 Adria Hasan
Adria Hasan Suroor Fatima Rizvi
Suroor Fatima Rizvi Sana Parveen
Sana Parveen Neelam Pathak
Neelam Pathak Aamir Nazir
Aamir Nazir Snober S. Mir
Snober S. Mir- 1Molecular Cell Biology Laboratory, Integral Information and Research Centre-4 (IIRC-4), Integral University, Lucknow, India
- 2Department of Bioengineering, Faculty of Engineering, Integral University, Lucknow, India
- 3Department of Biosciences, Faculty of Science, Integral University, Lucknow, India
- 4Department of Biochemistry, Dr. RML Avadh University, Faizabad, India
- 5Laboratory of Functional Genomics and Molecular Toxicology, Division of Neuroscience and Ageing Biology, CSIR-Central Drug Research Institute, Lucknow, India
Cancer formation is a highly regulated and complex process, largely dependent on its microenvironment. This complexity highlights the need for developing novel target-based therapies depending on cancer phenotype and genotype. Autophagy, a catabolic process, removes damaged and defective cellular materials through lysosomes. It is activated in response to stress conditions such as nutrient deprivation, hypoxia, and oxidative stress. Oxidative stress is induced by excess reactive oxygen species (ROS) that are multifaceted molecules that drive several pathophysiological conditions, including cancer. Moreover, autophagy also plays a dual role, initially inhibiting tumor formation but promoting tumor progression during advanced stages. Mounting evidence has suggested an intricate crosstalk between autophagy and ROS where they can either suppress cancer formation or promote disease etiology. This review highlights the regulatory roles of autophagy and ROS from tumor induction to metastasis. We also discuss the therapeutic strategies that have been devised so far to combat cancer. Based on the review, we finally present some gap areas that could be targeted and may provide a basis for cancer suppression.
Introduction
Autophagy, meaning “self-eating,” is a catabolic process where cytoplasmic organelles, proteins, and other macromolecules are degraded during starvation or other types of stress (1–3). It is vital in maintaining cellular homeostasis, helps eliminate pathogens, and is regulated by the autophagy-related (ATG) genes. The molecules/cargo to be degraded are sequestered in double-membrane vesicles (autophagosomes). Autophagosomes fuse to lysosomes, forming autolysosomes that lead to cargo degradation. The degraded molecules provide energy that can be used in anabolic and bioenergetic pathways (4). Apart from macroautophagy, there are two other forms of autophagy: microautophagy and chaperone-mediated autophagy (5). Any disruption in autophagic pathways has been shown to play a significant role in different diseases such as neurodegeneration, atherosclerosis, and cancer (6, 7).
Usually, autophagy acts as a tumor suppressor during initiation but promotes cancer cell proliferation in established tumors (8). Autophagy can be regulated by several factors, including starvation, infections, drugs, hypoxia, ATP/AMP ratio, and reactive oxygen species (ROS) levels (9). Cancer cells also exhibit high ROS levels (10) due to increased metabolism rate, incomplete oxidative phosphorylation, mitochondrial dysfunction, low nutrient levels, hypoxia, and low pH in their microenvironment (11–13). Under normal conditions, low ROS levels are generated to regulate signaling pathways, including autophagy, to maintain cellular homeostasis (14–16). Moreover, starvation conditions known to upregulate autophagy can also induce ROS. Consistently, studies have shown ROS-mediated regulation of autophagy as ROS scavengers or high expression of antioxidants can block stress-induced autophagy (17, 18).
ROS-induced autophagy can lead to cell death or survival (17, 19). High ROS levels can also activate several oncogenic pathways, such as mitogen-activated protein kinase (MAPK) and nuclear factor (NF)-κB signaling pathways. Contrarily, increased ROS can also promote cell death by activating the tumor suppressor p53 or apoptosis caused by excessive mitochondrial and DNA damage (20). Thus, an intricate cellular balance between autophagy and ROS is required to maintain cellular redox balance in normal and disease-related physiological conditions. Therefore, the exact role of autophagy and ROS in cancer cells is context-dependent and varies in different cancer phenotypes (21–24). This review describes the role of autophagy and ROS as tumor promoters and suppressors. We further discuss the intricate crosstalk between autophagy and ROS that can regulate tumor promotion, metastasis, and response to therapy and may ultimately decide the fate of cancer cells.
Regulation of Autophagy
Autophagy is moderately active at the basal level but becomes highly activated due to different cellular stresses, including chemotherapeutics and radiotherapy (25–27). To date, 35 different ATG genes have been identified in yeast that are also conserved in higher eukaryotes (28–31). The autophagy pathway can be divided into several steps: (a) initiation and nucleation, (b) autophagosome closure, (c) maturation through autophagosome–lysosome fusion, and (d) cargo degradation through lysosomal enzymes. Autophagy is regulated through a series of proteins, including mammalian target of rapamycin (mTOR) and 5' adenosine monophosphate-activated protein kinase (AMPK). Activated mTOR negatively regulates autophagy through phosphorylation of the Atg proteins. However, during stress conditions, mTOR is inhibited, and autophagy is enhanced. Conversely, AMPK negatively regulates mTOR and induces the autophagic process (32; 33). After mTOR inhibition, the Unc-51-like autophagy-activating kinase (ULK) complex is activated (34), which in turn activates the class III phosphoinositide 3 kinase (PI3K) (35). The class III PI3K complex consists of several proteins including VPS34, p150, Atg14, and Beclin-1, which initiates autophagosome formation. Beclin-1, a primary autophagy regulator, recruits different proteins involved in the maturation and elongation of the autophagosome. Subsequently, Atg9 protein mediates the trafficking of the source membrane for autophagosome elongation. These may include the Golgi complex, mitochondria, endoplasmic reticulum, endosome, and plasma membrane (36). The primary component required for autophagosome maturation is the ubiquitin-like protein lipidation system that conjugates phosphatidylethanolamine to the C terminus of Atg8 (LC-3) protein, thereby facilitating the incorporation of Atg8 protein into autophagosomal membranes (37, 38). The proteins Atg7 and Atg10 help in conjugating Atg12 protein to Atg5 protein. The Atg12–Atg5 protein complex then conjugates with Atg16L1 protein to promote Atg8 protein lipidation. Atg8 protein is present in the inactive pro-Atg8 form but is cleaved by Atg4B protein, leaving a C-terminal glycine residue (39). The lipidated form of Atg8 protein is strongly associated with the autophagosomal membranes. Yeast contain a single Atg8 protein, while mammals have seven Atg8 proteins in two structurally related subfamilies (MAP1LC3A, B, C and GABARAP, GABARAPL1, and GABARAPL2), signifying a complex diversification of their functions (37). During autophagy induction, damaged organelles, protein aggregates, and ubiquitinated proteins are sorted to the phagophore for degradation. The Atg5–Atg12–Atg16L protein complex localizes to the phagophore, forming a cup-shaped structure, and dissociates when LC3-II localizes to the phagophore to complete the autophagosome formation. The cargo adaptor proteins like p62, NBR1, or NIX are further recruited on the autophagosome to target ubiquitinated protein aggregates and damaged organelles for degradation (40–42). Furthermore, the autophagosome fuses with the lysosomes forming autolysosomes to degrade targeted contents (Figure 1). This fusion is mediated by lysosomal-associated membrane protein 2 (LAMP2), the small GTPase RAB7A and UVRAG. Finally, lysosomal hydrolases and cathepsins degrade the targeted proteins, while cathepsins degrade LC3-II on the inner autophagosomal surface (43).
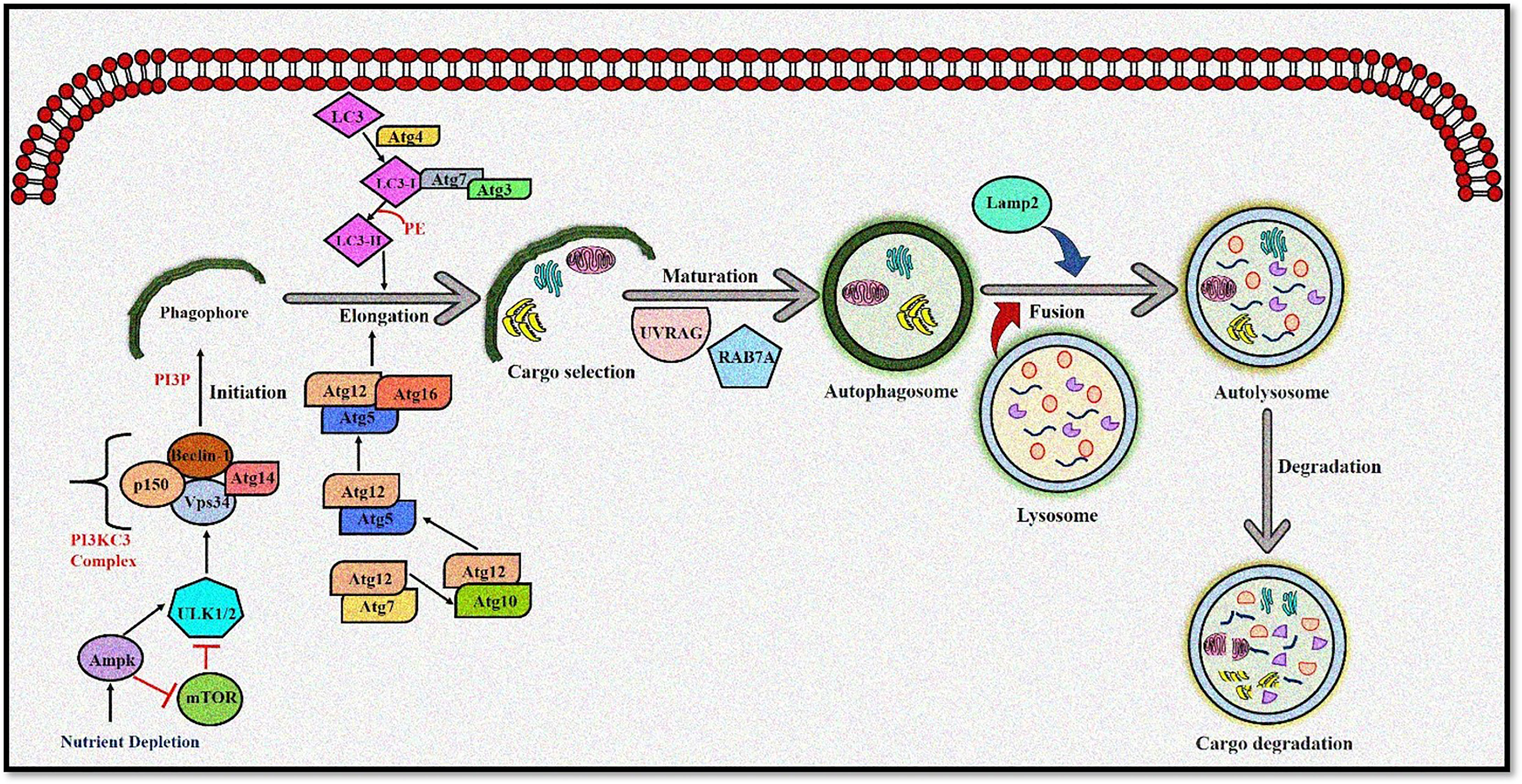
Figure 1 The autophagy pathway. AMPK and ULK1 kinase complex initiates autophagy. mTOR inhibition promotes phagophore formation through class III PI3K and Beclin 1 complex formation. Atg5-12 complex and LC3 are required to complete the autophagosome. After maturation, autophagosomes fuse with lysosomes to form autolysosomes where cargo degradation occurs. UVRAG, RAB7A, and LAMP2 mediate autophagosome maturation and fusion with lysosomes. AMPK, 5' adenosine monophosphate-activated protein kinase; ULK1, Unc-51 Like Autophagy Activating Kinase 1; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinase; ATG, Autophagy related; LC3, Microtubule-associated protein 1A/1B-light chain 3; UVRAG, UV radiation resistance-associated gene protein; RAB7A, Ras-related protein Rab-7a; LAMP2, lysosomal-associated membrane protein 2.
Reactive Oxygen Species and Autophagy
ROS include a reactive group of molecules such as hydroxyl radical, superoxide anion , and hydrogen peroxide (H2O2) (44). During normal physiological conditions, most intracellular ROS are produced in the mitochondria during oxidative phosphorylation due to the leaking of electrons from the electron transport chain (45, 46). However, an increase in intracellular ROS levels can promote mitochondrial dysfunction by accumulating high ROS levels, oxidation of lipids, proteins, and DNA damage (Table 1) (56, 57). The selective removal of damaged mitochondria by autophagy is called mitophagy. It is mediated by two signaling pathways, namely, NIX/BNIP3L and PARKIN (PARK2)/phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) (58–61). Targeting mitochondria toward mitophagy requires interaction between Nix/BNIP3L and GABARAP at the autophagosome (41, 62). PARKIN/PINK1 help remove dysfunctional mitochondria in response to ROS-induced mitochondrial membrane depolarization (63). Furthermore, the redox balance in a cell is maintained through the antioxidant defense system consisting of glutathione peroxide (GPx), catalase, glutathione reductase, glutathione S-transferase (GST), superoxide dismutase (SOD), and glutathione (64). Intracellular H2O2 is generated by SOD-catalyzed dismutation from formed within the mitochondria (46). Increased H2O2 levels were observed during tumorigenesis due to increased ROS production, high SOD levels, and inactivation of H2O2-scavenging enzymes (48). High H2O2 levels induce autophagic cell death in glioma cells after treatment with the polycyclic ammonium ion sanguinarine, which increases electron leakage from mitochondria and induces NADPH oxidases (NOXs) (65). NOXs, a membrane-bound enzyme complex, is another source of extracellular ROS (49) and are abnormally upregulated in cancer cells (66).
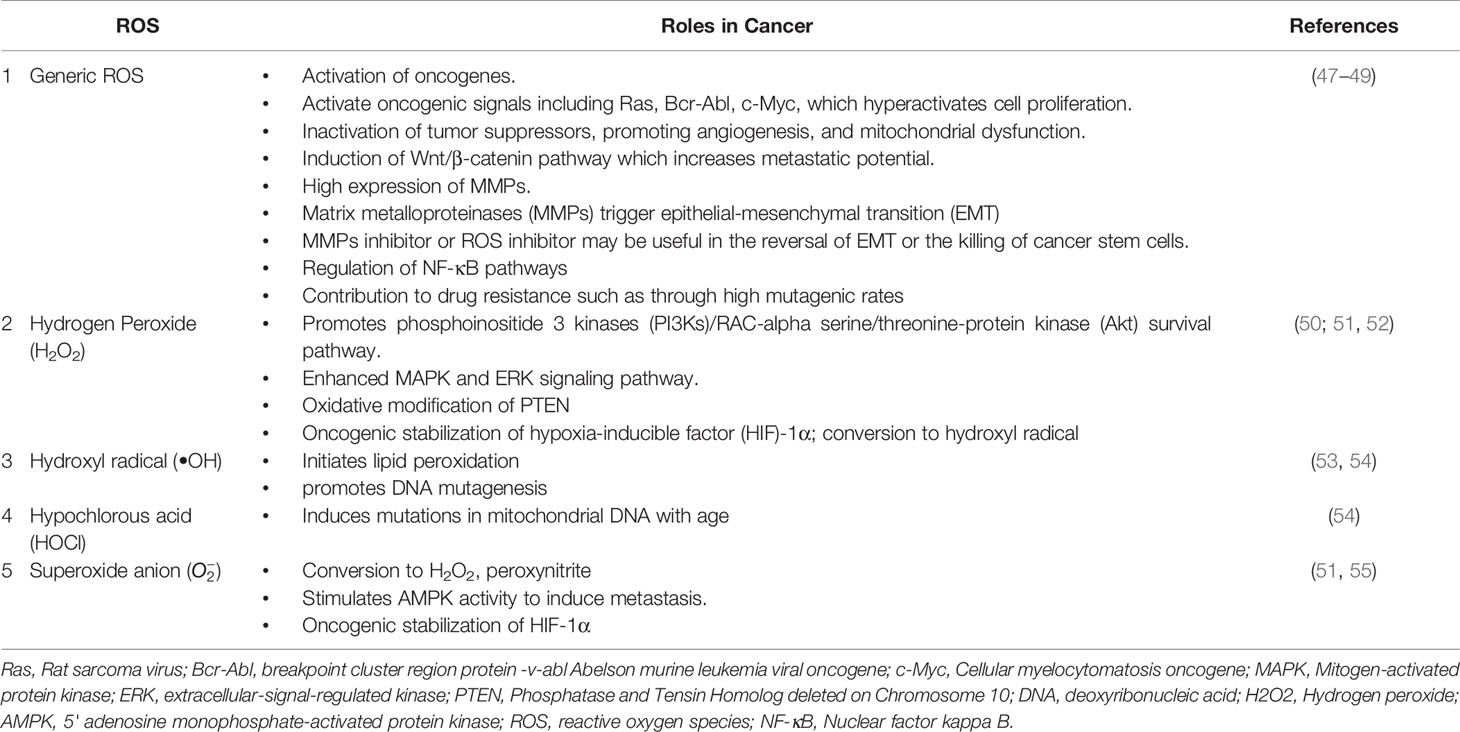
Table 1 Role of different reactive oxygen species in cancer.
Studies have demonstrated that several oncogenes, including K-RAS and c-MYC, induce intracellular ROS to promote cancer cell proliferation (67, 68). K-RAS also promotes extracellular ROS generation by increasing the activity of NOXs on the tumor cell membrane (69). In this regard, a study reports the tumor-promoting effect of autophagy in K-Ras [K-Ras(V12)]-induced malignant cell transformation, where inhibiting ROS with antioxidants reduced K-RasV12-induced induction of Atg5 protein and Atg7 protein, autophagy, and cancer growth (70). However, another study reports that rapamycin, an mTOR inhibitor, combined with (Hsp90) inhibitor IPI-504, causes tumor regression by promoting mitochondrial damage, oxidative stress, and autophagy in Kras/p53 mutant lung cancer and Nf1-deficient RAS-driven tumors (71).
Following another mechanism of action, ROS can also regulate autophagy through AMPK. AMPK induces autophagy during hypoxia or nutrient starvation by inhibiting mammalian target of rapamycin complex 1 (mTORC1 (72, 73). AMPK is phosphorylated by AMP-activated protein kinase kinase (AMPKK) following the accumulation of H2O2, which promotes its activation and autophagy induction (74). Inactivation of Atg4 protein increases autophagosomes and ATM-mediated oxidation of AMPK that inhibits mTORC1 in a H2O2-dependent manner (26, 75, 76). ROS can also mediate the induction of autophagy genes, including Beclin-1 or SQSTM1/p62, by regulating the activity of NF-κB in cancer cells (77–79).
The redox regulation of the proto-oncogene Akt provides another crucial point in the ROS-mediated regulation of autophagy. A well-described Akt-activating mechanism is PTEN oxidation (80, 81). In this regard, ROS production due to the growth factor stimulation promotes PTEN inactivation by forming a disulfide bridge between a cysteine in the catalytic site with a proximal cysteine residue. Consequently, Akt is activated due to increased PtdIns(3,4,5)P3 levels (81). However, disruption of mitochondrial membrane potential by an increase in H2O2 levels inhibits Akt, an upstream activator of mTOR, and induces autophagy (82; 83). This ROS-mediated signal transduction mechanism may also have a critical physiological role, as it may block catabolic pathways, like autophagy, in the presence of growth factors and may also induce the process of tumorigenesis.
Although ROS can promote autophagy induction, autophagy can also modulate ROS production. It was observed that caspase 8 inhibition and subsequent activation of JNK1 led to Atg6-Atg7 protein-dependent cell death when apoptosis was impaired (84). Moreover, caspase 8 inhibition promotes selective catalase degradation via autophagy that results in increased lipid peroxidation and autophagic cell death (85). Thus, it can be hypothesized that autophagy-mediated removal of catalase creates a self-sustaining loop, in which increased production of H2O2 by mitochondria may promote aberrant activation of autophagy, ultimately leading to autophagic cell death. However, catalase degradation was not observed under starvation conditions stimulating cytoprotective autophagy.
Furthermore, superoxides also modulate autophagy, as starvation-induced autophagy, mitochondrial electron transfer chain inhibitors, and the addition of exogenous H2O2 correlate with increased superoxide production and reduced H2O2 levels. Thus, overexpression of the SOD2 [manganese superoxide dismutase (Mn-SOD)] scavenges the superoxides, inhibits autophagy, and promotes H2O2 levels and starvation-induced cell death. In contrast, increasing superoxide levels by using the mitochondrial electron transfer chain inhibitors combined with SOD inhibitor 2-methoxyestradiol (2-ME) promoted both autophagy and cell death (86).
Thus, it can be concluded that autophagy and ROS-generating agents work in an unprecedented complex manner, as ROS-induced autophagy and vice versa can either be a cytoprotective mechanism that reduces oxidative stress or a self-destructing process promoting autophagic cell death (Figure 2). A clearer understanding of this intricate crosstalk between autophagy and ROS can help develop therapeutic strategies and open several opportunities to target the disease development process.
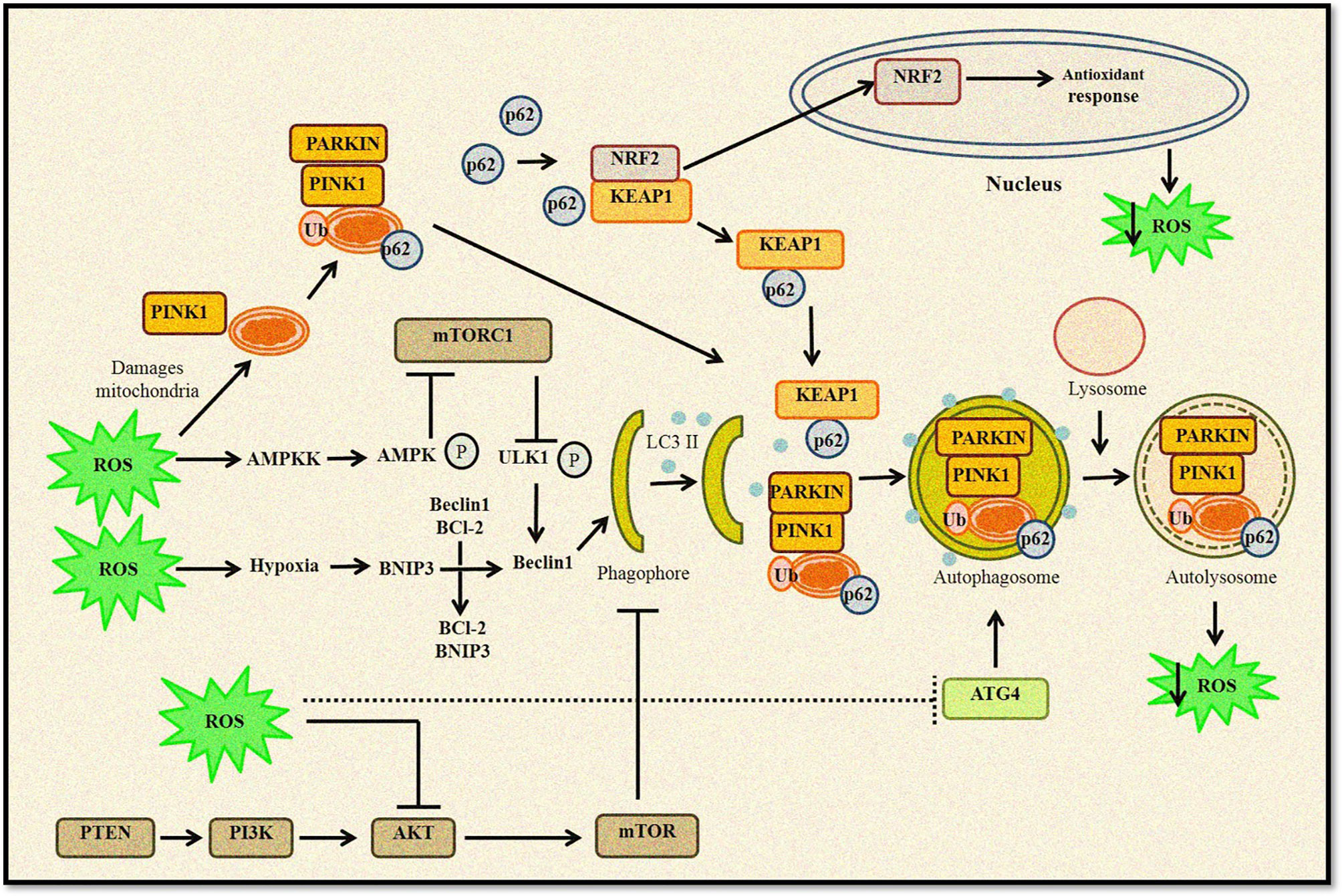
Figure 2 Relationship between ROS and autophagy. Increased ROS levels lead to oxidation of Atg4, which triggers autophagosome formation. ROS can regulate autophagy through AMPK activation that in turn phosphorylates ULK1 complex and promotes autophagy induction. Furthermore, disruption of Beclin 1–Bcl2 complex also induces autophagy. Any change in mitochondrial homeostasis promote ROS accumulation inducing mitophagy and removal of damaged mitochondria. Kelch-like ECH-associated protein 1 (KEAP1) degradation by p62-mediated selective autophagy leads to Nrf2-regulated antioxidant production and reduction in ROS levels. ROS can also inhibit the Akt/mTOR signaling cascade to induce autophagy.
Autophagy and Reactive Oxygen Species in Cancer: A Promoter or Suppressor
Autophagy usually acts as a survival pathway in normal and cancer cells exposed to various stresses like hypoxia, nutrient deprivation, or chemotherapeutics. These stress conditions also promote ROS generation that could aid in autophagy-mediated cell survival (25, 86). Indeed, ROS accumulation can activate several transcription factors like p53, hypoxia-inducible factor-1 (HIF-1), nuclear factor (erythroid-derived 2)-like 2 (NRF2), and forkhead box transcription factors (FOXO3), which can increase the transcription of several proteins involved in autophagy (87). The initial connection between autophagy and cancer was established when studies demonstrated that Beclin-1 is mono-allelically deleted in approximately 50% of breast, ovarian, and prostate cancers (88, 89). Other studies revealed that mice hemizygous for Beclin-1 show a high incidence of lymphoma, liver, and lung cancer (90, 91).
Thus, it was believed that autophagy acts as a tumor suppressor. It removes damaged mitochondria through mitophagy and prevents ROS accumulation, therefore limiting the tumor-promoting effect of ROS (92). Consequently, autophagy inhibition promotes ROS production, mitochondrial impairment, and DNA damage, all potentially pro-tumorigenic during tumor initiation (6) but deleterious at later stages (75, 93). Studies have shown that autophagy loss causes genomic instability and aneuploidy (94, 95). Furthermore, autophagy dysfunction can promote tumor cell-extrinsic effects, including a pro-tumorigenic inflammatory microenvironment (25).
ROS are also induced by several tumor-associated immune cells in the tumor microenvironment (TME) (96) that may trigger altered activation of macrophages and immunosuppression (97). Macrophages are the first host cells to enter the TME to kill cancer cells (98). However, tumor-associated macrophages (TAMs) infiltrate into the tumors and differentiate into mature pro-tumor macrophages (M1 and M2 macrophages) mediated by cytokines in the TME (99–101). Although the pro-tumorigenic role of M1 is context dependent based on tumor microenvironmental cues (102, 103). Macrophages also show phagocytotic activity toward damaged tumor cells (104). However, macrophages are recruited through chemokines during cancer initiation, amplifying an inflammatory response. Macrophages also produce redoxosomes (exosomes containing functional NOX complexes) in the TME, which generates extracellular ROS and is incorporated into neighboring cells through endocytosis (105). Thus, a supportive TME is essential for tumorigenesis, wherein ROS plays a significant role in creating immunosuppressive TME for cancer growth and metastasis. Hence, it is plausible that autophagy inhibition may promote pro-tumorigenic ROS, since dysregulated autophagy leads to mitochondrial damage and high ROS levels, and oxidative stress, all potentially pro-tumorigenic.
Several studies have demonstrated that dysregulated autophagy due to the deletion of proteins such as Atg16L1, Beclin-1, or LC-3B promotes the accumulation of damaged mitochondria and mitochondrial ROS. It also promotes inflammation linked to increased levels of IL-1β and IL-18 (106–109). ROS can also be induced by IL-1, whose high expression has been associated with a poor cancer prognosis (110). Moreover, increased ROS levels also activate pro-inflammatory factors such as the pyrin domain-containing 3 (NLRP3) inflammasome (109). Inflammation aids in cancer initiation and survival through vascularization and stimulating the TME through the IL-1 and IL-18 pathway. Inflammatory cells further produce ROS or reactive nitrogen species (RNS) via iNOS, xanthine oxidase (XO), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and myeloperoxidase (MPO). These oxidant-generating enzymes may promote damage to DNA damage. (111). Autophagy also plays a crucial role in inflammation by regulating the homeostasis, development, and survival of inflammatory cells (112). Inflammatory cells also release cytokines, activating NF-κB. NF-κB can help generate excess ROS or RNS by stimulating COX2, lipoxygenase (LOX), and inducible nitric oxide synthase (iNOS), that in turn may stimulate several oncogenes such as c-Jun and c-Fos and initiate tumorigenesis (113).
Another major regulator of both autophagy and ROS is the tumor suppressor p53 that plays a contrasting role in autophagy based on its subcellular localization (114). Nuclear p53 is suggested to activate autophagy through several transcriptional mechanisms. Many autophagy genes are said to be direct interacting partners of p53, and that autophagy helps in p53-dependent apoptosis and cancer suppression (115). In the nucleus, p53 activates the transcription of pro-autophagic molecules such as AMPK, damage-regulated autophagy modulator (DRAM), death-associated protein kinase 1 (DAPK-1), pro-apoptotic Bcl-2 proteins, sestrin 2, and Tuberous Sclerosis Complex 2 (TSC2) (116–120). However, cytoplasmic p53 inhibits autophagy, primarily through interactions with autophagic proteins (114). Cytoplasmic p53 mediates mitochondrial outer membrane permeabilization, promoting apoptosis and inhibiting autophagy (121, 122). Although the mechanism of cytoplasmic p53-mediated autophagy inhibition is not well elucidated, it was observed that cytoplasmic p53 inhibits AMPK and activates mTOR, leading to autophagy suppression (123).
p53 also can regulate autophagy by modulating ROS levels. During oxidative stress, basal p53 induces several antioxidants such as GPx1, MnSOD, ALDH4, and TPP53INP1 to remove oxidative stress (124–127). Additionally, p53 also exerts antioxidant effects by upregulating the expression of several p53 target genes in response to DNA damage and oxidative stress. This leads to inhibition of mTORC1 activity and autophagy induction. Sestrin1 and sestrin2 are the links between p53 activation and mTORC1 activity (119). Sestrins also regulate ROS (128) and inhibit mTORC1 activity by inducing the expression of the pro-autophagic AMPK, TSC1, and TSC2 (119).
However, p53 can also induce ROS. A study observed that silibinin, an active constituent extracted from Silybum marianum (milk thistle), induced ROS-mediated autophagy and apoptosis in HeLa cells (129). Furthermore, another study by the same group demonstrated that silibinin promotes p53-mediated ROS in HeLa cells. The study also observed that p53 inhibition decreased ROS generation and reversed silibinin’s growth-inhibitory effect. Moreover, silibinin was not able to induce ROS in the epithelial carcinoma cells (A431), as they lack p53 activity (p53His273mutation) (130). Another study reports that silibinin may upregulate p53-mediated autophagy by inhibiting MAPK and PI3K/Akt pathways and activating ROS/p38 and JNK pathways (131). Furthermore, upregulation in PI3K and AKT or downregulation in PTEN activates mTOR and inhibits autophagy. Thus, these oncogenic alterations suggest the importance of autophagy suppression during tumor initiation (132, 133).
Other studies also demonstrated that any defect in the autophagic machinery promotes tumor initiation, including liver and breast (114, 134). Tang etal. (114) demonstrated that low expression of Beclin-1 suggested poor prognosis in Her2, basal-like, and p53-mutant breast cancer. Autophagy also acts as a tumor suppressor through its role in cellular senescence, where cells undergo growth arrest (135). Kang etal. (136) demonstrated that GATA Binding Protein 4 (GATA4), a transcription factor regulating senescence, is degraded by p62-selective autophagy. Autophagic adapters, p62/SQSTM1, act as cargo receptors for autophagic degradation of ubiquitinated targets (137). p62 is upregulated under various stresses, including ROS, where ROS-induced p62 gene expression is mediated by NRF2. Furthermore, p62 protein activates NRF2 by interacting with the Nrf2-binding site on Keap1, a component of Cullin 3 (CUL3)-based E3 ubiquitin ligase for Nrf2, resulting in stabilization of Nrf2 and transcriptional activation of its target genes (138, 139). Another major autophagy regulator, Atg5 protein, also plays a dual role in the regulation of autophagy and apoptosis. Studies have indicated that overexpression of Atg5 protein can sensitize tumor cells to chemotherapy. In contrast, silencing the ATG5 gene with short interfering RNA made tumor cells partially resistant to chemotherapy. Atg5 protein is cleaved by calpains, a family of Ca2+-dependent cysteine proteases, producing an amino-terminal cleavage product. Calpain induction and subsequent Atg5 protein cleavage appear to be universal phenomena in apoptotic cells (140). Similarly, the Atg12 protein also has a dual function, participating in both autophagy and apoptosis, and is necessary for caspase activation in response to a range of apoptotic stress inducers. Non-conjugated Atg12 protein can bind to and inhibit Mcl-1 and Bcl-2 by a BH3-like motif, inducing mitochondria-dependent apoptosis (141). Knockout of ATG12 gene prevents Bax activation and cytochrome c in apoptotic cells.
Although autophagy functions as a tumor suppressor during the initiation of tumorigenesis (6), other studies have revealed that autophagy can also act as a tumor promoter (132; 142). Furthermore, autophagy can also promote resistance to many anticancer therapies (27). The pro-survival role of autophagy can be seen during stress conditions, including hypoxia and nutrient deprivation. Autophagy rapidly degrades unfolded proteins during stress and provides the substrate for ATP production (143, 144). Thus, autophagy is generally upregulated in hypoxic regions of a tumor and promotes cell survival (25).
During later stages of tumor initiation, autophagy is required for cell transformation by the RAS oncogene to promote cell tolerance to stress A high basal level of autophagy is observed in RAS-mutated cancers, including lung, colon, and pancreatic (145, 146). Furthermore, mutations in the RAS genes promote uncontrollable cell proliferation and apoptosis inhibition (147, 148). Herein, autophagy promotes cancer cell survival by providing nutrients during starvation or other stress conditions (149). Consequently, autophagy inhibition increases the accumulation of damaged mitochondria and promotes cell death (150). Thus, tumor cells utilize autophagy to survive metabolic stress, and autophagy mitigates cellular damage (151). Autophagy inhibition leads to slower tumor growth and increased sensitivity to cancer treatments. This has led researchers to assess the efficacy of autophagy inhibitors combined with chemotherapy to increase therapeutic responses in cancers.
Consistently, autophagy inhibition reduced malignant transformation and proliferation of mouse embryonic fibroblasts (MEFs) transformed with Harvey Rat Sarcoma Virus (HRAS) and MDA-MB-231 breast cancer cells presenting with KRAS expression (152). Other studies have shown that model systems such as immortalized baby mouse kidney (iBMK), MCF-10A, and pancreatic ductal adenocarcinoma (PDAC) cell lines harboring ectopic expression of the oncogenic KRAS has high basal autophagy levels. However, inhibiting autophagy by deleting the gene ATG5 or ATG7 prevented RAS-mediated cancer cell proliferation (145; 70, 153). It can be stated that mitochondrial respiration is required for RAS-induced tumorigenesis, and active autophagy maintains cellular homeostasis (154). Thus, RAS-mediated cancers are addicted to autophagy for survival, and dysregulated autophagy in these cancer types is proportional to decreased cancer cell survival, accumulation of damaged mitochondria, and oxidative stress that may ultimately promote cell death (155; 25). Furthermore, p62/SQSTM1 deficiency also reduces tumorigenicity and increases ROS levels following RAS activation (145, 156, 157). Another study also states that autophagy inhibition by FIP200 (FAK family-interacting protein of 200 kDa) deletion suppressed the breast cancer initiation in vivo driven by the polyoma virus middle T (PyMT) oncogene. The study demonstrated that FIP200 ablation promoted accumulation of p62/SQSTM1, ubiquitinated protein aggregates, and deficient LC3 conversion with an increased number of abnormal mitochondria confirming the pro-tumorigenic role of autophagy (158). Interestingly, FIP200 deletion did not affect apoptosis but significantly reduced the proliferation of breast cancer cells or Ras-transformed MEFs.
Taken together, these studies confirm the complex and paradoxical role of autophagy and ROS in cancer initiation and progression (Figure 3). However, this dual role also provides several therapeutic windows that could be exploited to develop targeted anticancer therapies.
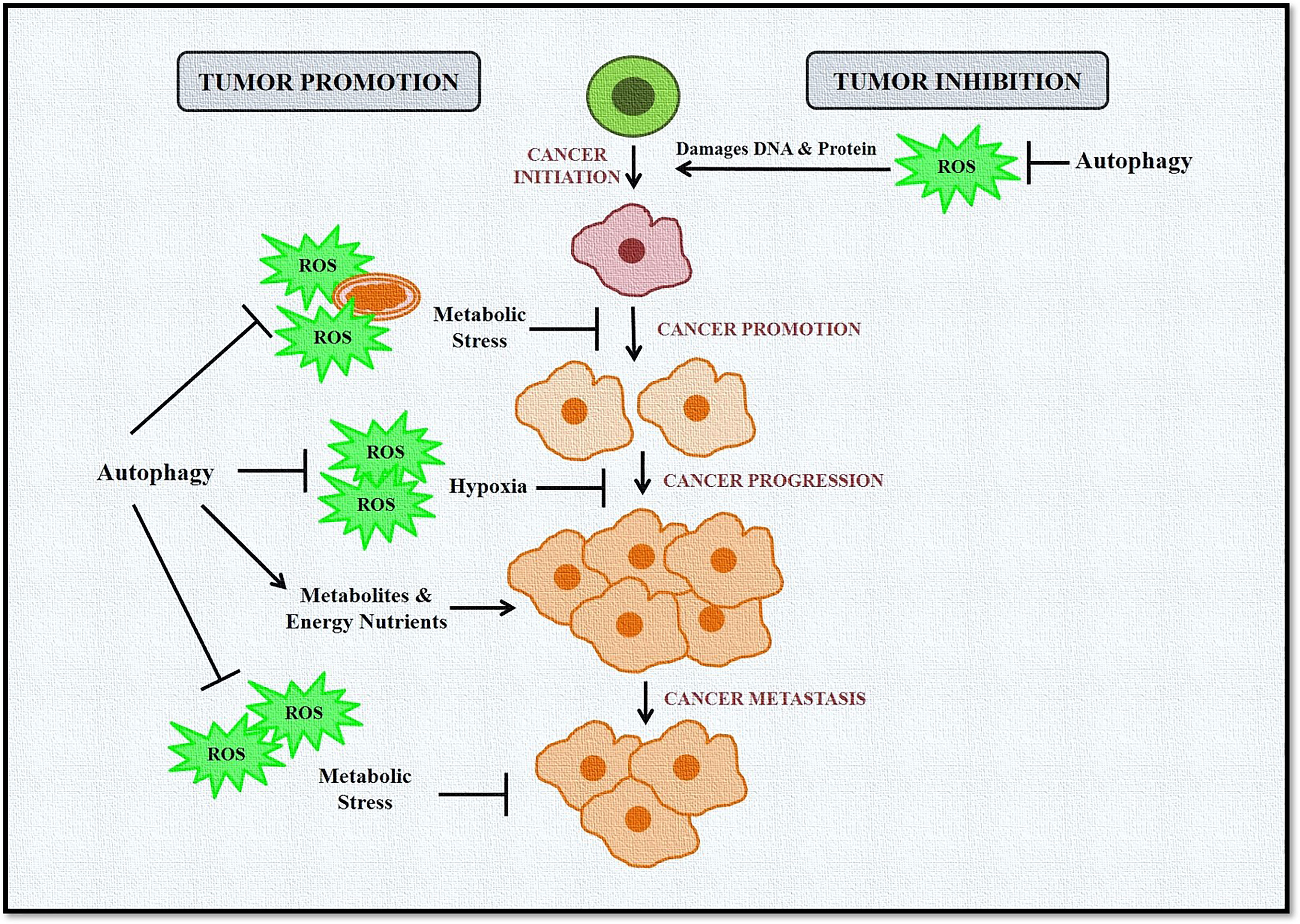
Figure 3 Role of autophagy and ROS in cancer promotion and suppression. Autophagy in cancer works in a context-dependent manner based on tumor type and stage. It acts as a suppressor during tumor initiation but plays a protective role in established tumors. During tumor initiation, autophagy targets ROS-damaged organelles, DNA, and protein toward degradation, leading to inhibition of tumorigenesis. Autophagy eliminates ROS-induced stress during tumor progression and metastasis and provides much-needed nutrients to cells, including cancer cells. ROS is also induced in cancer cells during hypoxic conditions, activating autophagy in stromal cells. These cells then provide high-energy nutrients for cancer cell survival.
Role of Autophagy and Reactive Oxygen Species in Epithelial to Mesenchymal Transition and Cancer Metastasis
Metastasis is a complex mechanism in which cancer cells undergo epithelial to mesenchymal transition (EMT) and spread from the tissue of origin to distant organs. It is the main reason behind high cancer mortality (159–161). EMT promotes contact inhibition in cancer cells, leading to invasive tumor epithelial phenotype (162). EMT can be regulated by several mechanisms, including epigenetics, transcriptional control, miRNAs, protein stability, alternative splicing, ROS, and autophagy (163, 164).
A study by Avivar-Valderas etal. (165) observed that in mammary tumor cells, autophagy was induced due to matrix detachment or integrin blockade in response to ROS-dependent upregulation of protein kinase R-like ER kinase (PERK1). Consistently, autophagy or PERK inhibition during matrix detachment or integrin signaling blockade induced cell death and reduced clonogenic recovery following detachment, highlighting the role of PERK-induced autophagy in mammary tumor cell survival during matrix detachment (165, 166). Furthermore, hepatocellular carcinoma and melanoma cells also require autophagy to survive following matrix detachment, leading to increased lung colonization during metastasis (167–169). Moreover, high ROS levels induced by matrix detachment may further promote autophagy activation through direct activation of Atg4 protein (26, 170).
One of the major contributors of EMT is transforming growth factor-beta 1 (TGF-β1) (171). Exogenous TGF-β1 regulates urokinase-type plasminogen activator (uPA) and Matrix metalloproteinase 9 (MMP9) to promote cell migration and invasion by activating NF-κB via the Rac1-NOXs-ROS-dependent mec`ism (172). ROS also regulates EMT via the non-canonical TGF-β1–TGF-β-activated kinase 1 (TAK1) pathway. TAK1 deficiency promotes integrin:Rac-induced ROS, further accelerating the EMT process. Consistently, low TAK1 expression was observed in invasive squamous cell carcinoma (SCC) but not in benign SCCs (173). ROS-mediated activation of Nrf2 also promotes Notch signaling and EMT induction (174). ROS can also activate TGF-β1 in response to ionizing radiation (175). Thus, these studies significantly highlight the role of ROS in EMT induction. Moreover, it is well characterized that cancer cells have a high metabolic rate. Therefore, to fulfill the bioenergetic needs of the cancer cells, an increase in ATP production and tricarboxylic acid (TCA) cycle is required. In turn, ROS is accumulated due to increased oxidative metabolism, disturbing the cellular homeostasis, dysregulating autophagy, inducing EMT, and promoting cancer cell survival and metastasis (6, 176, 177).
Furthermore, self-aggregation of TGF-β1-induced antiapoptotic factor (TIAF1) was observed in the cancer stroma and peritumor capsules of solid tumors, which is indicative of aggregation-dependent control of cancer progression and metastasis (178).
Autophagy also helps tumor cells adapt to hypoxic conditions before vascularization during in vivo tumor formation (179). High autophagy levels were observed in the hypoxic regions of the tumors. Autophagy can also be activated by ischemia to promote cancer cell survival and growth (25, 94, 95). Moreover, hypoxia can also induce ROS and stabilizes HIF-1α, the primary regulator of oxygen homeostasis (180). HIF-1α induces mitophagy via Bcl-2/adenovirus E1B 19-kDa-interacting protein 3 (BNIP3), along with a constitutive expression of Beclin-1 and Atg5 protein promotes cell survival during prolonged hypoxia by preventing increased ROS levels (181). BNIP3, a target gene for HIF-1α, induces autophagy by disrupting the Beclin 1–Bcl2 interaction (182). Autophagy dysregulation due to BECLIN-1, ATG5 gene, or ATG7 gene knockdown promotes hypoxia-induced cell death. Indeed, BNIP3-induced autophagy is required to prevent aberrant ROS levels during hypoxia and thus presents a survival mechanism (183–185). Autophagy is also induced in a HIF-1α-independent manner via AMPK and unfolded protein response (UPR) during hypoxia (186, 187).
Starvation-induced autophagy can also induce EMT and is required for HepG2 and BEL7402 HCC cell invasion in vitro. Thus, knockdown of autophagy genes like ATG7 or ATG3 in these cells suppressed EMT and invasion and decreased the expression of Fibronectin 1 (FN1), TGF-β1, and activated SMAD family member 3 (SMAD3) (188). Kim etal. (189) observed that another autophagy regulator, Unc-51 Like Autophagy Activating Kinase 2 (ULK2), promotes EMT by downregulating E-cadherin and increasing the invasiveness of lung cancer cells in vitro. Increased autophagy also promotes mesenchymal stem-like phenotype and invasion/migration of glioblastoma stem cell lines. Hence, autophagy dysregulation via ATG12 gene knockdown or p62/SQSTM1 deficiency reduced invasion and migration phenotypes in glioblastoma cells (190, 191).
Contrarily, another study argues that autophagy reduces migration of glioblastoma tumor cells via SNAIL and SLUG inhibition (192). Similarly, in hepatocytes, autophagy inhibition via liver-specific knockout of ATG7 gene (Alb-Cre;Atg7fl/fl) promoted the expression of vimentin and SNAIL. The study further reports that autophagy degraded Snail in a p62/SQSTM1-dependent manner. Moreover, treating wild-type MMH (murine hepatocytes) with TGF-β1 suppressed autophagy, whereas starvation-induced autophagy inhibited TGF-β1-mediated EMT (193).
Low basal autophagy levels also correlate with an increased propensity for migration and invasion in Skov-3 ovarian cancer cells compared to cells with high basal autophagy. Furthermore, a decrease in migration, invasion, and expression of the mesenchymal markers was observed due to starvation-induced autophagy, which was reversed following siRNA-mediated knockdown of ATG7 gene. Moreover, EMT transition in these cells was regulated via increased ROS and heme oxygenase 1 (HMOX1), highlighting a role of autophagy in the ROS–HMOX1–EMT signaling axis (194). Similarly, autophagy can also inhibit EMT by degrading SNAIL and TWIST, two major mesenchymal markers that promote the invasion phenotype in cancer cells (195). Apart from TGF-β1, EMT is also induced by IL-1, IL-6 that regulate SNAIL or TWIST. ROS also induces HIF-1α and lysyl oxidase (LOX), decreasing E-Cadherin levels and activating EMT and cancer cell migration. Thus, it is plausible that autophagy may also be detrimental to EMT by inhibiting inflammation and removing ROS (196).
The autophagy receptor p62/SQSTM1 stabilizes the transcription factor TWIST and induces EMT (197, 198). Autophagy inhibition also promotes p62/SQSTM1 accumulation and contributes to tumorigenesis. Autophagy loss promoted the expression of TWIST in a p62-dependent manner, where it directly binds to TWIST and prevents its proteasomal degradation, promoting EMT and metastasis in vivo (197). Another study also demonstrated that accumulation of p62/SQSTM1 stabilizes TWIST and activates TGF-β1–SMAD signaling, further promoting EMT-associated junction remodeling (198).
It is evident that a complex link exists between autophagy, ROS, and EMT (Figure 4). Thus, to design better treatment modalities, extensive knowledge of the interlinked cellular events would be necessary to regulate cellular homeostasis.
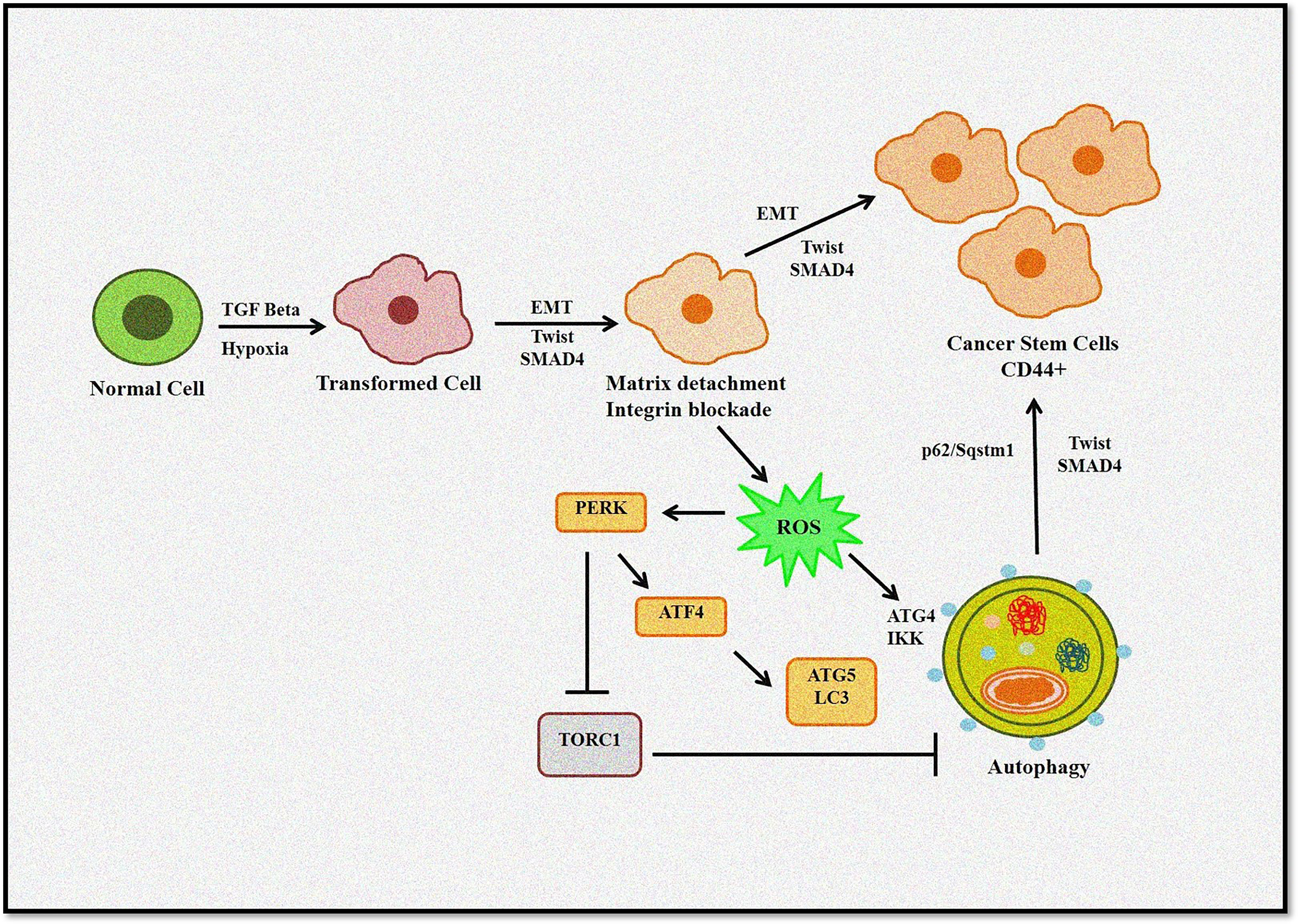
Figure 4 Role of autophagy and ROS in the EMT process. Autophagy induces tumor invasiveness by promoting stem cell phenotype linked to hypoxia and TGF-β. Matrix detachment leads to ROS-induced EMT transition and autophagy induction. Furthermore, p62/Sqstm1 autophagy cargo adapter interacts with Twist, an EMT regulator, preventing its proteasomal degradation and promoting invasion.
Role of Autophagy and Reactive Oxygen Species in Cancer Therapy
For the past two decades, autophagy has been an attractive target for researchers to develop better anticancer therapies. Several cancer drugs either induce cytoprotective autophagy or promote autophagic cell death or autophagy-mediated apoptosis in cancer cells. Indeed, the cytoprotective role of autophagy was observed against temozolomide (199), tamoxifen (200), the histone deacetylase inhibitor SAHA (201), cyclophosphamide (27), irradiation (202), imatinib mesylate (203), and cisplatin (204). Thus, autophagy inhibitors such as hydroxychloroquine were used combined with standard chemotherapeutics in clinical trials to increase the therapeutic potential of the drugs (205). However, it should be noted that the stage at which autophagy is inhibited may alter drug sensitivity and plays a critical role in deciding the fate of cancer cells.
Certain anticancer treatments also promote ROS-induced autophagy that can promote drug resistance. In this case, using autophagy inhibitors with the chemotherapy agents may help restore the sensitivity to the treatment. Moreover, the type and dosage of drugs used, along with the cancer genotype, are other factors that may decide the outcome of autophagy activation. Consistently, Beclin-1-dependent protective autophagy was induced when pancreatic cancer cells were exposed to sorafenib, a pan-kinase inhibitor combined with HDACI, a histone deacetylase inhibitor. However, Bcl-2 knockdown or inhibition conditioned Beclin 1-dependent autophagy to promote apoptosis into a toxic pathway promoting intrinsic apoptosis (206). Another study demonstrated that ROS-mediated activation of c-Jun N-terminal kinase (JNK) induced cytoprotective autophagy when human rhabdomyosarcoma (Rh30 and RD) cells were treated with ciclopirox olamine (CPX). However, inhibiting autophagy via chloroquine (CQ) promoted CPX-induced cell death (207).
Hahm et al. (208) reported that honokiol, derived from the bark of Magnolia officinalis, induced ROS-induced cytoprotective autophagy and promoted drug resistance in prostate cancer. However, inhibiting autophagy via 3-methyladenine (3-MA) or ATG5 gene siRNA sensitized cancer cells to apoptosis (208). Moreover, exposing breast and glioblastoma cancer cells to mitoquinone and quercetin, respectively, also promoted cytoprotective autophagy (209, 210). Hence, it can be hypothesized that any changes in the mitochondrial homeostasis would induce ROS and autophagy, which may lead to cell survival by autophagy-mediated degradation of damaged mitochondria. Therefore, autophagy inhibitors or siRNA-mediated silencing of ATG genes can turn protective ROS deleterious to cancer cells and promote apoptosis.
Another study showed that using 3-bromopyruvate (3-BrPA), a hexokinase II inhibitor, induced autophagy in breast cancer cells (MDA-MB-231435 and MDA-MB-435). However, ROS-mediated cell death was observed when 3-BrPA was used in combination with chloroquine, an autophagy inhibitor. The authors also concluded that autophagy induction was not dependent on ROS accumulation (211). Similar results were observed when A549 lung cancer cells were exposed to artemisinin, an antimalarial drug. Treatment with artemisinin induced autophagy that was attenuated by chloroquine. Autophagy inhibition promoted the accumulation of damaged mitochondria and ROS generation, resulting in apoptosis. Furthermore, apoptosis was ROS-dependent, as using a ROS scavenger N-acetyl-cysteine (NAC) rescued A549 cells from apoptosis via caspase-3 inhibition (212).
However, autophagy-induced apoptosis has also been reported. Carnosol, a polyphenol, inhibited the cell viability in MDA-MB-231 breast cancer cells. The study reported that carnosol caused DNA and mitochondrial damage and promoted ROS-dependent early autophagy and late apoptosis (213). Thus, this could be another mechanism of action that a drug could follow to induce cancer cell death. Some chemotherapy agents like 2-methoxyestradiol (2-ME) and arsenic trioxide (As2O3) also promote oxidative stress-mediated autophagic cell death (214). Nevertheless, ROS is essential for As2O3-mediated autophagic cell death in glioma cells (215). 2-ME also upregulates ROS levels by inhibiting complex I of the mitochondrial electron transport chain and mitochondrial SOD (77, 216, 217). Furthermore, 2-ME, a ROS-generating agent, induced autophagic cell death in a transformed cell line HEK293 and the cancer cell lines HeLa and U87 (77). However, both 2-ME and As2O3 can induce autophagy and apoptosis (17, 215).
Autophagy-induced apoptosis was also observed in A375, HT144, and Hs294T cells treated with the H1 histamine receptor antagonist terfenadine, which may increase ROS depending on culture condition (218). Similarly, in melanoma cancer cells (A375 and BLM), bortezomib, a proteasome inhibitor, at least in part via ROS-mitochondrial dysregulation-associated pathways (219). Another study revealed that sasanquasaponin III (SQS III) inhibited the viability of A375 cells by inducing apoptosis and autophagy. The authors further observed that both, apoptosis as well as autophagy induction was ROS dependent. (220). Moreover, resveratrol and psoralidin promoted ROS-triggered autophagy induction followed by apoptosis in colon and lung cancer cell death, respectively (221, 222).
Other studies also highlight the role of autophagy and ROS levels in cancer treatment. It was shown that 2-deoxy-D-glucose (2DG), when combined with cisplatin or staurosporine, promoted apoptosis but promoted cytoprotective autophagy and decreased ROS levels when combined with pyrimethamine. Moreover, 2DG alone promoted protective autophagy, inhibited ROS levels, and increased mitochondrial membrane potential in melanoma cells (8863 and 501) (223, 224).
Thus, several treatment studies can be used to induce cancer cell death. As cancer develops high resistance against apoptosis, causing autophagic cell death could be an option. Moreover, combining ROS and autophagy-inducing agents could also promote cancer cell death. Other strategies include combining apoptosis inducers with autophagy inhibitors in cancer cells harboring protective autophagy (Table 2). Taken together, choosing correct cancer treatment strategies is highly complex and should be based on tumor phenotype and genotype.
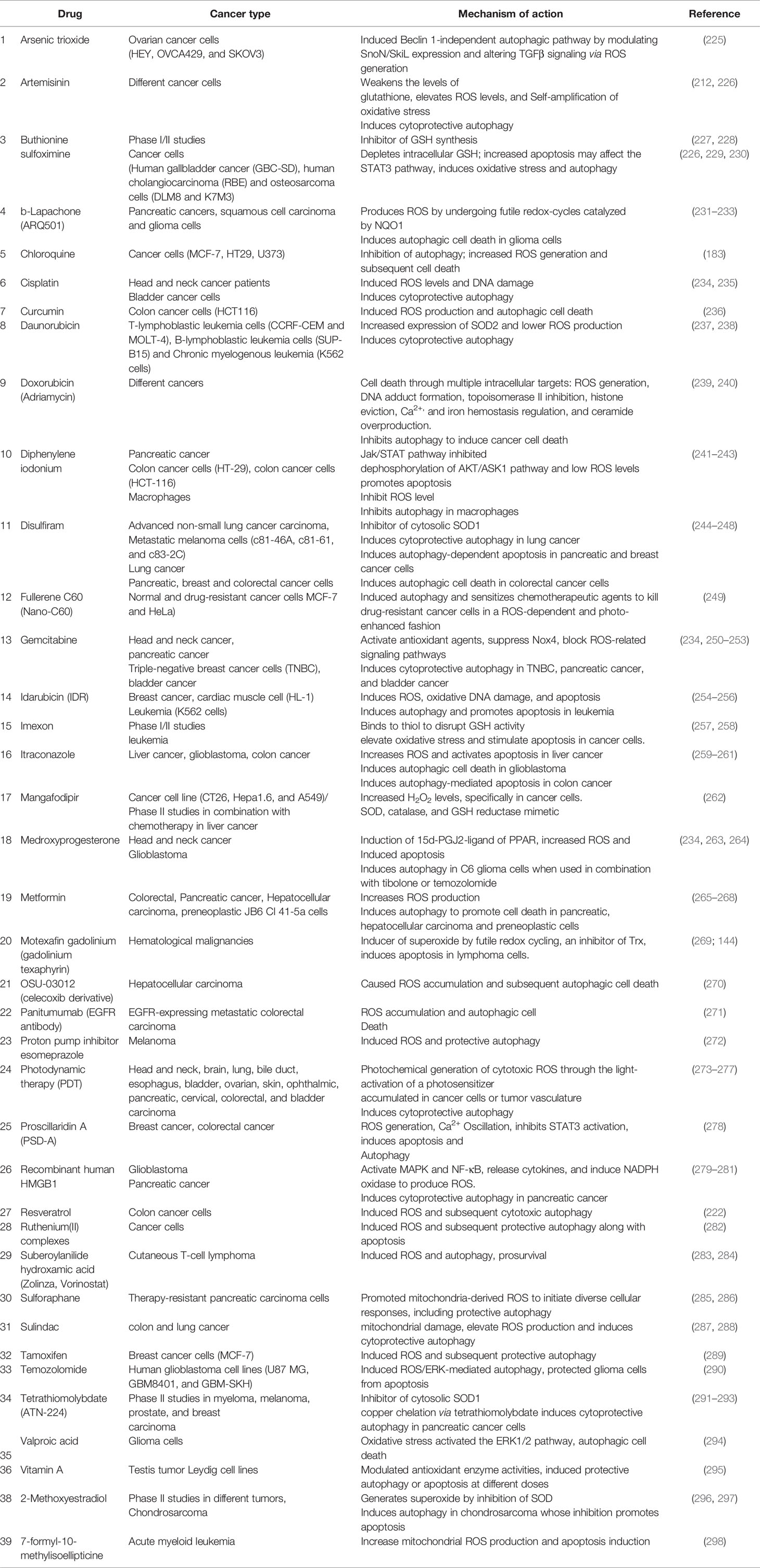
Table 2 ROS-inducing or -inhibiting chemotherapeutic agents and their effect on autophagy.
Conclusion
Thus, it can be concluded that ROS and autophagy work in a tight regulation with each other to maintain cellular homeostasis. They can either help cancer cells adapt to severe stress, which may otherwise be detrimental to cells, or induce cell death. This paradoxical role of ROS and autophagy in cancer is mainly dependent on the cancer types and their microenvironment. Therefore, it is imperative to decipher the crosslinked mechanisms in tumorigenesis with respect to ROS and autophagy so that autophagy modulators may be designed to target cancer.
This review highlights the role of ROS and autophagy in cancer survival and suppression mechanisms. The major mechanisms include response to hypoxia, turnover of antioxidant enzymes, oxidative damage-induced protein aggregation of regulatory molecules like TGF-β1, p53, enhanced survival in RAS-mutated cancers, EMT transition, and drug resistance. However, consistent with the role of autophagy and ROS in cancer, they provide large windows of opportunities to develop better treatment strategies that may help fulfill the unmet needs of cancer patients.
A better understanding of the molecular and chemical mechanisms of the redox regulation of autophagy is required. There are still some unanswered questions like 1) How does autophagy modulate the turnover of regulatory enzymes required for maintaining redox potential? 2) How do autophagy and ROS regulate the posttranslational modifications of specific tumor suppressors? 3) How does excessive ROS impair autophagy and dysregulate the cellular microenvironment to promote invasive phenotype? Answer to these questions may help develop better anticancer treatment options.
Author Contributions
AH: conceptualization, writing—original draft, writing—review and editing. SFR: preparation of figures. SP: preparation of tables. NP: critical revision of the article. AN: critical revision of the article. SSM: conceptualization, supervision, writing—original draft, writing—review and editing, critical revision of the article. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are thankful to Honorable Vice-Chancellor, Integral University, Lucknow, for providing infrastructural support and Dean Office, R&D, Integral University for providing article communication number (IU/R&D/2022-MCN0001357).
References
1. De Duve C, Wattiaux R. Functions of Lysosomes. Annu Rev Physiol (1966) 28:435–92. doi: 10.1146/annurev.ph.28.030166.002251
2. Rikiishi H. Novel Insights Into the Interplay Between Apoptosis and Autophagy. Int J Cell Biol (2012) 2012:317645. doi: 10.1155/2012/317645
3. Hu CAA, White K, Torres S, Ishak MA, Sillerud L, Miao Y, et al. Apoptosis and Autophagy: The Yin–Yang of Homeostasis in Cell Death in Cancer. In: Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging. Academic Press (2015). p. 161–81. doi: 10.1016/B978-0-12-801043-3.00010-8
4. Mizushima N, Komatsu M. Autophagy: Renovation of Cells and Tissues. Cell (2011) 147(4):728–41. doi: 10.1016/j.cell.2011.10.026
5. Li WW, Li J, Bao JK. Microautophagy: Lesser-Known Self-Eating. Cell Mol Life Sci (2012) 69(7):1125–36. doi: 10.1007/s00018-011-0865-5
6. White E. The Role for Autophagy in Cancer. J Clin Invest (2015) 125(1):42–6. doi: 10.1172/JCI73941
7. White E. Deconvoluting the Context-Dependent Role for Autophagy in Cancer. Nat Rev Cancer (2012) 12(6):401–10. doi: 10.1038/nrc3262
8. Chavez-Dominguez R, Perez-Medina M, Lopez-Gonzalez JS, Galicia-Velasco M, Aguilar-Cazares D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-Tumor Activity. Front Oncol (2020) 10:578418. doi: 10.3389/fonc.2020.578418
9. Weidberg H, Shvets E, Elazar Z. Biogenesis and Cargo Selectivity of Autophagosomes. Annu Rev Biochem (2011) 80:125–56. doi: 10.1146/annurev-biochem-052709-094552
10. Carew JS, Huang P. Mitochondrial Defects in Cancer. Mol Cancer (2002) 1:9. doi: 10.1186/1476-4598-1-9
11. Pelicano H, Carney D, Huang P. ROS Stress in Cancer Cells and Therapeutic Implications. Drug Resist Updat (2004) 7(2):97–110. doi: 10.1016/j.drup.2004.01.004
12. Jin S, White E. Role of Autophagy in Cancer: Management of Metabolic Stress. Autophagy (2007) 3(1):28–31. doi: 10.4161/auto.3269
13. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (New York N Y ) (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
14. Finkel T. Oxidant Signals and Oxidative Stress. Curr Opin Cell Biol (2003) 15(2):247–54. doi: 10.1016/s0955-0674(03)00002-4
15. Hamanaka RB, Chandel NS. Mitochondrial Reactive Oxygen Species Regulate Cellular Signaling and Dictate Biological Outcomes. Trends Biochem Sci (2010) 35(9):505–13. doi: 10.1016/j.tibs.2010.04.002
16. Scherz-Shouval R, Elazar Z. Regulation of Autophagy by ROS: Physiology and Pathology. Trends Biochem Sci (2011) 36(1):30–8. doi: 10.1016/j.tibs.2010.07.007
17. Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial Electron-Transport-Chain Inhibitors of Complexes I and II Induce Autophagic Cell Death Mediated by Reactive Oxygen Species. J Cell Sci (2007) 120(Pt 23):4155–66. doi: 10.1242/jcs.011163
18. Park SH, Chi GY, Eom HS, Kim GY, Hyun JW, Kim WJ, et al. Role of Autophagy in Apoptosis Induction by Methylene Chloride Extracts of Mori Cortex in NCI-H460 Human Lung Carcinoma Cells. Int J Oncol (2012) 40(6):1929–40. doi: 10.3892/ijo.2012.1386
19. Klionsky DJ, Emr SD. Autophagy as a Regulated Pathway of Cellular Degradation. Science (New York N Y) (2000) 290(5497):1717–21. doi: 10.1126/science.290.5497.1717
20. Wu WS. The Signaling Mechanism of ROS in Tumor Progression. Cancer Metastasis Rev (2006) 25(4):695–705. doi: 10.1007/s10555-006-9037-8
21. Farkas T, Daugaard M, Jäättelä M. Identification of Small Molecule Inhibitors of Phosphatidylinositol 3-Kinase and Autophagy. J Biol Chem (2011) 286(45):38904–12. doi: 10.1074/jbc.M111.269134
22. Zhuang W, Li B, Long L, Chen L, Huang Q, Liang Z. Induction of Autophagy Promotes Differentiation of Glioma-Initiating Cells and Their Radiosensitivity. Int J Cancer (2011) 129(11):2720–31. doi: 10.1002/ijc.25975
23. Bae H, Guan JL. Suppression of Autophagy by FIP200 Deletion Impairs DNA Damage Repair and Increases Cell Death Upon Treatments With Anticancer Agents. Mol Cancer Res (2011) 9(9):1232–41. doi: 10.1158/1541-7786.MCR-11-0098
24. Chen S, Rehman SK, Zhang W, Wen A, Yao L, Zhang J. Autophagy Is a Therapeutic Target in Anticancer Drug Resistance. Biochim Biophys Acta (2010) 1806(2):220–9. doi: 10.1016/j.bbcan.2010.07.003
25. Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy Promotes Tumor Cell Survival and Restricts Necrosis, Inflammation, and Tumorigenesis. Cancer Cell (2006) 10(1):51–64. doi: 10.1016/j.ccr.2006.06.001
26. Scherz-Shouval R, Elazar Z. ROS, Mitochondria and the Regulation of Autophagy. Trends Cell Biol (2007) 17(9):422–7. doi: 10.1016/j.tcb.2007.07.009
27. Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, et al. Autophagy Inhibition Enhances Therapy-Induced Apoptosis in a Myc-Induced Model of Lymphoma. J Clin Invest (2007) 117(2):326–36. doi: 10.1172/JCI28833
28. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a Mammalian Homologue of Yeast Apg8p, Is Localized in Autophagosome Membranes After Processing. EMBO J (2000) 19(21):5720–8. doi: 10.1093/emboj/19.21.5720
29. Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A Protein Conjugation System Essential for Autophagy. Nature (1998) 395(6700):395–8. doi: 10.1038/26506
30. Levine B, Kroemer G. Autophagy in the Pathogenesis of Disease. Cell (2008) 132(1):27–42. doi: 10.1016/j.cell.2007.12.018
31. Klionsky DJ, Cregg JM, Dunn WA Jr., Emr SD, Sakai Y, Sandoval IV, et al. A Unified Nomenclature for Yeast Autophagy-Related Genes. Dev Cell (2003) 5(4):539–45. doi: 10.1016/s1534-5807(03)00296-x
32. Scott RC, Schuldiner O, Neufeld TP. Role and Regulation of Starvation-Induced Autophagy in the Drosophila Fat Body. Dev Cell (2004) 7(2):167–78. doi: 10.1016/j.devcel.2004.07.009
33. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR Regulate Autophagy Through Direct Phosphorylation of Ulk1. Nat Cell Biol (2011) 13(2):132–41. doi: 10.1038/ncb2152
34. Torii S, Yoshida T, Arakawa S, Honda S, Nakanishi A, Shimizu S. Identification of PPM1D as an Essential Ulk1 Phosphatase for Genotoxic Stress-Induced Autophagy. EMBO Rep (2016) 17(11):1552–64. doi: 10.15252/embr.201642565
35. Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes With Mammalian Atg14 and UVRAG. Mol Biol Cell (2008) 19(12):5360–72. doi: 10.1091/mbc.e08-01-0080
36. Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, et al. Early Steps in Autophagy Depend on Direct Phosphorylation of Atg9 by the Atg1 Kinase. Mol Cell (2014) 53(3):471–83. doi: 10.1016/j.molcel.2013.12.011
37. Slobodkin MR, Elazar Z. The Atg8 Family: Multifunctional Ubiquitin-Like Key Regulators of Autophagy. Essays Biochem (2013) 55:51–64. doi: 10.1042/bse0550051
38. Klionsky DJ, Schulman BA. Dynamic Regulation of Macroautophagy by Distinctive Ubiquitin-Like Proteins. Nat Struct Mol Biol (2014) 21(4):336–45. doi: 10.1038/nsmb.2787
39. Noda NN, Inagaki F. Mechanisms of Autophagy. Annu Rev Biophys (2015) 44:101–22. doi: 10.1146/annurev-biophys-060414-034248
40. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. P62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J Biol Chem (2007) 282(33):24131–45. doi: 10.1074/jbc.M702824200
41. Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential Role for Nix in Autophagic Maturation of Erythroid Cells. Nature (2008) 454(7201):232–5. doi: 10.1038/nature07006
42. Johansen T, Lamark T. Selective Autophagy Mediated by Autophagic Adapter Proteins. Autophagy (2011) 7(3):279–96. doi: 10.4161/auto.7.3.14487
43. Mizushima N, Levine B. Autophagy in Mammalian Development and Differentiation. Nat Cell Biol (2010) 12(9):823–30. doi: 10.1038/ncb0910-823
44. Gough DR, Cotter TG. Hydrogen Peroxide: A Jekyll and Hyde Signalling Molecule. Cell Death Dis (2011) 2(10):e213. doi: 10.1038/cddis.2011.96
45. St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of Superoxide Production From Different Sites in the Mitochondrial Electron Transport Chain. J Biol Chem (2002) 277(47):44784–90. doi: 10.1074/jbc.M207217200
46. Murphy MP. How Mitochondria Produce Reactive Oxygen Species. Biochem J (2009) 417(1):1–13. doi: 10.1042/BJ20081386
47. Kumari S, Badana AK, Murali Mohan G, Shailender G, Malla R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark Insights (2018) 13:1177271918755391. doi: 10.1177/1177271918755391
48. Moloney JN, Cotter TG. ROS Signalling in the Biology of Cancer. Semin Cell Dev Biol (2018) 80:50–64. doi: 10.1016/j.semcdb.2017.05.023
49. Prasad S, Gupta SC, Tyagi AK. Reactive Oxygen Species (ROS) and Cancer: Role of Antioxidative Nutraceuticals. Cancer Lett (2017) 387:95–105. doi: 10.1016/j.canlet.2016.03.042
50. Chan JS, Tan MJ, Sng MK, Teo Z, Phua T, Choo CC, et al. Cancer-Associated Fibroblasts Enact Field Cancerization by Promoting Extratumoral Oxidative Stress. Cell Death Dis (2017) 8(1):e2562. doi: 10.1038/cddis.2016.492
51. Ježek J, Cooper KF, Strich R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants (Basel Switzerland) (2018) 7(1):13. doi: 10.3390/antiox7010013
52. Reczek CR, Chandel NS. The Two Faces of Reactive Oxygen Species in Cancer. Annu Rev Cancer Biol (2017) 1:79–98. doi: 10.1146/annurev-cancerbio-041916-065808
53. Barrera G. Oxidative Stress and Lipid Peroxidation Products in Cancer Progression and Therapy. ISRN Oncol (2012) 2012:137289. doi: 10.5402/2012/137289
54. Gurer-Orhan H, Ince E, Konyar D, Saso L, Suzen S. The Role of Oxidative Stress Modulators in Breast Cancer. Curr Med Chem (2018) 25(33):4084–101. doi: 10.2174/0929867324666170711114336
55. Wang Y, Branicky R, Noë A, Hekimi S. Superoxide Dismutases: Dual Roles in Controlling ROS Damage and Regulating ROS Signaling. J Cell Biol (2018) 217(6):1915–28. doi: 10.1083/jcb.201708007
56. Poli G, Leonarduzzi G, Biasi F, Chiarpotto E. Oxidative Stress and Cell Signalling. Curr Med Chem (2004) 11(9):1163–82. doi: 10.2174/0929867043365323
57. Fruehauf JP, Meyskens FL Jr. Reactive Oxygen Species: A Breath of Life or Death? Clin Cancer Res (2007) 13(3):789–94. doi: 10.1158/1078-0432.CCR-06-2082
58. Zhang J, Ney PA. Role of BNIP3 and NIX in Cell Death, Autophagy, and Mitophagy. Cell Death Differ (2009) 16(7):939–46. doi: 10.1038/cdd.2009.16
59. Novak I. Mitophagy: A Complex Mechanism of Mitochondrial Removal. Antioxid Redox Signal (2012) 17(5):794–802. doi: 10.1089/ars.2011.4407
60. Feng D, Liu L, Zhu Y, Chen Q. Molecular Signaling Toward Mitophagy and its Physiological Significance. Exp Cell Res (2013) 319(12):1697–705. doi: 10.1016/j.yexcr.2013.03.034
61. Youle RJ, Narendra DP. Mechanisms of Mitophagy. Nat Rev Mol Cell Biol (2011) 12(1):9–14. doi: 10.1038/nrm3028
62. Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX Is Required for Programmed Mitochondrial Clearance During Reticulocyte Maturation. Proc Natl Acad Sci USA (2007) 104(49):19500–5. doi: 10.1073/pnas.0708818104
63. Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, et al. PINK1 Stabilized by Mitochondrial Depolarization Recruits Parkin to Damaged Mitochondria and Activates Latent Parkin for Mitophagy. J Cell Biol (2010) 189(2):211–21. doi: 10.1083/jcb.200910140
64. Paulsen CE, Carroll KS. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem Rev (2013) 113(7):4633–79. doi: 10.1021/cr300163e
65. Li L, Ishdorj G, Gibson SB. Reactive Oxygen Species Regulation of Autophagy in Cancer: Implications for Cancer Treatment. Free Radical Biol Med (2012) 53(7):1399–410. doi: 10.1016/j.freeradbiomed.2012.07.011
66. Bauer G. Targeting Extracellular ROS Signaling of Tumor Cells. Anticancer Res (2014) 34(4):1467–82.
67. Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, et al. Mitogenic Signaling Mediated by Oxidants in Ras-Transformed Fibroblasts. Science (New York N Y ) (1997) 275(5306):1649–52. doi: 10.1126/science.275.5306.1649
68. Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, et al. C-Myc Can Induce DNA Damage, Increase Reactive Oxygen Species, and Mitigate P53 Function: A Mechanism for Oncogene-Induced Genetic Instability. Mol Cell (2002) 9(5):1031–44. doi: 10.1016/s1097-2765(02)00520-8
69. Du J, Nelson ES, Simons AL, Olney KE, Moser JC, Schrock HE, et al. Regulation of Pancreatic Cancer Growth by Superoxide. Mol Carcinog (2013) 52(7):555–67. doi: 10.1002/mc.21891
70. Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, et al. Involvement of Autophagy in Oncogenic K-Ras-Induced Malignant Cell Transformation. J Biol Chem (2011) 286(15):12924–32. doi: 10.1074/jbc.M110.138958
71. De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, et al. Exploiting Cancer Cell Vulnerabilities to Develop a Combination Therapy for Ras-Driven Tumors. Cancer Cell (2011) 20(3):400–13. doi: 10.1016/j.ccr.2011.08.014
72. Inoki K, Zhu T, Guan KL. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell (2003) 115(5):577–90. doi: 10.1016/s0092-8674(03)00929-2
73. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol Cell (2008) 30(2):214–26. doi: 10.1016/j.molcel.2008.03.003
74. Choi SL, Kim SJ, Lee KT, Kim J, Mu J, Birnbaum MJ, et al. The Regulation of AMP-Activated Protein Kinase by H(2)O(2). Biochem Biophys Res Commun (2001) 287(1):92–7. doi: 10.1006/bbrc.2001.5544
75. Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M. Interplay Between ROS and Autophagy in Cancer Cells, From Tumor Initiation to Cancer Therapy. Redox Biol (2015) 4:184–92. doi: 10.1016/j.redox.2014.12.003
76. Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, et al. ATM Signals to TSC2 in the Cytoplasm to Regulate Mtorc1 in Response to ROS. Proc Natl Acad Sci USA (2010) 107(9):4153–8. doi: 10.1073/pnas.0913860107
77. Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative Stress Induces Autophagic Cell Death Independent of Apoptosis in Transformed and Cancer Cells. Cell Death Differ (2008) 15(1):171–82. doi: 10.1038/sj.cdd.4402233
78. Djavaheri-Mergny M, Amelotti M, Mathieu J, Besançon F, Bauvy C, Souquère S, et al. NF-kappaB Activation Represses Tumor Necrosis Factor-Alpha-Induced Autophagy. J Biol Chem (2006) 281(41):30373–82. doi: 10.1074/jbc.M602097200
79. Boyer-Guittaut M, Poillet L, Liang Q, Bôle-Richard E, Ouyang X, Benavides GA, et al. The Role of GABARAPL1/GEC1 in Autophagic Flux and Mitochondrial Quality Control in MDA-MB-436 Breast Cancer Cells. Autophagy (2014) 10(6):986–1003. doi: 10.4161/auto.28390
80. Salmeen A, Barford D. Functions and Mechanisms of Redox Regulation of Cysteine-Based Phosphatases. Antioxid Redox Signal (2005) 7(5-6):560–77. doi: 10.1089/ars.2005.7.560
81. Leslie NR. The Redox Regulation of PI 3-Kinase-Dependent Signaling. Antioxid Redox Signal (2006) 8(9-10):1765–74. doi: 10.1089/ars.2006.8.1765
82. Byun YJ, Kim SK, Kim YM, Chae GT, Jeong SW, Lee SB. Hydrogen Peroxide Induces Autophagic Cell Death in C6 Glioma Cells via BNIP3-Mediated Suppression of the mTOR Pathway. Neurosci Lett (2009) 461(2):131–5. doi: 10.1016/j.neulet.2009.06.011
83. Zhang H, Kong X, Kang J, Su J, Li Y, Zhong J, et al. Oxidative Stress Induces Parallel Autophagy and Mitochondria Dysfunction in Human Glioma U251 Cells. Toxicol Sci (2009) 110(2):376–88. doi: 10.1093/toxsci/kfp101
84. Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7-Beclin 1 Program of Autophagic Cell Death by Caspase-8. Science (New York N Y ) (2004) 304(5676):1500–2. doi: 10.1126/science.1096645
85. Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, et al. Autophagic Programmed Cell Death by Selective Catalase Degradation. Proc Natl Acad Sci USA (2006) 103(13):4952–7. doi: 10.1073/pnas.0511288103
86. Chen Y, Azad MB, Gibson SB. Superoxide is the Major Reactive Oxygen Species Regulating Autophagy. Cell Death Differ (2009) 16(7):1040–52. doi: 10.1038/cdd.2009.49
87. Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell Mol Neurobiol (2015) 35(5):615–21. doi: 10.1007/s10571-015-0166-x
88. Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, et al. Cloning and Genomic Organization of Beclin 1, a Candidate Tumor Suppressor Gene on Chromosome 17q21. Genomics (1999) 59(1):59–65. doi: 10.1006/geno.1999.5851
89. Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and Tumour Suppressor Activity of a Novel Beclin1-Binding Protein UVRAG. Nat Cell Biol (2006) 8(7):688–99. doi: 10.1038/ncb1426
90. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of Tumorigenesis by Heterozygous Disruption of the Beclin 1 Autophagy Gene. J Clin Invest (2003) 112(12):1809–20. doi: 10.1172/JCI20039
91. Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an Autophagy Gene Essential for Early Embryonic Development, Is a Haploinsufficient Tumor Suppressor. Proc Natl Acad Sci U S A (2003) 100(25):15077–82. doi: 10.1073/pnas.2436255100
92. Morselli E, Galluzzi L, Kepp O, Mariño G, Michaud M, Vitale I, et al. Oncosuppressive Functions of Autophagy. Antioxid Redox Signal (2011) 14(11):2251–69. doi: 10.1089/ars.2010.3478
93. Imlay JA, Linn S. DNA Damage and Oxygen Radical Toxicity. Science (New York N Y ) (1988) 240(4857):1302–9. doi: 10.1126/science.3287616
94. Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, et al. Autophagy Mitigates Metabolic Stress and Genome Damage in Mammary Tumorigenesis. Genes Dev (2007) 21(13):1621–35. doi: 10.1101/gad.1565707
95. Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al. Autophagy Suppresses Tumor Progression by Limiting Chromosomal Instability. Genes Dev (2007) 21(11):1367–81. doi: 10.1101/gad.1545107
96. Karin M. NF-kappaB as a Critical Link Between Inflammation and Cancer. Cold Spring Harb Perspect Biol (2009) 1(5):a000141. doi: 10.1101/cshperspect.a000141
97. Ghosh S, Mukherjee S, Choudhury S, Gupta P, Adhikary A, Baral R, et al. Reactive Oxygen Species in the Tumor Niche Triggers Altered Activation of Macrophages and Immunosuppression: Role of Fluoxetine. Cell Signal (2015) 27(7):1398–412. doi: 10.1016/j.cellsig.2015.03.013
98. Munn LL. Cancer and Inflammation. Wiley Interdiscip Rev Syst Biol Med (2017) 9(2):10.1002/wsbm.1370. doi: 10.1002/wsbm.1370
99. Kitamura T, Qian BZ, Pollard JW. Immune Cell Promotion of Metastasis. Nat Rev Immunol (2015) 15(2):73–86. doi: 10.1038/nri3789
100. Komohara Y, Jinushi M, Takeya M. Clinical Significance of Macrophage Heterogeneity in Human Malignant Tumors. Cancer Sci (2014) 105(1):1–8. doi: 10.1111/cas.12314
101. Bingle L, Brown NJ, Lewis CE. The Role of Tumour-Associated Macrophages in Tumour Progression: Implications for New Anticancer Therapies. J Pathol (2002) 196(3):254–65. doi: 10.1002/path.1027
102. Bruna F, Scodeller P. Pro-Tumorigenic Macrophage Infiltration in Oral Squamous Cell Carcinoma and Possible Macrophage-Aimed Therapeutic Interventions. Front Oncol (2021) 11:675664. doi: 10.3389/fonc.2021.675664
103. Nasrollahzadeh E, Razi S, Keshavarz-Fathi M, Mazzone M, Rezaei N. Pro-Tumorigenic Functions of Macrophages at the Primary, Invasive and Metastatic Tumor Site. Cancer Immunol Immunother (2020) 69(9):1673–97. doi: 10.1007/s00262-020-02616-6
104. Jounai N, Kobiyama K, Takeshita F, Ishii KJ. Recognition of Damage-Associated Molecular Patterns Related to Nucleic Acids During Inflammation and Vaccination. Front Cell Infect Microbiol (2013) 2:168. doi: 10.3389/fcimb.2012.00168
105. Hervera A, De Virgiliis F, Palmisano I, Zhou L, Tantardini E, Kong G, et al. Reactive Oxygen Species Regulate Axonal Regeneration Through the Release of Exosomal NADPH Oxidase 2 Complexes Into Injured Axons. Nat Cell Biol (2018) 20(3):307–19. doi: 10.1038/s41556-018-0039-x
106. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the Autophagy Protein Atg16L1 Enhances Endotoxin-Induced IL-1beta Production. Nature (2008) 456(7219):264–8. doi: 10.1038/nature07383
107. Bensaad K, Cheung EC, Vousden KH. Modulation of Intracellular ROS Levels by TIGAR Controls Autophagy. EMBO J (2009) 28(19):3015–26. doi: 10.1038/emboj.2009.242
108. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat Immunol (2011) 12(3):222–30. doi: 10.1038/ni.1980
109. Zhou R, Yazdi AS, Menu P, Tschopp J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
110. Roy D, Sarkar S, Felty Q. Levels of IL-1 Beta Control Stimulatory/Inhibitory Growth of Cancer Cells. Front Biosci (2006) 11:889–98. doi: 10.2741/1845
111. Thannickal VJ, Fanburg BL. Reactive Oxygen Species in Cell Signaling. Am J Physiol Lung Cell Mol Physiol (2000) 279(6):L1005–28. doi: 10.1152/ajplung.2000.279.6.L1005
112. Qian M, Fang X, Wang X. Autophagy and Inflammation. Clin Trans Med (2017) 6(1):24. doi: 10.1186/s40169-017-0154-5
113. Aggarwal C. Targeted Therapy for Lung Cancer: Present and Future. Ann Palliat Med (2014) 3(3):229–35. doi: 10.3978/j.issn.2224-5820.2014.06.01
114. Tang J, Di J, Cao H, Bai J, Zheng J. P53-Mediated Autophagic Regulation: A Prospective Strategy for Cancer Therapy. Cancer Lett (2015) 363(2):101–7. doi: 10.1016/j.canlet.2015.04.014
115. Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, et al. Global Genomic Profiling Reveals an Extensive P53-Regulated Autophagy Program Contributing to Key P53 Responses. Genes Dev (2013) 27(9):1016–31. doi: 10.1101/gad.212282.112
116. Luo XJ, Li LL, Deng QP, Yu XF, Yang LF, Luo FJ, et al. Grifolin, a Potent Antitumour Natural Product Upregulates Death-Associated Protein Kinase 1 DAPK1 via P53 in Nasopharyngeal Carcinoma Cells. Eur J Cancer (Oxford England: 1990) (2011) 47(2):316–25. doi: 10.1016/j.ejca.2010.09.021
117. Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a P53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell (2006) 126(1):121–34. doi: 10.1016/j.cell.2006.05.034
118. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell (2005) 122(6):927–39. doi: 10.1016/j.cell.2005.07.002
119. Budanov AV, Karin M. P53 Target Genes Sestrin1 and Sestrin2 Connect Genotoxic Stress and mTOR Signaling. Cell (2008) 134(3):451–60. doi: 10.1016/j.cell.2008.06.028
120. Feng Z, Zhang H, Levine AJ, Jin S. The Coordinate Regulation of the P53 and mTOR Pathways in Cells. Proc Natl Acad Sci USA (2005) 102(23):8204–9. doi: 10.1073/pnas.0502857102
121. Green DR, Kroemer G. Cytoplasmic Functions of the Tumour Suppressor P53. Nature (2009) 458(7242):1127–30. doi: 10.1038/nature07986
122. Morselli E, Galluzzi L, Kroemer G. Mechanisms of P53-Mediated Mitochondrial Membrane Permeabilization. Cell Res (2008) 18(7):708–10. doi: 10.1038/cr.2008.77
123. Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, et al. Regulation of Autophagy by Cytoplasmic P53. Nat Cell Biol (2008) 10(6):676–87. doi: 10.1038/ncb1730
124. Budanov AV, Lee JH, Karin M. Stressin’ Sestrins Take an Aging Fight. EMBO Mol Med (2010) 2(10):388–400. doi: 10.1002/emmm.201000097
125. Pani G, Galeotti T. Role of MnSOD and P66shc in Mitochondrial Response to P53. Antioxid Redox Signal (2011) 15(6):1715–27. doi: 10.1089/ars.2010.3499
126. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a Novel P53 Target Gene Regulating Energy Metabolism and Antioxidant Function. Proc Natl Acad Sci USA (2010) 107(16):7455–60. doi: 10.1073/pnas.1001006107
127. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-Activated Glutaminase (GLS2), a P53-Inducible Regulator of Glutamine Metabolism and Reactive Oxygen Species. Proc Natl Acad Sci USA (2010) 107(16):7461–6. doi: 10.1073/pnas.1002459107
128. Budanov AV. Stress-Responsive Sestrins Link P53 With Redox Regulation and Mammalian Target of Rapamycin Signaling. Antioxid Redox Signal (2011) 15(6):1679–90. doi: 10.1089/ars.2010.3530
129. Fan S, Li L, Chen S, Yu Y, Qi M, Tashiro S, et al. Silibinin Induced-Autophagic and Apoptotic Death Is Associated With an Increase in Reactive Oxygen and Nitrogen Species in HeLa Cells. Free Radical Res (2011) 45(11-12):1307–24. doi: 10.3109/10715762.2011.618186
130. Fan S, Qi M, Yu Y, Li L, Yao G, Tashiro S, et al. P53 Activation Plays a Crucial Role in Silibinin Induced ROS Generation via PUMA and JNK. Free Radical Res (2012) 46(3):310–9. doi: 10.3109/10715762.2012.655244
131. Duan WJ, Li QS, Xia MY, Tashiro S, Onodera S, Ikejima T. Silibinin Activated P53 and Induced Autophagic Death in Human Fibrosarcoma HT1080 Cells via Reactive Oxygen Species-P38 and C-Jun N-Terminal Kinase Pathways. Biol Pharm Bull (2011) 34(1):47–53. doi: 10.1248/bpb.34.47
132. Kimmelman AC. The Dynamic Nature of Autophagy in Cancer. Genes Dev (2011) 25(19):1999–2010. doi: 10.1101/gad.17558811
133. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-Eating and Self-Killing: Crosstalk Between Autophagy and Apoptosis. Nat Rev Mol Cell Biol (2007) 8(9):741–52. doi: 10.1038/nrm2239
134. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-Deficient Mice Develop Multiple Liver Tumors. Genes Dev (2011) 25(8):795–800. doi: 10.1101/gad.2016211
135. Pérez-Mancera PA, Young AR, Narita M. Inside and Out: The Activities of Senescence in Cancer. Nat Rev Cancer (2014) 14(8):547–58. doi: 10.1038/nrc3773
136. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA Damage Response Induces Inflammation and Senescence by Inhibiting Autophagy of GATA4. Science (New York N Y ) (2015) 349(6255):aaa5612. doi: 10.1126/science.aaa5612
137. Rusten TE, Stenmark H. P62, an Autophagy Hero or Culprit? Nat Cell Biol (2010) 12(3):207–9. doi: 10.1038/ncb0310-207
138. Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A, et al. P62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-Driven Gene Transcription. J Biol Chem (2010) 285(29):22576–91. doi: 10.1074/jbc.M110.118976
139. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The Selective Autophagy Substrate P62 Activates the Stress Responsive Transcription Factor Nrf2 Through Inactivation of Keap1. Nat Cell Biol (2010) 12(3):213–23. doi: 10.1038/ncb2021
140. Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-Mediated Cleavage of Atg5 Switches Autophagy to Apoptosis. Nat Cell Biol (2006) 8(10):1124–32. doi: 10.1038/ncb1482
141. Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The Autophagy Protein Atg12 Associates With Antiapoptotic Bcl-2 Family Members to Promote Mitochondrial Apoptosis. Mol Cell (2011) 44(5):698–709. doi: 10.1016/j.molcel.2011.10.014
142. Guo JY, Xia B, White E. Autophagy-Mediated Tumor Promotion. Cell (2013) 155(6):1216–9. doi: 10.1016/j.cell.2013.11.019
143. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The Role of Autophagy During the Early Neonatal Starvation Period. Nature (2004) 432(7020):1032–6. doi: 10.1038/nature03029
144. Singh R, Cuervo AM. Autophagy in the Cellular Energetic Balance. Cell Metab (2011) 13(5):495–504. doi: 10.1016/j.cmet.2011.04.004
145. Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras Requires Autophagy to Maintain Oxidative Metabolism and Tumorigenesis. Genes Dev (2011) 25(5):460–70. doi: 10.1101/gad.2016311
146. Masliah-Planchon J, Garinet S, Pasmant E. RAS-MAPK Pathway Epigenetic Activation in Cancer: miRNAs in Action. Oncotarget (2016) 7(25):38892–907. doi: 10.18632/oncotarget.6476
147. Zhu D, Zhou J, Zhao J, Jiang G, Zhang X, Zhang Y, et al. ZC3H13 Suppresses Colorectal Cancer Proliferation and Invasion via Inactivating Ras-ERK Signaling. J Cell Physiol (2019) 234(6):8899–907. doi: 10.1002/jcp.27551
148. Su CC. Tanshinone IIA can Inhibit MiaPaCa-2 Human Pancreatic Cancer Cells by Dual Blockade of the Ras/Raf/MEK/ERK and PI3K/AKT/mTOR Pathways. Oncol Rep (2018) 40(5):3102–11. doi: 10.3892/or.2018.6670
149. Gonçalves PR, Rocha-Brito KJ, Fernandes MR, Abrantes JL, Durán N, Ferreira-Halder CV. Violacein Induces Death of RAS-Mutated Metastatic Melanoma by Impairing Autophagy Process. Tumour Biol (2016) 37(10):14049–58. doi: 10.1007/s13277-016-5265-x
150. Guo JY, White E. Autophagy Is Required for Mitochondrial Function, Lipid Metabolism, Growth, and Fate of KRAS(G12D)-Driven Lung Tumors. Autophagy (2013) 9(10):1636–8. doi: 10.4161/auto.26123
151. Yang ZJ, Chee CE, Huang S, Sinicrope FA. The Role of Autophagy in Cancer: Therapeutic Implications. Mol Cancer Ther (2011) 10(9):1533–41. doi: 10.1158/1535-7163.MCT-11-0047
152. Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy Facilitates Glycolysis During Ras-Mediated Oncogenic Transformation. Mol Biol Cell (2011) 22(2):165–78. doi: 10.1091/mbc.E10-06-0500
153. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic Cancers Require Autophagy for Tumor Growth. Genes Dev (2011) 25(7):717–29. doi: 10.1101/gad.2016111
154. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial Metabolism and ROS Generation Are Essential for Kras-Mediated Tumorigenicity. Proc Natl Acad Sci USA (2010) 107(19):8788–93. doi: 10.1073/pnas.1003428107
155. Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy Suppresses Progression of K-Ras-Induced Lung Tumors to Oncocytomas and Maintains Lipid Homeostasis. Genes Dev (2013) 27(13):1447–61. doi: 10.1101/gad.219642.113
156. Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The Signaling Adaptor P62 Is an Important NF-kappaB Mediator in Tumorigenesis. Cancer Cell (2008) 13(4):343–54. doi: 10.1016/j.ccr.2008.02.001
157. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy Suppresses Tumorigenesis Through Elimination of P62. Cell (2009) 137(6):1062–75. doi: 10.1016/j.cell.2009.03.048
158. Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of Autophagy by FIP200 Deletion Inhibits Mammary Tumorigenesis. Genes Dev (2011) 25(14):1510–27. doi: 10.1101/gad.2051011
159. Seyfried TN, Huysentruyt LC. On the Origin of Cancer Metastasis. Crit Rev Oncog (2013) 18(1-2):43–73. doi: 10.1615/critrevoncog.v18.i1-2.40
160. Clark AG, Vignjevic DM. Modes of Cancer Cell Invasion and the Role of the Microenvironment. Curr Opin Cell Biol (2015) 36:13–22. doi: 10.1016/j.ceb.2015.06.004
161. Lamouille S, Xu J, Derynck R. Molecular Mechanisms of Epithelial-Mesenchymal Transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi: 10.1038/nrm3758
162. Kalluri R, Weinberg RA. The Basics of Epithelial-Mesenchymal Transition. J Clin Invest (2009) 119(6):1420–8. doi: 10.1172/JCI39104
163. Polyak K, Weinberg RA. Transitions Between Epithelial and Mesenchymal States: Acquisition of Malignant and Stem Cell Traits. Nat Rev Cancer (2009) 9(4):265–73. doi: 10.1038/nrc2620
164. Yilmaz M, Christofori G. EMT, the Cytoskeleton, and Cancer Cell Invasion. Cancer Metastasis Rev (2009) 28(1-2):15–33. doi: 10.1007/s10555-008-9169-0
165. Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, et al. PERK Integrates Autophagy and Oxidative Stress Responses to Promote Survival During Extracellular Matrix Detachment. Mol Cell Biol (2011) 31(17):3616–29. doi: 10.1128/MCB.05164-11
166. Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of Autophagy During Extracellular Matrix Detachment Promotes Cell Survival. Mol Biol Cell (2008) 19(3):797–806. doi: 10.1091/mbc.e07-10-1092
167. Peng YF, Shi YH, Ding ZB, Ke AW, Gu CY, Hui B, et al. Autophagy Inhibition Suppresses Pulmonary Metastasis of HCC in Mice via Impairing Anoikis Resistance and Colonization of HCC Cells. Autophagy (2013) 9(12):2056–68. doi: 10.4161/auto.26398
168. Peng YF, Shi YH, Shen YH, Ding ZB, Ke AW, Zhou J, et al. Promoting Colonization in Metastatic HCC Cells by Modulation of Autophagy. PLoS One (2013) 8(9):e74407. doi: 10.1371/journal.pone.0074407
169. Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, et al. Tumor Vessel Normalization by Chloroquine Independent of Autophagy. Cancer Cell (2014) 26(2):190–206. doi: 10.1016/j.ccr.2014.06.025
170. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive Oxygen Species Are Essential for Autophagy and Specifically Regulate the Activity of Atg4. EMBO J (2019) 38(10):e101812. doi: 10.15252/embj.2019101812
171. Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFβ in Cancer. FEBS Lett (2012) 586(14):1959–70. doi: 10.1016/j.febslet.2012.02.037
172. Tobar N, Villar V, Santibanez JF. ROS-NFkappaB Mediates TGF-Beta1-Induced Expression of Urokinase-Type Plasminogen Activator, Matrix Metalloproteinase-9 and Cell Invasion. Mol Cell Biochem (2010) 340(1-2):195–202. doi: 10.1007/s11010-010-0418-5
173. Lam CR, Tan C, Teo Z, Tay CY, Phua T, Wu YL, et al. Loss of TAK1 Increases Cell Traction Force in a ROS-Dependent Manner to Drive Epithelial-Mesenchymal Transition of Cancer Cells. Cell Death Dis (2013) 4(10):e848. doi: 10.1038/cddis.2013.339
174. Matsuno Y, Matsuyama M, Kiwamoto T, Morishima Y, Ishii Y, Hizawa N. ROS-Nrf2 Pathway Mediates the Development of TGF-B1-Induced Epithelial-Mesenchymal Transition Through the Interaction With Notch Signaling. In: B61. Epithelial Cell Biology in Respiratory Disease. American Thoracic Society International Conference Abstracts (2018). p. A3805–5.
175. Robertson IB, Rifkin DB. Unchaining the Beast; Insights From Structural and Evolutionary Studies on TGFβ Secretion, Sequestration, and Activation. Cytokine Growth Factor Rev (2013) 24(4):355–72. doi: 10.1016/j.cytogfr.2013.06.003
176. Rabinowitz JD, White E. Autophagy and Metabolism. Science (New York N Y) (2010) 330(6009):1344–8. doi: 10.1126/science.1193497
177. Eng CH, Abraham RT. The Autophagy Conundrum in Cancer: Influence of Tumorigenic Metabolic Reprogramming. Oncogene (2011) 30(47):4687–96. doi: 10.1038/onc.2011.220
178. Chang JY, Chiang MF, Lin SR, Lee MH, He H, Chou PY, et al. TIAF1 Self-Aggregation in Peritumor Capsule Formation, Spontaneous Activation of SMAD-Responsive Promoter in P53-Deficient Environment, and Cell Death. Cell Death Dis (2012) 3(4):e302. doi: 10.1038/cddis.2012.36
179. Debnath J. The Multifaceted Roles of Autophagy in Tumors-Implications for Breast Cancer. J Mammary Gland Biol Neoplasia (2011) 16(3):173–87. doi: 10.1007/s10911-011-9223-3
180. Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive Oxygen Species Generated at Mitochondrial Complex III Stabilize Hypoxia-Inducible Factor-1alpha During Hypoxia: A Mechanism of O2 Sensing. J Biol Chem (2000) 275(33):25130–8. doi: 10.1074/jbc.M001914200
181. Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial Autophagy is an HIF-1-Dependent Adaptive Metabolic Response to Hypoxia. J Biol Chem (2008) 283(16):10892–903. doi: 10.1074/jbc.M800102200
182. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-Induced Autophagy Is Mediated Through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol Cell Biol (2009) 29(10):2570–81. doi: 10.1128/MCB.00166-09
183. Rouschop KM, Ramaekers CH, Schaaf MB, Keulers TG, Savelkouls KG, Lambin P, et al. Autophagy Is Required During Cycling Hypoxia to Lower Production of Reactive Oxygen Species. Radiother Oncol (2009) 92(3):411–6. doi: 10.1016/j.radonc.2009.06.029
184. Brahimi-Horn MC, Bellot G, Pouysségur J. Hypoxia and Energetic Tumour Metabolism. Curr Opin Genet Dev (2011) 21(1):67–72. doi: 10.1016/j.gde.2010.10.006
185. Mazure NM, Pouysségur J. Hypoxia-Induced Autophagy: Cell Death or Cell Survival? Curr Opin Cell Biol (2010) 22(2):177–80. doi: 10.1016/j.ceb.2009.11.015
186. Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia Signals Autophagy in Tumor Cells via AMPK Activity, Independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ (2008) 15(10):1572–81. doi: 10.1038/cdd.2008.84
187. Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, et al. The Unfolded Protein Response Protects Human Tumor Cells During Hypoxia Through Regulation of the Autophagy Genes MAP1LC3B and ATG5. J Clin Invest (2010) 120(1):127–41. doi: 10.1172/JCI40027
188. Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo Y, et al. Autophagy Promotes Hepatocellular Carcinoma Cell Invasion Through Activation of Epithelial-Mesenchymal Transition. Carcinogenesis (2013) 34(6):1343–51. doi: 10.1093/carcin/bgt063
189. Kim YH, Baek SH, Kim EK, Ha JM, Jin SY, Lee HS, et al. Uncoordinated 51-Like Kinase 2 Signaling Pathway Regulates Epithelial-Mesenchymal Transition in A549 Lung Cancer Cells. FEBS Lett (2016) 590(9):1365–74. doi: 10.1002/1873-3468.12172
190. Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A, et al. The Autophagy-Associated Factors DRAM1 and P62 Regulate Cell Migration and Invasion in Glioblastoma Stem Cells. Oncogene (2013) 32(6):699–712. doi: 10.1038/onc.2012.111
191. Macintosh RL, Timpson P, Thorburn J, Anderson KI, Thorburn A, Ryan KM. Inhibition of Autophagy Impairs Tumor Cell Invasion in an Organotypic Model. Cell Cycle (Georgetown Tex) (2012) 11(10):2022–9. doi: 10.4161/cc.20424
192. Catalano M, D’Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, et al. Autophagy Induction Impairs Migration and Invasion by Reversing EMT in Glioblastoma Cells. Mol Oncol (2015) 9(8):1612–25. doi: 10.1016/j.molonc.2015.04.016
193. Grassi G, Di Caprio G, Santangelo L, Fimia GM, Cozzolino AM, Komatsu M, et al. Autophagy Regulates Hepatocyte Identity and Epithelial-to-Mesenchymal and Mesenchymal-to-Epithelial Transitions Promoting Snail Degradation. Cell Death Dis (2015) 6(9):e1880. doi: 10.1038/cddis.2015.249
194. Zhao Z, Zhao J, Xue J, Zhao X, Liu P. Autophagy Inhibition Promotes Epithelial-Mesenchymal Transition Through ROS/HO-1 Pathway in Ovarian Cancer Cells. Am J Cancer Res (2016) 6(10):2162–77.
195. Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H, et al. DEDD Interacts With PI3KC3 to Activate Autophagy and Attenuate Epithelial-Mesenchymal Transition in Human Breast Cancer. Cancer Res (2012) 72(13):3238–50. doi: 10.1158/0008-5472.CAN-11-3832
196. Wang MC, Liang X, Liu ZY, Cui J, Liu Y, Jing L, et al. In Vitro Synergistic Antitumor Efficacy of Sequentially Combined Chemotherapy/Icotinib in Non-Small Cell Lung Cancer Cell Lines. Oncol Rep (2015) 33(1):239–49. doi: 10.3892/or.2014.3583
197. Qiang L, Zhao B, Ming M, Wang N, He TC, Hwang S, et al. Regulation of Cell Proliferation and Migration by P62 Through Stabilization of Twist1. Proc Natl Acad Sci USA (2014) 111(25):9241–6. doi: 10.1073/pnas.1322913111
198. Bertrand M, Petit V, Jain A, Amsellem R, Johansen T, Larue L, et al. SQSTM1/p62 Regulates the Expression of Junctional Proteins Through Epithelial-Mesenchymal Transition Factors. Cell Cycle (Georgetown Tex) (2015) 14(3):364–74. doi: 10.4161/15384101.2014.987619
199. Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of Autophagy in Temozolomide-Induced Cytotoxicity for Malignant Glioma Cells. Cell Death Differ (2004) 11(4):448–57. doi: 10.1038/sj.cdd.4401359
200. Qadir MA, Kwok B, Dragowska WH, To KH, Le D, Bally MB, et al. Macroautophagy Inhibition Sensitizes Tamoxifen-Resistant Breast Cancer Cells and Enhances Mitochondrial Depolarization. Breast Cancer Res Treat (2008) 112(3):389–403. doi: 10.1007/s10549-007-9873-4
201. Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, et al. Targeting Autophagy Augments the Anticancer Activity of the Histone Deacetylase Inhibitor SAHA to Overcome Bcr-Abl-Mediated Drug Resistance. Blood (2007) 110(1):313–22. doi: 10.1182/blood-2006-10-050260
202. Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy Delays Apoptotic Death in Breast Cancer Cells Following DNA Damage. Cell Death Differ (2007) 14(3):500–10. doi: 10.1038/sj.cdd.4402039
203. Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, et al. Targeting Autophagy Potentiates Tyrosine Kinase Inhibitor-Induced Cell Death in Philadelphia Chromosome-Positive Cells, Including Primary CML Stem Cells. J Clin Invest (2009) 119(5):1109–23. doi: 10.1172/JCI35660
204. Harhaji-Trajkovic L, Vilimanovich U, Kravic-Stevovic T, Bumbasirevic V, Trajkovic V. AMPK-Mediated Autophagy Inhibits Apoptosis in Cisplatin-Treated Tumour Cells. J Cell Mol Med (2009) 13(9B):3644–54. doi: 10.1111/j.1582-4934.2009.00663.x
205. White E, DiPaola RS. The Double-Edged Sword of Autophagy Modulation in Cancer. Clin Cancer Res (2009) 15(17):5308–16. doi: 10.1158/1078-0432.CCR-07-5023
206. Martin AP, Park MA, Mitchell C, Walker T, Rahmani M, Thorburn A, et al. BCL-2 Family Inhibitors Enhance Histone Deacetylase Inhibitor and Sorafenib Lethality via Autophagy and Overcome Blockade of the Extrinsic Pathway to Facilitate Killing. Mol Pharmacol (2009) 76(2):327–41. doi: 10.1124/mol.109.056309
207. Zhou H, Shen T, Shang C, Luo Y, Liu L, Yan J, et al. Ciclopirox Induces Autophagy Through Reactive Oxygen Species-Mediated Activation of JNK Signaling Pathway. Oncotarget (2014) 5(20):10140–50. doi: 10.18632/oncotarget.2471
208. Hahm ER, Sakao K, Singh SV. Honokiol Activates Reactive Oxygen Species-Mediated Cytoprotective Autophagy in Human Prostate Cancer Cells. Prostate (2014) 74(12):1209–21. doi: 10.1002/pros.22837
209. Gonzalez Y, Aryal B, Chehab L, Rao VA. Atg7- and Keap1-Dependent Autophagy Protects Breast Cancer Cell Lines Against Mitoquinone-Induced Oxidative Stress. Oncotarget (2014) 5(6):1526–37. doi: 10.18632/oncotarget.1715
210. Kim H, Moon JY, Ahn KS, Cho SK. Quercetin Induces Mitochondrial Mediated Apoptosis and Protective Autophagy in Human Glioblastoma U373MG Cells. Oxid Med Cell Longev (2013) 2013:596496. doi: 10.1155/2013/596496
211. Zhang Q, Zhang Y, Zhang P, Chao Z, Xia F, Jiang C, et al. Hexokinase II Inhibitor, 3-BrPA Induced Autophagy by Stimulating ROS Formation in Human Breast Cancer Cells. Genes Cancer (2014) 5(3-4):100–12. doi: 10.18632/genesandcancer.9
212. Ganguli A, Choudhury D, Datta S, Bhattacharya S, Chakrabarti G. Inhibition of Autophagy by Chloroquine Potentiates Synergistically Anti-Cancer Property of Artemisinin by Promoting ROS Dependent Apoptosis. Biochimie (2014) 107 Pt B:338–49. doi: 10.1016/j.biochi.2014.10.001
213. Al Dhaheri Y, Attoub S, Ramadan G, Arafat K, Bajbouj K, Karuvantevida N, et al. Carnosol Induces ROS-Mediated Beclin1-Independent Autophagy and Apoptosis in Triple Negative Breast Cancer. PLoS One (2014) 9(10):e109630. doi: 10.1371/journal.pone.0109630
214. Kondo Y, Kondo S. Autophagy and Cancer Therapy. Autophagy (2006) 2(2):85–90. doi: 10.4161/auto.2.2.2463
215. Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I. Induction of Autophagic Cell Death in Malignant Glioma Cells by Arsenic Trioxide. Cancer Res (2003) 63(9):2103–8.
216. Chen Y, Gibson SB. Is Mitochondrial Generation of Reactive Oxygen Species a Trigger for Autophagy? Autophagy (2008) 4(2):246–8. doi: 10.4161/auto.5432
217. Sweeney C, Liu G, Yiannoutsos C, Kolesar J, Horvath D, Staab MJ, et al. A Phase II Multicenter, Randomized, Double-Blind, Safety Trial Assessing the Pharmacokinetics, Pharmacodynamics, and Efficacy of Oral 2-Methoxyestradiol Capsules in Hormone-Refractory Prostate Cancer. Clin Cancer Res (2005) 11(18):6625–33. doi: 10.1158/1078-0432.CCR-05-0440
218. Nicolau-Galmés F, Asumendi A, Alonso-Tejerina E, Pérez-Yarza G, Jangi SM, Gardeazabal J, et al. Terfenadine Induces Apoptosis and Autophagy in Melanoma Cells Through ROS-Dependent and -Independent Mechanisms. Apoptosis (2011) 16(12):1253–67. doi: 10.1007/s10495-011-0640-y
219. Selimovic D, Porzig BB, El-Khattouti A, Badura HE, Ahmad M, Ghanjati F, et al. Bortezomib/proteasome Inhibitor Triggers Both Apoptosis and Autophagy-Dependent Pathways in Melanoma Cells. Cell Signal (2013) 25(1):308–18. doi: 10.1016/j.cellsig.2012.10.004
220. Liang QP, Xu TQ, Liu BL, Lei XP, Hambrook JR, Zhang DM, et al. Sasanquasaponin III From Schima Crenata Korth Induces Autophagy Through Akt/mTOR/p70S6K Pathway and Promotes Apoptosis in Human Melanoma A375 Cells. Phytomedicine (2019) 58:152769. doi: 10.1016/j.phymed.2018.11.029
221. Hao W, Zhang X, Zhao W, Chen X. Psoralidin Induces Autophagy Through ROS Generation Which Inhibits the Proliferation of Human Lung Cancer A549 Cells. PeerJ (2014) 2:e555. doi: 10.7717/peerj.555
222. Miki H, Uehara N, Kimura A, Sasaki T, Yuri T, Yoshizawa K, et al. Resveratrol Induces Apoptosis via ROS-Triggered Autophagy in Human Colon Cancer Cells. Int J Oncol (2012) 40(4):1020–8. doi: 10.3892/ijo.2012.1325
223. Giammarioli AM, Gambardella L, Barbati C, Pietraforte D, Tinari A, Alberton M, et al. Differential Effects of the Glycolysis Inhibitor 2-Deoxy-D-Glucose on the Activity of Pro-Apoptotic Agents in Metastatic Melanoma Cells, and Induction of a Cytoprotective Autophagic Response. Int J Cancer (2012) 131(4):E337–47. doi: 10.1002/ijc.26420
224. Antunes F, Corazzari M, Pereira G, Fimia GM, Piacentini M, Smaili S. Fasting Boosts Sensitivity of Human Skin Melanoma to Cisplatin-Induced Cell Death. Biochem Biophys Res Commun (2017) 485(1):16–22. doi: 10.1016/j.bbrc.2016.09.149
225. Raffoul F, Campla C, Nanjundan M. SnoN/SkiL, a TGFβ Signaling Mediator: A Participant in Autophagy Induced by Arsenic Trioxide. Autophagy (2010) 6(7):955–7. doi: 10.4161/auto.6.7.13041
226. Luo Y, Sun X, Huang L, Yan J, Yu BY, Tian J. Artemisinin-Based Smart Nanomedicines With Self-Supply of Ferrous Ion to Enhance Oxidative Stress for Specific and Efficient Cancer Treatment. ACS Appl Mater Interfaces (2019) 11(33):29490–7. doi: 10.1021/acsami.9b07390
227. Bailey HH, Mulcahy RT, Tutsch KD, Arzoomanian RZ, Alberti D, Tombes MB, et al. Phase I Clinical Trial of Intravenous L-Buthionine Sulfoximine and Melphalan: An Attempt at Modulation of Glutathione. J Clin Oncol (1994) 12(1):194–205. doi: 10.1200/JCO.1994.12.1.194
228. Maeda H, Hori S, Ohizumi H, Segawa T, Kakehi Y, Ogawa O, et al. Effective Treatment of Advanced Solid Tumors by the Combination of Arsenic Trioxide and L-Buthionine-Sulfoximine. Cell Death Differ (2004) 11(7):737–46. doi: 10.1038/sj.cdd.4401389
229. Li Q, Yin X, Wang W, Zhan M, Zhao B, Hou Z, et al. The Effects of Buthionine Sulfoximine on the Proliferation and Apoptosis of Biliary Tract Cancer Cells Induced by Cisplatin and Gemcitabine. Oncol Lett (2016) 11(1):474–80. doi: 10.3892/ol.2015.3879
230. Hollomon MG, Gordon N, Santiago-O’Farrill JM, Kleinerman ES. Knockdown of Autophagy-Related Protein 5, ATG5, Decreases Oxidative Stress and Has an Opposing Effect on Camptothecin-Induced Cytotoxicity in Osteosarcoma Cells. BMC Cancer (2013) 13:500. doi: 10.1186/1471-2407-13-500
231. Pardee AB, Li YZ, Li CJ. Cancer Therapy With Beta-Lapachone. Curr Cancer Drug Targets (2002) 2(3):227–42. doi: 10.2174/1568009023333854
232. Bey EA, Bentle MS, Reinicke KE, Dong Y, Yang CR, Girard L, et al. An NQO1- and PARP-1-Mediated Cell Death Pathway Induced in Non-Small-Cell Lung Cancer Cells by Beta-Lapachone. Proc Natl Acad Sci USA (2007) 104(28):11832–7. doi: 10.1073/pnas.0702176104
233. Park EJ, Choi KS, Kwon TK. β-Lapachone-Induced Reactive Oxygen Species (ROS) Generation Mediates Autophagic Cell Death in Glioma U87 MG Cells. Chem Biol Interact (2011) 189(1-2):37–44. doi: 10.1016/j.cbi.2010.10.013
234. Yen CJ, Hung CH, Tsai WM, Cheng HC, Yang HL, Lu YJ, et al. Effect of Exercise Training on Exercise Tolerance and Level of Oxidative Stress for Head and Neck Cancer Patients Following Chemotherapy. Front Oncol (2020) 10:3389/fonc.2020.01536. doi: 10.3389/fonc.2020.01536
235. Lin JF, Lin YC, Tsai TF, Chen HE, Chou KY, Hwang TI. Cisplatin Induces Protective Autophagy Through Activation of BECN1 in Human Bladder Cancer Cells. Drug Des Dev Ther (2017) 11:1517–33. doi: 10.2147/DDDT.S126464
236. Lee YJ, Kim NY, Suh YA, Lee C. Involvement of ROS in Curcumin-Induced Autophagic Cell Death. Korean J Physiol Pharmacol (2011) 15(1):1–7. doi: 10.4196/kjpp.2011.15.1.1
237. Al-Aamri HM, Ku H, Irving HR, Tucci J, Meehan-Andrews T, Bradley C. Time Dependent Response of Daunorubicin on Cytotoxicity, Cell Cycle and DNA Repair in Acute Lymphoblastic Leukaemia. BMC Cancer (2019) 19(1):179. doi: 10.1186/s12885-019-5377-y
238. Han W, Sun J, Feng L, Wang K, Li D, Pan Q, et al. Autophagy Inhibition Enhances Daunorubicin-Induced Apoptosis in K562 Cells. PLoS One (2011) 6(12):e28491. doi: 10.1371/journal.pone.0028491
239. Sritharan S, Sivalingam N. A Comprehensive Review on Time-Tested Anticancer Drug Doxorubicin. Life Sci (2021) 278:119527. doi: 10.1016/j.lfs.2021.119527
240. Aydinlik S, Erkisa M, Cevatemre B, Sarimahmut M, Dere E, Ari F, et al. Enhanced Cytotoxic Activity of Doxorubicin Through the Inhibition of Autophagy in Triple Negative Breast Cancer Cell Line. Biochim Biophys Acta Gen Subj (2017) 1861(2):49–57. doi: 10.1016/j.bbagen.2016.11.013
241. Fang B. Genetic Interactions of STAT3 and Anticancer Drug Development. Cancers (2014) 6(1):494–525. doi: 10.3390/cancers6010494
242. Doroshow JH, Juhasz A, Ge Y, Holbeck S, Lu J, Antony S, et al. Antiproliferative Mechanisms of Action of the Flavin Dehydrogenase Inhibitors Diphenylene Iodonium and Di-2-Thienyliodonium Based on Molecular Profiling of the NCI-60 Human Tumor Cell Panel. Biochem Pharmacol (2012) 83(9):1195–207. doi: 10.1016/j.bcp.2012.01.022
243. Ligeon LA, Pena-Francesch M, Vanoaica LD, Núñez NG, Talwar D, Dick TP, et al. Oxidation Inhibits Autophagy Protein Deconjugation From Phagosomes to Sustain MHC Class II Restricted Antigen Presentation. Nat Commun (2021) 12(1):1508. doi: 10.1038/s41467-021-21829-6
244. Cen D, Gonzalez RI, Buckmeier JA, Kahlon RS, Tohidian NB, Meyskens FL Jr. Disulfiram Induces Apoptosis in Human Melanoma Cells: A Redox-Related Process. Mol Cancer Ther (2002) 1(3):197–204.
245. Fruehauf JP, Trapp V. Reactive Oxygen Species: An Achilles’ Heel of Melanoma? Expert Rev Anticancer Ther (2008) 8(11):1751–7. doi: 10.1586/14737140.8.11.1751
246. Wu X, Xue X, Wang L, Wang W, Han J, Sun X, et al. Suppressing Autophagy Enhances Disulfiram/Copper-Induced Apoptosis in Non-Small Cell Lung Cancer. Eur J Pharmacol (2018) 827:1–12. doi: 10.1016/j.ejphar.2018.02.039
247. Zhang X, Hu P, Ding SY, Sun T, Liu L, Han S, et al. Induction of Autophagy-Dependent Apoptosis in Cancer Cells Through Activation of ER Stress: An Uncovered Anti-Cancer Mechanism by Anti-Alcoholism Drug Disulfiram. Am J Cancer Res (2019) 9(6):1266–81.
248. Hu Y, Qian Y, Wei J, Jin T, Kong X, Cao H, et al. The Disulfiram/Copper Complex Induces Autophagic Cell Death in Colorectal Cancer by Targeting ULK1. Front Pharmacol (2021) 12:3389/fphar.2021.752825. doi: 10.3389/fphar.2021.752825
249. Zhang Q, Yang W, Man N, Zheng F, Shen Y, Sun K, et al. Autophagy-Mediated Chemosensitization in Cancer Cells by Fullerene C60 Nanocrystal. Autophagy (2009) 5(8):1107–17. doi: 10.4161/auto.5.8.9842
250. Zhang L, Li J, Zong L, Chen X, Chen K, Jiang Z, et al. Reactive Oxygen Species and Targeted Therapy for Pancreatic Cancer. Oxid Med Cell Longev (2016) 2016:1616781. doi: 10.1155/2016/1616781
251. Chen M, He M, Song Y, Chen L, Xiao P, Wan X, et al. The Cytoprotective Role of Gemcitabine-Induced Autophagy Associated With Apoptosis Inhibition in Triple-Negative MDA-MB-231 Breast Cancer Cells. Int J Mol Med (2014) 34(1):276–82. doi: 10.3892/ijmm.2014.1772
252. Yang MC, Wang HC, Hou YC, Tung HL, Chiu TJ, Shan YS. Blockade of Autophagy Reduces Pancreatic Cancer Stem Cell Activity and Potentiates the Tumoricidal Effect of Gemcitabine. Mol Cancer (2015) 14:179. doi: 10.1186/s12943-015-0449-3
253. Yang X, Yin H, Zhang Y, Li X, Tong H, Zeng Y, et al. Hypoxia-Induced Autophagy Promotes Gemcitabine Resistance in Human Bladder Cancer Cells Through Hypoxia-Inducible Factor 1α Activation. Int J Oncol (2018) 53(1):215–24. doi: 10.3892/ijo.2018.4376
254. Mizutani H, Shiga C, Imai M, Ikemura K, Kitamura Y, Ohta K, et al. Idarubicin, an Anthracycline, Induces Oxidative DNA Damage in the Presence of Copper (II). Anticancer Res (2020) 40(10):5399–404. doi: 10.21873/anticanres.14548
255. Zhang Y, Li Q, Xu D, Li T, Gu Z, Huang P, et al. Idarubicin-Induced Oxidative Stress and Apoptosis in Cardiomyocytes: An In Vitro Molecular Approach. Hum Exp Toxicol (2021) 40(12_suppl):S553–62. doi: 10.1177/09603271211033774
256. Ristic B, Bosnjak M, Arsikin K, Mircic A, Suzin-Zivkovic V, Bogdanovic A, et al. Idarubicin Induces mTOR-Dependent Cytotoxic Autophagy in Leukemic Cells. Exp Cell Res (2014) 326(1):90–102. doi: 10.1016/j.yexcr.2014.05.021
257. Kim SJ, Kim HS, Seo YR. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid Med Cell Longev (2019) 2019:5381692. doi: 10.1155/2019/5381692
258. Graczyk-Jarzynka A, Zagozdzon R, Muchowicz A, Siernicka M, Juszczynski P, Firczuk M. New Insights Into Redox Homeostasis as a Therapeutic Target in B-Cell Malignancies. Curr Opin Hematol (2017) 24(4):393–401. doi: 10.1097/MOH.0000000000000351
259. Wang W, Dong X, Liu Y, Ni B, Sai N, You L, et al. Itraconazole Exerts Anti-Liver Cancer Potential Through the Wnt, PI3K/AKT/mTOR, and ROS Pathways. Biomed Pharmacother (2020) 131:110661. doi: 10.1016/j.biopha.2020.110661
260. Liu R, Li J, Zhang T, Zou L, Chen Y, Wang K, et al. Itraconazole Suppresses the Growth of Glioblastoma Through Induction of Autophagy: Involvement of Abnormal Cholesterol Trafficking. Autophagy (2014) 10(7):1241–55. doi: 10.4161/auto.28912
261. Deng H, Huang L, Liao Z, Liu M, Li Q, Xu R. Itraconazole Inhibits the Hedgehog Signaling Pathway Thereby Inducing Autophagy-Mediated Apoptosis of Colon Cancer Cells. Cell Death Dis (2020) 11(7):539. doi: 10.1038/s41419-020-02742-0
262. Alexandre J, Nicco C, Chéreau C, Laurent A, Weill B, Goldwasser F, et al. Improvement of the Therapeutic Index of Anticancer Drugs by the Superoxide Dismutase Mimic Mangafodipir. J Natl Cancer Inst (2006) 98(4):236–44. doi: 10.1093/jnci/djj049
263. Khanim FL, Hayden RE, Birtwistle J, Lodi A, Tiziani S, Davies NJ, et al. Combined Bezafibrate and Medroxyprogesterone Acetate: Potential Novel Therapy for Acute Myeloid Leukaemia. PLoS One (2009) 4(12):e8147. doi: 10.1371/journal.pone.0008147
264. Elmaci İ, Ozpinar A, Bilir A, Altinoz MA. Medroxyprogesterone Effects on Colony Growth, Autophagy and Mitochondria of C6 Glioma Cells Are Augmented With Tibolone and Temozolomide: Cell Kinetic and Electron Microscopical Studies With a Broad Review of the Literature. Clin Neurol Neurosurg (2019) 177:77–85. doi: 10.1016/j.clineuro.2018.12.022
265. Mogavero A, Maiorana MV, Zanutto S, Varinelli L, Bozzi F, Belfiore A, et al. Metformin Transiently Inhibits Colorectal Cancer Cell Proliferation as a Result of Either AMPK Activation or Increased ROS Production. Sci Rep (2017) 7(1):15992. doi: 10.1038/s41598-017-16149-z
266. Bharath LP, Agrawal M, McCambridge G, Nicholas DA, Hasturk H, Liu J, et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab (2020) 32(1):44–55.e6. doi: 10.1016/j.cmet.2020.04.015
267. Gao C, Fang L, Zhang H, Zhang WS, Li XO, Du SY. Metformin Induces Autophagy via the AMPK-mTOR Signaling Pathway in Human Hepatocellular Carcinoma Cells. Cancer Manag Res (2020) 12:5803–11. doi: 10.2147/CMAR.S257966
268. De Santi M, Baldelli G, Diotallevi A, Galluzzi L, Schiavano GF, Brandi G. Metformin Prevents Cell Tumorigenesis Through Autophagy-Related Cell Death. Sci Rep (2019) 9(1):66. doi: 10.1038/s41598-018-37247-6
269. Magda D, Miller RA. Motexafin Gadolinium: A Novel Redox Active Drug for Cancer Therapy. Semin Cancer Biol (2006) 16(6):466–76. doi: 10.1016/j.semcancer.2006.09.002
270. Gao M, Yeh PY, Lu YS, Hsu CH, Chen KF, Lee WC, et al. OSU-03012, a Novel Celecoxib Derivative, Induces Reactive Oxygen Species-Related Autophagy in Hepatocellular Carcinoma. Cancer Res (2008) 68(22):9348–57. doi: 10.1158/0008-5472.CAN-08-1642
271. Giannopoulou E, Antonacopoulou A, Matsouka P, Kalofonos HP. Autophagy: Novel Action of Panitumumab in Colon Cancer. Anticancer Res (2009) 29(12):5077–82.
272. Marino ML, Fais S, Djavaheri-Mergny M, Villa A, Meschini S, Lozupone F, et al. Proton Pump Inhibition Induces Autophagy as a Survival Mechanism Following Oxidative Stress in Human Melanoma Cells. Cell Death Dis (2010) 1(10):e87. doi: 10.1038/cddis.2010.67
273. Dolmans DE, Fukumura D, Jain RK. Photodynamic Therapy for Cancer. Nat Rev Cancer (2003) 3(5):380–7. doi: 10.1038/nrc1071
274. Brown SB, Brown EA, Walker I. The Present and Future Role of Photodynamic Therapy in Cancer Treatment. Lancet Oncol (2004) 5(8):497–508. doi: 10.1016/S1470-2045(04)01529-3
275. Martins WK, Belotto R, Silva MN, Grasso D, Suriani MD, Lavor TS, et al. Autophagy Regulation and Photodynamic Therapy: Insights to Improve Outcomes of Cancer Treatment. Front Oncol (2021) 10:610472. doi: 10.3389/fonc.2020.610472
276. Dewaele M, Martinet W, Rubio N, Verfaillie T, de Witte PA, Piette J, et al. Autophagy Pathways Activated in Response to PDT Contribute to Cell Resistance Against ROS Damage. J Cell Mol Med (2011) 15(6):1402–14. doi: 10.1111/j.1582-4934.2010.01118.x
277. Xiong L, Liu Z, Ouyang G, Lin L, Huang H, Kang H, et al. Autophagy Inhibition Enhances Photocytotoxicity of Photosan-II in Human Colorectal Cancer Cells. Oncotarget (2017) 8(4):6419–32. doi: 10.18632/oncotarget.14117
278. Saleem MZ, Alshwmi M, Zhang H, Din S, Nisar MA, Khan M, et al. Inhibition of JNK-Mediated Autophagy Promotes Proscillaridin A- Induced Apoptosis via ROS Generation, Intracellular Ca+2 Oscillation and Inhibiting STAT3 Signaling in Breast Cancer Cells. Front Pharmacol (2020) 11:1055. doi: 10.3389/fphar.2020.01055
279. Gdynia G, Keith M, Kopitz J, Bergmann M, Fassl A, Weber AN, et al. Danger Signaling Protein HMGB1 Induces a Distinct Form of Cell Death Accompanied by Formation of Giant Mitochondria. Cancer Res (2010) 70(21):8558–68. doi: 10.1158/0008-5472.CAN-10-0204
280. Sun X, Tang D. HMGB1-Dependent and -Independent Autophagy. Autophagy (2014) 10(10):1873–6. doi: 10.4161/auto.32184
281. Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, et al. HMGB1 Release and Redox Regulates Autophagy and Apoptosis in Cancer Cells. Oncogene (2010) 29(38):5299–310. doi: 10.1038/onc.2010.261
282. Tan C, Lai S, Wu S, Hu S, Zhou L, Chen Y, et al. Nuclear Permeable Ruthenium(II) β-Carboline Complexes Induce Autophagy to Antagonize Mitochondrial-Mediated Apoptosis. J Med Chem (2010) 53(21):7613–24. doi: 10.1021/jm1009296
283. Jazirehi AR. Regulation of Apoptosis-Associated Genes by Histone Deacetylase Inhibitors: Implications in Cancer Therapy. Anticancer Drugs (2010) 21(9):805–13. doi: 10.1097/CAD.0b013e32833dad91
284. Li J, Liu R, Lei Y, Wang K, Lau QC, Xie N, et al. Proteomic Analysis Revealed Association of Aberrant ROS Signaling With Suberoylanilide Hydroxamic Acid-Induced Autophagy in Jurkat T-Leukemia Cells. Autophagy (2010) 6(6):711–24. doi: 10.4161/auto.6.6.12397
285. Naumann P, Fortunato F, Zentgraf H, Büchler MW, Herr I, Werner J. Autophagy and Cell Death Signaling Following Dietary Sulforaphane Act Independently of Each Other and Require Oxidative Stress in Pancreatic Cancer. Int J Oncol (2011) 39(1):101–9. doi: 10.3892/ijo.2011.1025
286. Xiao D, Powolny AA, Antosiewicz J, Hahm ER, Bommareddy A, Zeng Y, et al. Cellular Responses to Cancer Chemopreventive Agent D,L-Sulforaphane in Human Prostate Cancer Cells Are Initiated by Mitochondrial Reactive Oxygen Species. Pharm Res (2009) 26(7):1729–38. doi: 10.1007/s11095-009-9883-5
287. Rai P, Young JJ, Burton DG, Giribaldi MG, Onder TT, Weinberg RA. Enhanced Elimination of Oxidized Guanine Nucleotides Inhibits Oncogenic RAS-Induced DNA Damage and Premature Senescence. Oncogene (2011) 30(12):1489–96. doi: 10.1038/onc.2010.520
288. Bauvy C, Gane P, Arico S, Codogno P, Ogier-Denis E. Autophagy Delays Sulindac Sulfide-Induced Apoptosis in the Human Intestinal Colon Cancer Cell Line HT-29. Exp Cell Res (2001) 268(2):139–49. doi: 10.1006/excr.2001.5285
289. de Medina P, Silvente-Poirot S, Poirot M. Tamoxifen and AEBS Ligands Induced Apoptosis and Autophagy in Breast Cancer Cells Through the Stimulation of Sterol Accumulation. Autophagy (2009) 5(7):1066–7. doi: 10.4161/auto.5.7.9820
290. Lin CJ, Lee CC, Shih YL, Lin TY, Wang SH, Lin YF, et al. Resveratrol Enhances the Therapeutic Effect of Temozolomide Against Malignant Glioma In Vitro and In Vivo by Inhibiting Autophagy. Free Radical Biol Med (2012) 52(2):377–91. doi: 10.1016/j.freeradbiomed.2011.10.487
291. Lowndes SA, Adams A, Timms A, Fisher N, Smythe J, Watt SM, et al. Phase I Study of Copper-Binding Agent ATN-224 in Patients With Advanced Solid Tumors. Clin Cancer Res (2008) 14(22):7526–34. doi: 10.1158/1078-0432.CCR-08-0315
292. Juarez JC, Manuia M, Burnett ME, Betancourt O, Boivin B, Shaw DE, et al. Superoxide Dismutase 1 (SOD1) Is Essential for H2O2-Mediated Oxidation and Inactivation of Phosphatases in Growth Factor Signaling. Proc Natl Acad Sci USA (2008) 105(20):7147–52. doi: 10.1073/pnas.0709451105
293. Yu Z, Zhou R, Zhao Y, Pan Y, Liang H, Zhang JS, et al. Blockage of SLC31A1-Dependent Copper Absorption Increases Pancreatic Cancer Cell Autophagy to Resist Cell Death. Cell Prolif (2019) 52(2):e12568. doi: 10.1111/cpr.12568
294. Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP. Autophagy Induced by Valproic Acid Is Associated With Oxidative Stress in Glioma Cell Lines. Neuro Oncol (2010) 12(4):328–40. doi: 10.1093/neuonc/nop005
295. Perri M, Pingitore A, Cione E, Vilardi E, Perrone V, Genchi G. Proliferative and Anti-Proliferative Effects of Retinoic Acid at Doses Similar to Endogenous Levels in Leydig MLTC-1/R2C/TM-3 Cells. Biochim Biophys Acta (2010) 1800(9):993–1001. doi: 10.1016/j.bbagen.2010.06.006
296. Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide Dismutase as a Target for the Selective Killing of Cancer Cells. Nature (2000) 407(6802):390–5. doi: 10.1038/35030140
297. Reumann S, Shogren KL, Yaszemski MJ, Maran A. Inhibition of Autophagy Increases 2-Methoxyestradiol-Induced Cytotoxicity in SW1353 Chondrosarcoma Cells. J Cell Biochem (2016) 117(3):751–9. doi: 10.1002/jcb.25360
Keywords: autophagy, ROS, tumor microenvironment, epithelial–mesenchymal transition, metastasis, anticancer therapy resistance
Citation: Hasan A, Rizvi SF, Parveen S, Pathak N, Nazir A and Mir SS (2022) Crosstalk Between ROS and Autophagy in Tumorigenesis: Understanding the Multifaceted Paradox. Front. Oncol. 12:852424. doi: 10.3389/fonc.2022.852424
Received: 11 January 2022; Accepted: 14 February 2022;
Published: 10 March 2022.
Edited by:
Abdelhabib Semlali, Laval University, CanadaReviewed by:
Sujit Kumar Bhutia, National Institute of Technology Rourkela, IndiaPaola Maycotte, Instituto Mexicano del Seguro Social, Mexico
Copyright © 2022 Hasan, Rizvi, Parveen, Pathak, Nazir and Mir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Snober S. Mir, c21pckBpdWwuYWMuaW4=, bWlyc3Nub2JlckBnbWFpbC5jb20=