 Gabriele Hintzen
Gabriele Hintzen Holger J. Dulat
Holger J. Dulat Erich Rajkovic
Erich Rajkovic
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 14 September 2022
Sec. Cancer Molecular Targets and Therapeutics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.892212
This article is part of the Research TopicWomen in Cancer Molecular Targets and Therapeutics, Volume II: 2022View all 23 articles
The epidermal growth factor receptor (EGFR) is a key player in the normal tissue physiology and the pathology of cancer. Therapeutic approaches have now been developed to target oncogenic genetic aberrations of EGFR, found in a subset of tumors, and to take advantage of overexpression of EGFR in tumors. The development of small-molecule inhibitors and anti-EGFR antibodies targeting EGFR activation have resulted in effective but limited treatment options for patients with mutated or wild-type EGFR-expressing cancers, while therapeutic approaches that deploy effectors of the adaptive or innate immune system are still undergoing development. This review discusses EGFR-targeting therapies acting through distinct molecular mechanisms to destroy EGFR-expressing cancer cells. The focus is on the successes and limitations of therapies targeting the activation of EGFR versus those that exploit the cytotoxic T cells and innate immune cells to target EGFR-expressing cancer cells. Moreover, we discuss alternative approaches that may have the potential to overcome limitations of current therapies; in particular the innate cell engagers are discussed. Furthermore, this review highlights the potential to combine innate cell engagers with immunotherapies, to maximize their effectiveness, or with unspecific cell therapies, to convert them into tumor-specific agents.
Epidermal growth factor receptor (EGFR, also known as ErbB1/HER1) is a cell-surface receptor tyrosine kinase that belongs to the ErbB family of receptors composed of four closely related members: EGFR (ErbB1/HER1), ErbB2 (HER2), ErbB3 (HER3) and ErbB4 (HER4) (1). Under normal physiological conditions, EGFR is expressed in nearly all cell types and tissues, with the exception of hematopoietic lineage cells, and those in bone marrow, spleen, soft tissues, adrenal gland and specific brain tissues, where the EGFR protein is undetectable (2).
EGFR exerts multifaceted functions in the maintenance of normal epithelial tissue homeostasis by driving cell proliferation, growth, differentiation, migration, and survival through a ligand-dependent activation of its kinase activity, required for the initiation of multiple signaling pathways within the cell (3, 4). The binding of ligands such as epidermal growth factor (EGF), amphiregulin, epiregulin, transforming growth factor-α and others to the extracellular domain of EGFR leads to the formation of EGFR homodimers or heterodimers with the ErbB2, ErbB3 or ErbB4 receptors, the activation of the kinase activity and the transphosphorylation of the key tyrosine residues in the intracellular kinase domain and the C-terminal tail (1). These phosphorylated residues act as a scaffold for the binding of numerous signaling proteins, which initiate the RAS-RAF-MEK-ERK, AKT-PI3K, PLCγ1-PKC, JNK, and JAK-STAT3 signaling pathways (3). Moreover, it has been shown that EGFR dimers with a perturbed catalytic activity can also sustain cell survival signals using the kinase activity-independent scaffolding function (5, 6).
Considering the fundamental role of EGFR in maintaining the homeostasis of healthy tissue, it is not surprising that EGFR gain-of-function mutations are often detected in some tumor types. EGFR activating mutations in the tyrosine kinase domain leading to the ligand-independent activation of EGFR are frequently detected in non-small cell lung cancer (NSCLC) and glioblastoma but are rarely found in other tumor types (7–11), providing a possible foundation for the tumor type-specific responses to EGFR-targeted therapies and immunotherapies. The frequency of EGFR-activating mutations in tumor tissue also varies among different global demographics, with 30–40% of patients with NSCLC from East Asia exhibiting these mutations, but only 5–15% of patients of non-Asian origin (7, 12). In glioblastoma, EGFR aberrations frequently found also include mutations and various deletions in the extracellular domain of EGFR, with the truncated variant EGFRvIII, which lacks exons 2–7, being the most common deletion (13). Interestingly, although the EGFRvIII variant does not require ligand for its activation, it has a relatively weak constitutive kinase activity (8, 13) and the resulting growth advantage is believed to be conferred by an impaired endocytic pathway preventing physiological downregulation of the receptor (14).
In cancers that may not necessarily show high rates of EGFR-activating mutations, deregulation can also occur through overexpression. Numerous studies have reported overexpression of EGFR in 27–100% of solid tumors, including NSCLC, colorectal cancer (CRC), squamous cell carcinoma of the head and neck (SCCHN), gastric-gastroesophageal junction cancer, urothelial cancer, clear cell renal cell carcinoma, hepatocellular carcinoma, glioblastoma, pancreatic cancer, and breast cancer, among others (4, 6, 10, 15). Overexpression of EGFR may result from an EGFR copy number gain due to amplification of the genomic region comprising the EGFR gene locus (16–19) and can lead to increased capability to form ligand-independent EGFR homodimers and heterodimers (20). In particular EGFR/ErbB2 heterocomplexes show a strong ligand-independent constitutive activity, resistance to the ubiquitin-dependent degradation of heterodimers and high levels of persistent signaling (21). In several cancer types, EGFR expression levels correlate with the disease prognosis (15). In patients with head and neck, ovarian, cervical, bladder and esophageal cancers, elevated EGFR levels were found to be a strong prognostic factor and were associated with reduced recurrence-free or overall survival (15), while in patients with gastric, breast, endometrial and colorectal cancers, increased EGFR levels correlated with poor survival rates, but were considered a modest prognostic factor (15).
The gain-of-function alterations in EGFR, such as substitution mutations, deletions and insertions, and high levels of cell-surface EGFR expression have been proven to have a crucial role in sustaining cancer cell proliferation, growth and survival, and cancer progression (12, 15). This, without doubt, has highlighted EGFR as an attractive therapeutic target for the treatment of patients with EGFR-expressing cancer.
Broadly, there are two main types of therapies approved to target EGFR: small molecule tyrosine kinase inhibitors (TKIs) and anti-EGFR antibodies. TKIs act to inhibit EGFR kinase activity, thus attenuating downstream signal transduction (22); whereas anti-EGFR antibodies serve to block ligand binding to the extracellular portion of EGFR leading to inhibition of downstream signaling, but also have the capacity to leverage cytotoxic immune cells to induce anti-tumor antibody-dependent cellular cytotoxicity (ADCC) (23, 24). Both types of therapy have demonstrated efficacy in subsets of patients with certain tumor types, with TKIs exhibiting efficacy in, for example, NSCLC with EGFR-activating mutations (25), and anti-EGFR antibodies in tumor types such as metastatic CRC (mCRC) (9), and SCCHN (26). Despite the effectiveness of these agents, however, several drawbacks have emerged, such as the development of drug resistance (26, 27). As such, there remains a significant unmet need in EGFR-expressing tumors for therapies which induce long-term remissions.
Promising novel therapeutic approaches exploit the frequent overexpression of EGFR in a broad range of different cancers and the tumor immune microenvironment. A number of therapeutic agents including CAR-T cells, CAR-NK cells, bispecific T-cell engagers, and bispecific innate cell engagers – all of which are currently being investigated in preclinical and clinical studies - exploit approaches to harness effectors of the adaptive or innate immune system in order to bridge them with the tumor cell-surface EGFR and prime them for destruction of the EGFR-expressing cancer cells (28, 29).
EGFR TKIs are orally available ATP-competitive compounds that reversibly or irreversibly bind the ATP-binding site in the EGFR tyrosine kinase domain, thus preventing EGFR activation, transphosphorylation of tyrosine residues and transduction of downstream signaling pathways (Figure 1). With the exception of erlotinib, TKIs targeting EGFR have only been approved by the U.S. Food and Drug Administration (FDA) for the treatment of patients with NSCLC, whose tumors frequently have activating EGFR mutations; several generations of these are available for clinical use (30) (Table 1). In contrast, TKIs have shown limited efficacy in tumors where mutations in EGFR are not present, such as NSCLC with wild-type EGFR (102), and where EGFR-activating mutations are less common, such as in mCRC and SCCHN (9–11, 103, 104). As such, EGFR TKIs will mostly be discussed below in the context of NSCLC with EGFR-activating mutations. Clinical efficacy data from key randomized trials mentioned below are given in Table 1.
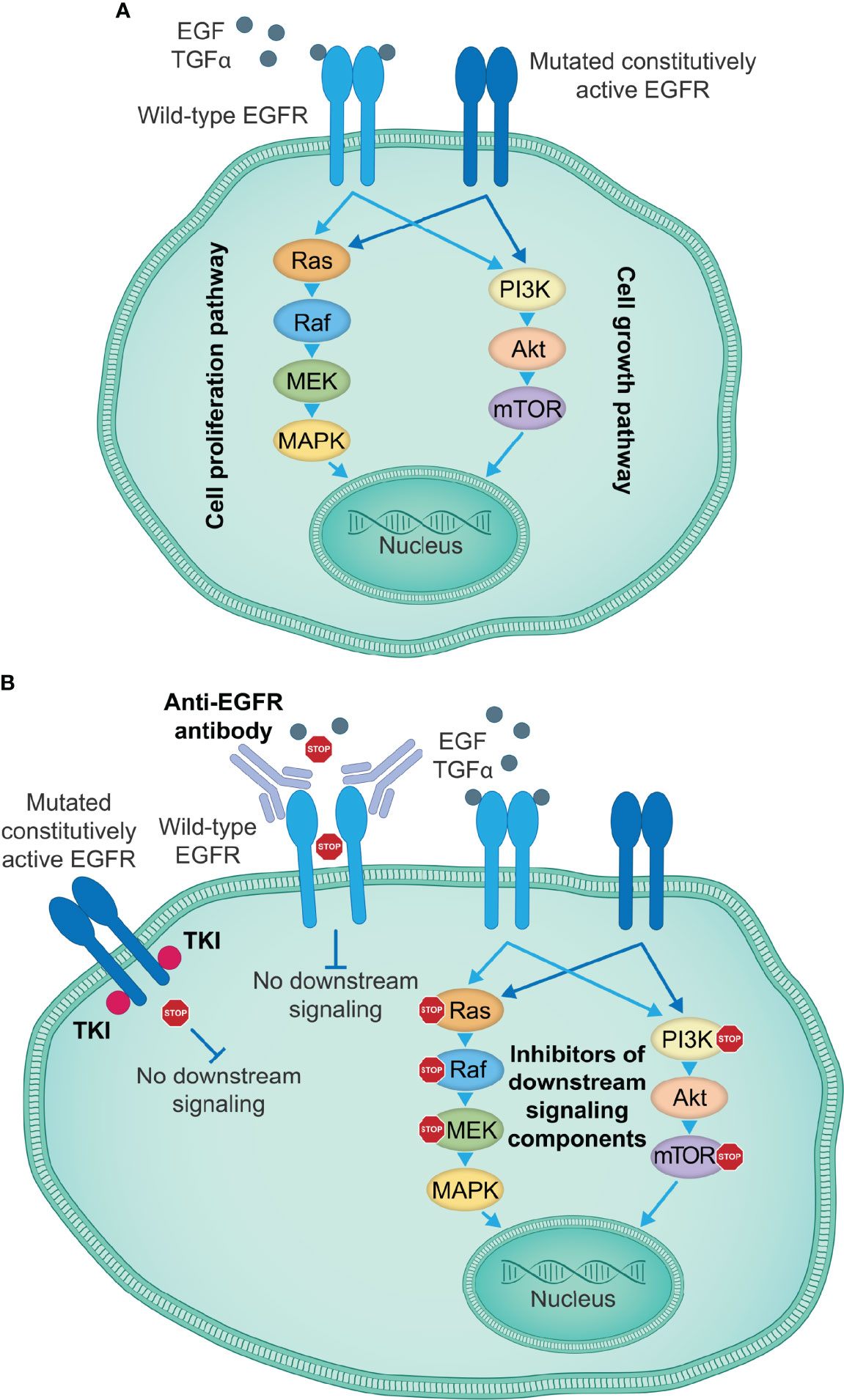
Figure 1 EGFR-targeting therapies inhibiting EGFR activation and signal transduction. (A) EGFR signaling induced by specific ligands, including EGF and TGFα among others, starts with the conformational switch upon binding of a ligand to the EGFR extracellular domain, the dimerization of EGFR monomers and transphosphorylation of intracellular tyrosine kinase domains, which creates docking sites for the adaptor molecules, leading to the activation of key downstream signaling pathways that govern cell proliferation and growth. Certain mutations in the EGFR kinase or extracellular domain induce a constitutively active state and ligand-independent oncogenic signaling downstream of activated EGFR, thus causing uncontrolled tumor cell proliferation and growth. (B) Currently approved therapeutic agents for the treatment of patients with lung or colorectal cancer, or SCCHN include TKIs targeting the EGFR kinase activity (e.g., first-generation small-molecule EGFR kinase inhibitors gefitinib, erlotinib and icotinib), which show particularly high efficacy in the presence of activating EGFR kinase domain mutations, and anti-EGFR antibodies (e.g., cetuximab) that prevent binding of a ligand to the wild-type EGFR and the activation of EGFR and the downstream signaling cascades. Other therapeutic approaches take advantage of individual signaling components downstream of EGFR to disrupt the EGFR signaling cascades and impair tumor cell proliferation and growth. EGF, epidermal growth factor; EGFR, EGF receptor; SCCHN, squamous cell carcinoma of the head and neck; TGFα, transforming growth factor α; TKIs, tyrosine kinase inhibitors.

Table 1 Clinically approved inhibitors of EGFR signaling, acquired resistance mechanisms, clinical efficacy, and safety profile identified in patients with cancer.
Erlotinib and gefitinib are two first-generation TKIs that bind reversibly to the active conformation of EGFR, blocking downstream signaling. Both are indicated for first-line use in patients with metastatic NSCLC carrying common EGFR sensitizing mutations in exons 18–21, such as the L858R missense mutation in exon 21 or exon 19 deletions, together accounting for more than 90% of exon 18–21 mutations in these patients (31, 105, 106). Erlotinib can also be indicated as a maintenance therapy or second- or subsequent-line therapy following disease progression and failure of at least one prior chemotherapy regimen (31). Several key phase III trials have demonstrated superior efficacy and progression-free survival (PFS) in patients treated with erlotinib or gefitinib versus standard chemotherapy regimens in patients with NSCLC exhibiting EGFR-sensitizing mutations, but no significant difference in overall survival (OS) was observed (33–37, 39, 42–45). These findings were confirmed by a meta-analysis of randomized trials comparing erlotinib/gefitinib monotherapy or the combination of erlotinib/gefitinib and chemotherapy with chemotherapy alone or placebo in patients with sensitizing EGFR mutation-positive-NSCLC, which showed a delayed disease progression when the treatment included gefitinib/erlotinib, but no effect on OS (107). Erlotinib is also indicated for the treatment of advanced or metastatic pancreatic cancer in combination with gemcitabine following a landmark phase III study, which demonstrated longer PFS and OS of the erlotinib combination versus gemcitabine alone (38). A third first-generation TKI, icotinib, is approved in China only for patients with advanced NSCLC carrying sensitizing EGFR mutations who have failed at least one prior chemotherapy regimen (108). The approval by the China National Medical Products Administration was based on the ICOGEN phase III study results that reported non-inferiority of icotinib versus gefitinib in terms of PFS and safety (46).
Despite proven efficacy of first-generation EGFR TKIs in subgroups of patients with EGFR-activating mutations in their tumors, resistance develops in most patients, with the median time to disease progression being around 12 months (34, 42, 109, 110). The underlying mechanism for acquired resistance is typically associated with the emergence of secondary EGFR mutations, that impair the binding of TKIs to EGFR, or alternatively, via mutations in other molecules that convey the EGFR-initiated signal transduction (34, 79, 109–111). The most common resistance mechanism, identified in approximately 50–70% of patients treated with first-generation TKIs, is the EGFR T790M mutation (32, 34, 41). The substitution of threonine with a much bulkier methionine leads to steric hindrance and the conformational change that prevents the binding of these TKIs to EGFR (112).
To improve TKI activity against common sensitizing EGFR mutations and the EGFR T790M resistance mutation, the second-generation irreversible EGFR inhibitors with a broader specificity to ErbB receptor family members have been developed (48, 55, 113). Preclinical studies demonstrated that this mode of action is more effective against EGFR sensitizing mutations and the EGFR T790M resistance mutation than inhibition by reversible first-generation EGFR inhibitors (114). Afatinib is a second-generation TKI that binds covalently and irreversibly to conserved cysteine residues in EGFR, ErbB2 and ErbB4 (48, 113). Afatinib is indicated for patients with NSCLC and exon 19 deletions (Ex19del), the L858R substitution mutation or other uncommon sensitizing EGFR mutations; it is also indicated for patients with squamous cell carcinoma of the lung after failure of first-line chemotherapy (48, 113). Key phase III trials have demonstrated superior efficacy and PFS with afatinib versus standard chemotherapy regimens in patients with NSCLC exhibiting Ex19del and L858R mutations in EGFR, with superior OS also observed in the Ex19del cohorts (52–54). A randomized phase IIb trial of afatinib versus gefitinib also demonstrated superior efficacy of afatinib with regards to overall response rate (ORR) and PFS, however, no significant OS benefit was observed (115, 116). In support of this, a meta-analysis of studies that investigated afatinib, gefitinib or erlotinib in patients with NSCLC showed an increase in PFS in patients treated with afatinib compared with those who were treated with erlotinib or gefitinib (117). However, analyses of tumor tissue from patients with NSCLC or lung adenocarcinoma who had received afatinib identified a T790M mutation in up to 70% of samples (49–51). Furthermore, a higher proportion of cells carrying the T790M allele was found in afatinib-resistant than erlotinib-resistant tumor cells (118). This suggested the T790M mutation was still a key mediator of resistance to afatinib in these tumors. Another second-generation irreversible inhibitor, dacomitinib, is also approved for first-line treatment of patients with metastatic NSCLC with the EGFR exon 19 deletion or the L858R substitution mutation (55) based on a study that has shown that first-line dacomitinib is superior over gefitinib in improving PFS in patients with NSCLC with sensitizing EGFR mutations (58). Previous studies also demonstrated superiority with regards to ORR and PFS over erlotinib (57, 119). Similar to afatinib, however, a mutation leading to the T790M change in the EGFR protein was present in about 50% of patient serum samples at the time of disease progression (56).
The discovery of a common acquired resistance mechanism to first- and second-generation EGFR TKIs through the T790M resistance mutation prompted the development of a third-generation TKI, osimertinib. This irreversible small-molecule EGFR TKI has been shown to be more active against EGFR carrying an activating/sensitizing mutation and T790M resistance mutation than against the wild-type EGFR (120, 121). Osimertinib is indicated for the first-line treatment of patients with metastatic NSCLC whose tumors have an EGFR exon 19 deletion or L858R mutation, or for those patients with T790M mutation-positive metastatic NSCLC who had progressed during or after first- or second-generation EGFR TKI therapy (60). Osimertinib extended PFS of treatment-naïve patients with NSCLC compared with gefitinib-treated patients (69). In addition, osimertinib in combination with chemotherapy showed a significant advantage in prolonging PFS when compared with chemotherapy alone in patients with T790M mutation-positive NSCLC who had progressed on prior systemic therapy, including EGFR TKIs (66, 68). Interestingly, osimertinib has also shown promising efficacy in in vitro and in vivo studies of glioblastomas exhibiting EGFRvIII mutations (122–124), and a tolerable safety profile in initial clinical studies (125). Previous attempts to inhibit EGFRvIII and glioblastoma in general with both first- and second-generation TKIs and monoclonal antibodies have been relatively unsuccessful, potentially due to the lack of TKI-sensitizing mutations present in NSCLC, the additional obstacle presented by the blood-brain-barrier, and only partial effectiveness of EGFR-targeting antibodies in blocking EGFRvIII (126–129). However, promising preclinical efficacy coupled with increased permeation of the blood-brain-barrier by osimertinib may improve outcomes in this context (124, 125).
Unfortunately, resistance to osimertinib also develops through further acquired mutations in EGFR (61). Analysis of patient plasma samples collected in clinical trials that investigated osimertinib as a first-line (61, 67) and a second-line therapy after failure of other first-line TKIs (67) identified an EGFR C797S mutation, which affects the critical site for osimertinib binding, in 7% and 14% of samples, respectively. The C797S mutation frequently occurs in the same allele as the T790M mutation, thus rendering osimertinib completely inactive (64). However, loss of the T790M mutation led to disease progression during treatment with osimertinib (63). A recent study reported that brigatinib, an ALK and EGFR inhibitor, when used in combination with cetuximab, was effective in patients with EGFR-sensitizing mutation/T790M/cis-C797S-positive NSCLC (72, 130). Also, fourth-generation TKIs that can overcome the C797S mutation-conferred resistance to osimertinib are currently undergoing preclinical and early (phase I) clinical development (30, 131–135).
Mechanisms of resistance to TKIs can also occur through compensatory signaling, which bypasses the requirement for signaling through EGFR by activating the same downstream effectors via alternative pathways (114). MET gene amplification and overexpression of MET protein occurs in approximately 3% of gefitinib-/erlotinib-resistant tumors (33) and results in compensatory signaling involving the ErbB3-dependent activation of the PI3K/AKT pathway (32, 136, 137). MET amplification has also been identified in 15% and 19% of plasma samples of patients treated in the first- and second-line setting with osimertinib, respectively (67). ERBB2 amplification is detected in around 6% of resistant tumors and can also contribute to the oncogenic signaling when EGFR is inhibited by first-generation TKIs (33). Moreover, amplification of ERBB2 is detected in 2% and 5% of patients with NSCLC that received first- and second-line osimertinib, respectively, and reduces sensitivity of tumors exhibiting T790M mutations to osimertinib (61, 67, 138). Preclinical studies using NSCLC cell lines have also demonstrated that activation of the JAK2/STAT3 pathway can also substitute for EGFR signaling, while increased levels of insulin growth factor-1 receptor (IGFR-1) and constitutive activation of the IGFR-1 pathway was reported in gefitinib- or erlotinib-resistant lung cancer cell lines and gefitinib-resistant tumors in patients with NSCLC (139).
Intercellular communication via exosomes is also emerging as a key mediator of resistance to TKIs in NSCLC by influencing cellular signaling (140). A recent study showed secretion of exosomes containing T790M-mutated EGFR by gefitinib-resistant NSCLC tumor cells could horizontally confer gefitinib resistance to sensitive recipient cells (141). Furthermore, transfer of non-coding RNAs via exosomes can also modulate response to TKIs, with miR-7 showing the ability to reverse gefitinib resistance through influencing YAP signaling (142), and miR-214 inducing gefitinib resistance through upregulation of signaling by PTEN and AKT (143). Moreover, circular RNA_102481 has been found to be significantly upregulated in NSCLC tumors resistant to EGFR-TKIs, and that silencing of this circular RNA could inhibit EGFR-mediated proliferation and sensitize cells to apoptosis (144). Knowledge of drug-resistance mediated by exosomes and non-coding RNA cargo may have clinical benefit beyond that of being a potential therapeutic target, in that sequencing of miRNAs within exosomes in patient biopsies could establish predictions of response to targeted therapies (144). There is an emerging role for integrative systems biology in identifying novel drug combinations, which may be able to help address the challenge of TKI resistance due to altered cell signaling. One study utilizing bioinformatic approaches to identify driver mutations in TKI-resistant NSCLC lines following RNA sequencing analysis yielded a novel combination of bosutinib and gefitinib that was able to inhibit proliferation and induce apoptosis in these cells (145). Emerging bioinformatic methods which can be used to identify novel drug combinations in cancer are extensively reviewed elsewhere (146).
Outside of the context of signaling, transformation of NSCLC to a different histologic type can also mediate resistance to TKIs. For example, conversion to a squamous cell histologic type was detected in 19% of tumor biopsies from patients, whose disease progressed on first-line treatment with osimertinib (62). Transformation of NSCLC into small-cell lung cancers and the process of epithelial-to-mesenchymal transition have also been cited as mechanisms of resistance to TKIs (30). Key mechanisms of resistance discussed in this section are summarized in (Table 1).
To date, four anti-EGFR monoclonal antibodies, namely cetuximab (chimeric immunoglobulin [Ig] G1), panitumumab (humanized IgG2), necitumumab and nimotuzumab (both humanized IgG1), have been granted approval for clinical use by regulatory bodies (147). These monoclonal antibodies inhibit EGFR signaling by interacting with the EGFR extracellular domain III, and preventing ligand binding to EGFR, receptor dimerization, and signal transduction, thus leading to the internalization of the receptor-antibody complexes and their destruction (147). The binding of cetuximab to EGFR creates steric hindrance prohibiting EGFR heterodimerization with other ErbB receptor family members (148). However, nimotuzumab, despite blocking the EGF-EGFR interaction, does not prevent the formation of an EGFR active conformation, and EGFR remains capable of conveying the basal ligand-independent signaling (149). In addition to this, key anti-EGFR+ tumor effects mediated by monoclonal antibodies such as cetuximab are independent of the inhibition of EGFR signaling and occur through the recruitment of cytotoxic natural killer (NK) cells, key effectors of the innate immune system which eradicate tumor cells through ADCC (23, 150). Efficacy data from key clinical trials for cetuximab, panitumumab, and necitumumab are provided in Table 1.
Cetuximab is approved for the treatment of patients with SCCHN and EGFR-expressing, KRAS wild-type mCRC (73). First-line treatment with cetuximab in combination with radiation therapy improved OS and locoregional disease control compared to radiotherapy alone in patients with locally or regionally advanced SCCHN (49.0 months versus 29.3 months) (95). The combination of cetuximab and platinum-based chemotherapy with fluorouracil, prolonged OS and PFS in patients with recurrent locoregional or metastatic SCCHN (94). Based on studies that have reported an improvement in OS with regimens containing cetuximab in patients with EGFR-expressing, KRAS wild-type mCRC (88, 90, 91), cetuximab is recommended in combination with different chemotherapy regimens as first- or second-line therapy, or as a single agent in patients who have failed or are resistant to certain chemotherapy regimens (73). In a recent study, the combination of cetuximab and the BRAF V600E inhibitor encorafenib prolonged OS in patients with BRAF V600E positive-mCRC compared with the combination of cetuximab and chemotherapy (OS= 9.0 months with combination therapy versus 5.4 months in the control group) (92); the combination of cetuximab and encorafenib was recently approved (September 2021) by the FDA for the treatment of patients, whose disease had progressed on one or (73) two prior regimens (151).
Panitumumab monotherapy increases PFS in patients with mCRC who progress during or following fluoropyrimidine-, oxaliplatin-, and irinotecan-containing chemotherapy regimens (97, 99). Interestingly, recent data from the phase III PARADIGM trial evaluating panitumumab in combination with chemotherapy in patients with mCRC have confirmed the results of previous studies (152, 153), that anti-EGFR antibody therapy in this context demonstrates superior efficacy in patients with left-sided tumors than right-sided tumors (96), perhaps reflecting differences in the genetic and molecular underpinnings of the disease highlighted in several previous studies (154, 155). Treatment with panitumumab, as with cetuximab, however, is ineffective in patients with mCRC carrying mutated KRAS or NRAS (98, 156). Necitumumab in combination with gemcitabine and cisplatin improves OS and PFS in patients with refractory metastatic squamous NSCLC and it has been approved for the first-line treatment in these patients (100, 101). Nimotuzumab has been approved for treatment of patients with SCCHN, glioma and nasopharyngeal cancer in some countries but it has not been recommended by the EMA and FDA for treatment of patients with glioma due to insufficient efficacy and high rates of adverse events (157). However, in some studies of patients with SCCHN, the combination of nimotuzumab and radiotherapy or chemoradiotherapy prolonged OS (60 month OS= 57% nimotuzumab + chemotherapy; 39% nimotuzumab + radiotherapy; 26% chemotherapy only; 26% radiotherapy only) (158).
Despite anti-EGFR inhibitory antibodies being efficacious in distinct subpopulations of patients with mCRC and SCCHN, inherent and acquired resistance to this class of therapy is also common. In patients with mCRC, activating KRAS and NRAS mutations are a biomarker of primary resistance to cetuximab and panitumumab and the use of these antibodies is not recommended in this setting (88, 90, 91, 156). Inherent resistance is also seen in patients with CRC whose tumors carry BRAF V600E (159), MAP2K1 (74) or PIK3CA (160) mutations, KRAS (161), ERBB2, MET or FGFR1 amplification (74), biallelic NF1 loss or aberrations in the non-canonical RAS/RAF pathway (111). The mechanisms for inherent and acquired resistance to anti-EGFR monoclonal antibodies seem to overlap in CRC, as KRAS, NRAS and EGFR ectodomain mutations [the latter have also been detected in patients with SCCHN (76, 77)], and MET and KRAS amplification have also been detected in circulating tumor DNA in patients with acquired resistance to anti-EGFR antibodies (78–83). A recent study evaluated transcriptomic profiles in tumor biopsy material collected from patients with CRC who had progressed on cetuximab monotherapy and showed that acquired resistance to cetuximab was largely mediated by the remodeling of the stromal compartment resulting in the cetuximab-resistant switch to the fibroblast- and growth factor-rich transcriptomic subtype (111). In addition, cetuximab resistance was associated with infiltration of cytotoxic immune cells and elevated expression of programmed death-ligand 1 (PD-L1) and lymphocyte-activation gene 3 (111).
In addition to the anti-EGFR monoclonal antibodies approved for the clinical use, several other anti-EGFR antibodies inhibiting EGFR signaling are currently undergoing clinical development and have been reviewed elsewhere (147).
Considering that amplification of MET and overexpression of the MET protein can compensate for the lack of EGFR signaling (74), it was hypothesized that inhibition of both EGFR and MET may be advantageous in combating acquired resistance to anti-EGFR antibodies. A bispecific antibody LY3164530 specific to both EGFR and MET was investigated in a phase I study in patients with different advanced or metastatic cancers, but was subsequently discontinued due to a high rate of adverse events, which is consistent with EGFR inhibition-related toxicities, and poor efficacy (162). Amivantamab (JNJ-61186372), another bispecific antibody targeting both EGFR and MET, was designed with an intention to treat patients with EGFR exon20ins-mutated NSCLC who currently have limited treatment options (163, 164). In an ongoing phase I/II trial in patients with NSCLC, amivantamab achieved a partial response in 36% of patients (164). These promising results in a population of patients with a poor prognosis has led to FDA breakthrough therapy designation for amivantamab (165). However, patients still experienced a high rate of adverse events, with grade ≥3 toxicities being reported in 36% of patients (164). Another bispecific antibody which is undergoing a phase I clinical evaluation is MCLA-158, an antibody-dependent cell-mediated cytotoxicity (ADCC)-enhanced human IgG1 targeting both EGFR and leucine-rich repeat-containing G-protein coupled receptor 5 (LGR5) (166). MCLA-158 showed antitumor activity against RAS mutated and wt CRC patient-derived organoids in vitro and induced either tumor regression or stasis in esophageal squamous and gastric adenocarcinoma patient-derived xenograft models expressing LGR5 and EGFR (166). In patients with mCRC, who progressed after receiving oxaliplatin, irinotecan and fluoropyrimidines, and EGFR monoclonal antibodies, MCLA-158 was well tolerated and no dose limiting toxicity was achieved (166). However, despite these promising data, there is a risk that therapies co-targeting EGFR and another cell-surface kinase receptor may experience similar issues with toxicity, due to the critical role of EGFR activity in healthy tissues (167).
One major concern relating to both EGFR TKIs and inhibitory anti-EGFR monoclonal antibodies is the high rate of adverse events and the frequently occurring cutaneous toxicities (167). EGFR plays a critical role in maintaining homeostasis of healthy mesenchymal, epithelial, and neurologic tissues, and the inhibition of the basal EGFR signaling can cause cell death, impaired cell proliferation and abnormal cell differentiation in these healthy tissues (167–169). Dermatologic toxicities occur in around 45–100% of patients treated with EGFR TKIs and monoclonal antibodies against EGFR (167), and the underlying cause for these toxicities is linked to a non-redundant role of EGFR in regulating normal keratinocyte growth, survival, differentiation, and migration, and maintenance of an adequate immune response in the skin (167, 169). Epithelia in normal tissues respond to injury by promoting proliferation of epithelial cells, and the inhibition of EGFR signaling interferes with the regeneration of epithelial surfaces, such as skin (168) and the gastrointestinal lining (169, 170). Persistent tissue damage destroys epithelial barriers, leading to pathogen invasion and acute inflammation, which perpetuates an even larger influx of immune cells leading to further tissue injury (171). As the barrier cannot be closed, the physiological balance between inflammation and tissue regeneration is disturbed.
A meta-analysis of toxicity data pooled from 28 randomized controlled trials, which investigated EGFR TKIs in patients with various cancers, reported diarrhea, rash, mucositis, alanine aminotransferase increased, and skin reaction as most common any grade adverse events, while the most frequently reported grade ≥3 toxicities included mucositis, pain, metabolism and nutrition disorders, diarrhea, dyspnea, and hypertension (172). Statistically significant differences in the risk ratios emerged for the different generation lines of TKIs (172). Second-generation TKIs (afatinib, dacomitinib, lapatinib, neratinib, and vandetanib) were associated with the highest risk of high-grade diarrhea compared with first- (gefitinib and erlotinib) or third-generation (osimertinib) TKIs and were more likely to cause any grade fatigue and nausea, and high-grade vascular disorders and fatigue than first-generation TKIs (172). Furthermore, in comparison to treatment with first-generation EGFR-TKIs (gefitinib or erlotinib), osimertinib had a higher rate of cardiac toxicity, manifesting in QT prolongation in 10% of patients who had received osimertinib compared with 4% of patients treated with EGFR-TKIs, and cardiac failure in 4% of patients treated with osimertinib and 2% with EGFR-TKIs (69). Effects of TKIs on hair growth include hypertrichosis, trichomegaly and a range of scalp hair changes (173).
Approximately 10–20% of patients treated with anti-EGFR antibodies experience grade 3/4 toxicities (73, 99, 174), including acneiform rash, radiation dermatitis enhancement, pruritus, mucositis, xerosis/fissures, paronychia, and gastrointestinal toxicity, all of which may lead to greatly reduced patient quality of life and antibody dose reduction, interruption or complete cessation of the treatment (73, 99, 174). There is also a risk of secondary infections occurring in these patients, which can be fatal (73, 99), and up to 96% of patients can also experience significant gastrointestinal disorders (73, 99). In clinical trials investigating cetuximab, cardiopulmonary arrest was reported in 2–3% of patients (73).
Taken together, these data demonstrate the critical role of EGFR kinase activity in normal tissue homeostasis and the response to injury, and the limitations of the use of TKIs and EGFR-targeting antibodies in patients with cancer due to their detrimental effect on normal tissue physiology. The necessity of management of dermatologic and gastrointestinal toxicities may require the EGFR-targeted treatment to be temporarily interrupted to allow patients to recover, and this may reduce the effectiveness of the therapy and potentially lead to acceleration of disease progression (167, 174).
The need for novel therapeutic approaches has emerged to circumvent shortcomings related to acquired resistance and on-target off-tumor toxicities induced by EGFR TKIs and anti-EGFR antibodies. One of these approaches is the development of therapeutic agents that exploit cell-surface EGFR as a decoy to direct the activity of key components of the adaptive immune system such as CD4+ T cells, B lymphocytes, and the cytotoxic CD8+ and γδ/αβ T-cell receptor positive (TCR+) T cells to EGFR-expressing cancer cells and to destroy them in a target-specific manner.
One such therapeutic strategy involves blocking inhibitory immune checkpoints using immune checkpoint inhibitors (CPIs). Several different molecules expressed on immune cells and cancer cells convey inhibitory and stimulatory signals, called immune checkpoints, to regulate cancer immunity. Cytotoxic T lymphocyte-associated protein-4 prevents activation of T lymphocytes, while programmed cell death protein-1 (PD-1) upon binding to its ligand PD-L1 inactivates the ability of cytotoxic T lymphocytes to destroy tumor cells (175). CPIs target these molecules and have revolutionized the treatment of many patients with different cancers (175). Two CPIs, nivolumab and pembrolizumab have been approved for the treatment of SCCHN following pivotal phase III trials. In the Checkmate-141 phase III trial, nivolumab demonstrated superior OS over standard-of-care therapy in patients with relapsed SCCHN (7.5 months vs. 5.1 months) (176). Similarly, in the Keynote-040 trial, patients treated with pembrolizumab monotherapy demonstrated superior OS over those treated with chemotherapy and cetuximab (8.4 months vs. 6.9 months) (177). This was observed to an even greater extent in the Keynote-048 trial in patients treated with pembrolizumab in combination with chemotherapy, versus chemotherapy plus cetuximab (13.0 months vs. 10.7 months) (178). Pembrolizumab with chemotherapy has also shown efficacy in mCRC, improving PFS over chemotherapy, however, no significant improvement in OS was observed (179).
In the context of NSCLC, PD-1 and PD-L1 inhibitors such as pembrolizumab, nivolumab, and atezolizumab in combination with chemotherapy have become the standard-of-care in frontline therapy after demonstrating significant improvements over chemotherapy with regards to ORR, PFS, and OS in patients without EGFR-activating mutations (180). However, much lower efficacy has been noted if EGFR-driver mutations are present (181). As EGFR signaling can induce a tumor-suppressive microenvironment through upregulation of factors such as IL-6, TGF-β and progranulin, and induces PD-L1 expression, and PD-L1 can mediate resistance to TKIs through upregulation of YAP1, there is a clear preclinical rationale for the use of combination therapies simultaneously targeting PD-L1 and EGFR signaling (181). Trials investigating combinations of TKIs with CPIs, however, have raised safety concerns, with one study of osimertinib in combination with durvalumab leading to a 38% incidence of pneumonitis, compared with 2.9% and 2% incidence of pneumonitis with osimertinib or durvalumab monotherapy, respectively (182). A high (39%) incidence of Grade 3 and 4 adverse events was also observed in patients receiving erlotinib and atezolizumab (183).
There is also a rationale for the use of CPIs in combination with EGFR-targeting antibodies such as cetuximab to treat other tumor types such as SCCHN and mCRC, with a predicted synergistic effect due to the ability of PD-L1 inhibitors to alleviate immunosuppression in the tumor microenvironment, and the ability of cetuximab to stimulate cells of the innate and adaptive immune system to induce anti-tumor ADCC (150). A pilot study of cetuximab plus radiotherapy and avelumab in patients with advanced SCCHN unfit for cisplatin treatment demonstrated manageable toxicity and transient immune-related toxicity, setting the scene for larger trials in this setting (184). Additionally, correlative science data from a phase I/II trial of cetuximab in combination with pembrolizumab demonstrated an increase in intratumoral CD3+ CD8+ cytotoxic T cells, a decrease in cytotoxic T cells in the peripheral blood, and decreased levels of PD1+ cytotoxic T cells in both the tumor and peripheral blood, consistent with therapy-related changes in the tumor microenvironment (185). Further combination regimens involving anti-EGFR antibody cetuximab and CPIs are currently being investigated in a number of phase I−III clinical trials in patients with EGFR-expressing tumors (150).
CAR-T cell therapies are T cells that have been modified ex vivo to target a specific tumor cell-surface antigen and thereby to use the adaptive immune system to destroy cancer cells. The first-generation CAR-T cells are engineered to express receptors comprising an extracellular single-chain variable fragment (scFv), which recognizes a specific tumor cell-surface antigen, a transmembrane domain, and an intracellular part containing immunoreceptor tyrosine-based activation motifs and a co-stimulatory domain, that is crucial for the T-cell activation, proliferation, persistence, and cytotoxicity (186). Two or more co-stimulatory domains are typically incorporated in the second- and third-generation CAR-T cell therapies (186).
In general, CAR-T therapies have been reported to be efficacious in small populations of patients with specific cancers, mainly those with hematologic malignancies, but in patients with solid tumors, their effectiveness has been limited (187, 188). The advantage of using therapeutic agents that engage the adaptive immune response is that the response is targeted to cells expressing specific antigens. However, the tumor microenvironment is usually not conducive to therapy, with poorly vascularized and hypoxic tumor regions preventing CAR-T cell homing to tumors and the anti-inflammatory tumor microenvironment being detrimental to the CAR-T cell survival (29). In addition, tumors often show heterogeneous expression of target antigens, and the lack of universally expressed cancer antigens significantly reduces the antitumor activity (186).
CAR-T therapy targeting EGFR is in early clinical development for numerous types of cancer (186, 189, 190). Several studies ongoing in patients with glioblastoma are investigating CAR-T cell therapies targeting the EGFRvIII mutant, which is identified in around 31% of glioblastomas (191), but not in healthy tissues, therefore, reducing the risk of off-target effects and toxicity (186) (Table 2). A phase I trial in patients with glioblastoma found that CAR-T cells targeting EGFRvIII specifically accumulated in tumors and showed a good safety profile (29). No EGFR-associated toxicities, such as rash and diarrhea, were reported, but clinically significant neurologic events occurred in three of 10 patients (29). Most patients showed a complete loss or reduced expression of EGFRvIII in their tumors, but the tumors also expressed anti-inflammatory markers and secreted cytokines, which reduced the effectiveness of the CAR-T-EGFRvIII cell therapy (29). Moreover, tumors showed intratumoral heterogeneity of EGFRvIII expression levels, suggesting that the lack of uniformity in target expression in tumor tissue may contribute to the suboptimal efficacy (13). In support of this, a pilot dose-escalation phase I trial investigated patients with EGFRvIII-expressing recurrent glioblastoma who were treated with a third-generation CAR-T-EGFRvIII cells and reported no clinically meaningful effect in these patients (194). Another phase I study investigated CAR-T therapy targeting EGFR in patients with EGFR-expressing biliary tract tumors (195). Ten of 17 treated patients had stable disease and 1 of 17 patients showed a complete response (195). CAR-T-EGFR cell therapy was well tolerated in this setting (195) (Table 2). Moreover, the enrichment of central memory T cells in the infused CAR-T-EGFR cells showed a good correlation with the persistence of CAR-T-EGFR cells in patients (195). In another phase I study, EGFR-targeted CAR-T cell infusions were well tolerated without severe toxicity in patients with NSCLC, and of 11 patients, two had partial response and five had stable disease for up to 8 months (196) (Table 2). CAR-T-EGFR cell therapy has also been shown to be well tolerated in patients with metastatic pancreatic cancer in a phase I trial, where out of 14 evaluable patients, four achieved partial response and eight had stable disease for 2–4 months (193) (Table 2).
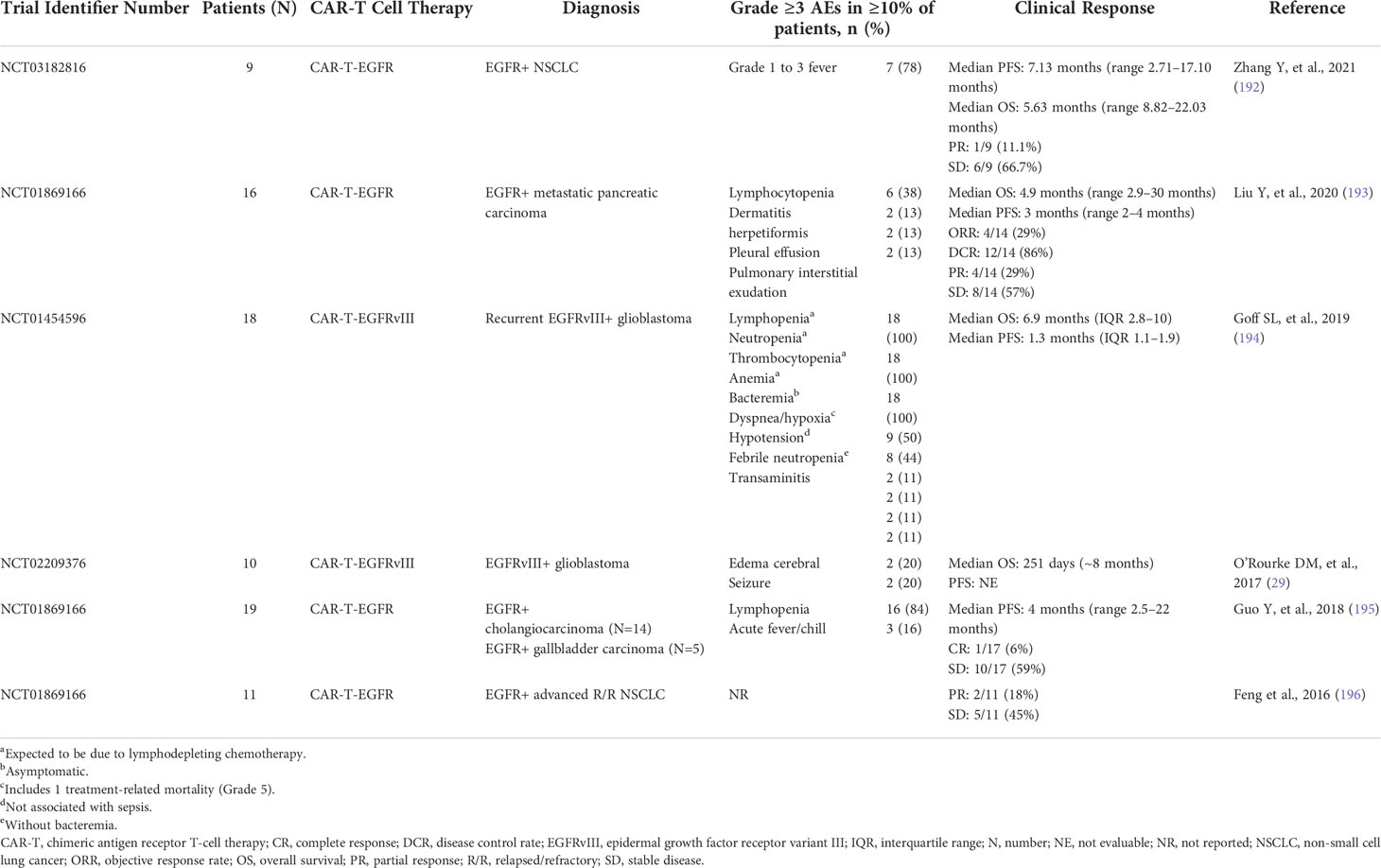
Table 2 Safety and clinical response to CAR-T therapies in phase I clinical trials.
However, other studies have shown CAR-T therapy to increase the risk of severe adverse events. Neurologic toxicities have been reported with CAR-T therapy (197, 198), with symptoms including encephalopathy, headache, tremor, aphasia and focal weakness (198). In one study, 20% of neurotoxicity events were of grade ≥3 severity (198). However, the most common toxicity experienced by patients is cytokine release syndrome (CRS) (197), which is a systemic inflammatory response to cytokines, that can lead to organ damage, and death and occurs when effector immune cells cross-react, triggering target-independent cytokine release. Therefore, patients receiving CAR-T therapy must be monitored for CRS, as treatment needs to be prompt and aggressive (197).
One novel EGFR-targeting CAR-T therapy for overcoming CAR-T-associated toxicity is currently in early preclinical development. The UniCAR system uses two separate modules; the first is the UniCAR-T cells that are inert, and the second is composed of a target module, containing an antigen binding domain linked to the E5B9 peptide epitope (199). The UniCAR T cells become activated only when the crosslinking to the target module via its E5B9 peptide epitope takes place (199). This system shows promise in cell models and can target the UniCAR-T cells effectively to tumor cells and only becomes active in the presence of a tumor antigen (199). Encouragingly, the UniCAR-T-EGFR cells also show activity against cancer cells expressing low levels of EGFR (199). However, the efficacy of this system in a clinical setting is currently unknown.
Another therapeutic modality that allows the coupling of specific tumor antigens with the immune response cells, namely T or NK cells, is bispecific antibodies. These antibodies bind concomitantly to two different antigens, one expressed on cancer cells and another on immune cells (191, 200). One bispecific antibody in preclinical development targets EGFR-expressing tumors by using an anti-EGFR IgG portion of the molecule, and engages the PD-1 on T cells via the scFv of an anti-PD-1 antibody (200). This antibody simultaneously inhibits EGFR signaling, activates T cells and initiates a tumor immune response by blocking the interaction between PD-1 on T cells and PD-L1 on tumor cells, and also induces strong ADCC (200). When tested in cellular cytotoxicity assays in vitro, the antibody induced EGFR-dependent cell death, and in tumor-bearing animals, recruited T cells to tumor xenografts (200).
Another bispecific antibody (hEGFRvIII-CD3) comprising two scFv fragments (one specific to the EGFRvIII antigen and another to the CD3 epitope) was designed to create a bridge between EGFR-expressing cancer cells and CD3+ T cells, and to prevent a non-specific targeting to EGFR-negative cells. The hEGFRvIII-CD3 antibody activated CD3+ T cells in a target-specific manner, induced the release of proinflammatory cytokines, prompted T-cell proliferation, and caused significant lysis of malignant glioma cell lines and patient-derived EGFRvIII-expressing malignant glioma samples in vitro (201). The hEGFRvIII-CD3 antibody also showed antitumor activity in several preclinical malignant glioma models, and significantly extended survival of experimental animals (201).
A novel bispecific T-cell engager AMG 596, which comprises two single-chain variable fragments with one being specific for EGFRvIII and another for CD3, was recently investigated in glioblastoma preclinical models (202). The simultaneous engagement of EGFRvIII expressed on glioblastoma cells and CD3 on T cells led to a potent antitumor activity against the EGFRvIII-expressing glioblastoma cells in vitro, while the treatment of mice bearing EGFRvIII-expressing orthotopic tumors significantly extended OS of experimental animals (202). Moreover, the treatment of cynomolgus monkeys with AMG 596 showed a good safety profile (202). A new type of a bispecific antibody that engages simultaneously EGFR on tumor cells and CD3 on T cells was designed to prevent its binding to EGFR expressed in healthy tissues (a masked form) but to become activated by proteases (an unmasked form) in the tumor microenvironment. Proteolytically activated, but not inactive, EGFR-CD3 T cell-engaging antibody showed specific EGFR-dependent tumor cell killing in vitro and caused tumor regression in preclinical tumor models, while a nonhuman primate study established that the maximum tolerated dose increased by 60-fold when the EGFR-CD3 antibody was administered in the masked form. Therefore, a localized activation of a bispecific antibody is likely to reduce on-target toxicity and increase its therapeutic index (203).
Although the preclinical investigation of these bispecific antibodies is showing promising results, they have not yet entered clinical development.
To overcome T-cell-associated toxicity issues, a novel approach is being developed that deploys immune cells mediating the innate immune responses to target specific cell-surface epitopes on cancer cells (204).
The innate immune system provides the immediate response to infection and foreign antigens and plays a key role in tumor immunosurveillance through recognition and destruction of transformed cells both prior to and following the establishment of a tumor (205–207). Innate immunity is also essential for the onset and maintenance of the adaptive immunity; by stimulating innate immunity it is possible to harness both sides of the immune system (204). The innate immune system comprises innate lymphoid cells, including a specialized population of NK cells, macrophages, neutrophils, dendritic cells, mast cells, basophils, eosinophils and γδ T cells (205). The key cells involved in tumor immunity are the NK cells, which induce ADCC, and scavenging macrophages, which phagocytose tumor cells via the antibody-dependent cellular phagocytosis (ADCP) mechanism (Figure 2); both can also stimulate the downstream activation of the adaptive immune response (205, 206, 208, 209). High levels of tumor-infiltrating NK cells have been shown to be associated with a favorable prognosis in numerous solid tumors, and several studies have also shown that intact cytolytic function of NK cells is important in protecting from the development of some types of malignancies (210–213). However, the more advanced tumors can upregulate the expression of inhibitory molecules inactivating NK cells, and escape from immunosurveillance (213–216).
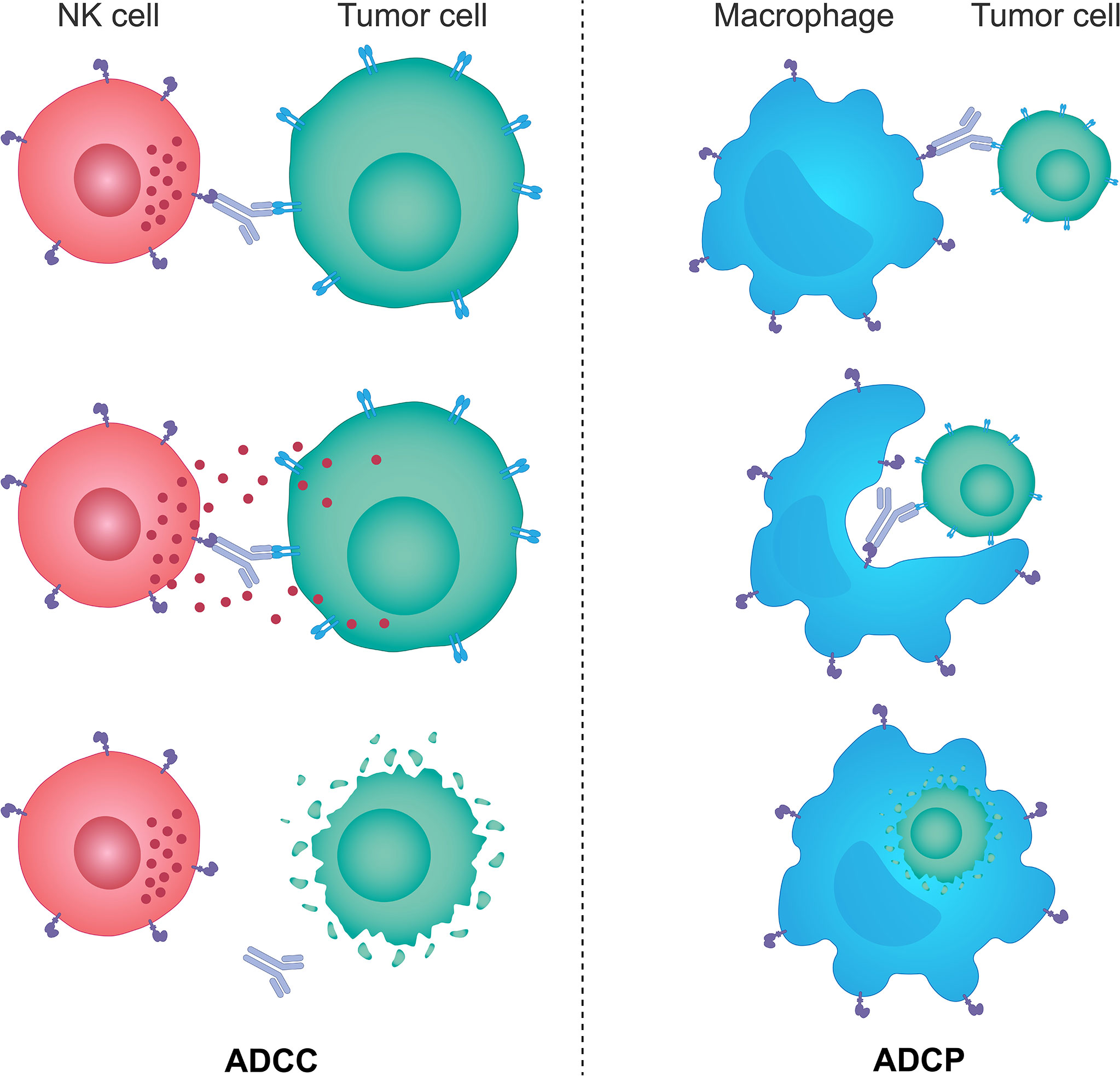
Figure 2 The mechanisms of ADCC and ADCP response. Monoclonal therapeutic antibodies designed to target specific tumor cell antigens can also use their Fc portion of the immunoglobulin to anchor NK cells and macrophages through specific Fc receptors expressed on the surface of these cells. Such interactions trigger activating signals downstream of Fc receptors in NK cells and macrophages and lead to NK cell-mediated ADCC and macrophage-mediated ADCP responses. NK cells brought in the vicinity of target tumor cells by monoclonal antibodies kill those cells predominantly through the perforin/granzyme cell death pathway, while activated macrophages engulf antibody-opsonized target tumor cells and degrade them through acidification of the phagosome. ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; Fc, fragment crystallizable; NK, natural killer.
ADCC is a process by which cells expressing specific antigens are recognized by antibodies, which also interact via a constant region (Fc) with the Fc-gamma receptors (FcγR) on the surface of immune effector cells, leading to the direct lysis of target cells (217). NK cells are considered to be the key effectors of the innate immune system mediating ADCC due to their unique expression of activating FcγRs, such as FcγR IIIa (CD16a) and FcγR Iic (CD32a) (217).
As previously alluded to, some EGFR-targeting antibodies, approved for the use in patients with mCRC and SCCHN, not only inhibit ligand-induced EGFR activity and signaling, but also elicit ADCC through the interaction between their IgG1 Fc region and the FcγR-expressing effector cells, typically NK cells, leading to the destruction of EGFR-expressing cancer cells (148, 217). Cetuximab has been shown to be capable of inducing NK cell mediated ADCC in preclinical, clinical, and ex vivo assays, which are extensively reviewed elsewhere (150). This is not the case for panitumumab, which contains an IgG2 Fc region and stimulates ADCC to a much lesser extent (23). This has been suggested to account for differences in the clinical efficacy of cetuximab and panitumumab in patients receiving either cetuximab or panitumumab in combination with chemotherapy regimens in SCCHN, where cetuximab has been shown to increase OS, but panitumumab does not (94, 95, 150, 218, 219). This again suggests that levels of ADCC and intratumoral immune activity play a key role in patient responses in these tumors. Interestingly, a study has shown that the FcγR genotype in patients with KRAS wild-type mCRC may correlate with the ADCC-mediated responsiveness to cetuximab, thus potentially providing a rationale for patient stratification (220). FcγRIIa H/H and H/R alleles of the FcγRIIa-H131R polymorphism elicited significantly higher ADCC compared with the R/R alleles, and the FcγRIIIa V/V and V/F alleles of the FcγRIIIa-V158F polymorphism induced stronger ADCC than the F/F alleles (220). Moreover, patients with the FcγRIIIa 158V allele had significantly longer PFS than those with the 158F/F allele (220). Another study showed that the combination of cetuximab and interleukin 12 in patients with unresectable primary or recurrent SCCHN resulted in higher ADCC and prolonged PFS (221). Similar to cetuximab, nimotuzumab, an IgG1 isotype antibody, has been shown to be capable of exerting a detrimental effect on EGFR-expressing cancer cells by NK cell-mediated ADCC in patients with SCCHN. Activation of NK cells led to dendritic cell maturation and priming of EGFR-specific CD8+ T cells (222).
Other EGFR-targeting antibodies, being tested in early stages of clinical development, have demonstrated superior ADCC responses when compared with cetuximab (163, 223). Imgatuzumab, a monoclonal antibody inhibiting EGFR signaling, induced a more robust ADCC response than cetuximab (223). However, in patients with KRASexon2-WT and KRASexon2-mutant-CRC, despite the ability to initiate a stronger ADCC response, the combination of imgatuzumab and FOLFIRI did not lead to improved PFS when compared with chemotherapy alone or cetuximab treatment (median PFS KRASe2-WT = 7.3 months with imgatuzumab + FOLFIRI, 6.1 months with cetuximab + FOLFIRI; median PFS KRASe2-mutant = 5.2 months imgatuzumab + FOLFIRI, 4.3 months with FOLFIRI only) (224, 225). The EGFR-MET targeting bispecific antibody amivantamab has also been shown to induce more robust ADCC than cetuximab, and a direct correlation was established between the ADCC activity and secreted interferon γ levels in preclinical NSCLC models with EGFR exon 20 insertions (163). Amivantamab has since been granted accelerated approval by the FDA for patients with NSCLC exhibiting EGFR exon 20 insertions who have progressed on platinum-based therapies, where its high-capacity for ADCC induction may be contributing to its favorable efficacy (226).
The capability for ADCC induction by anti-EGFR targeting antibodies may also be enhanced through combination with recently developed covalent inhibitors of KRAS G12C, AMG510 and MRTX849, in patients with tumors that harbor this mutation. Recent in vivo data has suggested that the inhibition of KRAS G12C using AMG510 creates a pro-inflammatory tumor microenvironment, promoting the anti-tumor activity of immune cells alone and in combination with immune CPIs (227). As the onset of the pro-inflammatory tumor microenvironment has been shown to be synonymous with the increased infiltration of immune cells such as T lymphocytes into the tissue, CD16+ subsets of which can mediate ADCC (228), with both AMG510 (227) and MRTX849 (229), combining KRAS G12C inhibitors with anti-EGFR antibodies, which stimulate ADCC, may have synergistic effects in these tumors. An ongoing phase III trial is currently investigating the impact on survival of MRTX849 in combination with cetuximab versus chemotherapy in patients with advanced CRC harboring KRAS G12C mutations (NCT04793958) (230).
NK cells, can be primed and modified ex vivo, in a similar manner to CAR-T cells (231), to express a CD38-CD3ξ domain required for NK cell signaling and scFv antibody fragments to introduce specificity to a range of diverse tumor antigens, including those targeting the cell-surface EGFR or/and EGFRvIII. The therapeutic use of CAR-NK cells may have significant advantages over the CAR-T cell therapy, as CAR-NK cells have a better safety profile due to a low potential to induce CRS, neurotoxicity and graft-versus-host disease, and the ability to exert the CAR-independent cytotoxicity (232).
Different CAR-NK-EGFR cells showed target-specific cytotoxicity in in vitro cell-based assays, reduced xenograft tumor growth in renal cell carcinoma and triple-negative breast cancer animal models, and significantly extended survival of intracranial tumor-bearing animals in metastatic breast cancer and glioblastoma models (231, 233–236). However, despite promising results in the preclinical setting, one study found that CAR-NK cell treatment of animals with glioblastoma failed to inhibit tumor progression and led to a pseudo-progression phenotype (237). In general, CAR-NK cell-based therapies face several challenges due to the short life-span of CAR-NK cells in the absence of cytokines, the need for expansion and activation ex vivo, inactivation by tumor cells and the tumor microenvironment (28), and cross-reactivity leading to NK cell fratricide (209). A number of clinical trials have been initiated to test CAR-NK cells, with specificity to antigens other than EGFR, in patients with hematologic malignancies and solid tumors (238). In heavily pretreated patients with relapsed or refractory B-cell hematologic malignancies, CAR-NK-CD19 therapy was found to be safe, and no CRS, immune effector cell-associated neurotoxicity syndrome or graft-versus-host disease were reported (239), suggesting that NK-CAR cells may have a superior toxicity profile compared with that shown by CAR-T cell therapies. However, although a promising method for targeting EGFR, these therapies are in an early stage of development and further work is needed to establish CAR-NK cells as effective therapies in solid tumors (28).
A novel therapeutic modality, the innate cell engager, has been developed to bind simultaneously to NK cells or macrophages via a distinct epitope on CD16A recognized by the CD16A-specific antibody variable domains, and to cancer cells via variable antibody domains specific to cancer epitopes in order to potentiate the NK cell- or macrophage-dependent destruction of cancer cells in solid tumors (209). This was hypothesized to prevent tumor escape from immunosurveillance, which depends on the balance between activating and inhibitory NK cell populations, due to increased cancer cell killing (209). Innate cell engagers can be engineered as bispecific or multispecific molecules and are derived from the fit-for-purpose redirected optimized cell killing antibody platform, comprising an array of bispecific and multispecific antibodies (209). These antibodies bind CD16A independently of the CD16A allotype, do not cross-react with the Fc binding site, thereby avoiding competition with the body’s own circulating serum IgG, do not exhibit NK cell fratricide, and bind to specific tumor cell antigens even when expressed at low levels. Consequently, innate cell engagers link tumor antigens, such as EGFR, to FcγRIIIa (CD16A) on NK cells or macrophages and activate ADCC and ADCP, resulting in tumor cell killing and phagocytosis of tumor cells, respectively (209), and potentially the reduction of a tumor mass.
AFM24 is a tetravalent bispecific innate cell engager that binds simultaneously to CD16A on NK cells and macrophages and EGFR that is expressed on the tumor cell surface (Figure 3). It has been designed to prevent cross-linking of effector cells, which is expected to reduce the risk of target-independent activation, cytokine release and subsequent CRS (209, 240, 241). AFM24 shows high specificity for CD16A and robust binding to NK cells and macrophages in in vitro assays (209, 240, 241). Preclinical studies have found that AFM24 binds EGFR-expressing tumors with high affinity and induces targeted, dose-dependent and potent lysis by NK cells and phagocytosis by macrophages (241). This was independent of the KRAS/BRAF mutation status and EGFR expression levels of tumor cells, thus suggesting that resistance mechanisms observed with the therapeutic agents targeting EGFR activity and signaling may not be relevant in this setting (Figure 4) (241).
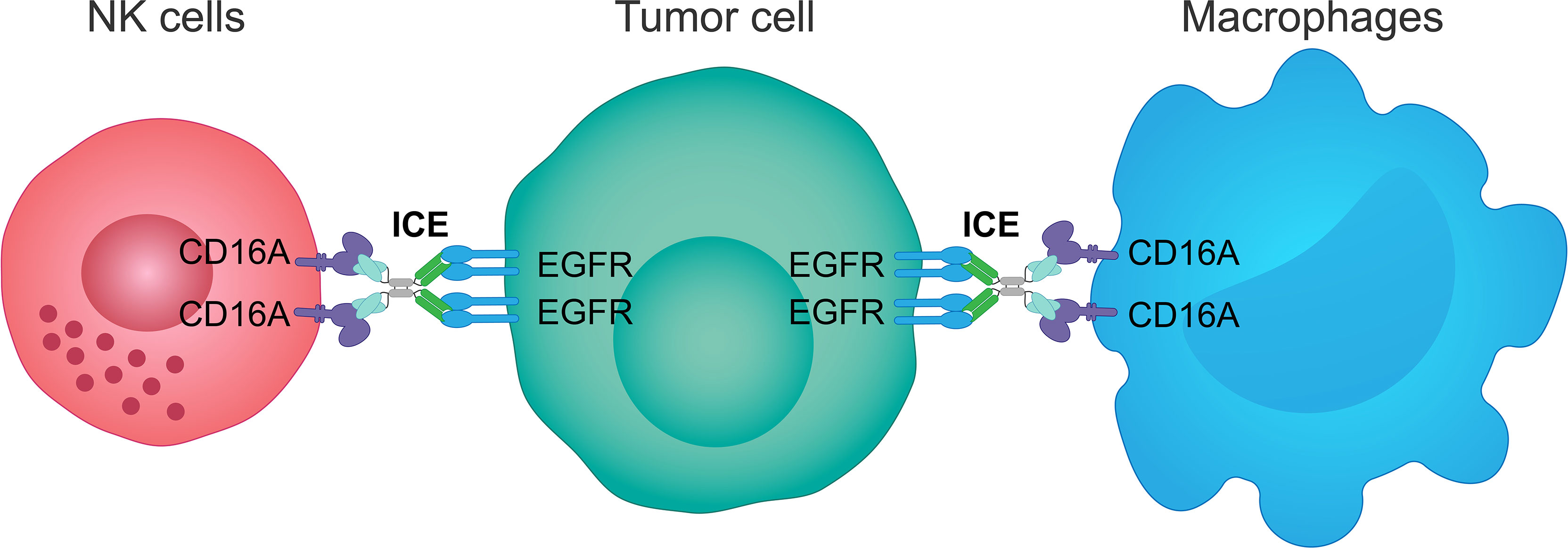
Figure 3 Mechanism of action of an innate cell engager targeting EGFR and CD16A. AFM24, a fully human tetravalent bispecific innate cell engager, binds simultaneously the CD16A receptor on NK cells or macrophages, with a much higher affinity than monoclonal antibodies, and the EGFR antigen on the surface of tumor cells. This creates a bridge between innate immune cells and EGFR-expressing tumor cells enabling ADCC mediated by NK cells and ADCP mediated by macrophages. ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; EGFR, epidermal growth factor receptor; ICE, innate cell engager; NK, natural killer.
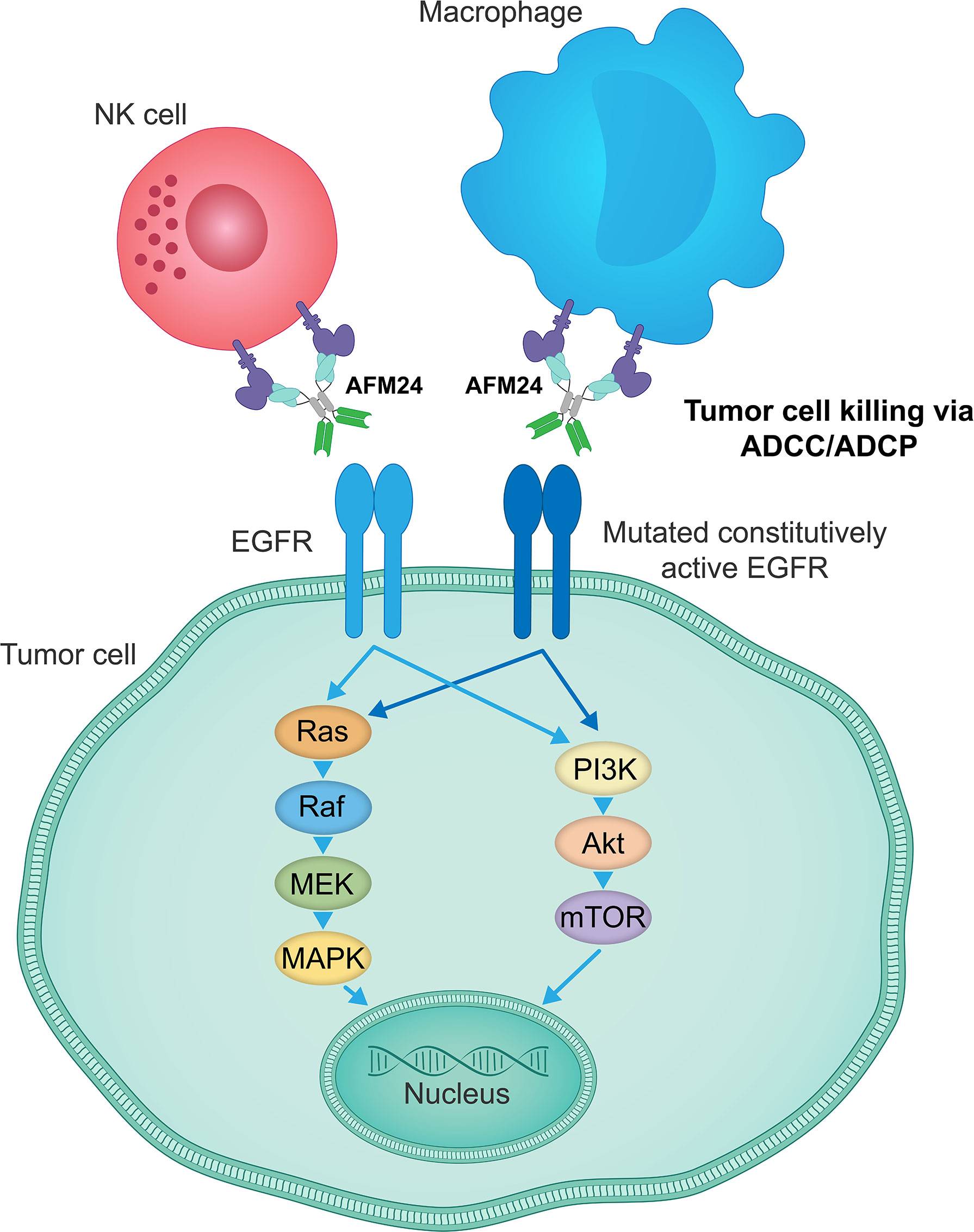
Figure 4 AFM24 activity is independent of EGFR signaling function. AFM24-mediated killing of EGFR-expressing tumor cells, by inducing ADCC and ADCP responses, does not rely on the EGFR activity, its mutational status or the disruption of downstream signaling pathways. ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; EGFR, epidermal growth factor receptor.
Preclinical data generated in cynomolgus monkeys have shown no off-target side effects, no evidence of CRS, and no dermatologic toxicities characteristic to EGFR signaling inhibitors (TKIs and anti-EGFR antibodies) (241). The observed improved toxicity profile in animal models may be determined by the inherent feature of the AFM24 design associated with a minimal effect on the EGFR signaling pathway. The tumor-associated microenvironment often shows persistent inflammation induced and maintained by a continuous production of different pro-inflammatory molecules, tumor vascularization, and infiltration of immune cells mediating the innate and adaptive immune responses (240). Considering that NK cells and macrophages are extremely abundant in tumor tissues, AFM24 can effectively utilize these cells to cause extensive tumor damage and to further promote inflammation and the antitumor immune response. Experiments using AFM24 at up to 75 mg/kg dose once weekly for 28 days in cynomolgus monkeys suggest that healthy tissues, including skin, can be spared as any tissue damage will be easily repaired by EGFR signaling due to its intact catalytic activity (240).
AFM24 has shown activity in several tumor cell lines of different origin, which suggests that it may be beneficial in patients with a range of solid cancers (240). This is reflected in the study design for the ongoing phase I/IIa trial (NCT04259450), which is investigating AFM24 in patients with advanced or metastatic EGFR-expressing solid cancers, including but not limited to colorectal, lung, gastric, esophageal, pancreatic, head and neck, breast, ovarian, cervical, urothelial, and renal cancer (242).
Conventional EGFR-targeting therapies that inhibit EGFR kinase activity and signaling have a detrimental effect on healthy tissues, particularly the skin and the gastrointestinal epithelial lining, where EGFR plays a crucial role in maintaining tissue homeostasis. On-target off-tumor effects limit the effectiveness of therapeutic interventions targeting EGFR signaling and combinations with therapeutic agents, which inflict deleterious effects on EGFR-expressing healthy tissues, would exacerbate the side effects and limit the therapeutic window of EGFR inhibitors even further. On the contrary, innate cell engagers, such as AFM24, show no effect on the regenerative capacity of healthy tissue and are only active in tissue areas with high immune cell content and pro-inflammatory milieu. Therefore, the combinations involving innate cell engagers can be more efficacious due to benefit from a further immune cell activation, without an increase in on-target off-tumor toxicity.
One promising approach for the enhancement of innate cell engager efficacy targeting EGFR is to combine these agents with allogeneic or autologous NK cell products in order to increase the proportion of effector cell/tumor cell pairings following the trafficking and homing of both therapeutic modalities to tumor tissue. To achieve this, NK cells can either be pre-complexed with innate cell engagers prior to administration or both therapies can be co-administered separately. This approach has recently been shown to be successful in improving the antitumor activity of AFM13, an innate cell engager targeting the cell-surface CD30, in preclinical CD30+ Hodgkin lymphoma models (243). This approach is being investigated in a clinical proof-of-concept study with CD30+ Hodgkin and Non-Hodgkin patients (NCT04074746) (244). To investigate the combination of AFM24 with NK cells and to provide further evidence of such a combination strategy, a clinical study has recently been initiated that combines AFM24 with an autologous NK cell product (NCT05099549) (245). Such combinations may overcome the limitation of sparse distribution of effector cells in tumor tissue and may lead to improved effectiveness of both treatments, without potentially introducing additive adverse events (246, 247).
Another approach that may potentially enhance the effectiveness of innate cell engagers targeting EGFR is their combination with CPIs, such as anti-PD-1 or anti-PD-L1 antibodies. CPIs act to enhance immune responses and to inhibit the tumor escape from immunosurveillance by preventing the tumor cell-induced T cell and NK cell suppression (248). The combination of AFM13 with anti-PD1 has demonstrated high response rates in HL (249). Recent studies provided evidence for PD-1/PD-L1 expression not only on T cells but also on NK cells, which suggests a new level of mechanistic complexity behind diminished anti-tumor NK cell responses (175, 249, 250). In support of these findings, PD-L1 engagement by atezolizumab was shown to directly activate NK cell functions (250). A phase I/IIa study of AFM24 in combination with atezolizumab has recently been initiated (NCT05109442) (251).
The excellent safety profile of innate cell engagers may allow to exploit the combinations involving multiple therapeutic agents with different mechanisms of action to fully leverage a targeted anti-tumor immune system.
EGFR has emerged as an oncogenic driver in a subset of patients with NSCLC, while widespread overexpression of EGFR protein has been found in a broad range of different types of cancer. Several different EGFR-targeting therapies have been developed and have entered the clinic but despite this, long-term survival rates of patients treated with these therapies have not significantly improved. Thus, there is a significant unmet need for therapies that are effective and safe in patients with EGFR-expressing solid tumors and can overcome currently documented inherent or acquired tumor resistance mechanisms to therapies targeting EGFR.
The response to TKIs in a subset of patients with NSCLC, whose tumors express EGFR carrying activating mutations, has been excellent but short-lived due to the inevitable evolution of cancer cells to acquire secondary resistance mutations, preventing the binding of TKIs to EGFR. Genetic alterations that activate signaling molecules downstream of EGFR or those in parallel signaling pathways can also create conditions where EGFR signaling, initially critical to the propagation of a tumor, is no longer required. Other EGFR-signaling inhibitors, such as anti-EGFR antibodies, have been approved in a subset of patients with KRAS wild-type mCRC, SCCHN and squamous NSCLC, but response rates are usually low and secondary resistance also develops through the redundancy of the EGFR signaling pathway or tumor transformation into another histologic type. On-target off-tumor and off-target toxicities to EGFR signaling inhibitors are of concern and may have a significant impact on patient quality of life and treatment effectiveness.
Overexpression of EGFR in many tumor types provides a basis for the design of therapies that use EGFR as a decoy to guide effectors of the adaptive or innate immune systems to EGFR-expressing cancers and to destroy EGFR-expressing cancer cells. Despite the promise of CAR-T cell therapies using cytotoxic T cells engineered to express constructs recognizing EGFR, these are still in early stages of development and present several serious complications that are currently difficult to overcome. Despite being effective in patients with hematologic malignancies, CAR-T cell therapies have not performed well in patients with solid tumors, potentially due to the hostile immunosuppressive tumor microenvironment and heterogeneity of cancer epitope expression. Serious neurotoxicity and CRS currently compromise the clinical use of these therapeutic agents.
To overcome the limitations of the T cell-based therapeutic approaches, target-specific innate cell engagers are undergoing preclinical and clinical evaluation and show high promise, in controlling on-target off-tumor toxicities. Based on preclinical investigations, the EGFR-specific innate immune system engager AFM24 activates potent mutation-independent antitumor immune responses by engaging NK cells and macrophages to mediate ADCC and ADCP and has shown a comparable activity to those therapies that utilize T cells. Importantly, the key advantage over therapies targeting EGFR signaling and those that use T cells is a much more favorable toxicity profile, with the lack of dermatologic toxicities and CRS, potentially determined by the lower proliferative potential of NK cells and macrophages and the EGFR signaling-independent mode of action of AFM24. These properties are key for delivering safer and more effective therapies to target tumors expressing EGFR. They also highlight the advantages of harnessing the innate immune system to address unmet needs in the treatment of patients with EGFR-expressing cancer.
GH, ER, and HD designed, wrote, reviewed, revised, and edited the manuscript. All authors contributed to the article and approved the submitted version.
This study received funding from Affimed GmbH.
Medical writing assistance was provided by Meridian HealthComms, Plumley, UK and was funded by Affimed GmbH, in accordance with Good Publication Practice (GPP3).
All authors are employees of Affimed GmbH.
The authors declare that this study received funding from Affimed GmbH. The funder had the following involvement with the study: funded the writing assistance.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wee P, Wang Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers (Basel) (2017) 9(5):52. doi: 10.3390/cancers9050052
2. The human protein atlas. Available at: https://www.proteinatlas.org/ENSG00000146648-EGFR/tissue (Accessed 8 February, 2022).
3. Yano S, Kondo K, Yamaguchi M, Richmond G, Hutchison M, Wakeling A, et al. Distribution and function of EGFR in human tissue and the effect of EGFR tyrosine kinase inhibition. Anticancer Res (2003) 23(5a):3639–50.
4. Real FX, Rettig WJ, Chesa PG, Melamed MR, Old LJ, Mendelsohn J. Expression of epidermal growth factor receptor in human cultured cells and tissues: relationship to cell lineage and stage of differentiation. Cancer Res (1986) 46(9):4726–31.
5. Thomas R, Srivastava S, Katreddy RR, Sobieski J, Weihua Z. Kinaseinactivated EGFR is required for the survival of wild-type EGFR-expressing cancer cells treated with tyrosine kinase inhibitors. Int J Mol Sci (2019) 20(10):2515. doi: 10.3390/ijms20102515
6. Thomas R, Weihua Z. Rethink of EGFR in cancer with its kinase independent function on board. Front Oncol (2019) 9:800. doi: 10.3389/fonc.2019.00800
7. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med (2008) 359(13):1367–80. doi: 10.1056/NEJMra0802714
8. Pelloski CE, Ballman KV, Furth AF, Zhang L, Lin E, Sulman EP, et al. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol (2007) 25(16):2288–94. doi: 10.1200/JCO.2006.08.0705
9. Chan DLH, Segelov E, Wong RS, Smith A, Herbertson RA, Li BT, et al. Epidermal growth factor receptor (EGFR) inhibitors for metastatic colorectal cancer. Cochrane Database System Rev (2017) 6(6):CD007047–CD. doi: 10.1002/14651858.CD007047.pub2
10. Xu MJ, Johnson DE, Grandis JR. EGFR-targeted therapies in the postgenomic era. Cancer Metastasis Rev (2017) 36(3):463–73. doi: 10.1007/s10555-017-9687-8
11. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature (2014) 505(7484):495–501. doi: 10.1038/nature12912
12. Zhang YL, Yuan JQ, Wang KF, Fu XH, Han XR, Threapleton D, et al. The prevalence of EGFR mutation in patients with non-small cell lung cancer: A systematic review and meta-analysis. Oncotarget (2016) 7(48):78985–93. doi: 10.18632/oncotarget.12587
13. An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene (2018) 37(12):1561–75. doi: 10.1038/s41388-017-0045-7
14. Gan HK, Cvrljevic AN, Johns TG. The epidermal growth factor receptor variant III (EGFRvIII): where wild things are altered. FEBS J (2013) 280(21):5350–70. doi: 10.1111/febs.12393
15. Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer (2001) 37(Suppl. 4):S9–15. doi: 10.1016/s0959-8049(01)00231-3
16. Birkman EM, Algars A, Lintunen M, Ristamaki R, Sundstrom J, Carpen O. EGFR gene amplification is relatively common and associates with outcome in intestinal adenocarcinoma of the stomach, gastro-oesophageal junction and distal oesophagus. BMC Cancer (2016) 16:406. doi: 10.1186/s12885-016-2456-1
17. Rossi E, Villanacci V, Danesino C, Donato F, Nascimbeni R, Bassotti G. Epidermal growth factor receptor overexpression/amplification in adenocarcinomas arising in the gastrointestinal tract. Rev Esp Enferm Dig (2011) 103(12):632–9. doi: 10.4321/s1130-01082011001200005
18. Huang SF, Cheng SD, Chien HT, Liao CT, Chen IH, Wang HM, et al. Relationship between epidermal growth factor receptor gene copy number and protein expression in oral cavity squamous cell carcinoma. Oral Oncol (2012) 48(1):67–72. doi: 10.1016/j.oraloncology.2011.06.511
19. Itakura Y, Sasano H, Shiga C, Furukawa Y, Shiga K, Mori S, et al. Epidermal growth factor receptor overexpression in esophageal carcinoma. An immunohistochemical study correlated with clinicopathologic findings and DNA amplification. Cancer (1994) 74(3):795–804. doi: 10.1002/1097-0142(19940801
20. Sawano A, Takayama S, Matsuda M, Miyawaki A. Lateral propagation of EGF signaling after local stimulation is dependent on receptor density. Dev Cell (2002) 3(2):245–57. doi: 10.1016/s1534-5807(02)00224-1
21. Schneider MR, Yarden Y. The EGFR-HER2 module: A stem cell approach to understanding a prime target and driver of solid tumors. Oncogene (2016) 35(23):2949–60. doi: 10.1038/onc.2015.372
22. Shah R, Lester JF. Tyrosine kinase inhibitors for the treatment of EGFR mutation-positive non–small-cell lung cancer: A clash of the generations. Clin Lung Cancer (2020) 21(3):e216–e28. doi: 10.1016/j.cllc.2019.12.003
23. Trivedi S, Srivastava RM, Concha-Benavente F, Ferrone S, Garcia-Bates TM, Li J, et al. Anti-EGFR targeted monoclonal antibody isotype influences antitumor cellular immunity in head and neck cancer patients. Clin Cancer Res (2016) 22(21):5229–37. doi: 10.1158/1078-0432.ccr-15-2971
24. Janani B, Vijayakumar M, Priya K, Kim JH, Prabakaran DS, Shahid M, et al. EGFR-based targeted therapy for colorectal cancer–promises and challenges. Vaccines (2022) 10(4):499. doi: 10.3390/vaccines10040499
25. Huang L, Jiang S, Shi Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J Hematol Oncol (2020) 13(1):143. doi: 10.1186/s13045-020-00977-0
26. Patel B, Saba NF. Current aspects and future considerations of EGFR inhibition in locally advanced and recurrent metastatic squamous cell carcinoma of the head and neck. Cancers (2021) 13(14):3545. doi: 10.3390/cancers13143545
27. Zhou J, Ji Q, Li Q. Resistance to anti-EGFR therapies in metastatic colorectal cancer: underlying mechanisms and reversal strategies. J Exp Clin Cancer Res (2021) 40(1). doi: 10.1186/s13046-021-02130-2
28. Pockley AG, Vaupel P, Multhoff G. NK cell-based therapeutics for lung cancer. Expert Opin Biol Ther (2020) 20(1):23–33. doi: 10.1080/14712598.2020.1688298
29. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9(399). doi: 10.1126/scitranslmed.aaa0984
30. Du X, Yang B, An Q, Assaraf YG, Cao X, Xia J. Acquired resistance to thirdgeneration EGFR-TKIs and emerging next-generation EGFR inhibitors. Innovation (Camb) (2021) 2(2):100103. doi: 10.1158/1078-0432.CCR-10-2277
31. U.S. Food and Drug Administration. TARCEVA® (erlotinib). Prescribing information. (2016). Available at: https://www.gene.com/download/pdf/tarceva_prescribing.pdf (Accessed 8 February, 2022).
32. Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res (2011) 17(5):1169–80. doi: 10.1158/1078-0432.CCR-10-2277
33. Nagano T, Tachihara M, Nishimura Y. Mechanism of resistance to epidermal growth factor receptor-tyrosine kinase inhibitors and a potential treatment strategy. Cells (2018) 7(11):212. doi: 10.3390/cells7110212
34. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PloS Med (2005) 2(3):e73–e. doi: 10.1371/journal.pmed.0020073
35. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutationpositive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol (2011) 12(8):735–42. doi: 10.1016/s1470-2045(11)70184-x
36. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol (2012) 13(3):239–46. doi: 10.1016/s1470-2045(11)70393-x
37. Wu YL, Zhou C, Liam CK, Wu G, Liu X, Zhong Z, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study. Ann Oncol (2015) 26(9):1883–9. doi: 10.1093/annonc/mdv270
38. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the national cancer institute of Canada clinical trials group. J Clin Oncol (2007) 25(15):1960–6. doi: 10.1200/JCO.2006.07.9525
39. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Final overall survival results from a randomised, phase III study of erlotinib versus chemotherapy as first-line treatment of EGFR mutation-positive advanced nonsmall-cell lung cancer (OPTIMAL, CTONG0802). Ann Oncol (2015) 26(9):1877–83. doi: 10.1093/annonc/mdv276
40. U.S. Food and Drug Administration. IRESSA® (gefitinib). Prescribing Information. (2018). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/206995s003lbl.pdf (Accessed 8 February, 2022)
41. Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med (2005) 352(8):786–92. doi: 10.1056/NEJMoa044238
42. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med (2009) 361(10):947–57. doi: 10.1056/NEJMoa0810699
43. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol (2010) 11(2):121–8. doi: 10.1016/s1470-2045(09)70364-x
44. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med (2010) 362(25):2380–8. doi: 10.1056/NEJMoa0909530
45. Yoshioka H, Shimokawa M, Seto T, Morita S, Yatabe Y, Okamoto I, et al. Final overall survival results of WJTOG3405, a randomized phase III trial comparing gefitinib versus cisplatin with docetaxel as the first-line treatment for patients with stage IIIB/IV or postoperative recurrent EGFR mutation-positive non-small-cell lung cancer. Ann Oncol (2019) 30(12):1978–84. doi: 10.1093/annonc/mdz399
46. Shi Y, Zhang L, Liu X, Zhou C, Zhang L, Zhang S, et al. Icotinib versus gefitinib in previously treated advanced non-small-cell lung cancer (ICOGEN): A randomised, double blind phase 3 non-inferiority trial. Lancet Oncol (2013) 14(10):953–61. doi: 10.1016/S14702045(13)70355-3
47. Shi YK, Wang L, Han BH, Li W, Yu P, Liu YP, et al. First-line icotinib versus cisplatin/pemetrexed plus pemetrexed maintenance therapy for patients with advanced EGFR mutation-positive lung adenocarcinoma (CONVINCE): A phase 3, open-label, randomized study. Ann Oncol (2017) 28(10):2443–50. doi: 10.1093/annonc/mdx359
48. U.S. Food and Drug Administration. GILOTRIF® (afatinib). Prescribing information. (2018). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/201292s014lbl.pdf (Accessed 8 February, 2022)
49. Hochmair MJ, Morabito A, Hao D, Yang CT, Soo RA, Yang JC, et al. Sequential treatment with afatinib and osimertinib in patients with EGFR mutation-positive non-small-cell lung cancer: An observational study. Future Oncol (2018) 14(27):2861–74. doi: 10.2217/fon-2018-0711
50. Tanaka K, Nosaki K, Otsubo K, Azuma K, Sakata S, Ouchi H, et al. Acquisition of the T790M resistance mutation during afatinib treatment in EGFR tyrosine kinase inhibitor-naive patients with non-small cell lung cancer harboring EGFR mutations. Oncotarget (2017) 8(40):68123–30. doi: 10.18632/oncotarget.19243
51. Wu SG, Liu YN, Tsai MF, Chang YL, Yu CJ, Yang PC, et al. The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients. Oncotarget (2016) 7(11):12404–13. doi: 10.18632/oncotarget.7189
52. Sequist LV, Yang JC, Yamamoto N, O'Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol (2013) 31(27):3327–34. doi: 10.1200/jco.2012.44.2806
53. Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-lung 6): An openlabel, randomised phase 3 trial. Lancet Oncol (2014) 15(2):213–22. doi: 10.1016/s1470-2045(13)70604-1
54. Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-lung 3 and LUX-lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol (2015) 16(2):141–51. doi: 10.1016/s1470-2045(14)71173-8
55. U.S. Food and Drug Administration. VIZIMPRO® (dacomitinib). Prescribing Information. (2018). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208065s008lbl.pdf (Accessed 8 February, 2022).
56. Janne PA, Ou SI, Kim DW, Oxnard GR, Martins R, Kris MG, et al. Dacomitinib as first-line treatment in patients with clinically or molecularly selected advanced non-small cell lung cancer: A multicentre, open-label, phase 2 trial. Lancet Oncol (2014) 15(13):1433–41. doi: 10.1016/S1470-2045(14)70461-9
57. Ramalingam SS, Jänne PA, Mok T, O'Byrne K, Boyer MJ, Von Pawel J, et al. Dacomitinib versus erlotinib in patients with advanced-stage, previously treated non-small cell lung cancer (ARCHER 1009): A randomised, double-blind, phase 3 trial. Lancet Oncol (2014) 15(12):1369–78. doi: 10.1016/s1470-2045(14)70452-8
58. Wu YL, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet Oncol (2017) 18(11):1454–66. doi: 10.1016/S1470-2045(17)30608-3
59. Mok TS, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Updated overall survival in a randomized study comparing dacomitinib with gefitinib as first-line treatment in patients with advanced non-small-cell lung cancer and EGFR-activating mutations. Drugs (2021) 81(2):257–66. doi: 10.1007/s40265-020-01441-6
60. U.S. Food and Drug Administration. TAGRISSO® (osimertinib). Prescribing Information.(2015). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/208065s000lbl.pdf (Accessed 8 February, 2022).
61. Cho BC, Cheng Y, Zhou C, Ohe Y, Imamura F, Lin M-C, et al. Mechanisms of acquired resistance to first-line osimertinib: preliminary data from the phase III FLAURA study. Ann Oncol (2018) 29(Suppl. 9):ix177.
62. Schoenfeld AJ, Chan JM, Kubota D, Sato H, Rizvi H, Daneshbod Y, et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin Cancer Res (2020) 26(11):2654–63. doi: 10.1158/1078-0432.CCR-19-3563
63. Zhao S, Li X, Zhao C, Jiang T, Jia Y, Shi J, et al. Loss of T790M mutation is associated with early progression to osimertinib in Chinese patients with advanced NSCLC who are harboring EGFR T790M. Lung Cancer (2019) 128:33–9. doi: 10.1016/j.lungcan.2018.12.010
64. Hidaka N, Iwama E, Kubo N, Harada T, Miyawaki K, Tanaka K, et al. Most T790M mutations are present on the same EGFR allele as activating mutations in patients with non-small cell lung cancer. Lung Cancer (2017) 108:75–82. doi: 10.1016/j.lungcan.2017.02.019
65. Ricordel C, Friboulet L, Facchinetti F, Soria JC. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann Oncol (2018) 29(suppl_1):i28–37. doi: 10.1093/annonc/mdx705
66. Papadimitrakopoulou VA, Mok TS, Han JY, Ahn MJ, Delmonte A, Ramalingam SS, et al. Osimertinib versus platinum-pemetrexed for patients with EGFR T790M advanced NSCLC and progression on a prior EGFR-tyrosine kinase inhibitor: AURA3 overall survival analysis. Ann Oncol (2020) 31(11):1536–44. doi: 10.1016/j.annonc.2020.08.2100
67. Papadimitrakopoulou VA, Wu Y-L, Han J-Y, Ahn M-J, Ramalingam SS, John T, et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann Oncol (2018) 29(Suppl 8):VIII741. doi: 10.1093/annonc/mdy424.064
68. Mok TS, Wu YL, Papadimitrakopoulou VA. Osimertinib in EGFR T790M positive lung cancer. N Engl J Med (2017) 376(20):1993–4. doi: 10.1056/NEJMc1703339
69. Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med (2018) 378(2):113–25. doi: 10.1056/NEJMoa1713137
70. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. New Engl J Med (2019) 382(1):41–50. doi: 10.1056/NEJMoa1913662
71. U.S. Food and Drug Administration. ALUNBRIG® (brigatinib). Prescribing Information. (2017). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208772lbl.pdf (Accessed 8 February, 2022).
72. Wang Y, Yang N, Zhang Y, Li L, Han R, Zhu M, et al. Effective treatment of lung adenocarcinoma harboring EGFR-activating mutation, T790M, and cis-C797S triple mutations by brigatinib and cetuximab combination therapy. J Thorac Oncol (2020) 15(8):1369–75. doi: 10.1016/j.jtho.2020.04.014
73. U.S. Food and Drug Administration. ERBITUX® (cetuximab). Prescribing Information. (2019). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125084s273lbl.pdf (Accessed 8 February, 2022).
74. Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature (2015) 526(7572):263–7. doi: 10.1038/nature14969
75. Bray SM, Lee J, Kim ST, Hur JY, Ebert PJ, Calley JN, et al. Genomic characterization of intrinsic and acquired resistance to cetuximab in colorectal cancer patients. Sci Rep (2019) 9(1):15365. doi: 10.1038/s41598-019-51981-5
76. Nair S, Trummell HQ, Rajbhandari R, Thudi NK, Nozell SE, Warram JM, et al. Novel EGFR ectodomain mutations associated with ligand-independent activation and cetuximab resistance in head and neck cancer. PloS One (2020) 15(2):e0229077. doi: 10.1371/journal.pone.0229077
77. Khattri A, Sheikh N, Acharya R, Tan Y-HC, Kochanny S, Lingen MW, et al. Mechanism of acquired resistance to cetuximab in head and neck cancer. J Clin Oncol (2018) 36(Suppl 15):e18061.
78. Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med (2014) 6(224):224ra24. doi: 10.1126/scitranslmed.3007094
79. Diaz LA Jr., Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature (2012) 486(7404):537–40. doi: 10.1038/nature11219
80. Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature (2012) 486(7404):532–6. doi: 10.1038/nature11156
81. Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discovery (2013) 3(6):658–73. doi: 10.1158/2159-8290.CD-12-0558
82. Mohan S, Heitzer E, Ulz P, Lafer I, Lax S, Auer M, et al. Changes in colorectal carcinoma genomes under anti-EGFR therapy identified by wholegenome wholegenome plasma DNA sequencing. PloS Genet (2014) 10(3):e1004271. doi: 10.1371/journal.pgen.1004271
83. Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med (2015) 21(7):827. doi: 10.1038/nm0715-827b
84. Price TJ, Newhall K, Peeters M, Kim TW, Li J, Cascinu S, et al. Prevalence and outcomes of patients (pts) with EGFR S492R ectodomain mutations in ASPECCT: Panitumumab (pmab) vs cetuximab (cmab) in pts with chemorefractory wild-type KRAS exon 2 metastatic colorectal cancer (mCRC). J Clin Oncol (2015) 33:e14623.
85. Arena S, Bellosillo B, Siravegna G, Martinez A, Canadas I, Lazzari L, et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res (2015) 21(9):2157–66. doi: 10.1158/1078-0432.CCR-14-2821
86. Wang D, Qian G, Zhang H, Magliocca KR, Nannapaneni S, Amin AR, et al. HER3 targeting sensitizes HNSCC to cetuximab by reducing HER3 activity and HER2/HER3 dimerization: Evidence from cell line and patient-derived xenograft models. Clin Cancer Res (2017) 23(3):677–86. doi: 10.1158/1078-0432.CCR-16-0558
87. Braig F, Marz M, Schieferdecker A, Schulte A, Voigt M, Stein A, et al. Epidermal growth factor receptor mutation mediates cross-resistance to panitumumab and cetuximab in gastrointestinal cancer. Oncotarget (2015) 6(14):12035–47. doi: 10.18632/oncotarget.3574
88. Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med (2009) 360(14):1408–17. doi: 10.1056/NEJMoa0805019
89. Qin S, Li J, Wang L, Xu J, Cheng Y, Bai Y, et al. Efficacy and tolerability of first-line cetuximab plus leucovorin, fluorouracil, and oxaliplatin (FOLFOX-4) versus FOLFOX-4 in patients with RAS wild-type metastatic colorectal cancer: The open-label, randomized, phase III TAILOR trial. J Clin Oncol (2018) 36(30):3031–9. doi: 10.1200/jco.2018.78.3183
90. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med (2004) 351(4):337–45. doi: 10.1056/NEJMoa033025
91. Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med (2007) 357(20):2040–8. doi: 10.1056/NEJMoa071834
92. Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med (2019) 381(17):1632–43. doi: 10.1056/NEJMoa1908075
93. Sobrero AF, Maurel J, Fehrenbacher L, Scheithauer W, Abubakr YA, Lutz MP, et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol (2008) 26(14):2311–9. doi: 10.1200/jco.2007.13.1193
94. Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med (2008) 359(11):1116–27. doi: 10.1056/NEJMoa0802656
95. Bonner JA, Harari PM, Giralt J, Cohen RB, Jones CU, Sur RK, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5- year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol (2010) 11(1):21–8. doi: 10.1016/s1470-2045(09)70311-0
96. Yoshino T, Watanabe J, Shitara K, Yasui H, Ohori H, Shiozawa M, et al. Panitumumab (PAN) plus mFOLFOX6 versus bevacizumab (BEV) plus mFOLFOX6 as first-line treatment in patients with RAS wild-type (WT) metastatic colorectal cancer (mCRC): Results from the phase 3 PARADIGM trial. J Clin Oncol (2022) 40(17_suppl):LBA1–LBA. doi: 10.1200/JCO.2022.40.17_suppl.LBA1
97. Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol (2007) 25(13):1658–64. doi: 10.1200/JCO.2006.08.1620
98. Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol (2010) 28(31):4697–705. doi: 10.1200/JCO.2009.27.4860
99. U.S. Food and Drug Administration. VECTIBIX® (panitumumab). Prescribing Information. (2021). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/125147s210lbl.pdf (Accessed 8 February, 2022).
100. U.S. Food and Drug Administration. PORTRAZZA® (necitumumab) injection. Prescribing Information. (2015). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125547s000lbl.pdf (Accessed 8 February, 2022)
101. Thatcher N, Hirsch FR, Luft AV, Szczesna A, Ciuleanu TE, Dediu M, et al. Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): An open-label, randomised, controlled phase 3 trial. Lancet Oncol (2015) 16(7):763–74. doi: 10.1016/S1470-2045(15)00021-2
102. Itchins M, Clarke S, Pavlakis N. Do EGFR tyrosine kinase inhibitors (TKIs) still have a role in EGFR wild-type pre-treated advanced non-small cell lung cancer (NSCLC)?-the shifting paradigm of therapeutics. Trans Lung Cancer Res (2018) 7(Suppl 1):S39–45. doi: 10.21037/tlcr.2018.01.06
103. Argiris A, Ghebremichael M, Gilbert J, Lee J-W, Sachidanandam K, Kolesar JM, et al. Phase III randomized, placebo-controlled trial of docetaxel with or without gefitinib in recurrent or metastatic head and neck cancer: An Eastern cooperative oncology group trial. J Clin Oncol (2013) 31(11):1405–14. doi: 10.1200/jco.2012.45.4272
104. Martins RG, Parvathaneni U, Bauman JE, Sharma AK, Raez LE, Papagikos MA, et al. Cisplatin and radiotherapy with or without erlotinib in locally advanced squamous cell carcinoma of the head and neck: A randomized phase II trial. J Clin Oncol (2013) 31(11):1415–21. doi: 10.1200/jco.2012.46.3299
105. Summary of product characteristics (SmPC) by AstraZeneca. In: Iressa 250mg film-coated tablets. Available at: https://www.medicines.org.uk/emc/medicine/22104#gref.
106. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer (2007) 7(3):169–81. doi: 10.1038/nrc2088
107. Lee CK, Brown C, Gralla RJ, Hirsh V, Thongprasert S, Tsai CM, et al. Impact of EGFR inhibitor in non-small cell lung cancer on progression-free and overall survival: A meta-analysis. J Natl Cancer Inst (2013) 105(9):595–605. doi: 10.1093/jnci/djt072
108. Shi Y, Sun Y, Ding C, Wang Z, Wang C, Wang Z, et al. China Experts consensus on icotinib for non-small cell lung cancer treatment (2015 version). Ann Transl Med (2015) 3(18):260. doi: 10.3978/j.issn.2305-5839.2015.10.30
109. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med (2011) 3(75):75ra26. doi: 10.1126/scitranslmed.3002003
110. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res (2013) 19(8):2240–7. doi: 10.1158/1078-0432.Ccr-12-2246
111. Woolston A, Khan K, Spain G, Barber LJ, Griffiths B, Gonzalez-Exposito R, et al. Genomic and transcriptomic determinants of therapy resistance and immune landscape evolution during anti-egfr treatment in colorectal cancer. Cancer Cell (2019) 36(1):35–50.e9. doi: 10.1016/j.ccell.2019.05.013
112. Lim SM, Syn NL, Cho BC, Soo RA. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat Rev (2018) 65:1–10. doi: 10.1016/j.ctrv.2018.02.006
113. Yang Z, Hackshaw A, Feng Q, Fu X, Zhang Y, Mao C, et al. Comparison of gefitinib, erlotinib and afatinib in non-small cell lung cancer: A meta-analysis. Int J Cancer (2017) 140(12):2805–19. doi: 10.1002/ijc.30691
114. Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol (2018). doi: 10.1093/annonc/mdx703
115. Paz-Ares L, Tan EH, O'Byrne K, Zhang L, Hirsh V, Boyer M, et al. Afatinib versus gefitinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: overall survival data from the phase IIb LUX-lung 7 trial. Ann Oncol (2017) 28(2):270–7. doi: 10.1093/annonc/mdw611
116. Park K, Tan EH, O'Byrne K, Zhang L, Boyer M, Mok T, et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol (2016) 17(5):577–89. doi: 10.1016/s1470-2045(16)30033-x
117. Popat S, Mok T, Yang JC, Wu YL, Lungershausen J, Stammberger U, et al. Afatinib in the treatment of EGFR mutation-positive NSCLC–a network metaanalysis. Lung Cancer (2014) 85(2):230–8. doi: 10.1016/j.lungcan.2014.05.007
118. Kohsaka S, Petronczki M, Solca F, Maemondo M. Tumor clonality and resistance mechanisms in EGFR mutation-positive non-small-cell lung cancer: implications for therapeutic sequencing. Future Oncol (2019) 15(6):637–52. doi: 10.2217/fon-2018-0736
119. Ramalingam SS, Blackhall F, Krzakowski M, Barrios CH, Park K, Bover I, et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol (2012) 30(27):3337–44. doi: 10.1200/jco.2011.40.9433
120. Roskoski R Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol Res (2019) 139:395–411. doi: 10.1016/j.phrs.2018.11.014
121. Dassonville O, Bozec A, Fischel JL, Milano G. EGFR targeting therapies: monoclonal antibodies versus tyrosine kinase inhibitors. Similarities differences. Crit Rev Oncol Hematol (2007) 62(1):53–61. doi: 10.1016/j.critrevonc.2006.12.008
122. Chagoya G, Kwatra SG, Nanni CW, Roberts CM, Phillips SM, Nullmeyergh S, et al. Efficacy of osimertinib against EGFRvIII+ glioblastoma. Oncotarget (2020) 11(22):2074–82. doi: 10.18632/oncotarget.27599
123. Liu X, Chen X, Shi L, Shan Q, Cao Q, Yue C, et al. The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma. J Exp Clin Cancer Res (2019) 38(1):219. doi: 10.1186/s13046-019-1235-7
124. Chen C, Cheng CD, Wu H, Wang ZW, Wang L, Jiang ZR, et al. Osimertinib successfully combats EGFR-negative glioblastoma cells by inhibiting the MAPK pathway. Acta Pharmacol Sin (2021) 42(1):108–14. doi: 10.1038/s41401-020-0418-2
125. Abousaud M, Faroqui NM, Lesser G, Strowd RE, Ramkissoon SH, Kwatra M, et al. Clinical experience using osimertinib in patients with recurrent malignant gliomas containing EGFR alterations. J Cancer Sci Clin Ther (2021) 5(2):210–20. doi: 10.26502/jcsct.5079114
126. Chakravarti A, Wang M, Robins HI, Lautenschlaeger T, Curran WJ, Brachman DG, et al. RTOG 0211: A phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. Int J Radiat Oncol Biol Phys (2013) 85(5):1206–11. doi: 10.1016/j.ijrobp.2012.10.008
127. Prados MD, Chang SM, Butowski N, DeBoer R, Parvataneni R, Carliner H, et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. J Clin Oncol (2009) 27(4):579–84. doi: 10.1200/jco.2008.18.9639
128. Reardon DA, Nabors LB, Mason WP, Perry JR, Shapiro W, Kavan P, et al. Phase i/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro Oncol (2015) 17(3):430–9. doi: 10.1093/neuonc/nou160
129. Sepúlveda-Sánchez JM, Vaz M, Balañá C, Gil-Gil M, Reynés G, Gallego Ó, et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. . Neuro Oncol (2017) 19(11):1522–31. doi: 10.1093/neuonc/nox105
130. Uchibori K, Inase N, Araki M, Kamada M, Sato S, Okuno Y, et al. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat Commun (2017) 8:14768. doi: 10.1038/ncomms14768
131. Wang S, Song Y, Liu D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett (2017) 385:51–4. doi: 10.1016/j.canlet.2016.11.008
132. Liu X, Zhang X, Yang L, et al. Abstract 1320: preclinical evaluation of TQB3804, a potent EGFR C797S inhibitor. Cancer Res (2019) 791320(13Supplement). doi: 10.1158/1538-7445.AM2019-1320
133. To C, Jang J, Chen T, Park E, Mushajiang M, De Clercq DJH, et al. Single and dual targeting of mutant EGFR with an allosteric inhibitor. Cancer Discovery (2019) 9(7):926–43. doi: 10.1158/2159-8290.CD-18-0903
134. Kashima K, Kawauchi H, Tanimura H, Tachibana Y, Chiba T, Torizawa T, et al. CH7233163 overcomes osimertinib-resistant EGFR-Del19/T790M/C797S mutation. Mol Cancer Ther (2020) 19(11):2288–97. doi: 10.1158/1535-7163.MCT-20-0229
135. Schalm SS, Dineen T, Lim SM, Park CW, Hsieh J, Woessner R, et al. 1296P BLU-945, a highly potent and selective 4th generation EGFR TKI for the treatment of EGFR T790M/C797S resistant NSCLC. Ann Oncol (2020) 31:S839. doi: 10.1016/j.annonc.2020.08.1610
136. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science (2007) 316(5827):1039–43. doi: 10.1126/science.1141478
137. Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA (2007) 104(52):20932–7. doi: 10.1073/pnas.0710370104
138. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br J Cancer (2019) 121(9):725–37. doi: 10.1038/s41416-019-0573-8
139. Lin Y, Wang X, Jin H. EGFR-TKI resistance in NSCLC patients: mechanisms and strategies. Am J Cancer Res (2014) 4(5):411–35.
140. Jouida A, McCarthy C, Fabre A, Keane MP. Exosomes: A new perspective in EGFR-mutated lung cancer. Cancer Metastasis Rev (2021) 40(2):589–601. doi: 10.1007/s10555-021-09962-6
141. Liu X, Jiang T, Li X, Zhao C, Li J, Zhou F, et al. Exosomes transmit T790M mutation-induced resistance in EGFR-mutant NSCLC by activating PI3K/AKT signalling pathway. J Cell Mol Med (2020) 24(2):1529–40. doi: 10.1111/jcmm.14838
142. Chen R, Qian Z, Xu X, Zhang C, Niu Y, Wang Z, et al. Exosomestransmitted miR-7 reverses gefitinib resistance by targeting YAP in non-small-cell lung cancer. Pharmacol Res (2021) 165:105442. doi: 10.1016/j.phrs.2021.105442
143. Wang YS, Wang YH, Xia HP, Zhou SW, Schmid-Bindert G, Zhou CC. MicroRNA-214 regulates the acquired resistance to gefitinib via the PTEN/AKT pathway in EGFR-mutant cell lines. Asian Pac J Cancer Prev (2012) 13(1):255–60. doi: 10.7314/apjcp.2012.13.1.255
144. Yang B, Teng F, Chang L, Wang J, Liu DL, Cui YS, et al. Tumor-derived exosomal circRNA_102481 contributes to EGFR-TKIs resistance via the miR-30a-5p/ROR1 axis in non-small cell lung cancer. Aging (Albany NY) (2021) 13(9):13264–86. doi: 10.18632/aging.203011
145. Kim J, Vasu VT, Mishra R, Singleton KR, Yoo M, Leach SM, et al. Bioinformatics-driven discovery of rational combination for overcoming EGFRmutant lung cancer resistance to EGFR therapy. Bioinformatics (2014) 30(17):2393–8. doi: 10.1093/bioinformatics/btu323
146. Ryall KA, Tan AC. Systems biology approaches for advancing the discovery of effective drug combinations. J Cheminform (2015) 7(1):7. doi: 10.1186/s13321-015-0055-9
147. Cai WQ, Zeng LS, Wang LF, Wang YY, Cheng JT, Zhang Y, et al. The latest battles between EGFR monoclonal antibodies and resistant tumor cells. Front Oncol (2020) 10:1249. doi: 10.3389/fonc.2020.01249
148. Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell (2005) 7(4):301–11. doi: 10.1016/j.ccr.2005.03.003
149. Talavera A, Friemann R, Gomez-Puerta S, Martinez-Fleites C, Garrido G, Rabasa A, et al. Nimotuzumab, an antitumor antibody that targets the epidermal growth factor receptor, blocks ligand binding while permitting the active receptor conformation. Cancer Res (2009) 69(14):5851–9. doi: 10.1158/0008-5472.CAN-08-4518
150. Ferris RL, Lenz H-J, Trotta AM, Garcı́a-Foncillas J, Schulten J, Audhuy F, et al. Rationale for combination of therapeutic antibodies targeting tumor cells and immune checkpoint receptors: Harnessing innate and adaptive immunity through IgG1 isotype immune effector stimulation. Cancer Treat Rev (2018) 63:48–60. doi: 10.1016/j.ctrv.2017.11.008
151. FDA Approves encorafenib in combination with cetuximab for metastatic colorectal cancer with a BRAF V600E mutation (2020). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-encorafenibcombination-cetuximab-metastatic-colorectal-cancer-braf-v600e-mutation (Accessed 16 February 2021).
152. Tejpar S, Stintzing S, Ciardiello F, Tabernero J, Van Cutsem E, Beier F, et al. Prognostic and predictive relevance of primary tumor location in patients with RAS wild-type metastatic colorectal cancer: Retrospective analyses of the CRYSTAL and FIRE-3 trials. JAMA Oncol (2017) 3(2):194–201. doi: 10.1001/jamaoncol.2016.3797
153. Venook AP, Niedzwiecki D, Innocenti F, Fruth B, Greene C, O'Neil BH, et al. Impact of primary (1°) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). Am Soc Clin Oncol (2016). doi: 10.1200/JCO.2016.34.15_suppl.3504
154. Missiaglia E, Jacobs B, D'Ario G, Di Narzo AF, Soneson C, Budinska E, et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann Oncol (2014) 25(10):1995–2001. doi: 10.1093/annonc/mdu275
155. Maus MK, Hanna DL, Stephens C, Grimminger PP, Epstein M, Astrow SH, et al. Gene expression profiles and tumor locations in colorectal cancer (left vs.right vs. rectum). Am Soc Clin Oncol (2013). doi: 10.1200/jco.2013.31.15_suppl.3527
156. Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med (2013) 369(11):1023–34. doi: 10.1056/NEJMoa1305275
157. Ramakrishnan MS, Eswaraiah A, Crombet T, Piedra P, Saurez G, Iyer H, et al. Nimotuzumab, a promising therapeutic monoclonal for treatment of tumors of epithelial origin. MAbs (2009) 1(1):41–8. doi: 10.4161/mabs.1.1.7509
158. Reddy BK, Lokesh V, Vidyasagar MS, Shenoy K, Babu KG, Shenoy A, et al. Nimotuzumab provides survival benefit to patients with inoperable advanced squamous cell carcinoma of the head and neck: A randomized, open-label, phase IIb, 5-year study in Indian patients. Oral Oncol (2014) 50(5):498–505. doi: 10.1016/j.oraloncology.2013.11.008
159. Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer (2009) 101(4):715–21. doi: 10.1038/sj.bjc.6605177
160. Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res (2009) 69(5):1851–7. doi: 10.1158/0008-5472.CAN-08-2466
161. Valtorta E, Misale S, Sartore-Bianchi A, Nagtegaal ID, Paraf F, Lauricella C, et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int J Cancer (2013) 133(5):1259–65. doi: 10.1002/ijc.28106
162. Patnaik A, Gordon M, Tsai F, Papadopoulos KP, Rasco D, Beeram M, et al. in patients with advanced or metastatic cancer. Cancer Chemother Pharmacol (2018) 82(3):407–18. doi: 10.1007/s00280-018-3623-7
163. Yun J, Lee S-H, Kim S-Y, Jeong S-Y, Kim J-H, Pyo K-H, et al. Antitumor activity of amivantamab (JNJ-61186372), an EGFR–MET bispecific antibody, in diverse models of EGFR exon 20 insertion–driven NSCLC. Cancer Discovery (2020) 10(8):1194–209. doi: 10.1158/2159-8290.Cd-20-0116
164. Park K, John T, Kim S-W, Lee JS, Shu CA, Kim D-W, et al. Amivantamab (JNJ-61186372), an anti-EGFR-MET bispecific antibody, in patients with EGFR exon 20 insertion (exon20ins)-mutated non-small cell lung cancer (NSCLC). J Clin Oncol (2020) 38(Suppl. 15):9512. doi: 10.1200/JCO.2020.38.15_suppl.9512
165. Janssen announces U.S. FDA breakthrough therapy designation granted for JNJ-6372 for the treatment of non-small cell lung cancer, in: (2020). Available at: https://www.jnj.com/janssen-announces-u-s-fda-breakthrough-therapy-designationgranted-for-jnj-6372-for-the-treatment-of-non-small-cell-lung-cancer (Accessed 17 February 2021).
166. Argiles G, Jungels C, Garcia-Carbonero R, Diez Garcia M, Bendell JC, Tabernero J, et al. Phase I dose-escalation study of MCLA-158, a first-in-class bispecific antibody targeting EGFR and LGR5, in metastatic colorectal cancer (CRC). J Clin Oncol 39(Suppl. 3). doi: 10.1200/JCO.2021.39.3_suppl.62
167. Lacouture ME. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat Rev Cancer (2006) 6(10):803–12. doi: 10.1038/nrc1970
168. Pastore S, Mascia F, Mariani V, Girolomoni G. The epidermal growth factor receptor system in skin repair and inflammation. J Invest Dermatol (2008) 128(6):1365–74. doi: 10.1038/sj.jid.5701184
169. Chen J, Zeng F, Forrester SJ, Eguchi S, Zhang MZ, Harris RC. Expression and function of the epidermal growth factor receptor in physiology and disease. Physiol Rev (2016) 96(3):1025–69. doi: 10.1152/physrev.00030.2015
170. Tang X, Liu H, Yang S, Li Z, Zhong J, Fang R. Epidermal growth factor and intestinal barrier function. Mediators Inflam (2016) 2016:1927348. doi: 10.1155/2016/1927348
171. Brazil JC, Quiros M, Nusrat A, Parkos CA. Innate immune cell-epithelial crosstalk during wound repair. J Clin Invest (2019) 129(8):2983–93. doi: 10.1172/JCI124618
172. Yin X, Zhao Z, Yin Y, Shen C, Chen X, Cai Z, et al. Adverse event profiles of epidermal growth factor receptor-tyrosine kinase inhibitors in cancer patients: A systematic review and meta-analysis. Clin Transl Sci (2020) 14(3):919–33. doi: 10.1111/cts.12957
173. Lacouture ME, Anadkat MJ, Bensadoun R-J, Bryce J, Chan A, Epstein JB, et al. Clinical practice guidelines for the prevention and treatment of EGFR inhibitor-associated dermatologic toxicities. Support Care Cancer (2011) 19(8):1079–95. doi: 10.1007/s00520-011-1197-6
174. Lacouture ME, Anadkat M, Jatoi A, Garawin T, Bohac C, Mitchell E. Dermatologic toxicity occurring during anti-EGFR monoclonal inhibitor therapy in patients with metastatic colorectal cancer: A systematic review. Clin Colorectal Cancer (2018) 17(2):85–96. doi: 10.1016/j.clcc.2017.12.004
175. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
176. Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. New Engl J Med (2016) 375(19):1856–67. doi: 10.1056/NEJMoa1602252
177. Cohen EEW, Soulières D, Le Tourneau C, Dinis J, Licitra L, Ahn M-J, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet (2019) 393(10167):156–67. doi: 10.1016/S0140-6736(18)31999-8
178. Burtness B, Harrington KJ, Greil R, Soulières D, Tahara M, de Castro G Jr, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet (2019) 394(10212):1915–28. doi: 10.1016/S0140-6736(19)32591-7
179. Diaz LA Jr., Shiu K-K, Kim T-W, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): final analysis of a randomised, open-label, phase 3 study. Lancet Oncol (2022) 23(5):659–70. doi: 10.1016/S1470-2045(22)00197-8
180. Denault MH, Melosky B. Immunotherapy in the first-line setting in wildtype NSCLC. Curr Oncol (2021) 28(6):4457–70. doi: 10.3390/curroncol28060378
181. Latif H, Liu SV. Combining immunotherapy and epidermal growth factor receptor kinase inhibitors: worth the risk? Ann Trans Med (2019). doi: 10.21037/atm.2019.03.6
182. Yang JC, Shepherd FA, Kim DW, Lee GW, Lee JS, Chang GC, et al. Osimertinib plus durvalumab versus osimertinib monotherapy in EGFR T790Mpositive NSCLC following previous EGFR TKI therapy: CAURAL brief report. J Thorac Oncol (2019) 14(5):933–9. doi: 10.1016/j.jtho.2019.02.001
183. Ma BBY, Rudin CM, Cervantes A, Dowlati A, Costa D, Schmid P, et al. 441O preliminary safety and clinical activity of erlotinib plus atezolizumab from a phase ib study in advanced NSCLC. Ann Oncol (2016) 27:ix141. doi: 10.1016/S0923-7534(21)00599-8
184. Elbers JBW, Al-Mamgani A, Tesseslaar MET, van den Brekel MWM, Lange CAH, van der Wal JE, et al. Immuno-radiotherapy with cetuximab and avelumab for advanced stage head and neck squamous cell carcinoma: Results from a phase-I trial. Radiother Oncol (2020) 142:79–84. doi: 10.1016/j.radonc.2019.08.007
185. Boland PM, Muhitch J, Abrams SI, Maguire O, Minderman H, Bajor DL, et al. Initial correlative studies from a trial of cetuximab and pembrolizumab in metastatic colorectal cancer (mCRC). J Clin Oncol (2020) 38(Suppl. 15):4062. doi: 10.1200/JCO.2020.38.15_suppl.4062
186. Yu S, Li A, Liu Q, Li T, Yuan X, Han X, et al. Chimeric antigen receptor T cells: A novel therapy for solid tumors. J Hematol Oncol (2017) 10(1):78. doi: 10.1186/s13045-017-0444-9
187. Helsen CW, Hammill JA, Lau VWC, Mwawasi KA, Afsahi A, Bezverbnaya K, et al. The chimeric TAC receptor co-opts the T cell receptor yielding robust antitumor activity without toxicity. Nat Commun (2018) 9(1):3049. doi: 10.1038/s41467-018-05395-y
188. Xia L, Zheng Z-Z, Liu J-Y, Chen Y-J, Ding J-C, Xia N-S, et al. EGFRtargeted CAR-T cells are potent and specific in suppressing triple-negative breast cancer both in vitro and in vivo. Clin Transl Immunol (2020) 9(5):e1135. doi: 10.1002/cti2.1135
189. Dong Y-H, Ding Y-M, Guo W, Huang J-W, Yang Z, Zhang Y, et al. The functional verification of EGFR-CAR T-cells targeted to hypopharyngeal squamous cell carcinoma. Onco Targets Ther (2018) 11:7053–9. doi: 10.2147/OTT.S175516
190. Ravanpay AC, Gust J, Johnson AJ, Rolczynski LS, Cecchini M, Chang CA, et al. EGFR806-CAR T cells selectively target a tumor-restricted EGFR epitope in glioblastoma. Oncotarget (2019) 10(66):7080–95. doi: 10.18632/oncotarget.27389
191. Gedeon PC, Choi BD, Hodges TR, Mitchell DA, Bigner DD, Sampson JH. An EGFRvIII-targeted bispecific T-cell engager overcomes limitations of the standard of care for glioblastoma. Expert Rev Clin Pharmacol (2013) 6(4):375–86. doi: 10.1586/17512433.2013.811806
192. Zhang Y, Zhang Z, Ding Y, Fang Y, Wang P, Chu W, et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J Cancer Res Clin Oncol (2021) 147(12):3725–34. doi: 10.1007/s00432-021-03613-7
193. Liu Y, Guo Y, Wu Z, Feng K, Tong C, Wang Y, et al. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: A phase I clinical trial. Cytotherapy (2020) 22(10):573–80. doi: 10.1016/j.jcyt.2020.04.088
194. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother (2019) 42(4):126–35. doi: 10.1097/CJI.0000000000000260
195. Guo Y, Feng K, Liu Y, Wu Z, Dai H, Yang Q, et al. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin Cancer Res (2018) 24(6):1277–86. doi: 10.1158/1078-0432.Ccr-17-0432
196. Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFRexpressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci (2016) 59(5):468–79. doi: 10.1007/s11427-016-5023-8
197. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood (2016) 127(26):3321–30. doi: 10.1182/blood-2016-04-703751
198. Rubin DB, Danish HH, Ali AB, Li K, LaRose S, Monk AD, et al. Neurological toxicities associated with chimeric antigen receptor T-cell therapy. Brain (2019) 142(5):1334–48. doi: 10.1093/brain/awz053
199. Jureczek J, Feldmann A, Bergmann R, Arndt C, Berndt N, Koristka S, et al. Highly efficient targeting of EGFR-expressing tumor cells with UniCAR T cells via target modules based on cetuximab(®). Onco Targets Ther (2020) 13:5515–27. doi: 10.2147/ott.S245169
200. Li L, Deng L, Meng X, Gu C, Meng L, Li K, et al. Tumor-targeting anti-EGFR x anti-PD1 bispecific antibody inhibits EGFR-overexpressing tumor growth by combining EGFR blockade and immune activation with direct tumor cell killing. Transl Oncol (2020) 14(1):100916. doi: 10.1016/j.tranon.2020.100916
201. Gedeon PC, Schaller TH, Chitneni SK, Choi BD, Kuan CT, Suryadevara CM, et al. A rationally designed fully human EGFRvIII:CD3-targeted bispecific antibody redirects human T cells to treat patient-derived intracerebral malignant glioma. Clin Cancer Res (2018) 24(15):3611–31. doi: 10.1158/1078-0432.CCR-17-0126
202. Sternjak A, Lee F, Thomas O, Balazs M, Wahl J, Lorenczewski G, et al. Preclinical assessment of AMG 596, a bispecific T-cell engager (BiTE) immunotherapy targeting the tumor-specific antigen EGFRvIII. Mol Cancer Ther (2021) 20(5):925–33. doi: 10.1158/1535-7163.MCT-20-0508
203. Boustany LM, Wong L, White CW, Diep L, Huang Y, Liu S, et al. EGFRCD3 bispecific probody™ therapeutic induces tumor regressions and increases maximum tolerated dose >60-fold in preclinical studies. Mol Cancer Ther (2018) 17(Suppl. 1):A164. doi: 10.1158/1535-7163.TARG-17-A164
204. Demaria O, Cornen S, Daeron M, Morel Y, Medzhitov R, Vivier E. Harnessing innate immunity in cancer therapy. Nature (2019) 574(7776):45–56. doi: 10.1038/s41586-019-1593-5
205. Gasteiger G, D'Osualdo A, Schubert DA, Weber A, Bruscia EM, Hartl D. Cellular innate immunity: An old game with new players. J Innate Immun (2017) 9(2):111–25. doi: 10.1186/s13223-018-0278-1
206. Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol (2018) 14(Suppl 2):49. doi: 10.1186/s13223-018-0278-1
207. Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol (2011) 29:235–71. doi: 10.18632/oncotarget.9149
208. Yong CS, John LB, Devaud C, Prince MH, Johnstone RW, Trapani JA, et al. A role for multiple chimeric antigen receptor-expressing leukocytes in antigenspecific responses to cancer. Oncotarget (2016) 7(23):34582–98. doi: 10.18632/oncotarget.9149
209. Ellwanger K, Reusch U, Fucek I, Wingert S, Ross T, Müller T, et al. Redirected optimized cell killing (ROCK®): A highly versatile multispecific fit-forpurpose antibody platform for engaging innate immunity. MAbs (2019) 11(5):899–918. doi: 10.1080/19420862.2019.161650
210. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet (2000) 356(9244):1795–9. doi: 10.1016/s0140-6736(00)03231-1
211. Cichocki F, Cooley S, Davis Z, DeFor TE, Schlums H, Zhang B, et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia (2016) 30(2):456–63. doi: 10.1038/leu.2015.260
212. Semeraro M, Rusakiewicz S, Minard-Colin V, Delahaye NF, Enot D, Vely F, et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Sci Transl Med (2015) 7(283):283ra55. doi: 10.1126/scitranslmed.aaa2327
213. Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front Immunol (2019) 10:3038. doi: 10.3389/fimmu.2019.03038
214. Reiners KS, Kessler J, Sauer M, Rothe A, Hansen HP, Reusch U, et al. Rescue of impaired NK cell activity in hodgkin lymphoma with bispecific antibodies in vitro and in patients. Mol Ther (2013) 21(4):895–903. doi: 10.1038/mt.2013.14
215. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol (2012) 12(4):239–52. doi: 10.1038/nri3174
216. Mamessier E, Sylvain A, Bertucci F, Castellano R, Finetti P, Houvenaeghel G, et al. Human breast tumor cells induce self-tolerance mechanisms to avoid NKG2D-mediated and DNAM-mediated NK cell recognition. Cancer Res (2011) 71(21):6621–32. doi: 10.1158/0008-5472.Can-11-0792
217. Lo Nigro C, Macagno M, Sangiolo D, Bertolaccini L, Aglietta M, Merlano MC. NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: biological evidence and clinical perspectives. Ann Transl Med (2019) 7(5):105. doi: 10.21037/atm.2019.01.42
218. Vermorken JB, Stöhlmacher-Williams J, Davidenko I, Licitra L, Winquist E, Villanueva C, et al. Cisplatin and fluorouracil with or without panitumumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck (SPECTRUM): An open-label phase 3 randomised trial. Lancet Oncol (2013) 14(8):697–710. doi: 10.1016/s1470-2045(13)70181-5
219. Giralt J, Trigo J, Nuyts S, Ozsahin M, Skladowski K, Hatoum G, et al. Panitumumab plus radiotherapy versus chemoradiotherapy in patients with unresected, locally advanced squamous-cell carcinoma of the head and neck (CONCERT-2): A randomised, controlled, open-label phase 2 trial. Lancet Oncol (2015) 16(2):221–32. doi: 10.1016/s1470-2045(14)71200-8
220. Trotta AM, Ottaiano A, Romano C, Nasti G, Nappi A, De Divitiis C, et al. Prospective evaluation of cetuximab-mediated antibody-dependent cell cytotoxicity in metastatic colorectal cancer patients predicts treatment efficacy. Cancer Immunol Res (2016) 4(4):366–74. doi: 10.1158/2326-6066.CIR-15-0184
221. McMichael EL, Benner B, Atwal LS, Courtney NB, Mo X, Davis ME, et al. A phase I/II trial of cetuximab in combination with interleukin-12 administered to patients with unresectable primary or recurrent head and neck squamous cell carcinoma. Clin Cancer Res (2019) 25(16):4955–65. doi: 10.1158/1078-0432.Ccr-18-21080432.CCR-16-0004
222. Mazorra Z, Lavastida A, Concha-Benavente F, Valdes A, Srivastava RM, Garcia-Bates TM, et al. Nimotuzumab induces NK cell activation, cytotoxicity, dendritic cell maturation and expansion of EGFR-specific T cells in head and neck cancer patients. Front Pharmacol (2017) 8:382. doi: 10.3389/fphar.2017.00382
223. Gerdes CA, Nicolini VG, Herter S, van Puijenbroek E, Lang S, Roemmele M, et al. GA201 (RG7160): A novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin Cancer Res (2013) 19(5):1126–38. doi: 10.1158/1078-0432.Ccr-12-0989
224. Delord JP, Tabernero J, Garcı́a-Carbonero R, Cervantes A, Gomez-Roca C, Bergé Y, et al. Open-label, multicentre expansion cohort to evaluate imgatuzumab in pre-treated patients with KRAS-mutant advanced colorectal carcinoma. Eur J Cancer (2014) 50(3):496–505. doi: 10.1016/j.ejca.2013.10.015
225. Bridgewater JA, Cervantes A, Markman B, Siena S, Cubillo A, Garcia Carbonero R, et al. GAIN-(C): Efficacy and safety analysis of imgatuzumab (GA201), a novel dual-acting monoclonal antibody (mAb) designed to enhance antibody-dependent cellular cytotoxicity (ADCC), in combination with FOLFIRI compared to cetuximab plus FOLFIRI in second-line KRAS exon 2 wild type (e2WT) or with FOLFIRI alone in mutated (e2MT) metastatic colorectal cancer (mCRC). J Clin Oncol (2015) 33(Suppl. 3):669. doi: 10.1200/jco.2015.33.3_suppl.669
226. Brazel D, Nagasaka M. Spotlight on amivantamab (JNJ-61186372) for EGFR exon 20 insertions positive non-small cell lung cancer. Lung Cancer (Auckland NZ) (2021) 12:133–8. doi: 10.2147/LCTT.S337861
227. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature (2019) 575(7781):217–23. doi: 10.1038/s41586-019-1694-1
228. Clemenceau B, Vivien R, Berthome M, Robillard N, Garand R, Gallot G, et al. Effector memory alphabeta T lymphocytes can express FcgammaRIIIa and mediate antibody-dependent cellular cytotoxicity. J Immunol (2008) 180(8):5327–34. doi: 10.4049/jimmunol.180.8.5327
229. Van Maldegem F, Valand K, Cole M, Patel H, Angelova M, Rana S, et al. Characterisation of tumour microenvironment remodelling following oncogene inhibition in preclinical studies with imaging mass cytometry. Nat Commun (2021) 12(1). doi: 10.1038/s41467-021-26214-x
230. Phase 3 study of MRTX849 with cetuximab vs chemotherapy in patients with advanced colorectal cancer with KRAS G12C mutation (KRYSTAL-10) (NCT04793958). Available at: https://clinicaltrials.gov/show/NCT04793958 (Accessed 8 February, 2022).
231. Genßler S, Burger MC, Zhang C, Oelsner S, Mildenberger I, Wagner M, et al. Dual targeting of glioblastoma with chimeric antigen receptor-engineerednatural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology (2016) 5(4):e1119354. doi: 10.1080/2162402x.2015.1119354
232. Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine (2020) 59:102975. doi: 10.1016/j.ebiom.2020.102975
233. Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget (2016) 7(19):27764–77. doi: 10.18632/oncotarget.8526
234. Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CARengineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep (2015) 5:11483. doi: 10.1038/srep11483
235. Liu Y, Zhou Y, Huang KH, Fang X, Li Y, Wang F, et al. Targeting epidermal growth factor-overexpressing triple-negative breast cancer by natural killer cells expressing a specific chimeric antigen receptor. Cell Prolif (2020) 53(8):e12858. doi: 10.1111/cpr.12858
236. Zhang Q, Tian K, Xu J, Zhang H, Li L, Fu Q, et al. Synergistic effects of cabozantinib and EGFR-specific CAR-NK-92 cells in renal cell carcinoma. J Immunol Res (2017) 2017:6915912. doi: 10.1155/2017/6915912
237. Nakazawa T, Murakami T, Natsume A, Nishimura F, Morimoto T, Matsuda R, et al. KHYG-1 cells with EGFRvIII-specific CAR induced a pseudoprogression-like feature in subcutaneous tumours derived from glioblastoma-like cells. Anticancer Res (2020) 40(6):3231–7. doi: 10.21873/anticanres.14304
238. Basar R, Daher M, Rezvani K. Next-generation cell therapies: the emerging role of CAR-NK cells. Blood Adv (2020) 4(22):5868–76. doi: 10.1182/bloodadvances.2020002547
239. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
240. Wingert S, Reusch U, Knackmuss S, Kluge M, Damrat M, Schniegler-Mattox U, et al. Preclinical evaluation of AFM24, a novel CD16A-specific innateimmune cell engager targeting EGFR-positive tumors. MAbs (2021) 13(1):1950264. doi: 10.1080/19420862.2021.1950264
241. Reusch U, Damrat M, Wingert S, Knackmuss SHJ, Mueller T, Fucek I, et al. AFM24, a bispecific EGFR/CD16A innate cell engager with the potential to overcome resistance to current targeted treatments for EGFR-positive malignancies. Cancer Res (2020) 80(Suppl. 16):5659.
242. Study to assess AFM24 in advanced solid cancers (NCT04259450). Available at: https://clinicaltrials.gov/ct2/show/NCT04259450. (Accessed 8 February, 2022)
243. Kerbauy LN, Marin ND, Kaplan M, Banerjee PP, Berrien-Elliott MM, Becker-Hapak M, et al. Combining AFM13, a bispecific CD30/CD16 antibody, with cytokine-activated blood and cord blood-derived NK cells facilitates CAR-like responses against CD30(+) malignancies. Clin Cancer Res (2021) 27(13):3744–56. doi: 10.1158/1078-0432.CCR-21-0164
244. Modified immune cells (AFM13-NK) and a monoclonal antibody (AFM13) in treating patients with recurrent or refractory CD30 positive Hodgkin or non-Hodgkin lymphomas (NCT04074776). Available at: https://clinicaltrials.gov/ct2/show/NCT04074746 (Accessed 8 February, 2022).
245. Safety, tolerability, and anti-tumor activity of AFM24 in combination with SNK01 in subjects with advanced/metastatic EGFR-expressing cancers (NCT05099549). Available at: https://clinicaltrials.gov/ct2/show/NCT05099549 (Accessed 8 February, 2022).
246. Levy E, Reger R, Segerberg F, Lambert M, Leijonhufvud C, Baumer Y, et al. Enhanced bone marrow homing of natural killer cells following mRNA transfection with gain-of-function variant CXCR4(R334X). Front Immunol (2019) 10:1262. doi: 10.3389/fimmu.2019.01262
247. Hervier B, Russick J, Cremer I, Vieillard V. NK cells in the human lungs. Front Immunol (2019) 10:1263. doi: 10.3389/fimmu.2019.01263
248. Bartlett NL, Herrera AF, Domingo-Domenech E, Mehta A, Forero-Torres A, Garcia-Sanz R, et al. A phase 1b study of AFM13 in combination with pembrolizumab in patients with relapsed or refractory Hodgkin lymphoma. Blood (2020) 136(21):2401–9. doi: 10.1182/blood.2019004701
249. Pesce S, Greppi M, Grossi F, Del Zotto G, Moretta L, Sivori S, et al. PD/1- PD-Ls checkpoint: insight on the potential role of NK cells. Front Immunol (2019) 10:1242. doi: 10.3389/fimmu.2019.01242
250. Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, et al. The mechanism of anti-PD-L1 antibody efficacy against PD-L1-negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discovery (2019) 9(10):1422–37. doi: 10.1158/2159-8290.CD-18-1259
251. Study to assess AFM24 in combination with atezolizumab in selected advanced/metastatic EGFR-expressing cancers (NCT05109442). Available at: https://clinicaltrials.gov/ct2/show/NCT05109442. (Accessed 8 February, 2022).
Keywords: epidermal growth factor receptor, innate immunity, adaptive immunity, cancer, tyrosine kinase inhibitor, bispecific antibody, CAR-T therapy, innate cell engager
Citation: Hintzen G, Dulat HJ and Rajkovic E (2022) Engaging innate immunity for targeting the epidermal growth factor receptor: Therapeutic options leveraging innate immunity versus adaptive immunity versus inhibition of signaling. Front. Oncol. 12:892212. doi: 10.3389/fonc.2022.892212
Received: 08 March 2022; Accepted: 28 July 2022;
Published: 14 September 2022.
Edited by:
Jeroen Hiltermann, University Medical Center Groningen, NetherlandsReviewed by:
Fiona Simpson, The University of Queensland, AustraliaCopyright © 2022 Hintzen, Dulat and Rajkovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriele Hintzen, Zy5oaW50emVuQGFmZmltZWQuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.