
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 14 April 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.891961
This article is part of the Research Topic Hybrid or Mixed Myelodyplastic / Myeloproliferative Disorders: Current Trends in Diagnosis and Treatment View all 10 articles
 Thomas P. Thomopoulos1
Thomas P. Thomopoulos1 Argiris Symeonidis2
Argiris Symeonidis2 Alexandra Kourakli2
Alexandra Kourakli2 Sotirios G. Papageorgiou1
Sotirios G. Papageorgiou1 Vasiliki Pappa1*
Vasiliki Pappa1*Chronic neutrophilic leukemia (CNL) represents a rare disease, that has been classified among the BCR/ABL-negative myeloproliferative neoplasms. The disease is characterized by marked leukocytosis with absolute neutrophilia and its clinical presentation may vary from asymptomatic to highly symptomatic with massive splenomegaly and constitutional symptoms. CNL prognosis remains relatively poor, as most patients succumb to disease complications or transform to acute myeloid leukemia. Recent studies have demonstrated that CSF3R mutations drive the disease, albeit the presence of other secondary mutations perplex the genetic landscape of the disease. Notably, the presence of CSF3R mutations has been adopted as a criterion for diagnosis of CNL. Despite the vigorous research, the management of the disease remains suboptimal. Allogeneic stem cell transplantation represents the only treatment that could lead to cure; however, it is accompanied by high rates of treatment-related mortality. Recently, ruxolitinib has shown significant responses in patients with CNL; however, emergence of resistance might perturbate long-term management of the disease. The aim of this review is to summarize the clinical course and laboratory findings of CNL, highlight its pathogenesis and complex genetic landscape, and provide the context for the appropriate management of patients with CNL.
Chronic neutrophilic leukemia (CNL) is a rare disease, with an annual incidence of about 1 new case per million people, a median age at diagnosis of about 65 years and a slight male preponderance. The disease was initially described in 1920 by Tuohy who reported the case of a 58-year-old female patient with persistent polymorphonuclear hyperleukocytosis and massive splenomegaly, who subsequently succumbed to the disease (1). Due to the rarity of the disease and the absence of clear diagnostic criteria, some early described cases are ill-defined; however, an additional well-documented case from France was published in 1932 (2), followed by a paucity of publications for almost 25 years, until the report of a case describing the disease as a myeloproliferative neoplasm (MPN) (3). After the identification of Philadelphia chromosome as a hallmark of chronic myelogenous leukemia (CML), Tanzer et al. were the first to report a patient without Philadelphia chromosome or increased marrow fibrosis but high Leucocyte- or Neutrophil- Alkaline Phosphatase (LAP or NAP) score. Notably, they used the term “Chronic Neutrophilic Leukemia” to describe the disease (4). Additional case reports or small series of patients were published thereafter that have delineated this syndrome, although in some of them the authors have described the disease as a variant of CML (5–12).
CNL is a clonal hematopoietic stem cell disorder, and this has been documented early, as soon as clonality assays with X-linked restriction fragment length polymorphisms (RFLP) and/or X-inactivation patterns were applied (13, 14). The clonal nature of the disease is also evident by the detection of cytogenetic abnormalities (15–17), although in most cases, patients with CNL have a normal karyotype. From the epidemiological aspect of view the precise etiology of the disease remains unknown and there is only one report, associating CNL with a previous exposure to thorotrast, in a patient who, eventually evolved to acute myeloid leukemia (AML) (18). Moreover, when applying strict molecular criteria for disease characterization, it appears that the median age at diagnosis might be younger (19). Recently, mutations of the CSF3R gene have been shown to constitute the driver genetic event for the manifestation of this disease (20), which according to the latest version of the WHO classification of Hematopoietic Neoplasms, is classified as a chronic myeloproliferative neoplasm (21) However, there has been a vigorous debate whether CNL should be classified instead among the myelodysplastic syndromes (MDS) or the overlap myelodysplastic/myeloproliferative (MDS/MPN) neoplasms, similarly to chronic myelomonocytic leukemia (CMML), as it might share several clinical, hematological and cytogenetic features, reminiscent of MDS or MDS/MPN. Recently, criteria for the precise diagnosis of this disease have been proposed by the latest revision of the WHO classification, although some variation in clinical parameters might exist.
CNL has substantial clinical and prognostic heterogeneity, which might correspond to the different driver mutations of the CSF3R gene. Most patients may go through an asymptomatic or minimally symptomatic period, characterized only by neutrophilic leukocytosis, with or without mild splenomegaly, lasting for several months or even years, before evolving to symptomatic disease. In many cases the disease may be discovered incidentally as unexplained persistent neutrophilia, after excluding any other potential causes as infections or inflammatory and neoplastic disorders, which occasionally can induce extreme hyperleukocytosis (22, 23). Less commonly, patients may complain for fatigue and restlessness, weight loss, night sweats, pruritus, and mild bone pain or may exhibit various hemorrhagic manifestations and recurrent episodes of gout (24). Fever is usually absent or minimal but persistent low-grade fever may be a prominent feature of advanced disease.
Bleeding diathesis, occasionally unrelated or disproportional to the degree of thrombocytopenia or splenomegaly/hypersplenism, is also a prominent feature of CNL, potentially reflecting platelet qualitative abnormalities (5, 7, 25, 26). Severe hemorrhagic manifestations, including recurrent episodes of epistaxis, blood oozing from the site of bone marrow aspiration, gastrointestinal tract hemorrhages and even intracranial hemorrhages have been reported throughout the disease course and may represent a common cause of death in these patients (5, 25, 26). One study of three patients with CNL, demonstrated prolonged bleeding time and abnormal platelet aggregation response to collagen, epinephrine and ADP, whereas in one patient, platelet adhesiveness and adenine content were found decreased (27). Moreover, abnormalities of the vascular compartment might contribute to the hemorrhagic manifestations. There have been several reports of cutaneous leucocytoclastic vasculitis or acute febrile neutrophilic dermatosis (Sweet’s syndrome), occurring in patients with CNL. Therefore, various types of erythematous and/or hemorrhagic skin lesions, occasionally affecting palms and soles, represent a common manifestation of CNL. In some of these cases it is unclear whether the described skin lesions represent an allergic reaction, vasculitis or frank skin infiltration by the leukemic neutrophils, but whatever might be the case, severity of skin lesions runs in parallel with disease activity and severity (28–32).
Despite leukocytosis/neutrophilia patients with CNL are prone to develop severe, life-threatening or even fatal infections from both, common and opportunistic pathogens, such as mycobacteria and various fungi. This could be explained by the defective function of neutrophils as described in the following section. Patients may present with a severe infection, sometimes inducing septic shock (33, 34) or infections may occur frequently, during the course of the disease, and ultimately, they may represent one of the main causes of death (35, 36). It is therefore, noteworthy that in such cases, suspicion for the diagnosis of CNL may be obscured by the concurrent presence of a severe infection and neutrophilic leukocytosis often marked, which might be attributed to the infection.
Hepato-splenomegaly is commonly present. Splenomegaly is found in about 40% of the patients at the time of initial diagnosis and in the majority of them is mild to moderate; however, as disease slightly and steadily progresses, splenomegaly may become severe in almost all patients (10–12). Several cases of massive splenomegaly have been described, rarely requiring palliative splenectomy. When disease is rapidly evolving and white blood cells (WBC) are fast increasing, splenomegaly might become painful. Hepatomegaly is less common and less prominent, whereas lymphadenopathy has occasionally been reported but it is unusual. In some cases, lymph node biopsy has clearly revealed neutrophilic infiltration (37), whereas in some autopsy studies, infiltration by immature neutrophils and by other hematopoietic cells were identified in retroperitoneal lymph nodes, spleen, liver and the kidneys, a finding supporting the proliferative nature of the disease (37, 38). Although, vein thrombotic episodes in association with CNL have been rarely reported (39, 40), a recent description of the clinical course of the disease in 19 patients, reported a history of thrombosis in about half of the patients (41).
The peripheral blood smear is characterized by persistent mature neutrophilic leukocytosis, with about 80-95% of the enumerating cells being neutrophils or bands. Left shift is absent or minimal and only occasional myelocytes-metamyelocytes are observed. Notably, CNL is characterized by the absence of monocytosis, eosinophilia or basophilia. In some cases, leukocytosis/neutrophilia might be severe, exceeding 100 x 109/L and reaching up to 500 x 109/L. Neutrophils appear morphologically normal by both, light and electron microscopy and in the majority of cases contain abundant primary and secondary granules (8). Presence of Döhle bodies and neutrophil inclusions have been reported rarely; however, dysgranulopoiesis is not consistent with the diagnosis of CNL (21). Notably, reported cases of CNL presenting with dysplastic morphological features, particularly in more advanced or long-standing cases, including abnormal chromatin clumping, poor granulation, cytoplasmic vacuolation, microtubular inclusions (42), ringed-shape nuclei (42, 43), are now considered to represent cases of atypical CML rather than genuine CNL cases
Neutrophils and granulocytes of patients with CNL usually have an activated phenotype, but they might also exhibit several functional abnormalities and are less viable in stress conditions. The enzymatic equipment of neutrophils is normal, as this can be revealed by the appropriate cytochemical stains. Myeloperoxidase is strongly positive as is also LAP, whose elevated score has been used to distinguish this disease from BCR/ABL positive CML, in which LAP is very low to completely absent (6, 7, 11, 12). Some studies have reported reduced lysozyme and β-glycuronidase content in the neutrophils (8, 37) but these enzymes are highly released from clonal neutrophils, following stimulation. Neutrophil motility, deformability and chemotaxis have been found abnormal and the respiratory burst is usually impaired, with reduced superoxide anion production in response to various stimuli, as well as decreased cytosolic C kinase activity (44, 45). Other studies have demonstrated normal phagocytosis and bactericidal activity and normal nitroblue tetrazolium reduction assays suggesting that these neutrophils are found in an abnormally activated status (46). Bactericidal activity, however, has also been reported to be decreased and the same patients exhibited decreased granulocytic clonogenic activity from both, peripheral blood and bone marrow, compared to normal subjects (47). In one study, several functional neutrophils tests were found normal, however neutrophils of CNL patients produced significantly lower amounts of leukotriene B4 (48). In another multifunctional study the authors found reduced serum G-CSF and GM-CSF levels and inadequate in vitro neutrophil stimulation, induced by these cytokines. They also demonstrated impaired STAT3 and MAP kinase downstream signal, despite the intact expression of both G-CSF- and GM-CSF receptor and they suggest a potential dysregulation of the intracellular part of the receptor(s), although they were unable to show any mutation of this domain (49).
Mild to moderate anemia might be present, in most cases normochromic-normocytic sharing the features of the anemia of chronic diseases (12), whereas cases with macrocytic anemia have also been reported, despite the very high serum B12 levels, reflecting underlying dyserythropoiesis, as shown by the report of impaired erythroblastic colony formation in patients with CNL (7).
Platelet count may be normal, reduced or less commonly, increased. Morphologically, platelets may appear normal, but giant platelets and platelets with poor granulation, reminiscent of storage pool disorders have been observed (11, 12). Deteriorating thrombocytopenia might be a feature of disease transformation towards AML. Rarely, nucleated red blood cells can also be identified. In a recent short report, numerous ringed sideroblasts were found in the marrow of a patient with CNL (50).
Bone marrow findings are more reminiscent of a myeloproliferative neoplasm, with almost 100% cellularity and fat disappearance, extreme granulocytic hyperplasia without maturation arrest and absence of monocytosis, monocyte precursors and basophilia (11, 12). The myeloid/erythroid ratio is usually higher than 10, but blast cells are not increased, unless the disease has entered an accelerated phase and evolves towards AML (51). Megakaryocytic hyperplasia is also common, and in most cases megakaryocytes appear morphologically normal and mainly mature and hyper-lobulated, i.e. with higher ploidy as compared to MDS or typical BCR/ABL-positive CML (27); however, megakaryocyte dysplasia is absent or minimal. Moreover, minimal fibrosis might be present in the bone marrow of patients with CNL but should not exceed a grade of 1+ (21). As a result of increased cellular turnover, pseudo-Gaucher cells may be found in the marrow (52).
The main findings of patients’ biochemical profile consist of elevated serum LDH and uric acid levels, reflecting the increased hematopoietic cell turnover. Serum B12 and transcobalamin-I and -III, that are released in circulation by the mature cells of granulopoietic lineage, are usually elevated. Serum alkaline phosphatase and γ-glutamyl-transpeptidase might also be found increased, reflecting liver infiltration by hematopoietic cells. Markers of acute phase reaction are usually normal at the time of initial diagnosis but may become abnormal in advanced stages, associated with marked leukocytosis or in cases of disease progression. Serum G-CSF levels are not routinely estimated, but if performed, they are found suppressed. In milder cases of CNL differential diagnosis should exclude a leukemoid reaction attributed to various underlying diseases and conditions, and other types of myeloproliferative disorders. A practical diagnostic approach together with the diagnostic criteria of CNL are shown in Table 1.
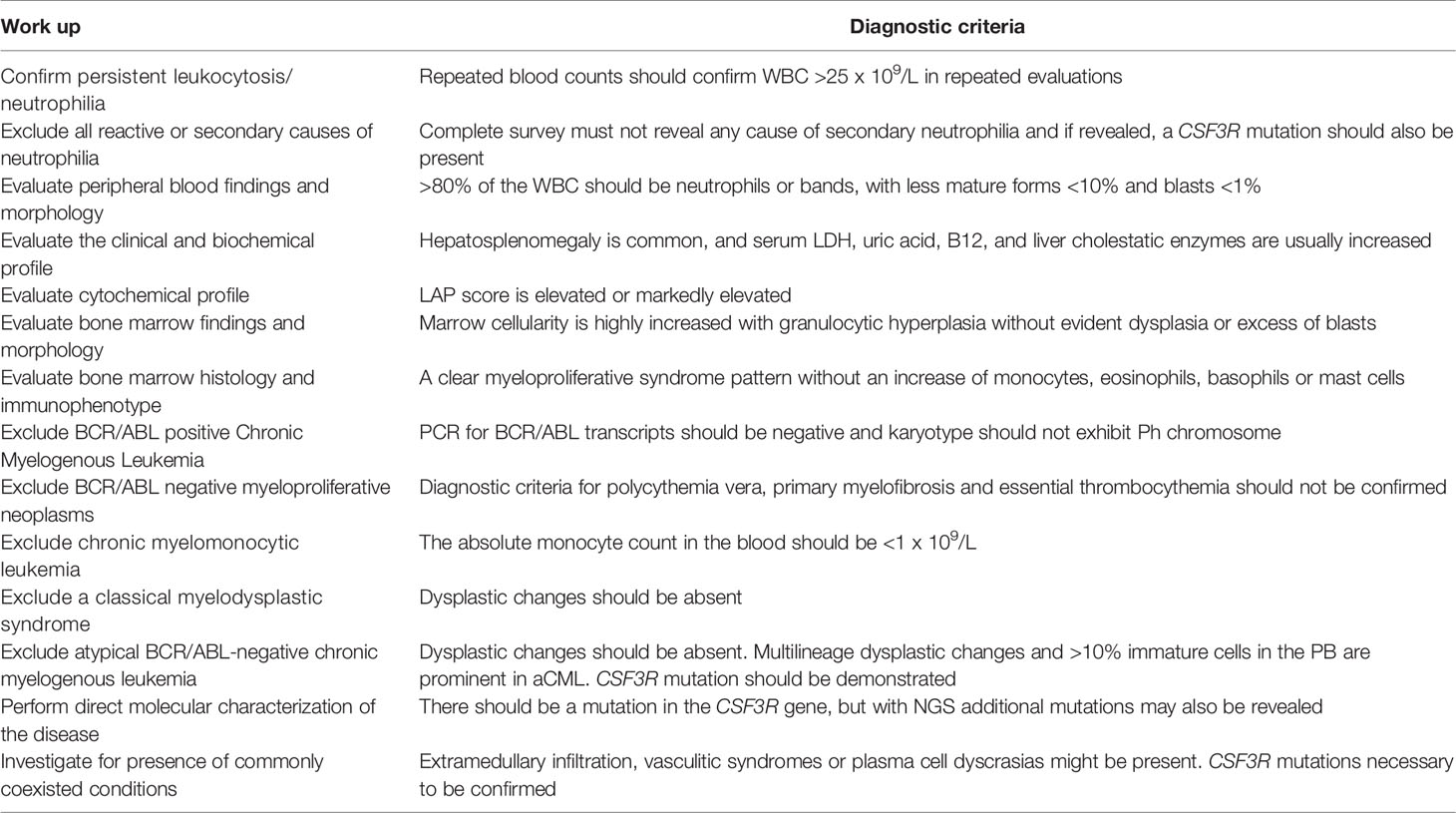
Table 1 Diagnostic work up and diagnostic criteria for CNL.
In order to establish the diagnosis of CNL, other cases of reactive leukocytosis should be excluded. Occasionally CNL has been reported to coexist with lymphoid neoplasms (53), but the most striking association has been with various plasma cell dyscrasias, such as monoclonal gammopathy of undetermined significance (MGUS) (39, 54–58), and mainly multiple myeloma. There have been several cases of patients reported to have concurrently or consecutively, these two, apparently different hematological dyscrasias (28, 59–71). However, most of these cases represent a neutrophilic leukemoid reaction, potentially mediated by cytokines produced by the clonal plasma cells. Notably, these cases cannot fulfill the diagnostic criteria for CNL (72). Nonetheless, in the presence of a plasma cell dyscrasia, clonality must be demonstrated by cytogenetic or molecular in order to establish the diagnosis of CNL the diagnosis of CNL (21).
As per the 2016 WHO classification, other MPN and MDS/MPN should also be excluded. Classical MPN, namely polycythemia vera, essential thrombocytosis, and primary myelofibrosis can be easily excluded by the absence of the characteristic morphological and molecular features of the latter. Similarly, the absence of BCR/ABL fusion gene invariably precludes the diagnosis of CML. Regarding chronic myelomonocytic leukemia, presence of persistent absolute monocytosis and dysplastic features distinguish this entity from CNL (21). Undoubtedly, the most challenging aspect in differential diagnosis is to distinguish CNL from atypical chronic myeloid leukemia (aCML). Presence of prominent dysplasia in >10% of cells, as well as, a more prominent immature granulocytosis favor the diagnosis of aCML. Notably, presence of CSF3R mutations cannot be used for differential diagnosis between aCML and CNL, as there have been several cases of aCML harboring CSF3R mutations (73).
The granulocyte colony stimulating factor (G-CSF), a cytokine primarily produced by endothelial cells, fibroblasts and macrophages, is the major regulator of both basal and emergency granulopoiesis. G-CSF is crucial for commitment of myeloid cells towards granulocytic differentiation; concomitantly, G-CSF accelerates maturation of metamyelocytes into mature neutrophils, prolongs the survival of neutrophils and their progenitors, and enhances neutrophil function (74).
G-CSF exerts its actions by binding to the receptor CSF3R on myeloid cells, which consists of a single polypeptide chain with great homology to other cytokine receptors. CSF3R extracellular domain, comprising of an immunoglobulin-like (IgG) domain followed by a cytokine receptor homology domain (CRH) and three fibronectin III (FNIII) domains, plays a crucial role in receptor activation. The intracellular domain contains two proximal motifs termed Box 1 and Box 2 that are critical for signal transduction as they bind to JAK, whereas the distal C-terminal domain drives differentiation and transduction of phagocytic signals in mature neutrophils. C-terminal domain also contains Box 3, which acts as a negative regulator of G-CSF signaling (75). A graphical representation of the CSF3R structure is provided in Figure 1.
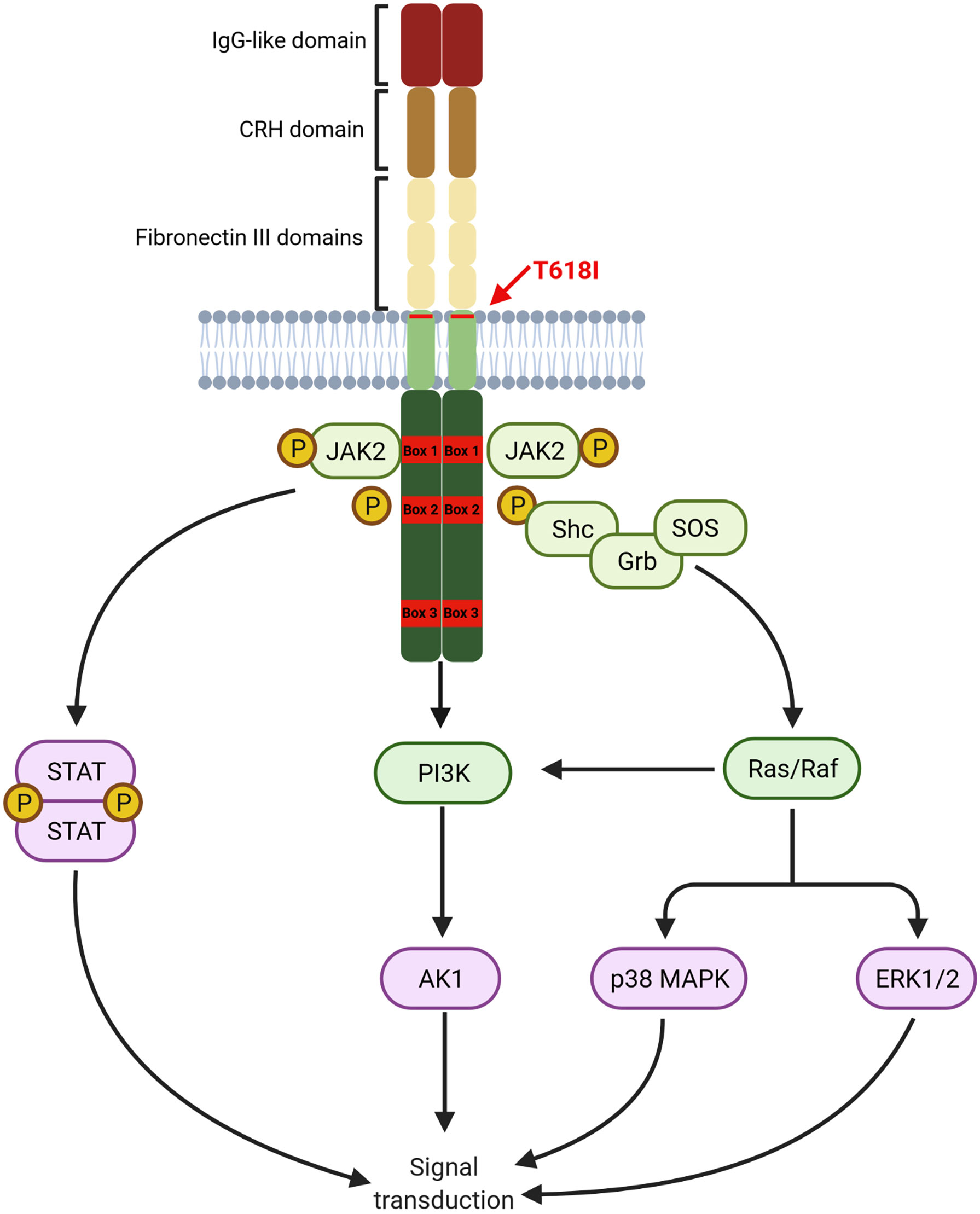
Figure 1 Structure of CSF3R. Proximal transmembrane T618I mutation induced a G-CSF independent activation of receptor homodimers leading to constant signal transduction primarily through the JAK/STAT pathway. Other pathways, namely PI3K/AKT and ERK1/2 are also constantly activated (Created with BioRender.com).
Binding of G-CSF to CSF3R induces homodimerization of the receptor leading to the activation of downstream pathways. The proximal domain activates mostly the JAK/STAT pathway leading to proliferation and differentiation. The PI3K/AKT pathway is also activated by a LYN kinase, also interacting with the proximal domain, leading to increased survival. Other pathways, particularly the MAPK/ERK are activated by interaction with more distal residues in the intracellular domain.
Given the importance of G-CSF in granulopoiesis, CSF3R mutations have been associated with the pathogenesis of various diseases. Extracellular domain missense mutations conferring refractoriness to G-CSF have been identified in severe congenital neutropenia (SCN) and chronic idiopathic neutropenia (CIN) (76). Nonsense or frameshift mutations in the extracellular domain, also found in SCN patients, promote ligand-independent binding to the full-length CSF3R that appears to suppress G-CSF-mediated signaling (77). On the other hand, intracellular domain mutations, mostly of nonsense nature, result in a truncated receptor with normal affinity for G-CSF, but a higher proliferation and lower differentiation potential in response to G-CSF. These mutations are almost exclusively found in patients with SCN who develop MDS or AML.
Proximal transmembrane mutations, namely T618I and T640N, promote the ligand-independent activation of pre-existing dimers of CSF3R and lead to low level, sustained activation of all downstream pathways, that cannot be terminated by internalization of the activated CSF3R, that usually follows the activation of wild-type CSF3R (78). These mutations are found to be particularly enriched in patients with CNL.
In their pivotal trial in 2013, Maxson et al. demonstrated that 8 of 9 CNL patients (89%) harbor CSF3R mutations. Notably, all of them had proximal transmembrane mutation, most commonly T618I, whereas an additional truncating intracellular mutation was found in three patients as a compound mutation (20, 79). The results of Maxson et al. have been subsequently validated by Pardanani et al. who demonstrated a CSF3R T618I mutation in 83% of patients with WHO-defined CNL. Most strikingly, this mutation was virtually absent from cases of atypical CML or other myeloproliferative neoplasms, highlighting that CSF3R T618I could serve as a highly sensitive and specific marker for CNL (80). The oncogenic potential of this mutation has been demonstrated in a murine model, as transplantation of hematopoietic cells harboring the T618I mutation sufficed for the development of a lethal myeloproliferative neoplasm resembling CNL (81). On the other hand, truncating mutations seem not to be oncogenic; however, they might act synergistically with proximal mutations to drive leukemogenesis, potentially through enhanced MAPK signaling, as shown by in vitro studies by Rohrabaugh et al. Notably, although compound mutations might confer resistance to JAK or SRC kinase inhibition by ruxolitinib and dasatinib respectively, cell lines harboring compound mutations remain sensitive to MEK inhibitors, such as trametinib.
Several recent studies have tried to elucidate the complex genetic landscape of CNL. In most cases, additional mutations in genes commonly affected in myeloid malignancies have been observed; however, their frequency has been highly variable among cohorts. The most commonly affected genes are those involved in epigenetic and transcriptional regulation (ASXL1, TET2), in the assembly of the spliceosome (SRSF2, U2AF1), as well as mutations in genes such as SETBP1. Notably, mutations in genes involved in the cell-signaling pathways, such as JAK2 and NRAS are scarcely found (82). The frequency of gene mutations in CNL is graphically represented in Figure 2.
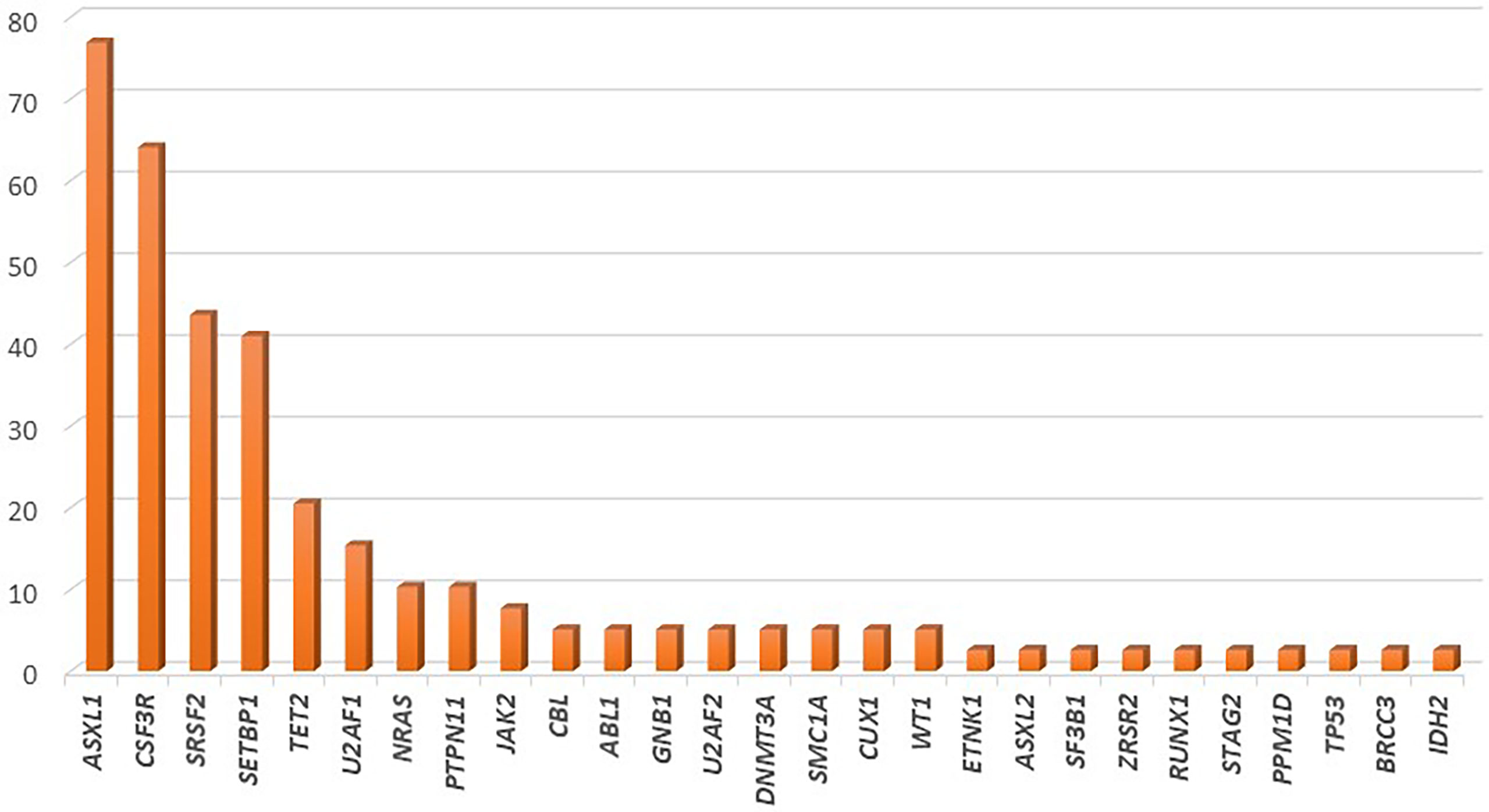
Figure 2 Frequency of gene mutations in chronic neutrophilic leukemia, as demonstrated by application of next generation sequencing in a cohort of 39 patients.
In the largest cohort of 39 CNL cases, mutations of ASXL1 were found in 77% of cases. This gene, which is involved in histone modification, was notably the most commonly affected gene in CNL, with frameshift or nonsense mutations. Further, ASXL1 mutations demonstrated variable variant allelic frequencies (VAF), indicating that this genetic event might occur early in the founder clone, or acquired in later subclones in other cases. Phenotypically, patients with ASXL1 mutations tended to be of older age and presented with higher WBC count, as well as increased needs for platelet transfusions; a trend for worse overall survival was also noted (83). In line with this finding, Elliot et al. demonstrated that ASXL1 mutations, present in 57% of cases, along with thrombocytopenia, were independent prognostic factors for adverse overall survival (84). TET2 mutations, have been observed less frequently in CNL (~21% of cases). In most cases these lesions might be acquired at later stages of clonal evolution. Regarding the disease phenotype, the presence of TET2 mutations correlated with low platelet count, low neutrophil percentage, high monocyte percentage, and bone marrow dysplasia (83). Of special interest is the fact that no cases with concomitant mutations of TET2 and SRSF2 have been observed in CNL, as the TET2mut/SRSF2mut genotype is highly specific of CMML (85). Furthermore, EZH2 mutations have been found relatively frequently in CNL cases, even though their significance remains unclear.
Mutations affecting the spliceosome have also been reported with variable frequency in patients with CNL. SRSF2 and U2AF1, mutated in up to 44% and 15% of patients respectively, are the most commonly affected genes within this group. Similarly to ASXL1, these mutations might be acquired in variable timepoints along the clonal evolution of CNL (83). The contribution of these mutations in disease phenotype and prognosis has yet to be assessed.
SETBP1 has been found to be mutated in approximately 41% of CNL cases. Notably, a high co-occurrence of SETBP1 mutations with CSF3R T618I has been consistently reported, reaching 67% in more recent studies (86, 87). A trend towards inferior overall survival for patients with CNL harboring SETBP1 mutations has been reported; however, a meta-analysis of the three available studies that assessed the prognostic significance of this gene in CNL demonstrated no association with overall survival (88). Interestingly, SETBP1 mutations have been associated with a more myeloproliferative phenotype with increased hemoglobin and platelet counts.
CNL demonstrates high genetic heterogeneity with frequent co-occurrence of mutations in different genes with variable VAF. Indeed, in the study of Zhang et al. the median number of mutations per patient was 3.6, indicating that more than three pathways were simultaneously affected in most patients (83). In an attempt to explain the genetic variability of CNL, Maxson et al. suggested that CNL pathogenesis might be a multistep process involving sequential genetic events giving rise to different subclones. At least two models of clonal evolution have been suggested (82). In the first model, a healthy hematopoietic stem cell acquires a mutation associated with epigenetic modification (ASXL1, TET2) or spliceosome assembly (SRSF2, U2A1), giving rise to a clonal hematopoiesis of indeterminate potential (CHIP) which remains asymptomatic or presents with minimal myelodysplasia. Acquisition of a signaling mutation, most commonly CSF3R T618I, offers a neutrophil differentiation bias leading to overt CNL. In the other model, the CSF3R T618I arises as a founder mutation in healthy stem cells, causing a highly myeloproliferative phenotype. Owing to the high replication potential, the cells rapidly acquire additional mutations in epigenetic/splicing genes, adding dysplastic features (82). These models are summarized in Figure 3.
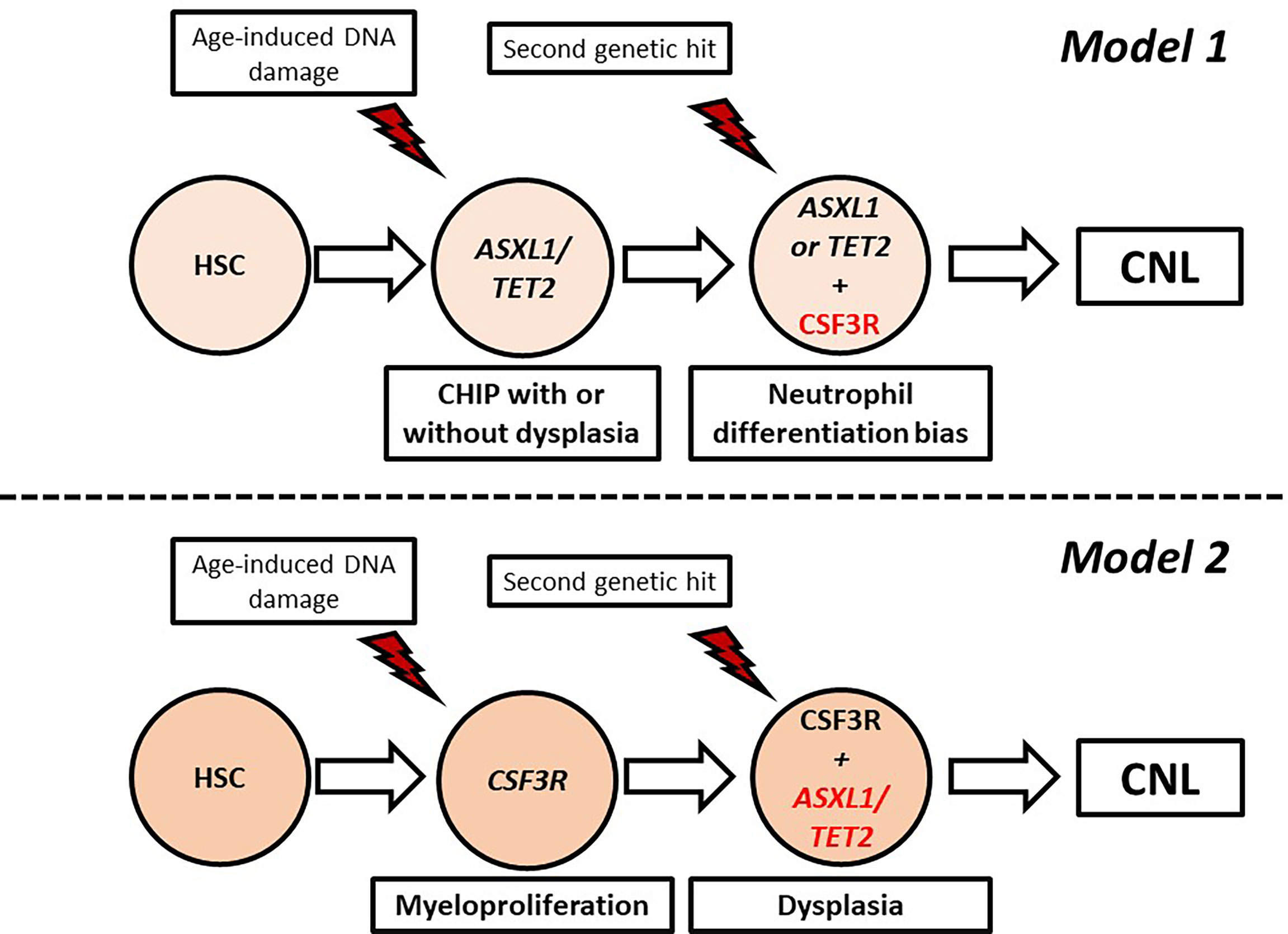
Figure 3 Proposed models of clonal evolution in chronic neutrophilic leukemia. CHIP, Clonal hematopoiesis of indeterminate potential; HSC, hematopoietic stem cell.
Most patients with CNL present with normal karyotype at diagnosis. In a series of 40 patients, cytogenetic abnormalities were reported in 32.5% patients. These abnormalities were detected at diagnosis in 20% of patients, or emerged as clonal evolution in the remaining patients. Given their scarcity, no definite conclusions can be drawn regarding the occurrence of cytogenetic abnormalities in CNL; however, deletion of 20q, 11q, 12p, and trisomy 21 might represent nonrandom abnormalities, as they present with increased frequency in other myeloid malignancies (89).
Overall survival of patients with CNL has been shown to be rather short. In a population-based study combining data from the Surveillance, Epidemiology, and End Results (SEER) program and the National Cancer Database (NCDB), an OS of 1.8 years in SEER and 2.2 years in NCDB was found (90). Similarly, in a series of 40 WHO-defined CNL, median overall survival was 23.5 months; causes of death included intracranial hemorrhage, progressive disease, infections, and leukemic transformation (89). Transformation to AML might occur in 16% of patients at a median of 21 months. Interestingly, several cases of transformation to CMML have been reported (41).
As CNL represents a rare disease, data pertaining to prognostic factors are relatively scarce. Cui et al. demonstrated that WBC count >50 x 109/L represent a negative prognostic factor for OS (86). As mentioned above, Elliot et al. showed that ASXL1 mutation and thrombocytopenia were independent prognostic factors of inferior OS (84). Most recently, presence of NRAS, ASXL1, GATA2, and DNMT3A mutations correlated with a trend of shorter OS, whereas CBL mutations predicted a more favorable OS (83). A prognostic scoring system has been suggested by Szuber et al. from Mayo Clinic, incorporating three variables, namely platelet count <160 × 109/L, leukocyte count >60 × 109/L, and presence of ASXL1 mutation. Thrombocytopenia has been assigned with two points, whereas the other variables with one point each. A two-tier stratification of patients into low-risk (0–1 points) and high-risk (2–4 points), yielded a statistically significant difference in OS (not reached for low-risk vs. 22.4 months for high-risk) (41).
Because of its exceptional rarity, there is no standard of care for CNL. Allogeneic hematopoietic stem cell transplantation (allo-HSCT) remains the only therapeutic approach with curative intent. Conventional treatments mainly aim to alleviate the disease burden, albeit they do not seem to affect the natural history of the disease and overall survival. Therefore, the development of potentially disease-modifying treatments constitutes a major unmet need. Currently, the available treatment options include pharmacological agents (hydroxyurea, interferons, JAK inhibitors, and other tyrosine kinase inhibitors) and allo-HSCT. A proposed algorithm of management of CNL is depicted in Figure 4.
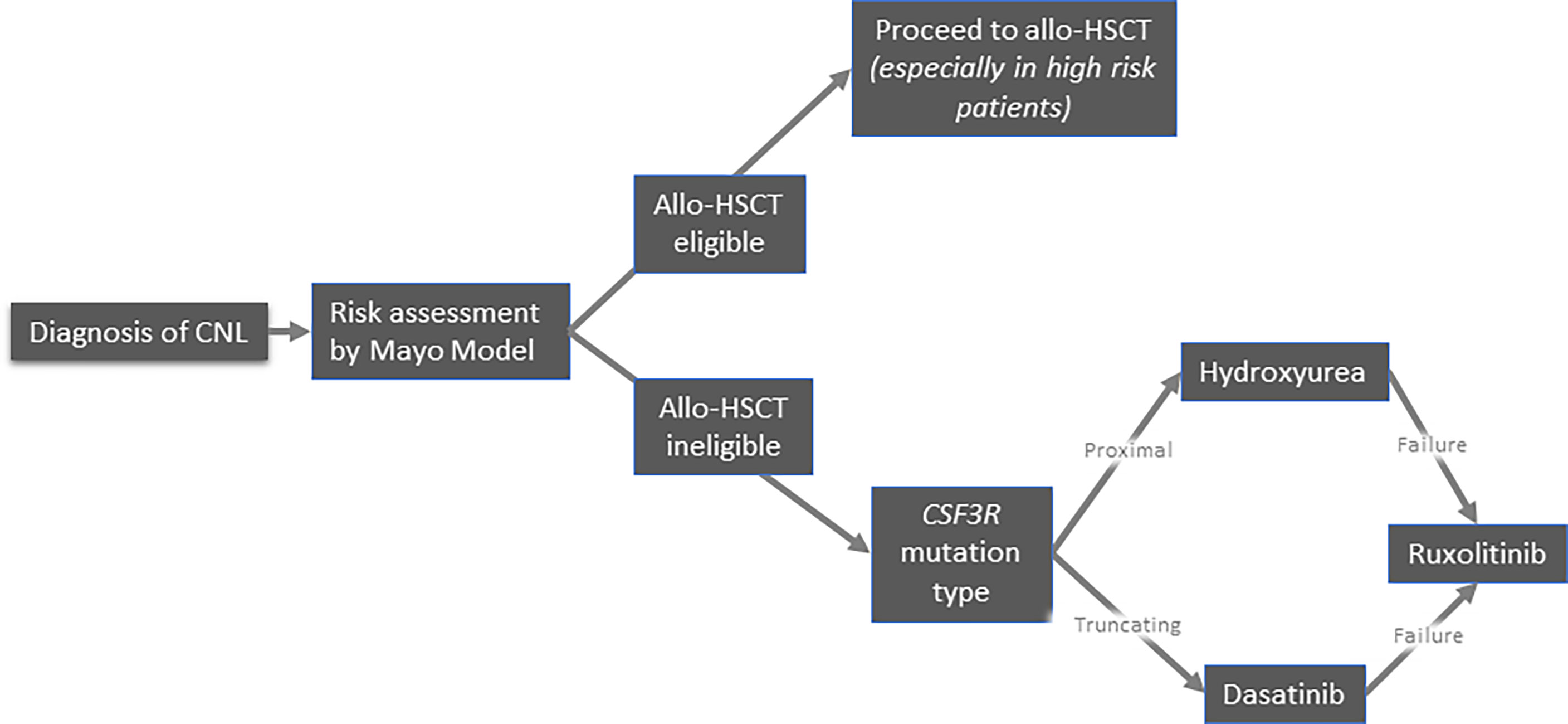
Figure 4 Suggested algorithm for treatment of patients with chronic neutrophilic leukemia.
Hydroxyurea, an oral cytoreductive agent, has traditionally been used in first-line treatment of CNL. Approximately 75% of patients are expected to demonstrate clinical response in terms of decrease in leukocytosis and/or splenomegaly; however, the responses are transient, as all patients are expected to demonstrate disease progression or transformation to AML within a median of 12 months (89).In this context, hydroxyurea should be considered palliative treatment, as it fails to halt clonal evolution and therefore disease progression.
Interferon-alpha (IFN-a) represents another therapeutic approach in CNL, based on published case reports; however, no clinical trial establishing its effectiveness has been reported. Although IFN-a might lead to long-term remission, even in patients primarily refractory to hydroxyurea, the impact of IFN-a to CNL clonal evolution has not been assessed. Recently, Yassin et al. reported the case of a woman with CNL achieving hematological response with pegylated interferon alpha-2a, albeit no data on duration of response have been disclosed (91). Despite the absence of solid evidence for the efficacy of IFN-a, it remains a valid option for CNL treatment.
Conventional chemotherapy with anthracycline and cytarabine (3 + 7) might represent a treatment option for patients transforming to AML; however, overall survival is relatively poor, as expected for secondary AML (92). Regarding CNL in chronic phase, intensive chemotherapy is not recommended as it is accompanied by high treatment-related mortality and the majority of patients are refractory to this approach (11, 89).
The discovery of the key role of CSF3R in CNL and the JAK/STAT signaling pathway in CNL pathobiology has impelled the investigation of ruxolitinib, a JAK1/2 inhibitor, for the treatment of CNL. Preclinical studies demonstrated that ruxolitinib could decrease WBC count and splenomegaly in mice with CSF3RT618I mutated CNL; however, initial case reports demonstrated variable responses to ruxolitinib (93–96). Similarly, Szuber et al. in a case series of 19 patients, noted that ruxolitinib, received by four patients, was associated with a temporary response in half of the patients (41).
A phase II trial of ruxolitinib including 21 patients with CNL has been published recently. Overall response rate (ORR) was 58% for CNL patients. Among them, 4 complete remissions and 9 partial remissions were noted. Further, ORR was restricted to 8% in the group with wild-type CSF3R, while no association between response and cytogenetics, number of mutations, and ASXL1 or SETBP1 mutations was observed. Median OS for all patients was 18.8 months. By response, median survival for non-responders and responders was 15.6 and 23.1 months respectively. Most importantly, ruxolitinib was shown to decrease the CSF3R T618I allelic burden in responders, particularly for those achieving CR (mean absolute VAF change: −0.26 for CR, −0.05 for PR).
The effect of compound mutations of CSF3R on sensitivity to ruxolitinib has been an object of debate. In vitro studies have demonstrated the resistance of cell lines harboring compound mutation to ruxolitinib; however, Gunawan et al. have reported on a case with compound CSF3R mutations that had a substantial yet short-lived response to ruxolitinib (94). Most recently, Hinze et al. reported a case of a compound-mutated CNL achieving long-term remission with ruxolitinib (97). The co-occurrence of CSF3R and SETBP1 mutations have shown to confer resistance to ruxolitinib; however, in the aforementioned phase II trial, no association between SETBP1 mutation and response to ruxolitinib was noted (98).
The mechanisms underlying clonal evolutions in patients with CNL undergoing treatment with ruxolitinib might be multifaceted. Stoner et al. evaluated seven patients with CNL demonstrating molecular or clinical progression under treatment with ruxolitinib. VAF reduction of CSF3R mutation was noted in 3 cases, whereas two patients demonstrated no changes in allelic burden overtime. Primary resistance to ruxolitinib could be attributed to co-occurring NOTCH2 and SRSF2 mutations in the founder clone of one case. Notably, subclones harboring STAT3, STAG2, and RUNX1 mutations emerged in three cases. Acting downstream of JAK kinases, STAT3 could bypass the inhibitory effects of ruxolitinib, whereas co-operation of RUNX1 with CSF3R mutations might be involved in CNL disease progression and AML transformation. On the contrary, the role of the STAG2 mutations in disease progression has not been elucidated yet (99).
Fedratinib, another JAK inhibitor is being evaluated in a phase II trial for patients with CNL (NCT05177211).
The rationale for the use of dasatinib in CNL stems from the demonstration of the in vitro sensitivity of CNL lines harboring truncating CSF3R mutations to SRC kinase inhibition (20); however, no data on the in vivo efficacy of dasatinib are available. On the contrary, a recent case report demonstrated that co-occurrence of truncating and proximal CSF3R mutations confer resistance to dasatinib in a patient with CNL (97). In this context, a short trial with dasatinib could be offered in patients harboring truncating mutations with close monitoring for disease progression.
Preclinical data have suggested that compound mutations of CSF3R might exert their leukemogenic potential through enhancement of MAPK signaling. In support of this, trametinib, a MEK1/2 inhibitor, has demonstrated in vitro activity in compound-mutant CNL models; however, this agent has not been tested in clinical practice.
Given the lack of therapeutic agents that could provide long-term disease control, allo-HSCT represents the only therapeutic option with curative potential; however, the published evidence remains scarce, consisting mostly of case reports and small case series. Elliot et al. reported of five patients with CNL who underwent allo-HSCT. Four of them achieved CR and remained disease-free up to six years from transplantation (89). In another case-series of 19 patients, two patients in blast transformation underwent allo-HSCT. One patient had a favorable outcome, remaining disease-free 40 months post-transplant, whereas the other patient died from transplantation-related complications (41).Ruan et al. in a population-based study reported that 2% of CNL patients underwent allo-HSCT. Notably, all of them were alive at five years post-treatment (90). Although these studies are informative of the curative role of allo-HSCT, they do not provide details pertaining to the transplantation procedures.
A retrospective nationwide study in Japan aimed to provide a more comprehensive overview of allo-HSCT in this rare myeloid neoplasm. Between 2003 and 2014, five patients with CNL underwent allo-HSCT. Three patients received hydroxyurea, one received dasatinib, and one was offered intensive chemotherapy, albeit none of the patients demonstrated response to treatment prior to transplantation. Notably, none of them received ruxolitinib. Four patients received transplantation from matched-unrelated donors, whereas one patient received transplantation from an HLA-haploidentical sibling donor. All but one patients received a myeloablative conditioning regimen. Neutrophil engraftment was achieved in all but one patient who died due to bleeding. Two of the patients who engrafted achieved CR that was retained without signs of relapse at day +362 and +441, whereas one patients, although in CR, died from sinusoidal obstruction syndrome at day +56; one patients showed no response to transplant and succumbed to the disease 76 days post-transplant.
Chronic neutrophilic leukemia represents an extremely rare myeloproliferative neoplasm that has been poorly characterized for over a century, owing to the wide variability of the clinical presentation and laboratory findings of patients. Recently, there has been an increase in the understanding of the molecular pathogenesis of the disease, as mutations in the CSF3R gene are believed to be the driver mutations in most cases, albeit other mutations are required for the development of overt disease. The prognosis of the disease remains poor even for patients that proceed to allo-HSCT, which represents the only therapeutic option with curative intent. Studies on other therapeutic modalities such as ruxolitinib, have demonstrated satisfactory response rates; however, clonal evolution and emergence of resistance to these agents might limit their efficacy. Therefore, there is an unmet need for the optimal treatment of patients with CNL, highlighting the need for more studies enrolling patients with WHO-defined CNL, that could disentangle the complex genetic landscape of the disease and provide novel potential targets for development of more potent therapeutic agents.
Conceptualization, investigation, data curation, writing-original draft preparation: TPT, AS, AK, SGP, and VP. Writing—review and editing: TPT, and VP. Supervision: VP All authors have read, reviewed, edited and agreed to the published version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tuohy EL. A Case of Splenomegaly With Polymorphonuclear Neutrophil Hyperleukocytosis. Am J Med Sci (1920) 160:18 24. doi: 10.1097/00000441-192007000-00003
2. Emile-Weil P, See G. La Leucémie Myélogène À Polynucléaires Neutrophiles. Presse Méd (1932) 40:1071–4.
3. Exton-Smith AN, Chazan AA. Myeloproliferative Syndrome Presenting as Neutrophilic Leukaemia. Proc R Soc Med (1957) 50(7):510–2. doi: 10.1177/003591575705000711
4. Tanzer J, Harel P, Boiron M, Bernard J. Cytochemical and Cytogenetic Findings in a Case of Chronic Neutriphilic Leukaemia of Mature Cell Type. Lancet (1964) 1(7329):387–8. doi: 10.1016/s0140-6736(64)92142-7
5. You W, Weisbrot IM. Chronic Neutrophilic Leukemia. Report of Two Cases and Review of the Literature. Am J Clin Pathol (1979) 72(2):233–42. doi: 10.1093/ajcp/72.2.233
6. Grinblat J, Mammon Z, Lewitus Z, Joshua H. Chronic Myelogenous Leukemia With Elevated Leukocyte Alkaline Phosphatase, Positive Indirect Coombs' Test, Neutrophilic Leukocytosis and Unusual Cytogenetical Findings. Acta Haematol (1977) 57(5):298–304. doi: 10.1159/000207894
7. Bareford D, Jacobs P. Chronic Neutrophilic Leukaemia. Am J Clin Pathol (1980) 73(6):837. doi: 10.1093/ajcp/73.6.837
8. Dotten DA, Pruzanski W, Wong D. Functional Characterization of the Cells in Chronic Neutrophilic Leukemia. Am J Hematol (1982) 12(2):157–65. doi: 10.1002/ajh.2830120208
9. Zittoun R, Réa D, Ngoc LH, Ramond S. Chronic Neutrophilic Leukemia. A Study of Four Cases. Ann Hematol (1994) 68(2):55–60. doi: 10.1007/bf01715131
10. Elliott MA, Dewald GW, Tefferi A, Hanson CA. Chronic Neutrophilic Leukemia (Cnl): A Clinical, Pathologic and Cytogenetic Study. Leukemia (2001) 15(1):35–40. doi: 10.1038/sj.leu.2401993
11. Böhm J, Schaefer HE. Chronic Neutrophilic Leukaemia: 14 New Cases of an Uncommon Myeloproliferative Disease. J Clin Pathol (2002) 55(11):862–4. doi: 10.1136/jcp.55.11.862
12. Elliott MA. Chronic Neutrophilic Leukemia and Chronic Myelomonocytic Leukemia: Who Defined. Best Pract Res Clin Haematol (2006) 19(3):571–93. doi: 10.1016/j.beha.2005.07.012
13. Kwong YL, Cheng G. Clonal Nature of Chronic Neutrophilic Leukemia. Blood (1993) 82(3):1035–6. doi: 10.1182/blood.V82.3.1035b.1035b
14. Standen GR, Steers FJ, Jones L. Clonality of Chronic Neutrophilic Leukaemia Associated With Myeloma: Analysis Using the X-Linked Probe M27 Beta. J Clin Pathol (1993) 46(4):297–8. doi: 10.1136/jcp.46.4.297
15. Lorente JA, Peña JM, Ferro T, Garcia-Alegria J, Gonzalez JJ, Barbado J, et al. A Case of Chronic Neutrophilic Leukemia With Original Chromosomal Abnormalities. Eur J Haematol (1988) 41(3):285–8. doi: 10.1111/j.1600-0609.1988.tb01194.x
16. Orazi A, Cattoretti G, Sozzi G. A Case of Chronic Neutrophilic Leukemia With Trisomy 8. Acta Haematol (1989) 81(3):148–51. doi: 10.1159/000205548
17. Di Donato C, Croci G, Lazzari S, Scarduelli L, Vignoli R, Buia M, et al. Chronic Neutrophilic Leukemia: Description of a New Case With Karyotypic Abnormalities. Am J Clin Pathol (1986) 85(3):369–71. doi: 10.1093/ajcp/85.3.369
18. Boggs DR, Kaplan SS. Cytobiologic and Clinical Aspects in a Patient With Chronic Neutrophilic Leukemia After Thorotrast Exposure. Am J Med (1986) 81(5):905–10. doi: 10.1016/0002-9343(86)90367-0
19. Ouyang Y, Qiao C, Chen Y, Zhang SJ. Clinical Significance of Csf3r, Srsf2 and Setbp1 Mutations in Chronic Neutrophilic Leukemia and Chronic Myelomonocytic Leukemia. Oncotarget (2017) 8(13):20834–41. doi: 10.18632/oncotarget.15355
20. Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, et al. Oncogenic Csf3r Mutations in Chronic Neutrophilic Leukemia and Atypical Cml. N Engl J Med (2013) 368(19):1781–90. doi: 10.1056/NEJMoa1214514
21. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
22. Tilak V, Rai M, Singh VP, Rai AK, Raman R. Hepatocellular Carcinoma Presenting as Neutrophilic Leukaemoid Reaction–a Rare Entity. J Indian Med Assoc (2007) 105(8):462, 4–5.
23. Mittal N, Gupta R, Rahman K, Singh P, Panda I, Nityanand S. A Case of Paraneoplastic Hyperleukocytosis Closely Mimicking Chronic Neutrophilic Leukemia. J Cancer Res Ther (2020) 16(1):186–8. doi: 10.4103/0973-1482.199434
24. Kakkar N, Joseph John M. Chronic Neutrophilic Leukemia Presenting as Secondary Gout: Report of a Rare Myeloproliferative Disorder. Indian J Cancer (2020) 57(2):201–4. doi: 10.4103/ijc.IJC_560_18
25. Hossfeld DK, Lokhorst HW, Garbrecht M. Neutrophilic Leukemia Accompanied by Hemorrhagic Diathesis: Report of Two Cases. Blut (1987) 54(2):109–13. doi: 10.1007/bf00321039
26. Silva PR, Ferreira C, Bizarro S, Cerveira N, Torres L, Moreira I, et al. Diagnosis, Complications and Management of Chronic Neutrophilic Leukaemia: A Case Report. Oncol Lett (2015) 9(6):2657–60. doi: 10.3892/ol.2015.3148
27. Mori H, Ikegami T, Shimizu T, Harada H, Miyoshi Y, Okada S, et al. [Changes in the Megakaryocyte-Platelet System in Chronic Neutrophilic Leukemia]. Rinsho Ketsueki (1991) 32(12):1574–6.
28. Gan GG, Pasagna JF, Eow GI, Nadarajan VS. Chronic Neutrophilic Leukaemia. Singapore Med J (2007) 48(3):e74-6.
29. Castanet J, Lacour JP, Garnier G, Perrin C, Taillan B, Fuzibet JG, et al. Neutrophilic Dermatosis Associated With Chronic Neutrophilic Leukemia. J Am Acad Dermatol (1993) 29(2 Pt 2):290–2. doi: 10.1016/0190-9622(93)70178-v
30. Kaur S, Bery A, Garg B, Sood N. Sweet's Syndrome Associated With Chronic Neutrophilic Leukemia. Indian J Dermatol Venereol Leprol (2015) 81(2):203–6. doi: 10.4103/0378-6323.152304
31. Kwon O, Park J, Chung H, Park KD. Leukemia Cutis in Chronic Neutrophilic Leukemia Associated With Colony Stimulating Factor 3 Receptor Mutation: Clinical Severity Paralleled With Hematologic Abnormality. Ann Dermatol (2019) 31(6):673–7. doi: 10.5021/ad.2019.31.6.673
32. Hobbs LK, Carr PC, Gru AA, Flowers RH. Case and Review: Cutaneous Involvement by Chronic Neutrophilic Leukemia Vs Sweet Syndrome- A Diagnostic Dilemma. J Cutan Pathol (2021) 48(5):644–9. doi: 10.1111/cup.13925
33. Mendiola VL, Qian YW, Jana B. Septic Shock Predisposed by an Underlying Chronic Neutrophilic Leukemia With an Atypical Presentation; A Case Report. Case Rep Oncol (2018) 11(3):871–9. doi: 10.1159/000495458
34. Sugino K, Gocho K, Ota H, Kobayashi M, Sano G, Isobe K, et al. Miliary Tuberculosis Associated With Chronic Neutrophilic Leukemia. Intern Med (2009) 48(15):1283–7. doi: 10.2169/internalmedicine.48.2080
35. Delgado Beltrán P, Rubio-Félix D, García Zueco JC, Capablo JL, Alvarez R. [Chronic Neutrophilic Leukemia (Cnl) and Meningeal Tuberculosis (Tbc): Chance or Cause?]. Sangre (Barc) (1995) 40(3):235–7.
36. Yasui H, Adachi Y, Ishii Y, Kato Y, Imai K. Mucormycosis as an Etiology of Cerebral Hemorrhage in Patients With Chronic Neutrophilic Leukemia. Am J Med (2003) 115(8):674–6. doi: 10.1016/s0002-9343(03)00426-1
37. Feremans W, Marcelis L, Ardichvili D. Chronic Neutrophilic Leukaemia With Enlarged Lymph Nodes and Lysozyme Deficiency. J Clin Pathol (1983) 36(3):324–8. doi: 10.1136/jcp.36.3.324
38. Nakamine H, Hirano K, Tsujimoto M, Nishino E, Takenaka T, Maeda J, et al. Lymph Node Involvement in Chronic Neutrophilic Leukemia. An Immunohistochemical Study. Virchows Arch A Pathol Anat Histopathol (1988) 412(3):241–5. doi: 10.1007/bf00737148
39. Watanabe A, Yoshida Y, Yamamoto H, Kimura S, Nagashima H, Kitajima K. A Case of Chronic Neutrophilic Leukemia With Paraproteinemia (Igg Type Lambda and Iga Type K). Jpn J Med (1984) 23(1):39–44. doi: 10.2169/internalmedicine1962.23.39
40. Luo Q, Shen J, Yang Y, Tang H, Shi M, Liu J, et al. Csf3r T618i, Asxl1 G942 Fs and Stat5b N642h Trimutation Co-Contribute to a Rare Chronic Neutrophilic Leukaemia Manifested by Rapidly Progressive Leucocytosis, Severe Infections, Persistent Fever and Deep Venous Thrombosis. Br J Haematol (2018) 180(6):892–4. doi: 10.1111/bjh.14456
41. Szuber N, Finke CM, Lasho TL, Elliott MA, Hanson CA, Pardanani A, et al. Csf3r-Mutated Chronic Neutrophilic Leukemia: Long-Term Outcome in 19 Consecutive Patients and Risk Model for Survival. Blood Cancer J (2018) 8(2):21. doi: 10.1038/s41408-018-0058-7
42. Takemori N, Hirai K, Onodera R, Saito N, Namiki M. Electron Microscopic Findings of Parallel Tubular Granules in a Case of Chronic Neutrophilic Leukaemia. J Clin Pathol (1994) 47(4):367–9. doi: 10.1136/jcp.47.4.367
43. Kanoh T, Saigo K, Yamagishi M. Neutrophils With Ring-Shaped Nuclei in Chronic Neutrophilic Leukemia. Am J Clin Pathol (1986) 86(6):748–51. doi: 10.1093/ajcp/86.6.748
44. Kaplan SS, Berkow RL, Joyce RA, Basford RE, Barton JC. Neutrophil Function in Chronic Neutrophilic Leukemia: Defective Respiratory Burst in Response to Phorbol Esters. Acta Haematol (1992) 87(1-2):16–21. doi: 10.1159/000204707
45. Okada S, Miyoshi Y, Takizawa Y, Hagiwara S, Mori H, Niikura H, et al. [Neutrophil Dysfunction in Chronic Neutrophilic Leukemia Without Rearrangements of Bcr and Immunoglobulin Heavy Chain Genes]. Rinsho Ketsueki (1989) 30(10):1881–5.
46. Ohtsuki T, Katsura Y, Mizukami H, Matsu-ura Y, Kimura F, Ohnishi M, et al. Elevated Neutrophil Function in Chronic Neutrophilic Leukemia. Am J Hematol (1992) 41(1):50–6. doi: 10.1002/ajh.2830410110
47. Mehrotra PK, Winfield DA, Fergusson LH. Cellular Abnormalities and Reduced Colony-Forming Cells in Chronic Neutrophilic Leukaemia. Acta Haematol (1985) 73(1):47–50. doi: 10.1159/000206273
48. Oogushi K, Shimamoto Y, Hamasaki Y, Koga H, Sano M, Yamaguchi M, et al. The Discrepancy Between Chemotaxis and Leukotriene B4 Production in a Patient With Chronic Neutrophilic Leukemia. Jpn J Med (1989) 28(6):717–21. doi: 10.2169/internalmedicine1962.28.717
49. Uemura Y, Taguchi T, Kubota T, Saito T, Bandobashi K, Yokoyama A. Neutrophil Function and Cytokine-Specific Signaling in Chronic Neutrophilic Leukemia. Int J Lab Hematol (2009) 31(1):36–47. doi: 10.1111/j.1751-553X.2007.01000.x
50. Feng Y, Feng W. Numerous Ring Sideroblasts in Chronic Neutrophilic Leukemia. Blood (2021) 138(18):1783. doi: 10.1182/blood.2021013310
51. Yin B, Chen X, Gao F, Li J, Wang HW. Analysis of Gene Mutation Characteristics in Patients With Chronic Neutrophilic Leukaemia. Hematology (2019) 24(1):538–43. doi: 10.1080/16078454.2019.1642554
52. Tang DJ, Yu ZF, Song JZ, Chen MC, Wang ZW. Gaucher-Like Cells in Chronic Neutrophilic Leukemia. A Case Report. Chin Med J (Engl) (1987) 100(12):988–9. doi: 10.5555/cmj.0366-6999.100.12.p988.01
53. Ito K, Usuki K, Iki S, Urabe A. Chronic Neutrophilic Leukemia Associated With Chronic Lymphocytic Leukemia. Int J Hematol (1998) 68(1):87–94. doi: 10.1016/s0925-5710(98)00031-0
54. Vorobiof DA, Benjamin J, Kaplan H, Dvilansky A. Chronic Granulocytic Leukemia, Neutrophilic Type, With Paraproteinemia (Iga Type K). Acta Haematol (1978) 60(5):316–20. doi: 10.1159/000207729
55. Naparstek Y, Zlotnick A, Polliack A. Coexistent Chronic Myeloid Leukemia and Iga Monoclonal Gammopathy: Report of a Case and Review of the Literature. Am J Med Sci (1980) 279(2):111–5. doi: 10.1097/00000441-198003000-00005
56. Kubo A, Kawanami M, Matsuyama E, Tamura T, Kanoh T. [Chronic Neutrophilic Leukemia Associated With Monoclonal Gammopathy (Iga, Kappa Type)]. Rinsho Ketsueki (1989) 30(6):858–62.
57. Florensa L, Woessner S, Vicente P, Roig Martí A, Solé F, Pérez A. Chronic Neutrophilic Leukemia Associated With Monoclonal Gammopathy of Undetermined Significance. A Multimethod Study. Ann Hematol (1993) 67(3):129–31. doi: 10.1007/bf01701736
58. Gao JP, Zhai LJ, Gao XH, Min FL. Chronic Neutrophilic Leukemia Complicated With Monoclonal Gammopathy of Undetermined Significance: A Case Report and Literature Review. J Clin Lab Anal (2022) 00:e24287. doi: 10.1002/jcla.24287
59. Franchi F, Seminara P, Giunchi G. Chronic Neutrophilic Leukemia and Myeloma. Report on Long Survival. Tumori (1984) 70(1):105–7. doi: 10.1177/030089168407000117
60. Lewis MJ, Oelbaum MH, Coleman M, Allen S. An Association Between Chronic Neutrophilic Leukaemia and Multiple Myeloma With a Study of Cobalamin-Binding Proteins. Br J Haematol (1986) 63(1):173–80. doi: 10.1111/j.1365-2141.1986.tb07507.x
61. Zoumbos NC, Chrysanthopoulos C, Starakis J, Kapatais-Zoumbos K. Kappa Light Chain Myeloma Developing in a Patient With Chronic Neutrophilic Leukaemia. Br J Haematol (1987) 65(4):504–5. doi: 10.1111/j.1365-2141.1987.tb04164.x
62. Rovira M, Cervantes F, Nomdedeu B, Rozman C. Chronic Neutrophilic Leukaemia Preceding for Seven Years the Development of Multiple Myeloma. Acta Haematol (1990) 83(2):94–5. doi: 10.1159/000205176
63. Standen GR, Jasani B, Wagstaff M, Wardrop CA. Chronic Neutrophilic Leukemia and Multiple Myeloma. An Association With Lambda Light Chain Expression. Cancer (1990) 66(1):162–6. doi: 10.1002/1097-0142(19900701)66:1<162::aid-cncr2820660129>3.0.co;2-z
64. Hidalgo C, Camacho J, Fernández J, Hernández A, Poveda F, Peña JM. [Chronic Neutrophilic Leukemia: Apropos of 2 Cases and Review of the Literature]. Med Clin (Barc) (1990) 95(11):421–3.
65. Troussard X, Lebrun E, Macro M, Galateau F, Reman O, Leporrier M. [Neutrophilic Leukemia and Multiple Myeloma. 2 Cases]. Ann Med Interne (Paris) (1992) 143(2):136–9.
66. Masini L, Salvarani C, Macchioni P, Baldi G, Rossi F, Croci G, et al. Chronic Neutrophilic Leukemia (Cnl) With Karyotypic Abnormalities Associated With Plasma Cell Dyscrasia: A Case Report. Haematologica (1992) 77(3):277–9.
67. Diéguez JC, Fernández Jurado A, Amián A, Rodríguez JN, Martino ML, Cañavate M, et al. [Chronic Neutrophilic Leukemia Associated With Myeloma. Simultaneous Presentation]. Sangre (Barc) (1992) 37(5):403–6.
68. Cehreli C, Undar B, Akkoc N, Onvural B, Altungoz O. Coexistence of Chronic Neutrophilic Leukemia With Light Chain Myeloma. Acta Haematol (1994) 91(1):32–4. doi: 10.1159/000204241
69. Dinçol G, Nalçaci M, Doğan O, Aktan M, Küçükkaya R, Ağan M, et al. Coexistence of Chronic Neutrophilic Leukemia With Multiple Myeloma. Leuk Lymphoma (2002) 43(3):649–51. doi: 10.1080/10428190290012218
70. Taiwo E, Wang H, Lewis R. Treatment of Coexisting Chronic Neutrophilic Leukemia and Light Chain Multiple Myeloma With Hydroxyurea, Bortezomib, and Dexamethasone. Case Rep Hematol (2014) 2014:869395. doi: 10.1155/2014/869395
71. Shi J, Ni Y, Li J, Qiu H, Miao K. Concurrent Chronic Neutrophilic Leukemia Blast Crisis and Multiple Myeloma: A Case Report and Literature Review. Oncol Lett (2015) 9(5):2208–10. doi: 10.3892/ol.2015.3043
72. Bain BJ, Ahmad S. Chronic Neutrophilic Leukaemia and Plasma Cell-Related Neutrophilic Leukaemoid Reactions. Br J Haematol (2015) 171(3):400–10. doi: 10.1111/bjh.13600
73. Dao KH, Tyner JW. What's Different About Atypical Cml and Chronic Neutrophilic Leukemia? Hematol Am Soc Hematol Educ Program (2015) 2015:264–71. doi: 10.1182/asheducation-2015.1.264
74. Roberts AW. G-Csf: A Key Regulator of Neutrophil Production, But That's Not All! Growth Factors (2005) 23(1):33–41. doi: 10.1080/08977190500055836
75. Mehta HM, Corey SJ. G-Csf, the Guardian of Granulopoiesis. Semin Immunol (2021) 54:101515. doi: 10.1016/j.smim.2021.101515
76. Liongue C, Ward AC. Granulocyte Colony-Stimulating Factor Receptor Mutations in Myeloid Malignancy. Front Oncol (2014) 4:93. doi: 10.3389/fonc.2014.00093
77. Druhan LJ, Ai J, Massullo P, Kindwall-Keller T, Ranalli MA, Avalos BR. Novel Mechanism of G-Csf Refractoriness in Patients With Severe Congenital Neutropenia. Blood (2005) 105(2):584–91. doi: 10.1182/blood-2004-07-2613
78. Mehta HM, Glaubach T, Long A, Lu H, Przychodzen B, Makishima H, et al. Granulocyte Colony-Stimulating Factor Receptor T595i (T618i) Mutation Confers Ligand Independence and Enhanced Signaling. Leukemia (2013) 27(12):2407–10. doi: 10.1038/leu.2013.164
79. Gotlib J, Maxson JE, George TI, Tyner JW. The New Genetics of Chronic Neutrophilic Leukemia and Atypical Cml: Implications for Diagnosis and Treatment. Blood (2013) 122(10):1707–11. doi: 10.1182/blood-2013-05-500959
80. Pardanani A, Lasho TL, Laborde RR, Elliott M, Hanson CA, Knudson RA, et al. Csf3r T618i Is a Highly Prevalent and Specific Mutation in Chronic Neutrophilic Leukemia. Leukemia (2013) 27(9):1870–3. doi: 10.1038/leu.2013.122
81. Fleischman AG, Maxson JE, Luty SB, Agarwal A, Royer LR, Abel ML, et al. The Csf3r T618i Mutation Causes a Lethal Neutrophilic Neoplasia in Mice That Is Responsive to Therapeutic Jak Inhibition. Blood (2013) 122(22):3628–31. doi: 10.1182/blood-2013-06-509976
82. Maxson JE, Tyner JW. Genomics of Chronic Neutrophilic Leukemia. Blood (2017) 129(6):715–22. doi: 10.1182/blood-2016-10-695981
83. Zhang H, Wilmot B, Bottomly D, Dao KT, Stevens E, Eide CA, et al. Genomic Landscape of Neutrophilic Leukemias of Ambiguous Diagnosis. Blood (2019) 134(11):867–79. doi: 10.1182/blood.2019000611
84. Elliott MA, Pardanani A, Hanson CA, Lasho TL, Finke CM, Belachew AA, et al. Asxl1 Mutations Are Frequent and Prognostically Detrimental in Csf3r-Mutated Chronic Neutrophilic Leukemia. Am J Hematol (2015) 90(7):653–6. doi: 10.1002/ajh.24031
85. Thomopoulos TP, Bouhla A, Papageorgiou SG, Pappa V. Chronic Myelomonocytic Leukemia - A Review. Expert Rev Hematol (2021) 14(1):59–77. doi: 10.1080/17474086.2021.1860004
86. Cui YJ, Jiang Q, Liu JQ, Li B, Xu ZF, Qin TJ, et al. The Clinical Characteristics, Gene Mutations and Prognosis of Chronic Neutrophilic Leukemia. Zhonghua Xue Ye Xue Za Zhi (2017) 38(1):28–32. doi: 10.3760/cma.j.issn.0253-2727.2017.01.006
87. Cui Y, Li B, Gale RP, Jiang Q, Xu Z, Qin T, et al. Csf3r, Setbp1 and Calr Mutations in Chronic Neutrophilic Leukemia. J Hematol Oncol (2014) 7:77. doi: 10.1186/s13045-014-0077-1
88. Shou LH, Cao D, Dong XH, Fang Q, Wu Y, Zhang Y, et al. Prognostic Significance of Setbp1 Mutations in Myelodysplastic Syndromes, Chronic Myelomonocytic Leukemia, and Chronic Neutrophilic Leukemia: A Meta-Analysis. PloS One (2017) 12(2):e0171608. doi: 10.1371/journal.pone.0171608
89. Elliott MA, Hanson CA, Dewald GW, Smoley SA, Lasho TL, Tefferi A. Who-Defined Chronic Neutrophilic Leukemia: A Long-Term Analysis of 12 Cases and a Critical Review of the Literature. Leukemia (2005) 19(2):313–7. doi: 10.1038/sj.leu.2403562
90. Ruan GJ, Smith CJ, Day C, Harmsen WS, Zblewski DL, Alkhateeb H, et al. A Population-Based Study of Chronic Neutrophilic Leukemia in the United States. Blood Cancer J (2020) 10(6):68. doi: 10.1038/s41408-020-0334-1
91. Yassin MA, Kohla S, Al-Sabbagh A, Soliman AT, Yousif A, Moustafa A, et al. A Case of Chronic Neutrophilic Leukemia Successfully Treated With Pegylated Interferon Alpha-2a. Clin Med Insights Case Rep (2015) 8:33–6. doi: 10.4137/CCRep.S22820
92. Martínez-Cuadrón D, Megías-Vericat JE, Serrano J, Martínez-Sánchez P, Rodríguez-Arbolí E, Gil C, et al. Treatment Patterns and Outcomes of 2310 Patients With Secondary Acute Myeloid Leukemia: A Pethema Registry Study. Blood Adv (2022) 6(4):1278–95. doi: 10.1182/bloodadvances.2021005335
93. Lasho TL, Mims A, Elliott MA, Finke C, Pardanani A, Tefferi A. Chronic Neutrophilic Leukemia With Concurrent Csf3r and Setbp1 Mutations: Single Colony Clonality Studies, In Vitro Sensitivity to Jak Inhibitors and Lack of Treatment Response to Ruxolitinib. Leukemia (2014) 28(6):1363–5. doi: 10.1038/leu.2014.39
94. Gunawan AS, McLornan DP, Wilkins B, Waghorn K, Hoade Y, Cross NCP, et al. Ruxolitinib, a Potent Jak1/Jak2 Inhibitor, Induces Temporary Reductions in the Allelic Burden of Concurrent Csf3r Mutations in Chronic Neutrophilic Leukemia. Haematologica (2017) 102(6):e238–40. doi: 10.3324/haematol.2017.163790
95. Nooruddin Z, Miltgen N, Wei Q, Schowinsky J, Pan Z, Tobin J, et al. Changes in Allele Frequencies of Csf3r and Setbp1 Mutations and Evidence of Clonal Evolution in a Chronic Neutrophilic Leukemia Patient Treated With Ruxolitinib. Haematologica (2017) 102(5):e207–9. doi: 10.3324/haematol.2016.163089
96. Stahl M, Xu ML, Steensma DP, Rampal R, Much M, Zeidan AM. Clinical Response to Ruxolitinib in Csf3r T618-Mutated Chronic Neutrophilic Leukemia. Ann Hematol (2016) 95(7):1197–200. doi: 10.1007/s00277-016-2664-4
97. Hinze A, Rinke J, Hochhaus A, Ernst T. Durable Remission With Ruxolitinib in a Chronic Neutrophilic Leukemia Patient Harboring a Truncation and Membrane Proximal Csf3r Compound Mutation. Ann Hematol (2021) 100(2):581–4. doi: 10.1007/s00277-020-04152-w
98. Dao KT, Gotlib J, Deininger MMN, Oh ST, Cortes JE, Collins RH Jr, et al. Efficacy of Ruxolitinib in Patients With Chronic Neutrophilic Leukemia and Atypical Chronic Myeloid Leukemia. J Clin Oncol (2020) 38(10):1006–18. doi: 10.1200/jco.19.00895
Keywords: chronic neutrophilic leukemia, myeloproliferative neoplasm, CSF3R, ruxolitinib, allogeneic HSCT
Citation: Thomopoulos TP, Symeonidis A, Kourakli A, Papageorgiou SG and Pappa V (2022) Chronic Neutrophilic Leukemia: A Comprehensive Review of Clinical Characteristics, Genetic Landscape and Management. Front. Oncol. 12:891961. doi: 10.3389/fonc.2022.891961
Received: 08 March 2022; Accepted: 28 March 2022;
Published: 14 April 2022.
Edited by:
Simona Soverini, University of Bologna, ItalyReviewed by:
Ibrahim C. Haznedaroglu, Hacettepe University Hospital, TurkeyCopyright © 2022 Thomopoulos, Symeonidis, Kourakli, Papageorgiou and Pappa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vasiliki Pappa, dmFzX3BhcHBhQHlhaG9vLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.