 Xuelian Song1
Xuelian Song1 Changran Wei
Changran Wei Xiangqi Li
Xiangqi Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 10 March 2022
Sec. Breast Cancer
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.855609
This article is part of the Research TopicFGFR signaling in breast cancer: state of art and future challengesView all 4 articles
Background: Breast cancer (BC) is now the leading cause of cancer in women, and bone is the primary site of distant BC metastasis. BC bone metastasis seriously affects the quality of life of patients and increases the mortality rate. However, the mechanism of BC bone metastasis is not fully understood.
Main Body: Paget’s “seed and soil” hypothesis led experts to explore the relationship between surface markers and receptors in breast tumors and various growth factors in bone. The relevant breast tumor markers serve as “seeds”, and the bone microenvironment that is suitable for the survival of the tumor serves as the “soil”. These factors interact to make up an entire system and form feedback pathways that accelerate the production of various cytokines, attracting BC cells to migrate to bone tissue, which worsens the development of BC and seriously affects the prognosis of patients. This process is a vicious cycle. At present, there are seven major signaling pathways involved in BC bone metastasis: the OPG/RANK/RANKL signaling pathway, TGF-β signaling pathway, IGF system, PI3K-AKT-mTOR signaling pathway, Wnt signaling pathway and Hippo signaling pathway. In addition, FGF-FGFR signaling pathway, androgen-AR/LSD1-target gene pathway, Notch signaling pathway, JAK-STAT signaling pathway and CaN/NFATC1 signaling pathway also seem to be associated with BC bone metastasis.
Conclusion: This review focuses on the signaling pathways related to BC bone metastasis and explores the interactions among these pathways, which will lay a solid theoretical foundation for further understanding the mechanism of BC bone metastasis and developing effective targeted therapeutic drugs.
Presently, breast cancer (BC) is the most common malignant tumor and the most important health burden, as well as the most common cause of cancer death among women worldwide. Recently, statistics have shown that the incidence of BC in various countries around the world is increasing at an accelerated rate, and the affected population is becoming younger (1). With the development of imaging technology, surgery and medical treatments, the diagnosis and treatment of BC have improved. The survival rate of BC patients is increased, and the recurrence rate and mortality rate have decreased correspondingly but remain high (2). Studies have shown that metastasis is found in approximately 5% of BC patients at initial diagnosis, and in 20% to 30% of local BC cases, distant metastasis occurs (3). Bone tissue is the most common metastatic site of advanced BC (4) and accounts for approximately 75% of metastasis cases (5), with a 5-year overall survival rate of 22.8% (6) and the highest rate of first recurrence in BC patients. BC bone metastases are mainly osteolytic metastases (7), which cause bone resorption and lead to osteolytic bone lesions. Metastatic bone disease is the result of the interaction between metastatic BC cells and some cells in the bone microenvironment, which indicates that the prognosis is poor and seriously affects the quality of life of these patients. In fact, BC patients with bone metastasis are extremely susceptible to bone-related complications, also known as skeletal-related events (SREs), including bone pain, hypercalcemia, pathological fractures, and spinal cord compression, are commonly used to assess the quality of life and status of patients with BC bone metastases (8). Bone is a site containing many growth factors and cytokines. These factors are released during bone resorption as part of normal bone remodeling or during bone abnormal diseases, such as BC bone metastasis, and affect the proliferation and differentiation of BC cells. Recognized factors on the BC cell surface can spread via the blood to any place that is suitable for growth (such as bone) and further development. BC metastasis to bone further promotes the release of bone growth factors and cytokines, accelerating the growth and development of tumor cells, resulting in a vicious cycle. In summary, this review focuses on the signaling pathways related to BC bone metastasis and explores the interactions among these pathways, which will lay a solid theoretical foundation for further understanding the mechanism of BC bone metastasis and developing effective targeted therapeutic drugs.
Langenbeck once said that “Every cancer cell must be viewed as a living organism capable of development. When plants seed, the seeds are carried in all directions, but they only can survive and grow if they land in the right soil.” In 1882, Fuchs argued that certain organs might be susceptible to secondary cancer. In 1889, Stephen Paget proposed the “seed and soil” hypothesis, which held that tumor cells (seeds) could only grow in fertile soil (bone microenvironment). He argued that in BC, bones suffer in a way that no embolic theory alone could explain (9). Therefore, bone tissue must have a specific microenvironment that supports BC cell growth and development. BC bone metastasis is a complex process involving many kinds of cells and cell growth factors (4). However, it is unclear which cells and cytokines are involved. Therefore, the first thing we need to do is to understand the “soil” and the “seed”.
Soil can be defined as a bone microenvironment, containing many cellular growth factors which are the essential factors for growth and metastasis of BC cells (Figure 1). Such as transforming growth factor-β (TGF-β) (10), insulin-like growth factor I and II (IGF-I and IGF-II) (11), fibroblast growth factor (FGF) (12), platelet-derived growth factor (PDGF) (13), bone morphogenetic protein (BMP) (14), interleukin-1 (IL-1) (15), interleukin-6 (IL-6) (16), interleukin-8 (IL-8) (17, 18), interleukin-11 (IL-11) (19), tumor necrosis factor-α, (TNF-α) (20), vascular cell adhesion factor-1 (VCAM-1) (21), vascular endothelial growth factor (VEGF) (22), bone sialoprotein (BSP) (23), osteopontin (OPN), and matrix metalloproteinases (MMPs) (24). These cellular growth factors are released by the bone matrix and can bind to receptors on the surface of BC cells and promote the chemotaxis, migration and adhesion of BC cells, allowing BC cells to successfully transfer to the bone.
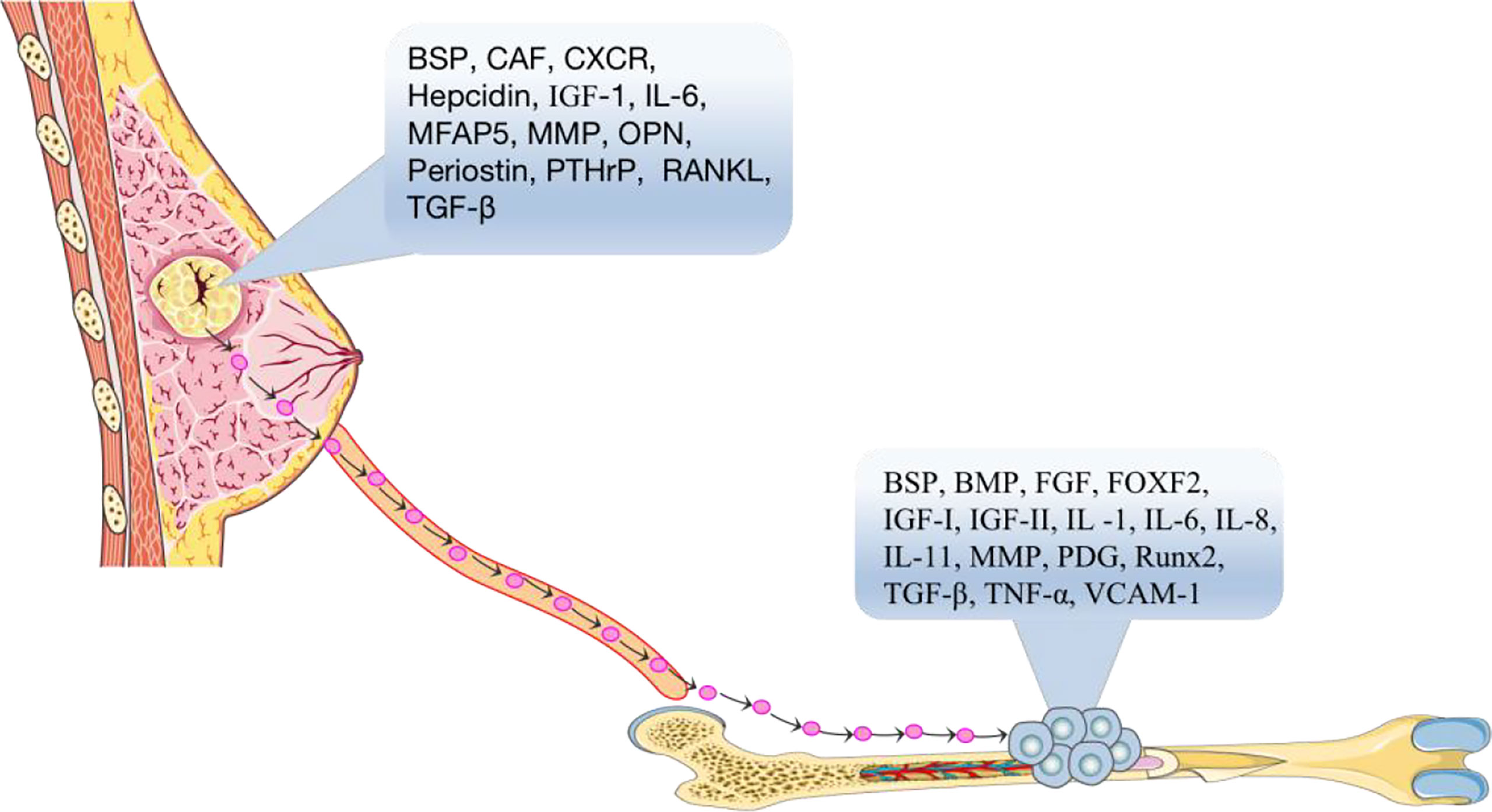
Figure 1 According to the seed and soil hypothesis, the primary breast tumor is the “seed” in the blood that spreads to the suitable growth environment of bone tissue through the actions of cytokines, BSP, CAF, CXCR, hepcidin, IGF-1, IL-6, MMPs, OPN, Periostin, PTHrP, RANKL, TGF-β, etc. BSP, BMP, FGF, FOXF2, IGF-I, IGF-II, IL-1, IL-6, IL-8, IL-11, MMPs, PDG, RUNX2, TGF-β, TNF-α, VCAM-1 and other “soil” factors in the bone microenvironment, which are suitable for tumor cell growth and development, accelerate the process of bone metastasis of tumor cells.
Seeds can be defined as the surface markers, which are the identifiable factors, of breast tumor cells (Figure 1). It has been reported that BC cells need to interact with receptors in an autocrine manner with the help of the chemokines, cell adhesion factors and growth factors before they can metastasize to bone, and then these cells can further grow and replicate. For example, the surface chemotactic factors on BC cells include CXC chemokine receptor (CXCR) (25), transforming growth factor-β (TGF-β) (10), insulin-like growth factor (IGF) (11), matrix metalloproteinases (MMPs) (26), parathyroid hormone related protein (PTHrP) (27), cancer-associated fibroblasts (CAFs) (28), BSP (29), IL-6 (30), hepcidin (31), periostin protein (32), OPN (29), MFAP5 (33), and RANKL (34, 35).
Bone metastases include the spread of cancer cells from the primary tumor via the blood to the bone marrow space (4). Notably, BC bone metastases are not directly caused by the destruction of bone but by the upregulation of osteoclasts via breast tumor cells, which disrupts the dynamic equilibrium between osteoclasts and osteoblasts. Osteolytic bone metastasis is the main cause of bone destruction. Various pathways have been investigated in the development of bone metastasis (Figure 2).
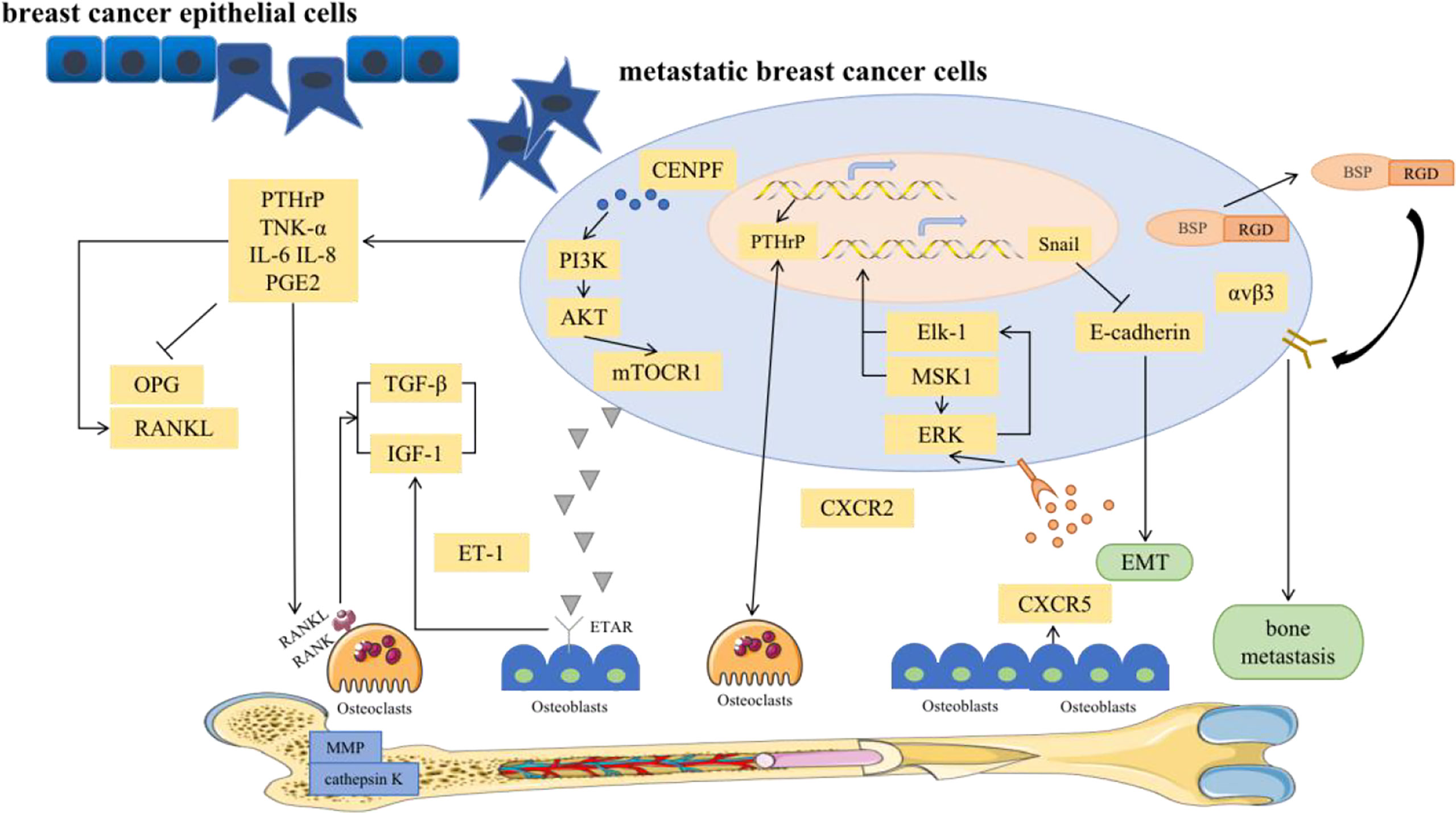
Figure 2 Major signal pathways for bone metastases in breast cancer.
The RANKL/RANK/OPG network was originally characterized as a regulator of bone remodeling (36). More and more studies have demonstrated that this network also plays a critical role in osteolysis in metastatic bone diseases, including BC bone metastases (37).
To elaborate, classic OPG, RANK, and RANKL systems provide growth and survival advantages for damaged breast epithelium, which is a prerequisite for BC initiation and a favorable pathway for BC bone metastasis (38, 39). RANK is highly expressed in breast cancer cells. Functionally, it has been shown that RANKL can stimulate the directed migration of mammary epithelial cells toward a source of RANKL (40). RANKL is involved in EMT within breast tumors through up-regulation of Snail. EMT describes a process in which epithelial shaped cells show a transformation into a mesenchymal phenotype. These changes enable cells to invade the surrounding microenvironment leading to cancer progression and an increased stemness of tumors. Stemness and transformation are the main points of action in which RANKL/RANK increases an aggressive behavior of a tumor bone metastasis (39). In addition, high expression of pathways related to T-cell proliferation. RANKL is known to enhance T-cell response and increase dendritic cell survival via binding to RANK. RANKL expression on the tumor was associated with downregulation of proliferation and cell cycle-related pathways. Furthermore, RANK expression was associated with activation of immune response and proliferation (41).
In fact, RANKL, its receptor RANK, and the decoy receptor OPG are the key regulators for osteoclast development and the activation of mature osteoclasts. Bone metabolism is thus regulated by a balance between RANKL/RANK signaling and OPG level. RANKL binds to RANK in the bone microenvironment, participates in the maturation of osteoclasts and mediates the activation of the nuclear factor kappa-B (NF-κB) and Jun N-terminal kinase (JNK) signaling pathways, leading to bone resorption. However, excessive absorption can be stopped by osteoprotegerin (42). In solid tumor bone metastasis, the balance between RANKL and osteoprotegerin is affected by many factors, such as PTHrP, vitamin D3 and prostaglandin (39). Among them, PTHrP is the most important osteophilic factor produced by breast tumor cells. PTHrP can reduce the expression of OPG and increase the expression of RANKL in osteoblasts to promote osteoclast formation (39). Thus, PTHrP changes the balance to promote the effects of RANKL. Higher RANKL expression leads to an increased likelihood that osteolysis will be the dominant process. Therefore, BC bone metastasis is more common in the osteolytic environment. In addition, RANKL activates bone resorption by osteoclasts and leads to the release of growth factors (such as TGF-β) from the bone matrix, which in turn stimulates the proliferation of tumor cells (43). Furthermore, metastatic tumor cells can directly secrete RANKL or stimulate osteoblasts to promote the production of RANKL, leading to bone matrix degradation and the release of many bone-derived growth factors and cytokines, which further stimulate the migration of tumor cells to bone (44). The absence of the RANKL/RANK system in breast epithelial cells prevents tumor growth. Importantly, RANKL was observed to be more accurate than conventional markers in the breast cancer patients, and was a good predictor of bone progression. It had been suggested that the RANKL could serve as an accurate marker of bone response in metastatic patients. High RANKL levels may identify patients with a shift in bone homeostasis towards bone resorption who could benefit from bone-targeted treatment aimed at inhibiting osteoclast action (45). Clinical trials have shown that RANKL inhibition significantly delays the occurrence of bone-related events in patients with confirmed bone metastases (46). Therefore, RANKL has become a key factor in the treatment of BC bone metastasis and may provide a unique opportunity for preventing bone metastasis in the future.
The transforming growth factor-β (TGF-β) family members signal via membrane-bound, heteromeric, serine-threonine kinase receptor complexes, whose activation by TGF-β ligands leads to phosphorylation of proteins of the SMAD family (Figure 3). The latter, in turn, accumulate in the nucleus and act as transcription factors to regulate target-gene expression, acting either directly on SMAD-specific cis-elements on DNA or via physical interaction with other transcription factors acting on their cognate DNA recognition sites. Negative control of the cell cycle drives the tumor suppressor functions of TGF-β in normal and premalignant tissues. On the other hand, TGF-β, which is secreted abundantly by tumor cells as well as by the local microenvironment, promotes invasion and metastases of various neoplasms through autocrine and paracrine mechanisms (47, 48). Of note, TGF-β induces epithelial-mesenchymal transition (EMT), whereby epithelial tumor cells acquire an invasive, mesenchymal-like phenotype accompanied by changes in the expression of cell-cell adhesion molecules and secretion of metalloproteinases, leading to metastasis (49). TGF-β is a critical mediator of bone metastasis, whereby complex bidirectional interactions between tumor cells and the bone microenvironment increase bone destruction and establishment of metastases in the bone. In fact, TGF-β signaling blockade by some modalities [i.e., over- expression of either a dominant-negative form of TGF-β receptor type II (50) or SMAD7 (51) in breast cancer cells (52, 53) was shown to be effective in reducing bone metastases.
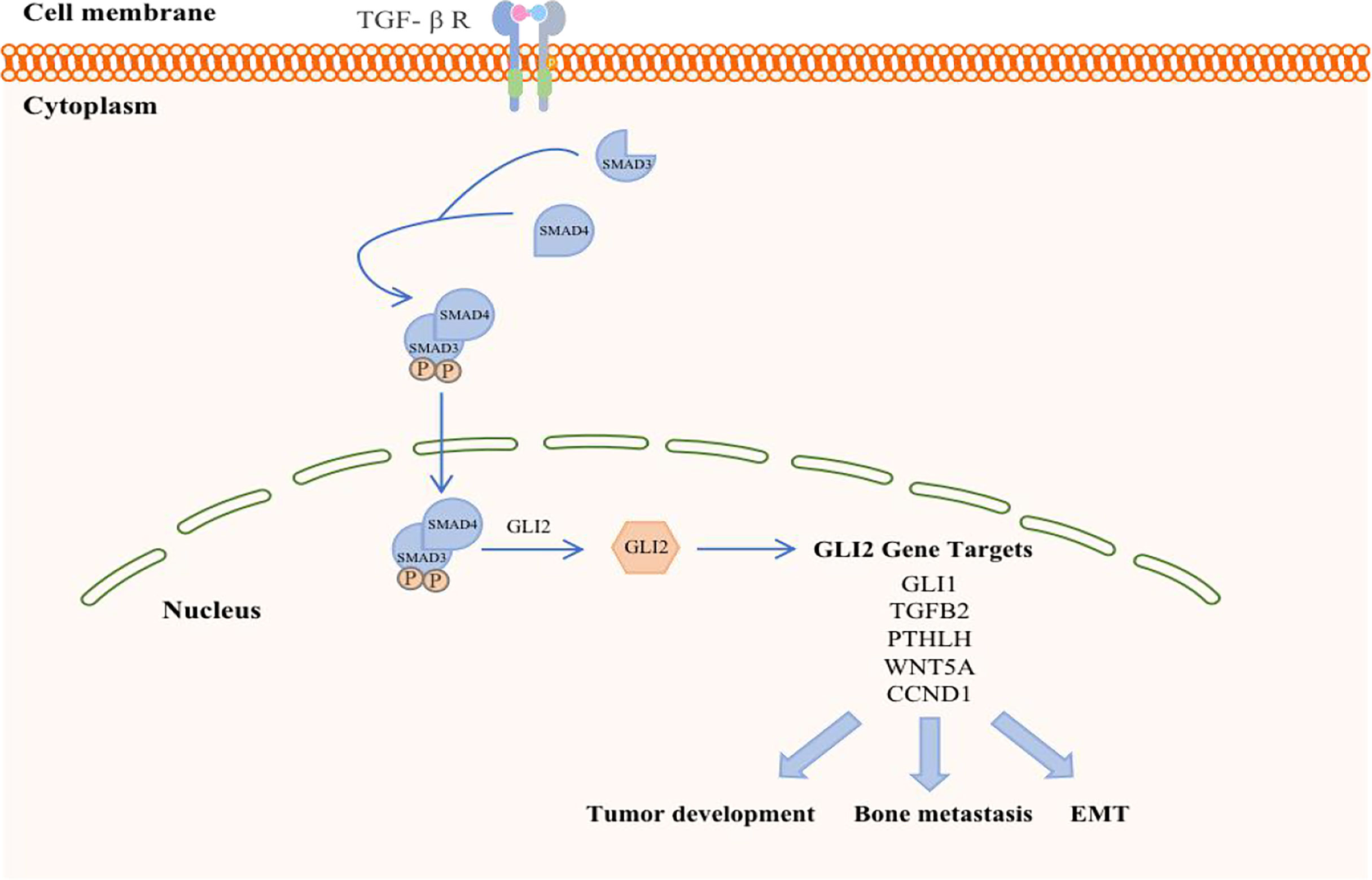
Figure 3 Schematic representation of TGF-β in breast cancer. TGF-β ligand activation of specific cell surface serine–threonine kinase receptors activates the SMAD cascade, resulting in transcriptional activation of the GLI2 gene. GLI2 protein may either regulate target gene expression and exert prooncogenic activities downstream of TGF-β signaling.
TGF-β signaling forms a feedback pathway in the occurrence and development of BC, accelerates the progression and metastasis of breast tumors, and participates in the regulation of other pathways to amplify tumor effects (54). First, the TGF-β signaling in BC cells and osteoclasts is activated in the context of BC bone metastasis. BC cells respond to TGF-β stimulation when they reach the bone and promote the maturation of osteoclasts by secreting cytokines such as parathyroid hormone (PTHLH). Mature osteoclasts in turn induce bone digestion, leading to the release of a variety of growth factors, including TGF-β embedded in the bone matrix, which further stimulates breast tumor cells (55). Therefore, the reactivity of TGF-β is a prerequisite for breast tumor cells to initiate osteolytic metastasis. The absence of TGF-β signal transduction in the myeloid system inhibits BC bone metastasis, especially in the development of breast cancer-induced osteolytic bone lesions. Furthermore, bFGF, which is a key molecule in myeloid-specific TGF-β signal transduction, can activate the downstream MAPK-ERK-cFOS pathway by binding to FGFR1 to mediate the development of BC bone damage (10). However, TGF-β signal transduction is not necessary for cellular autonomy in normal physiological conditions (56), and the loss of myeloid-specific TGF-β signaling in bones has other compensatory mechanisms (57).
In breast tissue, the basic function of IGF is to promote cell proliferation and differentiation through endocrine, paracrine and autocrine mechanisms, and IGF has an antiapoptotic effect. In fact, IGF acts not only on normal cells but also on tumor cells (58). IGF is also widely present in bones. However, the bone microenvironment is conducive for tumor survival because it harbors several growth factors (59), including insulin-like growth factors (IGFs). IGF promotes cancer progression, invasiveness and treatment resistance by activating the IGF receptor (IGFR) and various insulin receptors (11). In the context of bone biology, IGF contributes to the homing, dormancy, colonization and expansion of bone metastases. Preclinical evidence shows that IGF-1 and IGF-1R are the main factors associated with BC bone metastasis. IGF-1, a potent mitogen, activates IGF-1Rβ receptor tyrosine kinase during bone metastasis (60). The activation of IGF-1Rβ triggers downstream signaling including the PI3K/Akt pathway, a critical driver of tumor growth (61)
In primary tumors, a high level of IGF-1 in the environment can promote tumor cell metastasis to bone, suggesting that BC bone metastasis may be IGF-dependent (11). In addition, high expression of IGF-1R is important in malignant cell transformation. Jerome et al. found that IGF-1R could mediate the mitotic functions of normal and malignant breast cells, and high activity and expression of IGF-1R have been associated with BC (62). These results indicate that IGF-1R plays an important role in the pathogenesis of BC. Dunn et al. found that the adhesion, infiltration and metastasis of BC cells are also related to IGF-1R. Metastatic BC cell lines exhibited reduced collagen adhesion by 88% and invasion by 75% after the activation of IGF-1R was blocked by transfection, and when implanted in the breast, distant organ metastasis was also significantly reduced (63). furthermore, the IGF-1Rβ levels are increased in breast cancer metastasis and disruption of IGF-1/IGF-1R signaling inhibited tumorigenesis in preclinical models (64). Therefore, research on drugs related to blocking the activation of IGF-1R will be helpful in blocking BC bone metastasis.
Currently, there are many tumor treatments based on IGF, such as blocking the synthesis and secretion of IGF-1, blocking the binding of IGF-1 to its receptor, downregulating IGF-1, downregulating and antagonizing IGF-1R, and blocking receptor activity (65, 66). These drugs may not only be suitable for the treatment of carcinoma in situ but may be effective in patients with BC bone metastases.
The phosphoinositide 3-kinase (PI3K)-AKT-mTOR signal transduction pathway is present in a variety of cells in the body and participates in various physiological processes, such as cell metabolism, mitosis, and cell differentiation. This pathway is also one of the most common signaling pathways in a variety of cancers, regulating tumor cell proliferation, apoptosis, invasion and metastasis (67) (Figure 4). It is currently known that PI3K/mTOR proteins are highly expressed in human BC, which is associated with the development of breast cancer and osteolysis in vivo and in vitro. In addition, inhibition of the PI3K-AKT-mTOR signaling pathway can suppress breast cancer growth and breast cancer-induced osteoclast formation (68, 69).
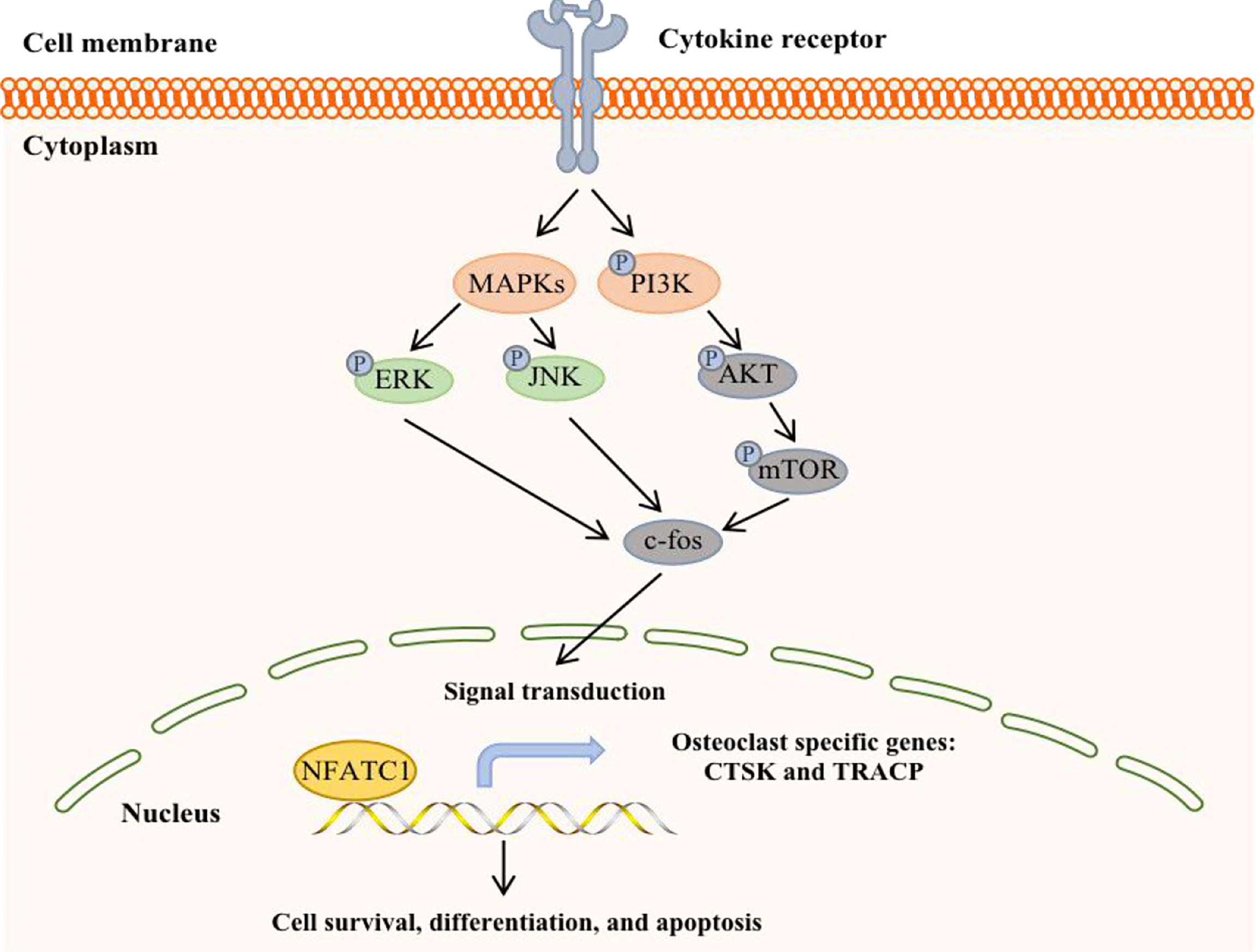
Figure 4 A potential working model in bone metastatic breast cancer.
Studies have shown that BC cells invade local lymph nodes via the PI3K-AKT-mTOR pathway and promote the expression of MMP-9 (70). mPRα promotes the angiogenesis and metastasis of BC cells by downregulating the connective tissue growth factor-mediated PI3K/AKT/ERK signaling pathway. Furthermore, the level of p-AKT in the spinal cord in BC bone metastasis pain model rats was significantly increased, indicating that the PI3K/AKT signaling pathway was activated. There was a study provides in vivo evidence that PI3K/mTOR activity is critical for the metastatic process in a model of bone metastasis. The highly bone metastatic variant of the MDA-MB-231 breast cancer model, 1833, showed PI3K/mTOR activation, high levels of p27pT157 and p27pT198, and p27-dependent motility/invasion in vitro (71). In fact, it has been found that BC bone metastasis could follow the activation of PI3K/protein kinase B (PKB)/AKT/ERK/cAMP response element-binding protein (CREB) signaling pathway, which induces EMT change and MMP2/MMP9 expression. In addition, hypoxia-inducible factor 1α (HIF-1α) and vascular endothelial growth factor (VEGF) are activated through activating the PI3K/AKT/mTOR signaling pathway, which promotes tumor angiogenesis. What is more, colony formation, migration, and invasion of BC cells by downregulating tumor suppressor (p53) and stimulating invasion-associated factor (MMP-9) (72).
In regards to the inhibitors, PKI-402 can induce osteoclast formation and reduce the expression of osteoclast-specific genes in RANKL-induced bone marrow-derived mouse macrophages and reduce the proliferation, migration and invasion of triple-negative breast cancer cell lines, such as MDA-MB-231 and MDA-MB-468 (67). In addition, PKI-402 can also prevent bone destruction caused by BC by inhibiting the PI3K-AKT-mTOR signaling pathway. In addition, GSK690693 is a specific inhibitor of AKT that can inhibit AKT signal transmission, reduce the expression of downstream effectors of p-AKT, and block the downstream signal cascade, thereby exerting biological effects (73). Intrathecal injection of GSK690693 significantly decreased the expression of p-AKT in the spinal cords of rats and inhibited the PI3K/AKT signaling pathway, thereby improving pain symptoms in rats. Furthermore, Asperolide A can inhibit PI3K/AKT/mTOR activation, leading to inhibited proliferation, inhibited migration, inhibited invasion, enhanced apoptosis, and arrest of G2M/S phase cell cycle in breast cancer cells as same as BEZ235 (inhibitor of PI3K/mTOR), and it hindered the phosphorylation of PI3K, AKT, mTOR, JNK, ERK, but not P65 or P38. It is also worth noting that the IGF-1R inhibitor AZD3463 can also inhibit BC bone metastasis by regulating the PI3K-AKT pathway (74). Therefore, there may be a beneficial relationship between the IGF system and the PI3K-AKT-mTOR signaling pathway in BC bone metastasis.
WNT signaling pathway, consisting of canonical and non-canonical branches, is believed to be responsible for control over various types of stem cells and may act as a niche factor to maintain stem cells in a self-renewing state (Figure 5). Moreover, dysregulated Wnt signaling pathway is strongly associated with several diseases including BC (75). Canonical WNT signalling is mediated by β-catenin and activates the TCF/LEF family of transcription factors, while non- canonical WNT/Ca2+ and WNT/planar cell polarity (PCP) pathways are independent of β-catenin. The WNT/Ca2+ pathway activates calcineurin, Ca2+/calmodulin-dependent protein kinase II (CaMKII) and protein kinase C (PKC), further leading to activation of multiple downstream signalling pathways. In fact, Wnt signaling has been shown to be overactivated in both breast cancers induced bone metastases. In particular, WNT signaling act as an important determinant in the cancer induced bone lesions, especially in controlling osteoblastic effect within tumor-harboring bone environment (76, 77).
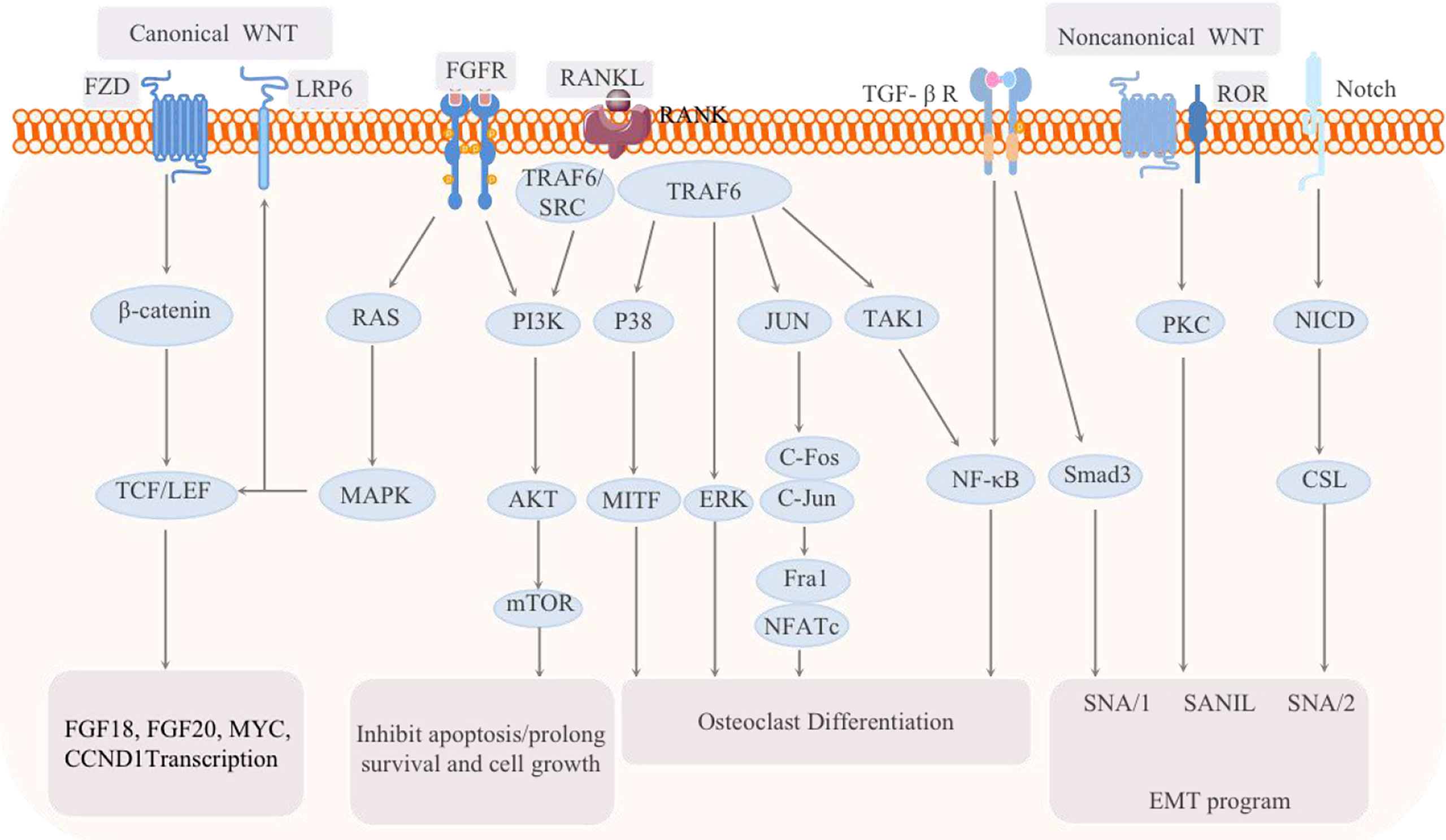
Figure 5 Schematic of the signaling pathways related to breast cancer bone metastasis: the classic WNT pathway and the noncanonical WNT pathway, as well as the FGF-FGFR, RANK-RANKL, TGF, and Notch pathways, are all involved in the regulation of breast cancer bone metastasis.
In vivo selection of metastatic cells from independent human breast cancer cell lines yields derivatives that harbor a hyperactive WNT signaling, in association with an enhanced ability to infiltrate and colonize the bones. Two WNT target genes, the transcription factors HOXB-9 and LEF-1, were identified as mediators of chemotactic invasion and colony outgrowth. These findings indicate activation of WNT signaling as a determinant of metastasis to bone during cancer progression. In fact, the overexpression of miR-574-5p seems to be associated with cancer bone metastasis involving aberrant WNT activation. The miR-574-5p, which functions to suppress Qki6/7, was reported to be upregulated in cancer samples that metastasized to bone when in comparison with the non-metastatic samples.
In addition, bone marrow-derived IL-1β stimulates the growth of BC cells in bone by inducing NF-κB and CREB signaling, leading to autocrine Wnt signaling and CSC colony formation (78). Some studies have shown that inhibiting this effect can prevent CSCs from forming colonies in the bone environment and prevent bone metastasis (78). Therefore, targeting IL-1β-NF-κB/CREB-Wnt signal transduction can be used as a therapy to prevent BC bone metastasis. It is currently known that IL-1β is a key cytokine produced by immune cells and nonimmune cells in human bone marrow that can enhance the ability of BC cells to form colonies (79, 80). Recently, it has been shown that tumor-derived Wnt can also promote the secretion of IL-1β by macrophages, which can accelerate the progression of BC bone metastasis; therefore, inhibiting this signal transduction pathway will delay disease progression. IL-1β-NF-κB/CREB Wnt signaling is a new way to promote BC cell colonization in the bone microenvironment. Drugs targeting this pathway can prevent not only bone metastasis in vivo but also colony formation of mammary stem cells in vitro (78).
The Hippo signaling pathway plays an important role in the occurrence, development and metastasis of BC. The core component of the Hippo signaling pathway is the transcriptional coactivator protein TAZ, and studies have shown that TAZ expression and activity was upregulated in highly metastatic breast cancer (81). Abelson tyrosine protein kinase (ABL) enhances the expression of TAZ and STAT5, which together promote BC bone metastasis (82). Similar to TAZ, YAP can also act as an oncogene to promote the occurrence and progression of BC in vivo and in vitro. YAP mainly promotes the growth, progression and metastasis of BC cells by interacting with transcription factors such as TEAD.
MST1/2 and LATS1/2, which are upstream signals of TAZ/YAP, are essential for the regulation of TAZ/YAP (83). The phosphorylation of Tyr1307 by HER3 can methylate the Lys59 site of MST1, which leads to the activation of YAP and TAZ in tumor cells and ultimately promotes bone metastasis (84). Bartucci et al. found that the nuclear expression of TAZ in bone metastases was significantly higher than that in primary tumors (85). It is believed that TAZ and YAP function as oncogenes in BC. However, notably, role of YAP as an oncogene is still controversial. Some researchers have pointed out that YAP can also act as a tumor suppressor gene and inhibit the development of BC. The mechanism remains to be studied in the future.
In addition, the hypoxic microenvironment in the bone marrow is conducive to tumor infiltration, which may regulate BC bone metastasis via the Hippo signaling pathway. HIF-1α is considered to be an indicator of hypoxia, and there is evidence that the transactivation of HIF-1 is regulated by the interaction between E-cadherin and Hippo pathway factors (86). Bendinelli et al. reported that HIF-1α interacts with TAZ and stimulates BC bone metastasis in a hypoxic microenvironment (87, 88). Hypoxia enhances the colocalization of TAZ and HIF-1α in the nuclei of human 1833 cells and interferes with the DNA binding activity of the HIF-1-dimer complex. In addition, oxidative stress/COX-2 may be a molecular connection between hypoxia stimulation, the Hippo pathway and the transcriptional regulator Snail. Blocking Cox-2 can downregulate the expression of HIF-1α and Snail in the nuclei of hypoxic 1833 cells and phosphorylate TAZ by interacting with LATS. The localization of LATS in the nucleus can promote the translocation of TAZ in the cytoplasm, mediate the phosphorylation and degradation of TAZ, and inhibit the entry of TAZ into the nucleus, modulating the transcriptional coactivation of TAZ and preventing the occurrence of tumors (88).
Therefore, YAP/TAZ inhibitors and COX-2 inhibitors can be studied in drug research and development and may serve as solutions to prevent tumor progression and reverse the tumor microenvironment to break the vicious cycle.
In fact, there are some other pathways associated with breast cancer bone metastasis. For example, bFGF is known to critically regulate self-renewal and proliferation of bone marrow derived MSCs. Therefore, activation of the bFGF pathway in breast cancer cells-bone marrow crosstalk can be speculated to foster a tumorigenic priming of the niche to harbor and support invading tumor cells (89, 90). In addition, some literature notes that there is a significant positive correlation between serum androgen levels and breast cancer risk (91). The androgen-AR/LSD1-target gene pathway seems to also be related to BC bone metastasis. Androgen can activate the estrogen receptor alpha (ERα, ESR1) signaling pathway in BC (92). However, some experts believe that AR is not an oncogene and may a tumor suppressor in some cases (93, 94), and further investigations are needed. The Notch pathway is also involved in BC bone metastasis, and it has been shown to have antagonistic or synergistic effects with the Ras pathway in different environments (95).
Distant metastasis of breast cancer is a complex pathological process that requires multiple steps and is controlled by multiple genes and signaling pathways, including the stages of adhesion, degradation, and migration (96). First, breast tumor cells have reduced adhesion and detach from the original site, and their surface receptors specifically bind to certain components of the basement membrane and extracellular matrix. Second, tumor cells secrete matrix metalloproteinases (MMPs) to degrade the protein components of the ECM, causing the ECM to lose its blocking effect. Eventually, the tumor cells spread from the original site, invade and enter lymphatic and blood vessels via defective areas in the ECM and metastasize to the distant site (29). As a nearby organ, bone tissue contains a large number of cytokines and provides conditions and an environment that supports for BC metastasis.
This article summarizes the current pathways related to BC bone metastasis, and the interactions are shown in Figure 5. It has been noted that the downstream p38, JNK, NF-κB, ERK and AKT pathways can all be activated during the activation and survival of osteoclasts via by the RANKL/RANK pathway (97). In BC T47D cells, RANKL activates the AKT and ERK1/2 pathways. The comprehensive signaling network composed of TGF-β, FGF, NF-κB, WNT, PI3K and JAK-STAT is also essential for the progression of BC bone metastasis. Key bone transfer factors, such as vascular cell adhesion molecule 1 (VCAM1), RANKL, PTHrP, and other important mediators of these pathways, have further confirmed this signaling network (97). It is worth noting that the RANKL/RANK system is an important molecular link between progesterone and epithelial carcinogenesis (35). Therefore, inhibition of this system can also be used to prevent and/or treat hormone-related BC. However, a new question must be considered. Is BC with progesterone receptor positivity more prone to bone metastasis than other subtypes?
In addition, a basic fibroblast growth factor (bFGF)-mediated, synergistic increase in proliferation of breast cancer cells and mesenchymal stromal cells (MSCs) in co-culture. The stromal induction was associated with elevated phosphoinositide-3 kinase (PI3K) signaling in the stroma, which coupled with elevated bFGF levels resulted in increased migration of breast cancer cells towards the MSCs. The perturbed cytokine profile in the stroma led to reduction in the osteogenic differentiation of MSCs via downregulation of platelet-derived growth factor-BB (PDGF-BB) (98). FGF and NF-κB are both important inducers of EMT and metastasis, and the NF-κB and TGF-β signaling pathways synergistically enhance EMT and cancer metastasis (12). Wnt signaling is one of the most important ways that induces EMT and breast stem cells (79). In addition, the Wnt and TGF-β signaling pathways may also induce a mutually reinforcing autocrine signaling network, which is essential for the sustained expression of EMT-related transcription factors and CSCs. The FGF15/19-FGFR4 signaling pathway can activate Wnt/GSK-3β/β-catenin (99, 100) and MST1/2 (101, 102). FGFs induce the dimerization, activation and tyrosine phosphorylation of FGFRs and the subsequent phosphorylation of FGFR substrate 2α (FRS2α) and phospholipase Cγ (PLCγ), thereby activating the RAS-ERK and PI3K-AKT-IP3-Ca2+, diacylglycerol (DAG)-protein kinase C (PKC) signaling pathways (103). Among them, FGF signal transduction to the RAS-MAPK branch and the typical Wnt signaling cascade mutually regulate transcription; FGF signal transduction to the PI3K-AKT, Hedgehog, Notch, TGF-β and noncanonical WNT signaling cascades regulates EMT and invasion (104). Therefore, there is crosstalk between the FGF-FGFR pathway and the WNT and Hippo signaling pathways. The PI3K signaling pathway also plays an important role in inducing EMT and metastasis (105).
Interesting, it has been found that applied mechanical tension can dramatically alter gene expression in breast cancer cells, leading to decreased proliferation, increased resistance to chemotherapeutic treatment and enhanced adhesion to inflamed endothelial cells and collagen I under fluidic shear stress. A mechanistic analysis of the pathways involved in these effects supported a complex signaling network that included Abl1, Lck, Jak2 and PI3K to regulate pro-survival signaling and enhancement of adhesion under flow. Studies using mouse xenograft models demonstrated reduced proliferation of breast cancer cells with orthotopic implantation and increased metastasis to the bones when the cancer cells were treated with mechanical load (75). Furthermore, hypoxia and hypoxia-inducible factors (HIFs) also play important roles in BC bone metastasis. HIF can directly regulate genes. In addition, some genes are directly regulated by hypoxia, such as TFF3, EGLN1, SNAI1, MMP9, TGFB3, SLC2A3 and CTGF (87). Therefore, hypoxia and HIFs may be important factors that regulate the EMT status of primary and secondary tumors.
In short, signaling pathways interact with each other to promote the development of BC bone metastasis. The secretion of various growth factors creates a vicious cycle, amplifying the signals associated with BC bone metastasis and promoting “seed” germination in the “soil”. It is not enough to understand the soil and seeds alone, and we should also increase efforts to thoroughly understand the various signaling pathways and their potential relationships. This understanding can truly prevent BC bone metastasis and lay a solid foundation for future treatment to relieve the pain and prolong the life of BC patients with bone metastases.
XS wrote this manuscript. XS and CW drew the pictures in this paper. In addition, XS, CW, and XL revised the manuscript. All authors contributed to the article and approved the submitted version.
The present study was supported by the National Natural Science Foundation of China (grant no. 81473687), the Academic Promotion Program of Shandong First Medical University (grant no. 2019QL017), the Natural Science Foundation of Shandong Province (grant no. ZR2020MH357, ZR2020MH312), Tai’an Science and Technology Innovation Development Project (grant no.2020NS092).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA: A Cancer J Clin (2020) 70(1):7–30. doi: 10.3322/caac.21590
2. Desantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Sauer AG, et al. Breast Cancer Statistics, 2019. CA: A Cancer J Clin (2019) 69(6):438–51. doi: 10.3322/caac.21583
3. Delrieu L, Pérol O, Fervers B, Friedenreich C, Vallance J, Febvey-Combes O, et al. A Personalized Physical Activity Program With Activity Trackers and a Mobile Phone App for Patients With Metastatic Breast Cancer: Protocol for a Single-Arm Feasibility Trial. JMIR Res Protoc (2018) 7(8):e10487. doi: 10.2196/10487
4. Tahara RK, Brewer TM, Theriault RL. Bone Metastasis of Breast Cancer. Adv Exp Med Biol (2019) 1152(1):105–29. doi: 10.1007/978-3-030-20301-6_7
5. Tulotta C, Ottewell P. The Role of IL-1B in Breast Cancer Bone Metastasis. Endocr Relat Cancer (2018) 25(7):421–34. doi: 10.1530/ERC-17-0309
6. Zhenchong X, Guangzheng D, Xinjian H, Xing L, Xie X, Wang J, et al. Bone Metastasis Pattern in Initial Metastatic Breast Cancer: A Population-Based Study. Cancer Manag Res (2018) 10(1):287–95. doi: 10.2147/CMAR.S155524
7. Liang Y, Zhang H, Song X, Yang Q. Metastatic Heterogeneity of Breast Cancer: Molecular Mechanism and Potential Therapeutic Targets. Semin Cancer Biol (2019) 60(1):14–27. doi: 10.1016/j.semcancer.2019.08.012
8. Wong KW, Ma WK, Wong CW, Wong MH, Tsang CF, Tsu HL, et al. Impact of Skeletal-Related Events on Survival in Patients With Metastatic Prostate Cancer Prescribed Androgen Deprivation Therapy. Hongkong Med J (2016) 22(2):106–15. doi: 10.12809/hkmj144449
9. Paget S. The Distribution of Secondary Growths in Cancer of the Breast.1889. Lancet (1989) 133(2):571–3. doi: 10.1016/S0140-6736(00)49915-0
10. Meng X, Ark AV, Lee P, Hostetter G, Bhowmick NA, Matrisian LM, et al. Myeloid-Specific TGF-β Signaling in Bone Promotes Basic-FGF and Breast Cancer Bone Metastasis. Oncogene (2016) 35(18):2370–85. doi: 10.1038/onc.2015.297
11. Rieunier G, Wu X, Macaulay VM, Lee AV, Weyer-Czernilofsky U, Bogenrieder T. Bad to the Bone: The Role of the Insulin-Like Growth Factor Axis in Osseous Metastasis. Clin Cancer Res (2019) 25(12):3479–85. doi: 10.1158/1078-0432.CCR-18-2697
12. Aukes K, Forsman C, Brady NJ, Astleford K, Blixt N, Sachdev D, et al. Breast Cancer Cell-Derived Fibroblast Growth Factors Enhance Osteoclast Activity and Contribute to the Formation of Metastatic Lesions. PloS One (2017) 12(10):e0185736. doi: 10.1371/journal.pone.0185736
13. Lev DC, Kim SJ, Onn A, Stone V, Nam D, Yazici S, et al. Inhibition of Platelet-Derived Growth Factor Receptor Signaling Restricts the Growth of Human Breast Cancer in the Bone of Nude Mice. Clin Cancer Res (2005) 11(1):306–14.
14. Zabkiewicz C, Resaul J, Hargest R, Jiang WG, Ye L. Bone Morphogenetic Proteins, Breast Cancer, and Bone Metastases: Striking the Right Balance. Endocr Relat Cancer (2017) 24(10):349–66. doi: 10.1530/ERC-17-0139
15. Holen I, Lefley DV, Francis SE, Rennicks S, Bradbury S, Coleman RE, et al. IL-1 Drives Breast Cancer Growth and Bone Metastasis In Vivo. Oncotarget (2016) 7(46):75571–84. doi: 10.18632/oncotarget.12289
16. Luyan Z, Xifa W, Yong F, Linlin Z, Jinbo J. Selenium Donors Inhibits Osteoclastogenesis Through Inhibiting IL-6 and Plays a Pivotal Role in Bone Metastasis From Breast Cancer. Toxicol Res (2020) 9(4):544–51. doi: 10.1093/toxres/tfaa053
17. Kamalakar A, Bendre MS, Washam CL, Fowler TW, Carver A, Dilley JD, et al. Circulating Interleukin-8 Levels Explain Breast Cancer Osteolysis in Mice and Humans. Bone (2014) 61(1):176–85. doi: 10.1016/j.bone.2014.01.015
18. Benoy IH. Increased Serum Interleukin-8 in Patients With Early and Metastatic Breast Cancer Correlates With Early Dissemination and Survival. Clin Cancer Res (2004) 10(21):7157–62. doi: 10.1158/1078-0432.CCR-04-0812
19. Liang M, Ma Q, Ding N, Luo F, Bai Y, Kang F, et al. IL-11 is Essential in Promoting Osteolysis in Breast Cancer Bone Metastasis via RANKL-Independent Activation of Osteoclastogenesis. Cell Death Dis (2019) 10(5):353–63. doi: 10.1038/s41419-019-1594-1
20. Hamaguchi T, Wakabayashi H, Matsumine A, Sudo A, Uchida A. TNF Inhibitor Suppresses Bone Metastasis in a Breast Cancer Cell Line. Biochem Bioph Res Co (2011) 407(3):525–30. doi: 10.1016/j.bbrc.2011.03.051
21. Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan Y, et al. VCAM-1 Promotes Osteolytic Expansion of Indolent Bone Micrometastasis of Breast Cancer by Engaging Alpha 4 Beta 1-Positive Osteoclast Progenitors. Cancer Cell (2011) 20(6):701–14. doi: 10.1016/j.ccr.2011.11.002
22. Goldhar AS, Vonderhaar BK, Trott JF, Hovey RC. Prolactin-Induced Expression of Vascular Endothelial Growth Factor via Egr-1. Mol Cell Endocrinol (2005) 232(1-2):9–19. doi: 10.1016/j.mce.2005.01.005
23. Wang J, Wang L, Xia B, Yang C, Lai H, Chen X. BSP Gene Silencing Inhibits Migration, Invasion, and Bone Metastasis of MDA-MB-231bo Human Breast Cancer Cells. PloS One (2013) 8(5):e62936. doi: 10.1371/journal.pone.0062936
24. Chimge NO, Frenkel B. The RUNX Family in Breast Cancer: Relationships With Estrogen Signaling. Oncogene (2013) 32(17):2121–30. doi: 10.1038/onc.2012.328
25. Tingting Q, Yancheng L, Yan D, Lei Z, Yining D, Yanhui S, et al. CXCR7 Regulates Breast Tumor Metastasis and Angiogenesis Invivo and Invitro. Mol Med Rep (2018) 17(1):3633–9. doi: 10.3892/mmr.2017.8286
26. Casimiro S, Mohammad KS, Pires R, Tato-Costa J, Alho I, Teixiera J, et al. RANKL/RANK/MMP-1 Molecular Triad Contributes to the Metastatic Phenotype of Breast and Prostate Cancer Cells In Vitro. PloS One (2013) 8(5):e63153. doi: 10.1371/journal.pone.0063153
27. Azim H, Azim HA. Targeting RANKL in Breast Cancer: Bone Metastasis and Beyond. Expert Rev Anticanc (2013) 13(2):195–201. doi: 10.1586/ERA.12.177
28. Guise TA. Breast Cancer Bone Metastases: It’s All About the Neighborhood. Cell (2013) 154(5):957–9. doi: 10.1016/j.cell.2013.08.020
29. Brook N, Brook E, Dharmarajan A, Dass CR, Chan A. Breast Cancer Bone Metastases: Pathogenesis and Therapeutic Targets. Int J Biochem Cell B (2018) 96(1):63–78. doi: 10.1016/j.biocel.2018.01.003
30. Tawara K, Oxford JT, Jorcyk CL. Clinical Significance of Interleukin (IL)-6 in Cancer Metastasis to Bone: Potential of Anti-IL-6 Therapies. Cancer Manag Res (2011) 3(21625400):177–89. doi: 10.2147/CMR.S1810
31. Shao X, Cao F, Tao M. The Clinical Value of Hepcidin in Breast Cancer and Its Bone Metastasis. Ann Clin Lab Sci (2017) 47(2):120–8.
32. Sasaki H, Yu CY, Dai M, Tam C, Loda M, Auclair D, et al. Elevated Serum Periostin Levels in Patients With Bone Metastases From Breast But Not Lung Cancer. Breast Cancer Res Treat (2003) 77(3):245–52. doi: 10.1023/A:1021899904332
33. Wu Z, Wang T, Fang M, Huang W, Sun Z, Xiao J, et al. MFAP5 Promotes Tumor Progression and Bone Metastasis by Regulating ERK/MMP Signaling Pathways in Breast Cancer. Biochem Bioph Res Co (2018) 498(3):495–501. doi: 10.1016/j.bbrc.2018.03.007
34. McGrath EE. OPG/RANKL/RANK Pathway as a Therapeutic Target in Cancer. J Thorac Oncol (2011) 6(9):1468–73. doi: 10.1097/JTO.0b013e318229421f
35. Hanada R, Hanada T, Sigl V, Schramek D, Penninger JM. RANKL/RANK-Beyond Bones. J Mol Med (2011) 89(7):647–56. doi: 10.1007/s00109-011-0749-z
36. Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, et al. Osteoprotegerin Ligand is a Cytokine That Regulates Osteoclast Differentiation and Activation. Cell (1998) 93(2):165–76. doi: 10.1016/s0092-8674(00)81569-x
37. Li XM, Liu YM, Wu B, Dong ZL, Wang YC, Lu JZ, et al. Potential Role of the OPG/RANK/RANKL Axis in Prostate Cancer Invasion and Bone Metastasis. Oncol Rep (2014) 32(6):2605–11. doi: 10.3892/or.2014.3511
38. Ferreira A, Alho I, Vendrell I, Melo M, Brás R, Costa AL, et al. The Prognostic Role of RANK SNP Rs34945627 in Breast Cancer Patients With Bone Metastases. Oncotarget (2016) 7(27):41380–9. doi: 10.18632/oncotarget.9356
39. Kiesel L, Kohl A. Role of the RANK/RANKL Pathway in Breast Cancer. Maturitas (2016) 86(1):10–6. doi: 10.1016/j.maturitas.2016.01.001
41. Azim HA Jr, Peccatori FA, Brohée S, Branstetter D, Loi S, Viale G, et al. RANK-Ligand (RANKL) Expression in Young Breast Cancer Patients and During Pregnancy. Breast Cancer Res (2015) 17:24. doi: 10.1186/s13058-015-0538-7
42. Drooger JC, van der Padt A, Sleijfer S, Jager A. Denosumab in Breast Cancer Treatment. Eur J Pharmacol (2013) 717(1-3):12–9. doi: 10.1016/j.ejphar.2013.03.034
43. Dougall WC, Chaisson M. The RANK/RANKL/OPG Triad in Cancer-Induced Bone Diseases. Cancer Metast Rev (2006) 25(4):541–9. doi: 10.1007/s10555-006-9021-3
44. Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, et al. The Osteoclast Differentiation Factor Osteoprotegerin-Ligand is Essential for Mammary Gland Development. Cell (2000) 103(1):41–50. doi: 10.1016/S0092-8674(00)00103-3
45. Ibrahim T, Ricci M, Scarpi E, Bongiovanni A, Ricci R, Riva N, et al. RANKL: A Promising Circulating Marker for Bone Metastasis Response. Oncol Lett (2016) 12(4):2970–5. doi: 10.3892/ol.2016.4977
46. Stopeck AT, Lipton A, Body JJ, Steger GG, Tonkin K, Boer RH, et al. Denosumab Compared Withzoledronic Acid for the Treatment of Bone Metastases in Patients Withadvanced Breast Cancer: A Randomized, Double-Blind Study. J Clin Oncol (2010) 28(35):5132–9. doi: 10.1200/JCO.2010.29.7101
47. Zhao Y, Ma J, Fan Y, Wang Z, Tian R, Ji W, et al. TGF-Beta Transactivates EGFR and Facilitates Breast Cancer Migration and Invasion Through Canonical Smad3 and ERK/Sp1 Signaling Pathways. Mol Oncol (2018) 12(3):305–21. doi: 10.1002/1878-0261.12162
48. Javelaud D, Alexaki VI, Mauviel A. Transforming Growth Factor-Beta in Cutaneous Melanoma. Pigment Cell Melanoma Res (2008) 21:123–32. doi: 10.1111/j.1755-148X.2008.00450.x
49. Meulmeester E, Ten Dijke P. The Dynamic Roles of TGF-Beta in Cancer. J Pathol (2011) 223:205–18. doi: 10.1002/path.2785
50. Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BJ, Wieser R, et al. TGF-Beta Signaling Blockade Inhibits PTHrP Secretion by Breast Cancer Cells and Bone Metastases Development. J Clin Invest (1999) 103:197–206. doi: 10.1172/JCI3523
51. Azuma H, Ehata S, Miyazaki H, Watabe T, Maruyama O, Imamura T, et al. Effect of Smad7 Expression on Metastasis of Mouse Mammary Carcinoma JygMC(A) Cells. J Natl Cancer Inst (2005) 97:1734–46. doi: 10.1093/jnci/dji399
52. Javelaud D, Delmas V, Mo€ller M, Sextius P, André J, Menashi S, et al. Stable Overexpression of Smad7 in Human Melanoma Cells Inhibits Their Tumorigenicity In Vitro and In Vivo. Oncogene (2005) 24:7624–9. doi: 10.1038/sj.onc.1208900
53. Javelaud D, Mohammad KS, McKenna CR, Fournier P, Luciani F, Niewolna M, et al. Stable Overexpression of Smad7 in Human Melanoma Cells Impairs Bone Metastasis. Cancer Res (2007) 67:2317–24. doi: 10.1158/0008-5472.CAN-06-3950
54. Novitskiy SV, Pickup MW, Chytil A, Polosukhina D, Owens P, Moses HL, et al. Deletion of TGF-β Signaling in Myeloid Cells Enhances Their Anti-Tumorigenic Properties. J Leukoc Biol (2012) 92(3):641–51. doi: 10.1189/jlb.1211639
55. Kang Y. Dissecting Tumor-Stromal Interactions in Breast Cancer Bone Metastasis. Endocrinol Metab (2016) 31(2):206–12. doi: 10.3803/EnM.2016.31.2.206
56. Maroni P, Bendinelli P, Matteucci E, Locatelli A, Nakamura T, Scita G, et al. Osteolytic Bone Metastasis is Hampered by Impinging on the Interplay Among Autophagy, Anoikis and Ossification. Cell Death Dis (2014) 5(1):e1005. doi: 10.1038/cddis.2013.465
57. Bendinelli P, Maroni P, Matteucci E, Desiderio MA. HGF and Tgfβ1 Differently Influenced Wwox Regulatory Function on Twist Program for Mesenchymal-Epithelial Transition in Bone Metastatic Versus Parental Breast Carcinoma Cells. Mol Cancer (2015) 14(1):112–29. doi: 10.1186/s12943-015-0389-y
58. Fukuda R. Insulin-Like Growth Factor 1 Induces Hypoxia-Inducible Factor 1-Mediated Vascular Endothelial Growth Factor Expression, Which is Dependent on MAP Kinase and Phosphatidylinositol 3-Kinase Signaling in Colon Cancer Cells. J Bio Chem (2002) 277(41):38205–11. doi: 10.1074/jbc.M203781200
59. Roodman GD. Mechanisms of Bone Metastasis. N Engl J Med (2004) 350:1655–64. doi: 10.1056/NEJMra030831
60. Hiraga T, Myoui A, Hashimoto N, Sasaki A, Hata K, Morita Y, et al. Bone-Derived IGF Mediates Crosstalk Between Bone and Breast Cancer Cells in Bony Metastases. Cancer Res (2012) 72:4238–49. doi: 10.1158/0008-5472
61. Riedemann J, Macaulay VM. IGF1R Signalling and its Inhibition. Endocr Relat Cancer (2006) 13(Suppl 1):S33–43. doi: 10.1677/erc.1.01280
62. Jerome L, Shiry L, Leyland-Jones B. Anti-Insulin-Like Growth Factor Strategies in Breast Cancer. Semin Oncol (2004) 31(3):54–63. doi: 10.1053/j.seminoncol.2004.01.007
63. Dunn SE, Ehrlich M, Sharp NJ, Reiss K, Solomon G, Hawkins R, et al. A Dominant Negative Mutant of the Insulin-Like Growth Factor-I Receptor Inhibits the Adhesion, Invasion, and Metastasis of Breast Cancer. Cancer Res (1998) 58(15):3353–61. doi: 10.1007/s00253-012-4439-8
64. Tandon M, Chen Z, Othman AH, Pratap J. Role of Runx2 in IGF-1rβ/Akt- and AMPK/Erk-Dependent Growth, Survival and Sensitivity Towards Metformin in Breast Cancer Bone Metastasis. Oncogene (2016) 35(36):4730–40. doi: 10.1038/onc.2015.518
65. Mohanraj L, Oh Y. Targeting IGF-I, IGFBPs and IGF-I Receptor System in Cancer: The Current and Future in Breast Cancer Therapy. Recent Pat Anti-Canc (2011) 6(2):166–77. doi: 10.2174/157489211795328512
66. Motallebnezhad M, Aghebati-Maleki L, Jadidi-Niaragh F, Nickho H, Samadi-Kafil H, Shamsasenjan H, et al. The Insulin-Like Growth Factor-I Receptor (IGF-IR) in Breast Cancer: Biology and Treatment Strategies. Tumour Biol (2016) 2016(9):1–11. doi: 10.1007/s13277-016-5176-x
67. Yuan G, Lian Z, Liu Q, Lin X, Xie D, Song F, et al. Phosphatidyl Inositol 3-Kinase (PI3K)-mTOR Inhibitor PKI-402 Inhibits Breast Cancer Induced Osteolysis. Cancer Lett (2019) 443(1):135–44. doi: 10.1016/j.canlet.2018.11.038
68. Courtney KD, Corcoran RB, Engelman JA. The PI3K Pathway As Drug Target in Human Cancer. J Clin Oncol (2010) 28(6):1075–83. doi: 10.1200/JCO.2009.25.3641
69. Jiang WL, Rixiati Y, Huang H, Shi YJ, Huang CG, Jiao BH. Asperolide A Prevents Bone Metastatic Breast Cancer via the PI3K/AKT/mTOR/c-Fos/NFATc1 Signaling Pathway. Cancer Med (2020) 9(21):8173–85. doi: 10.1002/cam4.3432
70. Wu X, Sun L, Wang X, Su P, Li Z, Zhang C, et al. Breast Cancer Invasion and Metastasis by Mpralpha Through the PI3K/Akt Signaling Pathway. Pathol Oncol Res (2016) 22(1):471–6. doi: 10.1007/s12253-015-0023-8
71. Wander SA, Zhao D, Besser AH, Hong F, Wei J, Ince TA, et al. PI3K/mTOR Inhibition can Impair Tumor Invasion and Metastasis In Vivo Despite a Lack of Antiproliferative Action In Vitro: Implications for Targeted Therapy. Breast Cancer Res Treat (2013) 138(2):369–81. doi: 10.1007/s10549-012-2389-6
72. Mei D, Zhu Y, Zhang L, Wei W. The Role of CTHRC1 in Regulation of Multiple Signaling and Tumor Progression and Metastasis. Mediators Inflammation (2020) 2020:9578701. doi: 10.1155/2020/9578701
73. Rhodes N, Heerding DA, Duckett DR, Eberwein DJ, Knick VB, Lansing TJ, et al. Characterization of an Akt Kinase Inhibitor With Potent Pharmacodynamic and Antitumor Activity. Cancer Res (2018) 68(7):2366–74. doi: 10.1158/0008-5472.CAN-07-5783
74. Hu GF, Wang C, Hu GX, Wu G, Zhang CJ, Zhu W, et al. AZD3463, an IGF-1R Inhibitor, Suppresses Breast Cancer Metastasis to Bone via Modulation of the PI3K-Akt Pathway. Ann Transl Med (2020) 8(6):336. doi: 10.21037/atm.2020.02.110
75. Spencer A, Sligar AD, Chavarria D, Lee J, Choksi D, Patil NP, et al. Biomechanical Regulation of Breast Cancer Metastasis and Progression. Sci Rep (2021) 11(1):9838. doi: 10.1038/s41598-021-89288-z
76. Katoh M, Katoh M. Molecular Genetics and Targeted Therapy of WNT-Related Human Diseases. Int J Mol Med (2017) 40(3):587–606. doi: 10.3892/ijmm.2017.3071
77. Anastas JN, Moon RT. WNT Signalling Pathways as Therapeutic Targets in Cancer. Nat Rev Cancer (2013) 13:11–26. doi: 10.1038/nrc3419
78. Eyre R, Alférez DG, Santiago-Gómez A, Spence K, McConnell JC, Hart C, et al. Microenvironmental Il1β Promotes Breast Cancer Metastatic Colonisation in the Bone via Activation of Wnt Signalling. Nat Commun (2019) 10(1):5016–31. doi: 10.1038/s41467-019-12807-0
79. Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell (2019) 177(7):1915–32. doi: 10.1016/j.cell.2019.04.040
80. Wellenstein MD. Loss of P53 Triggers WNT-Dependent Systemic Inflammation to Drive Breast Cancer Metastasis. Nature (2019) 572(7770):538–42. doi: 10.1038/s41586-019-1450-6
81. Zhao D, Zhi X, Zhou ZM, Chen CS. TAZ Antagonizes the WWP1-Mediated KLF5 Degradation and Promotes Breast Cell Proliferation and Tumorigenesis. Carcinogenesis (2012) 33(1):59–67. doi: 10.1093/carcin/bgr242
82. Wang J, Rouse C, Jasper JS, Pendergast AM. ABL Kinases Promote Breast Cancer Osteolytic Metastasis by Modulating Tumor-Bone Interactions Through TAZ and STAT5 Signalling. Sci Signal (2016) 9(413):ra12–2. doi: 10.1126/scisignal.aad3210
83. Heidary Arash E, Shiban A, Song S, Attisano L. MARK4 Inhibits Hippo Signaling to Promote Proliferation and Migration of Breast Cancer Cells. EMBO Rep (2017) 18(3):420–36. doi: 10.15252/embr.201642455
84. Li C, Wang S, Xing Z, Lin A, Liang K, Song J, et al. A ROR1-HER3-lncRNA Signalling Axis Modulates the Hippo-YAP Pathway to Regulate Bone Metastasis. Nat Cell Biol (2017) 19(2):106–19. doi: 10.1038/ncb3464
85. Bartucci M, Dattilo R, Moriconi C, Pagliuca A, Mottolese M, Federici G, et al. TAZ is Required for Metastatic Activity and Chemoresistance of Breast Cancer Stem Cells. Oncogene (2015) 34(6):681–90. doi: 10.1038/onc.2014.5
86. Maroni P, Matteucci E, Drago L, Banfi G, Bendinelli P, Desiderio MA. Hypoxia Induced E-Cadherin Involving Regulators of Hippo Pathway Due to HIF-1 Stabilization/Nuclear Translocation in Bone Metastasis From Breast Carcinoma. Exp Cell Res (2015) 330(2):287–99. doi: 10.1016/j.yexcr.2014.10.004
87. Xiang L, Gilkes DM, Hu H, Luo WB, Bullen JW, Liang HJ, et al. HIF-1α and TAZ Serve as Reciprocal Co-Activators in Human Breast Cancer Cells. Oncotarget (2015) 6(14):11768–78. doi: 10.1111/j.1445-2197.2010.05546.x
88. Gilkes DM. Implications of Hypoxia in Breast Cancer Metastasis to Bone. Int J Mol Sci (2016) 17(10):1669–79. doi: 10.3390/ijms17101669
89. Sharpe R, Pearson A, Herrera-Abreu MT, Johnson D, Mackay A, Welti JC, et al. FGFR Signaling Promotes the Growth of Triple-Negative and Basal-Like Breast Cancer Cell Lines Both In Vitro and In Vivo. Clin Cancer Res (2011) 17(16):5275–86. doi: 10.1158/1078-0432.CCR-10-2727
90. Katoh M. Fibroblast Growth Factor Receptors as Treatment Targets in Clinical Oncology. Nat Rev Clin Oncol (2019) 16(2):105–22. doi: 10.1038/s41571-018-0115-y
91. Dorgan JF, Stanczyk FZ, Kahle LL, Brinton LA. Prospective Case-Control Study of Premenopausal Serum Estradiol and Testosterone Levels and Breast Cancer Risk. Breast Cancer Res (2010) 12(6):1–8. doi: 10.1186/bcr2779
92. Shufelt CL, Braunstein GD. Testosterone and the Breast. Menopause Int (2008) 14(3):117–22. doi: 10.1258/mi.2008.008015
93. Feng J, Li L, Zhang N, Liu J, Zhang L, Gao H, et al. Androgen and AR Contribute to Breast Cancer Development and Metastasis: An Insight of Mechanisms. Oncogene (2017) 36(20):2775–90. doi: 10.1038/onc.2016.432
94. Narayanan R, Ahn S, Cheney MD, Yepuru M, Miller DD, Steiner MS, et al. Selective Androgen Receptor Modulators (SARMs) Negatively Regulate Triple-Negative Breast Cancer Growth and Epithelial: Mesenchymal Stem Cell Signaling. PloS One (2014) 9(7):e103202. doi: 10.1371/journal.pone.0103202
95. Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM, Zlobin A, et al. Activation of Notch-1 Signaling Maintains the Neoplastic Phenotype in Human Ras-Transformed Cells. Nat Med (2002) 8(9):979–86. doi: 10.1038/nm754
96. Fornetti J, Welm AL, Stewart SA. Understanding the Bone in Cancer Metastasis. J Bone Miner Res (2018) 33(12):2099–113. doi: 10.1002/jbmr.3618
97. Fazilaty H, Gardaneh M, Bahrami T, Salmaninejad A, Behnam B. Crosstalk Between Breast Cancer Stem Cells and Metastatic Niche: Emerging Molecular Metastasis Pathway. Tumour Biol (2013) 34(4):2019–30. doi: 10.1007/s13277-013-0831-y
98. Dhawan A, von Bonin M, Bray LJ, Freudenberg U, Bejestani EP, Werner C, et al. Functional Interference in the Bone Marrow Microenvironment by Disseminated Breast Cancer Cells. Stem Cells (2016) 34(8):2224–35. doi: 10.1002/stem.2384
99. Yu X, Yan N, Li Z, Hua Y, Chen W. FGF19 Sustains the High Proliferative Ability of Keratinocytes in Psoriasis Through the Regulation of Wnt/GSK-3β/β-Catenin Signalling via FGFR4. Clin Exp Pharmacol P (2019) 46(8):761–9. doi: 10.1111/1440-1681.13103
100. Tiong HK, Tan BS, Choo HL, Chung FF, Hii LW, Tan SH, et al. Fibroblast Growth Factor Receptor 4 (FGFR4) and Fibroblast Growth Factor 19 (FGF19) Autocrine Enhance Breast Cancer Cells Survival. Oncotarget (2016) 7(36):57633–50. doi: 10.18632/oncotarget.9328
101. Ji S, Liu Q, Zhang S, Chen Q, Wang C, Zhang W, et al. FGF15 Activates Hippo Signaling to Suppress Bile Acid Metabolism and Liver Tumorigenesis. Dev Cell (2019) 48(4):460–74. doi: 10.1016/j.devcel.2018.12.021
102. Turunen SP, von Nandelstadh P, Ohman T, Gucciardo E, Seashore-Ludlow B, Martins B, et al. FGFR4 Phosphorylates MST1 to Confer Breast Cancer Cells Resistance to MST1/2-Dependent Apoptosis. Cell Death Differ (2019) 26(12):2577–93. doi: 10.1038/s41418-019-0321-x
103. Katoh M. Therapeutics Targeting FGF Signaling Network in Human Diseases. Trends Pharmacol Sci (2016) 37(12):1081–96. doi: 10.1016/j.tips.2016.10.003
104. Katoh M, Nakagama H. FGF Receptors: Cancer Biology and Therapeutics. Med Res Rev (2013) 34(2):280–300. doi: 10.1002/med.21288
Keywords: breast cancer, bone metastasis, signaling pathway, cytokine, targeted therapy
Citation: Song X, Wei C and Li X (2022) The Signaling Pathways Associated With Breast Cancer Bone Metastasis. Front. Oncol. 12:855609. doi: 10.3389/fonc.2022.855609
Received: 15 January 2022; Accepted: 16 February 2022;
Published: 10 March 2022.
Edited by:
Alberto Farolfi, Scientific Institute of Romagna for the Study and Treatment of Tumors (IRCCS), ItalyReviewed by:
Bulent Erdogan, Trakya University, TurkeyCopyright © 2022 Song, Wei and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangqi Li, ZHJsaXhxaUBob3RtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.