 Henriett Butz
Henriett Butz József Lövey3,4†
József Lövey3,4† Anikó Bozsik
Anikó Bozsik- 1Department of Molecular Genetics, National Institute of Oncology, Budapest, Hungary
- 2Hereditary Tumours Research Group, Hungarian Academy of Sciences-Semmelweis University, Budapest, Hungary
- 3Department of Radiotherapy, National Institute of Oncology, Budapest, Hungary
- 4Department of Oncology, Faculty of Medicine, Semmelweis University, Budapest, Hungary
- 5Surgical and Molecular Tumor Pathology Centre, National Institute of Oncology, Budapest, Hungary
Since the introduction of next-generation sequencing, the frequency of germline pathogenic TP53 variants and the number of cases with unusual clinical presentations have been increasing. This has led to the expansion of the classical Li–Fraumeni syndrome concept to a wider cancer predisposition syndrome designated as the Li–Fraumeni spectrum. Here, we present a case with a malignant, metastatic perivascular epithelioid cell tumor (PEComa) of the thigh muscle and a sinonasal carcinoma harboring a novel TP53 germline splice mutation (NM_000546.5:c.97-2A>C). The classical presentation of LFS in the long-since deceased mother and the presence of a germline TP53 variant in the proband suggested a possible familial TP53-related condition. Complex pathological, molecular, and clinical genetic analyses (whole exome sequencing of germline variants, multigene panel sequencing of tumor DNA, Sanger validation, an in vitro functional test on splicing effect, 3D protein modeling, p53 immunohistochemistry, and pedigree analysis) were performed. The in vitro characterization of the splice mutation supported the pathogenic effect that resulted in exon skipping. A locus-specific loss of heterozygosity in the PEComa but not in the sinonasal carcinoma was identified, suggesting the causative role of the splice mutation in the PEComa pathogenesis, because we excluded known pathogenetic pathways characteristic to PEComas (TSC1/2, TFE3, RAD51B). However, the second hit affecting TP53 in the molecular pathogenesis of the sinonasal carcinoma was not identified. Although PEComa has been reported previously in two patients with Li–Fraumeni syndrome, to the best of our knowledge, this is the first report suggesting a relationship between the aberrant TP53 variant and PEComa.
Introduction
Germline pathogenic TP53 variants are associated with Li–Fraumeni syndrome (LFS), which is a rare, autosomal-dominant, hereditary tumor syndrome (1). Five cancer types account for the majority of LFS tumors, which are called “LFS core” tumors: adrenocortical carcinoma, breast cancer, central nervous system tumors, osteosarcomas, and soft-tissue sarcomas. However, LFS patients have an increased risk of several additional cancers, such as leukemia, lymphoma, gastrointestinal cancers, and cancers of head and neck, kidney, larynx, lung, skin, ovary, pancreas, prostate, testis, and thyroid (2). Also, many cases of germline TP53 pathogenic variants have been identified in children with cancers, or among adult females with breast cancers without a familial history of cancer. Hence the expansion of the LFS concept to a wider cancer predisposition syndrome: the terms “heritable TP53-related cancer (hTP53rc) syndrome” by the European Reference Network GENTURIS and “Li-Fraumeni spectrum” by Kratz et al. have been recently suggested (3, 4). Based on classic, familial cases, the cumulative cancer risk was initially given as 73%–100% by age 70, with risks close to 100% in women (5–7). However, based on population studies, and as a consequence of the increased availability for high-throughput testing, the overall cancer penetrance seems to be lower (3, 8). Still, based on a recent observational cohort study on cancer incidence, patterns, and genotype–phenotype associations, individuals with Li–Fraumeni syndrome had a nearly 24 times higher incidence of any cancer than the general population (9). Additionally, while the disease prevalence is not well established, the prevalence of the germline pathogenic TP53 carrier status in the general population was recently estimated to be approximately 1:4,500 (8).
In clinical genetics, testing criteria for the TP53 gene have been extensively discussed (3, 4), and for most tumors, based on the personal or family history suggestive of such a syndrome, germline testing is recommended (10). In addition, TP53 pathogenic/likely pathogenic (P/LP) variants are commonly detected somatically, and it is the most frequently mutated gene in tumor tissues (11–13). Therefore, it has been recently recommended that when only somatic testing is performed and a P/LP variant is identified in the TP53 gene, germline examination is indicated only when it is detected in sarcomas, breast cancer, or brain tumors (10).
In this current study, we report a peculiar case, where in the background of an unusual appearance of the Li–Fraumeni spectrum manifesting in a malignant perivascular epithelioid cell tumor (PEComa), a novel TP53 pathogenic variant was identified. While PEComa was described in two previous case reports of Li–Fraumeni patients (14, 15), PEComa was the first manifestation of the disease in our patient. Our molecular genetic assays suggest a potential relationship between the pathogenic TP53 variant and PEComa development.
Methods
Immunohistochemistry
Immunohistochemical characterization was performed on a Ventana Benchmark autostainer (Roche Tissue Diagnostics, Oro Valley, AZ, USA) using the ultraView Universal DAB Detection Kit. Antibodies used (in alphabetical order) and vendors were as follows: ERG, H-Caldesmon, MelanA, and SOX10 (Ventana, Oro Valley, AZ, USA). Further antibodies CD34 (Dako-Agilent, Santa Clara, CA, USA, 1:200), Desmin (Dako-Agilent, 1:200), EMA (Dako-Agilent, 1:800), H3K27me3 (Cell Signaling, Danvers, MA, USA, 1:50), HHF35 (Dako-Agilent, 1:50), HMB45 (Dako-Agilent, 1:50), S100 (Dako-Agilent, 1:4000), SMA (Dako-Agilent, 1:100), STAT6 (Santa Cruz, Dallas, TX, USA, 1:100), Vim (Dako-Agilent, 1:100), and p53 (Dako-Agilent, 1:200) were used.
Fluorescent In Situ Hybridization
Fluorescent in situ hybridization (FISH) was performed using the ZytoLight® SPEC EWSR1/FLI1 TriCheck™ and ZytoLight® SPEC TFE3 Dual Color Break Apart Probe.
Genetic Analysis
Germline genetic analysis of the proband and family members was performed following an informed consent based on the ethical approval by the Scientific and Research Committee of the Medical Research Council of the Ministry of Health, Hungary (ETT-TUKEB 53720-4/2019/EÜIG).
Nucleic Acid Isolation From Peripheral Blood and From Tumor Tissue
DNA purification from peripheral blood and formalin-fixed paraffin-embedded (FFPE) tissues was performed using the Gentra Puregene Blood Kit (Cat No.: 158389, Qiagen, Hilden, Germany) and the Maxwell RSC DNA FFPE Kit on a Maxwell RSC Instrument (Cat. No.: S1450, Madison, WI, USA) as part of the routine molecular pathology diagnostic workflow. For RNA analysis, blood was collected in Tempus™ Blood RNA Tubes (Thermo Fisher Scientific, Waltham, MA, USA) and RNA extraction was performed by using the Tempus™ Spin RNA Isolation Kit. Nucleic acid quality and quantity were determined by a NanoDrop® 1000 Spectrophotometer (NanoDrop Technologies, Thermo Fisher Scientific, Waltham, MA, USA).
Whole Exome Sequencing From Peripheral Blood
Whole exome sequencing was done as previously reported, using a Twist Human Core Exome library preparation with a Twist mitochondrial panel (Cat. No.: 102026, Twist Bioscience, San Francisco, CA, USA) on a NovaSeq Illumina platform (Illumina, San Diego, CA, USA) with an average coverage of 100x (16). Data were analyzed by applying the Genome Analysis Toolkit (GATK) Germline short variant discovery (SNPs + Indels) algorithm. Annotation of coding variants was performed, following the American College of Medical Genetics and Genomics (ACMG) recommendations (17).
Sanger Validation, Site-Specific LOH Analysis, and RNA Splicing Effect Test
Sanger validation and site-specific LOH analysis were performed as previously reported (16). Primers used for validation were as follows: TP53_ex04_FOR 5′-CTGGTAAGGACAAGGGTTGG-3′; TP53_ex04_REV: 5′-GCCAGGCATTGAAGTCTCAT-3′, and for LOH testing: TP53_int4ex4_F1: 5′-CTGGTAAGGACAAGGGTTGG-3′; TP53_int4ex4_F2: 5′-ACTTCCTGAAAACAACGTTCTG-3′; TP53_int4ex4_R1: 5′-TCATCTGGACCTGGGTCTTC-3′; TP53_int4ex4_R2: 5′-TCTGGACCTGGGTCTTCAGT-3′; TP53_int4ex4_R3: 5′-TCTGGGAGCTTCATCTGGAC-3′.
For testing the splicing effect, RNA extracted from whole blood was reverse transcribed using SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific, MA, USA). cDNA was then PCR-amplified with the following primers: TP53-C-e02_For: 5′-AGGAAACATTTTCAGACCTATGGA-3′, TP53-C-e06_Rev: 5′-CTGTCATCCAAATACTCCACACG-3′. PCR products were subjected to agarose gel electrophoresis next to controls and were then submitted for Sanger sequencing.
Multigene Panel Sequencing on FFPE Tumor DNA
Multigene panel sequencing of 161 genes related to personalized tumor therapy with Oncomine™ Comprehensive Assay v3M (Cat. No.: A35805, Thermo Fisher Scientific, Waltham, MA, USA) was performed as previously described on an Ion Torrent next-generation sequencing platform (Ion GeneStudio S5 System, Thermo Fisher Scientific, Waltham, MA, USA) (16). Data were analyzed using Oncomine Knowledge Reporter Software (Cat. No.: A34298, Thermo Fisher Scientific, Waltham, MA, USA).
Monogenic Mutation Analysis of Sinonasal Carcinoma
A real-time PCR test, a cobas 4800 KRAS Mutation test, and a BRAF/NRAS mutation test were used according to the manufacturer’s instructions.
3D Protein Modeling
For protein modeling, prediction, and analysis, Phyre2 software was used to compare wild-type and variant amino acid sequences (18). For assessing the variant protein function and disorder prediction, the Phyre Investigator algorithm was applied.
Variant Classification
Specifications of the ACMG/AMP variant interpretation guidelines for germline TP53 variants by Fortuno et al. were applied for variant classification (19). Accordingly, the ClinGen Sequence Variant Interpretation (SVI) Committee-approved decision tree (Abou Tayoun et al.) was used to determine the strength of PVS1 criteria, similarly to the TP53(NM_000546.5):c.97-1G>A variant (19, 20).
Results
Case Report
A 90 × 60 × 115 mm soft tissue tumor was observed in the medial part of the right thigh of a 38-year-old, Caucasian male patient. In addition to the right-thigh tumor, soft-tissue MRI and thoraco-abdominal and pelvic CT revealed three nodules in the chest that appeared suspicious for metastatic processes (15-mm nodule in the right-lobe S10; 102-mm nodule in the left-side S6 segment and a 10 × 15 mm nodule subcarinal). Following the surgical removal of the thigh soft-tissue tumor, which was diagnosed as a grade III myxofibrosarcoma, chemotherapy (6 series of EPI-ADM, parallel Lartruvo treatment from the second series) was started (Table 1).
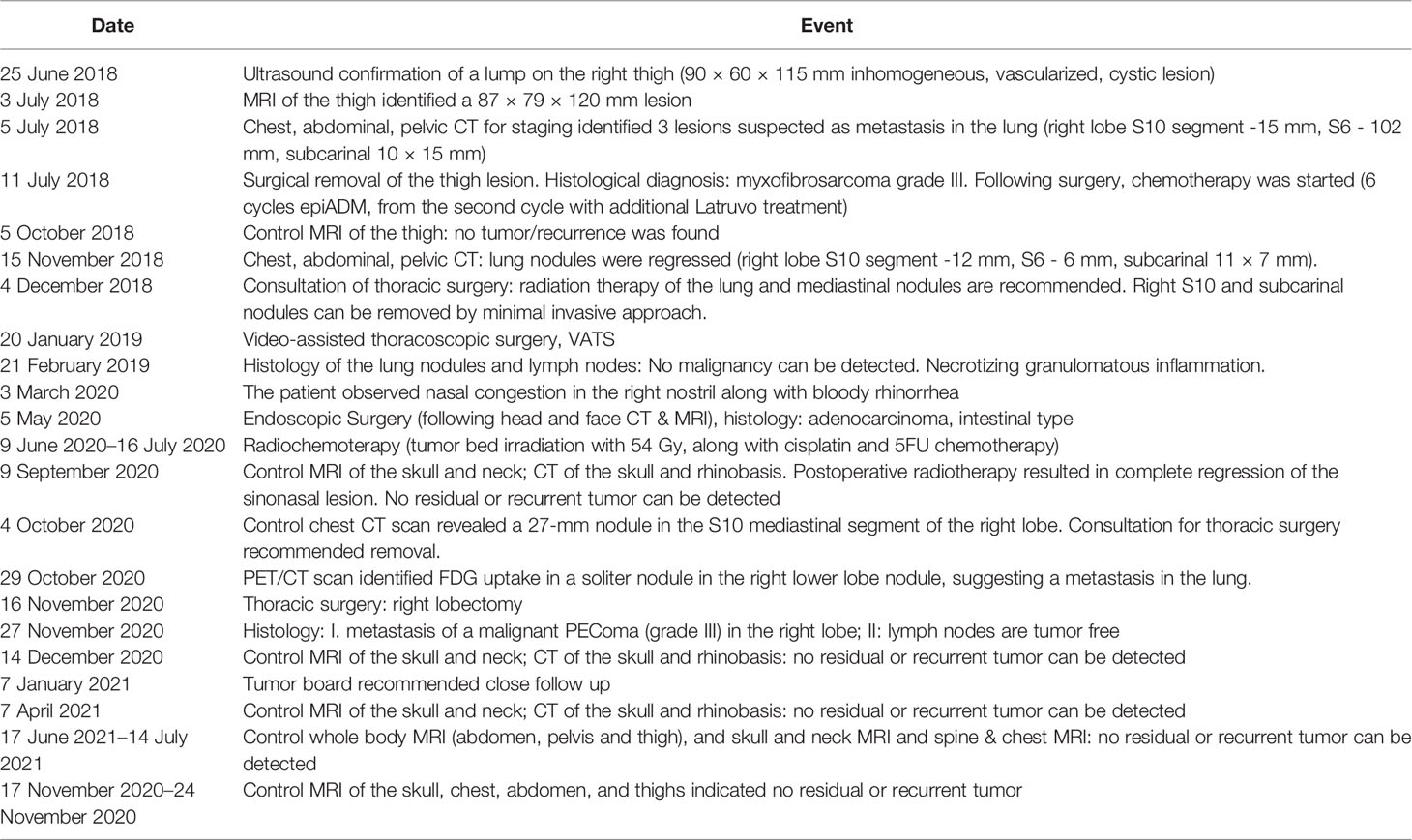
Table 1 Timeline of the patient history.
As the pulmonary nodules moderately regressed following chemotherapy, pulmonary surgery was performed to remove residual right-lobe nodules. Histology showed necrotizing granulomatous inflammation.
Three months later, the patient observed bloody rhinorrhea. Upon CT scanning, soft-tissue densities were observed in the sinonasal tract. After endoscopic surgery, intestinal-type sinonasal carcinoma showing typical histology and immunophenotype was diagnosed. Postoperative radiotherapy resulted in complete regression of the sinonasal tumor.
Ten months later, a control examination showed a right-lung nodule: therefore, a right lower lobectomy was performed. Pathological investigation revealed a cellular tumor showing a prominent perivascular arrangement. Tumor cells were pleomorphic with epithelioid or spindle-shaped character, and they had clear or abundant granular eosinophilic cytoplasm. Extensive necrotic areas and a very high mitotic rate (77/10HPF) were observed (Figure 1). Upon immunohistochemistry, tumor cells showed diffuse vimentin positivity. In the clear cell areas, tumor cells showed diffuse HMB45 positivity (Figure 1A). Focal but strong HMB45, desmin, H-Caldesmon, and smooth-muscle-actin expression were seen in the spindle cell areas (Figure 1A). Labeling for S100, SOX10, MelanA, cytokeratin (AE1–AE3), EMA, HHF35, CD34, STAT6, H3K27me3, and ERG was negative. Tumor cells were almost completely negative for p53 immunohistochemistry. Only scattered pleomorphic cells showed weak p53 expression. EWSR1 and TFE3 fluorescent in situ hybridization showed no rearrangement of the examined genes. The final diagnosis was metastasis of a malignant PEComa (grade III). In light of the histopathological results of the pulmonary lesion, the histological findings of the thigh tumor, which was originally diagnosed as a myxofibrosarcoma by another institute, were reevaluated by a specialist soft-tissue pathologist. Although the thigh tumor showed focal myxoid areas, probably resulting in the original diagnosis of myxofibrosarcoma, morphologically it was a similar mixture of epithelioid and spindle cells, as seen in the pulmonary lesion. Since only a limited panel of immunohistochemistry was performed at the time of the primary diagnosis, further immunohistochemistry including muscle markers and HMB45 was performed, which showed the same positive reaction as in the lung tumor (Figure 1B). As the thigh tumor showed a similar morphology and immunophenotype, it was reclassified as a primary PEComa, and the pulmonary tumor was considered as its metastasis.
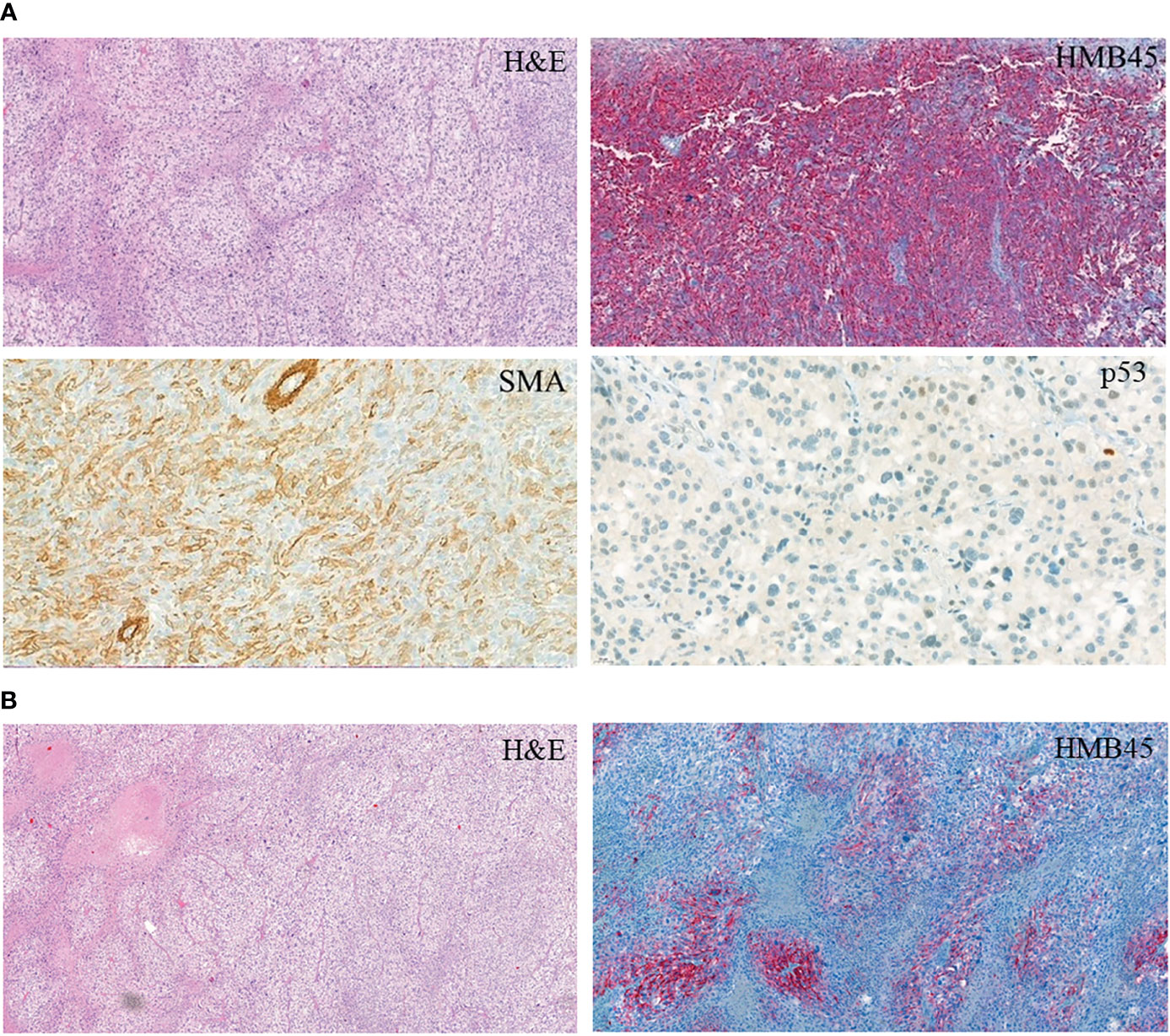
Figure 1 Immunohistochemistry of the PEComa tissue. (A) PEComa metastasis in the lung. (B) Primary tumor.
Following lobectomy, a control CT was negative. Twelve months after the lung surgery, the patient appeared to be tumor free and is under close clinical follow-up (Table 1).
Molecular and Clinical Genetic Findings
We had only a limited amount of tissue from the sinonasal carcinoma, which was unfortunately not sufficient for multigene analysis. Monogenic analysis using COBAS kits showed no evidence of KRAS, NRAS, or BRAF mutation.
Multigene panel (161 genes) sequencing was performed in the malignant PEComa and identified a TP53(NM_000546.5):c.97-2A>C variant with 66.21% allele frequency (variant allele frequency, VAF), but no other therapy-predictive pathogenic variant or gene fusion was detected. As VAF of the TP53 variant suggested a potential germline presence, the patient was referred for genetic consultation and molecular genetic analysis in our department. During the consultation and pedigree analysis, Li–Fraumeni core tumors in the long-since deceased mother of the proband were identified (osteosarcoma at age 14; breast cancer at the age of 33 and ovarian cancer at the age of 35). Based on the available information, no other relative was affected (Figure 2).
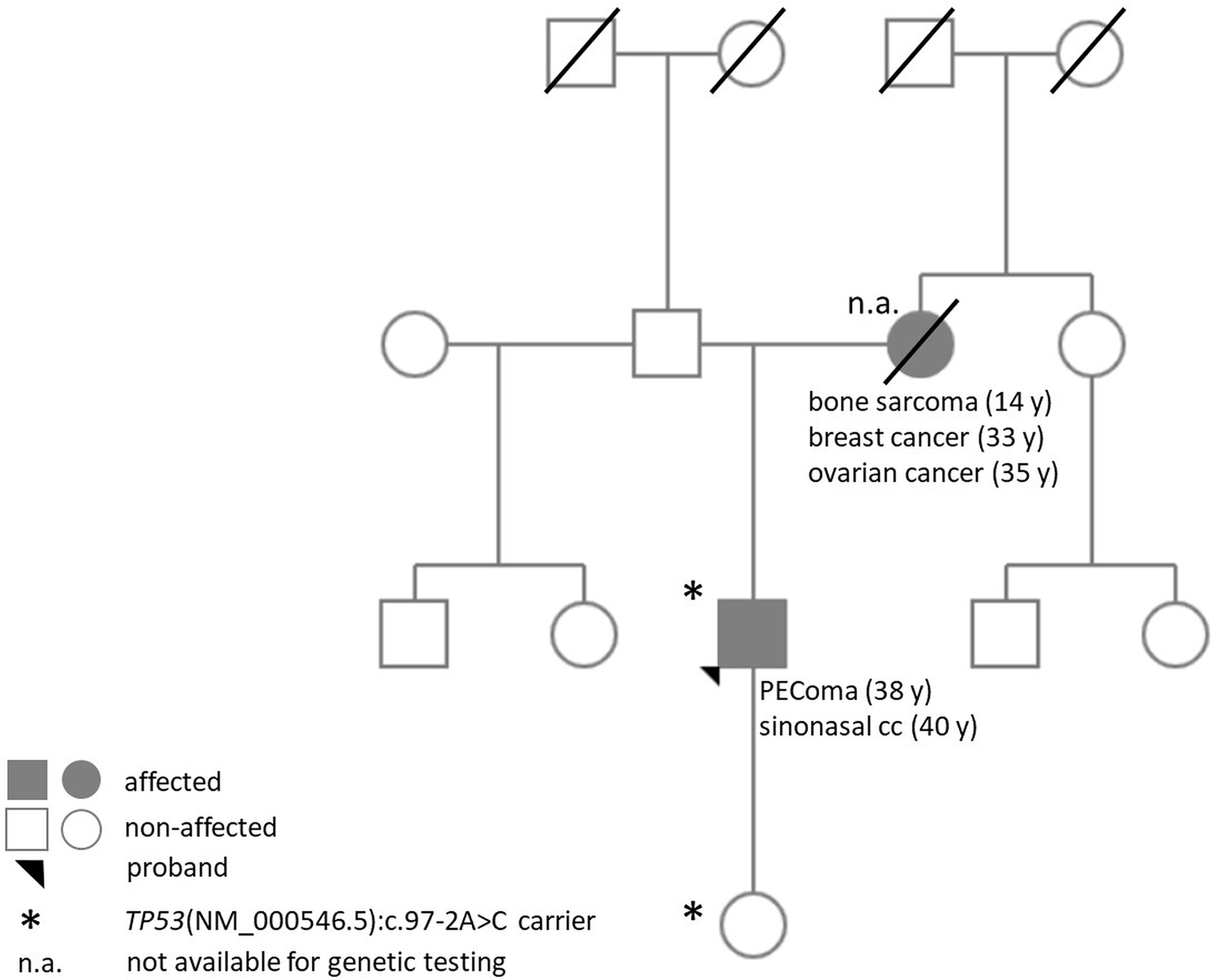
Figure 2 Pedigree of the proband.
We performed targeted Sanger sequencing of the identified variant and proved the germline presence of the TP53(NM_000546.5):c.97-2A>C variant (Figure 3A). Exome sequencing was also performed but was not able to identify other pathogenic variants in the compulsory gene list report or in potential hereditary cancer genes. Comparing the germline and the somatic (tumor types) variants, a partial locus-specific loss of heterozygosity (normalized reduction of the reference allele quantity was 0.3 relative to the variant allele) was observed in the PEComa, whereas no LOH was identified in the sinonasal carcinoma (Figures 3A, B). Additionally, in vitro RNA testing proved whole exon 4 skipping due to the TP53(NM_000546.5):c.97-2A>C variant (Figure 3A).
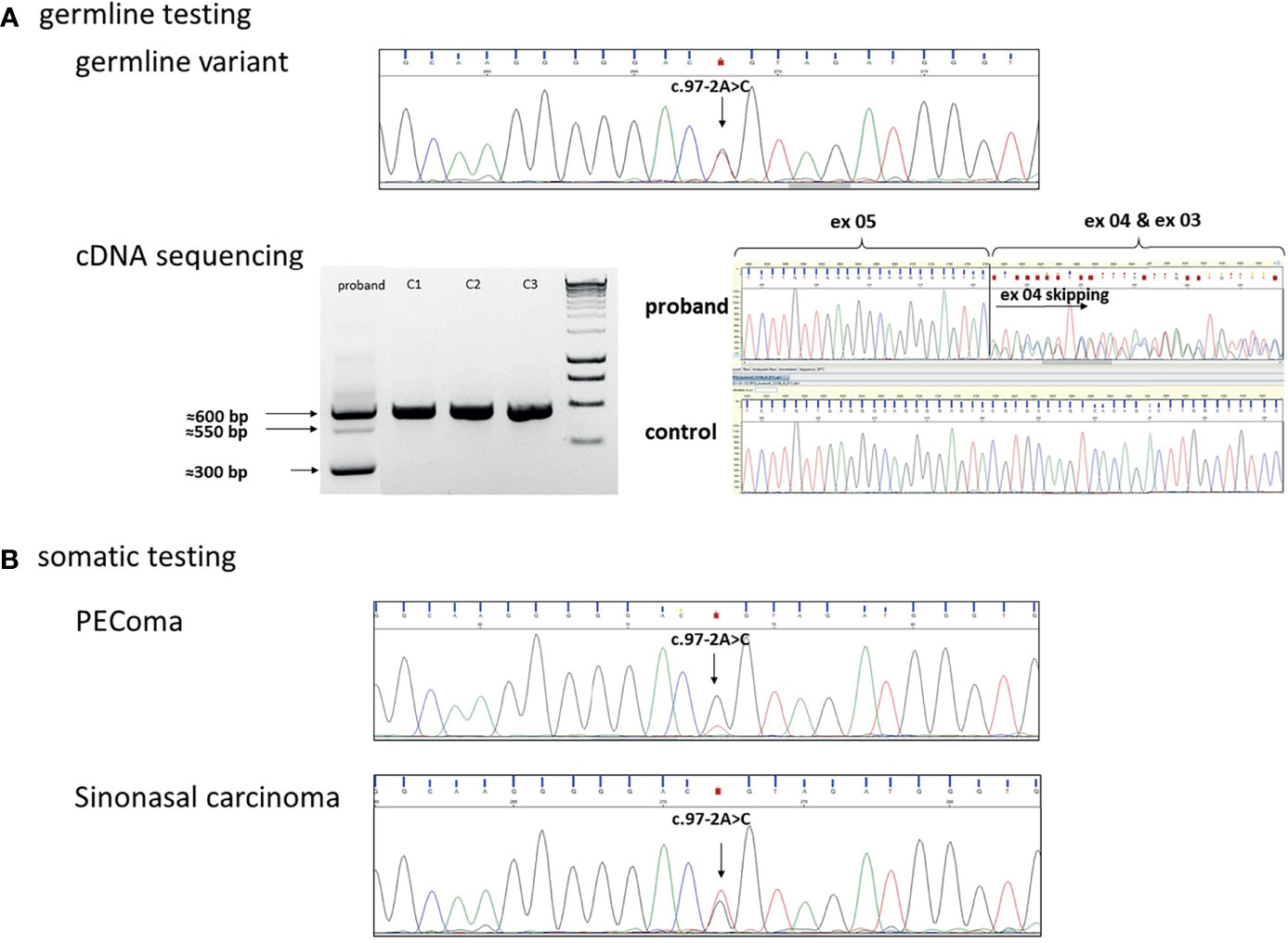
Figure 3 (A) Germline heterozygous TP53(NM_000546.5):c.97-2A>C variant in the DNA isolated from blood. cDNA sequencing identified exon 4 skipping, accordingly. On the electrophoresis gel, the 600-bp PCR product indicates the wild type, the 550-bp PCR product indicates a heteroduplex, and the 300-bp PCR product was confirmed as a skipped exon 4 transcript following Sanger sequencing. (C1, C2, C3 were used as controls). (B) Sanger sequencing in the PEComa tissue sample indicated the loss of the wild-type (wt) allele in the tumor: loss of heterozygosity (LOH) was detected. In sinonasal carcinoma, wild-type and variant alleles are presented in ~50%–50%: no LOH was detected.
While this exon skipping does not lead to a frame shift, it results in a loss of 93 amino acids (from amino acids 32 to 125) at the protein level. Based on protein modeling, the variant protein was predicted to have a different 3D structure (Figure 4). Additionally, 40.8% of the lost amino acids were predicted as “disordered” in the 3D structure, meaning that the change or loss might lead to damaged protein structure/function. This was also in line with the p53 immunohistological finding.
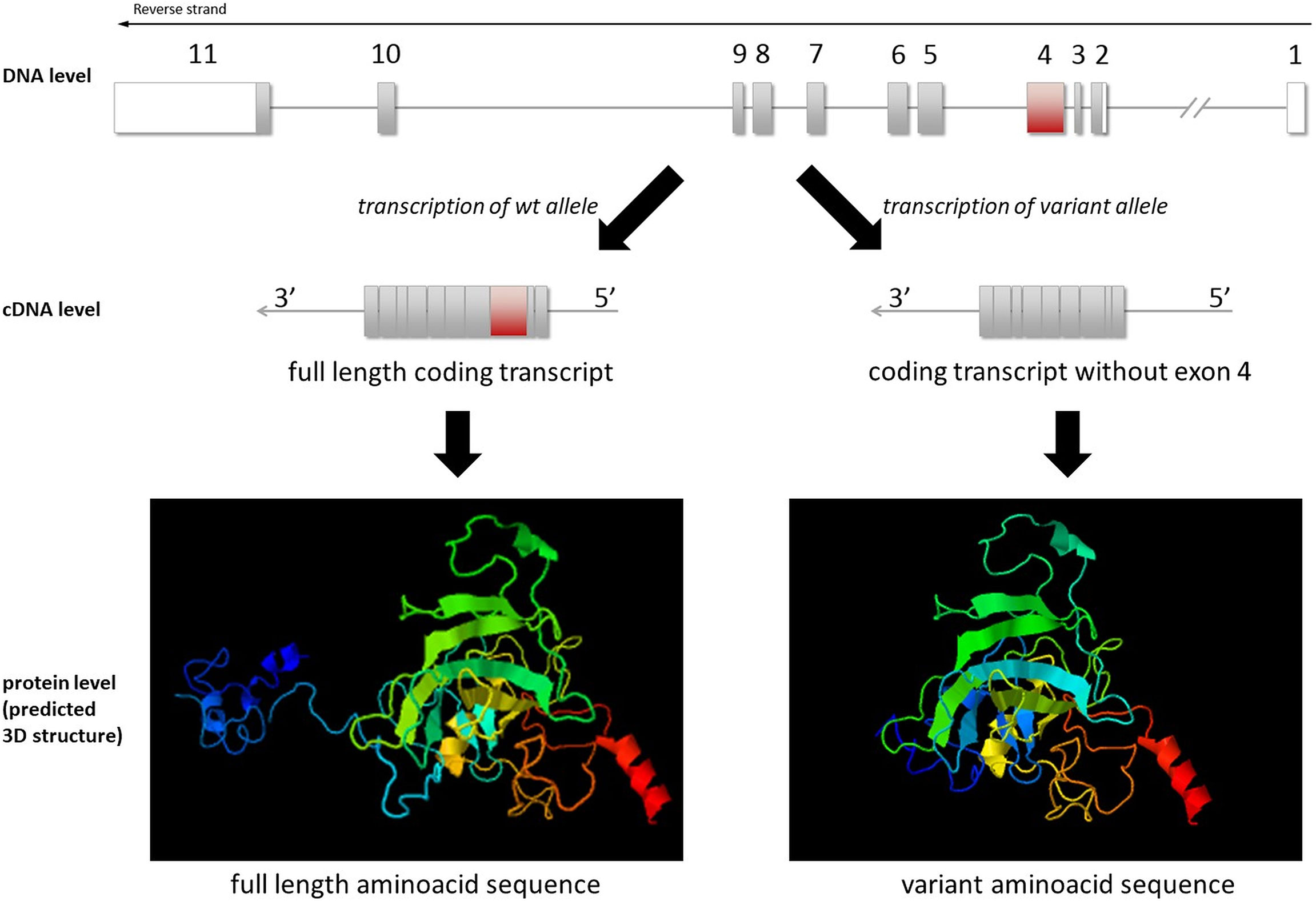
Figure 4 Illustration of TP53(NM_000546.5) exon 4 skipping at the DNA, cDNA, and protein levels. 3D modeling of the wild-type and variant protein indicated different structures (visualized by JSmol).
Based on the molecular and clinical findings, the TP53-specific ACMG classification of TP53(NM_000546.5):c.97-2A>C in this proband is Class 5, “pathogenic,” because it affects a splice site (PVS1_strong), is not found in gnomAD exomes or genomes (PM2_supporting), and matches computational predictions (PP3_mooderate). Furthermore, our additional evidence supports its pathogenicity: i) exon-skipping using an in vitro functional test and ii) the negative TP53 immunohistochemistry on the tumor tissue.
Discussion
We identified the TP53(NM_000546.5):c.97-2A>C variant as a novel, germline pathogenic alteration in the background a thigh-muscle PEComa. This variant, to the best of our knowledge, has not been previously reported in the literature. In the ClinVar database, a single submitter reported a different variant at the same localization (accession: VCV000246337.1; NM_000546.5:c.97-2A>G), but the molecular in vitro characterization of this variant has not been performed. Additionally, another variant affecting the same splice site at a different localization, NM_000546.5:c.97-1G>A, was identified in a patient meeting Chompret criteria and was found to cause abnormal splicing upon functional assay analysis (21, 22). We proved that the newly identified NM_000546.5:c.97-2A>C variant led to exon 4 skipping, potentially resulting in a different p53 protein structure that would be predicted to have decreased stability. This is supported by p53 immunohistochemistry, where tumor cells were predominantly negative, and only scattered, focal positivity could be seen in pleomorphic cells.
PEComas (perivascular epithelioid cell tumors) are rare, mesenchymal tumors of uncertain malignant potential, as recurrences may occur years after the initial diagnosis. Malignant metastasizing PEComas are very rare (23). The differential diagnosis can include carcinomas, smooth muscle tumors, and adipocytic neoplasms (23). Our case (first diagnosed as myxofibrosarcoma of the muscle) highlights the difficulties in the pathological diagnosis of malignant PEComa. Regarding PEComa pathogenesis, alterations in two, or recently three, main pathways have been described. Most commonly, a loss of function in the tuberous sclerosis complex subunit 1, TSC1 (~27%) or TSC2 (~73%), has been observed due to deletion or pathogenic missense variants, leading to activated mTOR signaling and increased cell growth (23, 24). TSC1 or TSC2 inactivation can appear somatically, or in individuals already harboring a germline TSC1/2 mutation. In both cases, mTOR inhibition can be a potential therapeutic option. The other main molecular feature behind PEComa pathogenesis (in approximately 23% of cases) is the rearrangement affecting TFE3 (transcription factor binding to IGHM enhancer 3), which is implicated in cell differentiation (23, 24). This has a significant clinical importance, as these tumors might be non-responsive to mTOR inhibition. Lately, rearrangements of RAD51B in uterine PEComas have also been identified (23). Similar to other tumors, somatic TP53 mutations have been described in PEComas, and they are potentially linked to malignancy (24–27).
Most PEComas are sporadic, and only a small subset is associated with the hereditary condition TSC. Recently, our group also identified a PTCH1 mutation in a patient with bilateral intra-abdominal PEComas suffering from Gorlin-Goltz syndrome (16). PEComas have been reported in only two Li–Fraumeni cases in the literature to date (14, 15). Contrary to these two examples, in our case the PEComa was the first manifestation in the LFS proband. In our case, we could not detect TSC1 or TSC2 sequence- or copy-number variants, either in the germline or somatically. The causative role of RAD51B was also excluded by multigene panel sequencing, copy number analysis, and fusion analysis. We did not detect TFE3 rearrangement by FISH analysis, which further reduces the likelihood of a causative role of the TFE3 pathway in the pathogenesis. However, we identified a site-specific LOH in the PEComa tissue regarding the novel TP53 pathogenic variant. The normal allele was lost in favor of the non-functional allele harboring the pathogenic variant, and this was supported by the immunohistochemical findings. This suggested a role for the defective TP53 pathway in the PEComa pathogenesis, which is also reported to be associated with the malignant, metastatic form of this tumor type in this patient.
While a sinonasal carcinoma can be part of the Li–Fraumeni spectrum, we were not able to identify the second hit affecting TP53 that causes the tumor development.
As TP53 pathogenic variants contribute to cancer proliferation and metastasis, targeting the signaling pathways that become altered by p53 mutation seems to be an attractive strategy (28). Whereas in the clinical practice there is currently no such drug available, several agents are under investigation in clinical trials (28). The prognostic and predictive role of TP53 pathogenic variants has been intensively investigated and reported in somatic settings (29). Currently, there are no special recommendations for treatment of the Li–Fraumeni spectrum; indeed, there are reports of treatment (chemo- and radiotherapy) failure (30). While the primary goal is always the treatment of the actual malignant disease, the radiation (both diagnostic and therapeutic) exposure should be minimized, as subsequent primary tumors, particularly within the radiotherapy field, often develop after the exposure (3). Therefore, avoiding radiotherapy when possible and instead using preferably non-genotoxic chemotherapies are recommended by recent guidelines (3).
The genetic counseling of patients carrying pathogenic TP53 variants is essential. Following international and national guidelines, the patients have to be informed of the disease, the risk of tumor development and localization, the potential options related to surveillance, and the screening of first-degree or at-risk relatives (3, 9). Accordingly, pre- and posttest genetic counseling and family screening were performed in our PEComa patient.
Conclusion
We identified a novel TP53 splice variant in an attenuated LFS patient manifesting with a malignant PEComa of unusual appearance. This rare, unexpected phenotype of the patient highlights the importance of the introduction of the Li–Fraumeni spectrum instead of the classic LFS concept. Additionally, using complex molecular genetic assays, we demonstrated the pathogenic role of a novel TP53 germline variant in the development of the PEComa. This may help with the interpretation of this variant in other patients identified in the future.
Data Availability Statement
The original datasets presented in this study are available in a publicly accessible repository: NCBI SRA database under accession number PRJNA815946.
Ethics Statement
The studies involving human participants were reviewed and approved by the Scientific and Research Committee of the Medical Research Council of the Ministry of Health, Hungary (ETT-TUKEB 53720-4/2019/EÜIG). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
Conceptualization, JL, HB, and AP. Germline genetic tests, AB, HB. Regular and molecular pathology tests, MS, ET. Genetic interpretation, HB. Writing—original draft preparation, HB. Writing—review and editing, AP, JL. Visualization, HB. Supervision, AP and JL. Project administration, HB. Funding acquisition, AP and HB. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Laboratories Excellence program (under the National Tumor biology Laboratory project (NLP-17) and the Hungarian Scientific Research Grant of National Research, Development and Innovation Office (NKFI FK 135065 to HB). HB is a recipient of Bolyai Research Fellowship of the Hungarian Academy of Sciences.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful for the proband agreeing to participate in this study. He read the manuscript and agreed to this submission, and we received his answer on January 2, 2022. The patient is regularly assessed by JL and is currently symptom-free. The patient is also in contact with the Hungarian Li-Fraumeni Advocacy Group.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.849004/full#supplementary-material
Supplementary Table | Germline variants detected by exome sequencing (mean allele frequency (MAF) <0.01). All data is available at GeneBank under Submission # 2537264.
References
1. Li FP. Soft-Tissue Sarcomas, Breast Cancer, and Other Neoplasms: A Familial Syndrome? Ann Intern Med (1969) 71:747. doi: 10.7326/0003-4819-71-4-747
2. Schneider K, Zelley K, Nichols KE, Garber J. Li-Fraumeni Syndrome (1993). University of Washington, Seattle: GeneReviews®. Seattle (WA. Available at: http://www.ncbi.nlm.nih.gov/books/NBK1311/ (Accessed December 18, 2021).
3. The European Reference Network GENTURIS, Frebourg T, Bajalica Lagercrantz S, Oliveira C, Magenheim R, Evans DG. Guidelines for the Li–Fraumeni and Heritable TP53-Related Cancer Syndromes. Eur J Hum Genet (2020) 28:1379–86. doi: 10.1038/s41431-020-0638-4
4. Kratz CP, Freycon C, Maxwell KN, Nichols KE, Schiffman JD, Evans DG, et al. Analysis of the Li-Fraumeni Spectrum Based on an International Germline TP53 Variant Data Set: An International Agency for Research on Cancer TP53 Database Analysis. JAMA Oncol (2021) 7:1800–5. doi: 10.1001/jamaoncol.2021.4398
5. Chompret A, Brugières L, Ronsin M, Gardes M, Dessarps-Freichey F, Abel A, et al. P53 Germline Mutations in Childhood Cancers and Cancer Risk for Carrier Individuals. Br J Cancer (2000) 82:1932–7. doi: 10.1054/bjoc.2000.1167
6. Mai PL, Best AF, Peters JA, DeCastro RM, Khincha PP, Loud JT, et al. Risks of First and Subsequent Cancers Among TP53 Mutation Carriers in the National Cancer Institute Li-Fraumeni Syndrome Cohort. Cancer (2016) 122:3673–81. doi: 10.1002/cncr.30248
7. Amadou A, Waddington Achatz MI, Hainaut P. Revisiting Tumor Patterns and Penetrance in Germline TP53 Mutation Carriers: Temporal Phases of Li–Fraumeni Syndrome. Curr Opin Oncol (2018) 30:23–9. doi: 10.1097/CCO.0000000000000423
8. de Andrade KC, Frone MN, Wegman-Ostrosky T, Khincha PP, Kim J, Amadou A, et al. Variable Population Prevalence Estimates of Germline TP53 Variants: A gnomAD-Based Analysis. Hum Mutat (2019) 40:97–105. doi: 10.1002/humu.23673
9. Andrade KC de, Khincha PP, Hatton JN, Frone MN, Wegman-Ostrosky T, Mai PL, et al. Cancer Incidence, Patterns, and Genotype–Phenotype Associations in Individuals With Pathogenic or Likely Pathogenic Germline TP53 Variants: An Observational Cohort Study. Lancet Oncol (2021) 22:1787–98. doi: 10.1016/S1470-2045(21)00580-5
10. El-Deiry WS, Goldberg RM, Lenz H, Shields AF, Gibney GT, Tan AR, et al. The Current State of Molecular Testing in the Treatment of Patients With Solid Tumors, 2019. CA A Cancer J Clin (2019) 69:305–43. doi: 10.3322/caac.21560
11. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational Landscape and Significance Across 12 Major Cancer Types. Nature (2013) 502:333–9. doi: 10.1038/nature12634
12. Leroy B, Fournier JL, Ishioka C, Monti P, Inga A, Fronza G, et al. The TP53 Website: An Integrative Resource Centre for the TP53 Mutation Database and TP53 Mutant Analysis. Nucleic Acids Res (2013) 41:D962–9. doi: 10.1093/nar/gks1033
13. Hainaut P, Pfeifer GP. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb Perspect Med (2016) 6:a026179. doi: 10.1101/cshperspect.a026179
14. Galera López M del M, Márquez Rodas I, Agra Pujol C, García Pérez Á, Velasco Sánchez E, Álvarez Álvarez R. Simultaneous Diagnosis of Liver PEComa in a Family With Known Li–Fraumeni Syndrome: A Case Report. Clin Sarcoma Res (2020) 10:24. doi: 10.1186/s13569-020-00143-7
15. Neofytou K, Famularo S, Khan AZ. PEComa in a Young Patient With Known Li-Fraumeni Syndrome. Case Rep Med (2015) 2015:1–4. doi: 10.1155/2015/906981
16. Igaz P, Toth G, Nagy P, Dezső K, Turai PI, Medvecz M, et al. Surprising Genetic and Pathological Findings in a Patient With Giant Bilateral Periadrenal Tumours: PEComas and Mutations of PTCH1 in Gorlin-Goltz Syndrome. J Med Genet (2021) 16:jmedgenet-2021-108082. doi: 10.1136/jmedgenet-2021-108082
17. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
18. Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat Protoc (2015) 10:845–58. doi: 10.1038/nprot.2015.053
19. Fortuno C, Lee K, Olivier M, Pesaran T, Mai PL, de Andrade KC, et al. Specifications of the ACMG/AMP Variant Interpretation Guidelines for Germline TP53 Variants. Hum Mutat (2021) 42:223–36. doi: 10.1002/humu.24152
20. Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, et al. Recommendations for Interpreting the Loss of Function PVS1 ACMG/AMP Variant Criterion. Hum Mutat (2018) 39:1517–24. doi: 10.1002/humu.23626
21. Varley JM, Chapman P, McGown G, Thorncroft M, White GRM, Greaves MJ, et al. Genetic and Functional Studies of a Germline TP53 Splicing Mutation in a Li–Fraumeni-Like Family. Oncogene (1998) 16:3291–8. doi: 10.1038/sj.onc.1201878
22. Varley JM, McGown G, Thorncroft M, White GR, Tricker KJ, Kelsey AM, et al. A Novel TP53 Splicing Mutation in a Li-Fraumeni Syndrome Family: A Patient With Wilms’ Tumour is Not a Mutation Carrier. Br J Cancer (1998) 78:1081–3. doi: 10.1038/bjc.1998.631
23. Thway K, Fisher C. PEComa: Morphology and Genetics of a Complex Tumor Family. Ann Diagn Pathol (2015) 19:359–68. doi: 10.1016/j.anndiagpath.2015.06.003
24. Agaram NP, Sung Y-S, Zhang L, Chen C-L, Chen H-W, Singer S, et al. Dichotomy of Genetic Abnormalities in PEComas With Therapeutic Implications. Am J Surg Pathol (2015) 39:813–25. doi: 10.1097/PAS.0000000000000389
25. Bing Z, Yao Y, Pasha T, Tomaszewski JE, Zhang PJ. P53 in Pure Epithelioid PEComa: An Immunohistochemistry Study and Gene Mutation Analysis. Int J Surg Pathol (2012) 20:113–20. doi: 10.1177/1066896912441829
26. Ma L, Kowalski D, Javed K, Hui P. Atypical Angiomyolipoma of Kidney in a Patient With Tuberous Sclerosis: A Case Report With P53 Gene Mutation Analysis. Arch Pathol Lab Med (2005) 129:676–9. doi: 10.5858/2005-129-0676-AAOKIA
27. Kawaguchi K, Oda Y, Nakanishi K, Saito T, Tamiya S, Nakahara K, et al. Malignant Transformation of Renal Angiomyolipoma: A Case Report. Am J Surg Pathol (2002) 26:523–9. doi: 10.1097/00000478-200204000-00017
28. Hu J, Cao J, Topatana W, Juengpanich S, Li S, Zhang B, et al. Targeting Mutant P53 for Cancer Therapy: Direct and Indirect Strategies. J Hematol Oncol (2021) 14:157. doi: 10.1186/s13045-021-01169-0
29. Huszno J, Grzybowska E. TP53 Mutations and SNPs as Prognostic and Predictive Factors in Patients With Breast Cancer. Oncol Lett (2018) 16:34–40. doi: 10.3892/ol.2018.8627
Keywords: Li–Fraumeni syndrome, heritable TP53-related cancer syndrome, TP53, p53, PEComa, germline mutation, Li-Fraumeni, Li-Fraumeni spectrum
Citation: Butz H, Lövey J, Szentkereszty M, Bozsik A, Tóth E and Patócs A (2022) Case Report: A Novel Pathomechanism in PEComa by the Loss of Heterozygosity of TP53. Front. Oncol. 12:849004. doi: 10.3389/fonc.2022.849004
Received: 05 January 2022; Accepted: 25 February 2022;
Published: 28 March 2022.
Edited by:
Arezou Ghazani, Brigham and Women’s Hospital and Harvard Medical School, United StatesReviewed by:
Kelvin Cesar De Andrade, National Cancer Institute (NIH), United StatesGilda Alves Brown, Rio de Janeiro State University, Brazil
Copyright © 2022 Butz, Lövey, Szentkereszty, Bozsik, Tóth and Patócs. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Attila Patócs, cGF0b2NzLmF0dGlsYUBvbmNvbC5odQ==
†These authors have contributed equally to this work