
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 28 October 2022
Sec. Cancer Genetics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.870863
This article is part of the Research TopicAdvances in Genetics and Molecular Diagnosis in Colorectal, Stomach, and Pancreatic Cancer.View all 11 articles
 Alisa Petriina Olkinuora1*
Alisa Petriina Olkinuora1* Andrea Constanza Mayordomo2,3
Andrea Constanza Mayordomo2,3 Anni Katariina Kauppinen1
Anni Katariina Kauppinen1 María Belén Cerliani3Mariana Coraglio4Ávila Karina Collia4Alejandro Gutiérrez4
María Belén Cerliani3Mariana Coraglio4Ávila Karina Collia4Alejandro Gutiérrez4 Karin Alvarez5Alessandra Cassana6Francisco Lopéz-Köstner5
Karin Alvarez5Alessandra Cassana6Francisco Lopéz-Köstner5 Federico Jauk7
Federico Jauk7 Hernán García-Rivello7Ari Ristimäki8,9
Hernán García-Rivello7Ari Ristimäki8,9 Laura Koskenvuo10
Laura Koskenvuo10 Anna Lepistö8,10
Anna Lepistö8,10 Taina Tuulikki Nieminen1
Taina Tuulikki Nieminen1 Carlos Alberto Vaccaro2,11
Carlos Alberto Vaccaro2,11 Walter Hernán Pavicic2,11
Walter Hernán Pavicic2,11 Päivi Peltomäki1
Päivi Peltomäki1Recently, biallelic germline variants of the DNA glycosylase genes MUTYH and NTHL1 were linked to polyposis susceptibility. Significant fractions remain without a molecular explanation, warranting searches for underlying causes. We used exome sequencing to investigate clinically well-defined adenomatous polyposis cases and families from Finland (N=34), Chile (N=21), and Argentina (N=12), all with known susceptibility genes excluded. Nine index cases (13%) revealed germline variants with proven or possible pathogenicity in the DNA glycosylase genes, involving NEIL1 (mono- or biallelic) in 3 cases, MUTYH (monoallelic) in 3 cases, NTHL1 (biallelic) in 1 case, and OGG1 (monoallelic) in 2 cases. NTHL1 was affected with the well-established, pathogenic c.268C>T, p.(Gln90Ter) variant. A recurrent heterozygous NEIL1 c.506G>A, p.(Gly169Asp) variant was observed in two families. In a Finnish family, the variant occurred in trans with a truncating NEIL1 variant (c.821delT). In an Argentine family, the variant co-occurred with a genomic deletion of exons 2 – 11 of PMS2. Mutational signatures in tumor tissues complied with biological functions reported for NEIL1. Our results suggest that germline variants in DNA glycosylase genes may occur in a non-negligible proportion of unexplained colon polyposis cases and may predispose to tumor development.
Familial Adenomatous Polyposis (FAP; OMIM#175100) is characterized by multiple adenomas in the colorectum and an increased risk of colorectal cancer (CRC). In FAP, more than 100 adenomas are typically present, whereas an attenuated form (AFAP) is characterized by 10 – 100 polyps and a generally milder disease (1). Profuse or attenuated adenomatous polyposis was first associated with germline APC (OMIM*611731) variants in families exhibiting autosomal dominant inheritance, but has since been linked to biallelic MUTYH variants (MAP for MUTYH-associated polyposis; OMIM#608456) in families with autosomal recessive inheritance (2). MUTYH encodes a DNA glycosylase that acts on oxidative DNA damage by removing adenine misincorporated opposite 8-oxoG (3).
Up to 11-25% of FAP cases arise de novo (4, 5), and a fifth of de novo adenomatous polyposis cases are attributable to APC mosaicism (6). As much as 20% of cases with clinical features of FAP show no pathogenic variants in APC. Moreover, 80% patients with attenuated polyposis are molecularly unexplained (7–9). Recently, new predisposition genes for adenomatous polyposis have been identified, including POLE and POLD1 (PPAP for polymerase proofreading associated polyposis; OMIM# 615083 and # 612591, respectively) (10), AXIN2 (11), and biallelic MSH3 (OMIM# 617100) (12) and MLH3 (13) variants. Moreover, biallelic NTHL1 (OMIM# 616415) variants have been associated with polyposis and multi-organ cancer predisposition (14). Except for AXIN2, all these genes contribute to DNA fidelity, through proofreading DNA after replication (POLE and POLD1), mismatch repair (MSH3 and MLH3), or base excision repair (NTHL1). Like MUTYH, NTHL1 targets oxidative DNA damage; it encodes a DNA glycosylase that repairs pyrimidine-derived oxidation products (3).
Inspired by recent findings of novel polyposis and cancer predisposition genes, we embarked on a study to uncover new molecular factors for unexplained polyposis cases across multiple populations by exome-wide screening. Our efforts revealed several families harboring potentially pathogenic germline variants in DNA glycosylase genes, including mono- and biallelic alterations of NEIL1 (OMIM *608844; Endonuclease VIII-Like 1).
This investigation was based on 67 index cases with attenuated or profuse adenomatous polyposis (34 from Finland, 21 from Chile, and 12 from Argentina) in which known genetic causes of polyposis had been excluded (APC, POLE, POLD1, PTEN and biallelic MUTYH; Figure 1). The cases were ascertained through the national polyposis research registries and local hospitals as described below. Most cases (47/66, 71%) exhibited attenuated polyposis. Detailed clinical data are available in Table S1. Patient DNA was extracted from blood or EBV-transformed lymphoblasts as described by Renkonen et al. (15) DNA from formalin-fixed paraffin-embedded (FFPE) samples was extracted as described by Isola et al. (16) Patient RNA was extracted from lymphoblastoid cells using the NucleoSpin RNA extraction kit (Macherey-Nagel, Düren, Germany).
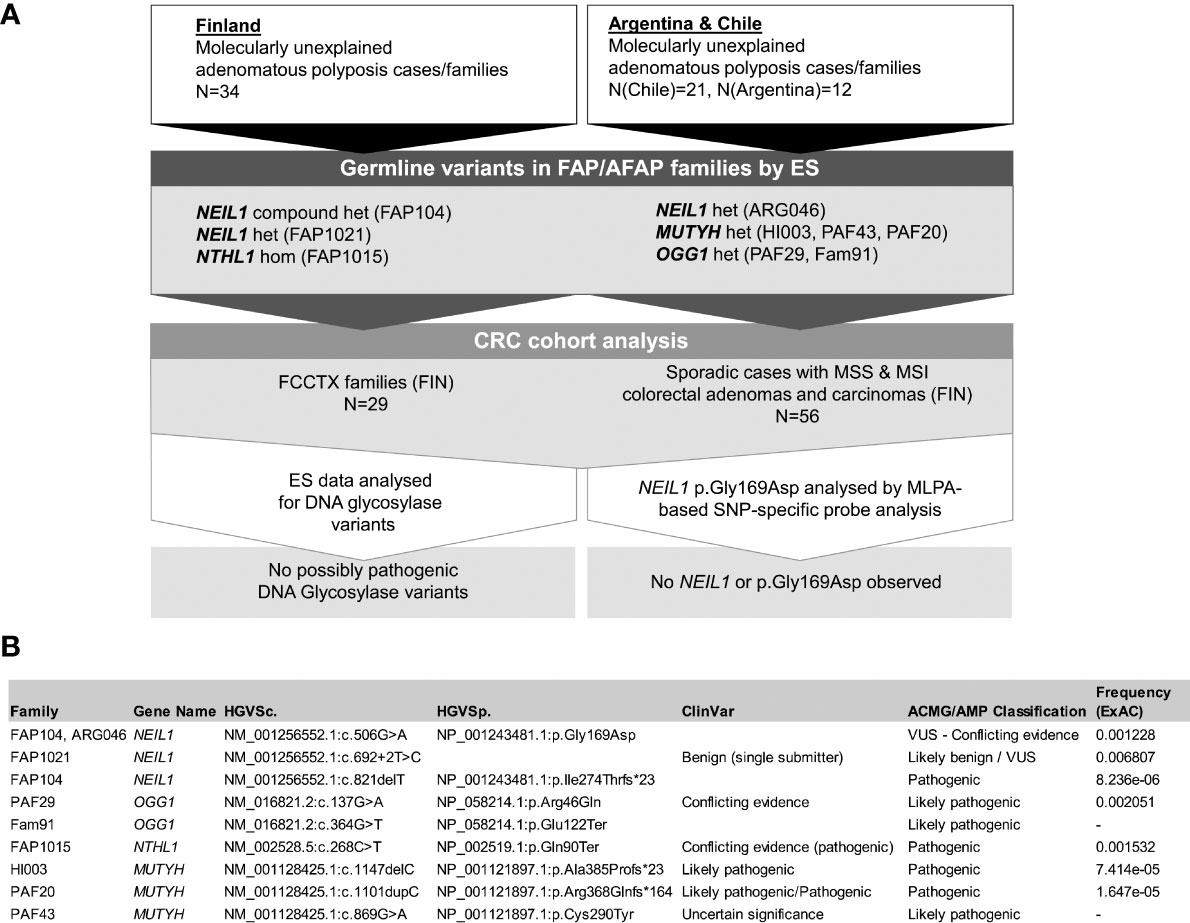
Figure 1 (A) Flowchart of the investigation including the Finnish and South American cohorts. Het denotes heterozygosity and hom homozygosity for the variants. (B) Detailed information of germline variants discovered in DNA glycosylase genes.
Most polyposis cases were attenuated (22/33, 67%; one family could not be classified) and sporadic (20/28, 71%; six families could not be classified) (Table S1). An additional cohort of 29 families representing molecularly unexplained cases with familial colorectal type X (FCCTX) (17) and a series of sporadic cases (56 individuals) with microsatellite-unstable (MSI) CRC (N=13) or microsatellite-stable (MSS) CRC (N=44) or MSS adenomas (N=15) (18) were available for comparison (Figure 1).
Thirty-three (21, Chile; 12, Argentina) unrelated families and index cases without known pathogenic variants in established colon polyposis-associated genes were screened as above. Most of the cases were attenuated (25/33, 76%) and had an apparent dominant mode of inheritance (16/28, 57%; five families could not be classified).
Written informed consent preceded study participation and sample donation. This study was approved by the institutional review board of the Helsinki University Central Hospital (Helsinki, Finland; Valvira/Dnro 10741/06.01.03.01/2015, 14.1.2016) and by the ethics committees of the Hospital de Gastroenterología “Dr. Carlos B. Udaondo” and Hospital Italiano de Buenos Aires (both from CABA, Argentina), and of the Clínica Las Condes (Santiago de Chile, Chile). The collection of archival specimens has been approved by the National Supervisory Authority for Welfare and Health (Valvira/Dnro 10741/06.01.03.01/2015, 14.1.2016).
ES was performed at the Institute for Molecular Medicine Finland (FIMM, Helsinki, Finland) on Illumina HiSeq 2000 platform. The sequencing coverage and quality statistics for each sample are summarized in Table S2. Reads were aligned to the human reference genome hg19 using the Burrows-Wheeler Aligner version 0.6.2. Quality control and primary and secondary analysis were carried out as described by Sulonen et al. (19) Tertiary analysis was carried out using VarSeq® software (Golden Helix). Variants with allele frequency <0.003, nonsynonymous (frameshift, stop gained/lost, missense, disrupting donor/acceptor site variants) and predicted pathogenic with at least five of six programs assessing protein function in silico (for missense changes) were selected. All variants in DNA glycosylase genes were confirmed by Sanger sequencing with primers listed in Table S3.
CNV analysis on ES data was carried out using the R package ExomeDepth (v1.1.10) (20). The patient ES data was run against appropriate patient samples with known pathogenic changes using default settings and annotated using common CNV data from the DECIPHER database (https://www.deciphergenomics.org/). All samples had a correlation score >0.99. Only CNVs with a BF score of 10 or above were considered as candidate CNVs.
To confirm that the two coding variants detected in NEIL1 in the index case of FAP104 affected different alleles (i.e., were in trans), cDNA was amplified with primers NEIL1_G83D_gcDNA_F and NEIL1_G83D_cDNA_R2 (Table S3) and cloned using the TOPO® TA Cloning® Kit for Subcloning (Thermo Fisher) according to manufacturer’s instructions. Transformed E. coli were then grown on selective plates (100 µg Ampicillin) overnight and white colonies were grown in LB (100 µg Ampicillin) overnight. Plasmids were extracted with GenElute™ Plasmid Miniprep Kit (Sigma-Aldrich) according to manufacturer’s instructions and Sanger sequenced using aforementioned primers as well as primers from Sjöblom et al. (21).
To evaluate allele-specific mRNA expression (ASE) in the lymphoblastoid cells from the index individual of FAP104, a Single Nucleotide Primer Extension (SNuPE) reaction was designed based on the heterozygous NEIL1 c.506G>A variant. PCR products specific for cDNA (generated with primers NEIL1_G83D_gcDNA_F + NEIL1_G83D_cDNA_R2, Table S3) and gDNA (NEIL1_G83D_gcDNA_F + NEIL1_G83D_gDNA_R) served as templates for primer extensions with NEIL1_G83D_SNuPE_ext as the extension primer and ddA as the stopping nucleotide. The expected extension products were 34 bp (wild-type allele) and 24 bp (variant allele). Allele peak area ratios R<0.6 or R>1.67 indicated ASE (22).
NEIL1 mRNA expression in the lymphoblastoid cells from the index individual of FAP104 and healthy controls was evaluated by quantitative reverse transcription PCR (qRT-PCR) with TaqMan® Gene Expression Assay (Applied Biosystems) for NEIL1 (Hs00908563_m1) and with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an endogenous reference. The NEIL1 reaction targeted exons 5 – 6 and covered the two main isoforms. The reactions were normalized against the NEIL1 expression of healthy controls and the relative quantities were calculated using the ΔΔCT analysis.
To evaluate the stability of NEIL1 protein, lymphoblastoid cells from the index of FAP104 and unrelated healthy controls were treated with MG132 (Selleck Chemicals, Houston, Texas, USA). MG132 is a cell-permeable, proteasome inhibitor which reduces degradation of ubiquitin-conjugated proteins. Briefly, 0.5 x 106 cells were incubated on 6 well plates for 8 hours and treated with 10, 30 or 50 µM MG132. Proteins from the cells were extracted in LAEMMLI extraction buffer.
NEIL1 protein expression in the treated and untreated lymphoblastoid cells from the index individual of FAP104 and healthy controls was assessed by Western blotting with the primary NEIL1 rabbit polyclonal antibody (12145-1-AP, RRID:AB_2251228; Proteintech, Rosemont, IL) targeting the NEIL1 short isoform (amino acids 1 – 390). The housekeeping protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control (ab128915, RRID:AB_11143050; Abcam, Cambridge, UK). P53 (#9282 RRID:AB_331476; Cell Signaling Technology, Danvers, Massachusetts, USA) was used as a technical control for MG132 experiments.
A custom assay utilizing Methylation-Specific Multiplex Ligation-Dependent Probe Amplification (MS-MLPA) was designed to evaluate NEIL1 promoter methylation in constitutional and tumor tissues. The NEIL1 promoter region was investigated with four MS-MLPA probe pairs (Table S3), of which NEIL1_1 is located just upstream of the area found to be the most informative for methylation by Chaisaingmongkol et al. (23, 24).
VarScan2 variant detection algorithm version 2.3.2 was applied to tumor-normal pairs to identify non-synonymous somatic variants from ES data. Annotation of the variants was done using SnpEff version 4.0 with the Ensembl v68 annotation database (https://www.ensembl.org). Variants with a somatic p-value less than 0.01 were selected for somatic mutational signature analysis, which was carried out using the R package MutationalPatterns (25). The signatures were mapped against the 30 single-base substitution (SBS) and 18 insertion-and-deletion (ID) signatures recognized by the COSMIC database (v2 for SBS and v3.1 for ID, respectively, cancer.sanger.ac.uk).
A colorectal tumor from ARG046 was investigated for MMR protein expression by standard immunohistochemical procedures (26). Primary antibodies used were as follows (Roche Ventana, Indiana, USA): Anti-MLH1 (M1; 790-4535, RRID:AB_2336022), anti-MSH6 (44; 790-4455, RRID:AB_2336020), anti-MSH2 (G219-1129; 760-4265, RRID:AB_2336002), and anti-PMS2 (EPR3947; 760-4531, RRID:AB_2336010). DNA from the same tumor was evaluated for MLH1 promoter methylation by MS-MLPA using the SALSA MLPA ME011-B3 probemix (MRC-Holland, Amsterdam, the Netherlands).
Blood DNAs from the index individuals from our polyposis cohorts were evaluated for large rearrangements in MMR genes and MUTYH by multiplex ligation-dependent probe amplification (MLPA) according to the manufacturer’s (MRC-Holland, Amsterdam, the Netherlands) instructions. SALSA MLPA P003-D1 and SALSA MLPA P072-D1 were used for MLH1/MSH2 and MSH6/MUTYH, respectively, whereas PMS2 was investigated by SALSA MLPA P008-C1. The results from fragment analysis were analyzed by Coffalyser™ (MRC-Holland, Amsterdam, the Netherlands).
Methylation ratios in sporadic tumors vs. matching normal tissues obtained from MS-MLPA analyses (Table S4) were compared using the Wilcoxon matched pairs test. IBM® SPSS® software (IBM SPSS Statistics 27, Armonk, NY: IBM Corp) was used for the analysis.
We investigated the exomes of 67 index cases with molecularly unexplained polyposis from two continents, focusing on genes from the DNA glycosylase family. Pathogenic and likely pathogenic germline variants as well as VUSes whose pathogenicity is unknown but that have the potential of being pathogenic are described in Figure 1 and Table S5. All germline variants fulfilling our selection criteria are listed for the DNA glycosylase-associated families in Table S6.
The European pathogenic founder variant NTHL1 c.268C>T, p.(Gln90Ter) (14) was detected in a homozygous state in the index individual from FAP1015 (Figure 1). This individual had attenuated polyposis and was the only member with colorectal tumor manifestations in the family (Table S1). The patient was additionally diagnosed with carcinomas of multiple organs characteristic of the tumor spectrum of NTHL1-associated polyposis (27).
NEIL1 variants were identified in FAP104 and FAP1021. The index of FAP104 with profuse polyposis (>200 polyps at 54 years of age) had two NEIL1 variants (Figure 1, Figure S1); a rare missense variant c.506G>A, p.(Gly169Asp), and a very rare frameshift variant c.821delT, p.(Ile274Thrfs*23), absent in the Finnish population. A subsequent cloning assay revealed that the variants affected different alleles. All three individuals with the NEIL1 c.506G>A variant had colorectal disease (cancer or polyps) and the same applied to the two individuals with the c.821delT variant (Figure 2).
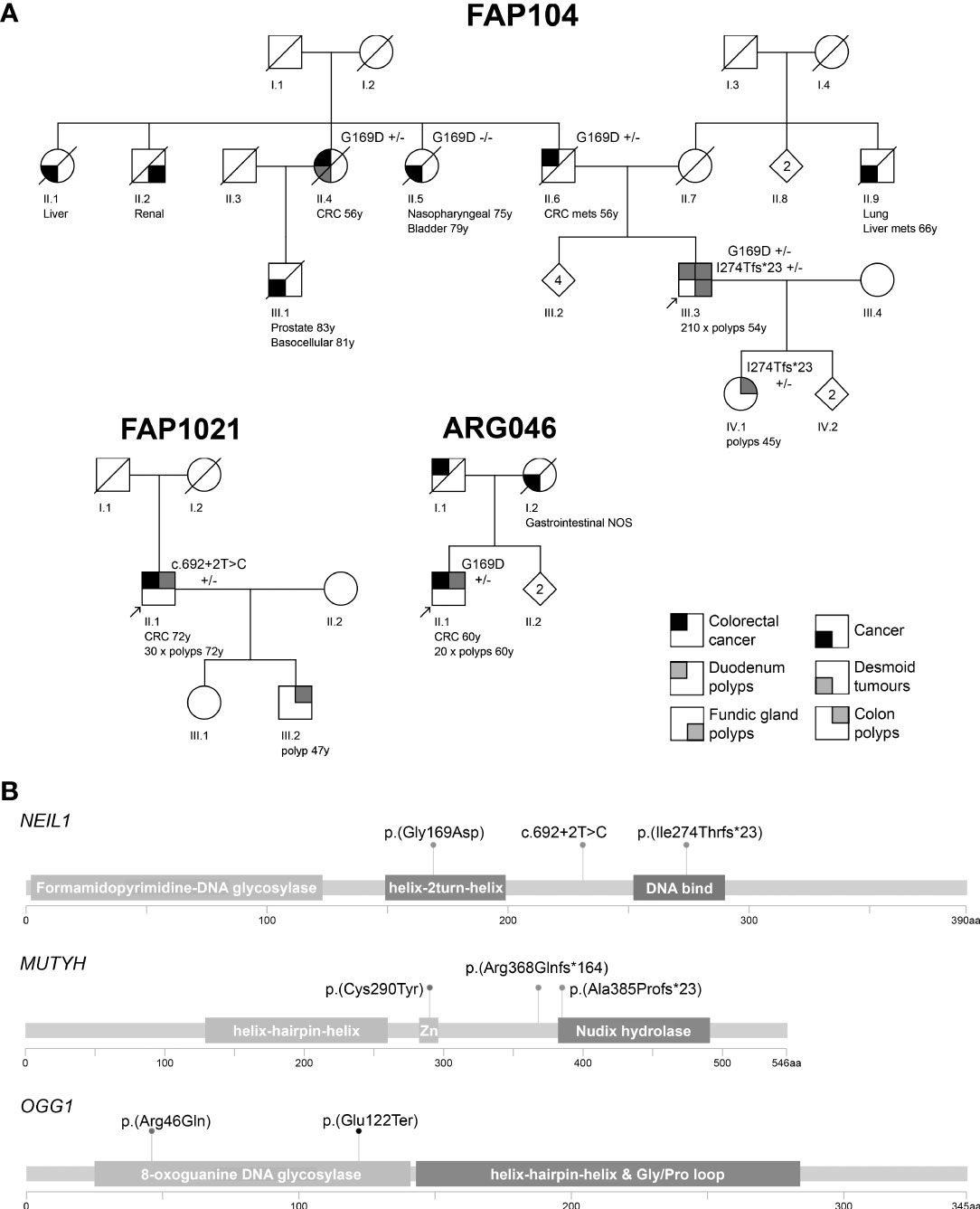
Figure 2 (A) Pedigrees of the polyposis families with NEIL1 variants. Numbers below the symbols are patient identifiers. Arrow denotes the index person. Zygosity of NEIL1 variants is shown (+/− heterozygous). Tumor manifestations and age at diagnosis (years) are given below the patient symbol. Mets refers to metastasis. Nonessential pedigree features were removed or modified to protect confidentiality. (B) Locations of the variants relative to the main functional domains of the DNA glycosylase genes. Zn denotes the metal binding sites in MUTYH as listed in the Uniprot database (www.uniprot.org; Q9UIF7). The pedigrees were generated with Pedigree Chart Designer and the lollipop diagrams with MutationMapper.
Conflicting evidence exists regarding the pathogenic significance of NEIL1 c.506G>A (Figure 1, Table S5). The variant allele frequency in the (global) population (0.001228) is higher than expected for a dominantly inherited disorder when comparing against NEIL1 variants reported through diagnostics. However, allele frequency of this variant in Finns (0.0001368) is almost ten times lower. Furthermore, previous functional studies conducted on NEIL1 c.506G>A consistently suggest pathogenicity (see Discussion). NEIL1 c.821delT is pathogenic according to the ACMG/AMP criteria (Figure 1, Table S5). Suitable biological specimens were available from the index of FAP104 to explore the consequences of the NEIL1 variants on RNA and protein level. We evaluated the relative mRNA expression from the two NEIL1 alleles by SNuPE and found that the frameshift variant containing transcripts were approximately twice less abundant than the missense variant containing transcripts in lymphoblastoid cells from the index individual of FAP104 (Figure 3A). By qRT-PCR, the total NEIL1 mRNA expression was essentially lower than in healthy controls studied for comparison (Figure 3B), suggesting that the ASE seen by SNuPE was more likely to reflect decreased expression from the frameshift allele than increased expression from the missense allele. Interestingly, Western blot analysis revealed a markedly elevated amount of normalized full-length NEIL1 protein compared to healthy controls, and no truncated protein was visible (Figure 3C). The abundant full-length protein likely originated from the missense allele, and no stable protein was apparently generated from the frameshift variant containing allele. In the absence of increased NEIL1 mRNA expression (Figure 3B), elevated NEIL1 protein in the Western blot was more likely to reflect aberrant protein stabilization than overexpression. The MG132 experiments (see Materials and Methods) did not reveal increased NEIL1 staining after treatment, indicating that regulation of NEIL1 protein expression is not MG132 mediated.
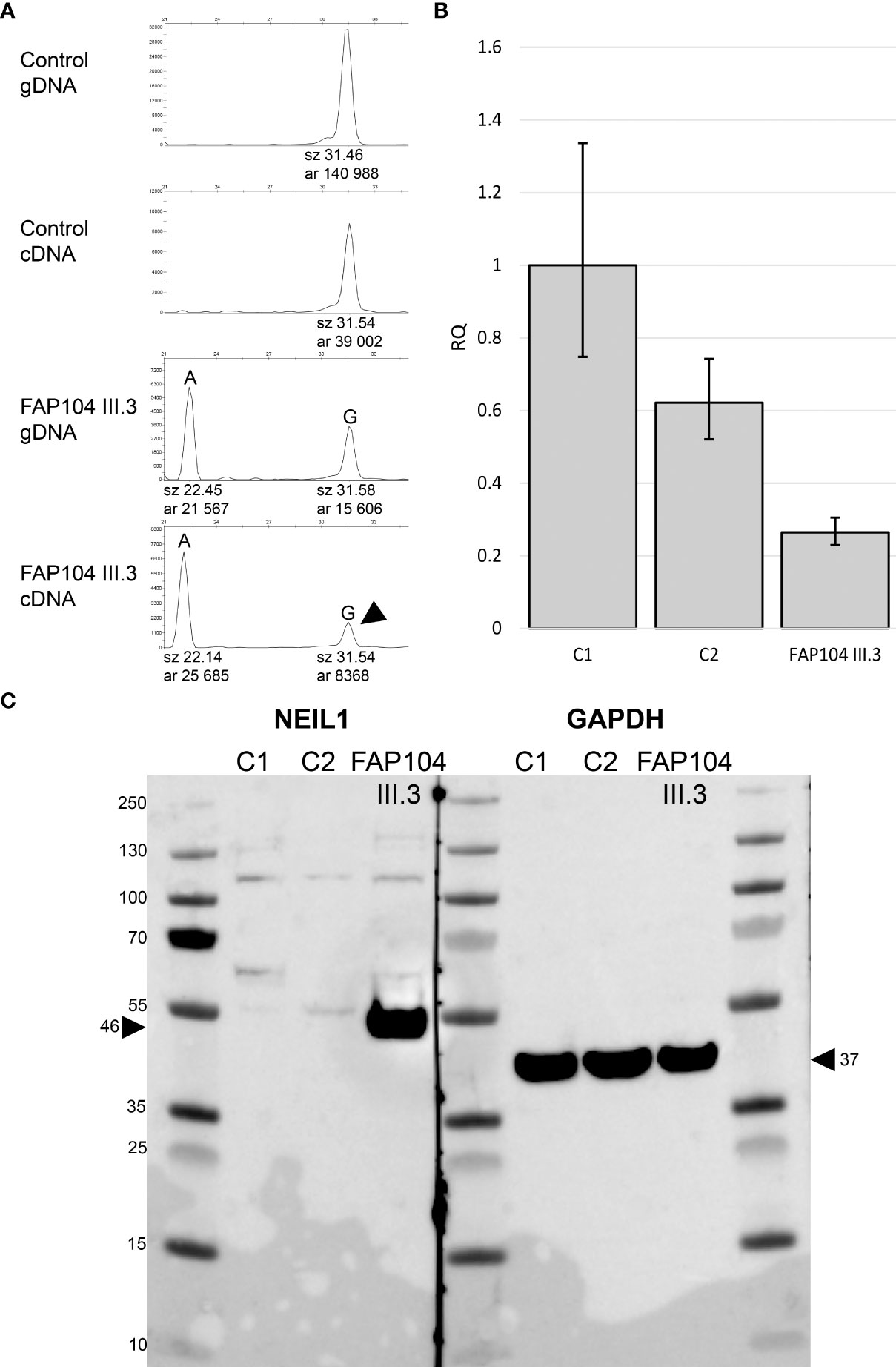
Figure 3 Expressional consequences of the NEIL1 c.506G>A, p.(Gly169Asp) variant. (A) ASE analysis based on the NEIL1 c.506G>A variant. Longer peak (G) represents the wild type sequence. The control individual is homozygous for the wild-type allele. The index individual of FAP104 is heterozygous: Allele A corresponds to the NEIL1 c.506A missense variant, whereas the G allele is known to have a frameshift variant (c.821delT) in a downstream position. This individual displays ASE with the peak area ratio of 0.45 for G to A in cDNA relative to gDNA. The result indicates that transcripts with G (arrowhead) having the frameshift variant are twice less abundant than transcripts containing the missense variant (A). (B) Relative quantity (RQ) from the qRT-PCR experiment targeting the two main isoforms of NEIL1 using the housekeeping gene GAPDH as an endogenous control. Whiskers indicate 95% confidence limits. The index of family FAP104 shows reduced NEIL1 RNA expression compared to the controls. (C) Western blot of two healthy control individuals and the index of FAP104. GAPDH was used as a loading control. FAP104 index displays elevated NEIL1 protein levels compared to the controls (arrowhead). No truncated NEIL1 protein is seen.
The index of FAP1021 with attenuated polyposis (30 polyps at 72 years of age) had a splice donor variant c.692+2T>C (Figure 1, Figure S1). In the literature, conflicting interpretations of pathogenicity for this splice variant exist (e.g., Dallosso AR et al. (28); Boldinova EO et al. (29)). In the absence of RNA, we were unable to experimentally verify splicing consequences of the variant. Based on available data, the ACMG/AMP classification is likely benign or VUS (Table S5). Available in silico software evaluated the splice donor variant highly likely to affect splicing (0.9918, 0.6039, 0.96, and 0.99683 for ADA, RF, SpliceAI, and SPiCE, respectively).
A patient from the Argentine family ARG046 with attenuated mixed polyposis and colorectal carcinoma at the age of 60 years was found to be heterozygous for the previously described NEIL1 c.506G>A variant (Figure 2). No other possibly pathogenic variants in NEIL1 were observed in the South American series.
Two families revealed likely deleterious OGG1 variants. The index of family PAF29 with attenuated polyposis had a heterozygous missense variant of OGG1, c.137G>A, p.(Arg46Gln). In the literature, the same variant was described in a patient with synchronous colorectal cancer at 36 years and adenomas (30). It was shown that the G to A change which affects the last nucleotide of exon 1 disrupts a splice donor sequence, resulting in extinct expression from the variant allele in cDNA from the patient (30). The authors classified the OGG1 c.137G>A variant pathogenic. Considering all available information, the ACMG/AMP criteria for likely pathogenic are fulfilled (Figure 1, Table S5). A heterozygous c.364G>T, p.(Glu122Ter) nonsense variant in the OGG1 gene, likely pathogenic by the ACMG/AMP criteria (Figure 1, Table S5), was detected in family 91. The variant was present in the index patient (ID 606) with attenuated polyposis but absent in the index patient’s brother (ID 657) with late-onset colorectal carcinoma (Table S1).
Three heterozygous MUTYH variants were observed in the South American series (Figure 1, Figure S2). By ES, a frameshift variant of MUTYH, c.1101dupC, p.(Arg368Glnfs*164), was present in three individuals (ID 47, 534, and 535) out of four with colorectal adenomas or carcinoma from the Chilean family PAF20 and affected two generations. MUTYH c.1147delC, p.(Ala385Profs*23) was observed in the index individual HI003 (no other affected members were known to exist in this family). Both MUTYH variants described above are pathogenic by the ACMG/AMP criteria (Table S5), with biallelic involvement linked to MAP. A missense variant, MUTYH c.869G>A, p.(Cys290Tyr), classified as likely pathogenic (Table S5), was found in the index individual of family PAF43 (carrier statuses of the remaining family members were unknown). This family showed features of MAP (over 100 polyps in the index individual and an apparent recessive transmission pattern, Figure S2), raising the possibility that the MUTYH allele currently considered wildtype might harbor a defect that had escaped detection. However, manual IGV analysis of the gene and MLPA (with MSH6-MUTYH and APC MLPA kits) for large genomic rearrangements provided no support for biallelic MUTYH involvement.
DNA was available from a colorectal tubular adenoma from the index of FAP104 (compound heterozygous NEIL1; c.506G>A and c.821delT), two desmoid tumors from the paternal aunt of the index of FAP104 (heterozygous NEIL1 c.506G>A), and a colorectal carcinoma from the index of ARG046 (heterozygous NEIL1 c.506G>A) for somatic mutational profiling. We first determined the total mutational loads, since elevated numbers of somatic variants may point to defects in DNA replication or repair (10, 15). The total numbers of somatic nonsynonymous variants were 281 (adenoma), 45 and 57 (desmoids), and 1146 (carcinoma) by VarScan2 analysis. Based on the commonly used threshold of 10 variants/Mb, only the carcinoma of ARG046 was hypermutated (35 somatic variants/Mb).
All somatic variants meeting our selection criteria (VarScan2 p<0.01) are listed in Table S7. No somatic variant or loss of heterozygosity of NEIL1 was observed in any sample. Thus, there was no evidence of a somatic “second hit” to the remaining wildtype allele in the monoallelic NEIL1 variant carriers. The adenoma from the index of FAP104 showed a truncating APC variant (c.4666dupA, p.Thr1556fs; variant allele frequency (VAF) 23%) and KRAS c. 35G>C, p.Gly12Ala (VAF 25%), both representing alterations typical of colorectal tumorigenesis. The two desmoid tumors revealed extensive sharing of somatic variants, suggesting a common origin for the tumors.
As the patterns of somatic variants can offer insights to the underlying biological processes, a mutational signature analysis was conducted on the tumors (Figures 4A, B). VarScan2-based somatic variants were included in this analysis. COSMIC (31) SBS signature 3 (defective homologous recombination) was prominent in all three tumors from FAP104 (Figure 4A). Desmoid tumors from individual II.4 additionally revealed SBS7 (ultraviolet radiation exposure) and a discernible SBS24 linked to aflatoxin-associated mutagenesis (32). Interestingly, the hypermutable colorectal carcinoma from ARG046 showed prominent MMR deficiency-associated signatures SBS6 and SBS26, together with SBS12 (unknown etiology). The ID signature 6 supported defective homologous recombination in tumors from FAP104, whereas ID7 was compatible with deficient MMR in the colorectal carcinoma from ARG046 (Figure 4B).
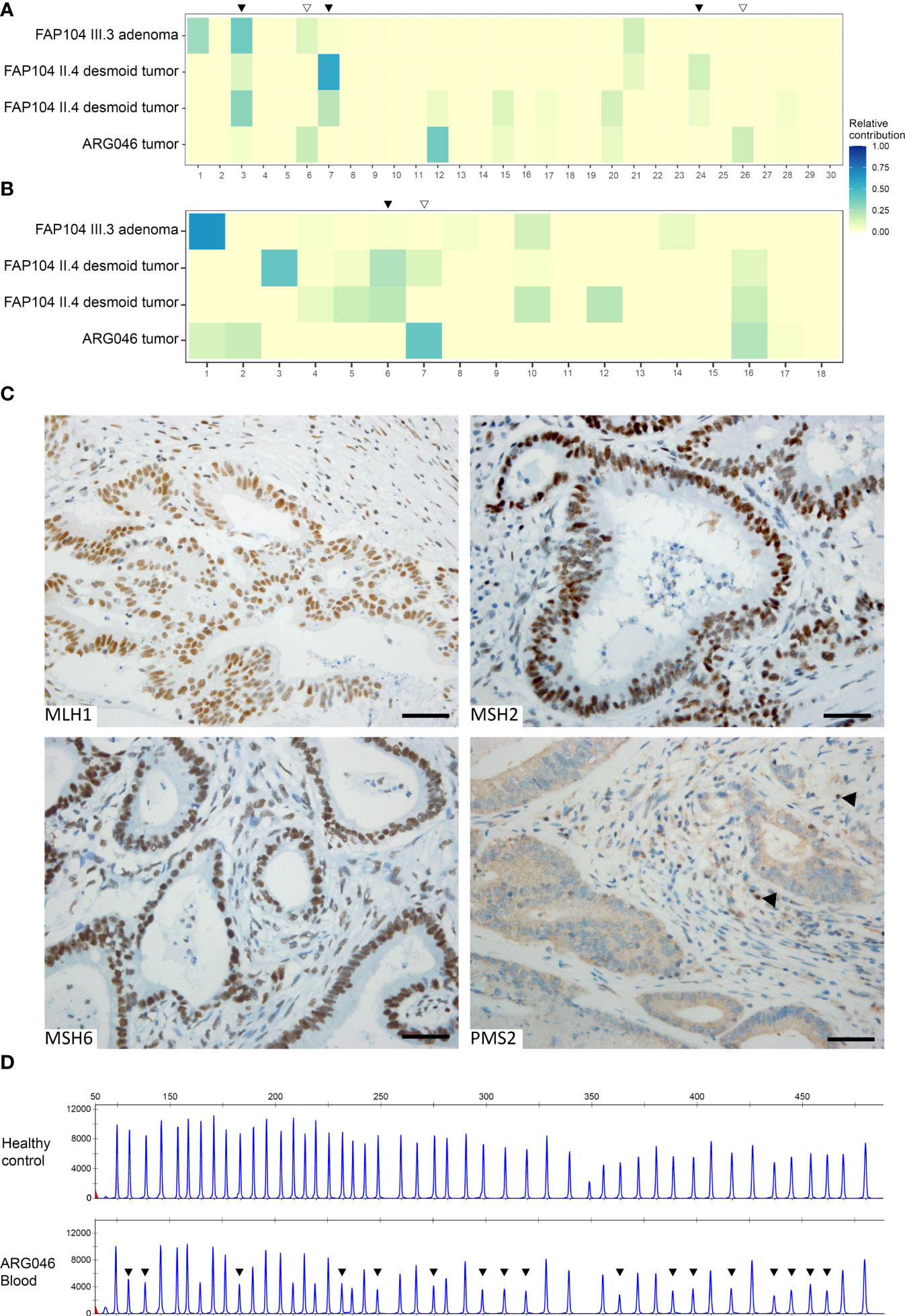
Figure 4 Somatic mutational signature analysis of four NEIL1-associated tumors (A, B). (A) Heatmap indicating the relative contribution of SBS signatures (COSMICv2) to the mutational landscape of each tumor. Black arrowheads indicate NEIL1-deficiency associated signatures prominent in tumors from FAP104, whereas open arrowheads represent MSI-signatures present in a colorectal carcinoma from ARG046. (B) Heatmap of the relative contributions of ID signatures (COSMICv3.1, GRCh37) to the mutational profiles of the tumors. Subsequent discovery of the PMS2 alteration in the ARG046 case (C, D). (C) Immunohistochemical analysis of the MMR proteins reveals a selective loss of PMS2 in the tumor cells. Normal cells retaining the PMS2 expression are indicated with arrowheads. Scale bar represents 50 μm. (D) PMS2-MLPA analysis of blood DNA of the ARG046 case as well as a healthy control. Arrowheads indicate reduced emission peaks at exons 2-11. The average probe ratios of exons 2-11 (0.54 ± 0.03) are indicative of a heterozygous deletion.
To resolve the MMR-deficient pattern of somatic alterations in the colorectal carcinoma from ARG046, the tumor was tested for MLH1 promoter methylation, but no hypermethylation was present. However, immunohistochemical analysis revealed selective absence of PMS2 protein (Figure 4C). Subsequent MLPA analysis of blood DNA showed a heterozygous deletion of PMS2 exons 2 - 11 (NM_000535.5:c.(23 + 1_24-1)-(2006 + 1_2007-1)del; Figure 4D). No additional cases with large rearrangements of MMR genes were detected when our entire polyposis series was evaluated by MLPA (and no small sequence alterations with possible pathogenicity existed in MMR genes by ES).
As NEIL1 is commonly hypermethylated in cancer (23, 24), we designed a MS-MLPA kit to determine constitutional and somatic methylation status of our patient samples. Of the four MS-MLPA probe pairs, NEIL1_1 interrogated a region previously shown to be informative for methylation (23, 24) and showed the best discrimination between normal and tumor tissues (Table S4). Blood and normal colonic mucosae even from reference individuals revealed considerable methylation, and examination of blood DNAs from our polyposis cases (with or without NEIL1 variants) raised no suspicion of constitutional NEIL1 epimutation in any case. Compared to paired normal tissues, tumors from individuals with NEIL1 variants occasionally displayed higher methylation dosage ratios, but no significant somatic hypermethylation of the promoter region was evident. Comparing paired tumor and normal tissues from sporadic cases with MSS or MSI carcinomas or adenomas revealed no significant difference by Wilcoxon matched pairs test (Z=-1.03, p=0.133 for MSI carcinomas vs matching normal tissues, and Z=-0.217, p=0.828 for MSS carcinomas vs matching normal tissues by NEIL1 I probe, respectively).
The DNA glycosylase family comprises eleven members, of which some (e.g., MUTYH) are monofunctional (capable of excising damaged or mispaired bases) and some (e.g., NTHL1, OGG1, and NEIL1) bifunctional (additionally having endonuclease activity to incise the modified strand) (33). The role of DNA glycosylases other than MUTYH and NTHL1 in (colon) tumor susceptibility is unknown and/or associated with conflicting evidence, which encouraged us to undertake the present study. In our exomic screen of 67 index cases from Finnish and South American cohorts, 9 (13%) revealed proven or potentially pathogenic germline variants affecting NEIL1 (3 cases), MUTYH (3 cases), NTHL1 (1 case), and OGG1 (2 cases). The findings suggest that germline variants in DNA glycosylase genes may explain a nontrivial proportion of unexplained cases of colorectal polyposis.
In our investigation, NTHL1 showed biallelic involvement, consistent with the recessive NTHL1-associated polyposis syndrome (14). Our OGG1 variants were monoallelic and suggested dominant transmission with reduced penetrance, which agrees with available literature (30), although a single case with a biallelic truncating OGG1 variant was recently reported in association with FCCTX (34). Biallelic germline variants of MUTYH underlying the well-defined recessive MAP syndrome (2) were excluded from our series at the outset; the significance of the observed monoallelic MUTYH variants will be addressed below. Finally, the transmission pattern of NEIL1-associated disease is unclear since no segregation studies for NEIL1 variants have been reported before. We detected one biallelic and two monoallelic NEIL1 cases that will be discussed in more detail below.
The NEIL1 missense variant c.506G>A, p.(Gly169Asp) occurred in two polyposis families, Finnish and Argentine (2/67, 3%). This variant was previously referred to as G83D according to annotation based on the short (390 amino acid) isoform. Forsbring et al. (35) found this variant in two patients among 37 with primary sclerosing cholangitis and cholangiocarcinoma (5%). Biochemical studies have provided consistent evidence that the variant is deleterious. Using 8-oxoG, thymine glycol, and 5-OHU as substrates, the NEIL1 Gly169Asp protein was found to be devoid of DNA glycosylase activity (35–38). Galick et al. (38) additionally showed that the variant NEIL1 protein acted as a dominant negative manner relative to the wild-type protein, being able to bind to damaged DNA but unable to repair it. Roy et al. (37) concluded that in individuals with the Gly169Asp variant, NEIL1 function is likely to be 50% compared to normal levels unless compensatory mechanisms exist. Our Western blot analysis on lymphoblastoid cells from the index of FAP104 with the c.506G>A variant revealed strikingly increased amount of NEIL1 protein, and we hypothesize that the c.506G>A variant is mutagenic due to the accumulation of functionally defective protein. Our result would comply with a possible oncogenic role proposed for NEIL1 in some studies (39).
The index individual of FAP104 was compound heterozygous for NEIL1 c.506G>A and c.821delT. We are not aware of the possible existence of any previous reports of biallelic constitutional NEIL1 involvement in association with human disease. Moreover, in FAP104, all five members who were verified to have either one of the NEIL1 variants (or both) had a colorectal tumor phenotype. The age at onset of disease (polyposis or cancer) of our heterozygous cases was relatively late with modest numbers of polyps (Figure 2, Table S1), which may indicate reduced penetrance. Apart from colon polyposis, profuse gastric fundic gland polyposis was apparent in the index of FAP104 as well as his daughter, both individuals with the NEIL1 frameshift variant (Figure 2). Stomach tissue is particularly prone to oxidative damage and some somatic NEIL1 variants and germline polymorphisms have been found in gastric cancer patients (40) indicating a possible role in stomach polyp formation.
SBS3 and ID6, which are associated with impaired homologous recombination (41), stood out among mutational signatures observed in our NEIL1-associated tumors (Figures 4A, B). This is compatible with observations that NEIL1 may participate in the repair of oxidized bases in D-loops (42) and R-loops (43) arising during homologous recombination or transcription. SBS7 which is connected to UV radiation was prominent in the desmoid tumors from a case with the NEIL1 c.506G>A variant. This is consistent with findings of Neil1-/- mice being sensitive to chronic UVB exposure (44). Our desmoid tumors also exhibited SBS24, the so-called aflatoxin signature. McCullough and Lloyd (32) demonstrated that NEIL1 is a major contributing factor to the repair of AFB1-N7-dG and AFB1-Fapy-dG adducts formed by aflatoxin mutagenesis. All in all, mutational signatures observed in tumors from our NEIL1 cases are well in agreement with the reported biological consequences of defective NEIL1 function.
Somatic hypermutability and MMR deficiency-associated signatures in a colorectal tumor from an Argentine case with the NEIL1 c.506G>A variant provided critical clues to discover a large genomic deletion of PMS2 as a concomitant germline alteration in this patient (Figures 4C, D). In analogy to DNA glycosylase genes, incomplete penetrance characterizes many pathogenic variants of PMS2 (45). Colonic polyposis commonly accompanies biallelic PMS2 variants, whereas monoallelic PMS2 variants typically manifest themselves as (late-onset) colorectal carcinoma (46). The PMS2 exon 2 – 11 deletion found in our ARG046 case was heterozygous (Figure 3D), and no other PMS2 sequence variants of suspected pathogenic significance were identified (Table S6). Modifying or additive effects of two or more defective genes may be necessary to explain the observed phenotypes of DNA glycosylase gene variants (30) and PMS2 variants (46), and base excision repair and MMR defects can potentiate each other’s effects (47). It is possible that the late-onset colorectal carcinoma in our ARG046 case mainly reflected the PMS2 defect, in agreement with available literature (see above), whereas NEIL1 c.506G>A might be necessary for the patient’s polyposis phenotype.
While biallelic germline variants of MUTYH cause predisposition to MAP (see Introduction), the clinical phenotype of monoallelic MUTYH variants remains unsettled. In our investigation, three families from the South American cohort revealed monoallelic MUTYH variants classified as pathogenic (two) and likely pathogenic (one). Among five individuals with monoallelic MUTYH variants, four exhibited polyposis with the polyp number ranging from below 20 (in three individuals) to over 100 (in one), and three had late-onset colorectal cancer (Table S1, Figure S2). Our findings together with published reports indicate that individuals with monoallelic MUTYH variants may be predisposed to colorectal polyposis of a variable degree and have a moderately increased risk of colorectal cancer (48, 49). The PAF43 index case manifested a phenotype akin to classical MAP, but no second MUTYH variant of possible pathogenic significance was identified. Since the ES runs included only about 300 bp flanking sequence, our approach does not exclude possible variants in regulatory regions (including deep intronic splice variants and pseudoexons).
In summary, we describe proven or possibly pathogenic germline variants of DNA glycosylase genes in 9/67 (13%) index cases with colon polyposis. Our study suggests a link between NEIL1 germline variants and colon polyposis. Because of the relatively limited number of individuals with NEIL1 variants in this investigation, our findings need to be confirmed in larger multinational cohorts.
The data analyzed in this study was obtained as described in Materials and Methods. The datasets are not readily available because our IRB approvals do not allow sharing raw exome sequencing data. However, all variants that fulfilled our selection criteria can be found in the Supplementary Tables. Requests to access these datasets should be directed to the corresponding author, APO, YWxpc2Eub2xraW51b3JhQGhlbHNpbmtpLmZp.
The studies involving human participants were reviewed and approved by Helsinki University Central Hospital (Helsinki, Finland; Valvira/Dnro 10741/06.01.03.01/2015, 14.1.2016) Hospital de Gastroenterología “Dr. Carlos B. Udaondo” and Hospital Italiano de Buenos Aires (both from CABA, Argentina) Clínica Las Condes (Santiago de Chile, Chile) by the National Supervisory Authority for Welfare and Health (Valvira/Dnro 10741/06.01.03.01/2015, 14.1.2016). The patients/participants provided their written informed consent to participate in this study.
Conceptualization, APO, PP, CAV, and WHP. Data curation, FIMM. Formal analysis, APO, AKK, PP, ACM, and MBC. Funding acquisition, PP, CAV, and WHP. Investigation, APO, AKK, AR, PP, ACM, MBC, FJ, HG-R, and WHP. Methodology, APO, AKK, TTN, PP, ACM, MBC, and WHP. Project administration, PP and WHP. Resources, LK, AL, PP, ACM, MBC, MC, AKC, AG, KA, AC, FL-K, CAV, and WHP. Software, APO. Supervision, TTN, PP, and WHP. Validation, APO, AKK, TTN, PP, ACM, MBC, and WHP. Visualization, APO, PP, and WHP. Writing-original draft, APO, PP, and WHP. Writing-review and editing, APO, PP, and WHP. All authors contributed to the article and approved the submitted version.
This work was supported by grants from Biomedicum Helsinki Foundation (to APO); the Mary and Georg Ehrnrooth’s Foundation (to LK); the Jalmari and Rauha Ahokas Foundation (to TTN); the Maud Kuistila Memorial Foundation (to TTN); the Jane and Aatos Erkko Foundation (to PP); the Academy of Finland (grant number 330606 to PP); Cancer Foundation Finland sr (to PP, AR and LK); and Helsinki University Central Hospital Research Funds (to AR); the Sigrid Juselius Foundation (to PP and AR); the HiLIFE Fellows 2017-2020 (to PP); the Agencia Nacional de Promoción Cientı́fica y Tecnológica (PICT_2017 -3210 to CAV and -3197 to WHP) and Instituto Nacional del Cáncer Argentina (AF_IV to CAV and WHP). The Doctoral Programme in Biomedicine of Helsinki University offered a paid doctoral student position to APO.
Saila Saarinen is thanked for expert laboratory assistance, and Maija Röntynen and Beatriz Alcala-Repo for collecting clinical data.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.870863/full#supplementary-material
1. Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis (2009) 4:22. doi: 10.1186/1750-1172-4-22
2. Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat Genet (2002) 30(2):227–32. doi: 10.1038/ng828
3. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J (2003) 17(10):1195–214. doi: 10.1096/fj.02-0752rev
4. Aretz S, Uhlhaas S, Caspari R, Mangold E, Pagenstecher C, Propping P, et al. Frequency and parental origin of de novo APC mutations in familial adenomatous polyposis. Eur J Hum Genet (2004) 12(1):52–8. doi: 10.1038/sj.ejhg.5201088
5. Aretz S, Stienen D, Friedrichs N, Stemmler S, Uhlhaas S, Rahner N, et al. Somatic APC mosaicism: A frequent cause of familial adenomatous polyposis (FAP). Hum Mutat (2007) 28(10):985–92. doi: 10.1002/humu.20549
6. Jansen AM, Crobach S, Geurts-Giele WR, van den Akker BE, Garcia MV, Ruano D, et al. Distinct patterns of somatic mosaicism in the APC gene in neoplasms from patients with unexplained adenomatous polyposis. Gastroenterology (2017) 152(3):546–9.e3. doi: 10.1053/j.gastro.2016.10.040
7. Spier I, Horpaopan S, Vogt S, Uhlhaas S, Morak M, Stienen D, et al. Deep intronic APC mutations explain a substantial proportion of patients with familial or early-onset adenomatous polyposis. Hum Mutat (2012) 33(7):1045–50. doi: 10.1002/humu.22082
8. Pavicic W, Nieminen TT, Gylling A, Pursiheimo JP, Laiho A, Gyenesei A, et al. Promoter-specific alterations of APC are a rare cause for mutation-negative familial adenomatous polyposis. Genes Chromosomes Cancer (2014) 53(10):857–64. doi: 10.1002/gcc.22197
9. Shirts BH, Salipante SJ, Casadei S, Ryan S, Martin J, Jacobson A, et al. Deep sequencing with intronic capture enables identification of an APC exon 10 inversion in a patient with polyposis. Genet Med (2014) 16(10):783–6. doi: 10.1038/gim.2014.30
10. Bellido F, Pineda M, Aiza G, Valdés-Mas R, Navarro M, Puente DA, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet Med (2016) 18(4):325–32. doi: 10.1038/gim.2015.75
11. Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet (2004) 74(5):1043–50. doi: 10.1086/386293
12. Adam R, Spier I, Zhao B, Kloth M, Marquez J, Hinrichsen I, et al. Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. Am J Hum Genet (2016) 99(2):337–51. doi: 10.1016/j.ajhg.2016.06.015
13. Olkinuora A, Nieminen TT, Mårtensson E, Rohlin A, Ristimäki A, Koskenvuo L, et al. Biallelic germline nonsense variant of MLH3 underlies polyposis predisposition. Genet Med (2018) 21(8):1868–73. doi: 10.1038/s41436-018-0405-x
14. Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, Spruijt L, et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet (2015) 47(6):668–71. doi: 10.1038/ng.3287
15. Renkonen E, Zhang Y, Lohi H, Salovaara R, Abdel-Rahman WM, Nilbert M, et al. Altered expression of MLH1, MSH2, and MSH6 in predisposition to hereditary nonpolyposis colorectal cancer. J Clin Oncol (2003) 21(19):3629–37. doi: 10.1200/JCO.2003.03.181
16. Isola J, DeVries S, Chu L, Ghazvini S, Waldman F. Analysis of changes in DNA sequence copy number by comparative genomic hybridization in archival paraffin-embedded tumor samples. Am J Pathol (2014) 145(6):1301–8. doi: 10.1016/S0165-4608(96)00076-3
17. Nieminen TT, Abdel-Rahman WM, Ristimäki A, Lappalainen M, Lahermo P, Mecklin JP, et al. BMPR1A mutations in hereditary nonpolyposis colorectal cancer without mismatch repair deficiency. Gastroenterology (2011) 141(1):e23–6. doi: 10.1053/j.gastro.2011.03.063
18. Joensuu EI, Abdel-Rahman WM, Ollikainen M, Ruosaari S, Knuutila S, Peltomäki P. Epigenetic signatures of familial cancer are characteristic of tumor type and family category. Cancer Res (2008) 68(12):4597–605. doi: 10.1158/0008-5472.CAN-07-6645
19. Sulonen AM, Ellonen P, Almusa H, Lepistö M, Eldfors S, Hannula S, et al. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol (2011) 12(9):R94. doi: 10.1186/gb-2011-12-9-r94
20. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics (2012) 28(21):2747–54. doi: 10.1093/bioinformatics/bts526
21. Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science (2006) 314(5797):268–74. doi: 10.1126/science.1133427
22. Nieminen TT, Pavicic W, Porkka N, Kankainen M, Järvinen HJ, Lepistö A, et al. Pseudoexons provide a mechanism for allele-specific expression of APC in familial adenomatous polyposis. Oncotarget (2016) 7(43):70685–98. doi: 10.18632/oncotarget.12206
23. Chaisaingmongkol J, Popanda O, Warta R, Dyckhoff G, Herpel E, Geiselhart L, et al. Epigenetic screen of human DNA repair genes identifies aberrant promoter methylation of NEIL1 in head and neck squamous cell carcinoma. Oncogene (2012) 31(49):5108–16. doi: 10.1038/onc.2011.660
24. Shinmura K, Kato H, Kawanishi Y, Igarashi H, Goto M, Tao H, et al. Abnormal expressions of DNA glycosylase genes NEIL1, NEIL2, and NEIL3 are associated with somatic mutation loads in human cancer. Oxid Med Cell Longev (2016) 2016:1546392. doi: 10.1155/2016/1546392
25. Blokzijl F, Janssen R, van Boxtel R, Cuppen E. MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med (2018) 10(1):33. doi: 10.1186/s13073-018-0539-0
26. Overbeek LI, Ligtenberg MJ, Willems RW, Hermens RP, Blokx WA, Dubois SV, et al. Interpretation of immunohistochemistry for mismatch repair proteins is only reliable in a specialized setting. Am J Surg Pathol (2008) 32(8):1246–51. doi: 10.1097/PAS.0b013e31816401bb
27. Grolleman JE, de Voer RM, Elsayed FA, Nielsen M, Weren RDA, Palles C, et al. Mutational signature analysis reveals NTHL1 deficiency to cause a multi-tumor phenotype. Cancer Cell (2019) 35(2):256–66.e5. doi: 10.1016/j.ccell.2018.12.011
28. Dallosso AR, Dolwani S, Jones N, Jones S, Colley J, Maynard J, et al. Inherited predisposition to colorectal adenomas caused by multiple rare alleles of MUTYH but not OGG1, NUDT1, NTH1 or NEIL 1, 2 or 3. Gut (2008) 57(9):1252–5. doi: 10.1136/gut.2007.145748
29. Boldinova EO, Khairullin RF, Makarova AV, Zharkov DO. Isoforms of base excision repair enzymes produced by alternative splicing. Int J Mol Sci (2019) 20(13):3279. doi: 10.3390/ijms20133279
30. Morak M, Massdorf T, Sykora H, Kerscher M, Holinski-Feder E. First evidence for digenic inheritance in hereditary colorectal cancer by mutations in the base excision repair genes. Eur J Cancer (2011) 47(7):1046–55. doi: 10.1016/j.ejca.2010.11.016
31. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500(7463):415–21. doi: 10.1038/nature12477
32. McCullough AK, Lloyd RS. Mechanisms underlying aflatoxin-associated mutagenesis - implications in carcinogenesis. DNA Repair (Amst) (2019) 77:76–86. doi: 10.1016/j.dnarep.2019.03.004
33. Weren RD, Ligtenberg MJ, Geurts van Kessel A, De Voer RM, Hoogerbrugge N, Kuiper RP. NTHL1 and MUTYH polyposis syndromes: Two sides of the same coin? J Pathol (2018) 244(2):135–42. doi: 10.1002/path.5002
34. de Oliveira Garcia FA, de Andrade ES, de Campos Reis Galvão H, da Silva Sábato C, Campacci N, de Paula AE, et al. New insights on familial colorectal cancer type X syndrome. Sci Rep (2022) 12(1):2846. doi: 10.1038/s41598-022-06782-8
35. Forsbring M, Vik ES, Dalhus B, Karlsen TH, Bergquist A, Schrumpf E, et al. Catalytically impaired hMYH and NEIL1 mutant proteins identified in patients with primary sclerosing cholangitis and cholangiocarcinoma. Carcinogenesis (2009) 30(7):1147–54. doi: 10.1093/carcin/bgp118
36. Prakash A, Carroll BL, Sweasy JB, Wallace SS, Doublié S. Genome and cancer single nucleotide polymorphisms of the human NEIL1 DNA glycosylase: Activity, structure, and the effect of editing. DNA Repair (Amst) (2014) 14:17–26. doi: 10.1016/j.dnarep.2013.12.003
37. Roy LM, Jaruga P, Wood TG, McCullough AK, Dizdaroglu M, Lloyd RS. Human polymorphic variants of the NEIL1 DNA glycosylase. J Biol Chem (2007) 282(21):15790–8. doi: 10.1074/jbc.M610626200
38. Galick HA, Marsden CG, Kathe S, Dragon JA, Volk L, Nemec AA, et al. The NEIL1 G83D germline DNA glycosylase variant induces genomic instability and cellular transformation. Oncotarget (2017) 8(49):85883–95. doi: 10.18632/oncotarget.20716
39. Xue W, Liu Y, Xin N, Miao J, Du J, Wang Y, et al. Nei endonuclease VIII-Like1 (NEIL1) inhibits apoptosis of human colorectal cancer cells. BioMed Res Int (2020) 2020:5053975. doi: 10.1155/2020/5053975
40. Shinmura K, Tao H, Goto M, Igarashi H, Taniguchi T, Maekawa M, et al. Inactivating mutations of the human base excision repair gene NEIL1 in gastric cancer. Carcinogenesis (2004) 25(12):2311–7. doi: 10.1093/carcin/bgh267
41. Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature (2020) 578(7793):94–101. doi: 10.1038/s41586-020-1943-3
42. Makasheva KA, Endutkin AV, Zharkov DO. Requirements for DNA bubble structure for efficient cleavage by helix-two-turn-helix DNA glycosylases. Mutagenesis (2020) 35(1):119–28. doi: 10.1093/mutage/gez047
43. Møllersen L, Rowe AD, Illuzzi JL, Hildrestrand GA, Gerhold KJ, Tveterås L, et al. Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Hum Mol Genet (2012) 21(22):4939–47. doi: 10.1093/hmg/dds337
44. Calkins MJ, Vartanian V, Owen N, Kirkali G, Jaruga P, Dizdaroglu M, et al. Enhanced sensitivity of Neil1(-/-) mice to chronic UVB exposure. DNA Repair (Amst) (2016) 48:43–50. doi: 10.1016/j.dnarep.2016.10.010
45. Dominguez-Valentin M, Sampson JR, Seppälä TT, Ten Broeke SW, Plazzer JP, Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the prospective lynch syndrome database. Genet Med (2020) 22(1):15–25. doi: 10.1038/s41436-019-0596-9
46. Goodenberger ML, Thomas BC, Riegert-Johnson D, Boland CR, Plon SE, Clendenning M, et al. PMS2 monoallelic mutation carriers: The known unknown. Genet Med (2016) 18(1):13–9. doi: 10.1038/gim.2015.27
47. Bridge G, Rashid S, Martin SA. DNA Mismatch repair and oxidative DNA damage: implications for cancer biology and treatment. Cancers (Basel) (2014) 6(3):1597–614. doi: 10.3390/cancers6031597
48. Peterlongo P, Mitra N, Sanchez de Abajo A, de la Hoya M, Bassi C, Bertario L, et al. Increased frequency of disease-causing MYH mutations in colon cancer families. Carcinogenesis (2006) 27(11):2243–9. doi: 10.1093/carcin/bgl093
Keywords: DNA glycosylase, NEIL1, OGG1, NTHL1, MUTYH, polyposis, germline variant, exome sequencing
Citation: Olkinuora AP, Mayordomo AC, Kauppinen AK, Cerliani MB, Coraglio M, Collia ÁK, Gutiérrez A, Alvarez K, Cassana A, Lopéz-Köstner F, Jauk F, García-Rivello H, Ristimäki A, Koskenvuo L, Lepistö A, Nieminen TT, Vaccaro CA, Pavicic WH and Peltomäki P (2022) Mono- and biallelic germline variants of DNA glycosylase genes in colon adenomatous polyposis families from two continents. Front. Oncol. 12:870863. doi: 10.3389/fonc.2022.870863
Received: 07 February 2022; Accepted: 03 October 2022;
Published: 28 October 2022.
Edited by:
Daniela Turchetti, University of Bologna, ItalyReviewed by:
Shirley V. Hodgson, St George’s, University of London, United KingdomCopyright © 2022 Olkinuora, Mayordomo, Kauppinen, Cerliani, Coraglio, Collia, Gutiérrez, Alvarez, Cassana, Lopéz-Köstner, Jauk, García-Rivello, Ristimäki, Koskenvuo, Lepistö, Nieminen, Vaccaro, Pavicic and Peltomäki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alisa Petriina Olkinuora, YWxpc2Eub2xraW51b3JhQGhlbHNpbmtpLmZp
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.