 Negesse Mekonnen
Negesse Mekonnen Hobin Yang1†
Hobin Yang1† Young Kee Shin
Young Kee Shin- 1Department of Pharmacy, Research Institute of Pharmaceutical Science, Seoul National University College of Pharmacy, Seoul, South Korea
- 2Department of Veterinary Science, School of Animal Science and Veterinary Medicine, Bahir Dar University, Bahir Dar, Ethiopia
- 3Bio-MAX/N-Bio, Seoul National University, Seoul, South Korea
- 4Department of Molecular Medicine and Biopharmaceutical Sciences, Seoul National University Graduate School of Convergence Science and Technology, Seoul, South Korea
- 5LOGONE Bio Convergence Research Foundation, Center for Companion Diagnostics, Seoul, South Korea
Homologous recombination (HR) is a highly conserved DNA repair mechanism that protects cells from exogenous and endogenous DNA damage. Breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) play an important role in the HR repair pathway by interacting with other DNA repair proteins such as Fanconi anemia (FA) proteins, ATM, RAD51, PALB2, MRE11A, RAD50, and NBN. These pathways are frequently aberrant in cancer, leading to the accumulation of DNA damage and genomic instability known as homologous recombination deficiency (HRD). HRD can be caused by chromosomal and subchromosomal aberrations, as well as by epigenetic inactivation of tumor suppressor gene promoters. Deficiency in one or more HR genes increases the risk of many malignancies. Another conserved mechanism involved in the repair of DNA single-strand breaks (SSBs) is base excision repair, in which poly (ADP-ribose) polymerase (PARP) enzymes play an important role. PARP inhibitors (PARPIs) convert SSBs to more cytotoxic double-strand breaks, which are repaired in HR-proficient cells, but remain unrepaired in HRD. The blockade of both HR and base excision repair pathways is the basis of PARPI therapy. The use of PARPIs can be expanded to sporadic cancers displaying the “BRCAness” phenotype. Although PARPIs are effective in many cancers, their efficacy is limited by the development of resistance. In this review, we summarize the prevalence of HRD due to mutation, loss of heterozygosity, and promoter hypermethylation of 35 DNA repair genes in ovarian, breast, colorectal, pancreatic, non-small cell lung cancer, and prostate cancer. The underlying mechanisms and strategies to overcome PARPI resistance are also discussed.
1 Introduction
Homologous recombination (HR) is one of the major pathways for the repair of DNA double-strand breaks (DSBs) in eukaryotic cells. Pathogenic mutations in genes encoding HR-related proteins are associated with the development of certain malignancies, including breast, ovarian, and other cancers (1). Normal and cancer cells rely on multiple DNA damage response pathways that specifically repair different forms of DNA damage. Key pathways include homologous recombination repair (HRR), base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), nonhomologous end-joining (NHEJ), translesion synthesis (TLS), and interstrand crosslink (ICL) repair (2, 3). However, these repair pathways are not equally effective in DNA repair, and some mechanisms are error-prone. For instance, non-canonical DNA repair systems such as NHEJ, single-strand annealing, and TLS are activated when the canonical pathways are deficient (4, 5). In response to DSBs, cells activate the HRR pathway, which relies on the undamaged sister chromatid as a template for repair. Because of this reliance on sister chromatids, HRR is active during the S and G2 phases of the cell cycle and is a high fidelity and error-free DNA repair pathway (3). By contrast, in NHEJ, the break ends are directly ligated without a homologous template, resulting in an error-prone repair pathway that can predispose to genetic instability (6, 7).
Genomic scars in relation to HR are caused by chromosomal and sub chromosomal aberrations. Genomic aberrations arise from mutation, structural copy number aberrations, or structural rearrangements. Mutations are substitutions (transversion & transition) or indel (insertion & deletion) mutations, and can inactivate tumor suppressor genes (TSGs). Structural copy number alterations can be copy number gain (leading to allelic imbalance) or copy number loss [leading to deletion, loss of heterozygosity (LOH), or haplo-insufficiency (one copy of a gene is deleted or contain loss of function mutation leading to insufficient level of proteins)]. Structural rearrangements can be inversion (paracentric), translocation (reciprocal), or recombination leading to copy neutral LOH events (8). Frequent copy number alterations are the hallmarks of homologous recombination deficiency (HRD) and can occur at the regional or whole chromosome level. Quantification of large-scale structural variants is used as an indicator of the HRD phenotype (presence of HRD in sporadic cancers other than BRCA1 and 2 inactivation), including telomeric allelic imbalance (TAI: large allelic imbalances extending to the telomere), large scale transition (LST: number of transitions between large regions of different allelic states or chromosomal breaks between adjacent regions of >10 MB), and LOH (large regions displaying somatic loss of one haplotype, which can be copy variable as in deletion or copy neutral LOH). Genes involved in HR, including tumor suppressor genes, can also be repressed by aberrant promoter hypermethylation, an epigenetic mechanism that contributes to HRD (9).
HR is a DNA repair pathway of clinical interest because of the sensitivity of HRD cells to poly-ADP-ribose polymerase (PARP) inhibitors (PARPIs) (10). DNA repair targeting therapies exploit DNA repair defects in cancer cells to generate synthetic lethality, and DNA repair defects vary according to cancer type. For example, approximately 50% of ovarian carcinomas exhibit dysfunctional HRR, whereas the rate of HRR dysfunction is lower in other cancer types such as colorectal cancer (CRC) (<5%) (11, 12). Hereditary mutations in one copy of the BRCA gene predispose patients to female breast cancer (85% lifetime risk), ovarian cancer (10%–40%), male breast cancer, pancreatic cancer (PC), prostate cancer, non-small cell lung cancer (NSCLC), CRC, and other cancer types. Precancerous cells deficient in BRCA1 and 2 cannot repair DSBs properly, resulting in genomic instability that eventually leads to cancer (8). These tumors are intrinsically sensitive to DNA damage response inhibitors (PARPIs), which induce synthetic lethality. Synthetic lethality can arise from the combined inactivation of HRR genes by (mutation, LOH and promoter hypermethylation) and PARP inhibition (13). The use of PARPIs in non-BRCA mutation carrier patients can be expanded to sporadic cancers that display “BRCAness” (cancers that have defective HR without germline BRCA1 and 2 mutations). Findings showed that TSGs with BRCAness phenotypes are often inactivated, for example, ATM, ATR, PALB2, RAD51, RAD51B, RAD51C, RAD51D, BARD1, and FANCM among others (14–17).
The use of PARPIs was recently expanded to other cancers in addition to breast and ovarian cancer, such as prostate and PC (18). Although PARPIs have shown beneficial effects in many other cancer types, the frequent development of resistance is challenging. For instance, in a phase II clinical trial, secondary resistance mutations were detected in circulating free tumor DNA in two patients with a germline BRCA2 mutation. These mutations were predicted to lead to the reversal of a somatic mutation (19). A comprehensive investigation of the underlying mechanisms is necessary to design strategies for overcoming PARPI resistance.
Another challenging issue is the development of effective biomarkers to identify patients who are more likely to respond to specific targeted therapy by using companion diagnosis (CDx). CDx is an in vitro medical device that uses biomarkers to provide information on the safe and effective use of drugs or biologicals. FDA-approved CDx includes BRACAnalysis CDx® and Myriad myChoice® CDx developed by Myriad Genetic Laboratories, and FoundationOne® CDx [F1CDx] and FoundationOne® Liquid CDx developed by Foundation medicine. The HRDetect test utilizes machine learning algorithm (20).
BRACAnalysis CDx® is an in vitro diagnostic method used for the detection and classification of DNA sequence variants in the protein-coding regions, intron or exon boundaries of the germline BRCA1 and 2 genes from whole blood sample. PCR and Sanger sequencing are used to detect small insertions and deletions (indels), and single nucleotide variants (SNVs). Large deletions and duplications are detected by multiplex PCR. The test results used as an aid to identify eligible patients for PARPIs in breast, ovarian, pancreatic and prostate cancers treatment (21). Myriad myChoice® CDx is NGS-based in vitro diagnostic test that evaluates the qualitative detection and classification of SNVs, indels and large rearrangements (LRs) in protein-coding regions and intron/exon boundaries of the BRCA1 and 2 genes, and determine Genomic Instability Score (GIS) by measuring [LOH, TAI, and LST] using DNA isolated from formalin-fixed paraffin-embedded (FFPE) tumor tissue. The test used to select eligible patients for ovarian cancer with positive HRD status for the treatment with Zejula® (niraparib) (22).
FoundationOne®CDx (F1CDx) is a qualitative NGS- and high throughput hybridization-based capture test for the detection of indels, substitutions and copy number alterations (CNAs) in 324 genes. It detects gene rearrangements, genomic signatures including microsatellite instability (MSI), tumor mutational burden (TMB) and positive HRD status (somatic BRCA-positive and/or LOH high) using DNA isolated from FFPE tumor tissue. It provides definite information for the identification of eligible patients for specific treatments of different class using specific biomarkers for many solid tumors (23).
FoundationOne® Liquid CDx is a qualitative NGS based test, which can identify indels, and substitutions in 311 genes, rearrangements in 4 genes and CNAs in 3 genes. It utilizes circulating cell-free DNA (cfDNA) isolated from plasma-driven peripheral whole blood collected in anti-coagulants. The test identifies patients that can benefit from different targeted treatments for NSCLC, breast, ovarian and prostate cancers based on specific biomarkers detected in each cancer. Negative result does not rule out the presence of an alteration in the patient’s tumor, in this case patients can opt for another tumor tissue-based CDx. The test analytical accuracy is not well demonstrated in all genes e.g., the test does not detect heterozygous deletions, and copy number losses/homozygous deletion in ATM (24).
HRDetect is a whole genome sequencing (WGS)-based classifier of HRD that can predict BRCA1 and 2 deficiency based on six mutational signatures (the HRD index [LOH + TAI + LST], microhomology-mediated indels, base-substitution signature 3 and 8, and rearrangement signature 3 and 5). It can also identify HRD in sporadic cancers (BRCAness) with and without any single detectable defect in HR genes (20, 25). HRDetect was shown highly sensitive method as compared to other HRD detection CDxs (20), but require clinical validation in independent set to avoid overfitting issue.
In this review, we did not classify mutation and hypermethylation data as bi-allelic or mono-allelic inactivation. Oftentimes, cases of pathogenic mutation in tumor suppressor genes lead to bi-allelic inactivation. Whole-exome sequencing analysis of breast cancer cases by Mutter et al. (26) revealed that 89% of bi-allelic inactivation results in HRD, whereas in cases of mono-allelic inactivation significant association existed between RAD51 functional status and LST. In a study by Li et al. (27) mono-allelic germline pathogenic mutation of PALB2 had predisposed to a high-risk breast cancer development, underscoring the role of PALB2 in HR repair. Moreover, protein-truncating variants and rare missense variants of DNA repair genes were significantly associated with the risk of breast cancer (16). Many findings confirmed the importance of haplo-insufficiency in tissue and gene specific manner; for instance, PTEN hypermorphic mice expressing 80% normal levels of PTEN protein was sufficient to predispose for different cancers development (28). Mono-allelic inactivation of TSGs (e.g., p53 and PTEN) leads to the inability to perform normal cellular functions which contributed to cancer development (29).
Here, we investigated the potential implications of pathogenic mutations, LOH, and promoter hypermethylation of HR-related genes using recent data in ovarian, breast, colorectal, pancreatic, non-small cell lung, and prostate cancers. Additionally, the mechanisms underlying PARPI resistance and possible strategies to overcome PARPI resistance are discussed.
1.1 BRCA1 and BRCA2 in Homologous Recombination Repair
BRCA1 and 2 interacts with a number of other DNA repair proteins to form a complex system for DNA damage repair, including ATM, RAD51, PALB2, MRE11A, RAD50, NBN, and the Fanconi anemia proteins (30). BRCA1 and BRCA2 are potential biomarkers for HRD in ovarian and breast cancer. In the presence of DNA DSBs, BRCA1 and 2 collaborate with other HR proteins to maintain the breaks. For instance, ATM is specifically activated in response to DSBs and is essential for phosphorylating many proteins involved in controlling cell cycle checkpoints and DNA repair. Three proteins are involved in recruiting ATM to DSBs, meiotic recombination 11 (MRE11A), RAD50, and NBS1 or MRN complex. Cells deficient in ATM and NBS1 are thus sensitive to PARPIs, similar to BRCA1- and BRCA2-deficient cells (7, 31). Germline pathogenic mutations of BRCA1 and BRCA2 suppress the HR mechanism and cause hereditary breast and ovarian cancer (HBOC) syndrome (32, 33). The functions of 35 HR-related genes are described briefly in Supplementary Table S1.
1. 2 Interaction Between FANC and BRCA Genes in Homologous Recombination Repair
Fanconi anemia (FA) is a clinically and genetically heterogeneous syndrome involving bone marrow failure (BMF), developmental/congenital abnormalities that may affect all organ systems (renal dysplasia, craniofacial malformations, endocrine dysfunction, developmental delay, VACTERL association, radial ray malformations, osteoporosis, progressive BMF, skin abnormalities, short stature, cardiac defects, decreased fertility, and genitourinary and gastrointestinal malformations), and cancer predisposition (34). This review focused only on the role of five FA genes in cancer predisposition. FA is a congenital defect that results from loss of function of any of 21 genes, which indicates their essential role in maintaining the chromosomal stability of hematopoietic stem cells. The main cause of FA Complementation Group (FANC) gene abnormality is mutation (95%). The unique clinical phenotype associated with FANC gene mutations implies that proteins encoded by these genes function in a common cellular pathway. This pathway, known as the FA/BRCA DNA repair pathway (Figure 1), preserves genomic homeostasis in response to specific types of DNA damage (35). The FA pathway serves to remove ICLs and shares components, such as BRCA2 and PALB2, with the HR and NER pathways (2).
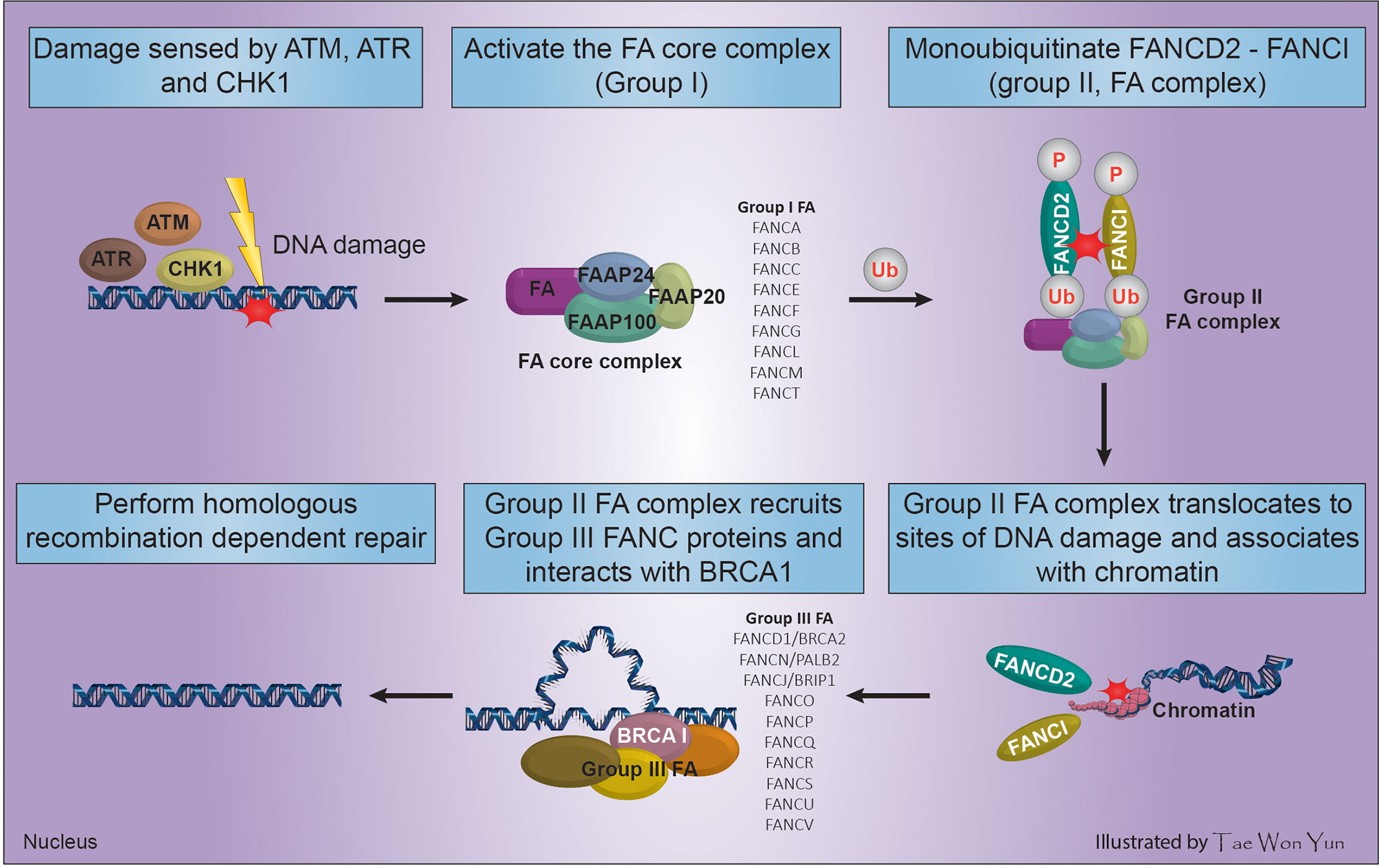
Figure 1 Process of FA complex formation and DNA interstrand cross-linking maintenance through the interaction of FANC and BRCA genes.
The main genome housekeeping function of the FA pathway in the DNA damage response necessitates multifactorial activation of HR. Cells use the error-prone NHEJ repair mechanism when FA-based repair systems are deficient, which may negatively affect genomic stability (36). In response to DNA damage caused by radiation, tobacco smoke, alcohol, or reactive oxygen species among others, FA proteins and three other Fanconi-associated proteins (FAAP100, FAAP24, and FAAP20) are activated by DNA damage response sensors such as ATM, ATR, and cell cycle check point (CHK1) to arrest the cell cycle, forming the FA core complex (group I) (34). The assembled FA core complex (group I) binds to the ubiquitin-conjugating enzyme UBE2T via the FANCL subunit and activates the FANCD2-FANCI complex (group II FA complex) through mono-ubiquitination and phosphorylation of FANCD2/FANCI (2). The ubiquitinated FANCD2-FANCI complex translocates to sites of DNA damage, associates with chromatin, and co-localizes with/recruits the downstream FA effector proteins (group III FA complex) including FANCD1/BRCA2, FANCN/PALB2, and FANCJ/BRIP1. Then, the group III FA complex perform HR-dependent DNA repair by interacting with BRCA1 (Figure 1) (35, 37).
2 Homologous Recombination Deficiency in Ovarian Cancer
The International Agency for Research on Cancer (IARC) (38) reports that ovarian cancer is the 8th and 9th cause of incidence and mortality worldwide in females, respectively. In 2020, the incidence and mortality of ovarian cancer were estimated at 3.4% (313,959 of all cancer types) and 4.68% (207,252 of all cancer types), respectively. Approximately 75% of epithelial ovarian cancer patients are diagnosed with advanced disease, which is curable in only a minority of cases, resulting in a modest 5-year survival rate of 20–30% (39). According to the cancer genome atlas (TCGA), high grade serous ovarian cancers (HGSOCs) are characterized by frequent genetic and epigenetic alterations of HR pathway genes, most commonly BRCA1 and BRCA2. In addition, approximately 50% of patients with HGSOC exhibit genetic and epigenetic alterations in the FANC-BRCA (Figure 1) pathway (11). Germline mutations in the BRCA1 and BRCA2 genes are well-known mechanisms of HRD, and loss of BRCA1 or BRCA2 thus poses a significant risk to genome integrity, leading not only to cancer predisposition, but also affecting the sensitivity to DNA-damaging agents and thus therapeutic approaches (40). Pathogenic variants of BRCA1 and BRCA2 only explain the genetic cause of approximately 10% of hereditary breast and ovarian cancers (transmitted to offspring), underscoring the clinical importance of testing other DNA repair genes (41). Findings showed that BRCA1 and 2 inactivation frequently led to higher HRD score in ovarian and breast cancers. This high HRD score has positive prognostic significance for platinum and PARPI therapy. In unclassified ovarian cancer patients who undertook germline BRCA1 and 2 test, 19% (44/235) were carriers of germline mutations, and somatic mutation test was done on 28 specimens, 42.9% (9/21) and 28.6% (2/7) were found to be BRCA1 and 2 positives, respectively (42). In another study by Pennington et al. (43), among 367 ovarian carcinomas tested for somatic mutation, 2.5% (19/367) and 1.63% (6/367) were positive for BRCA1 and 2, respectively. These carriers of somatic mutation have shown a positive impact on overall survival and platinum responsiveness as germline BRCA1 and 2 mutation carriers. Other factors such as germline and somatic mutations in HR genes (Table 1) and epigenetic alterations (promoter hypermethylation) are implicated in HRD (84).

Table 1 Prevalence of mutation, LOH, and promoter hypermethylation in ovarian cancer.
3 Homologous Recombination Deficiency in Breast Cancer
Breast cancer is the most common cancer among women and ranks first and fifth among causes of incidence and mortality, respectively, compared with all other cancers in both sexes in 2020 worldwide. According to the IARC (38), an estimated 2,261,419 (11.72% of all cancer types) new incidences and 684,996 (6.88% of all cancer types) deaths from breast cancer were recorded worldwide in 2020. Similar to ovarian cancer, DNA repair pathways are frequently anomalous in breast cancer, leading to the accumulation of DNA damage and genomic instability. The BRCA1 and BRCA2 genes are associated with hereditary breast and ovarian cancer (85). BRCA1 and BRCA2 play a significant role in DNA repair, especially as components of the FANC/BRCA DNA damage response pathway (Figure 1). This DNA repair pathway is a highly conserved system involved in the DSB response via HR and in the BER pathway for the repair of DNA single-strand breaks (SSBs) (9). The PARP enzyme plays a decisive role in this pathway and is critical for resolving the stalled replication forks. Inhibition of PARP during the base excision process requires BRCA-dependent HRR to resolve it. Targeting HR-related genes (Table 2) has potential for destabilizing tumor genomic integrity (13). Clinical trials confirmed that HRD is necessary for the sensitivity to DNA-damaging agents (e.g., cisplatin) and PARPIs (106).
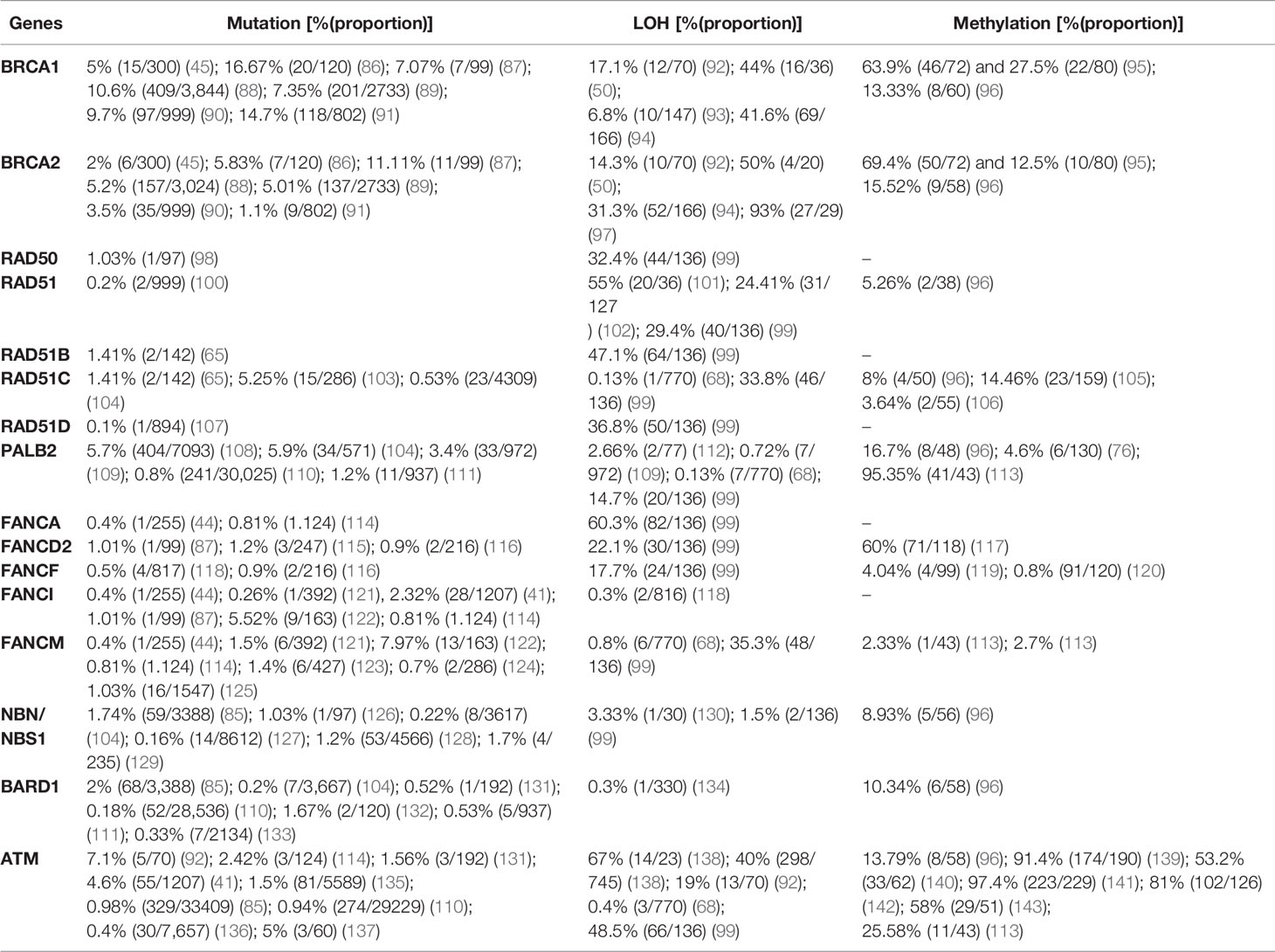
Table 2 Prevalence of mutation, LOH, and promoter hypermethylation in breast cancer.
Although BRCA1 and BRCA2 have been studied extensively, other genes are also involved in the occurrence of breast cancer. In 3,388 breast cancer patients who underwent genetic testing for 25 genes, nearly half of the pathogenic variants were in the BRCA1 (24%) and BRCA2 (24.5%) genes. The remaining 51.5% of pathogenic variants were detected in other genes tested including CHEK2 (11.7%), ATM (9.7%), PALB2 (9.3%), and Lynch syndrome genes (7%); other genes accounted for the remaining 13.8%. The same study showed that pathogenic mutations in BRCA1, PALB2, BARD1, BRIP1, and RAD51C are significantly more prevalent than those of other genes in triple negative breast cancer (TNBC) (85). In a study by Lang et al. (144) using NGS based sequencing, the prevalence of somatic BRCA1 and 2 mutation in sporadic breast cancer cases which carries germline-BRCA (gBRCA) mutations was 3.5% (15/416). Among these, 1.9% (8/416) and 1.7% (7/416) were BRCA1 and 2, respectively. In the same study, somatic BRCA mutation in gBRCA-negative cases was not detected; indicating somatic BRCA mutation in gBRCA-negative cases is rare. The prevalence of pathogenic mutations, LOH, and promoter hypermethylation in breast cancer is summarized in Table 2 based on recently published data.
4 Homologous Recombination Deficiency in Colorectal Cancer
CRC is the third most common cancer worldwide. In 2020 alone, the incidence of CRC was estimated at 1,931,590 (10% of all other cancers) worldwide, and it is the second most common cause of cancer-related deaths 935,173 (9.39% of all other cancers) in both sexes after lung cancer (38). Most CRC occurrences are sporadic and are not related to genetic predisposition or family history; however, 20–30% of patients with CRC have a positive family history, and 5% of these tumors arise from genetic predisposition (145). Mutation is a frequent event in CRC. According to TCGA (146), 16% of colorectal carcinomas are hyper-mutated; of these, 75% have high microsatellite instability, typically with hypermethylation and MLH1 silencing, and 25% have somatic mismatch repair gene and polymerase ϵ (POLE) mutations. The prevalence of mutations in APC, TP53, SMAD4, and PIK3CA was also reported, as well as KRAS mutations. Recurrent copy number alterations (e.g., ERBB2 amplification), chromosomal translocations, such as the fusion of NAV2 and the WNT pathway member TCF7L1, and biallelic inactivation of APC were among the common features leading to LOH in CRC. These and other findings confirmed the differences in both oncogenes and tumor suppressor genes involved in CRC compared with those in breast and ovarian cancers. However, a recent study from Japan revealed moderate genomic alterations in DNA repair genes including BRCA2, ATM, and NBN in CRC patients (147). High penetrance genes in HBOC differ from those in CRC; high penetrance genes in CRC are APC, MLH1, MSH2, MSH2/MUTYH, SMAD4, MAP, and APC/PMS2; moderate penetrance genes are MSH6 and PMS2; and low penetrance genes are APC p.I1307K and MUTYH mono-allelic (148).
The methylation profile of CRC differs from that of other cancers; it is characterized by global hypomethylation and promoter-specific DNA hypermethylation. At the whole genome level, CRC has 10–40% lower levels of absolute methylation than normal colonic tissue (149, 150). Global DNA hypomethylation, which is accompanied by genomic instability and tumor initiation, is primarily due to loss of methylation within repetitive elements such as long interspersed nuclear element-1 (LINE-1) and Arthrobacter luteus restriction endonucleases (Alu), and it is supposed to contribute to CRC initiation by enhancing genomic instability. On the other hand, genes methylated in CRC are established tumor suppressor genes, and 50% of these are also methylated in the normal colonic epithelium. As a result, methylation in CRC can have three phenotypes according to the CpG-island methylator phenotype (CIMP), i.e., CIMP-high, CIMP-low, and non-CIMP tumors (151). Similar to breast and ovarian cancers, the prevalence of mutation, LOH, and promoter hypermethylation in 35 HR-related genes is described based on recently published data (Table 3).
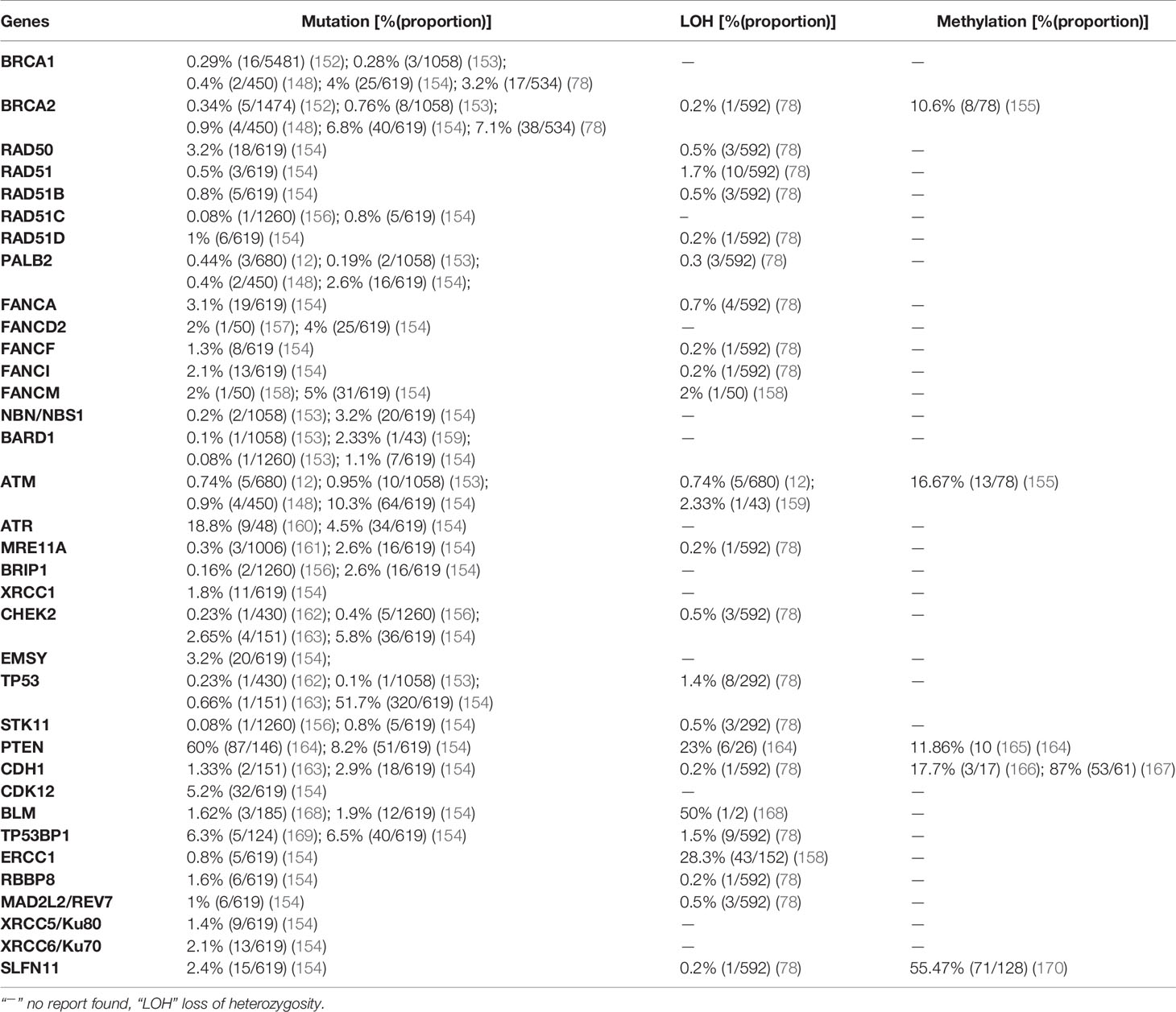
Table 3 Prevalence of mutation, LOH, and promoter hypermethylation in colorectal cancer.
5 Homologous Recombination Deficiency in Pancreatic Cancer
Pancreatic cancer (PC) is one of the most devastating types of cancer. PC has a low 5-year survival rate of 9%, and the development of new therapeutics is urgent. The global incidence and mortality of PC in 2020 were 2.6% (495,773 of all cancer types) and 4.68% (466,003 of all cancer types), respectively. PC is the 12th and 6th leading cause of cancer-related incidence and deaths in the world, respectively (38). Although most PC cases occur sporadically, familial (individual having two or more first-degree relatives diagnosed with PC) and hereditary syndrome PC account for 10% of cases. Hereditary cancer syndromes associated with increased risk of PC include Lynch syndrome, familial adenomatous polyposis, Peutz-Jeghers syndrome, atypical multiple mole melanoma, and familial and hereditary breast and ovarian cancer syndrome. Hereditary cancer syndrome accounts for 3% of PC cases, whereas familial PC accounts for 4–10% of cases (171, 172). Familial PC includes individuals with two or more affected first-degree relatives with PC excluding patients with hereditary syndrome. The risk of PC increases with the number of affected first-degree relatives. A study of 838 families including 5,179 individuals showed that the relative PC risk was 4.5 [95% confidence interval (CI): 0.5–16.3) among 1,253 cases with one affected first-degree relative, 6.4 (95% CI: 1.8–16.4) among 634 cases with two affected first-degree relatives, and 32 (95% CI: 10.4–74.7) in 106 cases with three or more affected first-degree relatives (173).
The specific hereditary pancreatic susceptibility genes are PRSS1, SPINK1, GGT1, CTRC, and CFTR, and mutations in these genes cause early onset PC. However, mutations in these genes are rare and account for a small proportion of PC cases, although the cumulative risk at 70 years age reaches 7–40% with early onset PC (174). Familial PC susceptibility genes include BRCA2, ATM, PALB2, CDKN2A, PRSS1, STK11, MLH1, and MSH2 (175). Recent findings showed the association of PC with genetic alterations in BRCA2, BRCA1, ATM, CHECK2, PALB2, FANCC, and CDKN2A genes (176–178). Mutations in BRCA2 are among the most common genetic mutations involved in familial pancreatic ductal carcinoma. BRCA mutation predisposes to PC, and PC more frequently affects BRCA2 mutation carriers than BRCA1 carriers. Among 204 BRCA mutation carriers with PC, 42.7% (87/204) had BRCA1 mutations and 57.3% (117/204) had BRCA2 mutations (179). The prevalence of mutation, LOH, and promoter hypermethylation of 35 HR genes is described according to recently published data in PC (Table 4).

Table 4 Prevalence of mutation, LOH, and promoter hypermethylation in pancreatic cancer.
6 Homologous Recombination Deficiency in Non-Small Cell Lung Carcinoma
Lung cancer is the leading cause of cancer-related morbidity and mortality in both genders worldwide. According to the IARC, the estimated number of incidences and deaths from lung cancer was 2,206,771 (11.44%) and 1,796,144 (18.04%) of all cancers worldwide in 2020, respectively (38). The majority of lung cancer cases are associated with smoking or the use of different tobacco products, although other factors such as asbestos, air pollution, radon gas exposure, and chronic infection also contribute to lung carcinogenesis (194). Inherited and acquired mechanisms of lung cancer susceptibility have been proposed, although they are rare. For instance, germline T790M mutation predisposes to a unique hereditary lung cancer syndrome that affects never-smokers and accounts for 31% of the estimated risks for lung cancer in never-smoker carriers (195, 196). Lung cancer is highly invasive, rapidly metastasizing, and broadly categorized into two histological groups, small-cell lung carcinomas (SCLCs) and NSCLCs, which grow and spread differently. NSCLCs account for 87% of cases and can be subdivided into three or four subtypes (adenocarcinoma, squamous cell carcinoma, large cell carcinoma, and undifferentiated NSCLCs), whereas SCLCs account for 12% of lung cancer cases (194).
Generally, lung adenocarcinoma is characterized by recurrent aberrations in multiple key pathways, including activation of RTK/RAS/RAF; activation of PI3K-mTOR; alterations of p53, cell cycle regulators, and the oxidative stress pathway; and mutation of various chromatin and RNA splicing factors. The research network of TCGA demonstrated the activation of oncogenes including KRAS (32%), EGFR (11%), MET (7%), BRAF (7%), MDM2 (8%), CDK4 (7%), PIK3C4 (4%), and CCND1 (4%) and the inactivation of tumor suppressor genes such as TP53 (46%), CDKN2A (43%), KEAP1 (19%), STK11 (17%), NF1 (11%), ATM (9%), RBM10 (9%), ARID1 (7%), ARID2 (7%), and RB1 (7%) in lung cancer (197). Analysis of DNA repair genes associated with squamous cell carcinoma showed a correlation between pathogenic mutations of DNA repair genes and tumor mutation burden. Among DNA repair genes BRCA1and BRCA2 showed the greatest mutation frequency and the tumor burden increased in correlation with the number of affected DNA repair genes (198). A study analyzing mutations in the BRCA 1 and 2 genes in different cancers showed that BRCA mutation is associated with increased incidence of non-breast and ovarian cancers in first- and second-degree relatives of high-risk breast cancer patients. Among 337 BRCA mutation carriers, the second highest BRCA mutation rate was recorded in lung cancer [8.8% (33/337)] after stomach cancer [13.8% (52/337)] (199). In this review, we displayed the prevalence of mutation, LOH, and promoter hypermethylation of 35 HR genes in NSCLC (Table 5).

Table 5 Prevalence of mutation, LOH, and promoter hypermethylation in NSCLC.
7 Homologous Recombination Deficiency in Prostate Cancer
Prostate cancer is the most prevalent cancer in men next to lung cancer, according to the IARC in 2020 (38), it ranked 2nd and 5th in its incidence and mortality among men, respectively. The estimated number of incidences and deaths from prostate cancer in 2020 were 1,414,259 (14.05%) and 375,304 (6.79%) worldwide, respectively. Prostate cancer is characterized by a high degree of heritability, and genetic components contribute significantly to disease incidence (210). A large cohort study conducted in the Nordic region that analyzed the different cancer heritability risks in monozygotic and dizygotic twins identified a risk of prostate cancer of 57% (95% CI: 51–63), which was higher than that of other cancer types such as ovarian cancer at 39% (95% CI: 23–55) and breast cancer at 31% (95% CI: 11–51) (210). Although the high rate of heritability of prostate cancer has been demonstrated in patients with a positive family history, candidate genes that contribute to prostate cancer heritability have not been identified except HOXB13 (211). Recurrent mutation of the HOXB13 gene at G84E was identified in many families. The HOXB13 G84E allele accounts for approximately 5% of hereditary prostate cancer (211). A study including 2,443 prostate cancer families of European descent detected at least one HOXB13 G84E mutation carrier, among 112 prostate cancer families (4.6%) (212). Moreover, a study conducted in unrelated subjects of European descent revealed, HOXB13 G84E mutation was detected in 1.4% (72/5083) and 0.07% (1/1401) of participants with- and without prostate cancer, respectively (P<0.05) (213). Another study comprising 9,012 men diagnosed with different cancers showed a rate of 0.54% (49/9012) of HOXB13 G84E mutation carriers, of whom 1.4% (19/1362) were positive for prostate cancer compared with 0.4%(23/5,898) of HOXB13 G84E mutation carriers without prostate cancer (p < 0.05) (214). Prostate cancer has a genetic origin in <5% of cases, and this risk becomes higher when high penetrance genes such as HOXB13 are involved (215). Recent gene linkage studies identified additional prostate cancer susceptibility genes such as HPC1, HPC2/ELAC2, MSR1, BRCA1, BRCA2, and BRIP1 (216).
An estimated 20% of patients with prostate cancer have a positive family history, which can be attributed not only to shared genes, but also to a shared pattern of exposure to environmental carcinogens and common lifestyle habits (216). Additional challenges in the management of prostate cancer include its genetic heterogeneity and a high rate of sporadic cases; many common genetic variants are associated with prostate cancer, explaining the familial clustering of the disease rather than hereditary causes (211). The importance of both germline and somatic alterations in DNA repair genes is suggested by the fact that carriers of mutations in these genes are at a high risk of developing aggressive or metastatic prostate cancer (211). Deleterious mutations of BRCA1 and BRCA2 are associated with increased risk of prostate cancer and experienced very aggressive course of the disease (215). However, studies focusing on families with only prostate cancer failed to identify a significant number of BRCA1 or BRCA2 mutations, indicating their minimal role in hereditary prostate cancer predisposition (211). A comprehensive genomic analysis of 1,013 prostate cancer patients revealed the indispensable role of alterations in DNA repair genes (78). Here, the prevalence of mutation, LOH, and promoter hypermethylation in 35 DNA repair genes in prostate cancer was described based on recently published data (Table 6).

Table 6 Prevalence of mutation, LOH, and promoter hypermethylation in prostate cancer.
8 Poly (ADP-Ribose) Polymerase Inhibitor Resistance Mechanism
8.1 The Role of PARP in DNA Repair
BER, HR, NHEJ, and micro homology-mediated end-joining (MMEJ) repair SSBs and DSBs. PARP1 is involved in all DNA repair mechanisms. SSBs are primarily repaired by BER (high fidelity DNA repair) using PARP1. DSBs are repaired by three mechanisms: HR, NHEJ, and MMEJ. HRR (high fidelity DNA repair mechanism) of DSBs is performed by recruiting BRCA1 and 2, RAD51, the MRN complex, and ATM, here PARP1 contributes to HR by recruiting MRE11 and NBS1 or by ribosylating BRCA. NHEJ (error-prone DNA repair mechanism) repair of DSBs involves the recruitment of Ku70, Ku80, and DNA-dependent protein kinase catalytic subunit (PKcs); PARP1 prevents the binding of Ku proteins to free DNA ends (first step of NHEJ) and thus inhibits NHEJ. MMEJ (error-prone DNA repair mechanism) repairs DSBs by recruiting Flap Structure-Specific Endonuclease-1 and NBN; PARP1 prevents binding of Ku proteins and directs DSBs to an alternate end-joining (MMEJ) repair pathway (225–227).
8.2 Mechanisms of PARP Inhibition
In the presence of SSBs, PARP1 binds to the SSB site and undergoes poly (ADP- ribosyl) ation, an important step for PARP1 activation; then, the poly(ADP-ribosyl)ated PARP1 recruits the DNA repair complexes BARD1-BRCA1 and MRN, which restore the integrity of DNA through a high fidelity DNA repair mechanism, resulting in cell survival. Inhibition of PARP by PARPIs drive to change the repair mechanism from SSBs to DSBs. PARPIs bind to PARP1 and inhibit its poly (ADP-ribosyl) ation as well as inhibiting BER. In addition, PARPIs prevent the release of PARP from the polymer form, thereby inhibiting the recruitment and binding of DNA damage repair proteins (PARP trapping), which further aggravates the inhibition of BER. Once BER is inhibited by PARPI, SSBs are converted into DSBs, forcing cells to opt for HRR. However, HR can only be used if cells are HR-proficient. HR defects (mutation, LOH, and hypermethylation) in HR-related genes such as BRCA1 and 2, RAD51 and its paralogs (RAD51B, RAD51C, and RAD51D), FA genes, PALB2, the MRN complex, BARD1, ATM, ATR, and BRIP1 cause cells to become HR-deficient and unable to repair DSBs. This causes the persistence of DNA DSBs, which leads to genomic instability and cell death. Cells need to activate alternative DNA repair mechanisms such as NHEJ and MMEJ, the only remaining repair mechanisms. Thus, cells are forced to use the two error-prone DSB repair mechanisms, which results in genomic instability and cell death (225–227). PARP1 and PARP2 are constitutively expressed enzymes that peak during the S-phase of the cell cycle and are activated by binding to DNA damage sites. PARPIs are particularly effective in the treatment of high grade ovarian and breast cancers with HR defects, which are characterized by frequent replication of tumor cells, and PARP expression and DNA damage recognition are highest during S-phase. However, the use of PARPIs is not limited to these two cancers. The concept of synthetic lethality or the BRCAness phenotype is wider, and most cancers with HR repair defects benefit from PARPI treatment including pancreatic and prostate cancers (227).
8.3 Mechanisms of Resistance to PARP Inhibitors
Despite the introduction of new drugs, the emergence of resistance to PARPIs remains a limiting factor (227, 228). Several mechanisms of PARPI resistance have been identified, including restoration of HRR proficiency, switching to alternate repair mechanisms such as NHEJ, replication fork stabilization, drug efflux, decreased PARP expression and binding, secondary mutations in HR-related genes and RAD51, regulation by microRNAs, phosphorylation of PARP by c-MET, loss of end resection regulation by 53BP1, epigenetic reversion of methylated promoters, and mutations in the shielding complex among others (Figure 2).
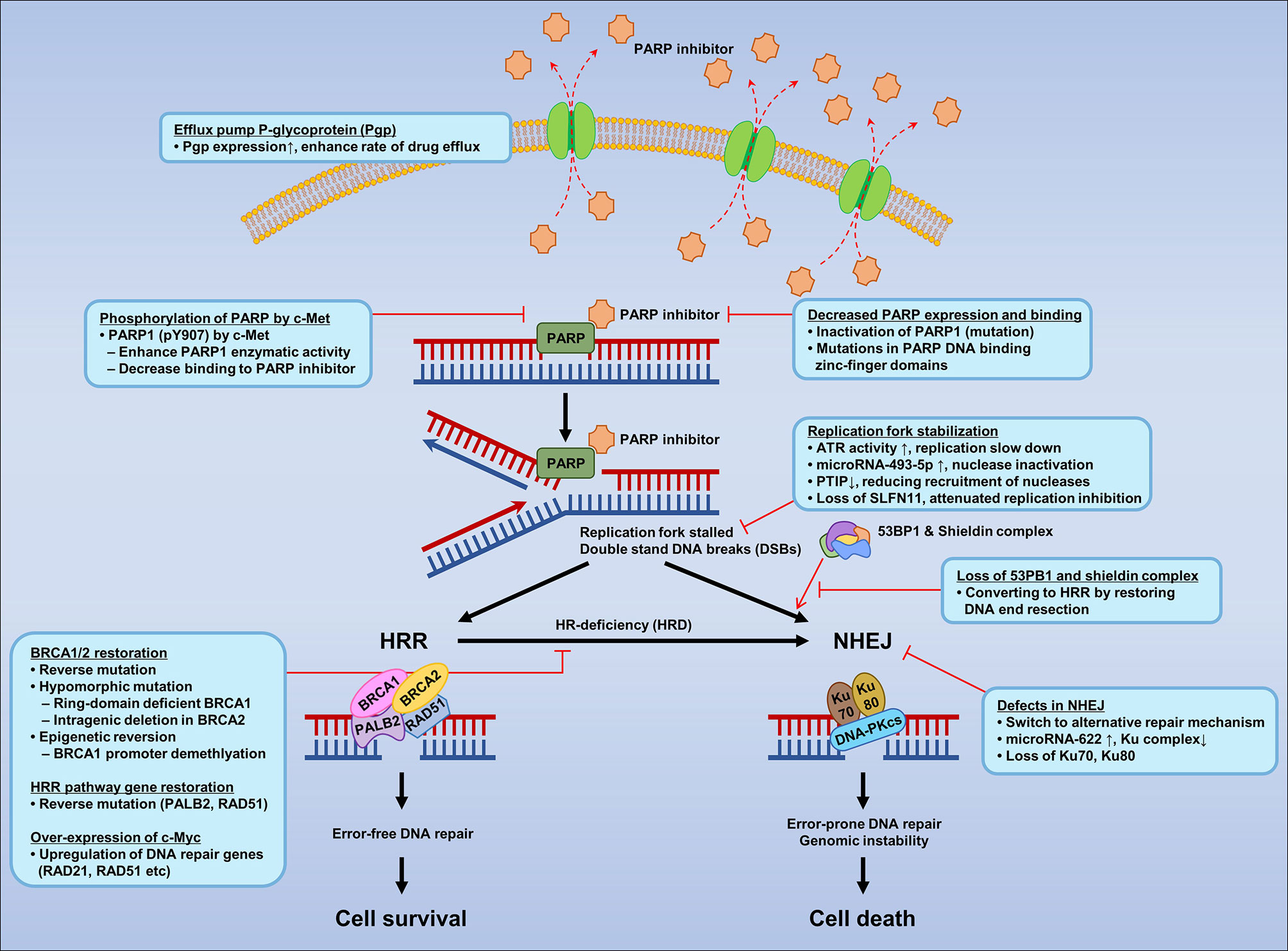
Figure 2 Mechanisms of resistance to PARP inhibitors according to mechanisms of PARPI in DNA repair.
8.3.1 Restoration of Homologous Recombination Proficiency
HR proficiency can be restored directly by reverse mutation of BRCA1 and BRCA2 mutants (229). Reverse mutation might be elicited by genomic instability due to BRCA loss. In addition, the presence of hypomorphic (partial loss of gene function) BRCA1 mutation leads to selection of cells with restored BRCA function, which confer resistance to PARPIs (230). In a recent study of high-grade ovarian carcinoma, BRCA reversion mutation was identified in 18% (2/11) and 13% (5/38) of pretreatment cell-free DNA extracts from platinum refractory and resistant cancers, respectively, compared with 2% (1/48) of platinum-sensitive cancers (p < 0.05) (231). Patients without BRCA reversion mutation detected in pretreatment circulation cell-free DNA extracts had significantly (p < 0.05) longer progression free survival than those with reversion mutation after treatment with rucaparib (9 versus 1.8 months), which decreased the clinical benefit from rucaparib (231). Although genetic reversion of BRCA1 and BRCA2 is one of the underlying mechanisms, it does not explain PARPI resistance in all cases. For example, loss of REV7 (MAD2L2) re-establishes CTIP-dependent end resection of DSBs in BRCA1-deficient cells, leading to HR restoration and PARPI resistance (232).
Secondary somatic mutations that restore BRCA1/2 in carcinomas from women with germline BRCA1/2 mutations predict the resistance to platinum and PARPIs. In cohorts of 64 primary and 46 recurrent ovarian carcinoma patients, secondary somatic mutation was detected in 3.1% (2/64) of primary carcinomas and in 28.3% (13/46) of secondary carcinomas (p < 0.05) due to secondary mutations in BRCA1/2. In the same study, 46.2% (12/26) of the platinum resistance recurrence cases had secondary mutations that restored BRCA1/2 function compared with 5.3% (1/19) of platinum-sensitive recurrence cases (p < 0.05) (165). Furthermore, the reversion mutations were not only detected in BRCA1/2, but also in other HRR pathway genes including RAD51C, RAD51D, and PALB2 in ovarian, prostate, and breast carcinomas as a mechanism of acquired resistance to platinum-based chemotherapies and PARPIs. Therefore, primary mutations of HR genes cause sensitivity to platinum and PARPI therapy, whereas secondary mutations cause resistance (231, 233). BRCA2 reversion mutations confer resistance to olaparib and talazoparib in prostate cancer patients. Analysis of circulating cell-free DNA provides information on reversion mutation heterogeneity that is not distinct from that of single solid tumor biopsy, as well as potential indications for monitoring the emergence of PARPI resistance (234).
8.3.2 RING Domain-Deficient BRCA1 and Intragenic Deletion in BRCA2
High expression levels of RING domain-deficient BRCA1 proteins promote cisplatin and PARPI resistance by reducing the DNA repair capacity of BRCA1 in breast cancer cell lines. The BRCA1 185delAG hypomorphic allele, a common inherited mutation located close to the protein translation start site, produces a shortened and nonfunctional peptide. In contrast to the full length BRCA1, the translation start site for the RING domain-deficient BRCA1 protein is located downstream of the frameshift mutation at the BRCA1-Met-297 codon and does not require interaction with BARD1 for stability unlike the full length BRCA1. Functionally, the RING domain-deficient BRCA1 supports RAD51 foci formation, which increases HRR and confers partial PARPI and cisplatin resistance (235). A recent case control association study and functional analysis of BRCA2 identified a hypomorphic missense variant (Y3035S) associated with a moderate risk of breast cancer. However, the role of this hypomorphic missense variant in the resistance to PARPIs and cisplatin remains to be investigated (236). Another study identified new BRCA2 isoforms that are expressed in resistant cell lines as a result of intragenic deletion of the c.6174delT mutation and restoration of the open reading frame. Reconstitution of BRCA2-deficient cells with these reverting BRCA2 alleles made resistant to PARPI and HR proficient (237). This finding indicates that resistance to PARPIs can arise by intragenic deletion mutations in BRCA2.
8.3.3 Epigenetic Reversion of Methylated Promoters
Epigenetic silencing of the promoter region of tumor suppressor genes is one of the mechanisms underlying HRD. For instance, BRCA1 promoter methylation is an important somatic driver in high grade serious ovarian carcinoma (238, 239). A patient with BRCA1 promoter methylation who was initially sensitive to PARPIs became resistant after loss of BRCA1 promoter methylation in the relapsed sample, and the gene was expressed at comparable levels to those in HR-proficient tumors (238). In the same study, analysis of the global methylation status of the primary and reverted samples revealed loss of BRCA1 promoter methylation. Another study showed BRCA1 promoter demethylation in therapy-resistant patients and patient-derived xenograft (PDX) tumors. Among 103 TNBC patients, 26 (25.24%) showed BRCA1 promoter methylation before treatment. Of these, 17 showed pathologic complete response and nine showed partial/no response; the three partial responders underwent post-treatment surgery. Post-treatment BRCA1 promoter methylation was 2.66-fold lower than that in pretreatment samples, and the mRNA expression of BRCA1 increased by 12–28-fold. Loss of BRCA1 promoter methylation was observed in 69.6% (16/23) of therapy-resistant PDX tumors with BRCA1 re-expression (240).
8.3.4 Switch to Alternate Repair Mechanisms
As discussed earlier, DNA DSBs in homologous recombination-deficient cancer cells can be repaired by alternative DNA repair mechanisms such as NHEJ and MMEJ. Thus, the shift from the canonical DNA repair mechanism to alternate repair mechanisms can affect the therapeutic efficacy of PARPIs (228). NHEJ functions throughout the cell cycle, and defects in NHEJ contribute to genomic instability and are associated with the development of chemo-resistance. NHEJ is crucial for determining the sensitivity to PARPIs, as confirmed recently (241). For example, ovarian cancer cells with a 40% deficiency in the NHEJ DSB repair pathway are resistant to PARP inhibition irrespective of HR status. Only NHEJ-competent and HRD cells are sensitive to the PARPI rucaparib, confirming the resistance observed in HRD tumors (241). Therefore defects in NHEJ, the lack of error-prone repair results in resistance to PARPIs. The role of NHEJ in PARPI resistance is related to the error proneness of NHEJ. The errors in repair cause lethal defects in DNA, and the absence of HR results in apoptosis, which is required for PARPI sensitivity (242).
8.3.5 Replication Fork Stabilization
HR-deficient cells are susceptible to replication fork degradation and are sensitive to PARPIs. However, cancer cells possess a mechanism to protect against replication fork degradation known as fork stabilization. Replication fork stabilization is a compensatory mechanism that protects the replication fork, which results in PARPI resistance in the absence of HR competency (105). Another mechanism to stabilize the replication fork is ATR activation in response to SSBs. In this mechanism, CHK1 is phosphorylated by ATR, and activated CHK1 phosphorylates WEE1 and inactivates the sCDC25A and CDC25C phosphatases. Activated WEE1 activates CDK1 and CDK2 to promote G1/S and G2/M cell cycle arrest (243, 244). Another mechanism of replication fork protection was identified by Meghani et al. (245). In this mechanism, miR-493-5p overexpression protects the replication fork from nuclease degradation, subsequently inducing PARPI and platinum resistance in BRCA2-mutated carcinomas. In addition, Pax2 transactivation domain-interacting protein (PTIP), which forms nuclear foci for DSBs, can destabilize the MRE11 nuclease installed replication forks. By contrast, loss of PTIP stabilizes nascent DNA strands by blocking degradation in BRCA1/2 deficient cells, a mechanism that rescues the stalled replication fork and causes PARPI and cisplatin resistance (246).
8.3.6 Decreased PARP Expression and Binding
Deletion of PARP1 using CRISPR/Cas9 gene editing tools in two ovarian cancer cells (one with BRCA1 mutation and one with BRCA1 promoter methylation) shows >90% reduction of PARP1 expression in BRCA1 mutant and promoter methylated cells as measured by immunofluorescence and western blot analysis. Therefore, loss of PARP1 by different mechanisms (e.g., mutation) results in resistance to PARPIs (247). Although BRCA1 mutant and promoter methylated ovarian cancer cells are synthetically lethal with PARPI, the loss of the target (PARP) results in PARPI resistance. Point mutations that interfere with the PARP1 DNA binding zinc-finger domains cause PARPI resistance and affect PARP1 trapping. PARP1 p.R591C mutation (c.1771C>T) was detected in ovarian cancer patient who showed resistant to olaparib (248). Other mutations that occur outside of the zinc-finger domain of PARP1 also reduce PARP trapping.
8.3.7 Efflux Pump P-Glycoprotein
The multidrug efflux pump P-glycoprotein (Pgp) contributes markedly to chemotherapy resistance by increasing rate of drug efflux. Long-term administration of PARPIs causes selective pressure-induced PARPI resistance mediated by the upregulation of a gene-encoding efflux pump. Pgp recognizes and transports a variety of chemical substrates with hydrophobic in nature (249). The expression of abcb1a and abcb1b, encoding murine Pgp were increased by 2 to 15 fold in 73.3% (11/15) of mice treated with AZD2281 (currently, Olaparib) (249). Upregulation of Pgp expression is considered a mechanism of resistance in BRCA1 and BRCA2 mutant cancers treated with PARPIs (250). Upregulation of multidrug resistance gene 1 increases the expression of Pgp and the rate of drug efflux, which decreases the therapeutic effect of PARPIs (251).
8.3.8 Loss of End Resection Regulation by 53BP1 and Mutation in the Shieldin Complex
The 53BP1 is an important regulator of the cellular response to DSBs, and it suppresses HR by stimulating NHEJ of the distal DNA end. Deletion of 53BP1 converts processing of damaged DNA ends into recombination of single-stranded DNA competent for HR. Loss of 53BP1 partially restores the error-free HR and reduces the sensitivity of BRCA1 mutant tumors to PARPIs (7, 252). 53BP1 is crucial for the control of DSB repair, as its presence promotes NHEJ and its loss promotes HR. Inhibiting 5′ end resection is necessary for HRD, and 53BP1 uses Rif1 to impair 5′ end resection. Rif1 inhibits end resection by recruiting CtIP, BLM, and Exo1, which restricts buildup of BRCA1/BARD1 complexes at sites of DNA damage. These mechanisms underlie the effect of 53BP1 on inducing chromosomal aberrations in BRCA1-deficient cells. Therefore, loss of 53BP1 favors HR and thus leads to PARPI resistance (253, 254).
The 53BP1 effector complex (shieldin) includes SHLD1, SHLD2, SHLD3, and REV7. Shieldin functions as a downstream effector of 53BP1-RIF1 in preventive DNA DSB repair, whereas deletion of the shieldin complex confers resistance to PARPIs in BRCA1-deficient cells. Binding of single-stranded DNA by SHLD2 is critical for shieldin function (255). BRCA1 mutant cancers show minimal resection of DSBs, which renders them deficient in homology-directed repair and sensitive to inhibitors of PARP1. In BRCA1 mutants, the resection of DSBs is inhibited by 53BP1, RIF1, and the shieldin complex, and loss/deletion of these factors reduces sensitivity to PARP1 inhibitors (256). Mutations in genes that encode shieldin subunits also cause resistance to PARPIs in BRCA1-deficient cells and tumors, resulting in restoration of HR (257). Silencing of shieldin components increases end resection, as extreme resection would make DNA ends unsuitable for repair by NHEJ; this may explain the defective NHEJ in shieldin-depleted cells. Downregulation of 53BP1, as well as that of REV7, confers resistance to PARPIs in BRCA1 mutant cells. An experiment in BRCA1-deficient and shieldin complex knockout cells confirmed the important role of the shieldin complex in controlling PARPI (olaparib) sensitivity. BRCA1-depleted cells are highly sensitive to olaparib; however, simultaneous depletion of shieldin components rescues cell viability similar to the effects of depletion of BRCA1 and 53BP1 (258). Therefore, loss of 53BP1, RIF1, and shieldin components is sufficient to bypass the HR function of BRCA1 and confer PARPI resistance.
8.3.9 Overexpression of MicroRNAs
Overexpression of miR-622 is implicated in the development of resistance to PARPIs and cisplatin by restoring HR and impairing NHEJ in BRCA1-deficient ovarian cancer. miR-622 suppresses NHEJ by downregulating the Ku complex, thus promoting HR-mediated repair of DNA DSBs in the S-phase of the cell cycle. In addition, overexpression of miRNA-622 in HGSOC patients is correlated with worse survival after platinum chemotherapy, associating miRNA-mediated resistance by rescuing HRR (259). Overexpression of miR-493-5p induces resistance to platinum and PARPIs in BRCA2 mutant patient-derived cells by targeting DNA repair pathways involved in genomic stability. MiR-493-5p induces resistance by downregulating R-loop processing genes, which increases the R-loop and decreases the single-strand repair pathway, and by downregulating nucleases, which protects the replication fork. HRR is not restored in relation to miR-493-5p mediated cisplatin and PARPI resistance. Overexpression of miR-493-5p is negatively correlated with disease-free survival, especially in BRCA2 mutant patients and specifically in platinum resistant or refractory disease (245).
8.3.10 Phosphorylation of PARP by C-Met
Phosphorylation of PARP1 at Tyr907 by the receptor tyrosine kinase c-Met causes PARPI resistance. The phosphorylation of PARP1 by c-Met (pY907) enhances PARP1 enzymatic activity and decreases binding to PARPI, resulting in resistance of cancer cells to PARPIs. PARPIs and c-Met inhibitors act synergistically in suppressing the growth of breast and lung cancer cells in vitro and in a xenograft model. Detection of pY907 is an indicator of PARPI resistance in combination with a poor response to PARPIs and high c-Met expression (260). PARPIs are commonly used for the treatment of ovarian and breast cancers. A recent study investigating the therapeutic efficacy of PARPIs against hepatocellular carcinoma (HCC) showed discouraging results. The mechanisms underlying the poor efficacy of PARPIs in HCC involve the formation of EGFR and MET heterodimer that interacts with and phosphorylates Y907 of PARP1 in the nucleus, which contributes to PARPI resistance. However, inhibition of both c-Met and EGFR sensitizes HCC cells to PARPIs, although both EGFR and c-Met are usually overexpressed in HCC (261). The use of c-Met and EGFR inhibitors in combination with PARPIs is a potential strategy for the treatment of HCC.
8.3.11 Overexpression of C-Myc
Overexpression of c-Myc increases cisplatin and PARPI resistance by reducing the production of the c-Myc inhibitor BIN1 (bridging integrator 1) which restores the intrinsic PARP-1 activity. Suppression of BIN1 releases the automodification domain of PARP1, which increases its intrinsic catalytic activity for DNA repair, thereby increasing resistance to PARPIs and cisplatin. Conversely, inhibition of c-Myc increases BIN1 abundance, which decreases PARP1 activity and reverses cisplatin and PARPI resistance (262). Myc amplification is accompanied by the upregulation of several DNA repair genes, including RAD21, RAD54L, and RAD51, in both breast and ovarian cancer. RAD51 is the third most significant DNA repair gene associated with Myc expression in TNBC tumor samples. c-Myc regulates PARPI resistance by upregulating RAD51 paralogs, which are important in HRR of damaged DNA. A recent study using TNBC cell lines confirmed that PARPI-resistant cells have increased RAD51 foci, whereas PARPI-sensitive cells show impaired RAD51 foci independent of BRCA mutation status. In the same study, pharmacological inhibition of c-Myc by dinaciclib reversed the resistance to PARPIs, confirming the induction of synthetic lethality and the role of c-Myc in drug resistance (263). RAD51C-deficient cancer cells are sensitive to the PARPI olaparib and undergo cell death by inducing G2/M cell cycle arrest and apoptosis. By contrast, silencing of RAD51C in resistant cancer cell lines increases the sensitivity to olaparib and decreases RAD51 foci (264).
8.3.12 Loss of SLFN11
High SLFN11 expression is associated with the response to DNA-damaging agents and the overall survival of patients with colorectal and ovarian cancer (265). Conversely, SLFN11 inactivation is a determinant of PARPI resistance. Cells that express SLFN11 are more sensitive to talazoparib and olaparib than cells with low SLFN11 expression. Genomic analysis confirmed the high correlation between treatment response and SLFN11, which is considered a biomarker of the response to PARPI treatment (266). PDXs and SCLC cell lines treated with cisplatin/PARPIs show down-regulation of SLFN11 associated with therapeutic resistance. This was confirmed by silencing SLFN11, which reduced the in vitro sensitivity to cisplatin and PARPIs as well as drug-induced DNA damage (267). SLFN11 was identified as a relevant predictive biomarker of sensitivity to PARPI monotherapy in SCLC, and loss of SLFN11 confers resistance to PARPIs. SCLC cell lines were treated with the PARPIs olaparib, rucaparib, and veliparib, and gene expression and the HRD genomic scar score were analyzed. SLFN11 was correlated with the response to olaparib, rucaparib, and veliparib treatment but not to the HRD genomic score scar. An in vivo PDX model and immunohistochemical staining confirmed that loss of SLNF11 confers resistance to PARPIs (268).
8.3.13 Loss of XRCC5 (Ku80) and XRCC6 (Ku70)
Loss of PARP activity leads to accumulation of SSBs, which are converted to DSBs by the cellular replication and/or transcription machinery. These DSBs can be repaired by HR in BRCA-proficient cells, whereas they accumulate in BRCA-deficient cells leading to cell death. NHEJ is initiated when free DNA ends are bound by XRCC5/Ku80 and XRCC6/Ku70 through the catalytic subunit of DNA-dependent protein kinases (DNA-PKcs). The DNA-PKc complex phosphorylates downstream targets and activates the DNA damage response, thereby initiating NHEJ (269). The NHEJ-mediated repair of DNA DSBs requires the formation of a Ku70/Ku80/DNA-PKc complex at the DSB sites. Simultaneous loss of HR and PARP1 activity results in deregulated/increased NHEJ activity, which increases the activation of DNA-PKcs leading to increased genomic instability (resulting from this error-prone pathway) (270). PARP1 plays a crucial role in suppressing NHEJ, which serves as a target of PARPI-induced lethality in HR-deficient cells. Conversely, inhibition or loss of multiple components of NHEJ such as XRCC5/Ku80, XRCC6/Ku70, and DNA-PK confer HR-deficient cells resistance to PARPIs by reducing NHEJ activity (242, 261). The activity of the error-prone NHEJ DSB repair pathway that causes genomic instability is required for PARPI sensitivity.
8.4 Strategies to Overcome PARP Inhibitor Resistance
Although PARPIs are likely to be beneficial for a large fraction of ovarian and breast cancer patients, the development of PARPI resistance brings challenges to their utility. As mentioned in this review, there are many mechanisms that can reverse HR deficiency to HR proficiency. Many strategies have been designed to reverse PARPI resistance (13, 105). For instance, replication fork stabilization is a compensatory mechanism for PARPI resistance. Cell cycle checkpoint (ATR, CHK1, WEE1) proteins that contribute to replication fork stabilization may be potential targets for combination therapy with PARPI by limiting the time for tumor cells to repair damaged DNA. The three proteins, ATR1, CHK1 and WEE1, play different roles in replication fork stabilization, indicating that different combination regimen may be effective for combating resistance. For example, ATR inhibitor AZD6738 sensitized BRCA2 mutant, BRCA2 reversion mutation, and BRCA1 wild-type ovarian cancer cells to olaparib more effectively than the CHK1 inhibitor MK8776 (271). WEE1 may have a critical role in cell cycle arrest compared to ATR and CHK1 because WEE1 is required to maintain ATR and CHK1 activity (243). WEE1 inhibitor AZD1775 had synergistic effect with olaparib in TNBC cells (272). Even inhibition of ATR, CHK1 and WEE1 proteins effectively abrogated G2 arrest, but not sufficient to overcome PARPI resistance caused by other mechanisms such as HR pathway.
Many strategies have been designed to selectively convert HR-proficient cells to HR-deficient status. The combined use of PARPIs with CDK1 inhibitors induces HRD in HR-proficient cells by inhibiting the phosphorylation of BRCA1 by CDK1. The reduction of CDK1 compromises the capacity of cells to repair DNA using HR because BRCA1-deficient cells do not efficiently form RAD51 foci (an essential component of HRR). In addition to checkpoint activation, CDK1-mediated phosphorylation of BRCA1 is required for HR (273). The PI3K/AKT/mTOR pathway is aberrantly dysregulated in certain cancers such as TNBC; therefore, direct inhibition of the PI3K/AKT/mTOR pathway in combination with PARPIs could be an effective strategy to overcome PARPI resistance. Under normal conditions, PI3K stabilizes and conserves DSB repair by interacting with the HR complex (274). mTOR inhibitors and PARPIs show strong synergism when used in combination, as indicated by the effect of mTOR inhibitors on suppressing HRR in BRCA-proficient TNBC cell lines (275). The combined use of PARPIs with histone deacetylase inhibitors (HDIs) can sensitize cancer cells to PARPIs because HDIs block the deacetylation of heatshock protein 90 (HSP90), which leads to the degradation of several proteins such as BRCA1, RAD52, ATR, and CHK1. Direct inactivation of HSP90 is another approach to the induction of BRCAness (276, 277).
Another mechanism to induce BRCAness is the combined use of PARP and EGFR inhibitors, which alters the DSB repair capacity and activates the intrinsic pathway of apoptosis. In vitro and in vivo findings show that inhibition of EGFR1 and 2 induces a transient DNA repair deficit and alters the interaction of EGFR with BRCA1 by increasing cytosolic BRCA1 and EGFR, pulling them away from their nuclear DNA repair substrates (277, 278). Another study showed that ATM depletion sensitizes breast cancer cell lines to the PARPI olaparib (279). PARPIs in combination with androgen receptor inhibitors promote DNA damage-induced cell death, which inhibits prostate cancer cell proliferation and the growth of tumor xenografts in mice, suggesting a potentially effective treatment combination for androgen-expressing breast cancers (280).
Recently, Johnson et al. (281) reported that BRCA-mutant TNBC cells with acquired PARPI resistance are resensitized to PARP inhibition by dinaciclib, a potent CDK12 inhibitor that disrupts HR. In BRCA-mutated cancer, de novo resistance to PARPIs is caused by residual HR. In addition, dinaciclib compromises HR repair and sensitizes BRCA wild-type TBNC cells to PARP inhibition. This study also showed that dinaciclib amplifies the response to PARPIs in HR-deficient cancers. MYC inhibitors induce PARPI sensitivity. The downstream oncogenic role of MYC relies on its heterodimerization with the basic loop helix protein MAX, which is essential in causing the transcriptional initiation of targets. For instance, the small molecule 10058-F4 inhibits MYC-MAX binding (282), resulting in the supression of RAD51 in MDA-MB-231 and SUM149 cell lines (263). CDK12 inhibitor dinaciclib which downregulates MYC expression resensitizes PARPI-resistant cells to PARP inhibition when used in combination with niraparib; the synergistic effect was observed in BRCA wild-type and mutant TNBC cell lines in associtaion with the down-regulation of the HR gene RAD51 (263). This finding indicates that targeting the c-Myc oncogene could be an effective strategy to induce synthetic lethality and reverse PARPI resistance in MYC-driven cancers.
Recent preclinical and clinical studies demonstrated that the efficacy of PARPI could be enhanced in combination with immune checkpoint inhibitors (ICIs) via a synergistic effect. In cancers with defective DNA repair, such as HRD, accumulated DNA damage by PARPI leaded to high tumor mutational burden resulting in neoantigen formation and an increased anti-cancer immune response (283, 284). In addition, these DNA damages might increase the exposure of double-strand DNA (dsDNA) in the cytoplasm and activate the stimulator of interferon genes (STING) pathway which upregulates cytokines like type I interferon, thereby promoting immune response and recruiting tumor-infiltrating lymphocytes (TILs), especially CD8+ T cells (285, 286). PARPI also increases PD-L1 expression, a biomarker for ICI response, through the STING pathway (287), the ATM-ATR-CHK1 pathway (288), and inactivation of glycogen synthase kinase 3-beta (GSK3β) (289). Upregulation of PD-L1 may be a resistance mechanism of PARPI. For these reasons, subsequent immune checkpoint blockade could sensitize PARPI-treated tumor regression. Clinical trials investigating combined regimen of PARPI and ICIs such as anti-PD1 (BGB-A317, nivolumab, pembrolizumab, TSR-042), anti-PD-L1 (atezolizumab, avelumab, durvalumab), and anti-CTLA4 (ipilimumab, tremelimumab) demonstrated promising results in patient outcomes in solid tumors (290, 291).
9 Conclusion
HRR is the guardian of the genome because of its role in repairing DSBs with high fidelity. Defects in HR due to mutation, LOH, and promoter hypermethylation of certain HR genes result in HRD, which confers sensitivity to DNA-damaging agents and PARPIs. This review demonstrated that HRD is higher in ovarian and breast cancers than in other cancer types such as CRC, PC, NSCLC, and prostate cancer. HRD is not limited to BRCA1 and 2, and comprises many DNA repair genes. The fundamental vulnerability of HRD has led to the design of a wide range of HRD-directed therapies. DNA repair targeted therapies exploit DNA repair defects because HR-deficient tumors are intrinsically sensitive to PARPIs. This highlights the concept of synthetic lethality associated with the concurrent inactivation of two or more HRR genes. The use of PARPIs in non-BRCA mutation carriers can be expanded to sporadic cancers that display DNA repair defects. PARPIs are essential for the treatment of ovarian and breast cancers. Recently, FDA approved olaparib for the treatment of prostate and pancreatic cancers characterized by HRD. However, the benefits of PARPIs are limited by the development of resistance, especially when used as monotherapy. Many mechanisms of resistance to PARPI have been identified in HR-deficient cancers, which are challenges to overcome. Numerous preclinical and clinical studies revealed that combination therapy of PARPI with targeted chemotherapy or ICIs improved the efficacy by overcoming PARPI resistance. Understanding the mechanisms of PARPIs resistance will be useful for designing strategies to overcome PARPI resistance as summarized in this review. Based on accumulated research, more potential PARPIs and more effective combined regimens targeting HR-deficient cancers would be developed in the future.
Author Contributions
NM, collected data, sketched the initial version, and reviewed the final manuscript. HY, reviewed and drafted the final version of the manuscript. YS, conceived the idea, guided the write up, and reviewed the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (grant number: HI17C2196).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Authors are extremely thankful to the financial support from Department of Molecular Medicine and Biopharmaceutical Sciences, Graduate School of Convergence Science and Technology, Seoul National University, Seoul, Republic of Korea, and the Department of Molecular Medicine and Biopharmaceutical Sciences, Graduate School of Convergence Science and Technology, and College of Medicine or College of Pharmacy, Seoul National University, Seoul, Republic of Korea. Authors are grateful for the financial support from the Research Institute of Pharmaceutical Sciences, College of Pharmacy, Seoul National University. Authors also extend thanks to Tae Won Yun for her contributions in preparing the illustrations.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.880643/full#supplementary-material
References
1. Liu JF, Konstantinopoulos PA. Chapter 6 - Homologous Recombination and BRCA Genes in Ovarian Cancer: Clinical Perspective of Novel Therapeutics. In: Birrer MJ, Ceppi L, Editors. Translational Advances in Gynecologic Cancers. Boston: Academic Press (2017). p. 111–28. doi: 10.1016/B978-0-12-803741-6.00006-9
2. Dietlein F, Thelen L, Reinhardt HC. Cancer-Specific Defects in DNA Repair Pathways as Targets for Personalized Therapeutic Approaches. Trends Genet TIG (2014) 30(8):326–39. doi: 10.1016/j.tig.2014.06.003
3. Haynes B, Saadat N, Myung B, Shekhar MP. Crosstalk Between Translesion Synthesis, Fanconi Anemia Network, and Homologous Recombination Repair Pathways in Interstrand DNA Crosslink Repair and Development of Chemoresistance. Mutat Res Rev Mutat Res (2015) 763:258–66. doi: 10.1016/j.mrrev.2014.11.005
4. Stover EH, Konstantinopoulos PA, Matulonis UA, Swisher EM. Biomarkers of Response and Resistance to DNA Repair Targeted Therapies. Clin Cancer Res (2016) 22(23):5651–60. doi: 10.1158/1078-0432.CCR-16-0247
5. Velic D, Couturier AM, Ferreira MT, Rodrigue A, Poirier GG, Fleury F, et al. DNA Damage Signalling and Repair Inhibitors: The Long-Sought-After Achilles' Heel of Cancer. Biomolecules (2015) 5(4):3204–59. doi: 10.3390/biom5043204
6. Stecklein SR, Kumaraswamy E, Behbod F, Wang W, Chaguturu V, Harlan-Williams LM, et al. BRCA1 and HSP90 Cooperate in Homologous and non-Homologous DNA Double-Strand-Break Repair and G2/M Checkpoint Activation. (2012) 109(34):13650–5. doi: 10.1073/pnas.1203326109
7. Gadducci A, Guerrieri ME. PARP Inhibitors Alone and in Combination With Other Biological Agents in Homologous Recombination Deficient Epithelial Ovarian Cancer: From the Basic Research to the Clinic. Crit Rev Oncol Hematol (2017) 114:153–65. doi: 10.1016/j.critrevonc.2017.04.006
8. Watkins JA, Irshad S, Grigoriadis A, Tutt ANJ. Genomic Scars as Biomarkers of Homologous Recombination Deficiency and Drug Response in Breast and Ovarian Cancers. (2014) 16(3):211. doi: 10.1186/bcr3670
9. den Brok WD, Schrader KA, Sun S, Tinker AV, Zhao EY, Aparicio S, et al. Homologous Recombination Deficiency in Breast Cancer: A Clinical Review. JCO Precis Oncol (2017) 1:1–13. doi: 10.1200/PO.16.00031
10. Hoppe MM, Sundar R, Tan DSP, Jeyasekharan AD. Biomarkers for Homologous Recombination Deficiency in Cancer. J Natl Cancer Inst (2018) 110(7):704–13. doi: 10.1093/jnci/djy085
11. TCGA. Integrated Genomic Analyses of Ovarian Carcinoma. Nature (2011) 474(7353):609–15. doi: 10.1038/nature10166
12. AlDubayan SH, Giannakis M, Moore ND, Han GC, Reardon B, Hamada T, et al. Inherited DNA-Repair Defects in Colorectal Cancer. Am J Hum Genet (2018) 102(3):401–14. doi: 10.1016/j.ajhg.2018.01.018
13. Papadimitriou M, Mountzios G, Papadimitriou CA. The Role of PARP Inhibition in Triple-Negative Breast Cancer: Unraveling the Wide Spectrum of Synthetic Lethality. Cancer Treat Rev (2018) 67:34–44. doi: 10.1016/j.ctrv.2018.04.010
14. Rose M, Burgess JT, O'Byrne K, Richard DJ, Bolderson E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front Cell Dev Biol (2020) 8:564601. doi: 10.3389/fcell.2020.564601
15. Westphalen B, Fine AD, Andre F, Ganesan S, Heinemann V, Rouleau E, et al. Pan-Cancer Analysis of Homologous Recombination Repair-Associated Gene Alterations and Genome-Wide Loss of Heterozygosity Score. Clin Cancer Res (2021). doi: 10.1158/1078-0432.CCR-21-2096
16. Dorling L, Carvalho S, Allen J, González-Neira A, Luccarini C, Wahlström C, et al. Breast Cancer Risk Genes - Association Analysis in More Than 113,000 Women. N Engl J Med (2021) 384(5):428–39. doi: 10.1056/NEJMoa1913948
17. Riaz N, Blecua P, Lim RS, Shen R, Higginson DS, Weinhold N, et al. Pan-Cancer Analysis of Bi-Allelic Alterations in Homologous Recombination DNA Repair Genes. Nat Commun (2017) 8(1):857. doi: 10.1038/s41467-017-00921-w
18. Chi J, Chung SY, Parakrama R, Fayyaz F, Jose J, Saif MW. The Role of PARP Inhibitors in BRCA Mutated Pancreatic Cancer. Therap Adv Gastroenterol (2021) 14:17562848211014818. doi: 10.1177/17562848211014818
19. Shroff RT, Hendifar A, McWilliams RR, Geva R, Epelbaum R, Rolfe L, et al. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis Oncol (2018) 2):1–15. doi: 10.1200/PO.17.00316
20. Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a Predictor of BRCA1 and BRCA2 Deficiency Based on Mutational Signatures. Nat Med (2017) 23(4):517–25. doi: 10.1038/nm.4292
21. BRACAnalysis CDx® Technical Information. FDA. Available at: https://www.accessdata.fda.gov/cdrh_docs/pdf14/P140020S020C.pdf.
22. Summary of Safety and Effectiveness Data (Ssed). FDA. Available at: https://www.accessdata.fda.gov/cdrh_docs/pdf19/P190014B.pdf.
23. FoundationOne®CDx Technical Information. FDA. Available at: https://info.foundationmedicine.com/hubfs/FMI%20Labels/FoundationOne_CDx_Label_Technical_Info.pdf.
24. FoundationOne® Liquid CDx Technical Information. FDA. Available at: https://info.foundationmedicine.com/hubfs/FMI%20Labels/FoundationOne_Liquid_CDx_Label_Technical_Info.pdf.
25. Chopra N, Tovey H, Pearson A, Cutts R, Toms C, Proszek P, et al. Homologous Recombination DNA Repair Deficiency and PARP Inhibition Activity in Primary Triple Negative Breast Cancer. Nat Commun (2020) 11(1):2662. doi: 10.1038/s41467-020-16142-7
26. Mutter RW, Riaz N, Ng CK, Delsite R, Piscuoglio S, Edelweiss M, et al. Bi-Allelic Alterations in DNA Repair Genes Underpin Homologous Recombination DNA Repair Defects in Breast Cancer. J pathology (2017) 242(2):165–77. doi: 10.1002/path.4890
27. Li A, Geyer FC, Blecua P, Lee JY, Selenica P, Brown DN, et al. Homologous Recombination DNA Repair Defects in PALB2-Associated Breast Cancers. NPJ Breast Cancer (2019) 5:23. doi: 10.1038/s41523-019-0115-9
28. Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, et al. Subtle Variations in Pten Dose Determine Cancer Susceptibility. Nat Genet (2010) 42(5):454–8. doi: 10.1038/ng.556
29. Venkatachalam S, Shi YP, Jones SN, Vogel H, Bradley A, Pinkel D, et al. Retention of Wild-Type P53 in Tumors From P53 Heterozygous Mice: Reduction of P53 Dosage can Promote Cancer Formation. EMBO J (1998) 17(16):4657–67. doi: 10.1093/emboj/17.16.4657
30. Moschetta M, George A, Kaye SB, Banerjee S. BRCA Somatic Mutations and Epigenetic BRCA Modifications in Serous Ovarian Cancer. Ann Oncol (2016) 27(8):1449–55. doi: 10.1093/annonc/mdw142
31. Gilardini Montani MS, Prodosmo A, Stagni V, Merli D, Monteonofrio L, Gatti V, et al. ATM-Depletion in Breast Cancer Cells Confers Sensitivity to PARP Inhibition. (2013) 32(1):95. doi: 10.1186/1756-9966-32-95
32. Petrucelli N, Daly MB, Feldman GL. Hereditary Breast and Ovarian Cancer Due to Mutations in BRCA1 and BRCA2. Genet In Med (2010) 12:245. doi: 10.1097/GIM.0b013e3181d38f2f
33. da Cunha Colombo Bonadio RR, Fogace RN, Miranda VC, Diz M. Homologous Recombination Deficiency in Ovarian Cancer: A Review of its Epidemiology and Management. Clinics (2018) 73(suppl 1):e450s. doi: 10.6061/clinics/2018/e450s
34. Nalepa G, Clapp DW. Fanconi Anaemia and Cancer: An Intricate Relationship. Nat Rev Cancer (2018) 18(3):168–85. doi: 10.1038/nrc.2017.116
35. Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V. Update of the Human and Mouse Fanconi Anemia Genes. Hum Genomics (2015) 9:32. doi: 10.1186/s40246-015-0054-y
36. Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, et al. Preventing Nonhomologous End Joining Suppresses DNA Repair Defects of Fanconi Anemia. Mol Cell (2010) 39(1):25–35. doi: 10.1016/j.molcel.2010.06.026
37. Yan Z, Delannoy M, Ling C, Daee D, Osman F, Muniandy PA, et al. A Histone-Fold Complex and FANCM Form a Conserved DNA-Remodeling Complex to Maintain Genome Stability. Mol Cell (2010) 37(6):865–78. doi: 10.1016/j.molcel.2010.01.039
38. Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The Long Tail of Oncogenic Drivers in Prostate Cancer. Nat Genet (2018) 50(5):645–51. doi: 10.1038/s41588-018-0078-z
39. Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D'Andrea AD. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov (2015) 5(11):1137–54. doi: 10.1158/2159-8290.CD-15-0714
40. Chen CC, Feng W, Lim PX, Kass EM, Jasin M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu Rev Cancer Biol (2018) 2:313–36. doi: 10.1146/annurev-cancerbio-030617-050502
41. Girard E, Eon-Marchais S, Olaso R, Renault AL, Damiola F, Dondon MG, et al. Familial Breast Cancer and DNA Repair Genes: Insights Into Known and Novel Susceptibility Genes From the GENESIS Study, and Implications for Multigene Panel Testing. Int J Cancer (2018). doi: 10.1002/ijc.31921
42. Hennessy BTJ, Timms KM, Carey MS, Gutin A, Meyer LA, Flake DD, et al. Somatic Mutations in BRCA1 and BRCA2 Could Expand the Number of Patients That Benefit From Poly (ADP Ribose) Polymerase Inhibitors in Ovarian Cancer. J Clin Oncol (2010) 28(22):3570–6. doi: 10.1200/JCO.2009.27.2997
43. Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin Cancer Res (2014) 20(3):764–75. doi: 10.1158/1078-0432.CCR-13-2287
44. Tedaldi G, Tebaldi M, Zampiga V, Danesi R, Arcangeli V, Ravegnani M, et al. Multiple-Gene Panel Analysis in a Case Series of 255 Women With Hereditary Breast and Ovarian Cancer. Oncotarget (2017) 8(29):47064–75. doi: 10.18632/oncotarget.16791
45. Quezada Urban R, Díaz Velásquez CE, Gitler R, Rojo Castillo MP, Sirota Toporek M, Figueroa Morales A, et al. Comprehensive Analysis of Germline Variants in Mexican Patients With Hereditary Breast and Ovarian Cancer Susceptibility. Cancers (2018) 10(10). doi: 10.3390/cancers10100361
46. Koczkowska M, Krawczynska N, Stukan M, Kuzniacka A, Brozek I, Sniadecki M, et al. Spectrum and Prevalence of Pathogenic Variants in Ovarian Cancer Susceptibility Genes in a Group of 333 Patients. Cancers (2018) 10(11). doi: 10.3390/cancers10110442
47. Harter P, Hauke J, Heitz F, Reuss A, Kommoss S, Marme F, et al. Prevalence of Deleterious Germline Variants in Risk Genes Including BRCA1/2 in Consecutive Ovarian Cancer Patients (AGO-TR-1). PLos One (2017) 12(10):e0186043. doi: 10.1371/journal.pone.0186043
48. Jean S, Li J, Katsaros D, Wubbenhorst B, Maxwell KN, Fishbein L, et al. Paclitaxel is Necessary for Improved Survival in Epithelial Ovarian Cancers With Homologous Recombination Gene Mutations. Oncotarget (2016) 7(30):48577–85. doi: 10.18632/oncotarget.9373
49. Yang SYC, Lheureux S, Karakasis K, Burnier JV, Bruce JP, Clouthier DL, et al. Landscape of Genomic Alterations in High-Grade Serous Ovarian Cancer From Exceptional Long- and Short-Term Survivors. Genome Med (2018) 10(1):81. doi: 10.1186/s13073-018-0590-x
50. Van Heetvelde M, Van Bockstal M, Poppe B, Lambein K, Rosseel T, Atanesyan L, et al. Accurate Detection and Quantification of Epigenetic and Genetic Second Hits in BRCA1 and BRCA2-Associated Hereditary Breast and Ovarian Cancer Reveals Multiple Co-Acting Second Hits. Cancer Lett (2018) 425:125–33. doi: 10.1016/j.canlet.2018.03.026
51. Brozek I, Ochman K, Debniak J, Morzuch L, Ratajska M, Stepnowska M, et al. Loss of Heterozygosity at BRCA1/2 Loci in Hereditary and Sporadic Ovarian Cancers. J Appl Genet (2009) 50(4):379–84. doi: 10.1007/BF03195697
52. Weren RDA, Mensenkamp AR, Simons M, Eijkelenboom A, Sie AS, Ouchene H, et al. Novel BRCA1 and BRCA2 Tumor Test as Basis for Treatment Decisions and Referral for Genetic Counselling of Patients With Ovarian Carcinomas. (2017) 38(2):226–35. doi: 10.1002/humu.23137
53. Lambrechts S, Smeets D, Moisse M, Braicu EI, Vanderstichele A, Zhao H, et al. Genetic Heterogeneity After First-Line Chemotherapy in High-Grade Serous Ovarian Cancer. Eur J Cancer (2016) 53:51–64. doi: 10.1016/j.ejca.2015.11.001
54. Zhu X, Zhao L, Lang J. The BRCA1 Methylation and PD-L1 Expression in Sporadic Ovarian Cancer. Int J Gynecol Cancer (2018) 28(8):1514–9. doi: 10.1097/IGC.0000000000001334
55. Losi L, Fonda S, Saponaro S, Chelbi ST, Lancellotti C, Gozzi G, et al. Distinct DNA Methylation Profiles in Ovarian Tumors: Opportunities for Novel Biomarkers. Int J Mol Sci (2018) 19(6). doi: 10.3390/ijms19061559
56. Lonning PE, Berge EO, Bjornslett M, Minsaas L, Chrisanthar R, Hoberg-Vetti H, et al. White Blood Cell BRCA1 Promoter Methylation Status and Ovarian Cancer Risk. Ann Intern Med (2018) 168(5):326–34. doi: 10.7326/M17-0101
57. Ruscito I, Dimitrova D, Vasconcelos I, Gellhaus K, Schwachula T, Bellati F, et al. BRCA1 Gene Promoter Methylation Status in High-Grade Serous Ovarian Cancer Patients–a Study of the Tumour Bank Ovarian Cancer (TOC) and Ovarian Cancer Diagnosis Consortium (OVCAD). Eur J Cancer (2014) 50(12):2090–8. doi: 10.1016/j.ejca.2014.05.001
58. Ignatov T, Eggemann H, Costa SD, Roessner A, Kalinski T, Ignatov A. BRCA1 Promoter Methylation is a Marker of Better Response to Platinum-Taxane-Based Therapy in Sporadic Epithelial Ovarian Cancer. J Cancer Res Clin Oncol (2014) 140(9):1457–63. doi: 10.1007/s00432-014-1704-5
59. Cunningham JM, Cicek MS, Larson NB, Davila J, Wang C, Larson MC, et al. Clinical Characteristics of Ovarian Cancer Classified by BRCA1, BRCA2, and RAD51C Status. Sci Rep (2014) 4:4026. doi: 10.1038/srep04026
60. Prieske K, Prieske S, Joosse SA, Trillsch F, Grimm D, Burandt E, et al. Loss of BRCA1 Promotor Hypermethylation in Recurrent High-Grade Ovarian Cancer. Oncotarget (2017) 8(47):83063–74. doi: 10.18632/oncotarget.20945
61. Ramalho EA, Silva-Filho JL, Cartaxo MF, Cavalcanti CB, Rego MJ, Oliveira MB, et al. Assessment of Changes in the BRCA2 and P53 Genes in Breast Invasive Ductal Carcinoma in Northeast Brazil. Biol Res (2014) 47:3. doi: 10.1186/0717-6287-47-3
62. Hjortkjaer M, Malik Aagaard Jorgensen M, Waldstrom M, Ornskov D, Sogaard-Andersen E, Jakobsen A, et al. The Clinical Importance of BRCAness in a Population-Based Cohort of Danish Epithelial Ovarian Cancer. Int J Gynecol Cancer (2019) 29(1):166–73. doi: 10.1136/ijgc-2018-000017
63. Coppa A, Nicolussi A, D'Inzeo S, Capalbo C, Belardinilli F, Colicchia V, et al. Optimizing the Identification of Risk-Relevant Mutations by Multigene Panel Testing in Selected Hereditary Breast/Ovarian Cancer Families. Cancer Med (2018) 7(1):46–55. doi: 10.1002/cam4.1251
64. Sung PL, Wen KC, Chen YJ, Chao TC, Tsai YF, Tseng LM, et al. The Frequency of Cancer Predisposition Gene Mutations in Hereditary Breast and Ovarian Cancer Patients in Taiwan: From BRCA1/2 to Multi-Gene Panels. PLos One (2017) 12(9):e0185615. doi: 10.1371/journal.pone.0185615
65. Golmard L, Caux-Moncoutier V, Davy G, Al Ageeli E, Poirot B, Tirapo C, et al. Germline Mutation in the RAD51B Gene Confers Predisposition to Breast Cancer. BMC Cancer (2013) 13:484. doi: 10.1186/1471-2407-13-484
66. Song H, Dicks E, Ramus SJ, Tyrer JP, Intermaggio MP, Hayward J, et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J Clin Oncol (2015) 33(26):2901–7. doi: 10.1200/JCO.2015.61.2408
67. Sanchez-Bermudez AI, Sarabia-Meseguer MD, Garcia-Aliaga A, Marin-Vera M, Macias-Cerrolaza JA, Henarejos PS, et al. Mutational Analysis of RAD51C and RAD51D Genes in Hereditary Breast and Ovarian Cancer Families From Murcia (Southeastern Spain). Eur J Med Genet (2018) 61(6):355–61. doi: 10.1016/j.ejmg.2018.01.015
68. Lu C, Xie M, Wendl MC, Wang J, McLellan MD, Leiserson MD, et al. Patterns and Functional Implications of Rare Germline Variants Across 12 Cancer Types. Nat Commun (2015) 6:10086. doi: 10.1038/ncomms10086
69. Bernards SS, Pennington KP, Harrell MI, Agnew KJ, Garcia RL, Norquist BM, et al. Clinical Characteristics and Outcomes of Patients With BRCA1 or RAD51C Methylated Versus Mutated Ovarian Carcinoma. Gynecol Oncol (2018) 148(2):281–5. doi: 10.1016/j.ygyno.2017.12.004
70. Abkevich V, Timms KM, Hennessy BT, Potter J, Carey MS, Meyer LA, et al. Patterns of Genomic Loss of Heterozygosity Predict Homologous Recombination Repair Defects in Epithelial Ovarian Cancer. Br J Cancer (2012) 107(10):1776–82. doi: 10.1038/bjc.2012.451
71. Singh J, Thota N, Singh S, Padhi S, Mohan P, Deshwal S, et al. Screening of Over 1000 Indian Patients With Breast and/or Ovarian Cancer With a Multi-Gene Panel: Prevalence of BRCA1/2 and Non-BRCA Mutations. Breast Cancer Res Treat (2018) 170(1):189–96. doi: 10.1007/s10549-018-4726-x
72. Barrington DA, Champion ML, Boitano TKL, Walters-Haygood CL, Farmer MB, Alvarez RD, et al. Characteristics of African American Women at High-Risk for Ovarian Cancer in the Southeast: Results From a Gynecologic Cancer Risk Assessment Clinic. Gynecol Oncol (2018) 149(2):337–40. doi: 10.1016/j.ygyno.2018.02.014
73. Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol (2016) 2(4):482–90. doi: 10.1001/jamaoncol.2015.5495
74. Nakagomi H, Sakamoto I, Hirotsu Y, Amemiya K, Mochiduki H, Omata M. Analysis of PALB2 Mutations in 155 Japanese Patients With Breast and/or Ovarian Cancer. Int J Clin Oncol (2016) 21(2):270–5. doi: 10.1007/s10147-015-0906-4
75. Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MD, Wendl MC, et al. Integrated Analysis of Germline and Somatic Variants in Ovarian Cancer. Nat Commun (2014) 5:3156. doi: 10.1038/ncomms4156
76. Potapova A, Hoffman AM, Godwin AK, Al-Saleem T, Cairns P. Promoter Hypermethylation of the PALB2 Susceptibility Gene in Inherited and Sporadic Breast and Ovarian Cancer. Cancer Res (2008) 68(4):998–1002. doi: 10.1158/0008-5472.CAN-07-2418
77. Ryland GL, Doyle MA, Goode D, Boyle SE, Choong DYH, Rowley SM, et al. Loss of Heterozygosity: What is it Good for? (2015) 8(1):45. doi: 10.1186/s12920-015-0123-z
78. Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors From 33 Types of Cancer. Cell (2018) 173(2):291–304.e6. doi: 10.1016/j.cell.2018.03.022
79. Ding JJ, Wang G, Shi WX, Zhou HH, Zhao EF. Promoter Hypermethylation of FANCF and Susceptibility and Prognosis of Epithelial Ovarian Cancer. Reprod Sci (2016) 23(1):24–30. doi: 10.1177/1933719115612136
80. Lim SL, Smith P, Syed N, Coens C, Wong H, van der Burg M, et al. Promoter Hypermethylation of FANCF and Outcome in Advanced Ovarian Cancer. Br J Cancer (2008) 98(8):1452–6. doi: 10.1038/sj.bjc.6604325
81. Schubert S, van Luttikhuizen JL, Auber B, Schmidt G, Hofmann W, Penkert J, et al. The Identification of Pathogenic Variants in BRCA1/2 Negative, High Risk, Hereditary Breast and/or Ovarian Cancer Patients: High Frequency of FANCM Pathogenic Variants. Int J Cancer (2018). doi: 10.1002/ijc.31992
82. Ramus SJ, Song H, Dicks E, Tyrer JP, Rosenthal AN, Intermaggio MP, et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J Natl Cancer Insti (2015) 107(11). doi: 10.1093/jnci/djv214
83. Ratajska M, Matusiak M, Kuzniacka A, Wasag B, Brozek I, Biernat W, et al. Cancer Predisposing BARD1 Mutations Affect Exon Skipping and are Associated With Overexpression of Specific BARD1 Isoforms. Oncol Rep (2015) 34(5):2609–17. doi: 10.3892/or.2015.4235
84. Hoeijmakers JH. Genome Maintenance Mechanisms for Preventing Cancer. Nature (2001) 411(6835):366–74. doi: 10.1038/35077232
85. Buys SS, Sandbach JF, Gammon A, Patel G, Kidd J, Brown KL, et al. A Study of Over 35,000 Women With Breast Cancer Tested With a 25-Gene Panel of Hereditary Cancer Genes. Cancer (2017) 123(10):1721–30. doi: 10.1002/cncr.30498
86. Silva FC, Lisboa BC, Figueiredo MC, Torrezan GT, Santos EM, Krepischi AC, et al. Hereditary Breast and Ovarian Cancer: Assessment of Point Mutations and Copy Number Variations in Brazilian Patients. BMC Med Genet (2014) 15:55. doi: 10.1186/1471-2350-15-55
87. Yang X, Wu J, Lu J, Liu G, Di G, Chen C, et al. Identification of a Comprehensive Spectrum of Genetic Factors for Hereditary Breast Cancer in a Chinese Population by Next-Generation Sequencing. PLos One (2015) 10(4):e0125571. doi: 10.1371/journal.pone.0125571
88. Kim YC, Zhao L, Zhang H, Huang Y, Cui J, Xiao F, et al. Prevalence and Spectrum of BRCA Germline Variants in Mainland Chinese Familial Breast and Ovarian Cancer Patients. Oncotarget (2016) 7(8):9600–12. doi: 10.18632/oncotarget.7144
89. Copson ER, Maishman TC, Tapper WJ, Cutress RI, Greville-Heygate S, Altman DG, et al. Germline BRCA Mutation and Outcome in Young-Onset Breast Cancer (POSH): A Prospective Cohort Study. Lancet Oncol (2018) 19(2):169–80. doi: 10.1016/S1470-2045(17)30891-4
90. Ryu JM, Choi HJ, Kim I, Nam SJ, Kim SW, Yu J, et al. Prevalence and Oncologic Outcomes of BRCA 1/2 Mutations in Unselected Triple-Negative Breast Cancer Patients in Korea. Breast Cancer Res Treat (2018). doi: 10.1007/s10549-018-5015-4
91. Engel C, Rhiem K, Hahnen E, Loibl S, Weber KE, Seiler S, et al. Prevalence of Pathogenic BRCA1/2 Germline Mutations Among 802 Women With Unilateral Triple-Negative Breast Cancer Without Family Cancer History. BMC Cancer (2018) 18(1):265. doi: 10.1186/s12885-018-4029-y
92. Jeong HM, Kim RN, Kwon MJ, Oh E, Han J, Lee SK, et al. Targeted Exome Sequencing of Korean Triple-Negative Breast Cancer Reveals Homozygous Deletions Associated With Poor Prognosis of Adjuvant Chemotherapy-Treated Patients. Oncotarget (2017) 8(37):61538–50. doi: 10.18632/oncotarget.18618
93. Deryusheva IV, Tsyganov M, Garbukov EY, Ibragimova MK, Kzhyshkovska JG, Slonimskaya E, et al. Genome-Wide Association Study of Loss of Heterozygosity and Metastasis-Free Survival in Breast Cancer Patients. Exp Oncol (2017) 39(2):145–50. doi: 10.31768/2312-8852.2017.39(2):145-150
94. Okada S, Tokunaga E, Kitao H, Akiyoshi S, Yamashita N, Saeki H, et al. Loss of Heterozygosity at BRCA1 Locus is Significantly Associated With Aggressiveness and Poor Prognosis in Breast Cancer. Ann Surg Oncol (2012) 19(5):1499–507. doi: 10.1245/s10434-011-2166-5
95. Vos S, van Diest PJ, Moelans CB. A Systematic Review on the Frequency of BRCA Promoter Methylation in Breast and Ovarian Carcinomas of BRCA Germline Mutation Carriers: Mutually Exclusive, or Not? Crit Rev Oncol/Hematol (2018) 127:29–41. doi: 10.1016/j.critrevonc.2018.05.008
96. Watanabe Y, Maeda I, Oikawa R, Wu W, Tsuchiya K, Miyoshi Y, et al. Aberrant DNA Methylation Status of DNA Repair Genes in Breast Cancer Treated With Neoadjuvant Chemotherapy. Genes Cells (2013) 18(12):1120–30. doi: 10.1111/gtc.12100
97. Pecuchet N, Popova T, Manie E, Lucchesi C, Battistella A, Vincent-Salomon A, et al. Loss of Heterozygosity at 13q13 and 14q32 Predicts BRCA2 Inactivation in Luminal Breast Carcinomas. Int J Cancer (2013) 133(12):2834–42. doi: 10.1002/ijc.28315
98. Goidescu IG, Caracostea G, Eniu DT, Stamatian FV. Prevalence of Deleterious Mutations Among Patients With Breast Cancer Referred for Multigene Panel Testing in a Romanian Population. Clujul Med (2018) 91(2):157–65. doi: 10.15386/cjmed-894
99. Takada M, Nagai S, Haruta M, Sugino RP, Tozuka K, Takei H, et al. BRCA1 Alterations With Additional Defects in DNA Damage Response Genes may Confer Chemoresistance to BRCA-Like Breast Cancers Treated With Neoadjuvant Chemotherapy. Genes Chromosomes Cancer (2017) 56(5):405–20. doi: 10.1002/gcc.22445
100. Silva MC, Bryan KE, Morrical MD, Averill AM, Dragon J, Wiegmans AP, et al. Defects in Recombination Activity Caused by Somatic and Germline Mutations in the Multimerization/BRCA2 Binding Region of Human RAD51 Protein. DNA Repair (Amst) (2017) 60:64–76. doi: 10.1016/j.dnarep.2017.10.008
101. Nowacka-Zawisza M, Brys M, Romanowicz-Makowska H, Kulig A, Krajewska WM. Loss of Heterozygosity in the RAD51 and BRCA2 Regions in Breast Cancer. Cancer Detect Prev (2008) 32(2):144–8. doi: 10.1016/j.cdp.2008.06.005
102. Gonzalez R, Silva JM, Dominguez G, Garcia JM, Martinez G, Vargas J, et al. Detection of Loss of Heterozygosity at RAD51, RAD52, RAD54 and BRCA1 and BRCA2 Loci in Breast Cancer: Pathological Correlations. Br J Cancer (1999) 81(3):503–9. doi: 10.1038/sj.bjc.6690722
103. Clague J, Wilhoite G, Adamson A, Bailis A, Weitzel JN, Neuhausen SL. RAD51C Germline Mutations in Breast and Ovarian Cancer Cases From High-Risk Families. PLos One (2011) 6(9):e25632. doi: 10.1371/journal.pone.0025632
104. Castera L, Harter V, Muller E, Krieger S, Goardon N, Ricou A, et al. Landscape of Pathogenic Variations in a Panel of 34 Genes and Cancer Risk Estimation From 5131 HBOC Families. Genet Med (2018). doi: 10.1038/s41436-018-0005-9
105. Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, et al. A Mutational Signature Reveals Alterations Underlying Deficient Homologous Recombination Repair in Breast Cancer. Nat Genet (2017) 49:1476. doi: 10.1038/ng.3934
106. Manie E, Popova T, Battistella A, Tarabeux J, Caux-Moncoutier V, Golmard L, et al. Genomic Hallmarks of Homologous Recombination Deficiency in Invasive Breast Carcinomas. Int J Cancer (2016) 138(4):891–900. doi: 10.1002/ijc.29829
107. TCGA. Comprehensive Molecular Portraits of Human Breast Tumours. Nature (2012) 490(7418):61–70. doi: 10.1038/nature11412
108. Momozawa Y, Iwasaki Y, Parsons MT, Kamatani Y, Takahashi A, Tamura C, et al. Germline Pathogenic Variants of 11 Breast Cancer Genes in 7,051 Japanese Patients and 11,241 Controls. Nat Commun (2018) 9(1):4083. doi: 10.1038/s41467-018-06581-8
109. Casadei S, Norquist BM, Walsh T, Stray S, Mandell JB, Lee MK, et al. Contribution of Inherited Mutations in the BRCA2-Interacting Protein PALB2 to Familial Breast Cancer. Cancer Res (2011) 71(6):2222–9. doi: 10.1158/0008-5472.CAN-10-3958
110. Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol (2017) 3(9):1190–6. doi: 10.1001/jamaoncol.2017.0424
111. Li JY, Jing R, Wei H, Wang M, Xiaowei Q, Liu H, et al. Germline Mutations in 40 Cancer Susceptibility Genes Among Chinese Patients With High Hereditary Risk Breast Cancer. Int J Cancer (2019) 144(2):281–9. doi: 10.1002/ijc.31601
112. Hartley T, Cavallone L, Sabbaghian N, Silva-Smith R, Hamel N, Aleynikova O, et al. Mutation Analysis of PALB2 in BRCA1 and BRCA2-Negative Breast and/or Ovarian Cancer Families From Eastern Ontario, Canada. Hereditary Cancer Clin Pract (2014) 12(1):19. doi: 10.1186/1897-4287-12-19
113. Scott CM, Joo JE, O'Callaghan N, Buchanan DD, Clendenning M, Giles GG, et al. Methylation of Breast Cancer Predisposition Genes in Early-Onset Breast Cancer: Australian Breast Cancer Family Registry. PLos One (2016) 11(11):e0165436. doi: 10.1371/journal.pone.0165436
114. Ellingson MS, Hart SN, Kalari KR, Suman V, Schahl KA, Dockter TJ, et al. Exome Sequencing Reveals Frequent Deleterious Germline Variants in Cancer Susceptibility Genes in Women With Invasive Breast Cancer Undergoing Neoadjuvant Chemotherapy. Breast Cancer Res Treat (2015) 153(2):435–43. doi: 10.1007/s10549-015-3545-6
115. Mantere T, Tervasmaki A, Nurmi A, Rapakko K, Kauppila S, Tang J, et al. Case-Control Analysis of Truncating Mutations in DNA Damage Response Genes Connects TEX15 and FANCD2 With Hereditary Breast Cancer Susceptibility. Sci Rep (2017) 7(1):681. doi: 10.1038/s41598-017-00766-9
116. Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria JC, et al. Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLos Med (2016) 13(12):e1002201. doi: 10.1371/journal.pmed.1002201
117. Dasgupta H, Mukherjee N, Islam S, Bhattacharya R, Alam N, Roy A, et al. Frequent Alterations of Homologous Recombination Repair Pathway in Primary and Chemotolerant Breast Carcinomas: Clinical Importance. Future Oncol (2017) 13(2):159–74. doi: 10.2217/fon-2016-0289
118. Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell (2015) 163(2):506–19. doi: 10.1158/1538-7445.SABCS14-S2-04
119. Tokunaga E, Okada S, Kitao H, Shiotani S, Saeki H, Endo K, et al. Low Incidence of Methylation of the Promoter Region of the FANCF Gene in Japanese Primary Breast Cancer. Breast Cancer (2011) 18(2):120–3. doi: 10.1007/s12282-009-0175-z
120. Wei M, Xu J, Dignam J, Nanda R, Sveen L, Fackenthal J, et al. Estrogen Receptor Alpha, BRCA1, and FANCF Promoter Methylation Occur in Distinct Subsets of Sporadic Breast Cancers. Breast Cancer Res Treat (2008) 111(1):113–20. doi: 10.1007/s10549-007-9766-6
121. Tavera-Tapia A, Perez-Cabornero L, Macias JA, Ceballos MI, Roncador G, de la Hoya M, et al. Almost 2% of Spanish Breast Cancer Families are Associated to Germline Pathogenic Mutations in the ATM Gene. Breast Cancer Res Treat (2017) 161(3):597–604. doi: 10.1007/s10549-016-4058-7
122. Garcia MJ, Fernandez V, Osorio A, Barroso A, Fernandez F, Urioste M, et al. Mutational Analysis of FANCL, FANCM and the Recently Identified FANCI Suggests That Among the 13 Known Fanconi Anemia Genes, Only FANCD1/BRCA2 Plays a Major Role in High-Risk Breast Cancer Predisposition. Carcinogenesis (2009) 30(11):1898–902. doi: 10.1093/carcin/bgp218
123. Nguyen-Dumont T, Myszka A, Karpinski P, Sasiadek MM, Akopyan H, Hammet F, et al. FANCM and RECQL Genetic Variants and Breast Cancer Susceptibility: Relevance to South Poland and West Ukraine. BMC Med Genet (2018) 19(1):12. doi: 10.1186/s12881-018-0524-x
124. Silvestri V, Rizzolo P, Zelli V, Valentini V, Zanna I, Bianchi S, et al. A Possible Role of FANCM Mutations in Male Breast Cancer Susceptibility: Results From a Multicenter Study in Italy. Breast (2018) 38:92–7. doi: 10.1016/j.breast.2017.12.013
125. Neidhardt G, Hauke J, Ramser J, Gross E, Gehrig A, Muller CR, et al. Association Between Loss-Of-Function Mutations Within the FANCM Gene and Early-Onset Familial Breast Cancer. JAMA Oncol (2017) 3(9):1245–8. doi: 10.1001/jamaoncol.2016.5592
126. Desjardins S, Beauparlant JC, Labrie Y, Ouellette G, Durocher F. Variations in the NBN/NBS1 Gene and the Risk of Breast Cancer in non-BRCA1/2 French Canadian Families With High Risk of Breast Cancer. BMC Cancer (2009) 9:181. doi: 10.1186/1471-2407-9-181
127. Shimelis H, LaDuca H, Hu C, Hart SN, Na J, Thomas A, et al. Triple-Negative Breast Cancer Risk Genes Identified by Multigene Hereditary Cancer Panel Testing. JNCI: J Natl Cancer Inst (2018) 110(8):855–62. doi: 10.1093/jnci/djy106
128. Huzarski T, Cybulski C, Jakubowska A, Byrski T, Gronwald J, Domagala P, et al. Clinical Characteristics of Breast Cancer in Patients With an NBS1 Mutation. Breast Cancer Res Treat (2013) 141(3):471–6. doi: 10.1007/s10549-013-2692-x
129. Kim H, Cho DY, Choi DH, Jung GH, Shin I, Park W, et al. Heterozygous Germline Mutations in NBS1 Among Korean Patients With High-Risk Breast Cancer Negative for BRCA1/2 Mutation. Familial Cancer (2015) 14(3):365–71. doi: 10.1007/s10689-015-9789-9
130. Suspitsin EN, Yanus GA, Sokolenko AP, Yatsuk OS, Zaitseva OA, Bessonov AA, et al. Development of Breast Tumors in CHEK2, NBN/NBS1 and BLM Mutation Carriers Does Not Commonly Involve Somatic Inactivation of the Wild-Type Allele. Med Oncol (2014) 31(2):828. doi: 10.1007/s12032-013-0828-9
131. Bonache S, Esteban I, Moles-Fernandez A, Tenes A, Duran-Lozano L, Montalban G, et al. Multigene Panel Testing Beyond BRCA1/2 in Breast/Ovarian Cancer Spanish Families and Clinical Actionability of Findings. J Cancer Res Clin Oncol (2018) 144(12):2495–513. doi: 10.1007/s00432-018-2763-9
132. Park JS, Lee ST, Nam EJ, Han JW, Lee JY, Kim J, et al. Variants of Cancer Susceptibility Genes in Korean BRCA1/2 Mutation-Negative Patients With High Risk for Hereditary Breast Cancer. BMC Cancer (2018) 18(1):83. doi: 10.1186/s12885-017-3940-y
133. Slavin TP, Maxwell KN, Lilyquist J, Vijai J, Neuhausen SL, Hart SN, et al. The Contribution of Pathogenic Variants in Breast Cancer Susceptibility Genes to Familial Breast Cancer Risk. NPJ Breast Cancer (2017) 3:22. doi: 10.1038/s41523-017-0024-8
134. Sabatier R, Adelaide J, Finetti P, Ferrari A, Huiart L, Sobol H, et al. BARD1 Homozygous Deletion, a Possible Alternative to BRCA1 Mutation in Basal Breast Cancer. Genes Chromosomes Cancer (2010) 49(12):1143–51. doi: 10.1002/gcc.20822
135. Hauke J, Horvath J, Gross E, Gehrig A, Honisch E, Hackmann K, et al. Gene Panel Testing of 5589 BRCA1/2-Negative Index Patients With Breast Cancer in a Routine Diagnostic Setting: Results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med (2018) 7(4):1349–58. doi: 10.1002/cam4.1376
136. Yang Z, Ouyang T, Li J, Wang T, Fan Z, Fan T, et al. Prevalence and Characterization of ATM Germline Mutations in Chinese BRCA1/2-Negative Breast Cancer Patients. Breast Cancer Res Treat (2019). doi: 10.1007/s10549-018-05124-5
137. Prodosmo A, Buffone A, Mattioni M, Barnabei A, Persichetti A, De Leo A, et al. Detection of ATM Germline Variants by the P53 Mitotic Centrosomal Localization Test in BRCA1/2-Negative Patients With Early-Onset Breast Cancer. J Exp Clin Cancer Res (2016) 35(1):135. doi: 10.1186/s13046-016-0410-3
138. Renault AL, Mebirouk N, Fuhrmann L, Bataillon G, Cavaciuti E, Le Gal D, et al. Morphology and Genomic Hallmarks of Breast Tumours Developed by ATM Deleterious Variant Carriers. Breast Cancer Res (2018) 20(1):28. doi: 10.1186/s13058-018-0951-9
139. Flanagan JM, Munoz-Alegre M, Henderson S, Tang T, Sun P, Johnson N, et al. Gene-Body Hypermethylation of ATM in Peripheral Blood DNA of Bilateral Breast Cancer Patients. Hum Mol Genet (2009) 18(7):1332–42. doi: 10.1093/hmg/ddp033
140. Delmonico L, Moreira Ados S, Franco MF, Esteves EB, Scherrer L, Gallo CV, et al. CDKN2A (P14(ARF)/p16(INK4a)) and ATM Promoter Methylation in Patients With Impalpable Breast Lesions. Hum Pathol (2015) 46(10):1540–7. doi: 10.1016/j.humpath.2015.06.016
141. Cao X, Tang Q, Holland-Letz T, Gundert M, Cuk K, Schott S, et al. Evaluation of Promoter Methylation of RASSF1A and ATM in Peripheral Blood of Breast Cancer Patients and Healthy Control Individuals. Int J Mol Sci (2018) 19(3). doi: 10.3390/ijms19030900
142. Bhattacharya N, Mukherjee N, Singh RK, Sinha S, Alam N, Roy A, et al. Frequent Alterations of MCPH1 and ATM are Associated With Primary Breast Carcinoma: Clinical and Prognostic Implications. Ann Surg Oncol (2013) 20 Suppl 3:S424–32. doi: 10.1245/s10434-012-2715-6
143. Begam N, Jamil K, Raju SG. Promoter Hypermethylation of the ATM Gene as a Novel Biomarker for Breast Cancer. Asian Pacific J Cancer Prev (2017) 18(11):3003–9. doi: 10.1016/j.genrep.2018.04.004
144. Lang GT, Shi JX, Hu X, Zhang CH, Shan L, Song CG, et al. The Spectrum of BRCA Mutations and Characteristics of BRCA-Associated Breast Cancers in China: Screening of 2,991 Patients and 1,043 Controls by Next-Generation Sequencing. Int J Cancer (2017) 141(1):129–42. doi: 10.1002/ijc.30692
145. Testa U, Pelosi E, Castelli G. Colorectal Cancer: Genetic Abnormalities, Tumor Progression, Tumor Heterogeneity, Clonal Evolution and Tumor-Initiating Cells. Med Sci (2018) 6(2):31. doi: 10.3390/medsci6020031
146. The Cancer Genome Atlas N, Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, et al. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature (2012) 487:330. doi: 10.1038/nature11252
147. Nagahashi M, Wakai T, Shimada Y, Ichikawa H, Kameyama H, Kobayashi T, et al. Genomic Landscape of Colorectal Cancer in Japan: Clinical Implications of Comprehensive Genomic Sequencing for Precision Medicine. Genome Med (2016) 8(1):136. doi: 10.1186/s13073-016-0387-8
148. Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol (2017) 3(4):464–71. doi: 10.1001/jamaoncol.2016.5194
149. Ehrlich M. DNA Methylation in Cancer: Too Much, But Also Too Little. Oncogene (2002) 21:5400. doi: 10.1038/sj.onc.1205651
150. Feinberg AP, Vogelstein B. Hypomethylation Distinguishes Genes of Some Human Cancers From Their Normal Counterparts. Nature (1983) 301(5895):89–92. doi: 10.1038/301089a0
151. Tse JWT, Jenkins LJ, Chionh F, Mariadason JM. Aberrant DNA Methylation in Colorectal Cancer: What Should We Target? Trends Cancer (2017) 3(10):698–712. doi: 10.1016/j.trecan.2017.08.003
152. Phelan CM, Iqbal J, Lynch HT, Lubinski J, Gronwald J, Moller P, et al. Incidence of Colorectal Cancer in BRCA1 and BRCA2 Mutation Carriers: Results From a Follow-Up Study. Br J Cancer (2014) 110(2):530–4. doi: 10.1038/bjc.2013.741
153. Yurgelun MB, Kulke MH, Fuchs CS, Allen BA, Uno H, Hornick JL, et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. J Clin Oncol (2017) 35(10):1086–95. doi: 10.1200/JCO.2016.71.0012
154. Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep (2016) 15(4):857–65. doi: 10.1016/j.celrep.2016.03.075
155. Rengucci C, De Maio G, Gardini AC, Zucca M, Scarpi E, Zingaretti C, et al. Promoter Methylation of Tumor Suppressor Genes in Pre-Neoplastic Lesions; Potential Marker of Disease Recurrence. J Exp Clin Cancer Res (2014) 33(1):65. doi: 10.1186/s13046-014-0065-x
156. Yurgelun MB, Allen B, Kaldate RR, Bowles KR, Judkins T, Kaushik P, et al. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology (2015) 149(3):604–13.e20. doi: 10.1053/j.gastro.2015.05.006
157. Smith CG, Naven M, Harris R, Colley J, West H, Li N, et al. Exome Resequencing Identifies Potential Tumor-Suppressor Genes That Predispose to Colorectal Cancer. (2013) 34(7):1026–34. doi: 10.1002/humu.22333
158. Smith DH, Christensen IJ, Jensen NF, Markussen B, Müller S, Nielsen HJ, et al. An Explorative Analysis of ERCC1-19q13 Copy Number Aberrations in a Chemonaive Stage III Colorectal Cancer Cohort. BMC Cancer (2013) 13:489–. doi: 10.1186/1471-2407-13-489
159. Esteban-Jurado C, Franch-Exposito S, Munoz J, Ocana T, Carballal S, Lopez-Ceron M, et al. The Fanconi Anemia DNA Damage Repair Pathway in the Spotlight for Germline Predisposition to Colorectal Cancer. Eur J Hum Genet (2016) 24(10):1501–5. doi: 10.1038/ejhg.2016.44
160. Lewis KA, Bakkum-Gamez J, Loewen R, French AJ, Thibodeau SN, Cliby WA. Mutations in the Ataxia Telangiectasia and Rad3-Related-Checkpoint Kinase 1 DNA Damage Response Axis in Colon Cancers. Genes Chromosomes Cancer (2007) 46(12):1061–8. doi: 10.1002/gcc.20486
161. Chubb D, Broderick P, Dobbins SE, Frampton M, Kinnersley B, Penegar S, et al. Rare Disruptive Mutations and Their Contribution to the Heritable Risk of Colorectal Cancer. Nat Commun (2016) 7:11883. doi: 10.1038/ncomms11883
162. Stoffel EM, Koeppe E, Everett J, Ulintz P, Kiel M, Osborne J, et al. Germline Genetic Features of Young Individuals With Colorectal Cancer. Gastroenterology (2018) 154(4):897–905.e1. doi: 10.1053/j.gastro.2017.11.004
163. You YN, Borras E, Chang K, Price BA, Mork M, Chang GJ, et al. Detection of Pathogenic Germline Variants Among Patients With Advanced Colorectal Cancer Undergoing Tumor Genomic Profiling for Precision Medicine. Dis Colon Rectum (2019) 62(4):429–37. doi: 10.1097/DCR.0000000000001322
164. Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L, et al. Frequent Inactivation of PTEN by Promoter Hypermethylation in Microsatellite Instability-High Sporadic Colorectal Cancers. Cancer Res (2004) 64(9):3014–21. doi: 10.1158/0008-5472.CAN-2401-2
165. Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary Somatic Mutations Restoring BRCA1/2 Predict Chemotherapy Resistance in Hereditary Ovarian Carcinomas. J Clin Oncol (2011) 29(22):3008–15. doi: 10.1200/JCO.2010.34.2980
166. Tan SH, Ida H, Lau QC, Goh BC, Chieng WS, Loh M, et al. Detection of Promoter Hypermethylation in Serum Samples of Cancer Patients by Methylation-Specific Polymerase Chain Reaction for Tumour Suppressor Genes Including RUNX3. Oncol Rep (2007) 18(5):1225–30. doi: 10.3892/or.18.5.1225
167. Michailidi C, Theocharis S, Tsourouflis G, Pletsa V, Kouraklis G, Patsouris E, et al. Expression and Promoter Methylation Status of Hmlh1, MGMT, APC, and CDH1 Genes in Patients With Colon Adenocarcinoma. Exp Biol Med (2015) 240(12):1599–605. doi: 10.1177/1535370215583800
168. de Voer RM, Hahn M-M, Mensenkamp AR, Hoischen A, Gilissen C, Henkes A, et al. Deleterious Germline BLM Mutations and the Risk for Early-Onset Colorectal Cancer. Sci Rep (2015) 5:14060. doi: 10.1038/srep14060
169. Yeon SY, Jo YS, Choi EJ, Kim MS, Yoo NJ, Lee SH. Frameshift Mutations in Repeat Sequences of ANK3, HACD4, TCP10L, TP53BP1, MFN1, LCMT2, RNMT, TRMT6, METTL8 and METTL16 Genes in Colon Cancers. Pathol Oncol Res (2018) 24(3):617–22. doi: 10.1007/s12253-017-0287-2
170. He T, Zhang M, Zheng R, Zheng S, Linghu E, Herman JG, et al. Methylation of SLFN11 is a Marker of Poor Prognosis and Cisplatin Resistance in Colorectal Cancer. Epigenomics (2017) 9(6):849–62. doi: 10.2217/epi-2017-0019
171. Ohmoto A, Yachida S, Morizane C. Genomic Features and Clinical Management of Patients With Hereditary Pancreatic Cancer Syndromes and Familial Pancreatic Cancer. Int J Mol Sci (2019) 20(3). doi: 10.3390/ijms20030561
172. Matsubayashi H, Takaori K, Morizane C, Maguchi H, Mizuma M, Takahashi H, et al. Familial Pancreatic Cancer: Concept, Management and Issues. World J Gastroenterol (2017) 23(6):935–48. doi: 10.3748/wjg.v23.i6.935
173. Klein AP, Brune KA, Petersen GM, Goggins M, Tersmette AC, Offerhaus GJ, et al. Prospective Risk of Pancreatic Cancer in Familial Pancreatic Cancer Kindreds. Cancer Res (2004) 64(7):2634–8. doi: 10.1158/0008-5472.CAN-03-3823
174. Zhan W, Shelton CA, Greer PJ, Brand RE, Whitcomb DC. Germline Variants and Risk for Pancreatic Cancer: A Systematic Review and Emerging Concepts. Pancreas (2018) 47(8):924–36. doi: 10.1097/MPA.0000000000001136
175. Zhen DB, Rabe KG, Gallinger S, Syngal S, Schwartz AG, Goggins MG, et al. BRCA1, BRCA2, PALB2, and CDKN2A Mutations in Familial Pancreatic Cancer: A PACGENE Study. Genet Med (2015) 17(7):569–77. doi: 10.1038/gim.2014.153
176. Brand R, Borazanci E, Speare V, Dudley B, Karloski E, Peters MLB, et al. Prospective Study of Germline Genetic Testing in Incident Cases of Pancreatic Adenocarcinoma. (2018) 124(17):3520–7. doi: 10.1002/cncr.31628
177. Lowery MA, Wong W, Jordan EJ, Lee JW, Kemel Y, Vijai J, et al. Prospective Evaluation of Germline Alterations in Patients With Exocrine Pancreatic Neoplasms. J Natl Cancer Inst (2018) 110(10):1067–74. doi: 10.1093/jnci/djy024
178. Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA (2018) 319(23):2401–9. doi: 10.1001/jama.2018.6228
179. Roch AM, Schneider J, Carr RA, Lancaster WP, House MG, Zyromski NJ, et al. Are BRCA1 and BRCA2 Gene Mutation Patients Underscreened for Pancreatic Adenocarcinoma? J Surg Oncol (2019). doi: 10.1002/jso.25376
180. Takeuchi S, Doi M, Ikari N, Yamamoto M, Furukawa T. Mutations in BRCA1, BRCA2, and PALB2, and a Panel of 50 Cancer-Associated Genes in Pancreatic Ductal Adenocarcinoma. Sci Rep (2018) 8(1):8105. doi: 10.1038/s41598-018-26526-x
181. Slavin TP, Banks KC, Chudova D, Oxnard GR, Odegaard JI, Nagy RJ, et al. Identification of Incidental Germline Mutations in Patients With Advanced Solid Tumors Who Underwent Cell-Free Circulating Tumor DNA Sequencing. J Clin Oncol (2018), Jco1800328. doi: 10.1200/JCO.18.00328
182. Shindo K, Yu J, Suenaga M, Fesharakizadeh S, Cho C, Macgregor-Das A, et al. Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. J Clin Oncol (2017) 35(30):3382–90. doi: 10.1200/JCO.2017.72.3502
183. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature (2016) 531(7592):47–52.
184. Lucas AL, Shakya R, Lipsyc MD, Mitchel EB, Kumar S, Hwang C, et al. High Prevalence of BRCA1 and BRCA2 Germline Mutations With Loss of Heterozygosity in a Series of Resected Pancreatic Adenocarcinoma and Other Neoplastic Lesions. Clin Cancer Res (2013) 19(13):3396–403. doi: 10.1158/1078-0432.CCR-12-3020
185. Waddell N, Pajic M, Patch A-M, Chang DK, Kassahn KS, Bailey P, et al. Whole Genomes Redefine the Mutational Landscape of Pancreatic Cancer. Nature (2015) 518:495.
186. Peng DF, Kanai Y, Sawada M, Ushijima S, Hiraoka N, Kitazawa S, et al. DNA Methylation of Multiple Tumor-Related Genes in Association With Overexpression of DNA Methyltransferase 1 (DNMT1) During Multistage Carcinogenesis of the Pancreas. Carcinogenesis (2006) 27(6):1160–8. doi: 10.1093/carcin/bgi361
187. Guo M, Jia Y, Yu Z, House MG, Esteller M, Brock MV, et al. Epigenetic Changes Associated With Neoplasms of the Exocrine and Endocrine Pancreas. Discov Med (2014) 17(92):67–73.
188. Takai E, Yachida S, Shimizu K, Furuse J, Kubo E, Ohmoto A, et al. Germline Mutations in Japanese Familial Pancreatic Cancer Patients. Oncotarget (2016) 7(45):74227–35. doi: 10.18632/oncotarget.12490
189. Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, et al. ATM Mutations in Patients With Hereditary Pancreatic Cancer. Cancer Discov (2012) 2(1):41–6. doi: 10.1158/2159-8290.CD-11-0194
190. Yurgelun MB, Chittenden AB, Morales-Oyarvide V, Rubinson DA, Dunne RF, Kozak MM, et al. Germline Cancer Susceptibility Gene Variants, Somatic Second Hits, and Survival Outcomes in Patients With Resected Pancreatic Cancer. Genet Med (2019) 21(1):213–23. doi: 10.1038/s41436-018-0009-5
191. Roberts NJ, Norris AL, Petersen GM, Bondy ML, Brand R, Gallinger S, et al. Whole Genome Sequencing Defines the Genetic Heterogeneity of Familial Pancreatic Cancer. (2016) 6(2):166–75. doi: 10.1158/2159-8290.CD-15-0402
192. Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-Exome Sequencing of Pancreatic Cancer Defines Genetic Diversity and Therapeutic Targets. Nat Commun (2015) 6:6744. doi: 10.1038/ncomms7744
193. House MG, Guo M, Iacobuzio-Donahue C, Herman JG. Molecular Progression of Promoter Methylation in Intraductal Papillary Mucinous Neoplasms (IPMN) of the Pancreas. Carcinogenesis (2003) 24(2):193–8. doi: 10.1093/carcin/24.2.193
194. Lemjabbar-Alaoui H, Hassan OU, Yang Y-W, Buchanan P. Lung Cancer: Biology and Treatment Options. Biochim Biophys Acta (2015) 1856(2):189–210. doi: 10.1016/j.bbcan.2015.08.002
195. Gazdar A, Robinson L, Oliver D, Xing C, Travis WD, Soh J, et al. Hereditary Lung Cancer Syndrome Targets Never Smokers With Germline EGFR Gene T790M Mutations. J Thorac Oncol (2014) 9(4):456–63. doi: 10.1097/JTO.0000000000000130
196. Cardona AF, Zatarain-Barron ZL, Rubio C, Martinez S, Ruiz-Patino A, Ricaurte L, et al. Probable Hereditary Familial Overlap Syndrome With Multiple Synchronous Lung Tumors. Lung Cancer (2018) 124:279–82. doi: 10.1016/j.lungcan.2018.08.022
197. TCGA, Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, et al. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature (2014) 511:543. doi: 10.1038/nature13385
198. Chae YK, Anker JF, Oh MS, Bais P, Namburi S, Agte S, et al. Mutations in DNA Repair Genes are Associated With Increased Neoantigen Burden and a Distinct Immunophenotype in Lung Squamous Cell Carcinoma. Sci Rep (2019) 9(1):3235. doi: 10.1038/s41598-019-39594-4
199. Kim H, Choi DH, Park W, Im YH, Ahn JS, Park YH, et al. The Association Between non-Breast and Ovary Cancers and BRCA Mutation in First- and Second-Degree Relatives of High-Risk Breast Cancer Patients: A Large-Scale Study of Koreans. Hereditary Cancer Clin Pract (2019) 17:1. doi: 10.1186/s13053-018-0103-3
200. Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular Determinants of Response to Anti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients With Non-Small-Cell Lung Cancer Profiled With Targeted Next-Generation Sequencing. J Clin Oncol (2018) 36(7):633–41. doi: 10.1200/JCO.2017.75.3384
201. Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, et al. Distinct Patterns of Somatic Genome Alterations in Lung Adenocarcinomas and Squamous Cell Carcinomas. Nat Genet (2016) 48(6):607–16. doi: 10.1038/ng.3564
202. Marsit CJ, Liu M, Nelson HH, Posner M, Suzuki M, Kelsey KT. Inactivation of the Fanconi Anemia/BRCA Pathway in Lung and Oral Cancers: Implications for Treatment and Survival. Oncogene (2004) 23(4):1000–4. doi: 10.1038/sj.onc.1207256
203. Leong TL, Gayevskiy V, Steinfort DP, De Massy MR, Gonzalez-Rajal A, Marini KD, et al. Deep Multi-Region Whole-Genome Sequencing Reveals Heterogeneity and Gene-by-Environment Interactions in Treatment-Naive, Metastatic Lung Cancer. Oncogene (2019) 38(10):1661–75. doi: 10.1038/s41388-018-0536-1
204. Kaluzna EM, Rembowska J, Ziolkowska-Suchanek I, Swiatek-Koscielna B, Gabryel P, Dyszkiewicz W, et al. Heterozygous P.I171V Mutation of the NBN Gene as a Risk Factor for Lung Cancer Development. Oncol Lett (2015) 10(5):3300–4. doi: 10.3892/ol.2015.3715
205. Scheffler M, Ihle MA, Hein R, Merkelbach-Bruse S, Scheel AH, Siemanowski J, et al. K-Ras Mutation Subtypes in NSCLC and Associated Co-Occuring Mutations in Other Oncogenic Pathways. J Thorac Oncol (2018). doi: 10.1016/j.jtho.2018.12.013
206. Liao L, Ji X, Ge M, Zhan Q, Huang R, Liang X, et al. Characterization of Genetic Alterations in Brain Metastases From Non-Small Cell Lung Cancer. FEBS Open Bio (2018) 8(9):1544–52. doi: 10.1002/2211-5463.12501
207. Gill RK, Yang SH, Meerzaman D, Mechanic LE, Bowman ED, Jeon HS, et al. Frequent Homozygous Deletion of the LKB1/STK11 Gene in Non-Small Cell Lung Cancer. Oncogene (2011) 30(35):3784–91. doi: 10.1038/onc.2011.98
208. Huang X, Wu C, Fu Y, Guo L, Kong X, Cai H. Methylation Analysis for Multiple Gene Promoters in Non-Small Cell Lung Cancers in High Indoor Air Pollution Region in China. Bull du Cancer (2018) 105(9):746–54. doi: 10.1016/j.bulcan.2018.05.004
209. Nogales V, Reinhold WC, Varma S, Martinez-Cardus A, Moutinho C, Moran S, et al. Epigenetic Inactivation of the Putative DNA/RNA Helicase SLFN11 in Human Cancer Confers Resistance to Platinum Drugs. Oncotarget (2016) 7(3):3084–97. doi: 10.18632/oncotarget.6413
210. Mucci LA, Hjelmborg JB, Harris JR, Czene K, Havelick DJ, Scheike T, et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA (2016) 315(1):68–76. doi: 10.1001/jama.2015.17703
211. Cooney KA. Inherited Predisposition to Prostate Cancer: From Gene Discovery to Clinical Impact. Trans Am Clin Climatolog Assoc (2017) 128:14–23.
212. Xu J, Lange EM, Lu L, Zheng SL, Wang Z, Thibodeau SN, et al. HOXB13 is a Susceptibility Gene for Prostate Cancer: Results From the International Consortium for Prostate Cancer Genetics (ICPCG). Hum Genet (2013) 132(1):5–14. doi: 10.1007/s00439-012-1229-4
213. Ewing CM, Ray AM, Lange EM, Zuhlke KA, Robbins CM, Tembe WD, et al. Germline Mutations in HOXB13 and Prostate-Cancer Risk. N Engl J Med (2012) 366(2):141–9. doi: 10.1056/NEJMoa1110000
214. Beebe-Dimmer JL, Hathcock M, Yee C, Okoth LA, Ewing CM, Isaacs WB, et al. The HOXB13 G84E Mutation Is Associated With an Increased Risk for Prostate Cancer and Other Malignancies. Cancer Epidemiol Biomarkers Prev (2015) 24(9):1366–72. doi: 10.1158/1055-9965.EPI-15-0247
215. Giri VN, Knudsen KE, Kelly WK, Abida W, Andriole GL, Bangma CH, et al. Role of Genetic Testing for Inherited Prostate Cancer Risk: Philadelphia Prostate Cancer Consensus Conference 2017. J Clin Oncol (2018) 36(4):414–24.
216. Rawla P. Epidemiology of Prostate Cancer. World J Oncol (2019) 10(2):63–89. doi: 10.14740/wjon1191
217. TCGA. The Molecular Taxonomy of Primary Prostate Cancer. Cell (2015) 163(4):1011–25. doi: 10.1016/j.cell.2015.10.025
218. Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis Oncol (2017) 2017. doi: 10.1200/PO.17.00029
219. Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic Correlates of Clinical Outcome in Advanced Prostate Cancer. Proc Natl Acad Sci (2019) 201902651.
220. Rabiau N, Thiam MO, Satih S, Guy L, Kemeny JL, Boiteux JP, et al. Methylation Analysis of BRCA1, RASSF1, GSTP1 and EPHB2 Promoters in Prostate Biopsies According to Different Degrees of Malignancy. In Vivo (2009) 23(3):387–91.
221. Gurioli G, Salvi S, Martignano F, Foca F, Gunelli R, Costantini M, et al. Methylation Pattern Analysis in Prostate Cancer Tissue: Identification of Biomarkers Using an MS-MLPA Approach. J Transl Med (2016) 14(1):249. doi: 10.1186/s12967-016-1014-6
222. Padar A, Sathyanarayana UG, Suzuki M, Maruyama R, Hsieh JT, Frenkel EP, et al. Inactivation of Cyclin D2 Gene in Prostate Cancers by Aberrant Promoter Methylation. Clin Cancer Res (2003) 9(13):4730–4.
223. Singal R, Ferdinand L, Reis IM, Schlesselman JJ. Methylation of Multiple Genes in Prostate Cancer and the Relationship With Clinicopathological Features of Disease. Oncol Rep (2004) 12(3):631–7. doi: 10.3892/or.12.3.631
224. Maruyama R, Toyooka S, Toyooka KO, Virmani AK, Zöchbauer-Müller S, Farinas AJ, et al. Aberrant Promoter Methylation Profile of Prostate Cancers and Its Relationship to Clinicopathological Features. Clin Cancer Res (2002) 8(2):514–9.
225. Konecny GE, Kristeleit RS. PARP Inhibitors for BRCA1/2-Mutated and Sporadic Ovarian Cancer: Current Practice and Future Directions. Br J Cancer (2016) 115:1157. doi: 10.1038/bjc.2016.311
226. Morales J, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, et al. Review of Poly (ADP-Ribose) Polymerase (PARP) Mechanisms of Action and Rationale for Targeting in Cancer and Other Diseases. Crit Rev Eukaryot Gene Expr (2014) 24(1):15–28. doi: 10.1615/CritRevEukaryotGeneExpr.2013006875
227. Lim JSJ, Tan DSP. Understanding Resistance Mechanisms and Expanding the Therapeutic Utility of PARP Inhibitors. Cancers (2017) 9(8). doi: 10.3390/cancers9080109
228. McCann KE. Poly-ADP-Ribosyl-Polymerase Inhibitor Resistance Mechanisms and Their Therapeutic Implications. Curr Opin Obstet gynecology (2019) 31(1):12–7. doi: 10.1097/GCO.0000000000000517
229. Ashworth A. A Synthetic Lethal Therapeutic Approach: Poly(ADP) Ribose Polymerase Inhibitors for the Treatment of Cancers Deficient in DNA Double-Strand Break Repair. J Clin Oncol (2008) 26(22):3785–90. doi: 10.1200/JCO.2008.16.0812
230. Drost R, Bouwman P, Rottenberg S, Boon U, Schut E, Klarenbeek S, et al. BRCA1 RING Function Is Essential for Tumor Suppression But Dispensable for Therapy Resistance. Cancer Cell (2011) 20(6):797–809. doi: 10.1016/j.ccr.2011.11.014
231. Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov (2019) 9(2):210–9. doi: 10.1158/2159-8290.CD-18-0715
232. Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, et al. REV7 Counteracts DNA Double-Strand Break Resection and Affects PARP Inhibition. Nature (2015) 521:541. doi: 10.1038/nature14328
233. Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated With Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov (2017) 7(9):984–98. doi: 10.1158/2159-8290.CD-17-0419
234. Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P, et al. Analysis of Circulating Cell-Free DNA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated With Resistance to PARP Inhibitors. Cancer Discov (2017) 7(9):999–1005. doi: 10.1158/2159-8290.CD-17-0146
235. Wang Y, Krais JJ, Bernhardy AJ, Nicolas E, Cai KQ, Harrell MI, et al. RING Domain-Deficient BRCA1 Promotes PARP Inhibitor and Platinum Resistance. J Clin Invest (2016) 126(8):3145–57. doi: 10.1172/JCI87033
236. Shimelis H, Mesman RLS, Von Nicolai C, Ehlen A, Guidugli L, Martin C, et al. BRCA2 Hypomorphic Missense Variants Confer Moderate Risks of Breast Cancer. Cancer Res (2017) 77(11):2789–99.
237. Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to Therapy Caused by Intragenic Deletion in BRCA2. Nature (2008) 451(7182):1111–5. doi: 10.1038/nature06548
238. Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, et al. Whole-Genome Characterization of Chemoresistant Ovarian Cancer. Nature (2015) 521(7553):489–94.
239. Kalachand RD, Stordal B, Madden S, Chandler B, Cunningham J, Goode EL, et al. BRCA1 Promoter Methylation and Clinical Outcomes in Ovarian Cancer: An Individual Patient Data Meta-Analysis. J Natl Cancer Instit (2020) 112(12):1190–203. doi: 10.1093/jnci/djaa070
240. Ter Brugge P, Kristel P, van der Burg E, Boon U, de Maaker M, Lips E, et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. J Natl Cancer Inst (2016) 108(11). doi: 10.1093/jnci/djw148
241. McCormick A, Donoghue P, Dixon M, O'Sullivan R, O'Donnell RL, Murray J, et al. Ovarian Cancers Harbor Defects in Nonhomologous End Joining Resulting in Resistance to Rucaparib. Clin Cancer Res (2017) 23(8):2050–60. doi: 10.1158/1078-0432.CCR-16-0564
242. Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous End Joining Drives Poly(ADP-Ribose) Polymerase (PARP) Inhibitor Lethality in Homologous Recombination-Deficient Cells. Proc Natl Acad Sci USA (2011) 108(8):3406–11. doi: 10.1073/pnas.1013715108
243. Haynes B, Murai J, Lee J-M. Restored Replication Fork Stabilization, a Mechanism of PARP Inhibitor Resistance, can be Overcome by Cell Cycle Checkpoint Inhibition. Cancer Treat Rev (2018) 71:1–7. doi: 10.1016/j.ctrv.2018.09.003
244. Rundle S, Bradbury A, Drew Y, Curtin NJ. Targeting the ATR-CHK1 Axis in Cancer Therapy. (2017) 9(5):41. doi: 10.3390/cancers9050041
245. Meghani K, Fuchs W, Detappe A, Drane P, Gogola E, Rottenberg S, et al. Multifaceted Impact of MicroRNA 493-5p on Genome-Stabilizing Pathways Induces Platinum and PARP Inhibitor Resistance in BRCA2-Mutated Carcinomas. Cell Rep (2018) 23(1):100–11. doi: 10.1016/j.celrep.2018.03.038
246. Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, et al. Replication Fork Stability Confers Chemoresistance in BRCA-Deficient Cells. Nature (2016) 535(7612):382–7. doi: 10.1038/nature18325
247. Makvandi M, Pantel A, Schwartz L, Schubert E, Xu K, Hsieh CJ, et al. A PET Imaging Agent for Evaluating PARP-1 Expression in Ovarian Cancer. J Clin Invest (2018) 128(5):2116–26. doi: 10.1172/JCI97992
248. Pettitt SJ, Krastev DB, Brandsma I, Drean A, Song F, Aleksandrov R, et al. Genome-Wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat Commun (2018) 9(1):1849. doi: 10.1038/s41467-018-03917-2
249. Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High Sensitivity of BRCA1-Deficient Mammary Tumors to the PARP Inhibitor AZD2281 Alone and in Combination With Platinum Drugs. Proc Natl Acad Sci USA (2008) 105(44):17079–84. doi: 10.1073/pnas.0806092105
250. Binkhathlan Z, Lavasanifar A. P-Glycoprotein Inhibition as a Therapeutic Approach for Overcoming Multidrug Resistance in Cancer: Current Status and Future Perspectives. Curr Cancer Drug Targets (2013) 13(3):326–46. doi: 10.2174/15680096113139990076
251. Choi YH, Yu AM. ABC Transporters in Multidrug Resistance and Pharmacokinetics, and Strategies for Drug Development. Curr Pharm Design (2014) 20(5):793–807. doi: 10.2174/138161282005140214165212
252. Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, et al. 53BP1 Loss Rescues BRCA1 Deficiency and is Associated With Triple-Negative and BRCA-Mutated Breast Cancers. Nat Struct Mol Biol (2010) 17:688. doi: 10.1038/nsmb.1831
253. Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 Regulates DSB Repair Using Rif1 to Control 5' End Resection. Science (2013) 339(6120):700–4. doi: 10.1126/science.1231573
254. Bouwman P, Jonkers J. Molecular Pathways: How can BRCA-Mutated Tumors Become Resistant to PARP Inhibitors? Clin Cancer Res (2014) 20(3):540–7. doi: 10.1158/1078-0432.CCR-13-0225
255. Dev H, Chiang T-WW, Lescale C, de Krijger I, Martin AG, Pilger D, et al. Shieldin Complex Promotes DNA End-Joining and Counters Homologous Recombination in BRCA1-Null Cells. Nat Cell Biol (2018) 20(8):954–65. doi: 10.1038/s41556-018-0140-1
256. Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, et al. 53bp1–RIF1–shieldin Counteracts DSB Resection Through CST- and Polα-Dependent Fill-in. Nature (2018) 560(7716):112–6. doi: 10.1038/s41586-018-0324-7
257. Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, et al. The Shieldin Complex Mediates 53BP1-Dependent DNA Repair. Nature (2018) 560(7716):117–21. doi: 10.1038/s41586-018-0340-7
258. Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell (2018) 173(4):972–88.e23. doi: 10.1016/j.cell.2018.03.050
259. Choi YE, Meghani K, Brault ME, Leclerc L, He YJ, Day TA, et al. Platinum and PARP Inhibitor Resistance Due to Overexpression of MicroRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell Rep (2016) 14(3):429–39. doi: 10.1016/j.celrep.2015.12.046
260. Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, et al. Blocking C-Met-Mediated PARP1 Phosphorylation Enhances Anti-Tumor Effects of PARP Inhibitors. Nat Med (2016) 22(2):194–201. doi: 10.1038/nm.4032
261. Dong Q, Du Y, Li H, Liu C, Wei Y, Chen MK, et al. EGFR and C-MET Cooperate to Enhance Resistance to PARP Inhibitors in Hepatocellular Carcinoma. Cancer Res (2019) 79(4):819–29. doi: 10.1158/0008-5472.CAN-18-1273
262. Pyndiah S, Tanida S, Ahmed KM, Cassimere EK, Choe C, Sakamuro D. C-MYC Suppresses BIN1 to Release Poly(ADP-Ribose) Polymerase 1: A Mechanism by Which Cancer Cells Acquire Cisplatin Resistance. Sci Signal (2011) 4(166):ra19. doi: 10.1126/scisignal.2001556
263. Carey JPW, Karakas C, Bui TY, Chen X, Vijayaraghavan S, Zhao Y, et al. Synthetic Lethality of PARP Inhibitors in Combination With MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res (2018) 78(3):742–57. doi: 10.1158/0008-5472.CAN-17-1494
264. Min A, Im S-A, Yoon Y-K, Song S-H, Nam H-J, Hur H-S, et al. RAD51C-Deficient Cancer Cells Are Highly Sensitive to the PARP Inhibitor Olaparib. (2013) 12(6):865–77. doi: 10.1158/1535-7163.MCT-12-0950
265. Deng Y, Cai Y, Huang Y, Yang Z, Bai Y, Liu Y, et al. High SLFN11 Expression Predicts Better Survival for Patients With KRAS Exon 2 Wild Type Colorectal Cancer After Treated With Adjuvant Oxaliplatin-Based Treatment. BMC Cancer (2015) 15:833. doi: 10.1186/s12885-015-1840-6
266. Murai J, Feng Y, Yu GK, Ru Y, Tang S-W, Shen Y, et al. Resistance to PARP Inhibitors by SLFN11 Inactivation can be Overcome by ATR Inhibition. Oncotarget (2016) 7(47):76534–50. doi: 10.18632/oncotarget.12266
267. Allison Stewart C, Tong P, Cardnell RJ, Sen T, Li L, Gay CM, et al. Dynamic Variations in Epithelial-to-Mesenchymal Transition (EMT), ATM, and SLFN11 Govern Response to PARP Inhibitors and Cisplatin in Small Cell Lung Cancer. Oncotarget (2017) 8(17):28575–87. doi: 10.18632/oncotarget.15338
268. Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, et al. PARP Inhibitor Activity Correlates With SLFN11 Expression and Demonstrates Synergy With Temozolomide in Small Cell Lung Cancer. Clin Cancer Res (2017) 23(2):523–35. doi: 10.1158/1078-0432.CCR-16-1040
269. Montoni A, Robu M, Pouliot E, Shah GM. Resistance to PARP-Inhibitors in Cancer Therapy. Front Pharmacol (2013) 4:18. doi: 10.3389/fphar.2013.00018
270. Scott CL, Swisher EM, Kaufmann SH. Poly (ADP-Ribose) Polymerase Inhibitors: Recent Advances and Future Development. J Clin Oncol (2015) 33(12):1397–406. doi: 10.1200/JCO.2014.58.8848
271. Kim Y, Kim A, Sharip A, Sharip A, Jiang J, Yang Q, et al. Reverse the Resistance to PARP Inhibitors. . Int J Biol Sci (2017) 13(2):198–208. doi: 10.7150/ijbs.17240
272. Ha DH, Min A, Kim S, Jang H, Kim SH, Kim HJ, et al. Antitumor Effect of a WEE1 Inhibitor and Potentiation of Olaparib Sensitivity by DNA Damage Response Modulation in Triple-Negative Breast Cancer. Sci Rep (2020) 10(1):9930. doi: 10.1038/s41598-020-66018-5
273. Johnson N, Li Y-C, Walton ZE, Cheng KA, Li D, Rodig SJ, et al. Compromised CDK1 Activity Sensitizes BRCA-Proficient Cancers to PARP Inhibition. Nat Med (2011) 17:875. doi: 10.1038/nm.2377
274. Kumar A, Fernandez-Capetillo O, Carrera AC. Nuclear Phosphoinositide 3-Kinase β Controls Double-Strand Break DNA Repair. Proc Natl Acad Sci (2010) 107(16):7491–6. doi: 10.1073/pnas.0914242107
275. Mo W, Liu Q, Lin CC-J, Dai H, Peng Y, Liang Y, et al. mTOR Inhibitors Suppress Homologous Recombination Repair and Synergize With PARP Inhibitors via Regulating SUV39H1 in BRCA-Proficient Triple-Negative Breast Cancer. Clin Cancer Res (2016) 22(7):1699–712. doi: 10.1158/1078-0432.CCR-15-1772
276. Kachhap SK, Rosmus N, Collis SJ, Kortenhorst MS, Wissing MD, Hedayati M, et al. Downregulation of Homologous Recombination DNA Repair Genes by HDAC Inhibition in Prostate Cancer is Mediated Through the E2F1 Transcription Factor. PLos One (2010) 5(6):e11208. doi: 10.1371/journal.pone.0011208
277. Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov (2017) 7(1):20–37. doi: 10.1158/2159-8290.CD-16-0860
278. Nowsheen S, Cooper T, Stanley JA, Yang ES. Synthetic Lethal Interactions Between EGFR and PARP Inhibition in Human Triple Negative Breast Cancer Cells. PLos One (2012) 7(10):e46614. doi: 10.1371/journal.pone.0046614
279. Gilardini Montani MS, Prodosmo A, Stagni V, Merli D, Monteonofrio L, Gatti V, et al. ATM-Depletion in Breast Cancer Cells Confers Sensitivity to PARP Inhibition. J Exp Clin Cancer Res (2013) 32(1):95. doi: 10.1186/1756-9966-32-95
280. Li L, Karanika S, Yang G, Wang J, Park S, Broom BM, et al. Androgen Receptor Inhibitor-Induced "BRCAness" and PARP Inhibition are Synthetically Lethal for Castration-Resistant Prostate Cancer. Sci Signaling (2017) 10(480). doi: 10.1126/scisignal.aam7479
281. Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep (2016) 17(9):2367–81. doi: 10.1016/j.celrep.2016.10.077
282. Follis AV, Hammoudeh DI, Daab AT, Metallo SJ. Small-Molecule Perturbation of Competing Interactions Between C-Myc and Max. Bioorg Med Chem Lett (2009) 19(3):807–10. doi: 10.1016/j.bmcl.2008.12.025
283. Higuchi T, Flies DB, Marjon NA, Mantia-Smaldone G, Ronner L, Gimotty PA, et al. CTLA-4 Blockade Synergizes Therapeutically With PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol Res (2015) 3(11):1257–68. doi: 10.1158/2326-6066.CIR-15-0044
284. Huang J, Wang L, Cong Z, Amoozgar Z, Kiner E, Xing D, et al. The PARP1 Inhibitor BMN 673 Exhibits Immunoregulatory Effects in a Brca1(-/-) Murine Model of Ovarian Cancer. Biochem Biophys Res Commun (2015) 463(4):551–6. doi: 10.1016/j.bbrc.2015.05.083
285. Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. PARP Inhibitor Efficacy Depends on CD8(+) T-Cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discovery (2019) 9(6):722–37. doi: 10.1158/2159-8290.CD-18-1218
286. Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and Prognostic Significance of BRCA1/2-Mutation Status With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes and Expression of PD-1/PD-L1 in High Grade Serous Ovarian Cancer. Oncotarget (2016) 7(12):13587–98. doi: 10.18632/oncotarget.7277
287. Meng J, Peng J, Feng J, Maurer J, Li X, Li Y, et al. Niraparib Exhibits a Synergistic Anti-Tumor Effect With PD-L1 Blockade by Inducing an Immune Response in Ovarian Cancer. J Transl Med (2021) 19(1):415. doi: 10.1186/s12967-021-03073-0
288. Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, et al. DNA Double-Strand Break Repair Pathway Regulates PD-L1 Expression in Cancer Cells. Nat Commun (2017) 8(1):1751. doi: 10.1038/s41467-017-01883-9
289. Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res (2017) 23(14):3711–20. doi: 10.1158/1078-0432.CCR-16-3215
290. Vikas P, Borcherding N, Chennamadhavuni A, Garje R. Therapeutic Potential of Combining PARP Inhibitor and Immunotherapy in Solid Tumors. Front Oncol (2020) 10:570. doi: 10.3389/fonc.2020.00570
291. Wu Z, Cui P, Tao H, Zhang S, Ma J, Liu Z, et al. The Synergistic Effect of PARP Inhibitors and Immune Checkpoint Inhibitors. Clin Med Insights Oncol (2021) 15:1179554921996288. doi: 10.1177/1179554921996288
292. Stafford JL, Dyson G, Levin NK, Chaudhry S, Rosati R, Kalpage H, et al. Reanalysis of BRCA1/2 Negative High Risk Ovarian Cancer Patients Reveals Novel Germline Risk Loci and Insights Into Missing Heritability. PLos One (2017) 12(6):e0178450. doi: 10.1371/journal.pone.0178450
293. Zhao Q, Yang J, Li L, Cao D, Yu M, Shen K. Germline and Somatic Mutations in Homologous Recombination Genes Among Chinese Ovarian Cancer Patients Detected Using Next-Generation Sequencing. J Gynecol Oncol (2017) 28(4):e39. doi: 10.3802/jgo.2017.28.e39
294. Beltrame L, Di Marino M, Fruscio R, Calura E, Chapman B, Clivio L, et al. Profiling Cancer Gene Mutations in Longitudinal Epithelial Ovarian Cancer Biopsies by Targeted Next-Generation Sequencing: A Retrospective Study. Ann Oncol (2015) 26(7):1363–71. doi: 10.1093/annonc/mdv164
295. Zighelboim I, Schmidt AP, Gao F, Thaker PH, Powell MA, Rader JS, et al. ATR Mutation in Endometrioid Endometrial Cancer is Associated With Poor Clinical Outcomes. J Clin Oncol (2009) 27(19):3091–6. doi: 10.1200/JCO.2008.19.9802
296. Caminsky NG, Mucaki EJ, Perri AM, Lu R, Knoll JH, Rogan PK. Prioritizing Variants in Complete Hereditary Breast and Ovarian Cancer Genes in Patients Lacking Known BRCA Mutations. Hum Mutat (2016) 37(7):640–52. doi: 10.1002/humu.22972
297. Minion LE, Dolinsky JS, Chase DM, Dunlop CL, Chao EC, Monk BJ. Hereditary Predisposition to Ovarian Cancer, Looking Beyond BRCA1/BRCA2. Gynecologic Oncol (2015) 137(1):86–92. doi: 10.1016/j.ygyno.2015.01.537
298. Kraus C, Hoyer J, Vasileiou G, Wunderle M, Lux MP, Fasching PA, et al. Gene Panel Sequencing in Familial Breast/Ovarian Cancer Patients Identifies Multiple Novel Mutations Also in Genes Others Than BRCA1/2. Int J Cancer (2017) 140(1):95–102. doi: 10.1002/ijc.30428
299. Tam KF, Liu VW, Liu SS, Tsang PC, Cheung AN, Yip AM, et al. Methylation Profile in Benign, Borderline and Malignant Ovarian Tumors. J Cancer Res Clin Oncol (2007) 133(5):331–41. doi: 10.1007/s00432-006-0178-5
300. Bogdanova N, Togo AV, Ratajska M, Kluzniak W, Takhirova Z, Tarp T, et al. Prevalence of the BLM Nonsense Mutation, P.Q548X, in Ovarian Cancer Patients From Central and Eastern Europe. Familial Cancer (2015) 14(1):145–9. doi: 10.1007/s10689-014-9748-x
301. de Souza Timoteo AR, Goncalves A, Sales LAP, Albuquerque BM, de Souza JES, de Moura PCP, et al. A Portrait of Germline Mutation in Brazilian at-Risk for Hereditary Breast Cancer. Breast Cancer Res Treat (2018) 172(3):637–46. doi: 10.1007/s10549-018-4938-0
302. Encinas G, Sabelnykova VY, de Lyra EC, Hirata Katayama ML, Maistro S, de Vasconcellos Valle PWM, et al. Somatic Mutations in Early Onset Luminal Breast Cancer. Oncotarget (2018) 9(32):22460–79. doi: 10.18632/oncotarget.25123
303. Marsh A, Healey S, Lewis A, Spurdle AB, Kedda MA, Khanna KK, et al. Mutation Analysis of Five Candidate Genes in Familial Breast Cancer. Breast Cancer Res Treat (2007) 105(3):377–89. doi: 10.1007/s10549-006-9461-z
304. De Mattos-Arruda L, Ng CKY, Piscuoglio S, Gonzalez-Cao M, Lim RS, De Filippo MR, et al. Genetic Heterogeneity and Actionable Mutations in HER2-Positive Primary Breast Cancers and Their Brain Metastases. Oncotarget (2018) 9(29):20617–30. doi: 10.18632/oncotarget.25041
305. Lolas Hamameh S, Renbaum P, Kamal L, Dweik D, Salahat M, Jaraysa T, et al. Genomic Analysis of Inherited Breast Cancer Among Palestinian Women: Genetic Heterogeneity and a Founder Mutation in TP53. Int J Cancer (2017) 141(4):750–6. doi: 10.1002/ijc.30771
306. Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, et al. Frequency of Mutations in Individuals With Breast Cancer Referred for BRCA1 and BRCA2 Testing Using Next-Generation Sequencing With a 25-Gene Panel. Cancer (2015) 121(1):25–33. doi: 10.1002/cncr.29010
307. Macias-Gomez NM, Peralta-Leal V, Meza-Espinoza JP, Gutierrez-Angulo M, Duran-Gonzalez J, Ramirez-Gonzalez JM, et al. Polymorphisms of the XRCC1 Gene and Breast Cancer Risk in the Mexican Population. Familial Cancer (2015) 14(3):349–54. doi: 10.1007/s10689-015-9787-y
308. Moran O, Nikitina D, Royer R, Poll A, Metcalfe K, Narod SA, et al. Revisiting Breast Cancer Patients Who Previously Tested Negative for BRCA Mutations Using a 12-Gene Panel. Breast Cancer Res Treat (2017) 161(1):135–42. doi: 10.1007/s10549-016-4038-y
309. Maatta KM, Nurminen R, Kankuri-Tammilehto M, Kallioniemi A, Laasanen SL, Schleutker J. Germline EMSY Sequence Alterations in Hereditary Breast Cancer and Ovarian Cancer Families. BMC Cancer (2017) 17(1):496. doi: 10.1186/s12885-017-3488-x
310. Liang X, Vacher S, Boulai A, Bernard V, Baulande S, Bohec M, et al. Targeted Next-Generation Sequencing Identifies Clinically Relevant Somatic Mutations in a Large Cohort of Inflammatory Breast Cancer. Breast Cancer Res (2018) 20(1):88. doi: 10.1186/s13058-018-1007-x
311. Kazim Z, Wahabi K, Perwez A, Lal P, Rizvi MA. PTEN Genetic and Epigenetic Alterations Define Distinct Subgroups in North Indian Breast Cancer Patients. Asian Pacific J Cancer Prev (2019) 20(1):269–76. doi: 10.31557/APJCP.2019.20.1.269
312. Yari K, Payandeh M, Rahimi Z. Association of the Hypermethylation Status of PTEN Tumor Suppressor Gene With the Risk of Breast Cancer Among Kurdish Population From Western Iran. Tumour Biol (2016) 37(6):8145–52. doi: 10.1007/s13277-015-4731-1
313. Brovkina OI, Shigapova L, Chudakova DA, Gordiev MG, Enikeev RF, Druzhkov MO, et al. The Ethnic-Specific Spectrum of Germline Nucleotide Variants in DNA Damage Response and Repair Genes in Hereditary Breast and Ovarian Cancer Patients of Tatar Descent. Front Oncol (2018) 8:421. doi: 10.3389/fonc.2018.00421
Keywords: pathogenic mutation, loss of heterozygosity, promoter hypermethylation, DNA repair genes, hereditary and familial cancer, PARP inhibitor, base excision repair
Citation: Mekonnen N, Yang H and Shin YK (2022) Homologous Recombination Deficiency in Ovarian, Breast, Colorectal, Pancreatic, Non-Small Cell Lung and Prostate Cancers, and the Mechanisms of Resistance to PARP Inhibitors. Front. Oncol. 12:880643. doi: 10.3389/fonc.2022.880643
Received: 21 February 2022; Accepted: 18 May 2022;
Published: 17 June 2022.
Edited by:
Ian Campbell, Peter MacCallum Cancer Centre, AustraliaReviewed by:
Guang Peng, University of Texas MD Anderson Cancer Center, United StatesPete Simpson, The University of Queensland, Australia
Copyright © 2022 Mekonnen, Yang and Shin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Young Kee Shin, eWtlZXNoaW5Ac251LmFjLmty
†These authors have contributed equally to this work