 Bryn Golesworthy1,2†
Bryn Golesworthy1,2† Yifan Wang1,2,3†
Yifan Wang1,2,3† Amanda Tanti1,2
Amanda Tanti1,2 Alain Pacis2,4
Alain Pacis2,4 Joan Miguel Romero1,2Adeline Cuggia1,2Celine Domecq1,2Guillaume Bourdel1,2Robert E. Denroche5Gun Ho Jang5
Joan Miguel Romero1,2Adeline Cuggia1,2Celine Domecq1,2Guillaume Bourdel1,2Robert E. Denroche5Gun Ho Jang5 Robert C. Grant5Ayelet Borgida6Barbara T. Grünwald7Anna Dodd7Julie M. Wilson5
Robert C. Grant5Ayelet Borgida6Barbara T. Grünwald7Anna Dodd7Julie M. Wilson5 Guillaume Bourque4,8Grainne M. O’Kane5,7
Guillaume Bourque4,8Grainne M. O’Kane5,7 Sandra E. Fischer5,9Chelsea Maedler Kron1,10
Sandra E. Fischer5,9Chelsea Maedler Kron1,10 Pierre-Olivier Fiset1,10Atilla Omeroglu1,10William D. Foulkes1,8Steven Gallinger5,6,7Marie-Christine Guiot2,10
Pierre-Olivier Fiset1,10Atilla Omeroglu1,10William D. Foulkes1,8Steven Gallinger5,6,7Marie-Christine Guiot2,10 Zu-Hua Gao1,10
Zu-Hua Gao1,10 George Zogopoulos1,2,3*
George Zogopoulos1,2,3*- 1The Research Institute of the McGill University Health Centre, Montreal, QC, Canada
- 2The Rosalind and Morris Goodman Cancer Institute of McGill University, Montreal, QC, Canada
- 3The Department of Surgery, McGill University, Montreal, QC, Canada
- 4The McGill Genome Center and Canadian Centre for Computational Genomics (C3G), Montreal, QC, Canada
- 5The Ontario Institute for Cancer Research, Toronto, ON, Canada
- 6The Lunenfeld Tanenbaum Research Institute, Mount Sinai Hospital, Toronto, ON, Canada
- 7The Wallace McCain Centre for Pancreatic Cancer, Princess Margaret Cancer Centre, University Health Network, Toronto, ON, Canada
- 8The Department of Human Genetics, McGill University, Montreal, QC, Canada
- 9The Division of Pathology, University Health Network, Toronto, ON, Canada
- 10The Department of Pathology, McGill University, Montreal, QC, Canada
The immune contexture of pancreatic ductal adenocarcinoma (PDAC) is generally immunosuppressive. A role for immune checkpoint inhibitors (ICIs) in PDAC has only been demonstrated for the rare and hypermutated mismatch repair (MMR) deficient (MMR-d) subtype. Homologous recombination repair (HR) deficient (HR-d) PDAC is more prevalent and may encompass up to 20% of PDAC. Its genomic instability may promote a T-cell mediated anti-tumor response with therapeutic sensitivity to ICIs. To investigate the immunogenicity of HR-d PDAC, we used multiplex immunohistochemistry (IHC) to compare the density and spatial distribution of CD8+ cytotoxic T-cells, FOXP3+ regulatory T-cells (Tregs), and CD68+ tumor-associated macrophages (TAMs) in HR-d versus HR/MMR-intact PDAC. We also evaluated the IHC positivity of programmed death-ligand 1 (PD-L1) across the subgroups. 192 tumors were evaluated and classified as HR/MMR-intact (n=166), HR-d (n=25) or MMR-d (n=1) based on germline testing and tumor molecular hallmarks. Intra-tumoral CD8+ T-cell infiltration was higher in HR-d versus HR/MMR-intact PDAC (p<0.0001), while CD8+ T-cell densities in the peri-tumoral and stromal regions were similar in both groups. HR-d PDAC also displayed increased intra-tumoral FOXP3+ Tregs (p=0.049) and had a higher CD8+:FOXP3+ ratio (p=0.023). CD68+ TAM expression was similar in HR-d and HR/MMR-intact PDAC. Finally, 6 of the 25 HR-d cases showed a PD-L1 Combined Positive Score of >=1, whereas none of the HR/MMR-intact cases met this threshold (p<0.00001). These results provide immunohistochemical evidence for intra-tumoral CD8+ T-cell enrichment and PD-L1 positivity in HR-d PDAC, suggesting that HR-d PDAC may be amenable to ICI treatment strategies.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the seventh leading cause of cancer death worldwide and its incidence is rising yearly (1). It is one of the most lethal malignancies, with a 5-year survival rate of less than 10% (1). Nearly 80% of patients are diagnosed with incurable locally advanced or metastatic disease and are treated with systemic chemotherapy (2). However, the effectiveness of empiric chemotherapeutic regimens, such as FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan and oxaliplatin) and gemcitabine plus nab-paclitaxel, remains poor (2). The 20% of patients who undergo curative-intent resection and receive adjuvant therapy reach a median overall survival of only 22.8 to 54.4 months, which highlights the systemic behavior of PDAC even at its earliest stage (3–5).
While empiric chemotherapy regimens remain the backbone of systemic treatment for PDAC, these treatment strategies have not led to marked improvements in survival [2]. Thus, guided by biomarker-driven treatment breakthroughs in other difficult-to-treat cancers and facilitated by the identification of PDAC molecular subtypes, precision medicine and immunotherapy strategies have emerged. The most characterized molecular PDAC subtypes are based on 1) genomic tumor alterations driven either by germline predisposition or somatic oncogenic aberrations and 2) transcriptomic expression patterns (6). Associations of these subtypes with PDAC progression and treatment responses have demonstrated their potential clinical value (7–9).
Homologous recombination repair (HR) deficiency (HR-d) PDAC is an actionable molecular subtype. It is primarily driven by germline mutations in BRCA2, BRCA1 and PALB2, which are present in 5-10% of incident PDAC cases (2, 10–13). In addition, 7-10% of PDAC cases without germline mutations in HR-associated genes harbor molecular hallmarks of HR-d driven by somatic or epigenetic events (14). HR-d PDAC is sensitive to platinum-based cytotoxic regimens and poly(ADP-ribose) polymerase (PARP) inhibitors, which exploit the intrinsic deficiency of HR-d tumor cells to repair DNA double strand breaks with high-fidelity (15). In addition, the genomic instability of HR-d tumors, characterized by specific genomic alterations that include deletions with flanking microhomology and the COSMIC Signature 3 pattern of base-substitution mutations, may lead to increased neoantigens and a T cell-mediated anti-tumor response with therapeutic sensitivity to immune checkpoint inhibitors (ICIs) (16, 17). Durable treatment responses to ICIs have been demonstrated in cancers with T cell-inflamed phenotypes, but the ability of HR-d PDAC to induce anti-tumor immunity has not been established (18). Moreover, the PDAC tumor microenvironment is generally immunosuppressive and a role for ICIs in PDAC has only been demonstrated for the rare and hypermutated mismatch repair (MMR) deficient (MMR-d) subtype, which exhibits inherent immunogenicity (19, 20). Thus, to assess the potential actionability of HR-d PDAC using ICIs, we characterized the spatial distribution of immune cells relative to tumor cells in HR-d versus HR/MMR-intact PDAC.
Materials and Methodology
Patient Cohorts
Tumor specimens from patients with pathological diagnoses of PDAC from two case series were evaluated. The first series included 141 PDAC cases from the Quebec Pancreas Cancer Study [QPCS, NCT04104230 (21)]. This series consisted of 130 consecutive patients enrolled in the QPCS between April 2012 and September 2018, with available primary resected PDAC specimens for construction of a tissue microarray (TMA). To compensate for the lower incidence of HR-d and MMR-d compared to HR/MMR-intact PDAC, we also included cases with germline mutations in HR or MMR genes (n=11) that were enrolled in the QPCS following construction of the TMA. These included primary (n=8) and metastatic (n=3) PDAC specimens from cases enrolled between October 2018 and December 2020. The second series consisted of 115 cases from the PanCuRx Translational Research Initiative, which were represented on a previously constructed TMA (22). Patient demographics, clinical characteristics and survival outcomes for both series were extracted from prospectively maintained study databases. Overall survival was calculated from the date of radiological diagnosis until death or censor date. Clinical staging was based on the 8th edition of the American Joint Committee on Cancer.
Construction of Tissue Microarrays
TMAs for the QPCS cases were created using an automated tissue microarrayer (TMA Grand Master, 3DHistech, RRID: SCR_021257). Representative tumor regions were identified on hematoxylin and eosin (H&E) stained slides by a board-certified pathologist (A.O., Z.H.G.) and marked on its corresponding formalin-fixed and paraffin-embedded (FFPE) block. Each case (n=130) was represented on a TMA by three 1.5-mm tumor cores. The PanCuRx TMA was previously constructed and included 115 cases that were each represented by at least two 1.5-mm cores (22).
Immunostaining and Spatial Analysis of Tumor Infiltrating Immune Cells
TMA and individual patient blocks were cut at a 4-micrometer (μm) thickness for immunohistochemistry (IHC). Multiplex chromogenic IHC was performed using the Discovery Ultra Autostaining Platform (Ventana Medical Systems, RRID: SCR_021254), with chromogenic detection kits from Ventana Medical Systems (No. 760-247, teal; No. 760-229, purple; No. 760-500, DAB, RRID: AB_2753116; No. 760-250, yellow; No. 760-271, green). QPCS and PanCuRx slides were stained for CD8 (Ventana, 790-4460, RRID: AB_2335985), Pan-cytokeratin (PanCK; Ventana, 760-2135, RRID: AB_2810237), Forkhead box P3 (FOXP3; 1:200, Abcam, ab20034, RRID: AB_445284), and programmed death-ligand 1 (PD-L1; E1L3N clone, 1:100, Cell Signaling, 13684S, RRID: AB_2687655) in combination with the DISCOVERY Amp HQ kit (Ventana, 760-4602). The QPCS series was also stained for CD68 (1:100, Abcam, ab125212, RRID: AB_10975465). Staining specificity was confirmed by board-certified pathologists (M.C.G., P-O.F.).
Immunostained slides were scanned using the Aperio AT2 ScanScope (Leica Biosystems, RRID: SCR_021256) at a 20x magnification. We trained a classifier algorithm on the HALO Image Analysis software (Indica Labs; RRID: SCR_018350) to use PanCK staining for assignment of tumor cell clusters. Regions of necrosis, blood vessels, acinar cells and islet cells were excluded. Tumor and immune cells were counted using the Multiplex-IHC v.3.0.4 package on HALO and averaged across replicate tumor cores for each case. Immune cell densities were calculated by normalizing immune cell counts to the total area of tumor clusters (mm2). For log10 transformation, cases with immune cell counts of zero were assigned a value corresponding to 90% of the lowest non-zero immune count in the cases evaluated.
To distinguish between immune cells infiltrating the tumor versus those encroaching the tumor perimeter, immune cells were classified as intra-tumoral if they were located within 10 μm from the edge of a tumor cell (Figure 1A). Immune cells were considered peri-tumoral if they were located between 10 μm and 50 μm from the tumor cell cluster perimeter. Immune cells that were more than 50 μm away from a tumor cell cluster perimeter were categorized as stromal. The CD8+:FOXP3+ ratio was calculated by dividing the total number of CD8+ cytotoxic T-lymphocytes by the total number of FOXP3+ regulatory T-lymphocytes (Tregs) in each core, and averaging across tumor replicates. The Combined Positive Score (CPS) was calculated for each core as previously described (23) and averaged across tumor replicates. A CPS of ≥1 was considered positive. The HALO analysis for PD-L1 staining was validated with manual scoring by a board-certified pathologist (M.C.G.).
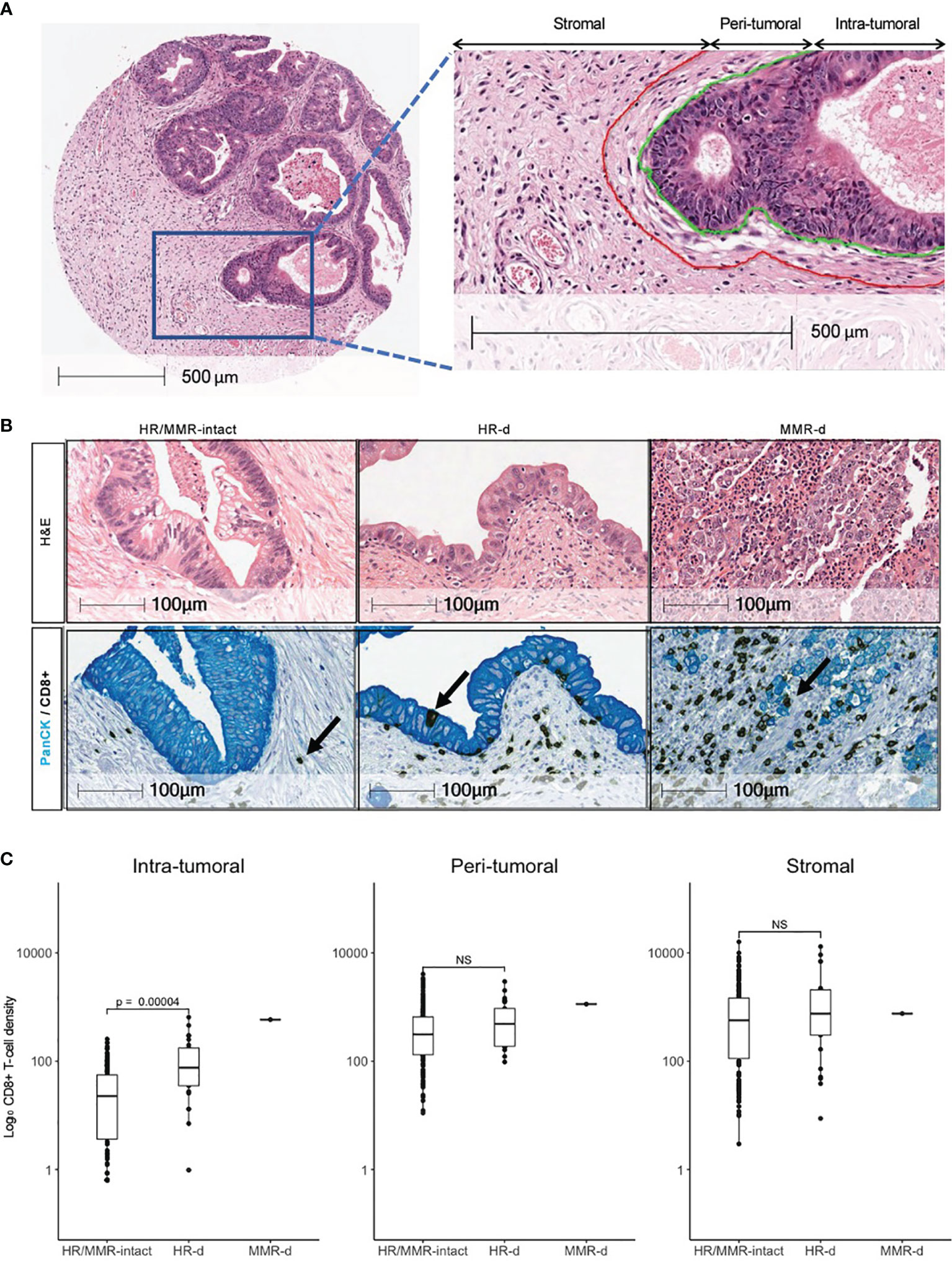
Figure 1 Distribution of CD8+ T-cells in PDAC. (A) Definitions of intra- tumoral, peri- tumoral and stromal regions. (B) Representative H&E images for HR/MMR-intact, HR-d and MMR-d PDAC with corresponding immunostaining for CD8 (brown) and Pan-cytokeratin (PanCK, teal). Black arrows show examples of CD8+ staining. (C) Comparison of CD8+ T-cell densities in HR/MMR-intact versus HR-d PDAC across the three tumor regions. The MMR-d case is shown as a reference for an immunogenic PDAC. NS, not significant.
Identification of HR-d and MMR-d Cases
Across the two series, 192 cases (QPCS n=114; PanCuRx n=78) were evaluable (Table 1 and Supplementary Table 1). These cases were classified into HR/MMR-intact, HR-d or MMR-d subgroups and their immune tumor microenvironment was compared (Supplementary Tables 2, 3). To identify HR-d cases in the QPCS series, we performed germline testing using lymphocyte DNA for BRCA1, BRCA2, and PALB2 mutations. There were 52 cases previously tested by whole genome sequencing (WGS, n=11), whole exome sequencing (WES, n=1), or targeted sequencing using genes panels that included BRCA1, BRCA2, and PALB2 (n=40, Supplementary Table 4) (13, 24, 25). The remaining 62 cases that had not undergone germline genetic testing were evaluated using the INVITAE Multi-Cancer gene panel (Supplementary Table 4). Germline mutations for the PanCuRx series were abstracted from previously reported WGS of lymphocyte DNA (22).

Table 1 Clinical characteristics of the 192 evaluable PDAC cases.
For cases that had undergone tumor whole genome sequencing (8, 17, 22), we calculated the HRDetect score using previously described methodology (26). Assignment of cases to the HR-d subgroup was based on the presence of a germline BRCA1, BRCA2 or PALB2 mutation and, if available, an HRDetect score of ≥0.9. If an HRDetect score was unavailable, cases with germline mutations were kept in the HR-d subgroup. Cases that carried a germline mutation, but did not meet the HRDetect threshold, were considered HR-intact. An HRDetect score of >0.7 for a low tumor cellularity case (303.001) was accepted to signify HD-d. In the absence of a germline HR-gene mutation, cases that had an HRDetect score >0.9 and evidence of somatic HR-gene inactivation were classified as HR-d.
We surveyed the QPCS and PanCuRx case series for germline MMR-gene mutations to identify potential MMR-d cases (Supplementary Tables 2, 3). However, assignment to the MMR-d subgroup was based on IHC for MMR protein deficiency (i.e., MLH1, MSH2, MSH6, PMS2) or an MSIsensor score ≥20 (https://github.com/niu-lab/msisensor2). For the QPCS series, IHC for MLH1 (G168-15, Biocare Medical, RRID: AB_1059376), MSH2 (G219-1129, Cell Marque, RRID: AB_1160591), MSH6 (EPR3945, Abcam, RRID: AB_2144959) and PMS2 (EPR3947, Cell Marque) was performed using a BenchMark ULTRA IHC Staining Module (Roche Diagnostics) with the OptiView DAB IHC Detection Kit. IHC staining was analyzed using the ImageScope software. Cases were considered MMR-intact if tumor cells displayed nuclear staining of all 4 MMR proteins. Cases were classified as MMR-d if tumor cells had complete loss of nuclear staining in one or more MMR proteins, with retained nuclear staining in adjacent stroma. Absence of MMR protein expression on a TMA core was confirmed by staining a whole section slide. MMR-d classification for the PanCuRx case series was based on previously reported IHC for MLH1, MSH2, MSH6 and PMS2 or MSIsensor scores (19, 22). Cases were assigned to the HR/MMR-intact subgroup if they did not meet criteria for HR-d or MMR-d subclassification or if they could not be evaluated for tumor molecular hallmarks due to biospecimen availability.
Statistical Analyses
All statistical analyses were performed using R Software (version 4.0.4, R Foundation for Statistical Computing). Continuous variables were expressed as mean ± standard deviation (SD), and differences were compared using the Wilcoxon test. Fisher’s Exact Test was used to compare the proportion of cases in the HR/MMR-intact versus HR-d groups meeting the PD-L1 CPS threshold of ≥1. Overall survival was estimated using the Kaplan–Meier method and compared between groups using a log-rank test.
Study Approval
All participants provided written informed consent. The study was conducted in accordance with the principles of the Declaration of Helsinki. The McGill University and the McGill University Health Centre (MUHC) Institutional Review Boards (#A02-M118-11A, #2018-3171, #2018-4139) approved the QPCS study, and the Institutional Review Board of the University Health Network (#15-9596) provided approval for the PanCuRx case series.
Results
Clinical Characteristics
Across the QPCS and PanCuRx case series, 192 PDAC cases were characterized, which included 166, 25, and 1 HR/MMR-intact, HR-d, and MMR-d cases, respectively (Table 1). The germline mutations and tumor genomic features of the HR-d and MMR-d cases are summarized in Table 2, and their clinical characteristics at tumor biospecimen acquisitions are detailed in Table 3. The HR-d group demonstrated a longer median overall survival (OS; 29.1 months versus 19.9 months, p=0.0073; Supplementary Figure 1). Notably, the HR-d group included a greater number of patients diagnosed with stage III/IV disease compared to the HR/MMR-intact group (52.0% versus 4.2%, p<0.0001).
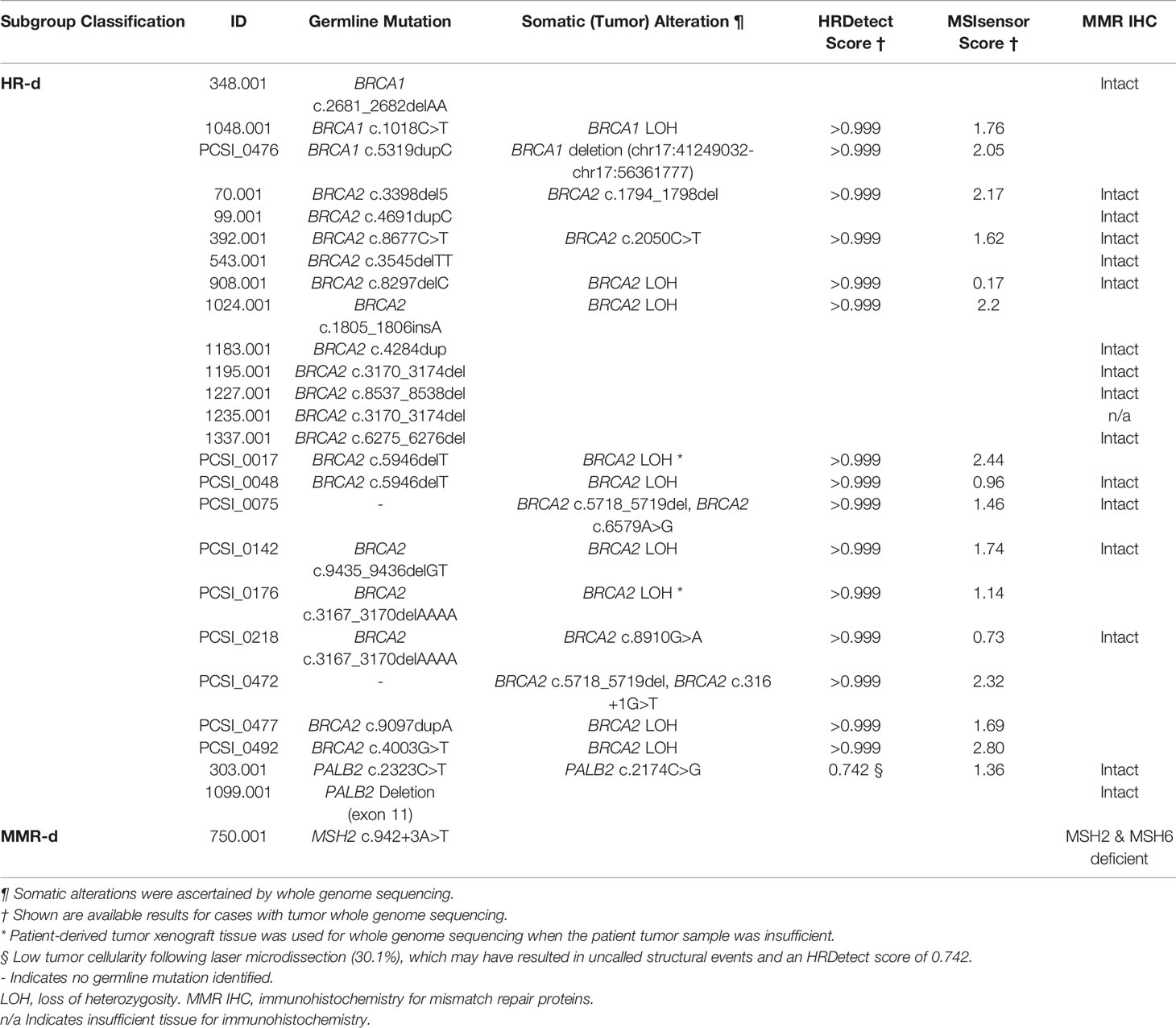
Table 2 Germline mutations and tumor genomic features of the HR-d and MMR-d cases.

Table 3 Clinical characteristics of the HR-d and MMR-d cases at tissue acquisition.
Increased Intra-tumoral Density of CD8+ T-cells and FOXP3+ Tregs in HR-d PDAC
HR-d tumors had a higher CD8+ T-cell intra-tumoral density compared to the HR/MMR-intact group (131.1 ± 154.9 cells/mm2 versus 40.5 ± 50.9 cells/mm2, p<0.0001; Figure 1). However, there was no difference in CD8+ T-cell infiltration in the peri-tumoral or stromal regions between the two groups. The CD8+ T-cell landscape for the MMR-d case is shown in parallel as a reference for a PDAC microenvironment responsive to immunotherapy. Following resection of the primary and adjuvant therapy with gemcitabine/capecitabine, this patient (750.001) developed a mesenteric recurrence, which responded to pembrolizumab (Supplementary Figure 2).
We subsequently evaluated the landscape of FOXP3+ Tregs and CD68+ tumor-associated macrophages (TAMs) in HR/MMR-intact versus HR-d tumors (Figure 2). FOXP3+ Tregs density was higher in the intra-tumoral region of HR-d versus HR/MMR-intact tumors (25.5 ± 27.3 cells/mm2 versus 13.6 ± 13.4 cells/mm2, p=0.049), while there was no difference in FOXP3+ staining in the peri-tumoral or stromal regions between the two groups. Moreover, the ratio of CD8+ to FOXP3+ cells was higher in HR-d versus HR/MMR-intact cases (23.9 ± 52.7 versus 9.8 ± 23.8, p=0.023). Similarly, the CD8+ to FOXP3+ ratio in the MMR-d tumor was also elevated. CD68+ TAM population densities were comparable across the three spatial compartments in HR-d and HR/MMR-intact PDAC.
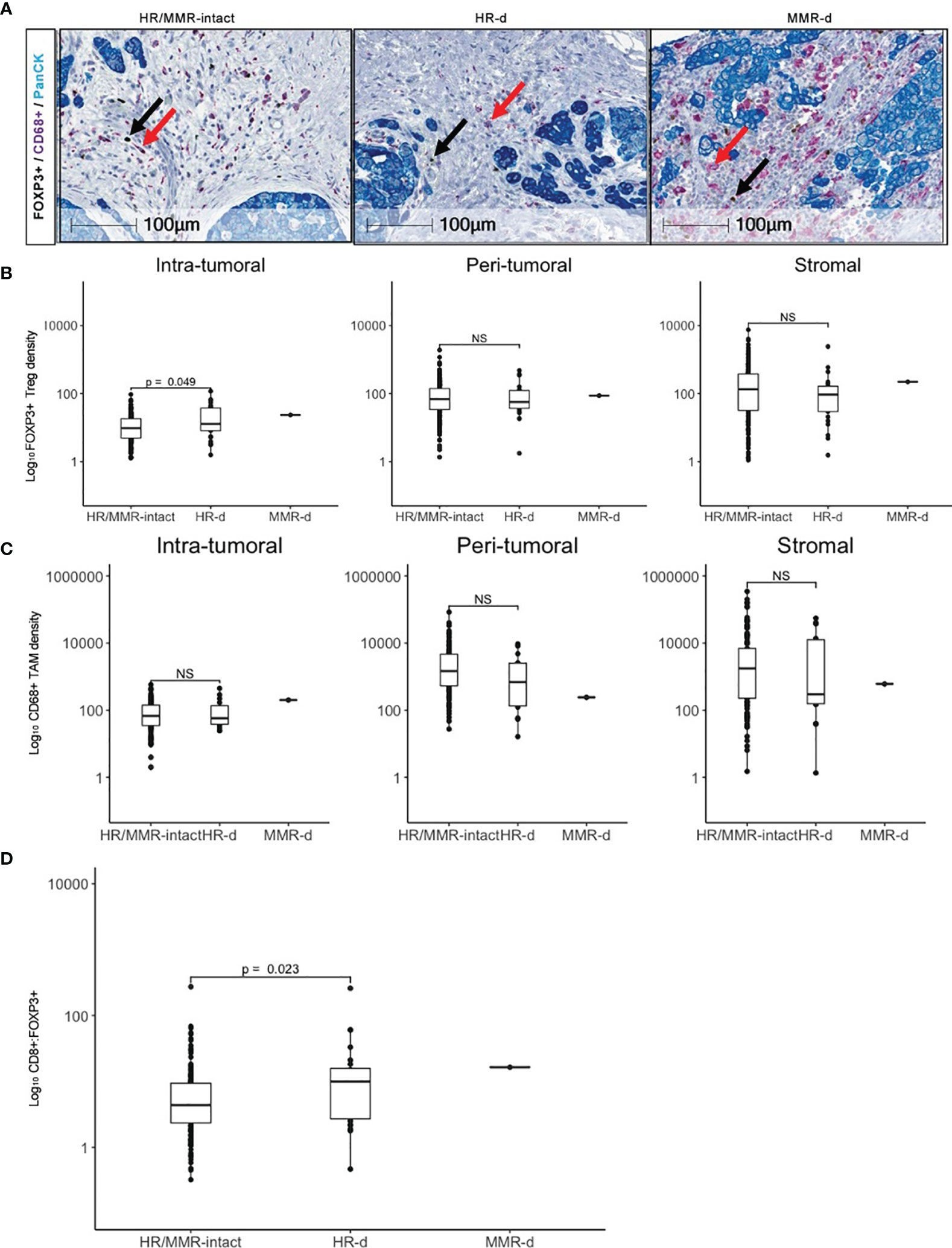
Figure 2 FOXP3+ Treg and CD68+ TAM infiltration in PDAC. (A) Representative FOXP3 (brown), CD68 (purple) and PanCK (teal) immunostaining for HR/MMR-intact, HR-d and MMR-d PDAC. Black and red arrows show examples of FOXP3+ and CD68+ staining, respectively. (B, C) Comparison of FOXP3+ Treg (B) and CD68+ TAM (C) densities in HR/MMR-intact versus HR-d PDAC across the intra-tumoral, peri- tumoral and stromal regions. (D) Comparison of overall CD8+:FOXP3+ ratios between HR/MMR-intact versus HR-d PDAC. The MMR-d case is shown as a reference. NS, not significant.
Increased PD-L1 Expression in HR-d PDAC
We observed higher PD-L1 expression in the HR-d versus the HR/MMR-intact group (5.1 ± 11.9 versus 0.03 ± 0.1, p=0.0025). Six of the 25 HR-d tumors met the PD-L1 Combined Positive Score (CPS) threshold of ≥1, whereas none of the 163 evaluable HR/MMR-intact tumors reached the positivity threshold (p<0.00001; Figures 3A, B). The 6 HR-d cases with a CPS score ≥1 consisted of 4 cases with treatment-naïve primary PDAC biopsies (1024.001, 1183.001, 1235.001, 1337.001) and 2 metastatic biopsies (543.001, liver metastasis; 1099.001, peritoneal metastasis). Case 543.001 was treatment naïve at the time of biopsy, whereas case 1099.001 had undergone a course of neoadjuvant FOLFIRINOX for stage III disease. The biopsied metastatic peritoneal nodule was identified at the time of surgical exploration and the curative-intent resection was aborted following the intra-operative finding of metastatic disease. Importantly, the MMR-d case with treatment sensitivity to pembrolizumab also met the CPS threshold (Figure 3B, Supplementary Figure 2).
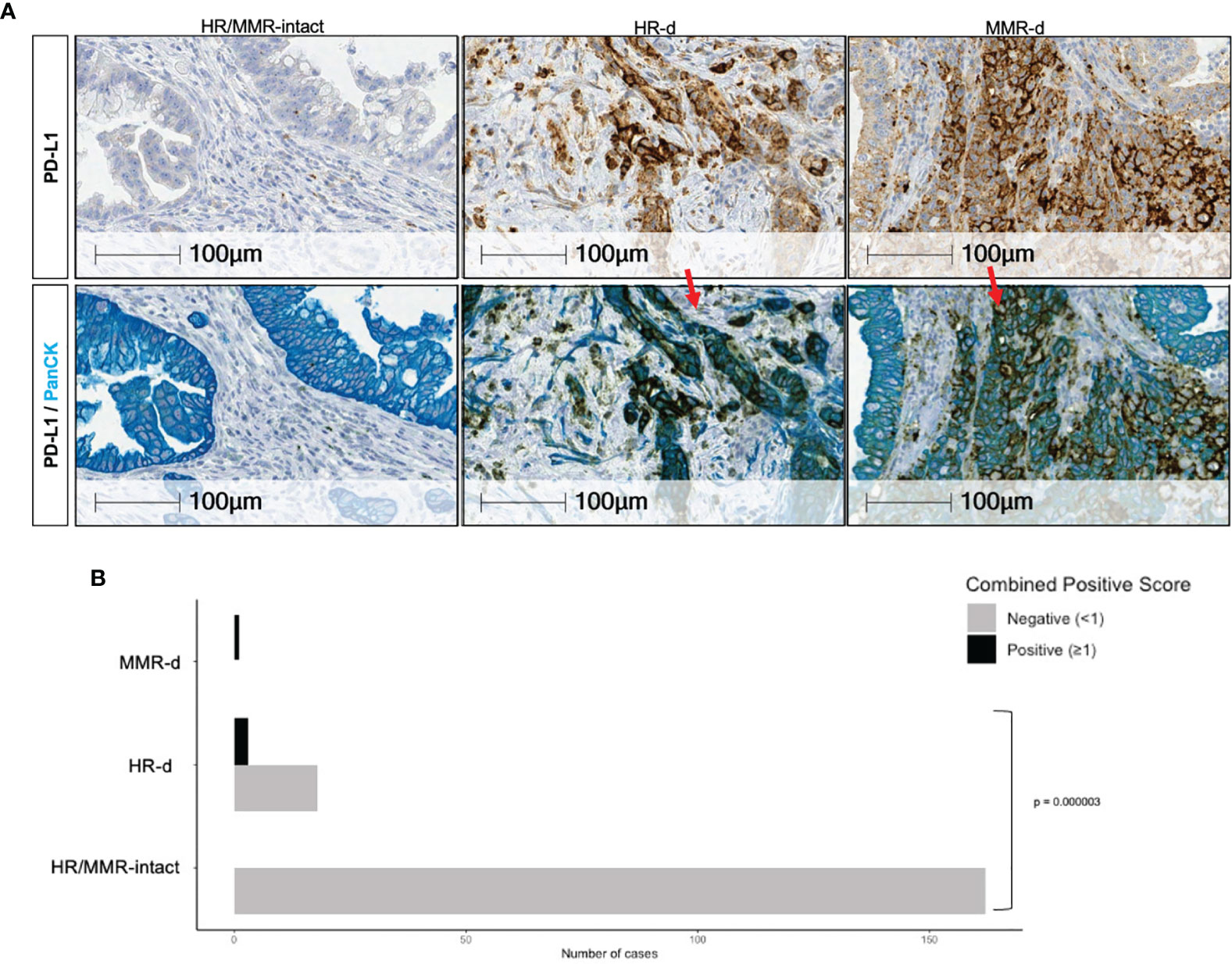
Figure 3 PD-L1 positivity in PDAC. (A) Representative PD-L1 immunostaining for HR/MMR- intact, HR-d and MMR-d PDAC. The top row shows tumors stained with PD-L1 (brown), while the bottom row shows the same tumor sections stained with PanCK (teal) following PD-L1 staining (brown). Red arrows in top panel show examples of PD-L1 staining. (B) Comparison of the proportion of cases in the HR/MMR-intact versus HR-d groups meeting the Combined Positivity Score (CPS) threshold of ≥ 1. Six of 25 HR-d cases had a CPS of ≥1, whereas none of the 163 evaluable HR/MMR-intact cases met the PD-L1 positivity threshold of ≥1. The MMR-d case scored >1.
Discussion
Genomic and transcriptomic profiling of PDAC has identified distinct molecular subclasses, such as HR-d (6). These advances have led to opportunities for targeted therapies and treatment stratification according to subclass assignment. Treatment responses to platinum-based therapies and PARP inhibitors have been demonstrated for HR-d PDAC (14, 27, 28). However, these therapies rarely lead to complete tumor responses and their durability is limited (17, 27). Therefore, there remains a clinical need to improve the current treatment strategies for HR-d PDAC. To this end, the genomic instability associated with HR-d PDAC may result in a tumor microenvironment amenable to ICI therapies. In the present study, we show increased intra-tumoral CD8+ T-lymphocyte infiltration and PD-L1 positivity by immunohistochemistry, corroborating previously reported transcriptomic evidence for T-cell inflammation in HR-d PDAC (22, 29). Although evaluation of the composition of cytotoxic, memory and exhausted intra-tumoral CD8+ T-lymphocyte populations in HR-d is subject to further investigation, there is evidence that infiltrating CD8+ T-lymphocytes are activated in HR-d. Indeed, Connor et al. showed that the cytolytic activity of infiltrating CD8+ T-lymphocytes, as measured by expression of granzyme A and perforin, and expression of co-regulatory genes, including cytotoxic T-lymphocyte antigen 4, programmed cell death 1 and indolamine 2,3-dioxygenase 1, was increased in HR-d and MMR-d PDAC (22). Moreover, these gene expression changes correlated with higher frequency of somatic mutations and tumor-specific neoantigens.
ICI therapies promote an anti-tumor immune response by disrupting signals that inhibit T-cell cytolytic activity (30). Cancers that exhibit immunogenicity, such as melanoma, have robust responses to ICI regimens with striking tumor regression and long-term survival (30). CD8+T-cell tumor infiltration has been associated with such potent and durable ICI treatment responses (31). Importantly, the proximity of cytolytic CD8 T-cells to tumor cell bed is predictive of ICI efficacy (32). However, while CD8+T-lymphocytes are adjacent to tumor cells in the microenvironment of melanoma tumors, the CD8+T-lymphocyte infiltrate in PDAC is primarily restricted to the stroma (33). Despite such observations suggesting that PDAC does not generally evoke an anti-tumor immune response, genomic and transcriptomic characterization of PDAC and its microenvironment has alluded to the existence of PDAC subclasses harboring immunogenicity (34, 35). The subclass prototype for PDAC with immunogenicity and responsiveness to ICIs is MMR-d, which exhibits elevated levels of tumor mutation burden (TMB) and tumor infiltrating CD8 T-lymphocytes compared to MMR-intact PDAC (19, 20, 36). Similarly, if the genomic instability that underlies HR-d PDAC leads to increased levels of neoantigens and tumor immunogenicity, the immune cell landscape of HR-d PDAC should also be distinguishable from the typical immune cold PDAC microenvironment. To this end, our results demonstrating elevated levels of intra-tumoral CD8+ T-cells in HR-d compared to HR/MMR-intact PDAC suggest that HR-d PDAC harbors a microenvironment with an inherent anti-tumor immune response. In support of the hypothesis that the genomic instability of HR-d tumors results in increased immunogenicity, a retrospective study of melanoma patients treated with ICI therapy found that tumors that regressed were enriched for BRCA2 loss of function mutations and harbored a higher TMB (37).
We also evaluated the tumor microenvironment topography of FOXP3+ Tregs and CD68+ TAMs, since their immunosuppressive properties counterpoise the anti-tumor effect of cytotoxic CD8+ T-lymphocytes (38, 39). Tregs and TAMs may lower ICI efficacy and contribute to ICI resistance (40). Moreover, elevated levels of tumor infiltrating Tregs and TAMs correlate with poor outcomes in PDAC and other malignancies (31). We detected Tregs and TAMs in both HR-d and HR/MMR-intact cases, but we only observed a difference in the intra-tumoral density of Tregs between the two groups. The increase in intra-tumoral Tregs suggests an immunosuppressive counterbalance response to the anti-tumoral effect of the CD8+ T-cell infiltration. The MMR-d case with a durable response to pembrolizumab exhibited comparable levels of Tregs and TAMs in its intra-tumoral, peri-tumoral and stromal regions.
HR-d cases had a significantly higher CD8+:FOXP3+ ratio compared to HR/MMR-intact cases. The CD8+:FOXP3+ ratio may be a more functional measure of anti-tumor immunity and a better indicator of ICI responsiveness compared to CD8+ and FOXP3+ measurements alone (41). As the proportion of CD8+ to FOXP3+ rises, escape of CD8+ cytotoxic activity is facilitated with the dilution of Tregs and their inhibitory effects. The resultant milieu may also enhance the efficacy of ICI therapies. However, the CD8+:FOXP3+ threshold that signifies ICI responsiveness remains to be defined.
Although Tregs are considered to be pro-tumoral and have been associated with poor clinical outcomes in PDAC (31, 41), their role may be more complex. Depletion of Tregs in a transgenic mouse model of exocrine pancreatic cancer did not relieve the Treg immunosuppressive effects, but instead led to tumor progression (42). TAMs are also peculiar immune cells that exhibit functional plasticity with their phenotypic polarization governed by environmental signals (40). The functional continuum of TAMs ranges from pro-tumor to anti-tumor activity. In PDAC, immunosuppressive polarization of TAMs has been proposed, which is likely promoted by the predominantly hypoxic PDAC microenvironment (39). Although the exact contributions of Tregs and TAMs to the pro-tumor versus anti-tumor counterforces in the PDAC microenvironment are not fully understood, their roles are likely largely immunosuppressive considering their abundance across all PDAC and the generally immune cold PDAC microenvironment (31, 40, 43). Moreover, their prevalence in MMR-d may underlie the lower efficacy of ICIs in MMR-d PDAC compared to other MMR-d cancers (44). Therefore, ICI therapies PDAC subtypes with immunogenicity, including MMR-d, may be enhanced with the addition of agents that target suppressive immune cells such as TAMs (45).
Expression of PD-L1 by tumor or tumor-infiltrating immune cells has emerged as a clinical biomarker for anti-tumor immunity that may be enhanced with ICI therapy (23, 46). However, PD-L1 status is not a predictive biomarker of ICI sensitivity across all cancer types. Indeed, Davis et al. reviewed 45 FDA ICI approvals and found that PD-L1 was only predictive in 29% of cases, with the remainder either not tested or not predictive (47). Despite these limitations, in addition to MMR testing, PD-L1 expression currently remains the only feasible clinical assay for ICI treatment selection across cancer types (30). As such, we compared PD-L1 positivity by IHC in HR-d versus HR/MMR-intact PDAC, and found increased PD-L1 immunostaining in the HR-d group. Since guidelines for interpreting PD-L1 staining in PDAC have not been established, we applied the Combined Positive Score (CPS) that is recognized as a clinically relevant scoring method of PD-L1 positivity in solid tumors (23, 48). Although tumors that do not meet the CPS positivity threshold of ≥1 may respond to ICIs, tumors that reach the CPS cutoff have a higher likelihood of responding effectively to anti-PD-1 therapy. Strikingly, 6 of 25 HR-d cases and the MMR-d case met the ≥1 CPS threshold, while none of the 163 evaluable HR/MMR-intact cases reached the benchmark. These observations support a role for anti-PD-1 therapy in HR-d PDAC. Moreover, considering the heterogeneity of treatment responses to platinums and PARP inhibitors across HR-d PDAC (14, 17), sensitivity to anti-PD-1 may also be variable in HR-d PDAC and may be correlated with PD-L1 positivity. Finally, a limitation of this research registry-based study is the inclusion of cases that were treatment naïve at the time of tissue acquisition as well as cases following pre-treatment with chemo- and/or radiation therapy that may have altered their tumor microenvironment (49).
In summary, we demonstrate that HR-d PDAC exhibits a distinguishable tumor microenvironment with enhanced immunogenicity and PD-L1 positivity compared to HR/MMR-intact PDAC. These findings provide immunohistochemical correlation of the previously reported genomic and transcriptomic results classifying HR-d PDAC as a molecular subtype with inherent immunogenicity, and provide motivation for clinical trials to evaluate the efficacy of immunotherapy regimens in HR-d PDAC.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The study was conducted in accordance with the principles of the Declaration of Helsinki. The McGill University and the McGill University Health Centre (MUHC) Institutional Review Boards (#A02-M118-11A, #2018-3171, #2018-4139) approved the QPCS study, and the Institutional Review Board of the University Health Network (#15-9596) provided approval for the PanCuRx case series. All patients provided their written informed consent to participate in this study.
Author Contributions
BGo: Conceptualization, Methodology, Software, Validation, Formal Analysis, Investigation, Data Curation, Writing – Original Draft, Writing – Review & Editing, Visualization YW: Conceptualization, Methodology, Validation, Investigation, Data Curation, Writing – Original Draft, Writing – Review & Editing, Visualization AT: Methodology, Formal Analysis, Investigation, Software, Writing – Review & Editing AP: Software, Data Curation, Writing – Review & Editing JR: Software, Writing – Review & Editing AC: Data Curation, Resources, Writing – Review & Editing CD: Data Curation, Resources, Writing – Review & Editing GBourd: Data Curation, Resources, Writing – Review & Editing RD: Software, Data Curation, Writing – Review & Editing GJ: Software, Data Curation, Writing – Review & Editing RG: Software, Data Curation, Writing – Review & Editing. AB Data Curation, Resources, Writing – Review & Editing BG Methodology, Validation, Resources, Writing – Review & Editing. AD: Methodology, Validation, Resources, Writing – Review & Editing JW: Methodology, Validation, Resources, Writing – Review & Editing GBourq: Software, Data Curation, Writing – Review & Editing GO’K: Methodology, Validation, Resources, Writing – Review & Editing SF: Methodology, Validation, Resources, Writing – Review & Editing CM-K: Resources, Validation, Writing – Review & Editing P-OF: Resources, Validation, Writing – Review & Editing AO: Resources, Validation, Writing – Review & Editing WF: Resources, Validation, Writing – Review & Editing SG: Resources, Conceptualization, Writing – Review & Editing M-CG: Resources, Validation, Writing – Review & Editing Z-HG: Resources, Validation, Writing – Review & Editing and GZ: Conceptualization, Resources, Writing – Original Draft, Writing – Review & Editing, Supervision, Project Administration.
Funding
This work was supported by the Terry Fox Research Institute (project no. 1078), the Pancreatic Cancer Canada Foundation, the Quebec Cancer Consortium with funding from the Ministère de l’Économie et de l’Innovation du Québec through the Fonds d’accélération des collaborations en santé, and the PanCuRx Translational Research Initiative with funding by the Government of Ontario through the Ontario Institute for Cancer Research. Germline testing for the QPCS case series was partially subsidized by Invitae (San Francisco, CA). BGo was supported by a Canderel Graduate Studentship and the Cedars Cancer Institute Fellowship. YW was supported by a Vanier Canada Graduate Scholarship, the Fonds de recherche du Québec–Santé/Ministère de la Santé et des Services sociaux training program, and the McGill University Surgical-Scientist Program. JR was supported by the Fonds de recherche du Québec–Santé/Ministère de la Santé et des Services sociaux training program and the Hilton J. McKeown Stipend from McGill University. GZ is a clinical research scholar of the Fonds de recherche du Québec–Santé and recipient of the Michal & Renata Hornstein Career Award from McGill University.
Conflict of Interest
P-OF has received honoraria from EMD Serono and consultation fees from Amgen, Bristol Myers Squibb, AstraZeneca Canada, Hoffmann La Roche, Merck Canada, Pfizer Canada and Roche Canada.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would also like to acknowledge Cleber Moraes and Dongmei Zuo for their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.860767/full#supplementary-material
Supplementary Figure 1 | Kaplan-Meier survival curves for the HR-d and HR/MMR-intact groups.
Supplementary Figure 2 | MMR-d PDAC. (A) IHC of QPCS case 750.001 showing intact nuclear MLH1 and PMS2 staining and absent nuclear MSH2 and MSH6 staining (red arrow) with intact stromal MSH2 and MSH6 staining (blue arrow) to indicate MMR-d. (B) Computed tomography showing mesenteric recurrence (orange arrow) following surgical resection of the primary. (C) Computed tomography following 18 months of pembrolizumab treatment showing a decrease in the mesenteric recurrence (orange arrow), indicating partial treatment response.
References
1. Ferlay J EM, Lam F, Colombet M, Mery L, Piñeros M, Znaor A, et al. Global Cancer Observatory: Cancer Today. Lyon, France: International Agency for Research on Cancer. (2020). Available at: https://gco.iarc.fr/today.
2. Wang Y, Lakoma A, Zogopoulos G. Building Towards Precision Oncology for Pancreatic Cancer: Real-World Challenges and Opportunities. Genes (2020) 11(9):1098. doi: 10.3390/genes11091098
3. Oettle H, Neuhaus P, Hochhaus A, Hartmann JT, Gellert K, Ridwelski K, et al. Adjuvant Chemotherapy With Gemcitabine and Long-Term Outcomes Among Patients With Resected Pancreatic Cancer: The CONKO-001 Randomized Trial. JAMA (2013) 310(14):1473–81. doi: 10.1001/jama.2013.279201
4. Neoptolemos JP, Palmer DH, Ghaneh P, Psarelli EE, Valle JW, Halloran CM, et al. Comparison of Adjuvant Gemcitabine and Capecitabine With Gemcitabine Monotherapy in Patients With Resected Pancreatic Cancer (ESPAC-4): A Multicentre, Open-Label, Randomised, Phase 3 Trial. Lancet (2017) 389(10073):1011–24. doi: 10.1016/S0140-6736(16)32409-6
5. Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N Engl J Med (2018) 379(25):2395–406. doi: 10.1056/NEJMoa1809775
6. Zheng-Lin B, O'Reilly EM. Pancreatic Ductal Adenocarcinoma in the Era of Precision Medicine. Semin Oncol (2021) 48:19–33. doi: 10.1053/j.seminoncol.2021.01.005
7. Martinelli P, Carrillo-de Santa Pau E, Cox T, Sainz B Jr., Dusetti N, Greenhalf W, et al. GATA6 Regulates EMT and Tumour Dissemination, and is a Marker of Response to Adjuvant Chemotherapy in Pancreatic Cancer. Gut (2017) 66(9):1665–76. doi: 10.1136/gutjnl-2015-311256
8. Aung KL, Fischer SE, Denroche RE, Jang GH, Dodd A, Creighton S, et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results From the COMPASS Trial. Clin Cancer Res (2018) 24(6):1344–54. doi: 10.1158/1078-0432.CCR-17-2994
9. O'Kane GM, Grünwald BT, Jang GH, Masoomian M, Picardo S, Grant RC, et al. GATA6 Expression Distinguishes Classical and Basal-Like Subtypes in Advanced Pancreatic Cancer. Clin Cancer Res (2020) 26(18):4901–10. doi: 10.1158/1078-0432.CCR-19-3724
10. Grant RC, Selander I, Connor AA, Selvarajah S, Borgida A, Briollais L, et al. Prevalence of Germline Mutations in Cancer Predisposition Genes in Patients With Pancreatic Cancer. Gastroenterology (2015) 148(3):556–64. doi: 10.1053/j.gastro.2014.11.042
11. Holter S, Borgida A, Dodd A, Grant R, Semotiuk K, Hedley D, et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J Clin Oncol (2015) 33(28):3124–9. doi: 10.1200/JCO.2014.59.7401
12. Hu C, Hart SN, Polley EC, Gnanaolivu R, Shimelis H, Lee KY, et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA (2018) 319(23):2401–9. doi: 10.1001/jama.2018.6228
13. Smith AL, Wong C, Cuggia A, Borgida A, Holter S, Hall A, et al. Reflex Testing for Germline BRCA1, BRCA2, PALB2, and ATM Mutations in Pancreatic Cancer: Mutation Prevalence and Clinical Outcomes From Two Canadian Research Registries. JCO Precis Oncol (2018) 2):1–16. doi: 10.1200/PO.17.00098
14. Golan T, O'Kane GM, Denroche RE, Raitses-Gurevich M, Grant RC, Holter S, et al. Genomic Features and Classification of Homologous Recombination Deficient Pancreatic Ductal Adenocarcinoma. Gastroenterology (2021) 160:2119–32. doi: 10.1053/j.gastro.2021.01.220
15. Lord CJ, Ashworth A. The DNA Damage Response and Cancer Therapy. Nature (2012) 481(7381):287–94. doi: 10.1038/nature10760
16. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, et al. Landscape of Somatic Mutations in 560 Breast Cancer Whole-Genome Sequences. Nature (2016) 534(7605):47–54. doi: 10.1038/nature17676
17. Wang Y, Park JYP, Pacis A, Denroche RE, Jang GH, Zhang A, et al. A Preclinical Trial and Molecularly Annotated Patient Cohort Identify Predictive Biomarkers in Homologous Recombination-Deficient Pancreatic Cancer. Clin Cancer Res (2020) 26(20):5462–76. doi: 10.1158/1078-0432.CCR-20-1439
18. Maleki Vareki S. High and Low Mutational Burden Tumors Versus Immunologically Hot and Cold Tumors and Response to Immune Checkpoint Inhibitors. J Immunother Cancer (2018) 6(1):157. doi: 10.1186/s40425-018-0479-7
19. Grant RC, Denroche R, Jang GH, Nowak KM, Zhang A, Borgida A, et al. Clinical and Genomic Characterisation of Mismatch Repair Deficient Pancreatic Adenocarcinoma. Gut (2020) 70:1894–903. doi: 10.1136/gutjnl-2020-320730
20. Luchini C, Brosens LAA, Wood LD, Chatterjee D, Shin JI, Sciammarella C, et al. Comprehensive Characterisation of Pancreatic Ductal Adenocarcinoma With Microsatellite Instability: Histology, Molecular Pathology and Clinical Implications. Gut (2021) 70(1):148–56. doi: 10.1136/gutjnl-2020-320726
21. Smith AL, Bascunana C, Hall A, Salman A, Andrei AZ, Volenik A, et al. Establishing a Clinic-Based Pancreatic Cancer and Periampullary Tumour Research Registry in Quebec. Curr Oncol (2015) 22(2):113–21. doi: 10.3747/co.22.2300
22. Connor AA, Denroche RE, Jang GH, Timms L, Kalimuthu SN, Selander I, et al. Association of Distinct Mutational Signatures With Correlates of Increased Immune Activity in Pancreatic Ductal Adenocarcinoma. JAMA Oncol (2017) 3(6):774–83. doi: 10.1001/jamaoncol.2016.3916
23. Kulangara K, Zhang N, Corigliano E, Guerrero L, Waldroup S, Jaiswal D, et al. Clinical Utility of the Combined Positive Score for Programmed Death Ligand-1 Expression and the Approval of Pembrolizumab for Treatment of Gastric Cancer. Arch Pathol Lab Med (2018) 143(3):330–7. doi: 10.5858/arpa.2018-0043-OA
24. Hansford S, Kaurah P, Li-Chang H, Woo M, Senz J, Pinheiro H, et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol (2015) 1(1):23–32. doi: 10.1001/jamaoncol.2014.168
25. Wong C, Chen F, Alirezaie N, Wang Y, Cuggia A, Borgida A, et al. A Region-Based Gene Association Study Combined With a Leave-One-Out Sensitivity Analysis Identifies SMG1 as a Pancreatic Cancer Susceptibility Gene. PloS Genet (2019) 15(8):e1008344. doi: 10.1371/journal.pgen.1008344
26. Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a Predictor of BRCA1 and BRCA2 Deficiency Based on Mutational Signatures. Nat Med (2017) 23(4):517–25. doi: 10.1038/nm.4292
27. Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N Engl J Med (2019) 381(4):317–27. doi: 10.1056/NEJMoa1903387
28. O'Reilly EM, Lee JW, Zalupski M, Capanu M, Park J, Golan T, et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J Clin Oncol (2020) 38(13):1378–88. doi: 10.1200/JCO.19.02931
29. Samstein RM, Krishna C, Ma X, Pei X, Lee KW, Makarov V, et al. Mutations in BRCA1 and BRCA2 Differentially Affect the Tumor Microenvironment and Response to Checkpoint Blockade Immunotherapy. Nat Cancer (2021) 1(12):1188–203. doi: 10.1038/s43018-020-00139-8
30. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
31. Orhan A, Vogelsang RP, Andersen MB, Madsen MT, Hölmich ER, Raskov H, et al. The Prognostic Value of Tumour-Infiltrating Lymphocytes in Pancreatic Cancer: A Systematic Review and Meta-Analysis. Eur J Cancer (2020) 132:71–84. doi: 10.1016/j.ejca.2020.03.013
32. Ku BM, Kim Y, Lee KY, Kim S-Y, Sun J-M, Lee S-H, et al. Tumor Infiltrated Immune Cell Types Support Distinct Immune Checkpoint Inhibitor Outcomes in Patients With Advanced non-Small Cell Lung Cancer. Eur J Immunol (2021) 51(4):956–64. doi: 10.1002/eji.202048966
33. Blando J, Sharma A, Higa MG, Zhao H, Vence L, Yadav SS, et al. Comparison of Immune Infiltrates in Melanoma and Pancreatic Cancer Highlights VISTA as a Potential Target in Pancreatic Cancer. Proc Natl Acad Sci (2019) 116(5):1692–7. doi: 10.1073/pnas.1811067116
34. Romero JM, Grünwald B, Jang G-H, Bavi PP, Jhaveri A, Masoomian M, et al. A Four-Chemokine Signature Is Associated With a T-Cell–Inflamed Phenotype in Primary and Metastatic Pancreatic Cancer. Clin Cancer Res (2020) 26(8):1997–2010. doi: 10.1158/1078-0432.CCR-19-2803
35. Puleo F, Nicolle R, Blum Y, Cros J, Marisa L, Demetter P, et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology (2018) 155(6):1999–2013.e3. doi: 10.1053/j.gastro.2018.08.033
36. Fraune C, Burandt E, Simon R, Hube-Magg C, Makrypidi-Fraune G, Kluth M, et al. MMR Deficiency is Homogeneous in Pancreatic Carcinoma and Associated With High Density of Cd8-Positive Lymphocytes. Ann Surg Oncol (2020) 27(10):3997–4006. doi: 10.1245/s10434-020-08209-y
37. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell (2016) 165(1):35–44. doi: 10.1016/j.cell.2016.02.065
38. Sahin IH, Askan G, Hu ZI, O'Reilly EM. Immunotherapy in Pancreatic Ductal Adenocarcinoma: An Emerging Entity? Ann Oncol (2017) 28(12):2950–61. doi: 10.1093/annonc/mdx503
39. Lin Y, Xu J, Lan H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J Hematol Oncol (2019) 12(1):76. doi: 10.1186/s13045-019-0760-3
40. Fares CM, Allen EMV, Drake CG, Allison JP, Hu-Lieskovan S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am Soc Clin Oncol Educ Book (2019) 39):147–64. doi: 10.1200/EDBK_240837
41. Kim H, Kwon HJ, Han YB, Park SY, Kim ES, Kim SH, et al. Increased CD3+ T Cells With a Low FOXP3+/CD8+ T Cell Ratio Can Predict Anti-PD-1 Therapeutic Response in Non-Small Cell Lung Cancer Patients. Mod Pathol (2019) 32(3):367–75. doi: 10.1038/s41379-018-0142-3
42. Zhang Y, Lazarus J, Steele NG, Yan W, Lee H-J, Nwosu ZC, et al. Regulatory T-Cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discovery (2020) 10(3):422–39. doi: 10.1158/2159-8290.CD-19-0958
43. Saleh R, Elkord E. FoxP3+ T Regulatory Cells in Cancer: Prognostic Biomarkers and Therapeutic Targets. Cancer Lett (2020) 490:174–85. doi: 10.1016/j.canlet.2020.07.022
44. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol (2020) 38(1):1–10. doi: 10.1200/JCO.19.02105
45. Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front Oncol (2020) 10:188. doi: 10.3389/fonc.2020.00188
46. Gibney GT, Weiner LM, Atkins MB. Predictive Biomarkers for Checkpoint Inhibitor-Based Immunotherapy. Lancet Oncol (2016) 17(12):e542–e51. doi: 10.1016/S1470-2045(16)30406-5
47. Davis AA, Patel VG. The Role of PD-L1 Expression as a Predictive Biomarker: An Analysis of All US Food and Drug Administration (FDA) Approvals of Immune Checkpoint Inhibitors. J ImmunoTherapy Cancer (2019) 7(1):278. doi: 10.1186/s40425-019-0768-9
48. Emancipator K. Keytruda and PD-L1: A Real-World Example of Co-Development of a Drug With a Predictive Biomarker. AAPS J (2020) 23(1):5. doi: 10.1208/s12248-020-00525-1
Keywords: pancreatic cancer, tumor microenvironment, T-cell inflammation, immunotherapy, PD-L1
Citation: Golesworthy B, Wang Y, Tanti A, Pacis A, Romero J M, Cuggia A, Domecq C, Bourdel G, Denroche R E, Jang GH, Grant RC, Borgida A, Grünwald BT, Dodd A, Wilson JM, Bourque G, O’Kane GM, Fischer SE, Kron C M, Fiset P-O, Omeroglu A, Foulkes W D, Gallinger S, Guiot M-C, Gao Z-H and Zogopoulos G (2022) Intra-Tumoral CD8+ T-Cell Infiltration and PD-L1 Positivity in Homologous Recombination Deficient Pancreatic Ductal Adenocarcinoma. Front. Oncol. 12:860767. doi: 10.3389/fonc.2022.860767
Received: 23 January 2022; Accepted: 23 March 2022;
Published: 25 April 2022.
Edited by:
Bruno Sainz, Jr., Autonomous University of Madrid, SpainReviewed by:
Mingfen Li, First Affiliated Hospital of Guangxi University of Chinese Medicine, ChinaEileen Parkes, University of Oxford, United Kingdom
Copyright © 2022 Golesworthy, Wang, Tanti, Pacis, Romero, Cuggia, Domecq, Bourdel, Denroche, Jang, Grant, Borgida, Grünwald, Dodd, Wilson, Bourque, O’Kane, Fischer, Kron, Fiset, Omeroglu, Foulkes, Gallinger, Guiot, Gao and Zogopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George Zogopoulos, Z2VvcmdlLnpvZ29wb3Vsb3NAbWNnaWxsLmNh
†These authors have contributed equally to this work and share first authorship