 Tiziana Annese
Tiziana Annese Roberto Tamma
Roberto Tamma Domenico Ribatti
Domenico Ribatti- 1Department of Medicine and Surgery, Libera Università del Mediterraneo (LUM) Giuseppe Degennaro University, Bari, Italy
- 2Department of Basic Medical Sciences, Neurosciences and Sensory Organs, Section of Human Anatomy and Histology, University of Bari Medical School, Bari, Italy
The in-depth characterization of cross-talk between tumor cells and T cells in solid and hematological malignancies will have to be considered to develop new therapeutical strategies concerning the reactivation and maintenance of patient-specific antitumor responses within the patient tumor microenvironment. Activation of immune cells depends on a delicate balance between activating and inhibitory signals mediated by different receptors. T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) is an inhibitory receptor expressed by regulatory T cells (Tregs), activated T cells, and natural killer (NK) cells. TIGIT pathway regulates T cell-mediated tumor recognition in vivo and in vitro and represents an exciting target for checkpoint blockade immunotherapy. TIGIT blockade as monotherapy or in combination with other inhibitor receptors or drugs is emerging in clinical trials in patients with cancer. The purpose of this review is to update the role of TIGIT in cancer progression, looking at TIGIT pathways that are often upregulated in immune cells and at possible therapeutic strategies to avoid tumor aggressiveness, drug resistance, and treatment side effects. However, in the first part, we overviewed the role of immune checkpoints in immunoediting, the TIGIT structure and ligands, and summarized the key immune cells that express TIGIT.
Introduction
Solid and hematological malignancies are complex ecosystems that arise from malfunctioning complex cellular mechanisms controlled by genetic and epigenetic factors that coordinate the cross-talk between tumor cells and the tumor microenvironment (TME) components. Among the cellular components of the TME, T cells are the second most abundant cell type after tumor-associated macrophages (TAMs) (1).
Following the development phase in the thymus, the diverse naïve T cells migrate to the secondary lymphoid organs, where they remain dormant until they are activated by recognition of the antigen-human leukocyte antigen (HLA) complex presented by the APC to their TCR (Figure 1). In addition to antigen recognition by TCR, naïve T cell activation is regulated by second signals known as co-stimulatory pathways, such as the well-noted CD28–CD80/CD86 and CTLA4–CD80/CD86 (3, 4). These co-stimulatory pathways have a lot of receptor/ligand pairs, also called immune checkpoints, which lead to positive signaling events, while other pathways send out negative signals (5).
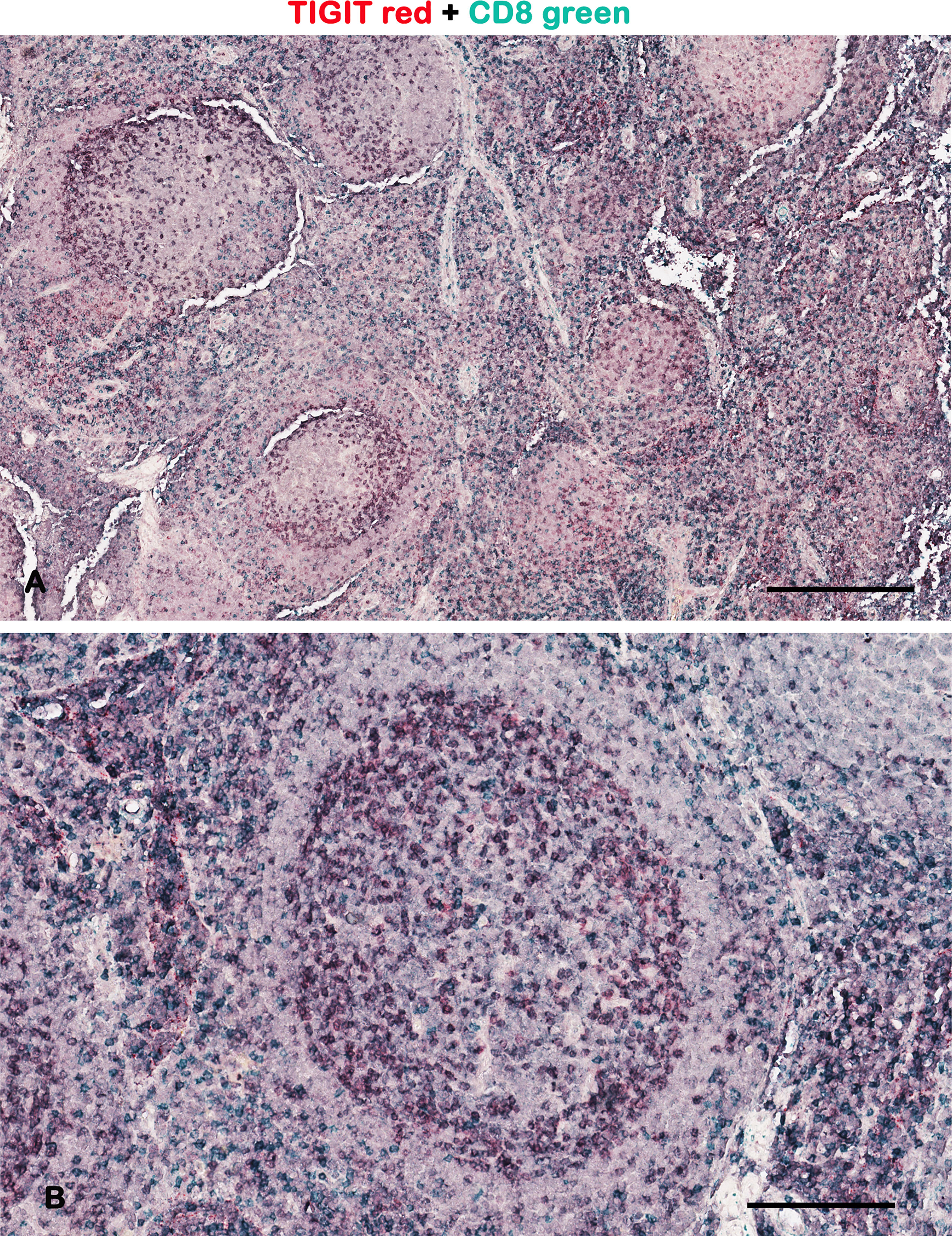
Figure 1 Representative brightfield images of double IHC for TIGIT and CD8 in a lymph node. Micrographs show TIGIT staining in red, CD8 staining in green, and the colocalized TIGIT+CD8+ signals in purple. As demonstrated by other authors, the TIGIT+ T cells are preferentially at the periphery of the germinal center (2). Scale bar: (A) 500 μm; (B) 165 μm.
CD28 is constitutively expressed on naïve CD4+ and CD8+ T cells, while CD80 and CD86 are inducible on APCs. CD28–CD80/CD86 pathway activates T cell responses. Other stimulatory immune checkpoints are members of the tumor necrosis factor (TNF) receptor superfamily (CD27, CD40, OX40, GITR, and CD137) or the B7–CD28 superfamily (ICOS) (6).
On the contrary, cytotoxic T-lymphocyte-associated protein 4 (CTLA4) is a negative regulatory inducible receptor for CD80/CD86 and has inhibitory effects on T cell responses, leading to T cell attenuation and tumor cell immune evasion (7). A considerable number of inhibitory immunoreceptors have been identified and studied in tumors, including but not limited to adenosine A2A receptor (A2AR), B7-H3, B7-H4, programmed death (PD-1), CTLA4, T cell immunoglobulin domain and mucin domain 3 (TIM3), T-cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT), and B and T lymphocyte attenuator (BTLA) (6, 8).
Interestingly, the immune system can constrain and promote tumor development and progression (5, 9, 10). Alterations in immune checkpoint pathways result in an imbalance of positive and negative co-stimulatory signals, which increases the risk of tumorigenesis and its progression. These signals are also involved in patients’ resistance to immunotherapies (9, 11).
Immune checkpoint inhibitors were developed to block checkpoints by making T cells free to attack cancer cells. These therapies are also referred to as checkpoint blockade therapies and are an emerging and attractive field to treat many cancers, but they do not work for all patients and can cause serious side effects (12). The failure of classical antitumor therapies could be attributed to the fact that most drugs currently in use primarily target tumor cells and not also TME cells. These cells are different cell types, including endothelial cells, stromal cells, and immune cells. Understanding the in situ cross-talk of heterogeneous tumor cells with various tumor-associated immune cells, such as T cells, will provide critical information for improving anticancer therapies.
The immune checkpoint inhibitors are most efficacious in patients with a TME enriched in tumor-infiltrating lymphocytes (TILs) (13). TILs are deputed to tumor immunoediting [“a dynamic process wherein immunity functions not only as an extrinsic tumor suppressor but also to shape tumor immunogenicity” (14)]. Immunoediting shapes tumor fate in three steps: elimination, equilibrium, and escape. The elimination step is the immunosurveillance step, in which a competent immune system (innate and adaptive immunity) recognizes and destroys transformed cells expressing highly immunogenic antigens long before they become clinically relevant (15).
If some cancer cells evade the elimination step, they will enter the equilibrium step, in which survived tumor cells and immune cells mutually edit each other. During this adaptation time, tumor cells undergo a complex process of natural selection [similar to that described by Darwin (16)] that presses on tumor cells with traits that are better suited to the environment than others.
These natural evolution-selected tumor cell variants develop resistance to elimination and put them in the escape step (17). A progressive establishment of an immunosuppressive TME characterizes the escape step (11, 18). This is the final step when aggressive-selected tumor clones develop diverse ways to escape the immune system that mimic peripheral tolerance (8, 19, 20): prevent the response of effector T cells, TAMs, natural killer (NK) cells, and tumor-associated neutrophils (TANs) (21); down-regulate their HLA (22); induce antigen presentation defects; eliminate neoantigens; inhibit immune cell chemoattraction to the tumor site; secrete or promote the secretion of immunosuppressive cytokines (23); modulate the recruitment and expansion of immunosuppressive cells, such as regulatory T cells (Tregs); orchestrate immune cell metabolism (24); and activate immune checkpoint pathways to inhibit the emerging antitumor immune response (25).
T cells immunoediting also occurs during tumorigenesis (26, 27). At first, in order to attack and eliminate tumor cells, APCs, via CD28-CD80/CD86 pathway, activate T cells, but at the same time regulate pro-inflammatory mechanisms, activating inhibitory pathways by immune checkpoints (28). Among immune checkpoint inhibitors, immediately after TCR engagement, CTLA4 is upregulated and competes with CD28 to bind to CD80/CD86 on APCs, limiting autoreactive T cells, decreasing T cell priming and proliferation, inducing immune tolerance, and preventing autoimmunity (29, 30).
In the immune response, PD-1 is also expressed on activated T cells, but it acts later and interferes with T cells that have already been activated (31). When the stimulating antigen is removed, PD-1 expression on responding T cells decreases, whereas it remains increased in the opposite scenario. Like CTLA4, the PD-1–PD-L1/PD-L2 pathway recruits phosphatases to block the stimulatory signals sent by TCR and CD28–CD80/CD86, resulting in decreased T cell activation, survival, cytokine generation, and metabolism (31). Overexpression of PD-1 on tumor cells or by the cellular component of the TME with its downstream pathway is a systematic strategy used by malignancies to increase exhausted T cells and to evade immunosurveillance. The fact that PD-1 overexpression happens later means that it will only be overexpressed and activated once an inflammatory process has begun (32).
Immune checkpoint activation and an immune infiltrate enriched in Tregs were identified as the primary tumor escape mechanisms in a mouse model of hypermutated and microsatellite-instable colorectal cancer (33). According to the same study, the expansion of the TCR following PD-1 blockade potentiates immunoediting (33).
In a subtype of advanced untreated primary colorectal cancer, immune checkpoints expression has been related to immune evasion via neo-antigen-related mechanisms (34). This subtype was called the “stealth subtype,” and immune evasion and poor prognosis have been associated with less clonal highly expressed neoantigens (HiNeo), high chromosomal instability, high immune checkpoint expression (PD-1, PD-L1, PD-L2), low neoantigen presentation (reduced HLAII), downregulation of functional CD8+ T cells, and a microenvironment poor in TAMs and B cells (34, 35).
After T cell activation, also TIGIT expression increases on T cells, where it competes with CD226 (DNAM-1) for binding to their shared ligands CD112 and CD155 (36). TIGIT expression is late in the cancer-immunity cycle. It is highly expressed on specialized CD4+ subsets, such as Treg and TFH, and lowly expressed on CD4+ and CD8+ exhausted T cells (37). Moreover, TIGIT+CD4+ T cells and TIGIT+CD8+ T cells displayed a memory phenotype (37).
The timing of immune checkpoint activation is currently under investigation because there is a debate about the reactivation of primed T cells and/or novel T cells. The former depends on memory T cells and presumes the existence of pre-existing cancer-specific T cells that recognize tumor-specific antigens. The second depends on novel T cells against neo-antigens and therefore assumes that T cells are primed and recruited to tumors after the initiation of therapy (38–40). This topic is interesting in patient stratification for immune checkpoint blockades therapy since the success of these therapies relies on antigen processing and presentation (41–43).
In this context, the TIGIT immune checkpoint is emerging as a promising target for anticancer therapy alone or combined with other immune blockade therapies (44). The purpose of this review is to update the role of TIGIT in cancer progression, looking at last year’s studies about its pathways that are often upregulated in immune cells and possible therapeutic strategies to avoid tumor aggressiveness, drug resistance, and treatment side effects. However, in the first part, we overviewed the TIGIT structure and ligands, and summarized the key immune cells that express TIGIT.
Overview Of TIGIT Structure and Ligands
TIGIT is also known as V-set and immunoglobulin domain-containing protein 9 (VSIG9) or V-set and transmembrane domain-containing protein 3 (VSTM3). Based on UniProt data resources, two alternatively spliced isoforms have been reported in humans.
It has an extracellular Ig-like V-type domain, a type I transmembrane domain, and a cytoplasmic domain with the immunoreceptor tyrosine-based inhibitor motif (ITIM) (45, 46) and the immunoglobulin tyrosine tail (ITT)-like motif (45, 47). ITIM modulates cellular responses by binding the SH2 domain of several SH2-containing tyrosine phosphatases, SHP1 (48) and SHP2, when phosphorylated (49).
After the extracellular ligand binding, the ITT-like domain is phosphorylated at Tyr225, binds the two cytosolic adaptor proteins Grb2 and β-arrestin2, and recruits the SH2-containing inositol phosphatase-1 (SHIP-1) that inhibits PI3K/MAPK signaling (via Grb2) (50) to reduce the NK cell effector functions (50), and TRAF6/NF-κB signaling (via β-arrestin2) to inhibit IFN-γ production (48).
Different studies show that phosphorylation of the tyrosine residue in either ITIM- (Y231) or ITT-like (Y225) motif is essential for signal transduction and the inhibitory function of TIGIT in humans (48, 50). When both tyrosine residues are mutated, the inhibitory activity of human TIGIT is completely lost (51).
TIGIT binds to nectin and nectin-like (NECL) adhesion molecules, including NECTIN-2 (CD112) (52, 53), NECTIN-3 (CD113) (45, 51), and NECL-5 (CD155) (54) to mediate cell adhesion and signaling.
TIGIT binds NECL in cis-trans, forming a receptor clustering. For instance, two TIGIT–CD155 dimers assemble into a heterotetramer with a core TIGIT–TIGIT cis-homodimer in which each TIGIT molecule binds one CD155 molecule (47).
CD112 is a cell adhesion protein involved in the modulation of T cell signaling. Two isoforms, delta and alpha, are annotated by alternative splicing. Depending on the receptor it binds to, CD112 can be either a co-stimulator or a co-inhibitor of T cell function: CD226 binding stimulates T cell proliferation and cytokine production (IL2, IL5, IL10, IL13, and IFN-γ) (55); PVRIG (also called CD112R) binding inhibits T cell proliferation (56). These interactions are competitive (57). CD112 binds with low affinity to TIGIT (46, 52). The TIGIT binds to CD112 destroys CD112–CD112 homodimer (52) and, as for TIGIT–CD155, homo- and heterodimers in the heterotetramer interact by a conserved “lock and key” binding (52). CD112 is highly expressed in bone marrow, kidney, pancreas, lung cells, and breast and ovarian cancer (58, 59).
CD113 is another cell adhesion protein that interacts with nectin and NECL molecules via heterophilic trans-interactions, such as CD112 at Sertoli-spermatid junctions (60). Through common signaling molecules such as SRC and RAP1, CD113 trans-interaction with CD155 activates CDC42 and RAC small G proteins (61). CD113 also establishes cell-cell junctions, such as adherens junctions and synapses (62, 63). It inhibits cell movement and proliferation by inducing endocytosis-mediated downregulation of CD155 on the cell surface (64). CD113 contributes to the morphology of the ciliary body (65). CD113 is highly expressed in the testis, placenta, kidney, liver, and lung (66). CD113, like CD112, has a low affinity for TIGIT, and their interaction prevents the self-destruction of normal cells by NK cells (46, 53).
CD155, the primary ligand for TIGIT, is also known as the Poliovirus receptor (PVR). CD155 has a “lock-and-key” motif that is essential for TIGIT binding and is highly conserved across PVR family members (47). CD155 is a glycoprotein with three extracellular immunoglobulin domains, transmembrane, and intracellular domains (67). Two splice forms, lacking the transmembrane region, have also been described as soluble or secreted isoforms that seem to compete with the membrane-anchored ones (68, 69). CD155 is highly expressed on CD11c+ human dendritic cells (DCs) (70, 71), macrophages (72, 73), T (74) and B cells (75), epithelial cells (74, 76), kidneys (76), nervous system (77), intestine (78, 79), and tumor cells (80, 81). In vivo, the CD155–TIGIT pathway suppresses immunological responses increasing IL-10 anti-inflammatory cytokine (82, 83) and decreasing IL-12 pro-inflammatory cytokine released by DCs (46, 84). This induces a tolerogenic phenotype in T cells (85). A more detailed description of the CD155–TIGIT pathway in cancer is present in the next section.
A new NECL that exclusively binds TIGIT was recently identified, NECTIN-4 (86). TIGIT binds NECTIN-4 with high affinity, comparable to CD155 (86). NECTIN-4 is involved in cell adhesion through trans-homophilic and -heterophilic interactions, including specific interactions with NECTIN-1 (CD111) (87), does not interact with CD226, CD96, or CD112 (86), and is overexpressed in several tumors of the breast (88, 89), bladder (90), lung (91, 92), and pancreas (93, 94).
TIGIT Pathways and Immune Cells Involved
TIGIT is expressed by a variety of immune cells. Its expression and related pathways have been discussed in this section.
In simple terms, TIGIT activation creates a tolerogenic microenvironment in both cell-intrinsic and cell-extrinsic ways (resumed in Figure 2 and discussed in the following). This means that TIGIT competes directly with CD226 for binding to CD155, CD112, or CD113 ligands in the former way, or that it is involved in events that indirectly induce immunosuppressive effects, such as TIGIT’s action on innate immunity cells in the second way.
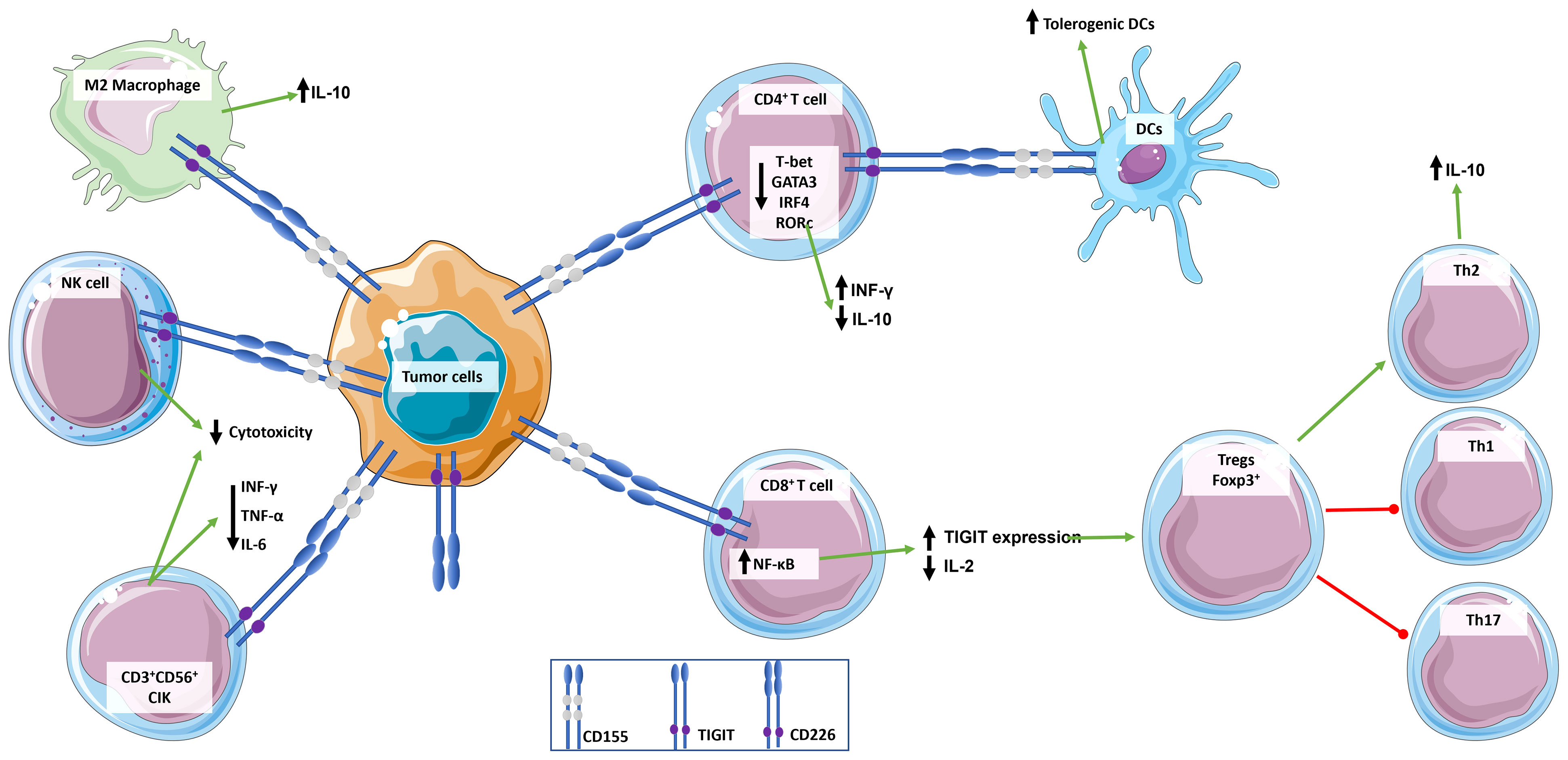
Figure 2 Role of TIGIT in the regulation of immune response. TIGIT transmits inhibitory signals via ITIM and immunoglobulin tyrosine tail (ITT)-like motifs in its cytoplasmic domain when it is engaged. TIGIT has multiple ligands, but it binds with greater affinity to CD155, which is widely expressed by immune cells and tumor cells. CD155 expressing tumor cells bind to TIGIT expressed by immune cells inducing an immunosuppressive and tolerogenic microenvironment: CD4+ T cells induce a tolerogenic phenotype in DCs, release the anti-inflammatory cytokine IL-10, and down-regulate INF-γ; CD8+ T cells up-express TIGIT and down-regulate the release of pro-inflammatory cytokine IL-2, which in turn promotes a T cell immunosuppressive phenotype characterized by increase in Foxp3+ Tregs and Th2 compared to pro-inflammatory Th1 and Th17; NKs cytotoxicity is suppressed; and macrophages switch to an M2 anti-inflammatory phenotype. This simplistic view does not integrate signals from the CD226/CD155 pathway.
CD226 is a member of the immunoglobulin superfamily and consists of an extracellular region with two IgV-like domains, a transmembrane region, a cytoplasmic region with ITT, and four putative tyrosine residues and one serine residue that are phosphorylated (95). It is mainly expressed on myeloid and lymphoid cells (96), through which promotes intercellular adhesion, lymphocyte communication, and lymphokine production, as well as enhances cellular cytotoxicity mechanisms (96).
TIGIT–CD155 in CD4+ T cells induces immunosuppression inhibiting T cell proliferation directly by inducing the down-expression of T-bet, GATA3, IFN regulatory factor 4 (IRF4), and retinoic acid-related orphan receptor c (RORc), which reduce the level of pro-inflammatory IFN-γ while increasing the level of anti-inflammatory IL-10 (97).
TIGIT–CD155 in NK cells reduces their cytotoxicity (51, 53), resulting in impaired granule polarization and IFN-γ production (50, 98). On the contrary, TIGIT blockade restored potent effector NK cells through CD226–LFA-1 signaling that increases adhesion to target cells, induces IFN-γ production by naïve CD4+ T cells, and enhances the cytotoxic function of NK cells (99, 100).
TIGIT–CD155 signaling was also observed in cytokine-induced killer (CIK) cells expressing CD3 and CD56 molecules (101, 102). As observed indirectly by Zhang et al., who analyzed the literature concerning the clinical trial ongoing on renal cell carcinoma patients enrolled in integrated CIK cell immunotherapy, the TIGIT blocked enhanced CIK proliferation and the release of pro-inflammatory cytokines, such as IFN-γ, IL-6, and TNF-α (102).
TIGIT–CD155 in CD8+ T cells induces immunosuppression via the NF-κB signaling pathway, promoting a tolerant state that is passed down across T cell generation. In this process, CD155+ naïve T cells trans-interact with TIGIT+ preceding tolerant T cells resulting in increased TIGIT expression and IL-2 suppression via Blimp1 increment (54, 103).
TIGIT–CD155 signaling was also observed in activated Foxp3+ Tregs, which suppress pro-inflammatory Th1 and Th17 but not Th2 cells via Akt repression and FoxO1 phosphorylation, IL-10 and fibrinogen-like protein 2 overexpression (104, 105). According to this shift in immunity from Th1 and Th17 to Th2 immunity and IL-10 release, CD226 is expressed on Th1 and Th17, but not on Th2 cells, and in the former, CD226–CD155 promotes IFN-γ and IL-17 production (106, 107).
Concerning TIGIT-mediated tolerogenic microenvironment by cell-extrinsic ways, it was observed that TIGIT suppresses T cell function by enhancing the immunosuppressive function of DCs and macrophages that express TIGIT ligands such as CD155 (46, 97, 108, 109).
TIGIT+CD4+ T cells exerted immunosuppressive effects indirectly by modulating the monocyte‐derived DCs cytokine production (97). TIGIT of CD4+ T cells interacts with CD155+ of DCs, modulating the Erk signaling pathway and increasing IL-10 production while decreasing IL-12p40 production and promoting tolerogenic DCs that suppress T cell responses (46, 97).
TIGIT was found to play a role in macrophages in an in vitro pig-to-human xenograft model (84). In this model, TIGIT is expressed by M2 macrophages but not by M1 macrophages or endothelial cells. At the same time, CD155 is expressed by both M1 and M2 macrophages. Here, the immunosuppressive effects of TIGIT are explained by reduced expression of pro-inflammatory cytokines, such as TNFα, IL-1β, and IL-12 in M1 via SHP-1 phosphorylation. In BALB/c mice, TIGIT immunomodulates CD155+ pro-inflammatory M1 into IL-10-secreting anti-inflammatory M2 (85).
All of this demonstrates the intricacy of the several targets and pathways that ani-TIGIT immunotherapies must consider.
TIGIT In Cancer Progression
Immune dysregulation may play a role in cancer progression (110). TIGIT overexpression has been found in the cellular microenvironment of several tumors, including lung (111), kidney (112), liver (113), glioma (114, 115), melanoma (116), colorectal carcinomas (117), gastric cancer (118), and neuroblastomas (119). TIGIT expression was found to be strongly associated with poor prognosis in colorectal cancer and positively correlated with pathological stages in renal clear cell carcinoma (120), kidney renal papillary cell carcinoma, and uveal melanoma (121, 122).
As explained in the introduction paragraph, immune cells interact with other microenvironment cells and the tumor cells in a cross-talk that determines the cancer features and heterogenicity (123, 124). Chronic antigen exposure, which characterizes the first part of tumorigenesis when tumor cells become detectable, stresses T cells, causing them to lose their effector function, become exhausted, and upregulate several immune inhibitor receptors (IRs) such as TIGIT (125, 126) (Figure 3). In various cancers, according to computational analyses, the TIGIT expression profile was related to the immune infiltration level, coupled with the expression of other IRs, including LAG3, CTLA4, PD-1, PD-L1, PD-L2, and it is related to tumor mutation burden (TMB), microsatellite instability (MSI), mismatch repair (MMR), and DNA methyltransferases (DNMTs) gene alterations in different tumors (122). Gene set enrichment analysis (GSEA) demonstrated a negative association among high TIGIT expression and cytokine-cytokine receptor interaction, chemokine signaling pathway, NK-mediated cytotoxicity, allograft rejection, INF-γ response, and IL6/JAK/STAT3 signaling (122). On the contrary, a low TIGIT expression was associated with oxidative phosphorylation and propanoate metabolism (122).
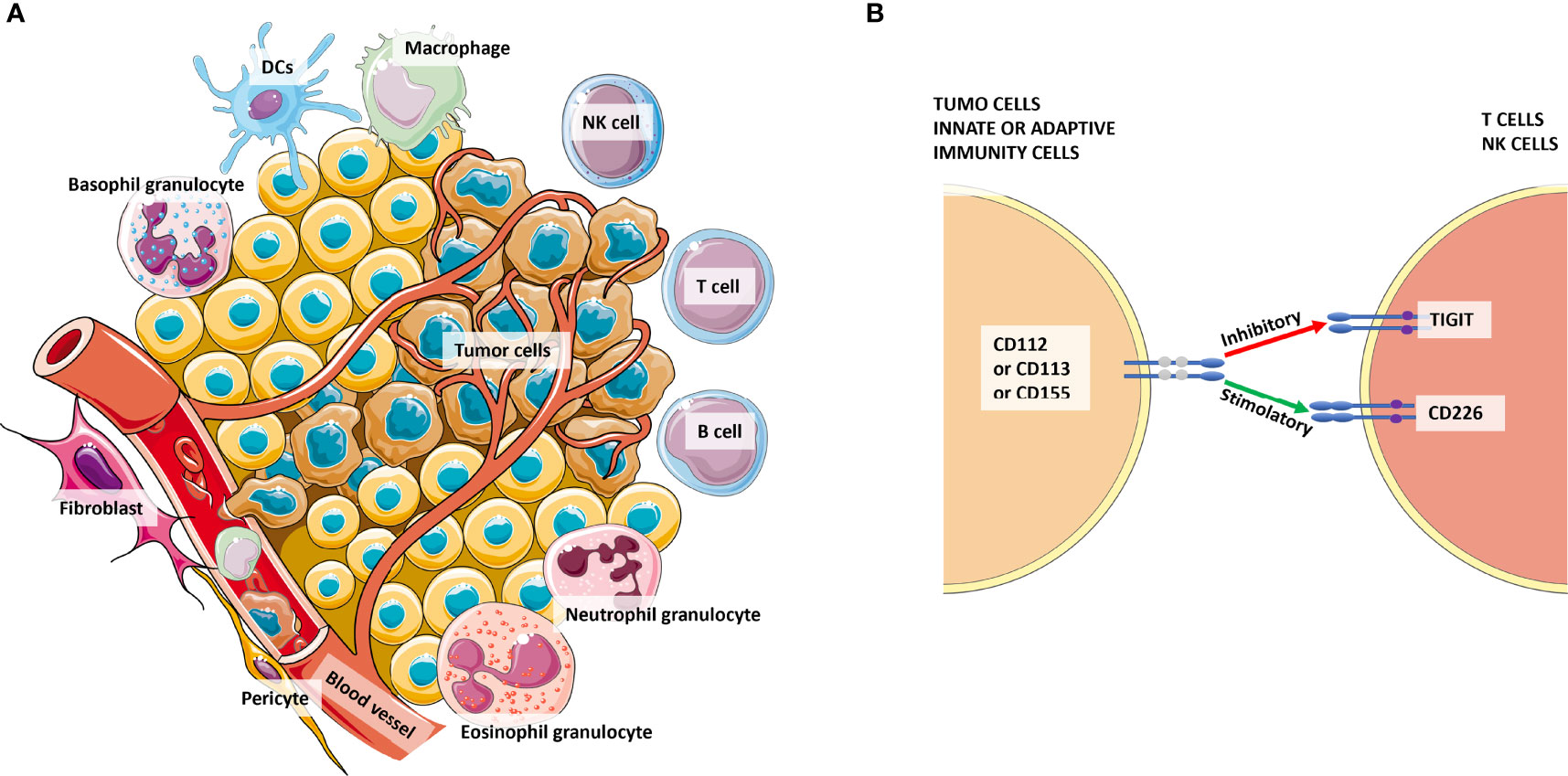
Figure 3 The complexity of the tumor microenvironment and focus on TIGIT+ cells. Panel (A) shows the major cellular components of the microenvironment that cross-talk with tumor cells. Panel (B) shows the competition among CD226 and TIGIT to bind their ligands CD112 or CD113 or CD155 expressed by tumor cells or antigen-presenting cells (APCs) from innate or adaptive immunity. Especially for CD155, the affinity for TIGIT is higher than its affinity for CD226. Thus, the signaling of the CD155-TIGIT synapse (red arrow) induces immunosuppression rather than effector cell activation and/or cytotoxicity.
Blocking the co-expression of IRs appears to be an excellent arm of immunotherapy. TIGIT co-expression with other IRs has been widely examined on CD8+ TILs and circulating T cells (116, 127). Li et al. demonstrated that distinct IRs are co-expressed on CD8+ TILs in T cell exhaustion of primary cancer treatment-naïve patients comprising breast, kidney, lung, liver, cervical, esophageal, gastric, and colorectal cancer (128). Almost 50% of CD8+ TILs were found PD-1+TIGIT+, indicating that TIGIT is preferentially co-expressed with PD-1 (128). Furthermore, in the same study for cervical cancer was observed that the advanced T cell differentiation (CD27–CCR7–CD45RA–) of PD-1+TIGIT+2B4+TIM3+KLRG-1–CTLA4– CD8+ TILs was associated with 60% of poorly differentiated cervical cancer (128). TIGIT mono-expression was also highly present in both TILs and circulating T cells, and this is probably the cause of the side effects after systemic treatment with TIGIT blockade (128).
TIGIT and PD-1 high co-expression was observed in PBLs (peripheral blood lymphocytes), MALs (malignant ascites lymphocytes), and TILs with increased frequency in tumor proximity in matched samples of patients at first diagnosis of ovarian cancer not treated (129). Moreover, the authors also observed TIGIT and TIM3 co-expression in PBLs, MALs, and TILs but with a decreased frequency in tumor proximity (129).
Multiple IRs expression, such as PD-1, PD-L1, TIGIT, and CTLA4, was reported in detail in TILs and circulating T cells in primary breast cancer and colorectal cancer in which immune checkpoint expression was correlated with promoter demethylation and post-translational histone modifications (130–134). For instance, TIGIT in colorectal cancer and TIGIT plus PD-L1 in colorectal cancer and breast cancer were found hypomethylated at the gene promoter level (134).
In esophageal squamous cell carcinoma, the analysis of the RNA-seq dataset from The Cancer Genome Atlas (TCGA) database and by multiplex-immunohistochemistry reactions on patient’s biopsies revealed a high expression of PD-L1 with TIM3 or TIGIT (135). This high IRs co-expression was positively correlated to a greater extent with CD8+ TILs and to a lesser extent with CD4+ TILs and was associated with poor overall survival (OS), TNM III/IV stage, and short restricted mean survival time (RMST) (135).
A TIGIT role in T cell exhaustion was also reported in chronic lymphocytic leukemia (CLL) (136). By flow cytometric and transcript expression analysis, Hajiasghar-Sharbaf et al. have observed a significantly high number of TIGIT+PD-1+CD8+ T cells (136), PD-1+TIM3+CD8 T cells (137), and PD-1+TIM3+CD4 T cells (138) in CLL patients compared with control, particularly in patients with advanced TNM stage.
In both myeloid leukemia and multiple myeloma, using flow cytometry, the bone marrow resident γδ T cells, a T cell subpopulation of non-MHC-restricted, have shown TIGIT, PD-1, TIM3, and the ectonucleoside triphosphate diphosphohydrolase-1 (CD39) co-expression at a high level compared to αβ T cell but similar to that expressed on CD8+ effector T cells (139). These markers were linked to signs of exhaustion, such as transcriptional reprogramming, decreased release of proinflammatory cytokines, decreased T cell proliferation, and lesser tumoricidal activity, and were associated with a lower OS for myeloid leukemia (139, 140).
In relapsed/refractory classic Hodgkin’s lymphoma, a TIGIT-mediated alternative system of immune escape was demonstrated to the classic PD-1/PD-L1 (141). TIGIT and PD-L1 were found to be mutually exclusively expressed and TIGIT+PD-1+CD3+CD4+T cells surrounding Hodgkin Reed-Sternberg (HRS) cells were associated with advanced TNM stages (141).
IRs blockades are mainly used for T cells, but also NK cells could be a valid target for immunotherapy (142, 143). The expression pattern of immune checkpoints on NK cells isolated from peripheral blood of patients affected by hepatitis B virus-related hepatocellular carcinoma (HBV-HCC) revealed a positive correlation among the co-expression of TIGIT and TIM3 in exhausted T cells, high rate of tumor progression, and poor clinical prognosis (144).
In melanoma patients, tumor-infiltrating NK cells were present at low frequencies in metastatic melanoma, had downregulated expression of both TIGIT and CD226, and in vitro experiments had shown their dysfunctional phenotype with higher lytic potential but lower lytic activity compared with TIGIT− NK cells against CD155+ MHC class I–deficient melanoma cells (145). Interestingly, in the same study, TIGIT blockade as a single treatment failed to reverse NK cells dysfunction, while together with IL-15 had reversed CD155-mediated NKs exhaustion and had inhibited experimental melanoma metastasis in vivo (145).
Despite their inhibitory effects on T cells, PD-1 and TIGIT co-expression were described in activated T cells with a cytotoxic effector phenotype and the CXCR5 overexpression (146–148). In Merkel cell carcinoma patients, the PD-1+TIGIT+ CD8+ T cells circulating population was significantly associated with clinical benefit (146). Moreover, a positive trend, but not significant, was observed in melanoma patients (146). In both diseases, the monitoring of PD-1+TIGIT+ CD8+ T cells was proposed as a predictive biomarker of clinical efficacy for PD-1 blockade (146).
Though under-investigated, TIGIT is also expressed in CD4+ Tregs in association with an increased hypomethylation state (149, 150). In melanoma patients, increased TIGIT/CD226 ratio was observed in CD4+ Tregs compared with CD4+ effector T cells and was associated with highly suppressive TME and poor clinical outcomes (149). TIGIT hypomethylation was found dependent by Foxp3. It is a marker of CD4+ Tregs and works as a transcriptional activator by binding to demethylated sequences containing a Forkhead-binding motif, as observed in TIGIT, MIR21, FOXP3, CTLA4, and CD25 (128, 150). Altogether, these data demonstrated that epigenetic regulators, such as demethylation inhibitors, together with immune checkpoint inhibitors, should be considered in new combined therapeutical approaches, and that the promoter methylation pattern of immune checkpoints could be a valid prognostic biomarker.
Here, we discern last year’s update concerning TIGIT’s role in cancer based on PubMed search [for an update concerning hematological malignances, see the review (144)]. We have also looked at studies investigating the correlation of TIGIT expression with the clinicopathological characteristics of such a tumor (such as grade, stage, and metastasis) to improve clinical diagnosis, the amount of surgical resection, prognosis determination, and target therapy. Indeed, a 2021 meta-analysis of TIGIT expression in the tumor microenvironment of various solid tumors revealed that it has prognostic value because it is associated with risk factors for OS and progression-free survival (PFS) (142). In Table 1, all the clinical trials evaluating anti-TIGIT immunotherapeutics started in 2021 are collected, while an in-depth discussion on TIGIT in clinical development is elegantly presented by Rotte et al. (143). (Note that there are now “new” cancers as glioblastoma and melanoma in the clinical trials and not only the “usual” lung cancers. This will give important clinical data on TIGIT blockade on different tumors).
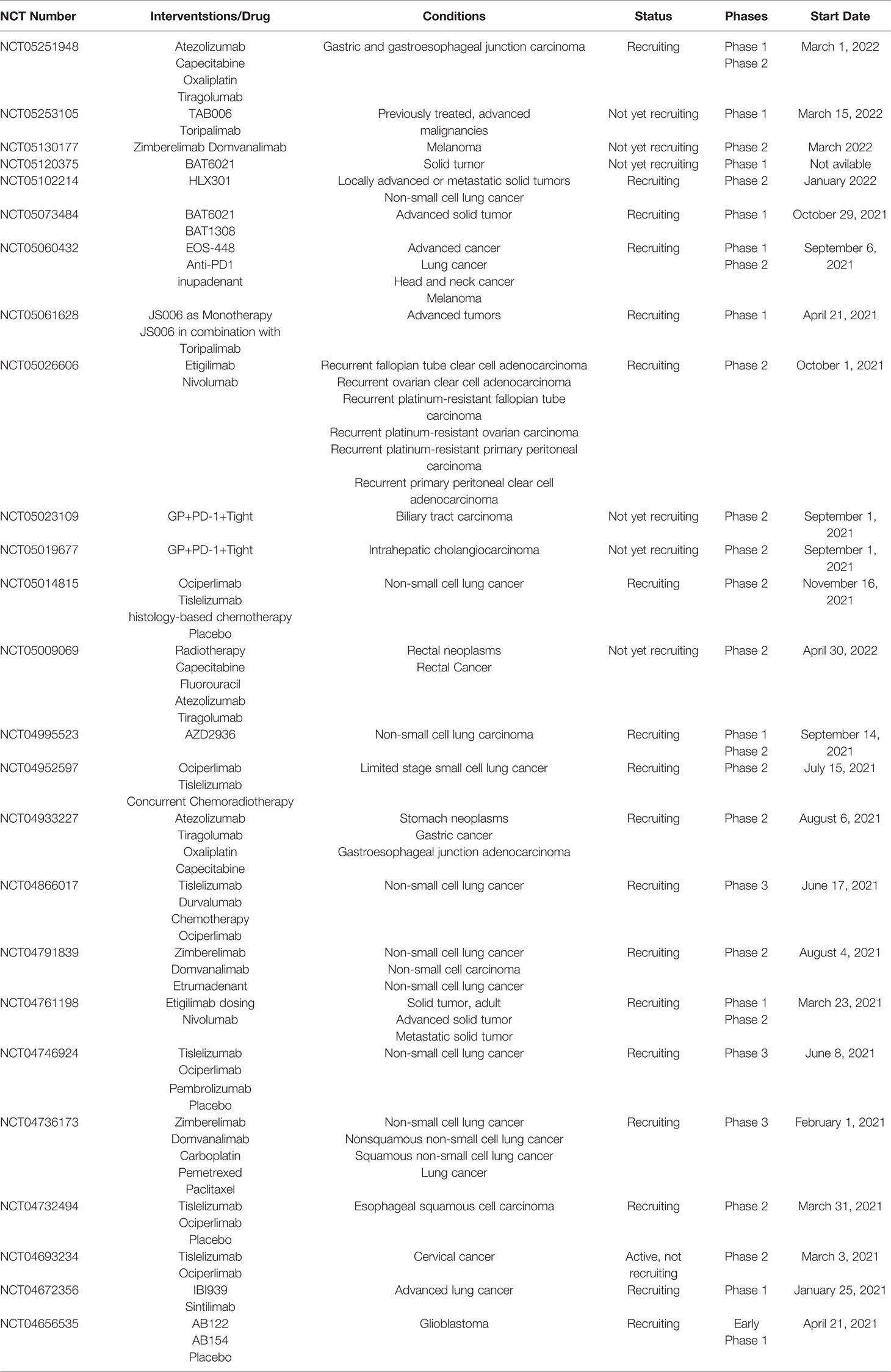
Table 1 Clinical trials evaluating anti-TIGIT immunotherapeutics started in 2021 (accessed on March 14, 2022).
Epigenetic modifications more and more play a role in the upregulation of immune checkpoints in cancer. Through qRT-PCR, CpG methylation, and repressive histone abundance experiments, TIGIT was found poorly expressed in primary breast cancer and adjacent non-cancerous tissues because its CpG islands at the promoter level were mostly hypermethylated (80-70%), while CpG islands of PD-L1 and LAG3 promoter were demethylated at 100% and 80-90%, respectively (130). In another study, using large-scale transcriptome data analysis of aggressive breast cancers, TIGIT was found to be highly and specifically expressed in aggressive breast cancer, and its pro-tumor activities were linked to immune-related genes (151). An in-depth analysis by the same authors revealed that TIGIT expression was positively correlated with T cells, CD8+ T cells, cytotoxic T cells, NK cells, B cells, DCs, and macrophages, but negatively correlated with neutrophils, endothelial cells, and fibroblasts (151). Furthermore, TIGIT expression was positively correlated with inflammation and immune response-related genes (LCK, HCK, MHC-I, MHC-II, STAT1, and interferon) (151). Accordingly, TIGIT expression seems closely related to higher malignant pathological types of breast cancer and might be a potential biomarker of breast cancer progression.
The role of epigenetics in TIGIT expression and immunotherapeutic sensitivity was also uncovered in gastric cancer. Increased TIGIT expression in gastric cancer appears to be a favorable event (152). TIGIT expression correlates with an active immune landscape, survival and immunotherapeutic sensitivity, and favorable prognosis, according to a bioinformatics-guided analysis. Patients with high TIGIT expression respond better to immunotherapy than those with low TIGIT expression (152).
The role of TIGIT in cancer progression was updated in bladder cancer. The failure of the antitumor immune response in bladder cancer was attributed to a subset of TIGIT+ Treg cells overexpressing interleukin IL-32 using single-cell sequencing technology on tissue and experiments in a mouse model (153). In support of this, the same study found that anti-TIGIT monoclonal antibodies, when used alone, have a dual effect: they boost the antitumor activities of T cells while decreasing IL-32, which in turn inhibits bladder cancer metastasis (153). Furthermore, in muscle-invasive bladder cancer, the worst clinical outcomes were attributed to a suppressive TME characterized by Th2 cells, Tregs, mast cells, neutrophils, and exhausted TIGIT+CD8+ T cells with low tumoricidal capacity that benefited from anti-PD-L1 and anti-TIGIT immunotherapy (154, 155). However, in patients with stage II of muscle-invasive bladder cancer with low TIGIT+ CD8+ T cell infiltrate, adjuvant chemotherapy prolongs their OS and recurrence-free survival (RFS) (155). Therefore, TIGIT+ T cells have a prognostic role in clinical outcomes in bladder cancer and seem to be a predictive biomarker for inferior adjuvant chemotherapy responsiveness.
The CD155–TIGIT pathway suppresses the immune system at different levels in colorectal cancer. In colorectal cancer patients and mouse models, the TME is populated by exhausted TIGIT+CD8+ T cells with co-expression of other IRs and low levels of pro-inflammatory cytokines (IFN-γ, IL-2, TNF-α) (103, 156). Furthermore, high TIGIT expression was linked to advanced disease, early recurrence, and lower survival rates (156), and with advanced TNM stage and better disease-free survival (DFS) in colorectal cancer patients with mismatch repair deficiency (157). Another study discovered a higher TIGIT+CD3+ T cell subpopulation in the peripheral blood and cancer tissue of colorectal cancer patients than in healthy donors (121). TIGIT+CD3+T cells were exhausted cells with decreased proliferation, cytokine production, and glucose metabolism (121). TIGIT blockade, combined with PD-1 blockade, reversed these pro-tumorigenic features in the human MC38 colorectal xenograft mouse model. According to this data, GSEA computational analysis revealed that TIGIT expression in colorectal cancer drives the negative regulation of cytokine-cytokine receptor interaction pathway, chemokine signaling, and cytotoxic function of NK cells (122). In vitro studies have revealed that CD155–TIGIT pathway suppresses the downstream effector NF-κB, which is usually involved in the production of IFN-γ by NK cells, which in turn would activate cytotoxic CD8+ T cells (103). On the contrary, the same authors demonstrated that knocking out CD155 in colorectal cancer cells promotes the effector function of tumor-infiltrating CD8+ T cells, and inhibition of the CD155–TIGIT pathway suppresses the tumor growth in an in vivo mouse model. Overall, TIGIT+ cells in colorectal cancer were linked to advanced disease, early recurrence, and lower survival rates (103).
In pancreatic cancer, the CD155–TIGIT pathway suppresses immunity and promotes immune evasion (158, 159). The cancer progression of a subset of patients with pancreatic adenocarcinoma in metastatic/advanced stages was related to high-affinity MHC-I-restricted neoepitopes expression and exhausted TILs in the intratumoral compartment. Functional studies using orthogonal preclinical models revealed a synergistically antitumor response when TIGIT/PD-1 co-blockade was combined with CD40 agonism because they had been reinvigorated tumor-reactive T cells (158).
TIGIT+ immune cells were also shown to play a role in cancer invasion and metastasis in esophageal carcinoma. A transcriptomic profile investigation followed by immunohistochemistry validation has identified the allograft inflammatory factor 1 (AIF1) gene as an unfavorable prognostic factor in this carcinoma and demonstrated that it is associated with immune infiltrates (160). In the tumor infiltrate, T cells and NK cells are affected by AIF1, which promotes TIGIT expression, and hence induces or strengthens immunotherapy resistance sustained by an immune infiltrate enriched in Th1 cells and exhausted T cells.
According to mRNA profiling of CD8+ T cells in a murine model of autochthonous liver cancer, TIGIT is a hallmark of T cell exhaustion in liver cancer at various stages of their differentiation (161). TILs from patients with primary hepatocellular carcinoma and intrahepatic cholangiocarcinoma had an increased TIGIT+CD8+ T cell subpopulation. However, two subsets of these patients were identified: one had significantly higher TIGIT and PD-1 expression levels in the tumor area than the surrounding peritumoral area; whereas the other had a similar level of expression for both IRs in the tumoral and peritumoral areas (161).
In renal cell carcinoma (RCC), immunohistochemistry and flow cytometry experiments to evaluate TIGIT and PD-1 expression in circulating immune cells and TILs revealed an increased TIGIT and PD-1 expression in the tumoral area compared with adjacent normal tissue, but TIGIT+ T cells and NK cells amount did not correlate with clinicopathological characteristics (age, sex, tumor diameter, Fuhrman grade, or TNM stage) (162). In contrast, a positive correlation with RCC clinicopathological characteristics was observed only for PD-1 (162).
CD155 and TIGIT were correlated with clinicopathological features in lung adenocarcinoma, in which CD155 expression was strongly associated with tumor staging and poor OS (111). TIGIT expression was associated with advanced TNM staging, which correlated with lymphatic metastasis and distant metastasis, with low antitumor immunity-related gene expression activation and poor PFS (111).
In oral squamous cell carcinoma, circulating T cells and TILs overexpressed TIGIT on CD4+ and CD8+ T cells, characterized by dysfunctional phenotype, including reduced proliferative capacity and low proinflammatory cytokine release (163). Higher TIGIT expression was also associated with higher T stage and nodal invasion but not with other clinicopathological variables such as age, gender, smoke/alcohol use, tumor site, and tumor differentiation (163).
Singer et al. proposed TIGIT expression as a predictive rather than prognostic biomarker for reactive tumor-infiltrating immune cells in soft sarcoma tissue in an elegant investigation on IL-15 and TIGIT blockade therapy to reactive tumor-infiltrating immune cells (164). The authors observed both activated and exhausted tumor-infiltrating NK cells and TILs and TIGIT upregulation in the TME, especially on NK cells, associated with superior distant disease recurrence-free and OS (165). Interestingly, activator and inhibitor pathways are not mutually exclusive and are a recent field of interest in targeted therapy (164, 165).
Under hypoxic conditions, HIF-1α transcript factor activation stimulates the expression of various IRs, including TIGIT. TIGIT and HIF-1α activity suppression experiments, using a siRNA carrier system, have revealed a critical role of these molecules in tumor growth, apoptosis, and metastasis in colorectal and breast cancer (166). In colorectal cancer cell line CT26 and breast cancer cell line 4T1 and in their in vivo mouse models, TIGIT and HIF-1α down-regulation diminished the colony formation ability and afflicted cancer cells’ angiogenesis and proliferation activities, suggesting simultaneous blocking of TIGIT and HIF-1α as a potential new treatment strategy (166).
Considering all these results, it is possible to speculate that later than tumorigenesis, when the tumor already presents an immune infiltrate, immune cells, particularly T cells, upregulate TIGIT, promoting an immunosuppressive microenvironment that leads to metastasis and unfavorable prognosis. The studies with an in-depth microenvironment characterization and association with clinicopathological features point out several diverse IRs expression combined analysis that might represent an effective outcome prediction panel in cancer. However, there is much work to be done to understand in more detail TIGIT’s role in the different tumor stages (e.g., initial diagnosis, progression, recurrence, metastases) in various cancers.
Therapeutic Strategies Targeting TIGIT Immune Checkpoint Expression
Cancer treatments are traditionally based on surgery, targeted therapies, chemotherapy, or radiation therapy (167). Immune inflammatory modulation-based therapy, or more simply immunotherapy, has lately emerged as a novel therapeutic arm with enormous potential, particularly in the treatment of cancer chemo-radiotherapy resistance (168, 169). Immunotherapy is a type of treatment that aids the immune system in fighting cancer and other diseases.
Immunotherapies have been shown to be effective against tumor-associated T cells that are dysfunctional. The rationale behind these therapies is that the cancer cells overexpress ligands for IRs, such as CD115, CD112, and others, to elude the immune system. Different immunotherapy strategies aim to boost the patient’s antitumor immune response against malignancies minimizing T cell exhaustion and providing protective effects against recurrence and metastasis with less toxicity when compared to traditional cancer therapy (170).
Here is an update on therapeutic strategies targeting TIGIT immune checkpoint expression.
Cancer immunotherapy strategies that boost innate and adaptive immunity are being developed to achieve long-lasting antitumor effects. Azelnidipine is a long-acting third-generation dihydropyridine calcium channel blocker that has been approved for the treatment of hypertension. However, using the molecular operating environment (MOE) by blocking and MST binding assays, molecular docking and structural analysis of CD172a and CD112 have indicated azelnidipine’s potential relevance in cancer immunotherapy (171). Azelnidipine inhibits the innate checkpoint CD47/CD172a and the adaptive checkpoint TIGIT–CD112 pathways and has anti-cancer effects by increasing the infiltration and function of CD8+ T cell and macrophage tumor cell phagocytosis in vivo and in vitro. This study extensively looked at the effect of TIGIT blockade on macrophages in the tumor. Tumor cells, like normal cells, can express CD47, a “do not eat me” signal that prevents CD172a+ macrophages from phagocytosing them. Zhou et al. demonstrated that azelnidipine blocks CD47–CD172a signaling, reactivates macrophage phagocytosis, and improves antitumor immunity even in combination with radiotherapy, as shown in the MC38 murine colon adenocarcinoma cell line (171). A cancer immunotherapy antibody targeting both CD47 and TIGIT has been patented (WO2020259535).
Alternative anticancer treatments with a systemic approach are being developed. In a mouse model of lung adenocarcinoma, triple therapy with the RadScopal approach (high-dose radiation to primary tumors plus low-dose radiation to secondary tumors) plus anti-TIGIT and plus anti-PD-1 prolong survival and block tumor growth while decreasing TIGIT+ exhausted T cells and TIGIT+ Tregs (104). This approach promotes a systemic antitumor response because low-dose radiation also reduces CD155 expression on TAMs and DCs (104). Combined therapies based on immunotherapy and radiation therapy promise to reset the TME.
TIGIT+ macrophages were also looked at in leukemia, in which the leukemia-associated macrophages (LAM) co-expressing TIGIT, TIM3, and LAG3 were identified as immunosuppressive M2 responsive to in vitro TIGIT blockade therapy that polarizes the M2 toward the M1 phenotype and improves phagocytosis of the CD47 expressing tumor cells (172, 173).
An in-depth characterization of TILs in bladder cancer using PBMC isolation and tumor single-cell isolation from fresh tumor tissue demonstrates that PD-1highTOX+ T cells play a key role in tumor evasion, which might be reversed by combining PD-1 and TIGIT inhibition (174).
pt?>Autophagy, a cell-intrinsic system that uses the lysosome to remove damaged organelles and proteins, plays a critical role in cellular immunity. Indeed, autophagic abnormalities linked with oncogenesis promote tumor escape by influencing cell immunogenicity, APC activity, and T cell activity (175). Artesunate, an anti-malaria drug, exerts anticancer activity by inhibiting proliferation, migration, and angiogenesis and inducing apoptosis and autophagy. Artesunate-induced autophagy was well demonstrated in human bladder cancer cells, upregulating ROS and activating the AMPK-mTOR-ULK1 axis and in uterine corpus endometrial carcinoma, enhancing NK cell cytotoxicity via interactions with tumor cells overexpressing CD155, and upregulating co-stimulator CD226 and downregulating co-inhibitor TIGIT (176–178).
Anti-TIGIT antibodies are used instead in more consolidated therapies. TIGIT blocked reduced tumor growth while promoting an immune infiltration enriched in effector cytokine-secreting CD8+ T cells (44, 116, 127, 179).
Vibostolimab is a humanized antibody that targets TIGIT preventing its binding with CD112 and CD155. Patients with advanced solid tumors who received vibostolimab alone or combined with the anti-PD-1 pembrolizumab in a phase I clinical trial (NCT02964013) showed controllable tolerance across escalating doses and all types of advanced solid tumors assessed. Increased NK cell activation of CD8+ T cells was found to have an anticancer effect in the study (180).
Etigilimab is another anti-TIGIT monoclonal antibody that is now being investigated in an open-label, multicenter, phase I/II clinical trial (NCT04761198) in patients with advanced or metastatic solid tumors for tolerance and pharmacokinetics with the anti-PD-1 nivolumab (181).
Combining anti-TIGIT and anti-PD-1 immunotherapy in metastatic melanoma has shown encouraging outcomes, with increased proliferation, cytokine generation, and degranulation of effector CD8+ T cells (116).
Mono- or dual TIGIT and PD-1–PD-L1 blockade aims to take advantage of the curative potential of pre-existing tumor-primed T cells in cancer treatment by promoting CD8+ T cell proliferation and function, resulting in protective memory T cells that ensure tumor rejection and avoid recurrence (182–184). Although several of these antibodies have received clinical approval, their effectiveness remains modest because immunological checkpoints and their signaling are regulated at multiple levels.
In addition to monoclonal antibodies, the most recent approach is to design T cells for TIGIT.
Hoogi et al. created a TIGIT : CD28 chimeric co-stimulatory switch receptor with the TIGIT exodomain fused to the CD28 signaling domain, which improved the activities of chimeric antigen receptor T cells by stimulating cytokine production and activating other T cell effector functions (185).
Efficacy And Toxicity of Anti-TIGIT Immune Checkpoint Therapy
Even though therapeutic strategies targeting immunological checkpoints have been approved for a variety of cancer types, patients continue to have poor prognoses and suffer from immune-related adverse events (irAEs) that affect numerous organs. irAEs are secondary to the infiltration of activated T cells and can affect any organ (186, 187). Skin, gastrointestinal tract, endocrine, lungs, thyroid, pituitary and adrenal glands, and the musculoskeletal system are the most usually impacted, while nervous, renal, hematologic, ophthalmic, and cardiovascular systems are less commonly affected (188–190). Four degrees of irAEs can be distinguished based on the organs involved and the severity: patients with grade 1 irAEs show skin toxicity (<10% body surface area) and no sign of toxicity for the gastrointestinal tract, liver, endocrine system, and lungs; patients with grade 4 of irAEs show elevated skin toxicity (> 30% body surface), hepatotoxicity, and severe symptoms of involvement of the cardiovascular, endocrine, and digestive apparatus (191). Grade 2 and 3 show intermediate signs. The management of irAEs is based on well-established clinical practice guidelines well reviewed by Barber in 2019 (192). Some irAEs are more common in immune therapy than chemotherapy, and their frequencies are positively associated with clinical efficacy, making them useful for clinical decisions (193).
To understand the efficacy and toxicity of immune checkpoint therapy, it should be noted that the types of antibodies used in anti-TIGIT therapies are very different and more or less tolerated. A murine, chimeric, humanized, or completely human IgG antibody could be used to suppress immunological checkpoints (194). The majority of anti-TIGIT antibodies in clinical trials are either humanized (such as ociperlimab, pembrolizumab, atezolizumab) or fully human (such as tiragolumab, etiligimab, ipilimumab, nivolumab, vibostolimab, domvanalimab) (195). Compared to other forms of IgG origin, humanized and completely human antibodies have increased in vivo tolerability but much-reduced immunogenicity (194).
Furthermore, blockade therapy efficacy depends on the antibody-dependent cellular cytotoxicity (ADCC) desired to destroy unfunctional T cells and tumor cells. ADCC is a non-phagocytic mechanism in which antibody-bound target cells are killed by innate immune cells such as NK cells, DCs, and macrophages (196). To activate ADCC, the targeted cell must express target antigens, the antibody must be preferentially IgG1 or IgG3 monoclonal because these two antibodies link any type of FcR, and the effector cell must have the Fc-gamma receptors (FcγR) (196). Concerning TIGIT, its FcγR is active in tiragolumab, ociperlimab, vibostolimab, EOS-448, etigilimab, and AGEN-1307, whereas it is inactive in domvanalimab, BMS-986207, and CASC-674 (195). However, FcγR presence or absence has not been tested for anti-TIGIT antibody clinical efficacy (197, 198).
Recently, TIGIT molecular was also used as Fc-fused protein in some reports demonstrating that TIGIT-Fc may act both as an immunosuppressor and as an immunostimulator in a microenvironment-dependent way (83, 85, 199–201). TIGIT-Fc is a dimer in which an Fc domain of an antibody is linked to the extracellular domain of TIGIT by covalent bonds. TIGIT-Fc has antibody-like features, such as a long serum half-life and efficient expression and purification in vitro, making it an ideal drug (46). Its action as an immunosuppressor was demonstrated in vitro and in a mouse model of acute allogeneic GVHD in which it decreased CD8+IFN-γ+ and CD8+ granzyme B+ T cells activation in a dendritic cell-dependent manner and reduced the release of IL-10 (83).
Moreover, TIGIT-Fc acts as a negative regulator of inflammation, inhibiting macrophage activation and imbalanced M1/M2 ratio in favor of M2 anti-inflammatory profile via c-Maf up-regulation, which promotes IL-10 transcription as demonstrated by in vivo and in vitro experiments using fibroblasts stably secreting TIGIT-Fc in the LPS shock model (85). In CLL, a tumor-supportive role of TIGIT+CD4+ T cells was observed in the presence of TIGIT-Fc via down-regulation of IFNγ and IL-10 production (201). Interestingly, this protumor activity of CD4+ T cells was dependent on CLL cell’s presence because in vitro experiments with CD4+ T cells alone did not show any effects (201).
On the contrary, TIGIT/ligand interactions using recombinant TIGIT-Fc molecule immunostimulatory functions were shown in xenograft mouse models containing different human tumor cells (A375, A431, SK-BR-3, SK-OV-3, and H2126) co-implanted with human T cells (200). The TIGIT-Fc treatment enhanced effector NK cell functions and activated an anti-tumor T cell immune response via CD4+ T cells preventing their exhaustion (200). Additionally, synergistic effects were observed in TIGIT-Fc plus anti-PD-L1 combined therapy (200).
Efficacy and toxicity of anti-TIGIT therapy were evaluated in the CITYSCAPE trial (NCT03563716), in which anti-TIGIT (tiragolumab) with anti-PD-L1 (atezolizumab) combined therapy were applied. The findings showed that this combined therapy in non-small cell lung cancer (NSCLC) is well tolerated when compared to CTLA4 with PD-L1 combined therapy, and that it improves responses and PFS in PD-L1–immune sensitive patients (202–205). Furthermore, despite the similar safety profiles of atezolizumab with placebo (AP) vs. atelozomab with tiragolumab (AT), 80.6% of patients in the AT group and 72% of patients in the AP group suffered irAEs. The irAEs included rash and thyroid issues, infusion reactions at the first dose, soft stool, diarrhea, and very few cases of more severe toxicities, like hepatitis (204, 205).
The anti-TIGIT vibostolimab was tested in patients with solid tumors as monotherapy or in combination with the anti-PD-L1 pembrolizumab in the phase I multicohort MK-7684-001 trial (NCT02964013). The ORR for vibostolimab monotherapy was more significant than for combination therapy in the sub-cohort of NSCLC patients with anti–PD-1–PD-L1–refractory disease (7% (95% CI, 2%-20%) vs. 5% (95% CI, <1%-18%)) (206). IrAEs were reported by 65% of patients in the same NSCLC sub-cohort, including pruritus, fatigue, rash, arthralgia, decreased appetite, and 13% also had lipase elevation and hypertension (206).
TIGIT blockade therapy may be more beneficial if it is evaluated as a first-line treatment. In February 2020, a multicenter, open-label, phase I/II study using the novel anti-TIGIT EOS884448 as monotherapy was launched in patients with previously treated advanced cancer (ovarian, head and neck, cervical, and colorectal) (NCT04335253) (207). Multiple mechanisms of action for EOS884448 were demonstrated: inhibition of TIGIT triggering activation of TIGITlow T cells and NK cells; depletion of immunosuppressive Treg and exhausted TIGIThigh T cells; and reverse activation via FcγR engagement (207). The pharmacokinetic and pharmacodynamic analysis demonstrated that exhausted Tregs and TIGIT+ T cells were depleted in a dose-dependent manner. Moreover, in this interventional study with multiple ascending-dose treatments, EOS884448 was generally well tolerated at all tested doses in patients with advanced cancer and had a promising antitumor activity as a single agent also in PD1-resistant patients. IrAEs were reported by 82% of patients, including pruritus, infusion-related reaction, fatigue pyrexia, rash macuolo-papular, eczema, and hypothyroidism (207).
Cancer patients’ stratification based on tumor response to immune checkpoint inhibitors is vital even if challenging to evaluate (208, 209). In fact, patients might experience clinical pseudoprogression that can be misinterpreted as disease progression because it cannot be evaluated with the existing response-evaluation criteria (210, 211). In tumor pseudoprogression, an increase in tumor size depends on infiltrating T cells, while in proper tumor progression, the increased tumor mass is due to proliferating tumor cells (210). In 2017, the Response Evaluation Criteria in Solid Tumors (RECIST) working group published a modified set of response criteria, the immune-related response criteria (iRECIST), adapted for immunotherapy because of the importance of a standardized strategy to evaluate its effects (212, 213).
Identifying prognostic biomarkers of response to TIGIT blockade alone or in combination with other IRs is needed to improve efficacy and reduce toxicity.
Challenges and Conclusions
To summarize, in cancer, the genetic and epigenetic alterations could initiate tumorigenesis, which activates T cells and NK cells, and TME gets infiltrated by immune cells. Following T cells and NK cells upregulate TIGIT expression, which leads to an immunosuppressive TME, promoting tumor progression, immune escape, and metastases that result in poor prognosis.
Immune inflammatory modulation-based therapy is a promising therapeutic strategy against solid and hematological malignancies, but the outcomes are not largely encouraging because some tumor types remain refractory primarily to these therapies (214). CD8+ T cells are extremely heterogeneous, while CD4+ T cells in immunosuppression and immunotherapy are under-investigated (44). Targeting only a part of the complicated tumor system is insufficient for most cancer therapies or only in the arm of immunotherapies, so patients cannot benefit for a long time. New combined multiple targets (other co-inhibitory receptors) for immunotherapy must be explored to improve treatment.
Guidelines should be set for immunotherapy research. The results of different studies are difficult to compare due to the different designs for types of cancer, sample size, and statistical analysis. Consequently, when the results of individual studies are analyzed, they are insufficient to adopt particular and successful therapeutic interventions.
Side effects of traditional and immune checkpoint blockade therapies should be evaluated in-depth. High cytokine release and effector cell infiltration into TME cause irAEs that sometimes lead to the death of patients (215–217). Skin, gastrointestinal tract, lung, or liver are all affected by irAEs. However, the TIGIT blockade seems to have fewer side effects compared with other IRs blockades, as demonstrated in TIGIT-/- mouse model (218–220). In this pre-clinical model, TIGIT blockade triggers fewer irAEs than anti-PD1 or anti -CTLA4 therapies (218–220).
Anti-TIGIT therapy is now being tested in 25 clinical trials, considering only those starting from 2021 (Table 1), but there is still considerable work to be done to discover new and safely targetable immune checkpoints that could be effective against various malignancies.
The immunological and stromal characterization of the TME cells and their amount and spatial distribution in relation to pathology and prognosis will help patient stratification, enhance personalized cancer therapy efficiency, and overcome tumor immune evasion mechanisms.
Author Contributions
This work was conceived and planned by TA. The original draft preparation and writing: TA. Review and editing: DR and RT. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Associazione “Il Sorriso di Antonio”, Corato, Italy and by ARES-Centro Salute Ambiente Progetto Ionico-Salentino.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
1. Speiser DE, Ho PC, Verdeil G. Regulatory Circuits of T Cell Function in Cancer. Nat Rev Immunol (2016) 16(10):599–611. doi: 10.1038/nri.2016.80
2. Blessin NC, Simon R, Kluth M, Fischer K, Hube-Magg C, Li W, et al. Patterns of TIGIT Expression in Lymphatic Tissue, Inflammation, and Cancer. Dis Markers (2019) 2019:5160565. doi: 10.1155/2019/5160565
3. Sharpe AH. Mechanisms of Costimulation. Immunol Rev (2009) 229(1):5–11. doi: 10.1111/j.1600-065X.2009.00784.x
4. Vijayakrishnan L, Slavik JM, Illes Z, Greenwald RJ, Rainbow D, Greve B, et al. An Autoimmune Disease-Associated CTLA-4 Splice Variant Lacking the B7 Binding Domain Signals Negatively in T Cells. Immunity (2004) 20(5):563–75. doi: 10.1016/s1074-7613(04)00110-4
5. Gonzalez H, Hagerling C, Werb Z. Roles of the Immune System in Cancer: From Tumor Initiation to Metastatic Progression. Genes Dev (2018) 32(19-20):1267–84. doi: 10.1101/gad.314617.118
6. O'Neill RE, Cao X. Co-Stimulatory and Co-Inhibitory Pathways in Cancer Immunotherapy. Adv Cancer Res (2019) 143:145–94. doi: 10.1016/bs.acr.2019.03.003
7. Gardner D, Jeffery LE, Sansom DM. Understanding the CD28/CTLA-4 (CD152) Pathway and Its Implications for Costimulatory Blockade. Am J Transplant (2014) 14(9):1985–91. doi: 10.1111/ajt.12834
8. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
9. Schreiber RD, Old LJ, Smyth MJ. Cancer Immunoediting: Integrating Immunity's Roles in Cancer Suppression and Promotion. Science (2011) 331(6024):1565–70. doi: 10.1126/science.1203486
10. Dunn GP, Old LJ, Schreiber RD. The Three Es of Cancer Immunoediting. Annu Rev Immunol (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
11. O'Donnell JS, Teng MWL, Smyth MJ. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat Rev Clin Oncol (2019) 16(3):151–67. doi: 10.1038/s41571-018-0142-8
12. Barrueto L, Caminero F, Cash L, Makris C, Lamichhane P, Deshmukh RR. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl Oncol (2020) 13(3):100738. doi: 10.1016/j.tranon.2019.12.010
13. Sharma P, Allison JP. The Future of Immune Checkpoint Therapy. Science (2015) 348(6230):56–61. doi: 10.1126/science.aaa8172
14. Borroni EM, Grizzi F. Cancer Immunoediting and Beyond in 2021. Int J Mol Sci (2021) 22(24):13275. doi: 10.3390/ijms222413275
15. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer Exome Analysis Reveals a T-Cell-Dependent Mechanism of Cancer Immunoediting. Nature (2012) 482(7385):400–4. doi: 10.1038/nature10755
16. On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life. Br Foreign Med Chir Rev (1860) 25(50):367–404.
17. Teng MW, Galon J, Fridman WH, Smyth MJ. From Mice to Humans: Developments in Cancer Immunoediting. J Clin Invest (2015) 125(9):3338–46. doi: 10.1172/JCI80004
18. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New Insights Into Cancer Immunoediting and Its Three Component Phases–Elimination, Equilibrium and Escape. Curr Opin Immunol (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
19. Wellenstein MD, de Visser KE. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity (2018) 48(3):399–416. doi: 10.1016/j.immuni.2018.03.004
20. Wu AA, Drake V, Huang HS, Chiu S, Zheng L. Reprogramming the Tumor Microenvironment: Tumor-Induced Immunosuppressive Factors Paralyze T Cells. Oncoimmunology (2015) 4(7):e1016700. doi: 10.1080/2162402X.2015.1016700
21. Palucka AK, Coussens LM. The Basis of Oncoimmunology. Cell (2016) 164(6):1233–47. doi: 10.1016/j.cell.2016.01.049
22. Cornel AM, Mimpen IL, Nierkens S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers (Basel) (2020) 12(7):1760. doi: 10.3390/cancers12071760
23. Kohli K, Pillarisetty VG, Kim TS. Key Chemokines Direct Migration of Immune Cells in Solid Tumors. Cancer Gene Ther (2021) 29(1):10–21. doi: 10.1038/s41417-021-00303-x
24. Kouidhi S, Elgaaied AB, Chouaib S. Impact of Metabolism on T-Cell Differentiation and Function and Cross Talk With Tumor Microenvironment. Front Immunol (2017) 8:270. doi: 10.3389/fimmu.2017.00270
25. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of Neoantigen Landscape During Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discovery (2017) 7(3):264–76. doi: 10.1158/2159-8290.CD-16-0828
26. Chen L, Flies DB. Molecular Mechanisms of T Cell Co-Stimulation and Co-Inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi: 10.1038/nri3405
27. Mpakali A, Stratikos E. The Role of Antigen Processing and Presentation in Cancer and the Efficacy of Immune Checkpoint Inhibitor Immunotherapy. Cancers (Basel) (2021) 13(1):134. doi: 10.3390/cancers13010134
28. Vigano S, Perreau M, Pantaleo G, Harari A. Positive and Negative Regulation of Cellular Immune Responses in Physiologic Conditions and Diseases. Clin Dev Immunol (2012) 2012:485781. doi: 10.1155/2012/485781
29. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) Bind With Similar Avidities But Distinct Kinetics to CD28 and CTLA-4 Receptors. Immunity (1994) 1(9):793–801. doi: 10.1016/s1074-7613(94)80021-9
30. Egen JG, Allison JP. Cytotoxic T Lymphocyte Antigen-4 Accumulation in the Immunological Synapse is Regulated by TCR Signal Strength. Immunity (2002) 16(1):23–35. doi: 10.1016/s1074-7613(01)00259-x
31. Sharpe AH, Pauken KE. The Diverse Functions of the PD1 Inhibitory Pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
32. Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. Role of the Tumor Microenvironment in PD-L1/PD-1-Mediated Tumor Immune Escape. Mol Cancer (2019) 18(1):10. doi: 10.1186/s12943-018-0928-4
33. Efremova M, Rieder D, Klepsch V, Charoentong P, Finotello F, Hackl H, et al. Targeting Immune Checkpoints Potentiates Immunoediting and Changes the Dynamics of Tumor Evolution. Nat Commun (2018) 9(1):32. doi: 10.1038/s41467-017-02424-0
34. Sugawara T, Miya F, Ishikawa T, Lysenko A, Nishino J, Kamatani T, et al. Immune Subtypes and Neoantigen-Related Immune Evasion in Advanced Colorectal Cancer. iScience (2022) 25(2):103740. doi: 10.1016/j.isci.2022.103740
35. Shen R, Li P, Li B, Zhang B, Feng L, Cheng S. Identification of Distinct Immune Subtypes in Colorectal Cancer Based on the Stromal Compartment. Front Oncol (2019) 9:1497. doi: 10.3389/fonc.2019.01497
36. Manieri NA, Chiang EY, Grogan JL. TIGIT: A Key Inhibitor of the Cancer Immunity Cycle. Trends Immunol (2017) 38(1):20–8. doi: 10.1016/j.it.2016.10.002
37. Yang ZZ, Kim HJ, Wu H, Jalali S, Tang X, Krull JE, et al. TIGIT Expression Is Associated With T-Cell Suppression and Exhaustion and Predicts Clinical Outcome and Anti-PD-1 Response in Follicular Lymphoma. Clin Cancer Res (2020) 26(19):5217–31. doi: 10.1158/1078-0432.CCR-20-0558
38. Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, et al. Anti-CTLA-4 Therapy Broadens the Melanoma-Reactive CD8+ T Cell Response. Sci Transl Med (2014) 6(254):254ra128. doi: 10.1126/scitranslmed.3008918
39. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal Replacement of Tumor-Specific T Cells Following PD-1 Blockade. Nat Med (2019) 25(8):1251–9. doi: 10.1038/s41591-019-0522-3
40. Pauken KE, Godec J, Odorizzi PM, Brown KE, Yates KB, Ngiow SF, et al. The PD-1 Pathway Regulates Development and Function of Memory CD8(+) T Cells Following Respiratory Viral Infection. Cell Rep (2020) 31(13):107827. doi: 10.1016/j.celrep.2020.107827
41. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated With Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med (2016) 375(9):819–29. doi: 10.1056/NEJMoa1604958
42. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to Checkpoint Blockade Therapy Through Inactivation of Antigen Presentation. Nat Commun (2017) 8(1):1136. doi: 10.1038/s41467-017-01062-w
43. Zhao X, Wangmo D, Robertson M, Subramanian S. Acquired Resistance to Immune Checkpoint Blockade Therapies. Cancers (Basel) (2020) 12(5):1161. doi: 10.3390/cancers12051161
44. Ge Z, Peppelenbosch MP, Sprengers D, Kwekkeboom J. TIGIT, the Next Step Towards Successful Combination Immune Checkpoint Therapy in Cancer. Front Immunol (2021) 12:699895. doi: 10.3389/fimmu.2021.699895
45. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 Is a Member of the CD28 Family and an Important Modulator of T-Cell Function. Eur J Immunol (2011) 41(4):902–15. doi: 10.1002/eji.201041136
46. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The Surface Protein TIGIT Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
47. Stengel KF, Harden-Bowles K, Yu X, Rouge L, Yin J, Comps-Agrar L, et al. Structure of TIGIT Immunoreceptor Bound to Poliovirus Receptor Reveals a Cell-Cell Adhesion and Signaling Mechanism That Requires Cis-Trans Receptor Clustering. Proc Natl Acad Sci USA (2012) 109(14):5399–404. doi: 10.1073/pnas.1120606109
48. Li M, Xia P, Du Y, Liu S, Huang G, Chen J, et al. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Receptor/Poliovirus Receptor (PVR) Ligand Engagement Suppresses Interferon-Gamma Production of Natural Killer Cells via Beta-Arrestin 2-Mediated Negative Signaling. J Biol Chem (2014) 289(25):17647–57. doi: 10.1074/jbc.M114.572420
49. Martinet L, Smyth MJ. Regulation of Immune Cell Functions Through Nectin and Nectin-Like Receptors. In: Ratcliffe MJH, editor. Encyclopedia of Immunobiology. Oxford: Academic Press (2016). p. 404–14.
50. Liu S, Zhang H, Li M, Hu D, Li C, Ge B, et al. Recruitment of Grb2 and SHIP1 by the ITT-Like Motif of TIGIT Suppresses Granule Polarization and Cytotoxicity of NK Cells. Cell Death Differ (2013) 20(3):456–64. doi: 10.1038/cdd.2012.141
51. Stanietsky N, Rovis TL, Glasner A, Seidel E, Tsukerman P, Yamin R, et al. Mouse TIGIT Inhibits NK-Cell Cytotoxicity Upon Interaction With PVR. Eur J Immunol (2013) 43(8):2138–50. doi: 10.1002/eji.201243072
52. Deuss FA, Gully BS, Rossjohn J, Berry R. Recognition of Nectin-2 by the Natural Killer Cell Receptor T Cell Immunoglobulin and ITIM Domain (TIGIT). J Biol Chem (2017) 292(27):11413–22. doi: 10.1074/jbc.M117.786483
53. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The Interaction of TIGIT With PVR and PVRL2 Inhibits Human NK Cell Cytotoxicity. Proc Natl Acad Sci USA (2009) 106(42):17858–63. doi: 10.1073/pnas.0903474106
54. Negishi N, Sato T, Yamashita-Kanemaru Y, Shibuya K, Uchida K, Kametani Y, et al. CD155-Transducing Signaling Through TIGIT Plays an Important Role in Transmission of Tolerant State and Suppression Capacity. Immunohorizons (2018) 2(10):338–48. doi: 10.4049/immunohorizons.1800033
55. Pende D, Bottino C, Castriconi R, Cantoni C, Marcenaro S, Rivera P, et al. PVR (CD155) and Nectin-2 (CD112) as Ligands of the Human DNAM-1 (CD226) Activating Receptor: Involvement in Tumor Cell Lysis. Mol Immunol (2005) 42(4):463–9. doi: 10.1016/j.molimm.2004.07.028
56. Samanta D, Ramagopal UA, Rubinstein R, Vigdorovich V, Nathenson SG, Almo SC. Structure of Nectin-2 Reveals Determinants of Homophilic and Heterophilic Interactions That Control Cell-Cell Adhesion. Proc Natl Acad Sci USA (2012) 109(37):14836–40. doi: 10.1073/pnas.1212912109
57. Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT, et al. Identification of CD112R as a Novel Checkpoint for Human T Cells. J Exp Med (2016) 213(2):167–76. doi: 10.1084/jem.20150785
58. Eberle F, Dubreuil P, Mattei MG, Devilard E, Lopez M. The Human PRR2 Gene, Related to the Human Poliovirus Receptor Gene (PVR), Is the True Homolog of the Murine MPH Gene. Gene (1995) 159(2):267–72. doi: 10.1016/0378-1119(95)00180-e
59. Lopez M, Aoubala M, Jordier F, Isnardon D, Gomez S, Dubreuil P. The Human Poliovirus Receptor Related 2 Protein Is a New Hematopoietic/Endothelial Homophilic Adhesion Molecule. Blood (1998) 92(12):4602–11. doi: 10.1182/blood.V92.12.4602
60. Inagaki M, Irie K, Ishizaki H, Tanaka-Okamoto M, Miyoshi J, Takai Y. Role of Cell Adhesion Molecule Nectin-3 in Spermatid Development. Genes Cells (2006) 11(9):1125–32. doi: 10.1111/j.1365-2443.2006.01006.x
61. Ogita H, Rikitake Y, Miyoshi J, Takai Y. Cell Adhesion Molecules Nectins and Associating Proteins: Implications for Physiology and Pathology. Proc Jpn Acad Ser B Phys Biol Sci (2010) 86(6):621–9. doi: 10.2183/pjab.86.621
62. Mizoguchi A, Nakanishi H, Kimura K, Matsubara K, Ozaki-Kuroda K, Katata T, et al. Nectin: An Adhesion Molecule Involved in Formation of Synapses. J Cell Biol (2002) 156(3):555–65. doi: 10.1083/jcb.200103113
63. Poon CE, Madawala RJ, Dowland SN, Murphy CR. Nectin-3 Is Increased in the Cell Junctions of the Uterine Epithelium at Implantation. Reprod Sci (2016) 23(11):1580–92. doi: 10.1177/1933719116648216
64. Fujito T, Ikeda W, Kakunaga S, Minami Y, Kajita M, Sakamoto Y, et al. Inhibition of Cell Movement and Proliferation by Cell-Cell Contact-Induced Interaction of Necl-5 With Nectin-3. J Cell Biol (2005) 171(1):165–73. doi: 10.1083/jcb.200501090
65. Inagaki M, Irie K, Ishizaki H, Tanaka-Okamoto M, Morimoto K, Inoue E, et al. Roles of Cell-Adhesion Molecules Nectin 1 and Nectin 3 in Ciliary Body Development. Development (2005) 132(7):1525–37. doi: 10.1242/dev.01697
66. Ozaki-Kuroda K, Nakanishi H, Ohta H, Tanaka H, Kurihara H, Mueller S, et al. Nectin Couples Cell-Cell Adhesion and the Actin Scaffold at Heterotypic Testicular Junctions. Curr Biol (2002) 12(13):1145–50. doi: 10.1016/s0960-9822(02)00922-3
67. Racaniello VR. Polio. In: Schaechter M, editor. Encyclopedia of Microbiology, 3rd ed. Oxford: Academic Press (2009). p. 459–68.
68. Baury B, Masson D, McDermott BM Jr., Jarry A, Blottiere HM, Blanchardie P, et al. Identification of Secreted CD155 Isoforms. Biochem Biophys Res Commun (2003) 309(1):175–82. doi: 10.1016/s0006-291x(03)01560-2
69. Iguchi-Manaka A, Okumura G, Kojima H, Cho Y, Hirochika R, Bando H, et al. Increased Soluble CD155 in the Serum of Cancer Patients. PloS One (2016) 11(4):e0152982. doi: 10.1371/journal.pone.0152982
70. Walwyn-Brown K, Guldevall K, Saeed M, Pende D, Onfelt B, MacDonald AS, et al. Human NK Cells Lyse Th2-Polarizing Dendritic Cells via NKp30 and DNAM-1. J Immunol (2018) 201(7):2028–41. doi: 10.4049/jimmunol.1800475
71. Pende D, Castriconi R, Romagnani P, Spaggiari GM, Marcenaro S, Dondero A, et al. Expression of the DNAM-1 Ligands, Nectin-2 (CD112) and Poliovirus Receptor (CD155), on Dendritic Cells: Relevance for Natural Killer-Dendritic Cell Interaction. Blood (2006) 107(5):2030–6. doi: 10.1182/blood-2005-07-2696
72. Huang WC, Kuo KT, Wang CH, Yeh CT, Wang Y. Cisplatin Resistant Lung Cancer Cells Promoted M2 Polarization of Tumor-Associated Macrophages via the Src/CD155/MIF Functional Pathway. J Exp Clin Cancer Res (2019) 38(1):180. doi: 10.1186/s13046-019-1166-3
73. Kamran N, Takai Y, Miyoshi J, Biswas SK, Wong JS, Gasser S. Toll-Like Receptor Ligands Induce Expression of the Costimulatory Molecule CD155 on Antigen-Presenting Cells. PloS One (2013) 8(1):e54406. doi: 10.1371/journal.pone.0054406
74. Maier MK, Seth S, Czeloth N, Qiu Q, Ravens I, Kremmer E, et al. The Adhesion Receptor CD155 Determines the Magnitude of Humoral Immune Responses Against Orally Ingested Antigens. Eur J Immunol (2007) 37(8):2214–25. doi: 10.1002/eji.200737072
75. Azzazi MO, Hegab HM, El-Ghammaz AMS, Saber HM, Afifi YA. Impact of Serum Soluble CD155 Level at Diagnosis on Interim Response to CHOP With or Without Rituximab in Diffuse Large B Cell Lymphoma. Clin Exp Med (2021) 3. doi: 10.1007/s10238-021-00741-9
76. Mueller S, Wimmer E. Recruitment of Nectin-3 to Cell-Cell Junctions Through Trans-Heterophilic Interaction With CD155, a Vitronectin and Poliovirus Receptor That Localizes to Alpha(V)Beta3 Integrin-Containing Membrane Microdomains. J Biol Chem (2003) 278(33):31251–60. doi: 10.1074/jbc.M304166200
77. Gromeier M, Solecki D, Patel DD, Wimmer E. Expression of the Human Poliovirus Receptor/CD155 Gene During Development of the Central Nervous System: Implications for the Pathogenesis of Poliomyelitis. Virology (2000) 273(2):248–57. doi: 10.1006/viro.2000.0418
78. Iwasaki A, Welker R, Mueller S, Linehan M, Nomoto A, Wimmer E. Immunofluorescence Analysis of Poliovirus Receptor Expression in Peyer's Patches of Humans, Primates, and CD155 Transgenic Mice: Implications for Poliovirus Infection. J Infect Dis (2002) 186(5):585–92. doi: 10.1086/342682
79. Ravens I, Seth S, Forster R, Bernhardt G. Characterization and Identification of Tage4 as the Murine Orthologue of Human Poliovirus Receptor/CD155. Biochem Biophys Res Commun (2003) 312(4):1364–71. doi: 10.1016/j.bbrc.2003.11.067
80. Sloan KE, Eustace BK, Stewart JK, Zehetmeier C, Torella C, Simeone M, et al. CD155/PVR Plays a Key Role in Cell Motility During Tumor Cell Invasion and Migration. BMC Cancer (2004) 4:73. doi: 10.1186/1471-2407-4-73
81. Gao J, Zheng Q, Xin N, Wang W, Zhao C. CD155, an Onco-Immunologic Molecule in Human Tumors. Cancer Sci (2017) 108(10):1934–8. doi: 10.1111/cas.13324
82. Wang FF, Wang Y, Wang L, Wang TS, Bai YP. TIGIT Expression Levels on CD4+ T Cells Are Correlated With Disease Severity in Patients With Psoriasis. Clin Exp Dermatol (2018) 43(6):675–82. doi: 10.1111/ced.13414
83. Zhang D, Hu W, Xie J, Zhang Y, Zhou B, Liu X, et al. TIGIT-Fc Alleviates Acute Graft-Versus-Host Disease by Suppressing CTL Activation via Promoting the Generation of Immunoregulatory Dendritic Cells. Biochim Biophys Acta Mol Basis Dis (2018) 1864(9 Pt B):3085–98. doi: 10.1016/j.bbadis.2018.06.022
84. Noguchi Y, Maeda A, Lo PC, Takakura C, Haneda T, Kodama T, et al. Human TIGIT on Porcine Aortic Endothelial Cells Suppresses Xenogeneic Macrophage-Mediated Cytotoxicity. Immunobiology (2019) 224(5):605–13. doi: 10.1016/j.imbio.2019.07.008
85. Chen X, Lu PH, Liu L, Fang ZM, Duan W, Liu ZL, et al. TIGIT Negatively Regulates Inflammation by Altering Macrophage Phenotype. Immunobiology (2016) 221(1):48–55. doi: 10.1016/j.imbio.2015.08.003
86. Reches A, Ophir Y, Stein N, Kol I, Isaacson B, Charpak Amikam Y, et al. Nectin4 Is a Novel TIGIT Ligand Which Combines Checkpoint Inhibition and Tumor Specificity. J Immunother Cancer (2020) 8(1):e000266. doi: 10.1136/jitc-2019-000266
87. Reymond N, Fabre S, Lecocq E, Adelaide J, Dubreuil P, Lopez M. Nectin4/PRR4, a New Afadin-Associated Member of the Nectin Family That Trans-Interacts With Nectin1/PRR1 Through V Domain Interaction. J Biol Chem (2001) 276(46):43205–15. doi: 10.1074/jbc.M103810200
88. MR M, Cabaud O, Josselin E, Finetti P, Castellano R, Farina A, et al. Nectin-4: A New Prognostic Biomarker for Efficient Therapeutic Targeting of Primary and Metastatic Triple-Negative Breast Cancer. Ann Oncol (2017) 28(4):769–76. doi: 10.1093/annonc/mdw678
89. Siddharth S, Goutam K, Das S, Nayak A, Nayak D, Sethy C, et al. Nectin-4 Is a Breast Cancer Stem Cell Marker That Induces WNT/beta-Catenin Signaling via Pi3k/Akt Axis. Int J Biochem Cell Biol (2017) 89:85–94. doi: 10.1016/j.biocel.2017.06.007
90. Hoffman-Censits JH, Lombardo KA, Parimi V, Kamanda S, Choi W, Hahn NM, et al. Expression of Nectin-4 in Bladder Urothelial Carcinoma, in Morphologic Variants, and Nonurothelial Histotypes. Appl Immunohistochem Mol Morphol (2021) 29(8):619–25. doi: 10.1097/PAI.0000000000000938
91. Li P, Hou F, Wang S, Luo N, Qi Y, Wang Y. A Novel NECTIN4-NTRK1 Fusion Identified in a Lung Squamous Cell Carcinoma Patient With MSI-H. J Cancer Res Clin Oncol (2021) 147(8):2483–6. doi: 10.1007/s00432-021-03622-6
92. Takano A, Ishikawa N, Nishino R, Masuda K, Yasui W, Inai K, et al. Identification of Nectin-4 Oncoprotein as a Diagnostic and Therapeutic Target for Lung Cancer. Cancer Res (2009) 69(16):6694–703. doi: 10.1158/0008-5472.CAN-09-0016
93. Challita-Eid PM, Satpayev D, Yang P, An Z, Morrison K, Shostak Y, et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res (2016) 76(10):3003–13. doi: 10.1158/0008-5472.CAN-15-1313
94. Nishiwada S, Sho M, Yasuda S, Shimada K, Yamato I, Akahori T, et al. Nectin-4 Expression Contributes to Tumor Proliferation, Angiogenesis and Patient Prognosis in Human Pancreatic Cancer. J Exp Clin Cancer Res (2015) 34:30. doi: 10.1186/s13046-015-0144-7
95. Zhang Z, Wu N, Lu Y, Davidson D, Colonna M, Veillette A. DNAM-1 Controls NK Cell Activation via an ITT-Like Motif. J Exp Med (2015) 212(12):2165–82. doi: 10.1084/jem.20150792
96. Shibuya A, Campbell D, Hannum C, Yssel H, Franz-Bacon K, McClanahan T, et al. DNAM-1, A Novel Adhesion Molecule Involved in the Cytolytic Function of T Lymphocytes. Immunity (1996) 4(6):573–81. doi: 10.1016/S1074-7613(00)70060-4
97. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 Axis Regulates Human T Cell Function. J Immunol (2012) 188(8):3869–75. doi: 10.4049/jimmunol.1103627
98. Sanchez-Correa B, Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Bergua JM, et al. DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell-Based Cancer Immunotherapy. Cancers (Basel) (2019) 11(6):877. doi: 10.3390/cancers11060877
99. Enqvist M, Ask EH, Forslund E, Carlsten M, Abrahamsen G, Beziat V, et al. Coordinated Expression of DNAM-1 and LFA-1 in Educated NK Cells. J Immunol (2015) 194(9):4518–27. doi: 10.4049/jimmunol.1401972
100. Martinet L, Ferrari De Andrade L, Guillerey C, Lee JS, Liu J, Souza-Fonseca-Guimaraes F, et al. DNAM-1 Expression Marks an Alternative Program of NK Cell Maturation. Cell Rep (2015) 11(1):85–97. doi: 10.1016/j.celrep.2015.03.006
101. Zhang B, Zhao W, Li H, Chen Y, Tian H, Li L, et al. Immunoreceptor TIGIT Inhibits the Cytotoxicity of Human Cytokine-Induced Killer Cells by Interacting With CD155. Cancer Immunol Immunother (2016) 65(3):305–14. doi: 10.1007/s00262-016-1799-4
102. Zhang Y, Ellinger J, Ritter M, Schmidt-Wolf IGH. Clinical Studies Applying Cytokine-Induced Killer Cells for the Treatment of Renal Cell Carcinoma. Cancers (Basel) (2020) 12(9):2471. doi: 10.3390/cancers12092471
103. Li S, Ding J, Wang Y, Wang X, Lv L. CD155/TIGIT Signaling Regulates the Effector Function of Tumor-Infiltrating CD8+ T Cell by NF-kappaB Pathway in Colorectal Cancer. J Gastroenterol Hepatol (2022) 37(1):154–63. doi: 10.1111/jgh.15730
104. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg Cells Expressing the Coinhibitory Molecule TIGIT Selectively Inhibit Proinflammatory Th1 and Th17 Cell Responses. Immunity (2014) 40(4):569–81. doi: 10.1016/j.immuni.2014.02.012
105. Lucca LE, Axisa PP, Singer ER, Nolan NM, Dominguez-Villar M, Hafler DA. TIGIT Signaling Restores Suppressor Function of Th1 Tregs. JCI Insight (2019) 4(3):e124427. doi: 10.1172/jci.insight.124427
106. Dardalhon V, Schubart AS, Reddy J, Meyers JH, Monney L, Sabatos CA, et al. CD226 is Specifically Expressed on the Surface of Th1 Cells and Regulates Their Expansion and Effector Functions. J Immunol (2005) 175(3):1558–65. doi: 10.4049/jimmunol.175.3.1558
107. Lozano E, Joller N, Cao Y, Kuchroo VK, Hafler DA. The CD226/CD155 Interaction Regulates the Proinflammatory (Th1/Th17)/Anti-Inflammatory (Th2) Balance in Humans. J Immunol (2013) 191(7):3673–80. doi: 10.4049/jimmunol.1300945
108. Wherry EJ, Kurachi M. Molecular and Cellular Insights Into T Cell Exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
109. Gorvel L, Olive D. Targeting the "PVR-TIGIT Axis" With Immune Checkpoint Therapies. F1000Res (2020) 9(F1000 Faculty Rev):354. doi: 10.12688/f1000research.22877.1
110. Zhang L, Pan J, Chen W, Jiang J, Huang J. Chronic Stress-Induced Immune Dysregulation in Cancer: Implications for Initiation, Progression, Metastasis, and Treatment. Am J Cancer Res (2020) 10(5):1294–307.
111. Sun Y, Luo J, Chen Y, Cui J, Lei Y, Cui Y, et al. Combined Evaluation of the Expression Status of CD155 and TIGIT Plays an Important Role in the Prognosis of LUAD (Lung Adenocarcinoma). Int Immunopharmacol (2020) 80:106198. doi: 10.1016/j.intimp.2020.106198
112. Dai S, Zeng H, Liu Z, Jin K, Jiang W, Wang Z, et al. Intratumoral Cxcl13(+)Cd8(+)T Cell Infiltration Determines Poor Clinical Outcomes and Immunoevasive Contexture in Patients With Clear Cell Renal Cell Carcinoma. J Immunother Cancer (2021) 9(2):e001823. doi: 10.1136/jitc-2020-001823
113. Liu X, Li M, Wang X, Dang Z, Jiang Y, Wang X, et al. PD-1(+) TIGIT(+) CD8(+) T Cells are Associated With Pathogenesis and Progression of Patients With Hepatitis B Virus-Related Hepatocellular Carcinoma. Cancer Immunol Immunother (2019) 68(12):2041–54. doi: 10.1007/s00262-019-02426-5
114. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 Dual Checkpoint Blockade Enhances Antitumor Immunity and Survival in GBM. Oncoimmunology (2018) 7(8):e1466769. doi: 10.1080/2162402X.2018.1466769
115. Xu J, Liu F, Li Y, Shen L. A 1p/19q Codeletion-Associated Immune Signature for Predicting Lower Grade Glioma Prognosis. Cell Mol Neurobiol (2020) 42(3):709–22. doi: 10.1007/s10571-020-00959-3
116. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 Impair Tumor Antigen-Specific CD8(+) T Cells in Melanoma Patients. J Clin Invest (2015) 125(5):2046–58. doi: 10.1172/JCI80445
117. Masson D, Jarry A, Baury B, Blanchardie P, Laboisse C, Lustenberger P, et al. Overexpression of the CD155 Gexne in Human Colorectal Carcinoma. Gut (2001) 49(2):236–40. doi: 10.1136/gut.49.2.236
118. Castriconi R, Dondero A, Corrias MV, Lanino E, Pende D, Moretta L, et al. Natural Killer Cell-Mediated Killing of Freshly Isolated Neuroblastoma Cells: Critical Role of DNAX Accessory Molecule-1-Poliovirus Receptor Interaction. Cancer Res (2004) 64(24):9180–4. doi: 10.1158/0008-5472.CAN-04-2682
119. Tahara-Hanaoka S, Shibuya K, Kai H, Miyamoto A, Morikawa Y, Ohkochi N, et al. Tumor Rejection by the Poliovirus Receptor Family Ligands of the DNAM-1 (CD226) Receptor. Blood (2006) 107(4):1491–6. doi: 10.1182/blood-2005-04-1684
120. Yin X, Zhang X, Liu Z, Sun G, Zhu X, Zhang H, et al. Assessment for Prognostic Value of Differentially Expressed Genes in Immune Microenvironment of Clear Cell Renal Cell Carcinoma. Am J Transl Res (2020) 12(9):5416–32.
121. Shao Q, Wang L, Yuan M, Jin X, Chen Z, Wu C. TIGIT Induces (CD3+) T Cell Dysfunction in Colorectal Cancer by Inhibiting Glucose Metabolism. Front Immunol (2021) 12:688961. doi: 10.3389/fimmu.2021.688961
122. Wen J, Mao X, Cheng Q, Liu Z, Liu F. A Pan-Cancer Analysis Revealing the Role of TIGIT in Tumor Microenvironment. Sci Rep (2021) 11(1):22502. doi: 10.1038/s41598-021-01933-9
123. Zhang Y, Song J, Zhao Z, Yang M, Chen M, Liu C, et al. Single-Cell Transcriptome Analysis Reveals Tumor Immune Microenvironment Heterogenicity and Granulocytes Enrichment in Colorectal Cancer Liver Metastases. Cancer Lett (2020) 470:84–94. doi: 10.1016/j.canlet.2019.10.016
124. Qian J, Olbrecht S, Boeckx B, Vos H, Laoui D, Etlioglu E, et al. A Pan-Cancer Blueprint of the Heterogeneous Tumor Microenvironment Revealed by Single-Cell Profiling. Cell Res (2020) 30(9):745–62. doi: 10.1038/s41422-020-0355-0
125. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T Cell Exhaustion by Multiple Inhibitory Receptors During Chronic Viral Infection. Nat Immunol (2009) 10(1):29–37. doi: 10.1038/ni.1679
126. Zarour HM. Reversing T-Cell Dysfunction and Exhaustion in Cancer. Clin Cancer Res (2016) 22(8):1856–64. doi: 10.1158/1078-0432.CCR-15-1849
127. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8(+) T Cell Effector Function. Cancer Cell (2014) 26(6):923–37. doi: 10.1016/j.ccell.2014.10.018
128. Li X, Wang R, Fan P, Yao X, Qin L, Peng Y, et al. A Comprehensive Analysis of Key Immune Checkpoint Receptors on Tumor-Infiltrating T Cells From Multiple Types of Cancer. Front Oncol (2019) 9:1066. doi: 10.3389/fonc.2019.01066
129. Weimer P, Wellbrock J, Sturmheit T, Oliveira-Ferrer L, Ding Y, Menzel S, et al. Tissue-Specific Expression of TIGIT, PD-1, TIM-3, and CD39 by Gammadelta T Cells in Ovarian Cancer. Cells (2022) 11(6):964. doi: 10.3390/cells11060964
130. Sasidharan Nair V, El Salhat H, Taha RZ, John A, Ali BR, Elkord E. DNA Methylation and Repressive H3K9 and H3K27 Trimethylation in the Promoter Regions of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, and PD-L1 Genes in Human Primary Breast Cancer. Clin Epigenet (2018) 10:78. doi: 10.1186/s13148-018-0512-1
131. Sasidharan Nair V, Toor SM, Taha RZ, Shaath H, Elkord E. DNA Methylation and Repressive Histones in the Promoters of PD-1, CTLA-4, TIM-3, LAG-3, TIGIT, PD-L1, and Galectin-9 Genes in Human Colorectal Cancer. Clin Epigenet (2018) 10(1):104. doi: 10.1186/s13148-018-0539-3
132. Syed Khaja AS, Toor SM, El Salhat H, Faour I, Ul Haq N, Ali BR, et al. Preferential Accumulation of Regulatory T Cells With Highly Immunosuppressive Characteristics in Breast Tumor Microenvironment. Oncotarget (2017) 8(20):33159–71. doi: 10.18632/oncotarget.16565
133. Wang W, Shen G, Wu S, Song S, Ni Y, Suo Z, et al. PD-1 mRNA Expression in Peripheral Blood Cells and its Modulation Characteristics in Cancer Patients. Oncotarget (2017) 8(31):50782–91. doi: 10.18632/oncotarget.15006
134. Elashi AA, Sasidharan Nair V, Taha RZ, Shaath H, Elkord E. DNA Methylation of Immune Checkpoints in the Peripheral Blood of Breast and Colorectal Cancer Patients. Oncoimmunology (2019) 8(2):e1542918. doi: 10.1080/2162402X.2018.1542918
135. Wang P, Chen Y, Long Q, Li Q, Tian J, Liu T, et al. Increased Coexpression of PD-L1 and TIM3/TIGIT Is Associated With Poor Overall Survival of Patients With Esophageal Squamous Cell Carcinoma. J Immunother Cancer (2021) 9(10):e002836. doi: 10.1136/jitc-2021-002836
136. Hajiasghar-Sharbaf R, Asgarian-Omran H, Valadan R, Hossein-Nattaj H, Shekarriz R, Zaboli E, et al. CD8+ T-Cells Co-Expressing PD-1 and TIGIT Are Highly Frequent in Chronic Lymphocytic Leukemia. Iran J Allergy Asthma Immunol (2021) 20(6):751–63. doi: 10.18502/ijaai.v20i6.8027
137. Taghiloo S, Allahmoradi E, Tehrani M, Hossein-Nataj H, Shekarriz R, Janbabaei G, et al. Frequency and Functional Characterization of Exhausted CD8(+) T Cells in Chronic Lymphocytic Leukemia. Eur J Haematol (2017) 98(6):622–31. doi: 10.1111/ejh.12880
138. Allahmoradi E, Taghiloo S, Tehrani M, Hossein-Nattaj H, Janbabaei G, Shekarriz R, et al. CD4+ T Cells are Exhausted and Show Functional Defects in Chronic Lymphocytic Leukemia. Iran J Immunol (2017) 14(4):257–69.
139. Brauneck F, Weimer P, Schulze Zur Wiesch J, Weisel K, Leypoldt L, Vohwinkel G, et al. Bone Marrow-Resident Vdelta1 T Cells Co-Express TIGIT With PD-1, TIM-3 or CD39 in AML and Myeloma. Front Med (Lausanne) (2021) 8:763773. doi: 10.3389/fmed.2021.763773
140. Jin Z, Ye W, Lan T, Zhao Y, Liu X, Chen J, et al. Characteristic of TIGIT and DNAM-1 Expression on Foxp3+ Gammadelta T Cells in AML Patients. BioMed Res Int (2020) 2020:4612952. doi: 10.1155/2020/4612952
141. Annibali O, Bianchi A, Grifoni A, Tomarchio V, Tafuri M, Verri M, et al. A Novel Scoring System for TIGIT Expression in Classic Hodgkin Lymphoma. Sci Rep (2021) 11(1):7059. doi: 10.1038/s41598-021-86655-8
142. Alfarra H, Weir J, Grieve S, Reiman T. Targeting NK Cell Inhibitory Receptors for Precision Multiple Myeloma Immunotherapy. Front Immunol (2020) 11:575609. doi: 10.3389/fimmu.2020.575609
143. Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front Immunol (2019) 10:3038. doi: 10.3389/fimmu.2019.03038
144. Yu L, Liu X, Wang X, Yan F, Wang P, Jiang Y, et al. TIGIT(+) TIM-3(+) NK Cells are Correlated With NK Cell Exhaustion and Disease Progression in Patients With Hepatitis B Virusrelated Hepatocellular Carcinoma. Oncoimmunology (2021) 10(1):1942673. doi: 10.1080/2162402X.2021.1942673
145. Chauvin JM, Ka M, Pagliano O, Menna C, Ding Q, DeBlasio R, et al. IL15 Stimulation With TIGIT Blockade Reverses CD155-Mediated NK-Cell Dysfunction in Melanoma. Clin Cancer Res (2020) 26(20):5520–33. doi: 10.1158/1078-0432.CCR-20-0575
146. Simon S, Voillet V, Vignard V, Wu Z, Dabrowski C, Jouand N, et al. PD-1 and TIGIT Coexpression Identifies a Circulating CD8 T Cell Subset Predictive of Response to Anti-PD-1 Therapy. J Immunother Cancer (2020) 8(2):e001631. doi: 10.1136/jitc-2020-001631
147. He R, Hou S, Liu C, Zhang A, Bai Q, Han M, et al. Follicular CXCR5- Expressing CD8(+) T Cells Curtail Chronic Viral Infection. Nature (2016) 537(7620):412–28. doi: 10.1038/nature19317
148. Leong YA, Chen Y, Ong HS, Wu D, Man K, Deleage C, et al. CXCR5(+) Follicular Cytotoxic T Cells Control Viral Infection in B Cell Follicles. Nat Immunol (2016) 17(10):1187–96. doi: 10.1038/ni.3543
149. Fourcade J, Sun Z, Chauvin JM, Ka M, Davar D, Pagliano O, et al. CD226 Opposes TIGIT to Disrupt Tregs in Melanoma. JCI Insight (2018) 3(14):e121157. doi: 10.1172/jci.insight.121157
150. Zhang Y, Maksimovic J, Naselli G, Qian J, Chopin M, Blewitt ME, et al. Genome-Wide DNA Methylation Analysis Identifies Hypomethylated Genes Regulated by FOXP3 in Human Regulatory T Cells. Blood (2013) 122(16):2823–36. doi: 10.1182/blood-2013-02-481788
151. Zhang Q, Gao C, Shao J, Wang Z. TIGIT-Related Transcriptome Profile and Its Association With Tumor Immune Microenvironment in Breast Cancer. Biosci Rep (2021) 41(3):BSR20204340. doi: 10.1042/BSR20204340
152. Ma J. Bioinformatics-Guided Analysis Uncovers TIGIT as an Epigenetically Regulated Immunomodulator Affecting Immunotherapeutic Sensitivity of Gastric Cancer. Cancer biomark (2021) 33(3):349–58. doi: 10.3233/CBM-210159
153. Wu K, Zeng J, Shi X, Xie J, Li Y, Zheng H, et al. Targeting TIGIT Inhibits Bladder Cancer Metastasis Through Suppressing IL-32. Front Pharmacol (2021) 12:801493. doi: 10.3389/fphar.2021.801493
154. Liu Z, Zeng H, Jin K, Yu Y, You R, Zhang H, et al. TIGIT and PD-1 Expression Atlas Predicts Response to Adjuvant Chemotherapy and PD-L1 Blockade in Muscle-Invasive Bladder Cancer. Br J Cancer (2022) 126(9):1310–17. doi: 10.1038/s41416-022-01703-y
155. Liu Z, Zhou Q, Wang Z, Zhang H, Zeng H, Huang Q, et al. Intratumoral TIGIT(+) CD8(+) T-Cell Infiltration Determines Poor Prognosis and Immune Evasion in Patients With Muscle-Invasive Bladder Cancer. J Immunother Cancer (2020) 8(2):e000978. doi: 10.1136/jitc-2020-000978
156. Liang R, Zhu X, Lan T, Ding D, Zheng Z, Chen T, et al. TIGIT Promotes CD8(+)T Cells Exhaustion and Predicts Poor Prognosis of Colorectal Cancer. Cancer Immunol Immunother (2021) 70(10):2781–93. doi: 10.1007/s00262-021-02886-8
157. Zhou X, Ding X, Li H, Yang C, Ma Z, Xu G, et al. Upregulation of TIGIT and PD-1 in Colorectal Cancer With Mismatch-Repair Deficiency. Immunol Invest (2021) 50(4):338–55. doi: 10.1080/08820139.2020.1758130
158. Freed-Pastor WA, Lambert LJ, Ely ZA, Pattada NB, Bhutkar A, Eng G, et al. The CD155/TIGIT Axis Promotes and Maintains Immune Evasion in Neoantigen-Expressing Pancreatic Cancer. Cancer Cell (2021) 39(10):1342–60 e14. doi: 10.1016/j.ccell.2021.07.007
159. Nishi K, Ishikura S, Umebayashi M, Morisaki T, Inozume T, Kinugasa T, et al. Mutant KRAS Promotes NKG2D(+) T Cell Infiltration and CD155 Dependent Immune Evasion. Anticancer Res (2020) 40(8):4663–74. doi: 10.21873/anticanres.14465
160. Xu X, Wang D, Li N, Sheng J, Xie M, Zhou Z, et al. The Novel Tumor Microenvironment-Related Prognostic Gene AIF1 May Influence Immune Infiltrates and is Correlated With TIGIT in Esophageal Cancer. Ann Surg Oncol (2021) 29(5):2930–40. doi: 10.1245/s10434-021-10928-9
161. Ostroumov D, Duong S, Wingerath J, Woller N, Manns MP, Timrott K, et al. Transcriptome Profiling Identifies TIGIT as a Marker of T-Cell Exhaustion in Liver Cancer. Hepatology (2021) 73(4):1399–418. doi: 10.1002/hep.31466
162. Hong X, Wang X, Wang T, Zhang X. Correlation of T Cell Immunoglobulin and ITIM Domain (TIGIT) and Programmed Death 1 (PD-1) With Clinicopathological Characteristics of Renal Cell Carcinoma May Indicate Potential Targets for Treatment. Med Sci Monit (2018) 24:6861–72. doi: 10.12659/MSM.910388
163. Liu X, Li Q, Zhou Y, He X, Fang J, Lu H, et al. Dysfunctional Role of Elevated TIGIT Expression on T Cells in Oral Squamous Cell Carcinoma Patients. Oral Dis (2021) 27(7):1667–77. doi: 10.1111/odi.13703
164. Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, et al. A Distinct Gene Module for Dysfunction Uncoupled From Activation in Tumor-Infiltrating T Cells. Cell (2016) 166(6):1500–11 e9. doi: 10.1016/j.cell.2016.08.052
165. Judge SJ, Darrow MA, Thorpe SW, Gingrich AA, O'Donnell EF, Bellini AR, et al. Analysis of Tumor-Infiltrating NK and T Cells Highlights IL-15 Stimulation and TIGIT Blockade as a Combination Immunotherapy Strategy for Soft Tissue Sarcomas. J Immunother Cancer (2020) 8(2):e001355. doi: 10.1136/jitc-2020-001355
166. Fathi M, Bahmanpour S, Barshidi A, Rasouli H, Karoon Kiani F, Mahmoud Salehi Khesht A, et al. Simultaneous Blockade of TIGIT and HIF-1alpha Induces Synergistic Anti-Tumor Effect and Decreases the Growth and Development of Cancer Cells. Int Immunopharmacol (2021) 101(Pt A):108288. doi: 10.1016/j.intimp.2021.108288
167. Debela DT, Muzazu SG, Heraro KD, Ndalama MT, Mesele BW, Haile DC, et al. New Approaches and Procedures for Cancer Treatment: Current Perspectives. SAGE Open Med (2021) 9:20503121211034366. doi: 10.1177/20503121211034366
168. Solomon BL, Garrido-Laguna I. TIGIT: A Novel Immunotherapy Target Moving From Bench to Bedside. Cancer Immunol Immunother (2018) 67(11):1659–67. doi: 10.1007/s00262-018-2246-5
169. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune Checkpoint Inhibitors: Recent Progress and Potential Biomarkers. Exp Mol Med (2018) 50(12):1–11. doi: 10.1038/s12276-018-0191-1
170. Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in Colorectal Cancer: Rationale, Challenges and Potential. Nat Rev Gastroenterol Hepatol (2019) 16(6):361–75. doi: 10.1038/s41575-019-0126-x
171. Zhou X, Jiao L, Qian Y, Dong Q, Sun Y, Zheng WV, et al. Repositioning Azelnidipine as a Dual Inhibitor Targeting CD47/SIRPalpha and TIGIT/PVR Pathways for Cancer Immuno-Therapy. Biomolecules (2021) 11(5):706. doi: 10.3390/biom11050706
172. Brauneck F, Fischer B, Wellbrock J, Bokemeyer C, Schulze zur Wiesch J, Haag F, et al. Blockade of Tigit on AML-Derived M2 Macrophages Results in Reprograming Into the M1 Phenotype and Enhances CD47-Mediated Phagocytosis. Blood (2021) 138:3351. doi: 10.1182/blood-2021-148075
173. Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 Antibody-Mediated Phagocytosis of Cancer by Macrophages Primes an Effective Antitumor T-Cell Response. Proc Natl Acad Sci USA (2013) 110(27):11103–8. doi: 10.1073/pnas.1305569110
174. Han HS, Jeong S, Kim H, Kim HD, Kim AR, Kwon M, et al. TOX-Expressing Terminally Exhausted Tumor-Infiltrating CD8(+) T Cells are Reinvigorated by Co-Blockade of PD-1 and TIGIT in Bladder Cancer. Cancer Lett (2021) 499:137–47. doi: 10.1016/j.canlet.2020.11.035
175. Ma Y, Galluzzi L, Zitvogel L, Kroemer G. Autophagy and Cellular Immune Responses. Immunity (2013) 39(2):211–27. doi: 10.1016/j.immuni.2013.07.017
176. Zhang J, Zhou L, Xiang JD, Jin CS, Li MQ, He YY. Artesunate-Induced ATG5-Related Autophagy Enhances the Cytotoxicity of NK92 Cells on Endometrial Cancer Cells via Interactions Between CD155 and CD226/TIGIT. Int Immunopharmacol (2021) 97:107705. doi: 10.1016/j.intimp.2021.107705
177. Zhou X, Chen Y, Wang F, Wu H, Zhang Y, Liu J, et al. Artesunate Induces Autophagy Dependent Apoptosis Through Upregulating ROS and Activating AMPK-mTOR-ULK1 Axis in Human Bladder Cancer Cells. Chem Biol Interact (2020) 331:109273. doi: 10.1016/j.cbi.2020.109273
178. Yin X, Liu Y, Qin J, Wu Y, Huang J, Zhao Q, et al. Artesunate Suppresses the Proliferation and Development of Estrogen Receptor-Alpha-Positive Endometrial Cancer in HAND2-Dependent Pathway. Front Cell Dev Biol (2020) 8:606969. doi: 10.3389/fcell.2020.606969
179. Zhang C, Wang Y, Xun X, Wang S, Xiang X, Hu S, et al. TIGIT Can Exert Immunosuppressive Effects on CD8+ T Cells by the CD155/TIGIT Signaling Pathway for Hepatocellular Carcinoma In Vitro. J Immunother (2020) 43(8):236–43. doi: 10.1097/CJI.0000000000000330
180. Niu J, Maurice-Dror C, Lee DH, Kim DW, Nagrial A, Voskoboynik M, et al. First-In-Human Phase 1 Study of the Anti-TIGIT Antibody Vibostolimab as Monotherapy or With Pembrolizumab for Advanced Solid Tumors, Including Non-Small-Cell Lung Cancer(). Ann Oncol (2022) 33(2):169–80. doi: 10.1016/j.annonc.2021.11.002
181. Mettu NB, Ulahannan SV, Bendell JC, Garrido-Laguna I, Strickler JH, Moore KN, et al. A Phase 1a/B Open-Label, Dose-Escalation Study of Etigilimab Alone or in Combination With Nivolumab in Patients With Locally Advanced or Metastatic Solid Tumors. Clin Cancer Res (2021) 28(5):882–92. doi: 10.1158/1078-0432.CCR-21-2780
182. Walsh SR, Simovic B, Chen L, Bastin D, Nguyen A, Stephenson K, et al. Endogenous T Cells Prevent Tumor Immune Escape Following Adoptive T Cell Therapy. J Clin Invest (2019) 129(12):5400–10. doi: 10.1172/JCI126199
183. Grapin M, Richard C, Limagne E, Boidot R, Morgand V, Bertaut A, et al. Optimized Fractionated Radiotherapy With Anti-PD-L1 and Anti-TIGIT: A Promising New Combination. J Immunother Cancer (2019) 7(1):160. doi: 10.1186/s40425-019-0634-9
184. Hansen K, Kumar S, Logronio K, Whelan S, Qurashi S, Cheng HY, et al. COM902, a Novel Therapeutic Antibody Targeting TIGIT Augments Anti-Tumor T Cell Function in Combination With PVRIG or PD-1 Pathway Blockade. Cancer Immunol Immunother (2021) 70(12):3525–40. doi: 10.1007/s00262-021-02921-8
185. Hoogi S, Eisenberg V, Mayer S, Shamul A, Barliya T, Cohen CJ. A TIGIT-Based Chimeric Co-Stimulatory Switch Receptor Improves T-Cell Anti-Tumor Function. J Immunother Cancer (2019) 7(1):243. doi: 10.1186/s40425-019-0721-y
186. Gordon R, Kasler MK, Stasi K, Shames Y, Errante M, Ciccolini K, et al. Checkpoint Inhibitors: Common Immune-Related Adverse Events and Their Management. Clin J Oncol Nurs (2017) 21(2 Suppl):45–52. doi: 10.1188/17.CJON.S2.45-52
187. Ventola CL. Cancer Immunotherapy, Part 2: Efficacy, Safety, and Other Clinical Considerations. P T (2017) 42(7):452–63.
188. Brahmer JR, Lacchetti C, Thompson JA. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline Summary. J Oncol Pract (2018) 14(4):247–9. doi: 10.1200/JOP.18.00005
189. Puzanov I, Diab A, Abdallah K, Bingham CO 3rd, Brogdon C, Dadu R, et al. Managing Toxicities Associated With Immune Checkpoint Inhibitors: Consensus Recommendations From the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer (2017) 5(1):95. doi: 10.1186/s40425-017-0300-z
190. Okiyama N, Tanaka R. Immune-Related Adverse Events in Various Organs Caused by Immune Checkpoint Inhibitors. Allergol Int (2022) 71(2):169–78. doi: 10.1016/j.alit.2022.01.001
191. Spain L, Diem S, Larkin J. Management of Toxicities of Immune Checkpoint Inhibitors. Cancer Treat Rev (2016) 44:51–60. doi: 10.1016/j.ctrv.2016.02.001
192. Barber FD. Adverse Events of Oncologic Immunotherapy and Their Management. Asia Pac J Oncol Nurs (2019) 6(3):212–26. doi: 10.4103/apjon.apjon_6_19
193. Zhou C, Li M, Wang Z, An D, Li B. Adverse Events of Immunotherapy in non-Small Cell Lung Cancer: A Systematic Review and Network Meta-Analysis. Int Immunopharmacol (2022) 102:108353. doi: 10.1016/j.intimp.2021.108353
194. Harding FA, Stickler MM, Razo J, DuBridge RB. The Immunogenicity of Humanized and Fully Human Antibodies: Residual Immunogenicity Resides in the CDR Regions. MAbs (2010) 2(3):256–65. doi: 10.4161/mabs.2.3.11641
195. Rotte A, Sahasranaman S, Budha N. Targeting TIGIT for Immunotherapy of Cancer: Update on Clinical Development. Biomedicines (2021) 9(9):1277. doi: 10.3390/biomedicines9091277
196. Lo Nigro C, Macagno M, Sangiolo D, Bertolaccini L, Aglietta M, Merlano MC. NK-Mediated Antibody-Dependent Cell-Mediated Cytotoxicity in Solid Tumors: Biological Evidence and Clinical Perspectives. Ann Transl Med (2019) 7(5):105. doi: 10.21037/atm.2019.01.42
197. Chand D, Waight JD, Paltrinieri E, Dietrich S, Bushell M, Costa M, et al. Abstract 2390: FcgR Co-Engagement by Anti-TIGIT Monoclonal Antibodies Enhances T Cell Functionality and Antitumor Immune Responses. Cancer Res (2019) 79(13_Supplement):2390–. doi: 10.1158/1538-7445.Am2019-2390
198. Chen X, Song X, Li K, Zhang T. FcgammaR-Binding Is an Important Functional Attribute for Immune Checkpoint Antibodies in Cancer Immunotherapy. Front Immunol (2019) 10:292. doi: 10.3389/fimmu.2019.00292
199. Wang J, Hou H, Mao L, Wang F, Yu J, Luo Y, et al. TIGIT Signaling Pathway Regulates Natural Killer Cell Function in Chronic Hepatitis B Virus Infection. Front Med (Lausanne) (2021) 8:816474. doi: 10.3389/fmed.2021.816474
200. Shen X, Fu W, Wei Y, Zhu J, Yu Y, Lei C, et al. TIGIT-Fc Promotes Antitumor Immunity. Cancer Immunol Res (2021) 9(9):1088–97. doi: 10.1158/2326-6066.CIR-20-0986
201. Catakovic K, Gassner FJ, Ratswohl C, Zaborsky N, Rebhandl S, Schubert M, et al. TIGIT Expressing CD4+T Cells Represent a Tumor-Supportive T Cell Subset in Chronic Lymphocytic Leukemia. Oncoimmunology (2017) 7(1):e1371399. doi: 10.1080/2162402X.2017.1371399
202. Chauvin JM, Zarour HM. TIGIT in Cancer Immunotherapy. J Immunother Cancer (2020) 8(2):e000957. doi: 10.1136/jitc-2020-000957
203. Rotte A, Jin JY, Lemaire V. Mechanistic Overview of Immune Checkpoints to Support the Rational Design of Their Combinations in Cancer Immunotherapy. Ann Oncol (2018) 29(1):71–83. doi: 10.1093/annonc/mdx686
204. Roche’s Novel Anti-TIGIT Tiragolumab Granted FDA Breakthrough Therapy Designation in Combination With Tecentriq for PD-L1-High non-Small Cell Lung Cancer. News Release. Roche. Available at: https://bit.ly/3hmWDm3 (Accessed April 26, 2021).
205. Rodriguez-Abreu D, Johnson ML, Hussein MA, Cobo M, Patel AJ, Secen NM, et al. Primary Analysis of a Randomized, Double-Blind, Phase II Study of the Anti-TIGIT Antibody Tiragolumab (Tira) Plus Atezolizumab (Atezo) Versus Placebo Plus Atezo as First-Line (1L) Treatment in Patients With PD-L1-Selected NSCLC (CITYSCAPE). J Clin Oncol (2020) 38(15_suppl):9503–. doi: 10.1200/JCO.2020.38.15_suppl.9503
206. Ahn MJ, Niu J, Kim DW, Rasco D, Mileham KF, Chung HC, et al. 1400p Vibostolimab, an Anti-TIGIT Antibody, as Monotherapy and in Combination With Pembrolizumab in Anti-PD-1/PD-L1-Refractory NSCLC. Ann Oncol (2020) 31:S887. doi: 10.1016/j.annonc.2020.08.1714
207. Van den Mooter TFA MA, Jungles C, Delafontaine BR, Nguyen TLA, Warot S, Truong C, et al. Preliminary Data From Phase I First-in-Human Study of EOS884448, a Novel Potent Anti-TIGIT Antibody, Monotherapy Shows Favorable Tolerability Profile and Early Signs of Clinical Activity in Immune-Resistant Advanced Cancers. In: American Association for Cancer Research (AACR) Annual Meeting 2021. Cancer Res (2021) 81(Suppl 13):CT118. doi: 10.1158/1538-7445.AM2021-CT118
208. Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring Immune-Checkpoint Blockade: Response Evaluation and Biomarker Development. Nat Rev Clin Oncol (2017) 14(11):655–68. doi: 10.1038/nrclinonc.2017.88
209. McCune JS. Rapid Advances in Immunotherapy to Treat Cancer. Clin Pharmacol Ther (2018) 103(4):540–4. doi: 10.1002/cpt.985
210. Somarouthu B, Lee SI, Urban T, Sadow CA, Harris GJ, Kambadakone A. Immune-Related Tumour Response Assessment Criteria: A Comprehensive Review. Br J Radiol (2018) 91(1084):20170457. doi: 10.1259/bjr.20170457
211. Chiou VL, Burotto M. Pseudoprogression and Immune-Related Response in Solid Tumors. J Clin Oncol (2015) 33(31):3541–3. doi: 10.1200/JCO.2015.61.6870
212. Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S, et al. iRECIST: Guidelines for Response Criteria for Use in Trials Testing Immunotherapeutics. Lancet Oncol (2017) 18(3):e143–e52. doi: 10.1016/S1470-2045(17)30074-8
213. Persigehl T, Lennartz S, Schwartz LH. iRECIST: How to do it. Cancer Imaging (2020) 20(1):2. doi: 10.1186/s40644-019-0281-x
214. Gun SY, Lee SWL, Sieow JL, Wong SC. Targeting Immune Cells for Cancer Therapy. Redox Biol (2019) 25:101174. doi: 10.1016/j.redox.2019.101174
215. Liu J, Blake SJ, Smyth MJ, Teng MW. Improved Mouse Models to Assess Tumour Immunity and irAEs After Combination Cancer Immunotherapies. Clin Transl Immunol (2014) 3(8):e22. doi: 10.1038/cti.2014.18
216. Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. Safety Profiles of Anti-CTLA-4 and Anti-PD-1 Antibodies Alone and in Combination. Nat Rev Clin Oncol (2016) 13(8):473–86. doi: 10.1038/nrclinonc.2016.58
217. Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse Effects of Immune-Checkpoint Inhibitors: Epidemiology, Management and Surveillance. Nat Rev Clin Oncol (2019) 16(9):563–80. doi: 10.1038/s41571-019-0218-0
218. Harjunpaa H, Blake SJ, Ahern E, Allen S, Liu J, Yan J, et al. Deficiency of Host CD96 and PD-1 or TIGIT Enhances Tumor Immunity Without Significantly Compromising Immune Homeostasis. Oncoimmunology (2018) 7(7):e1445949. doi: 10.1080/2162402X.2018.1445949
219. Guillerey C, Harjunpaa H, Carrie N, Kassem S, Teo T, Miles K, et al. TIGIT Immune Checkpoint Blockade Restores CD8(+) T-Cell Immunity Against Multiple Myeloma. Blood (2018) 132(16):1689–94. doi: 10.1182/blood-2018-01-825265
Keywords: cancer, immune-checkpoint, immune-therapy, TIGIT, tumor microenvironment
Citation: Annese T, Tamma R and Ribatti D (2022) Update in TIGIT Immune-Checkpoint Role in Cancer. Front. Oncol. 12:871085. doi: 10.3389/fonc.2022.871085
Received: 07 February 2022; Accepted: 14 April 2022;
Published: 17 May 2022.
Edited by:
Daniele Vergara, University of Salento, ItalyReviewed by:
Wenyan Fu, Shanghai Jiao Tong University, ChinaFranziska Brauneck, University Medical Center Hamburg-Eppendorf, Germany
Copyright © 2022 Annese, Tamma and Ribatti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Domenico Ribatti, ZG9tZW5pY28ucmliYXR0aUB1bmliYS5pdA==; Tiziana Annese, YW5uZXNlQGx1bS5pdA==