 Chen-Yun Wang1,2
Chen-Yun Wang1,2 Chi-Hong Chao1,2,3*
Chi-Hong Chao1,2,3*- 1Institute of Molecular Medicine and Bioengineering, National Yang Ming Chiao Tung University, Hsinchu, Taiwan
- 2Center For Intelligent Drug Systems and Smart Bio-devices (IDS2B), National Yang Ming Chiao Tung University, Hsinchu, Taiwan
- 3Department of Biological Science and Technology, National Yang Ming Chiao Tung University, Hsinchu, Taiwan
The transcription factor p53 is the most well-characterized tumor suppressor involved in multiple cellular processes, which has expanded to the regulation of metabolism in recent decades. Accumulating evidence reinforces the link between the disturbance of p53-relevant metabolic activities and tumor development. However, a full-fledged understanding of the metabolic roles of p53 and the underlying detailed molecular mechanisms in human normal and cancer cells remain elusive, and persistent endeavor is required to foster the entry of drugs targeting p53 into clinical use. This mini-review summarizes the indirect regulation of cellular metabolism by wild-type p53 as well as mutant p53, in which mechanisms are categorized into three major groups: through modulating downstream transcriptional targets, protein-protein interaction with other transcription factors, and affecting signaling pathways. Indirect mechanisms expand the p53 regulatory networks of cellular metabolism, making p53 a master regulator of metabolism and a key metabolic sensor. Moreover, we provide a brief overview of recent achievements and potential developments in the therapeutic strategies targeting mutant p53, emphasizing synthetic lethal methods targeting mutant p53 with metabolism. Then, we delineate synthetic lethality targeting mutant p53 with its indirect regulation on metabolism, which expands the synthetic lethal networks of mutant p53 and broadens the horizon of developing novel therapeutic strategies for p53 mutated cancers, providing more opportunities for cancer patients with mutant p53. Finally, the limitations and current research gaps in studies of metabolic networks controlled by p53 and challenges of research on p53-mediated indirect regulation on metabolism are further discussed.
Introduction
During the 40 years of discovery, TP53, encoding a transcription factor known as the guardian of the genome, has been well characterized as a pivotal tumor suppressor. Upon various stress signals, including DNA damage, metabolic stress, and induction of oncogenes, p53 is released from its core negative regulator MDM2 and activated through multiple post-translational modifications (PTMs), which subsequently results in the upregulation or downregulation of genes involved in DNA repair, cell-cycle arrest, senescence, and apoptosis. Furthermore, p53 has been identified as an important regulator of stemness, autophagy, redox homeostasis, cellular metabolism, as well as tumor microenvironments (TMEs) (1–3).
Being the most frequently mutated tumor suppressor gene in human cancers, TP53 mutation exists in more than 50% of human cancers. TP53 mutates variably in different types or subtypes of cancer (4), and the majority of TP53 mutations are missense mutations, a single-base substitution located in the DNA-binding domain (DBD), giving rise to a full-length p53 protein (5). The existence of hotspot mutations, which account for almost 30% of all the missense mutations in TP53, may confer maximal benefits on tumor cells (6). These mutations are classified into two main categories: DNA-contact mutations abrogate residues directly involved in DNA binding, such as R273H, while structural or conformational mutations disrupt the structure of the DBD, such as R175H (3). Typically, oncogenic effects of TP53 mutations might be exerted through loss of function (LOF) of wild-type p53 (WTp53), dominant-negative effect (DNE) over WTp53, and gain-of-function (GOF) independent of WTp53, which are not mutually exclusive (7, 8). Despite losing sequence-specific DNA binding (SSDB) activity, mutant p53 (MTp53) can regulate gene expression through both direct and indirect mechanisms (7, 9, 10). Direct binding of MTp53 to DNA can be achieved by DNA structure-selective binding (DSSB), in which MTp53 recognizes its target genes by selective binding to DNA secondary structures. On the other hand, MTp53 associates with various transcription factors to act as a transcriptional repressor or transcription cofactor. Moreover, cooperation between MTp53 and the SWI/SNF chromatin remodeling complex contributes to over 40% of MTp53-regulated gene expression (11). MTp53 also exerts its oncogenic functions via modulating non-coding RNAs (ncRNAs) (12). Taken together, all these facts reveal a significant contribution of the indirect mechanisms to MTp53-mediated biological effects.
As previously mentioned, p53 has emerged as a critical modulator of cellular metabolism. Actually, p53-mediated metabolic activities have been reported to involve in the development of several human diseases, including diabetes, ischemia, neurodegeneration, as well as cancer (13), which we will focus on in this review. In addition to inducing or repressing the expression of transcriptional target genes associated with p53-mediated metabolic pathways and directly interacting with metabolic enzymes to activate or inhibit their activities, p53 also regulates metabolism via indirect mechanisms. The indirect mechanisms which we define here are categorized into three groups. First, p53 regulates metabolic genes through modulating its direct targets, such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). Second, p53 activates or suppresses the expression of metabolic genes through interacting with other transcription factors. Third, p53 controls metabolism by affecting signaling pathways. These indirect mechanisms scale up the number of metabolism-associated genes regulated by p53, extend the influence of p53 to a wide variety of metabolic pathways, and expand the p53 regulatory networks of metabolism, further highlighting p53 as a master regulator of metabolism. The critical role of p53-mediated indirect regulation on metabolism in tumorigenesis and tumor progression is evident, for example, in our previous study that in spite of several direct transcriptional targets of p53 involved in mitochondrial respiration, restoration of the decreased oxidative phosphorylation (OXPHOS) caused by MTp53 could be achieved through ectopic expression of miR-200c, a downstream direct transcriptional target of p53, which enhances phosphoenolpyruvate carboxykinase 2 (PCK2) expression via downregulating ZEB1/BMI1 (14). Moreover, restoring miR-200c expression leads to suppression of tumor growth, whereas interference of PCK2, the key enzyme linking the TCA cycle and glycolysis (15), counteracts the miR-200c-mediated tumor-suppressing effect, which accentuates the vital role of the indirect mechanisms accounting for p53-regulated cellular metabolism, and provides a functional link between p53-regulated metabolism and p53-mediated biological functions. Furthermore, p53 is generally considered an important metabolic sensor by positioning at the center of several signaling pathways coordinating cellular metabolism (16). Notably, direct and indirect regulation of metabolism by p53 are not mutually exclusive but overlapped to some extent. This is exemplified by hexokinase 2 (HK2), the first rate-limiting enzyme of glycolysis, which is both a direct transcriptional target of p53 (17) and an indirect metabolic target of p53 through miR-34a (18) and miR-143 (19). Interestingly, glucose transporter 1 (GLUT1), transcriptionally repressed by p53 (20) is an indirect target of MTp53 through modulation of the RhoA/ROCK signaling pathway (21), indicating the flexibility of indirect mechanisms underlying p53-mediated metabolic activities.
Transcriptional targets of p53 in regulating metabolism have been delineated in widespread literature, especially in a review article that gives a clear summary of direct target genes activated or repressed by p53 in every aspect of metabolic pathways (16). Considering that the indirect mechanisms seem to play a critical role in contributing to p53-mediated control of cellular metabolisms and other biological functions, and a growing number of metabolic-associated genes and proteins have been reported to be indirectly modulated by p53, in the present work, we will briefly summarize the indirect regulation of cellular metabolism by WTp53 (Figure 1 and Table 1) and MTp53 (Figure 2 and Table 2), respectively.
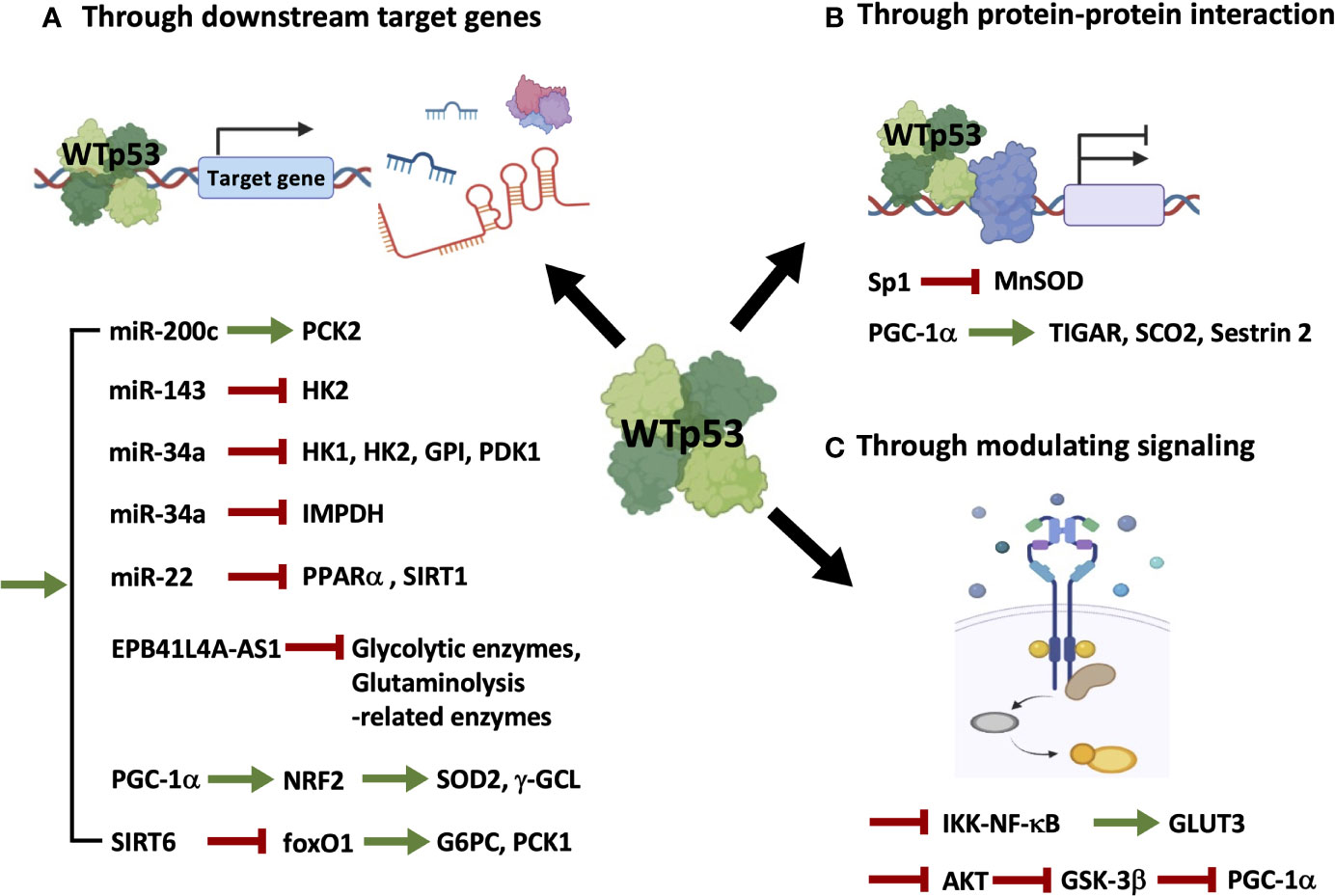
Figure 1 Wild-type p53 regulates cellular metabolism through indirect mechanisms. WTp53 indirectly regulates metabolism through inducing its direct targets, including miRNAs, lncRNAs, and proteins (A), associating with other transcription factors (B), or modulating signaling pathways (C).
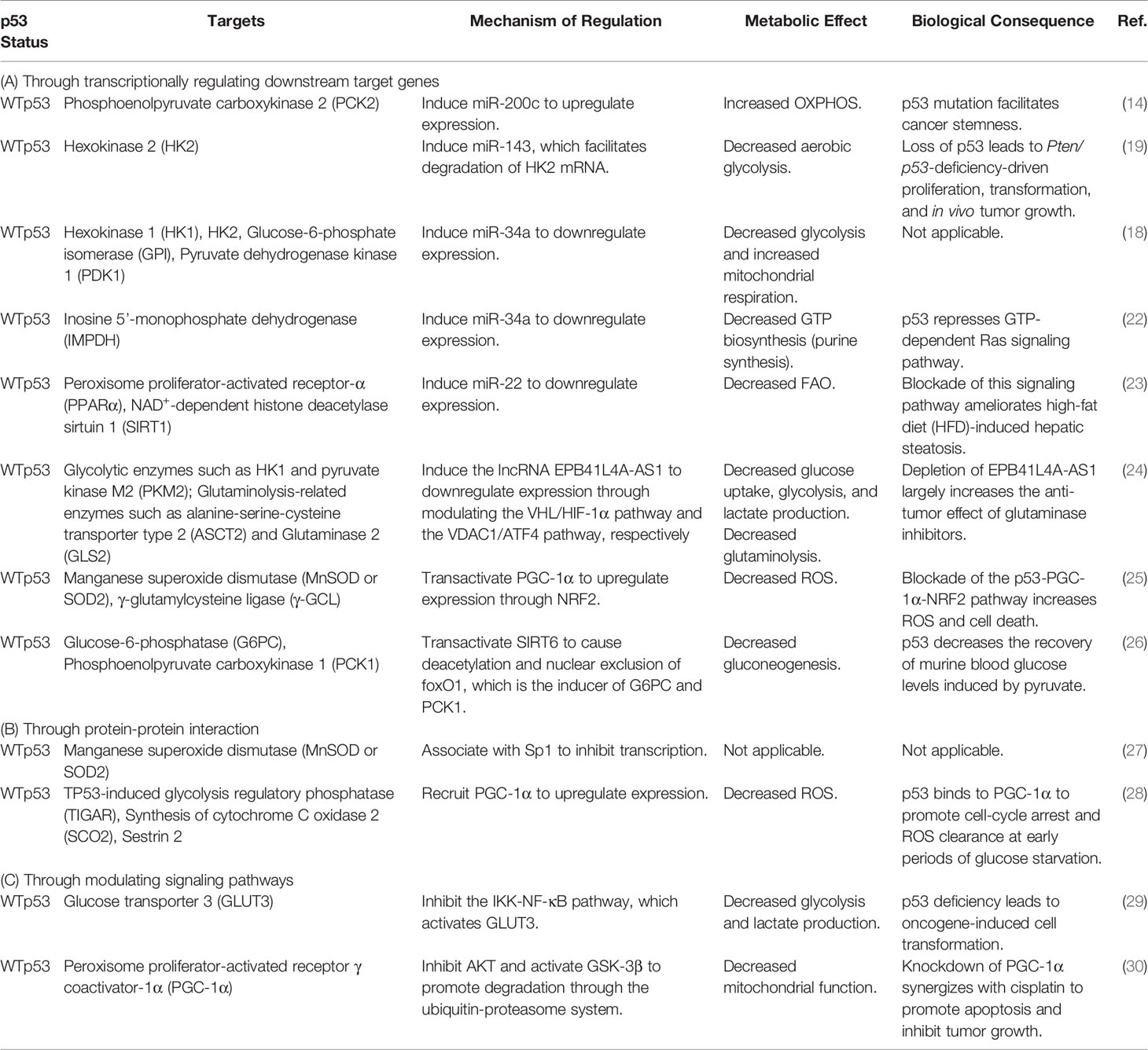
Table 1 Metabolic targets indirectly regulated by wild-type p53.
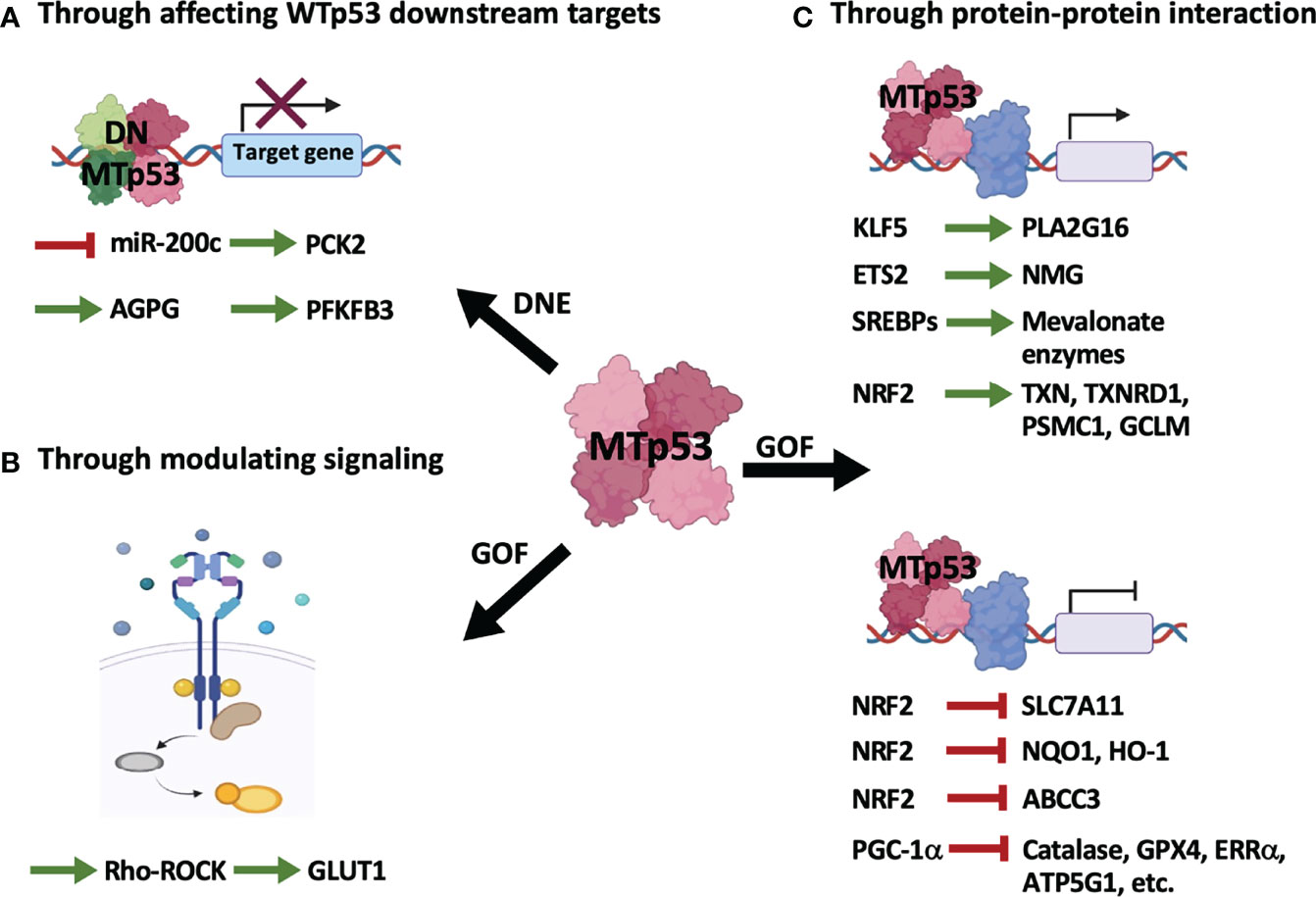
Figure 2 Mutant p53 regulates cellular metabolism through indirect mechanisms. MTp53 indirectly regulates metabolism by its dominant-negative effect (DNE) over WTp53 (A), and the gain-of-function (GOF) properties encompassing modulation of signaling pathways (B) or interaction with other transcription factors (C).
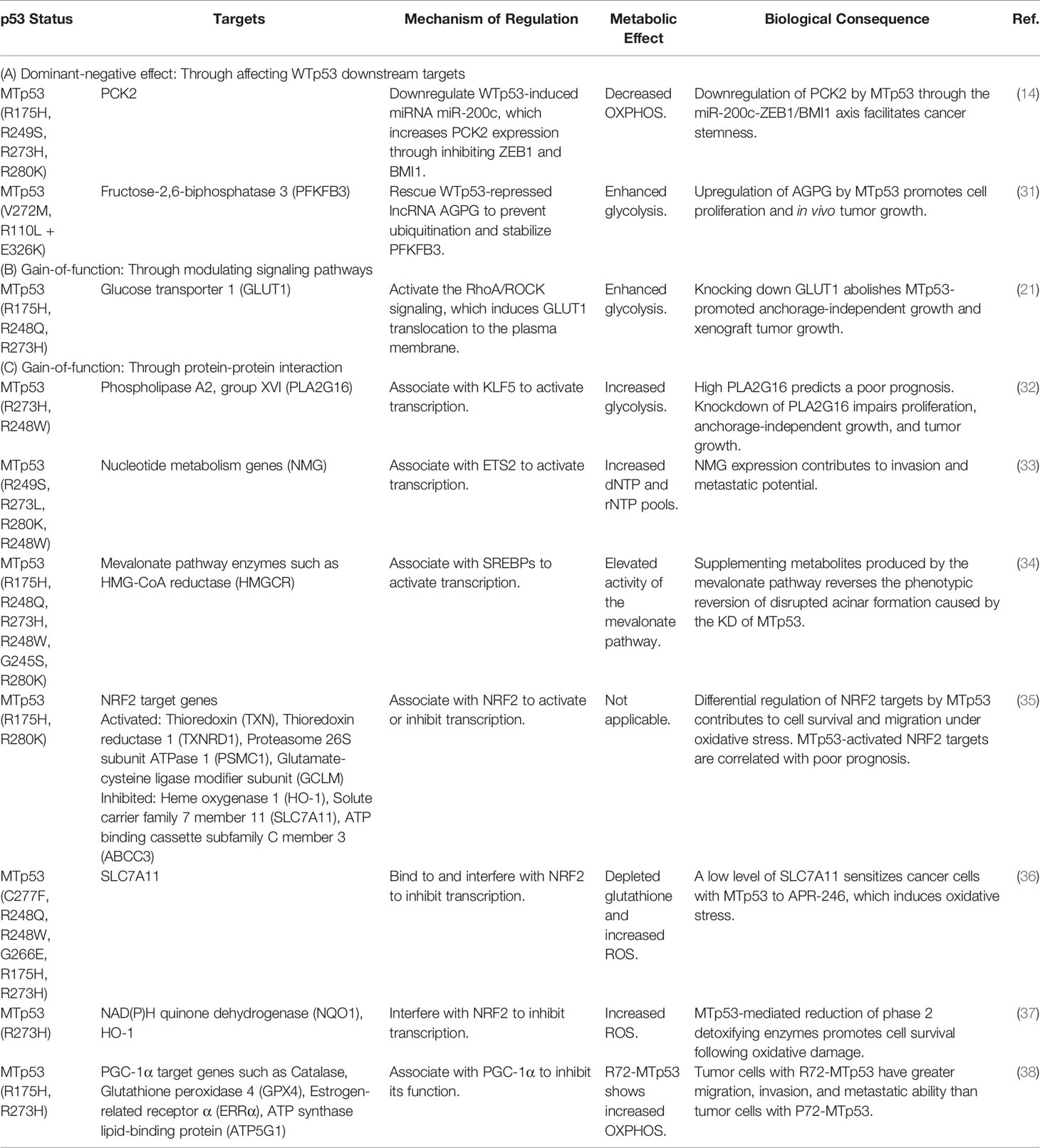
Table 2 Metabolic targets indirectly regulated by mutant p53.
Wild-Type p53-Mediated Indirect Regulation on Metabolism
As illustrated in Figure 1 and summarized in Table 1, mechanisms accounting for WTp53-mediated indirect regulation of metabolism can be categorized into three major types: through transcriptionally regulating downstream target genes (Figure 1A and Table 1A), through protein-protein interactions (Figure 1B and Table 1B), and through modulating signaling pathways (Figure 1C and Table 1C).
(A). WTp53 indirectly regulates the expression of metabolism-related genes by modulating its direct targets (Figure 1A and Table 1A). In our previous study (14), we demonstrate that miR-200c, a WTp53 target negatively regulating epithelial-mesenchymal transition (EMT) and stemness (39), promotes OXPHOS in basal-like breast cancer (BLBC) cells by inhibiting the downstream targets, ZEB1 and BMI1, which subsequently activates PCK2 expression. In prostate cancers, loss of p53 compromises p53-induced miR-143, facilitating degradation of HK2 mRNA, which is required for Pten/p53-deficiency-driven aerobic glycolysis, proliferation, transformation, and in vivo tumor growth (19). In another study, induction of miR-34a by p53 downregulates hexokinase 1 (HK1), HK2, glucose-6-phosphate isomerase (GPI), and pyruvate dehydrogenase kinase 1 (PDK1), potentiating mitochondrial respiration and decreasing glycolysis (18). In addition to regulating glucose metabolism, p53-induced miR-34a also represses the expression of inosine 5’-monophosphate dehydrogenase (IMPDH), the rate-limiting enzyme of GTP biosynthesis, leading to decreased Ras signaling (22). In the mouse model of diet-induced obesity (DIO)-related hepatic steatosis, activation of the hepatic cannabinoid-1 receptor (CB1R) induces expression of miR-22 through modulating the transcriptional activity of p53, which disturbs peroxisome proliferator-activated receptor-α (PPARα) and NAD+-dependent histone deacetylase sirtuin 1 (SIRT1), leading to decreased fatty acid oxidation (FAO) and increased fat accumulation in the liver (23). The lncRNA EPB41L4A-AS1, a transcriptional target of WTp53, decreases glycolysis and glutaminolysis via interacting with histone deacetylase 2 (HDAC2), in which interference of EPB41L4A-AS1 sensitizes tumor cells to glutaminase inhibitor (24). Aside from modulating non-coding RNAs, p53 transactivates peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), the master regulator of mitochondrial biogenesis and function (40), upon glutathione (GSH) depletion, which induces antioxidant response through nuclear factor E2-related factor 2 (NRF2)-mediated expression of manganese superoxide dismutase (MnSOD or SOD2) and γ-glutamylcysteine ligase (γ-GCL) (25). Furthermore, p53 inhibits the expression of the rate-limiting enzymes of gluconeogenesis, glucose-6-phosphatase (G6PC) and phosphoenolpyruvate carboxykinase 1 (PCK1), through transactivating NAD+-dependent histone deacetylase sirtuin 6 (SIRT6), which subsequently causes deacetylation and nuclear exclusion of forkhead box protein O1 (foxO1), the inducer of G6PC and PCK1 (26).
(B). WTp53 indirectly regulates metabolism through a protein-protein interaction with other transcription factors (Figure 1B and Table 1B). For example, p53 associates with the transcription factor, specificity protein 1 (Sp1), to repress transcription of MnSOD (27), an antioxidant enzyme differentially expressed in cancers (41). Interestingly, in addition to transcriptional control, p53 recruits PGC-1α to modulate its transactivation activity at early periods of metabolic stress, as indicated by upregulation of the proarrest and metabolic genes, including TP53-induced glycolysis regulatory phosphatase (TIGAR), synthesis of cytochrome C oxidase 2 (SCO2), and sestrin 2, which induces cell-cycle arrest and reactive oxygen species (ROS) clearance (28).
(C). WTp53 modulates signaling pathways to affect cellular metabolism (Figure 1C and Table 1C). An example of this is the study from Kawauchi et al. in which p53 deficiency in mouse embryonic fibroblasts (MEFs) leads to activation of the IKK-NF-κB pathway, which transactivates the Glut3 gene, and increases aerobic glycolysis and lactate production, contributing to oncogene-induced cell transformation (29). In contrast to transactivating or recruiting PGC-1α to serve as a coactivator, p53 destabilizes PGC-1α through activating the ubiquitin-proteasome system, mediated by AKT/GSK-3β-dependent phosphorylation of PGC-1α, which impairs mitochondrial function and increases chemosensitivity of non-small cell lung cancer (NSCLC) (30).
Mutant p53-Mediated Indirect Regulation on Metabolism
As illustrated in Figure 2 and summarized in Table 2, MTp53 indirectly regulates metabolism either by disturbing WTp53 function (Figure 2A and Table 2A) or by exerting its gain-of-function properties (Figures 2B, C and Tables 2B, C).
(A). MTp53-mediated metabolic reprogramming from perturbation of WTp53 downstream targets (Figure 2A and Table 2A) can be exemplified by our previous research that MTp53 attenuates OXPHOS through downregulating miR-200c, the positive regulator of PCK2, which facilitates cancer stemness in BLBC (14). In our study, the MTp53-exerted dominant-negative effect impedes the expression of miR-200c, which upregulates ZEB1 and BMI1 and subsequent downregulates PCK2. PCK2 deficiency not only leads to decreased OXPHOS in normal mammary epithelial cells but also compromises the increased OXPHOS by restoration of miR-200c in BLBC cells (14). Another example is a recent study that upregulation of the lncRNA Actin Gamma 1 Pseudogene (AGPG) by p53 deficiency leads to stabilization of fructose-2,6-biphosphatase 3 (PFKFB3), contributing to enhanced glycolysis, proliferation, and tumor growth (31).
(B). Independent of WTp53, the gain-of-function of MTp53 has a profound effect on various metabolic pathways. Similar to WTp53, MTp53 also regulates metabolism by affecting signaling pathways (Figure 2B and Table 2B). This is evidenced in a study that MTp53-boosted glycolysis promotes tumorigenesis through activation of the RhoA/ROCK signaling, subsequently inducing actin polymerization and translocation of GLUT1 to the plasma membrane (PM) (21).
(C). Distinct from WTp53-mediated indirect regulation on metabolism which is mainly mediated by its downstream target genes (Figure 1A and Table 1A), MTp53 gains novel abilities to interact with a variety of proteins, particularly transcription factors, to drive tumor-associated metabolic alterations (Figure 2C and Table 2C). Examples of metabolic genes activated by MTp53 encompass PLA2G16, encoding a phospholipase catalyzing the formation of free fatty acids and lysophospholipids. The association of MTp53 with Kruppel-like factor 5 (KLF5) transactivates PLA2G16, which leads to increased glycolytic rate and accelerates tumor growth (32). Besides, enhanced expression of nucleotide metabolism genes (NMG) can be induced by cooperation between MTp53 and ETS proto-oncogene 2 transcription factor (ETS2), which is associated with poor prognosis in breast cancer patients (33). Gain-of-function of MTp53 also confers its ability to interact with the sterol regulatory element binding proteins (SREBPs) and be recruited to the sterol regulatory elements (SRE-1), which then induces genes encoding enzymes in the mevalonate pathway, contributing to the disrupted acinar formation in breast cancer cells (34). Later research further demonstrates that MTp53 exerts a differential regulation on NRF2 targets regardless of whether cells are under unstressed or oxidative stress conditions, where expression of genes upregulated by MTp53, including thioredoxin (TXN), thioredoxin reductase 1 (TXNRD1), proteasome 26S subunit ATPase 1 (PSMC1), and glutamate-cysteine ligase modifier subunit (GCLM), correlates with worse overall survival in breast cancer patients (35).
On the other hand, accumulated MTp53 proteins interfere with NRF2 activity, resulting in decreased expression of SLC7A11, a cystine/glutamate antiporter. Downregulation of SLC7A11 leads to GSH depletion and increased ROS, which sensitizes cancer cells to APR-246, a MTp53 reactivator (36). In another study, MTp53 is found to protect cancer cells confronted with oxidative stress from death through diminishing NRF2-mediated phase 2 ROS detoxifying enzymes, NAD(P)H quinone dehydrogenase (NQO1) and heme oxygenase 1 (HO-1) (37). Like the aforementioned, MTp53 differentially regulates NRF2 targets, in which metabolic genes are suppressed by MTp53 including HO-1, SLC7A11, and ATP binding cassette subfamily C member 3 (ABCC3) (35). Surprisingly, not only WTp53 binds to PGC-1α, MTp53 also interacts with PGC-1α; though this association exerts an inhibitory effect on PGC-1α. Basu et al. demonstrate that the codon 72 polymorphism in TP53 impacts the binding and regulation of MTp53 to PGC-1α. The arginine 72 variant (R72) of MTp53, instead of the proline 72 variant (P72), shows decreased association with PGC-1α, resulting in increased PGC-1α function, enhanced OXPHOS, invasion, and metastasis (38).
Targeting Mutant p53: Recent Advances and Future Perspectives
Direct or Indirect?
Therapeutic strategies against tumors with p53 mutation are classified into direct and indirect approaches (42, 43). Direct targeting of MTp53 is attained by either restoring WTp53 function to MTp53 or depleting MTp53. Several MTp53 reactivators have been developed to refold MTp53 into the conformation of WTp53, prevent aggregation of MTp53 proteins, and restore DNA binding and transcriptional activity. In contrast, MTp53 destabilizers promote the degeneration of MTp53 and thus limit its expression, mainly through ubiquitin-proteasome degradation or autophagy-lysosome degradation pathways (44). Afterward, immunotherapies have emerged as promising strategies targeting tumors bearing MTp53 (45), including adoptive T cell therapy, the usage of antibody-drug conjugate (ADC) (46), and more recently, the development of the innovative bispecific antibody (BsAb) (47).
On the contrary, indirect approaches aim to disrupt connections between MTp53 and its synthetic lethal partners. Synthetic lethality is the concept that the simultaneous disturbance of two genes leads to cell death. In contrast, perturbation of an individual gene is tolerable, in which disruption of the two genes can be achieved by double mutations or single mutation of one gene combined with pharmacological inhibition of the other (48, 49). Due to the extensive influence of MTp53 on a variety of cellular processes and tumor development, synthetic lethality for cancer therapeutics against MTp53 is prospectively applicable to a wide range of cancers with TP53 mutation, and thus, the identification of synthetic lethal genes to MTp53 becomes attractive. The development of synthetic lethal strategies targeting MTp53 provides more opportunities and therapeutic options for cancer patients with TP53 mutation, which has advantages over approaches directly targeting MTp53, which are listed below.
(1). Synthetic lethality-based drugs are promising to broaden the strategies against cancers by their potential to overcome the major limitation of genetically targeted therapies. Not all cancer mutations are druggable, especially loss-of-function mutations (50, 51).
(2). It is quite challenging to develop MTp53 reactivators or inhibitors because of the diverse nature of MTp53 proteins. The structural diversity and the consequent distinct functional properties of every MTp53 make it practically infeasible to target all MTp53 with a single compound (42, 52). Synthetic lethal methods are more flexible because of the high dependency on MTp53-induced oncogenic effects rather than on MTp53 itself.
(3). MTp53 reactivators or inhibitors may induce severe toxicities and intolerable adverse effects. For example, MIRA-1, a MTp53 reactivator, could cause acute cytotoxicity to normal cells through induction of caspase-9-mediated apoptosis independent of p53 (53). APR-246 and COTI-2 are the only two drugs in clinical trials (42). Moreover, the relatively low-specificity inhibitors promoting degradation of MTp53, such as histone deacetylase inhibitors (HDACis), may elicit unfavorable adverse effects due to their widespread influence on normal cells (54).
(4). Restoration of WTp53 function to MTp53 might backfire because WTp53 has been paradoxically reported to favor tumor progression, particularly through metabolic regulation, which we highlight in this article. This notion is evident in a study that WTp53 potentiates glycolysis and decreases pyruvate uptake and thus OXPHOS in hepatocarcinoma (HCC) through inducing PUMA inhibition on mitochondrial pyruvate carrier (MPC) (55). Another study also substantiates that a low dosage of sulforaphane, the inducer of NRF2, could prevent apoptosis, promote proliferation, and enhance mitochondrial respiration in colorectal cancer (CRC) cells with WTp53 but not in p53-knockout CRC cells, suggesting a tumor-promoting role of WTp53 (56). Because of the potentially bi-faceted role of WTp53 in cancer, it is indispensable to ascertain the WTp53-mediated metabolic regulation and biological effects in different types of cancer.
Synthetic Lethality With Metabolism
Several candidate synthetic lethal genes to p53 involved in various cellular processes, such as DNA repair, cell-cycle control, cell growth and proliferation, and metabolism, have been identified (57). Among these, we suggest that synthetic lethality targeting MTp53 with metabolism is the most potentially feasible for clinical application for the reasons stated below.
(1). In comparison to targeting MTp53 with metabolism, synthetic lethality with other cellular processes like DNA damage response and cell-cycle arrest may not only kill tumor cells but also damage normal cells, which could lead to severe adverse effects. For example, UCN01, an inhibitor of checkpoint kinase 1 (Chk1), the critical regulator of intra-S and G2/M checkpoints (58), has been halted for further clinical development due to the worse efficacy and the induction of unacceptable toxicities (59). Similarly, BI 2536, an inhibitor of polo-like kinase 1 (Plk1), which plays a crucial role in mitosis (60), exhibited a poor response rate and short half-life and could induce severe adverse events, leading to discontinued clinical progress (61).
(2). The highly intertwined relationships among different biomolecular processes contribute to the formation of metabolic networks, which interact with each other to form the complex construct of metabolism (62). Perturbations of the connected molecules could lead to disconnection of these pathways and breakdown of the metabolic networks, consequently destroying the resilience, which could cause cellular dysfunction and diseases (62). For tumor cells displaying high metabolic plasticity, the above features make it a potential treatment strategy to disrupt the crosstalk between the interconnected metabolic pathways by dual inhibition of these pathways with metabolic drugs or targeting compensatory mechanisms with a combination of drugs inhibiting global regulators and the respective compensatory bioenergetic pathways (63). Dual targeting of metabolic pathways as a therapeutic strategy has been demonstrated in the case of Lewis lung carcinoma (LLA), in which 2-deoxy-D-glucose (2-DG), a non-metabolizable analog of glucose that competitively binds to hexokinase and inhibits glycolysis (64), significantly enhances the antitumor activity of dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase kinase (PDK) that activates pyruvate dehydrogenase (PDH) and boosts OXPHOS activity (65, 66), accompanied with inhibition of glycolysis and increased cytotoxicity of tumor-infiltrating monocytes (67). In the case of p53 mutated breast cancer cells, 2-DG increases the sensitivity of tumor cells to metformin, which indirectly activates AMP-activated protein kinase (AMPK) by inhibiting complex I of the mitochondrial electron transport chain (68), indicating co-treatment of 2-DG and metformin may be an effective therapeutic strategy (69). Alternatively, a combination of phenformin, an AMPK activator, and 2-DG induces metabolic stress, suppresses tumor growth, and promotes degradation of MTp53 proteins (70), foreshadowing the promising future of inhibiting compensatory mechanisms accounting for the metabolic flexibility of tumor cells.
(3). Nowadays, there have been various metabolic drugs approved for clinical use in cancers and other diseases, such as 5-fluorouracil (5-FU) targeting nucleotide metabolism and metformin inhibiting mitochondrial function. Besides, several metabolic inhibitors are currently in cancer clinical trials (71), which are prospectively applicable in cancer treatments in the imminent future. Uncovering the MTp53-induced metabolic alterations in cancers would assist the utilization of these metabolic drugs in combination therapies or adjuvant therapies.
Targeting p53-Mediated Indirect Regulation on Metabolism
In addition to targeting MTp53 with metabolism, mediators of MTp53-driven metabolic alterations are extremely potential synthetic lethal partners to MTp53. Specifically, the human genome comprises about 2% of protein-coding genes, leaving the vast majority to be non-coding RNAs (72). ncRNAs are critical regulators of many cellular processes, which indispensably coordinate the functional operation of complex networks in cells. Dysregulation of ncRNAs is detrimental, leading to pathological development like cancer (73). In viewing that p53 plays a pivotal role in regulating ncRNAs (12, 74), and that disturbance of ncRNAs contributes to cancer-associated metabolic reprogramming (75, 76), ncRNAs are expected to expand the synthetic lethal networks of MTp53 with metabolism. The examples below illustrate this point in detail.
As demonstrated in our previous work, MTp53 attenuates OXPHOS through disturbing the miR-200c-PCK2 axis, in which restoration of miR-200c rescues the decreased OXPHOS by MTp53 (14), implying therapeutic delivery of miR-200c through viral vectors or non-viral approaches, such as nanoparticles and liposomes (77), is a potential treatment strategy for intractable BLBC. It is noteworthy that restoring miR-200c expression not only boosts OXPHOS, but further nullifies MTp53-induced EMT and stemness, which may result from simultaneous activation of OXPHOS and direct inhibition of its downstream targets, ZEB1 and BMI1 (14). Likewise, miR-149-3p has been revealed to restore chemosensitivity of colorectal cancer cells through decreasing glycolytic activity via downregulating pyruvate dehydrogenase kinase 2 (PDK2), suggesting CRC patients with MTp53, in which the frequency of TP53 mutation is around 40-50%, might benefit from combined employment of chemotherapy and miR-149-3p-based therapy (78). In addition to miRNAs, targeting lncRNAs synthetic lethal to MTp53 is also a potential therapeutic intervention. In the case aforementioned in the section of MTp53-mediated indirect regulation on metabolism, the lncRNA AGPG is upregulated by MTp53, which enhances glycolysis through preventing PFKFB3 from degradation (31). Interference of AGPG shows significantly inhibitory effects on tumor cells, providing implications for developing an RNA interference-based strategy.
For the same reason, the concept of expanding the synthetic lethality to MTp53 through targeting its indirect regulation on metabolism can apply to other mechanisms, like the signaling pathways modulated by MTp53 (Figure 2B and Table 2B), or the transcription factors associated with MTp53 (Figure 2C and Table 2C). Furthermore, in addition to metabolic drugs, numerous small-molecule targeted drugs and therapeutic monoclonal antibodies are FDA-approved for cancer treatment (79–81). Consequently, further investigation of molecular mechanisms accounting for MTp53-mediated metabolic reprogramming offers opportunities for patients with MTp53 tumors, in which these targeted drugs might be available in medical treatments.
Together, this evidence unequivocally indicates that targeting MTp53-mediated indirect regulation rather than directly targeting MTp53-driven metabolic alteration may elicit more powerful tumoricidal effects.
Challenges on the Journey
Despite the accumulated myriad of research on p53 during the past four decades, a thorough understanding of the p53 regulatory network of cellular metabolism is still lacking. The progress of targeting MTp53 according to its regulation on cancer metabolism in practical clinical approaches remains stagnant, which could arise from the complexity of p53 and research limitations as following described.
(1). p53 regulation of metabolism is highly cell type-specific. A growing body of evidence has revealed that it is inappropriate to apply the general assumption of p53-regulated metabolism to all the types of cells, which is firmly supported by Kim et al. that WTp53 promotes glycolysis instead of OXPHOS in HCC (55). Furthermore, p53 regulates metabolism distinctly under stressed or unstressed conditions. For instance, p53 is demonstrated to increase OXPHOS in normoxia while decreasing OXPHOS in hypoxia in the cervix and breast cancer cells (82). As a result, the regulatory networks of p53 on cellular metabolism in different cells need to be more clearly defined.
(2). The exceedingly diverse nature of MTp53 proteins makes it a hurdle to precisely delineate MTp53-regulated metabolism in cancer cells. As shown in our previous study (14), stable expression of different MTp53 variants in normal mammary epithelial cells promotes glycolysis and suppresses mitochondrial respiration to varying extents. Erikson et al. also demonstrate that different hotspot mutations of p53 could have differential impacts on cellular metabolism like glycolysis and that endogenous and exogenous expression of even the same type of p53 mutation could exert opposite effects on OXPHOS (83).
(3). Murine cells and mouse models are frequently utilized in research exploring p53-regulated metabolism and the underlying molecular mechanisms; nevertheless, substantial differences in gene expression patterns and transcriptional regulatory programs have been observed between mice and humans (84). A more recent study corroborates that the p53 gene regulatory network (GRN) in mice differs from that in humans, in which the meta-analysis reveals extensive variation in p53-regulated gene expression profiles and high species-specificity of p53 transcriptional targets (85). This distinction can be exemplified by CPT1C, the brain isoform of the rate-limiting enzyme of FAO. CPT1C is generalized as a direct target of p53 (16, 86, 87); though, CPT1C is upregulated by p53 in mice but not in humans, reflecting the lack of p53 binding sites on human CPT1C promoter (85). In response to the differences in p53 GRN between mice and humans, more efforts are in need to confirm the metabolic roles of p53.
(4). Despite synthetic lethality targeting the mediators of MTp53-associated cancer metabolism portends a bright future for innumerable cancer patients with TP53 mutation, the diverse nature of MTp53, the intracellular intricate regulatory networks accounting for the modulation of metabolism, and the extrinsic environmental factors determine together with the specific synthetic lethal partners to a specific type of MTp53, making identifying the synthetic lethal partners a quite challenging work. Once a synthetic lethal partner has been identified, the following concern is whether there are drugs specific for this synthetic lethal partner, or whether this synthetic lethal partner could be delivered to tumor sites or transported into tumor cells. The challenges of research on p53-mediated indirect regulation of metabolism are summarized below.
(a). The mechanism accounting for MTp53 indirect regulation of metabolism by dominant-negative effect over WTp53 (Figure 2A and Table 2A) may conform to multiple forms of MTp53, whereas the mechanisms underlying MTp53 indirect regulation of metabolism by gain-of-function (Figures 2B, C and Tables 2B, C) may not. Different types of MTp53 proteins might have distinct synthetic lethal partners; therefore, more efforts are required to identify the particular synthetic lethal relationship between the mediators involved in MTp53-regulated cancer metabolism and the specific types of MTp53.
(b). The synthetic lethal partner in a cell type could cause distinct, even opposite effects in another. PGC-1α illustrates this point clearly. In ERBB2+ breast cancers, high expression of PGC-1α predicts poorer prognosis and is correlated with high expression of glutamine cluster, reflecting the increased expression of PGC-1α in ERBB2+ breast cancer cells potentiates glutamine metabolism and facilitates proliferation under low glucose and hypoxia (88). On the contrary, elevated expression of PGC-1α leads to increased FAO and TCA cycle, decreased glycolysis, and suppression of tumor growth and metastasis in prostate cancers (89).
(c). There might be no targeted drug for a specific synthetic lethal partner, or it is tricky to develop it. For example, to our knowledge, there is no specific inhibitor for PGC-1α currently, which is activated by decreased binding to R72-MTp53, contributing to migration, invasion, and metastasis (38).
(d). Although successes in miRNA-based anti-cancer therapy have been reported (90), such as the significantly reduced tumor burden and improved survival rate in Kras;Trp53 mutant NSCLC mice administered combinatorial treatment of miR-34a and let-7b using NOV340 liposomal nanoparticles (91), there are still challenges remained to be overcome in the clinical development of miRNA therapeutics. Maintaining stability and integrity of miRNA mimics or antagonists in the circulation, determining suitable dosages, delivering systems, and administration routes, increasing specificity and efficient penetration into tumors, as well as minimizing immunotoxicity and off-target effects all are issues needed to be concerned (90, 92).
In addition to the existing limitations to research on p53 regulatory networks of cellular metabolism, the links between p53-mediated cellular metabolism and p53-induced biological functions remain largely unknown since few studies have comprehensively deciphered the contribution of p53-regulated metabolic phenotype to tumor progression or tumor suppression. Deregulated metabolism has been designated as an emerging hallmark of cancer in the last decade (93), which implies uncovering the biological effects of metabolic alterations induced by p53 and the underlying molecular mechanisms is of great importance for designing the optimized treatment strategies. Like those mentioned above, our previous study reveals that MTp53 facilitates cancer stemness through attenuating OXPHOS by disturbance of the miR-200c-PCK2 axis (14), which fully addresses the molecular mechanism accounting for the connection between MTp53-induced metabolic alteration and traits of cancer. Notably, restoring miR-200c expression either in normal mammary epithelial cells overexpressing p53 mutants or in BLBC cells harboring endogenous MTp53 not only counteracts MTp53-induced EMT and stemness, but also recovers the decreased OXPHOS activity, foretelling the promising future of treating cancers with p53 mutation by modulating the mediators of metabolic reprogramming indirectly driven by MTp53. Moreover, metabostemness, describing cellular metabotypes as the driver to redirect normal cancer cells to less-differentiated cancer stem cell (CSC) states (94), interprets the significance of connecting metabolic features to traits of cancer cells, which opens a brand new way to cure cancer patients by circumventing resistance to therapies, metastasis, and tumor recurrence caused by CSCs (95) based on their metabolic dependencies.
Summary
Overall, in addition to activating or repressing target genes transcriptionally, p53 regulates metabolism-related gene expression through indirect mechanisms as well, either by regulating direct transcriptional targets, associating with other transcription factors, or modulating signaling pathways. The notion that perturbations of p53 regulatory networks of cellular metabolism are in relation to tumor initiation and progression accentuates the crucial role of p53 in shaping cancer-associated metabolic phenotypes. Besides, TP53, as the most frequently mutated tumor suppressor gene in cancers, renders MTp53 promising for treatments of p53 mutated tumors. The diversity of MTp53 proteins and the unfavorable non-specific toxicities of MTp53 reactivators or inhibitors to normal tissues not only make it challenging to develop drugs directly targeting MTp53, but also impede the entrance of these drugs into clinical trials. Focusing on the synthetic lethal partners to MTp53 with metabolism, especially targeting the mediators involved in MTp53-driven metabolic reprogramming, might help broaden the synthetic lethal networks of MTp53. This would provide more opportunities and treatment options for cancer patients with MTp53, avoid or alleviate the off-target effects or severe adverse events, and forge the clinical application due to the existence of various metabolic and targeted drugs with FDA approval. Regardless of the substantial literature on p53 and its regulation of cellular metabolism, the metabolic roles of p53 and the mechanistic relationships between p53 and p53-mediated metabotypes are still ambiguous. Several limitations should be taken into consideration when we devote ourselves to investigating the metabolic networks of p53: (1) cell type-specificity of p53; (2) the highly diverse nature of MTp53; (3) differences in p53 GRN between mice and humans. Furthermore, challenges of research concentrating on p53-mediated indirect regulation on metabolism, include: (1) different MTp53 proteins might have distinct synthetic lethal partners; (2) synthetic lethal partner-exerted effects might be cell type-specific; (3) there might be no targeted drugs for a specific synthetic lethal partner; and (4) difficulties in the clinical development of ncRNA-based therapies, should be considered and kept in mind. Further research is also desired to unveil the biological effects of p53-associated metabolic activities, which is particularly essential for elucidating the contribution of p53-induced metabolic changes to the onset and malignancy of cancers, providing important implications for the development of prevention and treatment strategies.
Author Contributions
C-YW and C-HC wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported in part by the following: Ministry of Science and Technology (109-2628-B-009-004 to C-HC). The “Smart Platform of Dynamic Systems Biology for Therapeutic Development” and “Center for Intelligent Drug Systems and Smart Bio-devices (IDS2B) from The Featured Areas Research Center Program” within the framework of the Higher Education Sprout Project of the Ministry of Education (MOE) in Taiwan.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Boutelle AM, Attardi LD. P53 and Tumor Suppression: It Takes a Network. Trends Cell Biol (2021) 31(4):298–310. doi: 10.1016/j.tcb.2020.12.011
2. Levine AJ. P53: 800 Million Years of Evolution and 40 Years of Discovery. Nat Rev Cancer (2020) 20(8):471–80. doi: 10.1038/s41568-020-0262-1
3. Joerger AC, Fersht AR. The P53 Pathway: Origins, Inactivation in Cancer, and Emerging Therapeutic Approaches. Annu Rev Biochem (2016) 85:375–404. doi: 10.1146/annurev-biochem-060815-014710
4. Leroy B, Anderson M, Soussi T. TP53 Mutations in Human Cancer: Database Reassessment and Prospects for the Next Decade. Hum Mutat (2014) 35(6):672–88. doi: 10.1002/humu.22552
5. Brosh R, Rotter V. When Mutants Gain New Powers: News From the Mutant P53 Field. Nat Rev Cancer (2009) 9(10):701–13. doi: 10.1038/nrc2693
6. Baugh EH, Ke H, Levine AJ, Bonneau RA, Chan CS. Why Are There Hotspot Mutations in the TP53 Gene in Human Cancers? Cell Death Differ (2018) 25(1):154–60. doi: 10.1038/cdd.2017.180
7. Weisz L, Oren M, Rotter V. Transcription Regulation by Mutant P53. Oncogene (2007) 26(15):2202–11. doi: 10.1038/sj.onc.1210294
8. Soussi T. P53 Alterations in Human Cancer: More Questions Than Answers. Oncogene (2007) 26(15):2145–56. doi: 10.1038/sj.onc.1210280
9. Kim E, Deppert W. Interactions of Mutant P53 With DNA: Guilt by Association. Oncogene (2007) 26(15):2185–90. doi: 10.1038/sj.onc.1210312
10. Alvarado-Ortiz E, de la Cruz-López K, Becerril-Rico J, Sarabia-Sánchez M, Ortiz-Sánchez E, García-Carrancá A. Mutant P53 Gain-Of-Function: Role in Cancer Development, Progression, and Therapeutic Approaches. Front Cell Dev Biol (2020) 8:607670–0. doi: 10.3389/fcell.2020.607670
11. Pfister NT, Fomin V, Regunath K, Zhou JY, Zhou W, Silwal-Pandit L, et al. Mutant P53 Cooperates With the SWI/SNF Chromatin Remodeling Complex to Regulate VEGFR2 in Breast Cancer Cells. Genes Dev (2015) 29(12):1298–315. doi: 10.1101/gad.263202.115
12. Di Agostino S. The Impact of Mutant P53 in the Non-Coding RNA World. Biomolecules (2020) 10(3):472. doi: 10.3390/biom10030472
13. Vousden KH, Ryan KM. P53 and Metabolism. Nat Rev Cancer (2009) 9(10):691–700. doi: 10.1038/nrc2715
14. Chao C-H, Wang C-Y, Wang C-H, Chen T-W, Hsu H-Y, Huang H-W, et al. Mutant P53 Attenuates Oxidative Phosphorylation and Facilitates Cancer Stemness Through Downregulating miR-200c–PCK2 Axis in Basal-Like Breast Cancer. Mol Cancer Res (2021) 19(11):1900–16. doi: 10.1158/1541-7786.MCR-21-0098
15. Yu S, Meng S, Xiang M, Ma H. Phosphoenolpyruvate Carboxykinase in Cell Metabolism: Roles and Mechanisms Beyond Gluconeogenesis. Mol Metab (2021) 53:101257–7. doi: 10.1016/j.molmet.2021.101257
16. Lacroix M, Riscal R, Arena G, Linares LK, Le Cam L. Metabolic Functions of the Tumor Suppressor P53: Implications in Normal Physiology, Metabolic Disorders, and Cancer. Mol Metab (2020) 33:2–22. doi: 10.1016/j.molmet.2019.10.002
17. Mathupala SP, Heese C, Pedersen PL. Glucose Catabolism in Cancer Cells: The Type II Hexokinase Promoter Contains Functionally Active Response Elements for the Tumor Suppressor P53. J Biol Chem (1997) 272(36):22776–80. doi: 10.1074/jbc.272.36.22776
18. Kim H-R, Roe J-S, Lee J-E, Cho E-J, Youn H-D. P53 Regulates Glucose Metabolism by miR-34a. Biochem Biophysl Res Commun (2013) 437(2):225–31. doi: 10.1016/j.bbrc.2013.06.043
19. Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, et al. Hexokinase 2-Mediated Warburg Effect Is Required for PTEN-And P53-Deficiency-Driven Prostate Cancer Growth. Cell Rep (2014) 8(5):1461–74. doi: 10.1016/j.celrep.2014.07.053
20. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The Tumor Suppressor P53 Down-Regulates Glucose Transporters GLUT1 and GLUT4 Gene Expression. Cancer Res (2004) 64(7):2627–33. doi: 10.1158/0008-5472.CAN-03-0846
21. Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-Associated Mutant P53 Drives the Warburg Effect. Nat Commun (2013) 4(1):1–15. doi: 10.1038/ncomms3935
22. Kim H-R, Roe J-S, Lee J-E, Hwang I-Y, Cho E-J, Youn H-D. A P53-Inducible microRNA-34a Downregulates Ras Signaling by Targeting IMPDH. Biochem Biophys Res Commun (2012) 418(4):682–8. doi: 10.1016/j.bbrc.2012.01.077
23. Azar S, Udi S, Drori A, Hadar R, Nemirovski A, Vemuri KV, et al. Reversal of Diet-Induced Hepatic Steatosis by Peripheral CB1 Receptor Blockade in Mice Is P53/miRNA-22/Sirt1/Pparα Dependent. Mol Metab (2020) 42:101087–7. doi: 10.1016/j.molmet.2020.101087
24. Liao M, Liao W, Xu N, Li B, Liu F, Zhang S, et al. LncRNA EPB41L4A-AS1 Regulates Glycolysis and Glutaminolysis by Mediating Nucleolar Translocation of HDAC2. EBioMedicine (2019) 41:200–13. doi: 10.1016/j.ebiom.2019.01.035
25. Aquilano K, Baldelli S, Pagliei B, Cannata SM, Rotilio G, Ciriolo MR. P53 Orchestrates the PGC-1α-Mediated Antioxidant Response Upon Mild Redox and Metabolic Imbalance. Antioxid Redox Signal (2013) 18(4):386–99. doi: 10.1089/ars.2012.4615
26. Zhang P, Tu B, Wang H, Cao Z, Tang M, Zhang C, et al. Tumor Suppressor P53 Cooperates With SIRT6 to Regulate Gluconeogenesis by Promoting FoxO1 Nuclear Exclusion. Proc Natl Acad Sci (2014) 111(29):10684–9. doi: 10.1073/pnas.1411026111
27. Dhar SK, Xu Y, Chen Y, Clair DKS. Specificity Protein 1-Dependent P53-Mediated Suppression of Human Manganese Superoxide Dismutase Gene Expression. J Biol Chem (2006) 281(31):21698–709. doi: 10.1074/jbc.M601083200
28. Sen N, Satija YK, Das S. PGC-1α, a Key Modulator of P53, Promotes Cell Survival Upon Metabolic Stress. Mol Cell (2011) 44(4):621–34. doi: 10.1016/j.molcel.2011.08.044
29. Kawauchi K, Araki K, Tobiume K, Tanaka N. P53 Regulates Glucose Metabolism Through an IKK-NF-κB Pathway and Inhibits Cell Transformation. Nat Cell Biol (2008) 10(5):611–8. doi: 10.1038/ncb1724
30. Deng X, Li Y, Gu S, Chen Y, Yu B, Su J, et al. P53 Affects PGC-1α Stability Through AKT/GSK-3β to Enhance Cisplatin Sensitivity in Non-Small Cell Lung Cancer. Front Oncol (2020) 10:1252–2. doi: 10.3389/fonc.2020.01252
31. Liu J, Liu Z-X, Wu Q-N, Lu Y-X, Wong C-W, Miao L, et al. Long Noncoding RNA AGPG Regulates PFKFB3-Mediated Tumor Glycolytic Reprogramming. Nat Commun (2020) 11(1):1–16. doi: 10.1038/s41467-020-15112-3
32. Xia W, Bai H, Deng Y, Yang Y. PLA2G16 Is a Mutant P53/KLF5 Transcriptional Target and Promotes Glycolysis of Pancreatic Cancer. J Cell Mol Med (2020) 24(21):12642–55. doi: 10.1111/jcmm.15832
33. Kollareddy M, Dimitrova E, Vallabhaneni KC, Chan A, Le T, Chauhan KM, et al. Regulation of Nucleotide Metabolism by Mutant P53 Contributes to Its Gain-Of-Function Activities. Nat Commun (2015) 6(1):1–13. doi: 10.1038/ncomms8389
34. Moon RR-B, Barsotti A, Chicas A, Li W, Polotskaia10 A, Bissell11 MJ, et al. Mutant P53 Disrupts Mammary Acinar Morphogenesis Via the Mevalonate Pathway. Cell (2012) 148(1-2):244–58. doi: 10.1016/j.cell.2011.12.017
35. Lisek K, Campaner E, Ciani Y, Walerych D, Del Sal G. Mutant P53 Tunes the NRF2-Dependent Antioxidant Response to Support Survival of Cancer Cells. Oncotarget (2018) 9(29):20508–23. doi: 10.18632/oncotarget.24974
36. Liu DS, Duong CP, Haupt S, Montgomery KG, House CM, Azar WJ, et al. Inhibiting the System Xc–/Glutathione Axis Selectively Targets Cancers With Mutant-P53 Accumulation. Nat Commun (2017) 8(1):1–14. doi: 10.1038/ncomms14844
37. Kalo E, Kogan-Sakin I, Solomon H, Bar-Nathan E, Shay M, Shetzer Y, et al. Mutant P53R273H Attenuates the Expression of Phase 2 Detoxifying Enzymes and Promotes the Survival of Cells With High Levels of Reactive Oxygen Species. J Cell Sci (2012) 125(22):5578–86. doi: 10.1242/jcs.106815
38. Basu S, Gnanapradeepan K, Barnoud T, Kung C-P, Tavecchio M, Scott J, et al. Mutant P53 Controls Tumor Metabolism and Metastasis by Regulating PGC-1α. Genes Dev (2018) 32(3-4):230–43. doi: 10.1101/gad.309062.117
39. Chang C-J, Chao C-H, Xia W, Yang J-Y, Xiong Y, Li C-W, et al. P53 Regulates Epithelial–Mesenchymal Transition and Stem Cell Properties Through Modulating miRNAs. Nat Cell Biol (2011) 13(3):317–23. doi: 10.1038/ncb2173
40. Jeninga EH, Schoonjans K, Auwerx J. Reversible Acetylation of PGC-1: Connecting Energy Sensors and Effectors to Guarantee Metabolic Flexibility. Oncogene (2010) 29(33):4617–24. doi: 10.1038/onc.2010.206
41. Dhar SK, Clair DKS. Manganese Superoxide Dismutase Regulation and Cancer. Free Radic Biol Med (2012) 52(11-12):2209–22. doi: 10.1016/j.freeradbiomed.2012.03.009
42. Hu J, Cao J, Topatana W, Juengpanich S, Li S, Zhang B, et al. Targeting Mutant P53 for Cancer Therapy: Direct and Indirect Strategies. J Hematol Oncol (2021) 14(1):1–19. doi: 10.1186/s13045-021-01169-0
43. Zhang C, Liu J, Xu D, Zhang T, Hu W, Feng Z. Gain-Of-Function Mutant P53 in Cancer Progression and Therapy. J Mol Cell Biol (2020) 12(9):674–87. doi: 10.1093/jmcb/mjaa040
44. Xu Z, Wu W, Yan H, Hu Y, He Q, Luo P. Regulation of P53 Stability as a Therapeutic Strategy for Cancer. Biochem Pharmacol (2021) 185:114407–04407. doi: 10.1016/j.bcp.2021.114407
45. Chasov V, Zaripov M, Mirgayazova R, Khadiullina R, Zmievskaya E, Ganeeva I, et al. Promising New Tools for Targeting P53 Mutant Cancers: Humoral and Cell-Based Immunotherapies. Front Immunol (2021) 12:707734–4. doi: 10.3389/fimmu.2021.707734
46. Low L, Goh A, Koh J, Lim S, Wang C-I. Targeting Mutant P53-Expressing Tumours With a T Cell Receptor-Like Antibody Specific for a Wild-Type Antigen. Nat Commun (2019) 10(1):1–14. doi: 10.1038/s41467-019-13305-z
47. Yang C, Lou G, Jin W. The Arsenal of TP53 Mutants Therapies: Neoantigens and Bispecific Antibodies. Signal Transduct Target Ther (2021) 6(1):1–2. doi: 10.1038/s41392-021-00635-y
48. O'Neil NJ, Bailey ML, Hieter P. Synthetic Lethality and Cancer. Nat Rev Genet (2017) 18(10):613–23. doi: 10.1038/nrg.2017.47
49. Topatana W, Juengpanich S, Li S, Cao J, Hu J, Lee J, et al. Advances in Synthetic Lethality for Cancer Therapy: Cellular Mechanism and Clinical Translation. J Hematol Oncol (2020) 13(1):1–22. doi: 10.1186/s13045-020-00956-5
50. Huang A, Garraway LA, Ashworth A, Weber B. Synthetic Lethality as an Engine for Cancer Drug Target Discovery. Nat Rev Drug Discovery (2020) 19(1):23–38. doi: 10.1038/s41573-019-0046-z
51. Li S, Topatana W, Juengpanich S, Cao J, Hu J, Zhang B, et al. Development of Synthetic Lethality in Cancer: Molecular and Cellular Classification. Signal Transduct Target Ther (2020) 5(1):1–14. doi: 10.1038/s41392-020-00358-6
52. Sabapathy K, Lane DP. Therapeutic Targeting of P53: All Mutants Are Equal, But Some Mutants Are More Equal Than Others. Nat Rev Clin Oncol (2018) 15(1):13–30. doi: 10.1038/nrclinonc.2017.151
53. Bou-Hanna C, Jarry A, Lode L, Schmitz I, Schulze-Osthoff K, Kury S, et al. Acute Cytotoxicity of MIRA-1/NSC19630, a Mutant P53-Reactivating Small Molecule, Against Human Normal and Cancer Cells Via a Caspase-9-Dependent Apoptosis. Cancer Lett (2015) 359(2):211–7. doi: 10.1016/j.canlet.2015.01.014
54. Borrero LJH, El-Deiry WS. Tumor Suppressor P53: Biology, Signaling Pathways, and Therapeutic Targeting. Biochim Biophys Acta Rev Cancer (2021) 1876(1):188556. doi: 10.1016/j.bbcan.2021.188556
55. Kim J, Yu L, Chen W, Xu Y, Wu M, Todorova D, et al. Wild-Type P53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell (2019) 35(2):191–203. doi: 10.1016/j.ccell.2018.12.012
56. Gwon Y, Oh J, Kim J-S. Sulforaphane Induces Colorectal Cancer Cell Proliferation Through Nrf2 Activation in a P53-Dependent Manner. Appl Biol Chem (2020) 63(1):1–11. doi: 10.1186/s13765-020-00578-y
57. Wang X, Simon R. Identification of Potential Synthetic Lethal Genes to P53 Using a Computational Biology Approach. BMC Med Genomics (2013) 6(1):1–10. doi: 10.1186/1755-8794-6-30
58. Neizer-Ashun F, Bhattacharya R. Reality CHEK: Understanding the Biology and Clinical Potential of CHK1. Cancer Lett (2021) 497:202–11. doi: 10.1016/j.canlet.2020.09.016
59. Morandell S, Yaffe MB. Exploiting Synthetic Lethal Interactions Between DNA Damage Signaling, Checkpoint Control, and P53 for Targeted Cancer Therapy. Prog Mol Biol Transl Sci (2012) 110:289–314. doi: 10.1016/B978-0-12-387665-2.00011-0
60. Lee S-Y, Jang C, Lee K-A. Polo-Like Kinases (Plks), a Key Regulator of Cell Cycle and New Potential Target for Cancer Therapy. Dev Reprod (2014) 18(1):65–71. doi: 10.12717/DR.2014.18.1.065
61. Vose JM, Friedberg JW, Waller EK, Cheson BD, Juvvigunta V, Fritsch H, et al. The Plk1 Inhibitor BI 2536 in Patients With Refractory or Relapsed Non-Hodgkin Lymphoma: A Phase I, Open-Label, Single Dose-Escalation Study. Leuk Lymphoma (2013) 54(4):708–13. doi: 10.3109/10428194.2012.729833
62. Gómez-Romero L, López-Reyes K, Hernández-Lemus E. The Large Scale Structure of Human Metabolism Reveals Resilience Via Extensive Signaling Crosstalk. Front Physiol (2020) 11:588012–2. doi: 10.3389/fphys.2020.588012
63. McGuirk S, Audet-Delage Y, St-Pierre J. Metabolic Fitness and Plasticity in Cancer Progression. Trends Cancer (2020) 6(1):49–61. doi: 10.1016/j.trecan.2019.11.009
64. Bere J, Jonnalagadda ND, Kappari L, Karangula J, Boggula N, Kappari V. 2-Deoxy-D-Glucose: An Update Review. J Innov Dev Pharm Tech Sci(JIDPTS) (2021) 4(5):68–78.
65. Michelakis E, Webster L, Mackey J. Dichloroacetate (DCA) as a Potential Metabolic-Targeting Therapy for Cancer. Br J Cancer (2008) 99(7):989–94. doi: 10.1038/sj.bjc.6604554
66. Tataranni T, Piccoli C. Dichloroacetate (DCA) and Cancer: An Overview Towards Clinical Applications. Oxid Med Cell Longev (2019) 2019:8201079–8201079. doi: 10.1155/2019/8201079
67. Pyaskovskaya O, Kolesnik D, Fedorchuk A, Prochorova I, Solyanik G. 2-Deoxy-D-Glucose Enhances Dichloroacetate Antitumor Action Against Lewis Lung Carcinoma. Exp Oncol (2016) 38(3):176–80. doi: 10.31768/2312-8852.2016.38(3):176-180
68. Pernicova I, Korbonits M. Metformin—Mode of Action and Clinical Implications for Diabetes and Cancer. Nat Rev Endocrinol (2014) 10(3):143–56. doi: 10.1038/nrendo.2013.256
69. Rajh M, Dolinar K, Miš K, Pavlin M, Pirkmajer S. Medium Renewal Blocks Anti-Proliferative Effects of Metformin in Cultured MDA-MB-231 Breast Cancer Cells. PloS One (2016) 11(5):e0154747–e0154747. doi: 10.1371/journal.pone.0154747
70. Jung CL, Mun H, Jo S-Y, Oh J-H, Lee C, Choi E-K, et al. Suppression of Gain-Of-Function Mutant P53 With Metabolic Inhibitors Reduces Tumor Growth In Vivo. Oncotarget (2016) 7(47):77664–682. doi: 10.18632/oncotarget.12758
71. Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting Cancer Metabolism in the Era of Precision Oncology. Nat Rev Drug Discovery (2022) 21(2):141–62. doi: 10.1038/s41573-021-00339-6
72. Wright MW, Bruford EA. Naming'junk': Human Non-Protein Coding RNA (ncRNA) Gene Nomenclature. Hum Genomics (2011) 5(2):1–9. doi: 10.1186/1479-7364-5-2-90
73. Anastasiadou E, Jacob LS, Slack FJ. Non-Coding RNA Networks in Cancer. Nat Rev Cancer (2018) 18(1):5–18. doi: 10.1038/nrc.2017.99
74. Chen S, Thorne RF, Zhang XD, Wu M, Liu L. Non-Coding RNAs, Guardians of the P53 Galaxy. Semin Cancer Biol (2021) 75:72–83. doi: 10.1016/j.semcancer.2020.09.002
75. Beltrán-Anaya FO, Cedro-Tanda A, Hidalgo-Miranda A, Romero-Cordoba SL. Insights Into the Regulatory Role of Non-Coding RNAs in Cancer Metabolism. Front Physiol (2016) 7:342–2. doi: 10.3389/fphys.2016.00342
76. Lin X, Wu Z, Hu H, Luo M-L, Song E. Non-Coding RNAs Rewire Cancer Metabolism Networks. Semin Cancer Biol (2021) 75:116–26. doi: 10.1016/j.semcancer.2020.12.019
77. Forterre A, Komuro H, Aminova S, Harada M. A Comprehensive Review of Cancer MicroRNA Therapeutic Delivery Strategies. Cancers (2020) 12(7):1852. doi: 10.3390/cancers12071852
78. Liang Y, Hou L, Li L, Li L, Zhu L, Wang Y, et al. Dichloroacetate Restores Colorectal Cancer Chemosensitivity Through the P53/Mir-149-3p/PDK2-Mediated Glucose Metabolic Pathway. Oncogene (2020) 39(2):469–85. doi: 10.1038/s41388-019-1035-8
79. Zhong L, Li Y, Xiong L, Wang W, Wu M, Yuan T, et al. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Signal Transduct Target Ther (2021) 6(1):1–48. doi: 10.1038/s41392-021-00572-w
80. Lu R-M, Hwang Y-C, Liu I-J, Lee C-C, Tsai H-Z, Li H-J, et al. Development of Therapeutic Antibodies for the Treatment of Diseases. J BioMed Sci (2020) 27(1):1–30. doi: 10.1186/s12929-019-0592-z
81. Ke X, Shen L. Molecular Targeted Therapy of Cancer: The Progress and Future Prospect. Front Lab Med (2017) 1(2):69–75. doi: 10.1016/j.flm.2017.06.001
82. Hernández-Reséndiz I, Román-Rosales A, García-Villa E, López-Macay A, Pineda E, Saavedra E, et al. Dual Regulation of Energy Metabolism by P53 in Human Cervix and Breast Cancer Cells. Biochim Biophys Acta Mol Cell Res (2015) 1853(12):3266–78. doi: 10.1016/j.bbamcr.2015.09.033
83. Eriksson M, Ambroise G, Ouchida AT, Lima Queiroz A, Smith D, Gimenez-Cassina A, et al. Effect of Mutant P53 Proteins on Glycolysis and Mitochondrial Metabolism. Mol Cell Biol (2017) 37(24):e00328–17. doi: 10.1128/MCB.00328-17
84. Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, et al. A Comparative Encyclopedia of DNA Elements in the Mouse Genome. Nature (2014) 515(7527):355–64. doi: 10.1038/nature13992
85. Fischer M. Conservation and Divergence of the P53 Gene Regulatory Network Between Mice and Humans. Oncogene (2019) 38(21):4095–109. doi: 10.1038/s41388-019-0706-9
86. Reilly PT, Mak TW. Molecular Pathways: Tumor Cells Co-Opt the Brain-Specific Metabolism Gene CPT1C to Promote Survival. Clin Cancer Res (2012) 18(21):5850–5. doi: 10.1158/1078-0432.CCR-11-3281
87. Goldstein I, Rotter V. Regulation of Lipid Metabolism by P53–Fighting Two Villains With One Sword. Trends Endocrinol Metab (2012) 23(11):567–75. doi: 10.1016/j.tem.2012.06.007
88. McGuirk S, Gravel S-P, Deblois G, Papadopoli DJ, Faubert B, Wegner A, et al. PGC-1α Supports Glutamine Metabolism in Breast Cancer. Cancer Metab (2013) 1(1):1–11. doi: 10.1186/2049-3002-1-22
89. Torrano V, Valcarcel-Jimenez L, Cortazar AR, Liu X, Urosevic J, Castillo-Martin M, et al. The Metabolic Co-Regulator PGC-1α Suppresses Prostate Cancer Metastasis. Nat Cell Biol (2016) 18(6):645–56. doi: 10.1038/ncb3357
90. Reda El Sayed S, Cristante J, Guyon L, Denis J, Chabre O, Cherradi N. MicroRNA Therapeutics in Cancer: Current Advances and Challenges. Cancers (2021) 13(11):2680. doi: 10.3390/cancers13112680
91. Kasinski AL, Kelnar K, Stahlhut C, Orellana E, Zhao J, Shimer E, et al. A Combinatorial MicroRNA Therapeutics Approach to Suppressing Non-Small Cell Lung Cancer. Oncogene (2015) 34(27):3547–55. doi: 10.1038/onc.2014.282
92. Chen Y, Gao D-Y, Huang L. In Vivo Delivery of miRNAs for Cancer Therapy: Challenges and Strategies. Adv Drug Del Rev (2015) 81:128–41. doi: 10.1016/j.addr.2014.05.009
93. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
94. Menendez JA, Alarcón T. Metabostemness: A New Cancer Hallmark. Front Oncol (2014) 4:262–2. doi: 10.3389/fonc.2014.00262
Keywords: wild-type p53, mutant p53, indirect regulation, metabolism, cancer treatment, synthetic lethality
Citation: Wang C-Y and Chao C-H (2022) p53-Mediated Indirect Regulation on Cellular Metabolism: From the Mechanism of Pathogenesis to the Development of Cancer Therapeutics. Front. Oncol. 12:895112. doi: 10.3389/fonc.2022.895112
Received: 13 March 2022; Accepted: 28 April 2022;
Published: 30 May 2022.
Edited by:
Thibaut Barnoud, Medical University of South Carolina, United StatesReviewed by:
Eytan Ruppin, Tel Aviv University, IsraelCopyright © 2022 Wang and Chao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chi-Hong Chao, Y2hhbzdAbnljdS5lZHUudHc=