 Adrian Chye
Adrian Chye India Allen
India Allen Megan Barnet
Megan Barnet Deborah L. Burnett
Deborah L. Burnett- 1Immunology Division, Garvan Institute of Medical Research, Darlinghurst, NSW, Australia
- 2St Vincent’s Clinical School, Faculty of Medicine, University of New South Wales, Darlinghurst, NSW, Australia
- 3Department of Medical Oncology, The Kinghorn Cancer Centre, Darlinghurst, NSW, Australia
Blockade of immune checkpoints transformed the paradigm of systemic cancer therapy, enabling substitution of a cytotoxic chemotherapy backbone to one of immunostimulation in many settings. Invigorating host immune cells against tumor neo-antigens, however, can induce severe autoimmune toxicity which in many cases requires ongoing management. Many immune-related adverse events (irAEs) are clinically and pathologically indistinguishable from inborn errors of immunity arising from genetic polymorphisms of immune checkpoint genes, suggesting a possible shared driver for both conditions. Many endocrine irAEs, for example, have analogous primary genetic conditions with varied penetrance and severity despite consistent genetic change. This is akin to onset of irAEs in response to immune checkpoint inhibitors (ICIs), which vary in timing, severity and nature despite a consistent drug target. Host contribution to ICI response and irAEs, particularly those of endocrine origin, such as thyroiditis, hypophysitis, adrenalitis and diabetes mellitus, remains poorly defined. Improved understanding of host factors contributing to ICI outcomes is essential for tailoring care to an individual’s unique genetic predisposition to response and toxicity, and are discussed in detail in this review.
Introduction
Monoclonal antibodies that block specific immune checkpoints enable durable anti-cancer response in a subset of patients with previously futile prognoses. Drugs targeting Programmed Cell Death Protein-1 (PD-1), its ligand (PD-L1) and Cytotoxic T-lymphocyte Antigen-4 (CTLA-4) are now considered part of standard of care for treatment of multiple cancers. Both targets represent immune checkpoints that contribute to physiological immune tolerance (as summarized here (1)). Despite remarkable response to PD-1/PD-L1 and/or CTLA-4 blockade in some patients, however, a substantial proportion do not gain benefit and mechanisms underlying individual variation in response are incompletely understood.
Immune-related adverse events (irAEs) are autoimmune sequelae of ICI therapy that can affect any organ in the body. Multiple endocrine irAEs including thyroiditis, hypophysitis, diabetes mellitus and adrenalitis, have been recognized with the use of ICI. Endocrinopathies are amongst the most common irAEs. Partly due to the large reserve capacity of many endocrine organs, they are distinct in their tendency to be diagnosed at the point of organ failure. In contrast, most non-endocrine irAEs are detected based on early clinical indications of inflammation, such as dyspnea or diarrhea. As such, the management of endocrine irAEs typically includes life-long hormone replacement (2). While non-endocrine irAEs are often treated with glucocorticoids, there is no clear evidence that immunosuppression is beneficial in preventing progression of endocrinopathy (2).
Successful checkpoint inhibition in cancer treatment requires a fundamental shift in immune homeostasis to a state of reduced immune tolerance. ICI therapy is thus inherently linked to irAEs, because lowering immune tolerance to cancer inextricably lowers tolerance to self. Through this lens, manifestation of irAEs may be a marker of successful immune activation. Accordingly, the occurrence and severity of irAEs, particularly those of endocrine origin, have been linked to improved cancer outcomes in studies that have corrected for immortal time bias in several tumors (3–6).
Significantly, many irAEs are clinically and pathologically indistinguishable from autoimmune disease associated with inherited variation in relevant checkpoint genes. Penetrance of irAEs following pharmacologic blockade of a checkpoint, however, is not equivalent to that of primary genetic blockade of the same gene (7, 8). Intriguingly, pharmacologic blockade in some cases causes more frequent endocrinopathy than genetic blockade, and in some cases less (Table 1). Clinical autoimmunity may manifest when acquired mutations layer on inherited mutations, stochastically bypassing tolerance to precipitate disease (19–22). Pharmacologic blockade of a checkpoint (such as PD-1) may be equivalent to an acquired functional genetic mutation, serving as an additional ‘switch’ weighing in favor of autoimmunity. Improved understanding of the mechanism for drug-induced irAEs and their similarities and differences to genetic autoimmune disease, therefore, has potential to improve patient selection and cancer outcomes.
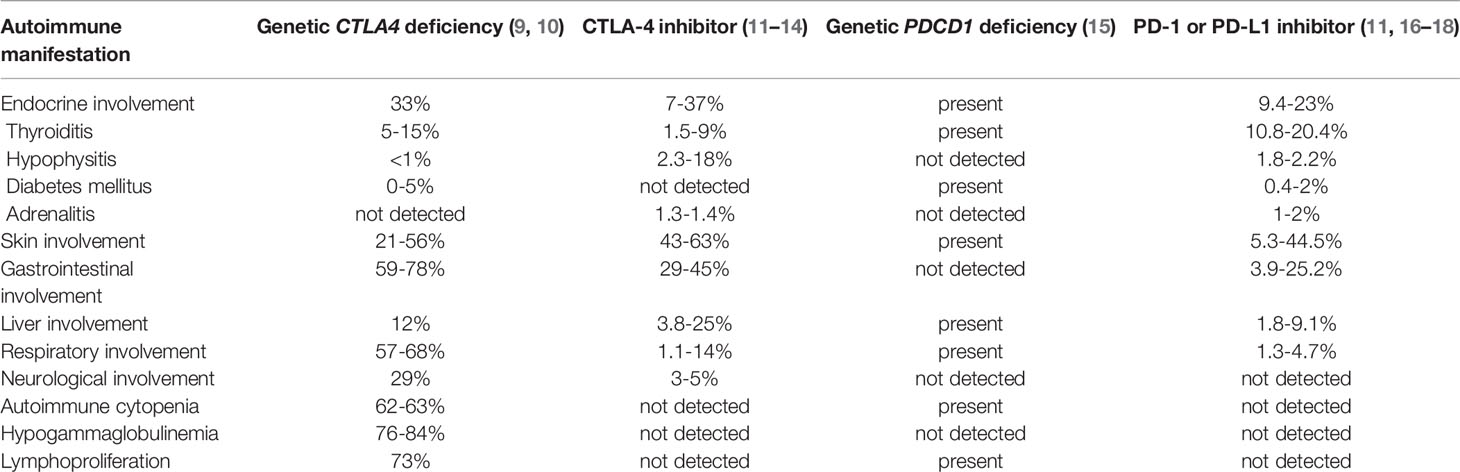
Table 1 Comparison of autoimmune manifestations in genetic deficiency versus pharmacological inhibition of CTLA-4 and PD-1/PD-L1.
In this review, we explore physiological mechanisms of immune tolerance that are leveraged in cancer immunotherapy and examine ICI-induced toxicity and analogous genetic disease, with a focus on endocrinopathies and host contribution to irAE response.
Physiological Immune Tolerance: Essential to Immune Homeostasis
Understanding tolerance in different cell types and physiological settings is an important foundation to understanding ICI response and toxicity. The immune system balances a challenging equilibrium of mounting appropriate response to pathogens whilst tolerating self and non-self commensals. Differing mechanisms of tolerance induction and maintenance for T and B cells, in central (bone marrow, thymus) and peripheral (spleen, lymph nodes) lymphoid organs, are summarized below. The majority of cancer immunotherapy research to date has focused on the role of T-cells, however B-cells play an important role in analogous primary autoimmune disease. Furthermore, auto-antibodies are a feature of many endocrine-related irAEs and may be key to understanding pathogenesis.
T-Cell Tolerance
Central T cell tolerance is established during T cell development in the thymus. Thymocytes express a repertoire of T cell receptors (TCRs), and undergo a process of positive and negative selection (23). During positive selection, thymocytes with TCRs that have sufficient affinity for peptide-Major Histocompatibility Complex (MHC) are selected for survival, while during negative selection thymocytes that bind to self-peptide are signaled to apoptose (24). T cells with a TCR that has sufficient affinity to self-peptide MHC molecules receive a survival signal and are released as naïve cells into the periphery, while T cells with insufficient MHC molecules undergo apoptosis (24). T cells with potentially hazardous self-reactive TCRs that have strong reactivity to self-peptides undergo clonal deletion or receptor editing in the thymus to reduce their reactivity (25). However, central tolerance is only partially effective in eliminating self-reactive T cell clones, allowing release of self-reactive T cell clones into the periphery (26).
Self-reactive naïve T cells that escape central tolerance are maintained by a number of peripheral T cell tolerance mechanisms including quiescence, ignorance, anergy, exhaustion, senescence and death (27). Quiescence is an active process that maintains naïve T cells at a lower metabolic state, in the G0 stage of the cell cycle, agnostic to TCR stimulation (28). Clonal ignorance is a mechanism maintaining self-reactive T cells in an inactive state despite the presence of self-antigen (29–31). T cell ‘anergy’ can result from unbalanced or deficient TCR stimulation without engagement of costimulatory receptors (32–34). When T cells are successfully stimulated and reach the effector stage, T cell exhaustion may arise, characterized by reduced effector function and sustained inhibitory receptor expression. In the context of chronic infections, T cell exhaustion avoids pathogenic inflammation and autoreactivity (35). T cells may eventually senesce and die, arresting growth when they reach their replicative potential or are exposed to various stressors (36). Additionally, peripheral deletional tolerance checkpoints serve a critical role in pruning the repertoire of peripheral T cells and terminating deleterious immune responses, preventing T cell mediated immunopathology (37).
B-Cell Tolerance
Highly reactive B Cell Receptors (BCRs) also experience negative selection and are actively eliminated in the bone marrow by central tolerance mechanisms including clonal deletion (38) and receptor editing (39, 40). Unlike T cells however, the extent to which B cells undergo positive selection to self-antigen in the bone marrow is still not well defined. Some mildly and moderately self-reactive B cells have been shown to undergo positive selection into the mature repertoire (41). Regardless of the mechanism, central B cell tolerance results in a significant proportion of auto reactive BCRs surviving and exiting the bone marrow into the periphery (42, 43). Assessment of mature peripheral B cells in healthy human controls show that 5-20% of cells encode for antibodies with poly- or self-reactivity (44). Self-reactive B cells tend to exist in a functionally silenced state of anergy (45). The benefit of preserving these potentially harmful auto-reactive cells was only recently shown to be the ability for “auto-antibody redemption”, wherein BCRs mutate away from self-reactivity to enable production of antibodies with high foreign antigen specificity (46–50).
Pathological Immune Tolerance: Immune Evasion by Cancer Cells
“Successful” cancer cells achieve unchecked proliferation by exploiting physiological mechanisms of immune tolerance to evade detection and/or destruction by the host immune system. These include downregulation of antigen-presenting machinery including MHC-1 (51), overexpression of immune-inhibitory molecules such as PD-L1 (52), induction of an immunosuppressive tumor microenvironment enriched for regulatory T cells (Tregs) and myeloid-derived suppressor cells (53–55) and secretion of directly immunosuppressive factors such as IL-10, Transforming Growth Factor-β (TGF-β), gangliosides, prostaglandin E2, and vascular endothelial growth factor (VEGF) (56–60).
Tregs play a critical role in tolerance to cancer cells through suppression of cytotoxic CD8+ T-cell proliferation, contributing to immune escape. Tregs express high levels of multiple checkpoint receptor molecules including CTLA-4, PD-1, T cell immunoreceptor with Ig and ITIM domains (TIGIT), Lymphocyte-Activation Gene 3 (LAG-3) and T-cell immunoglobulin domain and mucin domain 3 (TIM3) (61, 62).
Immune Checkpoint Inhibitors
Monoclonal antibodies targeting CTLA-4 and PD-1/PD-L1 are now considered part of standard of care in multiple cancer types, including melanoma, Non-Small Cell lung cancer (NSCLC), renal cell carcinoma, head and neck cancer, urothelial cancer, Merkel cell carcinoma, mesothelioma, and Hodgkin lymphoma.
CTLA-4 is a negative co-inhibitory receptor of T-cell activation and proliferation, playing a critical role in the maintenance of self-tolerance (63). It is constitutively upregulated in Tregs and transiently upregulated on the cell surface of conventional T cells following the engagement of co-stimulatory molecule CD28 with its ligands, CD80 and CD86, in order to curtail T cell responses (64). CTLA-4 has several mechanisms of action including competitively binding CD80 and CD86 and reducing CD28-dependent co-stimulation (65–67). Additionally, CTLA-4 is thought to strip the CD80 and CD86 ligands from APCs through a process of trans-endocytosis, further reducing potential for CD28 co-stimulation (65). It has been postulated that CTLA-4 may also directly restrain T-lymphocytes through down-regulating IL2 expression, as well as repression of T-cell proliferation (68).
PD-1 is a co-inhibitory receptor expressed on the surface of activated T-cells (including tumor infiltrating lymphocytes), B cells, NK cells, monocytes and dendritic cells (64, 69, 70). Ligation of PD-1 with its ligands, PD-L1 and PD-L2, activates inhibitory signaling cascades associated with exhaustion, resulting in the downregulation of T-cell response (69). PD-1 suppression of T cell activity occurs primarily within the peripheral tissues and tumor microenvironment (71).
The incidence and pattern of endocrine irAEs differs significantly between the major classes of checkpoint inhibitors, reflective of differing mechanisms of action (Table 1). ICI-induced thyroiditis is more frequent after PD-1 compared with CTLA-4 blockade (10-20.4% versus 1.5-9%). Whereas ICI-induced hypophysitis and colitis are more common following CTLA-4 compared with PD-1 inhibition (2.3-18% versus 1.2-2.2% and 29-45% versus 3.9-25% respectively). Combination checkpoint blockade, targeting both CTLA-4 and PD-1, increases rate and severity of irAEs, with tendency to earlier events and multi-organ autoimmunity (72, 73).
Endocrine irAEs: Prevalence and Significance
Accurate predictive models for type and severity of irAEs are lacking, although several potential biomarkers are being studied involving immune cells (74, 75), cytokines (76), autoantibodies (77), immunogenetics (78, 79) and microbiome (80, 81). Time of onset is also unpredictable; most irAEs develop within the first few weeks following commencement of an ICI, however they can manifest years after treatment initiation (82). IrAEs occur in up to 80% of patients receiving combination treatment (83), and are predominantly mild (CTCAE grade 1-2) (84), with less than 1% resulting in death (85).
Endocrinopathies are a subset of irAEs that affect endocrine organs, including thyroiditis, hypophysitis, diabetes mellitus and adrenalitis (86). Distinct to non-endocrine irAEs, endocrine-related irAEs are frequently irreversible, do not usually prompt treatment discontinuation and often require lifelong hormone replacement therapy (87). While numerous studies have examined the relationship between irAEs and treatment outcomes, most do not account for immortal time bias; an adjustment for shorter time on drug and less time available to develop irAEs for non-responders. Landmark analyses avert this bias and also support a correlation of irAEs with outcomes, particularly those involving endocrine organs and skin for patients with NSCLC and melanoma (3–6). The correlation of irAEs with survival in many settings suggests a possible shared driver of systemic and anti-cancer immune activation.
ICI-Induced Thyroiditis
ICI-induced thyroiditis is common in patients treated with ICI, with an incidence of up to 20% for single agent anti-PD-1 therapy (16). The clinical phenotype is painless thyroiditis, most often characterized by transient thyrotoxicosis followed by euthyroidism or progression to hypothyroidism without preceding thyrotoxicosis (88, 89). The true incidence of thyroiditis is likely underreported, in part because clinical trials define ICI-mediated hypothyroidism and hyperthyroidism as separate entities rather than a single pathology.
ICI-induced thyroiditis has much clinical and biochemical overlap with spontaneous autoimmune thyroid diseases (AITD), including Hashimoto’s thyroiditis and its variant form, subacute painless thyroiditis (also known as silent thyroiditis or subacute lymphocytic thyroiditis) (90). In AITD, antigen presenting cells and helper CD4+ T cells activate CD8+ T cells specific for thyroid parenchyma, causing direct tissue injury and thyroid antigen exposure. B cell activation from thyroid tissue leads to expression of anti-thyroid autoantibodies to thyroglobulin (Tg) and thyroid peroxidase (TPO) (91). It is unclear whether ICI-induced thyroiditis reflects exacerbation of an evolving AITD, or has a distinct mechanism with the shared endpoint of auto-reactive B cells. Anti-PD-1-induced destructive thyroiditis in preclinical modelling, however, was completely prevented by depletion of CD4+ T cells, partially prevented by depletion of CD8+ T cells but not affected by depletion of CD20+ B cells (92). This supports a more T-cell dominant process at play.
In patients with AITD, PD-1 positive T cells are significantly expanded in both the periphery and intrathyroidal lymphocytes compared to patients with (non-autoimmune) multi-nodular goiter (MNG) (93). PD-L1 positive parenchymal thyroid follicular cells are present in most AITD glands, but rarely in MNG glands, and have been hypothesized to serve as a peripheral tolerance mechanism induced to avoid recognition by self-reactive T lymphocytes (93).
Thyroid auto-antibodies develop in up to 70% of patients with ICI-induced thyroiditis (77). It is unclear, however, if these are generated in response to thyroid antigen released during destructive thyroiditis, or if ICI reactivates dormant thyroid auto-antibodies, therein initiating thyroiditis. Patients with pre-existing anti-thyroglobulin and anti-thyroid peroxidase antibodies have an increased propensity to ICI-induced thyroiditis compared with patients who do not harbor these autoantibodies (90, 94, 95). Moreover, there is shared genetic predisposition to AITD and ICI-induced thyroiditis, highlighting the direct contribution of host predisposition (96). Many patients who develop thyroid irAEs, however, do not have detectable thyroid auto-antibodies at baseline or following diagnosis of ICI-induced thyroiditis, suggesting an alternative pathogenesis distinct from AITD (97).
ICI-Induced Hypophysitis
ICI-induced hypophysitis is inflammation of the pituitary gland resulting in the decreased production of one or more pituitary hormones. It is more common following CTLA-4 blockade compared with PD-1 blockade (3.2% and 0.4% respectively), with incidence up to 6.4% in combination CTLA-4/PD-1 treatment (86). Symptoms may include headache, fatigue, anorexia or myalgias, but it is often diagnosed during investigation of low cortisol on routine bloods or on presentation in adrenal crisis. The diagnosis may be supported by radiographic pituitary enlargement. A uniform diagnostic criterion does not exist, therefore existing literature may inaccurately report case frequency (98).
There are key distinctions in the clinical phenotype between hypophysitis caused by CTLA-4 compared with PD-1 inhibitors. Firstly, in time of onset. CTLA-4 induced hypophysitis usually presents within weeks of ICI commencement (mean 9 weeks), whereas anti-PD-1-induced disease is more variable, but tends to occur later (mean 26 weeks) (99). Time to onset of disease in combination therapy falls between these two timeframes. Secondly, in symptomatology. Headache is more frequent in patients treated with CTLA-4-inhibitor or combination therapy, while the predominant symptoms of PD-1/PD-L1-inhibitor induced hypophysitis are fatigue, anorexia and myalgias (99, 100). Thirdly, axes involvement. Patients with anti-PD-1 related hypophysitis often have isolated ACTH-adrenal axis insufficiency, while CTLA-4-inhibitor related hypophysitis tends to involve multiple pituitary axes. Finally, in radiographic changes. Pituitary gland enlargement on MRI was almost universal in one multi-center review for patients with hypophysitis following CTLA-4 therapy, while this was only seen in a minority of cases (28%) resulting from anti-PD-1 therapy (99).
The underlying mechanism of ICI-mediated hypophysitis is largely unknown. One theory is that immune-related hypophysitis occurs more frequently in patients treated with CTLA-4 inhibitors because it directly targets pituitary gland cells, which express CTLA-4 and may activate antibody-dependent cell-mediated cytotoxicity and complement-mediated direct injury (101–103). In a murine model, CTLA-4 blockade resulted in complement deposition on endocrine cells including the pituitary (101). A human autopsy series showed disproportionately high CTLA-4 antigen expression within the pituitary of patients who had developed ICI-induced hypophysitis (104). The precise molecular basis for anti-PD-1 hypophysitis is unknown.
ICI-Induced Diabetes Mellitus
ICI-induced diabetes mellitus (ICI-DM) is defined by severe and persistent insulin deficiency caused by pancreatic β-islet cell failure following treatment with ICI (105). ICI-DM is rare, and occurs almost exclusively with anti-PD-1/PD-L1 therapy. A meta-analysis of 101 studies demonstrated the incidence of ICI-DM to be 0.4% for pembrolizumab, 2.0% for nivolumab, with no cases for patients treated with ipilimumab (11). A systematic review of the existing literature identified only 90 patients with ICI-induced DM, and observed that most patients were treated with anti-PD-1/PD-L1 monotherapy (71%) or anti-PD1/PD-L1 in combination with CTLA-4 blockade (15%), while anti-CTLA-4 alone accounted for only 3% of the patients (106).
The clinical presentation of ICI-DM is similar to that of classic type 1 diabetes mellitus (T1DM), ranging from asymptomatic hyperglycemia, polyuria, polydipsia, and fatigue, to life-threatening diabetic ketoacidosis. ICI-DM onset is also highly variable, with reported median time to diagnosis of between 7 to 17 weeks post treatment commencement, with potential to present years later (107, 108). As with other endocrine irAEs, ICI-DM is generally diagnosed at point of organ failure, and almost always requires life-long insulin replacement therapy (87).
ICI-DM is presumed to be caused by immune-mediated destruction of pancreatic β-cells, however may be pathologically distinct from T1DM (109). Analysis of pancreas tissue from a patient who developed ICI-DM following combination therapy showed marked peri-islet infiltration of CD8+ T cells, with very few residual pancreatic β cells. Destruction of β-cells was noted to be more severe than most cases of classic T1DM, and may reflect mechanistic differences (109). ICI-DM and classic T1DM also differ in prevalence of auto-antibodies. More than 90% of patients with T1DM have at least one of four common β-cells autoantibodies; to insulin, GAD (glutamic acid decarboxylase), ZnT8 (zinc transporter 8) or IA-2 (insulinoma-associated-2) (110). In contrast, only 50% of patients with ICI-DM are found to have a relevant autoantibody, with anti-GAD65 comprising the majority of these (105, 107). This may suggest that either autoantibodies play less of a role in the pathogenesis of ICI-DM compared to primary T1DM, or potentially that the autoantibodies still exist, but are sequestered in tissue.
ICI-Induced Adrenalitis
ICI-induced adrenalitis is a rare endocrine irAE and is defined by adrenal gland inflammation leading to adrenal cortex failure to generate hormones, including glucocorticoids, mineralocorticoids and sex steroids. A meta-analysis revealed an incidence of 0.7% across studies of monotherapy with anti-PD-1/PD-L1 or anti-CTLA-4, but up to 4.2% in studies of combination therapy (86). This shows some discordance with a WHO pharmacovigilance database review, which demonstrated that the majority of patients diagnosed with adrenalitis had been treated with PD-1 monotherapy (56%), and less commonly with CTLA-4 monotherapy (23.6%) or combination therapy (17.9%) (111). Primary adrenal insufficiency may be diagnosed on routine bloods showing reduced cortisol with appropriate elevation of pituitary-derived ACTH, but may present clinically with fatigue, orthostatic hypotension, or adrenal crisis (112). After acute management, long-term corticosteroid (with or without mineralocorticoid) replacement is usually required (113).
ICI-adrenalitis is presumed to be caused by immune-mediated destruction of the adrenal cortex (111). In published case reports, antibodies to 21-hydroxylase were detected in a handful of cases (114, 115), which may suggest a B-cell mediated process. The analogous autoimmune condition of primary adrenal insufficiency (Addison’s disease), is also characterized by antibodies against the steroidogenic enzymes; most commonly 21-hydroxylase, and less frequently a side-chain cleavage enzyme or 17-alpha-hydroxylase (116).
Pharmacological Inhibition Compared With Genetic Inhibition of Immune Checkpoints
Pharmacological inhibition of CTLA-4 and/or PD-1/PD-L1 has significant overlap, but also important differences, to autoimmune manifestations seen in humans with inborn errors of the corresponding genes, CTLA4 and PDCD1 (Table 1).
Mice homozygous for ctla-4 loss of function develop lymphoproliferative symptoms in multiple organs and die within weeks, but mice with ctla4 haploinsufficiency (one functional allele retained) remain healthy (117–119). In humans, inherited genetic heterozygosity for CTLA4 inactivating mutations results in immunodeficiency and autoimmunity (9, 10, 117, 120). In the largest study of CTLA4 haploinsufficiency in humans, 133 patients were characterized (10). The median age of onset of disease was 11 years and there was clinical penetrance (manifestation of disease) in 67% (10). The predominant clinical phenotypes included hypogammaglobulinemia (84%), lymphoproliferation (73%) and organ-specific autoimmunity such as hematological autoimmune cytopenia (62%), respiratory (68%), and gastrointestinal (59%) or neurological manifestations (29%) (10) (Table 1). Some auto-immune sequelae of germline CTLA-4 deficiency are almost indistinguishable from toxicity following its pharmacologic blockade, such as gastrointestinal inflammation (10, 12, 13). One factor contributing to observed differences may be distinct temporal dynamics between in-born genetic loss of CTLA-4 compared with acquired pharmacologic blockade, usually later in life. Mouse models of conditional CTLA-4 inhibition later in life, however, do not more closely resemble anti-CLTA-4 induced irAEs, suggesting other differences at play (121).
Key discrepancies between genetically and pharmacologically driven disease highlight the need to further understand both pathways. Firstly, clinical apparent autoimmunity, in general, appears to be more prevalent in humans with CTLA-4 haploinsufficiency compared with patients receiving CTLA-4 pharmacological blockade (10, 12, 13). This supports the notion that irAEs associated with CTLA-4 blockade are dose-dependent, and the monoclonal antibodies may only partially inhibit CTLA4 function leading to incomplete constellation of symptoms. Indeed, severe, life-threatening autoimmune toxicities are more frequent with higher doses of ipilimumab supporting this dose-dependent effect (122). Secondly, and somewhat counter to this, certain organ toxicities are significantly more common with CTLA-4 drug blockade compared with CTLA-4 germline haploinsufficiency. For example, severe hypophysitis is reported in up to 5% of patients treated with ipilimumab but is extremely rare (<1%) in genetic CTLA4 haploinsufficiency (10, 13) (Table 1). Finally, certain manifestations of primary CTLA4 deficiency are not seen in the drug-induced setting, significantly immunodeficiency, which is contributed to by autoantibody mediated cytopenia, lymphadenopathy and splenomegaly (9, 120, 123). The reason for this discord is not well understood.
In contrast to CTLA4, germline deficiencies in PD-1 more frequently lead to single organ effects, most commonly involving endocrine organs. Here, exogenous blockade appears to more closely match germline inactivation (124–126). Mice deficient in PD-1 initially develop normally but go on to manifest strain-dependent autoimmunity and heightened inflammation during infections (127–129). PD-1 deficiency also accelerates the onset and frequency of organ-specific endocrine autoimmunity such as T1DM in predisposed mouse models (130).
In humans, homozygous loss of function mutations in the gene encoding PD-1, PDCD1, is exceedingly rare (15). In one case study, homozygous frameshift (loss of function) mutation resulted in lymphoproliferative autoimmunity, autoimmune thyroiditis, T1DM, juvenile idiopathic arthritis, and abdominal tuberculosis. The patient died of pneumonitis at age 10 years (15). Though complete loss of PDCD1 is rare in humans and difficult to study, partial deficiency shows significant overlap with drug-induced irAEs (Table 1).
Further investigation is needed to explore similarities and discrepancies between genetic and drug-induced autoimmunity. As discussed above, timing and duration of exposure to pathway blockade is likely to play a role. Inherited genetic abnormalities may only manifest as autoimmunity when accumulation of acquired mutations leads to a break in tolerance (20). Treatment with anti-CTLA-4 and/or anti-PD-1 can be considered akin to an acquired mutation in terms of onset later in life, however is distinct from an acquired mutation in that blockade is maintained for a fixed period only. Comparison with analogous genetic disease is valuable because it can help elucidate mechanism, however genetic predisposition is likely to be just one of many factors contributing to outcome. Environmental factors including the microbiome are likely to play a major role (80, 81), but are beyond the scope of this review.
Shared Genetic Predisposition to Autoimmunity, irAE and ICI Response
Recent genetic association studies have revealed that polymorphisms in CTLA4 may modulate both autoimmunity and response and toxicity to ICIs (131, 132). Specific gene associations with autoimmune disease are outlined in Table 2. Many of these Genome-wide association studies (GWAS) single nucleotide polymorphisms (SNPs) highlight the dialectic nature of T-cell immune regulation in cancer and autoimmunity - the allele for cancer risk is often the opposite to that for autoimmune disease (132, 134, 157, 158, 166–170). Systemic testing for common autoimmune risk loci could provide a pathway for personalized biomarkers for ICI response and toxicity (171).
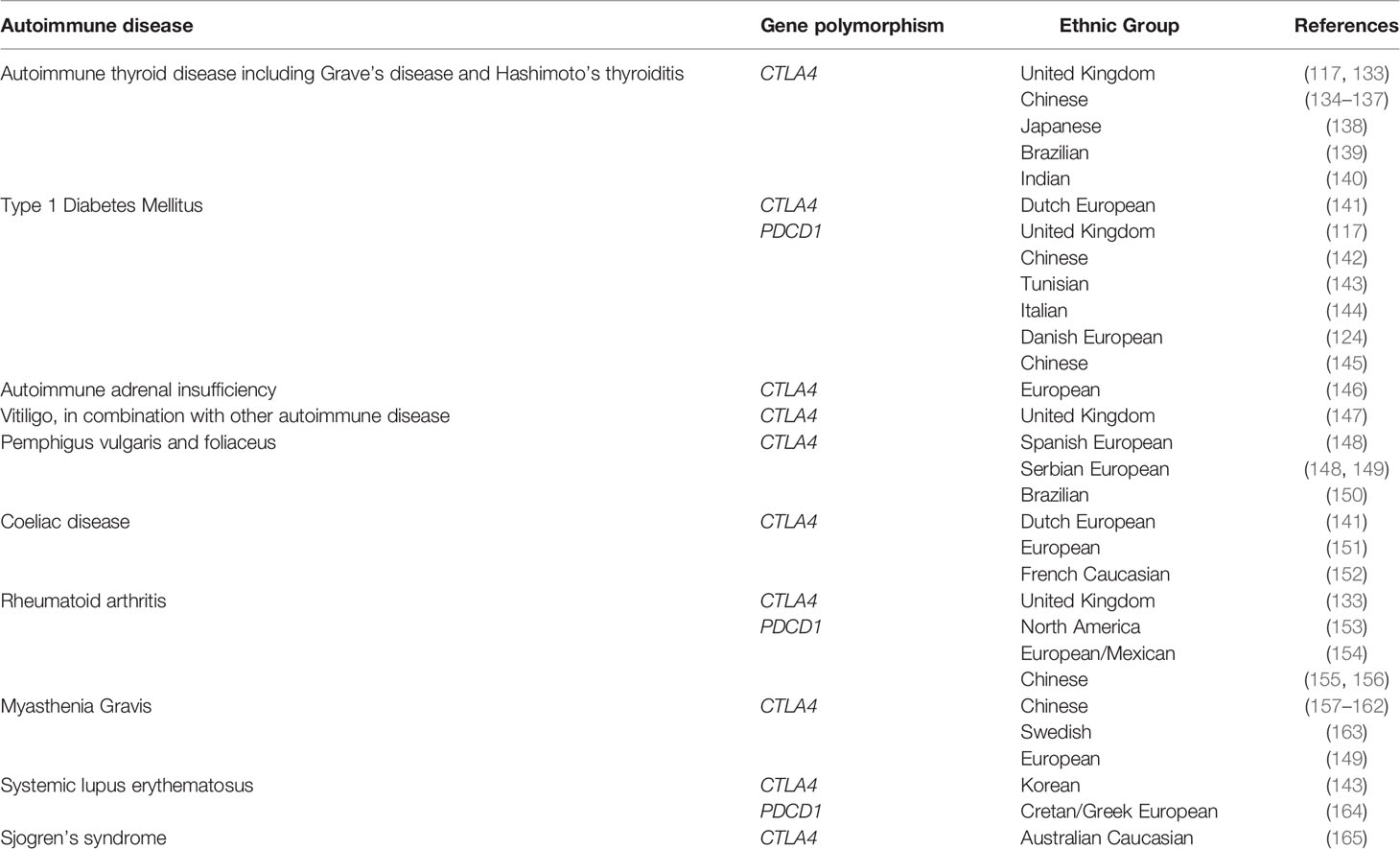
Table 2 Germline CTLA4 and PDCD1 gene polymorphisms are associated with autoimmune diseases.
Germline polymorphisms in CTLA4 have also been shown to influence susceptibility to irAEs. Refae et al. investigated a cohort of 94 patients with advanced cancer treated with anti-PD-1/PD-L1 and demonstrated that SNPs within a range of genes including UNG, IFNW1, CD274 and IFNL4 were predictive of irAEs corresponding to that gene function (78). Whole genome sequencing of 479 patients with bladder cancer treated with anti-PD-L1 therapy showed that high polygenic risk scores for vitiligo and psoriasis were associated with increased risk of skin-related irAEs (7).
A key question is whether the genetic risk for classical T1DM overlaps with ICI-DM. Polymorphisms of multiple genes are known to influence risk of T1DM, with the HLA-DQ, HLA-DR alleles having the largest effect, but also non-HLA genes such as PTPN22 and CTLA-4 (172). HLA polymorphisms linked to classic T1DM class II haplotypes – HLA-DR3-DQ2 and DR4-DQ8 in European populations, and DR4-DQ4 and DR9-DQ9 in Asian populations, have been demonstrated to be overrepresented patients with ICI-DM (106). Only one ICI-DM case has been evaluated for multiple HLA and non-HLA risk genes as part of a T1DM genetic risk score (GRS); this case had a GRS below the 5th percentile for T1DM (173), suggesting that ICI-DM genetic risk factors may be distinct from T1DM. Overall, HLA susceptibility haplotypes for T1DM may predispose to the development ICI-DM, but further studies are needed to understand the genetic contribution of HLA and non-HLA genes.
There is also known genetic susceptibility to spontaneous AITD, within which certain at-risk genes may also be certain at-risk genes may also be shared with ICI-induced thyroiditis. Data suggests there is an association of AITD with certain HLA alleles, particularly HLA-DR3, and with polymorphisms within CTLA4 and CD40 (174). GWAS have demonstrated key susceptibility genes implicated in both immune regulation and increased risk of ICI-induced thyroiditis, including CD69, CTLA4, PTPN22 and LPP (96). These loci and others were combined to calculate a polygenic risk score that could identify a cohort of patients with more than 6-fold increased risk of ICI-induced thyroiditis. Furthermore, germline susceptibility to AITD disease was found to be associated with improved survival, in a cohort of patients with triple-negative breast cancer treated with combination anti-PD-L1 inhibitor and chemotherapy (96).
Conclusion
Immune checkpoint inhibitors are now established as a pillar of cancer treatment, alongside chemotherapy, radiotherapy and surgery. Understanding mechanism of response and non-response to the drugs is paramount to broadening their potential for clinical benefit. Investigating drivers of ‘off-target’ effects, specifically irAEs, is key to this analysis, because anti-self responses are inextricably linked with anti-cancer responses in many settings due to lowering of immune tolerance thresholds. Furthermore, many irAEs are clinically and biologically analogous to genetic disease, for which we have significant insight into drivers and mitigating factors.
Autoimmunity of endocrine organs are common both as primary disease and as ICI-induced toxicity in patients treated for cancer. Endocrine autoimmunity driven by genetic variation in checkpoint genes can mimic drug-induced irAEs, however key differences exist in the pathophysiology for each. Both primary and drug-induced endocrine disease may involve underlying genetic susceptibility as part of a multifactorial pathogenesis. Further investigation into shared drivers of irAEs with analogous primary disease may lead to predictive models for genetic predisposition to ICI-induced toxicity. Furthermore, the correlation between irAEs and anti-cancer response highlights the potential for these genetic drivers to serve as novel therapeutic targets, with the caveat that ensuing toxicity may need aggressive management.
Current predictive markers (such as tumor and immune cell PD-L1 expression) can enrich for responders to immunotherapy but provide little insight into variation in cancer response or toxicity for the individual patient. There is increasing recognition that host factors, including genetic predisposition to both autoimmunity (for example to thyroid disease) and irAEs (for example to psoriasis and vitiligo), can be predictive of checkpoint inhibitor treatment outcomes. Genetic testing for common autoimmune risk loci could form part of a comprehensive personalized biomarker for ICI response and toxicity, leading to improved treatment selection and toxicity management for individual patients.
There is much to learn from host factors that contribute to maintenance and breakdown of physiological immune tolerance. Shared drivers of clinical autoimmunity and ICI-induced irAEs are pivotal to this analysis and have the potential to progress patient outcomes in cancer treatment significantly.
Author Contributions
AC, IA, MB and DB jointly wrote the manuscript. All authors contributed to the conception, design, and interpretation of the manuscript, and reviewed and wrote the final paper.
Funding
This work was supported by National Health and Medical Research Council (NHMRC) (Grants 10375), and by Bioplatforms Australia and The Kinghorn Foundation as part of the Australian Exceptional Responders Program (Grants PR10285).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Barnet MB, Blinman P, Cooper W, Boyer MJ, Kao S, Goodnow CC. Understanding Immune Tolerance of Cancer: Re-Purposing Insights From Fetal Allografts and Microbes. Bioessays (2018) 40(8):e1800050. doi: 10.1002/bies.201800050
2. Stelmachowska-Banaś M, Czajka-Oraniec I. Management of Endocrine Immune-Related Adverse Events of Immune Checkpoint Inhibitors: An Updated Review. Endocr Connect (2020) 9(10):R207–R28. doi: 10.1530/EC-20-0342
3. Fan Y, Xie W, Huang H, Wang Y, Li G, Geng Y, et al. Association of Immune Related Adverse Events With Efficacy of Immune Checkpoint Inhibitors and Overall Survival in Cancers: A Systemic Review and Meta-Analysis. Front Oncol (2021) 11:633032. doi: 10.3389/fonc.2021.633032
4. Street S, Chute D, Strohbehn I, Zhao S, Rengarajan M, Faje A, et al. The Positive Effect of Immune Checkpoint Inhibitor-Induced Thyroiditis on Overall Survival Accounting for Immortal Time Bias: A Retrospective Cohort Study of 6596 Patients. Ann Oncol (2021) 32(8):1050–1. doi: 10.1016/j.annonc.2021.05.357
5. Haratani K, Hayashi H, Chiba Y, Kudo K, Yonesaka K, Kato R, et al. Association of Immune-Related Adverse Events With Nivolumab Efficacy in Non-Small-Cell Lung Cancer. JAMA Oncol (2018) 4(3):374–8. doi: 10.1001/jamaoncol.2017.2925
6. Teraoka S, Fujimoto D, Morimoto T, Kawachi H, Ito M, Sato Y, et al. Early Immune-Related Adverse Events and Association With Outcome in Advanced Non-Small Cell Lung Cancer Patients Treated With Nivolumab: A Prospective Cohort Study. J Thorac Oncol (2017) 12(12):1798–805. doi: 10.1016/j.jtho.2017.08.022
7. Khan Z, Di Nucci F, Kwan A, Hammer C, Mariathasan S, Rouilly V, et al. Polygenic Risk for Skin Autoimmunity Impacts Immune Checkpoint Blockade in Bladder Cancer. Proc Natl Acad Sci U S A (2020) 117(22):12288–94. doi: 10.1073/pnas.1922867117
8. Hao Y, Cook MC. Inborn Errors of Immunity and Their Phenocopies: CTLA4 and PD-1. Front Immunol (2021) 12:806043. doi: 10.3389/fimmu.2021.806043
9. Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, et al. Autosomal Dominant Immune Dysregulation Syndrome in Humans With CTLA4 Mutations. Nat Med (2014) 20(12):1410–6. doi: 10.1038/nm.3746
10. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, Penetrance, and Treatment of 133 Cytotoxic T-Lymphocyte Antigen 4-Insufficient Subjects. J Allergy Clin Immunol (2018) 142(6):1932–46. doi: 10.1016/j.jaci.2018.02.055
11. de Filette J, Andreescu CE, Cools F, Bravenboer B, Velkeniers B. A Systematic Review and Meta-Analysis of Endocrine-Related Adverse Events Associated With Immune Checkpoint Inhibitors. Horm Metab Res (2019) 51(3):145–56. doi: 10.1055/a-0843-3366
12. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. New Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
13. Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, et al. Adjuvant Ipilimumab Versus Placebo After Complete Resection of High-Risk Stage III Melanoma (EORTC 18071): A Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol (2015) 16(5):522–30. doi: 10.1016/S1470-2045(15)70122-1
14. Ascierto PA, Del Vecchio M, Robert C, Mackiewicz A, Chiarion-Sileni V, Arance A, et al. Ipilimumab 10 Mg/Kg Versus Ipilimumab 3 Mg/Kg in Patients With Unresectable or Metastatic Melanoma: A Randomised, Double-Blind, Multicentre, Phase 3 Trial. Lancet Oncol (2017) 18(5):611–22. doi: 10.1016/S1470-2045(17)30231-0
15. Ogishi M, Yang R, Aytekin C, Langlais D, Bourgey M, Khan T, et al. Inherited PD-1 Deficiency Underlies Tuberculosis and Autoimmunity in a Child. Nat Med (2021) 27(9):1646–54. doi: 10.1038/s41591-021-01388-5
16. Weber J, Mandala M, Del Vecchio M, Gogas HJ, Arance AM, Cowey CL, et al. Adjuvant Nivolumab Versus Ipilimumab in Resected Stage III or IV Melanoma. New Engl J Med (2017) 377(19):1824–35. doi: 10.1056/NEJMoa1709030
17. Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S, et al. Adjuvant Pembrolizumab Versus Placebo in Resected Stage III Melanoma. New Engl J Med (2018) 378(19):1789–801. doi: 10.1056/NEJMoa1802357
18. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab Versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373(17):1627–39. doi: 10.1056/NEJMoa1507643
19. Goodnow CC. Multistep Pathogenesis of Autoimmune Disease. Cell (2007) 130(1):25–35. doi: 10.1016/j.cell.2007.06.033
20. Singh M, Jackson KJL, Wang JJ, Schofield P, Field MA, Koppstein D, et al. Lymphoma Driver Mutations in the Pathogenic Evolution of an Iconic Human Autoantibody. Cell (2020) 180(5):878–94.e19. doi: 10.1016/j.cell.2020.01.029
21. Yazar S, Alquicira-Hernandez J, Wing K, Senabouth A, Gordon MG, Andersen S, et al. Single-Cell eQTL Mapping Identifies Cell Type-Specific Genetic Control of Autoimmune Disease. Science (2022) 376(6589):eabf3041. doi: 10.1126/science.abf3041
22. Perez RK, Gordon MG, Subramaniam M, Kim MC, Hartoularos GC, Targ S, et al. Single-Cell RNA-Seq Reveals Cell Type-Specific Molecular and Genetic Associations to Lupus. Science (2022) 376(6589):eabf1970. doi: 10.1126/science.abf1970
23. Xing Y, Hogquist KA. T-Cell Tolerance: Central and Peripheral. Cold Spring Harb Perspect Biol (2012) 4(6):a006957. doi: 10.1101/cshperspect.a006957
24. Moran AE, Hogquist KA. T-Cell Receptor Affinity in Thymic Development. Immunology (2012) 135(4):261–7. doi: 10.1111/j.1365-2567.2011.03547.x
25. McGargill MA, Derbinski JM, Hogquist KA. Receptor Editing in Developing T Cells. Nat Immunol (2000) 1(4):336–41. doi: 10.1038/79790
26. Bouneaud C, Kourilsky P, Bousso P. Impact of Negative Selection on the T Cell Repertoire Reactive to a Self-Peptide: A Large Fraction of T Cell Clones Escapes Clonal Deletion. Immunity (2000) 13(6):829–40. doi: 10.1016/S1074-7613(00)00080-7
27. ElTanbouly MA, Noelle RJ. Rethinking Peripheral T Cell Tolerance: Checkpoints Across a T Cell’s Journey. Nat Rev Immunol (2021) 21(4):257–67. doi: 10.1038/s41577-020-00454-2
28. Michalek RD, Rathmell JC. The Metabolic Life and Times of a T-Cell. Immunol Rev (2010) 236:190–202. doi: 10.1111/j.1600-065X.2010.00911.x
29. Parish IA, Heath WR. Too Dangerous to Ignore: Self-Tolerance and the Control of Ignorant Autoreactive T Cells. Immunol Cell Biol (2008) 86(2):146–52. doi: 10.1038/sj.icb.7100161
30. Legoux FP, Lim JB, Cauley AW, Dikiy S, Ertelt J, Mariani TJ, et al. CD4+ T Cell Tolerance to Tissue-Restricted Self Antigens Is Mediated by Antigen-Specific Regulatory T Cells Rather Than Deletion. Immunity (2015) 43(5):896–908. doi: 10.1016/j.immuni.2015.10.011
31. Malhotra D, Linehan JL, Dileepan T, Lee YJ, Purtha WE, Lu JV, et al. Tolerance is Established in Polyclonal CD4(+) T Cells by Distinct Mechanisms, According to Self-Peptide Expression Patterns. Nat Immunol (2016) 17(2):187–95. doi: 10.1038/ni.3327
32. Goodnow CC. Pathways for Self-Tolerance and the Treatment of Autoimmune Diseases. Lancet (9274) 2001:2115–21:357. doi: 10.1016/s0140-6736(00)05185-0
33. Sloan-Lancaster J, Allen PM. Altered Peptide Ligand-Induced Partial T Cell Activation: Molecular Mechanisms and Role in T Cell Biology. Annu Rev Immunol (1996) 14:1–27. doi: 10.1146/annurev.immunol.14.1.1
34. Lamb JR, Skidmore BJ, Green N, Chiller JM, Feldmann M. Induction of Tolerance in Influenza Virus-Immune T Lymphocyte Clones With Synthetic Peptides of Influenza Hemagglutinin. J Exp Med (1983) 157(5):1434–47. doi: 10.1084/jem.157.5.1434
36. Campisi J, d'Adda di Fagagna F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat Rev Mol Cell Biol (2007) 8(9):729–40. doi: 10.1038/nrm2233
37. Herndon JM, Stuart PM, Ferguson TA. Peripheral Deletion of Antigen-Specific T Cells Leads to Long-Term Tolerance Mediated by CD8+ Cytotoxic Cells. J Immunol (2005) 174(7):4098. doi: 10.4049/jimmunol.174.7.4098
38. Burnet FM. The Clonal Selection Theory of Acquired Immunity ; the Abraham Flexner Lectures of Vanderbilt University. Nashville: Cambridge University Press (1959).
39. Nemazee D. Mechanisms of Central Tolerance for B Cells. Nat Rev Immunol (2017) 17(5):281–94. doi: 10.1038/nri.2017.19
40. Nemazee DA, Bürki K. Clonal Deletion of B Lymphocytes in a Transgenic Mouse Bearing Anti-MHC Class I Antibody Genes. Nature (1989) 337(6207):562–6. doi: 10.1038/337562a0
41. Noviski M, Tan C, Huizar J, Vykunta V, Mueller JL, Zikherman J. Optimal Development of Mature B Cells Requires Recognition of Endogenous Antigens. J Immunol (2019) 203(2):418. doi: 10.4049/jimmunol.1900175
42. Goodnow CC, Crosbie J, Jorgensen H, Brink RA, Basten A. Induction of Self-Tolerance in Mature Peripheral B Lymphocytes. Nature (1989) 342(6248):385–91. doi: 10.1038/342385a0
43. Merrell KT, Benschop RJ, Gauld SB, Aviszus K, Decote-Ricardo D, Wysocki LJ, et al. Identification of Anergic B Cells Within a Wild-Type Repertoire. Immunity (2006) 25(6):953–62. doi: 10.1016/j.immuni.2006.10.017
44. Yurasov S, Wardemann H, Hammersen J, Tsuiji M, Meffre E, Pascual V, et al. Defective B Cell Tolerance Checkpoints in Systemic Lupus Erythematosus. J Exp Med (2005) 201(5):703–11. doi: 10.1084/jem.20042251
45. Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, et al. Altered Immunoglobulin Expression and Functional Silencing of Self-Reactive B Lymphocytes in Transgenic Mice. Nature (1988) 334(6184):676–82. doi: 10.1038/334676a0
46. Burnett DL, Reed JH, Christ D, Goodnow CC. Clonal Redemption and Clonal Anergy as Mechanisms to Balance B Cell Tolerance and Immunity. Immunol Rev (2019) 292(1):61–75. doi: 10.1111/imr.12808
47. Nickerson KM, Christensen SR, Cullen JL, Meng W, Luning Prak ET, Shlomchik MJ. TLR9 Promotes Tolerance by Restricting Survival of Anergic Anti-DNA B Cells, Yet Is Also Required for Their Activation. J Immunol (2013) 190(4):1447–56. doi: 10.4049/jimmunol.1202115
48. Burnett DL, Langley DB, Schofield P, Hermes JR, Chan TD, Jackson J, et al. Germinal Center Antibody Mutation Trajectories Are Determined by Rapid Self/Foreign Discrimination. Science (2018) 360(6385):223–26. doi: 10.1126/science.aao3859
49. Burnett DL, Langley DB, Schofield P, Hermes JR, Chan TD, Jackson J, et al. Conformational Diversity Facilitates Antibody Mutation Trajectories and Discrimination Between Foreign and Self-Antigens. Proc Natl Acad Sci USA (2020) S117(36):22341–50. doi: 10.1073/pnas.2005102117
50. Sabouri Z, Schofield P, Horikawa K, Spierings E, Kipling D, Randall KL, et al. Redemption of Autoantibodies on Anergic B Cells by Variable-Region Glycosylation and Mutation Away From Self-Reactivity. Proc Natl Acad Sci USA (2014) 1110(25):2567–75. doi: 10.1073/pnas.2005102117
51. Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front Immunol (2021) 12:636568–. doi: 10.3389/fimmu.2021.636568
52. Staveley-O'Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, et al. Induction of Antigen-Specific T Cell Anergy: An Early Event in the Course of Tumor Progression. Proc Natl Acad Sci U S A (1998) 95(3):1178–83. doi: 10.1073/pnas.95.3.1178
53. Zou W. Regulatory T Cells, Tumour Immunity and Immunotherapy. Nat Rev Immunol (2006) 6(4):295–307. doi: 10.1038/nri1806
54. Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of Tumor-Derived Prostaglandin-E2 Blocks the Induction of Myeloid-Derived Suppressor Cells and Recovers Natural Killer Cell Activity. Clin Cancer Res an Off J Am Assoc Cancer Res (2014) 20(15):4096–106. doi: 10.1158/1078-0432.CCR-14-0635
55. Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol (2016) 37(3):208–20. doi: 10.1016/j.it.2016.01.004
56. Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of Vascular Endothelial Growth Factor by Human Tumors Inhibits the Functional Maturation of Dendritic Cells. Nat Med (1996) 2(10):1096–103. doi: 10.1038/nm1096-1096
58. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK Cells Stimulate Recruitment of Cdc1 Into the Tumor Microenvironment Promoting Cancer Immune Control. Cell (2018) 172(5):1022–37.e14. doi: 10.1016/j.cell.2018.01.004
59. Domínguez-Soto A, Sierra-Filardi E, Puig-Kröger A, Pérez-Maceda B, Gómez-Aguado F, Corcuera MT, et al. Dendritic Cell-Specific ICAM-3–Grabbing Nonintegrin Expression on M2-Polarized and Tumor-Associated Macrophages Is Macrophage-CSF Dependent and Enhanced by Tumor-Derived IL-6 and IL-10. J Immunol (2011) 186(4):2192–200. doi: 10.4049/jimmunol.1000475
60. McKallip R, Li R, Ladisch S. Tumor Gangliosides Inhibit the Tumor-Specific Immune Response. J Immunol (1999) 163(7):3718–26.
61. Driessens G, Kline J, Gajewski TF. Costimulatory and Coinhibitory Receptors in Anti-Tumor Immunity. Immunol Rev (2009) 229(1):126–44. doi: 10.1111/j.1600-065X.2009.00771.x
62. Sasidharan Nair V, Elkord E. Immune Checkpoint Inhibitors in Cancer Therapy: A Focus on T-Regulatory Cells. Immunol Cell Biol (2018) 96(1):21–33. doi: 10.1111/imcb.1003
63. Rudd CE, Schneider H. Unifying Concepts in CD28, ICOS and CTLA4 Co-Receptor Signalling. Nat Rev Immunol (2003) 3(7):544–56. doi: 10.1038/nri1131
64. Keir ME, Sharpe AH. The B7/CD28 Costimulatory Family in Autoimmunity. Immunol Rev (2005) 204:128–43. doi: 10.1111/j.0105-2896.2005.00242.x
65. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science (2011) 332(6029):600–3. doi: 10.1126/science.1202947
66. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) Bind With Similar Avidities But Distinct Kinetics to CD28 and CTLA-4 Receptors. Immunity (1994) 1(9):793–801. doi: 10.1016/S1074-7613(94)80021-9
67. Peach RJ, Bajorath J, Brady W, Leytze G, Greene J, Naemura J, et al. Complementarity Determining Region 1 (CDR1)- and CDR3-Analogous Regions in CTLA-4 and CD28 Determine the Binding to B7-1. J Exp Med (1994) 180(6):2049–58. doi: 10.1084/jem.180.6.2049
68. Appleman LJ, Berezovskaya A, Grass I, Boussiotis VA. CD28 Costimulation Mediates T Cell Expansion via IL-2-Independent and IL-2-Dependent Regulation of Cell Cycle Progression. J Immunol (2000) 164(1):144–51. doi: 10.4049/jimmunol.164.1.144
69. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol (2018) 8:86. doi: 10.3389/fonc.2018.00086
70. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor Antigen-Specific CD8 T Cells Infiltrating the Tumor Express High Levels of PD-1 and are Functionally Impaired. Blood (2009) 114(8):1537–44. doi: 10.1182/blood-2008-12-195792
71. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
72. Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. Safety Profiles of Anti-CTLA-4 and Anti-PD-1 Antibodies Alone and in Combination. Nat Rev Clin Oncol (2016) 13(8):473–86. doi: 10.1038/nrclinonc.2016.58
73. Sznol M, Ferrucci PF, Hogg D, Atkins MB, Wolter P, Guidoboni M, et al. Pooled Analysis Safety Profile of Nivolumab and Ipilimumab Combination Therapy in Patients With Advanced Melanoma. J Clin Oncol (2017) 35(34):3815–22. doi: 10.1200/JCO.2016.72.1167
74. Eun Y, Kim IY, Sun JM, Lee J, Cha HS, Koh EM, et al. Risk Factors for Immune-Related Adverse Events Associated With Anti-PD-1 Pembrolizumab. Sci Rep (2019) 9(1):14039. doi: 10.1038/s41598-019-50574-6
75. Diehl A, Yarchoan M, Hopkins A, Jaffee E, Grossman SA. Relationships Between Lymphocyte Counts and Treatment-Related Toxicities and Clinical Responses in Patients With Solid Tumors Treated With PD-1 Checkpoint Inhibitors. Oncotarget (2017) 8(69):114268–80. doi: 10.18632/oncotarget.23217
76. Oyanagi J, Koh Y, Sato K, Mori K, Teraoka S, Akamatsu H, et al. Predictive Value of Serum Protein Levels in Patients With Advanced Non-Small Cell Lung Cancer Treated With Nivolumab. Lung Cancer (2019) 132:107–13. doi: 10.1016/j.lungcan.2019.03.020
77. Osorio JC, Ni A, Chaft JE, Pollina R, Kasler MK, Stephens D, et al. Antibody-Mediated Thyroid Dysfunction During T-Cell Checkpoint Blockade in Patients With non-Small-Cell Lung Cancer. Ann Oncol (2017) 28(3):583–9. doi: 10.1093/annonc/mdw640
78. Refae S, Gal J, Ebran N, Otto J, Borchiellini D, Peyrade F, et al. Germinal Immunogenetics Predict Treatment Outcome for PD-1/PD-L1 Checkpoint Inhibitors. Investigat New Drugs (2020) 38(1):160–71. doi: 10.1007/s10637-019-00845-w
79. Bins S, Basak EA, El Bouazzaoui S, Koolen SLW, Oomen-de Hoop E, van der Leest CH, et al. Association Between Single-Nucleotide Polymorphisms and Adverse Events in Nivolumab-Treated Non-Small Cell Lung Cancer Patients. Br J Cancer (2018) 118(10):1296–301. doi: 10.1038/s41416-018-0074-1
80. Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L, et al. Intestinal Microbiome Analyses Identify Melanoma Patients at Risk for Checkpoint-Blockade-Induced Colitis. Nat Commun (2016) 7:10391. doi: 10.1038/ncomms10391
81. Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline Gut Microbiota Predicts Clinical Response and Colitis in Metastatic Melanoma Patients Treated With Ipilimumab. Ann Oncol (2017) 28(6):1368–79. doi: 10.1093/annonc/mdx108
82. Owen CN, Bai X, Quah T, Lo SN, Allayous C, Callaghan S, et al. Delayed Immune-Related Adverse Events With Anti-PD-1-Based Immunotherapy in Melanoma. Ann Oncol (2021) 32(7):917–25. doi: 10.1016/j.annonc.2021.03.204
83. Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab Plus Ipilimumab Versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med (2018) 378(14):1277–90. doi: 10.1056/NEJMoa1712126
84. Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse Effects of Immune-Checkpoint Inhibitors: Epidemiology, Management and Surveillance. Nat Rev Clin Oncol (2019) 16(9):563–80. doi: 10.1038/s41571-019-0218-0
85. De Velasco G, Je Y, Bossé D, Awad MM, Ott PA, Moreira RB, et al. Comprehensive Meta-Analysis of Key Immune-Related Adverse Events From CTLA-4 and PD-1/PD-L1 Inhibitors in Cancer Patients. Cancer Immunol Res (2017) 5(4):312–8. doi: 10.1158/2326-6066.CIR-16-0237
86. Barroso-Sousa R, Barry WT, Garrido-Castro AC, Hodi FS, Min L, Krop IE, et al. Incidence of Endocrine Dysfunction Following the Use of Different Immune Checkpoint Inhibitor Regimens: A Systematic Review and Meta-Analysis. JAMA Oncol (2018) 4(2):173–82. doi: 10.1001/jamaoncol.2017.3064
87. Chen C, Cohrs CM, Stertmann J, Bozsak R, Speier S. Human Beta Cell Mass and Function in Diabetes: Recent Advances in Knowledge and Technologies to Understand Disease Pathogenesis. Mol Metab (2017) 6(9):943–57. doi: 10.1016/j.molmet.2017.06.019
88. Orlov S, Salari F, Kashat L, Walfish PG. Induction of Painless Thyroiditis in Patients Receiving Programmed Death 1 Receptor Immunotherapy for Metastatic Malignancies. J Clin Endocrinol Metab (2015) 100(5):1738–41. doi: 10.1210/jc.2014-4560
89. Lee H, Hodi FS, Giobbie-Hurder A, Ott PA, Buchbinder EI, Haq R, et al. Characterization of Thyroid Disorders in Patients Receiving Immune Checkpoint Inhibition Therapy. Cancer Immunol Res (2017) 5(12):1133–40. doi: 10.1158/2326-6066.CIR-17-0208
90. Muir CA, Clifton-Bligh RJ, Long GV, Scolyer RA, Lo SN, Carlino MS, et al. Thyroid Immune-Related Adverse Events Following Immune Checkpoint Inhibitor Treatment. J Clin Endocrinol Metab (2021) 106(9):e3704–13. doi: 10.1210/clinem/dgab263
91. McLachlan SM, Rapoport B. Breaking Tolerance to Thyroid Antigens: Changing Concepts in Thyroid Autoimmunity. Endocrine Rev (2014) 35(1):59–105. doi: 10.1210/er.2013-1055
92. Yasuda Y, Iwama S, Sugiyama D, Okuji T, Kobayashi T, Ito M, et al. CD4+ T Cells are Essential for the Development of Destructive Thyroiditis Induced by Anti–PD-1 Antibody in Thyroglobulin-Immunized Mice. Sci Trans Med (2021) 13(593):eabb7495. doi: 10.1126/scitranslmed.abb7495
93. Álvarez-Sierra D, Marín-Sánchez A, Ruiz-Blázquez P, de Jesús Gil C, Iglesias-Felip C, González Ó, et al. Analysis of the PD-1/PD-L1 Axis in Human Autoimmune Thyroid Disease: Insights Into Pathogenesis and Clues to Immunotherapy Associated Thyroid Autoimmunity. J Autoimmun (2019) 103:102285. doi: 10.1016/j.jaut.2019.05.013
94. Kobayashi T, Iwama S, Yasuda Y, Okada N, Tsunekawa T, Onoue T, et al. Patients With Antithyroid Antibodies Are Prone To Develop Destructive Thyroiditis by Nivolumab: A Prospective Study. J Endocr Soc (2018) 2(3):241–51. doi: 10.1210/js.2017-00432
95. Kimbara S, Fujiwara Y, Iwama S, Ohashi K, Kuchiba A, Arima H, et al. Association of Antithyroglobulin Antibodies With the Development of Thyroid Dysfunction Induced by Nivolumab. Cancer Sci (2018) 109(11):3583–90. doi: 10.1111/cas.13800
96. Khan Z, Hammer C, Carroll J, Di Nucci F, Acosta SL, Maiya V, et al. Genetic Variation Associated With Thyroid Autoimmunity Shapes the Systemic Immune Response to PD-1 Checkpoint Blockade. Nat Commun (2021) 12(1):3355. doi: 10.1038/s41467-021-23661-4
97. Mazarico I, Capel I, Giménez-Palop O, Albert L, Berges I, Luchtenberg F, et al. Low Frequency of Positive Antithyroid Antibodies is Observed in Patients With Thyroid Dysfunction Related to Immune Check Point Inhibitors. J Endocrinol Invest (2019) 42(12):1443–50. doi: 10.1007/s40618-019-01058-x
98. Nguyen H, Shah K, Waguespack SG, Hu MI, Habra MA, Cabanillas ME, et al. Immune Checkpoint Inhibitor Related Hypophysitis: Diagnostic Criteria and Recovery Patterns. Endocr Relat Cancer (2021) 28(7):419–31. doi: 10.1530/ERC-20-0513
99. Faje A, Reynolds K, Zubiri L, Lawrence D, Cohen JV, Sullivan RJ, et al. Hypophysitis Secondary to Nivolumab and Pembrolizumab Is a Clinical Entity Distinct From Ipilimumab-Associated Hypophysitis. Eur J Endocrinol (2019) 181(3):211–9. doi: 10.1530/EJE-19-0238
100. Faje AT, Sullivan R, Lawrence D, Tritos NA, Fadden R, Klibanski A, et al. Ipilimumab-Induced Hypophysitis: A Detailed Longitudinal Analysis in a Large Cohort of Patients With Metastatic Melanoma. J Clin Endocrinol Metab (2014) 99(11):4078–85. doi: 10.1210/jc.2014-2306
101. Iwama S, De Remigis A, Callahan MK, Slovin SF, Wolchok JD, Caturegli P. Pituitary Expression of CTLA-4 Mediates Hypophysitis Secondary to Administration of CTLA-4 Blocking Antibody. Sci Transl Med (2014) 6(230):230ra45. doi: 10.1126/scitranslmed.3008002
102. Laurent S, Queirolo P, Boero S, Salvi S, Piccioli P, Boccardo S, et al. The Engagement of CTLA-4 on Primary Melanoma Cell Lines Induces Antibody-Dependent Cellular Cytotoxicity and TNF-α Production. J Transl Med (2013) 11:108. doi: 10.1186/1479-5876-11-108
103. Faje A. Immunotherapy and Hypophysitis: Clinical Presentation, Treatment, and Biologic Insights. Pituitary (2016) 19(1):82–92. doi: 10.1007/s11102-015-0671-4
104. Caturegli P, Di Dalmazi G, Lombardi M, Grosso F, Larman HB, Larman T, et al. Hypophysitis Secondary to Cytotoxic T-Lymphocyte-Associated Protein 4 Blockade: Insights Into Pathogenesis From an Autopsy Series. Am J Pathol (2016) 186(12):3225–35. doi: 10.1016/j.ajpath.2016.08.020
105. Quandt Z, Young A, Anderson M. Immune Checkpoint Inhibitor Diabetes Mellitus: A Novel Form of Autoimmune Diabetes. Clin Exp Immunol (2020) 200(2):131–40. doi: 10.1111/cei.13424
106. de Filette JMK, Pen JJ, Decoster L, Vissers T, Bravenboer B, van der Auwera BJ, et al. Immune Checkpoint Inhibitors and Type 1 Diabetes Mellitus: A Case Report and Systematic Review. Eur J Endocrinol (2019) 181(3):363–74. doi: 10.1530/EJE-19-0291
107. Stamatouli AM, Quandt Z, Perdigoto AL, Clark PL, Kluger H, Weiss SA, et al. Collateral Damage: Insulin-Dependent Diabetes Induced With Checkpoint Inhibitors. Diabetes (2018) 67(8):1471–80. doi: 10.2337/dbi18-0002
108. Wright JJ, Salem J-E, Johnson DB, Lebrun-Vignes B, Stamatouli A, Thomas JW, et al. Increased Reporting of Immune Checkpoint Inhibitor-Associated Diabetes. Diabetes Care (2018) 41(12):e150–e1. doi: 10.2337/dc18-1465
109. Yoneda S, Imagawa A, Hosokawa Y, Baden MY, Kimura T, Uno S, et al. T-Lymphocyte Infiltration to Islets in the Pancreas of a Patient Who Developed Type 1 Diabetes After Administration of Immune Checkpoint Inhibitors. Diabetes Care (2019) 42(7):e116–e8. doi: 10.2337/dc18-2518
110. Bingley PJ. Clinical Applications of Diabetes Antibody Testing. J Clin Endocrinol Metab (2010) 95(1):25–33. doi: 10.1210/jc.2009-1365
111. Grouthier V, Lebrun-Vignes B, Moey M, Johnson DB, Moslehi JJ, Salem JE, et al. Immune Checkpoint Inhibitor-Associated Primary Adrenal Insufficiency: WHO VigiBase Report Analysis. Oncologist (2020) 25(8):696–701. doi: 10.1634/theoncologist.2019-0555
112. Bornstein SR, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, et al. Diagnosis and Treatment of Primary Adrenal Insufficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2016) 101(2):364–89. doi: 10.1210/jc.2015-1710
113. Shi Y, Shen M, Zheng X, Yang T. Immune Checkpoint Inhibitor-Induced Adrenalitis and Primary Adrenal Insufficiency: Systematic Review and Optimal Management. Endocr Pract (2021) 27(2):165–9. doi: 10.1016/j.eprac.2020.09.016
114. Paepegaey AC, Lheure C, Ratour C, Lethielleux G, Clerc J, Bertherat J, et al. Polyendocrinopathy Resulting From Pembrolizumab in a Patient With a Malignant Melanoma. J Endocr Soc (2017) 1(6):646–9. doi: 10.1210/js.2017-00170
115. Hescot S, Haissaguerre M, Pautier P, Kuhn E, Schlumberger M, Berdelou A. Immunotherapy-Induced Addison's Disease: A Rare, Persistent and Potentially Lethal Side-Effect. Eur J Cancer (Oxford Engl 1990) (2018) 97:57–8. doi: 10.1016/j.ejca.2018.04.001
116. Chen S, Sawicka J, Betterle C, Powell M, Prentice L, Volpato M, et al. Autoantibodies to Steroidogenic Enzymes in Autoimmune Polyglandular Syndrome, Addison's Disease, and Premature Ovarian Failure. J Clin Endocrinol Metab (1996) 81(5):1871–6. doi; 10.1210/jcem.81.5.8626850
117. Ueda H, Howson JMM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-Cell Regulatory Gene CTLA4 With Susceptibility to Autoimmune Disease. Nature (2003) 423(6939):506–11. doi: 10.1038/nature01621
118. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative Disorders With Early Lethality in Mice Deficient in Ctla-4. Science (1995) 270(5238):985–8. doi: 10.1126/science.270.5238.985
119. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 Leads to Massive Lymphoproliferation and Fatal Multiorgan Tissue Destruction, Revealing a Critical Negative Regulatory Role of CTLA-4. Immunity (1995) 3(5):541–7. doi: 10.1016/1074-7613(95)90125-6
120. Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, et al. Immune Dysregulation in Human Subjects With Heterozygous Germline Mutations in CTLA4. Science (2014) 345(6204):1623–7. doi: 10.1126/science.1255904
121. Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD, et al. Deletion of CTLA-4 on Regulatory T Cells During Adulthood Leads to Resistance to Autoimmunity. J Exp Med (2015) 212(10):1603–21. doi: 10.1084/jem.20141030
122. Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab Monotherapy in Patients With Pretreated Advanced Melanoma: A Randomised, Double-Blind, Multicentre, Phase 2, Dose-Ranging Study. Lancet Oncol (2010) 11(2):155–64. doi: 10.1016/S1470-2045(09)70334-1
123. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients With LRBA Deficiency Show CTLA4 Loss and Immune Dysregulation Responsive to Abatacept Therapy. Science (2015) 349(6246):436–40. doi: 10.1126/science.aaa1663
124. Nielsen C, Hansen D, Husby S, Jacobsen BB, Lillevang ST. Association of a Putative Regulatory Polymorphism in the PD-1 Gene With Susceptibility to Type 1 Diabetes. Tissue Antigens (2003) 62(6):492–7. doi: 10.1046/j.1399-0039.2003.00136.x
125. Velázquez-Cruz R, Orozco L, Espinosa-Rosales F, Carreño-Manjarrez R, Solís-Vallejo E, López-Lara ND, et al. Association of PDCD1 Polymorphisms With Childhood-Onset Systemic Lupus Erythematosus. Eur J Hum Genet (2007) 15(3):336–41. doi: 10.1038/sj.ejhg.5201767
126. Gianchecchi E, Delfino DV, Fierabracci A. Recent Insights Into the Role of the PD-1/PD-L1 Pathway in Immunological Tolerance and Autoimmunity. Autoimmun Rev (2013) 12(11):1091–100. doi: 10.1016/j.autrev.2013.05.003
127. Nishimura H, Minato N, Nakano T, Honjo T. Immunological Studies on PD-1 Deficient Mice: Implication of PD-1 as a Negative Regulator for B Cell Responses. Int Immunol (1998) 10(10):1563–72. doi: 10.1093/intimm/10.10.1563
128. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of Lupus-Like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity (1999) 11(2):141–51. doi: 10.1016/S1074-7613(00)80089-8
129. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune Dilated Cardiomyopathy in PD-1 Receptor-Deficient Mice. Science (2001) 291(5502):319–22. doi: 10.1126/science.291.5502.319
130. Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD-Pdcd1-/- Mice as an Efficient Animal Model of Type I Diabetes. Proc Natl Acad Sci U S A (2005) 102(33):11823–8. doi: 10.1073/pnas.0505497102
131. Breunis WB, Tarazona-Santos E, Chen R, Kiley M, Rosenberg SA, Chanock SJ. Influence of Cytotoxic T Lymphocyte-Associated Antigen 4 (CTLA4) Common Polymorphisms on Outcome in Treatment of Melanoma Patients With CTLA-4 Blockade. J Immunother (2008) 31(6):586–90. doi: 10.1097/CJI.0b013e31817fd8f3
132. Queirolo P, Morabito A, Laurent S, Lastraioli S, Piccioli P, Ascierto PA, et al. Association of CTLA-4 Polymorphisms With Improved Overall Survival in Melanoma Patients Treated With CTLA-4 Blockade: A Pilot Study. Cancer Invest (2013) 31(5):336–45. doi: 10.3109/07357907.2013.793699
133. Vaidya B, Pearce SH, Charlton S, Marshall N, Rowan AD, Griffiths ID, et al. An Association Between the CTLA4 Exon 1 Polymorphism and Early Rheumatoid Arthritis With Autoimmune Endocrinopathies. Rheumatol (Oxford) (2002) 41(2):180–3. doi: 10.1093/rheumatology/41.2.180
134. Fang W, Zhang Z, Zhang J, Cai Z, Zeng H, Chen M, et al. Association of the CTLA4 Gene CT60/rs3087243 Single-Nucleotide Polymorphisms With Graves' Disease. BioMed Rep (2015) 3(5):691–6. doi: 10.3892/br.2015.493
135. Sun W, Zhang X, Wu J, Zhao W, Zhao S, Li M. Correlation of TSHR and CTLA-4 Single Nucleotide Polymorphisms With Graves Disease. Int J Genomics (2019) 2019:6982623–. doi: 10.1155/2019/6982623
136. Chen DP, Chu YC, Wen YH, Lin WT, Hour AL, Wang WT. Investigation of the Correlation Between Graves' Ophthalmopathy and CTLA4 Gene Polymorphism. J Clin Med (2019) 8(11):1842. doi: 10.3390/jcm8111842
137. Ting WH, Chien MN, Lo FS, Wang CH, Huang CY, Lin CL, et al. Association of Cytotoxic T-Lymphocyte-Associated Protein 4 (CTLA4) Gene Polymorphisms With Autoimmune Thyroid Disease in Children and Adults: Case-Control Study. PLoS One (2016) 11(4):e0154394. doi: 10.1371/journal.pone.0154394
138. Ikegami H, Awata T, Kawasaki E, Kobayashi T, Maruyama T, Nakanishi K, et al. The Association of CTLA4 Polymorphism With Type 1 Diabetes is Concentrated in Patients Complicated With Autoimmune Thyroid Disease: A Multicenter Collaborative Study in Japan. J Clin Endocrinol Metab (2006) 91(3):1087–92. doi: 10.1210/jc.2005-1407
139. Bufalo NE, Dos Santos RB, Rocha AG, Teodoro L, Romaldini JH, Ward LS. Polymorphisms of the Genes CTLA4, PTPN22, CD40, and PPARG and Their Roles in Graves' Disease: Susceptibility and Clinical Features. Endocrine (2021) 71(1):104–12. doi: 10.1007/s12020-020-02337-x
140. Ramgopal S, Rathika C, Padma MR, Murali V, Arun K, Kamaludeen MN, et al. Interaction of HLA-DRB1* Alleles and CTLA4 (+49 AG) Gene Polymorphism in Autoimmune Thyroid Disease. Gene (2018) 642:430–8. doi: 10.1016/j.gene.2017.11.057
141. Zhernakova A, Eerligh P, Barrera P, Wesoly JZ, Huizinga TW, Roep BO, et al. CTLA4 is Differentially Associated With Autoimmune Diseases in the Dutch Population. Hum Genet (2005) 118(1):58–66. doi: 10.1007/s00439-005-0006-z
142. Jin P, Xiang B, Huang G, Zhou Z. The Association of Cytotoxic T-Lymphocyte Antigen-4 + 49A/G and CT60 Polymorphisms With Type 1 Diabetes and Latent Autoimmune Diabetes in Chinese Adults. J Endocrinol Invest (2015) 38(2):149–54. doi: 10.1007/s40618-014-0162-x
143. Hudson LL, Rocca K, Song YW, Pandey JP. CTLA-4 Gene Polymorphisms in Systemic Lupus Erythematosus: A Highly Significant Association With a Determinant in the Promoter Region. Hum Genet (2002) 111(4-5):452–5. doi: 10.1007/s00439-002-0807-2
144. Nisticò L, Buzzetti R, Pritchard LE, van der Auwera B, Giovannini C, Bosi E, et al. The CTLA-4 Gene Region of Chromosome 2q33 Is Linked to, and Associated With, Type 1 Diabetes. Belgian Diabetes Registry. Hum Mol Genet (1996) 5(7):1075–80. doi: 10.1093/hmg/5.7.1075
145. Qian C, Guo H, Chen X, Shi A, Li S, Wang X, et al. Association of PD-1 and PD-L1 Genetic Polymorphyisms With Type 1 Diabetes Susceptibility. J Diabetes Res (2018) 2018:1614683. doi: 10.1155/2018/1614683
146. Wolff ASB, Mitchell AL, Cordell HJ, Short A, Skinningsrud B, Ollier W, et al. CTLA-4 as a Genetic Determinant in Autoimmune Addison's Disease. Genes Immunity (2015) 16(6):430–6. doi: 10.1038/gene.2015.27
147. Blomhoff A, Kemp EH, Gawkrodger DJ, Weetman AP, Husebye ES, Akselsen HE, et al. CTLA4 Polymorphisms Are Associated With Vitiligo, in Patients With Concomitant Autoimmune Diseases. Pigment Cell Res (2005) 18(1):55–8. doi: 10.1111/j.1600-0749.2004.00196.x
148. Tanasilovic S, Popadic S, Medenica L, Popadic D. Pemphigus Vulgaris and Pemphigus Foliaceus Determined by CD86 and CTLA4 Polymorphisms. Clinics Dermatol (2017) 35(2):236–41. doi: 10.1016/j.clindermatol.2016.05.021
149. Fernández-Mestre M, Sánchez K, Balbás O, Gendzekhzadze K, Ogando V, Cabrera M, et al. Influence of CTLA-4 Gene Polymorphism in Autoimmune and Infectious Diseases. Hum Immunol (2009) 70(7):532–5. doi: 10.1016/j.humimm.2009.03.016
150. Dalla-Costa R, Pincerati MR, Beltrame MH, Malheiros D, Petzl-Erler ML. Polymorphisms in the 2q33 and 3q21 Chromosome Regions Including T-Cell Coreceptor and Ligand Genes may Influence Susceptibility to Pemphigus Foliaceus. Hum Immunol (2010) 71(8):809–17. doi: 10.1016/j.humimm.2010.04.001
151. Song GG, Kim JH, Kim YH, Lee YH. Association Between CTLA-4 Polymorphisms and Susceptibility to Celiac Disease: A Meta-Analysis. Hum Immunol (2013) 74(9):1214–8. doi: 10.1016/j.humimm.2013.05.014
152. Djilali-Saiah I, Schmitz J, Harfouch-Hammoud E, Mougenot JF, Bach JF, Caillat-Zucman S. CTLA-4 Gene Polymorphism Is Associated With Predisposition to Coeliac Disease. Gut (1998) 43(2):187–9. doi: 10.1136/gut.43.2.187
153. Plenge RM, Padyukov L, Remmers EF, Purcell S, Lee AT, Karlson EW, et al. Replication of Putative Candidate-Gene Associations With Rheumatoid Arthritis in >4,000 Samples From North America and Sweden: Association of Susceptibility With PTPN22, CTLA4, and PADI4. Am J Hum Genet (2005) 77(6):1044–60. doi: 10.1086/498651
154. Prokunina L, Castillejo-López C, Oberg F, Gunnarsson I, Berg L, Magnusson V, et al. A Regulatory Polymorphism in PDCD1 Is Associated With Susceptibility to Systemic Lupus Erythematosus in Humans. Nat Genet (2002) 32(4):666–9. doi: 10.1038/ng1020
155. Kong EK, Prokunina-Olsson L, Wong WH, Lau CS, Chan TM, Alarcón-Riquelme M, et al. A New Haplotype of PDCD1 Is Associated With Rheumatoid Arthritis in Hong Kong Chinese. Arthritis Rheumatol (2005) 52(4):1058–62. doi: 10.1002/art.20966
156. Lin SC, Yen JH, Tsai JJ, Tsai WC, Ou TT, Liu HW, et al. Association of a Programmed Death 1 Gene Polymorphism With the Development of Rheumatoid Arthritis, But Not Systemic Lupus Erythematosus. Arthritis Rheumatol (2004) 50(3):770–5. doi: 10.1002/art.20040
157. Cai GM, Gao Z, Yue YX, Xie YC, Gao X, Hao HJ, et al. Association Between CTLA-4 Gene Polymorphism and Myasthenia Gravis in a Chinese Cohort. J Clin Neurosci (2019) 69:31–7. doi: 10.1016/j.jocn.2019.08.079
158. Sun L, Meng Y, Xie Y, Zhang H, Zhang Z, Wang X, et al. CTLA4 Variants and Haplotype Contribute Genetic Susceptibility to Myasthenia Gravis in Northern Chinese Population. PLoS One (2014) 9(7):e101986. doi: 10.1371/journal.pone.0101986
159. Huang D, Liu L, Norén K, Xia S, Trifunovic J, Pirskanen R, et al. Genetic Association of Ctla-4 to Myasthenia Gravis With Thymoma. J Neuroimmunol (1998) 88(1):192–8. doi: 10.1016/S0165-5728(98)00119-2
160. Zheng K, Zhang J, Zhang P, Guo Y. PTPN22 and CTLA-4 Gene Polymorphisms in Resected Thymomas and Thymus for Myasthenia Gravis. Thorac Cancer (2012) 3(4):307–12. doi: 10.1111/j.1759-7714.2012.00121.x
161. Chuang WY, Ströbel P, Gold R, Nix W, Schalke B, Kiefer R, et al. A CTLA4high Genotype Is Associated With Myasthenia Gravis in Thymoma Patients. Ann Neurol (2005) 58(4):644–8. doi: 10.1002/ana.20577
162. Li HF, Hong Y, Zhang X, Xie Y, Skeie GO, Hao HJ, et al. Gene Polymorphisms for Both Auto-Antigen and Immune-Modulating Proteins Are Associated With the Susceptibility of Autoimmune Myasthenia Gravis. Mol Neurobiol (2017) 54(6):4771–80. doi: 10.1007/s12035-016-0024-y
163. Wang XB, Kakoulidou M, Qiu Q, Giscombe R, Huang D, Pirskanen R, et al. CDS1 and Promoter Single Nucleotide Polymorphisms of the CTLA-4 Gene in Human Myasthenia Gravis. Genes Immun (2002) 3(1):46–9. doi: 10.1038/sj.gene.6363816
164. Bertsias GK, Nakou M, Choulaki C, Raptopoulou A, Papadimitraki E, Goulielmos G, et al. Genetic, Immunologic, and Immunohistochemical Analysis of the Programmed Death 1/Programmed Death Ligand 1 Pathway in Human Systemic Lupus Erythematosus. Arthritis Rheumatol (2009) 60(1):207–18. doi: 10.1002/art.24227
165. Downie-Doyle S, Bayat N, Rischmueller M, Lester S. Influence of CTLA4 Haplotypes on Susceptibility and Some Extraglandular Manifestations in Primary Sjögren's Syndrome. Arthritis Rheumatol (2006) 54(8):2434–40. doi: 10.1002/art.22004
166. Mahmoudi H, Ebrahimi E, Daneshpazhooh M, Balighi K, Mirzazadeh A, Elikaei Behjati S, et al. Single-Nucleotide Polymorphisms Associated With Pemphigus Vulgaris: Potent Markers for Better Treatment and Personalized Medicine. Int J Immunogenet (2020) 47(1):41–9. doi: 10.1111/iji.12451
167. Westra HJ, Martinez-Bonet M, Onengut-Gumuscu S, Lee A, Luo Y, Teslovich N, et al. Fine-Mapping and Functional Studies Highlight Potential Causal Variants for Rheumatoid Arthritis and Type 1 Diabetes. Nat Genet (2018) 50(10):1366–74. doi: 10.1038/s41588-018-0216-7
168. Fang M, Huang W, Mo D, Zhao W, Huang R. Association of Five Snps in Cytotoxic T-Lymphocyte Antigen 4 and Cancer Susceptibility: Evidence From 67 Studies. Cell Physiol Biochem (2018) 47(1):414–27. doi: 10.1159/000489953
169. Geng R, Song F, Yang X, Sun P, Hu J, Zhu C, et al. Association Between Cytotoxic T Lymphocyte Antigen-4 +49A/G, -1722t/C, and -1661A/G Polymorphisms and Cancer Risk: A Meta-Analysis. Tumour Biol (2014) 35(4):3627–39. doi: 10.1007/s13277-013-1480-x
170. Sun T, Hu Z, Shen H, Lin D. Genetic Polymorphisms in Cytotoxic T-Lymphocyte Antigen 4 and Cancer: The Dialectical Nature of Subtle Human Immune Dysregulation. Cancer Res (2009) 69(15):6011–4. doi: 10.1158/0008-5472.CAN-09-0176
171. Maillet D, Corbaux P, Stelmes JJ, Dalle S, Locatelli-Sanchez M, Perier-Muzet M, et al. Association Between Immune-Related Adverse Events and Long-Term Survival Outcomes in Patients Treated With Immune Checkpoint Inhibitors. Eur J Cancer (Oxford Engl 1990) (2020) 132:61–70. doi: 10.1016/j.ejca.2020.03.017
172. Concannon P, Rich SS, Nepom GT. Genetics of Type 1A Diabetes. N Engl J Med (2009) 360(16):1646–54. doi: 10.1056/NEJMra0808284
173. Lowe JR, Perry DJ, Salama AKS, Mathews CE, Moss LG, Hanks BA. Genetic Risk Analysis of a Patient With Fulminant Autoimmune Type 1 Diabetes Mellitus Secondary to Combination Ipilimumab and Nivolumab Immunotherapy. J Immunother Cancer (2016) 4:89–. doi: 10.1186/s40425-016-0196-z
Keywords: cancer, immune checkpoint inhibitor (ICI), immune related adverse events (irAE), immunotherapy, autoimmunity, genetic biomarkers
Citation: Chye A, Allen I, Barnet M and Burnett DL (2022) Insights Into the Host Contribution of Endocrine Associated Immune-Related Adverse Events to Immune Checkpoint Inhibition Therapy. Front. Oncol. 12:894015. doi: 10.3389/fonc.2022.894015
Received: 11 March 2022; Accepted: 10 June 2022;
Published: 14 July 2022.
Edited by:
Marijo Bilusic, University of Miami Health System, United StatesReviewed by:
Lisa Cordes, National Institutes of Health (NIH), United StatesMichelle Rengarajan, Harvard Medical School, United States
Copyright © 2022 Chye, Allen, Barnet and Burnett. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Megan Barnet, bS5iYXJuZXRAZ2FydmFuLm9yZy5hdQ==; Deborah L. Burnett, ZC5idXJuZXR0QGdhcnZhbi5vcmcuYXU=
†These authors have contributed equally to this work and share last authorship