 Li Yang
Li Yang Xin Chen1†
Xin Chen1† Aijun Yu
Aijun Yu- 1Department of Gynecological Oncology, The Cancer Hospital of the University of Chinese Academy of Sciences (Zhejiang Cancer Hospital), Institute of Basic Medicine and Cancer (IBMC), Chinese Academy of Sciences, Hangzhou, China
- 2Department of Traditional Chinese Medicine, The Cancer Hospital of the University of Chinese Academy of Sciences (Zhejiang Cancer Hospital), Institute of Basic Medicine and Cancer (IBMC), Chinese Academy of Sciences, Hangzhou, China
- 3Department of Pathology, The Cancer Hospital of the University of Chinese Academy of Sciences (Zhejiang Cancer Hospital), Institute of Basic Medicine and Cancer (IBMC), Chinese Academy of Sciences, Hangzhou, China
Emerging studies have revealed that N6-methyladenosine modification is involved in the development of various cancers. However, the m6A modification pattern of endometrioid ovarian cancer (EOC) has not been demonstrated. In the present study, high-throughput sequencing combined with methylated RNA immunoprecipitation (MeRIP-seq) and RNA sequencing were used to obtain the transcriptome-wide m6A modifications of endometrioid ovarian cancer for the first time. The roles of methyltransferase-like 3 (METTL3) in EOC cell line COV362 were explored. In total, 39,237 m6A-modified peaks related to 17,082 genes were identified in the EOC group, and 52,848 m6A peaks representing 19,349 genes were detected in endometriosis group. Functional enrichment analysis revealed that m6A enriched genes were associated with tight junctions, cell adhesion molecules, platinum drug resistance, adherens junction, and more. METTL3 knockdown in the COV362 cells significantly decreased cell proliferation, promoted cell apoptosis, and induced cell cycle arrest at the G0/G1 phase. Our study presented the transcriptome-wide m6A modifications of endometrioid ovarian cancer for the first time and revealed various differentially expressed genes with methylated m6A modifications. This study may provide new directions for in-depth research of the underlying molecular mechanisms and signaling pathways of EOC development and progression.
Introduction
Ovarian cancer is the third most common cancer worldwide and it has the highest mortality rate in females. According to the Global Cancer Statistics, there were 313,959 new cases diagnosed and 207,252 women died of the disease in 2020 (1). Epithelial ovarian cancer is the most common type of ovarian cancer. Endometrioid ovarian cancer (EOC) accounts for 10-15% of epithelial ovarian cancer (2, 3). Ovarian endometriosis (OE) is associated with EOC and is commonly considered a direct precursor lesion to endometrioid and clear cell ovarian cancer types (4). Most of the patients with EOC are diagnosed at an early stage with good prognosis. However, some patients still have early recurrence and metastasis with poor prognosis.
N6-methyladenosine (m6A) is the most abundant RNA modification types which accounts for nearly 80% of all methylation modifications in eukaryotic cells (5). It has been reported that multiple protein complexes were involved in the metabolism of m6A RNA methylation. It is catalyzed by a multicomponent methyltransferase complex, called “writers” and is removed by demethylases called “erasers”. “Writers” are mainly composed of three subunits: METTL3 functions as a catalytic subunit, methyltransferase-like 14 (METTL14) functions as a structural subunit, and Wilms’ tumor 1-associated protein (WTAP) works as an adaptor molecule (6–10). The identification of m6A demethylase indicates that m6A methylation in RNA is reversible and dynamic. Only two m6A “erasers”, fat mass and obesity-associated protein (FTO) and α-ketoglutarate-dependent dioxygenase homolog 5 (ALKBH5) have been reported so far (11, 12). “Readers” serve as binding proteins so that m6A group can exhibit biological functions. They consist of proteins that contain a YT521-B homology (YTH) domain, including YTHDF1-3, and YTHDC1-2 and insulin-like growth factor 2 mRNA-binding proteins 1, 2, and 3 (IGF2BP1, IGF2BP2, IGF2BP3) (13). Some other m6A “readers” have also been identified, such as fragile X mental retardation 1 (FMR1) and eukaryotic initiation factor 3 (eIF3), and the list is still growing (14).
Evidences suggest that m6A methylation affects all most every aspect of RNA metabolism, for example, RNA structure, RNA stability, mRNA nuclear export, translation, mRNA decay and noncoding RNA and miroRNA processing (15, 16). Accumulating data have indicated that m6A participates in many fundamental biological processes, such as embryogenesis, neurogenesis, stress responses, and DNA damage responses (17). Furthermore, there are emerging studies that have revealed that altered m6A modification participates in the development of cancers (18).
The detection of m6A modification patterns at the transcriptome-wide level helps to better understand the potential biological effects of m6A modification in RNA. With the development of MeRIP-seq in 2012 (19, 20), the transcriptome-wide distribution of m6A of several cancers have been reported. However, the profiles of transcriptome-wide m6A distribution in many cancers are still unknown.
To further investigate the functions of m6A, in this study, the combination of MeRIP-seq and RNA sequencing was applied to acquire the transcriptome-wide m6A modifications of EOC for the first time. Furthermore, the roles and potential mechanisms of METTL3 were studied in EOC cells in vitro.
Materials and Methods
Clinical Specimen
Three pairs of EOC samples and endometriosis samples were collected at the time of surgery in Zhejiang cancer hospital. All tissues were histopathologically reviewed by two gynecological pathologists independently. Samples were stored at -80°C in separated centrifuge tubes before use. The current study was approved by the Institutional Review Board of Zhejiang cancer hospital. All the patients recruited in the study provided a written informed consent.
MeRIP-Seq and RNA Sequencing
Trizol reagent (Invitrogen, CA, USA) was used to extract total RNA from each sample according to the instruction. Bioanalyzer 2100 and RNA 6000 Nano LabChip Kit (Agilent, CA, USA) were used to conduct total RNA quality control and quantity assessment based on the threshold of RIN (RNA integrity number) values > 7.0. Approximately more than 200 μg of total RNA was obtained to isolate Poly (A) mRNA with poly-T oligo attached magnetic beads (Invitrogen). Then the fragmented RNA of about 100-nt-long oligonucleotides was incubated with m6A-specific antibody (No. 202003, Synaptic Systems, Germany) in IP buffer solution(50 mM Tris-HCl, 750 mM NaCl, and 0.5% Igepal CA-630) and with BSA (0.5 μg μl−1) for 2h at 4°C. Then, the mixture was incubated with protein-A beads. Bound RNA was eluted using elution buffer containing 1 × IP buffer and 6.7mM m6A. Eluted RNA was precipitated by 75% ethanol solution. Final cDNA libraries of eluted m6A-containing fragments (IP) and untreated input control fragments (input) were prepared using dUTP method in accordance with a strand-specific library preparation. The average insert length of the paired-end libraries was ~100 ± 50 bp. Subsequently, the IP samples and input samples were submitted to conduct 150bp paired-end sequencing on the Illumina Novaseq™ 6000 platform (LC-BIO Bio, Hangzhou, China) according to the recommended protocol.
Data Analysis
After reads that contained adaptor contamination, low quality bases, and undetermined bases were removed using Cutadapt and perl scripts in-house, the sequence quality was verified by Fastp. The high-quality clean reads were mapped to the genome of homo sapiens (Version: v96) with default parameters using HISAT2 software. The mapping reads were used for peaking calling by R package exomePeak, while identified m6A peaks with bed or bam formats were visualized using IGV software (http://www.igv.org/). MEME and HOMER were used for de novo and known motif finding followed by localization of the motif involving peak summit by perl scripts in-house. Called peaks annotation and intersection with gene architecture were conducted using ChIPseeker. Next, expression level for all mRNAs from input libraries were calculated as FPKM (FPKM = [total_exon_fragments/mapped_reads (millions) × exon_length (kB)]) using StringTie. The differentially expressed peaks or mRNAs were identified with log2 (fold change) > 1 or log2 (fold change) < -1 and P value < 0.05 by R package edgeR.
Cell Culture
Human EOC cell line COV362 was cultured in Dulbecco’s modified Eagle’s medium (DMEM, Hyclone, Tauranga, New Zealand) containing 10% fetal bovine serum (FBS, Gibco, NY, USA), 100 U/ml penicillin and 100 ug/ml streptomycin (Invitrogen, CA, USA) at 37°C incubator with a humidified atmosphere with 5% CO2.
SiRNA Transfection
METTL3 down-regulation was achieved using sequence-specific small interfering RNA (siRNA). METTL3 siRNA and negative control was from GenePharma (Shanghai, China). COV362 cells grown in 6-well plates were transfected with 1 ug/ml sequence-specific siRNA or vector siRNA prepared in a mix with lipo3000 reagent and DMEM containing 10% serum. The cells were then cultured for 6 hours before use.
Real Time PCR
Real time PCR was performed to detect whether METTL3 was successfully knockdown by siRNA, as described previously (21). The primer pairs of METTL3 used were 5’-CCCTATGGGACCCTGACAG-3’ (forward primer) and 5’-CTGGTTGAAGCCTTGGGGAT-3’ (reverse primer), and the primer pairs of GAPDH were 5’-CGGAGTCAACGGATTTGGTCGTAT-3’ (forward primer) and 5’- AGCCTTCTCCATGGTGGTGAAGAC-3’ (reverse primer).
The MTT Assay
We planted COV362 cells at a concentration of 1×105/ml in 96-well plates. Then cells were incubated in 200µl medium which contains 20 µl MTT (5 mg/mL) at 37°C for 4 h. Next, we discarded the medium and added 150 µl dimethyl sulfoxide (DMSO) into each well. After shaking for 10 minutes, the absorbance at 570nm were measured by Microplate Reader (BIO-RAD550, Hercules, CA) in each well. The cell viability rate was calculated as experimental OD value/control OD value × 100%.
The TUNEL Assay
We used 4% paraformaldehyde solution to fix cells at room temperature for 30 minutes. Equilibration buffer were added to cover samples and incubated for 10 minutes. Then, enough TUNEL reagent (Roche, Basel, Switzerland) were added to cover samples and incubated at 37°C for 60 minutes. DAPI solution (Beyotime, Shanghai, China) was added to stain the nuclei. At last, the samples were analyzed using a fluorescent microscope (Olympus, Tokyo, Japan).
Cell Cycle Assay
Flow cytometry was performed using the PI single staining method to conduct cell cycle analysis, as described previously (21). In general, we collected cells and suspended them in 1×PBS and then we fixed the cells in ethanol at 4°C overnight. Next, the cells were centrifuged for 5 min at 2000 rpm and resuspended 1×PBS and were centrifuged again. The cells were mixed with 500 μL of propidium (Beyotime, Shanghai, China) and kept in the dark for 10 min. Then flow cytometry (BD Biosciences, CA, USA) was used to analyze the samples.
Western Blotting
Western blotting analysis was used to detect the alterations of protein levels, as previously described (21). The primary antibodies used in the experiments were against METTL3 (Abcam, ab195352), Cytokeratin 8 (KRT8, Abcam, ab53280), FAS (Abcam, ab133619) and GAPDH (Abcam, ab9485). The corresponding IgG-HRP secondary antibody was from Abcam (ab205718). The bend density was analyzed by Image J software (NIH, USA). The experiments were done at least three independent times for final analyses.
Statistical Methods
All values were expressed as means ± standard error of the mean (SEM). One-way analysis of variance (ANOVA) was used to evaluate significant of differences in relative METTL3 mRNA expression in COV362 cells, cell viability and cell cycle analysis among multiple groups (Control, Vector and cells with METTL3 siRNA), and Student’s t-test was performed to compare relative METTL3 and KRT8 mRNA expression between EOC and OE groups. P value less than 0.05 was considered statistically significant. We performed all statistical analyses using SPSS (version 22.0, SPSS Inc., Chicago, IL, USA).
Results
Transcriptome-Wide Detection of m6A Modifications in EOC
Three pairs of human EOC and endometriosis samples were selected for MeRIP-seq and RNA sequencing assays.
A total of 39,237 m6A modified peaks were discovered in the EOC group, representing 17,082 genes. In the endometriosis group, 52,848 m6A peaks were identified, representing 19,349 genes. Among them, 19,365 m6A peaks and 14,874 genes were identified in both groups (Figures 1A, B). Table 1 showed the top 30 altered m6A peaks in EOC group. The results indicate the significant difference in global m6A distribution in EOC and OE groups.
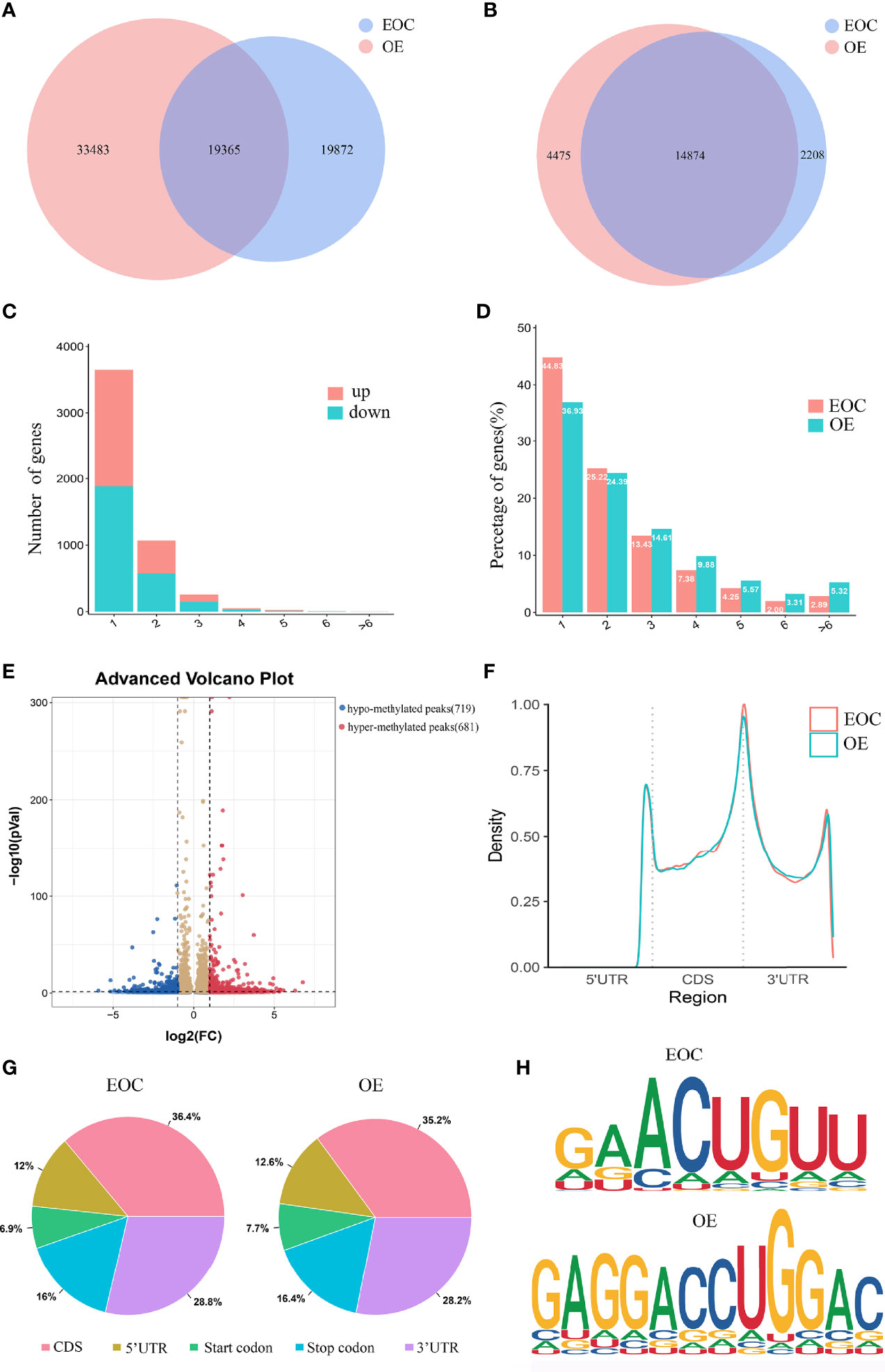
Figure 1 Characteristics of m6A distributions in EOC and OE. (A) Venn diagrams of overlaps of m6A peaks and (B) genes in EOC and OE tissues. (C) The distribution of differentially modified m6A peaks with significance per gene. (D) Number of peaks per gene in EOC and OE tissues. (E) Volcano plots showing the significant differential m6A peaks in EOC samples compared with OE samples. (F) The distribution and difference in the density of m6A peaks along all the transcripts in EOC and OE. (G) Pie charts showing the distributions of m6A peaks in EOC and OE tissues. (H) Representative m6A motifs enriched from the altered m6A peaks in EOC and OE groups, with P-values of 1*e-166 and 1*e-247, respectively.
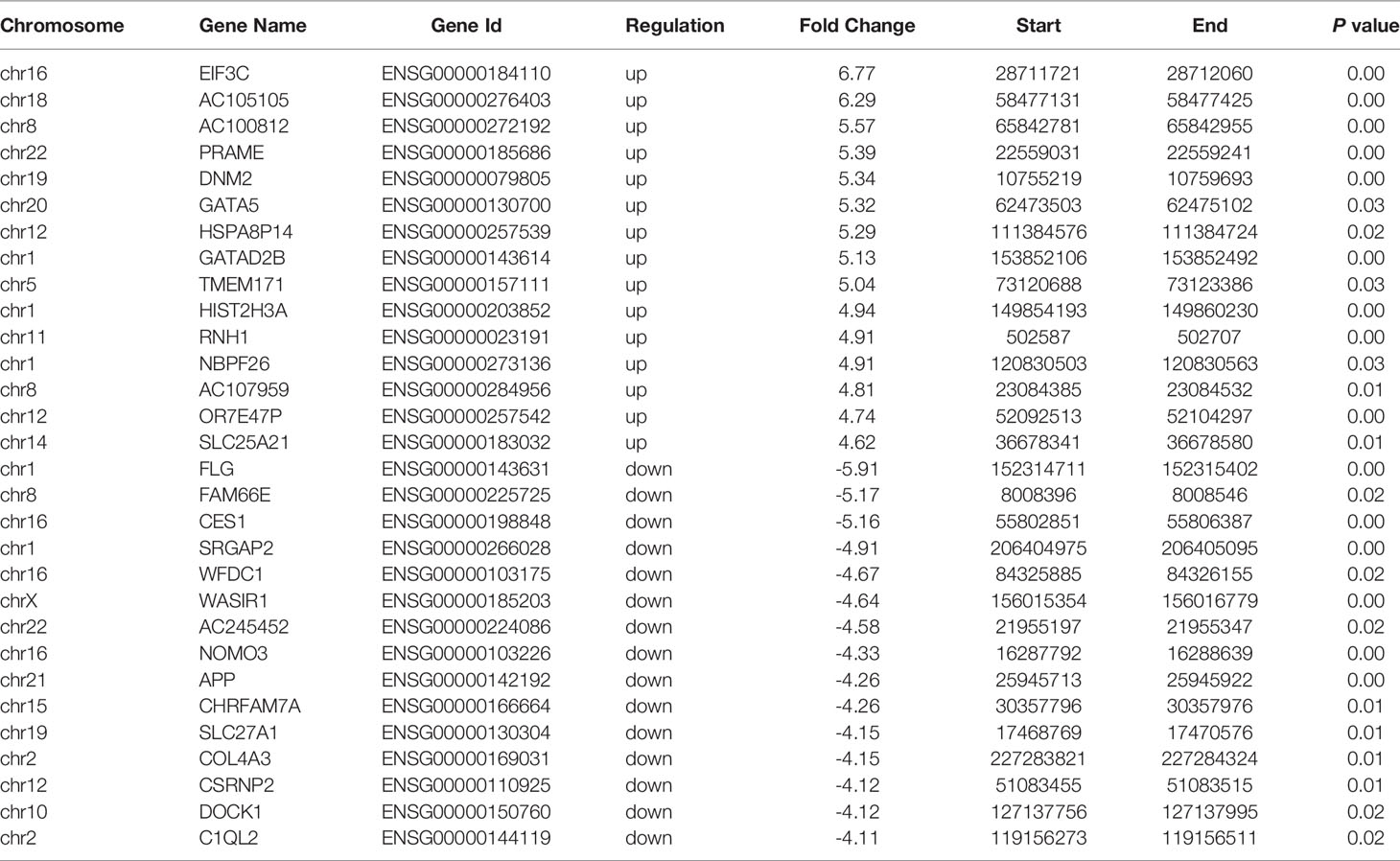
Table 1 The top 30 differently methylated m6A peaks (EOC vs OE).
By analyzing the alteration in the number of m6A peaks per gene, we found that most genes had one m6A peak. Approximately 45% of genes in EOC harbored one m6A peak, and about 83% of genes had 1-3 m6A modified sites. (Figures 1C, D)
Then we compared the abundance of the m6A peaks between EOC and OE samples. As shown in Figure 1E, 681 hyper-methylated m6A sites were discovered in EOC group compared with OE group, and a total of 719 hypo-methylated m6A sites were found.
Next, we investigated the distribution of differentially methylated m6A sites in transcriptome-wide scale of EOC and OE tissues. Each transcript was divided into start codon, 5’-UTR, CDS, 3’-UTR and stop codon. Figure 1F showed the distribution of m6A peaks in the two groups according to their locations in RNA transcripts. In general, the m6A peaks were especially enriched in CDS, 3’UTR, and stop codon in both groups (Figure 1F, G). Enrichment analysis results revealed m6A RRACH (R represents for G/A; A for m6A; H for A/C/U) consensus sequences, which reinforced the authenticity of the data (Figure 1H).
Alterations of m6A Peaks Are Related to Important Signaling Pathways
We performed GO analysis and KEGG analysis in hyper-methylated genes to evaluate the biological significance of m6A-enriched genes in EOC. GO analysis included three functional domains: biological process (BP), cellular component (CC) and molecular function (MF). The top 25 enriched BP terms, top 15 enriched CC terms and top 10 enriched MF terms of the hyper-methylated genes are shown in Figure 2A. The top 20 GO terms of genes with upregulated m6A peaks are shown in Figure 2B. Figure 2C shows that in KEGG analysis, hyper-methylated genes were associated with tight junction, pathogenic Escherichia coli infection, glutathione metabolism, cell adhesion molecules (CAMs), platinum drug resistance, adherens junction, etc.
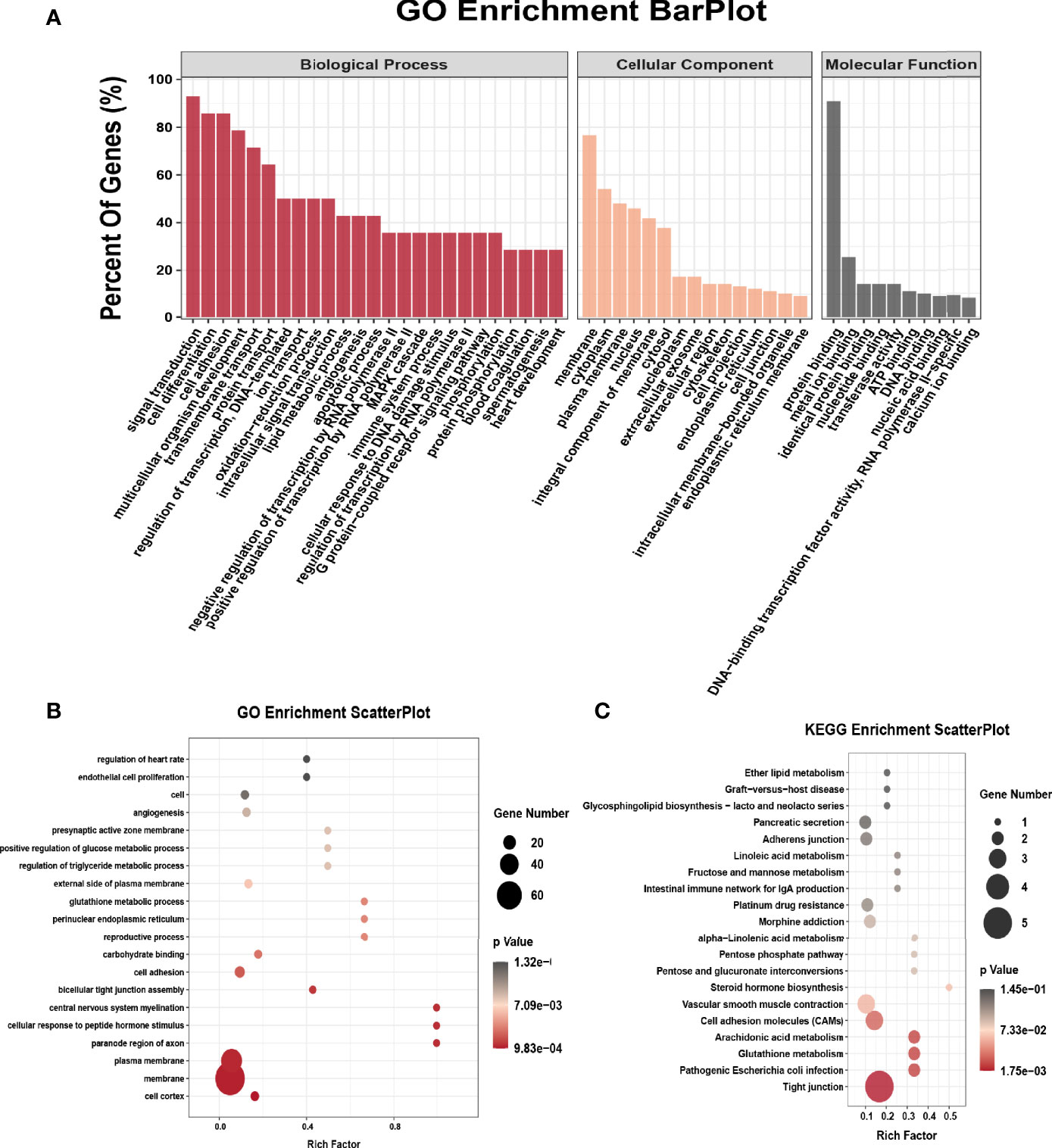
Figure 2 GO and KEGG pathway enrichment of hyper-methylated m6A genes. (A) Major enrichment and meaningful GO terms of hyper-methylated m6A genes in EOC. (B) The top 20 significant GO enrichment terms with m6A hypermethylation. (C) The top 20 significant KEGG pathways with m6A hypermethylation.
The Conjoint Analysis of MeRIP-Seq and RNA Sequencing
Compared to OE group, EOC group had 2876 significantly upregulated genes and 2640 significantly downregulated genes (Figure 3A).
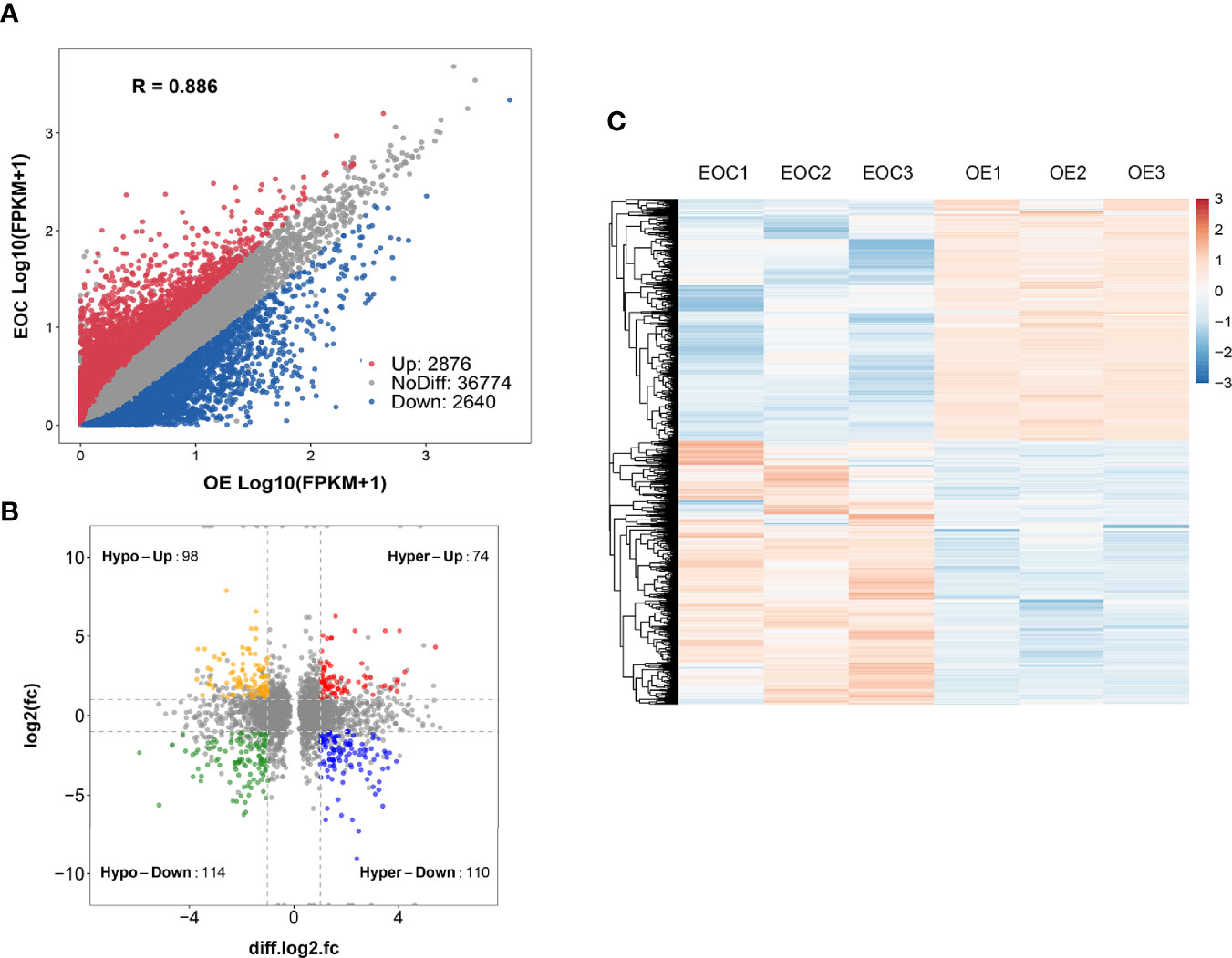
Figure 3 Conjoint analysis of MeRIP-seq and RNA sequencing data. (A) Scatter plot presenting the differentially expressed genes in EOC. (B) Four-quadrant plots presenting the distribution of genes with significant alterations in both the m6A modification and mRNA levels. (C) Heatmap plots exhibiting the differentially expressed genes of EOC and OE groups.
All genes were classified into 4 parts in conjoint analysis of MeRIP-seq and RNA sequencing data: a total of 184 hyper-methylated m6A peaks in which 74 mRNA transcripts were upregulated (hyper-up) and 110 mRNA transcripts were downregulated (hyper-down), and 212 hypo-methylated m6A peaks in 98 upregulated mRNA transcripts (hypo-up) or 114 down-regulated transcripts (hypo-down) (Figure 3B). A heatmap is created to investigate the differentially expression profiles of genes in EOC and OE (Figure 3C). GO analysis and KEGG pathway analysis were performed in those genes with differential expression and methylated m6A peaks synchronously. Figure 4A shows the top 10 terms of BPs, CCs, and MFs. Figure 4B shows the top 20 GO terms of these genes. KEGG pathway analysis showed that genes were enriched in human papillomavirus infection, focal adhesion, tight junction, and ECM−receptor interaction, etc. (Figure 4C).
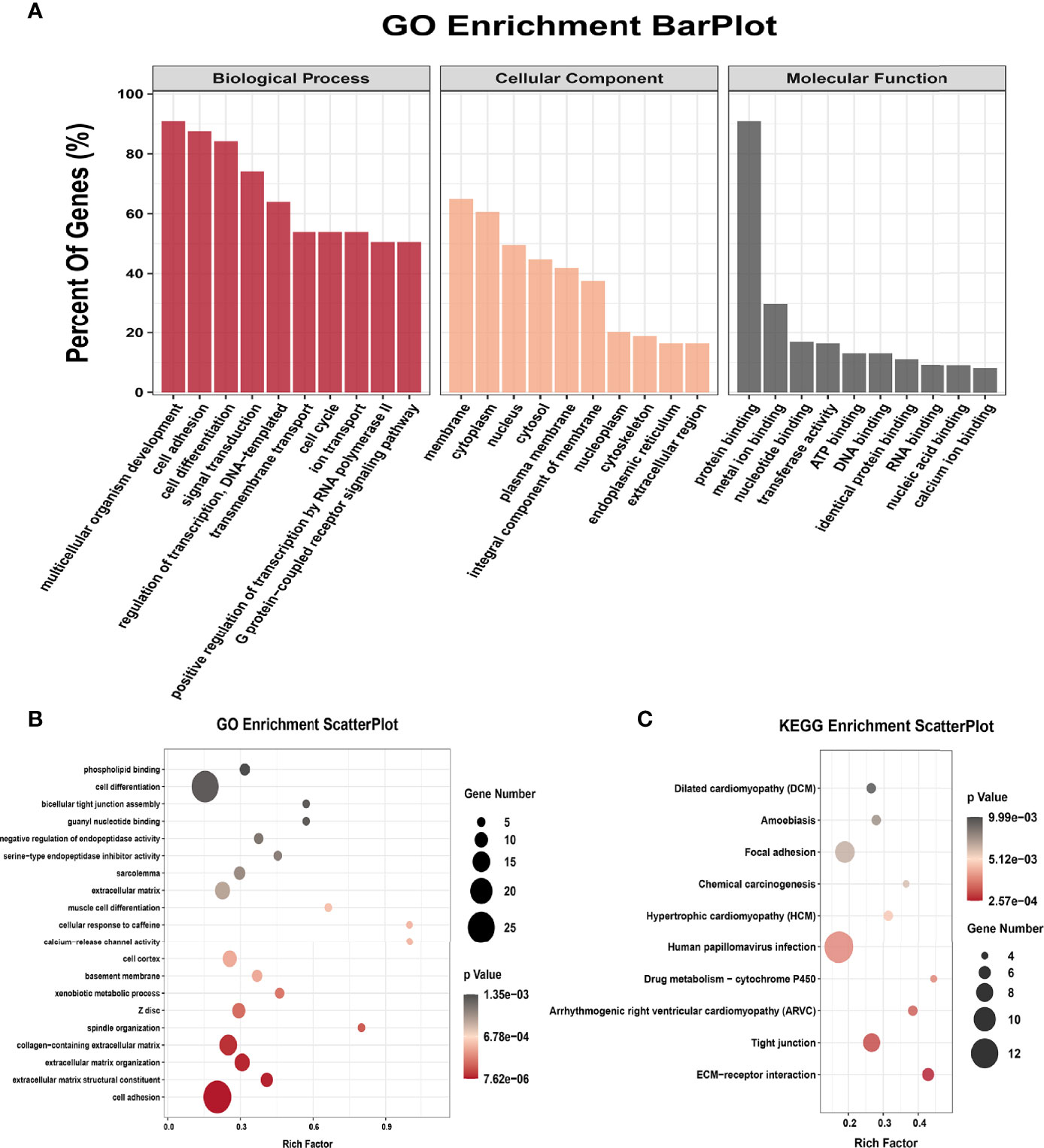
Figure 4 GO terms and KEGG pathways of genes with significant alterations in both the m6A modification and mRNA levels. (A) Major enrichment and meaningful GO terms of genes with significant alterations in both the m6A modification and mRNA levels. (B) The top 20 GO terms of genes with significant alterations in both the m6A modification and mRNA levels. (C) The top 10 KEGG pathways of genes with significant alterations in both the m6A modification and mRNA levels.
Functional Analysis of METTL3 in EOC Cell Line COV362
We further explored the data and found that METTL3 mRNA were highly expressed in the EOC group (Figure 5A). Also, the mRNA expression of KRT8 were significantly higher in the EOC group (Figure 5B). We then transfected the EOC cell line COV362 with METTL3 siRNA and successfully established a METTL3 knockdown cell line (Figure 5C). The MTT assays showed that the knockdown of METTL3 significantly led to the decreasing of cell proliferation (Figure 5D). The TUNEL assays revealed that knockdown of METTL3 increased cell apoptosis (Figure 5E). Furthermore, cell cycle analysis by flow cytometry showed that the percentage of cells in G0/G1 phase increased and S and G2/M phase decreased in cells with METTL3 knockdown (Figure 5F). Western blot data demonstrated that the expression level of KRT8 markedly decreased, and FAS increased in METTL3 knockdown cells (Figure 5G).
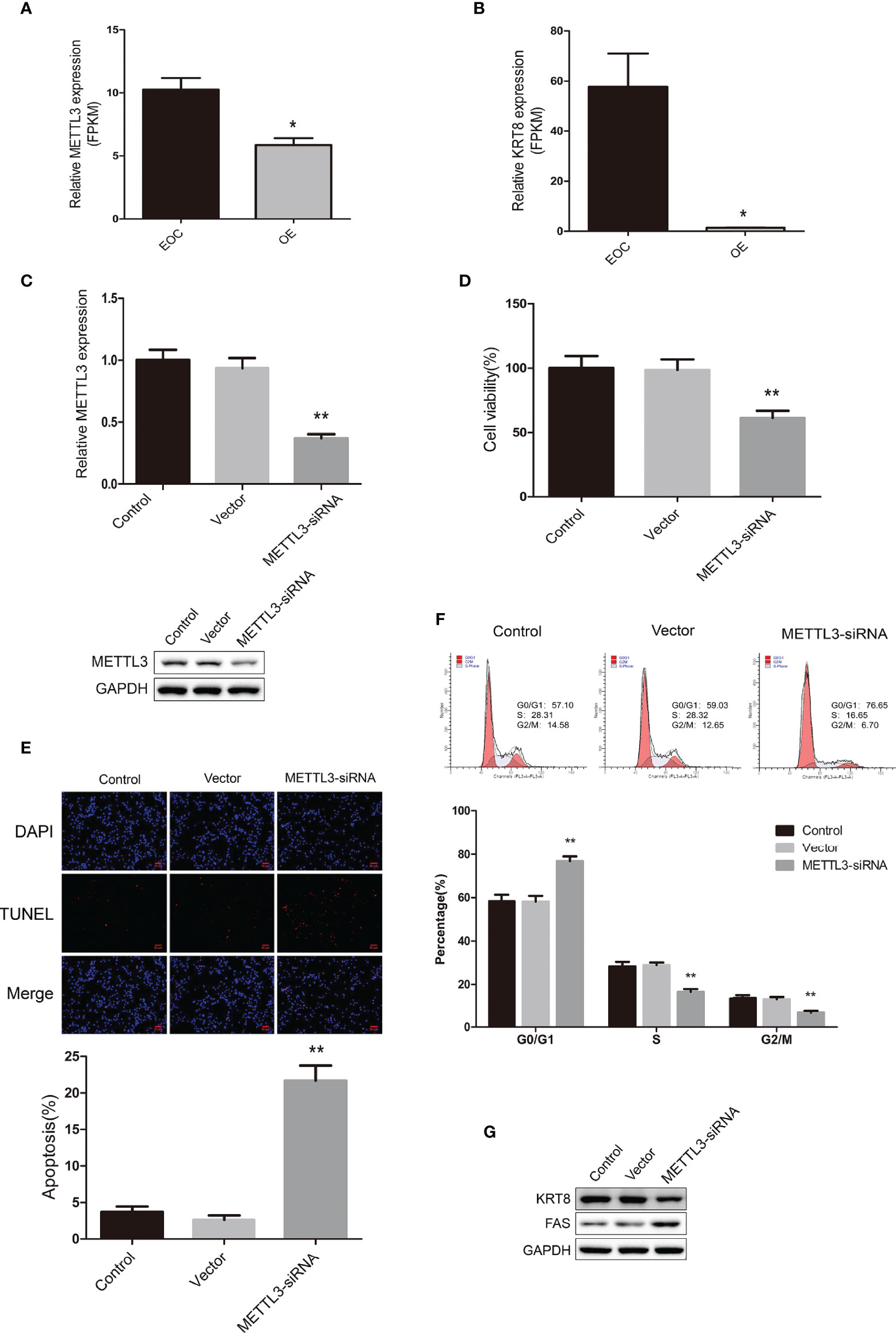
Figure 5 (A) The relative mRNA expression level of METTL3 in EOC and OE groups. (B) The relative mRNA expression level of KRT8 in EOC and OE groups. (C) Real-time PCR analysis and western blot analysis for METTL3 expression in COV362 untreated cells (Control), cells transfected with empty vector (Vector), and cells transfected with METTL3-siRNA (METTL3-siRNA). (D) MTT assay showing cell viability of Control, Vector and METTL3-siRNA cells. (E) The TUNEL assay showing the apoptosis rates of Control, Vector and METTL3-siRNA cells. (F) Cell cycle analysis of Control, Vector and METTL3-siRNA cells. (G) Western blot showing the expression levels of KRT8 and FAS in Control, Vector and METTL3-siRNA cells. * P < 0.05, ** Compared with the other groups, P < 0.01.
Discussion
There are more than 170 modifications in RNA, and m6A is the most abundant modification in mRNA. M6A plays important roles in various aspects of RNA regulation, including RNA stability, splicing, degradation and translation. Increasing evidence suggests that m6A modification is involved in the tumorigenesis, proliferation, invasion, and progression of many kinds of malignancies (22, 23). M6A modification not only affects cancer functions such as cancer metabolism, cancer stem cell formation, epithelial–mesenchymal transition, but also participates in multiple signaling pathways (24). Although the transcriptome-wide m6A profiling of several cancers have been identified, the m6A methylation of EOC has not been reported. The present study identified the transcriptome-wide m6A distribution of EOC using a combination of MeRIP-seq and RNA sequencing for the first time.
In this study, we performed MeRIP-seq to determine the genome-wide profiling of m6A modification in EOC. Then the alterations of gene expression were also analyzed. Furthermore, GO analysis and KEGG pathway analysis were performed to evaluate the biological significance and pathophysiological roles of m6A-enriched genes and genes with differential expression and methylated m6A peaks synchronously in EOC by a conjoint analysis of MeRIP-seq and RNA-sequencing data. Additionally, we further explored the functions of METTL3 gene in EOC cells.
In the current study, we found that the difference in m6A modification between EOC and OE was significant. Briefly, 681 m6A peaks were upregulated and 719 m6A peaks were downregulated differentially in the EOC group. Furthermore, cancer-related biological pathways associated with hyper-methylated genes were significantly enriched. This suggests that m6A modification may contribute to EOC pathogenesis.
GO analysis revealed that cell adhesion-related hyper-methylated genes were enriched. There are four major groups of cell adhesion molecules: cadherins, integrins, selectins, and immunoglobulins (25). The integrin-mediated adhesion of epithelial cells to extracellular matrix proteins are lost and multiple signal transduction events are induced in the classic view of malignant transformation in the epithelium. This can be due to decreased E-cadherin expression resulting from transcriptional repression, its promoter methylation, or, uncommonly, a genetic or sporadic mutation (26–29). KEGG pathway analysis revealed that tight junctions are related to genes with up-methylated sites. Many researches have shown that tight junctions are closely related to the genesis and progression of many tumors. For instance, claudins, a group of membrane proteins that play a critical role in maintaining the proper function of epithelial tight junctions, were reported to participate in the carcinogenesis and metastasis of various cancers. Previous studies showed that claudin-1 overexpression is associated with the malignant behavior of colorectal cancer (30). Cytoplasmic expression of claudin-1 enhances the migratory ability of melanoma cells (31). Claudin-1 promotes the invasive ability of oral squamous cell carcinoma OSC-4 and NOS-2 cell lines through MT1-MMP and MMP-2 activation (32). E-cadherin expression was reduced by Claudin-1 via upregulating of ZEB-1 in colon cancer cells (33). Also, cell survival, invasion, and motility were increased in human ovarian surface epithelial cells with overexpression of claudin-3 and -4 increased (34). Hence, together with our data, we suggest that abnormal m6A methylation of genes participate in change of cell adhesion and tight junction, which may promote ovarian tumorigenesis and metastasis. Thus, modulating the m6A modification of genes in these pathways may provide new directions for the treatment of EOC.
Accumulating evidence has identified that METTL3 is implicated in human cancers either as an oncogene or a cancer suppressor in recent years. The controversial role of METTL3 in human cancer cells may be due to the different mechanisms of origin in various cancers. For instance, previous reports showed that METTL3 expression was significantly higher in hepatocellular carcinoma (HCC) than normal controls and it was correlated with shorter recurrence-free survival and overall survival (35). METTL3 knockout suppressed HCC oncogenicity and lung metastasis in vivo notably (36). It was demonstrated that suppressor of cytokine signaling 2 was the target of METTL3-mediated m6A modification and METTL3 repressed its expression through an m6A-YTHDF2-dependent pathway in liver cancer (36). Another study revealed that the epithelial-mesenchymal transition (EMT) was regulated by METTL3 in HCC cells through the methylating of CDS of Snail and then triggering polysome-mediated translation of Snail mRNA in cancer cells (37). It was reported that high expression of METTL3 was related with a poor prognosis in gastric cancer patients. In this study, Zinc finger MYM-type containing 1 (ZMYM1) was found to be a bona fide target of METTL3, which bound to and mediated the repression of E-cadherin promoter, thus driving the EMT program and leading to metastasis (38). Another study revealed that METTL3 level was elevated in colorectal cancer metastatic tissues and it was predictive of poor prognosis. SRY-box 2 (SOX2) was a downstream gene of METTL3, and the m6A of SOX2 transcripts was recognized by IGF2BP2 to prevent mRNA degradation (39). It was also reported that METTL3 can act as a tumor suppressor in some type of cancers. Liu et al. reported that about 70% of endometrial tumors exhibit reduced m6A levels than normal endometrium. The differences were likely due to a METTL14 mutation or reduced expression of METTL3, which promotes cell proliferation and the tumorigenicity of endometrial cancer through the activation of AKT signaling pathway (40).
A previous study showed that the expression of METTL3 and overall m6A level were elevated in EOC tissues, and the high expression of METTL3 was associated with poor malignancy and survival of EOC by mediating aberrant m6A modification (41). In our study, we found that METTL3 mRNA was highly expressed in the EOC group. Furthermore, the mRNA expression of KRT8 was significantly higher in the EOC group. We found that METTL3 knockdown in the EOC cell line significantly decreased cell proliferation, increased cell apoptosis, and induced cell cycle arrest at the G0/G1 phase. In addition, the expression level of KRT8 protein markedly decreased and FAS protein increased. As previous reports indicated KRT8 provide resistance to FAS-mediated apoptosis (42, 43), we deduced that knockdown of METTL3 may downregulate KRT8 expression, thus inducing FAS expression. However, whether METTL3 has an oncogenic role in EOC and is it mediated by KRT8 needs to be further explored. Also, more in-depth research should be done in the future to determine whether the function of METTL3 is related to its role as a methyltransferase.
In summary, we presented the transcriptome-wide m6A modifications of EOC for the first time using MeRIP-seq and discovered differentially expressed genes with various methylated m6A modifications. This study may provide new directions for in-depth research of the underlying molecular mechanisms and signaling pathways of EOC development and progression thus indicating new approaches for the treatment of EOC. Although some inhibitors of m6A methylation have been developed with promising effects, new regulators or inhibitors of m6A modifications should be further explored to provide novel therapeutic strategies for cancers.
Data Availability Statement
The data presented in the study are deposited in the The Gene Expression Omnibus (GEO) repository. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE196748.
Ethics Statement
The studies involving human participants were reviewed and approved by The Institutional Review Board of Zhejiang cancer hospital. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
AY and MW designed and supervised the whole study. LY and XC performed experiments and wrote the manuscript. XQ and JZ revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Basic Public Welfare Research Program of Zhejiang Province (LGF19H160011) and Medical and Health Science and Technology Project of Zhejiang Province (2019ZD001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Prat J. Ovarian Carcinomas: Five Distinct Diseases With Different Origins, Genetic Alterations, and Clinicopathological Features. Virchows Archiv (2012) 460(3):237–49. doi: 10.1007/s00428-012-1203-5
3. Seidman JD, Horkayne-Szakaly I, Haiba M, Boice CR, Kurman RJ, Ronnett BM. The Histologic Type and Stage Distribution of Ovarian Carcinomas of Surface Epithelial Origin. Int J Gynecol Pathol (2004) 23(1):41–4. doi: 10.1097/01.pgp.0000101080.35393.16
4. Anglesio MS, Yong PJ. Endometriosis-Associated Ovarian Cancers. Clin Obstet Gynecol (2017) 60(4):711–27. doi: 10.1097/GRF.0000000000000320
5. Tuck MT. The Formation of Internal 6-Methyladenine Residues in Eucaryotic Messenger RNA. Int J Biochem (1992) 24(3):379–86. doi: 10.1016/0020-711X(92)90028-Y
6. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell (2017) 169(7):1187–200. doi: 10.1016/j.cell.2017.05.045
7. Wang P, Doxtader KA, Nam Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell (2016) 63(2):306–17. doi: 10.1016/j.molcel.2016.05.041
8. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Corrigendum: Structural Basis of N(6)-Adenosine Methylation by the METTL3-METTL14 Complex. Nature (2017) 542(7640):260. doi: 10.1038/nature21073
9. Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP Is a Regulatory Subunit of the RNA N6-Methyladenosine Methyltransferase. Cell Res (2014) 24(2):177–89. doi: 10.1038/cr.2014.3
10. Schöller E, Weichmann F, Treiber T, Ringle S, Treiber N, Flatley A, et al. Interactions, Localization, and Phosphorylation of the M(6)A Generating METTL3-METTL14-WTAP Complex. RNA (New York NY) (2018) 24(4):499–512. doi: 10.1261/rna.064063.117
11. Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-Methyladenosine in Nuclear RNA Is a Major Substrate of the Obesity-Associated FTO. Nat Chem Biol (2011) 7(12):885–7. doi: 10.1038/nchembio.687
12. Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 Is a Mammalian RNA Demethylase That Impacts RNA Metabolism and Mouse Fertility. Mol Cell (2013) 49(1):18–29. doi: 10.1016/j.molcel.2012.10.015
13. Cao G, Li HB, Yin Z, Flavell RA. Recent Advances in Dynamic M6a RNA Modification. Open Biol (2016) 6(4):160003. doi: 10.1098/rsob.160003
14. Berlivet S, Scutenaire J, Deragon JM, Bousquet-Antonelli C. Readers of the M(6)A Epitranscriptomic Code. Biochim Biophys Acta Gene Regul Mech (2019) 1862(3):329–42. doi: 10.1016/j.bbagrm.2018.12.008
15. Liu Q, Gregory RI. RNAmod: An Integrated System for the Annotation of mRNA Modifications. Nucleic Acids Res (2019) 47(W1):W548–w55. doi: 10.1093/nar/gkz479
16. Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-Methyladenosine Alters RNA Structure to Regulate Binding of a Low-Complexity Protein. Nucleic Acids Res (2017) 45(10):6051–63. doi: 10.1093/nar/gkx141
17. Yang C, Hu Y, Zhou B, Bao Y, Li Z, Gong C, et al. The Role of M(6)A Modification in Physiology and Disease. Cell Death Dis (2020) 11(11):960. doi: 10.1038/s41419-020-03143-z
18. Xu J, Zhu C, Yu Y, Wu W, Cao J, Li Z, et al. Systematic Cancer-Testis Gene Expression Analysis Identified CDCA5 as a Potential Therapeutic Target in Esophageal Squamous Cell Carcinoma. EBioMedicine (2019) 46:54–65. doi: 10.1016/j.ebiom.2019.07.030
19. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3’ UTRs and Near Stop Codons. Cell (2012) 149(7):1635–46. doi: 10.1016/j.cell.2012.05.003
20. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the Human and Mouse M6a RNA Methylomes Revealed by M6a-Seq. Nature (2012) 485(7397):201–6. doi: 10.1038/nature11112
21. Yang L, Shi T, Liu F, Ren C, Wang Z, Li Y, et al. REV3L, a Promising Target in Regulating the Chemosensitivity of Cervical Cancer Cells. PloS One (2015) 10(3):e0120334. doi: 10.1371/journal.pone.0120334
22. Chen M, Wong CM. The Emerging Roles of N6-Methyladenosine (M6a) Deregulation in Liver Carcinogenesis. Mol Cancer (2020) 19(1):44. doi: 10.1186/s12943-020-01172-y
23. Wang T, Kong S, Tao M, Ju S. The Potential Role of RNA N6-Methyladenosine in Cancer Progression. Mol Cancer (2020) 19(1):88. doi: 10.1186/s12943-020-01204-7
24. Tan F, Zhao M, Xiong F, Wang Y, Zhang S, Gong Z, et al. N6-Methyladenosine-Dependent Signalling in Cancer Progression and Insights Into Cancer Therapies. J Exp Clin Cancer Res: CR (2021) 40(1):146. doi: 10.1186/s13046-021-01952-4
25. Freemont AJ, Hoyland JA. Cell Adhesion Molecules. Clin Mol Pathol (1996) 49(6):M321–30. doi: 10.1136/mp.49.6.M321
26. Vleminckx K, Vakaet L Jr., Mareel M, Fiers W, van Roy F. Genetic Manipulation of E-Cadherin Expression by Epithelial Tumor Cells Reveals an Invasion Suppressor Role. Cell (1991) 66(1):107–19. doi: 10.1016/0092-8674(91)90143-M
27. Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A Causal Role for E-Cadherin in the Transition From Adenoma to Carcinoma. Nature (1998) 392(6672):190–3. doi: 10.1038/32433
28. Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, et al. The Transcription Factor Snail Is a Repressor of E-Cadherin Gene Expression in Epithelial Tumour Cells. Nat Cell Biol (2000) 2(2):84–9. doi: 10.1038/35000034
29. Auerkari EI. Methylation of Tumor Suppressor Genes P16(INK4a), P27(Kip1) and E-Cadherin in Carcinogenesis. Oral Oncol (2006) 42(1):5–13. doi: 10.1016/j.oraloncology.2005.03.016
30. Huo Q, Kinugasa T, Wang L, Huang J, Zhao J, Shibaguchi H, et al. Claudin-1 Protein Is a Major Factor Involved in the Tumorigenesis of Colorectal Cancer. Anticancer Res (2009) 29(3):851–7.
31. French AD, Fiori JL, Camilli TC, Leotlela PD, O’Connell MP, Frank BP, et al. PKC and PKA Phosphorylation Affect the Subcellular Localization of Claudin-1 in Melanoma Cells. Int J Med Sci (2009) 6(2):93–101. doi: 10.7150/ijms.6.93
32. Oku N, Sasabe E, Ueta E, Yamamoto T, Osaki T. Tight Junction Protein Claudin-1 Enhances the Invasive Activity of Oral Squamous Cell Carcinoma Cells by Promoting Cleavage of Laminin-5 Gamma2 Chain via Matrix Metalloproteinase (MMP)-2 and Membrane-Type MMP-1. Cancer Res (2006) 66(10):5251–7. doi: 10.1158/0008-5472.CAN-05-4478
33. Singh AB, Sharma A, Smith JJ, Krishnan M, Chen X, Eschrich S, et al. Claudin-1 Up-Regulates the Repressor ZEB-1 to Inhibit E-Cadherin Expression in Colon Cancer Cells. Gastroenterology (2011) 141(6):2140–53. doi: 10.1053/j.gastro.2011.08.038
34. Agarwal R, D’Souza T, Morin PJ. Claudin-3 and Claudin-4 Expression in Ovarian Epithelial Cells Enhances Invasion and Is Associated With Increased Matrix Metalloproteinase-2 Activity. Cancer Res (2005) 65(16):7378–85. doi: 10.1158/0008-5472.CAN-05-1036
35. Liu GM, Zeng HD, Zhang CY, Xu JW. Identification of METTL3 as an Adverse Prognostic Biomarker in Hepatocellular Carcinoma. Digest Dis Sci (2021) 66(4):1110–26. doi: 10.1007/s10620-020-06260-z
36. Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-Methyladenosine Methyltransferase-Like 3 Promotes Liver Cancer Progression Through YTHDF2-Dependent Posttranscriptional Silencing of SOCS2. Hepatol (Baltimore Md) (2018) 67(6):2254–70. doi: 10.1002/hep.29683
37. Lin X, Chai G, Wu Y, Li J, Chen F, Liu J, et al. RNA M(6)A Methylation Regulates the Epithelial Mesenchymal Transition of Cancer Cells and Translation of Snail. Nat Commun (2019) 10(1):2065. doi: 10.1038/s41467-019-09865-9
38. Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, et al. METTL3-Mediated N6-Methyladenosine Modification Is Critical for Epithelial-Mesenchymal Transition and Metastasis of Gastric Cancer. Mol Cancer (2019) 18(1):142. doi: 10.1186/s12943-019-1065-4
39. Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, et al. METTL3 Facilitates Tumor Progression via an M(6)A-IGF2BP2-Dependent Mechanism in Colorectal Carcinoma. Mol Cancer (2019) 18(1):112. doi: 10.1186/s12943-019-1038-7
40. Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, et al. M(6)A mRNA Methylation Regulates AKT Activity to Promote the Proliferation and Tumorigenicity of Endometrial Cancer. Nat Cell Biol (2018) 20(9):1074–83. doi: 10.1038/s41556-018-0174-4
41. Ma Z, Li Q, Liu P, Dong W, Zuo Y. METTL3 Regulates M6a in Endometrioid Epithelial Ovarian Cancer Independently of METTl14 and WTAP. Cell Biol Int (2020) 44(12):2524–31. doi: 10.1002/cbin.11459
42. Trisdale SK, Schwab NM, Hou X, Davis JS, Townson DH. Molecular Manipulation of Keratin 8/18 Intermediate Filaments: Modulators of FAS-Mediated Death Signaling in Human Ovarian Granulosa Tumor Cells. J Ovarian Res (2016) 9:8. doi: 10.1186/s13048-016-0217-z
Keywords: endometrioid ovarian cancer, N6-methyladenosine, MeRIP-seq, METTL3, modification patterns
Citation: Yang L, Chen X, Qian X, Zhang J, Wu M and Yu A (2022) Comprehensive Analysis of the Transcriptome-Wide m6A Methylome in Endometrioid Ovarian Cancer. Front. Oncol. 12:844613. doi: 10.3389/fonc.2022.844613
Received: 28 December 2021; Accepted: 01 February 2022;
Published: 23 February 2022.
Edited by:
Dong-Hua Yang, St. John’s University, United StatesCopyright © 2022 Yang, Chen, Qian, Zhang, Wu and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aijun Yu, eWFqMTk5M0AxMjYuY29t; Meijuan Wu, d3VtakB6amNjLm9yZy5jbg==
†These authors have contributed equally to this work