
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 28 March 2022
Sec. Thoracic Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.856132
This article is part of the Research Topic Influence of Potential Diagnostic Biomarkers in Lung Cancer View all 26 articles
 Brendan Knapp1
Brendan Knapp1 Laura Mezquita2
Laura Mezquita2 Siddhartha Devarakonda3Mihaela Aldea2Saiama N. Waqar3Kym Pepin3Jeffrey P. Ward3
Siddhartha Devarakonda3Mihaela Aldea2Saiama N. Waqar3Kym Pepin3Jeffrey P. Ward3 Angela Botticella4Karen Howarth5Charlene Knape6Clive Morris5Ramaswamy Govindan3
Angela Botticella4Karen Howarth5Charlene Knape6Clive Morris5Ramaswamy Govindan3 Benjamin Besse2
Benjamin Besse2 Daniel Morgensztern3*
Daniel Morgensztern3*Introduction: Circulating tumor DNA (ctDNA) testing may identify patients at high risk for recurrence following chemoradiation (CRT) for locally advanced non-small cell lung cancer (LA-NSCLC). We evaluated the feasibility of ctDNA testing on a readily available commercial fixed-gene panel to predict outcomes in patients with LA-NSCLC.
Methods: Plasma of 43 patients was collected at CRT initiation (pre-CRT), completion (post-CRT1), quarterly follow up for 12 months (post-CRT2, 3, 4, 5 respectively) after CRT, and at disease progression. ctDNA analysis was performed using InVisionFirst®-Lung to detect mutations in 36 cancer-related genes. ctDNA clearance was defined as absence of pre-CRT variants at post-CRT1. Patients without detectable pre-CRT variants or no post-CRT1 samples were excluded.
Results: Twenty eight of 43 patients (65%) had detectable variants pre-CRT. Nineteen of 43 patients (44%) had detectable pre-CRT variants and post-CRT1 samples and were included in analysis. Median age at diagnosis was 65 years (43-82), and most patients had stage IIIB disease (10/19, 53%). Two patients died from non-cancer related causes before post-CRT2 and were excluded from further analysis. All three patients who did not clear ctDNA had tumor relapse with a median time to relapse of 74 days (30-238), while 50% (7/14) of those who cleared ctDNA have remained disease free. Progression free survival was longer in patients who cleared ctDNA compared to those who did not (median 567 vs 74 d, p = 0.01).
Conclusions: Although it is feasible to use ctDNA testing on a limited gene panel to identify patients with LA-NSCLC who are at high risk for disease recurrence following CRT, further studies will be necessary to optimize these assays before they can be used to inform clinical care in patients with lung cancer.
Lung cancer remains the leading cause of cancer deaths worldwide (1). Non-small cell lung cancer (NSCLC) accounts for 85% of lung cancer diagnoses (2). Approximately 27% of patients with NSCLC present with locally advanced disease (3), which is typically managed with concurrent chemoradiotherapy (CRT) (4). The PACIFIC trial showed improved overall survival in this patient population with the addition of consolidation durvalumab. However, a substantial proportion of patients with locally advanced NSCLC still recur with metastatic disease despite receiving CRT and consolidation durvalumab with curative intent (5). Additionally, a subset of patients may be cured by CRT alone without the need for consolidation durvalumab (6), which is associated with a 30.5% grade three or higher adverse event rate (5). Prognostic biomarkers therefore have the potential to identify patients who are likely to benefit from treatment intensification or de-escalation.
Tumor cells release circulating tumor DNA (ctDNA) into the bloodstream (7), and it is possible to identify mutations relevant for tumor development, progression, and resistance to therapy through ctDNA analyses. Additionally, the utility of ctDNA testing in informing prognosis in patients with early stage and locally advanced NSCLC (LA-NSCLC) has previously been described (8, 9). Since ctDNA testing has the potential to identify patients at high risk for recurrence following completion of concurrent CRT for LA-NSCLC, it may help identify patients that are likely to benefit more from escalation of treatment intensity (10). Past studies with ctDNA have, however, used large fixed hybrid gene capture panels that require prior knowledge of genomic alterations in a tumor (9, 10). Since this approach requires upfront sequencing of a tumor and construction of customized gene panels, its large-scale clinical application is limited. Here, we investigate the feasibility of using a commercial fixed gene panel based ctDNA testing approach, which does not require a priori knowledge of a tumor’s genomic profile, to predict outcomes in patients with locally advanced NSCLC. Based on results from previous studies, we hypothesized that outcomes in patients with locally advanced CRT are poorer when treatments fail to clear variants detected through pre-treatment ctDNA testing.
Patients with histologically confirmed NSCLC with stage II or III NSCLC, who were not surgical candidates, and were eligible for curative intent treatment with concurrent chemotherapy and radiation were eligible for participation in this study. Patients were staged by the AJCC 8th edition TNM system. Patients receiving induction or consolidation chemotherapy and consolidation immune checkpoint inhibitors (ICIs) were also included. Progression following CRT was defined based on imaging, and when required, a biopsy at the discretion of the treating provider.
Plasma for ctDNA testing was collected within 2 weeks of CRT initiation (pre-CRT), CRT completion (post-CRT1), quarterly follow up appointments for 12 months (post-CRT2, 3, 4, and 5 respectively) after CRT completion, and at the time of disease progression (PD) (Figure 1 and Supplementary Table). All samples were collected with informed consent in accordance with the Declaration of Helsinki of 2013. Twenty mL of blood was collected in cell-free DNA blood collection (Streck) tubes. Tubes were labeled with specimen ID, subject ID, collection date and time point and shipped to Inivata (7020 Kit Creek Road, Suite 140, Research Triangle Park, NC 27560). ctDNA analysis was performed using the InVisionFirst®-Lung panel, to detect the presence of single nucleotide variations, insertions, deletions, and copy number alterations in 36 cancer-related genes, which are frequently altered in lung cancer (Supplementary Figure 1). Full assay details have been described previously (11–13). In brief, 36 cancer related genes were sequenced using gene-specific primers designed for hotspots and entire coding regions of interest. Next generation sequencing libraries were prepared from 2,000–16,000 amplifiable copies of the genome using a two-step PCR amplification process incorporating replicate and patient-specific barcodes and Illumina sequencing adaptors. Samples were pooled to generate a library of 12nM. 1.8 pM libraries were sequenced on the Illumina NextSeq 500, with sequencing files analyzed using Inivata’s proprietary Somatic Mutation Analysis (ISoMA) and FUSP pipelines.
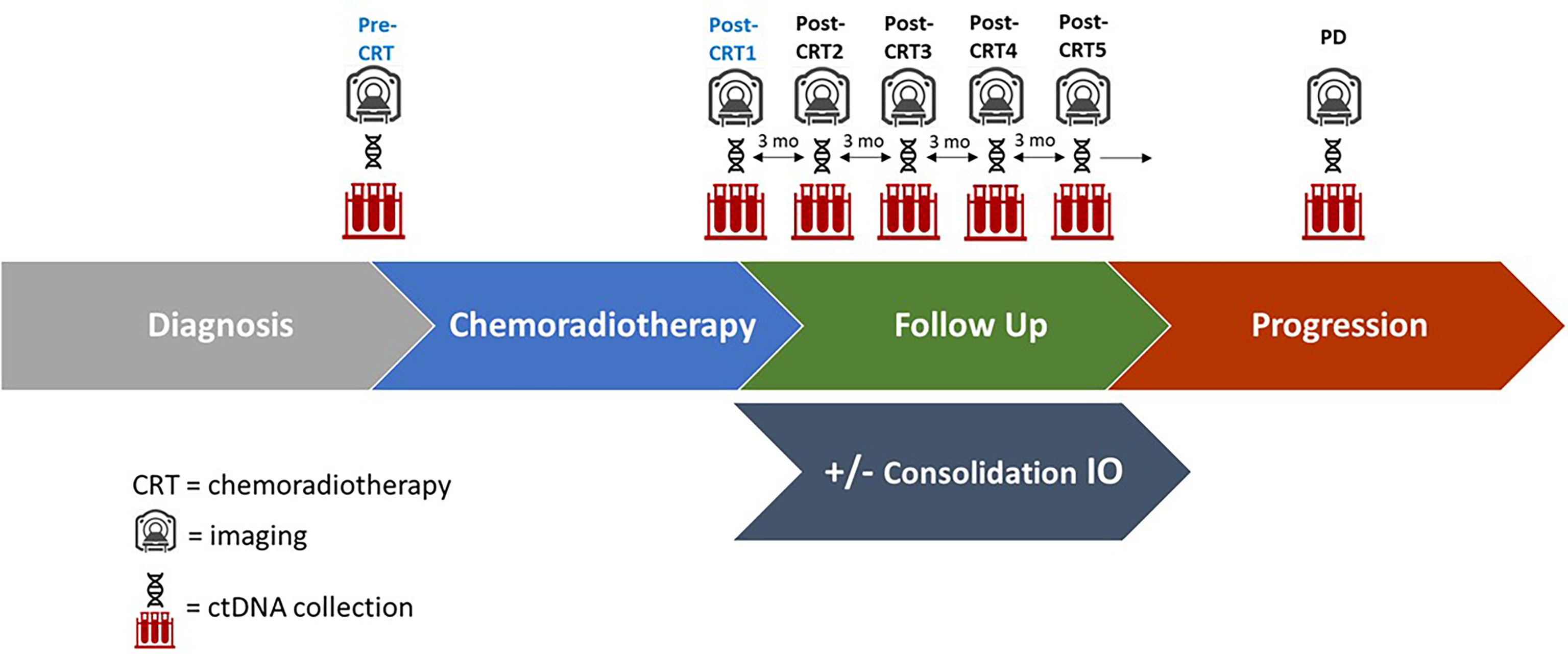
Figure 1 Study Schema of ctDNA collection, imaging, and follow up. Plasma for ctDNA was collected at the time of chemoradiotherapy initiation (pre-CRT), at completion of chemoradiotherapy (post-CRT1), and at quarterly follow up appointments for 12 months (post-CRT2,3,4, and 5 respectively), as well as at the time of disease progression (PD) if it occurred. A subset of patients received consolidation immune checkpoint inhibitors (IO). Imaging was performed every three months following completion of CRT.
Clearance of ctDNA was defined as the absence of non-germline pre-CRT variants at post-CRT1. Germline mutations were defined as a mutation allelic frequency (AF) greater than 40% detected at all time points (14, 15). Patients without detectable pre-CRT variants or no post-CRT1 samples were excluded from analysis. Maximum, mean, and median AF for all mutations detected in a sample were described, at each time point. All studies were undertaken in compliance with ethical principles established in the International Conference on Harmonization Good Clinical Practice and the Declaration of Helsinki. The study was approved by the Washington University in St. Louis Institutional Review Board. All statistical analyses were performed using GraphPad Prism (San Diego, CA; RRID : SCR_002798).
A total of 43 patients were prospectively enrolled between September 2017 and October 2019. Ten patients were enrolled from Institut Gustave Roussy and 33 patients from Washington University School of Medicine in St. Louis. Twenty eight of 43 patients (65%) had detectable variants on pre-CRT ctDNA testing. Nineteen of 43 patients (44%) had detectable pre-CRT variants and post-CRT1 samples collected and were included in the final analysis (Figure 2). In this cohort of 19 patients (Table 1), the median age at diagnosis was 65 years (range 43 - 82). The majority of patients were smokers (16/19, 84%), male (12 of 19, 63%), and were diagnosed with stage IIIB disease (10 of 19, 53%). Nine patients (47%) were diagnosed with squamous cell carcinoma, seven (37%) with adenocarcinoma, and three (16%) with poorly differentiated or NSCLC not otherwise specified. All 19 patients received weekly carboplatin and paclitaxel chemotherapy. Ten of 19 (53%) received no consolidation ICIs, while 8 (42%) patients received an ICI (6 patients received durvalumab and 2 received atezolizumab).
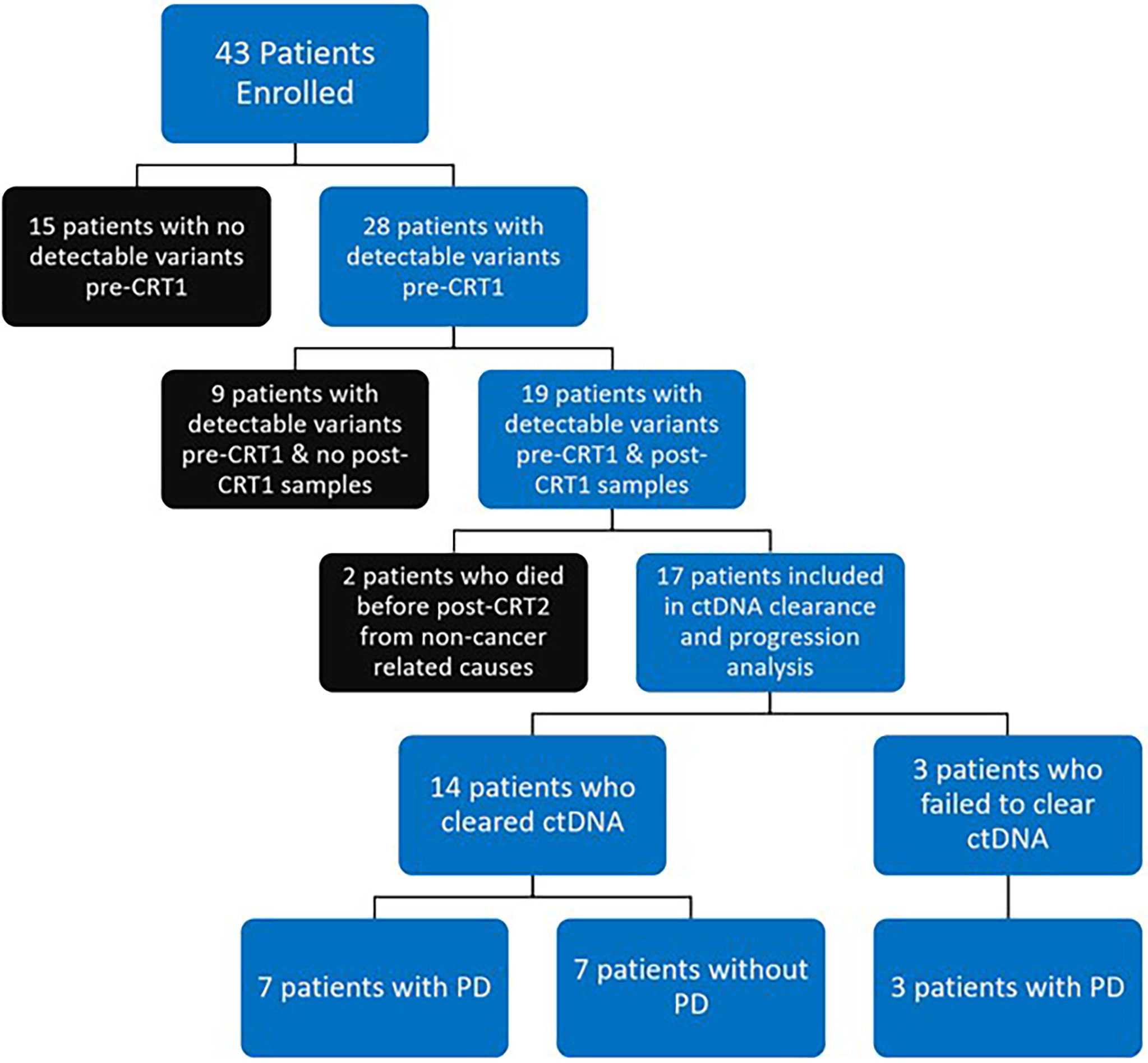
Figure 2 Flow Diagram of Patient Disposition: 43 patients were prospectively enrolled. 28 of 43 patients (65%) had detectable variants at time of chemoradiotherapy (CRT) initiation (pre-CRT). 19 of 43 patients (44%) had detectable variants at pre-CRT and had samples collected at completion of CRT (post-CRT1) and were included in final analysis. 2 of 19 patients (one who cleared ctDNA, one who did not) died from non-cancer related causes before the first visit post completion of CRT (post-CRT2) and were excluded from analysis on clearance. Of the 17 patients who were included in analysis of ctDNA clearance and disease progression (PD), 14 (82%) had clearance of ctDNA, with 7 having PD and 7 remaining free from progression. All 3 patients who did not clear ctDNA had PD.
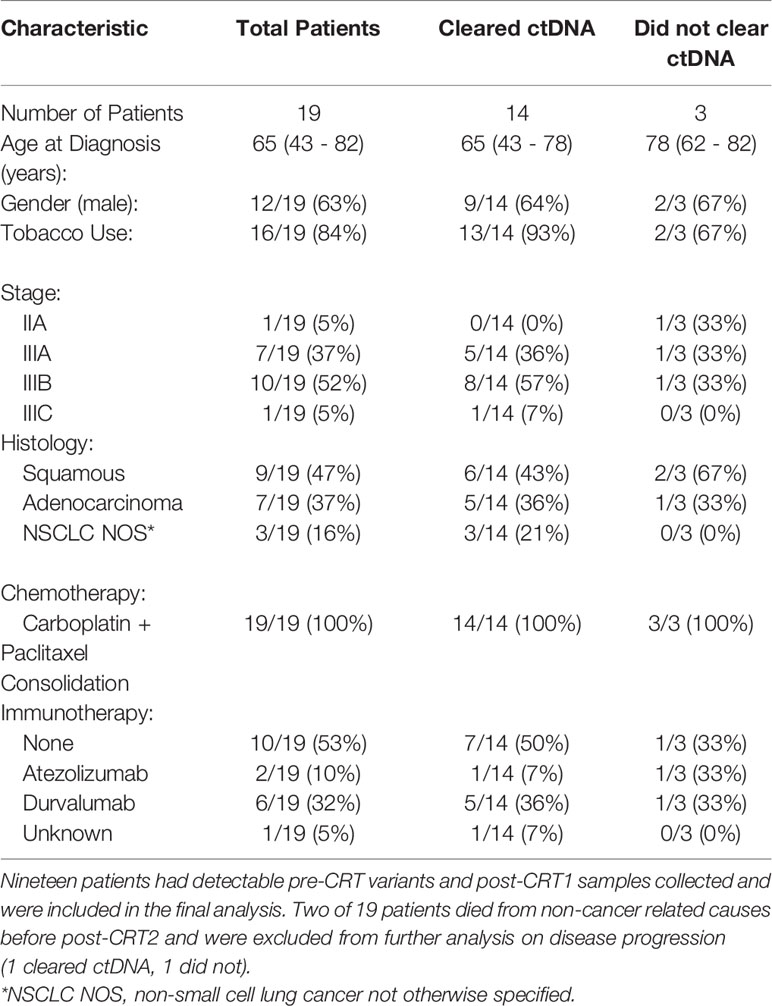
Table 1 Baseline Characteristics.
A median of two mutations per sample (range 1-5) were detected with a median mean AF of 0.53% (range 0.05-16.28%) in pre-CRT samples. 100% (46 of 46) of mutations in the cohort of interest were non-synonymous. In pre-CRT samples from the 19 patients included in the final analysis, mutations in TP53 were the most frequently detected (17/19, 89%), followed by mutations in PIK3CA (5/19, 26%), CDKN2A (4/19, 21%), and EGFR (3/19, 16%) (Figure 3).
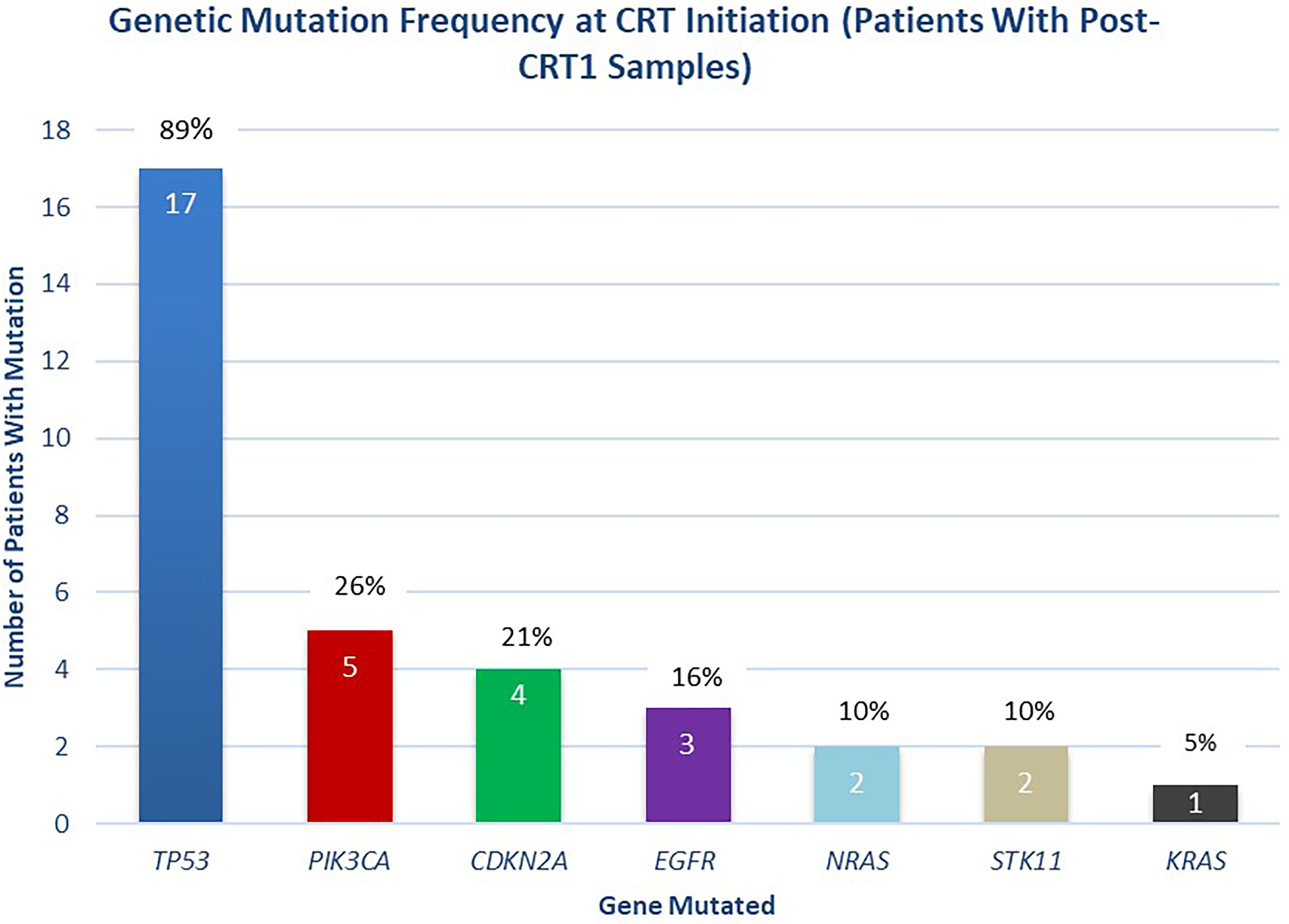
Figure 3 Distribution of Mutations Detected at Initiation of Chemoradiotherapy. Distribution of mutations detected at the initiation of chemoradiotherapy (pre-CRT), in patients with post-CRT1 samples collected (n = 19 patients).
Two of 19 patients died from non-cancer related causes (myocardial infarction, acute respiratory distress syndrome) before post-CRT2 and were excluded from further analysis (1 cleared ctDNA, another did not). Three patients failed to clear ctDNA at post-CRT1 and 14 patients cleared ctDNA. All the 3 patients that failed to clear eventually progressed with a median time to progression of 74 days (range 30-238). In contrast, 7 of the 14 patients that cleared ctDNA at post-CRT1, remained disease free at the time of this analysis with a median follow-up of 469 days (range 130-710). The median time to progression in the seven patients who cleared ctDNA and later progressed was 217 days (range 53-587 days). In all patients who cleared ctDNA, the median PFS was 567 days, compared to a median PFS of 74 days in patients who did not clear (p = 0.01) (Figure 4). Of the 24 patients who did not have complete biomarker data, 1 died from non-cancer related causes before post-CRT2 and 2 patients were lost to follow up. Of the remaining 21 patients, 10 had disease progression (median time to progression 182 days, range 29 - 522) and 11 have not (median follow up 453 days, range 147 – 1099).
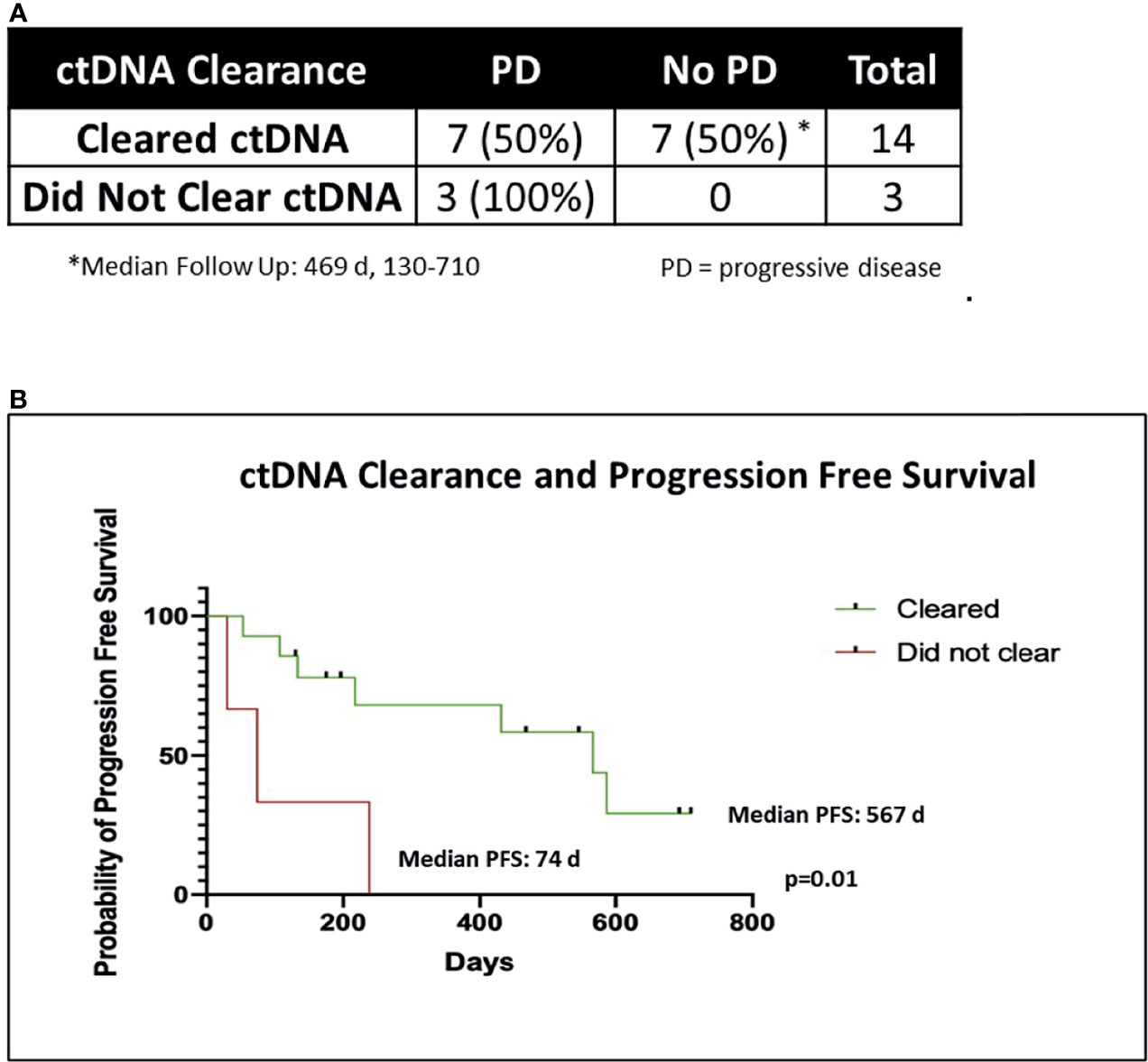
Figure 4 ctDNA clearance and freedom from progression. (A) Chi-Square table of ctDNA clearance and disease progression (PD). Two of 19 patients died of non-cancer related causes before post-CRT2 and were excluded from analysis of disease progression, leaving 17 patients. (B) Kaplan-Meier graph of progression free survival (PFS) in patients who cleared ctDNA (n = 14) versus those who did not (n = 3), using the Mantel Cox log rank test.
Of the 14 patients who cleared ctDNA, one patient’s consolidation therapy status was unknown, leaving 13 with known consolidation status in this group. Six of 13 patients who cleared ctDNA received consolidation ICIs, of which two had disease progression, with four remaining free from progression. In the seven of 13 patients who cleared ctDNA and did not receive consolidation ICIs, five had had disease progression, with two remaining progression free.
In the 14 patients who cleared ctDNA at post-CRT1, the pre-CRT median mean AF was 0.71% (range, 0.06 - 16.28%), with a median maximum AF of 0.90% (0.06 - 51.95%). In the seven of 14 patients that cleared ctDNA and later progressed, the median mean AF at pre-CRT was 2.11% (0.06 - 16.28%), with a median maximum AF of 2.80% (0.06 - 51.95%). In the seven of 14 patients that cleared and have not progressed, the median mean AF at pre-CRT was 0.52% (0.10 - 2.33%), and the median maximum AF was 0.63% (0.10 - 2.35%). There was no statistically significant difference in mean or maximum AF at pre-CRT in patients who progressed versus those who did not progress (p >0.05) (Figure 5). In the three patients who did not clear at post-CRT1, the pre-CRT median mean AF was 0.54% (0.26 - 0.72%) and the median maximum AF was 1.27% (0.47 - 1.61%). At post-CRT1, the median mean AF was 0.26% (0.11 - 1.89%) and the median maximum AF was 0.42% (0.17 - 5.55%).
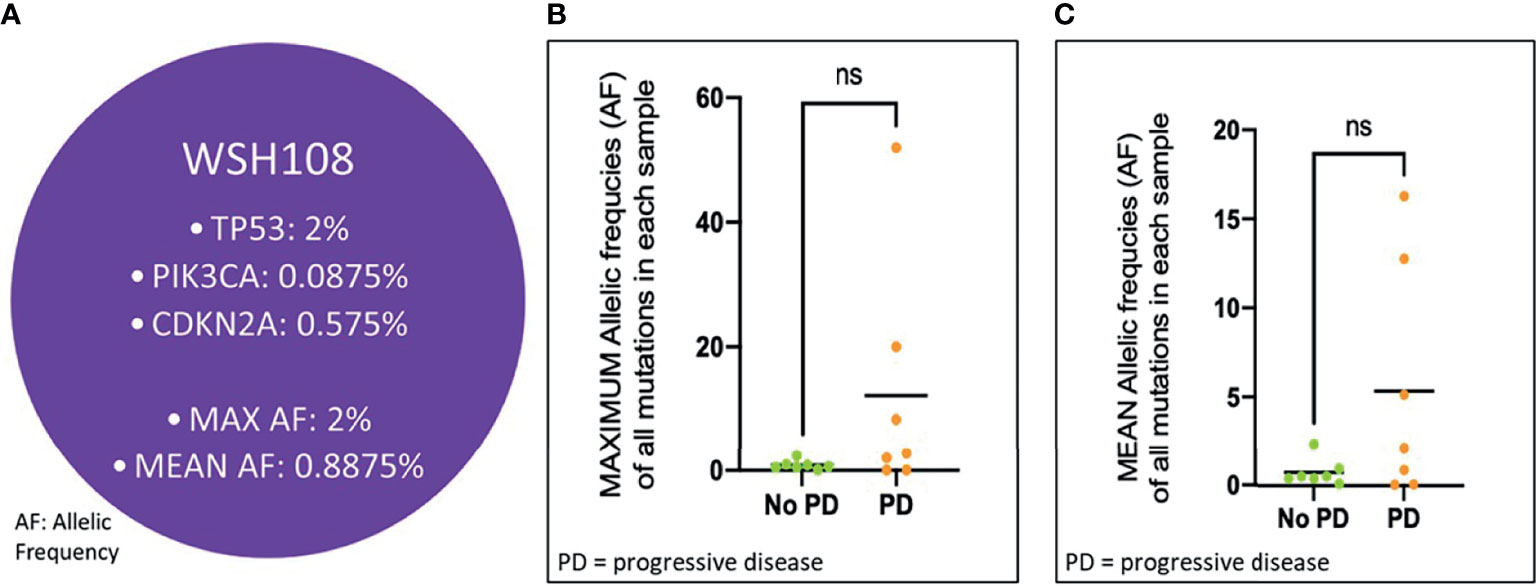
Figure 5 Mean and Max Allelic Frequency in Patients at CRT initiation who Cleared ctDNA, Stratified by Progression. (A) Schematic representation of a patient’s (WSH108) pre-CRT sample. Allelic frequency (AF) for each mutation detected in the sample was used for determining maximum and mean AF values. Each of these values across patients who progressed vs. did not progress were compared in the cohort of patients clearing ctDNA using a Student’s t-test, as depicted in (B, C) ns, not significant.
New variants (non-pre-CRT1) were observed in eight (42%) of patients during follow-up with a median AF of 0.32% (range 0.03 - 15.14%). Of the eight patients with new mutations, six progressed and two did not. Of the seven of 14 patients who cleared ctDNA that later progressed, three had new mutations with a median AF of 0.07% (range 0.03 - 0.46%). In the three patients that did not clear ctDNA, all patients demonstrated new mutations at post-CRT1 or later, in addition to the pre-CRT mutations, with a median AF of 3.66% (range 0.17 - 15.14%) in the new variants.
To our knowledge, this is the first study utilizing commercially available ctDNA testing on a limited gene panel, instead of customized patient-specific gene panels, to show that detection of residual ctDNA following concurrent CRT in patients with locally advanced NSCLC is associated with a poor prognosis. All patients that failed to clear ctDNA following CRT in our analysis demonstrated disease progression. In addition, time to progression was much shorter in patients that failed to clear ctDNA, compared to patients that cleared ctDNA and eventually progressed (median 74 days vs 217 days). Furthermore, half the patients that cleared ctDNA following CRT continue to remain disease free at the time of this reporting.
Clearance of ctDNA following treatment, whether surgical or CRT, has been associated with improved prognosis in several other studies including lung cancer (10, 16–18). These studies however, used customized gene panels, the design of which required a priori tumor sequencing. In the study by Moding et al., which utilized personalized profiling by deep sequencing (CAPP-Seq) ctDNA analysis of 65 patients receiving CRT for locally advanced NSCLC, patients with undetectable ctDNA following CRT had improved outcomes relative to those with detectable ctDNA, regardless of whether they received consolidation ICI (10). Patients with detectable ctDNA following CRT who received consolidation ICIs had significantly improved outcomes than those who did not receive an ICI. The results from these studies, in addition to ours, support ctDNA clearance as a predictive biomarker in patients with locally advanced NSCLC. While previous studies have demonstrated the prognostic value of ctDNA clearance using customized gene panels, our study suggests that it could be feasible to utilize commercially available gene panels for the same purpose, but systematic testing through well-designed clinical trials are needed to further evaluate this.
The advantage of utilizing such commercial gene panel tests is that they are easy to incorporate into clinical practice and are not associated with the challenges and high costs associated with upfront tumor sequencing and design of customized gene panels. However, the successful translation of assays that utilize ctDNA sequencing on a limited gene panel to the clinic will require addressing certain crucial limitations that we identified in our analysis. For instance, the limited sequencing space and number of genes included on limited panels can potentially misclassify patients not clearing ctDNA as having cleared ctDNA. In this regard, the 65% of patients with mutations detected post-CRT in our study could be erroneously low due to the decreased sensitivity of our limited gene sequencing platform. Additionally, as is the limitation with all ctDNA assays that lack sequencing data from tumor tissue or peripheral blood mononuclear cells, it is difficult to distinguish tumor derived ctDNA from DNA originating from clonal hematopoiesis of indeterminate potential (CHIP).
Nevertheless, despite these limitations, the most commonly detected mutations in our study were in genes that are frequently mutated in lung cancer, which supports the conclusions reported. Mutations in TP53, PIK3CA, CDKN2A, KRAS, and EGFR were the most commonly detected, which is similar to known frequencies of mutations in lung cancer from tumor biopsies (19–22). Eight of the 17 patients (47%) had new emerging variants, defined as not present in the pre-CRT samples, with six of these patients (75%) developing relapse. It is possible that some of these variants were present in the pre-CRT and post-CRT1 samples, but at a level of detection that is lower than the limit of detection for the assay, leading to the possible misclassification of these patients into the “ctDNA cleared” category. We anticipate that advances in ctDNA sequencing technology in the near future may help address these limitations of liquid biopsies and improve the performance of this assay. Since it is also possible that such emerging variants could have been acquired by a tumor during the course of treatment, assays with fixed panels comprising of several frequently mutated genes in lung cancer may have an inherent advantage over assays using customized panels guided by tissue sequencing for identifying patients with emerging variants early in the course of their treatment, which may potentially be used for treatment changes.
The findings of our study are best viewed as exploratory given the limited sample size. Apart from the small sample size, the lack of longitudinal post-CRT variant data at all time points for all participants, and the fact that only a limited number of patients received consolidation immunotherapy in our study, which is currently the standard of care - limited our ability to investigate whether ctDNA clearance has the ability to identify patients most likely to benefit from immunotherapy.
In summary, results from this study demonstrate that it is feasible to employ ctDNA testing utilizing an “off the shelf” gene panel assay to identify LA-NSCLC patients who are at high risk for disease recurrence following CRT. Our results suggest that the failure to clear ctDNA after CRT is a poor prognostic factor for early progression. However, this approach is not without certain limitations that can potentially be circumvented by advances in sequencing technology, utilizing matched peripheral blood cell sequencing, and using expanded gene panels. Whether such assays can be used to inform clinical practice merits future investigation. Prospective validation of our results in larger studies is necessary before these findings can be translated to the clinic.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by Washington University in St. Louis Institutional Review Board. The patients/participants provided their written informed consent to participate in this study.
BK: Formal Analysis, Investigation, Data Curation, Writing – Original Draft, Writing – Review & Editing, Visualization. LM: Conceptualization, Methodology, Validation, Formal Analysis, Investigation, Resources, Data Curation, Writing – Review & Editing, Project Administration, Funding Acquisition. SD: Methodology, Validation, Formal analysis, Investigation, Data Curation, Writing – Original Draft, Writing – Review and Editing, Visualization, Project Administration. MA: Formal Analysis, Investigation, Data Curation, Writing – Review & Editing. SW: Methodology, Investigation, Writing – Review & Editing. KP: Formal Analysis, Investigation, Data Curation, Writing – Review & Editing. JW: Methodology, Investigation, Writing – Review & Editing. AB: Methodology, Investigation, Writing – Review & Editing. KH: Methodology, Writing – Review & Editing, Project Administration, Funding Acquisition. CK: Methodology, Writing – Review & Editing, Funding Acquisition. CM: Methodology, Writing – Review & Editing, Project Administration, Funding Acquisition. RG: Methodology, Validation, Investigation, Resources, Writing – Review & Editing. BB: Conceptualization, Methodology, Validation, Investigation, Data Curation, Writing – Review & Editing, Supervision, Project Administration. DM: Conceptualization, Methodology, Validation, Investigation, Resources, Data Curation, Writing – Original Draft, Writing – Review & Editing, Visualization, Supervision, Project Administration, Funding Acquisition. All authors contributed to the article and approved the submitted version.
LM: Lectures and educational activities: Bristol-Myers Squibb, Tecnofarma, AstraZeneca, Roche, Takeda. Consulting/advisory role: Roche, Takeda. Research Grants: Bristol-Myers Squibb, Boehringer Ingelheim. Travel, Accommodations, Expenses: Bristol-Myers Squibb, Roche. Others: International Mentorship Program funded by AstraZeneca. SW: Research grant support from -2% effort on “Duke-UNC Wash U Partnership for Early Phase Clinical Trials in Cancer” 1UM1 CA186704-01 grant as mentored faculty, DSMB Chair for Hoosier Cancer Research Network study, and institutional PI for studies with support from Hoffmann-La Roche Ltd, Ariad, Pfizer Pharmaceuticals, Inc., Hengrui Therapeutics, Xcovery, EMD Serono Research & Development Institute, Inc., Checkpoint Therapeutics, Inc., Genentech, Inc., Lilly, Stemcentrx, Inc., Ignyta, Inc., Bristol-Myers Squibb Pharmaceutical, Synermore Biologics Co., Ltd., Novartis Pharmaceuticals Corporation, Merck & Company, Inc., NewLink Genetics Corporation, and Celgene.
JW: Advisory board for Novocure, Consultant for Novoure, Employment for Millipore (Spouse), Travel/Expenses from Halozyme. KH: employee and stockholder for Inivata. CK: employee and stockholder for Inivata. CM: employee and stockholder for Inivata.
RG: Advisory Board for Achilles, Consulting for GenePlus, Horizon Pharmaceuticals (Spouse). BB: Research support from 4D Pharma, Abbvie, Amgen, Aptitude Health, AstraZeneca, BeiGene, Blueprint Medicines, BMS, Boehringer Ingelheim, Celgene, Cergentis, Cristal Therapeutics, Daiichi-Sankyo, Eli Lilly, GSK, Inivata, Janssen, Onxeo, OSE immunotherapeutics, Pfizer, Roche-Genentech, Sanofi, Takeda, Tolero Pharmaceuticals. DM: Advisory board for Abbvie, Takeda, PharmaMar, Gilead, Boehringer Ingelheim; Consultant for Abbvie, Takeda, Boehringer Ingelheim, PharmaMar, Gilead.
The study received funding from Inivata. The funder had the following involvement in the study: study design, analysis and data interpretation, the writing of this article, and the decision to submit it for publication.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.856132/full#supplementary-material
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Duma N, Santana-Davila R, Molina JR. Non–Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin Proc (2019) 94:1623–40. doi: 10.1016/j.mayocp.2019.01.013
3. Morgensztern D, Ng SH, Gao F, Govindan R. Trends in Stage Distribution for Patients With non-Small Cell Lung Cancer: A National Cancer Database Survey. J Thorac Oncol (2010) 5:29–33. doi: 10.1097/JTO.0b013e3181c5920c
4. Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman J, Chirieac LR, et al. Non-Small Cell Lung Cancer, Version 5.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw (2017) 15:504–35. doi: 10.6004/jnccn.2017.0050
5. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall Survival With Durvalumab After Chemoradiotherapy in Stage III NSCLC. N Engl J Med (2018) 379:2342–50. doi: 10.1056/NEJMoa1809697
6. Liang J, Bi N, Wu S, Chen M, Lv C, Zhao L, et al. Etoposide and Cisplatin Versus Paclitaxel and Carboplatin With Concurrent Thoracic Radiotherapy in Unresectable Stage III non-Small Cell Lung Cancer: A Multicenter Randomized Phase III Trial. Ann Oncol (2017) 28:777–83. doi: 10.1093/annonc/mdx009
7. Corcoran RB, Chabner BA. Application of Cell-Free DNA Analysis to Cancer Treatment. N Engl J Med (2018) 379:1754–65. doi: 10.1056/NEJMra1706174
8. Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA Analysis Depicts Early-Stage Lung Cancer Evolution. Nature (2017) 545:446–51. doi: 10.1038/nature22364
9. Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov (2017) 7:1394–403. doi: 10.1158/2159-8290.CD-17-0716
10. Moding EJ, Liu Y, Nabet BY, Chabon JJ, Chaudhuri AA, Hui AB, et al. Circulating Tumor DNA Dynamics Predict Benefit From Consolidation Immunotherapy in Locally Advanced non-Small-Cell Lung Cancer. Nat Cancer (2020) 1:176–83. doi: 10.1038/s43018-019-0011-0
11. Plagnol V, Woodhouse S, Howarth K, Lensing S, Smith M, Epstein M, et al. Analytical Validation of a Next Generation Sequencing Liquid Biopsy Assay for High Sensitivity Broad Molecular Profiling. PloS One (2018) 13:e0193802. doi: 10.1371/journal.pone.0193802
12. Guibert N, Jones G, Beeler JF, Plagnol V, Morris C, Mourlanette J, et al. Targeted Sequencing of Plasma Cell-Free DNA to Predict Response to PD1 Inhibitors in Advanced non-Small Cell Lung Cancer. Lung Cancer (2019) 137:1–6. doi: 10.1016/j.lungcan.2019.09.005
13. Pritchett MA, Camidge DR, Patel M, Khatri J, Boniol S, Friedman EK, et al. Prospective Clinical Validation of the InVisionFirst-Lung Circulating Tumor DNA Assay for Molecular Profiling of Patients With Advanced Nonsquamous Non-Small-Cell Lung Cancer. JCO Precis Oncol (2019) 3:1–15. doi: 10.1200/PO.18.00299
14. Slavin TP, Banks KC, Chudova D, Oxnard GR, Odegaard JI, Nagy RJ, et al. Identification of Incidental Germline Mutations in Patients With Advanced Solid Tumors Who Underwent Cell-Free Circulating Tumor DNA Sequencing. J Clin Oncol (2018) 36:3459. doi: 10.1200/JCO.18.00328
15. Stout LA, Kassem N, Hunter C, Philips S, Radovich M, Schneider BP. Identification of Germline Cancer Predisposition Variants During Clinical ctDNA Testing. Sci Rep (2021) 11:13624. doi: 10.1038/s41598-021-93084-0
16. Goldberg SB, Narayan A, Kole AJ, Decker RH, Teysir J, Carriero NJ, et al. Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin Cancer Res (2018) 24:1872–80. doi: 10.1158/1078-0432.CCR-17-1341
17. Lee Y, Park S, Kim WS, Lee JC, Jang SJ, Choi J, et al. Correlation Between Progression-Free Survival, Tumor Burden, and Circulating Tumor DNA in the Initial Diagnosis of Advanced-Stage EGFR-Mutated non-Small Cell Lung Cancer. Thorac Cancer (2018) 9:1104–10. doi: 10.1111/1759-7714.12793
18. Raja R, Kuziora M, Brohawn PZ, Higgs BW, Gupta A, Dennis PA, et al. Early Reduction in ctDNA Predicts Survival in Patients With Lung and Bladder Cancer Treated With Durvalumab. Clin Cancer Res (2018) 24:6212–22. doi: 10.1158/1078-0432.CCR-18-0386
19. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic Mutations Affect Key Pathways in Lung Adenocarcinoma. Nature (2008) 455:1069–75. doi: 10.1038/nature07423
20. Cancer Genome Atlas Research Network. Comprehensive Genomic Characterization of Squamous Cell Lung Cancers. Nature (2012) 489:519–25. doi: 10.1038/nature11404
21. Cancer Genome Atlas Research Network. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature (2014) 511:543–50. doi: 10.1038/nature13385
Keywords: ctNDA, fixed gene panel, prognostication, non-small cell lung cancer, commercially available
Citation: Knapp B, Mezquita L, Devarakonda S, Aldea M, Waqar SN, Pepin K, Ward JP, Botticella A, Howarth K, Knape C, Morris C, Govindan R, Besse B and Morgensztern D (2022) Exploring the Feasibility of Utilizing Limited Gene Panel Circulating Tumor DNA Clearance as a Biomarker in Patients With Locally Advanced Non-Small Cell Lung Cancer. Front. Oncol. 12:856132. doi: 10.3389/fonc.2022.856132
Received: 16 January 2022; Accepted: 28 February 2022;
Published: 28 March 2022.
Edited by:
Idris Bahce, Academic Medical Center, NetherlandsReviewed by:
Alberto Pavan, Azienda ULSS 3 Serenissima, ItalyCopyright © 2022 Knapp, Mezquita, Devarakonda, Aldea, Waqar, Pepin, Ward, Botticella, Howarth, Knape, Morris, Govindan, Besse and Morgensztern. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Morgensztern, RGFuaWVsbW9yZ2Vuc3p0ZXJuQHd1c3RsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.