 Maulee Sheth1
Maulee Sheth1 Leyla Esfandiari1,2,3*
Leyla Esfandiari1,2,3*- 1Department of Biomedical Engineering, University of Cincinnati, Cincinnati, OH, United States
- 2Department of Electrical Engineering and Computer Science, University of Cincinnati, Cincinnati, OH, United States
- 3Department of Environmental and Public Health Sciences, University of Cincinnati, Cincinnati, OH, United States
Cancer is primarily a disease of dysregulation – both at the genetic level and at the tissue organization level. One way that tissue organization is dysregulated is by changes in the bioelectric regulation of cell signaling pathways. At the basis of bioelectricity lies the cellular membrane potential or Vmem, an intrinsic property associated with any cell. The bioelectric state of cancer cells is different from that of healthy cells, causing a disruption in the cellular signaling pathways. This disruption or dysregulation affects all three processes of carcinogenesis – initiation, promotion, and progression. Another mechanism that facilitates the homeostasis of cell signaling pathways is the production of extracellular vesicles (EVs) by cells. EVs also play a role in carcinogenesis by mediating cellular communication within the tumor microenvironment (TME). Furthermore, the production and release of EVs is altered in cancer. To this end, the change in cell electrical state and in EV production are responsible for the bioelectric dysregulation which occurs during cancer. This paper reviews the bioelectric dysregulation associated with carcinogenesis, including the TME and metastasis. We also look at the major ion channels associated with cancer and current technologies and tools used to detect and manipulate bioelectric properties of cells.
Introduction
Carcinogenesis, also termed oncogenesis or tumorigenesis, is rooted in two major theories or hypotheses, both significantly different from one another. The somatic mutation theory (SMT), which has been prevailing in cancer research for more than sixty years proposes that the origin of cancer can be explained by an accumulation of several DNA mutations in a single somatic cell. Tumor development is then a multistep process where successive mutations produce advantageous biological compatibilities (1). The SMT explains many features of cancer such as hereditary cancers and the success of gene-targeting cancer therapies (2). However, non-genotoxic carcinogens which induce cancer without any DNA modifications (3) and the absence of mutations in some tumors (4) contradict this theory. Alternatively, the tissue organization field theory (TOFT) proposed in 1999, hypothesizes that carcinogenesis is a problem of tissue organization instead of having a cellular level origin. Here, the carcinogenic agents disrupt the reciprocal interactions between cells that maintain tissue organization, repair, and homeostasis, hence creating altered microenvironments which allow the parenchymal cells to exercise their ability to proliferate and migrate (5).
Bioelectric regulation is an important mechanism of cell communication and dysregulation of this mechanism can result into alterations in tissue organization, fitting the tissue organization field theory of carcinogenesis. While bioelectricity has been extensively studied in cells with neural origins, its role in non-neural cell activity and functionality has only emerged more recently. With advances in understanding the underlying bioelectric mechanisms of cancer and development of molecular tools to measure and control these electric fields, we are now able to better identify the role of bioelectric signaling in carcinogenesis. Another important mechanism that facilitates intercellular communication for the maintenance of tissue homeostasis is the production and release of extracellular vesicles (EVs) by cells of different tissue types. Cancer-derived EVs play a role in all steps of carcinogenesis by mediating the communication between cancer cells and non-cancer cells as well as malignant cells and non-malignant cells within the tumor microenvironment (TME) (6). Furthermore, the production of EVs is aberrant during cancer which in turn plays an important role in disturbing the bioelectrical signaling pathways between cells.
Several review papers (7–10) focusing on the bioelectric control of one or the other aspect of cancer, such as migration or metastasis, have been published. In this paper, we provide a more extensive review of bioelectric regulation in multiple cancer processes including initiation, promotion, the tumor microenvironment, and metastasis. We also look at the major ion channels implicated in cancer and current technologies and tools used to measure and manipulate bioelectric properties of cells in vivo.
Bioelectricity and Endogenous Electric Fields – An Overview
Membrane potential (Vmem) is an electrical property associated with any cell, specific to its origin and function. The electric nature of the membrane potential produces endogenous electric fields (EFs) due to the segregation of charges by molecular machines such as pumps, transporters and ion channels that are primarily located in the plasma membrane of the cell (11). These transmembrane voltage gradients have been established to control not only neural signaling via gap junctions, but also cell proliferation, migration, differentiation, and orientation in both, excitable and non-excitable cells (12, 13).
Depending on the presence of relative charges, all excitable and non-excitable cells possess an electric gradient across their plasma membrane (Figure 1A). When the cytoplasm becomes more positively charged relative to the extracellular space, the cell is said to be depolarized and will have a less negative Vmem. When the cytoplasm becomes more negatively charged relative to the extracellular space, the cell in said to be hyperpolarized and will have a more negative Vmem (Figure 1B). It is worthwhile to note that Vmem is not only a key intrinsic cellular property, but also an integral part of the microenvironment where it acts both, spatially and temporally, to guide cellular behavior (9). It does so by enabling the cells to make decisions based on the states of their neighbors (14). Physiological Vmem can range from -90 to -10 mV, depending on the cell type and physiological state (13, 15). Furthermore, as Vmem is primarily established by ion channels that are gated post-translationally, two cells that are in the exact same genetic and transcriptional states could theoretically be in very different bioelectric states (16).
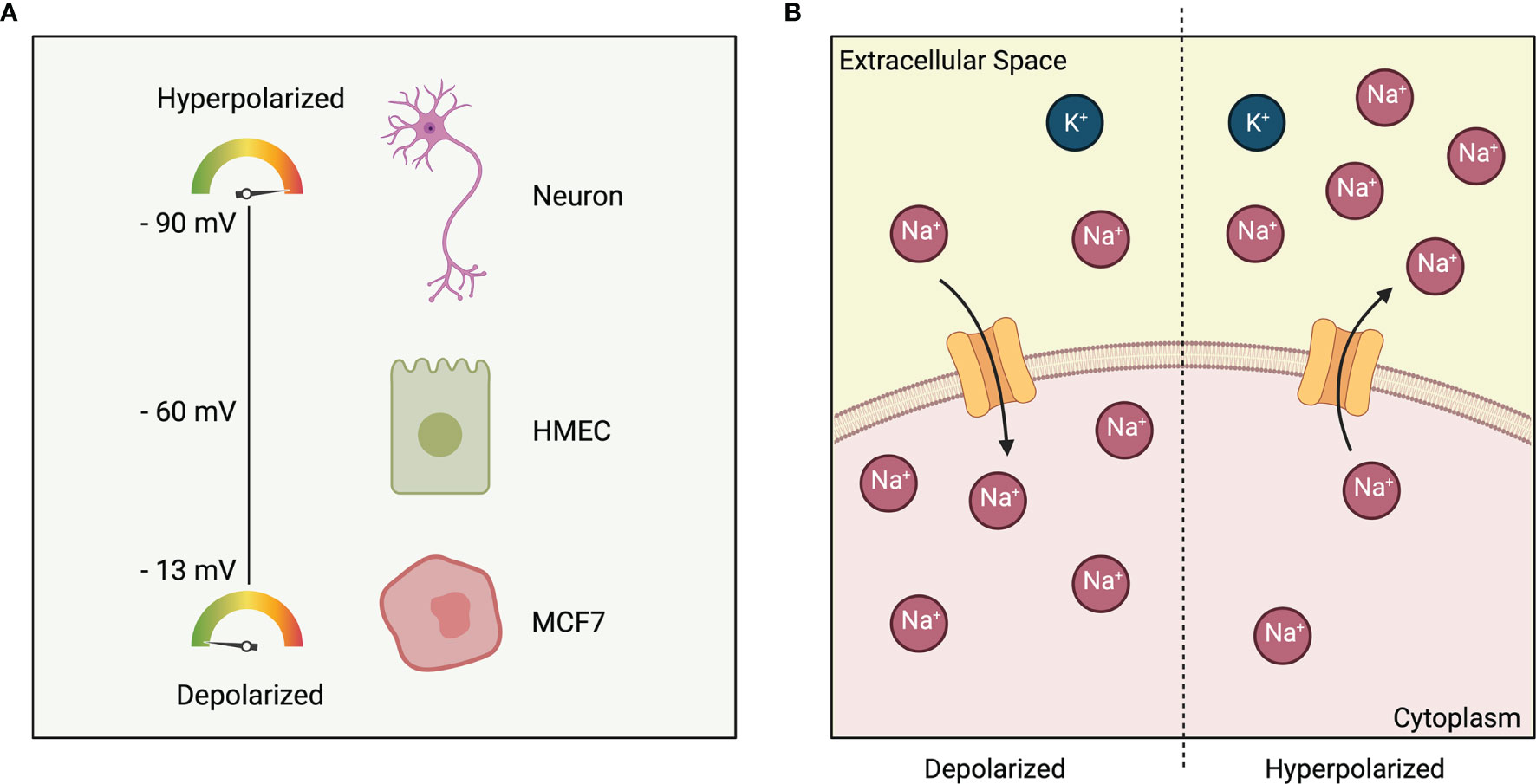
Figure 1 (A) Polarization of cells based on cell type. Excitable cell such as neurons have a membrane potential of -90 mV. Non-excitable cells such as HMEC: Human Mammary Epithelial Cell and MCF7: Estrogen-receptor-positive breast cancer cell line are at -60 mV and -13 mV respectively. (B) Depolarized cell state (left) indicated by a more positive charge in the cytoplasm relative to the extracellular space. Hyperpolarized cell state (right) indicated by a less positive charge in the cytoplasm relative to the extracellular space.
Bioelectricity in Cancer Processes
Bioelectric properties of cells and the electrical state of cells in the microenvironment are known to control several key behaviors of relevance to cancer (17–24). Here we first introduce some major ion channels implicated in cancer. Then we look at the role of bioelectricity in cancer initiation and progression, the tumor microenvironment, and migration and metastasis.
Ion Channels and Cancer
Ion channels are membrane proteins that create ionic concentration gradients by regulating the flow of ions across the plasma membrane. The primary function of ion channels is to maintain cellular homeostasis by regulating the inward and outward ion flux, but they are also higher order regulators of many downstream molecular signaling pathways (7). The four main ions that play a role in establishing the resting Vmem of a cell are: Ca2+, Na+, K+, and Cl-. The Goldman equation links the overall transmembrane potential to the concentrations and permeabilities of various ion species. The resting potential Vmem depends on the internal and external K+, Na+, and Cl- concentrations, ambient temperature, and permeability of each ion specie. Alterations in ion channel expression and activity are associated with the initiation, proliferation, and metastasis of cancer cells (21, 25). For instance, there is a host of ion channels whose expression is dysregulated in cancer cells and have been found to be associated with a metastatic phenotype (7). Here, we summarize major ion channels responsible for the disruption of homeostasis and aberrant activation of downstream signaling pathways in cancer including voltage-gated cation channels (CaV, NaV, KV), mechanosensitive cation channels, transient receptor potential (TRP) channels, and chloride channels (CLCs). Several review papers focusing extensively on ion channels implicated in cancer can be found in literature (26–29). It is worthwhile to note that disruption in expression of these ion channels leads to deregulation in a host of different signaling pathways in cancer (27–30). Prominent ones include the mitogen-activated protein kinase (MAPK) pathways, ERK and JNK signaling pathways, Wnt/ß-catenin pathway, PI3K/Akt pathway, Notch signaling, and the Rac and Rho pathways.
Calcium Channels
Voltage-gated calcium channels (VGCCs) and transient receptor potential (TRP) ion channels are primary channels facilitating Ca2+ ion diffusion. VGCCs are present in human breast cancer cells but not in normal human mammary epithelial cells (HMECs) (31). Berzingi et al. studied the effect of calcium ions on cell proliferation. Upon 5 days of culture, it was found that MCF7 breast cancer cells showed almost no growth in a culture medium without Ca2+ ions compared with cells growing to nearly 100% confluence in a medium containing 2 mM Ca2+ ions. Furthermore, blocking external Ca2+ ions from entering the cell through voltage-gated calcium channels using Verapamil indicated that cell growth was substantially inhibited in MDA-MB-231, breast cancer cells (32). The intracellular calcium concentration is also integral for cancer cell metastasis since it regulates the cell cytoskeletal dynamics, protease activity, cell volume, and pH – all of which play a role in migration and invasion of cancer cells (33–36). Calcium is also involved in driving ECM degradation and cell invasion by promoting epithelial-mesenchymal transition (EMT) pathways and the activity of matrix metalloproteinases (37, 38). Furthermore, multiple TRP channels are regulated differently in various cancers. Expression levels of TRPC3 in some breast and ovarian tumors (39) and TRPC6 in breast, liver, stomach cancers and in glioma are elevated (40). In non-small-cell lung carcinoma cells, Ca2+ entry mediated by TRPC1 and its associated signaling was found to activate the Pl3K/Akt and MAPK downstream pathways and simulate proliferation (41). Some TRP channels including TRPC1 (42), TRPC3 (43), TRPC6 (44–46), TRPM2 (47–49), and TRPM8 (50, 51) also simulate apoptosis by increasing Ca2+ activity. Consequent increase in TRPC6-mediated Ca2+ entry has also been found to alter the Notch pathway, leading to tumorigenesis in human glioblastoma multiforme (GBM) and GBM-derived cell lines (52). TRPV4 is also a critical regulator of the Rho signaling pathway involved in cancer cell adhesion and migration (53).
Sodium Channels
Cancer cells can effectively use Na+ flux to indirectly promote a metastatic phenotype. For instance, changes in Na+ flux can create localized areas of depolarization that can drive the movement of Ca2+ and H+ ions. Activity of Na+/Ca2+ exchangers located in the plasma membrane of cells has also been linked to favor ECM degradation and cell invasion, as has been demonstrated in MDA-MB-231 breast cancer cells that overexpress a voltage gated sodium channel (VGSC) (54). The expression of NaV1.7 also promotes cellular invasion at the transcriptional level by epidermal growth factor (EGF) and EGF receptor (EGFR) signaling via the ERK1/2 pathway (55). In colon cancer cells, NaV1.5 activity and the subsequent depolarization have been found to play a role in the induction of invasion-related genes through the MEK, ERK1/2 pathway (56, 57). Furthermore, a sodium-channel SCN5A has been identified as a key regulator of a genetic network that controls colon cancer invasion (57). The activity of some sodium channels has also been shown to further simulate the expression of more sodium channels in prostate and breast cancer cell lines. This allows the cells to substantially increase ion flux by creating a positive feedback loop of channel activity-induced channel expression (58). Finally, changes in the intracellular Na+ concentration can also alter cellular pH (10). A decrease in the pH surrounding a tumor is known to influence cell adhesion via the formation of integrin-mediated focal adhesion contacts (59–61).
Potassium Channels
K+ ions predominantly move from the intracellular to extracellular space through their channels to maintain the steady state resting potential of a cell. K+ indirectly affects the Vmem by driving the entry of Ca2+ into the cell. At the same time, the proliferation of some tumor cells is dependent on voltage-gated potassium channels (62–67) that alter cell volume by affecting K+ flow. A variety of tumor cells express KV10.1 (68, 69) or KV11.1 (HERG) (70) or both channels. The K+ channel EAG has been found to be expressed in 100% of cervical cancer biopsies analyzed and overexpression of EAG in human cells has been shown to increase cell proliferation in culture (71, 72). Furthermore, overexpressing K+ channels in breast cancer cells has been found to drive cell migration mediated by cadherin-11 and MAPK signaling (73). Calcium-dependent K+ channel KCa3.1 also promoted proliferation by directly interacting with ERK1/2 and JNK signaling pathways (74). Finally, Ca2+ flow through TRPM8 regulates activity of Ca2+-sensitive K+ channels such as KCa1.1, which plays a role in migration (75, 76). In breast cancer cells, overexpression of TRPM8 increased the metastatic potential via activation of the AKT glycogen synthase kinase-3 ß (GSK-3ß) pathway (77).
Chloride Channels
Chloride is the main anion that accompanies the transport of cations such as calcium, sodium, and potassium. Chloride channels play an important role in cancer cell migration due to their role in maintaining cell volume (78). Cl- channels have been revealed to have a role in glioblastomas from studies in glioma cell lines (79, 80). Studies of human prostate cancer cell lines have also shown chloride channels to play a role as key regulators of proliferation through cell size regulation (81). Chloride ion channel-4 Cl-/H+ exchanger has been found to enhance migration, invasion, and metastasis of glioma and colon cancer cells by regulating the cell volume (65). For instance, genetic knockdown of ClC-3 has been found to substantially reduce migration in glioma cells (82).
Piezo Channels
Piezo channels are non-selective Ca2+-permeable channels whose gating can be simulated by several mechanical stimuli affecting the plasma membrane, including compression, stretching, poking, shear stress, membrane tension, and suction (83–85). A recent study has also demonstrated that Piezo channels show significant sensitivity to voltage cues and thus can also be viewed as important members of the voltage-gated ion channel family (86). Two major piezo channels – Piezo1 and Piezo2 have been identified which are mainly expressed in different tissues. Piezo channels are overexpressed in several cancers, such as breast, gastric, and bladder, whereas in other cancers, their downregulation has been described. Several studies conducted in vitro and in vivo have demonstrated that the activation of Piezo channels can drive a Ca2+ influx, thus modulating key Ca2+-dependent signaling pathways associated with cancer cell migration, proliferation, and angiogenesis (87). Overexpression of Piezo1 has also been found to promote prostate cancer development through the activation of the Akt/mTOR pathway (88). Furthermore, the mechanistic effects of Piezo2 are associated with a Ca2+-dependent upregulation of Wnt11 expression which enhances the angiogenic potential of endothelial cells in cancer via ß-catenin-dependent signaling (89).
Cancer Initiation and Promotion
Resting potential established by ion channel and pump proteins is important for determination of differentiation state and proliferation. One way that carcinogenesis occurs is due to the disruption of electrical gradients, or the mechanisms by which they are perceived by cells (24). Vmem is an important non-genetic biophysical aspect of the microenvironment that regulates the balance between normally patterned growth and carcinogenesis (7). Cancerous and proliferative tissues are generally more positively charged or depolarized than non-proliferative cells (90, 91). Vmem values from -10 to -30 mV correspond to more undifferentiated, proliferative, and stem-like cells (92). For instance, the resting membrane potential in normal human mammary epithelial cells (HMEC) is -60 mV. This value goes up to -13 mV in breast cancer cells isolated from patients (93). Berzingi et al. experimentally compared Vmem in HMEC and two different invasive ductal human carcinoma cell lines, MCF7 (estrogen-receptor-positive) and MDA-MB-231 (estrogen-receptor-negative). The results indicated that MCF7 and MDA-MB-231 cells are 30.4 mV and 27.3 mV more depolarized in comparison to HMEC cells, respectively. It was also seen that HMEC grew at a much slower rate compared to MCF7 and MDA-MB-231 (32).
Lobikin et al. used a Xenopus tadpole model to confirm the role of ion flow in oncogenesis in vivo by investigating the consequences of depolarizing select cell groups (67). Embryos were exposed to glycine-gated chloride channel (GlyCl) activator ivermectin to control the membrane potential of a widely distributed, sparse population of cells expressing the GlyCl channel. The membrane potential of these specific cells could be set to any desired level by manipulating external chloride levels following ivermectin treatment. Tadpoles whose cells were depolarized were seen to exhibit excess melanocytes with a much more arborized appearance and colonize areas normally devoid of melanocytes, such as around the eyes and mouth. It was also shown that depolarization induces the up regulation of cancer relevant genes such as Sox10 and SLUG (94). Furthermore, susceptibility to oncogene-induced tumorigenesis was shown to be significantly reduced by forced prior expression of hyperpolarizing ion channels indicating that bioelectric signaling of the cellular microenvironment can both, induce and suppress, cancer-like cell behavior.
Vmem has been suggested as a cancer biomarker due to its role as an early indicator of tumorigenesis and is associated with tumors of diverse molecular origin (95–98). Induced tumor like structures (ITLS) can be formed in Xenopus and zebrafish embryos by mis-expressing mammalian oncogenes (Gli1, Xrel3 and KRASG12D) and mutant tumor suppressors (p53Trp248). ITLS’s are formed as a result of genetic interference with signaling pathways altered in several types of cancer including basal cell carcinoma, lung cancer, leukemia, melanoma, and rhabdomyosarcoma (99–102). Fluorescence reporters of Vmem in the injected animals have been found to reveal unique depolarization of tumors and increased sodium content compared to healthy tissues (7, 103). Moreover, depolarized foci have a higher success rate in predicting tumor formation as compared to cancer specific antigen level in the serum. For instance, Chernet and Levin found that depolarized foci, while present in only 19-30% of oncogene-injected embryos, predict tumor formation with 50-56% success rate as compared to prostate specific antigen level in the serum, which when used as a biomarker for prostate cancer, has a 29% predictive value (104, 105).
Recently, Carvalho developed a computational model of cancer initiation, including the propagation of a cell depolarization wave in the tissue under consideration (106). This model looks at an electrically connected single layer tissue in two and three dimensions and simulates ion exchange between cells as well as between cells and the extracellular environment. It was seen that a polarized tissue with cells in quiescent state tends to change state if a large enough perturbation changes its homeostatic conditions, such as a carcinogenic event. The induced depolarized state is able to then propagate to neighboring cells in a wave like manner. The developed model shows the importance of community effects associated with cell electrical communication leading to both, short- and long-range influences and ultimately, cancer.
The Tumor Microenvironment
The microenvironment functions to guide the cell through space and to direct tissue growth through time. It also plays a significant role in the physiological outcome of a given Vmem input. The tumor microenvironment (TME) is a complex entity and consists of multiple cell types embedded in the extracellular matrix (ECM), including immune cells, endothelial cells and cancer associated fibroblasts (CAFs) which communicate with cancer cells and with other CAFs during tumor progression (107). One way this communication is mediated is by a plethora of bioactive molecules, including proteins, lipids, coding and non-coding RNAs, and metabolites, which are secreted into extracellular vesicles (EVs) (108, 109).
The mechanical microenvironment impacts bioelectric regulation and cell proliferation (9). An early indication of this were studies in the late 1900s which found that cells within a low cell density (fewer cell-cell contacts) exhibited reduced proliferation (110) and that cells in a confluent monolayer are more hyperpolarized than individual cells (111, 112). Similarly, chemical components of the cellular microenvironment have the ability to impact cell phenotype. Factors such as hypoxia (113) and pH (114) have been demonstrated to drive cancer progression. Moreover, hypoxic tumors exhibit more aggressive phenotypes. Tumor cells under hypoxia can produce a secretion partly in the form of EVs that modulates the microenvironment to facilitate tumor angiogenesis and metastasis (115). Vmem thus functions at the interface of chemical and mechanical signals by creating an electrical gradient across cells, which in turn gates voltage-sensitive channels. This creates a tightly connected communication pathway between a cell and its microenvironment (9, 116–119).
The key components of the mechanical microenvironment (Figure 2) are solid and fluid pressure, substratum stiffness (120–128) tissue geometry, and mechanical stress (129–131). These components of the physical microenvironment are primarily dependent on mechanosensitive calcium channels CaV3.3 (132, 133). Cells have the ability to sense the surrounding substratum by applying force through actomyosin motors in stress fibers linked to focal adhesions (134). Varying the substratum stiffness has been demonstrated to influence cellular behaviors including differentiation (122), apoptosis (126), proliferation (125), gene expression (135–137), migration (138), cell stiffness (139), and epithelial-mesenchymal transition (EMT) (127). Along with microenvironments of varying rigidity, cells also experience mechanical stress due to the dense packing of neighboring cells. Cell-cell contacts are critical for propagation of bioelectric signals via the transport of ions through gap junctions (140–142). The normal breast epithelium cell line MCF10A was demonstrated to respond differently to an EF in vitro depending on the confluency of the cell culture (143). The study observed that clustered cells are more sensitive to an EF due to increased cell-cell contacts.
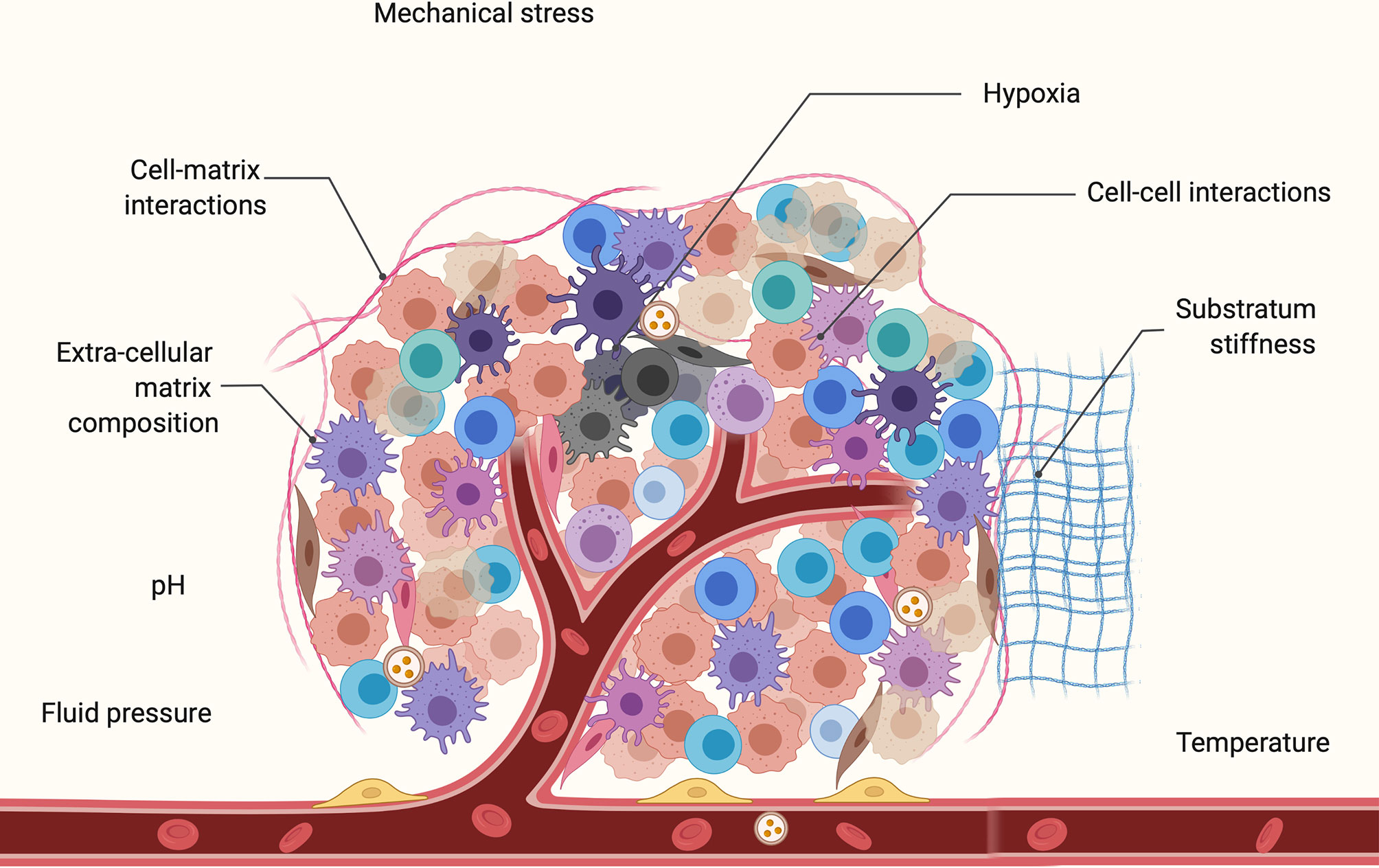
Figure 2 Key components of the tumor microenvironment (TME) which comprises of multiple cell types including cancer cells, immune cells, endothelial cells, and cancer associated fibroblasts. This includes mechanical components such as fluid pressure, substratum stiffness, mechanical stress, cell-cell, and cell-matrix interactions. Also shown are some chemical components of the microenvironment such as pH, temperature, and hypoxic core of the tumor. Cell-cell and cell-TME communication is mediated by a variety of bioactive molecules during carcinogenesis.
Physical signals from the Vmem of the microenvironment also contribute to tumorigenesis (9). Furthermore, pressure activates oncogenic factors such as p38, ERK, and c-Src which are involved in the regulation of cell proliferation, differentiation, and apoptosis (132). Tumors in vivo are under higher pressure and are also stiffer than the surrounding tissue which creates a microenvironment that promotes cell proliferation (133). Increased pressure also enhances the invasiveness of tumor cells (121). Additionally, a key communication pathway between cells and their ECM is Integrin signaling pathway which regulates cytosolic Ca2+ levels (144). These cytosolic Ca2+ concentrations play an important role in cancer-related processes such as EMT (38), metastasis (21), and apoptosis (126, 145). For instance, inducing EMT in human breast cancer cells has been shown to upregulate cytosolic calcium levels (38).
Cell Migration and Metastasis
The dissemination of primary tumor cells to secondary organs is called metastasis. This involves cancer cells breaking away from the primary tumor, traveling through blood or lymphatic systems, and forming secondary or metastatic tumors in other parts of the body. Metastasis is a multi-step process (Figure 3) and involves the following events: local invasion to surrounding tissues, intravasation into the vasculature or lymphatics (where they are called circulating tumor cells or CTCs), survival and circulation in the vessels, and extravasation and colonization in a secondary organ (where they are called disseminated tumor cells or DTCs) (146, 147). Bioelectricity mediates many of the normal cell functions which are disrupted in metastasis. Factors such as ion channel expression, Vmem, and external EFs have been determined to regulate invasion and metastasis. Furthermore, the migration of cancer cells out of the primary tumors into local tissues through various physical barriers is driven by components of the local tumor microenvironment and executed by complex signaling pathways in the cell (10).
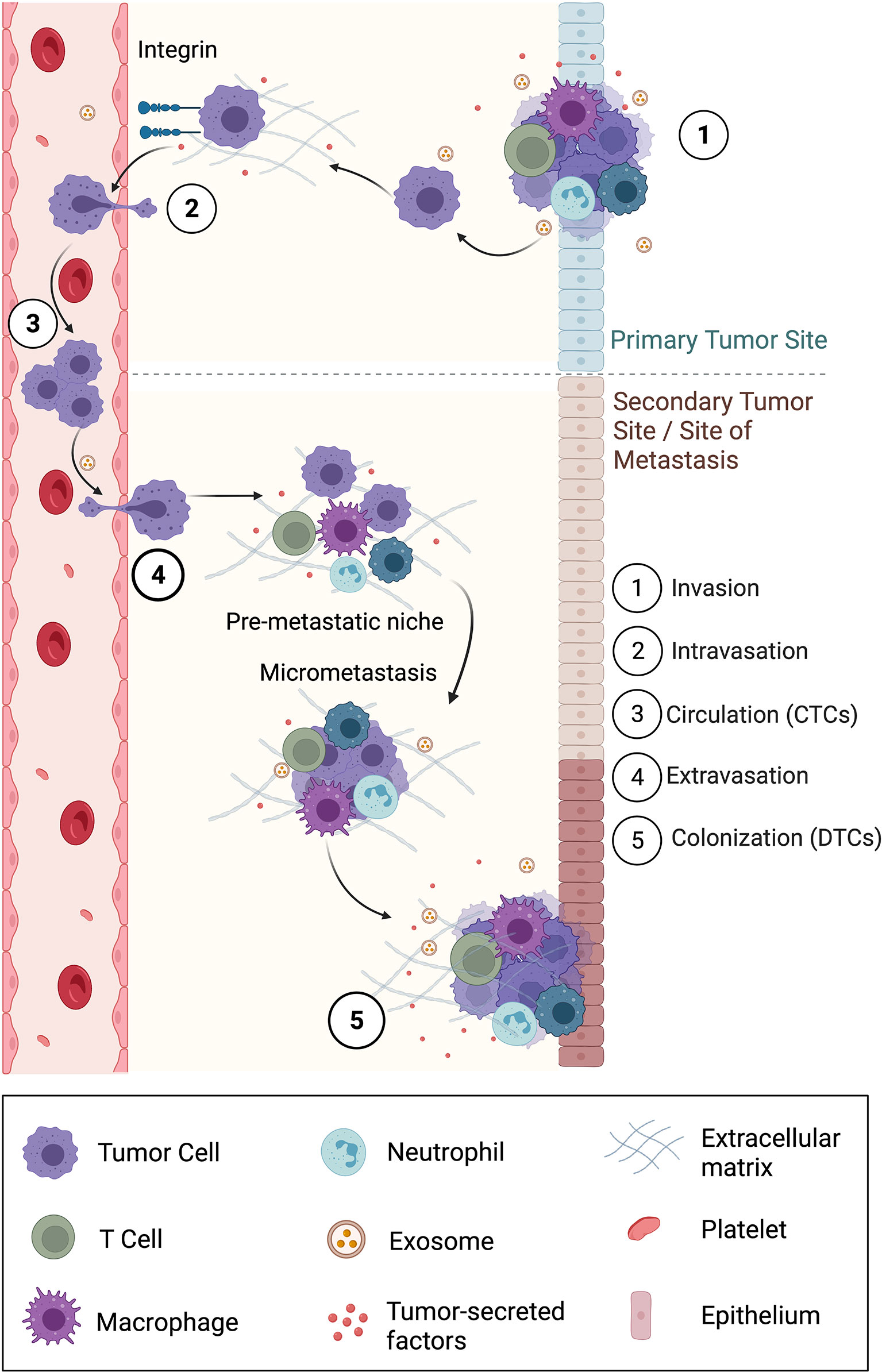
Figure 3 Overview of the five-step metastatic cascade involving local invasion, intravasation into surrounding vasculature, circulation, extravasation, and finally colonization in a secondary location. Also shown is the formation of a pre-metastatic niche that supports the survival of disseminated tumor cells (DTCs) into a successful metastasis. Exosomes, a subpopulation of EVs play a primary role in carrying information from the primary site to the secondary site or site of metastasis, especially to form the pre-metastatic niche.
Cues within the TME can promote local invasion (148). For instance, an ECM protein fibronectin can attract breast cancer tumor cells to the vasculature via haptotaxis (directional migration in response to substrate-bound cues) to promote dissemination. During tumor invasion, constant communication occurs between tumor cells and surrounding stromal cells via extracellular vesicles (EVs) (115). Even upon entering a secondary tissue, the transition of a DTC into an overt metastasis is highly dependent on the local microenvironment of this organ (10). Hence, the formation of a supportive premetastatic niche, composed of ECM and resident immune cells is essential to provide nutrients and survival signals that drive DTC survival and outgrowth. Recent work suggests that tumor cells may be able to prime the premetastatic site from a distance before colonization to create a more favorable niche, for example, by secreting exosomes, a subpopulation of EVs (6). Furthermore, even within cancer cells, there is variability in the amount of depolarization. A more depolarized Vmem is associated with a higher metastatic potential and forced hyperpolarization of cells can reduce their migration and invasiveness (24, 141, 149, 150).
To study the effect of EFs on cell galvanotaxis, Zhu et al. employed unique probe systems to characterize cancer cell electrical properties and their migratory ability under an EF (151). It was found that tumors established from 4T1, a triple-negative murine breast cancer cell line, produced heterogeneous intratumor potentials causing a flow of endogenous EFs inside and outside of the tumors, which may in turn affect cell migration behavior and ultimately contribute to cancer metastasis. Moreover, tumor electric potentials were found to increase with increase in tumor size, which is an important factor since the primary tumor size has been reported to be linked to the metastatic potential (152, 153). Finally, it was also found that metastatic sublines (m4T1) from lung, heart, axillary lymph node and spleen showed different galvanotaxis thresholds. For instance, parental 4T1 and lung metastatic cell lines were found to respond to EFs as low as 50 mV/mm, while other metastatic sublines showed an anodal migration in a field of 100 mV/mm or higher. Additionally, the migration speeds also varied among different metastatic sublines. Cancer cell monolayers were found to have a higher migration persistence (defined as the ratio of displacement to trajectory length) under EFs than that of isolated cells, suggesting that cancer cells migrated more linearly in a certain direction when responding to EFs collectively.
Interestingly, bioelectric factors override most chemical gradients and other cues in a multi-cue environment during cell migration (8). Lobikin et al. investigated a cell population termed as “instructor” cells which when depolarized, is able to direct the activity of an entirely different set of cells (7). The “instructor” cells induce metastatic phenotype in normal melanocytes by serotonergic signaling, a mechanism which mediates long-range bioelectric signaling. Furthermore, instructor cells also disrupt blood vessel patterning upon depolarization. The melanocytes were then found to acquire three properties commonly associated with metastasis – hyper-proliferation, a highly dendritic morphology, and invasion into tissues such as blood vessels, gut, and neural tube. This data illustrated the power of depolarized Vmem as an epigenetic initiator of widespread metastatic behavior in the absence of a centralized tumor.
Applications
Current Devices, Materials and Technologies
Molecular-resolution tools have recently been developed for real-time detection and manipulation of bioelectric properties in vivo (91, 154). An important component of such investigations is the ability to track spatio-temporal distribution of Vmem gradients in vivo, over significant periods of time.
Detection of Bioelectric Properties
Microelectrodes are a common tool used to measure the electrophysiological characteristics of cells and are extremely powerful for single cell measurements. For instance, Zhu et al. used glass microelectrodes to measure intratumor potentials in subcutaneous tumors established from a triple-negative murine cancer cell line (4T1) (151). However, measurements corresponding to multicellular areas and volumes are constrained by the smaller size of these electrodes. Furthermore, the sample under study needs to be kept perfectly still (154). Fluorescent bioelectricity reporters are a more recent development which has facilitates measurement of electrophysiological properties when it is not feasible to use microelectrodes. These dyes can be used to achieve subcellular resolution, measure many cells simultaneously in vivo, and to track bioelectric gradients over long period of time despite cell movements and divisions (154). Chernet and Levin utilized voltage-sensitive fluorescent dyes to non-invasively detect areas of depolarization in oncogene-induced tumor structures in Xenopus larvae (24). A few other tools for the characterization of bioelectrical events are highly sensitive ion-selective extracellular electrode probes (105, 155) that reveal ion flux at the cell membrane, reporter proteins (156–159) and techniques that report individual ion species content such as protons (160) and sodium (161). Bioelectronic sensors or biosensors can also be used to sense electric fields, ionic concentrations, and biological markers (162–167). Based on the type of sensor, both intracellular and extracellular recordings of a single cell or a group of cells can be measured. A common transistor biosensor platform used for extracellular recordings is the organic electrochemical transistor (OECT) which is inherently sensitive to ionic species and external electric fields (14). The OECT is typically made of a poly(3,4-ethylenedioxythiophene): polystyrene sulfonate (PEDOT : PSS) mixture and has been implemented for recording of electrochemical gradients in non-excitable cells such as Caco-2 as well as excitable cells (168). Meanwhile, silicon nanowires are suitable for crossing the cell membrane and are commonly used for intracellular readings. These nanowires are synthesized with spatially controlled electrical properties. A nanoscale field effect transistor (NFET) is then created on an individual nanowire by varying the doping levels. NFETs allow localized and tuneable 3D sensing and recording of single cells and even 3D cellular networks. By having a three-dimensional probe presentation, NFETs overcome a major limitation of most traditional nanoelectronic devices which have a more planar design. Tian et al. used three-dimensional NFETs as localized bioprobes for intracellular readings in cardiomyocytes (169). While these methods are excellent tools for measuring cell electrical properties, tools that can manipulate these properties are essential to study the effects of altering cell states.
Manipulation of Bioelectric Properties
Bioactuators are a class of devices that can be used to modify cell behavior by delivering directly biophysical signals such as electrophoretic delivery of ions and small molecules targeting specific cell locations (14). Additionally, a variety of nanomaterials have been developed for reading and writing bioelectric cues in tissue. These include biocompatible piezoelectric materials and nanoparticles that alter the resting potential of cells by contact, without the use of transgenes (170–173). Warren and Payne determined that nanoparticles with amine-modified surfaces induced significant depolarization in both, Chinese Hamster Ovary (CHO) cells and HeLa cells (173). Conductive polymers are another class of materials that can stimulate cells or tissue cultured upon them (174–176) by applying an electrical signal. Conductive polymers used in tissue engineering include conductive nanofibers, conductive hydrogels, conductive composite films, and conductive composite scaffolds fabricated using methods such as electrospinning, coating, or deposition by in situ polymerization (177). For instance, Jayaram et al. used PEDOT : PSS conducting polymer microwires to depolarize cells and achieve a more positive membrane potential in E. coli cells (170). Thourson and Payne also demonstrated the use of PEDOT : PSS microwires to control action potentials of cardiomyocytes (178). Conductive polymer microwires thus provide a minimally invasive platform to control electrical properties of cells with high spatial precision. Detailed reviews on conductive polymers have been previously published (177, 179).
As mentioned previously, treatment with ivermectin is another way to control the transmembrane potential of a select group of cells by manipulating of endogenous chloride channels (Figure 4A). Ivermectin targets GlyR-expressing cells and hence opens their chloride channels. Chloride ions can then be made to enter or exit the GlyR-expressing cells by manipulating the external chloride levels, thus controlling their transmembrane potential (7). For instance, a low level of chloride in the external medium would cause chloride ions to exit the cell and into the medium, hence depolarizing the cell. Lobikin et al. employed this method in frog models to regulate the membrane potential of a specific group of cells expressing GlyCl channels to desired levels and study the consequences on metastasis and tumorigenesis in vivo.
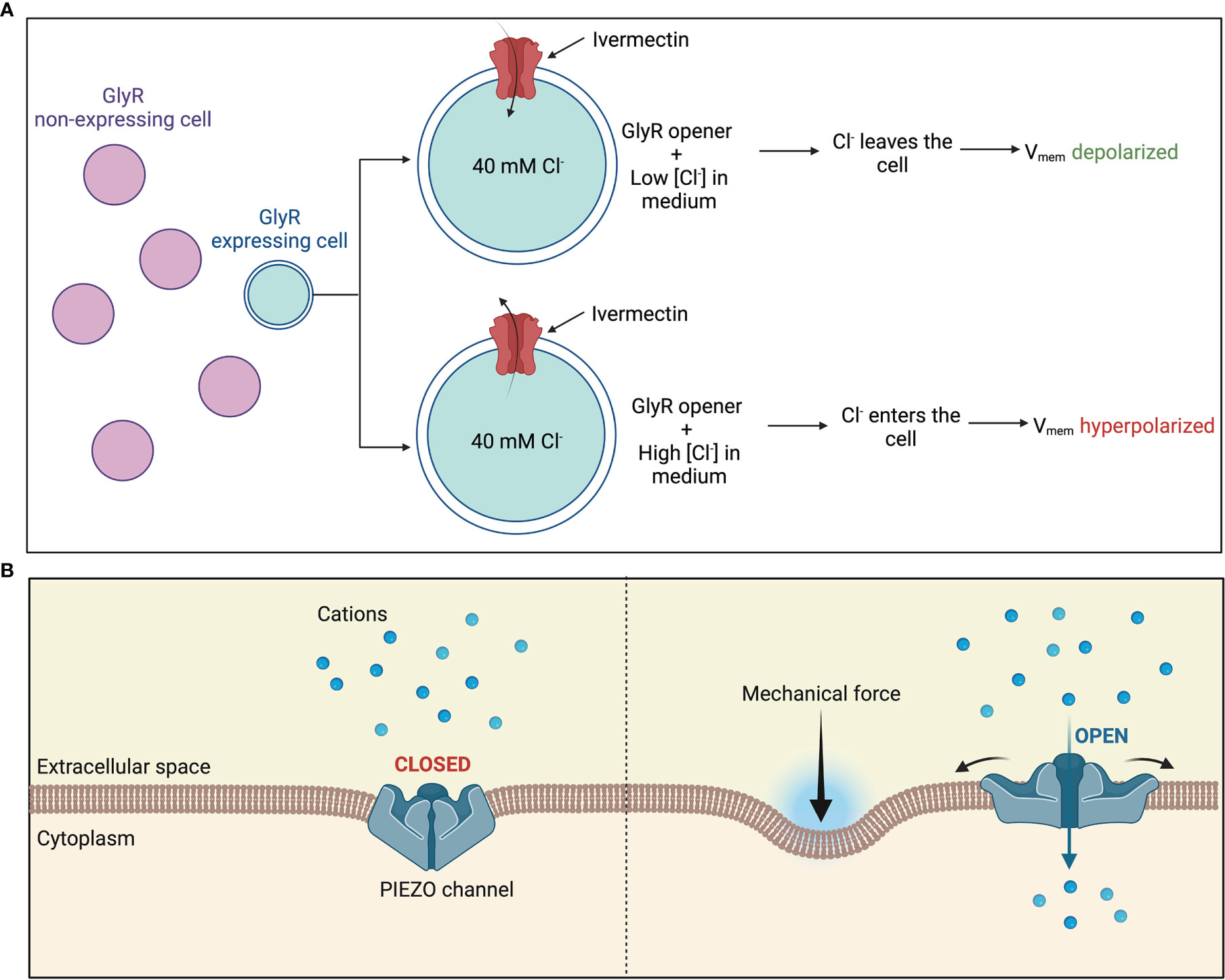
Figure 4 Manipulating bioelectric properties of cells (A) Manipulation of endogenous chloride channels as a means of manipulating Vmem of a select group of cells. Treatment with ivermectin causes chloride channels in GlyR-expressing cells to open. External chloride levels are then manipulated to regulate movement of chloride flux into or out of the cytoplasm (B) Piezo1 and Piezo2 are mechanically activated cation channels. Application of a mechanical force causes the central pore to open, allowing an influx of positive charge into the cell.
Another potential way to manipulate the bioelectric properties of cells is by controlling the mechanosensitive Ca2+-permeable Piezo channels which have emerged as major transducers of mechanical stress into Ca2+ dependent signals. These mechanosensitive Piezo channels expressed on the plasma membrane are gated by various mechanical stimuli such as stiffness, compression, tension forces, and shear stress. Channel activation then allows a Ca2+ influx into the cytoplasm which then mediates the cell polarity (Figure 4B). Piezo1 may also be pharmacologically activated by agonists such as Jedi1, Jedi2 and Yoda1 or inhibited by channel pore blockers, competitive antagonists, and peptides such as Ruthenium Red, GsMTx-4, Dooku1 and Aß peptides which distort the membrane mechanical properties (87). Han et al. demonstrated that activation of Piezo1 via mechanical stimuli in 1 μm using a heat-polished glass probe controlled by a piezo electric device or via agonist Yoda1 mediated Ca2+ influx in pancreatic cancer cells, resulting into a more depolarized state (88).
Extracellular Vesicles and Electricity
Extracellular vesicles (EVs) facilitate inter-cellular communication via delivery of proteins and nucleic acids, including microRNA (miRNA) and mRNA (180). EVs-mediated communication is vital during the establishment of planar cell polarity and the developmental patterning of tissues (181). EVs are particularly enriched in the tumor microenvironment (182, 183) and as mentioned in the previous sections of this paper, they play a special role in cancer development and progression. In a recent study, Fukuta et al. demonstrated that external stimuli such as low levels of electric field treatment that activate intracellular signaling would likely increase exosome secretion from the cells. It was seen that an electric field of 0.34 mA/cm2 increases the secretion of these EVs from cultured cells of murine melanoma B16F1 and murine fibroblast 3T3 Swiss Albino without compromising their quality (180). These results together indicate that the bioelectric dysregulation or depolarization of cells that occurs during cancer may be responsible for the upregulation of EVs in the cancer tumor microenvironment. At the same time, the increase in production of EVs plays a role in disrupting the bioelectric homeostasis, forming a feedback loop. The change in cell state and EV production along with the interdependence of the two are major mechanisms responsible for the bioelectric dysregulation of cancer.
Conclusions
Bioelectric signaling is a growing field of study that takes us a step closer to understanding cancer as a disease, all the way from initiation to metastasis. A lot is known about cancer and its biology as per the somatic mutation theory. On the other hand, the role of electric fields in cancer processes, while strongly established over the last few decades, needs further investigation. Understanding the bioelectric mechanisms underlying cancer is especially important since it will allow us to develop new biomedical and bioengineering tools and techniques as per the tissue organization field theory. These new engineering tools, along with the existing biological knowledge will enhance our understanding of cancer and enable the development of novel treatments for patients.
Another exciting area of study is the interplay between the bioelectric dysregulation and enhancement of extracellular vesicles (EVs) within the context of the cancer microenvironment. It has been well established that EVs play a significant role in facilitating the signaling pathways involved in all processes of carcinogenesis. This paper provides a detailed review of the current knowledge about bioelectric dysregulation that underlies different processes of cancer. However, little is known about the interdependence of these two mechanisms. Furthermore, EVs, especially exosomes, have been proven to have a role in therapeutic strategies for cancer. Understanding this crosstalk will not only enhance our knowledge of cancer, but also help develop efficient exosome-based cancer immunotherapies and drug delivery vehicles.
Author Contributions
MS wrote the main manuscript text and prepared all figures. LE gave suggestions and ideas on the literature search and manuscript writing. All authors contributed to the article and approved the submitted version.
Funding
This work has been funded by the National Science Foundation NSF CAREER ECCS (2046037).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Figures 1 and 4A were created with BioRender.com. Figure 2 was adapted from “Tumor Microenvironment 2”, by BioRender.com (2021). Figure 3 was adapted from “Overview of Metastatic Cascade”, by BioRender.com (2021). Figure 4B was adapted from “PIEZO Channels: How Do They Allow Mechanosensation?”, by BioRender.com. (2021). Figures 2, 3, and 4B were retrieved from https://app.biorender.com/biorender-templates.
Abbreviations
EV, extracellular vesicle; TME, tumor microenvironment; Vmem, membrane potential; EF, electric field; SMT, somatic mutation theory; TOFT, tissue organization field theory; HMEC, human mammary epithelial cell; MCF7, estrogen-receptor-positive breast cancer cell line; MDA-MB-231, estrogen-receptor-negative breast cancer cell line; ECM, extracellular matrix; EMT, epithelial mesenchymal transition; CAF, cancer associated fibroblast; GlyCl, glycine-gated chloride channel; VGCC, voltage gated calcium channel; VGSC, voltage gated sodium channel; EAG, ether-à-go-go K+ channel; TRP, transient receptor potential; MAPK, mitogen-activated protein kinase; JNK, c-Jun N-terminal kinase; ERK, extracellular signal-regulated kinase; Pl3K/Akt, phosphatidylinositol 3-kinase/protein kinase B; MEK, mitogen-activated ERK kinase; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; GBM, glioblastoma multiforme; GSK-3ß, glycogen synthase kinase-3 ß; ITLS, induced tumor like structure; MCF10A, normal breast epithelium cell line; CTC, circulating tumor cell; DTC, disseminated tumor cell; mRNA, messenger RNA; miRNA, micro RNA; B16F1, murine melanoma cell line; 3T3, murine fibroblast cell line; OECT, organic electrochemical transistor; CHO, Chinese hamster ovary; PEDOT : PSS, poly(3,4-ethylenedioxythiophene): polystyrene sulfonate.
References
1. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Vaux DL. In Defense of the Somatic Mutation Theory of Cancer. Bioessays (2011) 33:341–3. doi: 10.1002/bies.201100022
3. Mally A, Chipman JK. Non-Genotoxic Carcinogens: Early Effects on Gap Junctions, Cell Proliferation and Apoptosis in the Rat. Toxicology (2002) 180:233–48. doi: 10.1016/s0300-483x(02)00393-1
4. Versteeg R. Cancer: Tumours Outside the Mutation Box. Nature (2014) 506:438–9. doi: 10.1038/nature13061
5. Sonnenschein C, Soto AM. Theories of Carcinogenesis: An Emerging Perspective. Semin Cancer Biol (2008) 18:372–7. doi: 10.1016/j.semcancer.2008.03.012
6. Wortzel I, Dror S, Kenific CM, Lyden D. Exosome-Mediated Metastasis: Communication From a Distance. Dev Cell (2019) 49:347–60. doi: 10.1016/j.devcel.2019.04.011
7. Lobikin M, Chernet B, Lobo D, Levin M. Resting Potential, Oncogene-Induced Tumorigenesis, and Metastasis: The Bioelectric Basis of Cancer In Vivo. Phys Biol (2012) 9:65002. doi: 10.1088/1478-3975/9/6/065002
8. Funk RH. Endogenous Electric Fields as Guiding Cue for Cell Migration. Front Physiol (2015) 6:143. doi: 10.3389/fphys.2015.00143
9. Silver BB, Nelson CM. The Bioelectric Code: Reprogramming Cancer and Aging From the Interface of Mechanical and Chemical Microenvironments. Front Cell Dev Biol (2018) 6:21. doi: 10.3389/fcell.2018.00021
10. Payne SL, Levin M, Oudin MJ. Bioelectric Control of Metastasis in Solid Tumors. Bioelectricity (2019) 1:114–30. doi: 10.1089/bioe.2019.0013
11. Simanov D, Mellaart-Straver I, Sormacheva I, Berezikov E. The Flatworm Macrostomum Lignano Is a Powerful Model Organism for Ion Channel and Stem Cell Research. Stem Cells Int (2012) 2012:167265. doi: 10.1155/2012/167265
12. Sundelacruz S, Levin M, Kaplan DL. Role of Membrane Potential in the Regulation of Cell Proliferation and Differentiation. Stem Cell Rev Rep (2009) 5:231–46. doi: 10.1007/s12015-009-9080-2
13. Blackiston DJ, McLaughlin KA, Levin M. Bioelectric Controls of Cell Proliferation: Ion Channels, Membrane Voltage and the Cell Cycle. Cell Cycle (2009) 8:3527–36. doi: 10.4161/cc.8.21.9888
14. Levin M, Selberg J, Rolandi M. Endogenous Bioelectrics in Development, Cancer, and Regeneration: Drugs and Bioelectronic Devices as Electroceuticals for Regenerative Medicine. iScience (2019) 22:519–33. doi: 10.1016/j.isci.2019.11.023
15. Al Ahmad M, Al Natour Z, Mustafa F, Rizvi T. Electrical Characterization of Normal and Cancer Cells. IEEE Access PP (2018) 1:25979–86. doi: 10.1109/ACCESS.2018.2830883
16. Levin M. Molecular Bioelectricity: How Endogenous Voltage Potentials Control Cell Behavior and Instruct Pattern Regulation In Vivo. Mol Biol Cell (2014) 25:3835–50. doi: 10.1091/mbc.E13-12-0708
17. McCaig CD, Song B, Rajnicek AM. Electrical Dimensions in Cell Science. J Cell Sci (2009) 122:4267–76. doi: 10.1242/jcs.023564
18. Levin M. Bioelectric Mechanisms in Regeneration: Unique Aspects and Future Perspectives. Semin Cell Dev Biol (2009) 20:543–56. doi: 10.1016/j.semcdb.2009.04.013
19. Rajnicek AM, Foubister LE, McCaig CD. Growth Cone Steering by a Physiological Electric Field Requires Dynamic Microtubules, Microfilaments and Rac-Mediated Filopodial Asymmetry. J Cell Sci (2006) 119:1736–45. doi: 10.1242/jcs.02897
20. Becchetti A. Ion Channels and Transporters in Cancer. 1. Ion Channels and Cell Proliferation in Cancer. Am J Physiol Cell Physiol (2011) 301:C255–265. doi: 10.1152/ajpcell.00047.2011
21. Prevarskaya N, Skryma R, Shuba Y. Ion Channels and the Hallmarks of Cancer. Trends Mol Med (2010) 16:107–21. doi: 10.1016/j.molmed.2010.01.005
22. Pardo LA, Contreras-Jurado C, Zientkowska M, Alves F, Stühmer W. Role of Voltage-Gated Potassium Channels in Cancer. J Membr Biol (2005) 205:115–24. doi: 10.1007/s00232-005-0776-1
23. Kunzelmann K. Ion Channels and Cancer. J Membr Biol (2005) 205:159–73. doi: 10.1007/s00232-005-0781-4
24. Chernet B, Levin M. Endogenous Voltage Potentials and the Microenvironment: Bioelectric Signals That Reveal, Induce and Normalize Cancer. J Clin Exp Oncol Suppl (2013) 1:S1–002. doi: 10.4172/2324-9110.S1-002
25. Arcangeli A, Crociani O, Lastraioli E, Masi A, Pillozzi S, Becchetti A. Targeting Ion Channels in Cancer: A Novel Frontier in Antineoplastic Therapy. Curr Med Chem (2009) 16:66–93. doi: 10.2174/092986709787002835
26. Jiang LH, Adinolfi E, Roger S. Editorial: Ion Channel Signalling in Cancer: From Molecular Mechanisms to Therapeutics. Front Pharmacol (2021) 12:711593. doi: 10.3389/fphar.2021.711593
27. Lang F, Stournaras C. Ion Channels in Cancer: Future Perspectives and Clinical Potential. Philos Trans R Soc Lond B Biol Sci (2014) 369:1638. doi: 10.1098/rstb.2013.0108
28. Litan A, Langhans SA. Cancer as a Channelopathy: Ion Channels and Pumps in Tumor Development and Progression. Front Cell Neurosci (2015) 9:86. doi: 10.3389/fncel.2015.00086
29. Prevarskaya N, Skryma R, Shuba Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol Rev (2018) 98(2):559–621. doi: 10.1152/physrev.00044.2016
30. Dhillon AS, Hagan S, Rath O, Kolch W. MAP Kinase Signalling Pathways in Cancer. Oncogene (2007) 26(22):3279–90. doi: 10.1038/sj.onc.1210421
31. Taylor JT, Huang L, Pottle JE, Pottle K, Yang Y, Zeng X, et al. Selective Blockade of T-Type Ca2+ Channels Suppresses Human Breast Cancer Cell Proliferation. Cancer Lett (2008) 267:116–24. doi: 10.1016/j.canlet.2008.03.032
32. Berzingi S, Newman M, Yu HG. Altering Bioelectricity on Inhibition of Human Breast Cancer Cells. Cancer Cell Int (2016) 16:72. doi: 10.1186/s12935-016-0348-8
33. Guéguinou M, Harnois T, Crottes D, Uguen A, Deliot N, Gambade M, et al. SK3/TRPC1/Orai1 Complex Regulates SOCE-Dependent Colon Cancer Cell Migration: A Novel Opportunity to Modulate Anti-EGFR mAb Action by the Alkyl-Lipid Ohmline. Oncotarget (2016) 7:36168–84. doi: 10.18632/oncotarget.8786
34. Jacquemet G, Baghirov H, Georgiadou M, Sihto H, Peuhu E, Cettour-Janet P, et al. L-Type Calcium Channels Regulate Filopodia Stability and Cancer Cell Invasion Downstream of Integrin Signaling. Nat Commun (2016) 7:13297. doi: 10.1038/ncomms13297
35. Hegle AP, Marble DD, Wilson GF. A Voltage-Driven Switch for Ion-Independent Signaling by Ether-À-Go-Go K+ Channels. Proc Natl Acad Sci USA (2006) 103:2886–91. doi: 10.1073/pnas.0505909103
36. Ouadid-Ahidouch H, Dhennin-Duthille I, Gautier M, Sevestre H, Ahidouch A. TRP Calcium Channel and Breast Cancer: Expression, Role and Correlation With Clinical Parameters. Bull Cancer (2012) 99:655–64. doi: 10.1684/bdc.2012.1595
37. Kohn EC, Jacobs W, Kim YS, Alessandro R, Stetler-Stevenson WG, Liotta LA, et al. Calcium Influx Modulates Expression of Matrix Metalloproteinase-2 (72-kDa Type IV Collagenase, Gelatinase a). J Biol Chem (1994) 269:21505–11. doi: 10.1016/S0021-9258(17)31833-1
38. Davis FM, Azimi I, Faville RA, Peters AA, Jalink K, Putney JW Jr, et al. Induction of Epithelial-Mesenchymal Transition (EMT) in Breast Cancer Cells is Calcium Signal Dependent. Oncogene (2014) 33:2307–16. doi: 10.1038/onc.2013.187
39. Aydar E, Yeo S, Djamgoz M, Palmer C. Abnormal Expression, Localization, and Interaction of Canonical Transient Receptor Potential Ion Channels in Human Breast Cancer Cell Lines and Tissues: A Potential Target for Breast Cancer Diagnosis and Therapy. Cancer Cell Int (2009) 9:23. doi: 10.1186/1475-2867-9-23
40. Ding X, He Z, Zhou K, Cheng J, Yao H, Lu D, et al. Essential Role of TRPC6 Channels in G2/M Phase Transition and Development of Human Glioma. J Natl Cancer Inst (2010) 102(14):1052–68. doi: 10.1093/jnci/djq217
41. Tajeddine N, Gailly P. TRPC1 Protein Channel is Major Regulator of Epidermal Growth Factor Receptor Signaling. J Biol Chem (2012) 287(20):16146–57. doi: 10.1074/jbc.M112.340034
42. Kusaba T, Okigaki M, Matui A, Murakami M, Ishikawa K, Kimura T, et al. Klotho is Associated With VEGF Receptor-2 and the Transient Receptor Potential Canonical-1 Ca2+ Channel to Maintain Endothelial Integrity. Proc Natl Acad Sci USA (2010) 107(45):19308–13. doi: 10.1073/pnas.1008544107
43. Shan D, Marchase RB, Chatham JC. Overexpression of TRPC3 Increases Apoptosis But Not Necrosis in Response to Ischemia-Reperfusion in Adult Mouse Cardiomyocytes. Am J Physiol Cell Physiol (2008) 294(3):C833–41. doi: 10.1152/ajpcell.00313.2007
44. Mukerji N, Damodaran TV, Winn MP. TRPC6 and FSGS: The Latest TRP Channelopathy. Biochim Biophys Acta (2007) 1772(8):859–68. doi: 10.1016/j.bbadis.2007.03.005
45. Sun YH, Li Q, Feng SL, Li BX, Pan ZW, Xu CQ, et al. Calcium-Sensing Receptor Activation Contributed to Apoptosis Stimulates TRPC6 Channel in Rat Neonatal Ventricular Myocytes. Biochem Biophys Res Commun (2010) 394(4):955–61. doi: 10.1016/j.bbrc.2010.03.096
46. Yu L, Lin Q, Liao H, Feng J, Dong X, Ye J, et al. TGF-β1 Induces Podocyte Injury Through Smad3-ERK-NF-κb Pathway and Fyn-Dependent TRPC6 Phosphorylation. Cell Physiol Biochem (2010) 26(6):869–78. doi: 10.1159/000323996
47. Gao Y, Lei Z, Lu C, Roisen FJ, El-Mallakh RS. Effect of Ionic Stress on Apoptosis and the Expression of TRPM2 in Human Olfactory Neuroepithelial-Derived Progenitors. World J Biol Psychiatry (2010) 11(8):972–84. doi: 10.3109/15622975.2010.507784
48. Hecquet CM, Malik AB. Role of H(2)O(2)-Activated TRPM2 Calcium Channel in Oxidant-Induced Endothelial Injury. Thromb Haemost (2009) 101(4):619–25. doi: 10.1160/TH08-10-0641
49. Massullo P, Sumoza-Toledo A, Bhagat H, Partida-Sánchez S. TRPM Channels, Calcium and Redox Sensors During Innate Immune Responses. Semin Cell Dev Biol (2006) 17(6):654–66. doi: 10.1016/j.semcdb.2006.11.006
50. Li Q, Wang X, Yang Z, Wang B, Li S. Menthol Induces Cell Death via the TRPM8 Channel in the Human Bladder Cancer Cell Line T24. Oncology (2009) 77(6):335–41. doi: 10.1159/000264627
51. Prevarskaya N, Zhang L, Barritt G. TRP Channels in Cancer. Biochim Biophys Acta (2007) 1772(8):937–46. doi: 10.1016/j.bbadis.2007.05.006
52. Chigurupati S, Venkataraman R, Barrera D, Naganathan A, Madan M, Paul L, et al. Receptor Channel TRPC6 is a Key Mediator of Notch-Driven Glioblastoma Growth and Invasiveness. Cancer Res (2010) 70(1):418–27. doi: 10.1158/0008-5472.CAN-09-2654
53. Thoppil RJ, Cappelli HC, Adapala RK, Kanugala AK, Paruchuri S, Thodeti CK. TRPV4 Channels Regulate Tumor Angiogenesis via Modulation of Rho/Rho Kinase Pathway. Oncotarget (2016) 7(18):25849–6. doi: 10.18632/oncotarget.8405
54. Gillet L, Roger S, Besson P, Lecaille F, Gore J, Bougnoux P, et al. Voltage-Gated Sodium Channel Activity Promotes Cysteine Cathepsin-Dependent Invasiveness and Colony Growth of Human Cancer Cells. J Biol Chem (2009) 284:8680–91. doi: 10.1074/jbc.M806891200
55. Campbell TM, Main MJ, Fitzgerald EM. Functional Expression of the Voltage-Gated Na+ Channel Nav1.7 is Necessary for EGF-Mediated Invasion in Human non-Small Cell Lung Cancer Cells. J Cell Sci (2013) 126:4939 – 4949. doi: 10.1242/jcs.130013
56. House CD, Wang BD, Ceniccola K, Williams R, Simaan M, Olender J, et al. Voltage-Gated Na+ Channel Activity Increases Colon Cancer Transcriptional Activity and Invasion Via Persistent MAPK Signaling. Sci Rep (2015) 5:11541. doi: 10.1038/srep11541
57. House CD, Vaske CJ, Vaske AM, Obias V, Frank B, Luu T, et al. Voltage-Gated Na+ Channel SCN5A is a Key Regulator of a Gene Transcriptional Network That Controls Colon Cancer Invasion. Cancer Res (2010) 70:6957–67. doi: 10.1158/0008-5472.CAN-10-1169
58. Chioni AM, Shao D, Grose R, Djamgoz MB. Protein Kinase A and Regulation of Neonatal Nav1.5 Expression in Human Breast Cancer Cells: Activity-Dependent Positive Feedback and Cellular Migration. Int J Biochem Cell Biol (2010) 42:346–58. doi: 10.1016/j.biocel.2009.11.021
59. Schwab A, Fabian A, Hanley PJ, Stock C. Role of Ion Channels and Transporters in Cell Migration. Physiol Rev (2012) 92:1865–913. doi: 10.1152/physrev.00018.2011
60. Cardone RA, Casavola V, Reshkin SJ. The Role of Disturbed pH Dynamics and the Na+/H+ Exchanger in Metastasis. Nat Rev Cancer (2005) 5:786–95. doi: 10.1038/nrc1713
61. Kaverina I, Krylyshkina O, Small JV. Regulation of Substrate Adhesion Dynamics During Cell Motility. Int J Biochem Cell Biol (2002) 34:746–61. doi: 10.1016/s1357-2725(01)00171-6
63. Fraser SP, Grimes JA, Djamgoz MB. Effects of Voltage-Gated Ion Channel Modulators on Rat Prostatic Cancer Cell Proliferation: Comparison of Strongly and Weakly Metastatic Cell Lines. Prostate (2000) 44:61–76. doi: 10.1002/1097-0045(20000615)44:1<61::aid-pros9>3.0.co;2-3
64. Fukushiro-Lopes DF, Hegel AD, Rao V, Wyatt D, Baker A, Breuer EK, et al. Preclinical Study of a Kv11.1 Potassium Channel Activator as Antineoplastic Approach for Breast Cancer. Oncotarget (2018) 9:3321–37. doi: 10.18632/oncotarget.22925
65. Lallet-Daher H, Roudbaraki M, Bavencoffe A, Mariot P, Gackière F, Bidaux G, et al. Intermediate-Conductance Ca2+-Activated K+ Channels (IKCa1) Regulate Human Prostate Cancer Cell Proliferation Through a Close Control of Calcium Entry. Oncogene (2009) 28:1792–806. doi: 10.1038/onc.2009.25
66. Zhang P, Yang X, Yin Q, Yi J, Wenzhuang S, Zhao L, et al. Inhibition of SK4 Potassium Channels Suppresses Cell Proliferation, Migration and the Epithelial-Mesenchymal Transition in Triple-Negative Breast Cancer Cells. PloS One (2016) 11:e0154471. doi: 10.1371/journal.pone.0154471
67. Blackiston D, Adams DS, Lemire JM, Lobikin M, Levin M. Transmembrane Potential of GlyCl-Expressing Instructor Cells Induces a Neoplastic-Like Conversion of Melanocytes via a Serotonergic Pathway. Dis Model Mech (2011) 4:67–85. doi: 10.1242/dmm.005561
68. Ufartes R, Schneider T, Mortensen LS, de Juan Romero C, Hentrich K, Knoetgen H, et al. Behavioral and Functional Characterization of Kv10.1 (Eag1) Knockout Mice. Hum Mol Genet (2013) 22(11):2247–62. doi: 10.1093/hmg/ddt076
69. Agarwal JR, Griesinger F, Stühmer W, Pardo LA. The Potassium Channel Ether À Go-Go is a Novel Prognostic Factor With Functional Relevance in Acute Myeloid Leukemia. Mol Cancer (2010) 9:18. doi: 10.1186/1476-4598-9-18
70. Jehle J, Schweizer PA, Katus HA, Thomas D. Novel Roles for hERG K(+) Channels in Cell Proliferation and Apoptosis. Cell Death Dis (2011) 2:e193. doi: 10.1038/cddis.2011.77
71. Farias LM, Ocaña DB, Díaz L, Larrea F, Avila-Chávez E, Cadena A, et al. Ether a Go-Go Potassium Channels as Human Cervical Cancer Markers. Cancer Res (2004) 64:6996–7001. doi: 10.1158/0008-5472.CAN-04-1204
72. Pardo LA, del Camino D, Sánchez A, Alves F, Brüggemann A, Beckh S, et al. Oncogenic Potential of EAG K(+) Channels. EMBO J (1999) 18:5540–7. doi: 10.1093/emboj/18.20.5540
73. Payne SL, Ram P, Srinivasan DH, Le TT, Levin M, Oudin MJ. Potassium Channel-Driven Bioelectric Signaling Regulates Metastasis in Triple-Negative Breast Cancer. EBioMedicine (2022) 75:103767. doi: 10.1016/j.ebiom.2021.103767
74. Millership JE, Devor DC, Hamilton KL, Balut CM, Bruce JI, Fearson IM, et al. Calcium- Activated K+ Channels Increase Cell Proliferation Independent of K+ Conductance. Am J Physiol Cell Physiol (2011) 300:C792–802. doi: 10.1152/ajpcell.00274.2010
75. Turner KL, Sontheimer H. Cl- and K+ Channels and Their Role in Primary Brain Tumour Biology. Philos Trans R Soc London Ser B Biol Sci (2014) 369(1638):20130095. doi: 10.1098/rstb.2013.0095
76. Wondergem R, Ecay TW, Mahieu F, Owsianik G, Nilius B. HGF/SF and Menthol Increase Human Glioblastoma Cell Calcium and Migration. Biochem Biophys Res Commun (2008) 372(1):210–5. doi: 10.1016/j.bbrc.2008.05.032
77. Liu J, Chen Y, Shuai S, Ding D, Li R, Luo R, et al. TRPM8 Promotes Aggressiveness of Breast Cancer Cells by Regulating EMT via Activating AKT/GSK-3ß Pathway. Tumor Biol (2014) 35:8969 – 8977. doi: 10.1007/s13277-014-2077-8
78. Jirsch J, Deeley RG, Cole SP, Stewart AJ, Fedida D. Inwardly Rectifying K+ Channels and Volume-Regulated Anion Channels in Multidrug-Resistant Small Cell Lung Cancer Cells. Cancer Res (1993) 53:4156–60.
79. Habela CW, Olsen ML, Sontheimer H. ClC3 is a Critical Regulator of the Cell Cycle in Normal and Malignant Glial Cells. J Neurosci (2008) 28:9205–17. doi: 10.1523/JNEUROSCI.1897-08.2008
80. Volkov A, Waite AJ, Wooten JD, Markin VS. Circadian Rhythms in Biologically Closed Electrical Circuits of Plants. Plant Signal Behav (2012) 7:282–4. doi: 10.4161/psb.18798
81. Shuba YM, Prevarskaya N, Lemonnier L, Van Coppenolle F, Kostyuk PG, Mauroy B, et al. Volume-Regulated Chloride Conductance in the LNCaP Human Prostate Cancer Cell Line. Am J Physiol Cell Physiol (2000) 279:C1144–1154. doi: 10.1152/ajpcell.2000.279.4.C1144
82. Cuddapah VA, Sontheimer H. Molecular Interaction and Functional Regulation of ClC-3 by Ca2+/calmodulin-Dependent Protein Kinase II (CaMKII) in Human Malignant Glioma. J Biol Chem (2010) 285(15):11188–96. doi: 10.1074/jbc.M109.097675
83. Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, et al. Piezo1 and Piezo2 are Essential Components of Distinct Mechanically Activated Cation Channels. Science (2010) 330:55–60. doi: 10.1126/science.1193270
84. Cox CD, Bae C, Ziegler L, Hartley S, Nikolova-Krstevski V, Rohde PR, et al. Removal of the Mechanoprotective Influence of the Cytoskeleton Reveals PIEZO1 is Gated by Bilayer Tension. Nat Commun (2016) 7:10366. doi: 10.1038/ncomms10366
85. Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, Cahalan SM, et al. Piezo1, a Mechanically Activated Ion Channel, is Required for Vascular Development in Mice. Proc Natl Acad Sci USA (2014) 111:10347–52. doi: 10.1073/pnas.1409233111
86. Moroni M, Servin-Vences MR, Fleischer R, Sánchez-Carranza O, Lewin GR. Voltage Gating of Mechanosensitive PIEZO Channels. Nat Commun (2018) 9:1096. doi: 10.1038/s41467-018-03502-7
87. De Felice D, Alaimo A. Mechanosensitive Piezo Channels in Cancer: Focus on Altered Calcium Signaling in Cancer Cells and in Tumor Progression. Cancers (Basel) (2020) 12:1780. doi: 10.3390/cancers12071780
88. Han Y, Liu C, Zhang D, Men H, Huo L, Geng Q, et al. Mechanosensitive Ion Channel Piezo1 Promotes Prostate Cancer Development Through the Activation of the Akt/mTOR Pathway and Acceleration of Cell Cycle. Int J Oncol (2019) 55(3):629–44. doi: 10.3892/ijo.2019.4839
89. Yang H, Liu C, Zhou RM, Yao J, Li XM, Shen Y, et al. Piezo2 Protein: A Novel Regulator of Tumor Angiogenesis and Hyperpermeability. Oncotarget (2016) 7:44630–44643. doi: 10.18632/oncotarget.10134
90. Levin M. Molecular Bioelectricity in Developmental Biology: New Tools and Recent Discoveries: Control of Cell Behavior and Pattern Formation by Transmembrane Potential Gradients. Bioessays (2012) 34:205–17. doi: 10.1002/bies.201100136
91. Adams DS, Levin M. Endogenous Voltage Gradients as Mediators of Cell-Cell Communication: Strategies for Investigating Bioelectrical Signals During Pattern Formation. Cell Tissue Res (2013) 352:95–122. doi: 10.1007/s00441-012-1329-4
92. Levin M, Stevenson CG. Regulation of Cell Behavior and Tissue Patterning by Bioelectrical Signals: Challenges and Opportunities for Biomedical Engineering. Annu Rev BioMed Eng (2012) 14:295–323. doi: 10.1146/annurev-bioeng-071811-150114
93. Marino AA, Iliev IG, Schwalke MA, Gonzalez E, Marler KC, Flanagan CA, et al. Association Between Cell Membrane Potential and Breast Cancer. Tumour Biol (1994) 15:82–9. doi: 10.1159/000217878
94. Morokuma J, Blackiston D, Adams DS, Seebohm G, Trimmer B, Levin M. Modulation of Potassium Channel Function Confers a Hyperproliferative Invasive Phenotype on Embryonic Stem Cells. Proc Natl Acad Sci USA (2008) 105:16608–13. doi: 10.1073/pnas.0808328105
95. Cone CD. Unified Theory on the Basic Mechanism of Normal Mitotic Control and Oncogenesis. J Theor Biol (1971) 30:151–81. doi: 10.1016/0022-5193(71)90042-7
96. Cone CD. The Role of the Surface Electrical Transmembrane Potential in Normal and Malignant Mitogenesis. Ann N Y Acad Sci (1974) 238:420–35. doi: 10.1111/j.1749-6632.1974.tb26808.x
97. Binggeli R, Weinstein RC. Membrane Potentials and Sodium Channels: Hypotheses for Growth Regulation and Cancer Formation Based on Changes in Sodium Channels and Gap Junctions. J Theor Biol (1986) 123:377–401. doi: 10.1016/s0022-5193(86)80209-0
98. Binggeli R, Cameron IL. Cellular Potentials of Normal and Cancerous Fibroblasts and Hepatocytes. Cancer Res (1980) 40:1830–5.
99. Stratton MR, Fisher C, Gusterson BA, Cooper CS. Detection of Point Mutations in N-Ras and K-Ras Genes of Human Embryonal Rhabdomyosarcomas Using Oligonucleotide Probes and the Polymerase Chain Reaction. Cancer Res (1989) 49:6324–7.
100. Gilmore TD, Kalaitzidis D, Liang MC, Starczynowski DT. The C-Rel Transcription Factor and B-Cell Proliferation: A Deal With the Devil. Oncogene (2004) 23:2275–86. doi: 10.1038/sj.onc.1207410
101. McNulty SE, del Rosario R, Cen D, Meyskens FL, Yang S. Comparative Expression of NFkappaB Proteins in Melanocytes of Normal Skin vs. Benign Intradermal Naevus and Human Metastatic Melanoma Biopsies. Pigment Cell Res (2004) 17:173–80. doi: 10.1111/j.1600-0749.2004.00128.x
102. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 Signaling Regulates Human Glioma Growth, Cancer Stem Cell Self-Renewal, and Tumorigenicity. Curr Biol (2007) 17:165–72. doi: 10.1016/j.cub.2006.11.033
103. Chernet BT, Levin M. Transmembrane Voltage Potential is an Essential Cellular Parameter for the Detection and Control of Tumor Development in a Xenopus Model. Dis Model Mech (2013) 6:595–607. doi: 10.1242/dmm.010835
104. Thompson IM, Pauler IM, Goodman IM, Tangen IM, Lucia IM, Parnes IM, et al. Prevalence of Prostate Cancer Among Men With a Prostate-Specific Antigen Level < or =4.0 Ng Per Milliliter. N Engl J Med (2004) 350:2239–46. doi: 10.1056/NEJMoa031918
105. Smith DS, Humphrey PA, Catalona WJ. The Early Detection of Prostate Carcinoma With Prostate Specific Antigen: The Washington University Experience. Cancer (1997) 80:1852–6. doi: 10.1002/(SICI)1097-0142(19971101)80:9<1852::AID-CNCR25>3.0.CO;2-3
106. Carvalho J. A Bioelectric Model of Carcinogenesis, Including Propagation of Cell Membrane Depolarization and Reversal Therapies. Sci Rep (2021) 11:13607. doi: 10.1038/s41598-021-92951-0
107. Li C, Teixeira AF, Zhu HJ, Ten Dijke P. Cancer Associated-Fibroblast-Derived Exosomes in Cancer Progression. Mol Cancer (2021) 20:154. doi: 10.1186/s12943-021-01463-y
108. Maia J, Caja S, Strano Moraes MC, Couto N, Costa-Silva B. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front Cell Dev Biol (2018) 6:18. doi: 10.3389/fcell.2018.00018
109. Wu Q, Zhou L, Lv D, Zhu X, Tang H. Exosome-Mediated Communication in the Tumor Microenvironment Contributes to Hepatocellular Carcinoma Development and Progression. J Hematol Oncol (2019) 12:53. doi: 10.1186/s13045-019-0739-0
110. Todaro GJ, Green H. Quantitative Studies of the Growth of Mouse Embryo Cells in Culture and Their Development Into Established Lines. J Cell Biol (1963) 17:299–313. doi: 10.1083/jcb.17.2.299
111. Blennerhassett MG, Kannan MS, Garfield RE. Density-Dependent Hyperpolarization in Cultured Aortic Smooth Muscle Cells. Am J Physiol (1989) 256:C644–651. doi: 10.1152/ajpcell.1989.256.3.C644
112. Bossu JL, Elhamdani A, Feltz A. Voltage-Dependent Calcium Entry in Confluent Bovine Capillary Endothelial Cells. FEBS Lett (1992) 299:239–42. doi: 10.1016/0014-5793(92)80123-x
113. Pang MF, Siedlik MJ, Han S, Stallings-Mann M, Radisky DC, Nelson CM. Tissue Stiffness and Hypoxia Modulate the Integrin-Linked Kinase ILK to Control Breast Cancer Stem-Like Cells. Cancer Res (2016) 76:5277–87. doi: 10.1158/0008-5472.CAN-16-0579
114. Damaghi M, Wojtkowiak JW, Gillies RJ. pH Sensing and Regulation in Cancer. Front Physiol (2013) 4:370. doi: 10.3389/fphys.2013.00370
115. Yu S, Cao H, Shen B, Feng J. Tumor-Derived Exosomes in Cancer Progression and Treatment Failure. Oncotarget (2015) 6:37151–68. doi: 10.18632/oncotarget.6022
117. Ohkubo T, Yamazaki J. T-Type Voltage-Activated Calcium Channel Cav3.1, But Not Cav3.2, is Involved in the Inhibition of Proliferation and Apoptosis in MCF-7 Human Breast Cancer Cells. Int J Oncol (2012) 41:267–75. doi: 10.3892/ijo.2012.1422
118. Rothberg BS. The BK Channel: A Vital Link Between Cellular Calcium and Electrical Signaling. Protein Cell (2012) 3:883–92. doi: 10.1007/s13238-012-2076-8
119. Martinac B. The Ion Channels to Cytoskeleton Connection as Potential Mechanism of Mechanosensitivity. Biochim Biophys Acta (2014) 1838:682–91. doi: 10.1016/j.bbamem.2013.07.015
120. Discher DE, Janmey P, Wang YL. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science (2005) 310:1139–43. doi: 10.1126/science.1116995
121. Piotrowski-Daspit AS, Tien J, Nelson CM. Interstitial Fluid Pressure Regulates Collective Invasion in Engineered Human Breast Tumors via Snail, Vimentin, and E-Cadherin. Integr Biol (Camb) (2016) 8:319–31. doi: 10.1039/c5ib00282f
122. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell (2006) 126:677–89. doi: 10.1016/j.cell.2006.06.044
123. Kim TJ, Seong J, Ouyang M, Sun J, Lu S, Hong JP, et al. Substrate Rigidity Regulates Ca2+ Oscillation via RhoA Pathway in Stem Cells. J Cell Physiol (2009) 218:285–93. doi: 10.1002/jcp.21598
124. Kostic A, Lynch CD, Sheetz MP. Differential Matrix Rigidity Response in Breast Cancer Cell Lines Correlates With the Tissue Tropism. PloS One (2009) 4:e6361. doi: 10.1371/journal.pone.0006361
125. Tilghman RW, Cowan CR, Mih JD, Koryakina Y, Gioeli D, Slack-Davis JK, et al. Matrix Rigidity Regulates Cancer Cell Growth and Cellular Phenotype. PloS One (2010) 5:e12905. doi: 10.1371/journal.pone.0012905
126. Zhang YH, Zhao CQ, Jiang LS, Dai LY. Substrate Stiffness Regulates Apoptosis and the mRNA Expression of Extracellular Matrix Regulatory Genes in the Rat Annular Cells. Matrix Biol (2011) 30:135–44. doi: 10.1016/j.matbio.2010.10.008
127. Lee K, Chen QK, Lui C, Cichon MA, Radisky DC, Nelson CM. Matrix Compliance Regulates Rac1b Localization, NADPH Oxidase Assembly, and Epithelial-Mesenchymal Transition. Mol Biol Cell (2012) 23:4097–108. doi: 10.1091/mbc.E12-02-0166
128. Pathak MM, Nourse JL, Tran T, Hwe J, Arulmoli J, Le DT, et al. Stretch-Activated Ion Channel Piezo1 Directs Lineage Choice in Human Neural Stem Cells. Proc Natl Acad Sci USA (2014) 111:16148–53. doi: 10.1073/pnas.1409802111
129. Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric Control of Cell Life and Death. Science (1997) 276:1425–8. doi: 10.1126/science.276.5317.1425
130. Dike LE, Chen CS, Mrksich M, Tien J, Whitesides GM, Ingber DE. Geometric Control of Switching Between Growth, Apoptosis, and Differentiation During Angiogenesis Using Micropatterned Substrates. In Vitro Cell Dev Biol Anim (1999) 35:441–8. doi: 10.1007/s11626-999-0050-4
131. Vogel V, Sheetz M. Local Force and Geometry Sensing Regulate Cell Functions. Nat Rev Mol Cell Biol (2006) 7:265–75. doi: 10.1038/nrm1890
132. Walsh MF, Woo RK, Gomez R, Basson MD. Extracellular Pressure Stimulates Colon Cancer Cell Proliferation via a Mechanism Requiring PKC and Tyrosine Kinase Signals. Cell Prolif (2004) 37:427–41. doi: 10.1111/j.1365-2184.2004.00324.x
133. Basson MD, Zeng B, Downey C, Sirivelu MP, Tepe JJ. Increased Extracellular Pressure Stimulates Tumor Proliferation by a Mechanosensitive Calcium Channel and PKC-β. Mol Oncol (2015) 9:513–26. doi: 10.1016/j.molonc.2014.10.008
134. Kobayashi T, Sokabe M. Sensing Substrate Rigidity by Mechanosensitive Ion Channels With Stress Fibers and Focal Adhesions. Curr Opin Cell Biol (2010) 22:669–76. doi: 10.1016/j.ceb.2010.08.023
135. Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix Density-Induced Mechanoregulation of Breast Cell Phenotype, Signaling and Gene Expression Through a FAK-ERK Linkage. Oncogene (2009) 28:4326–43. doi: 10.1038/onc.2009.299
136. Bordeleau F, Califano JP, Negrón Abril YL, Mason BN, LaValley DJ, Shin SJ, et al. Tissue Stiffness Regulates Serine/Arginine-Rich Protein-Mediated Splicing of the Extra Domain B-Fibronectin Isoform in Tumors. Proc Natl Acad Sci USA (2015) 112:8314–9. doi: 10.1073/pnas.1505421112
137. Branco da Cunha C, Klumpers DD, Koshy ST, Weaver JC, Chaudhuri O, Seruca R, et al. CD44 Alternative Splicing in Gastric Cancer Cells is Regulated by Culture Dimensionality and Matrix Stiffness. Biomaterials (2016) 98:152–62. doi: 10.1016/j.biomaterials.2016.04.016
138. Lo CM, Wang HB, Dembo M, Wang YL. Cell Movement is Guided by the Rigidity of the Substrate. Biophys J (2000) 79:144–52. doi: 10.1016/S0006-3495(00)76279-5
139. Tee SY, Fu J, Chen CS, Janmey PA. Cell Shape and Substrate Rigidity Both Regulate Cell Stiffness. Biophys J (2011) 100:L25–27. doi: 10.1016/j.bpj.2010.12.3744
140. Nogi T, Levin M. Characterization of Innexin Gene Expression and Functional Roles of Gap-Junctional Communication in Planarian Regeneration. Dev Biol (2005) 287:314–35. doi: 10.1016/j.ydbio.2005.09.002
141. Chernet BT, Fields C, Levin M. Long-Range Gap Junctional Signaling Controls Oncogene-Mediated Tumorigenesis in Xenopus Laevis Embryos. Front Physiol (2014) 5:519. doi: 10.3389/fphys.2014.00519
142. Mathews J, Levin M. Gap Junctional Signaling in Pattern Regulation: Physiological Network Connectivity Instructs Growth and Form. Dev Neurobiol (2017) 77:643–73. doi: 10.1002/dneu.22405
143. Lalli ML, Asthagiri AR. 2014 40th Annual Northeast Bioengineering Conference (NEBEC). Boston, MA: NEBEC (2014). p. 1–2.
144. Kwon MS, Park CS, Choi KR, Park CS, Ahnn J, Kim JI, et al. Calreticulin Couples Calcium Release and Calcium Influx in Integrin-Mediated Calcium Signaling. Mol Biol Cell (2000) 11:1433–43. doi: 10.1091/mbc.11.4.1433
145. Orrenius S, Zhivotovsky B, Nicotera P. Regulation of Cell Death: The Calcium-Apoptosis Link. Nat Rev Mol Cell Biol (2003) 4:552–65. doi: 10.1038/nrm1150
146. Chitty JL, Filipe EC, Lucas MC, Herrmann D, Cox TR, Timpson P. Recent Advances in Understanding the Complexities of Metastasis. F1000Res (2018) 7:F1000 Faculty Rev-1169. doi: 10.12688/f1000research.15064.2
147. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell (2017) 168:670–91. doi: 10.1016/j.cell.2016.11.037
148. Oudin MJ, Weaver VM. Physical and Chemical Gradients in the Tumor Microenvironment Regulate Tumor Cell Invasion, Migration, and Metastasis. Cold Spring Harb Symp Quant Biol (2016) 81:189–205. doi: 10.1101/sqb.2016.81.030817
149. Chernet BT, Levin M. Transmembrane Voltage Potential of Somatic Cells Controls Oncogene-Mediated Tumorigenesis at Long-Range. Oncotarget (2014) 5:3287–306. doi: 10.18632/oncotarget.1935
150. Chernet BT, Adams DS, Lobikin M, Levin M. Use of Genetically Encoded, Light-Gated Ion Translocators to Control Tumorigenesis. Oncotarget (2016) 7:19575–88. doi: 10.18632/oncotarget.8036
151. Zhu K, Hum NR, Reid B, Sun Q, Loots GG, Zhao M. Electric Fields at Breast Cancer and Cancer Cell Collective Galvanotaxis. Sci Rep (2020) 10:8712. doi: 10.1038/s41598-020-65566-0
152. Sivaramakrishna R, Gordon R. Detection of Breast Cancer at a Smaller Size can Reduce the Likelihood of Metastatic Spread: A Quantitative Analysis. Acad Radiol (1997) 4:8–12. doi: 10.1016/s1076-6332(97)80154-7
153. Laura S, Coombs NJ, Ung O, Boyages J. Tumour Size as a Predictor of Axillary Node Metastases in Patients With Breast Cancer. ANZ J Surg (2006) 76:1002–6. doi: 10.1111/j.1445-2197.2006.03918.x
154. Adams DS, Levin M. General Principles for Measuring Resting Membrane Potential and Ion Concentration Using Fluorescent Bioelectricity Reporters. Cold Spring Harb Protoc (2012) 2012:385–97. doi: 10.1101/pdb.top067710
155. Reid B, Nuccitelli R, Zhao M. Non-Invasive Measurement of Bioelectric Currents With a Vibrating Probe. Nat Protoc (2007) 2:661–9. doi: 10.1038/nprot.2007.91
156. Baker BJ, Mutoh H, Dimitrov D, Akemann W, Perron A, Iwamoto Y, et al. Genetically Encoded Fluorescent Sensors of Membrane Potential. Brain Cell Biol (2008) 36:53–67. doi: 10.1007/s11068-008-9026-7
157. Mutoh H, Perron A, Akemann W, Iwamoto Y, Knöpfel T. Optogenetic Monitoring of Membrane Potentials. Exp Physiol (2011) 96:13–8. doi: 10.1113/expphysiol.2010.053942
158. Shen B, Xiang Z, Miller B, Louie G, Wang W, Noel JP, et al. Genetically Encoding Unnatural Amino Acids in Neural Stem Cells and Optically Reporting Voltage-Sensitive Domain Changes in Differentiated Neurons. Stem Cells (2011) 29:1231–40. doi: 10.1002/stem.679
159. Tsutsui H, Karasawa S, Okamura Y, Miyawaki A. Improving Membrane Voltage Measurements Using FRET With New Fluorescent Proteins. Nat Methods (2008) 5:683–5. doi: 10.1038/nmeth.1235
160. Tantama M, Hung YP, Yellen G. Imaging Intracellular pH in Live Cells With a Genetically Encoded Red Fluorescent Protein Sensor. J Am Chem Soc (2011) 133:10034–7. doi: 10.1021/ja202902d
161. Tseng AS, Beane WS, Lemire JM, Masi A, Levin M. Induction of Vertebrate Regeneration by a Transient Sodium Current. J Neurosci (2010) 30:13192–200. doi: 10.1523/JNEUROSCI.3315-10.2010
162. Fang Y, Li X, Fang Y. Organic Bioelectronics for Neural Interfaces. J Mater Chem C (2015) 3:6424–30. doi: 10.1039/C5TC00569H
163. Löffler S, Melican K, Nilsson KPR, Richter-Dahlfors A. Organic Bioelectronics in Medicine. J Intern Med (2017) 282:24–36. doi: 10.1111/joim.12595
164. Rivnay J, Owens RM, Malliaras GG. The Rise of Organic Bioelectronics. Chem Mater (2014) 26:679–85. doi: 10.1021/cm4022003
165. Simon DT, Gabrielsson EO, Tybrandt K, Berggren M. Organic Bioelectronics: Bridging the Signaling Gap Between Biology and Technology. Chem Rev (2016) 116:13009–41. doi: 10.1021/acs.chemrev.6b00146
166. Someya T, Bao Z, Malliaras GG. The Rise of Plastic Bioelectronics. Nature (2016) 540:379–85. doi: 10.1038/nature21004
167. Tian B, Xu S, Rogers JA, Cestellos-Blanco S, Yang P, Carvalho-de-Souza JL, et al. Roadmap on Semiconductor-Cell Biointerfaces. Phys Biol (2018) 15:031002. doi: 10.1088/1478-3975/aa9f34
168. Strakosas X, Bongo M, Owens RM. The Organic Electrochemical Transistor for Biological Applications. J Appl Polymer Sci (2015) 132(15):41735. doi: 10.1002/app.41735
169. Tian B, Cohen-Karni T, Qing Q, Duan X, Xie P, Lieber CM, et al. Three-Dimensional, Flexible Nanoscale Field-Effect Transistors as Localized Bioprobes. Science (2010) 329:830–4. doi: 10.1126/science.1192033
170. Jayaram DT, Luo Q, Thourson SB, Finlay AH, Payne CK. Controlling the Resting Membrane Potential of Cells With Conducting Polymer Microwires. Small (2017) 13:10.1002. doi: 10.1002/smll.201700789
171. Rana MA, Yao N, Mukhopadhyay S, Zhang F, Warren E, Payne C. 2016 American Control Conference (ACC). Boston, MA: Proc. of 2016 American Control Conference (ACC2016) (2016). p. 400–5.
172. Shin EH, Li Y, Kumar U, Sureka HV, Zhang X, Payne CK, et al. Membrane Potential Mediates the Cellular Binding of Nanoparticles. Nanoscale (2013) 5:5879–86. doi: 10.1039/c3nr01667f
173. Warren EA, Payne CK. Cellular Binding of Nanoparticles Disrupts the Membrane Potential. RSC Adv (2015) 5:13660–6. doi: 10.1039/C4RA15727C
174. Liu X, Gilmore KJ, Moulton SE, Wallace GG. Electrical Stimulation Promotes Nerve Cell Differentiation on Polypyrrole/Poly (2-Methoxy-5 Aniline Sulfonic Acid) Composites. J Neural Eng (2009) 6:65002. doi: 10.1088/1741-2560/6/6/065002
175. Lee JY, Lee JW, Schmidt CE. Neuroactive Conducting Scaffolds: Nerve Growth Factor Conjugation on Active Ester-Functionalized Polypyrrole. J R Soc Interface (2009) 6:801–10. doi: 10.1098/rsif.2008.0403
176. Jeong SI, Jun ID, Choi MJ, Nho YC, Lee YM, Shin H, et al. Development of Electroactive and Elastic Nanofibers That Contain Polyaniline and Poly(L-Lactide-Co-Epsilon-Caprolactone) for the Control of Cell Adhesion. Macromol Biosci (2008) 8:627–37. doi: 10.1002/mabi.200800005
177. Guo B, Ma PX. Conducting Polymers for Tissue Engineering. Biomacromolecules (2018) 19:1764–82. doi: 10.1021/acs.biomac.8b00276
178. Thourson SB, Payne CK. Modulation of Action Potentials Using PEDOT:PSS Conducting Polymer Microwires. Sci Rep (2017) 7:10402. doi: 10.1038/s41598-017-11032-3
179. Balint R, Cassidy NJ, Cartmell SH. Conductive Polymers: Towards a Smart Biomaterial for Tissue Engineering. Acta Biomater (2014) 10:2341–53. doi: 10.1016/j.actbio.2014.02.015
180. Fukuta T, Nishikawa A, Kogure K. Low Level Electricity Increases the Secretion of Extracellular Vesicles From Cultured Cells. Biochem Biophys Rep (2020) 21:100713. doi: 10.1016/j.bbrep.2019.100713
181. Lakkaraju A, Rodriguez-Boulan E. Itinerant Exosomes: Emerging Roles in Cell and Tissue Polarity. Trends Cell Biol (2008) 18:199–209. doi: 10.1016/j.tcb.2008.03.002
182. Rabinowits G, GerÁel-Taylor C, Day JM, Taylor DD, Kloecker GH. Exosomal microRNA: A Diagnostic Marker for Lung Cancer. Clin Lung Cancer (2009) 10:42–6. doi: 10.3816/CLC.2009.n.006
Keywords: bioelectricity, extracellular vesicles, tumor microenvironment, membrane potential, carcinogenesis, piezo channels
Citation: Sheth M and Esfandiari L (2022) Bioelectric Dysregulation in Cancer Initiation, Promotion, and Progression. Front. Oncol. 12:846917. doi: 10.3389/fonc.2022.846917
Received: 31 December 2021; Accepted: 21 February 2022;
Published: 14 March 2022.
Edited by:
Daniele Vergara, University of Salento, ItalyCopyright © 2022 Sheth and Esfandiari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leyla Esfandiari, ZXNmYW5kbGFAdWNtYWlsLnVjLmVkdQ==