
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 22 March 2022
Sec. Cancer Molecular Targets and Therapeutics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.853484
This article is part of the Research Topic Megakaryocytes as Regulators of Tumor Microenvironments View all 9 articles
 Paola Verachi1
Paola Verachi1 Francesca Gobbo1,2
Francesca Gobbo1,2 Fabrizio Martelli3
Fabrizio Martelli3 Andrea Martinelli4Giuseppe Sarli2Andrew Dunbar5,6
Andrea Martinelli4Giuseppe Sarli2Andrew Dunbar5,6 Ross L. Levine5,6,7
Ross L. Levine5,6,7 Ronald Hoffman8Maria Teresa Massucci9
Ronald Hoffman8Maria Teresa Massucci9 Laura Brandolini9Cristina Giorgio9
Laura Brandolini9Cristina Giorgio9 Marcello Allegretti9*
Marcello Allegretti9* Anna Rita Migliaccio10,11* on behalf of Myeloproliferative Neoplasm Research Consortium
Anna Rita Migliaccio10,11* on behalf of Myeloproliferative Neoplasm Research ConsortiumA major role for human (h)CXCL8 (interleukin-8) in the pathobiology of myelofibrosis (MF) has been suggested by observations indicating that MF megakaryocytes express increased levels of hCXCL8 and that plasma levels of this cytokine in MF patients are predictive of poor patient outcomes. Here, we demonstrate that, in addition to high levels of TGF-β, the megakaryocytes from the bone marrow of the Gata1low mouse model of myelofibrosis express high levels of murine (m)CXCL1, the murine equivalent of hCXCL8, and its receptors CXCR1 and CXCR2. Treatment with the CXCR1/R2 inhibitor, Reparixin in aged-matched Gata1low mice demonstrated reductions in bone marrow and splenic fibrosis. Of note, the levels of fibrosis detected using two independent methods (Gomori and reticulin staining) were inversely correlated with plasma levels of Reparixin. Immunostaining of marrow sections indicated that the bone marrow from the Reparixin-treated group expressed lower levels of TGF-β1 than those expressed by the bone marrow from vehicle-treated mice while the levels of mCXCL1, and expression of CXCR1 and CXCR2, were similar to that of vehicle-treated mice. Moreover, immunofluorescence analyses performed on bone marrow sections from Gata1low mice indicated that treatment with Reparixin induced expression of GATA1 while reducing expression of collagen III in megakaryocytes. These data suggest that in Gata1low mice, Reparixin reduces fibrosis by reducing TGF-β1 and collagen III expression while increasing GATA1 in megakaryocytes. Our results provide a preclinical rationale for further evaluation of this drug alone and in combination with current JAK inhibitor therapy for the treatment of patients with myelofibrosis.
Primary Myelofibrosis (MF) is due to both a primary clonal myeloproliferation as a result of activating mutations of the JAK/STAT pathway and a secondary inflammatory response characterized by micro-environmental changes and aberrant release of multiple pro-inflammatory cytokines (1). Abnormal megakaryocytes (MKs) play a crucial role in the development of the MF stromal reaction (2), which includes bone marrow (BM) reticulin fibrosis, osteosclerosis, increased microvessel density, a proinflammatory milieu, anemia, splenomegaly, and extramedullary hematopoiesis (1, 3). The current JAK1/2 inhibitor therapy improves clinical symptoms but does not alter the clinical progression to more overt phases of MF or blast phase (4, 5). Therefore, novel therapeutic strategies aiming to reduce the inflammatory microenvironment that contributes to the sustained proliferation of the malignant hematopoietic stem cells (HSCs) are currently being investigated (6).
Human (h)CXCL8 (C-X-C Motif Chemokine Ligand 8) is a member of the chemokine family and exerts its biological activities by signaling through the CXCR1 and CXCR2 receptors. The chemokine family also includes CXCL1, CXCL2, CXCL3, CXCL5, CXCL6, and CXCL7, and together they share an ELR (glutamic, leucine, and arginine) motif that mediates CXCR1/2 binding. Most of the studies published until now have investigated the biological effects exerted by hCXCL8 and its receptors on polymorphonuclear leukocytes. However, the effects exerted by hCXCL8 on other cell types, such as endothelial, epithelial and fibroblasts, known also to express CXCR1/CXCR2, are still poorly defined (7). hCXCL8 is produced by several bone marrow cells, namely, megakaryocytes (MKs) (8), and exhibits many biological functions in inflammation, hematopoietic stem cell (HSC) proliferation, mobilization, and neo-angiogenesis. A previous study demonstrated that hCXCL8 contributes to altered MK proliferation, differentiation, and ploidy in myeloid metaplasia with MF (9). Moreover, high levels of circulating hCXCL8 were detected in patients with MF and were predictive of inferior survival (3). In addition, Dunbar et al. have demonstrated that the malignant CD34+ clones from a subset of MF patients secrete high levels of hCXCL8 in vitro which is associated with adverse clinical outcome and increased marrow fibrosis (10).
Full understanding of the pathophysiological role of hCXCL8 has been limited by known differences between human and rodents. In humans, hIL-8/CXCL8 (and CXCL6, also known as GCP-2) exerts its activity by activating both CXCR1 and CXCR2 (11, 12), whereas the other ELR-CXC chemokines selectively bind CXCR2 (13). For many years, only one functional ELR-CXC receptor was identified in mice and was characterized as the homologue of human CXCR2 (14, 15). This receptor, showing high affinity for the murine counterparts of hCXCL8, mCXCL1(KC) and MIP-2, was believed to be responsible for the functions attributed to the two human receptors. Consistent with this, gene ablation of mouse CXCR2 impairs neutrophil responses to murine MIP-2 and to hIL-8/CXCL8 (16, 17) confirming a critical role for CXCR2 in neutrophil recruitment and activation. The orthologous murine CXCR1 (mCXCR1) (18, 19) was subsequently identified and found to be expressed by BM, peripheral mononuclear cells, CD4+ and CD8+ T cells, and certain lymphoid cell types but was first considered to be a non-functional receptor due to the repeated failure of any attempt to identify its cognate ligand/s. More recently, mCXCR1 has been confirmed to behave as a functional receptor (20), specifically activated by mGCP-2, hGCP-2/CXCL6, and hIL-8/CXCL8, and to play a key role in collagen-induced arthritis (mCIA). This discovery paved the way for novel studies on the biology of CXCR1/2 using mouse models. These studies support the notion that mCXCR1 is the functional murine orthologue of hCXCR1 and that hIL-8/CXCL8 is functionally replaced, in addition to mCXCL1, by CXCL6 in mice (21).
Reparixin is a dual, non-competitive allosteric antagonist of the hCXCL1 receptors hCXCR1/R2 with a marked selectivity for hCXCR1 (IC50 = 1 nM for hCXCR1; IC50 = 400 nM for hCXCR2) and cross-reactivity with mCXCR1/2 (22, 23). In particular, the allosteric modulation exerted by Reparixin inhibits human and murine CXCR1/R2 activation independently of the cognate ligand (24) and without blocking the binding of the ligand to its receptors (25). The mechanism of action of Reparixin accounts for the functional selectivity of the drug that allows it to switch off the G-protein mediated pathway activation without impairing ligand-induced internalization and scavenging thus not affecting the extracellular levels of mCXCL1. The in vivo activity of Reparixin was originally evaluated in animal models of ischemia/reperfusion injury (22) and the therapeutic potential of CXCR1/R2 inhibition was further investigated in the airway inflammation of several mouse models. In particular, Reparixin was shown to ameliorate pulmonary fibrosis caused by the administration of particulate matter and bleomycin in mice (26). Similarly, the CXCR1/R2 inhibition exerted by ladarixin, a dual allosteric blocker of CXCR1/R2 structurally similar to Reparixin, has been shown to reduce neutrophil infiltration and collagen deposition in the bleomycin-induced mouse model of pulmonary fibrosis (27). These data suggest CXCR1/2 inhibition might have anti-fibrotic effects across numerous organs.
Mice carrying the hypomorphic Gata1low mutation express the same MKs alterations observed in MF patients and develop progressive MF closely resembling human disease, namely, a BM failure syndrome and development of extramedullary hematopoiesis (28–30). Previous studies have demonstrated that MKs from the BM of Gata1low mice express not only high levels of TGF-β1 (31), but also high levels of mCXCL1, the murine equivalent of hCXCL8 (32). Using these data as a foundation, in the current study we have evaluated whether MKs from Gata1low mice express CXCR1 and CXCR2 and tested whether treatment with Reparixin affects the development of MF in this mouse model.
Gata1low mice were originally obtained from Dr. S. Orkin and bred in the animal facility of the Istituto Superiore di Sanità as described (30). Littermates were genotyped at birth by PCR as previously described (33) and those found not to carry the mutation were used as wild-type (WT) controls. All the experiments were performed according to the protocols approved by the institutional animal care committee according to the European Directive 86/609/EEC.
Sixteen eight-month-old Gata1low mice were then anesthetized with 2–3% isoflurane and implanted subcutaneously with an ALZET® Osmotic Pump (model 2002) pre-filled with 200 μl of vehicle (sterile saline) or Reparixin (7.5 mg/h/kg in sterile saline) as described by the instructions of the manufacturer. The concentration of Reparixin was chosen on the basis of previous concentration–response and efficacy studies in which the selected concentration of 7.5 mg/h/kg administered by continuous infusion was proven to be able to reduce pathological outcomes in preclinical models of liver ischemia and reperfusion, neuropathic pain, and acquired epilepsy (34–36) Before treatment, the mouse genotype had been confirmed by PCR as described (33). The scheme of the two experiments and of their end-points is outlined in Figure 1.
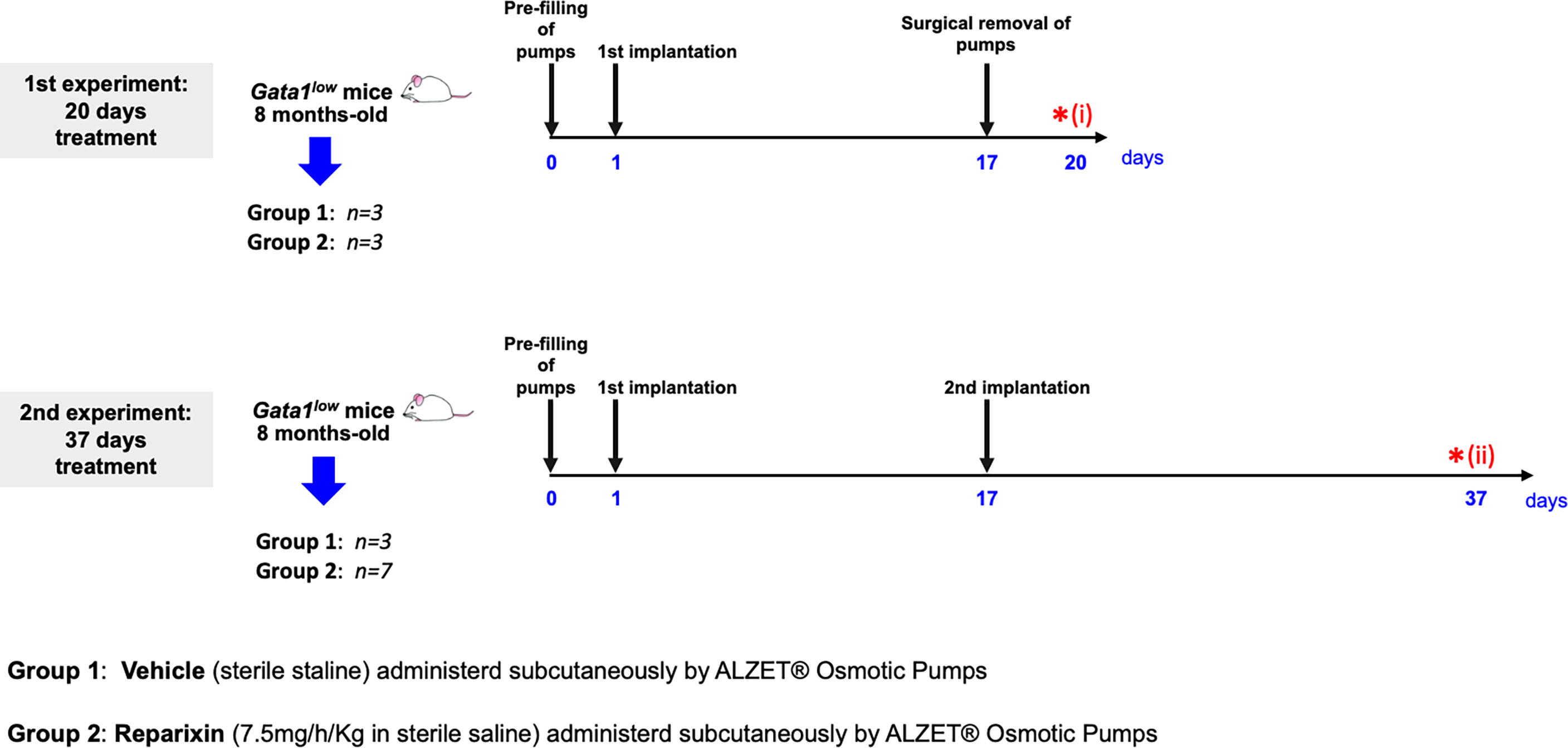
Figure 1 Scheme of the two treatments of Gata1low mice with either vehicle or Reparixin for 20 (experiment 1) or 37 (experiment 2) days. In experiment 1, mini-pumps were removed at day 17 and three mice per experimental group were sacrificed at day 17 for end-point determination. In experiment 2, mini-pumps were removed at day 17 from three vehicles and six Reparixin-treated mice and the mini-pumps were replaced with a second implant. These mice were then treated for 17 additional days and sacrificed at day 37. Red asterisks indicate the timing of the sacrifice in the first (i) and second (ii) experiment.
In the first experiment, mini-pumps were removed on day 17, and three mice per experimental group were weighed, bled for blood count determinations and plasma collection, and sacrificed for histopathological evaluation of the BM and spleen at day 20. Since the manufacturer guarantees that the Alzet model 2002 mini-pumps deliver the drug with the predicted rates only for 14 days, to mimic the clinical situation in which Reparixin is likely to be administered to patients for long time, in the second experiment, mini-pumps were removed from three mice treated with vehicle and seven mice treated with Reparixin and replaced with newly filled devices. These mice were then treated for 17 additional days and analyzed on day 37, when they were weighed, bled for blood count determinations and plasma collection, and sacrificed for histopathological analyses.
Mice were topically anesthetized with lidocaine (one drop/eye) and blood was collected from the retro-orbital plexus into microcapillary tubes using sodium citrate 3.2% (ratio 1:9) as an anticoagulant. Hematocrit (Htc), platelet (Plt), and white blood cell (WBC) counts were evaluated by an accredited commercial laboratory which provides diagnostic services on laboratory animals (Plaisant Laboratory). For drug concentration determinations, plasma was separated from whole blood by centrifugation for 20 min at 10,000 rpm and stored at −20°.
Determinations of Reparixin levels in plasma samples involved protein precipitation by the addition of acetonitrile (Sigma-Aldrich, S. Louis, MO, USA) (ratio 1:3). Samples were then centrifuged (20,000×g for 15 min at 4°C) and supernatants were analyzed by high-performance liquid chromatography (HPLC, Dionex-Thermo Fisher Scientific, Sunnivale, CA, USA) using an electrospray ionization (ESI) source for detection. The chromatographic column was a Gemini C18 100 × 2.0 mm, 5 µm (Phenomenex, Torrance, CA, USA) and the lower limit of quantification is 0.05 µg/ml.
Femurs were fixed in formaldehyde (10% v/v with neutral buffer), treated for 1 h with a decalcifying solution (Osteodec; Bio-Optica, Milan, Italy) and paraffin embedded. Spleens were fixed in formaldehyde as previously described (32) and embedded in paraffin. Paraffin-embedded tissues were cut into consecutive 3 μm sections and stained either with Hematoxylin–Eosin (H&E; Hematoxylin cat no. 01HEMH2500; Eosin cat no. 01EOY101000; Histo-Line Laboratories, Pantigliate, MI, Italy) or Gomori silver (cat no. 04-040801; Bio-Optica) or Reticulin (cat no. 04-040802; Bio-Optica) staining. These two last stainings both reveal the presence of reticulin fibers and were used as independent evaluations of fibrosis to increase the rigor of the assessment of the findings. BM sections were immune-stained with anti-CXCL1 (cat# ab86436, Abcam, Cambridge, UK), anti-CXCR1 (cat# GTX100389, Genetex, Irvine, CA, USA), anti-CXCR2 (cat# catalog ab14935, Abcam) or anti-TGF-β1 (cat no. sc-130348, Santa Cruz Biotechnology, Santa Cruz, CA, USA, from now on we use TGF-β1 or TGF-β, depending whether results were obtained with reagents which recognize the TGF-β1 isoform or all the three TGF-β isoforms), antibodies. Immunoreactions were detected with avidin–biotin immune-peroxidase (Vectastain Elite ABC Kit, Vector Laboratories, Burlingame, CA, USA) and the chromogen 3,3′-diaminobenzidine (0.05% w/v). Slides were counterstained with Papanicolaou’s hematoxylin (Histo-Line Laboratories). Images were acquired with an optical microscope (Eclipse E600; Nikon, Shinjuku, Japan) equipped with the Imaging Source “33” Series USB 3.0 Camera (cat no. DFK 33UX264; Bremen, DE). Images were processed and the intensity of the immunostaining quantified with the software ImageJ (version 1.52t) (National Institutes of Health).
Three micron-thick BM sections were dewaxed in xylene and treated with EDTA buffer pH = 8 for 20’ in a pressure cooker (110–120°C, high pressure) for antigen retrieval. Sections were labeled with antibody against CD42b (a rat monoclonal antibody that recognizes the alpha chain of platelet glycoprotein I, cat no. ab183345, Abcam), GATA1 (rat monoclonal, cat no. sc-265, Santa Cruz), and Collagen III (rabbit polyclonal, cat no. ab7778, Abcam) overnight at 4°C. Detection of primary antibodies was visualized with a secondary antibody goat anti rat Alexa Fluor 488 (cat no. ab150165, Abcam) and goat anti rabbit Alexa Fluor 555 (cat no. ab150078, Abcam). Sections were counterstained with DAPI (cat no. D9542-5MG, Sigma-Aldrich, Darmstadt, DE) and mounted with Fluor-shield histology mounting medium (Catalog F6182-10MG, Sigma-Aldrich). Slides were examined using a Nikon Eclipse Ni microscope equipped with filters appropriate for the fluorochrome to be analyzed. Images were recorded with a Nikon DS-Qi1Nc digital camera and NIS Elements software BR 4.20.01. Confocal microscopy determinations were performed at ×40 magnification on at least 120 CD42b positive cells (MKs) per area (0.720 mm2) of bone marrow section per mouse. We have analyzed a total of 6 mice treated with vehicle, three mice treated with Reparixin for three days and 7 mice treated with Reparixin for 37 days.
Data were analyzed and plotted using GraphPad Prism 8.0.2 software (GraphPad Software, San Diego, California United States) and presented as Mean (± SD) or as box charts, when appropriate. Shapiro–Wilk test was used to confirm that the data are normally distributed. Comparisons between the two groups were performed with one way ANOVA while comparisons between multiple groups were performed with the Tukey’s multiple comparisons test. Linear correlations were calculated by the Pearson R test. Differences were considered statistically significant with a p <0.05.
The body weight of the mice included in the two experiments and its variations during treatment are shown in Figure 2. Probably due to sampling bias, before treatment the weight of the mice included in the vehicle and Reparixin group is statistically different (p <0.01). The difference in weight of the Reparixin-treated mice remains statistically different from that of the vehicle mice at day 20 (p <0.05). However, there is no statistically significant difference between the Reparixin-treated mice and the corresponding vehicle group both at days 20 and 37 (Figure 2). The wellbeing of the treated mice was monitored daily by a veterinarian. Death was not recorded and the mice remained active with no significant changes in behavior (no lethargy, no excessive grooming, no change in coat luster) during all the period of observation. The Hct and WBC counts remained within normal ranges in all experimental groups, while Plt counts were low as has been previously observed in Gata1low mice (Table 1). In-depth analyses of the WBC populations, revealed significant greater neutrophil counts at day 37 versus day 20 (Table 1), while the lymphocyte and monocyte counts remained similar among groups (Figure S1).
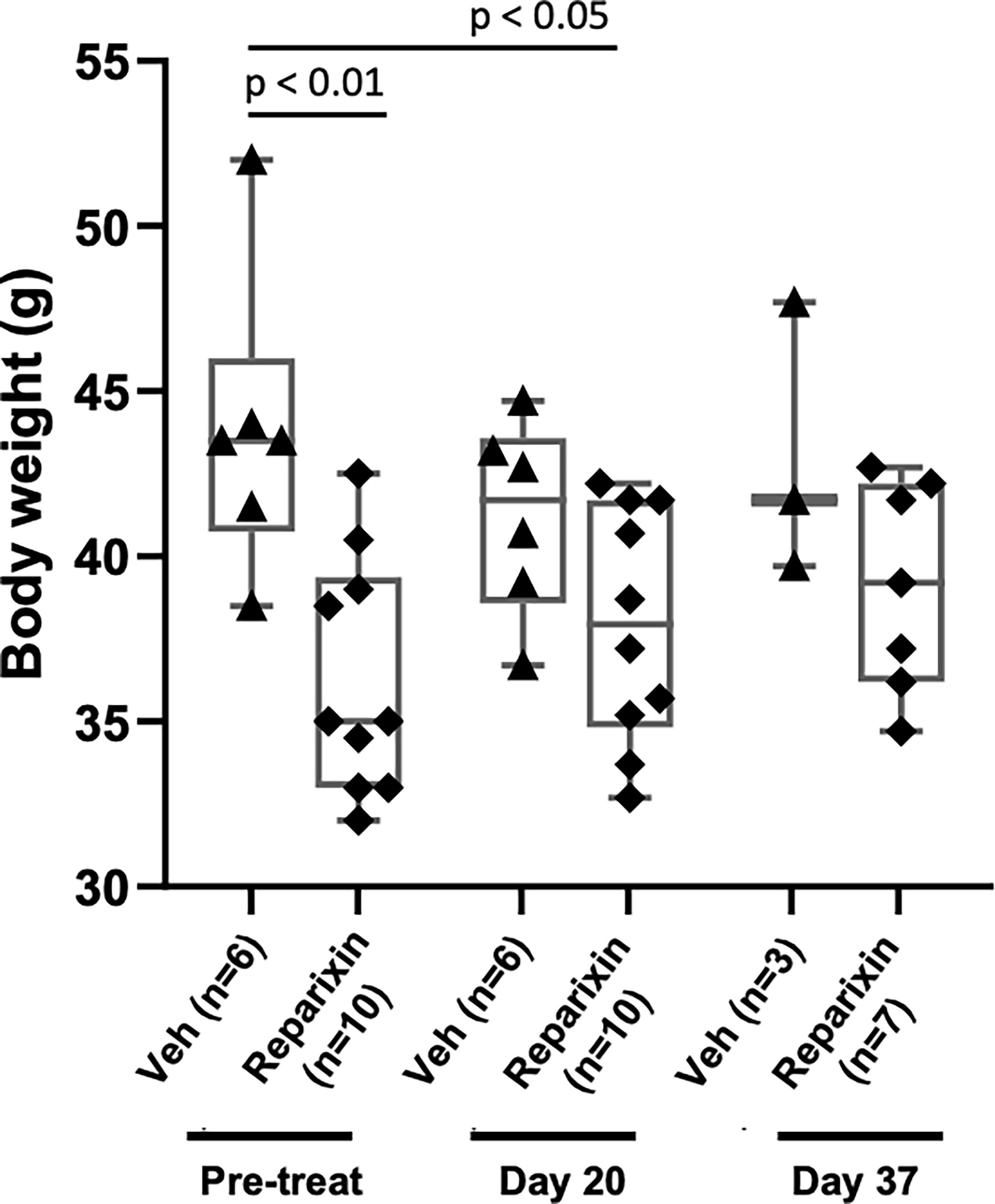
Figure 2 Treatment with Reparixin did not affect animal weights. Body weight determinations performed before (Pre-treat) and after 20 or 37 days of treatment with Reparixin. Data are presented as box charts and as value per individual mouse (each symbol a different mouse). The data are normally distributed by Shapiro–Wilk test and the Reparixin pre-treatment (p < 0.01) and at day 20 (p < 0.05) group are statistically different from the vehicle group by Turkey’s multiple comparison test.
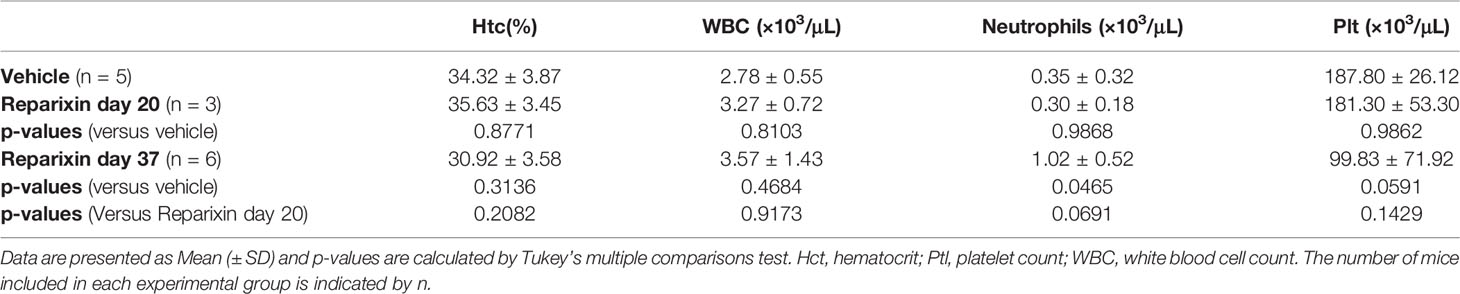
Table 1 Blood parameters observed in mice treated with vehicle or with Reparixin for 20 or 37 days.
According to the specification provided by the manufacturer, Alzet model 2002 mini-pumps have an approximate 0.2 ml reservoir that delivers a preloaded drug or vehicle solutions continuously for at least 14 days at a rate of 5 μl/h. To confirm that mice remained to the drug at the time of sacrifice, the plasma levels of Reparixin at days 20 and 37 were determined. Plasma levels ranged from 3.24 to 17.87 μg/ml. These levels are similar to those observed in previous experiments using the same device (36). Notably, plasma levels of Reparixin were significantly higher in mice treated for 20 days (13.90 ± 4.18) compared to those that underwent a second implantation and were treated for 17 additional days (6.71 ± 4.18) (Figure 3). Although the levels of drug detected at the second time-point were similar to those determined in prior studies (36), the observation that mice treated with the same concentration for a longer time have a plasma level of the drug lower than those treated for lower times is puzzling. It is possible that changes in the cell composition of the longer treated mice lead to greater amounts of tissue-bound Reparixin reducing the free levels of the drug found in plasma. However, it is also possible that the plasma levels of Reparixin at the two time points reflect differences in the efficiency of the derma to absorb the drug. This alternative hypothesis is supported by the observation that, since GATA1 regulates the differentiation of dermal mast cells, derma of Gata1low mice contains great numbers of these cells (37). In addition, wild-type CD1 mice, the background in which we harbor the Gata1low mutations express a systemic pro-inflammatory signature which determines chronic dermatitis with dermal fibrosis (32). This baseline dermal fibrosis is also present in Gata1low mice (ARM, unpublished observations) and it is possibly exacerbated once the mast cells are activated by the mechanical stress induced by the mini-pumps implanted subcutaneously, reducing the efficiency of the dermal absorption and of the plasma levels of the drug. Therefore, the plasma levels of Reparixin are a true reflection of the concentration of the drug delivered by the mini-pumps to the animals.
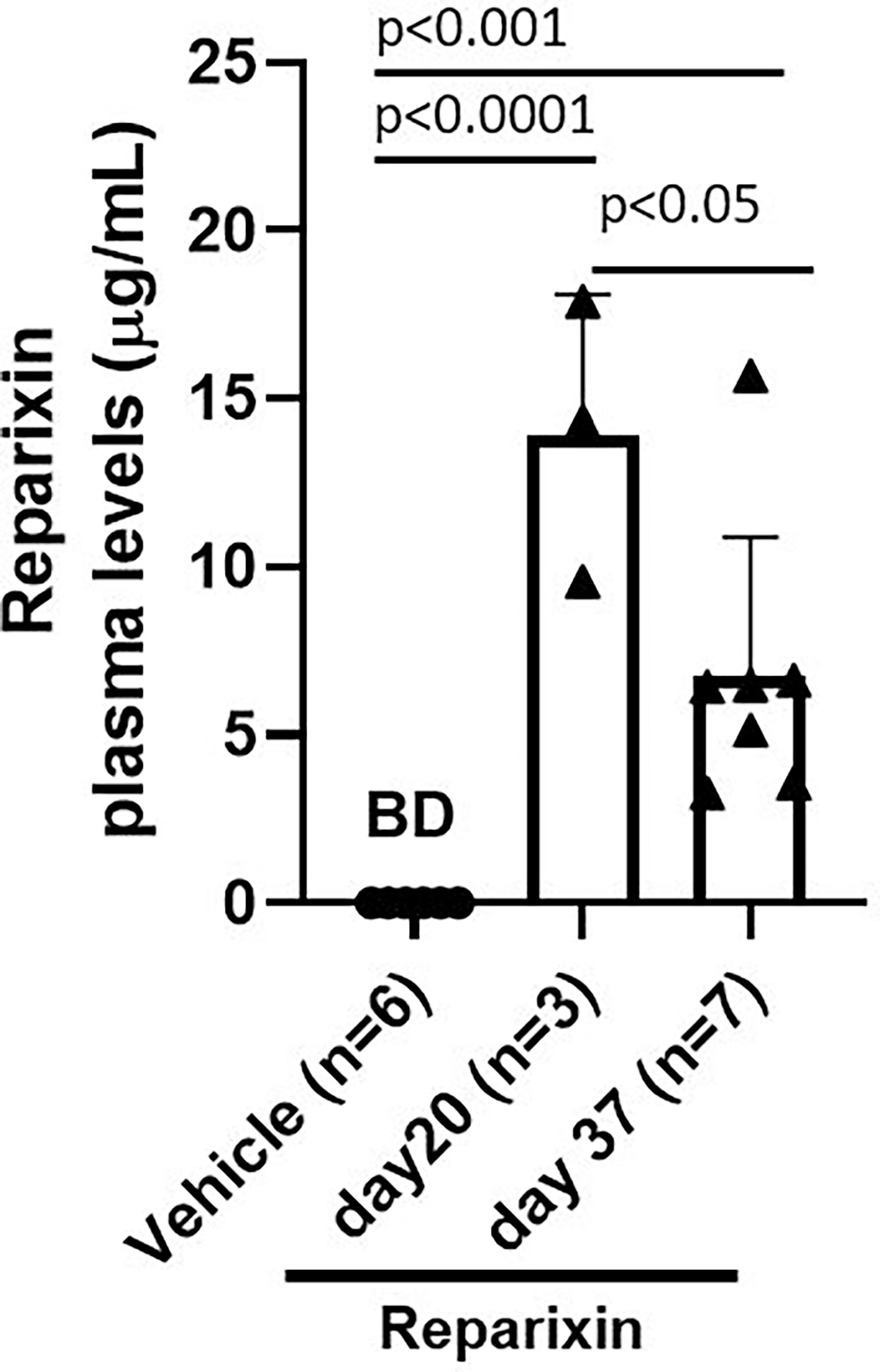
Figure 3 Plasma levels (μg/ml) of Reparixin detected at days 20 and 37. Plasma levels of Reparixin were significantly higher in Gata1low mice scarified at day 20 as compared to those sacrificed at day 37. Data are presented as Mean (± SD) and are analyzed by Tukey’s multiple comparisons test. Dots represent values observed in the individual mice. P < 0.05 was considered statistically significant. Abbreviations: BD, below detectable levels (i.e., <20% of the lower limit of quantitation (0.2 µg/ml).
The BM from Gata1low mice is characterized by reduced cellularity due to accumulation of reticulin fibers with age (30). Overall, the treatment had modest effects on BM cellularity (Table 2).

Table 2 Bone marrow cellularity and spleen weight determined in Gata1low mice treated either with vehicle or with Reparixin for 20 or 37 days.
However, a reduction in BM fibrosis was observed by both Gomori and Reticulin stainings in mice treated with Reparixin compared to those treated with vehicle alone to a statistically significant degree on day 20 (Figures 4A, B). Since mice treated for 20 days had higher concentrations of the drug in the plasma, we assessed whether the effects exerted by Reparixin on fibrosis was concentration-dependent rather than time-dependent by performing concentration/effect correlation analyses. Notably, this analysis revealed that the levels of fibrosis were inversely correlated with the plasma levels of the drug in individual mice (Figure 4C).
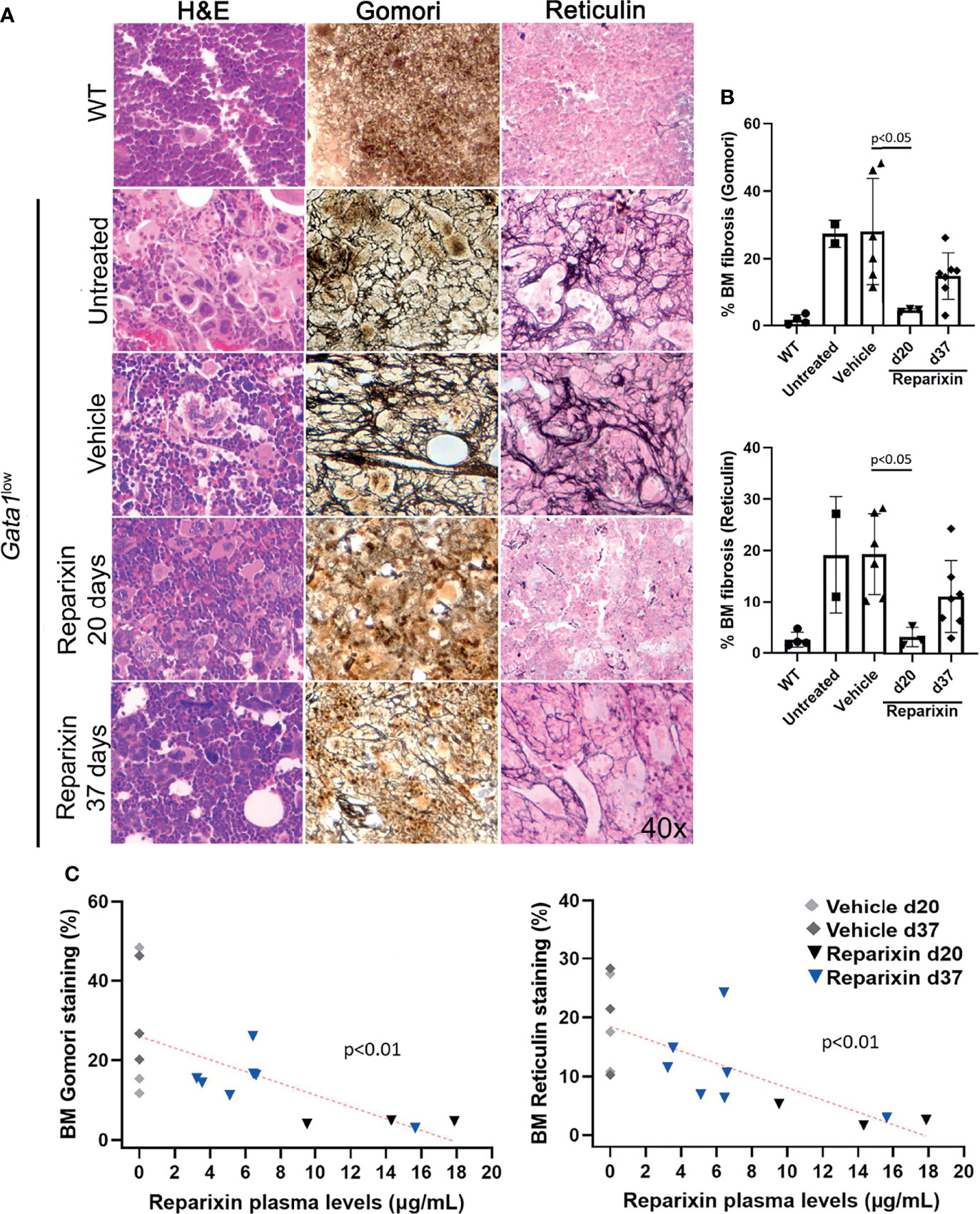
Figure 4 Reparixin decreases in a concentration-dependent fashion the fibrosis present in the BM from Gata1low mice. (A) H&E, Reticulin, and Gomori staining of BM sections from representative Gata1low mice treated either with vehicle or Reparixin for 20 or 37 days, as indicated. Untreated Gata1low and WT littermates are presented as positive and negative controls, respectively. Magnification ×400. (B) Levels of fibrosis quantified by computer image analyses on BM sections stained with Gomori or Reticulin from multiple Gata1low mice treated either with vehicle or Reparixin, as indicated. Data are presented as Mean (± SD) and are analyzed by Tukey’s multiple comparisons test. P < 0.05 was considered statistically significant. (C) Linear regression analyses between fibrosis plasma concentration or Reparixin in individual mice (Pearson R = −0.66, p < 0.01 for Gomori staining; Pearson R = −0.71, p < 0.01 for Reticulin staining). Each dot represents an individual mouse.
Gata1low mice developed extramedullary hematopoiesis with fibrosis in the spleen and associated splenomegaly. Despite the fact that the treatment with Reparixin did not induce significant changes in spleen volumes (Table 2), histological analyses indicated a remarkable reduction in the fibrosis expressed by the spleen in the Reparixin-treated mice (Figure 5A). As for BM, the reductions observed in the spleen were statistically significant only at day 20 (Figure 5B) but analyses of all the time points revealed a significant inverse correlation between fibrosis detected by reticulin staining and plasma levels of Reparixin in individual mice (Figure 5C).
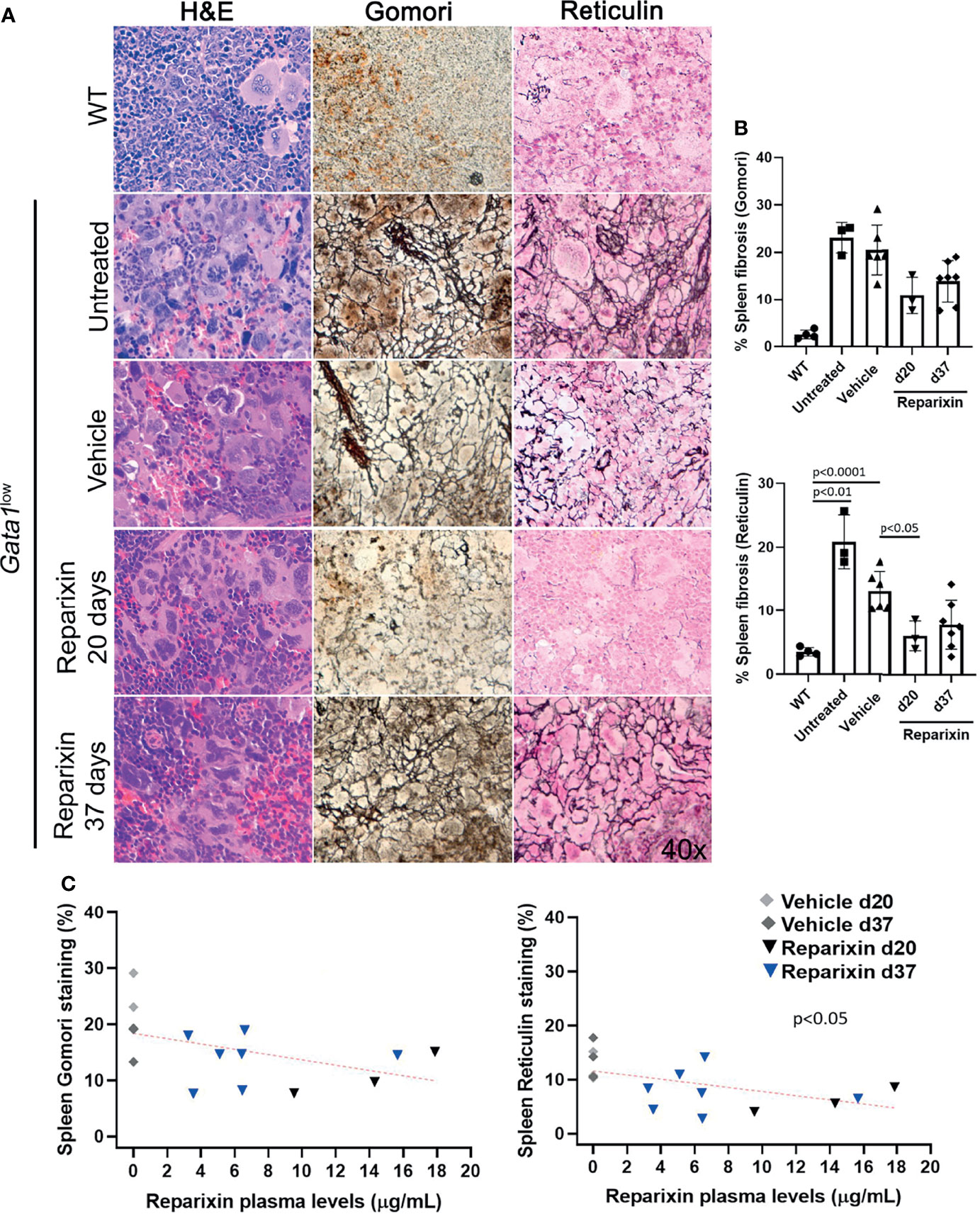
Figure 5 Reparixin decreased in a concentration-dependent fashion the degree of fibrosis present in the spleen of Gata1low mice. (A) H&E, Reticulin, Gomori and reticulin stainings of spleen sections from representative Gata1low mice treated either with vehicle or Reparixin for 20 or 37 days, as indicated. Untreated Gata1low and WT littermates are presented as positive and negative controls, respectively. Magnification ×400. (B) Levels of fibrosis quantified by image analyses in spleen sections stained with Gomori or Reticulin from Gata1low mice treated either with vehicle or Reparixin, as indicated. Data were presented as Mean (± SD) and were analyzed by Tukey’s multiple comparisons test. P < 0.05 was considered statistically significant. (C) Linear regression analyses of fibrosis and plasma levels of Reparixin in individual mice (Pearson R = −0.48, not significant for Gomori staining; Pearson R = −0.53, p < 0.05 for Reticulin staining). Each dot represents a single mouse.
To gain greater insights on the effects of Reparixin on fibrosis reduction in Gata1low mice, we determined by immunohistochemistry the content of CXCL1, and of its receptors CXCR1 and CXCR2, in the BM of Gata1low mice treated either with Reparixin or with vehicle. We had previously demonstrated that the BM from Gata1low mice contain high levels of TGF-β and express an altered TGF-β signature which was thought to be the cause of the marrow fibrosis since treatment of the mice with the receptor-1(R1) kinase inhibitor SB431542 (31) or with a TGF-β trap (38) reversed both the abnormal TGF-β signature and the increase in marrow fibrosis, restoring hematopoiesis in BM, of these mice. On the basis of these data, the content of TGF-β in the BM of Gata1low mice treated either with Reparixin or with vehicle was determined. These analyses indicated that the MKs from the Reparixin-treated mice express significantly lower levels of TGF-β1 than the corresponding cells from animals receiving vehicle alone. By contrast the levels of CXCL1, CXCR1, and CXCR2 expressed by the Reparixin-treated MKs remained comparable to that of the vehicle-treated cells (Figure 6). Although the reductions in TGF-β1 content were not correlated with the plasma levels of Reparixin of individual mice (data not shown), these results suggest that Reparixin may decrease fibrosis by reducing the levels of TGF-β1.
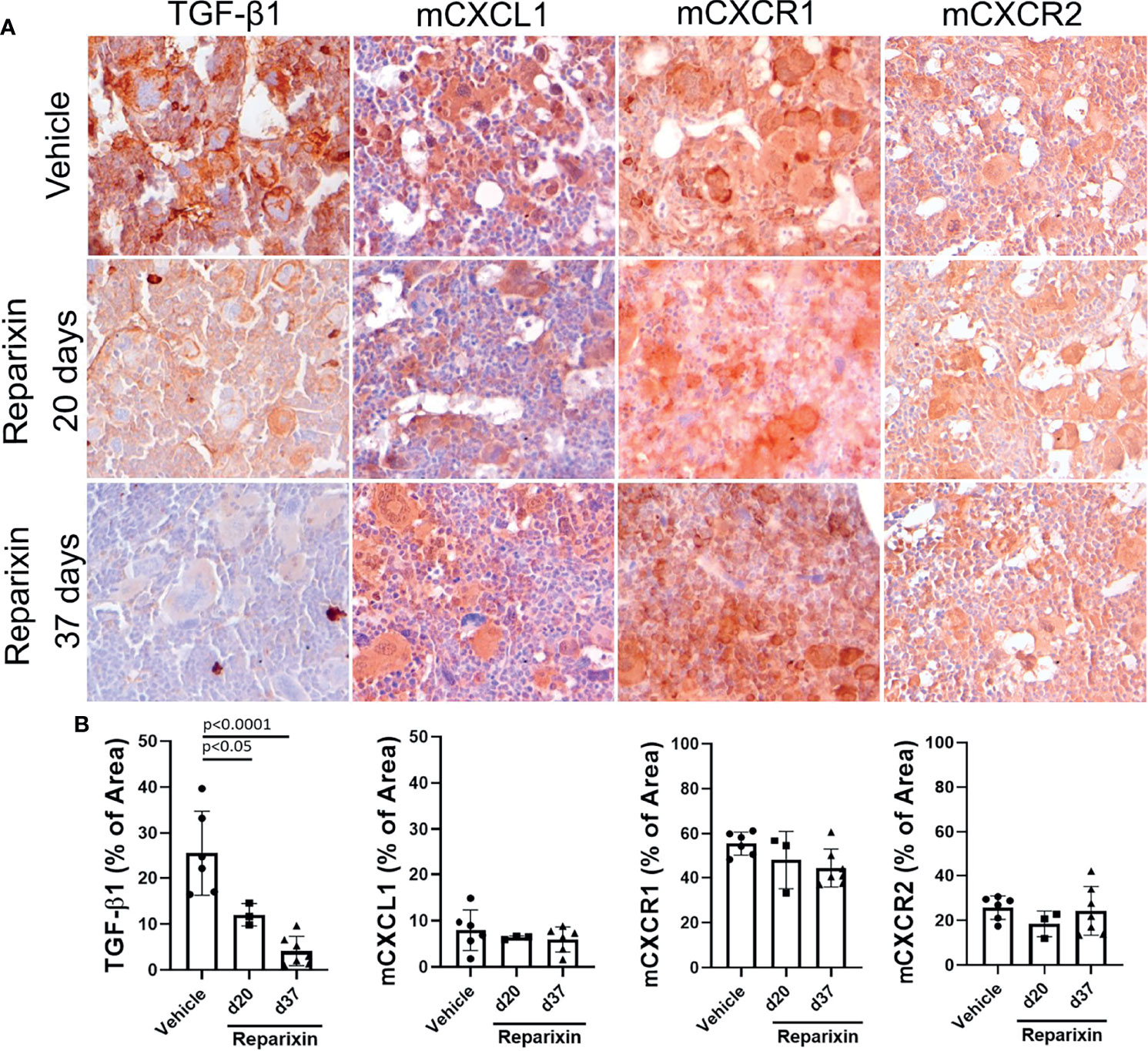
Figure 6 Treatment with Reparixin decreases the TGF-β1 content of the BM from Gata1low mice. (A) Immunohistochemical staining for TGF-β1, mCXCL1, mCXCR1, and mCXCR2 of BM sections from representative Gata1low mice treated either with vehicle or Reparixin for 20 or 37 days, as indicated. Magnifications ×40. (B) Quantification by computer assisted imaging of the TGF-β1, mCXCL1, mCXCR1, and mCXCR2 content in the BM from Gata1low mice treated with either with vehicle or Reparixin for 20 or 37 days, as indicated. Data are presented as Mean (± SD) and were analyzed by Tukey’s multiple comparisons test. Values observed in individual mice are presented as dots. P < 0.05 were considered statistically significant.
Previous studies have established that in both MF patients and in MPN driver mutation animal models of myelofibrosis the reduced levels of GATA1 is due to a ribosomopathy that reduces the GATA1 content in the malignant MKs (2, 39). In addition, MK-restricted expression of JAK2V617F, the most common driver mutation of MF, is sufficient to induce myelofibrosis in mice (40). The finding from the Balduini laboratory that a large proportion of the malignant MKs in the BM of MF patients express collagen, suggests that hypomorphic GATA1 MKs are directly responsible for the fibrosis observed in these patients (41). This hypothesis is supported by recent single cell profiling of BM cells that has identified a previously unrecognized population of MKs poised to exert niche-functions by secreting collagen and other extracellular matrix proteins. These niche supporting MKS morphologically resemble immature MKs (low ploidy levels with reduced presence of granules and platelet-territories in their cytoplasm) the maturation of which is sustained by low levels of GATA1 and high levels of TGF-β signaling (42–44).
Since the hypomorphic Gata1low mutation is characterized by reduced expression of GATA1 in MKs and the BM from these mice express high levels of TGF-β (32, 44), we hypothesized that this fibrosis may be sustained by increased numbers of niche-poised MKs with low levels of GATA1 due to the increase in BM TGF-β and that the reduced levels of TGF-β induced by Reparixin would reduce the degree of marrow fibrosis by reducing the frequency of this niche-poised MKs present in BM. To test this hypothesis, we first analyzed the content of GATA1 in MKs from BM sections of mice treated either with vehicle or with Reparixin (Figure 7). This analysis confirmed that the BM from Gata1low mice contained greater numbers of MKs and that the GATA1 content of these MKs was reduced (Figure 7A). Although the number of MKs in both vehicle- and Reparixin-treated mice remained greater than normal, the GATA1 content of the MKs from the Reparixin-treated mice was greatly increased (Figures 7B, C). Of note, the percentage of MKs expressing high GATA1 levels was directly correlated with the plasma levels of Reparixin detected in individual mice (Figure 7D).
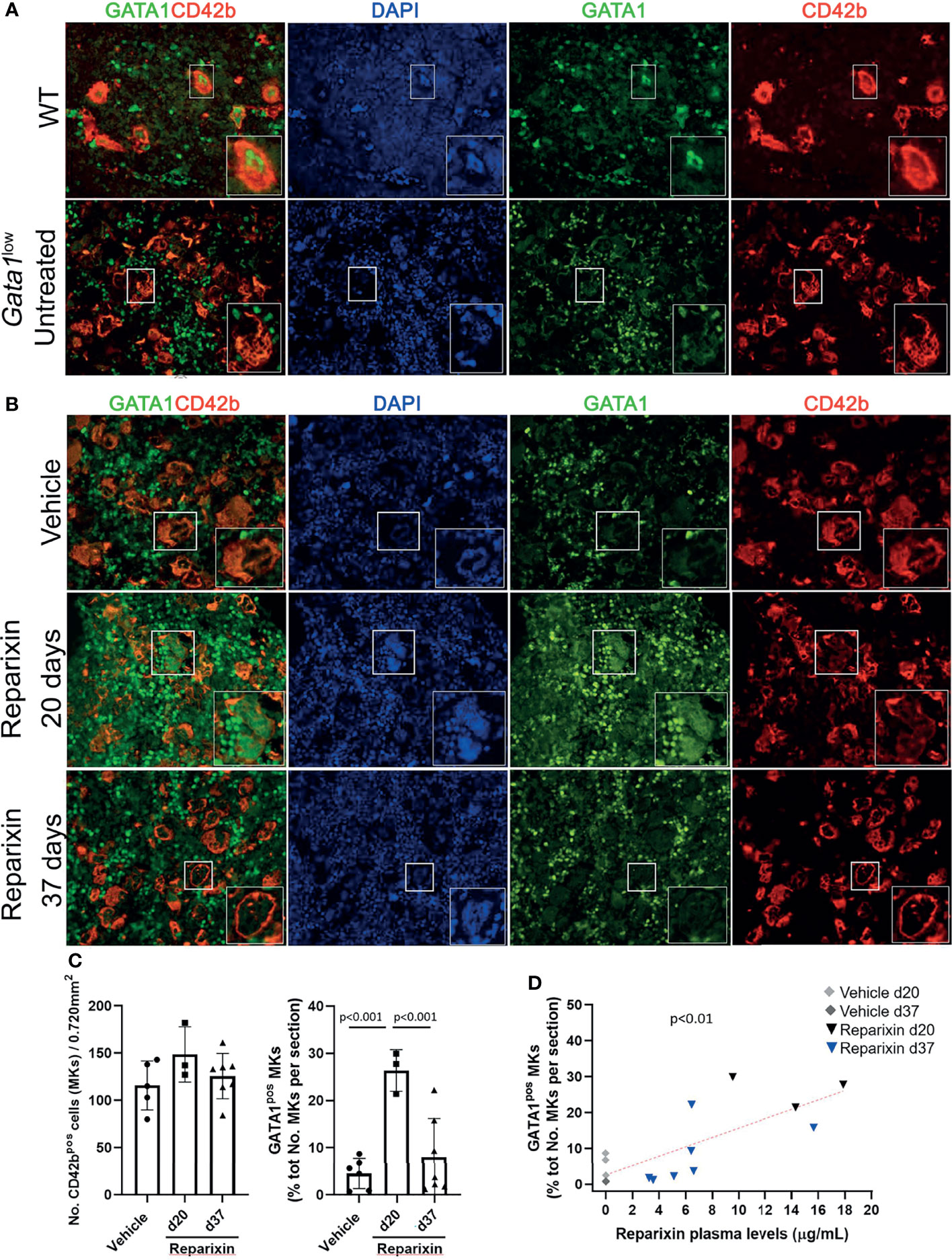
Figure 7 Reparixin restores in a concentration-dependent fashion the GATA1 content of the MKs in the BM of Gata1low mice. (A) Double immunofluorescence staining with GATA1 (FITCH-green) and CD42b (TRITCH-red), as a marker of MKs (45), antibodies of BM sections from representative wild-type (WT) and untreated Gata1low mice, as indicated. The hypomorphic Gata1low mutation selectively reduces expression of GATA1 in the MKs from the BM of Gata1low mice compared to the correspondent cells from WT littermates. Magnification ×400. (B) Double immunofluorescence staining with GATA1 (FITCH-green) and CD42b (TRITCH-red) of BM sections from representative mice treated either with vehicle or Reparixin for 20 or 37 days, as indicated. Magnification ×400. The boxes in (A) and (B)_ indicate the MKs shown at greater magnification on the bottom of the panels. (C) Frequency of MKs and percentage of MKs positive for GATA1 in BM sections from multiple treated mice. Data are presented as Mean (± SD) and as values in individual mice (each symbol a mouse). Results were analyzed by Tukey’s multiple comparisons test. P < 0.05 were considered statistically significant. (D) Correlation between the percent of MKs positive for GATA1and plasma levels of the Reparixin in individual mice (Pearson R = 0.76, p < 0.01). Each dot represents a single mouse.
We then compared the percent of MKs expressing Collagen III in the BM of Gata1low and WT mice (Figures 8A, B). Since the anti-ColIII antibody is conjugated with the same fluorochrome that labels most of the commercially available mouse CD42 antibodies, in these studies MKs were recognized on the basis of their large size and their polylobulated nuclei as revealed by DAPI staining. Greater number of the MKs in the BM from Gata1low expressed increased levels of collagen III while these cells were rare in WT mice, suggesting that the BM from Gata1low mice was enriched for niche-poised MKs.
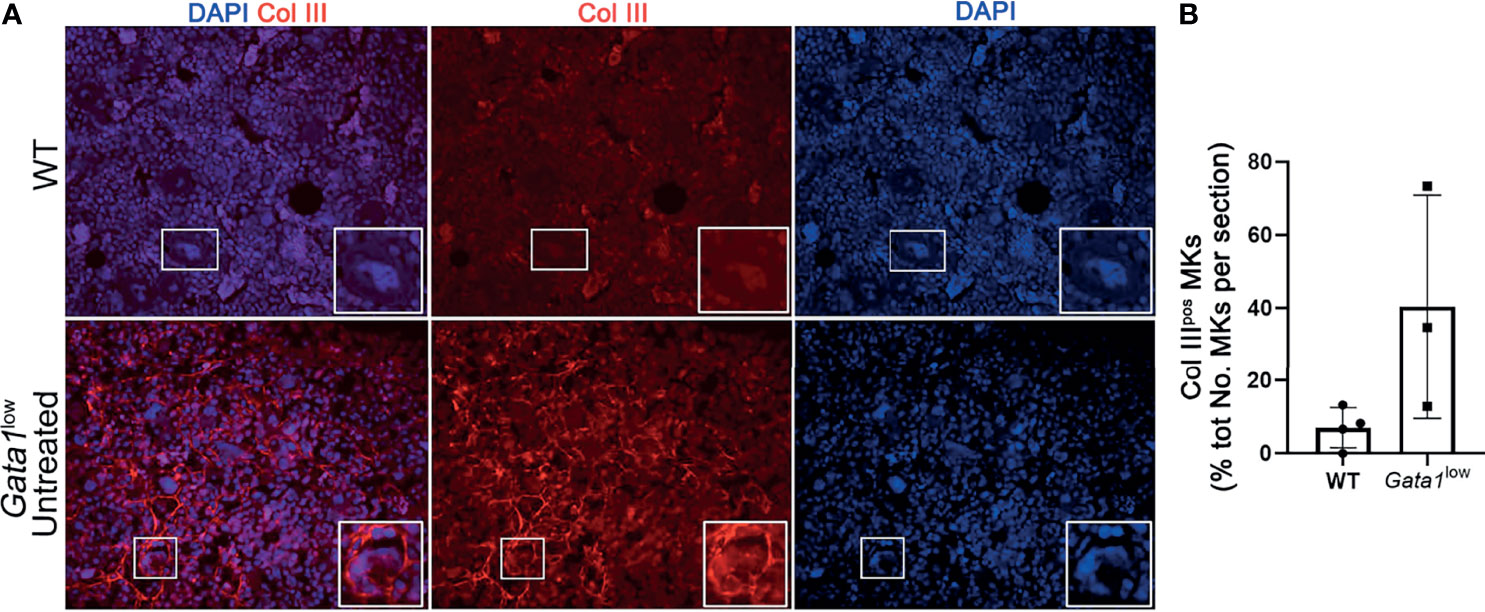
Figure 8 The bone marrow from Gata1low mice contains great numbers of megakaryocytes that express Collagen III. (A) Representative immunofluorescence staining with an anti-Col III (TRITCH-red) antibody showing that the MKs from Gata1low mice express higher levels of Col III than those from their WT littermates. Megakaryocytes were recognized on the basis of their morphology (size and morphology of the nucleus). Magnification ×400. The white boxes indicate representative MKs shown at greater magnification on the right bottom side of each panel. (B) Percent of MKs positive for CollIII (Col IIIpos MKs) over the total numbers of MKs observed in BM sections from WT and Gata1low littermates. Data are presented as Mean (± SD) and as frequency per individual mouse (dots) and were analyzed by One-way ANOVA. Each dot represents a single mouse. P < 0.05 was considered statistically significant.
Notably, we then demonstrated that treatment with Reparixin reduced in a concentration-dependent fashion the frequency of MKs expressing collagen III in the BM from Gata1low mice while the frequency of these cells in the BM of the vehicle treated mice remained increased (Figure 9).
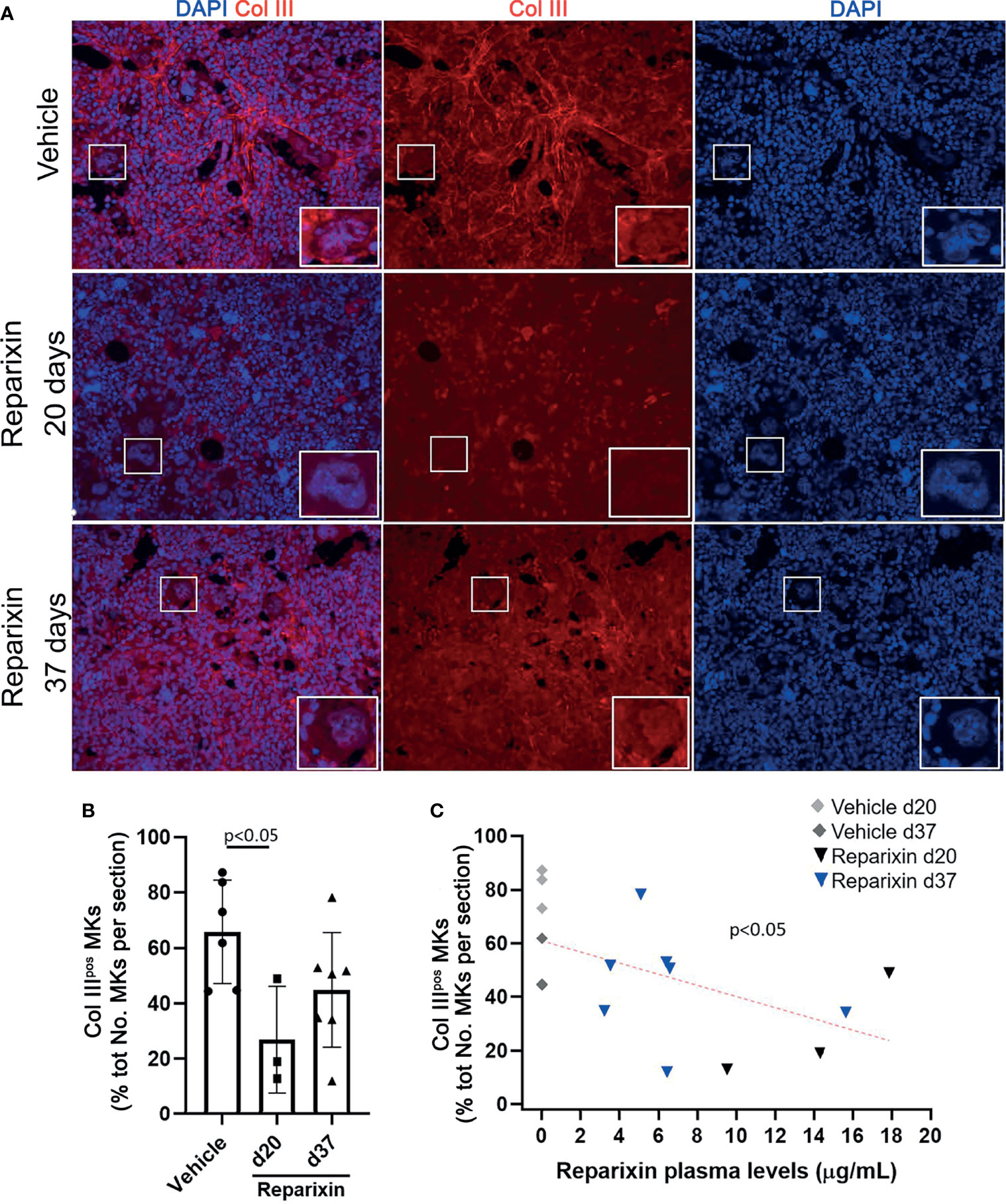
Figure 9 Reparixin reduces in a concentration-dependent fashion the frequency of Col IIIpos MKs in the bone marrow of Gata1low mice. (A) Immunofluorescence analyses with an anti-Col III (TRITCH-red) antibody of BM sections from representative Gata1low mice treated either with vehicle or with Reparixin for 20 or 37 days, as indicated. Megakaryocytes were recognized by morphology. Magnification ×400. The white rectangles indicate representative MKs shown at greater magnification on the bottom right of each panel. (B) Frequency of MKs positive for Col III over the total number of MKs per sections quantified by computer image analyses. Data are presented as Mean (± SD) and as values per single mouse (each dot a mouse) and were analyzed by Tukey’s test multiple comparisons test. P < 0.05 was considered statistically significant. (C) Linear regression analyses between the frequency of CollIIIpos MKs and plasma levels of Reparixin in individual mice (Pearson R = −0.53, p < 0.05). Each dot represents a single mouse.
On average, niche-poised MKs are smaller than platelet-poised cells and their nuclei contains lower number of lobi (43). The analyses of the morphology of the MKs shown in Figure 7 at greater magnification revealed that the MKs from the Reparixin-treated mice that contained GATA1 are on average smaller and with less lobated nuclei than those that were negative for GATA1 (Figure S2). These results suggest that Reparixin may have specifically increased the GATA1 content in the niche-poised MKs, explaining why, in spite of the increase of GATA1 in the MKs, the Plt numbers of the treated mice did not increase (Table 1). This hypothesis was tested by performing immunofluorescence studies to assess whether Reparixin increased the GATA1 content in the same MK population that expresses collagen (Figure 10). This analysis was performed only on the mice that had been treated for day 20 which exhibited the greatest reduction of fibrosis. The frequency of MKs expressing collagen III which were also positive for GATA1 was significantly greater in mice treated with Reparixin suggesting that Reparixin had specifically increased GATA1 content in niche-poised MKs, possibly hampering their pro-fibrotic functions.
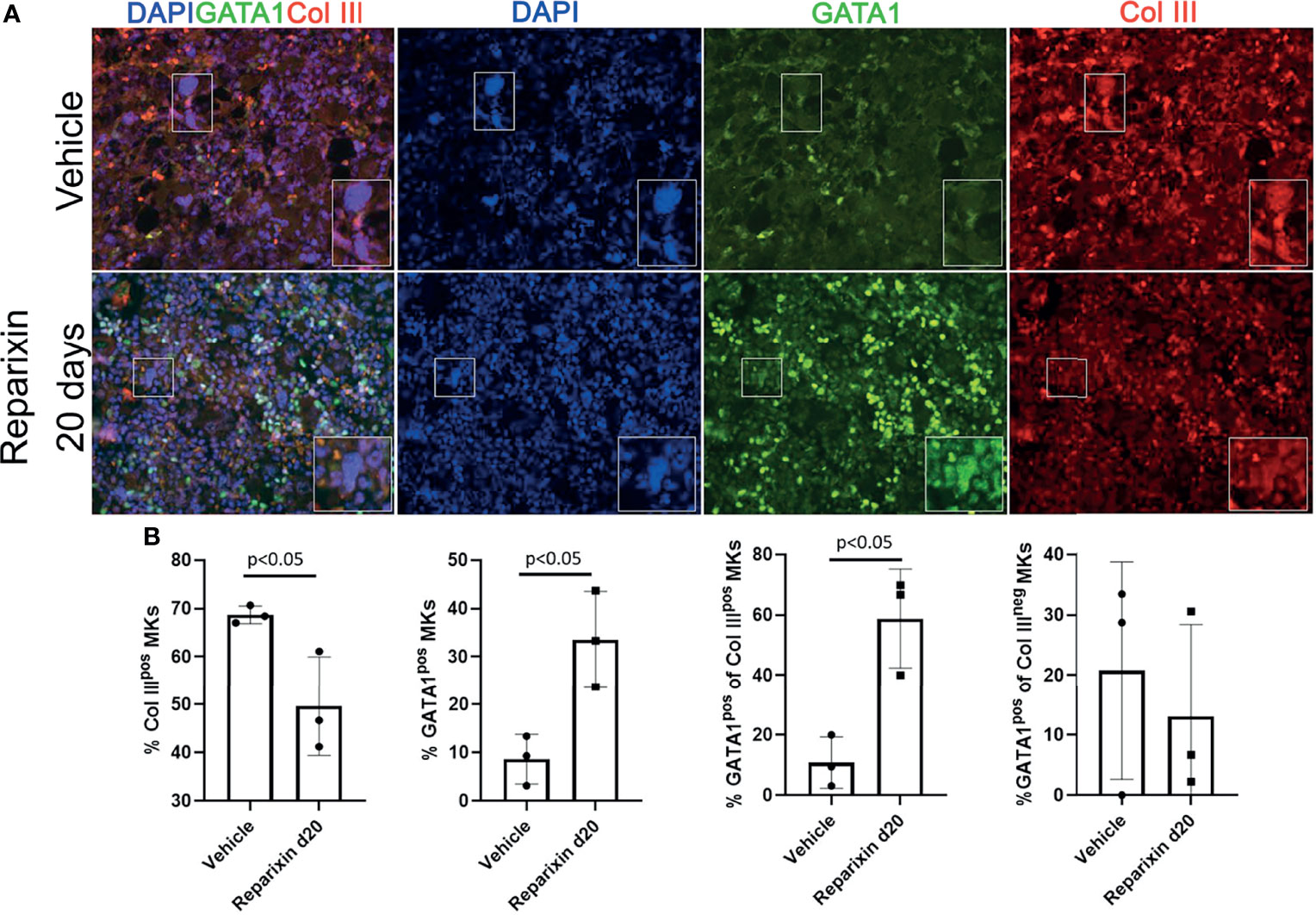
Figure 10 Reparixin increases the GATA1 content in the MK subpopulation that express high levels of Col III. (A) Double immunofluorescence staining with anti-GATA1 (FITCH-green) and Col III (TRITCH-red) antibodies of BM sections from representative Gata1low mice treated with either vehicle or Reparixin for 20 days. Magnification ×400. The rectangles indicate a representative MK shown at greater magnification on the bottom right of each panel. (B) Frequency of MKs positive for Col III (Col IIIpos MKs), for GATA1 (Gata1pos MKs) and of the subsets of MKs positive or negative for Col III that were also positive for GATA1, respectively. Data are presented as Mean (± SD) and as values for individual mouse (each mouse a dot). Statistical analyses were performed by One-way ANOVA. P < 0.05 was considered statistically significant.
hCXCL8 is one of the pro-inflammatory cytokines expressed at high levels in MF and is associated with the poorest patient outcomes (3). In addition, malignant CD34+ cells and MKs from MF patients express high levels of hCXCL8 (10). We have now determined that the BM from the Gata1low MF mouse model contains MKs that not only express increased levels of TGF-β, but also of mCXCL1, the murine equivalent of hCXCL8, and that the MKs from these mice express higher levels of CXCR1 and CXCR2 receptors. The recent consideration of CXCR1/R2 allosteric inhibitors for therapy of inflammatory lung diseases associated with fibrosis (26, 27) suggests that these inhibitors might be effective in treating BM fibrosis. This hypothesis was tested in the present study that assessed the effects of the CXCR1/R2 inhibitor Reparixin on the myelofibrotic phenotype expressed by Gata1low mice.
In accordance with the observations that allosteric modulation of CXCR1/2 is well tolerated (46–48), treatment with Reparixin showed no significant changes in body weight and in blood parameters in Gata1low mice, and no deaths were recorded during the entire period of observation.
Having found that an intrinsic variability in circulating drug levels was associated with prolonged administration of Reparixin by mini-pumps (36 and this manuscript), we performed correlative analyses to assess concentration-dependent treatment effects. Notably, by histological analysis we observed that Reparixin reduced in a concentration-dependent fashion the degree of BM and splenic fibrosis of Gata1low mice. Since CXCL1 and its CXCR1 and CXCR2 receptors contribute to the control of megakaryocytic proliferation, differentiation, and ploidy in MF (9), we evaluated whether inhibition of the CXCR1/R2 signaling might reduce the fibrosis by restoring the MKs abnormalities observed in Gata1low mice.
First, immunohistochemistry staining showed for the first time that mCXCR1, the recently characterized murine orthologue of hCXCR1 is highly expressed, together with mCXCR2 specifically in the MKs from Gata1low mice. Consistent with the fact that allosteric modulation does not block the binding of the endogenous ligand to its receptors or alter its constitutive activity (22), we observed that Reparixin treatment did not alter the expression of CXCL1 or CXCR1/R2 receptors by MKs from the BM of Gata1low mice. Since CXCR1/CXCR2 are also activated by CXCL6 and MIP2, we may not formally exclude that altered levels of these two chemokines are also involved in the development of myelofibrosis in our model and that their levels where normalized by treatment with Reparixin. This hypothesis will be tested as part of a separate study.
We observed, however, that the BM of Reparixin-treated mice expressed lower levels of TGF-β as compared to vehicle treated mice. The effect of Reparixin on TGF-β expression was independent of the plasma concentration of Reparixin. Since Reparixin is a >100 fold more potent inhibitor of CXCR1 (both h and m) than CXCR2 (both h and m), (22, 23), we hypothesize that TGF-β production by murine MKs is primarily regulated by mCXCR1 and therefore is not influenced by the variability in circulating drug levels. The suggested role of mCXCR1 in the regulation of TGF-β content is supported by recent data highlighting the prominent role of the CXCL6/CXCR1 axis in the release of TGF-β by liver Kupffer cells both in patients with liver fibrosis and in a mouse model of carbon tetrachloride-induced chronic liver injury and fibrosis (49). We may not exclude however, that Reparixin reduced TGF-β content in the bone marrow indirectly by reducing the frequency of pathological MKs emperipolesis with the neutrophils which triggers the release of this factor in the bone marrow. In fact, since it is well known that CXCL1 induces neutrophil chemotaxis (50), it is possible that Reparixin, by decreasing neutrophil chemotaxis toward the MKs reduces their pathological emperipolesis with these cells reducing the amount of TGF-β they release in the microenvironment. This hypothesis is currently under investigation as part of a separate study (Dunbar et al., under revision). In conclusion, our results indicate that, in addition to CXCR2 indicated by loss of function studies in mouse models, (Dunbar et al., under review), CXCR1 is also involved in the regulation of profibrotic signaling at least in Gata1low mice.
We previously reported that pharmacological inhibition of TGF-β signaling in Gata1low mice restored the maturation of MKs and increased their content of GATA1 (31). Since we observed a significant decrease of the TGF-β expressed by MKs following Reparixin-treatment, we hypothesized that Reparixin might restore MK functions by reducing TGF-β content. This hypothesis is counterintuitive since Reparixin did not increase platelets numbers, which are typically low in Gata1low mice. This apparent paradox was explained by recent single cell profiling analyses indicating that murine BM contains three distinctive MK populations, each one exerting a different function. In addition to MKs poised to generate platelets, the BM contains MK poised to exert immune functions in the lungs (51) or niche functions during the embryogenesis but possibly also in adult organs undergoing tissue repair (42–44, 51). The BMs from Gata1low mice, and also that of MF patients, are characterized by an increased proportion of immature MKs that express increased levels of collagen (41, 52 and this manuscript). By immunostaining, we observed that Reparixin increased GATA1 expression while reducing the collagen III content in Gata1low mouse MKs. Notably, we demonstrated that the increase of GATA1 levels was most evident in the subpopulation of MKs expressing collagen, suggesting that Reparixin may target the niche-poised MKs reducing their ability to mediate collagen deposition and fibrosis in this animal model. At the moment, there is little experimental indication on the mechanism(s) that increased GATA1 content in the MKs from the Reparixin-treated mice. On the basis of published data, we suggest that this increase is mediated by the decreased TGF-β levels observed in these mice. In fact, it is well known that TGF-β, through a mechanism still poorly identified, retains MK immature (53). Since MK maturation requires GATA1 upregulation [with consequently downregulation of GATA2, a maturation mechanism defined the GATA1 to GATA2 switch (54)], it is conceivable that TGF-β reduces MK maturation by reducing expression of GATA1 and increasing that of GATA2. This hypothesis has been directly tested by the finding that a small TGF-β inhibitor rescues the abnormal maturation (and myelofibrosis) in Gata1low mice by increasing the levels of Gata1 mRNA while reducing those of Gata2 (31, 55). Due to the hypomorphic mutation, Gata1low cells lacks one of the three major hypersensitive sites of the gene but still contain two other important regulatory sequences (56, 57), which may be the target of the TGF-β inhibitors which have been demonstrated to be capable to upregulate its expression (31). Although drugs that increase GATA1 in MKs, such as Aurora kinase inhibitors, have been shown to be effective in reduce fibrosis in myelofibrosis patients (58), it may be debated whether drugs, such as Reparixin, that increase GATA1 protein in a mouse model in which the transcription of the gene is reduced by deletion of its regulatory sequence, will be effective in patients where the GATA1 content is reduced by inefficient translation of GATA1 mRNA (39). However, since Reparixin appears to act via TGF-β and the TGF-β TRAP AVID200 is capable to downregulate GATA2 (and therefore presumably to upregulates GATA1), restoring the maturation of MKs expanded in vitro from CD34+ cells in the subset of patients who are responsive to the drug (38), we believe that Reparixin will be effective also in patients.
In conclusion, these results indicate that treatment with Reparixin rescues the MF phenotype of Gata1low mice and provides a rationale for considering Reparixin as a therapeutic option to treat MF patients.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by the Institutional animal care committee according to the European Directive 86/609/EEC.
PV, FG, FM, MTM, LB and CG performed experiments and analyzed the data. PV and FG performed statistical analyses. GS reviewed all the histopathogical determinations. ARM and MA designed the study, interpreted the data and wrote the manuscript. All the authors read the manuscript and concur with its content. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This study was supported by grants from the National Cancer Institute (P01-CA108671), the National Heart, Lung and Blood Institute (1R01-HL134684), the Associazione Italiana Ricerca Cancro (AIRC, IG23525), by funds from the Italian Ministry of Economic Development DM 02/08/2019 and DD 02/10/2019 grant proposal number 1410, and by the Dompé Farmaceutici Spa R&D.
MTM, LB, CG and MA are employees of Dompè Farmaceutici SPA and ARM received research funds from Dompè Farmaceutici SPA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that this study received funding from Dompè Farmaceutici SpA. The funder had the following involvement with the study: writing of the article and interpretation of the data.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The technical support of Mrs. Yvan Gilardi, Enrico Cardarelli and Andrea Giovannelli, of the Animal Facility of the Istituto Superiore di Sanità and the editorial assistance of Dr. Gisella Gaspari are gratefully acknowledged.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.853484/full#supplementary-material
1. Tefferi A. Pathogenesis of Myelofibrosis With Myeloid Metaplasia. J Clin Oncol (2005) 23(33):8520–30. doi: 10.1200/JCO.2004.00.9316
2. Vannucchi AM, Pancrazzi A, Guglielmelli P, Di Lollo S, Bogani C, Baroni G, et al. Abnormalities of GATA-1 in Megakaryocytes From Patients With Idiopathic Myelofibrosis. Am J Pathol (2005) 167(3):849–58. doi: 10.1016/S0002-9440(10)62056-1
3. Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating Interleukin (IL)-8, IL-2r, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. J Clin Oncol (2011) 29(10):1356–63. doi: 10.1200/JCO.2010.32.9490
4. Verstovsek S, Mesa RA, Gotlib J, Gupta V, DiPersio J, Catalano JV, et al. Long-Term Treatment With Ruxolitinib for Patients With Myelofibrosis: 5-Year Update From the Randomized, Double-Blind, Placebo-Controlled, Phase 3 COMFORT-I Trial. J Hematol Oncol (2017) 10(1):55. doi: 10.1186/s13045-017-0417-z
5. Harrison CN, Vannucchi AM, Kiladjian J-J, Al-Ali HK, Gisslinger H, Knoops L, et al. Long-Term Findings From COMFORT-II, A Phase 3 Study of Ruxolitinib vs Best Available Therapy for Myelofibrosis. Leukemia (2016) 30:1701. doi: 10.1038/leu.2016.323
6. Zimran A, Durán G, Giraldo P, Rosenbaum H, Giona F, Petakov M, et al. Long-Term Efficacy and Safety Results of Taliglucerase Alfa Through 5years in Adult Treatment-Naïve Patients With Gaucher Disease. Blood Cells Mol Dis (2019) 78:14–21. doi: 10.1016/j.bcmd.2016.07.002
7. Russo RC, Garcia CC, Teixeira MM, Amaral FA. The CXCL8/IL-8 Chemokine Family and its Receptors in Inflammatory Diseases. Expert Rev Clin Immunol (2014) 10(5):593–619. doi: 10.1586/1744666X.2014.894886
8. Takeuchi K, Higuchi T, Yamashita T, Koike K. Chemokine Production by Human Megakaryocytes Derived From CD34-Positive Cord Blood Cells. Cytokine (1999) 11:424–34. doi: 10.1006/cyto.1998.0455
9. Emadi S, Clay D, Desterke C, Guerton B, Maquarre E, Charpentier A. IL-8 and its CXCR1 and CXCR2 Receptors Participate in the Control of Megakaryocytic Proliferation, Differentiation, and Ploidy in Myeloid Metaplasia With Myelofibrosis. Blood (2005) 105:464. doi: 10.1182/blood-2003-12-4415
10. Dunbar A, Lu M, Farina M, Park Y, Yang J, Kim D, et al. Increased Interleukin-8 (IL8)-CXCR2 Signaling Promotes Progression of Bone Marrow Fibrosis in Myeloproliferative Neoplasms. Blood (2020) 136(Supplement 1):6–7. doi: 10.1182/blood-2020-138843
11. Murphy PM, Tiffany HL. Cloning of Complementary DNA Encoding a Functional Human Interleukin-8 Receptor. Science (1991) 253:1280–3. doi: 10.1126/science.1891716
12. Holmes WE, Lee J, Kuang WJ, Rice GC, Wood WI. Structure and Functional Expression of a Human Interleukin-8 Receptor. Science (1991) 253:1278–80. doi: 10.1126/science.1840701
13. Wuyts A, Van Osselaer N, Haelens A, Samson I, Herdewijn P, Ben-Baruch A, et al. Characterization of Synthetic Human Granulocyte Chemotactic Protein 2: Usage of Chemokine Receptors CXCR1 and CXCR2 and In Vivo Inflammatory Properties. Biochemistry (1997) 36:2716–23. doi: 10.1021/bi961999z
14. Bozic CR, Gerard NP, von Uexkull-Guldenband C, Kolakowski LF Jr., Conklyn MJ, Breslow R, et al. The Murine Interleukin 8 Type B Receptor Homologue and its Ligands. Expression and Biological Characterization. J Biol Chem (1994) 269:29355–8. doi: 10.1016/S0021-9258(18)43882-3
15. Cerretti DP, Nelson N, Kozlosky CJ, Morrissey PJ, Copeland NG, Gilbert DJ, et al. The Murine Homologue of the Human Interleukin-8 Receptor Type B Maps Near the Ity-Lsh-Bcg Disease Resistance Locus. Genomics (1993) 18:410–3. doi: 10.1006/geno.1993.1486
16. Cacalano G, Lee J, Kikly K, Ryan AM, Pitts-Meek S, Hultgren B, et al. Neutrophil and B Cell Expansion in Mice That Lack the Murine IL-8 Receptor Homolog. Science (1994) 265:682–4. doi: 10.1126/science.8036519
17. Lee J, Cacalano G, Camerato T, Toy K, Moore MW, Wood WI. Chemokine Binding and Activities Mediated by the Mouse IL-8 Receptor. J Immunol (1995) 155:2158–64.
18. Fu W, Zhang Y, Zhang J, Chen WF. Cloning and Characterization of Mouse Homolog of the CXC Chemokine Receptor CXCR1. Cytokine (2005) 31:9–17. doi: 10.1016/j.cyto.2005.02.005
19. Moepps B, Nuesseler E, Braun M, Gierschik P. A Homolog of the Human Chemokine Receptor CXCR1 is Expressed in the Mouse. Mol Immunol (2006) 43:897–914. doi: 10.1016/j.molimm.2005.06.043
20. Fan X, Patera AC, Pong-Kennedy A, Deno G, Gonsiorek W, Manfra DJ, et al. Murine CXCR1 is a Functional Receptor for GCP-2/CXCL6 and Interleukin-8/CXCL8. J Biol Chem (2007) 282(16):11658–66. doi: 10.1074/jbc.M607705200
21. Wuyts A, Haelens A, Proost P, Lenaerts JP, Conings R, Opdenakker G, et al. Identification of Mouse Granulocyte Chemotactic Protein-2 From Fibroblasts and Epithelial Cells. Functional Comparison With Natural KC and Macrophage Inflammatory Protein-2. J Immunol (1996) 157:1736–43.
22. Bertini R, Allegretti M, Bizzarri C, Moriconi A, Locati M, Zampella G, et al. Noncompetitive Allosteric Inhibitors of the Inflammatory Chemokine Receptors CXCR1 and CXCR2: Prevention of Reperfusion Injury. PNAS (2004) 101(32):11791–6. doi: 10.1073/pnas.0402090101
23. Citro A, Valle A, Cantarelli E, Mercalli A, Pellegrini S, Liberati D. CXCR1/2 Inhibition Blocks and Reverses Type 1 Diabetes in Mice. Diabetes (2015) 64(4):1329–40. doi: 10.2337/db14-0443
24. Allegretti M, Bertini R, Bizzarri C, Beccari A, Mantovani A, Locati M. Allosteric Inhibitors of Chemoattractant Receptors: Opportunities and Pitfalls. Trends Pharmacol Sci (2008) 29(6):280–6. doi: 10.1016/j.tips.2008.03.005
25. Jurcevic S, Humfrey C, Uddin M, Warrington S, Larsson B, Keen C. The Effect of a Selective CXCR2 Antagonist (AZD5069) on Human Blood Neutrophil Count and Innate Immune Functions. Br J Clin Pharmacol (2015) 80(6):1324–36. doi: 10.1111/bcp.12724
26. Cheng IY, Liu CC, Lin JH, Hsu TW, Hsu JW, Li AF, et al. Particulate Matter Increases the Severity of Bleomycin-Induced Pulmonary Fibrosis Through KC-Mediated Neutrophil Chemotaxis. Int J Mol Sci (2019) 21(1):227. doi: 10.3390/ijms21010227
27. Mattos MS, Ferrero MR, Kraemer L, Lopes GAO, Reis DC, Cassali GD, et al. CXCR1 and CXCR2 Inhibition by Ladarixin Improves Neutrophil-Dependent Airway Inflammation in Mice. Front Immunol (2020) 11:566953. doi: 10.3389/fimmu.2020.566953
28. Centurione L, Di Baldassarre A, Zingariello M, Bosco D, Gatta V, Rana RA, et al. Increased and Pathologic Emperipolesis of Neutrophils Within Megakaryocytes Associated With Marrow Fibrosis in GATA-1low Mice. Blood (2004) 104:3573–80. doi: 10.1182/blood-2004-01-0193
29. Vyas P, Ault K, Jackson CW, Orkin SH, Shivdasani RA. Consequences of GATA-1 Deficiency in Megakaryocytes and Platelets. Blood (1999) 93:2867–75. doi: 10.1182/blood.V93.9.2867
30. Vannucchi AM, Bianchi L, Cellai C, Paoletti F, Rana RA, Lorenzini R, et al. Development of Myelofibrosis in Mice Genetically Impaired for GATA-1 Expression (GATA-1low Mice). Blood (2002) 100(4):1123–32. doi: 10.1182/blood-2002-06-1913
31. Zingariello M, Martelli F, Ciaffoni F, Masiello F, Ghinassi B, D’Amore E, et al. Characterization of the TGF-β1 Signaling Abnormalities in the Gata1low Mouse Model of Myelofibrosis. Blood (2013) 121(17):3345–63. doi: 10.1182/blood-2012-06-439661
32. Zingariello M, Verachi P, Gobbo F, Martelli F, Falchi M, Mazzarini M, et al. Resident Self-Tissue of Proinflammatory Cytokines Rather Than Their Systemic Levels Correlates With Development of Myelofibrosis in Gata1low Mice. Biomolecules (2022) 12(2):234. doi: 10.3390/biom12020234
33. Martelli F, Ghinassi B, Panetta B, Alfani E, Gatta V, Pancrazzi, et al. Variegation of the Phenotype Induced by the Gata1low Mutation in Mice of Different Genetic Backgrounds. Blood (2005) 106:4102–13. doi: 10.1182/blood-2005-03-1060
34. Cavalieri B, Mosca M, Ramadori P, Perrelli M-G, De Simone L, Colotta F, et al. Neutrophil Recruitment in the Reperfused-Injured Rat Liver was Effectively Attenuated by Repertaxin, a Novel Allosteric Noncompetitive Inhibitor of CXCL8 Receptors: A Therapeutic Approach for the Treatment of Post-Ischemic Hepatic Syndromes. Int J Immunopathol Pharmacol (2005) 18(3):475–86. doi: 10.1177/039463200501800307
35. Brandolini L, Benedetti E, Ruffini PA, Russo R, Cristiano L, Antonosante A, et al. CXCR1/2 Pathways in Paclitaxel-Induced Neuropathic Pain. Oncotarget (2017) 8(14):23188–201. doi: 10.18632/oncotarget.15533
36. Di Sapia R, Zimmer TS, Kebede V, Balosso S, Ravizza T, Sorrentino D, et al. CXCL1-CXCR1/2 Signaling is Induced in Human Temporal Lobe Epilepsy and Contributes to Seizures in a Murine Model of Acquired Epilepsy. Neurobiol Dis (2021) 158:105468. doi: 10.1016/j.nbd.2021.105468
37. Migliaccio AR, Rana RA, Sanchez M, Lorenzini R, Centurione L, Bianchi L, et al. GATA-1 as a Regulator of Mast Cell Differentiation Revealed by the Phenotype of the GATA-1low Mouse Mutant. J Exp Med (2003) 197(3):281–96. doi: 10.1084/jem.20021149
38. Varricchio L, Iancu-Rubin C, Upadhyaya B, Zingariello M, Martelli F, Verachi P, et al. TGF-β1 Protein Trap AVID200 Beneficially Affects Hematopoiesis and Bone Marrow Fibrosis in Myelofibrosis. JCI Insight (2021) 6(18):e145651. doi: 10.1172/jci.insight.145651
39. Gilles L, Arslan AD, Marinaccio C, Wen QJ, Arya P, McNulty M, et al. Downregulation of GATA1 Drives Impaired Hematopoiesis in Primary Myelofibrosis. J Clin Invest (2017) 127(4):1316–20. doi: 10.1172/jci82905
40. Zhan H, Ma Y, Lin CHS, Kaushansky K. JAK2V617F-Mutant Megakaryocytes Contribute to Hematopoietic Stem/progenitor Cell Expansion in a Model of Murine Myeloproliferation. Leukemia (2016) 30(12):2332–41. doi: 10.1038/leu.2016.114
41. Abbonante V, Di Buduo CA, Gruppi C, Malara A, Gianelli U, Celesti G, et al. Thrombopoietin/TGF-β1 Loop Regulates Megakaryocyte Extracellular Matrix Component Synthesis. Stem Cells (2016) 34(4):1123–33. doi: 10.1002/stem.2285
42. Migliaccio AR, Hoffman R. An Outline of the Outset of Thrombopoiesis in Human Embryos at Last. Cell Stem Cell (2021) 28(3):363–5. doi: 10.1016/j.stem.2021.02.007
43. Sun S, Jin C, Si J, Lei Y, Chen K, Cui Y, et al. Single-Cell Analysis of Ploidy and the Transcriptome Reveals Functional and Spatial Divergency in Murine Megakaryopoiesis. Blood (2021) 138(14):1211–24. doi: 10.1182/blood.2021010697
44. Wang H, He J, Xu C, Chen X, Yang H, Shi S, et al. Decoding Human Megakaryocyte Development. Cell Stem Cell (2021) 28(3):535–49.e8. doi: 10.1016/j.stem.2020.11.006
45. Sim X, Jarocha D, Hayes V, Hanby HA, Marks MS, Camire RM, et al. Identifying and Enriching Platelet-Producing Human Stem Cell-Derived Megakaryocytes Using Factor V Uptake. Blood (2017) 130(2):192–204. doi: 10.1182/blood-2017-01-761049
46. Goldstein LJ, Perez RP, Yardley D, Han LK, Reuben JM, Gao H, et al. A Window-of-Opportunity Trial of the CXCR1/2 Inhibitor Reparixin in Operable HER-2-Negative Breast Cancer. Breast Cancer Res (2020) 22(1):4. doi: 10.1186/s13058-019-1243-8
47. Maffi P, Lundgren T, Tufveson G, Rafael E, Shaw JAM, Liew A, et al. Targeting CXCR1/2 Does Not Improve Insulin Secretion After Pancreatic Islet Transplantation: A Phase 3, Double-Blind, Randomized, Placebo-Controlled Trial in Type 1 Diabetes. Diabetes Care (2020) 43(4):710–8. doi: 10.2337/dc19-1480
48. Opfermann P, Derhaschnig U, Felli A, Wenisch J, Santer D, Zuckermann A, et al. A Pilot Study on Reparixin, a CXCR1/2 Antagonist, to Assess Safety and Efficacy in Attenuating Ischaemia-Reperfusion Injury and Inflammation After on-Pump Coronary Artery Bypass Graft Surgery. Clin Exp Immunol (2015) 180(1):131–42. doi: 10.1111/cei.12488
49. Cai X, Li Z, Zhang Q, Qu Y, Xu M, Wan X, et al. CXCL6-EGFR-Induced Kupffer Cells Secrete TGF-β1 Promoting Hepatic Stellate Cell Activation via the SMAD2/BRD4/C-MYC/EZH2 Pathway in Liver Fibrosis. J Cell Mol Med (2018) 22(10):5050–61. doi: 10.1111/jcmm.13787
50. Teijeira A, Garasa S, Ochoa MDC, Cirella A, Olivera I, Glez-vaz J, et al. Differential Interleukin-8 Thresholds for Chemotaxis and Netosis in Human Neutrophils. Eur J Immunol (2021) 51(9):2274–80. doi: 10.1002/eji.202049029
51. Pariser DN, Hilt ZT, Ture SK, Blick-Nitko SK, Looney MR, Cleary SJ, et al. Lung Megakaryocytes are Immune Modulatory Cells. J Clin Invest (2021) 131(1):e137377. doi: 10.1172/JCI137377
52. Malara A, Currao M, Gruppi C, Celesti G, Viarengo G, Buracchi C, et al. Megakaryocytes Contribute to the Bone Marrow-Matrix Environment by Expressing Fibronectin, Type IV Collagen, and Laminin. Stem Cells (2014) 32(4):926–37. doi: 10.1002/stem.1626
53. Kuter DJ, Gminski DM, Rosenberg RD. Transforming Growth Factor Beta Inhibits Megakaryocyte Growth and Endomitosis. Blood (1992) 79(3):619–26. doi: 10.1182/blood.V79.3.619.bloodjournal793619
54. Bresnick EH, Lee HY, Fujiwara T, Johnson KD, Keles S. GATA Switches as Developmental Drivers. J Biol Chem (2010) 285(41):31087–93. doi: 10.1074/jbc.R110.159079
55. Yue L, Bartenstein M, Zhao W, Ho WT, Han Y, Murdun C, et al. Efficacy of ALK5 Inhibition in Myelofibrosis. JCI Insight (2017) 2(7):e90932. doi: 10.1172/jci.insight.90932
56. McDevitt MA, Shivdasani RA, Fujiwara Y, Yang H, Orkin SH. A "Knockdown" Mutation Created by Cis-Element Gene Targeting Reveals the Dependence of Erythroid Cell Maturation on the Level of Transcription Factor GATA-1. Proc Natl Acad Sci (1997) 94(13):6781–5. doi: 10.1073/pnas.94.13.6781
57. Migliaccio AR, Martelli F, Verrucci M, Sanchez M, Valeri M, Migliaccio G, et al. Gata1 Expression Driven by the Alternative HS2 Enhancer in the Spleen Rescues the Hematopoietic Failure Induced by the Hypomorphic Gata1low Mutation. Blood (2009) 114(10):2107–20. doi: 10.1182/blood-2009-03-211680
Keywords: myelofibrosis, TGF-β, megakaryocytes, GATA1, CXCL8 (interleukin-8)
Citation: Verachi P, Gobbo F, Martelli F, Martinelli A, Sarli G, Dunbar A, Levine RL, Hoffman R, Massucci MT, Brandolini L, Giorgio C, Allegretti M and Migliaccio AR (2022) The CXCR1/CXCR2 Inhibitor Reparixin Alters the Development of Myelofibrosis in the Gata1low Mice. Front. Oncol. 12:853484. doi: 10.3389/fonc.2022.853484
Received: 12 January 2022; Accepted: 16 February 2022;
Published: 22 March 2022.
Edited by:
Elena Levantini, Beth Israel Deaconess Medical Center and Harvard Medical School, United StatesReviewed by:
Lorena Figueiredo-Pontes, University of São Paulo, BrazilCopyright © 2022 Verachi, Gobbo, Martelli, Martinelli, Sarli, Dunbar, Levine, Hoffman, Massucci, Brandolini, Giorgio, Allegretti and Migliaccio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Rita Migliaccio, YW1pZ2xpYWNjaW9AYWx0aXVzLm9yZw==; Marcello Allegretti, bWFyY2VsbG8uYWxsZWdyZXR0aUBkb21wZS5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.