 Keely Tan
Keely Tan
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 13 May 2022
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.876451
This article is part of the Research Topic Stromal and Immune Microenvironment in Breast Cancer Metastasis View all 6 articles
Breast cancer is a complex, dynamic disease that acquires heterogeneity through various mechanisms, allowing cancer cells to proliferate, survive and metastasise. Heterogeneity is introduced early, through the accumulation of germline and somatic mutations which initiate cancer formation. Following initiation, heterogeneity is driven by the complex interaction between intrinsic cellular factors and the extrinsic tumour microenvironment (TME). The TME consists of tumour cells and the subsequently recruited immune cells, endothelial cells, fibroblasts, adipocytes and non-cellular components of the extracellular matrix. Current research demonstrates that stromal-immune cell interactions mediated by various TME components release environmental cues, in mechanical and chemical forms, to communicate with surrounding and distant cells. These interactions are critical in facilitating the metastatic process at both the primary and secondary site, as well as introducing greater intratumoral heterogeneity and disease complexity by exerting selective pressures on cancer cells. This can result in the adaptation of cells and a feedback loop to the cancer genome, which can promote therapeutic resistance. Thus, targeting TME and immune-stromal cell interactions has been suggested as a potential therapeutic avenue given that aspects of this process are somewhat conserved between breast cancer subtypes. This mini review will discuss emerging ideas on how the interaction of various aspects of the TME contribute to increased heterogeneity and disease progression, and the therapeutic potential of targeting the TME.
Breast cancer is a complex and heterogeneous disease that accounted for 24.5% of cancer diagnoses and 15.5% of cancer-related deaths in women in 2020 alone, making it the most commonly diagnosed and most lethal cancer in women worldwide (1). In Australia, the 5-year survival rate for those diagnosed with early-stage breast cancer is 91%, but this dramatically decreases to 32% in patients with invasive metastatic disease (2). Metastasis is a primary hallmark of cancer and is a multi-step process that begins with intravasation of cancer cells from the primary tumour site, migration and survival in the vascular or lymphatic systems, and is completed following extravasation and colonisation at a distal site (3, 4). The ability of cancer cells to survive, progress and metastasise is largely determined by intratumoral heterogeneity (ITH), which is dependent on both genetic and environmental influences (5). Whilst environmental influences encompasses both external and internal factors, this review will focus on the internal tumor microenvironment (TME).
The formation of the TME is initiated in response to metabolic needs of rapidly proliferating cancer cells. At the heart of this complex network are the tumor cells themselves, which manipulate surrounding cells through signaling networks activated by biochemical and biomechanical mechanisms. The cell types present in the TME include a variety of non-malignant stromal cells, such as endothelial cells, adipocytes, fibroblasts, immune cells, and extracellular matrix (ECM) proteins (6) (Figure 1). They influence cancer progression by contributing to cellular genomic and biological variations, increasing clonal evolution, and thus increasing ITH (7, 8). It is thought that increased ITH is a key feature of cancer cell survival and progression (6, 9). This mini review will discuss emerging ideas on how key players of the breast cancer TME influence ITH, metastasis and therapeutic resistance. Whilst the focus of this review will be on the tumor-promoting functions of the TME, it is important to acknowledge that these factors exist in a relatively plastic state, and thus are only pro-tumorigenic in optimal conditions (10).
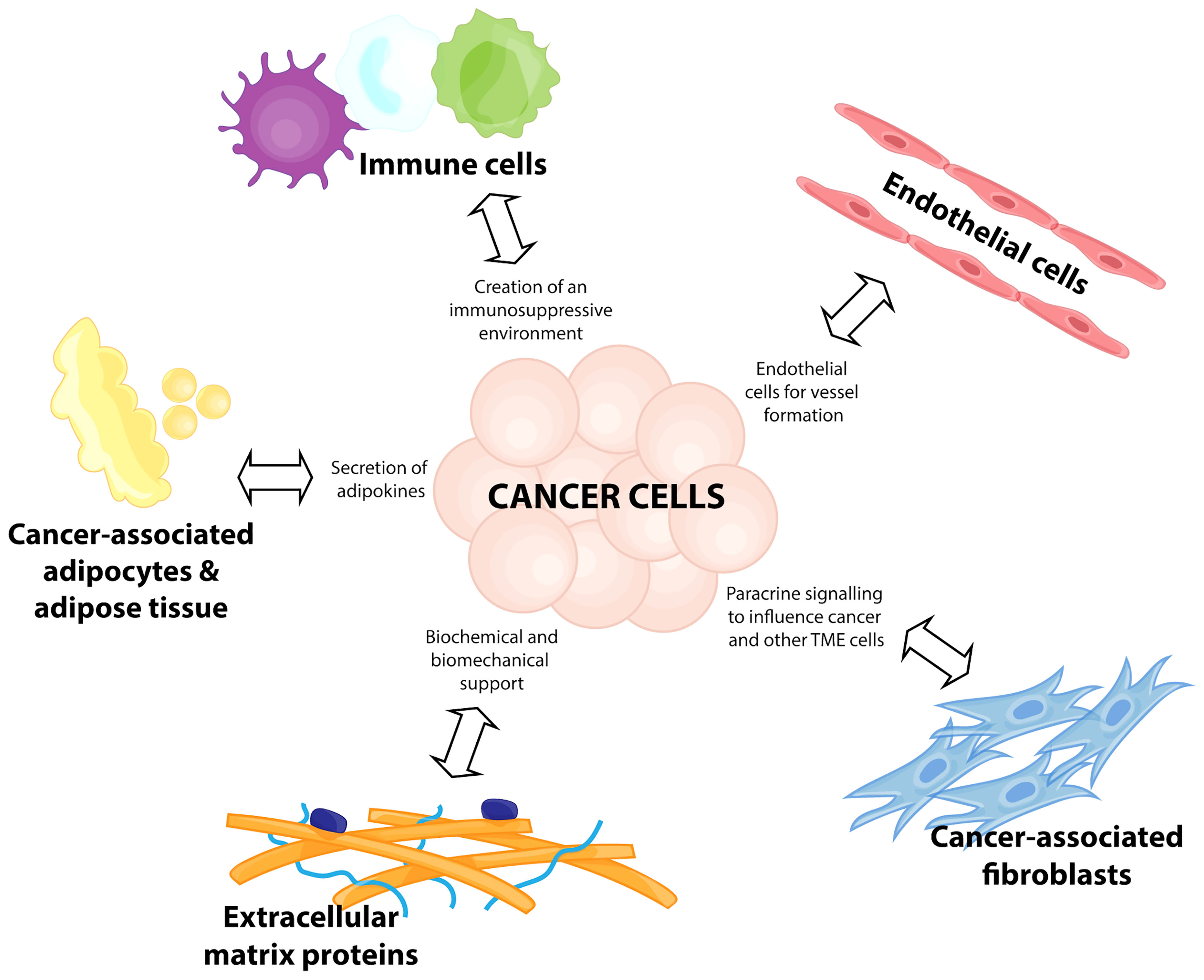
Figure 1 Components of a breast cancer tumor microenvironment. Breast cancer disease progression relies on a complex network of cells and interactions, termed the tumor microenvironment (TME). This consists of immune cells for the creation of an immunosuppressive environment, recruitment of endothelial cells for vessel formation, transformation of cancer-associated fibroblasts to participate in paracrine signalling to influence the cancer cells and other TME cells, release of adipokines from cancer-associated adipocytes and adipose tissue, and the biochemical and biomechanical support from extracellular matrix proteins.
ITH is defined as the genomic and biological variations acquired from exposure to various microenvironmental elements, contributing to the phenotypic characteristics of the cancer and increasing its survival capabilities (7, 8). Whilst it is known that intrinsic cell heterogeneity exists within a tumor cell population, greater heterogeneity can be introduced through environmental influence. Increasing the diversity of TME components in the peritumor space provides a plethora of signals to cancer cells, contributing to increased ITH through genetic and reversible epigenetic modulation (7, 10). For example, hypoxia has been linked to the regulation of epigenetic modulators, including G9a histone H3 lysine 9 (H3K9me) methyltransferase (11) and histone demethylase jumonji domain containing protein 2C (JMJD2C) (12). This increases the activity of hypoxia inducible transcription factor (HIF)-1, which contributes to breast cancer progression. Furthermore, cancer associated fibroblasts (CAFs) and their associated ECM products also trigger epigenetic modifications that influence cancer phenotype.
Initially, tumor-secreted factors recruit and transform surrounding non-malignant cells into cancer-associated cells, but these cancer-associated cells also possess their own ability to influence other TME cell types through dynamic, bi-directional communication (6). Typically, a range of immune cells are involved in launching an anti-tumor immune response, including natural killer (NK) cells, CD8+ and CD4+ T cells and dendritic cells (DCs) (13, 14). However, these cells can be exploited by various TME elements to become pro-tumorigenic. As a tumor begins to expand, its growth is limited by the increasing hypoxic microenvironment and decreasing metabolic substrates, triggering an angiogenic switch via HIF-1 and -2 activation. This results in a range of physiological effects, including the production of pro-angiogenic growth factors, such as vascular endothelial growth factor (VEGF) (15). These factors encourage recruitment and proliferation of endothelial cells from pre-existing capillary beds, resulting in the formation of a disorganised and highly permeable tumor vascular network, allowing for the transport of more oxygen and nutrients (16). In addition, a VEGF gradient and interstitial flow from leaky tumor vasculature promotes lymphangiogenesis, resulting in lympathic endothelial cell migration in the direction of flow and gradient (17, 18). The presence of these leaky vessels and the mechanical stress caused by increasing ECM production by TME components, results in the transportation of antigen-presenting cells (APCs) from adjacent tissue. The presence of these antigens stimulates the recruitment of macrophages and DCs to the tumor site (19). Whilst normally anti-tumorigenic, secretion of factors by TME components, such as transforming growth factor (TGF)-ß by CAFs, has the potential to neutralize the anti-tumor response of NK cells, neutrophils and macrophages (20), enhancing immune evasion. Concurrently, the recruitment and enhancement of immunosuppressive cells, such as regulatory T cells (Tregs) and tumor-associated macrophages (TAMs), can create an immunosuppressive TME (21, 22).
Hypoxic breast cancer cells also secrete a variety of paracrine signaling molecules that can reprogram progenitor cells into CAFs (23). CAFs represent a major portion of the breast tumor stroma and are a highly heterogeneous population derived from various cell types, such as resident fibroblasts, bone marrow cells and adipocytes (24–26). CAF-secreted factors such as TGF-ß, act in an autocrine manner to further promote differentiation of fibroblasts into CAFs (27). These CAF-secreted factors can also influence the functions of other cell types within the TME (28). Secretion of stimulating factors, such as IL-6, results in ECM remodeling and matrix stiffening, providing a physical barrier to immune cells, particularly T lymphocytes (28), while conversely increasing the number of infiltrating TAMs (29). Whilst TME components can reduce infiltration of anti-tumorigenic immune cells, the presence of extracellular vesicles (EVs) has also been shown to decrease proliferation of CD8+ and CD4+ T cells (30). In addition to being a CAF precursor, cancer-associated adipocytes (CAAs) have been associated with breast cancer progression. The breast is rich in adipose tissue, and the interaction between the adipocytes and breast cancer cells is significant in disease progression (31). CAAs exhibit a unique morphology and phenotype, with smaller and dispersed lipid droplets, a decrease in volume with a dilated interstitial space, and alterations in shape (32, 33). These CAAs can be peritumoral or intratumoral and release various adipokines, such as IL-6 and leptin. Interestingly, the effect of CAAs differs depending on the location, with intratumorally located CAAs reflective of decreased cancer cell proliferation and improved patient survival (34), whilst peritumoral CAAs are associated with poor prognosis (33). However, both intra- and peritumoral CAAs are associated with increased inflammation and metastasis. It is postulated that the release of adipokines attracts immune cells, such as monocytes, to the primary tumor site, which facilitates immune evasion (35, 36). Ultimately, the recruitment and reprogramming of various non-malignant cell types creates a diverse TME, which functions to support and facilitate breast cancer progression.
Once the TME framework has been established to support the growth and survival of the primary breast tumor, these various components can promote the metastatic dissemination of cancer cells (37, 38). For breast cancer, the major sites of metastasis are lungs, liver, brain and bone (39), however prior to undergoing metastasis breast cancer cells release chemokines and cytokines into the circulation. The premetastatic release of these chemokine and cytokines creates a favourable premetastatic niche (PMN), which recruits circulating tumor cells (CTCs) to the secondary site (37, 40). Thus, it is the combination of both the primary TME and the PMN that drives metastasis. These two microenvironments support breast cancer cells in the process of invasion and intravasation, survival in the lymphatic and vascular systems, extravasation and successful colonisation. Once CTCs have reached the distal site, the formation of a metastatic niche is initiated, which supports the growth of the secondary lesion (41) (Figure 2).
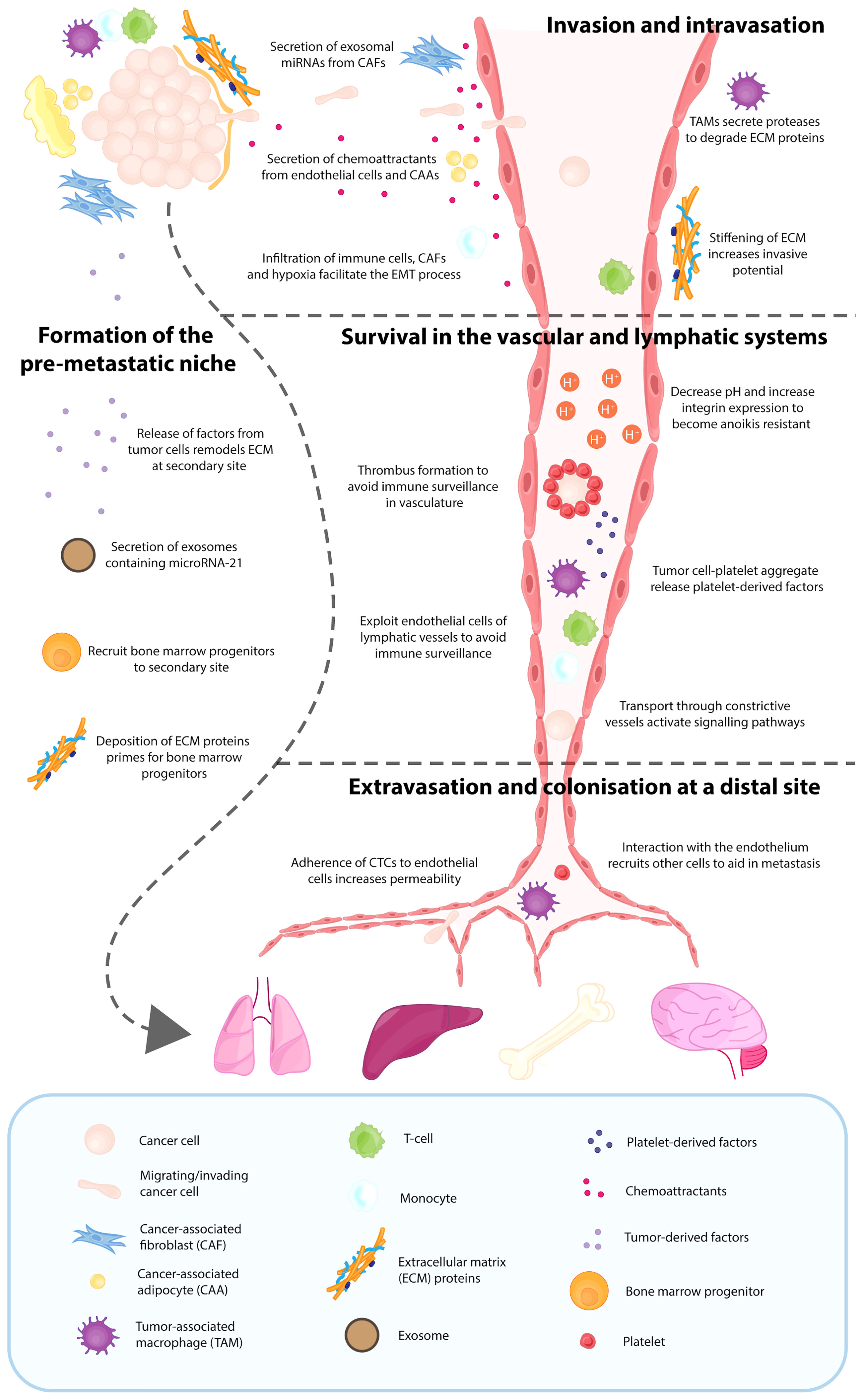
Figure 2 Contribution of various tumor microenvironment components in the process of metastasis. Breast cancer metastasis relies on various components of the tumor microenvironment, with the major sites of metastasis being the lungs, liver, brain and bone. Even before breast cancer cells escape from the primary site, several factors and exosomes have been released into the circulation by the breast cancer cells, which results in deposition of extracellular matrix (ECM) proteins at the distal site and recruitment of bone marrow progenitors. This creates a favourable pre-metastatic niche (PMN), which is crucial for metastatic dissemination. The TME and PMN support breast cancer cells in the process of invasion and intravasation, survival in the lymphatic and vascular systems, and the eventual extravasation and successful colonisation. Once these CTCs have reached the distal organ, the formation of a metastatic niche is initiated, which supports the growth of the secondary lesion.
The formation of the PMN is initiated with local changes and followed by systemic changes. Initially, vascular leakiness occurs because of the disorganised tumor-derived vasculature, resulting in alterations to resident cells, such as fibroblasts, and the recruitment of non-resident cells, such as bone marrow progenitors (40). In breast cancer, HIF signalling has been demonstrated to result in the secretion of lysyl oxidase (LOX), LOX-like proteins (LOXL2 and 4) and exosomes, which has previously been shown to be crucial in establishing a premetastatic niche in the lungs and bones (42). In contrast, Hif1α deletion in a PyMT mammary cancer model reduced bone metastasis but was associated with increased pulmonary metastasis (43). Previous research has also correlated a hypoxia transcriptome with both bone (44) and lung (45) metastasis signatures, suggesting that although there are genetic changes reflected in the transcriptome, there may also be distinctive signals within the TME that cells respond to in order to determine the phenotypic response (43). The release of these factors by the primary tumor can result in ECM remodeling at the secondary metastatic site, priming the location for bone marrow progenitor cell colonisation. VEGFR1+ haematopoietic bone marrow progenitors, which are mobilised during the tumor’s angiogenic switch, have also been demonstrated to be crucial in the initiation of the PMN. The homing of these VEGFR1+ bone marrow progenitors to the premetastatic site is dependent on the prior increased deposition of ECM proteins, such as fibronectin, which binds to its receptor integrin α4ß1 on progenitor cells (40). More recently, the contribution of exosomes released from breast cancer cells has been demonstrated to be critical in the formation of the premetastatic niche for bone metastasis. This was through the activity of the exosomal derived microRNA-21, which is involved in the process of bone remodeling (46), which may suggest that site specific adaptations determine the organ of breast cancer metastasis.
Whilst the formation of this secondary TME is critical for the deposition of CTCs, the cells must first escape from the primary site. To successfully intravasate, cancer cells rely on the contribution of intrinsic cell properties, microenvironmental factors and mechanical cues (47). A fundamental aspect of metastasis is the ability of cancer cells to undergo EMT, and this process is facilitated by various elements of the TME, such as the presence of hypoxia, infiltration of immune cells and activity of CAFs (43, 48). These TME factors are also able to further promote the migratory and invasive ability of these transformed cancer cells. Cancer cell migration has been demonstrated to be supported by the expression of focal adhesion kinase (FAK) in CAFs through the secretion of exosomal miRNAs (49). In addition, various TME components, such as CAAs and endothelial cells, can secrete chemoattractants to promote the movement of cancer cells in the direction of the gradient (17, 36). For example, a study by Cho and colleagues used a 3D macrofluidic device to demonstrate the communication between lymphatic vessels and tumor cells through the CXCL21/CXCR7 axis, with lymphatic endothelial cells secreting CXCL21, a chemoattract for breast cancer cells expressing CXCR7, causing invasion of MDA-MB-231 towards the lymphatic vessels (17). The secretion of degradative enzymes by TAMs and EVs, function to lyse surrounding ECM, further supporting the process of invasion (50, 51). These microenvironmental factors also have the potential to modify ECM proteins, which results in ECM stiffening. The stiffening of the ECM sends mechanical cues to further perpetuate the activation of CAFs in a positive feedback loop, and this tissue rigidity is associated with more aggressive breast cancers, with worse patient prognosis (52). Rigidity has been associated with upregulation of Mammalian-enabled (Mena), particularly the MenaINV isoform, which is a protein that has been shown to be upregulated in invasive breast cancer cells and associated with invadopodia maturation (52, 53). Furthermore, the direct contact between an MenaINV overexpressing breast cancer cell, an endothelial cell and a Tie2high/VEGFhigh perivascular macrophage is crucial in forming the TME of metastasis (TMEM), a process required for intravasation (54).
Whilst the newly formed vascular and lymphatic networks can act as a transport route for cancer cells to secondary distal sites, travelling through these harsh environments exerts selective pressures that cancer cells must overcome (55). Upon exiting the primary tumor site, cancer cells experience anoikis, a form of cell death initiated from the detachment of cells from the ECM (56). In order to overcome this and become anoikis resistant, cancer cells can employ various mechanisms, such as reprograming the metabolism of the TME, decreasing the pH (57) and upregulating the expression of specific integrins (58). Once tumor cells enter the vascular system, they activate platelets to form a thrombus around the cells, protecting cancer cells from immune surveillance (59, 60). It is thought that this tumor cell platelet aggregate releases a plethora of platelet-derived factors, such as TGF-ß1, which has the potential to decrease the antitumor activity of NK cells (61). Tumor cells that disseminate through the lymphatic system initially experience a hypoxic microenvironment, which is overcome by exploiting the native vasculature of the lymph node (62). To survive in the lymphatic system, cancer cells must also evade immune surveillance, which can be achieved through exploitation of the lymphatic endothelial cell’s ability to scavenge and cross-present lymph antigens, resulting in removal of autoreactive naive CD8+ T cells (62). Furthermore, the migration of these CTCs through highly constrictive vessels can via mechanotransduction mechanisms, result in the activation of various signalling pathways which may contribute to increased cancer cell survival, heterogeneity and motility (63, 64).
The process of metastasis is inefficient, with <0.01% of cells disseminated from the primary tumor successfully colonising at a distal site. This process generally occurs within the capillaries, where they are able to lodge and interact with the endothelium (55). This interaction with the endothelium can promote the recruitment of various other cells, such as macrophages, platelets and neutrophils which aid in the successful metastasis of cancer cells (65, 66). Furthermore, components of the TME, such as hypoxia-associated HIF activity of cancer cells, have been implicated in the process of extravasation. This involves stimulating the release of factors such as L1 cell adhesion molecule and angiopoietin-like 4 that promote the adherence of CTCs to the endothelium of lungs and interferes with endothelial cell adhesion molecules, respectively, increasing vascular permeability (67). It is important to note that the optimum metastatic conditions are tissue specific, an important consideration when attempting to identify new treatment options for metastatic disease. For example, CTCs honing towards the brain secrete factors, such as α-crystallin, neuroserpin and cathepsin S (68), whereas CTCs with an affinity for the lungs tend to depend on myeloid cell populations (69). During the initial formation of any metastatic foci, the cancer cells remain close to existing vasculature before stimulating angiogenesis for its own blood supply (68). Once the cells have successfully colonised, the microenvironment is again altered by the newly deposited tumor cells, and this is then termed the metastatic niche (41).
In addition to its contribution to breast cancer progression and metastasis, various components of the TME have been implicated in the therapeutic resistance of breast tumors (70, 71). Brechbuhl and colleagues identified that the contribution of CAF subpopulations to therapeutic resistance varies, with CD146+ CAFs in estrogen receptor (ER) positive breast cancer cells displaying sensitivity to tamoxifen treatment, while CD146- CAFs were correlated with decreased ER expression in the same cells and thus increased resistance to tamoxifen treatment (71). A recent study has identified that there is a novel relationship between ductal carcinoma in situ and fibroblasts that express platelet-derived growth factor receptor (PDGFR)α(low)/PDGFRß(high) through elevated Notch signalling in fibroblasts (72). This may be due to the paracrine interactions of PDGF-CC released from breast cancer cells interacting with fibroblasts through PDGFRs, orchestrating an ERα-negative phenotype through stimulation of hepatocyte growth factor (HGF), insulin-like growth factor binding protein (IGFBP) 3 and stanniocalcin (STC) 1 secretion from CAFs (73). Subsequent treatment of PeRo-Lum1 cells, a luminal mammary cancer cell line, with CAF-conditioned medium resulted in decreased cell sensitivity to tamoxifen through the decrease in ERα expression, suggesting that the interactions between CAFs and cancer cells can alter the molecular subtype of breast cancers and ultimately, endocrine therapy resistance. Furthermore, exosomes can promote therapeutic resistance by enabling interaction between TME components as well as through direct interaction with breast cancer cells (70, 74). Given the major contribution of TME components to breast cancer progression, some TME targeting therapeutics have been identified.
Breast cancer was previously viewed as a disease with low immunogenicity, however recent research demonstrates the possibility of immunotherapy in the treatment of breast cancer (75). This is further supported with atezolizumab and pembrolizumab being recently FDA-approved for use in combination with chemotherapy for programmed death ligand 1-positive triple negative breast cancers (76). In addition, the most recent WHO histopathological assessment of breast tumor guide now includes investigation of tumor-infiltrating leukocytes and fibrotic foci (77), highlighting the increasing recognition of the TME in determining disease severity. A recent study by Harney and colleagues also demonstrated the effects of a potent Tie2 inhibitor, rebastinib, on a mammary cancer model (78). Independent treatment resulted in reduced Tie2+ macrophages, TMEM function and angiogenesis, which presented as decreased mammary tumor growth, metastasis and overall increased survival, however it was the combination of rebastinib and paclitaxel that was most effective. Most of the available studies highlight the success in combining therapies, whether that be traditional therapies such as radiotherapy and chemotherapy, with a TME-targeting agent, such as anti-angiogenics and immunotherapy (76, 78) or two TME-targeting agents (79). Whilst these studies are a step in the right direction, there is still much unknown with respect to translating TME targets into the clinic and thus this should be a focus of breast cancer research, particularly metastatic breast cancer.
It is only within the past 3 decades that cancer research has shifted from primarily focusing on cancer cells to appreciating that cancer is a complex system with many biological aspects that contribute to tumor growth and progression (80). Without the structural integrity of the TME, breast cancer cells would not successfully grow and metastasise. It is factors released from the primary TME that establish a premetastatic niche at a distal secondary site, thus targeting aspects of the TME has the potential to decrease the prevalence of metastasis, the greatest contributor to breast cancer mortality rates. In addition, the components of the TME are somewhat conserved between breast cancer subtypes, offering a broad-spectrum therapeutic target for breast cancer.
KT designed the study, was responsible for writing the article and the creation of all figures. MN designed the study and was responsible for writing and revising the manuscript. All authors contributed to the generation of the concepts and ideas provided.
This work was supported by Cancer Council NSW Research Project Grant (RG 20-08) and Priority-driven Collaborative Cancer Research Scheme (Grant #1130499), funded by the National Breast Cancer Foundation Australia with the assistance of Cancer Australia awarded to MN.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. NBCF. Breast Cancer Stats 2021. Available at: https://nbcf.org.au/about-breast-cancer/breast-cancer-stats/.
3. Hanahan D, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
4. Gupta GP, Massagué J. Cancer Metastasis: Building a Framework. Cell (2006) 127(4):679–95. doi: 10.1016/j.cell.2006.11.001
5. Marusyk A, Almendro V, Polyak K. Intra-Tumour Heterogeneity: A Looking Glass for Cancer? Nat Rev Cancer (2012) 12(5):323–34. doi: 10.1038/nrc3261
6. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun Signal (2020) 18:1–19. doi: 10.1186/s12964-020-0530-4
7. Liu J, Dang H, Wang XW. The Significance of Intertumor and Intratumor Heterogeneity in Liver Cancer. Exp Mol Med (2018) 50(1):e416. doi: 10.1038/emm.2017.165
8. Hinohara K, Polyak K. Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol (2019) 29(7):569–79. doi: 10.1016/j.tcb.2019.03.003
9. Hanahan D, Coussens LM. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell (2012) 21(3):309–22. doi: 10.1016/j.ccr.2012.02.022
10. Vitale I, Shema E, Loi S, Galluzzi L. Intratumoral Heterogeneity in Cancer Progression and Response to Immunotherapy. Nat Med (2021) 27(2):212–24. doi: 10.1038/s41591-021-01233-9
11. Casciello F, Al-Ejeh F, Kelly G, Brennan DJ, Ngiow SF, Young A, et al. G9a Drives Hypoxia-Mediated Gene Repression for Breast Cancer Cell Survival and Tumorigenesis. Proc Natl Acad Sci USA (2017) 114(27):7077–82. doi: 10.1073/pnas.1618706114
12. Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone Demethylase JMJD2C Is a Coactivator for Hypoxia-Inducible Factor 1 That Is Required for Breast Cancer Progression. Proc Natl Acad Sci USA (2012) 109(49):E3367–E76. doi: 10.1073/pnas.1217394109
13. Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front Immunol (2020) 11:940. doi: 10.3389/fimmu.2020.00940
14. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and Tumor Progression: Signaling Pathways and Targeted Intervention. Signal Transduct Tar Ther (2021) 6(1):263. doi: 10.1038/s41392-021-00658-5
15. Carmeliet P, Jain RK. Angiogenesis in Cancer and Other Diseases. Nature. (2000) 407(6801):249–57. doi: 10.1038/35025220
16. Azzi S, Hebda JK, Gavard J. Vascular Permeability and Drug Delivery in Cancers. Front Oncol (2013) 3:211. doi: 10.3389/fonc.2013.00211
17. Cho Y, Na K, Jun Y, Won J, Yang JH, Chung S. Three-Dimensional In Vitro Lymphangiogenesis Model in Tumor Microenvironment. Front Bioeng Biotechnol (2021) 9:697657. doi: 10.3389/fbioe.2021.697657
18. Choi D, Park E, Jung E, Seong YJ, Yoo J, Lee E, et al. Laminar Flow Downregulates Notch Activity to Promote Lymphatic Sprouting. J Clin Invest (2017) 127(4):1225–40. doi: 10.1172/JCI87442
19. Swartz MA, Lund AW. Lymphatic and Interstitial Flow in the Tumour Microenvironment: Linking Mechanobiology With Immunity. Nat Rev Canc (2012) 12(3):210–9. doi: 10.1038/nrc3186
20. Bierie B, Moses HL. Transforming Growth Factor Beta (TGF-Beta) and Inflammation in Cancer. Cytokine Growth Factor Rev (2010) 21(1):49–59. doi: 10.1016/j.cytogfr.2009.11.008
21. Bates JP, Derakhshandeh R, Jones L, Webb TJ. Mechanisms of Immune Evasion in Breast Cancer. BMC Canc (2018) 18(1):556. doi: 10.1186/s12885-018-4441-3
22. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune Evasion in Cancer: Mechanistic Basis and Therapeutic Strategies. Semin Cancer Biol (2015) 35 Suppl:S185–S98. doi: 10.1016/j.semcancer.2015.03.004
23. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The Hypoxic Tumour Microenvironment. Oncogenesis (2018) 7(1):10. doi: 10.1038/s41389-017-0011-9
24. Bochet L, Lehuede C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte-Derived Fibroblasts Promote Tumor Progression and Contribute to the Desmoplastic Reaction in Breast Cancer. Cancer Res (2013) 73(18):5657–68. doi: 10.1158/0008-5472.CAN-13-0530
25. Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell (2011) 19(2):257–72. doi: 10.1016/j.ccr.2011.01.020
26. Lappano R, Rigiracciolo DC, Belfiore A, Maggiolini M, De Francesco EM. Cancer Associated Fibroblasts: Role in Breast Cancer and Potential as Therapeutic Targets. Expert Opin Ther Tar (2020) 24(6):559–72. doi: 10.1080/14728222.2020.1751819
27. Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-Beta and Stromal Cell-Derived Factor-1 (SDF-1) Signaling Drives the Evolution of Tumor-Promoting Mammary Stromal Myofibroblasts. Proc Natl Acad Sci USA (2010) 107(46):20009–14. doi: 10.1073/pnas.1013805107
28. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk Between Cancer-Associated Fibroblasts and Immune Cells in the Tumor Microenvironment: New Findings and Future Perspectives. Mol Cancer (2021) 20(1):1–30. doi: 10.1186/s12943-021-01428-1
29. Seager RJ, Hajal C, Spill F, Kamm RD, Zaman MH. Dynamic Interplay Between Tumour, Stroma and Immune System can Drive or Prevent Tumour Progression. Converg Sci Phys Oncol (2017) 3:1–34. doi: 10.1088/2057-1739/aa7e86
30. Wen SW, Sceneay J, Lima LG, Wong CS, Becker M, Krumeich S, et al. The Biodistribution and Immune Suppressive Effects of Breast Cancer-Derived Exosomes. Cancer Res (2016) 76(23):6816–27. doi: 10.1158/0008-5472.CAN-16-0868
31. Kothari C, Diorio C, Durocher F. The Importance of Breast Adipose Tissue in Breast Cancer. Int J Mol Sci (2020) 21(16):1–34. doi: 10.3390/ijms21165760
32. Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res (2011) 71(7):2455–65. doi: 10.1158/0008-5472.CAN-10-3323
33. Zhao C, Wu M, Zeng N, Xiong M, Hu W, Lv W, et al. Cancer-Associated Adipocytes: Emerging Supporters in Breast Cancer. J Exp Clin Cancer Res (2020) 39(1):156. doi: 10.1186/s13046-020-01666-z
34. Tokumaru Y, Oshi M, Katsuta E, Yan L, Huang JL, Nagahashi M, et al. Intratumoral Adipocyte-High Breast Cancer Enrich for Metastatic and Inflammation-Related Pathways But Associated With Less Cancer Cell Proliferation. Int J Mol Sci (2020) 21(16):1–17. doi: 10.3390/ijms21165744
35. Wu Q, Li B, Li J, Sun S, Yuan J, Sun S. Cancer-Associated Adipocytes as Immunomodulators in Cancer. Biomark Res (2021) 9:1–21. doi: 10.1186/s40364-020-00257-6
36. Wu Q, Li B, Li Z, Li J, Sun S, Sun S. Cancer-Associated Adipocytes: Key Players in Breast Cancer Progression. J Hematol Oncol (2019) 12(1):95. doi: 10.1186/s13045-019-0778-6
37. Place AE, Jin Huh S, Polyak K. The Microenvironment in Breast Cancer Progression: Biology and Implications for Treatment. Breast Cancer Res (2011) 13(6):227. doi: 10.1186/bcr2912
38. Zhuyan J, Chen M, Zhu T, Bao X, Zhen T, Xing K, et al. Critical Steps to Tumor Metastasis: Alterations of Tumor Microenvironment and Extracellular Matrix in the Formation of Pre-Metastatic and Metastatic Niche. Cell Biosci (2020) 10:89. doi: 10.1186/s13578-020-00453-9
39. Bale R, Putzer D, Schullian P. Local Treatment of Breast Cancer Liver Metastasis. Cancers (Basel) (2019) 11(9):1–9. doi: 10.3390/cancers11091341
40. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-Positive Haematopoietic Bone Marrow Progenitors Initiate the Pre-Metastatic Niche. Nature (2005) 438(7069):820–7. doi: 10.1038/nature04186
41. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-Metastatic Niches: Organ-Specific Homes for Metastases. Nat Rev Canc (2017) 17(5):302–17. doi: 10.1038/nrc.2017.6
42. Wong CC-L, Gilkes DM, Zhang H, Chen J, Wei H, Chaturvedi P, et al. Hypoxia-Inducible Factor 1 Is a Master Regulator of Breast Cancer Metastatic Niche Formation. Proc Natl Acad Sci USA (2011) 108(39):16369–74. doi: 10.1073/pnas.1113483108
43. Todd VM, Vecchi LA 3rd, Clements ME, Snow KP, Ontko CD, Himmel L, et al. Hypoxia Inducible Factor Signaling in Breast Tumors Controls Spontaneous Tumor Dissemination in a Site-Specific Manner. Commun Biol (2021) 4(1):1122. doi: 10.1038/s42003-021-02648-3
44. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. A Multigenic Program Mediating Breast Cancer Metastasis to Bone. Cancer Cell (2003) 3(6):537–49. doi: 10.1016/S1535-6108(03)00132-6
45. Min AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes That Mediate Breast Cancer Metastasis to Lung. Nature (2005) 436:518–24. doi: 10.1038/nature03799
46. Yuan X, Qian N, Ling S, Li Y, Sun W, Li J, et al. Breast Cancer Exosomes Contribute to Pre-Metastatic Niche Formation and Promote Bone Metastasis of Tumor Cells. Theranostics (2021) 11(3):1429–45. doi: 10.7150/thno.45351
47. Sznurkowska MK, Aceto N. The Gate to Metastasis: Key Players in Cancer Cell Intravasation. FEBS J (2021) 1–19. doi: 10.1111/febs.16046
48. Neophytou CM, Panagi M, Stylianopoulos T, Papageorgis P. The Role of Tumor Microenvironment in Cancer Metastasis: Molecular Mechanisms and Therapeutic Opportunities. Cancers (Basel) (2021) 13(9):1–22. doi: 10.3390/cancers13092053
49. Wu HJ, Hao M, Yeo SK, Guan JL. FAK Signaling in Cancer-Associated Fibroblasts Promotes Breast Cancer Cell Migration and Metastasis by Exosomal miRNAs-Mediated Intercellular Communication. Oncogene. (2020) 39(12):2539–49. doi: 10.1038/s41388-020-1162-2
50. Qiu SQ, Waaijer SJH, Zwager MC, de Vries EGE, van der Vegt B, Schroder CP. Tumor-Associated Macrophages in Breast Cancer: Innocent Bystander or Important Player? Cancer Treat Rev (2018) 70:178–89. doi: 10.1016/j.ctrv.2018.08.010
51. Brena D, Huang MB, Bond V. Extracellular Vesicle-Mediated Transport: Reprogramming a Tumor Microenvironment Conducive With Breast Cancer Progression and Metastasis. Transl Oncol (2022) 15(1):101286. doi: 10.1016/j.tranon.2021.101286
52. Berger AJ, Renner CM, Hale I, Yang X, Ponik SM, Weisman PS, et al. Scaffold Stiffness Influences Breast Cancer Cell Invasion via EGFR-Linked Mena Upregulation and Matrix Remodeling. Matrix Biol (2020) 85–86:80–93. doi: 10.1016/j.matbio.2019.07.006
53. Weidmann MD, Surve CR, Eddy RJ, Chen X, Gertler FB, Sharma VP, et al. Mena(INV) Dysregulates Cortactin Phosphorylation to Promote Invadopodium Maturation. Sci Rep (2016) 6:36142. doi: 10.1038/srep36142
54. Borriello L, Karagiannis GS, Duran CL, Coste A, Oktay MH, Entenberg D, et al. The Role of the Tumor Microenvironment in Tumor Cell Intravasation and Dissemination. Eur J Cell Biol (2020) 99(6):151098. doi: 10.1016/j.ejcb.2020.151098
55. Paizal JP, Au SH, Bakal C. Squeezing Through the Microcirculation: Survival Adaptations of Circulating Tumour Cells to Seed Metastasis. Br J Canc (2021) 124:58–65. doi: 10.1038/s41416-020-01176-x
56. Lim GE, Sung JY, Yu S, Kim Y, Shim J, Kim HJ, et al. Pygenic Acid A (PA) Sensitizes Metastatic Breast Cancer Cells to Anoikis and Inhibits Metastasis In Vivo. Int J Mol Sci (2020) 21(22):1–19. doi: 10.3390/ijms21228444
57. Adeshakin FO, Adeshakin AO, Afolabi LO, Yan D, Zhang G, Wan X. Mechanisms for Modulating Anoikis Resistance in Cancer and the Relevance of Metabolic Reprogramming. Front Oncol (2021) 11:626577. doi: 10.3389/fonc.2021.626577
58. Hamidi H, Ivaska J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat Rev Canc (2018) 18(9):533–48. doi: 10.1038/s41568-018-0038-z
59. Schlesinger M. Role of Platelets and Platelet Receptors in Cancer Metastasis. J Hematol Oncol (2018) 11(1):125. doi: 10.1186/s13045-018-0669-2
60. Konstantopoulos K, Thomas SN. Cancer Cells in Transit: The Vascular Interactions of Tumor Cells. Annu Rev BioMed Eng (2009) 11:177–202. doi: 10.1146/annurev-bioeng-061008-124949
61. Kopp HG, Placke T, Salih HR. Platelet-Derived Transforming Growth Factor-Beta Down-Regulates NKG2D Thereby Inhibiting Natural Killer Cell Antitumor Reactivity. Cancer Res (2009) 69(19):7775–83. doi: 10.1158/0008-5472.CAN-09-2123
62. Padera TP, Meijer EF, Munn LL. The Lymphatic System in Disease Processes and Cancer Progression. Annu Rev BioMed Eng (2016) 18:125–58. doi: 10.1146/annurev-bioeng-112315-031200
63. Lee HJ, Diaz MF, Price KM, Ozuna JA, Zhang S, Sevick-Muraca EM, et al. Fluid Shear Stress Activates YAP1 to Promote Cancer Cell Motility. Nat Commun (2017) 8:14122. doi: 10.1038/ncomms14122
64. Rianna C, Radmacher M, Kumar S. Direct Evidence That Tumor Cells Soften When Navigating Confined Spaces. Mol Biol Cell (2020) 31(16):1726–34. doi: 10.1091/mbc.E19-10-0588
65. Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A Distinct Macrophage Population Mediates Metastatic Breast Cancer Cell Extravasation, Establishment and Growth. PloS One (2009) 4(8):e6562. doi: 10.1371/journal.pone.0006562
66. Reymond N, d'Agua BB, Ridley AJ. Crossing the Endothelial Barrier During Metastasis. Nat Rev Canc (2013) 13(12):858–70. doi: 10.1038/nrc3628
67. Zhang H, Wong CC, Wei H, Gilkes DM, Korangath P, Chaturvedi P, et al. HIF-1-Dependent Expression of Angiopoietin-Like 4 and L1CAM Mediates Vascular Metastasis of Hypoxic Breast Cancer Cells to the Lungs. Oncogene. (2012) 31(14):1757–70. doi: 10.1038/onc.2011.365
68. Cacho-Díaz B, García-Botello DR, Wegman-Ostrosky T, Reyes-Soto G, Ortiz-Sánchez E, Herrera-Montalvo LA. Tumor Microenvironment Differences Between Primary Tumor and Brain Metastases. J Trans Med (2020) 18:1–12. doi: 10.1186/s12967-019-02189-8
69. Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, et al. Visualization of Immediate Immune Responses to Pioneer Metastatic Cells in the Lung. Nature. (2016) 531(7595):513–7. doi: 10.1038/nature16985
70. Wu P, Gao W, Su M, Nice EC, Zhang W, Lin J, et al. Adaptive Mechanisms of Tumor Therapy Resistance Driven by Tumor Microenvironment. Front Cell Dev Biol (2021) 9:641469. doi: 10.3389/fcell.2021.641469
71. Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, et al. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin Cancer Res (2017) 23(7):1710–21. doi: 10.1158/1078-0432.CCR-15-2851
72. Strell C, Paulsson J, Jin SB, Tobin NP, Mezheyeuski A, Roswall P, et al. Impact of Epithelial-Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. J Natl Cancer Inst (2019) 111(9):983–95. doi: 10.1093/jnci/djy234
73. Roswall P, Bocci M, Bartoschek M, Li H, Kristiansen G, Jansson S, et al. Microenvironmental Control of Breast Cancer Subtype Elicited Through Paracrine Platelet-Derived Growth Factor-CC Signaling. Nat Med (2018) 24:463–73. doi: 10.1038/nm.4494
74. Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, et al. Exosome Transfer From Stromal to Breast Cancer Cells Regulates Therapy Resistance Pathways. Cell. (2014) 159(3):499–513. doi: 10.1016/j.cell.2014.09.051
75. Gatti-Mays ME, Balko JM, Gameiro SR, Bear HD, Prabhakaran S, Fukui J, et al. If We Build it They Will Come: Targeting the Immune Response to Breast Cance. NPJ Breast Canc (2019) 5:1–13. doi: 10.1038/s41523-019-0133-7
76. Schreiber AR, Kagihara JA, Weiss JA, Nicklawsky A, Gao D, Borges VF, et al. Clinical Outcomes for Patients With Metastatic Breast Cancer Treated With Immunotherapy Agents in Phase I Clinical Trials. Front Oncol (2021) 11:640690. doi: 10.3389/fonc.2021.640690
77. Cancer IAfRo. WHO Classification of Tumours. Breast Tumours. Lyon, France: International Agency for Research on Cancer (2019).
78. Harney AS, Karagiannis GS, Pignatelli J, Smith BD, Kadioglu E, Wise SC, et al. The Selective Tie2 Inhibitor Rebastinib Blocks Recruitment and Function of Tie2(Hi) Macrophages in Breast Cancer and Pancreatic Neuroendocrine Tumors. Mol Cancer Ther (2017) 16(11):2486–501. doi: 10.1158/1535-7163.MCT-17-0241
79. Chen W, Shen L, Jiang J, Zhang L, Zhang Z, Pan J, et al. Antiangiogenic Therapy Reverses the Immunosuppressive Breast Cancer Microenvironment. biomark Res (2021) 9(1):59. doi: 10.1186/s40364-021-00312-w
Keywords: breast cancer, tumour microenvironment, immune cell, stroma, metastasis, disease progression
Citation: Tan K and Naylor MJ (2022) Tumour Microenvironment-Immune Cell Interactions Influencing Breast Cancer Heterogeneity and Disease Progression. Front. Oncol. 12:876451. doi: 10.3389/fonc.2022.876451
Received: 15 February 2022; Accepted: 18 April 2022;
Published: 13 May 2022.
Edited by:
Guangwen "Gary" Ren, Jackson Laboratory, United StatesReviewed by:
Sarah Boyle, Centre for Cancer Biology (CCB), AustraliaCopyright © 2022 Tan and Naylor. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew J. Naylor, matthew.naylor@sydney.edu.au
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.