 Nawar Al Nasrallah
Nawar Al Nasrallah Benjamin M. Wiese
Benjamin M. Wiese Catherine R. Sears
Catherine R. Sears- 1Division of Pulmonary, Critical Care, Sleep and Occupational Medicine, Department of Medicine, Indiana University School of Medicine, Indianapolis, IN, United States
- 2Division of Pulmonary Medicine, Richard L. Roudebush Veterans Affairs Medical Center, Indianapolis, IN, United States
Xeroderma pigmentosum complementation group C (XPC) is a DNA damage recognition protein essential for initiation of global-genomic nucleotide excision repair (GG-NER). Humans carrying germline mutations in the XPC gene exhibit strong susceptibility to skin cancer due to defective removal via GG-NER of genotoxic, solar UV-induced dipyrimidine photoproducts. However, XPC is increasingly recognized as important for protection against non-dermatologic cancers, not only through its role in GG-NER, but also by participating in other DNA repair pathways, in the DNA damage response and in transcriptional regulation. Additionally, XPC expression levels and polymorphisms likely impact development and may serve as predictive and therapeutic biomarkers in a number of these non-dermatologic cancers. Here we review the existing literature, focusing on the role of XPC in non-dermatologic cancer development, progression, and treatment response, and highlight possible future applications of XPC as a prognostic and therapeutic biomarker.
Introduction
Genomic instability from altered DNA repair processes is a hallmark of cancer, playing an important role in both tumor development and treatment response (1). Importantly, the therapeutic efficacy of many chemotherapy drugs and radiation relies on the induction of DNA damage as a means of selectively eliminating rapidly proliferating tumor cells. (2).
Daily DNA damage comes from a variety of different sources exogenous to the cell, such as ultraviolet (UV) light, tobacco smoking, and other chemicals, as well as endogenous sources such as oxidative stress caused by normal cellular metabolism (3). The nucleotide excision repair (NER) pathway is the primary DNA repair pathway involved in repair of bulky, helix distorting intrastrand DNA crosslinks caused by UV or platinum chemotherapeutics, as well as bulky monoadducts induced by environmental carcinogens including B[a]P-7,8-dihydrodiol-9,10-epoxide (BPDE) and aflatoxin B1 (AFB1). Much of our understanding of NER comes from studying the repair of UV-induced lesions, such as pyrimidine-pyrimidone (6-4) photoproduct (6–4PPs) and cyclobutane pyrimidine dimers (CPDs), for which NER serves as the primary repair pathway (4). Critical to its role in cancer therapeutic response, NER is the primary repair pathway for 1,2-d(GpG) and 1,3-d(GpTpG) intrastrand platinum crosslinking lesions, the predominant DNA adducts produced by the commonly used chemotherapeutic drugs cisplatin and carboplatin (5). The NER pathway consists of 4 essential steps: recognition, incision/excision, re-synthesis, and ligation (2). Differing in the mechanism of DNA damage recognition, NER is divided into two subpathways: global genomic NER (GG-NER) and transcription-coupled NER (TC-NER). Both NER subpathways repair helix-destabilizing DNA lesions, with TC-NER rapidly repairing damage in actively transcribed genes. TC-NER is initiated when the RNA polymerase II complex is physically stalled at the site of a DNA damaging lesion; this subsequently triggers recruitment of CSB and coordinated recruitment of other TC-NER recognition proteins including CSA, XAB2, UVSSA, USP7 and others (6, 7). Initiated by the xeroderma pigmentosum group C (XPC) complex, GG-NER recognizes helix-distorting lesions anywhere throughout the genome but is primarily responsible for the slower repair of damage on non-transcribed portions (8, 9). Following damage recognition, subsequent NER repair then progresses identically between both NER subpathways. XPC is critical to damage recognition and initiation of GG-NER, but dispensable for TC-NER (9).
There is a clear and established association between defective NER and tumor development, as illustrated by the rare autosomal recessive congenital syndrome xeroderma pigmentosum (XP). XP patients are characterized by defective nucleotide excision repair (NER) of sunlight-induced dipyrimidine photoproducts (10). Depending on the mutated NER protein, XP patients present with a spectrum of disease, which consists of various neurological degenerative disorders and even developmental defects, but all XP patients present with extreme photosensitivity and a strong predisposition to skin cancer (10, 11). Those with a mutation in XPC (XP-C), a common cause of XP in Europe, the United States and North Africa, present with classical XP skin manifestations, including photosensitivity and early dermatologic malignancies, without neurological or developmental defects (11, 12). Indeed, both non-melanomatous skin cancers and melanomas develop more often (10,000 and 2,000-fold increased incidence) and at a much younger age in XP compared to non-XP populations, with a median age at diagnosis of 9 and 22 years respectively (13). Importantly, although XP patients most commonly die of skin cancers or of progressive neurologic diseases, internal malignancies are frequently described in XP patients, with a 39-year prospective cohort study finding internal cancers as the cause of death in 17%, highlighting an important role of NER in non-dermatologic malignancies as well (13, 14).
XPC is increasingly recognized as an important player in solid organ cancer development and response to cancer therapeutics, both through its canonical role in GG-NER and through other repair pathways. Here we review the most recent updates on the role of XPC in non-dermatologic malignancies.
XPC Role in DNA Repair
XPC in GG-NER
XPC is essential to GG-NER, serving as the primary initiating factor. XPC scans the genome in a 5’-to-3’ directionality until it detects strand distortion caused by DNA damaging lesions, binding the opposite strand in a sequence-independent manner (8, 15, 16). The XPC protein in vivo is found in a heterodimeric form with RAD23B (human orthologue HR23B) which further stimulates XPC’s role in NER repair (17). Centrin2 forms a heterotrimer with XPC/HR23B, which has been found to augment binding to DNA damage sites (18). While the XPC complex is typically sufficient to identify NER-repaired DNA lesions, some minimally strand-distorting lesions, such as UV-induced CPD, require recognition by DDB2 and DDB1, which then recruit XPC to the damage site (7).
After the initial recognition of a helix distorting lesion by either XPC or RNA polymerase II, NER proceeds in a stepwise sequence that involves recruitment of several proteins. Transcription factor IIH complex (TFIIH) partially unwinds the DNA duplex at the site of DNA damage, creating an opened bubble (16, 19). TFIIH further coordinates repair by interacting with XPA, stabilizing the bubble along with the single stranded binding protein RPA, and finally engaging with the nuclease (XPF/ERCC1) that makes an incision 5’ of the lesion. Subsequent repair involves coordination of repair synthesis by DNA polymerases δ, ϵ or κ, subsequent incision 3’ of the DNA lesion by XPG to remove the damaged strand, and finally repair of the nick by DNA ligases. Several excellent reviews are available which expand upon and provide excellent graphical representation of the steps involved in NER (7, 16, 20).
A number of recent studies highlight that post-translational modifications of XPC, including polyubiquitination, SUMOylation and phosphorylation, likely impact XPC efficiency to detect DNA damage and initiate NER (21–25). Polyubiquitination of XPC appears to aid in repair of UV-damaged DNA, by allowing XPC to replace DDB1/DDB2 proteins and in promoting XPC binding to the site of DNA damage (21, 25). Tight control of XPC ubiquitination is likely required to ensure DNA repair and may be dysregulated in human cancers. For instance, overexpression of ubiquitin ligases, such as Cullin-RING ubiquitin ligase 4 A (CUL4A), is common in cigarette smoke-related lung cancer, and inversely correlates to XPC expression (26). SUMOylation of XPC appears to stabilize the protein, preventing proteasome degradation and enhancing GG-NER in the setting of UV-induced DNA damage (22). XPC phosphorylation is closely regulated after DNA damage, with phosphorylation at serine 982 likely mediated by the DNA damage response proteins ATM and ATR, and dephosphorylation mediated by wild-type p53-induced phosphatase 1 (WIP1) (27, 28). Following UVB exposure, serine/threonine casein kinase 2 (CK2) phosphorylates XPC at serine 94, leading to recruitment of ubiquitinated XPC and downstream NER factors to DNA damage sites (24). Phosphorylation of XPC at serine 892 seems to decrease repair of UVB-induced DNA damage, including CPD and 6-4PP, while serine 94 phosphorylation promotes GG-NER repair (24). However, whether these modifications impact the role of XPC on other repair pathways, or how they affect XPC’s role in repair of DNA damage from other sources, such as cisplatin, is not well-studied. Further, modification of other proteins may impact XPC function. For instance, histone acetylation may decrease NER through attenuated XPC interaction at sites of DNA damage (23). These modifications, which regulate XPC function in GG-NER repair and the downstream DNA damage response, are likely to impact cancer risk and response to therapy, although this specific link requires more study.
XPC in Other DNA Repair Processes
It is important to note the mounting evidence highlighting an important link between the role of XPC in DNA repair, DNA damage response and transcriptional regulation and cancer development. These are summarized in Figure 1. In particular, the impact of XPC DNA damage repair extends beyond its canonical role in GG-NER. XPC may play a role as a more global DNA damage sensor. Recent in vitro studies have elucidated a role of Rad4, the yeast homolog of XPC, in the recognition and repair of multiple contiguous mismatched base pairs (29). Specifically, in vitro binding and conformational studies suggest that Rad4/XPC interacts with the nucleotides directly across from the mismatched bases (on the complementary strand), leading to subsequent unwinding, DNA bending, and flipping out of the mismatched nucleotides and stabilization of this conformation to allow for subsequent DNA repair (29, 30). These studies suggest a mechanism by which XPC acts as a universal DNA damage sensor, recognizing sites of DNA distortion and binding in a lesion-agnostic fashion (“non-specific binding”). Indeed, recent studies suggest that the Rad4/XPC-DNA binding leads to different conformational changes based on the lesion type, such that XPC bound at the site of UV-induced DNA damage (“specific binding”) facilitates recruitment and initiation of NER while “non-specific” binding to minimally strand-distorting lesions facilitates non-NER repair (29, 30). Extensive structural analysis has been done to understand sequence and structural changes of DNA lesions sensitive and resistant to Rad4/XPC binding and subsequent GG-NER efficiency (31).
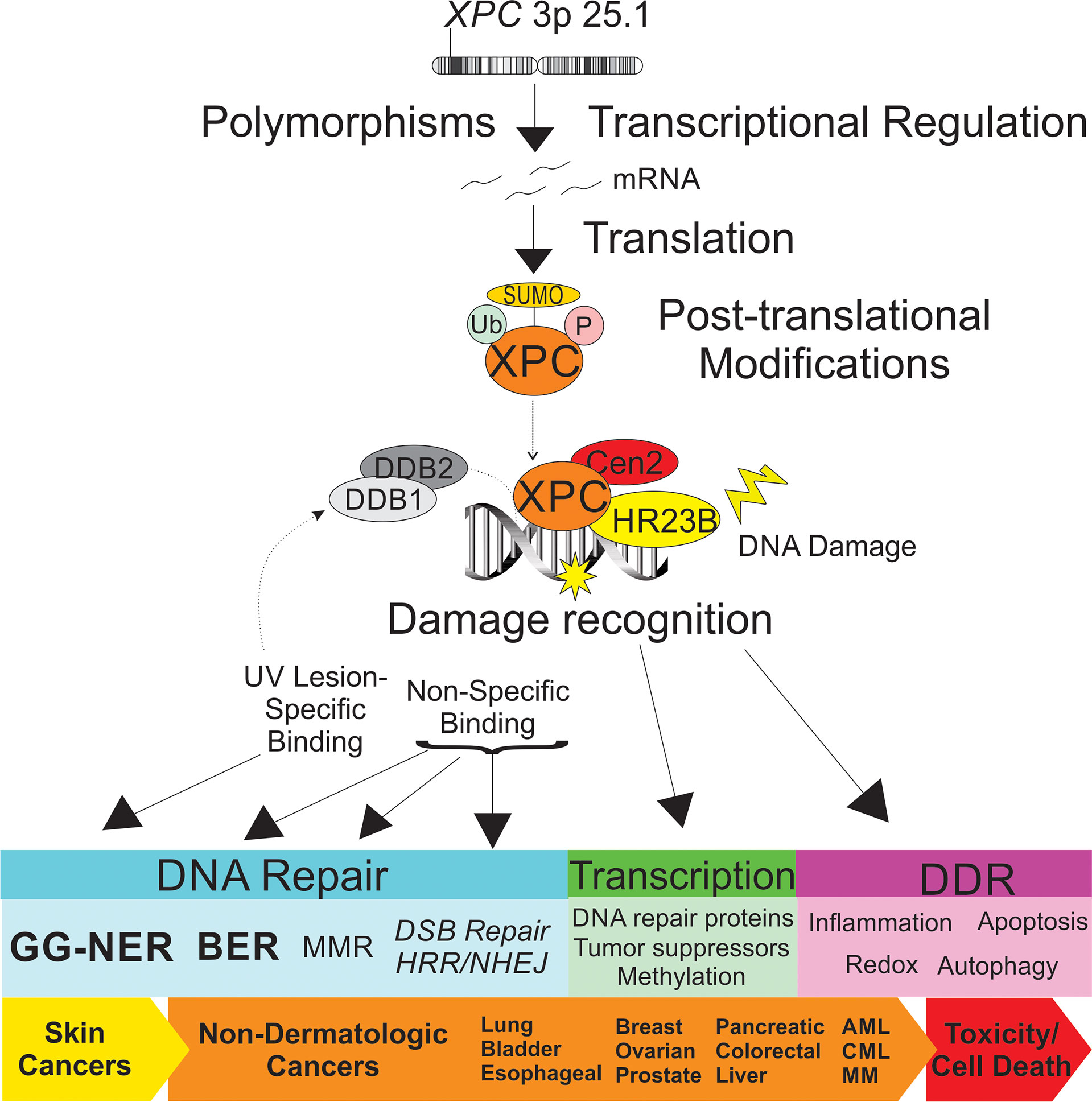
Figure 1 Schematic representation of the impact of XPC in dermatologic and non-dermatologic malignancies. Both XPC mutations and transcriptional regulation of XPC expression levels are described as impacting risk of the cancer development and response to treatment. Post-translational modifications of XPC include ubiquitination, SUMOylation and phosphorylation, which impact XPC expression levels and XPC function. XPC is a versatile DNA damage sensor, leading to differing binding affinities and DNA-XPC conformational changes for UV-induced DNA damage (“specific binding”, in concert with the UV-DDB complex, leading to GG-NER) and other DNA damage (“non-specific binding”, leading to other DNA repair pathways). Differential response of XPC to DNA damage leads to classical GG-NER or alternate DNA repair, altered transcriptional regulation, and DNA damage response ultimately impacting cancer risk and tumor cell toxicity. XPC, xeroderma pigmentosum group c; Ub, ubiquitin; SUMO, small ubiquitin-like modifier; P, phosphorylation site; DDB1, DNA damage-binding 1; DDB2, DNA damage binding 2; Cen2, centrin 2; HR23B, human UV excision repair protein RAD23; GG-NER, global genomic nucleotide excision repair; BER, base excision repair; MMR, mismatch repair; DSB, double strand break; HRR, homologous recombination repair; DDR, DNA damage response; AML, acute myeloid leukemia; CML, chronic myeloid leukemia; MM, multiple myeloma.
Mounting evidence points to a role of XPC in base excision repair (BER). BER is the primary repair mechanism of small, base modifications that do not distort the DNA helical structure. Fibroblasts obtained from XP-C patients displayed increased oxidative DNA damage after UVB-irradiation compared to fibroblasts without an XPC defect. These UV-treated XPC deficient fibroblasts had decreased gene expression of a number of factors involved in BER, including OGG1, MYH, APE1, LIG3, XRCC1, and Polβ, and this correlated with decreased protein expression in three BER-glycosylases: OGG1, MYH, and APE1 (32). Likewise, XPC deficient fibroblast cell lines show lower levels of APE1 and OGG1 mRNA compared to XPC proficient cells, however transiently complementing these cells with XPC only augmented the level and function of OGG1 but not APE1, suggesting a differential impact of XPC on OGG1 glycosylase activity (33). Numerous in vitro studies support a role of XPC in augmenting BER activity, particularly through augmentation of the glycosylase activities of OGG1, SMUG1, 3-methyladenine DNA glycosylase (MPG) and thymine DNA glycosylase (TDG) (34–37). XPC may also augment BER through DNA damage recognition. Interestingly, live cell imaging studies show a rapid recruitment of both cockayne syndrome protein B (CSB, involved in TC-NER) and XPC to the BER-repaired 8-dihydro-8-oxodeoxyguanosine (8-OHdG) DNA lesion, suggesting a role of XPC in early recognition of BER-repaired lesions, even though these do not cause significant strand distortion (38). This may be further explained by the recent finding that DDB2 rapidly localizes to 8-OHdG lesions, preceding and augmenting XPC and subsequent OGG1 recruitment (39). This role of DDB2 in recruiting XPC to minimally helix-distorting lesions is similar to that modeled in GG-NER repair. Interestingly, this recent study suggested a specific role of XPC and DDB2 in augmenting OGG1-mediated BER repair of 8-OHdG lesions in non-transcribed, heavily chromatin-bound genomic regions, which differed from the mechanism observed for repair of 8-OHdG lesions in actively transcribed regions, which ultimately involved recruitment of XPA by OGG1 but was independent of XPC and DDB2 (39). In vivo studies further support a supportive role of XPC in BER. Xpc deficient mice had increased oxidative stress and mutation load over time with treatment with pro-oxidant agents, which was not observed in Xpa deficient and wild type mice (40). However, there was a comparable increase of 8-OHdG lesions by liquid chromatography electrospray tandem mass spectrometry in the uterus of both Xpc deficient and Xpc proficient mice after treatment with equine estrogen, suggesting the effect may be specific to the damaging agent, duration of treatment or tissue-specific (41). Urethane-treated Xpc-/- mice developed an increase in lung adenocarcinomas compared to their wild-type counterparts, but treatment with the anti-oxidant N-acetylcysteine (NAC) decreased tumor development, further supporting a link between XPC, oxidative damage and cancer development (42). Although modified base recognition and augmentation of BER glycosylase and APE1 endonuclease activity have all been proposed, exactly how XPC is involved in BER of oxidized DNA lesions and the subsequent cancer development remain areas of active research.
Mismatched DNA nucleotides, particularly those occurring during replication, are repaired by DNA mismatch repair (MMR). In humans, deficient MMR, through both sporadic and inherited genetic disease, is linked to aging and cancer by promoting genomic instability (43, 44). In particular, defective MMR leads to Lynch syndrome, characterized by a high lifetime risk of colon and other cancer, and MMR defects are associated with ~10-20% of sporadic colon cancers (45, 46). Increasingly, cooperative and possibly overlapping roles of both MMR and NER proteins have been implicated in the recognition and repair of some DNA interstrand crosslinks (ICLs), one of the most cytotoxic types of DNA damage. ICLs are caused by a number of environmental toxins as well as commonly used chemotherapeutic agents, including cisplatin, carboplatin and oxaliplatin commonly used to treat solid-organ tumors (47). Repair of these lesions requires cooperation between different DNA repair pathways, including the Fanconi anemia (FA), NER, homologous recombination repair (HRR) and translesion synthesis (TLS) pathways (47). XPC, along with other NER proteins, were found to be essential for repair of site-specific ICLs caused by psoralen and mitomycin C in vitro using a host-cell reactivation assay (48). Further, both the MMR and NER pathways have been implicated in the repair of triplex-forming oligonucleotide (TFO)-directed psoralen ICLs (Tdp-ICLs) (49–52). Specifically, in MSH2-deficient human cell-free extracts, both binding by the XPC complex and repair of Tdp-ICLs were decreased, further highlighting a cooperative role between NER and MMR ICL repair (53, 54). Additionally, two NER protein complexes, XPC-Rad23B and XPA-RPA can bind psoralen ICLs in cells and in vitro, forming a complex with the MMR complex MutSβ, without which cell toxicity to psoralen increases (55). Further evidence of a connection between XPC and MMR is evidenced in cisplatin-treated XPC deficient cells, in which altered expression was noted in three MMR genes: MLH1, MSH2, and MSH6 (56). Cells deficient in Xpa and Msh2 are less sensitive to UV-induced cellular toxicity compared to Xpa-/- cells with normal Msh2 expression, suggesting a role of MSH2 in the DNA damage response but not necessarily in NER repair of UV-induced DNA damage (57). Finally, combined defects in NER and MMR have been associated with increased UV-induced skin cancers. Combined Xpa and Msh2 deficiencies in mice are associated with an increase in UV-induced skin cancers, and similarly Xpc-/-; Msh2-/- mice developed UV-induced skin cancers earlier than their wild-type counterparts or those deficient in either Xpc or Msh2 alone, suggesting cooperative but non-overlapping roles in UV-induced DNA damage repair (57, 58). An XPC-deficient lymphoblastoid cell line modified by acquired tolerance to the MMR-dependent chemical N-methyl-N-nitrosourea (MNU) exhibited decreased MSH6 expression and MMR efficiency (59). These XPC-deficient, MSH6-low cells effectively repaired UV- and cisplatin-induced lesions by TC-NER, suggesting that the previously observed MMR-NER interactions may rest in interactions with proteins involved in GG-NER, particularly in cancer development. Of interest, the authors of this study noted unusual difficulty in producing MMR deficient variants in two XPC-deficient lymphoblastoid cell lines, further suggesting possible yet still undefined interactions between XPC and MMR functions. Overall, these findings suggest that XPC may cooperate with MMR proteins in the identification and repair of strand-distorting configurations of mismatched nucleotides and ICLs and may serve a role in regulation of the MMR pathway for some types of DNA damage, impacting of mutagenesis.
Additionally, XPC may play a role in DNA double strand break (DSB) repair. Long-term XPC knock-down in HeLa cells was associated with increased sensitivity to the chemotherapeutic drug, etoposide, the cytotoxicity of which is dependent on replication-induced DSB; gamma-irradiation of these cells lead to cell cycle alterations without altered clonogenic survival (60). Furthermore, the increased somatic and germ line mutation rates, as measured by expanded simple tandem repeat (ESTR), were increased in Xpc deficient mice exposed to whole body irradiation (61). More direct evaluation of NHEJ activity in vitro using Manley extracts from XPC knock-down HeLa cells showed a capacity of NHEJ rejoining with linear but not circular DNA (60). XPC deficiency has also been associated with inhibition of BRCA1 expression on bladder cancer cells treated with cisplatin, resulting in accumulation of DNA damage and pointing to a potential indirect role of XPC in homologous recombination or, more likely, replication-induced double strand breaks (62). Overall, this suggests a complex, likely indirect role of XPC in the repair of multiple types of DNA damage.
The impact of XPC in DNA damage is not solely associated with its roles in DNA repair but has been implicated in altered downstream DNA damage response (Figure 1). For instance, at sites of UV-induced DNA damage, XPC attracts and physically interacts with Ataxia telangiectasia- and Rad3- related (ATR) and Ataxia telangiectasia mutated (ATM) proteins, two kinases important in DNA damage- and replication stress-induced checkpoint activations. Both DDB2 and XPC facilitate ATR and ATM phosphorylation and subsequent activation, leading to phosphoactivation of ATR- and ATM- substrates involved in cell cycle regulation (including Chk1 and Chk2) (63). Additionally, XPC facilitates ATR- and ATM- recruitment to sites of DNA damage as well as two proteins, BRCA1 and RAD51, known to be involved in replication and HRR (63). XPC has been implicated in enhancing DNA damage–induced apoptosis through inhibition of caspase-2 transcription (64), and both increased apoptosis and altered autophagy are observed in cells exposed to carcinogenic cigarette smoke and arsenic trioxide in vitro and in vivo (65, 66). Independent of DNA damage, XPC silencing and overexpression in mouse and human embryonic stem cell models support a role of XPC in global DNA demethylation through augmentation of TDG avidity (37, 67). XPC may have an even broader role on transcriptional regulation through coordination with other transcription factors and has been linked with regulation of a number of genes, including tumor suppressor genes, even in the absence of DNA damage (37, 68, 69). XPC involvement in the DNA damage response may also impact cell redox homeostasis and also in local inflammation. For instance, silencing of XPC in arsenic trioxide-treated human glioma cells was associated with decreased anti-oxidant factors and subsequent increase in oxidative damage, including 8-OHdG (65). Melis and colleagues described the glutathione anti-oxidant response as deficient in Xpc-/- mice, and most recently, Mori and colleagues describe a redox imbalance due to compromised mitochondrial function and reduced glutathione peroxidase activity (70, 71). Lung fibroblasts exposed to both the carcinogen BPDE and to the chemotherapeutic drug cisplatin produced higher levels of the pro-inflammatory, tumor promoting cytokine interleukin-6 (IL-6) through the p38-SAPK pathway (72). As the local tumor immune response is increasingly recognized as critical to solid organ cancer development, the role of XPC in local tumor microenvironment, including immune escape, warrants further investigation.
XPC in Hematologic Cancers
The role of XPC in hematopoietic malignancy has been explored over the last several years, both in mouse models and observations in various patient populations. XPC deficient mice (Xpc-/-) have a significantly higher frequency of spontaneous mutations in the hprt gene in splenic T lymphocytes as compared to Xpa-/- and Csb-/- mice; this was also enhanced with aging (73). Similarly, long-term exposure to paraquat in Xpc-/- mice leads to an increase in lymphoid hyperplasia (40). XPC deficient mice had hypocellular bone marrow associated with a 10-fold increased sensitivity to carboplatin and decreased cell and overall mouse survival as compared to wild type mice, suggesting an important role of XPC in hematopoietic cell response to treatment with platinum-containing drugs (74). Importantly, these studies suggest that XPC expression may impact bone marrow suppression and altered hematopoiesis, common treatment-limiting adverse events associated with platinum-based chemotherapeutic agents.
Alterations in DNA repair processes, including those associated with XPC deficiency, have been linked to hematologic malignancies in a human population (75). While overshadowed by the recognition of skin malignancies early after identification of the XP phenotype, early case reports include pediatric and young adult XP-C patients who develop hematologic malignancies (14). More recent studies have shown an increased propensity for hematologic malignancies and sarcomas in populations of individuals with xeroderma pigmentosum deficient in XPC (XP-C). Individuals with XP-C are at an increased risk of leukemia and other hematologic malignancies, as well as alterations in genotoxic effects due to treatment of these cancers (76, 77). Sarasin et al. examined a cohort of 161 patients with XP-C and found that 13 of these individuals developed either overt myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) with a median age of 22 years at diagnosis (Table 1). This finding of MDS/AML was specific for the most common homozygous frameshift XPC mutation delTG (c.1643_1644delTG; p.Val548Ala>fsX25) and has not been observed with an increased frequency in other XP patients (77). Similarly, a cohort of 117 individuals with XP-C were followed from 1971 to 2018 and four patients were found to develop hematologic malignancies, including MDS, acute leukemias and high grade lymphoma (110). More recently, a shared mutational profile was identified by whole genome sequencing in leukemias from six XP-C patients, which differed from the mutational patterns in non-XP-C spontaneous AML samples and corresponded to a pattern described with altered GG-NER (111). Single nucleotide polymorphisms (SNPs) of the XPC gene have been studied in a number of malignancies, many of which may modify disease risk, prognosis or alter treatment response (Figure 2). Of these, several have been studied in leukemias (Table 1). In AML treated with induction chemotherapy, the XPC Ala499Val SNP was associated with lower overall disease-free survival, particularly when combined with an XPD codon 751 AC/CC polymorphism (78), and two XPC SNPs (Ala499Val and Lys939Gln) were associated with variable responses to imatinib in BCR-ABL driven chronic myelogenous leukemia (CML) (79). In regard to tolerating induction chemotherapy or hematopoietic stem cell transplantation in the setting of XPC abnormalities, there is little data.
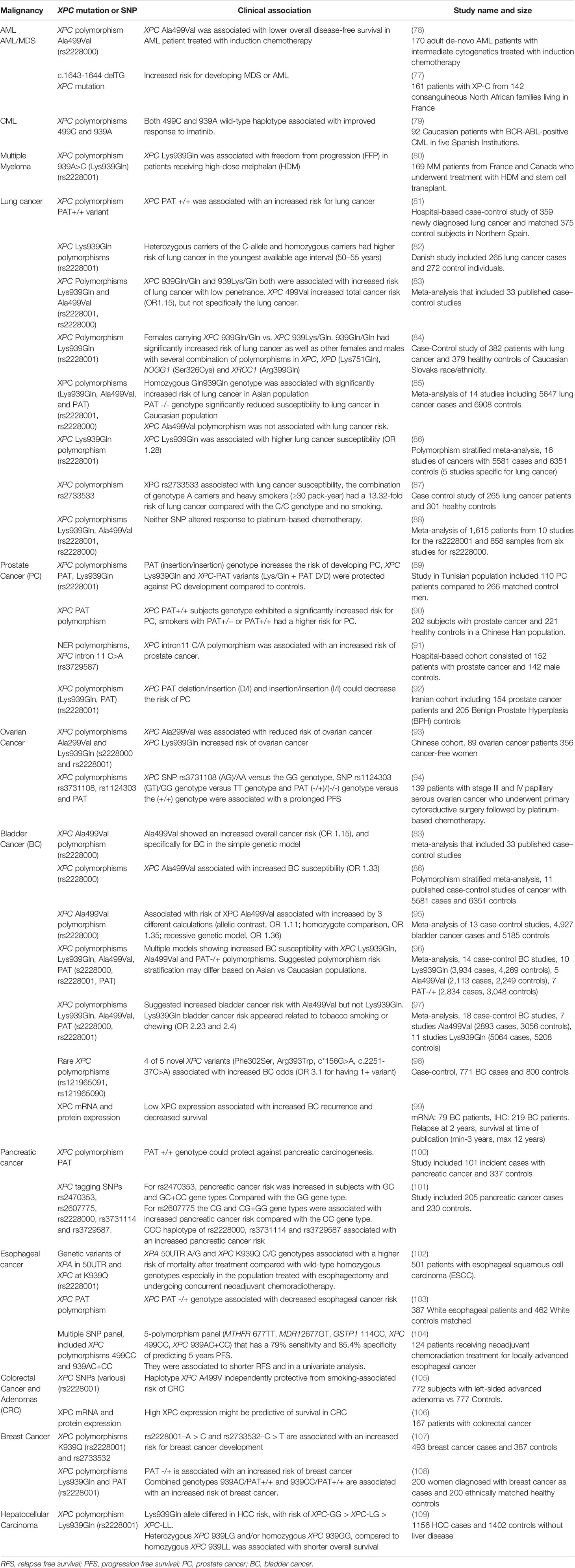
Table 1 Summary of clinical studies evaluating XPC polymorphisms and epigenetic alterations by malignancy.
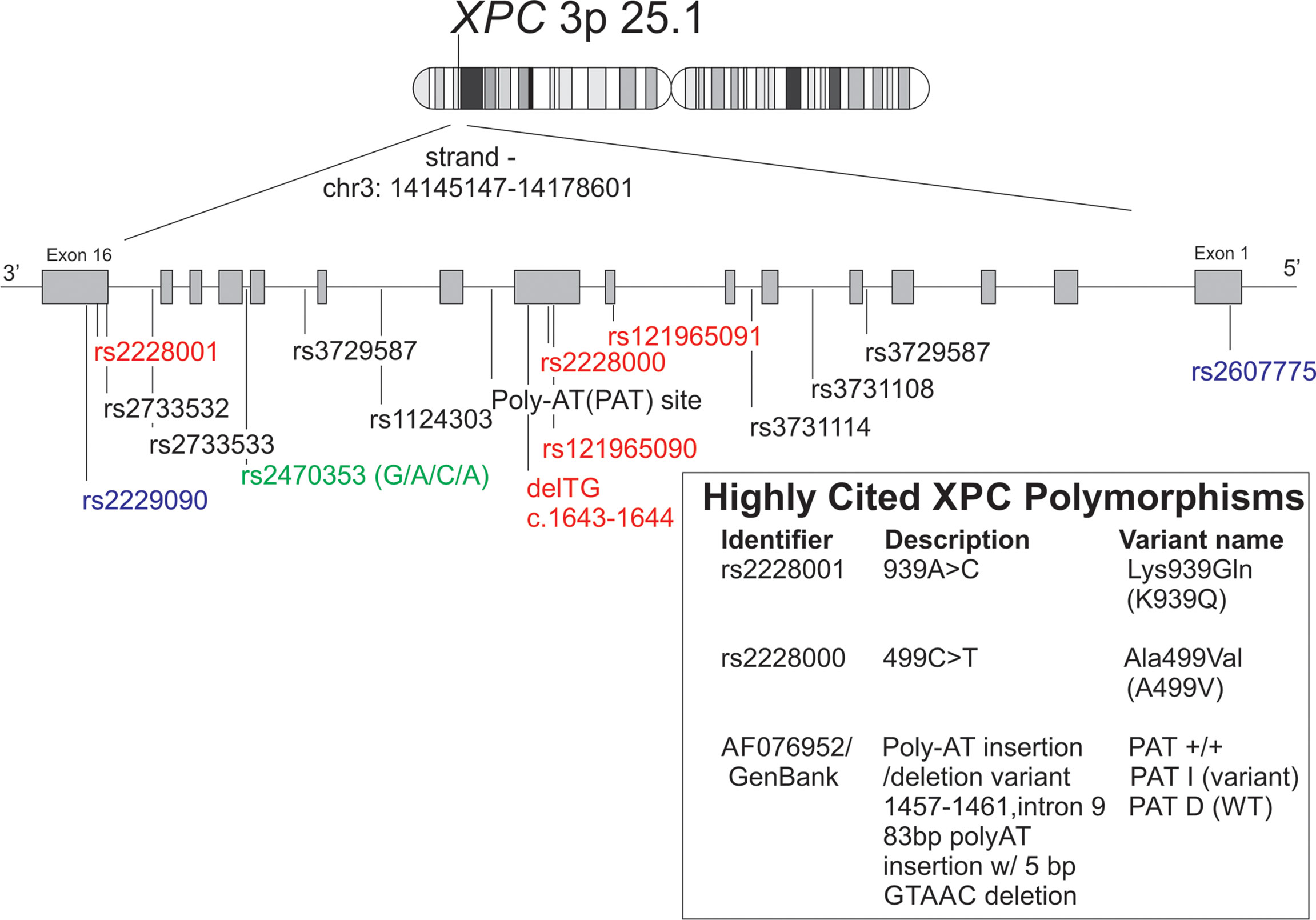
Figure 2 Schematic representation of XPC polymorphisms discussed in this manuscript along with alternate names/identifiers for the XPC polymorphisms most commonly studied in non-dermatologic cancers. Reference XPC gene (chr 3:p25.1) with polymorphisms was reproduced using the GRCh38 (hg38) sequencing using the UCSC genome browser tool. [(112) http://genome.ucsc.edu. Accessed 1/30/22]. Red = missense mutations, blue = 5’ or 3’ UTR variants, green = upstream of transcript variant.
Recently, the NER pathway has been studied in the setting of multiple myeloma (MM) due to the reliance on alkylating agents in the treatment of this malignancy; DNA damage caused by alkylating agents are typically repaired by NER. Dumontet et al. found that SNPs in multiple genes, including XPC, were associated with longer time to progression- in individuals treated with vincristine-adriamycin-dexamethasone followed by high dose melphalan and stem cell transplantation (80). Similarly, inhibition of the NER pathway in multiple myeloma increases the sensitivity to alkylating agents and overcomes resistance to these alkylating agents (113). Though XPC has not been explicitly implicated in these latter studies, it warrants further investigation given the role of the NER pathway and reliance on alkylating agents in multiple myeloma.
XPC in Solid Cancers
Lung Cancer
Lung cancer is characterized by some of the highest levels of genomic diversity, and alterations in DNA repair pathways, including NER, have been proposed to play a role in lung cancer development (114, 115). Although dominated by dermatologic malignancies, early series of XP-C patients reveal cases of bronchogenic lung carcinomas (14, 116). Germline mutations causing XP-C are rare, however, more common XPC polymorphisms and variations in gene expression have been studied in lung cancer (Table 1). In the most common subset of lung cancers, non-small cell lung cancer (NSCLC), decreased tumor XPC mRNA level has been associated with poor outcomes (117).
Numerous studies associate various XPC SNP polymorphisms with lung cancer development, which, among other factors, may be influenced by gender and cigarette smoking status (Table 1) (81–87), and many XPC polymorphisms have been found to functionally modulate DNA repair capacity (118). It is likely that epigenetic regulation leads to decreased XPC gene expression. Decreased XPC mRNA expression has been identified in human specimens from lung adenocarcinoma and lung squamous cell carcinoma, the two most common NSCLC histologic subtypes (119–121). Pre-clinical studies support epigenetic regulation of XPC with different environmental exposures, possibly due to promoter hypermethylation or histone-related transcriptional regulation (122). For instance, exposure of C57Bl/6 mice to 6 months of cigarette smoke led to decreased Xpc mRNA expression without altered expression of other studied BER and NER genes, including Xpa and Ogg1 (66). XPC protein expression is decreased in lung fibroblast and bronchial epithelial cell lines treated in culture with cigarette smoke extract, but not other NER proteins including XPA, and may be due to protein turnover by ubiquitination (123). Tight control of XPC ubiquitination is likely required to ensure DNA repair but may be dysregulated in human cancers, including lung cancers, which have been shown to have high levels of ubiquitin ligases, such as Cullin-RING ubiquitin ligase 4 A (CUL4A), overexpression of which is common in cigarette smoke-related lung cancer, and which is inversely proportionate to XPC expression (26). Additionally, murine exposure to side-stream smoke (up to 4 months) and nicotine-containing e-cigarette vape (12 weeks) led to increased DNA adduct formation and decreased Xpc and Ogg1 mRNA expression in the lungs (124, 125). Importantly, these studies also show decreased in vitro BER and NER repair using lysates from e-cigarette vape exposed mouse lungs, correlating decreased gene expression to decreased repair function.
The strongest evidence supporting a critical role of XPC in lung cancer comes from translational animal studies. Two mouse models of global Xpc deficiency have been created, both of which are associated with complete loss of functional XPC and cause characteristic skin cancers with exposure to UV light (126, 127). Increased DNA damage has been observed in the lungs of Xpc deficient mice upon exposure to oxidizing agents, but not in mice deficient in another NER protein, Xpa, although both show increased mutational frequency in the liver (40, 128). Exposure to urban air pollution led to increased lung inflammation and DNA damage in Xpc deficient mice (129). Mice homozygous deficient in Xpc develop lung tumors (primarily adenomas) with advanced age (16-17 months), although development of adenocarcinomas were rare without a co-existing loss of another tumor suppressor gene (130). However, exposure of Xpc deficient mice to chronic cigarette smoke and carcinogens, including urethane, MCA-BHT, 2-acetylaminofluorene (AAF) and NOH-AAF leads to lung adenocarcinoma development (42, 131), and with advanced age and chronic cigarette smoke, Xpc deficient mouse lungs develop an increase in lung compliance and alveolar rarefication similar to that seen in emphysema, a lung disease which predisposes to lung cancer (66). Importantly, mice heterozygous in Xpc (Xpc+/-) exposed to the carcinogen, urethane, developed an intermediate number of lung tumors when to compared to urethane-treated Xpc deficient and proficient littermate mice, suggesting a gene-dose effect and further supporting a role for intermediate levels of XPC expression, either through polymorphisms or epigenetic regulation, in lung cancer development (42).
Other more recently proposed mechanisms for XPC involvement in NSCLC development include regulation of cell proliferation and migration, and transcriptional regulation of p53. For instance, XPC, complexed with HR23B, impacts p53 transcriptional regulation of MMP1, low expression of which was associated with increased tumor size and metastasis (132). Cui and colleagues studied the impact of XPC on NSCLC cell lines in vitro, finding that XPC knock-down led to increased NSCLC cell growth and migration due to decreased surface e-cadherin expression through regulation of the SNAIL pathway (133). Although strong evidence supports an important role of XPC in lung cancer development, more research is needed to understand the link between alterations in XPC expression levels and XPC function on lung carcinogenesis and oncogenic development of characteristic genomic and transcriptomic alterations.
Prostate Cancer
Prostate cancer (PC) is the most common malignancy in males (134), and XPC polymorphisms have been correlated to an increased risk of PC development in several studies (Table 1). For instance, the XPC polymorphism, XPC PAT (PAT I/I genotype) was associated with an increased odds of prostate cancer, associated with a 3.83-fold increased risk in a Tunisian population. In contrast, other XPC polymorphisms, including those heterozygous for Lys939Gln (939Lys/Gln) along with the PAT D/D haplotype are considered protective of prostate cancer (89). One more study reported an increased risk of developing PC in those with the XPC PAT polymorphism (PAT +/+ or PAT +/-) along with tobacco smoking in a Chinese population (90). Other studies have shown varied increases in PC risk with other XPC polymorphisms (91, 135) (Table 1). It does not appear that XPC polymorphisms are associated with more advanced disease in PC, and similarly, studies did not find an association between XPC gene polymorphisms and Gleason score (a measure of histologic PC staging which correlates to prognosis) (89, 92). However, using TCGA data, low XPC expression was associated with worse overall survival in PC, similar to analyses in many other solid organ tumors (135). These studies suggest that XPC polymorphisms may serve as a tool to identify those at the highest risk for developing PC, which can help in targeting high and low-risk individuals to appropriate screening and clinical evaluations.
Ovarian Cancer
Like other solid organ tumors, XPC polymorphisms have been identified as one factor that may increase or decrease the risk of ovarian cancer as summarized in Table 1. Along with SNPs in two other NER proteins, XRCC1 and XRCC2, the XPC Ala499Val polymorphism was found to correlate to a decreased odds of ovarian cancer (OR 0.35) while the XPC Lys939Gln polymorphism was associated with an increased risk of ovarian cancer (OR 1.72) in a dominant genetic model (93). XPC polymorphisms may also serve as a biomarker in response to platinum-based chemotherapies as some specific SNP polymorphisms were associated with prolonged progression-free survival (PFS) (94). Going further, in ovarian cancer, overexpression of the eukaryotic translation initiation factor 3a (eIF3a) was associated with decreased response to cisplatin through downregulating XPC mRNA expression (136). This further supports an important role of XPC in predicting response to platinum-based chemotherapy through its canonical involvement in GG-NER.
Bladder Cancer
As with several other cancers, DNA damage due to carcinogen exposure, including cigarette smoking, is strongly associated with bladder cancer. In this, as in several other cancers, XPC polymorphisms were associated with low penetrance susceptibility to bladder cancer (Table 1) (83, 86, 95–97). Several rare XPC mutations, identified in patients with bladder cancer, were studied in vitro and were associated with decreased XPC mRNA and protein expression (98). Supporting their likely role in bladder cancer development, XPC mRNA and protein expression is decreased in bladder cancer tumors and may portend a worse prognosis (99, 137, 138). A variable impact of factors such as cigarette smoking have been correlated to XPC expression in bladder cancers, and more recently, studies have suggested a role of both promoter hypermethylation and histone deacetylation by HDACs in regulation of XPC mRNA expression in bladder cancer (138, 139), the latter of which is supported by previously studies reporting SIRT-1 deacetylase regulation of XPC expression in other (skin) cancers (140). Overall, these studies support a role of XPC expression in variable risk and outcomes of bladder cancer, although the exact mechanisms of epigenetic regulation, and the specific mechanisms by which risk is altered in low XPC, remains less clear.
Pancreatic Cancer
XPC may play a role as a risk factor for developing pancreatic cancer. As summarized in Table 1, some XPC polymorphisms have been described as increasing pancreatic cancer risk, particularly in smokers with the rs2470353 and rs2607775 variants (101). However, one study suggested a protective role of the XPC-PAT polymorphism (PAT +/+) in pancreatic cancer risk (100). Other studies suggested a role for genetic variants of other NER associated proteins, including ERCC1, but not necessarily XPC as a risk factor for developing pancreatic cancer (141). None-the-less, the specific role of NER, and specifically of XPC expression and epigenetic regulation, still need to be further explored in pancreatic cancer development.
Other Solid Organ Cancers
In esophageal cancer XPC may play a role as a risk factor for developing malignancy. XPC genetic variants, specifically the XPC K939Q C/C genotypes were found to be associated with a higher mortality after treatment compared with patients with a wild-type homozygous genotype; particularly in those who were post-treatment with esophagectomy or neoadjuvant chemoradiation (102). Another polymorphism, XPC PAT +/+, was associated with decreased risk for esophageal cancer (103). The prognostic value of XPC is further supported by having two XPC polymorphisms, XPC 499CC and XPC 939AC+CC (939 Lys and Gln), as part of a 5-polymorphism panel (high risk genotype) that has a 79% sensitivity and 85.4% specificity of predicting 5 years progression free survival (104), indicating a potential prognostic role of XPC polymorphisms in esophageal cancer risk.
XPC may also play a role as a risk factor for other cancers including advanced colorectal cancer. The XPC polymorphism Ala499Val was found to play a protective role in developing advanced colorectal adenomas in smokers (105), and others have suggested a protective role of higher XPC mRNA and protein expression levels on colorectal survival, possibly related to an improved response to chemoradiation (106). A recent case-control association study using tissue from 493 breast cancer and 387 control cases suggested an association between two XPC polymorphisms, rs2228001-A>C (Lys939Gln) and rs2733532-C>T, with an increased odds of breast cancer (107), and another study with 200 cases and controls suggested an association between the XPC PAT+ allele and higher odds of breast cancer (108).
Finally, some evidence supports a role of XPC in liver (hepatocellular) carcinoma development. In a case-control study of hepatocellular carcinoma HCC related to aflatoxin B1 exposure, XPC polymorphism codon 939Gln allele, whether heterozygous (XPC-LG) or homozygous (XPC-GG), is associated with increased risk of HCC; these genotype variants correlated with decreased XPC tumor protein expression by IHC as well as a shorter overall survival (109).
XPC as Tumor Suppressor and an Emerging Biomarker of Cancer Development
Numerous cancers are associated with decreased XPC expression, but the mechanism by which this occurs is less clear. The XPC gene, along with several other tumor suppressor genes, is located on chromosome 3p, a frequently site of chromosomal deletion in human tumors (130, 142). However, various modes of transcriptional regulation have been implicated in altered tumor XPC expression as well, and XPC expression may be altered in cells outside of the tumor itself. While studies have suggested decreased XPC expression in NSCLC tumor cells compared to surrounding lung (119), in 21 patients with NSCLC in which blood, tumor and lung tissue were collected, XPC mRNA expression was found to strongly correlate between blood and NSCLC tumor tissue, supporting the potential use of a minimally invasive blood draw as a prognostic and therapeutic biomarker (143).
The impact of low XPC mRNA expression may extend beyond alterations in DNA damage response and repair. Interestingly, XPC deficiency may also cause a mutational hot spot in the tumor suppressor p53 when treated with UV light, mediated by non-dipyrimidine base damage (144). Furthermore, there is evidence that XPC regulates a p53 post-ubiquitylation event and that XPC deficiency compromises p53 degradation, which may play a role in developing malignancy (145). These later two studies were performed in skin fibroblast cells and in vitro cell culture models, and whether XPC is involved in p53 regulation and mutations in other malignancies has not been well studied. In addition to its role in a number of DNA repair pathways, XPC has been implicated in transcriptional regulation both in response and independent of DNA damage. In the setting of DNA damage, studies have supported E2F1 transcriptional regulation of XPC expression (146). Recently, XPC itself has been implicated in post-translational histone modification and recruitment of transcription factors such as E2F1 to gene promoter sites independent of its regulatory role in DNA repair (69). High expression of miRNA-346, commonly elevated in NSCLC and other cancers, was associated with lower XPC mRNA and protein expression, indicating another potential mechanism for XPC downregulation in human cancers (147).
XPC as a Biomarker of Response to Therapy
In addition to XPC polymorphisms and expression levels as potential biomarkers associated with risk for many malignancies, XPC may predict disease progression. In patients with NSCLC, low tumor XPC mRNA expression is associated with advanced stage at diagnosis and an increased rate of cancer relapse after treatment in never-smokers (148). Similarly in colorectal cancer, increased XPC expression was associated with longer 5 year survival in treated patients compared to patients with low XPC expression (106). XPC polymorphisms have been described as predicting response to platinum-based chemotherapy. For instance, DNA samples from whole blood cells showed that XPC rs2229090 GC/CC genotypes were associated with longer progressive free survival compared to the AA and GG genotypes (149). These findings are consistent with translational and in vitro studies inversely linking XPC mRNA expression with response to cisplatin, particularly in lung adenocarcinoma where cisplatin chemotherapy treatment remains a mainstay in locally advanced disease (150). However, a link between XPC polymorphisms and response to cisplatin therapy has not been clearly shown, with a recent meta-analysis (88). High mutational burden has been associated with improved response to the immune checkpoint inhibitors. Typically, angiosarcomas have poor response to immunotherapy, but a recent report highlights an angiosarcomas that developed in an XP-C patient which had the features suggestive of a good response to immunotherapy and ultimately benefitted from a good response to the immune checkpoint inhibitor pembrolizumab (151). This report provides a preliminary but intriguing potential link between XPC, high tumor mutation burden and response to immunotherapies.
In the last few years, more attention has been paid to targeting DNA repair as a modality to augment cancer therapy. For instance, in a micro-RNA (miR) screen of prostate cancer, miR-890, which directly inhibited transcription of XPC along with other DNA repair proteins, led to increased sensitivity to ionizing radiation, although further mechanistic testing indicated that IR-sensitization by miR-890 persisted in XPC knock-down cells, suggesting an indirect role of XPC in double-strand break repair and overlapping gene-functionality in IR-sensitization (152). However, most studies show a predictive role in response to chemotherapies, especially platinum-based agents, which cause DNA lesions that are primarily repaired by NER, requiring XPC for recognition (2). Since increased NER repair could mean increased resistance to platinum-based therapy, inhibiting XPC could be a viable option to overcome platinum resistance in cancer cells. For instance modulation of XPC by hyperthermia or by treatment with sodium arenite was found to suppress XPC-induced cisplatin toxicity and sensitize tumors to platinum based therapy in a mouse ovarian cancer xenograft model (153). However, others have found a seemingly contradictory impact of histone deacetylase (HDAC) inhibitors in bladder cancer, showing a correlation between HDAC inhibition, increased XPC expression and higher cisplatin-induced activation of the pro-apoptotic protein, caspase 3 (139). Additionally, it is unclear if described decreases in XPC expression are in cancer cells alone or found in other cells within the tumor microenvironment, such as fibroblasts, in which XPC inhibition could be expected to decrease the tumor promoting cytokine IL-6 (72). On the other hand, this inhibition may help to sensitize tumor cells to other therapies due to the involvement of XPC in other DNA repair pathways and in checkpoint activation. Future studies should explore XPC targeting by small molecular inhibitors to investigate these possibilities, especially given conflicting data regarding XPC expression levels and therapeutic response to chemotherapeutic agents.
Conclusion
XPC is increasingly recognized as playing an important role in the development of non-dermatologic malignancies. Decreased XPC mRNA and protein expression has been described in a number of cancers, with gene polymorphisms, deletions, and transcriptional regulation all active areas of research in the regulation of XPC expression. Additionally, research supports a role of XPC in the prognosis and treatment response in several of these cancers. Although XPC’s essential role in the recognition of bulky DNA lesions and subsequent activation of GG-NER, when altered, is a leading mechanism for development of UV-induced dermatologic malignancies and in modifications of cancer response to chemotherapies including cisplatin, recent data support a non-canonical role of XPC in DNA damage response and repair mechanisms, tumor suppressor transcriptional regulation, and in the development of non-dermatologic malignancies. Future studies would benefit from studying XPC as a biomarker of cancer prognosis and response to treatment in non-dermatologic malignancies.
Author Contributions
Conceptualization, NN and CRS. Writing—original draft preparation, NN, BMW and CRS. Writing—review and editing, NN and CRS. Funding acquisition, CRS. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the U.S. Department of Veterans Affairs BLR&D, Merit Review grant I01-BX005353 to CRS and in part by the National Institutes of Health, U.S.A. T32HL091816 to BMW.
Author Disclaimer
CRS is employed by the U.S. Department of Veterans Affairs. The contents of this manuscript do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Conflict of Interest
CRS has served on Scientific and Medical Advisory Boards for Biodesix, Inc., bioAffinity Technologies, and as a scientific medical consultant for Bristol-Myers Squibb Company; these are not relevant to the topic of this manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Gavande NS, Vandervere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, et al. DNA Repair Targeted Therapy: The Past or Future of Cancer Treatment? Pharmacol Ther (2016) 160:65–83. doi: 10.1016/j.pharmthera.2016.02.003
3. Jalal S, Earley JN, Turchi JJ. DNA Repair: From Genome Maintenance to Biomarker and Therapeutic Target. Clin Cancer Res (2011) 17:6973–84. doi: 10.1158/1078-0432.CCR-11-0761
4. Nemzow L, Lubin A, Zhang L, Gong F. XPC: Going Where No DNA Damage Sensor has Gone Before. DNA Repair (Amst) (2015) 36:19–27. doi: 10.1016/j.dnarep.2015.09.004
5. Zamble DB, Mu D, Reardon JT, Sancar A, Lippard SJ. Repair of Cisplatin-DNA Adducts by the Mammalian Excision Nuclease. Biochemistry (1996) 35:10004–13. doi: 10.1021/bi960453+
6. Fousteri M, Mullenders LH. Transcription-Coupled Nucleotide Excision Repair in Mammalian Cells: Molecular Mechanisms and Biological Effects. Cell Res (2008) 18:73–84. doi: 10.1038/cr.2008.6
7. Spivak G. Nucleotide Excision Repair in Humans. DNA Repair (Amst) (2015) 36:13–8. doi: 10.1016/j.dnarep.2015.09.003
8. Shell SM, Hawkins EK, Tsai MS, Hlaing AS, Rizzo CJ, Chazin WJ. Xeroderma Pigmentosum Complementation Group C Protein (XPC) Serves as a General Sensor of Damaged DNA. DNA Repair (Amst) (2013) 12:947–53. doi: 10.1016/j.dnarep.2013.08.013
9. Zebian A, Shaito A, Mazurier F, Rezvani HR, Zibara K. XPC Beyond Nucleotide Excision Repair and Skin Cancers. Mutat Res Rev Mutat Res (2019) 782:108286. doi: 10.1016/j.mrrev.2019.108286
10. Lehmann AR, Mcgibbon D, Stefanini M. Xeroderma Pigmentosum. Orphanet J Rare Dis (2011) 6:70. doi: 10.1186/1750-1172-6-70
11. Digiovanna JJ, Kraemer KH. Shining a Light on Xeroderma Pigmentosum. J Invest Dermatol (2012) 132:785–96. doi: 10.1038/jid.2011.426
12. Chavanne F, Broughton BC, Pietra D, Nardo T, Browitt A, Lehmann AR, et al. Mutations in the XPC Gene in Families With Xeroderma Pigmentosum and Consequences at the Cell, Protein, and Transcript Levels. Cancer Res (2000) 60:1974–82.
13. Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, Boyle J, et al. Cancer and Neurologic Degeneration in Xeroderma Pigmentosum: Long Term Follow-Up Characterises the Role of DNA Repair. J Med Genet (2011) 48:168–76. doi: 10.1136/jmg.2010.083022
14. Kraemer KH, Lee MM, Scotto J. Xeroderma Pigmentosum: Cutaneous, Ocular, and Neurologic Abnormalities in 830 Published Cases. Arch Dermatol (1987) 123:241–50. doi: 10.1001/archderm.123.2.241
15. Sugasawa K, Akagi J, Nishi R, Iwai S, Hanaoka F. Two-Step Recognition of DNA Damage for Mammalian Nucleotide Excision Repair: Directional Binding of the XPC Complex and DNA Strand Scanning. Mol Cell (2009) 36:642–53. doi: 10.1016/j.molcel.2009.09.035
16. Mu H, Geacintov NE, Broyde S, Yeo JE, Scharer OD. Molecular Basis for Damage Recognition and Verification by XPC-RAD23B and TFIIH in Nucleotide Excision Repair. DNA Repair (Amst) (2018) 71:33–42. doi: 10.1016/j.dnarep.2018.08.005
17. Sugasawa K, Masutani C, Uchida A, Maekawa T, van der Spek PJ, Bootsma D, et al. HHR23B, a Human Rad23 Homolog, Stimulates XPC Protein in Nucleotide Excision Repair In Vitro. Mol Cell Biol (1996) 16:4852–61. doi: 10.1128/MCB.16.9.4852
18. Renaud E, Miccoli L, Zacal N, Biard DS, Craescu CT, Rainbow AJ, et al. Differential Contribution of XPC, RAD23A, RAD23B and CENTRIN 2 to the UV-Response in Human Cells. DNA Repair (2011) 10:835–47. doi: 10.1016/j.dnarep.2011.05.003
19. Friedberg EC. How Nucleotide Excision Repair Protects Against Cancer. Nat Rev Cancer (2001) 1:22–33. doi: 10.1038/35094000
20. Scharer OD. Nucleotide Excision Repair in Eukaryotes. Cold Spring Harb Perspect Biol (2013) 5:a012609. doi: 10.1101/cshperspect.a012609
21. Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, et al. UV-Induced Ubiquitylation of XPC Protein Mediated by UV-DDB-Ubiquitin Ligase Complex. Cell (2005) 121:387–400. doi: 10.1016/j.cell.2005.02.035
22. Wang QE, Zhu Q, Wani G, El-Mahdy MA, Li J, Wani AA. DNA Repair Factor XPC is Modified by SUMO-1 and Ubiquitin Following UV Irradiation. Nucleic Acids Res (2005) 33:4023–34. doi: 10.1093/nar/gki684
23. Kakumu E, Nakanishi S, Shiratori HM, Kato A, Kobayashi W, Machida S, et al. Xeroderma Pigmentosum Group C Protein Interacts With Histones: Regulation by Acetylated States of Histone H3. Genes Cells (2017) 22:310–27. doi: 10.1111/gtc.12479
24. Shah P, Zhao B, Qiang L, He YY. Phosphorylation of Xeroderma Pigmentosum Group C Regulates Ultraviolet-Induced DNA Damage Repair. Nucleic Acids Res (2018) 46:5050–60. doi: 10.1093/nar/gky239
25. Chauhan AK, Sun Y, Zhu Q, Wani AA. Timely Upstream Events Regulating Nucleotide Excision Repair by Ubiquitin-Proteasome System: Ubiquitin Guides the Way. DNA Repair (Amst) (2021) 103:103128. doi: 10.1016/j.dnarep.2021.103128
26. Jia L, Yan F, Cao W, Chen Z, Zheng H, Li H, et al. Dysregulation of CUL4A and CUL4B Ubiquitin Ligases in Lung Cancer. J Biol Chem (2017) 292:2966–78. doi: 10.1074/jbc.M116.765230
27. Matsuoka S, Ballif BA, Smoogorzewska A, Mcdonald ER, Hurov KE, Luo J, et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science (2007) 316:5828. doi: 10.1126/science.1140321
28. Nguyen TA, Slattery SD, Moon SH, Darlington YF, Lu X, Donehower LA. The Oncogenic Phosphatase WIP1 Negatively Regulates Nucleotide Excision Repair. DNA Repair (Amst) (2010) 9:813–23. doi: 10.1016/j.dnarep.2010.04.005
29. Chakraborty S, Steinbach PJ, Paul D, Mu H, Broyde S, Min JH, et al. Enhanced Spontaneous DNA Twisting/Bending Fluctuations Unveiled by Fluorescence Lifetime Distributions Promote Mismatch Recognition by the Rad4 Nucleotide Excision Repair Complex. Nucleic Acids Res (2018) 46:1240–55. doi: 10.1093/nar/gkx1216
30. Panigrahi A, Vemuri H, Aggarwal M, Pitta K, Krishnan M. Sequence Specificity, Energetics and Mechanism of Mismatch Recognition by DNA Damage Sensing Protein Rad4/XPC. Nucleic Acids Res (2020) 48:2246–57. doi: 10.1093/nar/gkaa078
31. Geacintov NE, Broyde S. Repair-Resistant DNA Lesions. Chem Res Toxicol (2017) 30:1517–48. doi: 10.1021/acs.chemrestox.7b00128
32. Fayyad N, Kobaisi F, Beal D, Mahfouf W, Ged C, Morice-Picard F, et al. Xeroderma Pigmentosum C (XPC) Mutations in Primary Fibroblasts Impair Base Excision Repair Pathway and Increase Oxidative DNA Damage. Front Genet (2020) 11:561687. doi: 10.3389/fgene.2020.561687
33. De Melo JT, De Souza Timoteo AR, Lajus TB, Brandao JA, De Souza-Pinto NC, Menck CF, et al. XPC Deficiency Is Related to APE1 and OGG1 Expression and Function. Mutat Res (2016) 784-785:25–33. doi: 10.1016/j.mrfmmm.2016.01.004
34. Miao F, Bouziane M, Dammann R, Masutani C, Hanaoka F, Pfeifer G, et al. 3-Methyladenine-DNA Glycosylase (MPG Protein) Interacts With Human RAD23 Proteins. J Biol Chem (2000) 275:28433–8. doi: 10.1074/jbc.M001064200
35. Shimizu Y, Iwai S, Hanaoka F, Sugasawa K. Xeroderma Pigmentosum Group C Protein Interacts Physically and Functionally With Thymidine DNA Glycosylase. EMBO J (2003) 22:164–73. doi: 10.1093/emboj/cdg016
36. Parlanti E, D’errico M, Degan P, Calcagnile A, Zijno A, van der Pluijm I, et al. The Cross Talk Between Pathways in the Repair of 8-Oxo-7,8-Dihydroguanine in Mouse and Human Cells. Free Radical Biol Med (2012) 53:2171–7. doi: 10.1016/j.freeradbiomed.2012.08.593
37. Ho JJ, Cattoglio C, Mcswiggen DT, Tjian R, Fong YW. Regulation of DNA Demethylation by the XPC DNA Repair Complex in Somatic and Pluripotent Stem Cells. Genes Dev (2017) 31:830–44. doi: 10.1101/gad.295741.116
38. Menoni H, Hoeijmakers JH, Vermeulen W. Nucleotide Excision Repair-Initiating Proteins Bind to Oxidative DNA Lesions In Vivo. J Cell Biol (2012) 199:1037–46. doi: 10.1083/jcb.201205149
39. Kumar N, Theil AF, Roginskaya V, Ali Y, Calderon M, Watkins SC, et al. Global and Transcription-Coupled Repair of 8-oxoG is Initiated by Nucleotide Excision Repair Proteins. Nat Commun (2022) 13:974. doi: 10.1038/s41467-022-28642-9
40. Melis JP, Kuiper RV, Zwart E, Robinson J, Pennings JL, Van Oostrom CT, et al. Slow Accumulation of Mutations in Xpc Mice Upon Induction of Oxidative Stress. DNA Repair (2013) 12:1081–6. doi: 10.1016/j.dnarep.2013.08.019
41. Okamoto Y, Chou PH, Kim SY, Suzuki N, Laxmi YR, Okamoto K, et al. Oxidative DNA Damage in XPC-Knockout and Its Wild Mice Treated With Equine Estrogen. Chem Res Toxicol (2008) 21:1120–4. doi: 10.1021/tx700428m
42. Zhou H, Saliba J, Sandusky GE, Sears CR. XPC Protects Against Smoking and Carcinogen-Induced Lung Adenocarcinoma. Carcinogenesis (2019) 40:403–11. doi: 10.1093/carcin/bgz003
43. Modrich P, Lahue R. Mismatch Repair in Replication Fidelity, Genetic Recombination, and Cancer Biology. Annu Rev Biochem (1996) 65:101–33. doi: 10.1146/annurev.bi.65.070196.000533
44. Kolodner RD, Marsischky GT. Eukaryotic DNA Mismatch Repair. Curr Opin Genet Dev (1999) 9:89–96. doi: 10.1016/S0959-437X(99)80013-6
45. Poynter JN, Siegmund KD, Weisenberger DJ, Long TI, Thibodeau SN, Lindor N, et al. Molecular Characterization of MSI-H Colorectal Cancer by MLHI Promoter Methylation, Immunohistochemistry, and Mismatch Repair Germline Mutation Screening. Cancer Epidemiol Biomarkers Prev (2008) 17:3208–15. doi: 10.1158/1055-9965.EPI-08-0512
46. Li X, Liu G, Wu W. Recent Advances in Lynch Syndrome. Exp Hematol Oncol (2021) 10:37. doi: 10.1186/s40164-021-00231-4
47. Deans AJ, West SC. DNA Interstrand Crosslink Repair and Cancer. Nat Rev Cancer (2011) 11:467–80. doi: 10.1038/nrc3088
48. Noll DM, Mason TM, Miller PS. Formation and Repair of Interstrand Cross-Links in DNA. Chem Rev (2006) 106:277–301. doi: 10.1021/cr040478b
49. Mchugh PJ, Spanswick VJ, Hartley JA. Repair of DNA Interstrand Crosslinks: Molecular Mechanisms and Clinical Relevance. Lancet Oncol (2001) 2:483–90. doi: 10.1016/S1470-2045(01)00454-5
50. Chen Z, Xu XS, Yang J, Wang G. Defining the Function of XPC Protein in Psoralen and Cisplatin-Mediated DNA Repair and Mutagenesis. Carcinogenesis (2003) 24:1111–21. doi: 10.1093/carcin/bgg051
51. Christensen LA, Finch RA, Booker AJ, Vasquez KM. Targeting Oncogenes to Improve Breast Cancer Chemotherapy. Cancer Res (2006) 66:4089–94. doi: 10.1158/0008-5472.CAN-05-4288
52. Jain A, Wang G, Vasquez KM. DNA Triple Helices: Biological Consequences and Therapeutic Potential. Biochimie (2008) 90:1117–30. doi: 10.1016/j.biochi.2008.02.011
53. Thoma BS, Wakasugi M, Christensen J, Reddy MC, Vasquez KM. Human XPC-Hhr23b Interacts With XPA-RPA in the Recognition of Triplex-Directed Psoralen DNA Interstrand Crosslinks. Nucleic Acids Res (2005) 33:2993–3001. doi: 10.1093/nar/gki610
54. Wu Q, Christensen LA, Legerski RJ, Vasquez KM. Mismatch Repair Participates in Error-Free Processing of DNA Interstrand Crosslinks in Human Cells. EMBO Rep (2005) 6:551–7. doi: 10.1038/sj.embor.7400418
55. Zhao J, Jain A, Iyer RR, Modrich PL, Vasquez KM. Mismatch Repair and Nucleotide Excision Repair Proteins Cooperate in the Recognition of DNA Interstrand Crosslinks. Nucleic Acids Res (2009) 37:4420–9. doi: 10.1093/nar/gkp399
56. Wang G, Chuang L, Zhang X, Colton S, Dombkowski A, Reiners J, Diakiw A, Xu XS. (2004). The Initiative Role of XPC Protein in Cisplatin DNA Damaging Treatment-Mediated Cell Cycle Regulation. Nucleic Acids Res 32(7):2231–40. doi: 10.1093/nar/gkh541
57. Yoshino M, Nakatsuru Y, Te Riele H, Hirota S, Kitamura Y, Tanaka K. Additive Roles of XPA and MSH2 Genes in UVB-Induced Skin Tumorigenesis in Mice. DNA Repair (Amst) (2002) 1(11):935–40. doi: 10.1016/S1568-7864(02)00144-1
58. Meira LB, Reis AMC, Cheo DL, Nahari D, Burns DK, Friedberg EC. Cancer Predisposition in Mutant Mice Defective in Multiple Genetic Pathways: Uncovering Important Genetic Interactions. Mutat Res (2001) 477:51–8. doi: 10.1016/S0027-5107(01)00097-5
59. Kobayashi K, O’driscoll M, Macpherson P, Mullenders L, Vreeswijk M, Karran P. XPC Lymphoblastoid Cells Defective in the Hmutsalpha DNA Mismatch Repair Complex Exhibit Normal Sensitivity to UVC Radiation and Normal Transcription-Coupled Excision Repair of DNA Cyclobutane Pyrimidine Dimers. DNA Repair (Amst) (2004) 3:649–57. doi: 10.1016/j.dnarep.2004.02.007
60. Despras E, Pfeiffer P, Salles B, Calsou P, Kuhfittig-Kulle S, Angulo JF, et al. Long-Term XPC Silencing Reduces DNA Double-Strand Break Repair. Cancer Res (2007) 67:2526–34. doi: 10.1158/0008-5472.CAN-06-3371
61. Miccoli L, Burr KL, Hickenbotham P, Friedberg EC, Angulo JF, Dubrova YE. The Combined Effects of Xeroderma Pigmentosum C Deficiency and Mutagens on Mutation Rates in the Mouse Germ Line. Cancer Res (2007) 67:4695–9. doi: 10.1158/0008-5472.CAN-06-3844
62. Wang H, Huang Y, Shi J, Zhi Y, Yuan F, Yu J, et al. XPC Deficiency Leads to Centrosome Amplification by Inhibiting BRCA1 Expression Upon Cisplatin-Mediated DNA Damage in Human Bladder Cancer. Cancer Lett (2019) 444:136–46. doi: 10.1016/j.canlet.2018.12.004
63. Ray A, Milum K, Battu A, Wani G, Wani AA. NER Initiation Factors, DDB2 and XPC, Regulate UV Radiation Response by Recruiting ATR and ATM Kinases to DNA Damage Sites. DNA Repair (Amst) (2013) 12:273–83. doi: 10.1016/j.dnarep.2013.01.003
64. Wang QE, Han C, Zhang B, Sabapathy K, Wani AA. Nucleotide Excision Repair Factor XPC Enhances DNA Damage-Induced Apoptosis by Downregulating the Antiapoptotic Short Isoform of Caspase-2. Cancer Res (2012) 72:666–75. doi: 10.1158/0008-5472.CAN-11-2774
65. Liu SY, Wen CY, Lee YJ, Lee TC. XPC Silencing Sensitizes Glioma Cells to Arsenic Trioxide via Increased Oxidative Damage. Toxicol Sci (2010) 116:183–93. doi: 10.1093/toxsci/kfq113
66. Sears CR, Zhou H, Justice MJ, Fisher AJ, Saliba J, Lamb I, et al. Xeroderma Pigmentosum Group C Deficiency Alters Cigarette Smoke DNA Damage Cell Fate and Accelerates Emphysema Development. Am J Respir Cell Mol Biol (2018) 58:402–11. doi: 10.1165/rcmb.2017-0251OC
67. Ito S, Yamane M, Ohtsuka S, Niwa H. The C-Terminal Region of Xpc Is Dispensable for the Transcriptional Activity of Oct3/4 in Mouse Embryonic Stem Cells. FEBS Lett (2014) 588:1128–35. doi: 10.1016/j.febslet.2014.02.033
68. Le May N, Mota-Fernandes D, Velez-Cruz R, Iltis I, Biard D, Egly JM. NER Factors Are Recruited to Active Promoters and Facilitate Chromatin Modification for Transcription in the Absence of Exogenous Genotoxic Attack. Mol Cell (2010) 38:54–66. doi: 10.1016/j.molcel.2010.03.004
69. Bidon B, Iltis I, Semer M, Nagy Z, Larnicol A, Cribier A, et al. XPC is an RNA Polymerase II Cofactor Recruiting ATAC to Promoters by Interacting With E2F1. Nat Commun (2018) 9:2610. doi: 10.1038/s41467-018-05010-0
70. Melis JP, Luijten M, Mullenders LH, Van Steeg H. The Role of XPC: Implications in Cancer and Oxidative DNA Damage. Mutat Res (2011) 728:107–17. doi: 10.1016/j.mrrev.2011.07.001
71. Mori MP, Costa RA, Soltys DT, Freire TS, Rossato FA, Amigo I, et al. Lack of XPC Leads to a Shift Between Respiratory Complexes I and II But Sensitizes Cells to Mitochondrial Stress. Sci Rep (2017) 7:155. doi: 10.1038/s41598-017-00130-x
72. Schreck I, Grico N, Hansjosten I, Marquardt C, Bormann S, Seidel A, et al. The Nucleotide Excision Repair Protein XPC Is Essential for Bulky DNA Adducts to Promote Interleukin-6 Expression via the Activation of P38-SAPK. Oncogene (2016) 35:908–18. doi: 10.1038/onc.2015.145
73. Wijnhoven SWP, Kool HJM, Mullenders LHF, Van Zeeland AA, Friedberg EC, van der Horst GTJ, et al. Age-Dependent Spontaneous Mutagenesis in Xpc Mice Defective in Nucleotide Excision Repair. Oncogene (2000) 19:5034–7. doi: 10.1038/sj.onc.1203844
74. Fischer JL, Kumar MA, Day TW, Hardy TM, Hamilton S, Besch-Williford C, et al. The Xpc Gene Markedly Affects Cell Survival in Mouse Bone Marrow. Mutagenesis (2009) 24:309–16. doi: 10.1093/mutage/gep011
75. Furutani E, Shimamura A. Germline Genetic Predisposition to Hematologic Malignancy. J Clin Oncol (2017) 35:1018–28. doi: 10.1200/JCO.2016.70.8644
76. El-Zein R, Monroy CM, Etzel CJ, Cortes AC, Xing Y, Collier AL, et al. Genetic Polymorphisms in DNA Repair Genes as Modulators of Hodgkin Disease Risk. Cancer (2009) 115:1651–9. doi: 10.1002/cncr.24205
77. Sarasin A, Quentin S, Droin N, Sahbatou M, Saada V, Auger N, et al. Familial Predisposition to TP53/complex Karyotype MDS and Leukemia in DNA Repair-Deficient Xeroderma Pigmentosum. Blood (2019) 133:2718–24. doi: 10.1182/blood-2019-01-895698
78. Strom SS, Estey E, Outschoorn UM, Garcia-Manero G. Acute Myeloid Leukemia Outcome: Role of Nucleotide Excision Repair Polymorphisms in Intermediate Risk Patients. Leuk Lymphoma (2010) 51:598–605. doi: 10.3109/10428190903582804
79. Guillem VM, Cervantes F, Martinez J, Alvarez-Larran A, Collado M, Camos M, et al. XPC Genetic Polymorphisms Correlate With the Response to Imatinib Treatment in Patients With Chronic Phase Chronic Myeloid Leukemia. Am J Hematol (2010) 85:482–6. doi: 10.1002/ajh.21726
80. Dumontet C, Landi S, Reiman T, Perry T, Plesa A, Bellini I, et al. Genetic Polymorphisms Associated With Outcome in Multiple Myeloma Patients Receiving High-Dose Melphalan. Bone Marrow Transplant (2010) 45:1316–24. doi: 10.1038/bmt.2009.335
81. Marin MS, Lopez-Cima MF, Garcia-Castro L, Pascual T, Marron MG, Tardon A. Poly (AT) Polymorphism in Intron 11 of the XPC DNA Repair Gene Enhances the Risk of Lung Cancer. Cancer Epidemiol Biomarkers Prev (2004) 13:1788–93. doi: 10.1158/1055-9965.1788.13.11
82. Vogel U, Overvad K, Wallin H, Tjonneland A, Nexo BA, Raaschou-Nielsen O. Combinations of Polymorphisms in XPD, XPC and XPA in Relation to Risk of Lung Cancer. Cancer Lett (2005) 222:67–74. doi: 10.1016/j.canlet.2004.11.016
83. Francisco G, Menezes PR, Eluf-Neto J, Chammas R. XPC Polymorphisms Play a Role in Tissue-Specific Carcinogenesis: A Meta-Analysis. Eur J Hum Genet (2008) 16:724–34. doi: 10.1038/ejhg.2008.6
84. Letkova L, Matakova T, Musak L, Sarlinova M, Krutakova M, Slovakova P, et al. DNA Repair Genes Polymorphism and Lung Cancer Risk With the Emphasis to Sex Differences. Mol Biol Rep (2013) 40:5261–73. doi: 10.1007/s11033-013-2626-z
85. Jin B, Dong Y, Zhang X, Wang H, Han B. Association of XPC Polymorphisms and Lung Cancer Risk: A Meta-Analysis. PloS One (2014) 9:e93937. doi: 10.1371/journal.pone.0093937
86. Qiu L, Wang Z, Shi X. Associations Between XPC Polymorphisms and Risk of Cancers: A Meta-Analysis. Eur J Cancer (2008) 44:2241–53. doi: 10.1016/j.ejca.2008.06.024
87. Mei C, Hou M, Guo S, Hua F, Zheng D, Xu F, et al. Polymorphisms in DNA Repair Genes of XRCC1, XPA, XPC, XPD and Associations With Lung Cancer Risk in Chinese People. Thorac Cancer (2014) 5:232–42. doi: 10.1111/1759-7714.12073
88. Xie C, Zhao J, Hua W, Tan P, Chen Y, Rui J, et al. Effect of XPC Polymorphisms on the Response to Platinum-Based Chemotherapy: A Meta-Analysis. Onco Targets Ther (2019) 12:3839–48. doi: 10.2147/OTT.S202617
89. Said R, Bougatef K, Setti Boubaker N, Jenni R, Derouiche A, Chebil M, et al. Polymorphisms in XPC Gene and Risk for Prostate Cancer. Mol Biol Rep (2019) 46:1117–25. doi: 10.1007/s11033-018-4572-2
90. Liu Y, Chen Z, Wei Q, Yuan F, Zhi Y, Song B, et al. Poly (AT) Polymorphism in the XPC Gene and Smoking Enhance the Risk of Prostate Cancer in a Low-Risk Chinese Population. Cancer Genet (2012) 205:205–11. doi: 10.1016/j.cancergen.2012.01.013
91. Yoshino Y, Takeuchi S, Katoh T, Kuroda Y. XPC Intron11 C/A Polymorphism as a Risk Factor for Prostate Cancer. Environ Health Prev Med (2016) 21:100–4. doi: 10.1007/s12199-015-0505-z
92. Kahnamouei SA, Narouie B, Sotoudeh M, Mollakouchekian MJ, Simforoosh N, Ziaee SA, et al. Association of XPC Gene Polymorphisms With Prostate Cancer Risk. Clin Lab (2016) 62:1009–15. doi: 10.7754/Clin.Lab.2015.150914
93. Zhao Z, Zhang A, Zhao Y, Xiang J, Yu D, Liang Z, et al. The Association of Polymorphisms in Nucleotide Excision Repair Genes With Ovarian Cancer Susceptibility. Biosci Rep (2018) 38:BSR20180114. doi: 10.1042/BSR20180114
94. Fleming ND, Agadjanian H, Nassanian H, Miller CW, Orsulic S, Karlan BY, et al. Xeroderma Pigmentosum Complementation Group C Single-Nucleotide Polymorphisms in the Nucleotide Excision Repair Pathway Correlate With Prolonged Progression-Free Survival in Advanced Ovarian Cancer. Cancer (2012) 118:689–97. doi: 10.1002/cncr.26329
95. Zhang Y, Wang X, Zhang W, Gong S. An Association Between XPC Lys939Gln Polymorphism and the Risk of Bladder Cancer: A Meta-Analysis. Tumor Biol (2013) 34:973–82. doi: 10.1007/s13277-012-0633-7
96. Dai Q-S, Hua R-X, Zeng R-F, Long J-T, Peng Z-W. XPC Gene Polymorphisms Contribute to Bladder Cancer Susceptibility: A Meta-Analysis. Tumor Biol (2014) 35:447–53. doi: 10.1007/s13277-013-1062-y
97. Sankhwar M, Sankhwar SN, Bansal SK, Gupta G, Rajender S. Polymorphisms in the XPC Gene Affect Urinary Bladder Cancer Risk: A Case-Control Study, Meta-Analyses and Trial Sequential Analyses. Sci Rep (2016) 6:27018. doi: 10.1038/srep27018
98. Qiao B, Ansari AH, Scott GB, Sak SC, Chambers PA, Elliott F, et al. In Vitro Functional Effects of XPC Gene Rare Variants From Bladder Cancer Patients. Carcinogenesis (2011) 32:516–21. doi: 10.1093/carcin/bgr005
99. Qiu J, Wang X, Meng X, Zheng Y, Li G, Ma J, et al. Attenuated NER Expressions of XPF and XPC Associated With Smoking Are Involved in the Recurrence of Bladder Cancer. PloS One (2014) 9:e115224. doi: 10.1371/journal.pone.0115224
100. Wang L, Lin DX, Lu XH, Miao XP, Li H. [Polymorphisms of the DNA Repair Genes XRCC1 and XPC: Relationship to Pancreatic Cancer Risk]. Wei Sheng Yan Jiu (2006) 35:534–6.
101. Liang XH, Yan D, Zhao JX, Ding W, Xu XJ, Wang XY. Interaction of Polymorphisms in Xeroderma Pigmentosum Group C With Cigarette Smoking and Pancreatic Cancer Risk. Oncol Lett (2018) 16:5631–8. doi: 10.3892/ol.2018.9350
102. Yang PW, Hsieh CY, Kuo FT, Huang PM, Hsu HH, Kuo SW, et al. The Survival Impact of XPA and XPC Genetic Polymorphisms on Patients With Esophageal Squamous Cell Carcinoma. Ann Surg Oncol (2013) 20:562–71. doi: 10.1245/s10434-012-2622-x
103. Pan J, Lin J, Izzo JG, Liu Y, Xing J, Huang M, et al. Genetic Susceptibility to Esophageal Cancer: The Role of the Nucleotide Excision Repair Pathway. Carcinogenesis (2009) 30:785–92. doi: 10.1093/carcin/bgp058
104. Gusella M, Giacopuzzi S, Bertolaso L, Zanoni A, Pezzolo E, Modena Y, et al. Genetic Prediction of Long-Term Survival After Neoadjuvant Chemoradiation in Locally Advanced Esophageal Cancer. Pharmacogenomics J (2017) 17:252–7. doi: 10.1038/tpj.2016.9
105. Huang WY, Berndt SI, Kang D, Chatterjee N, Chanock SJ, Yeager M, et al. Nucleotide Excision Repair Gene Polymorphisms and Risk of Advanced Colorectal Adenoma: XPC Polymorphisms Modify Smoking-Related Risk. Cancer Epidemiol Biomarkers Prev (2006) 15:306–11. doi: 10.1158/1055-9965.EPI-05-0751
106. Hu LB, Chen Y, Meng XD, Yu P, He X, Li J. Nucleotide Excision Repair Factor XPC Ameliorates Prognosis by Increasing the Susceptibility of Human Colorectal Cancer to Chemotherapy and Ionizing Radiation. Front Oncol (2018) 8:290. doi: 10.3389/fonc.2018.00290
107. Malik SS, Zia A, Rashid S, Mubarik S, Masood N, Hussain M, et al. XPC as Breast Cancer Susceptibility Gene: Evidence From Genetic Profiling, Statistical Inferences and Protein Structural Analysis. Breast Cancer (2020) 27:1168–76. doi: 10.1007/s12282-020-01121-z
108. Qazvini MG, Salehi Z, Mashayekhi F, Saedi HS. A33512C and Intronic Poly(AT) Insertion/Deletion (PAT-/+) Polymorphisms of the XPC Gene and Their Association With the Risk of Breast Cancer. Clin Breast Cancer (2020) 20:e771–7. doi: 10.1016/j.clbc.2020.05.014
109. Long XD, Ma Y, Zhou YF, Ma AM, Fu GH. Polymorphism in Xeroderma Pigmentosum Complementation Group C Codon 939 and Aflatoxin B1-Related Hepatocellular Carcinoma in the Guangxi Population. Hepatology (2010) 52:1301–9. doi: 10.1002/hep.23807
110. Oetjen KA, Levoska MA, Tamura D, Ito S, Douglas D, Khan SG, et al. Predisposition to Hematologic Malignancies in Patients With Xeroderma Pigmentosum. Haematologia (2020) 105:e146. doi: 10.3324/haematol.2019.223370
111. Yurchenko AA, Padioleau I, Matkarimov BT, Soulier J, Sarasin A, Nikolaev S. XPC Deficiency Increases Risk of Hematologic Malignancies Through Mutator Phenotype and Characteristic Mutational Signature. Nat Commun (2020) 11:5834. doi: 10.1038/s41467-020-19633-9
112. Kent WJ, Hsu F, Karolchik D, Kuhn RM, Clawson H, Trumbower H, Haussler D. Exploring relationships and mining data with the UCSC Gene Sorter. Genome Res. (2005) 15(5):737–41. doi: 10.1101/gr.3694705
113. Szalat R, Samur MK, Fulciniti M, Lopez M, Nanjappa P, Cleynen A, et al. Nucleotide Excision Repair Is a Potential Therapeutic Target in Multiple Myeloma. Leukemia (2018) 32:111–9. doi: 10.1038/leu.2017.182
114. Kandoth C, Mclellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational Landscape and Significance Across 12 Major Cancer Types. Nature (2013) 502:333–9. doi: 10.1038/nature12634
115. Sears CR. DNA Repair as an Emerging Target for COPD-Lung Cancer Overlap. Respir Invest (2019) 57:111–21. doi: 10.1016/j.resinv.2018.11.005
116. Kraemer KH, Lee MM, Scotto J. DNA Repair Protects Agains Cutaneous and Internal Neoplasia: Evidence From Xeroderma Pigmentosum. Carcinogenesis (1984) 5:511–4. doi: 10.1093/carcin/5.4.511
117. Wu YH, Cheng YW, Chang JT, Wu TC, Chen CY, Lee H. Reduced XPC Messenger RNA Level may Predict a Poor Outcome of Patients With Nonsmall Cell Lung Cancer. Cancer (2007) 110:215–23. doi: 10.1002/cncr.22743
118. Zhu Y, Yang H, Chen Q, Lin J, Grossman HB, Dinney CP, et al. Modulation of DNA Damage/DNA Repair Capacity by XPC Polymorphisms. DNA Repair (Amst) (2008) 7:141–8. doi: 10.1016/j.dnarep.2007.08.006
119. Stearman RS, Dwyer-Nield L, Zerbe L, Blaine SA, Chan Z, Bunn PA, et al. Analysis of Orthologous Gene Expression Between Human Pulmonary Adenocarcinoma and a Carcinogen-Induced Murine Model. Am J Pathol (2005) 167:1763–75. doi: 10.1016/S0002-9440(10)61257-6
120. Saviozzi S, Ceppi P, Novello S, Ghio P, Lo Iacono M, Borasio P, et al. Non-Small Cell Lung Cancer Exhibits Transcript Overexpression of Genes Associated With Homologous Recombination and DNA Replication Pathways. Cancer Res (2009) 69:3390–6. doi: 10.1158/0008-5472.CAN-08-2981
121. De Feraudy S, Ridd K, Richards LM, Kwok PY, Revet I, Oh D, et al. The DNA Damage-Binding Protein XPC Is a Frequent Target for Inactivation in Squamous Cell Carcinomas. Am J Pathol (2010) 177:555–62. doi: 10.2353/ajpath.2010.090925
122. Wu YH, Tsai Chang JH, Cheng YW, Wu TC, Chen CY, Lee H. Xeroderma Pigmentosum Group C Gene Expression Is Predominantly Regulated by Promoter Hypermethylation and Contributes to P53 Mutation in Lung Cancers. Oncogene (2007) 26:4761–73. doi: 10.1038/sj.onc.1210284
123. Holcomb N, Goswami M, Han SG, Clark S, Orren DK, Gairola CG, et al. Exposure of Human Lung Cells to Tobacco Smoke Condensate Inhibits the Nucleotide Excision Repair Pathway. PloS One (2016) 11:e0158858. doi: 10.1371/journal.pone.0158858
124. Lee H-W, Wang H-T, Weng M-W, Chin C, Huang W, Lepor H, et al. Cigarette Side-Stream Smoke Lung and Bladder Carcinogenesis: Inducing Mutagenic Acrolein-DNA Adducts, Inhibiting DNA Repair and Enhancing Anchorage-Independence-Growth Cell Transformation. Oncotarget (2015) 6:33226–36. doi: 10.18632/oncotarget.5429
125. Lee HW, Park SH, Weng MW, Wang HT, Huang WC, Lepor H, et al. E-Cigarette Smoke Damages DNA and Reduces Repair Activity in Mouse Lung, Heart, and Bladder as Well as in Human Lung and Bladder Cells. Proc Natl Acad Sci USA (2018) 115:E1560–9. doi: 10.1073/pnas.1718185115
126. Sands AT, Abuin A, Sanchez A, Conti CJ, Bradley A. High Susceptibility to Ultraviolet-Induced Carcinogenesis in Mice Lacking XPC. Nature (1995) 377:162–5. doi: 10.1038/377162a0
127. Friedberg EC, Bond JP, Burns DK, Cheo DL, Greenblatt MS, Meira LB, et al. Defective Nucleotide Excision Repair in XPC Mutant Mice and its Association With Cancer Predisposition. Mutat Res (2000) 459:99–108. doi: 10.1016/S0921-8777(99)00068-3
128. Melis JP, Wijnhoven SW, Beems RB, Roodbergen M, Van Den Berg J, Moon H, et al. Mouse Models for Xeroderma Pigmentosum Group A and Group C Show Divergent Cancer Phenotypes. Cancer Res (2008) 68:1347–53. doi: 10.1158/0008-5472.CAN-07-6067
129. De Oliveira Alves N, Martins Pereira G, Di Domenico M, Costanzo G, Benevenuto S, De Oliveira Fonoff AM, et al. Inflammation Response, Oxidative Stress and DNA Damage Caused by Urban Air Pollution Exposure Increase in the Lack of DNA Repair XPC Protein. Environ Int (2020) 145:106150. doi: 10.1016/j.envint.2020.106150
130. Hollander MC, Philburn RT, Patterson AD, Velasco-Miguel S, Friedberg EC, Linnoila RI, et al. Deletion of XPC Leads to Lung Tumors in Mice and Is Associated With Early Events in Human Lung Carcinogenesis. Proc Natl Acad Sci USA (2005) 102:13200–5. doi: 10.1073/pnas.0503133102
131. Cheo DL, Burns DK, Meira LB, Houle JF, Friedberg EC. Mutational Inactivation of the Xeroderma Pigmentosum Group C Gene Confers Predisposition to 2-Acetylaminofluorene-Induced Liver and Lung Cancer and to Spontaneous Testicular Cancer in Trp53-/- Mice. Cancer Res (1999) 59:771–5.
132. Wu YH, Wu TC, Liao JW, Yeh KT, Chen CY, Lee H. P53 Dysfunction by Xeroderma Pigmentosum Group C Defects Enhance Lung Adenocarcinoma Metastasis via Increased MMP1 Expression. Cancer Res (2010) 70:10422–32. doi: 10.1158/0008-5472.CAN-10-2615
133. Cui T, Srivastava AK, Han C, Yang L, Zhao R, Zou N, et al. XPC Inhibits NSCLC Cell Proliferation and Migration by Enhancing E-Cadherin Expression. Oncotarget (2015) 6:10060–72. doi: 10.18632/oncotarget.3542
134. Islami F, Ward EM, Sung H, Cronin KA, Tangka FKL, Sherman RL, et al. Annual Report to the Nation on the Status of Cancer, Part 1: National Cancer Statistics. J Natl Cancer Inst (2021) 113(12):1648-69. doi: 10.1093/jnci/djab131
135. Qin F, Gao SL, Xu K, Su QX, Zhang Z, Shi L, et al. XPC Exon15 Lys939Gln Variant Increase Susceptibility to Prostate Adenocarcinoma: Evidence Based on 4306 Patients and 4779 Controls. Medicine (Baltimore) (2020) 99:e21160. doi: 10.1097/MD.0000000000021160
136. Zhang Y, Yu JJ, Tian Y, Li ZZ, Zhang CY, Zhang SF, et al. Eif3a Improve Cisplatin Sensitivity in Ovarian Cancer by Regulating XPC and p27Kip1 Translation. Oncotarget (2015) 6:25441–51. doi: 10.18632/oncotarget.4555
137. Chen Z, Yang J, Wang G, Song B, Li J, Xu Z. Attenuated Expression of Xeroderma Pigmentosum Group C Is Associated With Critical Events in Human Bladder Cancer Carcinogenesis and Progression. Cancer Res (2007) 67:4578–85. doi: 10.1158/0008-5472.CAN-06-0877
138. Yang J, Xu Z, Li J, Zhang R, Zhang G, Ji H, et al. XPC Epigenetic Silence Coupled With P53 Alteration Has a Significant Impact on Bladder Cancer Outcome. J Urol (2010) 184:336–43. doi: 10.1016/j.juro.2010.03.044
139. Xu XS, Wang L, Abrams J, Wang G. Histone Deacetylases (HDACs) in XPC Gene Silencing and Bladder Cancer. J Hematol Oncol (2011) 4:17. doi: 10.1186/1756-8722-4-17
140. Ming M, Shea CR, Guo X, Li X, Soltani K, Han W, et al. Regulation of Global Genome Nucleotide Excision Repair by SIRT1 Through Xeroderma Pigmentosum C. Proc Natl Acad Sci USA (2010) 107:22623–8. doi: 10.1073/pnas.1010377108
141. Zhao F, Shang Y, Zeng C, Gao D, Li K. Association of Single Nucleotide Polymorphisms of DNA Repair Genes in NER Pathway and Susceptibility to Pancreatic Cancer. Int J Clin Exp Pathol (2015) 8:11579–86.
142. Zabarovsky ER, Lerman MI, Minna JD. Tumor Suppressor Genes on Chromosome 3p Involved in the Pathogenesis of Lung and Other Cancers. Oncogene (2002) 21:6915–35. doi: 10.1038/sj.onc.1205835
143. Schena M, Guarrera S, Buffoni L, Salvadori A, Voglino F, Allione A, et al. DNA Repair Gene Expression Level in Peripheral Blood and Tumour Tissue From Non-Small Cell Lung Cancer and Head and Neck Squamous Cell Cancer Patients. DNA Repair (Amst) (2012) 11:374–80. doi: 10.1016/j.dnarep.2012.01.003
144. Nahari D, Mcdaniel LD, Task LB, Daniel RL, Velasco-Miguel S, Friedberg EC. Mutations in the Trp53 Gene of UV-Irradiated Xpc Mutant Mice Suggest a Novel Xpc-Dependent DNA Repair Process. DNA Repair (2004) 3:379–86. doi: 10.1016/j.dnarep.2003.03.001
145. Krzeszinski JY, Choe V, Shao J, Bao X, Cheng H, Luo S, et al. XPC Promotes MDM2-Mediated Degradation of the P53 Tumor Suppressor. Mol Biol Cell (2014) 25:213–21. doi: 10.1091/mbc.e13-05-0293
146. Lin PS, Mcpherson LA, Chen AY, Sage J, Ford JM. The Role of the Retinoblastoma/E2F1 Tumor Suppressor Pathway in the Lesion Recognition Step of Nucleotide Excision Repair. DNA Repair (Amst) (2009) 8:795–802. doi: 10.1016/j.dnarep.2009.03.003
147. Sun C-C, Li S-J, Yuan Z-P, Li D-J. MicroRNA-346 Facilitates Cell Growth and Metastasis, and Supresses Cell Apoptosis in Human Non-Small Cell Lung Cancer by Regulation of XPC/ERK/Snail/E-Cadherin Pathway. Aging (2016) 8:2509–24. doi: 10.18632/aging.101080
148. Yeh KT, Wu YH, Lee MC, Wang L, Li CT, Chen CY, et al. XPC mRNA Level may Predict Relapse in Never-Smokers With Non-Small Cell Lung Cancers. Ann Surg Oncol (2012) 19:734–42. doi: 10.1245/s10434-011-1992-9
149. Zhang R, Jia M, Xue H, Xu Y, Wang M, Zhu M, et al. Genetic Variants in ERCC1 and XPC Predict Survival Outcome of non-Small Cell Lung Cancer Patients Treated With Platinum-Based Therapy. Sci Rep (2017) 7:10702. doi: 10.1038/s41598-017-10800-5
150. Lai TC, Chow KC, Fang HY, Cho HC, Chen CY, Lin TY, et al. Expression of Xeroderma Pigmentosum Complementation Group C Protein Predicts Cisplatin Resistance in Lung Adenocarcinoma Patients. Oncol Rep (2011) 25:1243–51. doi: 10.3892/or.2011.1184
151. Lehmann AR, Fassihi H. Molecular Analysis Directs the Prognosis, Management and Treatment of Patients With Xeroderma Pigmentosum. DNA Repair (Amst) (2020) 93:102907. doi: 10.1016/j.dnarep.2020.102907
152. Hatano K, Kumar B, Zhang Y, Coulter JB, Hedayati M, Mears B, et al. A Functional Screen Identifies miRNAs That Inhibit DNA Repair and Sensitize Prostate Cancer Cells to Ionizing Radiation. Nucleic Acids Res (2015) 43:4075–86. doi: 10.1093/nar/gkv273
153. Muenyi CS, States VA, Masters JH, Fan TW, Helm CW, States JC. Sodium Arsenite and Hyperthermia Modulate Cisplatin-DNA Damage Responses and Enhance Platinum Accumulation in Murine Metastatic Ovarian Cancer Xenograft After Hyperthermic Intraperitoneal Chemotherapy (HIPEC). J Ovarian Res (2011) 4:9. doi: 10.1186/1757-2215-4-9
Keywords: nucleotide excision repair (NER), base excision repair (BER), lung cancer, biomarker, bladder cancer, chemotherapy, xeroderma pigmentosum (XP)
Citation: Nasrallah NA, Wiese BM and Sears CR (2022) Xeroderma Pigmentosum Complementation Group C (XPC): Emerging Roles in Non-Dermatologic Malignancies . Front. Oncol. 12:846965. doi: 10.3389/fonc.2022.846965
Received: 31 December 2021; Accepted: 09 March 2022;
Published: 21 April 2022.
Edited by:
Michael Weinfeld, University of Alberta, CanadaReviewed by:
Elliot Drobetsky, Université de Montréal, CanadaBennett Van Houten, University of Pittsburgh, United States
Copyright © 2022 Nasrallah, Wiese and Sears. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine R. Sears, Y3J1ZmF0dG9AaXUuZWR1