
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 18 May 2022
Sec. Cancer Genetics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.844527
 Juan Du1
Juan Du1 Xin Qiu2
Xin Qiu2 Changchang Lu2
Changchang Lu2 Yahui Zhu1Weiwei Kong1
Yahui Zhu1Weiwei Kong1 Mian Xu3Xin Zhang3Min Tang4Jun Chen5Qi Li5
Mian Xu3Xin Zhang3Min Tang4Jun Chen5Qi Li5 Aimei Li6
Aimei Li6 Jian He6Qing Gu7Lei Wang8Yudong Qiu9
Jian He6Qing Gu7Lei Wang8Yudong Qiu9 Baorui Liu1*
Baorui Liu1*Purpose: Although mutational analysis of pancreatic cancer has provided valuable clinical information, it has not significantly changed treatment prospects. The purpose of this study is to further investigate molecular alterations in locally advanced pancreatic cancer and identify predictors of the efficacy of nab-paclitaxel plus gemcitabine (AG) chemotherapy.
Experimental design: Tumor samples from 118 pancreatic cancer patients who received AG chemotherapy as first-line treatment were sequenced and genomic profile was generated. Molecular alterations and the involved signaling pathways were analyzed. Genes with a significant difference in mutation frequency between primary and metastatic tumors were identified, and prognostic-related mutant genes were screened using SPSS version 22.0.
Results: The most common altered genes in the patients were KRAS (94.9%), TP53 (81.4%), CDKN2A (36.4%), and SMAD4 (22.9%). The mutational frequencies of CDKN2B (14.8% vs. 0%, p = 0.001), FAT3 (7.4% vs. 0%, p = 0.041), MTAP (13% vs. 1.6%, p = 0.023), and SMAD4 (31.4% vs. 15.6%, p = 0.049) in metastatic tumors were significantly higher than that in primary tumors. TP35 and KRAS mutations were significantly correlated with objective response rate, while EPHA7, RNF43, and HMGA2 mutations were significantly correlated with disease control rate. Additionally, patients with TGFR2B, FGF23, EPHA7, SMARCA4, CARD11, ADGRA2, CCNE1, and ACVR2A alterations had a worse overall survival. Further, EPHA7, CARD11, NOTCH1, GATA6, ACVR2A, and HMGA2 mutations indicated undesirable progression-free survival.
Conclusions: CDKN2B, FAT3, MTAP, and SMAD4 may be biomarkers that distinguish primary tumors from metastases. EPHA7 mutation may serve as a prognostic biomarker to predict the treatment efficacy of AG chemotherapy in locally advanced pancreatic cancer.
Pancreatic cancer is a highly invasive malignant tumor and has the worst prognosis of all human cancers, with a 5 year survival rate of 11% (1). Despite great advances in treatments like radiotherapy and chemotherapy and improvements in surgical techniques, pancreatic cancer will likely become the second leading cause of cancer-related deaths by 2030 (2). So far, surgical resection is the only curative treatment. Unfortunately, only approximately 15–20% of pancreatic cancer patients have resectable disease at diagnosis (3). Even worse, among these patients the median survival is about 8.5 to 11.1 months owing to early metastasis (4, 5). Indeed, aggressive and cytotoxic chemotherapy is still the standard treatment for pancreatic cancer. The National Comprehensive Cancer Network (NCCN) lists nab-paclitaxel plus gemcitabine (AG chemotherapy) as the preferred first-line therapy for the treatment of locally advanced pancreatic cancer and metastatic pancreatic cancer. However, the median progression-free survival of metastatic pancreatic cancer treated with AG chemotherapy was only 5.5 months, and the median overall survival of metastatic pancreatic cancer was 8.5 months (6). Moreover, due to the absence of effective prognostic and predictive biomarkers and the limited clinical success of preferred therapies, the successful treatment of pancreatic cancer is still elusive (7). Hence, to improve existing treatments and develop novel therapeutic strategies, a better understanding of the molecular pathology of pancreatic cancer is vital.
Next-generation sequencing (NGS) is now widely available for examination of tumor samples in commercial laboratories, and advances in NGS-based genomics have greatly increased our understanding of the molecular basis of pancreatic cancer. Based on comprehensive integrated genomic and transcriptomic analysis, a study in 2016 classified pancreatic cancer into four subtypes (stable, locally rearranged, scattered, and unstable) that are associated with specific histopathological characteristics (3). Genomic analyses revealed four commonly mutated genes (KRAS, TP53, SMAD4, and CDKN2A) in pancreatic cancer. These mutations cluster in core molecular pathways, including DNA damage repair, cell cycle regulation, TGF-β signaling, chromatin regulation, and axonal guidance (8, 9). Further, several molecular profiling studies revealed that up to 48% of pancreatic cancers harbor therapeutically relevant genomic alterations, such as ERBB2, MET, FGFR1, CDK6, PIK3R3, and PIK3CA at low individual patient prevalence (3, 8, 10). In addition, a recent study showed that tumor fraction was remarkably higher in metastatic than localized tumors, and genetic heterogeneity was found between distinct metastatic tumors, especially in different organs (11). However, our knowledge of the genomic landscape of metastatic pancreatic cancer remains limited.
In this study we performed genomic analysis of 118 pancreatic cancer patients using NGS. Primary tumors were obtained from 64 patients and metastatic tumors from 54 patients, and the mutational landscapes and mutated-gene related pathways of the primary and metastatic tumors were mapped and compared. Furthermore, mutated genes associated with prognosis were screened for the potential prediction of treatment response and development of new targeted treatments.
A total of 118 pancreatic cancer patients from the Nanjing Drum Tower Hospital, the Affiliated Hospital of Nanjing University Medical School, were enrolled for this study from 2018 to 2020. Tumor samples or biopsy tissues were collected from these patients for the detection of genomic alterations. Pancreatic cancer patients with locally advanced tumors or metastases were included. Relevant basic information, family history, clinical and pathological features, and treatment history were collected. All of the patients received AG chemotherapy as a first-line treatment, and some received second-line (and third-line therapy) according their own will. Treatment response was assessed with abdominal enhanced computed tomography and magnetic resonance imaging following the criteria of Response Evaluation Criteria in Solid Tumors (RECIST). Treatment response was quantitatively classified into four categories: complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD). The information for the 118 patients has not been previously published.
The project was approved by the Ethics Committee of Nanjing Drum Tower Hospital (approval number: EXPLORE-PC101). Informed consent was provided by all patients or their immediate families. This work was conducted in accordance with the guidelines of the Declaration of Helsinki. All methods used in this protocol were carried out in accordance with the relevant guidelines and regulations.
The genomic alterations were identified using the NGS-based YuanSuTM450 gene panel (OrigiMed, Shanghai, China), which covers all the coding exons of 450 cancer-related genes that are frequently rearranged in solid tumors. The genes were captured and sequenced with a mean depth of 800× using Illumina NextSeq 500. The procedures followed those described by Frampton et al. (12). GAs) were identified as previously described methods (13): single nucleotide variants (SNV) were identified using MuTect (v1.7). Insertion-deletions (indels) were identified using PINDEL (V0.2.5). The functional impacts of GAs were annotated using SnpEff3.0. Copy number variation (CNV) regions were identified using Control-FREEC (v9.7) with the following parameters: window = 50,000 and step = 10,000. Gene fusions were detected through an in-house developed pipeline. Gene rearrangements were assessed using Integrative Genomics Viewer (IGV). TMB was estimated by counting the coding somatic mutations, including SNV and indels, per megabase of the sequence examined in each patient. The TMB value was further divided into two groups: high TMB (TMB-H), defined as ≥10 mutations/Mb, and low TMB (TMB-L), defined as <10 mutations/Mb.
Statistical analyses were performed using SPSS version 22.0 (SPSS Inc., Chicago, IL, USA). Overall survival (OS) was defined as the time from the start of AG therapy to death from any cause. Progression-free survival (PFS) was defined as the time from the start of AG therapy to disease progression. The objective response rate (ORR) was defined as the percentage of patients that achieved PR and SD. The disease control rate (DCR) was defined as the percentage of patients that achieved PR. Fisher’s exact test was used to analyze associations between categorical variables. The Student’s t−test and the Wilcoxon rank test were used to analyze associations between normally distributed data and non-normally distributed data, respectively. The Kruskal test was used to analyze associations between multiple groups of nonparametric data. A value of p < 0.05 was considered statistically significant.
A total of 118 pancreatic cancer patients were enrolled in this study. The median age of these patients was 61 years old (range = 33−85 years old); 67 of them were male (56.8%) and 51 were female (43.2%). The tumor tissues were divided into primary tumors (n = 64, 54.2%) and metastatic tumors (n = 54, 45.8%). Based on tumor stage, there were 36 patients at stage III (30.5%) and 82 patients at stage IV (69.5%). The median TMB of the 118 patients was 3.1 Mut/Mb (range = 0.0−69.5). In the cohort, 4 patients were identified as TMB-H (3.4%) while 114 patients were identified as TMB-L (96.6%). Microsatellite stability (MSS) status was also measured in all patients; 1 patient had high microsatellite instability (MSI-H), and 117 patients had MSS. Detailed clinical characteristics of each patient are listed in Table 1.
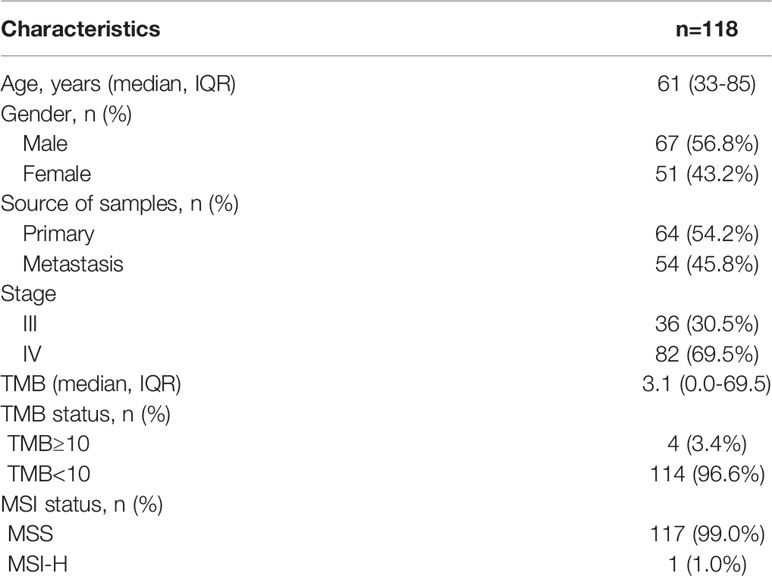
Table 1 Clinical characteristics of patients.
A total of 792 variations in 267 genes, including 447 (56.4%) substitution/indels, 157 (19.8%) gene amplification, 135 (17.0%) truncation, 18 (2.3%) fusions/rearrangements, 35 (4.4%) gene homozygous deletions, and 23 (2.9%) gene homozygous deletions, were detected in the 118 pancreatic cancer patients. The most common gene alterations in the study group were KRAS (94.9%), TP53 (81.4%), CDKN2A (36.4%), and SMAD4 (22.9%) (Figure 1A). Notably, the most common KRAS and TP53 variants observed were substitution/indels (Figure 1). The most frequent mutations were in codon 12 of KRAS (84%), including KRAS G12D (50%) and G12V (34%) (Figures 1B, C).
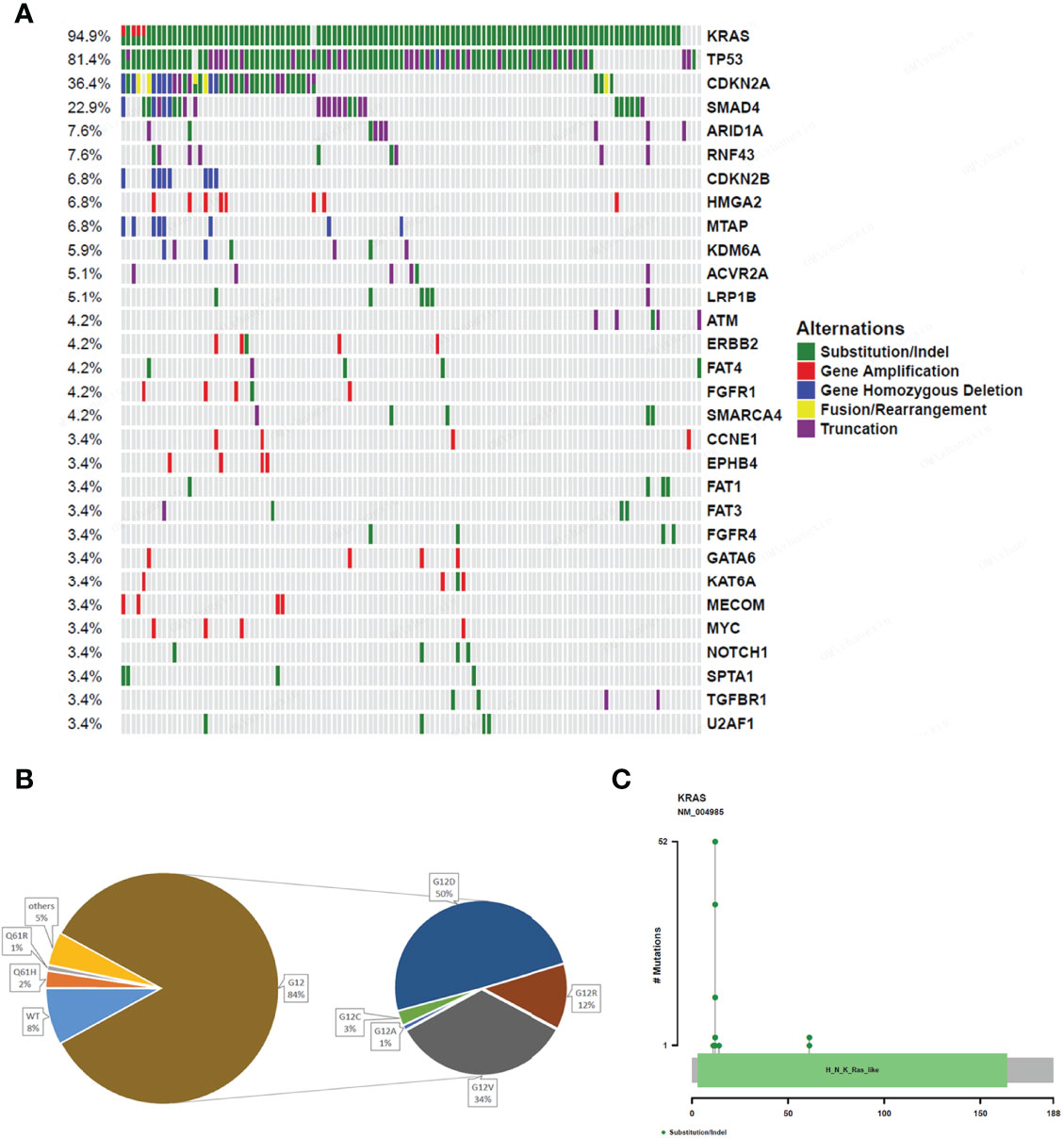
Figure 1 Gene alterations of 118 pancreatic cancer patients. (A) The mutation profiles. The panel shows the matrix of mutations with each type of mutation in a different color. Each column denotes an individual tumor and each row represents a gene. The right panel shows the name of mutations and the left panel shows the proportion of mutations. Green; Substitution/Indel, Red; Gene amplification, Blue; Gene homozygous deletion, Yellow; Fusion/Rearrangement, Purple; Truncation. (B) Ratio of different mutation types in KRAS gene. (C) The distribution map of KRAS gene mutation sites showing the distribution of mutation sites.
In order to identify the functions of the mutated genes and understand how these mutations contribute to selective advantages during tumorigenesis, their involved regulatory pathways were analyzed. All the mutated genes were mainly involved in the following signaling pathways: RTK.RAS (94.9%), TP53 (83.9%), cell cycle (41.5%), TGF-β (35.6%), DDR (16.9%), WNT (13.6%), HIPPO (11.9%), NOTCH (11.9%), PI3K (11%), HR (9.3%), MYC (5.9%), and NFR2 (0.8%) (Figures 2A, B). RTK.RAS pathway alterations were present in most patients, predominantly due to KRAS mutations (94%). In patients lacking KRAS mutations, other RTK.RAS genes were often mutated, including BRAF, EGFR, ERBB2/3/4, FGFR1/3/4, and ROS1 (Figure 2A). There was no significant difference in the involved regulatory pathways between primary and metastatic tumors (Figure 2C).
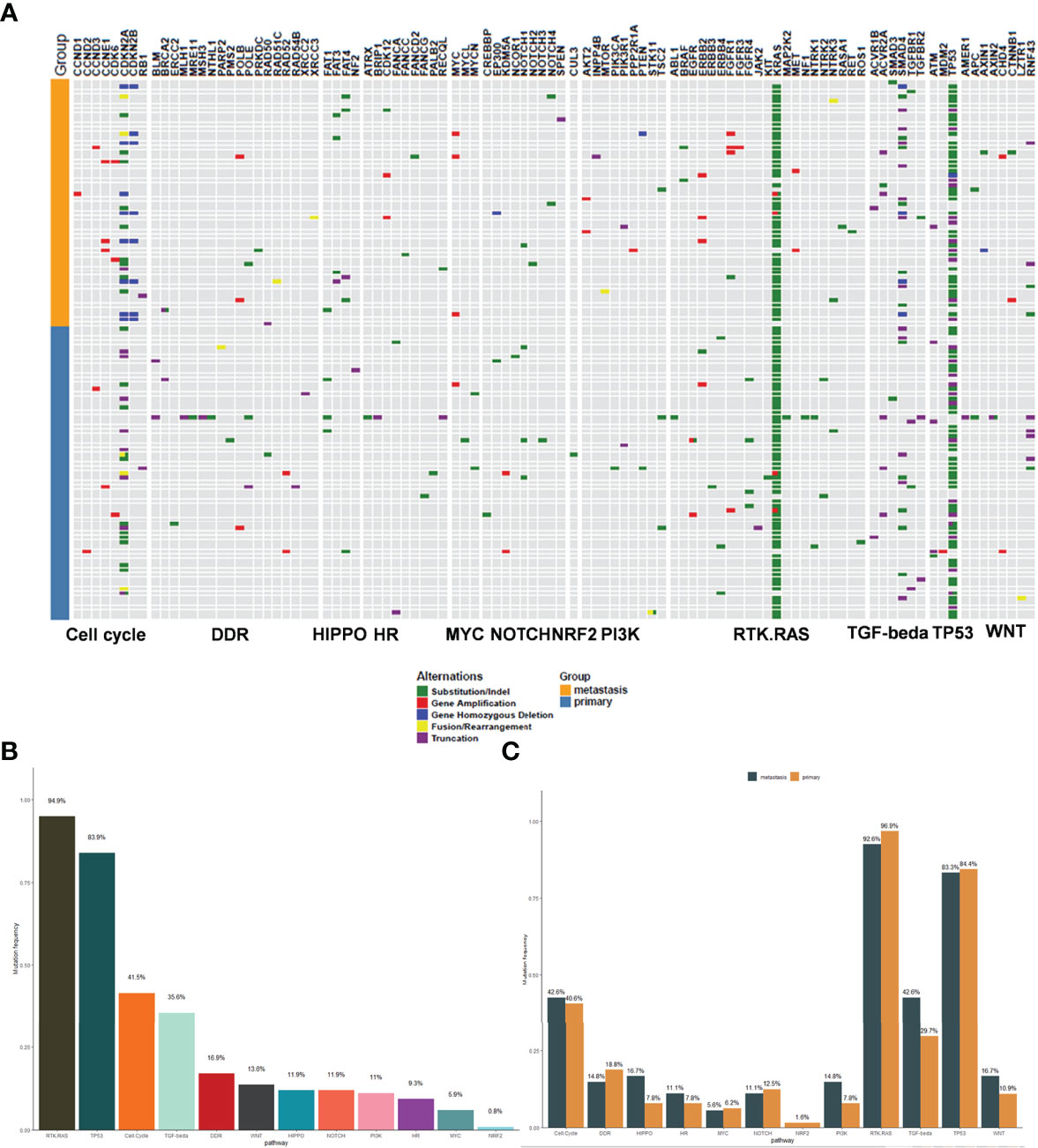
Figure 2 The signaling pathway involved in the mutant genes. (A) The map of mutant genes and the involved signal pathways. Light blue represents the primary tumors and orange represents the metastatic tumors. The upper panel shows the mutant genes and the lower panel shows the signal pathways. Green; Substitution/Indel, Red; Gene amplification, Blue; Gene homozygous deletion, Yellow; Fusion/Rearrangement, Purple; Truncation. (B) The mutation frequency of the signaling pathways involved in the mutant genes. (C) The comparison of mutation frequency of the signaling pathways between primary and metastatic tumors.
Tumor metastasis of pancreatic cancers is an important prognostic factor. To explore the molecular mechanisms involved, we performed genomic analysis of the primary and metastatic tumors in this study and analyzed their mutational characteristics individually. The most common gene alterations in the 64 primary pancreatic cancer were KRAS (96.9%), TP53 (81.2%), CDKN2A (35.9%), and SMAD4 (15.6%) (Figure 3A). The most common gene alterations in the 54 metastatic pancreatic cancer were KRAS (92.6%), TP53 (81.5%), CDKN2A (37%), SMAD4 (31.5%), CDKN2B (14.8%), and MTAP (13%) (Figure 3B). Similarly, the most common changes in KRAS and TP53 were substitution/indels in primary and metastatic tumors (Figures 3A, B). KRAS G12D and G12V showed the highest variation frequency (43% and 22% in primary tumors, and 47% and 45% in metastatic tumors, respectively) (Figures 3C–F). To further explore the association and compare the molecular differences between primary and metastatic tumors, the frequencies of the most common mutations were compared. As shown in Figure 3G, the mutational frequencies (metastatic vs. primary) of CDKN2B (14.8% vs. 0%, p = 0.001), FAT3 (7.4% vs. 0%, p = 0.041), MTAP (13% vs. 1.6%, p = 0.023), and SMAD4 (31.4% vs. 15.6%, p = 0.049) in metastatic tumors were significantly higher than in primary tumors. Detailed numbers and mutational frequencies are listed in Table 2. Notably, CDKN2B and FAT3 alterations occurred only in patients with metastatic tumors.
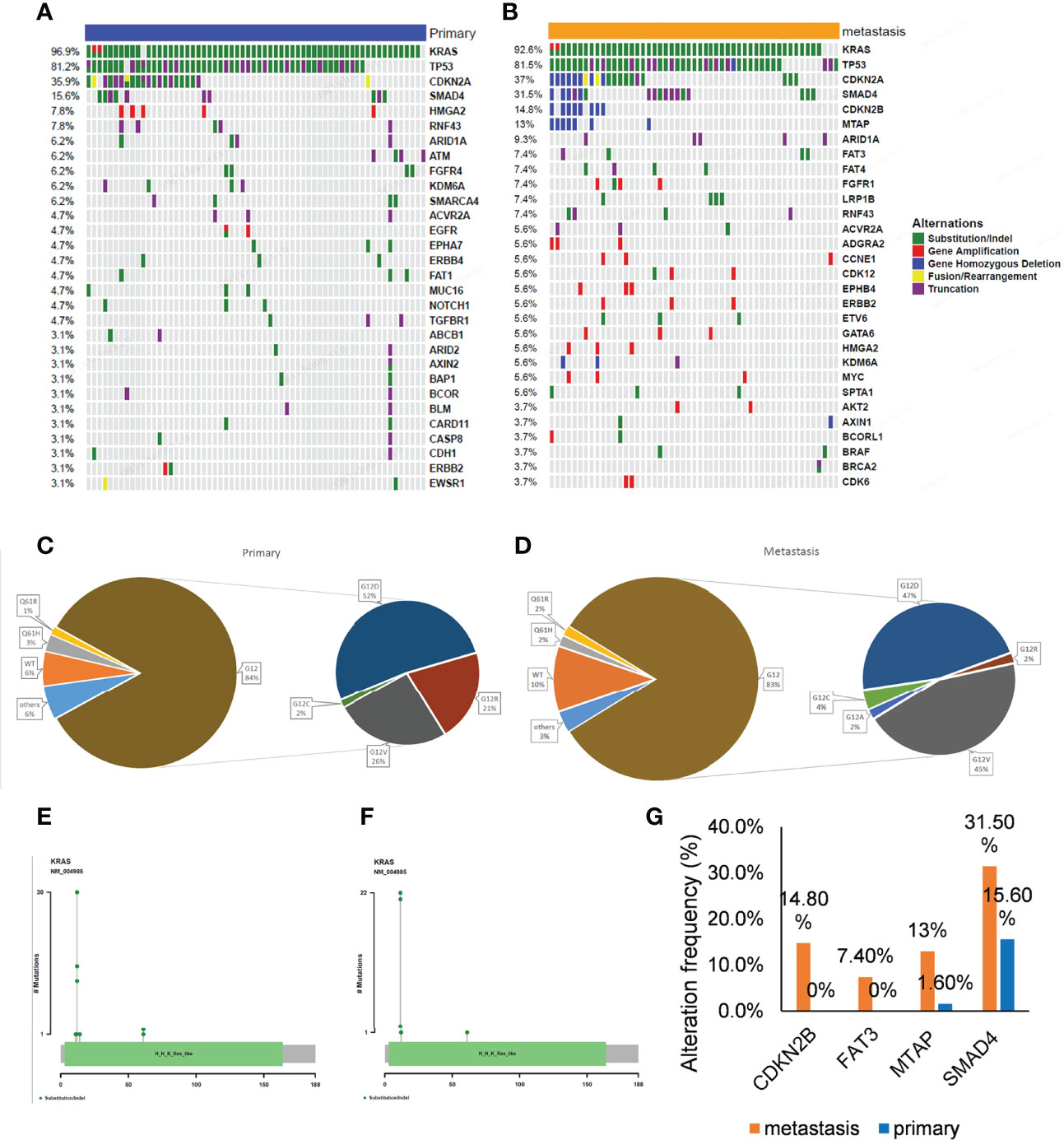
Figure 3 Gene alterations of 64 primary and 54 metastatic tumors. (A, B) The mutation profiles. The panel shows the matrix of mutations with each type of mutation in a different color. Each column denotes an individual tumor and each row represents a gene. The right panel shows the name of mutations and the left panel shows the proportion of mutations. Green; Substitution/Indel, Red; Gene amplification, Blue; Gene homozygous deletion, Yellow; Fusion/Rearrangement, Purple; Truncation. (C, D) Ratio of different mutation types in KRAS gene. (E, F) The distribution map of KRAS gene mutation sites showed the distribution of mutation sites. (G) Genes with a significant difference in mutation frequency between primary and metastatic tumors.

Table 2 Detailed patient number and the mutational frequency.
Of the 118 patients, 89 had a complete follow-up with the best objective response assessment and PFS and OS evaluations. Hence, the mutational and follow-up information of these 89 patients was used to analyze the influence of mutated genes on clinical response rate and prognosis. The results revealed that the ORR of TP53 mutated patients (46.3%) was significantly higher than that of TP53 wild type patients (18.2%) and that the ORR of KRAS mutated patients (43.8%) was significantly higher than that of KRAS wild type patients (0%), demonstrating that TP35 (p = 0.02) and KRAS (p = 0.01) mutations were significantly correlated to better ORR (Figures 4A, B). At the same time, the DCR of EPHA7 mutated patients (0%), ERNF43 mutated patients (20%), and HMGA2 mutation patients (25%) was dramatically lower than that of EPHA7 wild type patients (79.1%), ERNF43 wild type patients (79.8%), and HMGA2 wild type patients (78.8%) (Figures 4C, D), verifying that EPHA7 (p = 0.01), RNF43 (p = 0.01), and HMGA2 (p = 0.04) mutations were significantly correlated to worse DCR. In addition, patients with TGFR2B, FGF23, EPHA7, SMARCA4, CARD11, ADGRA2, CCNE1, and ACVR2A alterations had a worse OS (Figures 5A–H), and EPHA7, CARD11, NOTCH1, GATA6, ACVR2A, and HMGA2 mutations indicated undesirable PFS (Figures 5I–N).
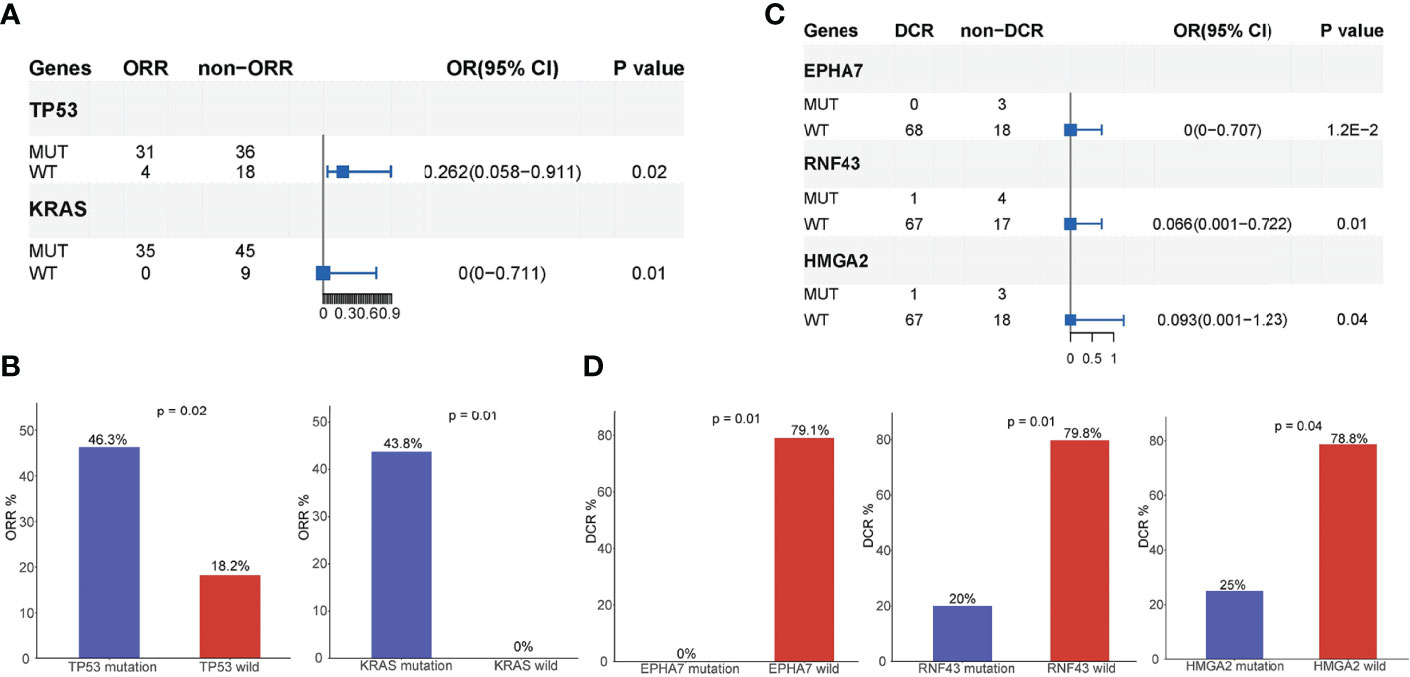
Figure 4 Analysis of mutated genes associated with prognosis. (A, B) TP35 and KRAS mutations were significantly correlated to ORR of AG chemotherapy. The ORR of TP53 mutant patients is 46.3%, while the ORR of TP53 wild type patients is 18.2%.The ORR of KRAS mutant patients is 43.8%, while the ORR of KRAS wild type patients is 0%. (C, D) EPHA7, RNF43, and HMGA2 mutations were significantly correlated to the DCR of AG chemotherapy. The DCR of EPHA7 mutant patients is 0%, while the DCR of EPHA7 wild type patients is 79.1%.
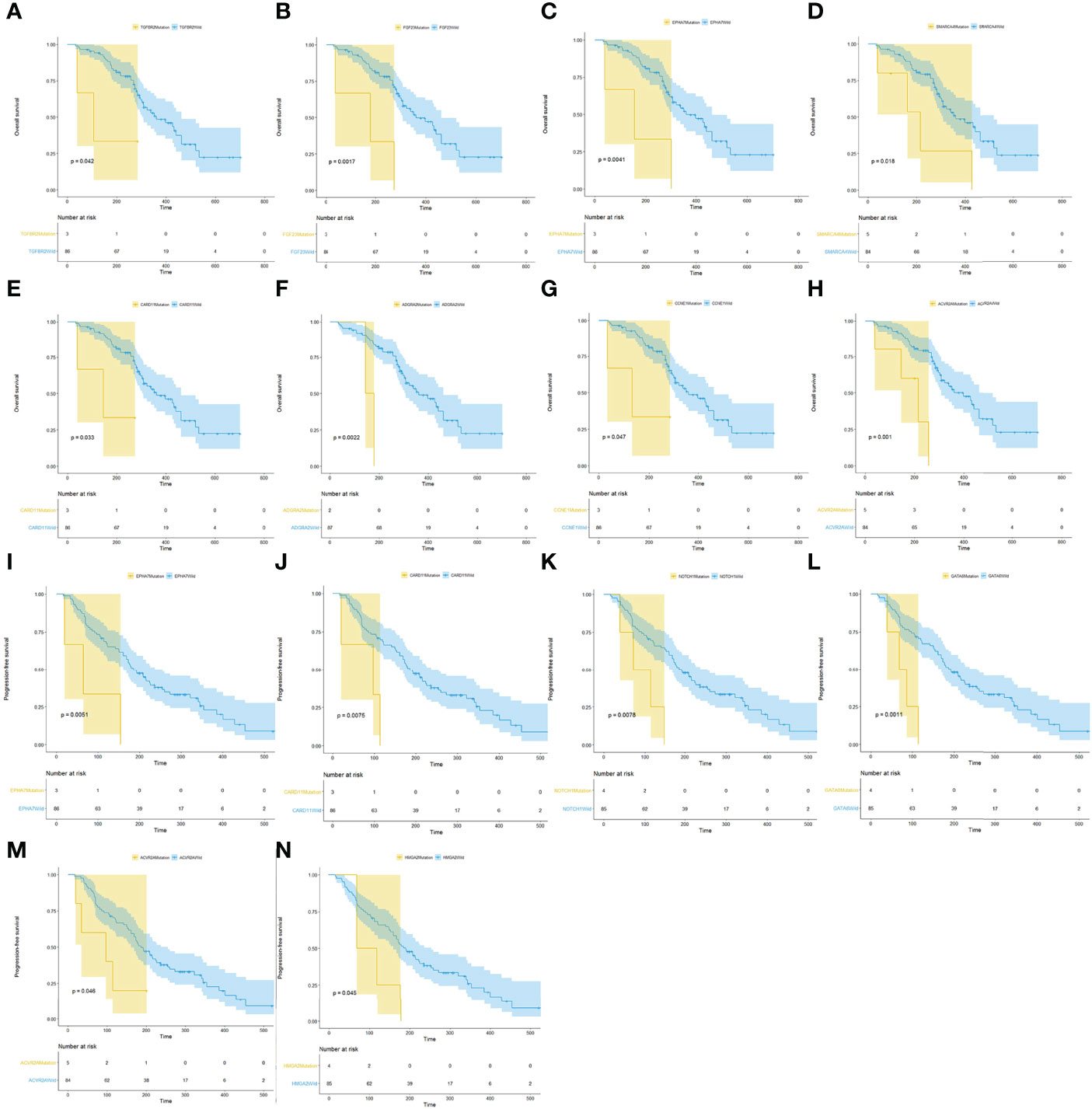
Figure 5 Analysis of mutated genes associated with prognosis. (A–H) Survival analysis showed that patients with TGFR2B, FGF23, EPHA7, SMARCA4, CARD11, ADGRA2, CCNE1, and ACVR2A alterations had a worse OS. (I–N) EPHA7, CARD11, NOTCH1, GATA6, ACVR2A, and HMGA2 S mutations indicated undesirable PFS.
Pancreatic cancer remains one of the most fatal malignancies with a very poor prognosis. In fact, the primary clinical treatment for advanced and/or metastatic pancreatic cancer remains conventional chemotherapy, such as AG chemotherapy (5, 6) and the FOLFIRINOX regimen, but the treatment results are unsatisfactory. Currently, the NCCN guidelines for pancreatic cancer have no guidance for individualized chemotherapy, save for a recommendation that patients with germline BRCA1/2 gene mutations choose platinum chemotherapy. Hence, it is essential to be able to select populations that aremore likely to respond to existing treatments, such as AG chemotherapy, and to develop novel targeted therapeutic strategies for pancreatic cancer patients.
In recent years, many researchers have examined the pancreatic cancer genome using NGS to lay down the molecular foundation for targeted therapy. Waddell et al. revealed that KRAS and CDKN2A were mutated in over 90% of pancreatic cancers, while the TP53 and SMAD4 genes were mutated in 75% and 55% of the patients, respectively (8). In addition, Kamisawa et al. showed that pancreatic cancer cells were characterized by a hypermutated landscape, with the KRAS oncogene, TP53, CDKN2A, and SMAD4 tumor suppressor genes the four most commonly mutated genes (14). Similarly, our results showed that the most common gene alterations in the 118 pancreatic cancer patients studied were in KRAS (94.9%), TP53 (81.4%), CDKN2A (36.4%), and SMAD4 (22.9%). In addition, KRAS mutations in codon 12 were the most common (84%), with KRAS G12D and G12V the highest variation frequency (50% and 34%, respectively). These results are in agreement with the study done by Shinichi et al., which showed activating mutations in KRAS in 92% of pancreatic cancer patients, with mutations in codon 12 being the most common (90%) (15).
Metastasis is a vital risk factor for poor prognosis in pancreatic cancer patients because patients with metastases are usually not suitable candidates for operative treatment. However, the specific molecular mechanism of pancreatic cancer metastasis is not clear and may involve the abnormal expression of cancer-related genes and the disorder of related signaling pathways. A study of 2552 pancreatic ductal adenocarcinoma (PDAC) patients found that the most common gene alterations were mutations in KRAS and PTEN (59% and 62%, respectively), with differences in prevalence by site of metastasis (p = 0.042 and p = 0.037, respectively). KRAS mutations were more commonly found in metastases in the lung (72%) than other sites (59%, p = 0.042). Low expression of ERCC1 was found in 49% of lung metastases from PDAC but only 15% in PDAC in the pancreas (p < 0.001) (16). In another study, 109 differentially expressed genes related with pancreatic cancer metastasis were screened by analyzing the pancreatic cancer metastasis-related gene chip data in the GEO database. Of these, 49 were upregulated and 60 were downregulated (17).
In our study, the most common gene alterations in the 66 primary pancreatic cancers were KRAS (96.9%), TP53 (81.2%), CDKN2A (35.9%), and SMAD4 (15.6%) (Figure 2A). Further, the most common gene alterations in the 54 metastatic pancreatic cancers were KRAS (92.6%), TP53 (81.5%), CDKN2A (37%), SMAD4 (31.5%), CDKN2B (14.8%), and MTAP (13%) (Figure 2B). Importantly, the mutational frequency of CDKN2B (14.8% vs. 0%, p = 0.001), FAT3 (7.4% vs. 0%, p = 0.041), MTAP (13% vs. 1.6%, p = 0.023), and SMAD4 (31.4% vs. 15.6%, p = 0.049) in metastatic tumors were significantly higher than those in primary tumors. Notably, CDKN2B and FAT3 alterations occurred only in metastatic tumors. Other studies have shown that loss of SMAD4 expression is associated with metastasis in pancreatic cancer (18–20). Evidently, the relationship between metastatic pancreatic cancer and other significantly mutated genes, such as CDKN2B, FAT3, and MTAP, remains obscure.
All detected mutations are involved in the dysregulation of core signaling pathways that affect not only the specific features of tumor cells, such as proliferation and migration, but also the tumor microenvironment (14). Approximately 32 recurrently mutated genes in pancreatic cancer that aggregate into 10 core pathways have been reported: KRAS, TGF-β, WNT, NOTCH, ROBO/SLIT signaling, G1/S transition, SWI-SNF, chromatin modification, DNA repair, and RNA processing (3). Another study revealed that MAPK (87%), PI3K/AKT/mTOR (19%), DNA repair (15%), cell cycle (11%), and AKT/mTOR (19%) pathway alterations were present in pancreatic cancer patients (21). In this study, all the mutated genes were mainly involved in the following signaling pathways: RTK.RAS, TP53, cell cycle, TGF-β, DDR, WNT, HIPPO, NOTCH, PI3K, HR, MYC, and NFR2. We found RTK.RAS (94.9%) pathway alterations in most pancreatic cancer patients. These were predominantly due to KRAS mutations, but in patients lacking KRAS mutations, other RTK.RAS genes were often mutated, such as BRAF, EGFR, ERBB2/3/4, FGFR1/3/4, and ROS1. This RTK.RAS signaling pathways dysregulation is consistent with previous studies (3, 8, 9). Additionally, 11.9% of patients had some molecular defect in the PI3K pathway, including mutations in STK11, PIK3CA, TSC2, and AKT2. These frequencies were comparable to those from other published studies (21, 22). Furthermore, in a retrospective analysis, 13.9% (94/677) of patients with actionable mutations had mutations in the DDR pathway (23–26). Similarly, our data showed that 16.9% of pancreatic cancer patients had DDR pathway alterations, with actionable mutations such as BRCA2, and POLE. However, our study showed no significant difference in the regulatory pathways affected in primary and metastatic tumors.
Up to 85% of pancreatic cancer patients are diagnosed with already locally advanced and/or metastatic tumors because of a lack of specific symptoms and early biomarkers for this highly aggressive disease (14). Therefore, it is particularly important to explore molecular biomarkers for early prediction and prognosis. An activating point mutation of the KRAS oncogene in codon 12 (exon 2) is the initiating event in the majority of PDAC patients (27, 28). The point mutation of KRAS damages the intrinsic GTPase activity of RAS and prevents GAPs from promoting the conversion of GTP (active) to GDP (inactive). Thus, KRAS is permanently bound to GTP and activates downstream signaling pathways, increasing cellular proliferation and survival (28, 29). Several studies have investigated whether KRAS mutations influence the prognosis of PDAC, and the results showed that the KRAS mutation has a negative influence on the prognosis of PDAC patients, especially G12D, G12R, or a combination of the two mutants (30, 31). Our study results are consistent with these findings, where 94.9% of our study population had KRAS mutations, most frequently in codon 12 (84%), predominantly KRAS G12D and G12V (50% and 34%, respectively). Additionally, patients with KRAS mutations had a shorter ORR. In addition, our data also revealed that EPHA7 mutations were significantly correlated to DCR, and patients with EPHA7 alterations had a worse OS and undesirable PFS. Combined with a recent study suggesting that EPHA7 mutations could serve as a potential predictive biomarker for immune checkpoint inhibitors in multiple cancers (32), our results suggest that EPHA7 mutation indicate a poor prognosis, and the use of immune checkpoint inhibitors in EPHA7-mutated pancreatic cancer patients may result in better clinical outcomes. HMGA2 acts as an independent prognostic marker associated with lymph node metastasis (33) and a significantly shorter OS in PDAC (34). Consistently, our results revealed that HMGA2 mutations were significantly correlated to DCR, and patients with HMGA2 alterations had an undesirable PFS. Studies have classified PDAC into classical, quasimesenchymal, and exocrine-like subtypes. Classical PDAC showed that higher GATA6 mRNA expression is associated with a better prognosis (35, 36). Additionally, low GATA6 expression is associated with shorter OS (37). Our data showed that GATA6 mutations were associated with a shorter PFS. Other prognostic factors detected in this study, such as TP53, RNF43, FGFBR2, FGF23, SMARCA4, CARD11, ADGRA2, CCNE1, ACVR2A, NOTCH1, and TGFR2, have not been associated with the prognosis of pancreatic cancer in other studies. A larger sample size is needed for further analysis.
In conclusion, this study used NGS to comprehensively analyze the mutation landscape of pancreatic cancer patients, including primary tumors and metastatic tumors, and the involved signaling pathways. The most commonly mutated genes were KRAS, TP53, CDKN2A and SMAD4. In addition, CDKN2B, FAT3, MTAP, and SMAD4 may be molecular markers that distinguish primary tumors from metastases. Furthermore, KRAS, EPHA7, TP53, HMGA2, GATA6, RNF43, FGFBR2, FGF23, SMARCA4, CARD11, ACVR2A, NOTCH1, and TGFR2 may serve as potential prognostic biomarkers for pancreatic cancer patients who received AG chemotherapy for first-line treatment. Particularly, KRAS and EPHA7 are highly likely to serve as prognostic factors and therapeutic targets in the management of locally advanced pancreatic cancer.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by The ethics committee of Nanjing Drum Tower Hospital. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Conception/Design: JD and BL. Provision of study material or patients: JD, XQ, CL, YZ and WK. Collection and/or assembly of data: MT, JC, QL, AL, JH, QG, LW and YQ. Data analysis and interpretation: XZ. Manuscript writing: JD and MX. Final approval of manuscript: JD and BL. All authors have read and approved the submitted version of the manuscript.
National Natural Science Foundation of China (No: 82072926), Natural Science Foundation of Jiangsu Province (No: BK20191114), Chen Xiaoping Foundation for the Development of Science and Technology of Hubei Province (No. CXPJJH11900001-2019101), Nanjing Medical Science and technology development project (No: YKK20080)
Author MX and XZ were employed by Shanghai OrigiMed Co, Ltd, Shanghai, China.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2022. CA Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res (2014) 74(11):2913–21. doi: 10.1158/0008-5472.CAN-14-0155
3. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature (2016) 531(7592):47–52. doi: 10.1038/nature16965
4. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX Versus Gemcitabine for Metastatic Pancreatic Cancer. N Engl J Med (2011) 364(19):1817–25. doi: 10.1056/NEJMoa1011923
5. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased Survival in Pancreatic Cancer With Nab-Paclitaxel Plus Gemcitabine. N Engl J Med (2013) 369(18):1691–703. doi: 10.1056/NEJMoa1304369
6. Philip P, Lacy J, Portales F, Sobrero A, Pazo-Cid R., Manzano Mozo J, et al. Nab-Paclitaxel Plus Gemcitabine in Patients With Locally Advanced Pancreatic Cancer (LAPACT): A Multicentre, Open-Label Phase 2 Study. Lancet Gastroenterol Hepatol (2020) 5(3):285–94. doi: 10.1016/S2468-1253(19)30327-9
7. Herbst B, Zheng L. Precision Medicine in Pancreatic Cancer: Treating Every Patient as an Exception. Lancet Gastroenterol Hepatol (2019) 4(10):805–10. doi: 10.1016/S2468-1253(19)30175-X
8. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole Genomes Redefine the Mutational Landscape of Pancreatic Cancer. Nature (2015) 518(7540):495–501. doi: 10.1038/nature14169
9. Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic Cancer Genomes Reveal Aberrations in Axon Guidance Pathway Genes. Nature (2012) 491(7424):399–405. doi: 10.1038/nature11547
10. Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, et al. Real-Time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov (2018) 8(9):1096–111. doi: 10.1158/2159-8290.CD-18-0275
11. Wei T, Zhang J, Li J, Chen Q, Zhi X, Tao W, et al. Genome-Wide Profiling of Circulating Tumor DNA Depicts Landscape of Copy Number Alterations in Pancreatic Cancer With Liver Metastasis. Mol Oncol (2020) 14(9):1966–77. doi: 10.1002/1878-0261.12757
12. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and Validation of a Clinical Cancer Genomic Profiling Test Based on Massively Parallel DNA Sequencing. Nat Biotechnol (2013) 31(11):1023–31. doi: 10.1038/nbt.2696
13. Cao J, Chen L, Li H, Chen H, Yao J, Mu S, et al. An Accurate and Comprehensive Clinical Sequencing Assay for Cancer Targeted and Immunotherapies. Oncologist (2019) 24(12):e1294–302. doi: 10.1634/theoncologist.2019-0236
14. Kamisawa T, Wood LD, Itoi T, Takaori K, et al. Pancreatic Cancer. Lancet (2016) 388(10039):73–85. doi: 10.1016/S0140-6736(16)00141-0
15. Yachida S, White CM, Naito Y, Zhong Y, Brosnan JA, Macgregor-Das AM, et al. Clinical Significance of the Genetic Landscape of Pancreatic Cancer and Implications for Identification of Potential Long-Term Survivors. Clin Cancer Res (2012) 18(22):6339–47. doi: 10.1158/1078-0432.CCR-12-1215
16. Ferguson MD, Dong L, Wan J, Deneve JL, Dickson PV, Behrman SW, et al. Molecular Alterations Associated With DNA Repair in Pancreatic Adenocarcinoma Are Associated With Sites of Recurrence. J Gastrointest Cancer (2019) 50(2):285–91. doi: 10.1007/s12029-018-0073-8
17. Xu JS, Liao KL, Wang X, He J, Wang XZ. Combining Bioinformatics Techniques to Explore the Molecular Mechanisms Involved in Pancreatic Cancer Metastasis and Prognosis. J Cell Mol Med (2020) 24(24):14128–38. doi: 10.1111/jcmm.16023
18. Boone BA, Sabbaghian S, Zenati M, Marsh JW, Moser AJ, Zureikat AH, et al. Loss of SMAD4 Staining in Pre-Operative Cell Blocks Is Associated With Distant Metastases Following Pancreaticoduodenectomy With Venous Resection for Pancreatic Cancer. J Surg Oncol (2014) 110(2):171–5. doi: 10.1002/jso.23606
19. Kang CM, Hwang HK, Park J, Kim C, Cho SK, Yun M, et al. Maximum Standard Uptake Value as a Clinical Biomarker for Detecting Loss of SMAD4 Expression and Early Systemic Tumor Recurrence in Resected Left-Sided Pancreatic Cancer. Med (Baltimore) (2016) 95(17):e3452. doi: 10.1097/MD.0000000000003452
20. Wang C, Zhang T, Liao Q, Dai M, Guo J, Yang X, et al. Metformin Inhibits Pancreatic Cancer Metastasis Caused by SMAD4 Deficiency and Consequent HNF4G Upregulation. Protein Cell (2021) 12(2):128–44. doi: 10.1007/s13238-020-00760-4
21. Pishvaian MJ, Bender RJ, Halverson D, Rahib L, Hendifar AE, Mikhail S, et al. Molecular Profiling of Patients With Pancreatic Cancer: Initial Results From the Know Your Tumor Initiative. Clin Cancer Res (2018) 24(20):5018–27. doi: 10.1158/1078-0432.CCR-18-0531
22. Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-Exome Sequencing of Pancreatic Cancer Defines Genetic Diversity and Therapeutic Targets. Nat Commun (2015) 6:6744. doi: 10.1038/ncomms7744
23. Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, et al. Overall Survival in Patients With Pancreatic Cancer Receiving Matched Therapies Following Molecular Profiling: A Retrospective Analysis of the Know Your Tumor Registry Trial. Lancet Oncol (2020) 21(4):508–18. doi: 10.1016/S1470-2045(20)30074-7
24. Lowery MA, Jordan EJ, Basturk O, Ptashkin RN, Zehir A, Berger MF, et al. Real-Time Genomic Profiling of Pancreatic Ductal Adenocarcinoma: Potential Actionability and Correlation With Clinical Phenotype. Clin Cancer Res (2017) 23(20):6094–100. doi: 10.1158/1078-0432.CCR-17-0899
25. Dhir M, Choudry HA, Holtzman MP, Pingpank JF, Ahrendt SA, Zureikat AH, et al. Impact of Genomic Profiling on the Treatment and Outcomes of Patients With Advanced Gastrointestinal Malignancies. Cancer Med (2017) 6(1):195–206. doi: 10.1002/cam4.992
26. Chantrill LA, Nagrial AM, Watson C, Johns AL, Martyn-Smith M, Simpson S, et al. Precision Medicine for Advanced Pancreas Cancer: The Individualized Molecular Pancreatic Cancer Therapy (IMPaCT) Trial. Clin Cancer Res (2015) 21(9):2029–37. doi: 10.1158/1078-0432.CCR-15-0426
27. Bournet B, Buscail C, Muscari F, Cordelier P, Buscail L. Targeting KRAS for Diagnosis, Prognosis, and Treatment of Pancreatic Cancer: Hopes and Realities. Eur J Cancer (2016) 54:75–83. doi: 10.1016/j.ejca.2015.11.012
28. di Magliano MP, Logsdon CD. Roles for KRAS in Pancreatic Tumor Development and Progression. Gastroenterology (2013) 144(6):1220–9. doi: 10.1053/j.gastro.2013.01.071
29. Jonckheere N, Vasseur R, Van Seuningen I. The Cornerstone K-RAS Mutation in Pancreatic Adenocarcinoma: From Cell Signaling Network, Target Genes, Biological Processes to Therapeutic Targeting. Crit Rev Oncol Hematol (2017) 111:7–19. doi: 10.1016/j.critrevonc.2017.01.002
30. Ogura T, Yamao K, Hara K, Mizuno N, Hijioka S, Imaoka H, et al. Prognostic Value of K-Ras Mutation Status and Subtypes in Endoscopic Ultrasound-Guided Fine-Needle Aspiration Specimens From Patients With Unresectable Pancreatic Cancer. J Gastroenterol (2013) 48(5):640–6. doi: 10.1007/s00535-012-0664-2
31. Rachakonda PS, Bauer AS, Xie H, Campa D, Rizzato C, Canzian F, et al. Somatic Mutations in Exocrine Pancreatic Tumors: Association With Patient Survival. PloS One (2013) 8(4):e60870. doi: 10.1371/journal.pone.0060870
32. Zhang Z, Wu HX, Lin WH, Wang ZX, Yang LP, Zeng ZL, et al. EPHA7 Mutation as a Predictive Biomarker for Immune Checkpoint Inhibitors in Multiple Cancers. BMC Med (2021) 19(1):26. doi: 10.1186/s12916-020-01899-x
33. Gundlach J, Hauser C, Schlegel F, Willms A, Halske C, Röder C, et al. Prognostic Significance of High Mobility Group A2 (HMGA2) in Pancreatic Ductal Adenocarcinoma: Malignant Functions of Cytoplasmic HMGA2 Expression. J Cancer Res Clin Oncol (2021) 147(11):3313–24. doi: 10.1007/s00432-021-03745-w
34. Strell C, Norberg K, Mezheyeuski A, Schnittert J, Kuninty P, Moro C, et al. Stroma-Regulated HMGA2 Is an Independent Prognostic Marker in PDAC and AAC. Br J Cancer (2017) 117(1):65–77. doi: 10.1038/bjc.2017.140
35. Eyres M, Lanfredini S, Xu H, Burns A, Blake A, Willenbrock F, et al. TET2 Drives 5hmc Marking of GATA6 and Epigenetically Defines Pancreatic Ductal Adenocarcinoma Transcriptional Subtypes. Gastroenterology (2021) 161(2):653–68.e16. doi: 10.1101/2020.10.22.342436
36. Collisson E, Sadanandam A, Olson P, Gibb W, Truitt M, Gu S, et al. Subtypes of Pancreatic Ductal Adenocarcinoma and Their Differing Responses to Therapy. Nat Med (2011) 17(4):500–3. doi: 10.1038/nm.2344
Keywords: mutational analysis, advanced pancreatic cancer, metastasis, prognostic biomarker, AG chemotherapy
Citation: Du J, Qiu X, Lu C, Zhu Y, Kong W, Xu M, Zhang X, Tang M, Chen J, Li Q, Li A, He J, Gu Q, Wang L, Qiu Y and Liu B (2022) Molecular Landscape and Prognostic Biomarker Analysis of Advanced Pancreatic Cancer and Predictors of Treatment Efficacy of AG Chemotherapy. Front. Oncol. 12:844527. doi: 10.3389/fonc.2022.844527
Received: 28 December 2021; Accepted: 13 April 2022;
Published: 18 May 2022.
Edited by:
Heather Cunliffe, University of Otago, New ZealandReviewed by:
Fabienne Portales, Institut du Cancer de Montpellier (ICM), FranceCopyright © 2022 Du, Qiu, Lu, Zhu, Kong, Xu, Zhang, Tang, Chen, Li, Li, He, Gu, Wang, Qiu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baorui Liu, YmFvcnVpbGl1QG5qdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.