
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 22 February 2022
Sec. Cancer Molecular Targets and Therapeutics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.840843
This article is part of the Research Topic Circulating Tumor DNA in Cancer: A Role as a Response and Monitoring “Next-Generation” Biomarker in Cancer therapy View all 11 articles
 Tun Kiat Ko1,2
Tun Kiat Ko1,2 Elizabeth Lee1,2
Elizabeth Lee1,2 Cedric Chuan-Young Ng1,2Valerie Shiwen Yang3,4,5Mohamad Farid3,4
Cedric Chuan-Young Ng1,2Valerie Shiwen Yang3,4,5Mohamad Farid3,4 Bin Tean Teh1,4,5,6
Bin Tean Teh1,4,5,6 Jason Yongsheng Chan2,3,4*Nagavalli Somasundaram3,4*
Jason Yongsheng Chan2,3,4*Nagavalli Somasundaram3,4*Liquid biopsy circulating tumor DNA (ctDNA)-based approaches may represent a non-invasive means for molecular interrogation of gastrointestinal stromal tumors (GISTs). We deployed a customized 29-gene Archer® LiquidPlex™ targeted panel on 64 plasma samples from 46 patients. The majority were known to harbor KIT mutations (n = 41, 89.1%), while 3 were PDGFRA exon 18 D842V mutants and the rest (n = 2) were wild type for KIT and PDGFRA. In terms of disease stage, 14 (30.4%) were localized GISTs that had undergone complete surgical resection while the rest (n = 32) were metastatic. Among ten patients, including 7 on tyrosine kinase inhibitors, with evidence of disease progression at study inclusion, mutations in ctDNA were detected in 7 cases (70%). Known somatic mutations in KIT (n = 5) or PDGFRA (n = 1) in ctDNA were identified only among 6 of the 10 patients. These KIT mutants included duplication, indels, and single-nucleotide variants. The median mutant AF in ctDNA was 11.0% (range, 0.38%–45.0%). In patients with metastatic progressive KIT-mutant GIST, tumor burden was higher with detectable KIT ctDNA mutation than in those without (median, 5.97 cm vs. 2.40 cm, p = 0.0195). None of the known tumor mutations were detected in ctDNA for localized cases (n = 14) or metastatic cases without evidence of disease progression (n = 22). In patients with serial samples along progression of disease, secondary acquired mutations, including a potentially actionable PIK3CA exon 9 c.1633G>A mutation, were detected. ctDNA mutations were not detectable when patients responded to a switch in TKI therapy. In conclusion, detection of GIST-related mutations in ctDNA using a customized targeted NGS panel represents an attractive non-invasive means to obtain clinically tractable information at the time of disease progression.
Gastrointestinal stromal tumor (GIST) is the commonest mesenchymal neoplasm originating from the gastrointestinal tract. GISTs are classically defined by activating oncogenic mutations in KIT (KIT proto-oncogene receptor tyrosine kinase) (80%) or PDGFRA (platelet-derived growth factor receptor alpha) (10%) genes (1–3). In advanced stages, most patients benefit from targeted therapy using the tyrosine kinase inhibitor (TKI) imatinib, though acquired resistance and disease progression associated with the development of secondary mutations usually occur after 18 to 24 months. Under pharmacological pressure, secondary mutations can develop, conferring resistance to imatinib. In imatinib-resistant GISTs, secondary mutations typically occur in the ATP-binding pocket (exon 13) or in the kinase activation loop (exon 17) (4).
In the contemporary management of patients with advanced GISTs, radiological response evaluation following TKI treatment remains standard of care, and there is currently no specific blood-based biomarker for the purposes of monitoring disease progression or to determine mechanisms responsible for acquired TKI resistance. At the time of disease progression, knowledge of the specific secondary mutations could enable tailoring of treatment, though this requires invasive tumor biopsy at the site of progression. Understandably, such an approach does not comprehensively capture the evolving global tumor landscape and may not yield viable results that reflect tumor heterogeneity (5). In addition to defining resistance pathways, the secondary mutations also have therapeutic implications. Previous preclinical studies have suggested that sunitinib is preferentially more active against mutations in the ATP-binding pocket, while regorafenib has increased activity against mutations in the kinase activation loop (6).
The optimal approach to determine mechanisms of acquired resistance during therapy and guide personalized approach to subsequent management remains to be elucidated. Non-invasive tumor mutation profiling using several liquid biopsy cell-free circulating tumor DNA (ctDNA)-based approaches has been explored in patients with GISTs (7). While some of these approaches have been shown to capture the molecular heterogeneity of the whole tumor, their utility is limited to patients with high tumor burden as a result of suboptimal assay sensitivity, and their practical use in the clinic remains in question. In this study, we investigated a liquid biopsy approach for the detection of primary and secondary acquired mutations in patients with GISTs, using a customized Archer® LiquidPlex™ targeted panel. The advantage of Archer® LiquidPlex™ includes the Anchored Multiplex PCR (AMP™) enrichment chemistry, in which ctDNA fragments are ligated to molecular barcodes that allow for error correction for confident variant reporting. Furthermore, Archer® LiquidPlex™ is able to capture a fragment size that is smaller than 160 base pairs, a size that can be missed by other platforms. Finally, Archer® LiquidPlex™ can detect variants at 0.3% allele frequency with ctDNA input that is as low as 1 ng.
Blood samples were collected from patients who were diagnosed with GISTs and seen at the National Cancer Centre Singapore between April 1998 and September 2021. A total of 64 plasma samples and 7 whole blood samples from 46 patients were included in the final analysis. Relevant demographical and clinical information were collected and utilized for the analysis. For all GISTs diagnosed at our center, Sanger sequencing was routinely performed to detect KIT and PDGFRA gene mutations. Selected cases may undergo panel testing via next-generation sequencing at the discretion of the managing physician. All data were obtained at the time of diagnosis or subsequent follow-up. Written informed consent for use of biospecimens and clinical data was obtained in accordance with the Declaration of Helsinki. The research study was carried out with approval from the SingHealth Centralised Institutional Review Board (CIRB 2018/3182). The datasets created and analysed during this study are available from the corresponding authors upon reasonable request.
Whole blood samples were collected in 10-ml EDTA-coated tubes (BD, Cat. 368589) and processed within 2 h of collection using an in-house protocol. The samples were centrifuged for 10 min at 300 × g at room temperature to separate plasma from red blood cells. Plasma layer was harvested in 1-ml aliquots in 1.5-ml Microcentrifuge Tubes (Axygen, MCT-150-C) and spun again in a microcentrifuge at 9,720 × g at 4°C. Plasma was collected and stored at −80°C until use. QIAamp Circulating Nucleic Acid Kit (Catalog #55114, Qiagen) was used to isolate ctDNA from plasma by following the manufacturer’s instruction. Purified ctDNA was stored at −20°C.
The following is the protocol for extracting ctDNA from frozen whole blood. DNA were isolated by using DNeasy Blood & Tissue Kit (Qiagen) with the following modification to the protocol. Briefly, proteinase K was added to whole blood at the following volume ratio (proteinase K:whole blood, 1:10). Subsequently, Buffer AL (with no ethanol added) was added into thawed blood, containing proteinase K, at a volume ratio of 6:1 before incubating at 56°C for at least 10 min. Absolute ethanol was added at a volume that was equal to that of Buffer AL used. The number of DNA purification columns used was dependent on the volume of whole blood. The ratio was 1 column per 200 µl of thawed whole blood. Subsequent steps followed the manufacturer’s instruction.
Once eluted, DNA was obtained from whole blood, Mag-Bind® Total Pure NGS (Omega Bio-tek) paramagnetic beads were used to separate the genomic DNA from ctDNA; 0.6× bead volume (calculation was based on the volume of the DNA eluent) was added to the DNA eluent, and the beads were used to remove the genomic DNA. The beads containing the genomic DNA was isolated from the supernatant by a magnetic column. Subsequently, the leftover supernatant was moved to a clean microfuge tube where 2× bead volume (calculation was based on the volume of the original DNA eluent) was added. This volume of beads would isolate out the ctDNA from the supernatant. The beads were isolated by magnet, washed, and ctDNA was eluted according to the manufacturer’s instructions. The profile of the eluted ctDNA fraction was visualized on Agilent TapeStation by using Agilent genomic DNA ScreenTape (Agilent Technologies, CA, USA) to ensure that there was no contaminating genomic DNA. If genomic DNA was still present in the eluted ctDNA fraction, the DNA clean-up would be repeated. Purified ctDNA, from plasma or whole blood, were quantified by using Qubit dsDNA assay kit (Thermo Fisher Scientific). Purified ctDNA profile was also visualized on Agilent TapeStation by using Agilent High Sensitivity D100 ScreenTape.
NGS libraries were made from, depending on individual sample ctDNA yield, 5 ng to 37 ng (median: 10.1 ng) of ctDNA input. The NGS libraries were constructed by following the Archer® LiquidPlex™ Protocol for Illumina® (Invitae). We are using a customized Archer® LiquidPlex™ targeted panel, dubbed LiquidPlex NCCS GIST 18265 v1.0, that consists of probes that target specific exons of 29 genes that are known to associate with cancer and encompass known somatic mutations of GISTs (Supplementary Table 1). The libraries were paired-end sequenced (2 × 150 bp) for 5–20 million raw reads with at least 5% PhiX by using the standard Illumina NGS protocol on the NovaSeq platform (Novogene).
The raw sequencing data were analyzed with Archer Analysis pipeline (version 6.2.7; https://archerdx.com/technology-platform/analysis) as previously reported (8). We used the default settings for detecting variants that were statistically significant [in our case, the variant must have an allele frequency (AF) outlier p-value < 0.05]. For ctDNA samples where no significant variants were detected by the default setting, we would manually check all the mapped NGS reads for the presence of KIT, PDGFRA, or other variants that were previously detected by Sanger sequencing of the corresponding tumor. If such variant was detected, it would only be reported as a significant variant if AF outlier p-value < 0.05.
Comparisons of tumor size with detectable ctDNA were performed using Mann–Whitney U test. All statistical evaluations were made assuming a two-sided test with a significance level of 0.05 unless otherwise stated. All tests were performed using MedCalc statistical Software for Windows version 19.0.4 (MedCalc Software, Ostend, Belgium).
A total of 46 patients were included in the study (Supplementary Figure 1). The median age at study enrolment was 63.7 years (range, 30.3 to 89.1 years). Twenty-six (56.5%) were male and 20 (43.5%) were female. Primary tumor locations were stomach (n = 24, 52.2%), small bowel (n = 19, 41.3%), and rectum (n = 3, 6.5%). The majority were known to harbor KIT mutations (n = 41, 89.1%), while 3 patients (NCCS-GIST-06, NCCS-GIST-43, and NCCS-GIST-44) were PDGFRA exon 18 D842V mutants and KIT wild type. Two cases (NCCS-GIST-45 and NCCS-GIST-46) were wild type for both KIT and PDGFRA, one of which harbored KRAS exon2 G12V mutation. In terms of disease stage, 14 (30.4%) were localized GISTs that had undergone complete surgical resection. The rest (n = 32) were metastatic GISTs at various points of their treatment trajectories. Importantly, 10 patients, including 7 on TKI treatment, had evidence of disease progression at study inclusion and were the focus of the study. Clinical and demographic characteristics of all patients are summarized in Table 1.

Table 1 Characteristics of GIST patients at enrolment.
Blood samples of the 46 patients were drawn at study inclusion. Targeted exon panel sequencing was performed to identify mutations in plasma ctDNA. Among the 10 patients with metastatic GIST with evidence of disease progression, mutations in ctDNA were detected in 7 cases (70%). Known somatic mutations in KIT (n = 5) or PDGFRA (n = 1) in ctDNA were identified only among 6 of the 10 patients (Figure 1). These KIT mutants included short sequence tandem duplication, indels, and single-nucleotide variants. The median mutant AF in ctDNA was 11.0% (range 0.38%–45.0%). In patients with metastatic progressive KIT-mutant GIST, tumor burden (as measured by the average diameter of the 3 largest lesions) was higher with detectable KIT ctDNA mutation than in those without (median, 5.97 cm vs. 2.40 cm, p = 0.0195) (Figure 2). The 4 cases with undetectable primary KIT mutations include GISTs with exon 11 c.1708_1728del (NCCS-GIST-07), exon 11 c.1669_1674del (NCCS-GIST-08), exon 11 c.1654_1659del and exon 13 c.1961T>C (NCCS-GIST-09), and exon 9 c.1504_1509dup (NCCS-GIST-10). None of the known tumor mutations were detected in ctDNA for localized cases (n = 14) or metastatic cases without evidence of disease progression (n = 22). In patient NCCS-GIST-02, only the known KIT exon 11 c.1727T>C mutation, but not the exon 17 c.2466T>A mutation was detected in ctDNA. On the other hand, in patient NCCS-GIST-03, ctDNA identified KIT exon 17 c.2467T>G on top of the known exon 11 c.1653_1670del mutation—this patient had prior imatinib-resistant GIST (no molecular evaluation done apart from time of diagnosis) and was progressing despite the 4th-line treatment with pazopanib. For patient NCCS-GIST-07 with small bowel GIST progressing on 1st-line treatment with imatinib, ctDNA detected TP53 c.879_880del and SETD2 c.2238del mutations, but not the known KIT exon 11 c.1708_1728del mutation (Table 2).
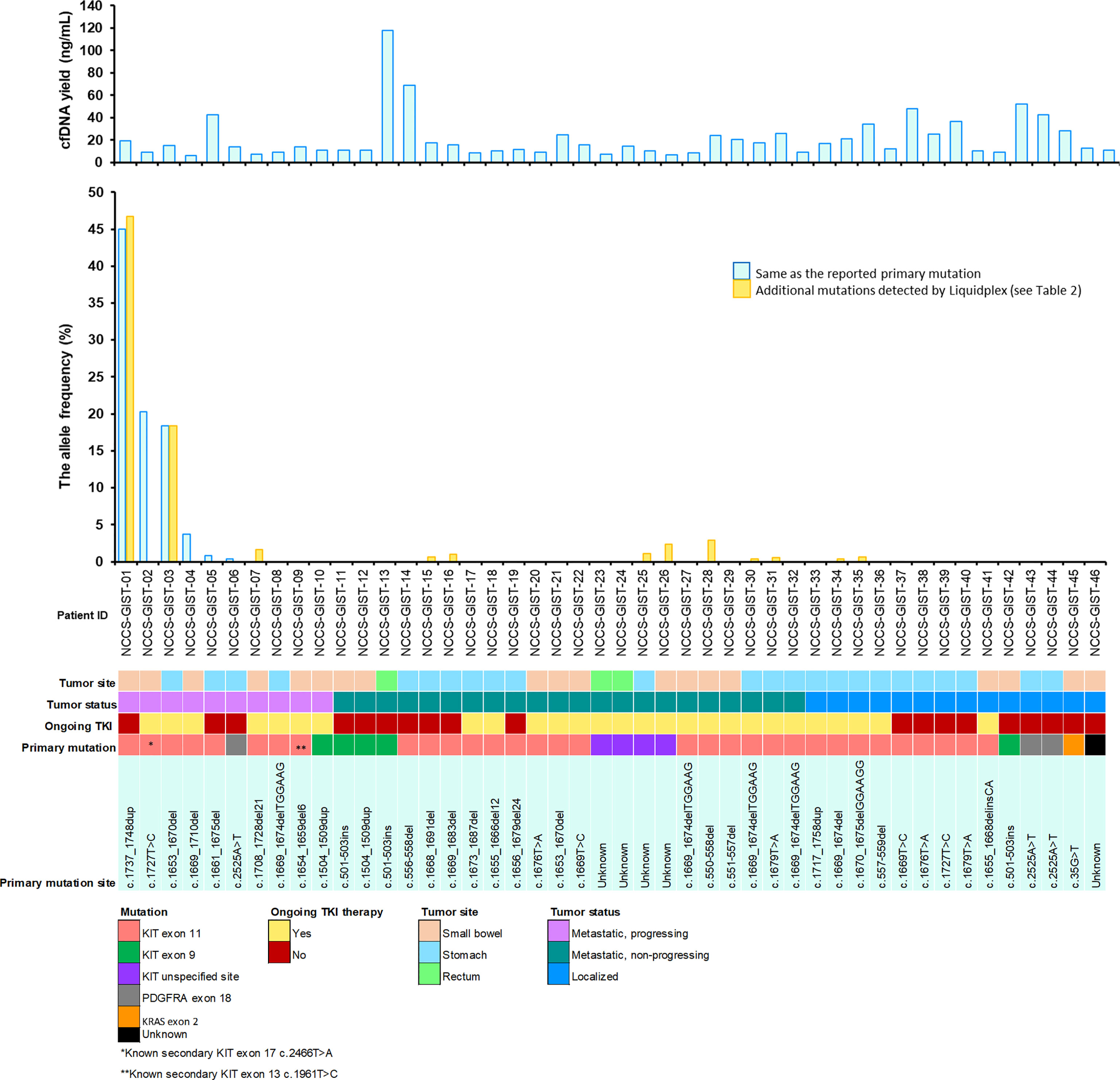
Figure 1 Overview of study cohort and plasma ctDNA mutation detection. Clinical and molecular characteristics of the 46 patients included in the study are shown. For the mutant allele frequency bar chart, the light blue bar  indicates mutants that were detected in ctDNA are the same as that reported for the corresponding primary tumor. The gold-colored bar
indicates mutants that were detected in ctDNA are the same as that reported for the corresponding primary tumor. The gold-colored bar  indicates additional mutants detected in ctDNA that is not reported in the corresponding primary tumor. See Table 2 for more information on the mutation profiles of both the primary tumor and the corresponding ctDNA.
indicates additional mutants detected in ctDNA that is not reported in the corresponding primary tumor. See Table 2 for more information on the mutation profiles of both the primary tumor and the corresponding ctDNA.
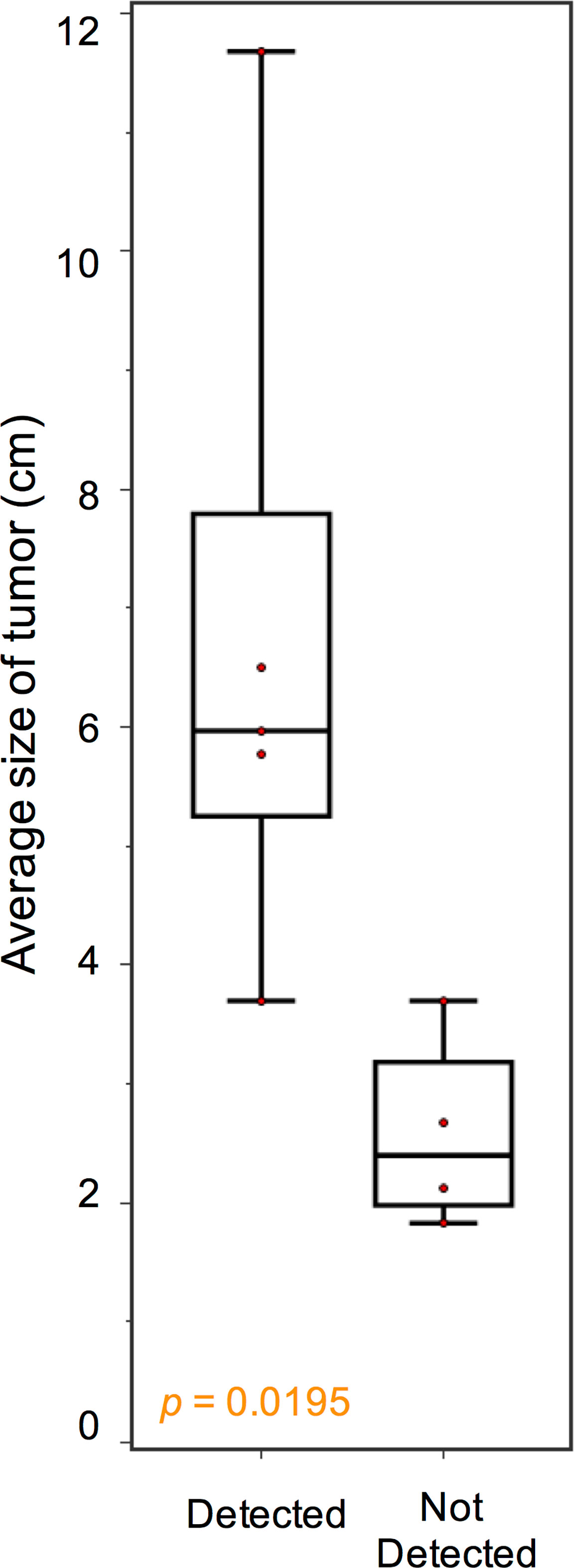
Figure 2 Association between KIT ctDNA mutation detection and tumor burden. In patients with advanced progressive GIST, tumor burden, as measured by the average diameter of the 3 largest lesions, was higher with detectable KIT ctDNA mutation than in those without (median, 5.97 cm vs. 2.40 cm, p = 0.0195).
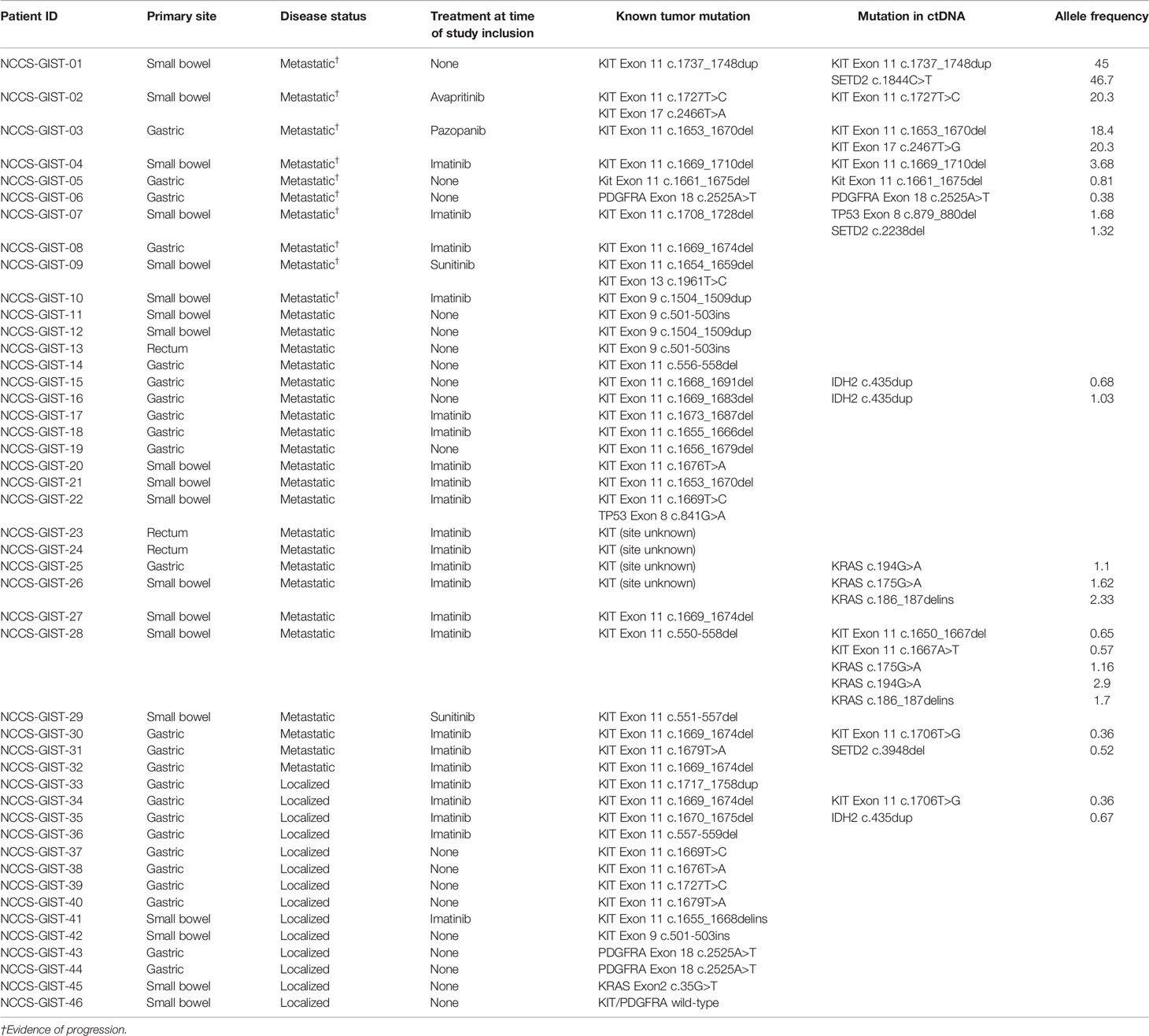
Table 2 Mutational profiles of tumor and plasma ctDNA.
Mutations in SETD2, previously reported to confer worse prognosis in GIST (9), were detected in ctDNA in 3 patients with metastatic GIST. In patients with localized GIST following surgical resection (n = 14), including 5 on adjuvant imatinib, no known tumor mutations could be detected from ctDNA. Likewise, none could be detected from ctDNA from patients with metastatic GIST without evidence of disease progression (n = 22), including 15 patients on TKI treatment. In this group of patients, mutations in IDH2 (n = 3), KRAS (n =3), KIT (n = 3), and SETD2 (n = 1) were identified at low AF (median, 0.86%; range, 0.68 to 2.9%), though the corresponding mutation in the primary tumor was not known.
Serial plasma samples were available for 6 patients with metastatic GIST who underwent TKI therapy. For NCCS-GIST-17 (Figure 3A), the KIT exon 11 c.1673_1687del variant, as detected in the tumor, was not detected in the ctDNA from the first plasma sampling when no disease progression was evident (stable disease, SD) (week 0). In the ctDNA from the second plasma sampling at the time of disease progression in the primary stomach tumor (week 26), 2 KIT variants were detected, namely, exon 11 c.1673_1687del (AF = 3.1%) and exon 17 c.2485G>C (AF = 3.4%), the latter of which was a new variant not previously detected in the tumor. Additionally, a PIK3CA c.1633G>A variant was also detected (AF = 2.6%). Interestingly, in the ctDNA from the third plasma sampling (week 42, with progressive liver metastases), the PIK3CA c.1633G>A variant AF increased by nearly 32-fold (AF = 82.5%) while those KIT variants were reduced by at least 14.2-fold (exon 11 c.1673_1687del, AF = 0.15%; exon 17 c.2485G>C, AF = 0.24%). Generally, the detection of the KIT and PIK3CA variants in the ctDNA positively correlated with disease progression as evaluated by computed tomography (CT) scans.
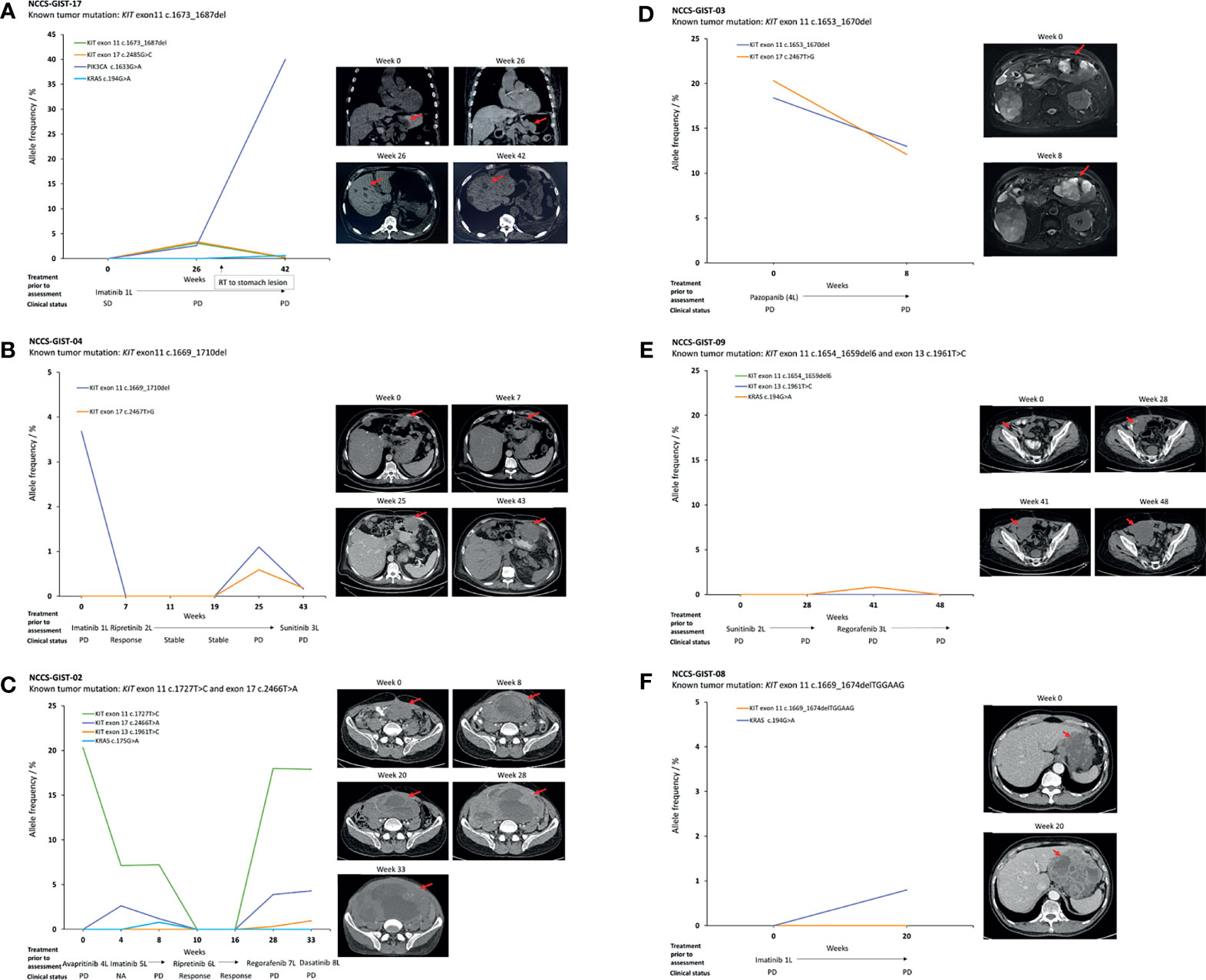
Figure 3 Temporal correlation of ctDNA mutations and patients with advanced GIST progressing while on TKI therapy. (A) Detectable KIT exon 11 and 17 mutations, as well as a potentially actionable PI3K3CA c.1633G>A mutation upon disease progression. The PI3K3CA variant was dominant over other mutations in ctDNA on further disease progression. (B–D) Trajectory and correlation of ctDNA variants with disease status. (E, F) Non-detection of KIT mutations in ctDNA despite disease progression. Each figure is accompanied with the CT scans taken at the indicated time points. The red arrows refer to the location of the tumor. SD, stable disease; PD progressive disease.
For patients NCCS-GIST-04, NCCS-GIST-02, and NCCS-GIST-09, multiple plasma samplings at different time points (4–7 samples) have allowed for the monitoring of dynamic changes to the different KIT variants detected in the ctDNA in response to different types of TKI used. In NCCS-GIST-04 (Figure 3B) and NCCS-GIST-02 (Figure 3C), KIT variants detected in ctDNA progressively decreased and were not detected in ctDNA when patients were responsive to TKI treatments. However, we observed that after a period of positive drug response, known KIT variants re-emerged in ctDNA with additional new variants that were not previously detected in tumor. The emergence of additional new KIT variant correlated with resistance to corresponding TKI treatment and disease progression.
Patient NCCS-GIST-04 switched from imatinib to ripretinib (at week 0) at the time of disease progression based on imaging assessment (Figure 3B). Imaging at week 7 confirmed response to ripretinib therapy—correspondingly, the AF for the KIT exon 11 c.1669_1710del variant fell from 3.68% at week 0 to 0% at week 7. Subsequently from week 7 to week 19, no KIT variant was detected in the ctDNA, and this coincided with a state of continued disease stability for the patient. At week 25, disease progression was detected on CT scan, and this coincided with the re-emergence of the KIT variant exon 11 c.1669_1710del (AF = 1.1%) and the detection of a new KIT variant exon 17 c.2467T>G (AF = 0.59%). At week 43, the AF values for both KIT variants were still detectable, albeit reduced (exon 11 c.1669_1710del, AF = 0.16%; exon 17 c.2467T>G, AF = 0.18%). This coincided with the patient switching to sunitinib. Despite the reduction in AF values for the 2 KIT variants, the CT scan showed that the disease had progressed with increased tumor size.
On the other hand, NCCS-GIST-02 switched from avapritinib to imatinib and subsequently ripretinib as a result of lack of response in terms of tumor reduction in the first 8 weeks of monitoring; the KIT variants from exon 11 c.1727T>C and exon 17 c.2466T>A became undetectable from week 10 to week 16 (Figure 3C), correlating with positive response to ripretinib treatment. Subsequently, ctDNA collected from week 28 and week 33 contain not only the previously detected KIT variants from exon 11 c.1727T>C and exon 17 c.2466T>A, but also a new exon 13 c.1961T>C variant. This indicated the emergence of a novel TKI-resistant variant that correlated with disease progression. Conversely, for NCCS-GIST-09 (Figure 3E), despite disease progression, no KIT variant, as previously detected in tumor, was detected in ctDNA collected at the different time points. There was, however, a KRAS c.194G>A variant that was detected at week 41 albeit at low AF (0.84%), but it was not detected at the last sampling at week 48.
There were 2 patients with 2 serial samplings, namely, NCCS-GIST-03 and NCCS-GIST-08. For NCCS-GIST-03, 2 KIT variants were detected (Figure 3D). The exon 11 c.1653_1670del variant was previously detected in tumor, while the exon 17 c.2467T>G variant was only found in plasma ctDNA. Although the variant AF for both variants decreased from week 0 to week 8, it did not correlate with decrease in tumor size. For NCCS-GIST-08 (Figure 3F), the KIT variant previously reported in tumor was not detected in ctDNA from both samplings (week 0 and week 20). A KRAS c.194G>A variant was detected, albeit at low AF (0.8%), in the week 20 sampling.
In an exploratory analysis, we attempted to detect known variants (previously found in the tumor) from ctDNA extracted from whole blood rather than from plasma. ctDNA was extracted from whole blood from 7 patients with metastatic GIST (Supplementary Table 2). NGS libraries were subsequently constructed from these extracted ctDNA. We compared the result from whole blood with those corresponding plasma-derived ctDNA as well as tumor. KIT variant was detected in 1 whole blood sample from NCCS-GIST-02. The KIT variant, exon 11 c.1727T>C, was the same as that reported for the corresponding plasma-derived ctDNA and tumor. The AF for this KIT variant in whole blood-derived ctDNA was 1.61%. This AF value was approximately 1/12 of the AF value found in plasma ctDNA (20.3%).
Our results showed that known tumor mutations, including both KIT and PDGFRA, were detectable in ctDNA only among patients with metastatic GIST with measurable disease progression. However, even in this group of patients, the detection of these mutations may depend on tumor burden at the time of progression. In some cases (NCCS-GIST-08 and NCCS-GIST-09), KIT mutations in ctDNA remain undetectable despite continued disease progression across serial samplings, suggesting that not all GISTs shed sufficient ctDNA for detection of mutations. Interestingly, as demonstrated by case NCCS-GIST-02 at week 0, despite the primary KIT exon 11 c.1727T>C being detected at high AF, the secondary KIT exon 17 c.2466T>A was not detected, implying that these mutations may not be shed at the same rates. Subsequent sampling upon disease progression simultaneously identified KIT exon 17 c.2466T>A and exon 13 c.1961T>C variants. Regardless, any detected ctDNA mutation became undetectable upon disease response to a subsequent line of TKI therapy. These results are generally consistent with previous reports (10–13). Taken together, these findings highlight the value of ctDNA mutation testing in the setting of progressive disease, enabling the detection of secondary acquired mutations, and facilitating treatment response assessment.
Among the GIST patients who progressed on TKI therapy evaluated in our study, we identified a potentially actionable acquired PIK3CA exon 9 c.1633G>A variant in NCCS-GIST-17 upon resistance to imatinib. In a previous study on 529 imatinib-naïve GISTs, only eight primary and two metastatic cases harbored PIK3CA mutations, though these cases tended to be large (>10 cm) (14). These results suggest that PIK3CA mutations may confer growth advantages in GISTs and may form the dominant clone in the setting of imatinib resistance. Consequently, this offers an opportunity for therapeutic intervention using PI3K inhibitors (15). Interestingly, we also observed the occurrence of mutations in the histone modifier gene SETD2 in three patients. Previously, Huang et al. demonstrated somatic alterations of SETD2 in 10 out of 89 (11.2%) high-risk/metastatic GIST cases but not low-/intermediate-risk cases. In gastric GISTs, SETD2 mutations were associated with hypomethylated heterochromatin and worse relapse-free survival (9). The ability to identify predictive and prognostic biomarkers in a non-invasive manner is an attractive feature of liquid biopsy-based testing, though the actual clinical utility will require validation in a larger cohort.
Several liquid biopsy-based assays deployed for detection of KIT or PDGFRA mutations have previously been reported in GISTs, including allele-specific ligation PCR (10), digital droplet PCR (11), BEAMing (16), and targeted amplicon sequencing (12, 17). Although assays targeting single mutations are highly sensitive, they are not easily generalizable in GISTs as they are characterized by a range of primary and secondary mutations. In our study, we deployed a customized targeted panel (Archer® LiquidPlex™) comprising 29 cancer and/or GIST-associated genes, emphasizing the utility of a larger sequencing footprint in picking up clinically relevant mutations beyond KIT and PDGFRA. Similar to a previous report (18), the sensitivity of our assay depends on tumor size, and is most applicable in the setting of progressive disease and TKI resistance. An additional limiting factor in the detection of ctDNA in our study may include the extent of vascularization of individual tumors, which would determine the ease of accessibility into the blood system for the ctDNA. We do take note that some of our plasma samples have been in storage at −80°C for more than the latest recommended storage period of 9 months (19). Among the 7 out of 10 patients with progressing metastatic GIST with significant detectable ctDNA mutations, 4 of the plasma samples were stored between 8 and 10 years, while the rest (n = 3) were stored for less than 1 year. In this case, storage period may not be the main issue. It is likely that when stored properly at the right temperature and repeated freeze–thaw is not allowed, plasma can be stored for a longer period. A previous report suggests the feasibility of using small volumes of dried whole blood spots for ctDNA mutation detection (20), and our exploratory investigation showed that this approach is clearly less sensitive than using plasma ctDNA, and the reagent costs also implies lower cost-effectiveness.
In conclusion, detection of GIST-related mutations in ctDNA using a customized targeted NGS panel represents an attractive non-invasive means to obtain clinically tractable information.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by SingHealth Centralised Institutional Review Board. The patients/participants provided their written informed consent to participate in this study.
TK and JC analyzed the data and drafted the manuscript. TK, EL, CN, and BT provided technical advice and experimental support. VY, MF, and JC obtained patient data and contributed samples. JC and NS designed the study, interpreted the results, and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the ArcherDX Merit Grant Award (ArcherDX LLC is a subsidiary of Invitae Corporation), Singapore Ministry of Health’s National Medical Research Council Research Training Fellowship (NMRC/Fellowship/0054/2017), SHF-Foundation Research Grant (SHF/FG653P/2017), Agency for Science, Technology and Research (A*STAR) IAF-PP Grant (H18/01/a0/019), as well as the SingHealth Duke-NUS Academic Medical Centre and Oncology ACP (08-FY2017/P1/14-A28, 08/FY2020/EX/08-A43, and 08/FY2020/EX/75-A151). The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank all patients for their participation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.840843/full#supplementary-material
1. Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-Of-Function Mutations of C-Kit in Human Gastrointestinal Stromal Tumors. Science (1998) 279(5350):577–80. doi: 10.1126/science.279.5350.577
2. Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA Activating Mutations in Gastrointestinal Stromal Tumors. Science (2003) 299(5607):708–10. doi: 10.1126/science.1079666
3. Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, Shinomura Y, et al. Gain-Of-Function Mutations of Platelet-Derived Growth Factor Receptor Alpha Gene in Gastrointestinal Stromal Tumors. Gastroenterology (2003) 125(3):660–7. doi: 10.1016/s0016-5085(03)01046-1
4. Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular Correlates of Imatinib Resistance in Gastrointestinal Stromal Tumors. J Clin Oncol (2006) 24(29):4764–74. doi: 10.1200/JCO.2006.06.2265
5. Liegl B, Kepten I, Le C, Zhu M, Demetri GD, Heinrich MC, et al. Heterogeneity of Kinase Inhibitor Resistance Mechanisms in GIST. J Pathol (2008) 216(1):64–74. doi: 10.1002/path.2382
6. Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and Secondary Kinase Genotypes Correlate With the Biological and Clinical Activity of Sunitinib in Imatinib-Resistant Gastrointestinal Stromal Tumor. J Clin Oncol (2008) 26(33):5352–9. doi: 10.1200/JCO.2007.15.7461
7. Liu X, Chu KM. Molecular Biomarkers for Prognosis of Gastrointestinal Stromal Tumor. Clin Transl Oncol (2019) 21(2):145–51. doi: 10.1007/s12094-018-1914-4
8. Namløs HM, Boye K, Mishkin SJ, Barøy T, Lorenz S, Bjerkehagen B, et al. Noninvasive Detection of ctDNA Reveals Intratumor Heterogeneity and Is Associated With Tumor Burden in Gastrointestinal Stromal Tumor. Mol Cancer Ther (2018) 17(11):2473–80. doi: 10.1158/1535-7163.MCT-18-0174
9. Huang KK, McPherson JR, Tay ST, Das K, Tan IB, Ng CC, et al. SETD2 Histone Modifier Loss in Aggressive GI Stromal Tumours. Gut (2016) 65(12):1960–72. doi: 10.1136/gutjnl-2015-309482
10. Maier J, Lange T, Kerle I, Specht K, Bruegel M, Wickenhauser C, et al. Detection of Mutant Free Circulating Tumor DNA in the Plasma of Patients With Gastrointestinal Stromal Tumor Harboring Activating Mutations of CKIT or PDGFRA. Clin Cancer Res (2013) 19(17):4854–67. doi: 10.1158/1078-0432.CCR-13-0765
11. Boonstra PA, Ter Elst A, Tibbesma M, Bosman LJ, Mathijssen R, Atrafi F, et al. A Single Digital Droplet PCR Assay to Detect Multiple KIT Exon 11 Mutations in Tumor and Plasma From Patients With Gastrointestinal Stromal Tumors. Oncotarget (2018) 9(17):13870–83. doi: 10.18632/oncotarget.24493
12. Kang G, Bae BN, Sohn BS, Pyo JS, Kang GH, Kim KM. Detection of KIT and PDGFRA Mutations in the Plasma of Patients With Gastrointestinal Stromal Tumor. Target Oncol (2015) 10(4):597–601. doi: 10.1007/s11523-015-0361-1
13. Wada N, Kurokawa Y, Takahashi T, Hamakawa T, Hirota S, Naka T, et al. Detecting Secondary C-KIT Mutations in the Peripheral Blood of Patients With Imatinib-Resistant Gastrointestinal Stromal Tumor. Oncology (2016) 90(2):112–7. doi: 10.1159/000442948
14. Lasota J, Felisiak-Golabek A, Wasag B, Kowalik A, Zięba S, Chłopek M, et al. Frequency and Clinicopathologic Profile of PIK3CA Mutant GISTs: Molecular Genetic Study of 529 Cases. Mod Pathol (2016) 29(3):275–82. doi: 10.1038/modpathol.2015.160
15. Floris G, Wozniak A, Sciot R, Li H, Friedman L, Van Looy T, et al. A Potent Combination of the Novel PI3K Inhibitor, GDC-0941, With Imatinib in Gastrointestinal Stromal Tumor Xenografts: Long-Lasting Responses After Treatment Withdrawal. Clin Cancer Res (2013) 19(3):620–30. doi: 10.1158/1078-0432.CCR-12-2853
16. Demetri GD, Jeffers M, Reichardt P, Kang YK, Blay JY, Rutkowski P, et al. Mutational Analysis of Plasma DNA From Patients in the Phase III GRID Study of Regorafenib vs Placebo in Tyrosine Kinase Inhibitor Refractory GIST: Correlating Genotype With Clinical Outcome. J Clin Oncol (2013) 31(15_suppl):10503. doi: 10.1200/jco.2013.31.15_suppl.10503
17. Kang G, Sohn BS, Pyo JS, Kim JY, Lee B, Kim KM. Detecting Primary KIT Mutations in Presurgical Plasma of Patients With Gastrointestinal Stromal Tumor. Mol Diagn Ther (2016) 20(4):347–51. doi: 10.1007/s40291-016-0203-6
18. Xu H, Chen L, Shao Y, Zhu D, Zhi X, Zhang Q, et al. Clinical Application of Circulating Tumor DNA in the Genetic Analysis of Patients With Advanced GIST. Mol Cancer Ther (2018) 17(1):290–6. doi: 10.1158/1535-7163.MCT-17-0436
19. Greytak SR, Engel KB, Parpart-Li S, Murtaza M, Bronkhorst AJ, Pertile MD, et al. Harmonizing Cell-Free DNA Collection and Processing Practices Through Evidence-Based Guidance. Clin Cancer Res (2020) 26(13):3104–9. doi: 10.1158/1078-0432.CCR-19-3015
Keywords: liquid biopsy, imatinib, non-invasive, KIT, ctDNA (circulating tumor DNA)
Citation: Ko TK, Lee E, Ng CC-Y, Yang VS, Farid M, Teh BT, Chan JY and Somasundaram N (2022) Circulating Tumor DNA Mutations in Progressive Gastrointestinal Stromal Tumors Identify Biomarkers of Treatment Resistance and Uncover Potential Therapeutic Strategies. Front. Oncol. 12:840843. doi: 10.3389/fonc.2022.840843
Received: 21 December 2021; Accepted: 28 January 2022;
Published: 22 February 2022.
Edited by:
Lixuan Wei, Mayo Clinic, United StatesReviewed by:
Hirotoshi Kikuchi, Hamamatsu University School of Medicine, JapanCopyright © 2022 Ko, Lee, Ng, Yang, Farid, Teh, Chan and Somasundaram. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jason Yongsheng Chan, amFzb24uY2hhbi55LnNAbmNjcy5jb20uc2c=; Nagavalli Somasundaram, bmFnYXZhbGxpLnNvbWFzdW5kYXJhbUBzaW5naGVhbHRoLmNvbS5zZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.