 Meimei Wu
Meimei Wu Yanfang Liang2
Yanfang Liang2 Xin Zhang
Xin Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 26 May 2022
Sec. Breast Cancer
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.860932
This article is part of the Research Topic Exploring the Breast Tumor Microenvironment: Association to Metastasis, Novel Risk Factors and Novel Treatments and Immunotherapies View all 31 articles
Breast cancer has become the most common malignant disease in the world according to the International Agency for Research on Cancer (IARC), and the most critical cause of death is distant metastasis. The lung is the extremely common visceral site for breast cancer metastasis. Lung metastasis of breast cancer is not only dependent on the invasive ability of the tumor itself, but also closely relates to the pulmonary microenvironment. In the progression of breast cancer, the formation of specific microenvironment in lungs can provide suitable conditions for the metastasis of breast cancer. Pulmonary inflammatory response, angiogenesis, extracellular matrix remodeling, some chemotherapeutic agents and so on all play important roles in the formation of the pulmonary microenvironment. This review highlights recent findings regarding the alterations of pulmonary microenvironment in lung metastasis of breast cancer, with a focus on various cells and acellular components.
Among female cancers, breast cancer had awfully high morbidity and mortality (1, 2), and was highly malignant, poor prognosis and prone to local recurrence and distant metastasis (3–5). Lung metastasis was a very common distant metastasis of breast cancer, the incidence of which was about 21%~32% (6), and the prognosis of patients with lung metastasis was greatly poor, with a median survival of only 25 months (7). In the process of lung metastasis of breast cancer, the breast cancer cells would undergo several steps, such as leaving the primary lesion, invading the circulatory system, reaching the lung tissues, colonizing in lungs and forming clinically visible metastasis (8, 9). In this series of activities, metastatic breast cancer cells were considered as “seeds”, and the microenvironment in metastatic niche was considered as “soil”. Besides, the formation of metastatic niche was the result of the interaction between “seed” and “soil”. However, previous studies focused on the molecular and functional changes of metastatic cancer cells themselves, ignoring the induction or interference of the microenvironment in the process of metastasis. In recent years, the study of lung microenvironment of breast cancer metastasis showed that the dynamic changes of “soil” in this process were as important as the ability of “seed” itself. Therefore, this review mainly discussed the role of various cells, particularly neutrophils, and acellular elements in pulmonary microenvironment targeting breast cancer lung metastasis (Figure 1).
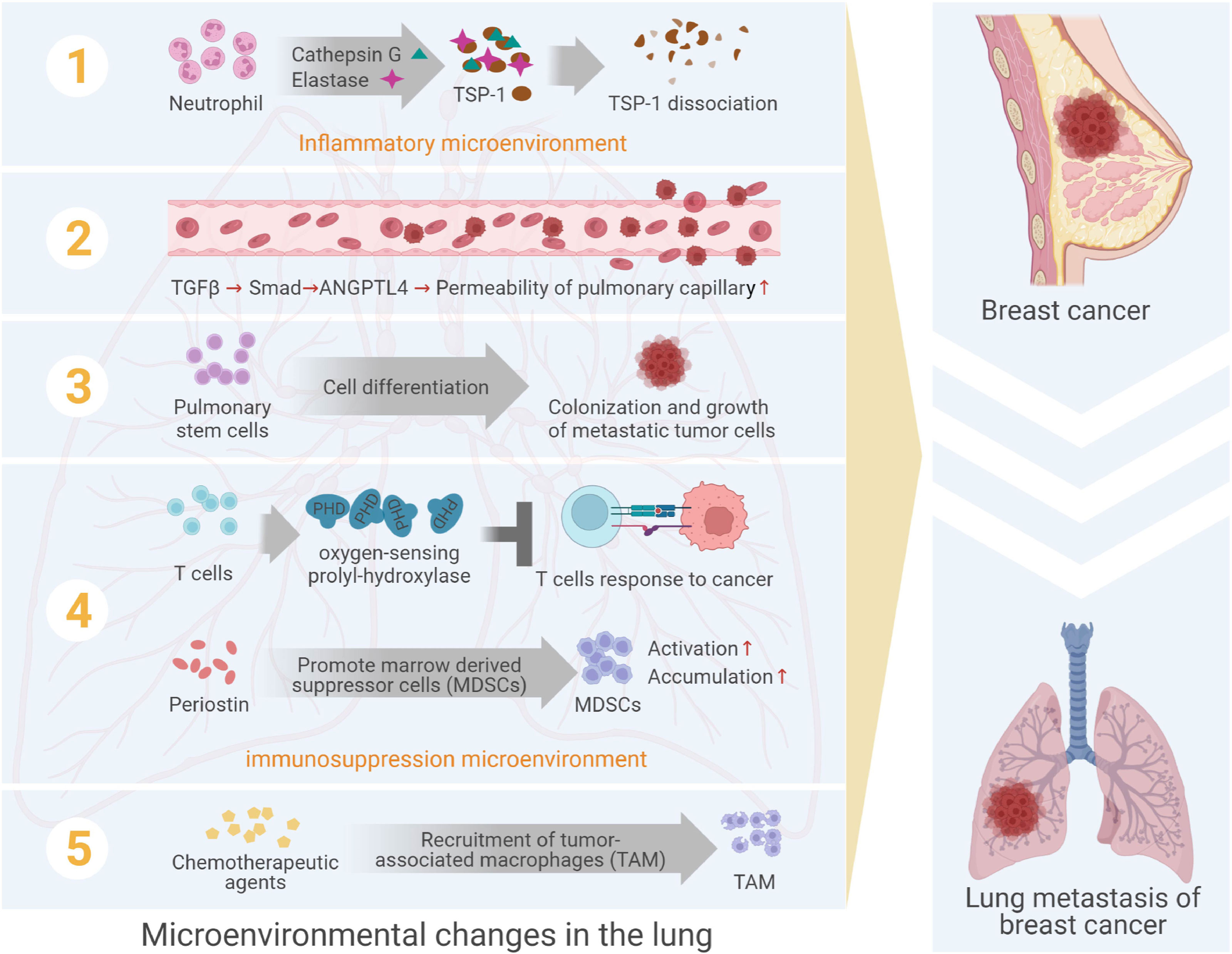
Figure 1 The formation of pulmonary microenvironment in lung metastasis from breast cancer. The formation of sites adapted to the survival of metastatic cancer cells is regulated by acellular components and a variety of cells, including neutrophils, vascular endothelial cells, stem cell-like lung cells, immune cells, etc.
By the 1860s, Virchow (10) had found a large number of leukocytes in tumor tissue and established a link between inflammation and cancer. With the further research, the close relationship between inflammation and cancer had gradually been confirmed (10–12). Existing studies displayed that the lung was a relatively open organ of the body to the external environment and was susceptible to various external stimuli to induce inflammation (13–15), and the lung inflammation enhanced tumor metastatic outgrowth in lungs (16). Bacterial lipopolysaccharide (LPS) and Nicotine in cigarettes were well-characterized inducer of lung inflammation (13, 15, 17).
LPS-mediated and cigarette smoke-induced lung inflammation were closely related to breast cancer metastasis to the lungs (18, 19), which was marked by increased neutrophils influx and up-regulation of pro-inflammatory cytokines (16, 20). Also, smoking promoted lung metastasis of breast cancer precisely because lungs continued to be exposed to nicotine, which generated inflammatory microenvironment in lungs (20). Pulmonary inflammatory microenvironment would result in the influx of a vast number of activated neutrophils and the formation of appropriate tumor metastasis niche in lungs. Furthermore, some tumor-promoting neutrophils released lipocalin 2 (LCN2) protein activated by STAT3 to induce tumor metastatic growth (20). Thus, neutrophils might play an important role in pulmonary metastatic microenvironment.
Neutrophils were the major circulating leukocytes in human body. Besides, neutrophils were recruited as the first line of defense in the immune response to fight off acute inflammation (21). If pathogens broke through the physical barrier formed by the skin and mucous membranes to enter the tissues, pathogens at the site of infection and macrophages produced signals to activate endothelial cells, which captured circulating neutrophils and induced them to cross the endothelium to bind to the pathogen (21–23). After migration from the blood to the tissues, neutrophil immune function was enhanced, while activated neutrophils produced specific active substances that attracted more inflammatory cells including neutrophils, macrophages and T cells to the site of inflammation and regulate their activity (21, 24). The activated neutrophils delivered lethal blows to pathogens through various mechanisms such as degranulation and release of active proteins. Nevertheless, hyperactivated neutrophils often lead to significant damage to normal tissue (21, 25).
Lung tissues were the main site where many pathogenic microorganisms invaded the body, thus lung tissues were particularly dependent on the surveillance of innate immune cells dominated by neutrophils (26, 27). Besides, lungs were the major neutrophil reservoir. When the lungs were infected, neutrophils leaked from pulmonary circulating capillaries into the lungs, and the neutrophils then activated and engulfed pathogens and released various active substances (28). Neutrophils killed pathogens in lungs by two main mechanisms: oxidative and non-oxidative. The oxidative pathway was the primary mode of pathogen killing by neutrophils through the production of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system (29). Non-oxidative pathways included various antimicrobial proteins and proteases, such as neutrophil elastase (NE), myeloperoxidase (MPO), alkaline phosphatase (ALP), defensins, lysozyme and neutrophil extracellular traps (NETs), which promoted neutrophils phagocytosis of pathogens, neutrophils migration, and regulated the inflammatory response (30–32).
Although neutrophils played an important role in fighting infections with lung pathogens such as bacteria, fungi and viruses, continuous neutrophil infiltration could facilitate lung metastasis. Neutrophils in lungs could be “hijacked” by cancer cells and helped them spread into lungs.
Neutrophils were recruited in the niche and altered their polarization state in the tumor-bearing host, switching from suppressing to promoting roles in tumor metastasis (33, 34). Neutrophils could elicit a metastatic inflammatory microenvironment by suppressing innate and adaptive anti-tumor immunity (35–37). With the increased focus on neutrophils accelerating lung metastasis in recent years, there was accumulating evidence indicating that neutrophils in lungs played a key role in the formation of the metastatic niche in lungs. Signaling factors derived from cancer cells activate neutrophils stored in lungs or recruited neutrophils to migrate into lungs, thereby neutrophils promoted tumor metastasis into lungs and colonize in lungs.
It was found that tumor exosomal RNAs promoted lung metastatic niche formation by activating alveolar epithelial Toll-like receptor 3 (TLR3) to recruit neutrophils (38). TLR3 in host alveolar epithelial cells was critical for neutrophil recruitment and niche formation before lung metastasis (38, 39). In mice with TLR3 deficiency, the incidence of lung metastasis was significantly reduced, and the survival of tumor-bearing mice was significantly prolonged (38). TLR3 was highly expressed in lung epithelial cells (40), and the lung epithelial TLR3 pathway was activated on exposure to virus, fungus, and even cigarettes (41, 42). Interestingly, high epithelial TLR3 expression was correlated with smoke-induced chronic inflammation in lungs (43), which might explain why smoking facilitated lung metastasis of breast cancer as mentioned above.
Liu et al. revealed that the natural immune receptor TLR3 expressed by type II lung epithelial cells could identify the exosomal RNA secreted by tumor into the blood circulation (44–46), thus triggering the expressions of chemokines (CXCL1, CXCL2, CXCL5, and CXCL12) and recruiting neutrophils into the lungs (38). Neutrophils in the bone marrow showed no significant differences in the mice with or without tumor inoculation. It was known that chemokine and its receptor contributed to neutrophil mobilization and recruitment. Moreover, Liu et al. discovered a marked increase in the level of chemokines (CXCL1, CXCL2, CXCL5, and CXCL12) in serum and bronchoalveolar lavage fluid after administration of exosomes, at which time there were no cancer cells in lungs (38). Accordingly, neutrophils might accumulate in the pre-metastatic lung induced by tumor exosomal RNAs.
Additionally, Xiao et al. showed that breast cancer cells secreted cathepsin C exciting neutrophils in lungs and recruiting more neutrophils into lungs to promote lung metastasis of breast cancer (47). Cathepsin C, a significant lysosomal cysteine protease, mediated the maturation process of neutrophil serine proteases, and participated in the inflammation and immune regulation process associated with neutrophils (48). Cathepsin C, derived from breast cancer cells, promoted lung metastasis of breast cancer cells by regulating neutrophil infiltration and the formation of NETs in lungs metastasis microenvironment (47).
Mechanically, tumor-secreted cathepsin C promoted the maturation and release of IL-1β in pulmonary neutrophils by activating the neutrophils membrane localization proteinase 3 (PR3) through enzyme digestion (49, 50). PR3 up-regulated the neutrophils IL-1β/nuclear factor kappa B (NF-κB) signaling pathway and their well-known downstream targets of cytokines such as IL-6 and chemokine (C-C motif) ligand 3 (CCL3), resulting in attracting more neutrophils to migrate into the lungs (47, 51). At the same time, tumor-secreted cathepsin C also up-regulated neutrophils reactive oxygen species (ROS) levels through PR3-IL-1β-P38/mitogen-activated protein kinase (MAPK), inducing the formation of NETs to degrade the anti-tumor factor thrombospondin-1 (Tsp-1) in lungs metastasis microenvironment. NETs enhanced tumor progression by affecting the endothelium, platelets, and the extracellular matrix and even the tumor cells themselves (52–54). Besides, the production of ROS and the formation of NETs supported lung metastasis of breast cancer by downregulating the thrombospondin-1 (Tsp-1) (16, 47). Tsp-1 had been found to protect lung tissues from tumor development (55). Apart from ROS and NETs, Ser proteases, NE and cathepsin G released by neutrophils could target and destroy Tsp-1, thus promoting breast cancer lung metastasis (16, 56, 57).
Bioluminescent imaging analyses showed that cathepsin C-induced elevation of cancer cell signal in lungs was already prominent at the first week after intravenous inoculation, indicating a role of cathepsin C in the early stage of metastatic colonization (47). Nevertheless, within 7 days after inoculation, cathepsin C-enhanced cancer cells had been existed in lungs at the early time points. Thus, it was not confirmed that whether the cathepsin C released by cancer cells reached lungs via blood circulation prior to the migration of cancer cells to lungs and recruit neutrophils, or neutrophils were recruited by releasing cathepsin C after the migration of cancer cells to lungs and established the “congenial soil” for lung metastasis of breast cancer.
In addition to promoting breast cancer cell metastasis through NETs or ROS, neutrophil-derived leukotrienes also promoted cancer cell colonization. Leukotrienes were a natural chemical that promoted inflammation. When leukotrienes released in the body, it could cause constriction of the airways, muscle tightness, and excess mucus and fluid (58). It was known that cancer cells within tumors were heterogeneous and retained different tumorigenic potentials. Metastasis-initiating cells depended on a favorable microenvironment to efficiently growed at the distant site (59–61). Wculek et al. revealed that neutrophil-derived leukotrienes facilitated the colonization of lung tissues by selectively expanding the sub-pool of breast cancer cells that retained high tumorigenic potential (62). Consequently, genetic or pharmacological suppression of arachidonic acid 5-lipooxygenase (ALOX5), which was responsible for leukotrienes synthesis in neutrophils, eliminated the carcinogenic activity of neutrophils and inhibited metastasis formation.
In lungs, not only do pathogens induced the recruitment of neutrophils, but also tumor cells with pulmonary metastasis also released signals to recruit neutrophils. Concordantly, they all drived the recruitment of neutrophils to form the metastatic niche in lungs.
The lung microenvironment was a very complex and changeable environment, and the formation of sites adapted to the survival of metastatic cancer cells was regulated by pulmonary inflammatory response, angiogenesis, immune response, extracellular matrix remodeling and so on. Among them, neutrophils played an extremely important part in lung inflammation. Beyond that, there were many other cells involved in the regulation of the reactions described above, including vascular endothelial cells, stem cell-like lung cells, immune cells, etc.
The endothelial cells of the capillaries in lungs were tightly attached (63, 64), unlike the discontinuous endothelial cells in the vascular system of bone, which were easy for other cells to pass through (65). Breast cancer cells must first break through the barrier of capillaries in lungs to colonize and grow. It was found that TGF-β promoted the metastasis of estrogen receptor (ER)-negative breast cancer to the lungs by upregulating the expression of a cytokine called angiopoietin-like protein 4 (ANGPTL4) (66, 67), which blocked the contact between pulmonary vascular endothelial cells (68). Within the tumor microenvironment, TGF-β was produced by macrophages, mesenchymal cells and the cancer cells themselves (66). In breast carcinoma, selective losses of growth inhibitory responses often accrued through alterations downstream of Smad, leaving the rest of the TGFβ pathway operational and open to co-option for tumor progression advantage (69). Cancer cell-derived ANGPTL4 was a TGFβ target in breast cancer, which was involved in the regulation of cancer growth, metastasis and angiogenesis (70). ANGPTL4 was induced by TGF-β through Smad signal transduction pathway (71, 72), which resisted the tight connection and adherent connection of vascular endothelia, destroyed the integrity of capillary wall, increased the permeability of pulmonary capillary, as well as induced tumor cells to exudate from blood vessels and entered the lung tissues for colonization and growth (66, 67).
In other words, disruption of the tight junctions of vascular endothelial cells in lungs provided favorable conditions for the metastasis of cancer cells to the lung. Therefore, factors that damage pulmonary vascular endothelial cells should be taken into account. They might be used as a drug target to prevent lung metastasis of breast cancer, or as a predictive method for patients at high risk for lung metastasis of breast cancer, so that patients could be more closely monitored and more aggressively treated with drugs.
After successfully entering the lung tissues, breast cancer cells needed the assistance of lung cells for colonization and metastatic growth. Such a cluster of cells existed in lungs, alveolar type 1 (AT1) cells and AT2 cells. They mainly formed the lung’s respiratory units and alveoli (73, 74). During development, AT1 and AT2 cells arose directly from a bipotent progenitor, whereas after birth new AT1 cells derived from rare, self-renewing, long-lived, mature AT2 cells that produced slowly expanding clonal foci of alveolar renewal (74, 75). There was a switch after birth, when AT2 cells function as stem cells that contributed to alveolar renewal, repair and cancer progression (76, 77).
It was reported that breast cancer cells entered the lung tissues in the early phase, the function of AT2 cells as stem cell-like lung cells were enhanced, and various types of lung cells were differentiated, finally forming the metastatic niche suitable for the growth and colonization of breast cancer (78). Ombrato et al. designed a version of a secreted monomeric Cherry red fluorescent protein (mCherry) containing a modified lipo-permeable Transactivator of Transcription peptide (sLP-mCherry) (79, 80), to develop a labelling system where metastatic cancer cells directly identified their neighboring cells in vivo (78). They engineered 4T1 breast cancer cells to express the sLP-mCherry protein alongside a canonical cell-retained Green Fluorescent Protein, which referred to as Labelling-4T1. Notably, when metastasis was formed, the number of mCherry+ niche cells in the tissue was still proportional to the growing metastatic cells. Cherry-niche epithelial cells had a higher proliferative activity compared to their normal lung counterpart. Furthermore, lineage-labelled AT2 cells showed no plasticity in co-culture with CD31+ cells. when exposed to cancer cells, lineage-labelled AT2 cells generated a notable amount of multi-lineage bronchioalveolar organoids, supporting the idea of a reprogramming activity driven by cancer cell-derived factors ex vivo. Concordantly, there were alveolar cell clusters with increased proliferative activity at the metastatic borders of human breast cancer lung metastasis, which suggested that the response of lung parenchyma to metastatic growth might occur in both mice and humans (78). However, it was not known how AT2 cells differentiate into large numbers of cells to support the growth of cancer cells.
Cancer cells that had metastasized to targeted organs must also evade their immune response in order to colonize and grow. Metastasis of breast cancer cells to lungs and colonization of growth must also create an immune-permitting environment in lungs. T cells had an important impact in regulating immune function. Whereas effector T (Teff) cells promoted immune activation and would drive clearance of infections and cancer, regulatory T (Treg) cells, dependent upon the transcription factor forkhead box protein 3 (Foxp3), suppressed their function, preventing excessive autoimmune and allergic reactions (81, 82).
It was reported that expression of a protein called oxygen-sensing prolyl-hydroxylase (PHD) in T cells inhibited the immune response to cancer cells by interfering with T cells, allowing circulating cancer cells to colonize in lungs (83). In the normal body, the oxygen-sensitive PHD protein in lungs was designed to prevent the inflammatory responses that occurred when innocuous substances were inhaled during daily life (84, 85).
In breast cancer patients, however, the immunosuppressive effect of the oxygen-sensitive PHD protein in the highly oxygenated lung microenvironment opened the door to cancer cells and created a “fertile soil” for their growth. PHD proteins limited pulmonary type helper (Th)-1 responses, promoted CD4+ regulatory T (Treg) cell induction, and restrained CD8+ T cell effector function (83). Clever et al. engineered mice harboring a T cell-specific deletion of all three PHD proteins. Cd4-driven Cre recombinase expression resulted in significant reduction of Egln1, Egln2, and Egln3 mRNA transcripts, which encoded PHD2, PHD1, and PHD3 proteins respectively, in CD4+, CD8+, and NKT T cells, but not in other lymphoid cell subsets. Upon tumor colonization, PHD proteins promoted Treg cell expansion and restrained IFN-γ-dependent clearance of tumors.
The researchers also found that using a drug inhibitor or knocking out the PHD protein in the T-cells of inbred mice stimulated an immune response to cancer cells and inhibited metastasis to the lungs (83). These findings provided a new immunological basis for explaining the susceptibility of breast cancer to lung metastasis.
There was increasing evidence that MDSCs were key components of the pre-metastatic niche and played an important role in tumor cell implantation and metastasis by crafting pre-metastatic niches for proliferation, immunosuppression, and vascular remodeling (86, 87). More importantly, blocking the aggregation of these cells in target organs had been shown to prevent tumor metastasis (88). The initial events responsible for the metastasis process were the expansion of hematopoietic stem cells and progenitor cells in the bone marrow and their differentiation into MDSCs at the site of early metastasis (89, 90). MDSCs accumulated in the pre-metastatic niche also inhibited anti-tumor T cells through Arginase 1 and ROS production (91). In the presence of Breg cells, MDSCs produced more ROS and NO and became more suppressive to CD8+ T cells (92–94).
Carbonic anhydrase IX (CAIX) was one of the most highly expressed genes in the hypoxic environment of solid tumors. CAIX promoted intracellular pH buffering through extracellular CO2 hydration to produce bicarbonate and protons (95). Bicarbonate was transferred into cells by bicarbonate transporters to maintain alkaline intracellular pH conducive to cell survival, while the protons produced contributed to the acidification of the extracellular space and increased the migratory and invasive behavior of the tumor (96). Consistently, CAIX played an important role in the cellular invasion of breast cancer cells (97). Furthermore, CAIX was required for tumor growth and metastasis and maintenance of the stemness phenotype within the hypoxic niche of breast tumors (97, 98). It was found that hypoxia-induced CAIX expression in primary tumors derived from 4T1 breast cancer cells was needed for the production of chemokines and cytokines required for the mobilization of granulocytic MDSCs to a functional metastatic niche in a syngeneic preclinical model of spontaneous breast cancer lung metastasis (95). CAIX promoted the production of known soluble mediators of breast cancer metastasis, CXCL10, CCL5, and G-CSF, by hypoxic breast cancer cells (95). Besides, hypoxia-induced CAIX facilitated the activation of the NF-kB pathway causing the stimulation of G-CSF production to trigger MDSCs infiltration in the lung. Thus, during the early stages of breast cancer metastasis, CAIX promoted the development of the breast cancer lung metastatic niche via accelerating the production of G-CSF to recruit MDSCs.
Macrophages were the principal immune cells within the tumor microenvironment and were obligate partners for tumor cell migration and metastasis (99). Normal lung tissue contains an abundance of alveolar and interstitial macrophages (100). At metastatic sites a distinct population of metastasis associated macrophages promoted tumor cell extravasation, seeding and persistent growth (101). It was showed that breast tumor cell-released microparticles (T-MPs) from the primary tumor site played a critical role in the metastatic process. The T-MPs remodeled the lung parenchyma via a macrophage-dependent pathway to create an altered inflammatory and mechanical response to tumor cell invasion (102). Mechanistically, circulating T-MPs readily entered the lung parenchyma where they were taken up by local macrophages and induced CCL2 production. CCL2 recruited CD11b+Ly6Chigh inflammatory monocytes to the lungs where they matured into F4/80+CD11b+Ly6C- macrophages that not only produced IL6 but also triggered fibrin deposition. IL6 and the deposited fibrin facilitated the survival and growth of tumor-repopulating cells in the lungs by providing chemical and mechanical signals, respectively, thus setting the stage for lung metastasis of breast cancer (103).
For years the extracellular matrix proteins was mainly considered to serve as a scaffold, but now it was evident that the extracellular matrix proteins was a critical part of the tumor microenvironment (104). The extracellular matrix proteins regulated cancer cell behavior such as proliferation, adhesion, migration and differentiation (105) and its importance in cancer and metastasis had been extensively reviewed (106, 107).
Periostin was a multifunctional extracellular matrix protein, which was expressed by fibroblasts in the normal tissue and in the stroma of the primary tumor (59). Besides, periostin was closely related to the occurrence and development of a variety of tumors and provided an appropriate site for the metastatic growth of cancer cells by regulating the formation of neovascularization and immune regulation at the metastatic site (108, 109). It was reported that periostin protein derived from tumor fibroblasts determined the lung metastasis efficiency of breast cancer and the size of metastatic cancer. Wnt signaling activity in metastasis was abrogated in the absence of periostin. Wnt was known to control tumor stem cell maintenance in a variety of tissues (110, 111). Similarly, the levels of the general Wnt target gene Axin2 in metastasis in wild-type hosts was higher than in mutant hosts. Periostin acted as a niche component that promoted tumor stem cell maintenance and metastatic colonization by augmenting Wnt signaling (59).
In addition, periostin protein played an important role in the metastasis microenvironment and perivascular niche (112). Periostin promoted the pulmonary accumulation of MDSCs during the early stage of breast tumor metastasis. Periostin deletion decreased neutrophil and monocytic cell populations in the bone marrow of mice and suppressed the accumulation of MDSCs to metastatic sites. Periostin-deficient MDSCs displayed reduced activation of ERK, AKT and STAT3 and periostin deficiency decreased the immunosuppressive functions of MDSCs during tumor progression. Moreover, lysyl oxidase contributed to periostin-promoted metastatic niche formation and tumor metastasis. However, the metastatic role of periostin was largely limited to ER-negative breast cancer patients (113).
The metastatic growth of cancer cells required not only the regulation of angiogenesis and immune regulation at the metastatic site, but also the supply of nutrients to satisfy its proliferation and growth (114, 115). It had been found that under the induction of signals released by tumor cells, neutrophils could not only infiltrate into lung tissues to play the role of immune regulation, but also stored a large amount of lipids (116, 117). Once tumor cells migrated to the lung, these lipids could be used as reserve food for tumor cells to promote their colonization and growth. These lipids were not inherent to the neutrophils, but were induced by CD140a+ mesenchymal cells once they reached the lungs (118). Lung mesenchymal cells significantly up-regulated the expression of neutrophil lipid droplet related genes, including Hilpda, Cidec and Atgl (119, 120). These up-regulated triglyceride lipase inhibitors in turn inhibited ATGL enzyme activity, leading to the accumulation of triglycerides in pulmonary neutrophils (118). Neutrophil-specific knockout of Atgl enhanced lipid accumulation in cells and significantly promoted lung metastasis of breast cancer in vivo. In contrast, knockout of Hilpda genes in neutrophils reduced lipid storage and significantly inhibited metastatic colonization of breast tumors (118).
In the process of disease progression of cancer, chemotherapy drugs were like a double-edged sword. Although they had contributed to21 adjuvant chemotherapy after surgery or preoperative neoadjuvant chemotherapy, but some chemotherapeutic drugs promote metastasis while killing cancer cells (121, 122). It was found that breast cancer cells spread through recombinant TEK tyrosine kinase (TIE2)/Mammalian-enabled (MENA) pathway dependent infiltrating sites in cancer cells, namely, tumor microenvironment of metastasis (TMEM) (123, 124). Using fixed tissue and in vivo imaging of PyMT mice models and patient-derived xenografts (125), it was found that paclitaxel increased the density and activity of the TMEM site and MENA expression and promoted distant metastasis. Besides, in residual breast cancer patients treated with neoadjuvant paclitaxel after doxorubicin and cyclophosphamide, TMEM score and expression of mechanism related MENAINV subtypes were increased, suggesting that although chemotherapy reduced tumor size, it increased the risk of metastasis and spread (126).
In addition, although paclitaxel was clearly beneficial in reducing tumor size, it increased the circulation of tumor cells in the blood, helping breast cancer cells escape from the primary focus, and directly acted on the lungs, altering the lung microenvironment and helping cancer cells colonize the lungs (127). In metastatic lung, paclitaxel improved the tissue microenvironment in which cancer cells grew, which changed include an increase in inflammatory monocytes and a decrease in cytotoxicity (121, 128, 129). Importantly, these changes in both primary and metastatic lung were dependent on ATF3 stress-inducing genes in non-cancer host cells (127).
After that, exosomes produced by paclitaxel treated cancer cells could promote the release of exosomes from cancer cells, change the lung microenvironment and promote lung metastasis of breast cancer (130). Paclitaxel not only promoted the production of more exosomes in cancer cells, but also promoted the entry of annexin A6 (ANXA6) protein, a non-glycosylated polypeptide chain into exosomes by increasing calcium levels in cancer cells, which contributed to paclitaxel resistance in breast cancer (131). After ANXA6-carrying exosomes were transported to the lungs through blood circulation, they promoted the expression of chemokines in lung tissues and recruited mononuclear macrophages, which were the “agent provocateurs” that helped lung metastasis of breast cancer (132). Macrophages had been shown to play important roles in cancer metastasis. Macrophages were generally categorized into either of two functionally contrasting subtypes, namely classical activated M1 macrophages and alternatively activated M2 macrophages (133, 134). M2 macrophages promoted the occurrence and metastasis of tumor cells, inhibited T cell-mediated anti-tumor immune response, promoted tumor angiogenesis, and leaded to tumor progression (135). Both M1 and M2 macrophages had high degree of plasticity and thus could be converted into each other upon tumor microenvironment changes or therapeutic interventions (133).
In order to verify the effect of ANXA6, the researchers knocked out the ANXA6 gene in breast cancer cells and found that exosomes secreted by paclitaxel-induced cancer cells did not carry ANXA6 and no longer had the function of promoting lung metastasis of tumor (130). Blocking the metastasis promoting function of pulmonary mononuclear macrophages could also prevent paclitaxel-induced lung metastasis of breast cancer (130). In addition to paclitaxel, adriamycin also promoted the entry of ANXA6 into exosomes and facilitated lung metastasis of breast cancer (130). Paclitaxel and adriamycin could be said to be two completely different chemotherapy drugs, both of which promoted tumor metastasis through exosome and ANXA6, perhaps this effect of chemotherapy in promoting metastasis was widespread.
The vast majority of cancer deaths were caused by metastasis (136). Current research suggested that tumor metastasis depended not only on the ability of tumor cells to invade themselves, but also on the formation of a metastatic niche. The construction of the metastatic microenvironment determined whether the tumor cells invading the circulatory system could attach, survived, proliferated and eventually formed metastatic cancer at the distant site of metastasis. Before metastasis of tumor cells, functional domestication of cells and matrix components in target organs of metastasis could be performed to form a microenvironment conducive to tumor cell colonization (137).
With the continuous development of the research on the metastatic microenvironment, results of more and more research on the metastatic lung microenvironment of breast cancer had been reported. Currently, there were in-depth studies on the neutrophils-dependent inflammatory microenvironment in lungs, and the inflammation induced oxidative stress response and promoted angiogenesis, which contributed to the metastatic growth of tumors (138). In addition, in the initial stage of inflammation, the activation of inflammatory factors also triggered vascular dilation, which increased the permeability of the vascular wall, and tumor cells exuded from the blood vessels and colonized as well as grew in the metastatic organs (66, 139). In addition, many cells or soluble factors in lungs were affected by the signals released by breast cancer cells, and then modified the lung microenvironment to make it suitable for the growth of breast cancer. Or cytokines in lungs microenvironment attracted chemotaxis, migration, and colonization of breast cancer cells, thereby promoting lung metastasis of breast cancer.
Although some progress had been made in the study of metastatic lung microenvironment of breast cancer, many mechanisms remained unclear due to the complexity and variability of metastatic microenvironment and high heterogeneity. Many questions needed to be further explored and solved: were these metastatic microenvironments induced by breast cancer or existed before the remodeling of the pulmonary microenvironment? How to optimize in vitro and in vivo experimental model to more accurately simulate the human lung microenvironment? In such a variable lung microenvironment, neutrophils, which were the most deeply studied, not only promoted tumor metastasis by inducing inflammatory lung microenvironment, but also stored nutrients for newly metastasis to lung tumor cells in advance to provide energy for their colonization and growth in lung. In addition, neutrophils could be directly measured in peripheral blood. Could corresponding kits be developed to detect markers of peripheral blood neutrophils, which were used as a monitoring indicator for the prognosis of breast cancer with lung metastasis? In sum, the research results of pulmonary microenvironment would be another major breakthrough in the treatment of lung metastasis of breast cancer and provided a new perspective for the suppression of lung metastasis of breast cancer.
The lung was a common site for metastasis of malignant tumors such as primary bladder cancer, breast cancer, colon cancer, kidney cancer, melanoma, ovarian cancer, pancreatic cancer, osteosarcoma, rectal cancer, gastric cancer, thyroid cancer and endometrial cancer (140). These findings above-mentioned encouraged the design of pre-clinical and clinical studies to examine the benefits of targeting the formation of pulmonary microenvironment in lung metastasis from bladder cancer, colon cancer, kidney cancer and so on, which might be effective in blocking metastatic relapse.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This study was supported by the grants from the Dongguan Social Science and Technology Development Project (201950715025192), the grants from the National Natural Science Foundation of China (81802918), the China Postdoctoral Science Foundation Grant (2019M660206), the Science and Technology Project of Guangdong Province (2019A1515011565, 2018A030310007), and the Science and Technology Project of Jiangmen (2020030103140008978, 2019030102430012905).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
LPS, lipopolysaccharide; LCN2, lipocalin 2; NADPH, nicotinamide adenine dinucleotide phosphate; NE, neutrophil elastase; MPO, myeloperoxidase; ALP, alkaline phosphatase; NETs, neutrophil extracellular traps; NF-κB, nuclear factor kappa B; CCL3, chemokine (C-C motif) ligand 3; ROS: reactive oxygen species; Tsp-1, thrombospondin-1; ALOX5, arachidonic acid 5-lipooxygenase; CXCL8, chemokine (C-X-C motif) ligand 8; DKK1, Dickkopf-1; PCP, planar cell polarity; CaMKII, calmodulin-dependent protein kinase II; TGF-β, transforming growth factor-β; PR3, proteinase 3; MAPK, mitogen-activated protein kinase; TLR3, Toll-like receptor 3; ER, estrogen receptor; ANGPTL4, angiopoietin-like protein 4; AT1, alveolar type 1; mCherry, monomeric Cherry red fluorescent protein; Foxp3, forkhead box protein 3; PHD, prolyl-hydroxylase; DAMPs, damp-associated molecular patterns; NLRP3, NOD-like receptor protein 3; MDSCs, marrow derived suppressor cells; CAIX, Carbonic anhydrase IX; MMTV, mammary tumor virus; TIE2, TEK tyrosine kinase; MENA, mammalian-enabled; ANXA6, annexin A6.
1. Young RC. Cancer Statistics, 2002: Progress or Cause for Concern? CA: Cancer J Clin (2002) 52(1):6–7. doi: 10.3322/canjclin.52.1.6
2. Zhang X, Ren D, Guo L, Wang Lan, Wu Shu, Lin Chuyong, et al. Thymosin Beta 10 Is a Key Regulator of Tumorigenesis and Metastasis and a Novel Serum Marker in Breast Cancer. Breast Cancer Res (2017) 19(1):15. doi: 10.1186/s13058-016-0785-2
3. DeSantis CE, Miller KD, Dale W, Mohile SG, Cohen HJ, Leach CR, et al. Cancer Statistics for Adults Aged 85 Years and Older, 2019. CA: Cancer J Clin (2019) 69(6):452–67. doi: 10.3322/caac.21577
5. Lin C, Liao W, Jian Y, Peng Y, Zhang X, Ye L, et al. CGI-99 Promotes Breast Cancer Metastasis via Autocrine Interleukin-6 Signaling. Oncogene (2017) 36(26):3695–705. doi: 10.1038/onc.2016.525
6. Wu Q, Li J, Zhu S, Wu J, Chen C, Liu Q, et al. Breast Cancer Subtypes Predict the Preferential Site of Distant Metastases: A SEER Based Study. Oncotarget (2017) 8(17):27990–6. doi: 10.18632/oncotarget.15856
7. Xiao W, Zheng S, Liu P, Zou Y, Xie X, Yu P, et al. Risk Factors and Survival Outcomes in Patients With Breast Cancer and Lung Metastasis: A Population-Based Study. Cancer Med (2018) 7(3):922–30. doi: 10.1002/cam4.1370
8. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic Reprogramming and Cancer Progression. Sci (New York NY) (2020) 368(6487):eaaw5473. doi: 10.1126/science.aaw5473
9. Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, et al. E-Cadherin Is Required for Metastasis in Multiple Models of Breast Cancer. Nature (2019) 573(7774):439–44. doi: 10.1038/s41586-019-1526-3
10. Balkwill F, Mantovani A. Inflammation and Cancer: Back to Virchow? J Lancet (London England) (2001) 357(9255):539–45. doi: 10.1016/S0140-6736(00)04046-0
11. Coussens LM, Werb Z. Inflammation and Cancer. Nature (2002) 420(6917):860–7. doi: 10.1038/nature01322
12. Jiang L, Yu L, Zhang X, Lei F, Wang L, Liu X, et al. miR-892b Silencing Activates NF-κb and Promotes Aggressiveness in Breast Cancer. Cancer Res (2016) 76(5):1101–11. doi: 10.1158/0008-5472.CAN-15-1770
13. Murin S, Pinkerton KE, Hubbard NE, Erickson K. The Effect of Cigarette Smoke Exposure on Pulmonary Metastatic Disease in a Murine Model of Metastatic Breast Cancer. Chest (2004) 125(4):1467–71. doi: 10.1378/chest.125.4.1467
14. Smith HA, Kang Y. Acute Infection Induces a Metastatic Niche: A Double Menace for Cancer Patients. Clin Cancer Res (2013) 19(17):4547–9. doi: 10.1158/1078-0432.CCR-13-1524
15. Murin S, Inciardi J. Cigarette Smoking and the Risk of Pulmonary Metastasis From Breast Cancer. Chest (2001) 119(6):1635–40. doi: 10.1378/chest.119.6.1635
16. El Rayes T, Catena R, Lee S, Stawowczyk M, Joshi N, Fischbach C, et al. Lung Inflammation Promotes Metastasis Through Neutrophil Protease-Mediated Degradation of Tsp-1. Proc Natl Acad Sci USA (2015) 112(52):16000–5. doi: 10.1073/pnas.1507294112
17. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140(6):805–20. doi: 10.1016/j.cell.2010.01.022
18. Yan L, Cai Q, Xu Y. The Ubiquitin-CXCR4 Axis Plays an Important Role in Acute Lung Infection-Enhanced Lung Tumor Metastasis. Clin Cancer Res (2013) 19(17):4706–16. doi: 10.1158/1078-0432.CCR-13-0011
19. Jiang M, Xu X, Bi Y, Xu J, Qin C, Han M, et al. Systemic Inflammation Promotes Lung Metastasis via E-Selectin Upregulation in Mouse Breast Cancer Model. Cancer Biol Ther (2014) 15(6):789–96. doi: 10.4161/cbt.28552
20. Tyagi A, Sharma S, Wu K, Wu S-Y, Xing F, Liu Y, et al. Nicotine Promotes Breast Cancer Metastasis by Stimulating N2 Neutrophils and Generating Pre-Metastatic Niche in Lung. Nat Commun (2021) 12(1):474. doi: 10.1038/s41467-020-20733-9
21. Kolaczkowska E, Kubes P. Neutrophil Recruitment and Function in Health and Inflammation. Nat Rev Immunol (2013) 13(3):159–75. doi: 10.1038/nri3399
22. Hsu LC, Enzler T, Seita J, Timmer AM, Lee C-Y, Lai T-Y, et al. IL-1β-Driven Neutrophilia Preserves Antibacterial Defense in the Absence of the Kinase Ikkβ. Nat Immunol (2011) 12(2):144–50. doi: 10.1038/ni.1976
23. Filippi MD. Neutrophil Transendothelial Migration: Updates and New Perspectives. Blood (2019) 133(20):2149–58. doi: 10.1182/blood-2018-12-844605
24. Oliveira C, Navarro-Xavier RA, Anjos-Vallota EA, Martins JO, Silveira VLF, Gonçalves LRC, et al. Effect of Plant Neutrophil Elastase Inhibitor on Leucocyte Migration, Adhesion and Cytokine Release in Inflammatory Conditions. Br J Pharmacol (2010) 161(4):899–910. doi: 10.1111/j.1476-5381.2010.00924.x
25. Kumar V, Sharma A. Neutrophils: Cinderella of Innate Immune System. Int Immunopharmacol (2010) 10(11):1325–34. doi: 10.1016/j.intimp.2010.08.012
26. Cortjens B, van Woensel JBM, Bem RA. Neutrophil Extracellular Traps in Respiratory Disease: Guided Anti-Microbial Traps or Toxic Webs? Paediatric Respir Rev (2017) 21:54–61. doi: 10.1016/j.prrv.2016.03.007
27. Aulakh GK. Neutrophils in the Lung: "the First Responders". Cell Tissue Res (2018) 371(3):577–88. doi: 10.1007/s00441-017-2748-z
28. Giacalone VD, Margaroli C, Mall MA, Tirouvanziam R. Neutrophil Adaptations Upon Recruitment to the Lung: New Concepts and Implications for Homeostasis and Disease. Int J Mol Sci (2020) 21(3):851. doi: 10.3390/ijms21030851
29. El-Benna J, Hurtado-Nedelec M, Marzaioli V, Marie J-C, Gougerot-Pocidalo M-A, Dang PM-C. Priming of the Neutrophil Respiratory Burst: Role in Host Defense and Inflammation. Immunol Rev (2016) 273(1):180–93. doi: 10.1111/imr.12447
30. Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, et al. Myeloperoxidase is Required for Neutrophil Extracellular Trap Formation: Implications for Innate Immunity. Blood (2011) 117(3):953–9. doi: 10.1182/blood-2010-06-290171
31. Okun DB, Tanaka KR. Leukocyte Alkaline Phosphatase. Am J Hematol (1978) 4(3):293–9. doi: 10.1002/ajh.2830040312
32. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil Elastase and Myeloperoxidase Regulate the Formation of Neutrophil Extracellular Traps. J Cell Biol (2010) 191(3):677–91. doi: 10.1083/jcb.201006052
33. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-Beta: "N1" Versus "N2" TAN. Cancer Cell (2009) 16(3):183–94. doi: 10.1016/j.ccr.2009.06.017
34. Liu Y, Gu Y, Cao X. The Exosomes in Tumor Immunity. Oncoimmunology (2015) 4(9):e1027472. doi: 10.1080/2162402X.2015.1027472
35. Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau C-S, et al. IL-17-Producing γδ T Cells and Neutrophils Conspire to Promote Breast Cancer Metastasis. Nature (2015) 522(7556):345–8. doi: 10.1038/nature14282
36. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil Extracellular Traps Sequester Circulating Tumor Cells and Promote Metastasis. J Clin Invest (2013) 123(8):3446–58. doi: 10.1172/JCI67484
37. Wu CF, Andzinski L, Kasnitz N, Kröger A, Klawonn F, Lienenklaus S, et al. The Lack of Type I Interferon Induces Neutrophil-Mediated Pre-Metastatic Niche Formation in the Mouse Lung. Int J Cancer (2015) 137(4):837–47. doi: 10.1002/ijc.29444
38. Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, et al. Tumor Exosomal RNAs Promote Lung Pre-Metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell (2016) 30(2):243–56. doi: 10.1016/j.ccell.2016.06.021
39. Kimura G, Ueda K, Eto S, Watanabe Y, Masuko T, Kusama T, et al. Toll-Like Receptor 3 Stimulation Causes Corticosteroid-Refractory Airway Neutrophilia and Hyperresponsiveness in Mice. Chest (2013) 144(1):99–105. doi: 10.1378/chest.12-2610
40. Handke W, Oelschlegel R, Franke R, Krüger DH, Rang A. Hantaan Virus Triggers TLR3-Dependent Innate Immune Responses. J Immunol (2009) 182(5):2849–58. doi: 10.4049/jimmunol.0802893
41. Babiceanu MC, Howard BA, Rumore AC, Kita H, Lawrence CB. Analysis of Global Gene Expression Changes in Human Bronchial Epithelial Cells Exposed to Spores of the Allergenic Fungus, Alternaria Alternata. Front Microbiol (2013) 4:196. doi: 10.3389/fmicb.2013.00196
42. Wortham BW, Eppert BL, Flury JL, Garcia SM, Borchers MT. TLR and NKG2D Signaling Pathways Mediate CS-Induced Pulmonary Pathologies. PloS One (2013) 8(10):e78735. doi: 10.1371/journal.pone.0078735
43. Milara J, Morell A, Ballester B, Sanz C, Freire J, Qian X, et al. Roflumilast Improves Corticosteroid Resistance COPD Bronchial Epithelial Cells Stimulated With Toll Like Receptor 3 Agonist. Respir Res (2015) 16(1):12. doi: 10.1186/s12931-015-0179-5
44. Wagner WR, Griffith BP. Reconstructing the Lung. Science (2010) 329(5991):520–2. doi: 10.1126/science.1194087
45. Whitsett JA, Alenghat T. Respiratory Epithelial Cells Orchestrate Pulmonary Innate Immunity. Nat Immunol (2015) 16(1):27–35. doi: 10.1038/ni.3045
46. Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of Double-Stranded RNA and Activation of NF-kappaB by Toll-Like Receptor 3. Nature (2001) 413(6857):732–8. doi: 10.1038/35099560
47. Xiao Y, Cong M, Li J, He D, Wu Q, Tian P, et al. Cathepsin C Promotes Breast Cancer Lung Metastasis by Modulating Neutrophil Infiltration and Neutrophil Extracellular Trap Formation. Cancer Cell (2021) 39(3):423–37.e7. doi: 10.1016/j.ccell.2020.12.012
48. Shen XB, Chen X, Zhang ZY, Wu FF, Liu XH. Cathepsin C Inhibitors as Anti-Inflammatory Drug Discovery: Challenges and Opportunities. Eur J Med Chem (2021) 225:113818. doi: 10.1016/j.ejmech.2021.113818
49. Korkmaz B, Jaillet J, Jourdan ML, Gauthier A, Gauthier F, Attucci S, et al. Catalytic Activity and Inhibition of Wegener Antigen Proteinase 3 on the Cell Surface of Human Polymorphonuclear Neutrophils. J Biol Chem (2009) 284(30):19896–902. doi: 10.1074/jbc.M901471200
50. Korkmaz B, Attucci S, Juliano MA, Kalupov T, Jourdan M-L, Juliano L, et al. Measuring Elastase, Proteinase 3 and Cathepsin G Activities at the Surface of Human Neutrophils With Fluorescence Resonance Energy Transfer Substrates. Nat Protoc (2008) 3(6):991–1000. doi: 10.1038/nprot.2008.63
51. Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, et al. Converting Enzyme-Independent Release of Tumor Necrosis Factor Alpha and IL-1beta From a Stimulated Human Monocytic Cell Line in the Presence of Activated Neutrophils or Purified Proteinase 3. Proc Natl Acad Sci U.S.A. (1999) 96(11):6261–6. doi: 10.1073/pnas.96.11.6261
52. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil Extracellular Traps Produced During Inflammation Awaken Dormant Cancer Cells in Mice. Science (2018) 361(6409):eaao4227. doi: 10.1126/science.aao4227
53. Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H, et al. Neutrophils Facilitate Ovarian Cancer Premetastatic Niche Formation in the Omentum. J Exp Med (2019) 216(1):176–94. doi: 10.1084/jem.20181170
54. Pieterse E, Rother N, Garsen M, Hofstra JM, Satchell SC, Hoffmann M, et al. Neutrophil Extracellular Traps Drive Endothelial-To-Mesenchymal Transition. Arterioscler Thromb Vasc Biol (2017) 37(7):1371–9. doi: 10.1161/ATVBAHA.117.309002
55. Catena R, Bhattacharya N, El Rayes T, Wang S, Choi H, Gao D, et al. Bone Marrow-Derived Gr1+ Cells Can Generate a Metastasis-Resistant Microenvironment via Induced Secretion of Thrombospondin-1. Cancer Discovery (2013) 3(5):578–89. doi: 10.1158/2159-8290.CD-12-0476
56. Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl Peptidase I Activates Neutrophil-Derived Serine Proteases and Regulates the Development of Acute Experimental Arthritis. J Clin Invest (2002) 109(3):363–71. doi: 10.1172/JCI0213462
57. Korkmaz B, Lesner A, Letast S, Mahdi YK, Jourdan ML, Dallet-Choisy S, et al. Neutrophil Proteinase 3 and Dipeptidyl Peptidase I (Cathepsin C) as Pharmacological Targets in Granulomatosis With Polyangiitis (Wegener Granulomatosis). Semin Immunopathol (2013) 35(4):411–21. doi: 10.1007/s00281-013-0362-z
58. Kanaoka Y, Boyce JA. Cysteinyl Leukotrienes and Their Receptors: Cellular Distribution and Function in Immune and Inflammatory Responses. J Immunol (2004) 173(3):1503–10. doi: 10.4049/jimmunol.173.3.1503
59. Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr HA, Delaloye JF, et al. Interactions Between Cancer Stem Cells and Their Niche Govern Metastatic Colonization. Nature (2011) 481(7379):85–9. doi: 10.1038/nature10694
60. Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Céspedes MV, et al. Dependency of Colorectal Cancer on a TGF-β-Driven Program in Stromal Cells for Metastasis Initiation. Cancer Cell (2012) 22(5):571–84. doi: 10.1016/j.ccr.2012.08.013
61. Oskarsson T, Batlle E, Massagué J. Metastatic Stem Cells: Sources, Niches, and Vital Pathways. Cell Stem Cell (2014) 14(3):306–21. doi: 10.1016/j.stem.2014.02.002
62. Wculek SK, Malanchi I. Neutrophils Support Lung Colonization of Metastasis-Initiating Breast Cancer Cells. Nature (2015) 528(7582):413–7. doi: 10.1038/nature16140
63. Wettschureck N, Strilic B, Offermanns S. Passing the Vascular Barrier: Endothelial Signaling Processes Controlling Extravasation. Physiol Rev (2019) 99(3):1467–525. doi: 10.1152/physrev.00037.2018
64. Jones JH, Friedrich E, Hong Z, Minshall RD, Malik AB. PV1 in Caveolae Controls Lung Endothelial Permeability. Am J Respir Cell Mol Biol (2020) 63(4):531–9. doi: 10.1165/rcmb.2020-0102OC
65. Sipkins DA, Wei X, Wu JW, Runnels JM, Côté D, Means TK, et al. In Vivo Imaging of Specialized Bone Marrow Endothelial Microdomains for Tumour Engraftment. Nature (2005) 435(7044):969–73. doi: 10.1038/nature03703
66. Padua D, Zhang XHF, Wang Q, Nadal C, Gerald WL, Gomis RR, et al. TGFbeta Primes Breast Tumors for Lung Metastasis Seeding Through Angiopoietin-Like 4. Cell (2008) 133(1):66–77. doi: 10.1016/j.cell.2008.01.046
67. Welm AL. TGFbeta Primes Breast Tumor Cells for Metastasis. Cell (2008) 133(1):27–8. doi: 10.1016/j.cell.2008.03.012
68. Huang RL, Teo Z, Chong HC, Zhu P, Tan MJ, Tan CK, et al. ANGPTL4 Modulates Vascular Junction Integrity by Integrin Signaling and Disruption of Intercellular VE-Cadherin and Claudin-5 Clusters. Blood (2011) 118(14):3990–4002. doi: 10.1182/blood-2011-01-328716
69. Massagué J, Seoane J, Wotton D. Smad Transcription Factors. Genes Dev (2005) 19(23):2783–810. doi: 10.1101/gad.1350705
70. Cai YC, Yang H, Wang KF, Chen TH, Jiang WQ, Shi YX, et al. ANGPTL4 Overexpression Inhibits Tumor Cell Adhesion and Migration and Predicts Favorable Prognosis of Triple-Negative Breast Cancer. BMC Cancer (2020) 20(1):878. doi: 10.1186/s12885-020-07343-w
71. Gong X, Hou Z, Endsley MP, Gronseth EI, Rarick KR, Jorns JM, et al. Interaction of Tumor Cells and Astrocytes Promotes Breast Cancer Brain Metastases Through TGF-β2/ANGPTL4 Axes. NPJ Precis Oncol (2019) 3:24. doi: 10.1038/s41698-019-0094-1
72. Keklikoglou I, Koerner C, Schmidt C, Zhang JD, Heckmann D, Shavinskaya A, et al. MicroRNA-520/373 Family Functions as a Tumor Suppressor in Estrogen Receptor Negative Breast Cancer by Targeting NF-κb and TGF-β Signaling Pathways. Oncogene (2012) 31(37):4150–63. doi: 10.1038/onc.2011.571
73. Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ, et al. Single-Cell Wnt Signaling Niches Maintain Stemness of Alveolar Type 2 Cells. Science (2018) 359(6380):1118–23. doi: 10.1126/science.aam6603
74. Desai TJ, Brownfield DG, Krasnow MA. Alveolar Progenitor and Stem Cells in Lung Development, Renewal and Cancer. Nature (2014) 507(7491):190–4. doi: 10.1038/nature12930
75. Adamson IY, Bowden DH. Derivation of Type 1 Epithelium From Type 2 Cells in the Developing Rat Lung. Lab Invest (1975) 32(6):736–45.
76. Rock JR, Hogan BL. Epithelial Progenitor Cells in Lung Development, Maintenance, Repair, and Disease. Annu Rev Cell Dev Biol (2011) 27:493–512. doi: 10.1146/annurev-cellbio-100109-104040
77. Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, et al. Integrin α6β4 Identifies an Adult Distal Lung Epithelial Population With Regenerative Potential in Mice. J Clin Invest (2011) 121(7):2855–62. doi: 10.1172/JCI57673
78. Ombrato L, Nolan E, Kurelac I, Mavousian A, Bridgeman VL, Heinze I, et al. Metastatic-Niche Labelling Reveals Parenchymal Cells With Stem Features. Nature (2019) 572(7771):603–8. doi: 10.1038/s41586-019-1487-6
79. Barash S, Wang W, Shi Y. Human Secretory Signal Peptide Description by Hidden Markov Model and Generation of a Strong Artificial Signal Peptide for Secreted Protein Expression. Biochem Biophys Res Commun (2002) 294(4):835–42. doi: 10.1016/S0006-291X(02)00566-1
80. Flinterman M, Farzaneh F, Habib N, Malik F, Gäken J, Tavassoli M, et al. Delivery of Therapeutic Proteins as Secretable TAT Fusion Products. Mol Ther (2009) 17(2):334–42. doi: 10.1038/mt.2008.256
81. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-Dependent Programme of Regulatory T-Cell Differentiation. Nature (2007) 445(7129):771–5. doi: 10.1038/nature05543
82. Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T, et al. Organ-Specific Autoimmune Diseases Induced in Mice by Elimination of T Cell Subset. I. Evidence for the Active Participation of T Cells in Natural Self-Tolerance; Deficit of a T Cell Subset as a Possible Cause of Autoimmune Disease. J Exp Med (1985) 161(1):72–87. doi: 10.1084/jem.161.1.72
83. Clever D, Roychoudhuri R, Constantinides MG, Askenase MH, Sukumar M, Klebanoff CA, et al. Oxygen Sensing by T Cells Establishes an Immunologically Tolerant Metastatic Niche. Cell (2016) 166(5):1117–31.e14. doi: 10.1016/j.cell.2016.07.032
84. Walmsley SR, Chilvers ER, Thompson AA, Vaughan K, Marriott HM, Parker LC, et al. Prolyl Hydroxylase 3 (PHD3) Is Essential for Hypoxic Regulation of Neutrophilic Inflammation in Humans and Mice. J Clin Invest (2011) 121(3):1053–63. doi: 10.1172/JCI43273
85. Ahmad T, Kumar M, Mabalirajan U, Pattnaik B, Aggarwal S, Singh R, et al. Hypoxia Response in Asthma: Differential Modulation on Inflammation and Epithelial Injury. Am J Respir Cell Mol Biol (2012) 47(1):1–10. doi: 10.1165/rcmb.2011-0203OC
86. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front Immunol (2018) 9:398. doi: 10.3389/fimmu.2018.00398
87. Wei H, Guo C, Zhu R, Zhang C, Han N, Liu R, et al. Shuangshen Granules Attenuate Lung Metastasis by Modulating Bone Marrow Differentiation Through mTOR Signalling Inhibition. J Ethnopharmacol (2021) 281:113305. doi: 10.1016/j.jep.2020.113305
88. Liu Y, Cao X. Immunosuppressive Cells in Tumor Immune Escape and Metastasis. J Mol Med (Berl) (2016) 94(5):509–22. doi: 10.1007/s00109-015-1376-x
89. Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-Colony Stimulating Factor Promotes Lung Metastasis Through Mobilization of Ly6G+Ly6C+ Granulocytes. Proc Natl Acad Sci U.S.A. (2010) 107(50):21248–55. doi: 10.1073/pnas.1015855107
90. Giles AJ, Reid CM, Evans JD, Murgai M, Vicioso Y, Highfill SL, et al. Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression Within the Pre-Metastatic Niche. Cancer Res (2016) 76(6):1335–47. doi: 10.1158/0008-5472.CAN-15-0204
91. Tacke RS, Lee HC, Goh C, Courtney J, Polyak SJ, Rosen HR, et al. Myeloid Suppressor Cells Induced by Hepatitis C Virus Suppress T-Cell Responses Through the Production of Reactive Oxygen Species. Hepatology (2012) 55(2):343–53. doi: 10.1002/hep.24700
92. Bodogai M, Moritoh K, Lee-Chang C, Hollander CM, Sherman-Baust CA, Wersto RP, et al. Immunosuppressive and Prometastatic Functions of Myeloid-Derived Suppressive Cells Rely Upon Education From Tumor-Associated B Cells. Cancer Res (2015) 75(17):3456–65. doi: 10.1158/0008-5472.CAN-14-3077
93. Liu Y, Cao X. Characteristics and Significance of the Pre-Metastatic Niche. Cancer Cell (2016) 30(5):668–81. doi: 10.1016/j.ccell.2016.09.011
94. Trovato R, Canè S, Petrova V, Sartoris S, Ugel S, De Sanctis F. The Engagement Between MDSCs and Metastases: Partners in Crime. Front Oncol (2020) 10:165. doi: 10.3389/fonc.2020.00165
95. Chafe SC, Lou Y, Sceneay J, Vallejo M, Hamilton MJ, McDonald PC, et al. Carbonic Anhydrase IX Promotes Myeloid-Derived Suppressor Cell Mobilization and Establishment of a Metastatic Niche by Stimulating G-CSF Production. Cancer Res (2015) 75(6):996–1008. doi: 10.1158/0008-5472.CAN-14-3000
96. Gatenby RA, Gillies RJ. Why Do Cancers Have High Aerobic Glycolysis? Nat Rev Cancer (2004) 4(11):891–9. doi: 10.1038/nrc1478
97. Lock FE, McDonald PC, Lou Y, Serrano I, Chafe SC, Ostlund C, et al. Targeting Carbonic Anhydrase IX Depletes Breast Cancer Stem Cells Within the Hypoxic Niche. Oncogene (2013) 32(44):5210–9. doi: 10.1038/onc.2012.550
98. Lou Y, McDonald PC, Oloumi A, Chia S, Ostlund C, Ahmadi A, et al. Targeting Tumor Hypoxia: Suppression of Breast Tumor Growth and Metastasis by Novel Carbonic Anhydrase IX Inhibitors. Cancer Res (2011) 71(9):3364–76. doi: 10.1158/0008-5472.CAN-10-4261
99. Condeelis J, Pollard JW. Macrophages: Obligate Partners for Tumor Cell Migration, Invasion, and Metastasis. Cell (2006) 124(2):263–6. doi: 10.1016/j.cell.2006.01.007
100. Tan SY, Krasnow MA. Developmental Origin of Lung Macrophage Diversity. Development (2016) 143(8):1318–27. doi: 10.1242/dev.129122
101. Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, et al. A Distinct Macrophage Population Mediates Metastatic Breast Cancer Cell Extravasation, Establishment and Growth. PloS One (2009) 4(8):e6562. doi: 10.1371/journal.pone.0006562
102. Ma R, Ji T, Chen D, Dong W, Zhang H, Yin X, et al. Tumor Cell-Derived Microparticles Polarize M2 Tumor-Associated Macrophages for Tumor Progression. Oncoimmunology (2016) 5(4):e1118599. doi: 10.1080/2162402X.2015.1118599
103. Zhang H, Yu Y, Zhou L, Ma J, Tang K, Xu P, et al. Circulating Tumor Microparticles Promote Lung Metastasis by Reprogramming Inflammatory and Mechanical Niches via a Macrophage-Dependent Pathway. Cancer Immunol Res (2018) 6(9):1046–56. doi: 10.1158/2326-6066.CIR-17-0574
104. Nyström H. Extracellular Matrix Proteins in Metastases to the Liver - Composition, Function and Potential Applications. Semin Cancer Biol (2021) 71:134–42. doi: 10.1016/j.semcancer.2020.06.004
105. Bonnans C, Chou J, Werb Z. Remodelling the Extracellular Matrix in Development and Disease. Nat Rev Mol Cell Biol (2014) 15(12):786–801. doi: 10.1038/nrm3904
106. Pickup MW, Mouw JK, Weaver VM. The Extracellular Matrix Modulates the Hallmarks of Cancer. EMBO Rep (2014) 15(12):1243–53. doi: 10.15252/embr.201439246
107. Kai F, Drain AP, Weaver VM. The Extracellular Matrix Modulates the Metastatic Journey. Dev Cell (2019) 49(3):332–46. doi: 10.1016/j.devcel.2019.03.026
108. Liu AY, Zheng H, Ouyang G. Periostin, A Multifunctional Matricellular Protein in Inflammatory and Tumor Microenvironments. Matrix Biol (2014) 37:150–6. doi: 10.1016/j.matbio.2014.04.007
109. Ma H, Wang J, Zhao X, Wu T, Huang Z, Chen D, et al. Periostin Promotes Colorectal Tumorigenesis Through Integrin-FAK-Src Pathway-Mediated YAP/TAZ Activation. Cell Rep (2020) 30(3):793–806.e796. doi: 10.1016/j.celrep.2019.12.075
110. Malanchi I, Peinado H, Kassen D, Hussenet T, Metzger D, Chambon P, et al. Cutaneous Cancer Stem Cell Maintenance is Dependent on β-Catenin Signalling. Nature 2(008) 452(7187):650–3. doi: 10.1038/nature06835
111. Barker N, Ridgway RA, van Es JH, van deWetering M, Begthel H, van denBorn M, et al. Crypt Stem Cells as the Cells-of-Origin of Intestinal Cancer. Nature (2009) 457(7229):608–11. doi: 10.1038/nature07602
112. Wang Z, Ouyang G. Periostin: A Bridge Between Cancer Stem Cells and Their Metastatic Niche. Cell Stem Cell (2012) 10(2):111–2. doi: 10.1016/j.stem.2012.01.002
113. Wang Z, Xiong S, Mao Y, Chen M, Ma X, Zhou X, et al. Periostin Promotes Immunosuppressive Premetastatic Niche Formation to Facilitate Breast Tumour Metastasis. J Pathol (2016) 239(4):484–95. doi: 10.1002/path.4747
114. Sun L, Suo C, Li ST, Zhang H, Gao P. Metabolic Reprogramming for Cancer Cells and Their Microenvironment: Beyond the Warburg Effect. Biochim Biophys Acta Rev Cancer (2018) 1870(1):51–66. doi: 10.1016/j.bbcan.2018.06.005
115. Campbell SL, Wellen KE. Metabolic Signaling to the Nucleus in Cancer. Mol Cell (2018) 71(3):398–408. doi: 10.1016/j.molcel.2018.07.015
116. Perego M, Tyurin VA, Tyurina YY, Yellets J, Nacarelli T, Lin C, et al. Reactivation of Dormant Tumor Cells by Modified Lipids Derived From Stress-Activated Neutrophils. Sci Transl Med (2020) 12(572):eabb5817. doi: 10.1126/scitranslmed.abb5817
117. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty Acid Transport Protein 2 Reprograms Neutrophils in Cancer. Nature (2019) 569(7754):73–8. doi: 10.1038/s41586-019-1118-2
118. Li P, Lu M, Shi J, Gong Z, Hua L, Li Q, et al. Lung Mesenchymal Cells Elicit Lipid Storage in Neutrophils That Fuel Breast Cancer Lung Metastasis. Nat Immunol (2020) 21(11):1444–55. doi: 10.1038/s41590-020-0783-5
119. Missaglia S, Coleman RA, Mordente A, Tavian D. Neutral Lipid Storage Diseases as Cellular Model to Study Lipid Droplet Function. Cells (2019) 8(2):187. doi: 10.3390/cells8020187
120. Nielsen TS, Jessen N, Jørgensen JO, Møller N, Lund S. Dissecting Adipose Tissue Lipolysis: Molecular Regulation and Implications for Metabolic Disease. J Mol Endocrinol (2014) 52(3):R199–222. doi: 10.1530/JME-13-0277
121. Ran S. The Role of TLR4 in Chemotherapy-Driven Metastasis. Cancer Res (2015) 75(12):2405–10. doi: 10.1158/0008-5472.CAN-14-3525
122. Butti R, Gunasekaran VP, Kumar TVS, Banerjee P, Kundu GC. Breast Cancer Stem Cells: Biology and Therapeutic Implications. Int J Biochem Cell Biol (2019) 107:38–52. doi: 10.1016/j.biocel.2018.12.001
123. Harney AS, Arwert EN, Entenberg D, Wang Y, Guo P, Qian BZ, et al. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discovery (2015) 5(9):932–43. doi: 10.1158/2159-8290.CD-15-0012
124. Karagiannis GS, Goswami S, Jones JG, Oktay MH, Condeelis JS. Signatures of Breast Cancer Metastasis at a Glance. J Cell Sci (2016) 129(9):1751–8. doi: 10.1242/jcs.183129
125. Attalla S, Taifour T, Bui T, Muller W. Insights From Transgenic Mouse Models of PyMT-Induced Breast Cancer: Recapitulating Human Breast Cancer Progression In Vivo. Oncogene (2021) 40(3):475–91. doi: 10.1038/s41388-020-01560-0
126. Karagiannis GS, Pastoriza JM, Wang Y, Harney AS, Entenberg D, Pignatelli J, et al. Neoadjuvant Chemotherapy Induces Breast Cancer Metastasis Through a TMEM-Mediated Mechanism. Sci Trans Med (2017) 9(397):eaan0026. doi: 10.1126/scitranslmed.aan0026
127. Chang YS, Jalgaonkar SP, Middleton JD, Hai T. Stress-Inducible Gene Atf3 in the Noncancer Host Cells Contributes to Chemotherapy-Exacerbated Breast Cancer Metastasis. Proc Natl Acad Sci USA (2017) 114(34):E7159–68. doi: 10.1073/pnas.1700455114
128. Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune Microenvironments in Solid Tumors: New Targets for Therapy. Genes Dev (2011) 25(24):2559–72. doi: 10.1101/gad.169029.111
129. Emmenegger U, Kerbel RS. Cancer: Chemotherapy Counteracted. Nature (2010) 468(7324):637–8. doi: 10.1038/468637a
130. Keklikoglou I, Cianciaruso C, Güç E, Squadrito ML, Spring LM, Tazzyman S, et al. Chemotherapy Elicits Pro-Metastatic Extracellular Vesicles in Breast Cancer Models. Nat Cell Biol (2019) 21(2):190–202. doi: 10.1038/s41556-018-0256-3
131. Guo Z, Guo A, Zhou C. Breast Cancer Stem Cell-Derived ANXA6-Containing Exosomes Sustain Paclitaxel Resistance and Cancer Aggressiveness in Breast Cancer. Front Cell Dev Biol (2021) 9:718721. doi: 10.3389/fcell.2021.718721
132. Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 Recruits Inflammatory Monocytes to Facilitate Breast-Tumour Metastasis. Nature (2011) 475(7355):222–5. doi: 10.1038/nature10138
133. Morrissey SM, Zhang F, Ding C, Montoya-Durango DE, Hu X, Yang C, et al. Tumor-Derived Exosomes Drive Immunosuppressive Macrophages in a Pre-Metastatic Niche Through Glycolytic Dominant Metabolic Reprogramming. Cell Metab (2021) 33(10):2040–2058.e2010. doi: 10.1016/j.cmet.2021.09.002
134. Cassetta L, Pollard JW. Targeting Macrophages: Therapeutic Approaches in Cancer. Nat Rev Drug Discovery (2018) 17(12):887–904. doi: 10.1038/nrd.2018.169
135. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P, et al. Tumour-Associated Macrophages as Treatment Targets in Oncology. Nat Rev Clin Oncol (2017) 14(7):399–416. doi: 10.1038/nrclinonc.2016.217
136. Wu X, Zhang X, Yu L, Zhang C, Ye L, Ren D, et al. Zinc Finger Protein 367 Promotes Metastasis by Inhibiting the Hippo Pathway in Breast Cancer. Oncogene (2020) 39(12):2568–82. doi: 10.1038/s41388-020-1166-y
137. Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, et al. Pre-Metastatic Niches: Organ-Specific Homes for Metastases. Nat Rev Cancer (2017) 17(5):302–17. doi: 10.1038/nrc.2017.6
138. West XZ, Malinin NL, Merkulova AA, Tischenko M, Kerr BA, Borden EC, et al. Oxidative Stress Induces Angiogenesis by Activating TLR2 With Novel Endogenous Ligands. Nature (2010) 467(7318):972–6. doi: 10.1038/nature09421
139. Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Vascular Permeability, Vascular Hyperpermeability and Angiogenesis. Angiogenesis (2008) 11(2):109–19. doi: 10.1007/s10456-008-9099-z
Keywords: breast cancer, lung metastasis, pulmonary microenvironment, inflammation, angiogenesis, extracellular matrix remodeling, neutrophils
Citation: Wu M, Liang Y and Zhang X (2022) Changes in Pulmonary Microenvironment Aids Lung Metastasis of Breast Cancer. Front. Oncol. 12:860932. doi: 10.3389/fonc.2022.860932
Received: 24 January 2022; Accepted: 25 April 2022;
Published: 26 May 2022.
Edited by:
Karen Elizabeth Nava-Castro, National Autonomous University of Mexico, MexicoReviewed by:
Alicia Bolt, University of New Mexico, United StatesCopyright © 2022 Wu, Liang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Zhang, emhhbmd4NDVAbWFpbDMuc3lzdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.