu H, Miao Z-H, Ding J. DNA Damage, C-Myc Suppression and Apoptosis Induced by the Novel Topoisomerase II Inhibitor, Salvicine, in Human Breast Cancer MCF-7 Cells. Cancer Chemother Pharmacol (2005) 55:286–94. doi: 10.1007/s00280-004-0877-z
72. Zhou J, Chen Y, Lang J-Y, Lu J-J, Ding J. Salvicine Inactivates β1 Integrin and Inhibits Adhesion of MDA-MB-435 Cells to Fibronectin Via Reactive Oxygen Species Signaling. Mol Cancer Res (2008) 6:194–204. doi: 10.1158/1541-7786.MCR-07-0197
73. Yao J, Feng F, Lin C, Zhang X, Fu M, Liang X, et al. The Mechanism of Resistanceto 2', 2'-Difluorodeoxycytidine (Gemcitabine) in a Pancreatic Cancer Cell Line. Zhonghua Zhong Liu Za Zhi [Chin J Oncol] (2005) 27:721–6.
74. Cai Y-J, Lu J-J, Zhu H, Xie H, Huang M, Lin L-P, et al. Salvicine Triggers DNA Double-Strand Breaks and Apoptosis by GSH-Depletion-Driven H2O2 Generation and Topoisomerase II Inhibition. Free Radical Biol Med (2008) 45:627–35. doi: 10.1016/j.freeradbiomed.2008.05.017
75. Lang JY, Chen H, Zhou J, Zhang YX, Zhang XW, Li MH, et al. Antimetastatic Effect of Salvicine on Human Breast Cancer MDA-MB-435 Orthotopic Xenograft Is Closely Related to Rho-Dependent Pathway. Clin Cancer Res Off J Am Assoc Cancer Res (2005) 11:3455–64. doi: 10.1158/1078-0432.Ccr-04-2026
76. Meng LH, Zhang JS, Ding J. Salvicine, a Novel DNA Topoisomerase II Inhibitor, Exerting its Effects by Trapping Enzyme-DNA Cleavage Complexes. Biochem Pharmacol (2001) 62:733–41. doi: 10.1016/s0006-2952(01)00732-8
77. Lichota A, Gwozdzinski K. Anticancer Activity of Natural Compounds From Plant and Marine Environment. Int J Mol Sci (2018) 19(11):3533. doi: 10.3390/ijms19113533
78. MENG LH, Ding J. Salvicine, A Novel Topoisomerase II Inhibitor, Exerts its Potent Anticancer Activity by ROS Generation 1. Acta Pharmacol Sin (2007) 28:1460–5. doi: 10.1111/j.1745-7254.2007.00698.x
79. Hu C-X, Zuo Z-L, Xiong B, Ma J-G, Geng M-Y, Lin L-P, et al. Salvicine Functions as Novel Topoisomerase II Poison by Binding to ATP Pocket. Mol Pharmacol (2006) 70:1593–601. doi: 10.1124/mol.106.027714
80. Deng F, Lu JJ, Liu HY, Lin LP, Ding J, Zhang JS. Synthesis and Antitumor Activity of Novel Salvicine Analogues. Chin Chem Lett (2011) 22:25–8. doi: 10.1016/j.cclet.2010.07.009
81. Commoner B, Townsend J, Pake GE. Free Radicals in Biological Materials. Nature (1954) 174:689–91. doi: 10.1038/174689a0
82. Dahlgren C, Karlsson A. Respiratory Burst in Human Neutrophils. J Immunol Methods (1999) 232:3–14. doi: 10.1016/s0022-1759(99)00146-5
83. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. ROS and ROS-Mediated Cellular Signaling. Oxid Med Cell Longevity (2016) 2016:4350965. doi: 10.1155/2016/4350965
84. Perry JJ, Shin DS, Getzoff ED, Tainer JA. The Structural Biochemistry of the Superoxide Dismutases. Biochim Biophys Acta (2010) 1804:245–62. doi: 10.1016/j.bbapap.2009.11.004
85. Meitzler JL, Antony S, Wu Y, Juhasz A, Liu H, Jiang G, et al. NADPH Oxidases: A Perspective on Reactive Oxygen Species Production in Tumor Biology. Antioxid Redox Signal (2014) 20:2873–89. doi: 10.1089/ars.2013.5603
86. Fransen M, Nordgren M, Wang B, Apanasets O. Role of Peroxisomes in ROS/RNS-Metabolism: Implications for Human Disease. Biochim Biophys Acta (2012) 1822:1363–73. doi: 10.1016/j.bbadis.2011.12.001
87. Ziech D, Franco R, Pappa A, Panayiotidis MI. Reactive Oxygen Species (ROS)–induced Genetic and Epigenetic Alterations in Human Carcinogenesis. Mutat Res (2011) 711:167–73. doi: 10.1016/j.mrfmmm.2011.02.015
88. Zulato E, Ciccarese F, Agnusdei V, Pinazza M, Nardo G, Iorio E, et al. LKB1 Loss is Associated With Glutathione Deficiency Under Oxidative Stress and Sensitivity of Cancer Cells to Cytotoxic Drugs and γ-Irradiation. Biochem Pharmacol (2018) 156:479–90. doi: 10.1016/j.bcp.2018.09.019
89. Fan PC, Zhang Y, Wang Y, Wei W, Zhou YX, Xie Y, et al. Quantitative Proteomics Reveals Mitochondrial Respiratory Chain as a Dominant Target for Carbon Ion Radiation: Delayed Reactive Oxygen Species Generation Caused DNA Damage. Free Radical Biol Med (2019) 130:436–45. doi: 10.1016/j.freeradbiomed.2018.10.449
90. Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-Dose Parenteral Ascorbate Enhanced Chemosensitivity of Ovarian Cancer and Reduced Toxicity of Chemotherapy. Sci Trans Med (2014) 6(222):222ra18. doi: 10.1126/scitranslmed.3007154
91. Wright ME, Virtamo J, Hartman AM, Pietinen P, Edwards BK, Taylor PR, et al. Effects of Alpha-Tocopherol and Beta-Carotene Supplementation on Upper Aerodigestive Tract Cancers in a Large, Randomized Controlled Trial. Cancer (2007) 109:891–8. doi: 10.1002/cncr.22482
92. Curtin NJ. DNA Repair Dysregulation From Cancer Driver to Therapeutic Target. Nat Rev Cancer (2012) 12:801–17. doi: 10.1038/nrc3399
93. Goldstein M, Kastan MB. The DNA Damage Response: Implications for Tumor Responses to Radiation and Chemotherapy. Annu Rev Med (2015) 66:129–43. doi: 10.1146/annurev-med-081313-121208
94. McNally JP, Millen SH, Chaturvedi V, Lakes N, Terrell CE, Elfers EE, et al. Manipulating DNA Damage-Response Signaling for the Treatment of Immune-Mediated Diseases. Proc Natl Acad Sci USA (2017) 114:E4782–e4791. doi: 10.1073/pnas.1703683114
95. Rahman M, Zilani M, Hasan N, Islam M, Hasan M, Yasmin F, et al. In Vivo Neuropharmacological Potential of Gomphandra Tetrandra (Wall.) Sleumer and In-Silico Study Against β-Amyloid Precursor Protein. Processes (2021) 9:1449. doi: 10.3390/pr9081449
96. Zilani MNH, Islam MA, Biswas P, Anisuzzman M, Hossain H, Shilpi JA, et al. Metabolite Profiling, Anti-Inflammatory, Analgesic Potentials of Edible Herb Colocasia Gigantea and Molecular Docking Study Against COX-II Enzyme. J Ethnopharmacol (2021) 281:114577. doi: 10.1016/j.jep.2021.114577
97. Polo SE, Jackson SP. Dynamics of DNA Damage Response Proteins at DNA Breaks: A Focus on Protein Modifications. Genes Dev (2011) 25:409–33. doi: 10.1101/gad.2021311
98. Lord CJ, Ashworth A. The DNA Damage Response and Cancer Therapy. Nature (2012) 481:287–94. doi: 10.1038/nature10760
99. Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS, et al. Cisplatin Induces a Mitochondrial-ROS Response That Contributes to Cytotoxicity Depending on Mitochondrial Redox Status and Bioenergetic Functions. PloS One (2013) 8:e81162. doi: 10.1371/journal.pone.0081162
100. Salehi F, Behboudi H, Kavoosi G, Ardestani SK. Oxidative DNA Damage Induced by ROS-Modulating Agents With the Ability to Target DNA: A Comparison of the Biological Characteristics of Citrus Pectin and Apple Pectin. Sci Rep (2018) 8:13902. doi: 10.1038/s41598-018-32308-2
101. Cannan WJ, Tsang BP, Wallace SS, Pederson DS. Nucleosomes Suppress the Formation of Double-Strand DNA Breaks During Attempted Base Excision Repair of Clustered Oxidative Damages. J Biol Chem (2014) 289:19881–93. doi: 10.1074/jbc.M114.571588
102. Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev M. Oxidative Stress Induces Degradation of Mitochondrial DNA. Nucleic Acids Res (2009) 37:2539–48. doi: 10.1093/nar/gkp100
103. Kotsantis P, Petermann E, Boulton SJ. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling Into Place. Cancer Discov (2018) 8:537–55. doi: 10.1158/2159-8290.Cd-17-1461
104. Zeman MK, Cimprich K. Causes and Consequences of Replication Stress. Nat Cell Biol (2014) 16:2–9. doi: 10.1038/ncb2897
105. Halazonetis TD, Gorgoulis VG, Bartek J. An Oncogene-Induced DNA Damage Model for Cancer Development. Sci (New York NY) (2008) 319:1352–5. doi: 10.1126/science.1140735
106. Maya-Mendoza A, Ostrakova J, Kosar M, Hall A, Duskova P, Mistrik M, et al. Myc and Ras Oncogenes Engage Different Energy Metabolism Programs and Evoke Distinct Patterns of Oxidative and DNA Replication Stress. Mol Oncol (2015) 9:601–16. doi: 10.1016/j.molonc.2014.11.001
107. Meng Y, Chen CW, Yung MMH, Sun W, Sun J, Li Z, et al. DUOXA1-Mediated ROS Production Promotes Cisplatin Resistance by Activating ATR-Chk1 Pathway in Ovarian Cancer. Cancer Lett (2018) 428:104–16. doi: 10.1016/j.canlet.2018.04.029
108. Somyajit K, Gupta R, Sedlackova H, Neelsen KJ, Ochs F, Rask MB, et al. Redox-Sensitive Alteration of Replisome Architecture Safeguards Genome Integrity. Sci (New York NY) (2017) 358:797–802. doi: 10.1126/science.aao3172
109. Ito K, Takubo K, Arai F, Satoh H, Matsuoka S, Ohmura M, et al. Regulation of Reactive Oxygen Species by Atm is Essential for Proper Response to DNA Double-Strand Breaks in Lymphocytes. J Immunol (Baltimore Md 1950) (2007) 178:103–10. doi: 10.4049/jimmunol.178.1.103
110. Zegura B, Lah TT, Filipic M. The Role of Reactive Oxygen Species in Microcystin-LR-Induced DNA Damage. Toxicology (2004) 200:59–68. doi: 10.1016/j.tox.2004.03.006
111. Nowicki MO, Falinski R, Koptyra M, Slupianek A, Stoklosa T, Gloc E, et al. BCR/ABL Oncogenic Kinase Promotes Unfaithful Repair of the Reactive Oxygen Species-Dependent DNA Double-Strand Breaks. Blood (2004) 104:3746–53. doi: 10.1182/blood-2004-05-1941
112. Sallmyr A, Fan J, Rassool FV. Genomic Instability in Myeloid Malignancies: Increased Reactive Oxygen Species (ROS), DNA Double Strand Breaks (DSBs) and Error-Prone Repair. Cancer Lett (2008) 270:1–9. doi: 10.1016/j.canlet.2008.03.036
113. Karanjawala ZE, Murphy N, Hinton DR, Hsieh CL, Lieber MR. Oxygen Metabolism Causes Chromosome Breaks and is Associated With the Neuronal Apoptosis Observed in DNA Double-Strand Break Repair Mutants. Curr Biol CB (2002) 12:397–402. doi: 10.1016/s0960-9822(02)00684-x
114. Wiseman H, Halliwell B. Damage to DNA by Reactive Oxygen and Nitrogen Species: Role in Inflammatory Disease and Progression to Cancer. Biochem J (1996) 313( Pt 1):17–29. doi: 10.1042/bj3130017
115. Schneider CC, Hessenauer A, Götz C, Montenarh M. DMAT, an Inhibitor of Protein Kinase CK2 Induces Reactive Oxygen Species and DNA Double Strand Breaks. Oncol Rep (2009) 21:1593–7. doi: 10.3892/or_00000392
116. Kim KT, Levis M, Small D. Constitutively Activated FLT3 Phosphorylates BAD Partially Through Pim-1. Br J Haematol (2006) 134:500–9. doi: 10.1111/j.1365-2141.2006.06225.x
117. Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, Smith BD, et al. FLT3-Targeted Tyrosine Kinase Inhibitor is Cytotoxic to Leukemia Cells In Vitro and In Vivo. Blood (2002) 99:3885–91. doi: 10.1182/blood.v99.11.3885
118. Tse KF, Allebach J, Levis M, Smith BD, Bohmer FD, Small D. Inhibition of the Transforming Activity of FLT3 Internal Tandem Duplication Mutants From AML Patients by a Tyrosine Kinase Inhibitor. Leukemia (2002) 16:2027–36. doi: 10.1038/sj.leu.2402674
119. Vinkemeier U, Cohen SL, Moarefi I, Chait BT, Kuriyan J, Darnell JE Jr. DNA Binding of In Vitro Activated Stat1 Alpha, Stat1 Beta and Truncated Stat1: Interaction Between NH2-Terminal Domains Stabilizes Binding of Two Dimers to Tandem DNA Sites. EMBO J (1996) 15:5616–26. doi: 10.1002/j.1460-2075.1996.tb00946.x
120. Piloto O, Wright M, Brown P, Kim KT, Levis M, Small D. Prolonged Exposure to FLT3 Inhibitors Leads to Resistance via Activation of Parallel Signaling Pathways. Blood (2007) 109:1643–52. doi: 10.1182/blood-2006-05-023804
121. Degirmenci U, Wang M, Hu J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells (2020) 9(1):198. doi: 10.3390/cells9010198
122. Padua RA, Guinn BA, Al-Sabah AI, Smith M, Taylor C, Pettersson T, et al. RAS, FMS and P53 Mutations and Poor Clinical Outcome in Myelodysplasias: A 10-Year Follow-Up. Leukemia (1998) 12:887–92. doi: 10.1038/sj.leu.2401044
123. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA Damage: Mechanisms, Mutation, and Disease. FASEB J Off Publ Fed Am Soc Exp Biol (2003) 17:1195–214. doi: 10.1096/fj.02-0752rev
124. Mills KD, Ferguson DO, Alt FW. The Role of DNA Breaks in Genomic Instability and Tumorigenesis. Immunol Rev (2003) 194:77–95. doi: 10.1034/j.1600-065x.2003.00060.x
125. Islam MA, Zilani MNH, Biswas P, Khan DA, Rahman MH, Nahid R, et al. Evaluation of In Vitro and in Silico Anti-Inflammatory Potential of Some Selected Medicinal Plants of Bangladesh Against Cyclooxygenase-II Enzyme. J Ethnopharmacol (2022) 285:114900. doi: 10.1016/j.jep.2021.114900
126. Coutts AS, La Thangue N. The P53 Response During DNA Damage: Impact of Transcriptional Cofactors. Biochem Soc Symp (2006) 73:181–9. doi: 10.1042/bss0730181
127. Hurley PJ, Bunz F. ATM and ATR: Components of an Integrated Circuit. Cell Cycle (Georgetown Tex) (2007) 6:414–7. doi: 10.4161/cc.6.4.3886
128. Riballo E, Kühne M, Rief N, Doherty A, Smith GC, Recio MJ, et al. A Pathway of Double-Strand Break Rejoining Dependent Upon ATM, Artemis, and Proteins Locating to Gamma-H2AX Foci. Mol Cell (2004) 16:715–24. doi: 10.1016/j.molcel.2004.10.029
129. Nussenzweig A, Nussenzweig MC. A Backup DNA Repair Pathway Moves to the Forefront. Cell (2007) 131:223–5. doi: 10.1016/j.cell.2007.10.005
130. Wang H, Rosidi B, Perrault R, Wang M, Zhang L, Windhofer F, et al. DNA Ligase III as a Candidate Component of Backup Pathways of Nonhomologous End Joining. Cancer Res (2005) 65:4020–30. doi: 10.1158/0008-5472.Can-04-3055
131. Fernandes MS, Reddy MM, Gonneville JR, DeRoo SC, Podar K, Griffin JD, et al. BCR-ABL Promotes the Frequency of Mutagenic Single-Strand Annealing DNA Repair. Blood (2009) 114:1813–9. doi: 10.1182/blood-2008-07-172148
132. Fan J, Li L, Small D, Rassool F. Cells Expressing FLT3/ITD Mutations Exhibit Elevated Repair Errors Generated Through Alternative NHEJ Pathways: Implications for Genomic Instability and Therapy. Blood (2010) 116:5298–305. doi: 10.1182/blood-2010-03-272591
133. Stanicka J, Russell EG, Woolley JF, Cotter TG. NADPH Oxidase-Generated Hydrogen Peroxide Induces DNA Damage in Mutant FLT3-Expressing Leukemia Cells. J Biol Chem (2015) 290:9348–61. doi: 10.1074/jbc.M113.510495
134. Wang JC. Cellular Roles of DNA Topoisomerases: A Molecular Perspective. Nat Rev Mol Cell Biol (2002) 3:430–40. doi: 10.1038/nrm831
135. Pommier Y. Topoisomerase I Inhibitors: Camptothecins and Beyond. Nat Rev Cancer (2006) 6:789–802. doi: 10.1038/nrc1977
136. Matias-Barrios VM, Radaeva M, Song Y, Alperstein Z, Lee AR, Schmitt V, et al. Discovery of New Catalytic Topoisomerase II Inhibitors for Anticancer Therapeutics. Front Oncol (2021) 10:633142. doi: 10.3389/fonc.2020.633142
137. Lin KW, Lin WH, Su CL, Hsu HY, Lin CN. Design, Synthesis and Antitumour Evaluation of Novel Anthraquinone Derivatives. Bioorg Chem (2021) 107:104395. doi: 10.1016/j.bioorg.2020.104395
138. Pendleton M, Lindsey RH Jr, Felix CA, Grimwade D, Osheroff N. Topoisomerase II and Leukemia. Ann New Y Acad Sci (2014) 1310:98–110. doi: 10.1111/nyas.12358
139. Pommier Y, Leo E, Zhang H, Marchand C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem Biol (2010) 17:421–33. doi: 10.1016/j.chembiol.2010.04.012
140. Kimura K, Saijo M, Ui M, Enomoto T. Growth State- and Cell Cycle-Dependent Fluctuation in the Expression of Two Forms of DNA Topoisomerase II and Possible Specific Modification of the Higher Molecular Weight Form in the M Phase. J Biol Chem (1994) 269:1173–6. doi: 10.1016/S0021-9258(17)42238-1
141. Yang X, Li W, Prescott ED, Burden SJ, Wang JC. DNA Topoisomerase IIbeta and Neural Development. Sci (New Y NY) (2000) 287:131–4. doi: 10.1126/science.287.5450.131
142. Matias-Barrios VM, Radaeva M, Song Y, Alperstein Z, Lee AR, Schmitt V, et al. Discovery of New Catalytic Topoisomerase II Inhibitors for Anticancer Therapeutics. Front Oncol (2020) 10:633142. doi: 10.3389/fonc.2020.633142
143. Aplan PD, Chervinsky DS, Stanulla M, Burhans WC. Site-Specific DNA Cleavage Within the MLL Breakpoint Cluster Region Induced by Topoisomerase II Inhibitors. Blood (1996) 87:2649–58. doi: 10.1182/blood.V87.7.2649.bloodjournal8772649
144. Matias Barrios VM. New Catalytic Topoisomerase II Inhibitors Discovered for Anticancer Therapeutics. University of British Columbia: University of British Columbia (2021).
145. McClendon AK, Osheroff N. DNA Topoisomerase II, Genotoxicity, and Cancer. Mutat Res (2007) 623:83–97. doi: 10.1016/j.mrfmmm.2007.06.009
146. Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res (2018) 46:D1074–d1082. doi: 10.1093/nar/gkx1037
147. Wu CC, Li YC, Wang YR, Li TK, Chan NL. On the Structural Basis and Design Guidelines for Type II Topoisomerase-Targeting Anticancer Drugs. Nucleic Acids Res (2013) 41:10630–40. doi: 10.1093/nar/gkt828
148. Li TK, Chen AY, Yu C, Mao Y, Wang H, Liu LF. Activation of Topoisomerase II-Mediated Excision of Chromosomal DNA Loops During Oxidative Stress. Genes Dev (1999) 13:1553–60. doi: 10.1101/gad.13.12.1553
149. Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, et al. Phosphorylation of Histone H2AX and Activation of Mre11, Rad50, and Nbs1 in Response to Replication-Dependent DNA Double-Strand Breaks Induced by Mammalian DNA Topoisomerase I Cleavage Complexes. J Biol Chem (2003) 278:20303–12. doi: 10.1074/jbc.M300198200
150. Arefin A, Ema TI, Islam T, Hossen MS, Islam T, Al Azad S, et al. Target Specificity of Selective Bioactive Compounds in Blocking α-Dystroglycan Receptor to Suppress Lassa Virus Infection: An in Silico Approach. J Biomed Res (2021) 35:459. doi: 10.7555/JBR.35.20210111
151. Adachi N, Suzuki H, Iiizumi S, Koyama H. Hypersensitivity of Nonhomologous DNA End-Joining Mutants to VP-16 and ICRF-193: Implications for the Repair of Topoisomerase II-Mediated DNA Damage. J Biol Chem (2003) 278:35897–902. doi: 10.1074/jbc.M306500200
152. Jackson SK, Thomas MP, Smith S, Madhani M, Rogers SC, James PE. In Vivo EPR Spectroscopy: Biomedical and Potential Diagnostic Applications. Faraday Discuss (2004) 126:103–17. doi: 10.1039/b307162f
153. Khan RA, Hossain R, Siyadatpanah A, Al-Khafaji K, Khalipha ABR, Dey D, et al. Diterpenes/Diterpenoids and Their Derivatives as Potential Bioactive Leads Against Dengue Virus: A Computational and Network Pharmacology Study. Molecules (2021) 26:6821. doi: 10.3390/molecules26226821
154. Qing C, Zhang J, Ding J. In Vitro Cytotoxicity of Salvicine, a Novel Diterpenoid Quinone. Zhongguo Yao Li Xue Bao Acta Pharmacol Sin (1999) 20:297–302.
155. Elmore S. Apoptosis: A Review of Programmed Cell Death. Toxicol Pathol (2007) 35:495–516. doi: 10.1080/01926230701320337
156. Liebermann DA, Gregory B, Hoffman B. AP-1 (Fos/Jun) Transcription Factors in Hematopoietic Differentiation and Apoptosis. Int J Oncol (1998) 12:685–700. doi: 10.3892/ijo.12.3.685
157. Shang X, Shiono Y, Fujita Y, Oka S, Yamazaki Y. Synergistic Enhancement of Apoptosis by DNA- and Cytoskeleton-Damaging Agents: A Basis for Combination Chemotherapy of Cancer. Anticancer Res (2001) 21:2585–9.
158. Miao ZH, Ding J. Transcription Factor C-Jun Activation Represses Mdr-1 Gene Expression. Cancer Res (2003) 63:4527–32.
159. Kong FB, Wang XT, Xie YB, Xiao Q. [Inhibitory Effect of E2F-1-Silencing Lentivirus Vector on Chemoresistance of Subcutaneous Human Gastric Cancer in Nude Mice]. Zhonghua Zhong Liu Za Zhi [Chin J Oncol] (2013) 35:655–9.
160. Boreddy SR, Srivastava SK. Pancreatic Cancer Chemoprevention by Phytochemicals. Cancer Lett (2013) 334:86–94. doi: 10.1016/j.canlet.2012.10.020
161. Xu B, Ding J, Chen K-X, Miao Z-H, Huang H, Liu H, et al. Therapy. Advances in Cancer Chemotherapeutic Drug Research in China. Recent Adv Cancer Res Ther (2012) 287:287. doi: 10.1016/B978-0-12-397833-2.00012-1
162. Hsu S, Singh B, Schuster G. Induction of Apoptosis in Oral Cancer Cells: Agents and Mechanisms for Potential Therapy and Prevention. Oral Oncol (2004) 40:461–73. doi: 10.1016/j.oraloncology.2003.09.012
163. Ohtani T, Hatori M, Ito H, Takizawa K, Kamijo R, Nagumo M. Involvement of Caspases in 5-FU Induced Apoptosis in an Oral Cancer Cell Line. Anticancer Res (2000) 20:3117–21.
164. Tong D, Poot M, Hu D, Oda D. 5-Fluorouracil-Induced Apoptosis in Cultured Oral Cancer Cells. Oral Oncol (2000) 36:236–41. doi: 10.1016/s1368-8375(99)00079-2
165. Desai SD, Li TK, Rodriguez-Bauman A, Rubin EH, Liu LF. Ubiquitin/26S Proteasome-Mediated Degradation of Topoisomerase I as a Resistance Mechanism to Camptothecin in Tumor Cells. Cancer Res (2001) 61:5926–32.
166. Heyder C, Gloria-Maercker E, Hatzmann W, Niggemann B, Zänker KS, Dittmar T. Role of the Beta1-Integrin Subunit in the Adhesion, Extravasation and Migration of T24 Human Bladder Carcinoma Cells. Clin Exp Metastasis (2005) 22:99–106. doi: 10.1007/s10585-005-4335-z
167. Wartenberg M, Ling FC, Schallenberg M, Bäumer AT, Petrat K, Hescheler J, et al. Down-Regulation of Intrinsic P-Glycoprotein Expression in Multicellular Prostate Tumor Spheroids by Reactive Oxygen Species. J Biol Chem (2001) 276:17420–8. doi: 10.1074/jbc.M100141200
168. Fiore G, Nencini C, Cavallo F, Capasso A, Bader A, Giorgi G, et al. ;In Vitro Antiproliferative Effect of Six Salvia Species on Human Tumor Cell Lines. (2006) 20:701–3. doi: 10.1002/ptr.1911
169. Chen Y, Zheng L, Liu J, Zhou Z, Cao X, Lv X, et al. Shikonin Inhibits Prostate Cancer Cells Metastasis by Reducing Matrix Metalloproteinase-2/-9 Expression via AKT/mTOR and ROS/ERK1/2 Pathways. Int Immunopharmacol (2014) 21:447–55. doi: 10.1016/j.intimp.2014.05.026
170. Bendas G, Borsig L. Cancer Cell Adhesion and Metastasis: Selectins, Integrins, and the Inhibitory Potential of Heparins. Int J Cell Biol (2012) 2012:676731. doi: 10.1155/2012/676731
171. Borsig L. Selectins in Cancer Immunity. Glycobiology (2018) 28:648–55. doi: 10.1093/glycob/cwx105
172. Casal JI, Bartolomé RA. RGD Cadherins and α2β1 Integrin in Cancer Metastasis: A Dangerous Liaison. Biochim Biophys Acta Rev Cancer (2018) 1869:321–32. doi: 10.1016/j.bbcan.2018.04.005
173. Huang Q, Shen HM, Shui G, Wenk MR, Ong CN. Emodin Inhibits Tumor Cell Adhesion Through Disruption of the Membrane Lipid Raft-Associated Integrin Signaling Pathway. Cancer Res (2006) 66:5807–15. doi: 10.1158/0008-5472.Can-06-0077
174. Hood JD, Cheresh DA. Role of Integrins in Cell Invasion and Migration. Nat Rev Cancer (2002) 2:91–100. doi: 10.1038/nrc727
175. Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J Signal Transduct (2011) 2011:792639. doi: 10.1155/2011/792639
176. Keshari RS, Verma A, Barthwal MK, Dikshit M. Reactive Oxygen Species-Induced Activation of ERK and P38 MAPK Mediates PMA-Induced NETs Release From Human Neutrophils. J Cell Biochem (2013) 114:532–40. doi: 10.1002/jcb.24391
177. Fidler IJ. Critical Determinants of Metastasis. Semin Cancer Biol (2002) 12:89–96. doi: 10.1006/scbi.2001.0416
178. Monteiro J, Fodde R. Cancer Stemness and Metastasis: Therapeutic Consequences and Perspectives. Eur J Cancer (Oxf Engl 1990) (2010) 46:1198–203. doi: 10.1016/j.ejca.2010.02.030
179. Guan X. Cancer Metastases: Challenges and Opportunities. Acta Pharm Sinica B (2015) 5:402–18. doi: 10.1016/j.apsb.2015.07.005
180. Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, Snyder N, et al. Drug Resistance in Cancer: An Overview. Cancers (2014) 6:1769–92. doi: 10.3390/cancers6031769
181. Mansoori B, Mohammadi A, Davudian S, Shirjang S, Baradaran B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv Pharm Bull (2017) 7:339–48. doi: 10.15171/apb.2017.041
182. Fromm MF. The Influence of MDR1 Polymorphisms on P-Glycoprotein Expression and Function in Humans. Adv Drug Deliv Rev (2002) 54:1295–310. doi: 10.1016/s0169-409x(02)00064-9
183. Dhooge CR, De Moerloose BM, Benoit YC, Van Roy N. Philippé; Laureys, G.G. Expression of the MDR1 Gene Product P-Glycoprotein in Childhood Neuroblastoma. Cancer (1997) 80:1250–7. doi: 10.1002/(sici)1097-0142(19971001)80:7<1250::aid-cncr8>3.0.co;2-o
184. Nanayakkara AK, Follit CA, Chen G, Williams NS, Vogel PD, Wise JG. Targeted Inhibitors of P-Glycoprotein Increase Chemotherapeutic-Induced Mortality of Multidrug Resistant Tumor Cells. Sci Rep (2018) 8:967. doi: 10.1038/s41598-018-19325-x
Keywords: diterpenoid quinone, multidrug-resistant (MDR), topoisomerase II, ROS signaling, DNA damage response (DDR), anticancer properties
Citation: Dey D, Hasan MM, Biswas P, Papadakos SP, Rayan RA, Tasnim S, Bilal M, Islam MJ, Arshe FA, Arshad EM, Farzana M, Rahaman TI, Baral SK, Paul P, Bibi S, Rahman MA and Kim B (2022) Investigating the Anticancer Potential of Salvicine as a Modulator of Topoisomerase II and ROS Signaling Cascade. Front. Oncol. 12:899009. doi: 10.3389/fonc.2022.899009
Received: 18 March 2022; Accepted: 02 May 2022;
Published: 01 June 2022.
Edited by:
Valeria P. Sulsen, University of Buenos Aires, ArgentinaReviewed by:
Mohammad Imran Khan, King Abdulaziz University, Saudi ArabiaVaishali Aggarwal, University of Pittsburgh, United States
Copyright © 2022 Dey, Hasan, Biswas, Papadakos, Rayan, Tasnim, Bilal, Islam, Arshe, Arshad, Farzana, Rahaman, Baral, Paul, Bibi, Rahman and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Md. Ataur Rahman, YXRhdXIxOTgxcmFobWFuQGhvdG1haWwuY29t; Bonglee Kim, Ym9uZ2xlZWtpbUBraHUuYWMua3I=
†These authors have contributed equally to this work