 Yuewen Zhang1
Yuewen Zhang1 Lei Wu1
Lei Wu1 Zhao Wang1Jinpeng Wang1Shrabasti Roychoudhury2
Zhao Wang1Jinpeng Wang1Shrabasti Roychoudhury2 Bartlomiej Tomasik3
Bartlomiej Tomasik3 Gang Wu1,4Geng Wang5
Gang Wu1,4Geng Wang5 Xinrui Rao1,4*
Xinrui Rao1,4* Rui Zhou1,4*
Rui Zhou1,4*- 1Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2Division of Radiation and Genome Stability, Department of Radiation Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, United States
- 3Department of Oncology and Radiotherapy, Medical University of Gdansk, Gdansk, Poland
- 4Institute of Radiation Oncology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 5Department of Gastrointestinal Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
DNA replication is a process fundamental in all living organisms in which deregulation, known as replication stress, often leads to genomic instability, a hallmark of cancer. Most malignant tumors sustain persistent proliferation and tolerate replication stress via increasing reliance to the replication stress response. So whilst replication stress induces genomic instability and tumorigenesis, the replication stress response exhibits a unique cancer-specific vulnerability that can be targeted to induce catastrophic cell proliferation. Radiation therapy, most used in cancer treatment, induces a plethora of DNA lesions that affect DNA integrity and, in-turn, DNA replication. Owing to radiation dose limitations for specific organs and tumor tissue resistance, the therapeutic window is narrow. Thus, a means to eliminate or reduce tumor radioresistance is urgently needed. Current research trends have highlighted the potential of combining replication stress regulators with radiation therapy to capitalize on the high replication stress of tumors. Here, we review the current body of evidence regarding the role of replication stress in tumor progression and discuss potential means of enhancing tumor radiosensitivity by targeting the replication stress response. We offer new insights into the possibility of combining radiation therapy with replication stress drugs for clinical use.
Background
Although radiation therapy (RT) is used to treat ~50% of malignant tumors (1), it accounts for only 5% of the total cost of cancer patient care, making it the most cost-effective cancer treatment (2). RT is also an effective treatment for patients exhibiting a poor performance status who cannot tolerate surgery (3). Although new technologies, such as CyberKnife®, Tomotherapy®, and proton and heavy ion radiotherapy have been developed, radioresistance remains a crucial factor limiting our ability to cure cancer (4). Primary radioresistance can be caused by genomic or epigenetic changes in tumor cells, and radiation-induced genomic changes lead to secondary radioresistance, which is the most common cause of treatment failure and disease recurrence (5). Owing to limitations associated with normal tissue tolerance, increasing radiosensitivity in only cancer cells remains challenging.
Replication stress (RS) is the slowing or stalling of replication fork progression and is a major cause of genomic instability in cancer cells, which induces the accumulation of mutated and damaged DNA (6). In normal tissues, RS is a factor in the natural aging process (7). Cellular response to RS activates checkpoints to arrest cell cycle and repair DNA damage. Importantly, RS is selectively higher in cancer cells than in normal cells, and makes cancer cells more dependent on RS response pathways to survive (8, 9). Oncogene activation drives continuous proliferation, which is the basis for the generation of RS known as oncogene-induced RS. It is an important source of genome instability and might therefore be the basis of intratumor heterogeneity (10). Moreover, RS-induced DNA damage in tumors activates specific DNA damage repair pathways due to different genomic background cancer types. It also causes cells to enter mitosis with under-replicated regions that can cause genomic instability, thus potentially enhancing malignant behaviors (11). If the cellular response to RS is ineffective, then cells enter mitosis with an excess of damaged DNA, resulting in genomic instability or cell death due to mitotic catastrophe (12). These differences between normal and tumor cells suggest that targeting RS may contribute to the specific elimination of tumors (13).
RS has been highlighted as a hallmark of malignant tumor radiosensitivity (6, 14). Impaired responses to RS sensitize tumors to radiation (15), highlighting the importance of RS-aimed therapy for radiation treatment. Here, we summarize the current body of evidence concerning RS in cancer radiosensitivity, including known inhibitors and other potential targets. Treatments targeting RS-related pathways are suggested as an ideal radiosensitizer for cancer treatment.
RS
Accurate DNA information is crucial for ensuring genomic stability. Conserving DNA integrity during DNA replication requires coordination between multiple cis- and trans-acting factors, such as regulating fork movement, nucleotide supply, transcription machinery, cellular checkpoints, and DNA repair pathways (16, 17). Here, we briefly summarize how RS occurs in malignant cells and the differences between cancer and normal cells, and then reason why RS is an ideal target for cancer treatment.
Sources
Several major exogenous and endogenous factors that cause RS are listed here. Endogenous factors include alternative structures of DNA, centromeres, telomeres, DNA binding non-histones, replication, and transcription conflicts. All replication stressors affect the replication fork timing, causing the replication fork to slow down or even stall. Exogenous factors including DNA damage caused by radiation or cytotoxic substances, nucleotide loss, and abnormal replication, which activate DNA damage response (DDR) (Figure 1) (18).
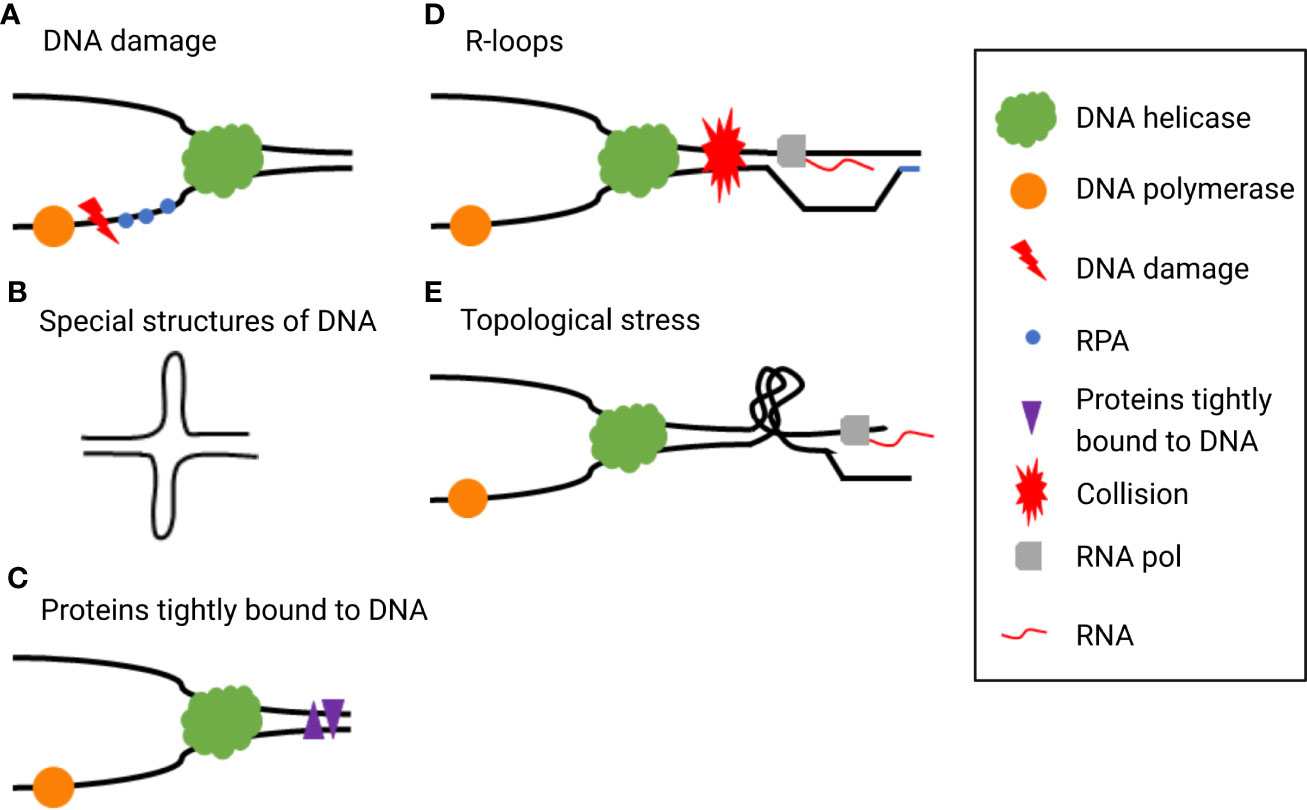
Figure 1 Typical exogenous and endogenous sources cause replication stress (RS), such as (A) DNA damage, (B) special DNA structures, (C) proteins tightly bound to DNA, (D) R-loops, and (E) topological stress.
RS Responses
Cells have several strategies for dealing with RS called “RS responses”, including re-priming, fork reversal and restart, translation synthesis, template switching, and break-induced replication (16). RS response dysregulation is a typical characteristic of tumors, which may be caused by the loss of tumor suppressor factor or abnormal oncogene expression. Chronic RS increases the chance of breakage or gap formation in fragile sites, resulting in genomic instability, promoting further activation of oncogenes, and inducing malignant tumors in the early stage (8). Although mild or moderate levels of RS may induce tumorigenesis and promote tumor progression by accumulation genomic instability, in the event of severe and persistent RS, cells will finally develop mitotic disaster, senescence, or apoptosis (19). In the absence of active ataxia telangiectasia and rad3-related (ATR) and checkpoint kinase 1 (CHK1), replication forks cannot be stalled and thus continue to trigger dormant replication origins, leading to deoxynucleotide triphosphate pool depletion as well as slowing and stalling replication fork progression (12). When single-stranded DNA (ssDNA) is no longer protected by replication protein A (RPA), the replication fork collapses, resulting in double-strand breaks (DSBs). When these cells enter mitosis, unduplicated chromosomes trigger cell death through mitotic disasters (20, 21). Moreover, mutations produced during cancer development enhance RS and cause tumor cells to be hyper-dependent on RS response (18), which may be a potential target for cancer therapy (Figure 2).
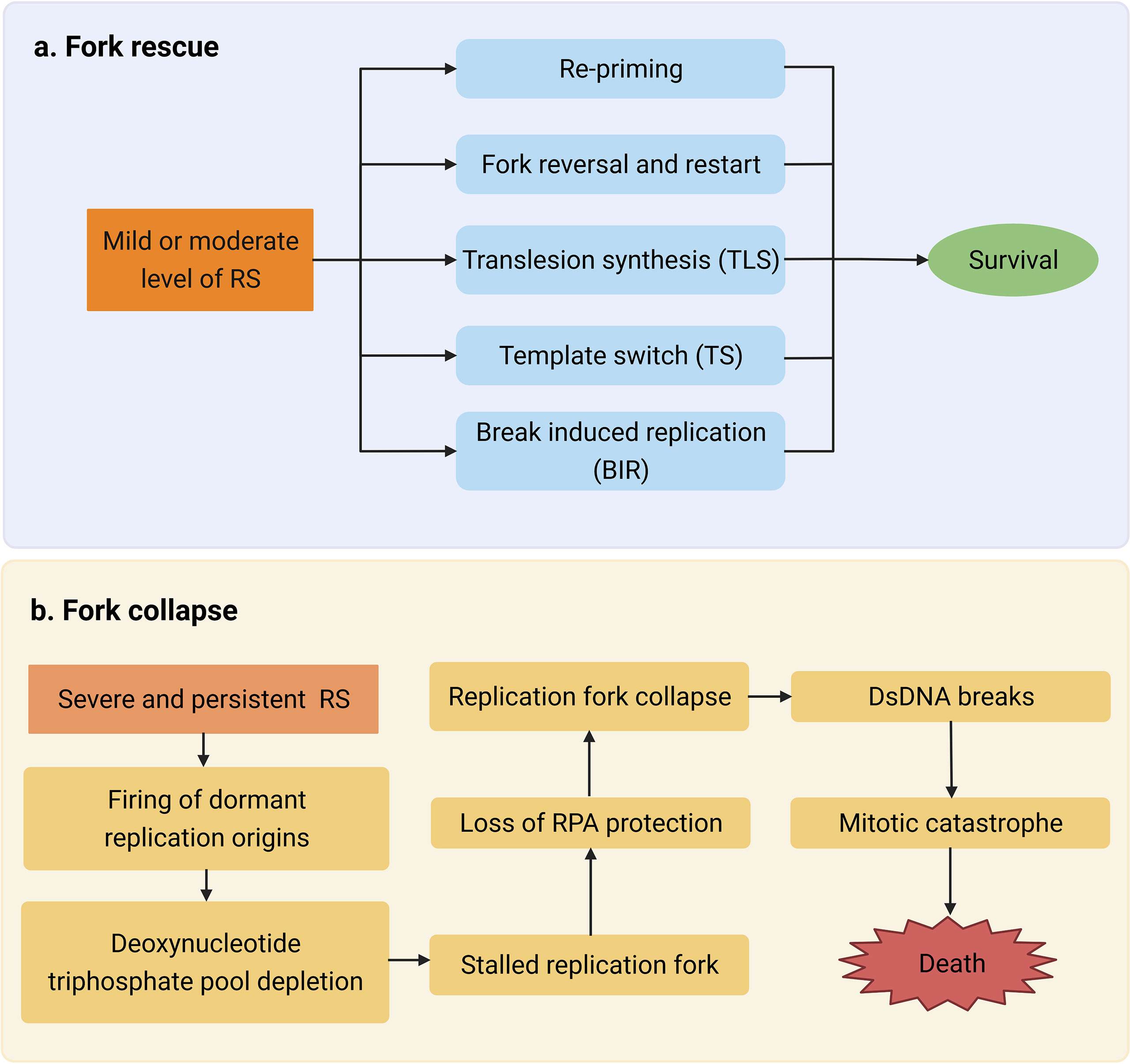
Figure 2 (A) Mild or moderate level of replication stress (RS) activates multiple mechanisms such as re-priming to repair DNA damage. (B) Severe and persistent RS leads to double-stranded DNA (dsDNA) break accumulation and eventually causes mitotic catastrophe which triggers cell death.
RS and Radioresistance in Cancer
It is well-established that tumor radiation sensitivity greatly varies among individuals. As a result, some drugs have been reported to target multiple sensitivity or resistance factors (18, 22–24). Tumor radiosensitivity is mainly related to the intrinsic sensitivity of tumor cells and the cancer microenvironment (25). Here, we summarize the well-known mechanisms of radiation resistance and analyze the relationship between RS and the resistance factors (Figure 3).
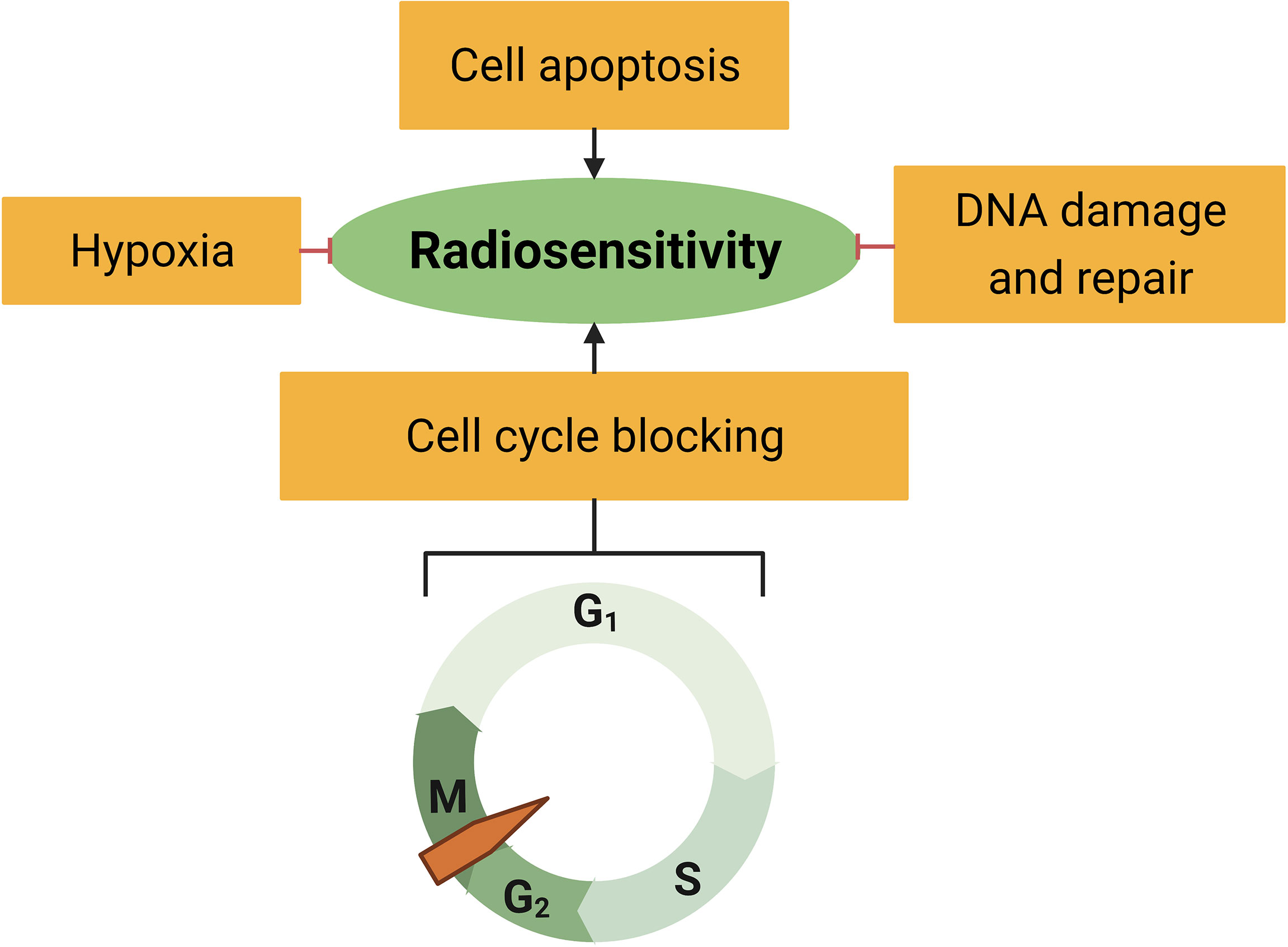
Figure 3 Radiosensitivity is associated with hypoxia, cell apoptosis, cell cycle distribution, and DNA damage response.
Hypoxia
Hypoxia is a common feature of malignant tumors resulting from rapid cell proliferation coupled with abnormal vasculature formation (26) and plays a pivotal role in tumor progression and treatment resistance (27). Hypoxia inducible factor (HIF), especially HIF-1, is the key regulator response to hypoxia. Clinical data have shown that eliminating the hypoxic state of tumors is an effective radiosensitizer (28, 29). Preclinical research has shown that NVX-108 increases tumor oxygen levels by 400%, significantly enhancing radio sensitivity (30). Phase I/II clinical trials have indicated the safety of NVX-108, and studies evaluating its efficacy are ongoing (29). Hypoxia also alters cell cycle response to ensure survival and minimal errors throughout cell division (31). Recent research claimed that hypoxia-induced RS was linked to the unfolded protein response (UPR) (32). There are few proteins that link hypoxic DDR and UPR, which suggests that they could be novel therapeutic targets to improve radiotherapy response (33, 34).
Cell Apoptosis
Apoptosis is a key part of the intrinsic tumor suppression mechanism, which is triggered when proliferation becomes aberrant (35, 36). Targeting tumor cell apoptosis also contributes to radiosensitization. A high proportion of cells die through apoptosis, which is a positive indicator of radiosensitivity (37), and enhancing apoptosis effectively enhances tumor radiosensitivity. Knocking down remodeling and spacing factor-1 (RSF-1) enhanced the radiosensitivity of cervical cancer cells by redistributing the cell cycle, inducing cell apoptosis, and eventually inhibiting cell proliferation (38). Astaxanthin enhances irradiation-induced apoptosis in esophageal squamous cell carcinoma cells (39). Deficient RS response also leads to cell apoptosis, which suggests a role as a synergistic factor to RT (40).
Cell Cycle Distribution
The cell cycle distribution of cancer cells affects radio sensitization, especially for some cancer types that depend more on other DDR pathways rather than homologous repair (HR) (41). In different cell cycles, the differences in chromosome structure lead to unequal radiosensitivity. Clinicians believe G2/M is the most sensitive phase since the radiation induces more complex damage that induce longer cell cycle arrest and therefore need proficient HR for repair (42). Meanwhile, the damage that occurs during G2/M can more easily cause premature entry into mitosis, which can lead to a higher possibility of passing incorrect genomic information to the next generation, or even cause mitotic catastrophe directly (43). Eurycomalactone, an active quassinoid isolated from Eurycoma longifolia, has been shown to sensitize non-small cell lung cancer cells to X-rays through a G2/M block (44). Further studies have focused on the G2/M arrest after receiving radiation. When DDR is activated, it temporarily stops the cell cycle to provide more time for repair, or if the damage is too severe, induces apoptosis. Eliminating the radiation-induced G2/M arrest or forcing damage cells to enter into mitosis both sensitizes cancer cells to radiation treatment (45, 46). This cell-cycle-dependent radiosensitization mechanism provides potential directions for further research into radiosensitizers.
DNA Damage and Repair
Cells respond to DNA damage by activating the DDR pathway. Abnormal activation of DDR in tumor cells leads to the generation of radiotherapy resistance (46). High RS also leads to DNA damage and activate the DDR pathway. The five major DNA repair pathways are base excision repair, nucleotide excision repair, mismatch repair, HR, and non-homologous end joining (NHEJ). Any impaired pathway can be compensated for by the overactivation of other pathways (47). These compensatory mechanisms in tumor cells lead to different responses to treatment with DNA damage agents, as well as RT (1, 48). Both RS and radiation activate a similar DDR pathway, providing the possibility of a synergistic effect of targeting RS with RT (49, 50).
Targeting RS as Radiation Sensitizer
Cancer cells relying more on RS response than normal cells to survive provides a potential target of anti-tumor treatment sensitization (51). In this section, we summarized and discussed the specific application of reagents targeting the RS response or RS-induced DDR that have already been demonstrated to be effective or have the potential to enhance tumor radiosensitization (Table 1, Figure 4).
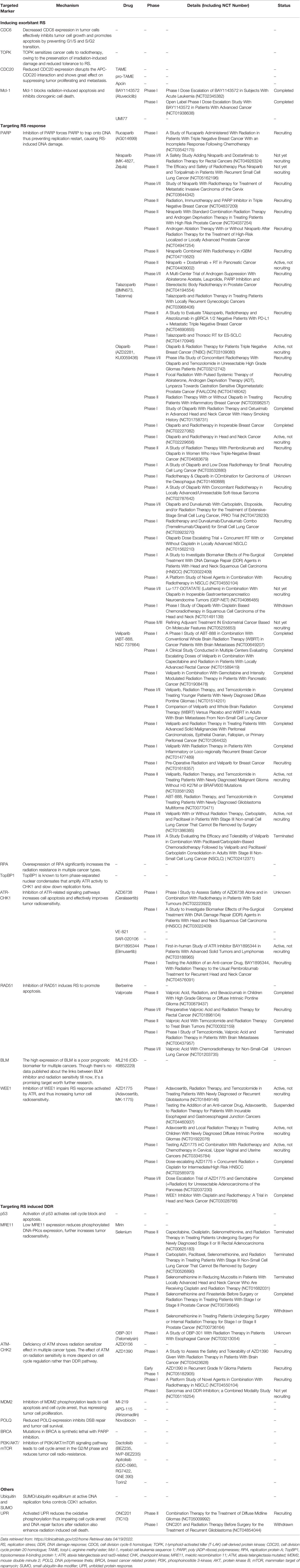
Table 1 Targeting replication stress as radiation sensitizer.
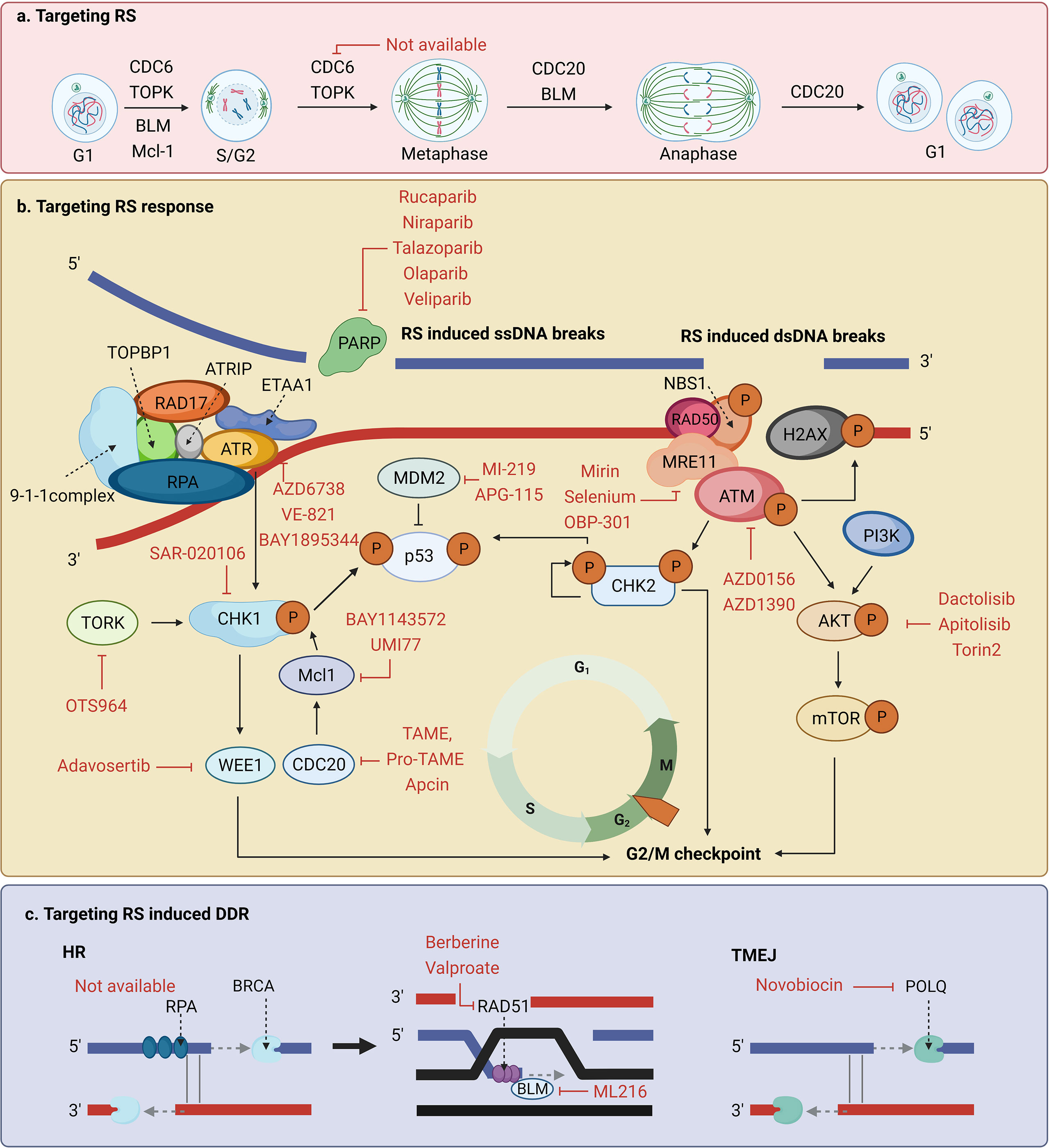
Figure 4 Potential targets and corresponding inhibitors of (A) the replication stress (RS), (B) the RS response, or (C) RS-induced DNA damage response (DDR) that have been previously reported.
Inducing Exorbitant RS
In this section, we summarized and discussed the known factors that contribute to the normal DNA replication process. Losing control of them triggers RS thus synthetically sensitizing radiation.
CDC6
Cell division cycle 6 homologue (CDC6) is an important regulator of DNA replication in eukaryotic cells (52, 53) involved in replication complex assembly during G1 phase. Replication fork stall accumulation caused by RS triggers G2/M checkpoint activation. CDC6 promotes the response of the G2/M checkpoint (54) and is positively correlated with tumor progression. Decreased CDC6 expression in tumor cells effectively inhibits tumor cell growth and promotes apoptosis by preventing G1/S and S/G2 transition (55). CDC6 overexpression has been observed in radiation-resistant cells, contributing to an increase in radiation resistance in cancer cells (56). CDC6 downregulation enhanced cisplatin-resistant bladder cancer cell sensitivity in a clinical trial, which is also related to DSB damage (57). Therefore, CDC6 inhibition in tumor cells might be an effective target for enhancing tumor radiosensitivity. Although CDC6 has druggable sites for a chemical molecular, it is an essential protein in most cell lines that makes it difficult for clinical transformation (58). Thus, further study on the regulatory mechanism of CDC6 in radiation resistance will help to develop clinical practical drugs in the future.
TOPK
T-lymphoid-activated killer (T-LAK) cell-derived protein kinase (TOPK) is a mitogen-activated protein kinase kinase-like kinase that plays an important role in cell cycle regulation. TOPK overexpression is a pathophysiological feature in different tumors (59).
TOPK knockdown does not change the radiation response of normal tissues but significantly enhances cancer cell radiosensitivity, and TOPK disruption may lead to tumor-specific radiosensitivity (60). Thus, TOPK, as a cancer-specific biomarker and biochemical target, may enhance the efficacy of cancer treatment while causing minimal damage to normal tissues (59). TOPK was found to enhance tumor radiosensitivity by enhancing intratumor RS (61). Further experiments demonstrated that TOPK helps to restart the stopped replication fork. However, when TOPK was depleted, increased levels of stalled replication forks were observed, with or without external DNA damage (61). Therefore, TOPK suppression increases internal replication damage. Owing to the preservation of irradiation-induced damage and reduced tolerance to RS, TOPK sensitizes cancer cells to radiotherapy.
TOPK interacts with CHK1 and cell division cycle 25 homologue C (CDC25C) complex (key participants in the replication of the damage induced) (61). It facilitates mitotic progression at the G2/M checkpoint via cyclin-dependent kinase 1 (CDK1), and also occurs in response to replication stressors (such as irradiation) by influencing the action of key intermediates such as CHK1 (61). Therefore, the synergistic effect of TOPK inhibition and radiotherapy is likely to produce DSBs after replication. However, unlike CHK1, the toxicity of TOPK inhibitors is limited in normal tissues due to low expression. Therefore, TOPK appears to be a promising target for further research.
CDC20
Cell division cycle 20 homologue (CDC20) has important functions in chromosome segregation and mitotic exit. It is the target of the spindle assembly checkpoint (SAC) and the key cofactor of the anaphase-promoting complex or cyclosome (APC/C) E3 ubiquitin ligase, thus regulating APC/C ubiquitin activity on specific substrates for their subsequent degradation by the proteasome (62). CDC20 is overexpressed in tumor cells and acts as a poor prognostic factor in multiple cancers (63, 64). It further increased after radiation and has been reported to increase radiation resistance via regulating B-cell lymphoma-2 (Bcl-2)/Bcl-2-associated X protein (Bax), forkhead box proteins O1 (FoxO1), or myeloid cell leukemia sequence 1 (Mcl-1)/p-CHK1 in different cancer types (65–67). Suppression of CDC20 expression reverses the radioresistance (65–67). There are multiple available inhibitors of CDC20, including tosyl-L-arginine methyl ester (TAME), Pro-TAME, and apcin. Their main effects involve disrupting the APC-CDC20 interaction (68, 69). Some of them showed great efficacy in suppress tumor proliferating and metastasis (70, 71). However, there has been no evidence on the effects of the CDC20 inhibitors on radiosensitivity. Therefore, CDC20 could be a potential target as a radiosensitizer, but more evidence in future studies is needed.
Mcl-1
As the first anti-apoptotic protein in the Bcl-2 family, Mcl-1 is regulated by the cell cycle and reach peak expression levels in the S/G2 phase. It acts as a functional switch in selecting between HR and NHEJ pathways after DNA damage (72). It blocks radiation-induced apoptosis and inhibits clonogenic cell death (73). Targeting Mcl-1 by a small molecule enhances RS sensitivity to cancer therapy (72). BAY1143572 downregulated Mcl-1 by inhibiting binding of HIF-1α to the Mcl-1 promoter (74). UMI77 is a selective inhibitor of Mcl-1 that dissociates Mcl-1 from the pro-apoptotic protein Bak and produced significant radiosensitization in pancreas cancers (75).
Targeting RS Response
Here, we summarize important RS response factors that are essential for cells to survive. Inhibition of these factors leads to uncontrolled replication collapse and even mitotic catastrophe, which makes them ideal targets for radiosensitization.
PARP
Poly (ADP-ribose) polymerases (PARPs) are involved in DDR and recruit DNA repair proteins to damaged sites by catalyzing ADP-ribosylation, leading to the formation of poly (ADP-ribose) polymers (76). PARP1, the most abundant PARP, plays a similar role to PARP2 in the DDR process and is an important regulator of fork reversal (77). Inhibition of PARP directly increases the speed of fork elongation and does not cause fork stalling, which contrasts with the accepted model in which inhibitors of PARP induce fork stalling and collapse. Aberrant acceleration of fork progression by 40% above the normal velocity leads to DNA damage (78).
However, the effects of PARP inhibitor do not directly decrease the expression of PARP. Rather, the inhibitor forces PARP to become stuck on DNA, thus preventing replication restart and causing RS-induced DNA damage (79). It was also linked to decreased replication fork length with greater ssDNA gaps, which in turn cause more genomic instability at G2/M (80). With all the evidence of PARP inhibitors in RS-induced DNA damage, researchers have reported on various preclinical models of combination therapy with PARP inhibitors and ionizing radiation (IR) (81). Olaparib, a PARP inhibitor that has been widely used in cancer treatment, has been reported to have strong tumor-specific radiosensitization effects (82, 83).
RPA
The RPA complex is one of the first responders to coordinate DNA replication (18, 84). It consists of three subunits, RPA1 (RPA70), RPA2 (RPA32), and RPA3 (RPA14), which are essential to protect ssDNA at replication forks and recruits DNA polymerases α, δ, and ϵ for the initiation and elongation steps of DNA replication (84). It has been reported that RPA1 phosphorylation upon RS decreases the ubiquitination of chromatin-loaded RPA1, leading to an accumulation of RPA1 on stalled replication forks. This helps the DNA-binding domains of RPA2 to bind with RPA1-coated ssDNA, thus contributing to increased RPA2 binding stability (85). Loss of RPA accelerates fork breakage, whereas overexpression of RPA is sufficient to delay a “replication catastrophe” (86). It also plays an important role in DDR in relation to the HR pathway (87). Furthermore, overexpression of RPA significantly increases the radiation resistance in multiple cancer types (88–90). However, there has been no reported inhibitors of RPA because it is an essential protein to all cells. Furthermore, it is a downstream factor of ATR, and thus the regulation of ATR may produce similar effects (86). RING finger and WD repeat domain 3 (RFWD3)-mediated ubiquitination of RPA helps to remove RPA from the damage site, which is a crucial step for HR (91), and thus provides a possible target for increasing radiation sensitivity via ubiquitination regulation.
TopBP1
DNA topoisomerase II-binding protein 1 (TopBP1) serves as a scaffold to assemble protein complexes in a phosphorylation-dependent manner via its multiple breast cancer C-terminal (BRCT) repeats. It is repurposed to scaffold different processes dependent on cell cycle-regulated changes in phosphorylation of target proteins (92). It is known to form phase-separated nuclear condensates that amplifies ATR activity to CHK1 and slow down replication forks (93). TopBP1 also stabilized bloom syndrome helicase (BLM) to maintain genome stability (94). It is often overexpressed in cancer and can bypass control by CDK2 to interact with treslin, leading to enhanced DNA replication (95).
However, it has been reported that at low levels, TopBP1 activates ATR/CHK1, but once TopBP1 protein accumulates above an optimal level, it paradoxically leads to lower activation of ATR/CHK1. This is due to the perturbation of ATR-TopBP1 interaction and ATR chromatin loading by excessive TopBP1. Depletion of TopBP1 in some specific cancer cells enhanced ATR/CHK1 activation and S-phase checkpoint response after RS (96). Thus, simply inhibiting TopBP1 may lead to unexpected results, which makes it not an ideal target for radiation sensitization.
ATR-CHK1
ATR of the phosphoinositide 3-kinase (PI3K) family is a central regulator of RS. After ssDNA fragments are coated with PRA, ATR and ATR-interacting protein are recruited and activated. It further phosphorylates various proteins, including CHK1 kinase, which inhibits mitotic entry and dormant origin activation. Mitotic entry is inhibited by CDC25 phosphatase phosphorylation, which prevents subsequent mitotic CDK activation (97). Cancer genome sequencing showed a very low ATR or CHK1 mutation or deletion frequency. Instead, these genes are often amplified in cancer cells, probably because they need to process high levels of RS to survive. ATR and CHK1 inhibition can increase RS, leading to mitotic catastrophes that trigger cell death (98). Furthermore, inhibition of ATR-related signaling pathways can increase cell apoptosis and effectively improve tumor radiosensitivity (99). ATR inhibitors, such as AZD6738 and VE-821 as well as the CHK1 inhibitor SAR-020106 were effective radiosensitizers in preclinical studies (100–102). An ongoing phase I clinical trial (NCT03188965) is assessing the safety profile of ATR inhibitors (BAY1895344) (103).
RAD51
RAD51 is a master regulator of DNA replication and plays important roles in DSB repair, RS, and mitosis (104). RAD51 is a core factor in overcoming RS by slowing or stalling replication forks, which threatens replication integrity (105). It facilitates fork inversion, protects reverse forks, repairs and restarts broken replication forks, and post-replication gap filling (104, 106).
RAD51 inhibition may lead to increased tumor radiosensitivity, and it has been reported as a potential target of berberine in osteosarcoma radiosensitization (107). Valproate was found to increase tumor tissue cell radiosensitivity by increasing levels of RFWD3 and inhibiting RAD51 (108). The inhibition of nucleophosmin1 (NPM1) by YTR107, a small molecule that binds with NPM1, inhibits pentamer formation and represses RAD51 formation after IR. The synergistic effect of YTR107 and the PARP1/2 inhibitor ABT-888 increased RS and radiation-induced cell mortality (109).
BLM
BLM is a 3’-5’ ATP-dependent RecQ DNA helicase that is one of the most essential genome stabilizers involved in the regulation of DNA replication, recombination, and both homologous and non-homologous pathways of DSB repair (110). It interacts with topoisomerase IIIα (TOP3A), RecQ-mediated genome instability (RMI) 1, and RMI2 to form the BLM-Topoisomerase IIIα-RMI1-RMI2 (BTR) complex, which dissolves double Holliday junctions to produce non-crossover HR products. It also promotes DNA-end resection, restart of stalled replication forks, and processing of ultra-fine DNA bridges in mitosis (111). BLM helicase-deficient cells exhibit multiple defects in DNA replication, including accumulation of abnormal DNA replication intermediates, slower replication fork velocity, and excessive firing of dormant origins, thus exhibit increased levels of chromatid breakage and HR (112). It interacts directly with both RAD51 and RPA, and the function in DNA replication is regulated by sumoylation (113).
The high expression of BLM is a poor prognostic biomarker for multiple cancers (114, 115). Biallelic pathogenic variants in BLM cause bloom syndrome with severe pre- and postnatal growth deficiency, immune abnormalities, sensitivity to sunlight, insulin resistance, and a high risk for many cancers that occur at an early age (116). The symptoms of bloom syndrome including sensitivity to ultraviolet damage, which is similar to radiation, provide the possibility of transforming this genomic defect into a treatment sensitizer. ML216 is a small molecule inhibitor of BLM, and inhibits cell proliferation of BLM-proficient cells and increases the frequency of sister chromatid exchanges (117). Though there has been no data published on the links between a BLM inhibitor and radiation sensitivity till now, it is a promising target worth further research.
WEE1
When ssDNA accumulation at stalled replication forks activates ATR, it phosphorylates CHK1, which in turn activates WEE1 kinase and inhibits CDC25 phosphatase. While WEE1 inhibits CDKs, the key drivers of cell cycle progression, by phosphorylating the conserved threonine 14 (Thr14) and tyrosine 15 (Tyr15) residues, CDC25 activates CDKs by dephosphorylating the same residues (118). Elevated WEE1 expression reduces RS and activates G2/M checkpoints, conferring cell resistance to CHK1 inhibitors (98). Recently, it has been reported that the ATR-WEE1 module inhibits the MOS4-associated complex (MAC) to regulate RS responses (118).
The evidence suggests that WEE1 inhibition impairs the RS response activated by ATR, and thus increases tumor cell radiosensitivity (119). WEE1 kinase inhibitors sensitize tumor cells to proton and X-ray irradiation by inducing RS, independent of TP53 mutation status, such as AZD1775 (120–122). Clinical trials have shown that the WEE1 inhibitor adavosertib could potentiate the efficacy of RT; however, its clinical application is limited by its unfavorable safety profile (123).
Targeting RS-Induced DDR
The RS response shares many biological pathways with DDR. They are widely intertwined and thus hard to completely distinguish (124). Here, we grouped the proteins that are typically related to the DDR pathway but are not necessarily involved in the RS response. Targeting these proteins usually impairs the DDR processing to enhance radiosensitivity, which makes them the most promising targets.
p53
The p53 signaling pathway plays a key role in determining radiosensitivity in normal tissues but is often inactivated during cancer. Loss of p53 in tumor cells allows them to escape cell cycle arrest and apoptosis checkpoints and promotes the growth of early-stage cancer cells by skipping the cell cycle checkpoint caused by RS (125). During DNA replication, IR-induced DNA damage stalls replication forks, and single-strand breaks (SSBs) can be transformed into DSBs, thereby activating the ataxia telangiectasia mutated (ATM)/ATR pathway. ATM and ATR phosphorylate p53 to increase its stability and activate target genes. RS induced by chemotherapy drugs such as trifluridine leads to cell senescence or apoptosis of tumor cells according to the state of p53 (126). Acetylation of p53 may modulate cancer cell radiosensitivity, which provides a promising strategy for radiosensitization (127).
MRE11
Meiotic recombination 11 (MRE11), the core of the MRE11/RAD50/NBS1 (MRN) complex, is involved in DNA break end detection, phosphorylation-dependent signal amplification, and DSB repair (128). The complex is critical for ATM activation of DSBs and downstream activation of G2/M and p53-dependent G1/S cell cycle checkpoints (129, 130). MRE11 also has endonuclease and exonuclease activities residing in the phosphodiesterase domain. These nuclease activities are crucial for the pathway choice of HR and NHEJ (131). Cancer cells rely on DNA repair for survival during cancer therapies, and thus MRE11 might be a promising synergistic therapeutic target.
Dysfunction of it in neoplastic breast tumors results in the accumulation of R-loops, replication-associated DSB, abundance of genomic deletions, and uncontrolled proliferation (132). Evidence suggests that its expression in cancer cells is critical for radioresistance (133). Low MRE11 expression in colorectal cancer cells reduced phosphorylated DNA-PKcs expression and further increases tumor radiosensitivity (134). There are different small and large molecular inhibitors targeting MRE11. Mirin is the first inhibitor found to specifically target MRE11 exonuclease activity with radiosensitizing properties (135). Lung cancer cells treated with Selenium, which is an essential trace element, showed decreased expression of MRE11 and significantly reduced colony formation relative to IR (136). OBP-301, with the insertion of the human telomerase reverse transcriptase (hTERT) promotor, also showed reduced MRE11 expression and thus enhanced radiosensitivity of lung cancer cells (137). Therefore, MRE11 inhibitors are clinically significant for enhancing radiosensitivity, and several clinical trials investigating their potential are ongoing (131).
ATM-CHK2
ATM kinase is a member of the PI3K-like protein kinase (PIKK) family with extensive roles in DDR signaling (138). Upon recruitment by the MRN complex to DSBs, ATM autophosphorylates at different serine sites resulting in the activation of CHK2, p53, and H2AX, which are involved in DNA repair processes and cell cycle arrest (139). The most important transducer of ATM signaling is CHK2, a kinase that signals to DNA repair, cell cycle arrest, and apoptosis. ATM phosphorylates CHK2 on threonine 68 (Thr68), thereby causing CHK2 dimerization and autophosphorylation of the kinase domain and is required for full activation (140).
ATM orchestrates the cellular DDR to cytotoxic DNA DSBs induced by radiation (141, 142). Overexpression of ATM indicates radiation resistance in breast cancer cells (143), whereas deficiency of ATM showed radiation sensitizer effects in multiple cancer types (144–147). Interestingly, more studies have focused on the radiation sensitizer effects that are dependent on the cell cycle and proliferation status (148, 149). After inhibition of proliferation, ATM status did not alter cell death or micronucleus formation after radiation, which suggests that ATM in endothelial cells was immaterial if a cell cycle block was present at the time of irradiation. It is consistent with other data showing that the effect of ATM on radiation sensitivity is more dependent on cell cycle regulation rather than the DDR pathway (148,149). Considering that ATM is a large protein with extensive regions of unknown function, the inhibition of its kinase activity may produce better synergistic effect on treatment. AZD0156, as a potent and selective bioavailable inhibitor of ATM, showed strong radiosensitizer effects in vitro and in a lung xenograft model (150). Specially, the ATM inhibitor AZD1390 is optimized for penetration of the blood-brain barrier with radiosensitizing effects on glioma and lung cancer cell lines, even in a brain metastasis model (141, 151). All of the evidence suggests that treatments targeting ATM may be promising in clinical trials.
MDM2
Mouse double minute 2 (MDM2) protein is a major negative regulator of p53 (152). When activated, p53 suppresses tumors in response to cell damage by mediating cell proliferation, cell cycle arrest, DNA repair, metabolism, angiogenesis, senescence, and apoptosis (153). In normal cells, the self-regulating feedback loop between MDM2 and p53 controls p53 expression (154, 155). The rescue of p53 function in cancer cells by inhibiting the interaction between p53 and MDM2 restored cycle arrest and apoptosis (156). Furthermore, inhibition of MDM2 phosphorylation leads to cell apoptosis and cell cycle arrest, thus repressing tumor cell proliferation in esophageal cancer cells (157). Additionally, MDM2 inhibitors, such as MI-219, increase tumor cell radiosensitivity in a p53-dependent manner. MI-219 combined with radiation resulted in increased p53-dependent DNA damage (158). A novel small-molecule inhibitor, APG-115, was found to enhance gastric adenocarcinoma cell radiosensitivity by blocking the interaction between MDM2 and p53 (159). Therefore, blocking the MDM2/p53 pathway has broad application prospects for treating tumors and enhancing tumor radiosensitivity, especially for tumors with low TP53 mutation levels, such as those of myeloid leukemia.
POLQ
DNA polymerase theta (POLQ) is a DNA polymerase that protects against error-prone transduction DNA synthesis and error-prone DSB (160). It is involved in a major DNA repair pathway that was initially named as alternative end-joining or microhomology-mediated end joining, and was later termed polymerase theta-mediated end joining because POLQ is indispensable in this process (161). POLQ overexpression reduces replication fork speed and impairs cell cycle progression (162). Furthermore, breast cancer related protein (BRCA) 2 and POLQ co-inhibition significantly improves tumor cell sensitivity to cisplatin (163). Reduced POLQ expression inhibits DSB repair and tumor cell survival. Several hepatocellular carcinoma cell lines (Huh7, HepG2, MHCC-92L, SK-HEP-1, and BEL-7404) with low POLQ expression after knockdown were found to be significantly sensitive to chemotherapeutic drugs (160). Depletion of POLQ in POLQ-dependent cancers (i.e., malignancies deficient in HR) leads to synthetic lethality. Furthermore, POLQ depletion was shown to synergize with PARP inhibition (164, 165), and the antibiotic novobiocin was recently reported as a selective POLQ inhibitor (166). Thus, combining novobiocin with radiotherapy should be a new research direction for targeting radioresistance.
BRCA
BRCAs (including BRCA1 and BRCA2) are thought to be the predominant proteins involved in HR in the DDR pathway. In addition, as a master regulator of HR, BRCA1 and BRCA2 also mediate fork protection (167, 168). BRCA mutations increase the susceptibility to various cancer types, including breast, ovarian, prostate, and pancreatic cancers (167). It is also well known that mutations in BRCA result in synthetic lethality with PARP inhibition. The underlying mechanism includes HR deficiency and increasing replication gaps. PARP inhibition results in replication fork collapse, chromosomal instability, cell cycle arrest in G2, and subsequent apoptosis in BRCA-deficient cells (169). Therefore, targeting PARP has become a reliable therapeutic strategy for eliminating BRCA1/2-mutated malignancies at diverse sites including the breast, primary peritoneum, fallopian tubes, ovaries, and pancreas (also see section 4.1.2) (170).
It has been reported that BRCA-deficient tumors are more sensitive to chemotherapeutic agents that induce RS (171). Furthermore, mutations in BRCA1/2 enhance radiosensitivity, indicating the possibility of BRCA as a biomarker of radiation sensitivity (172, 173). Since BRCA1/2 are both large proteins and have complex multiple functions, the development of inhibitors directly targeting BRCA1/2 is difficult to achieve. Therefore, PARPi has been suggested to patients with BRCA1/2 mutations for the synergistic lethal effects. The function of PARPi in radiosensitization are summarized in 4.2.1. Further research may focus on inhibitors that specifically affect the function of BRCA.
PI3K/AKT/mTOR
The PI3K/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway activates the downstream mediator mTOR to translate specific mRNA transcripts (174, 175). They synergistically work with CHK1 to repress DSB-induced RAD51 foci, thus impairing the HR process and enhancing RS in tumor cells. In addition, PI3K/mTORi slows the fork speed by increasing cell division cycle 45 homologue (CDC45) to promote a new origin of replication, thus enhancing CHK1-induced RS (176). The PI3K/AKT/mTOR signaling pathway is hyperactivated or altered in many cancer types (177). Inhibition of the pathway reduces tumor cell radioresistance (178, 179). For example, dactolisib, a dual PI3K/mTOR inhibitor, causes cell cycle arrest in the G2/M phase and improves the radiosensitivity of DU145 cell lines. Dactolisib also inhibits radiation-induced DSB repair in glioblastoma (GBM) cell lines by inhibiting DNA-PKcs and ATM and improves the radiosensitivity of radioresistant prostate cancer cell lines (180).
Torin 2 is a special class of PI3K pathway drugs, which not only inhibits the cell cycle at G1/S but also interferes with S phase progression, causing ssDNA accumulation, DNA damage, and increased checkpoint signaling in triple-negative BRCA cells (181). Furthermore, the dual PI3K/mTOR inhibitor apitolisib (GDC-0980) was demonstrated to inhibit growth and induce apoptosis in human GBM cells (182).
Others
Despite all the classic proteins we discussed above, several novel concepts have been suggested in recent research. A large number of accessory factors involved in the assembly of replisomes have been reported, which includes multi-protein complexes that monitor replication fork progression, generate checkpoint and damage signals, and coordinate DNA synthesis with chromatin assembly (183). We list below several newly identified processes that may be related to RS and radiation sensitivity that may provide ideas for translating basic research into clinical trials.
Ubiquitin and SUMO
Post-translational modification of the DNA replication machinery by ubiquitin and small ubiquitin-like modifier (SUMO) plays key roles in cell division, DNA replication/repair, signal transduction, and cellular metabolism (184). Recent research revealed that ubiquitin/SUMO pathways are essential regulators of DNA replication during initiation, the S phase or elongation, and DNA replication termination (185). SUMO/ubiquitin equilibrium at active DNA replication forks controls CDK1 activation. An increase in ubiquitination of the replisome results in premature disassembly of the replication machinery and generation of CDK1-dependent DNA damage in the S phase (186).
Our group has identified ubiquitination factors that affect radiation sensitivity. We showed that ubiquitin-specific protease 9X (USP9X) mediates lysine-specific demethylase 4C (KDM4C) deubiquitination, which activates transforming growth factor-β2 (TGF-β2)/Smad/ATM signaling to promote radioresistance in lung cancer (142). Furthermore, ubiquitin-conjugating enzyme E2O (UBE2O) facilitates tumorigenesis and radioresistance by promoting MAX interactor 1 (Mxi1) ubiquitination and degradation (187). The SUMO-specific protease (SENP) pathway is also involved in tumor radiation sensitization (188). SUMO E3 ligase PIAS4, which is an essential signal for p53-binding protein 1 (53BP1) loading to the damage site, promote radiation resistance by increasing DDR (189). Ring finger protein 4 (Rnf4), an E3 ubiquitin ligase that targets SUMO-modified proteins, target SUMOylated mediators of DNA damage checkpoint protein 1 (MDC1) and SUMOylated BRCA1 loading at sites of DNA damage. Rnf4-deficient cells and mice exhibit increased sensitivity to IR by suppressing DDR (190). These findings identify ubiquitylation/SUMO as possible radiosensitization targets, but further research is needed.
UPR
UPR is the master regulator of endoplasmic reticulum (ER) stress. A deficiency in UPR results in apoptosis (191). Recent research revealed the link between hypoxia-induced RS and UPR (192). The induction of RNA/DNA helicase senataxin (SETX) in hypoxia is reliant on the protein kinase R (PKR)-like ER kinase (PERK)/activating transcription factor 4 (ATF4) arm of the UPR (32). Hypoxia is present in the majority of human tumors and is associated with poor prognosis due to the protection it affords to radiotherapy and chemotherapy (27). As we described earlier (section 3.1), anti-hypoxia treatments provide additional radiation benefits through cell apoptosis, which establishes a link between UPR and radiation sensitivity.
UPR is widely involved in the establishment and progression of cancers, including BRCA, prostate cancer, and GBM multiforme (193). Elevated mitochondrial UPR markers (mtHSP70 and HSP60) are associated with poor prognosis in patients with lung adenocarcinoma, which is activated by Maf1 through ATF5. Suppressing IR-induced mitochondrial UPR activation by rapamycin resulted in increased sensitivity to IR-mediated cytotoxicity (194). ONC201, an UPR activator, reduced oxidative phosphorylation and thus impairs cell cycle arrest, and the inhibition of DNA repair factors after radiation also enhanced radiation-induced cell death (34). As a new concept of radiosensitization, the clinical significance of UPR still requires further studies.
RS-Induced Innate Immune Response in Radiation Sensitivity
Nowadays, the immune microenvironment is the hotspot in cancer research. It involves all processes of tumorigenesis, cancer progression, and treatment resistance. Innate immunity refers to nonspecific defense mechanisms that act immediately after antigen appearance. The activation of innate immune responses relies on pattern recognition receptors (PRRs). These PRRs detect endogenous damage-associated molecular patterns (DAMPs) or exogenous conserved pathogen-associated molecular patterns (PAMPs) to initiate a signaling cascade resulting in the production of interferons (IFNs) and inflammatory mediators (195, 196).
RS-Induced Innate Immune Activation
As research progressed, some evidence revealed the relationship between RS and innate immune response, which plays a key role in cancer treatment resistance (197). In this study, we have summarized and discussed the potential relationship between targeting RS and innate immune activation.
Innate Immunity Activation by RS in Immune Cells
Excessive RS or RS deficiency leads to the accumulation of replication blockage-derived DNA in the cytoplasm or the formation of micronuclei, resulting in activating the cyclic GMP-AMP (cGAMP) and the cGAMP receptor stimulator of the interferon gene (STING) pathway. cGAMP synthase (cGAS) is a DNA sensor that recognizes and binds with DNA fragments in the cytoplasm, enabling cGAMP synthesis. cGAMP subsequently activates STING. The activation of STING further increases interferon regulatory factor 3 (IRF3) and nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) levels (198). IRF3 and NF-κB act as transcription factors to trigger the transcription of IFN-I and cytokines (199). Apart from cGAS, γ-interferon-inducible protein-16, a cytosolic DNA sensor, can detect both self and non-self dsDNA to promote IRF3 and NF-κB-dependent interferon production via STING (195, 200, 201). IFN-1 plays a crucial role in both basal and therapeutic-induced immune responses to cancer. It is a potent immune cell activator, resulting in the activation and maturation of antigen presenting cells (198). The promotion of dendritic cell migration to the tumor site and their maturation depends on IFN-1 signaling (202, 203). Innate immune cells respond to IFN-1 by increasing antigen presentation and the production of immune response mediators, such as cytokines and chemokines. These events help in antigen presentation and chemokine production in innate cells as well as induce antibody production and enhance T-cell responses (198).
Innate Immunity Activation by RS in Tumor Cells
Innate immunity activation by tumor cells is a complex phenomenon. As we mentioned above, cancer cells usually experience higher RS, leading to more cytoplasmic DNA and micronuclei formation. They activate innate immunity by secreting IFN-1 via the cGAS-STING pathway, exocrine exosomes, or extracellular vesicles (EVs), which can be captured by immune cells for inducing a further immune response.
Cancer cells exposed to RS-inducing agents or deficient in RS response show the increased production of IFN-1 and proinflammatory cytokines that can foster an innate immune response (204, 205). One study found that the inhibition of the ATM/CHK2 DNA damage checkpoint axis led to excessive RS and cytosolic DNA accumulation, which subsequently activated the DNA sensor STING-mediated innate immune response in ARID1A-deficient tumors (206). Cytosolic DNA can also be released in exosomes or EVs (207, 208). Exosomes/EVs containing DNA works as DAMPs to innate immune cells. Study found that EVs and exosome dsDNA promoted inflammation via activating the STING pathway in macrophages (209).
The activation of STING in dendritic cells is essential for radiation-induced antitumor immunity (210). In contrast, cGAS-STING activation in tumor cells impairs HR in DDR, which promotes tumorigenesis (211). Moreover, cGAS can act as a decelerator of DNA replication forks, suppressing replication-associated DNA damage (212). The complex network mechanism made it hard to simply target or enhance cGAS-STING to reverse cancer treatment resistance. In contrast, high RS or RS-response deficiency always leads to simultaneous cell damage and immune activation. Hence, it would be a better choice for cancer treatment sensitization.
Targeting RS Response Enhances Radiation Sensitivity by Innate Immunity
The immune response caused by RT remains controversial. The inflammatory responses caused by RT are different depending on the RT pattern (213). Immune cells are highly radiosensitive compared with tumor cells (214). Conventional RT-induced myeloid-derived suppressor cell filtering leads to the suppressive tumor microenvironment (TME) rather than the active TME (215). Though the hypothesis that the damage signal released from tumor cells alone can activate a systemic antitumor immune response called the abscopal effect has been observed in a small-sample study (203), confirming the hypothesis without combining the signal with checkpoint inhibitors is difficult. The basic research revealed that RT may increase programmed death ligand 1 (PD-L1) levels in tumor and immune cells, contributing to immunosuppression and in part explaining the clinical success of the combination of RT with programmed cell death protein 1 (PD-1)/PD-L1 immunotherapy (216). As basic research data are available, more clinical trials regarding the combination of anti-PD-1/PD-L1 antibody with radiation are going on (217–220). Polymorphonuclear neutrophils recruited in the TME post-RT can facilitate tumor progression by forming neutrophil extracellular traps (221). Taken together, the tumor immune microenvironment is thought to be suppressed rather than activated after radiation, which plays a key role in radioresistance. Promoting immune response activation of TME is the key to enhance radiosensitivity (Figure 5).
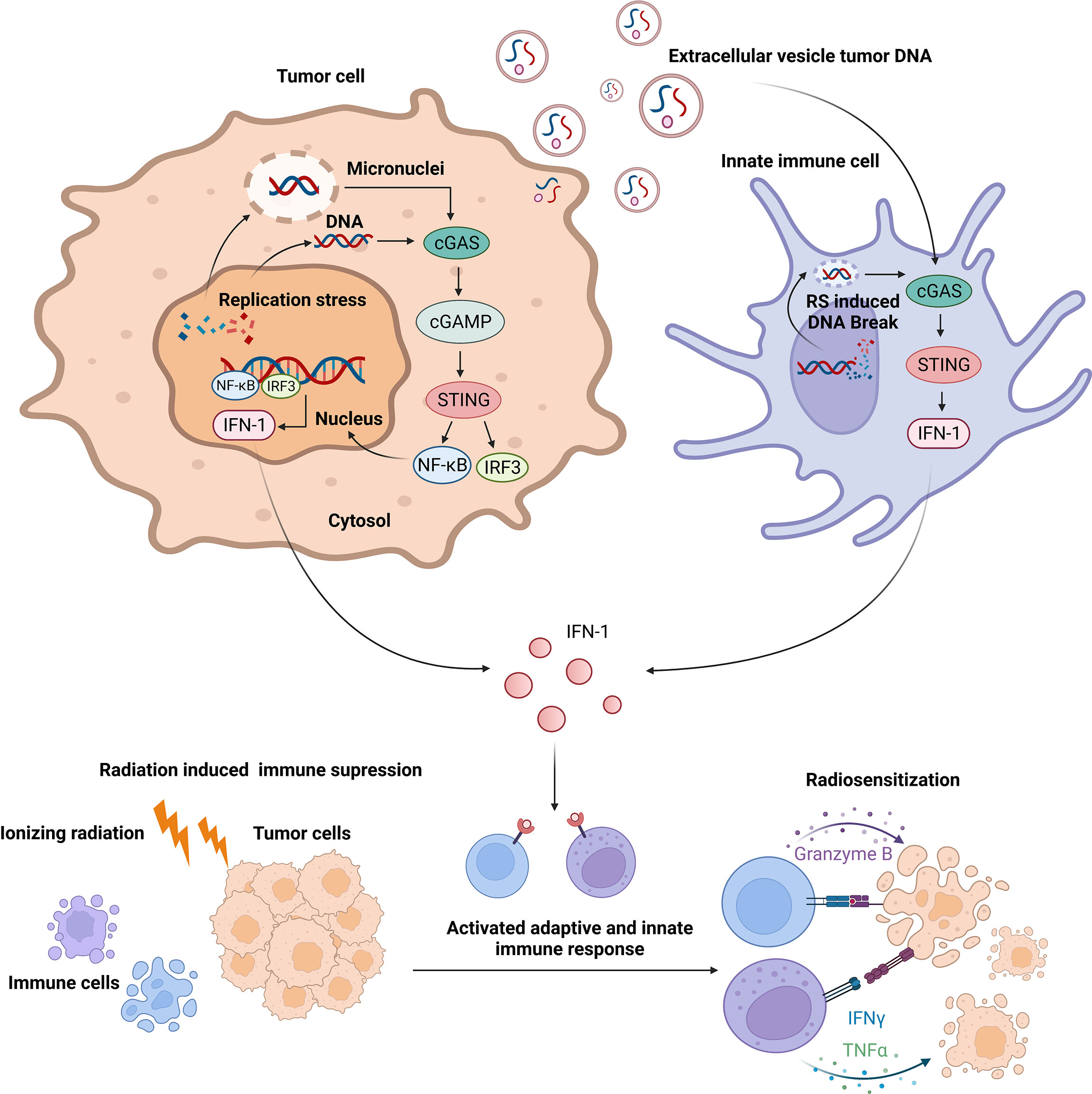
Figure 5 Replication stress-induced activation of innate immune response enhances radiosensitivity via cyclic GMP-AMP synthase–STING signaling.
Enhancing RS and targeting RS response are good choices to manage tumors. As mentioned above, excessive RS or RS response deficiency results in more DNA damage, which is the synergy effect of RS and RT from the direct tumor side. As they lead to dsDNA accumulation in the cytoplasm and cell apoptosis, which activate innate immunity, they may enhance radiation sensitivity from the indirect immune side (215, 222).
RAD51-depleted cells accumulate more cytosolic DNA after radiation, activating the STING pathway to increase innate immune response (223). ATR inhibition and radiation drive immune cell infiltration via tumor cell-intrinsic cytokine release to boost immunogenic response to radiotherapy and modulate the radiation-induced inflammatory TME (224). PARP inhibitor and radiation work synergistically to kill lung cancer cells by activating antitumor immunity in the form of increased CD8+ T lymphocytes and the activated STING/TANK-binding kinase 1/IRF3 pathway (225). WEE1 inhibitor increases tumor-specific cytotoxicity and shows a positive effect on immune response after radiation by dendritic cell activation, which can be combined with immune therapy (226, 227).
These studies indicate that the RS-induced activation of innate immune response may be crucial to enhance the radiosensitivity of tumor cells. However, more evidence is needed to draw a general conclusion. Moreover, further studies are needed on the interaction between the effect of RS-induced innate immune response on tumor-cell radiosensitivity and radiation-induced antitumor immunity to achieve the optimal radiotherapy efficacy.
Conclusion
We have summarized the mechanisms of how RS response affects tumor radiosensitivity from the direct tumor side and indirect innate immune side and have further discussed potential targets and drugs to increase radiosensitization. We have reviewed several strategies including directly increasing RS, targeting RS response or RS-induced DDR, and other novel pathways. Although these strategies are predominantly based on preclinical evidence, they provide promising new ideas for enhancing radiosensitivity. As the relationship between RS and tumor radiosensitivity will be explored in the future, we expect these new strategies to bring substantial benefits to patients suffering from radioresistant malignancies.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81802287 to RZ), College Student Innovation and Entrepreneurship Training Program (No. DYLC2021076 and S202110487427 to LW).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Graphs were created with BioRender.com.
Abbreviations
RT, radiation therapy; RS, replication stress; DDR, DNA damage response; ATR, ataxia telangiectasia and rad3-related; CHK1, checkpoint kinase 1; ssDNA, single-stranded DNA; RPA, replication protein A; DSBs, double-strand breaks; UPR, unfolded protein response; HR, homologous recombination; T-LAK, T-lymphoid-activated killer; TOPK, T-LAK cell-derived protein kinase; Mcl-1, myeloid cell leukemia sequence 1; PARPs, poly (ADP-ribose) polymerases; IR, ionizing radiation; ATM, ataxia telangiectasia mutated; MRE11, meiotic recombination 11; MDM2, mouse double minute 2; POLQ, DNA polymerase theta; BRCA, breast cancer related protein; mTOR, mammalian target of the rapamycin; SUMO, small ubiquitin-like modifier; HIF, hypoxia inducible factor; RSF-1, spacing factor-1; NHEJ, non-homologous end joining; CDC, cell division cycle; SAC, spindle assembly checkpoint; APC/C, anaphase-promoting complex or cyclosome; PI3K, phosphoinositide 3-kinase; Bcl-2, B-cell lymphoma-2; Bax, Bcl-2-associated X protein; TAME, tosyl-L-arginine methyl ester; TOP3A, topoisomerase IIIα; RMI, RecQ-mediated genome instability; BTR, BLM-Topoisomerase IIIα-RMI1-RMI2; BLM, bloom syndrome helicase; MAC, MOS4-associated complex; MRN, MRE11/RAD50/NBS1; hTERT, human telomerase reverse transcriptase; RFWD3, RING finger and WD repeat domain 3; CHK1, checkpoint kinase 1; AKT, protein kinase B; mTOR, mammalian target of rapamycin; GBM, glioblastoma; USP9X, ubiquitin-specific protease 9X; KDM4C, lysine-specific demethylase 4C; TGF-β2, transforming growth factor-β2; UBE2O, ubiquitin-conjugating enzyme E2O; Mxi1, MAX interactor 1; SENP, SUMO-specific protease; 53BP1, p53-binding protein 1; Rnf4, Ring finger protein 4; MDC1, mediators of DNA damage checkpoint protein 1; ER, endoplasmic reticulum; SETX, senataxin; PKR, protein kinase R; PERK, PKR-like ER kinase; ATF4, activating transcription factor 4; PRRs, pattern recognition receptors; DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; IFNs, interferons; cGAMP, cyclic GMP-AMP; STING, stimulator of the interferon gene; cGAS, cGAMP synthase; IRF3, interferon regulatory factor 3; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cell; EVs, extracellular vesicles; TME, tumor microenvironment; PD-L1, programmed death ligand 1; PD-1, programmed cell death protein 1.
References
1. Buckley AM, Lynam-Lennon N, O'Neill H, O'Sullivan J. Targeting Hallmarks of Cancer to Enhance Radiosensitivity in Gastrointestinal Cancers. Nat Rev Gastroenterol Hepatol (2020) 17(5):298–313. doi: 10.1038/s41575-019-0247-2
2. Baskar R, Itahana K. Radiation Therapy and Cancer Control in Developing Countries: Can We Save More Lives? Int J Med Sci (2017) 14(1):13–7. doi: 10.7150/ijms.17288
3. O'Donovan A, Leech M, Gillham C. Assessment and Management of Radiotherapy Induced Toxicity in Older Patients. J Geriatr Oncol (2017) 8(6):421–7. doi: 10.1016/j.jgo.2017.07.001
4. Kim BM, Hong Y, Lee S, Liu P, Lim JH, Lee YH, et al. Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy. Int J Mol Sci (2015) 16(11):26880–913. doi: 10.3390/ijms161125991
5. Olivares-Urbano MA, Griñán-Lisón C, Marchal JA, Núñez MI. CSC Radioresistance: A Therapeutic Challenge to Improve Radiotherapy Effectiveness in Cancer. Cells (2020) 9(7):1651. doi: 10.3390/cells9071651
6. Gaillard H, García-Muse T, Aguilera A. Replication Stress and Cancer. Nat Rev Cancer (2015) 15(5):276–89. doi: 10.1038/nrc3916
7. Billard P, Poncet DA. Replication Stress at Telomeric and Mitochondrial DNA: Common Origins and Consequences on Ageing. Int J Mol Sci (2019) 20(19):4959. doi: 10.3390/ijms20194959
8. Irony-Tur Sinai M, Kerem B. DNA Replication Stress Drives Fragile Site Instability. Mutat Res (2018) 808:56–61. doi: 10.1016/j.mrfmmm.2017.10.002
9. Kitao H, Iimori M, Kataoka Y, Wakasa T, Tokunaga E, Saeki H, et al. DNA Replication Stress and Cancer Chemotherapy. Cancer Sci (2018) 109(2):264–71. doi: 10.1111/cas.13455
10. Macheret M, Halazonetis TD. DNA Replication Stress as a Hallmark of Cancer. Annu Rev Pathol (2015) 10:425–48. doi: 10.1146/annurev-pathol-012414-040424
11. Terradas M, Martín M, Genescà A. Detection of Impaired DNA Replication and Repair in Micronuclei as Indicators of Genomic Instability and Chromothripsis. Methods Mol Biol (Clifton NJ) (2018) 1769:197–208. doi:10.1007/978-1-4939-7780-2_13
12. Zhu H, Swami U, Preet R, Zhang J. Harnessing DNA Replication Stress for Novel Cancer Therapy. Genes (Basel) (2020) 11(9):990. doi: 10.3390/genes11090990
13. Baillie KE, Stirling PC. Beyond Kinases: Targeting Replication Stress Proteins in Cancer Therapy. Trends Cancer (2021) 7(5):430–46. doi: 10.1016/j.trecan.2020.10.010
14. Meyer F, Engel AM, Krause AK, Wagner T, Poole L, Dubrovska A, et al. Efficient DNA Repair Mitigates Replication Stress Resulting in Less Immunogenic Cytosolic DNA in Radioresistant Breast Cancer Stem Cells. Front Immunol (2022) 13:765284. doi: 10.3389/fimmu.2022.765284
15. Yang L, Shen C, Pettit CJ, Li T, Hu AJ, Miller ED, et al. Wee1 Kinase Inhibitor AZD1775 Effectively Sensitizes Esophageal Cancer to Radiotherapy. Clin Cancer Res (2020) 26(14):3740–50. doi: 10.1158/1078-0432.CCR-19-3373
16. Muñoz S, Méndez J. DNA Replication Stress: From Molecular Mechanisms to Human Disease. Chromosoma (2017) 126(1):1–15. doi: 10.1007/s00412-016-0573-x
17. Ngoi NYL, Pham MM, Tan DSP, Yap TA. Targeting the Replication Stress Response Through Synthetic Lethal Strategies in Cancer Medicine. Trends Cancer (2021) 7(10):930–57. doi: 10.1016/j.trecan.2021.06.002
18. Mazouzi A, Velimezi G, Loizou JI. DNA Replication Stress: Causes, Resolution and Disease. Exp Cell Res (2014) 329(1):85–93. doi: 10.1016/j.yexcr.2014.09.030
19. Ubhi T, Brown GW. Exploiting DNA Replication Stress for Cancer Treatment. Cancer Res (2019) 79(8):1730–9. doi: 10.1158/0008-5472.CAN-18-3631
20. Berti M, Vindigni A. Replication Stress: Getting Back on Track. Nat Struct Mol Biol (2016) 23(2):103–9. doi: 10.1038/nsmb.3163
21. Kotsantis P, Petermann E, Boulton SJ. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling Into Place. Cancer Discovery (2018) 8(5):537–55. doi: 10.1158/2159-8290.CD-17-1461
22. Bouleftour W, Rowinski E, Louati S, Sotton S, Wozny AS, Moreno-Acosta P, et al. A Review of the Role of Hypoxia in Radioresistance in Cancer Therapy. Med Sci Monit (2021) 27:e934116. doi: 10.12659/MSM.934116
23. Cao K, Chen Y, Zhao S, Huang Y, Liu T, Liu H, et al. Sirt3 Promoted DNA Damage Repair and Radioresistance Through ATM-Chk2 in Non-Small Cell Lung Cancer Cells. J Cancer (2021) 12(18):5464–72. doi: 10.7150/jca.53173
24. Chen K, Li L, Qu S, Pan X, Sun Y, Wan F, et al. Silencing hTERT Attenuates Cancer Stem Cell-Like Characteristics and Radioresistance in the Radioresistant Nasopharyngeal Carcinoma Cell Line CNE-2r. Aging (2020) 12(24):25599–613. doi: 10.18632/aging.104167
25. Linam J, Yang LX. Recent Developments in Radiosensitization. Anticancer Res (2015) 35(5):2479–85.
26. Bader SB, Dewhirst MW, Hammond EM. Cyclic Hypoxia: An Update on Its Characteristics, Methods to Measure It and Biological Implications in Cancer. Cancers (Basel) (2020) 13(1):23. doi: 10.3390/cancers13010023
27. Horsman MR, Overgaard J. The Impact of Hypoxia and its Modification of the Outcome of Radiotherapy. J Radiat Res (2016) 57)Suppl 1:i90–i8. doi: 10.1093/jrr/rrw007
28. Koukourakis MI, Giatromanolaki A, Sivridis E, Simopoulos C, Turley H, Talks K, et al. Hypoxia-Inducible Factor (HIF1A and HIF2A), Angiogenesis, and Chemoradiotherapy Outcome of Squamous Cell Head-and-Neck Cancer. Int J Radiat Oncol Biol Phys (2002) 53(5):1192–202. doi: 10.1016/S0360-3016(02)02848-1
29. Rockwell S, Dobrucki IT, Kim EY, Marrison ST, Vu VT. Hypoxia and Radiation Therapy: Past History, Ongoing Research, and Future Promise. Curr Mol Med (2009) 9(4):442–58. doi: 10.2174/156652409788167087
30. Johnson JL, Leos RA, Baker AF, Unger EC. Radiosensitization of Hs-766t Pancreatic Tumor Xenografts in Mice Dosed With Dodecafluoropentane Nano-Emulsion-Preliminary Findings. J Biomed Nanotechnol (2015) 11(2):274–81. doi: 10.1166/jbn.2015.1903
31. Druker J, Wilson JW, Child F, Shakir D, Fasanya T, Rocha S. Role of Hypoxia in the Control of the Cell Cycle. Int J Mol Sci (2021) 22(9):4874. doi: 10.3390/ijms22094874
32. Ramachandran S, Ma TS, Griffin J, Ng N, Foskolou IP, Hwang MS, et al. Hypoxia-Induced SETX Links Replication Stress With the Unfolded Protein Response. Nat Commun (2021) 12(1):3686. doi: 10.1038/s41467-021-24066-z
33. Bolland H, Ma TS, Ramlee S, Ramadan K, Hammond EM. Links Between the Unfolded Protein Response and the DNA Damage Response in Hypoxia: A Systematic Review. Biochem Soc Trans (2021) 49(3):1251–63. doi: 10.1042/BST20200861
34. Amoroso F, Glass K, Singh R, Liberal F, Steele RE, Maguire S, et al. Modulating the Unfolded Protein Response With ONC201 to Impact on Radiation Response in Prostate Cancer Cells. Sci Rep (2021) 11(1):4252. doi: 10.1038/s41598-021-83215-y
35. Kaczanowski S. Apoptosis: Its Origin, History, Maintenance and the Medical Implications for Cancer and Aging. Phys Biol (2016) 13(3):031001. doi: 10.1088/1478-3975/13/3/031001
36. Lowe SW, Cepero E, Evan G. Intrinsic Tumour Suppression. Nature (2004) 432(7015):307–15. doi: 10.1038/nature03098
37. Soliman AM, Khalil A, Ramadan E, Ghorab MM. Induction of Apoptosis, Cytotoxicity and Radiosensitization by Novel 3,4-Dihydroquinazolinone Derivatives. Bioorg Med Chem Lett (2021) 49:128308. doi: 10.1016/j.bmcl.2021.128308
38. Tian J, Kong E, Wang X, Xie Z, Chang CY, Sheu JJ, et al. RSF-1 siRNA Enhances Tumor Radiosensitivity in Cervical Cancer via Enhanced DNA Damage, Cell Cycle Redistribution, and Promotion of Apoptosis. Onco Targets Ther (2020) 13:3061–71. doi: 10.2147/OTT.S246632
39. Qian X, Tan C, Yang B, Wang F, Ge Y, Guan Z, et al. Astaxanthin Increases Radiosensitivity in Esophageal Squamous Cell Carcinoma Through Inducing Apoptosis and G2/M Arrest. Dis Esophagus (2017) 30(6):1–7. doi: 10.1093/dote/dox027
40. Lindemann A, Patel AA, Tang L, Tanaka N, Gleber-Netto FO, Bartels MD, et al. Combined Inhibition of Rad51 and Wee1 Enhances Cell Killing in HNSCC Through Induction of Apoptosis Associated With Excessive DNA Damage and Replication Stress. Mol Cancer Ther (2021) 20(7):1257–69. doi: 10.1158/1535-7163.MCT-20-0252
41. Hintelmann K, Berenz T, Kriegs M, Christiansen S, Gatzemeier F, Struve N, et al. Dual Inhibition of PARP and the Intra-S/G2 Cell Cycle Checkpoints Results in Highly Effective Radiosensitization of HPV-Positive HNSCC Cells. Front Oncol (2021) 11:683688. doi: 10.3389/fonc.2021.683688
42. De Marco Zompit M, Stucki M. Mechanisms of Genome Stability Maintenance During Cell Division. DNA Repair (Amst) (2021) 108:103215. doi: 10.1016/j.dnarep.2021.103215
43. Branigan TB, Kozono D, Schade AE, Deraska P, Rivas HG, Sambel L, et al. MMB-FOXM1-Driven Premature Mitosis is Required for CHK1 Inhibitor Sensitivity. Cell Rep (2021) 34(9):108808. doi: 10.1016/j.celrep.2021.108808
44. Dukaew N, Konishi T, Chairatvit K, Autsavapromporn N, Soonthornchareonnon N, Wongnoppavich A. Enhancement of Radiosensitivity by Eurycomalactone in Human NSCLC Cells Through G₂/M Cell Cycle Arrest and Delayed DNA Double-Strand Break Repair. Oncol Res (2020) 28(2):161–75. doi: 10.3727/096504019X15736439848765
45. Nikolakopoulou A, Soni A, Habibi M, Karaiskos P, Pantelias G, Terzoudi GI, et al. G2/M Checkpoint Abrogation With Selective Inhibitors Results in Increased Chromatid Breaks and Radiosensitization of 82-6 hTERT and RPE Human Cells. Front Public Health (2021) 9:675095. doi: 10.3389/fpubh.2021.675095
46. Esposito F, Giuffrida R, Raciti G, Puglisi C, Forte S. Wee1 Kinase: A Potential Target to Overcome Tumor Resistance to Therapy. Int J Mol Sci (2021) 22(19):10689. doi: 10.3390/ijms221910689
47. Chatterjee N, Walker GC. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ Mol Mutagen (2017) 58(5):235–63. doi: 10.1002/em.22087
48. Siqi Y, Yi Y, Qibin S, Sihao Z, Yanjun G, Bin W. DNA Damage Repair and Radiosensitivity of Tumor. Chin J Radiol Health (2020) 29(4):442–5. doi: 10.13491/j.issn.1004-714X.2020.04.030
49. Mohapatra D, Das B, Suresh V, Parida D, Minz AP, Nayak U, et al. Fluvastatin Sensitizes Pancreatic Cancer Cells Toward Radiation Therapy and Suppresses Radiation- and/or TGF-β-Induced Tumor-Associated Fibrosis. Lab Invest J Tech Methods Pathol (2022) 102(3):298–311. doi: 10.1038/s41374-021-00690-7
50. Xie Y, Liu C, Zhang Y, Li A, Sun C, Li R, et al. PKI-587 Enhances Radiosensitization of Hepatocellular Carcinoma by Inhibiting the PI3K/AKT/mTOR Pathways and DNA Damage Repair. PloS One (2021) 16(10):e0258817. doi: 10.1371/journal.pone.0258817
51. Dreyer SB, Upstill-Goddard R, Paulus-Hock V, Paris C, Lampraki EM, Dray E, et al. Targeting DNA Damage Response and Replication Stress in Pancreatic Cancer. Gastroenterology (2021) 160(1):362–77.e13. doi: 10.1053/j.gastro.2020.09.043
52. Lim N, Townsend PA. Cdc6 as a Novel Target in Cancer: Oncogenic Potential, Senescence and Subcellular Localisation. Int J Cancer (2020) 147(6):1528–34. doi: 10.1002/ijc.32900
53. Sun S, Zhang X, Xu M, Zhang F, Tian F, Cui J, et al. Berberine Downregulates CDC6 and Inhibits Proliferation via Targeting JAK-STAT3 Signaling in Keratinocytes. Cell Death Dis (2019) 10(4):274. doi: 10.1038/s41419-019-1510-8
54. Borlado LR, Méndez J. CDC6: From DNA Replication to Cell Cycle Checkpoints and Oncogenesis. Carcinogenesis (2008) 29(2):237–43. doi: 10.1093/carcin/bgm268
55. Zhang X, Zhang M, Guo Q, Hu X, Zhao Z, Ni L, et al. MicroRNA-1297 Inhibits Proliferation and Promotes Apoptosis in Gastric Cancer Cells by Downregulating CDC6 Expression. Anti Cancer Drugs (2019) 30(8):803–11. doi: 10.1097/CAD.0000000000000776
56. Yu X, Liu Y, Yin L, Peng Y, Peng Y, Gao Y, et al. Radiation-Promoted CDC6 Protein Stability Contributes to Radioresistance by Regulating Senescence and Epithelial to Mesenchymal Transition. Oncogene (2019) 38(4):549–63. doi: 10.1038/s41388-018-0460-4
57. Chen S, Chen X, Xie G, He Y, Yan D, Zheng D, et al. Cdc6 Contributes to Cisplatin-Resistance by Activation of ATR-Chk1 Pathway in Bladder Cancer Cells. Oncotarget (2016) 7(26):40362–76. doi: 10.18632/oncotarget.9616
58. Cocker JH, Piatti S, Santocanale C, Nasmyth K, Diffley JF. An Essential Role for the Cdc6 Protein in Forming the Pre-Replicative Complexes of Budding Yeast. Nature (1996) 379(6561):180–2. doi: 10.1038/379180a0
59. Herbert KJ, Ashton TM, Prevo R, Pirovano G, Higgins GS. T-LAK Cell-Originated Protein Kinase (TOPK): An Emerging Target for Cancer-Specific Therapeutics. Cell Death Dis (2018) 9(11):1089. doi: 10.1038/s41419-018-1131-7
60. Pirovano G, Ashton TM, Herbert KJ, Bryant RJ, Verrill CL, Cerundolo L, et al. TOPK Modulates Tumour-Specific Radiosensitivity and Correlates With Recurrence After Prostate Radiotherapy. Br J Cancer (2017) 117(4):503–12. doi: 10.1038/bjc.2017.197
61. Herbert KJ, Puliyadi R, Prevo R, Rodriguez-Berriguete G, Ryan A, Ramadan K, et al. Targeting TOPK Sensitises Tumour Cells to Radiation-Induced Damage by Enhancing Replication Stress. Cell Death Differ (2021) 28(4):1333–46. doi: 10.1038/s41418-020-00655-1
62. Piano V, Alex A, Stege P, Maffini S, Stoppiello GA, Huis In 't Veld PJ, et al. CDC20 Assists its Catalytic Incorporation in the Mitotic Checkpoint Complex. Science (2021) 371(6524):67–71. doi: 10.1126/science.abc1152
63. Zhang X, Zhang X, Li X, Bao H, Li G, Li N, et al. Connection Between CDC20 Expression and Hepatocellular Carcinoma Prognosis. Med Sci Monit Int Med J Exp Clin Res (2021) 27:e926760. doi: 10.12659/MSM.926760
64. Dai L, Song ZX, Wei DP, Zhang JD, Liang JQ, Wang BB, et al. CDC20 and PTTG1 are Important Biomarkers and Potential Therapeutic Targets for Metastatic Prostate Cancer. Adv Ther (2021) 38(6):2973–89. doi: 10.1007/s12325-021-01729-3
65. Zhao S, Zhang Y, Lu X, Ding H, Han B, Song X, et al. CDC20 Regulates the Cell Proliferation and Radiosensitivity of P53 Mutant HCC Cells Through the Bcl-2/Bax Pathway. Int J Biol Sci (2021) 17(13):3608–21. doi: 10.7150/ijbs.64003
66. Wang L, Yang C, Chu M, Wang ZW, Xue B. Cdc20 Induces the Radioresistance of Bladder Cancer Cells by Targeting FoxO1 Degradation. Cancer Lett (2021) 500:172–81. doi: 10.1016/j.canlet.2020.11.052
67. Gao Y, Wen P, Chen B, Hu G, Wu L, Xu A, et al. Downregulation of CDC20 Increases Radiosensitivity Through Mcl-1/P-Chk1-Mediated DNA Damage and Apoptosis in Tumor Cells. Int J Mol Sci (2020) 21(18):6692. doi: 10.3390/ijms21186692
68. Wang Z, Wan L, Zhong J, Inuzuka H, Liu P, Sarkar FH, et al. Cdc20: A Potential Novel Therapeutic Target for Cancer Treatment. Curr Pharm Design (2013) 19(18):3210–4. doi: 10.2174/1381612811319180005
69. Wang L, Zhang J, Wan L, Zhou X, Wang Z, Wei W. Targeting Cdc20 as a Novel Cancer Therapeutic Strategy. Pharmacol Ther (2015) 151:141–51. doi: 10.1016/j.pharmthera.2015.04.002
70. Sungwan P, Lert-Itthiporn W, Silsirivanit A, Klinhom-On N, Okada S, Wongkham S, et al. Bioinformatics Analysis Identified CDC20 as a Potential Drug Target for Cholangiocarcinoma. PeerJ (2021) 9:e11067. doi: 10.7717/peerj.11067
71. Song C, Lowe VJ, Lee S. Inhibition of Cdc20 Suppresses the Metastasis in Triple Negative Breast Cancer (TNBC). Breast Cancer (Tokyo Japan) (2021) 28(5):1073–86. doi: 10.1007/s12282-021-01242-z
72. Chen G, Magis AT, Xu K, Park D, Yu DS, Owonikoko TK, et al. Targeting Mcl-1 Enhances DNA Replication Stress Sensitivity to Cancer Therapy. J Clin Invest (2018) 128(1):500–16. doi: 10.1158/1538-7445.AM2018-328
73. Skvara H, Thallinger C, Wacheck V, Monia BP, Pehamberger H, Jansen B, et al. Mcl-1 Blocks Radiation-Induced Apoptosis and Inhibits Clonogenic Cell Death. Anticancer Res (2005) 25(4):2697–703. doi: 10.1086/316541
74. Tong Z, Mejia A, Veeranki O, Verma A, Correa AM, Dokey R, et al. Targeting CDK9 and MCL-1 by a New CDK9/p-TEFb Inhibitor With and Without 5-Fluorouracil in Esophageal Adenocarcinoma. Ther Adv Med Oncol (2019) 11:1758835919864850. doi: 10.1177/1758835919864850
75. Wei D, Zhang Q, Schreiber JS, Parsels LA, Abulwerdi FA, Kausar T, et al. Targeting Mcl-1 for Radiosensitization of Pancreatic Cancers. Trans Oncol (2015) 8(1):47–54. doi: 10.1016/j.tranon.2014.12.004
76. Slade D. PARP and PARG Inhibitors in Cancer Treatment. Genes Dev (2020) 34(5-6):360–94. doi: 10.1101/gad.334516.119
77. Ferrara R, Simionato F, Ciccarese C, Grego E, Cingarlini S, Iacovelli R, et al. The Development of PARP as a Successful Target for Cancer Therapy. Expert Rev Anticancer Ther (2018) 18(2):161–75. doi: 10.1080/14737140.2018.1419870
78. Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, Bartek J. High Speed of Fork Progression Induces DNA Replication Stress and Genomic Instability. Nature (2018) 559(7713):279–84. doi: 10.1038/s41586-018-0261-5
79. Wang F, Zhu S, Fisher LA, Wang L, Eurek NJ, Wahl JK 3rd, et al. Phosphatase 1 Nuclear Targeting Subunit Mediates Recruitment and Function of Poly (ADP-Ribose) Polymerase 1 in DNA Repair. Cancer Res (2019) 79(10):2526–35. doi: 10.1158/0008-5472.CAN-18-1673
80. Waissi W, Amé JC, Mura C, Noël G, Burckel H. Gemcitabine-Based Chemoradiotherapy Enhanced by a PARP Inhibitor in Pancreatic Cancer Cell Lines. Int J Mol Sci (2021) 22(13):6825. doi: 10.3390/ijms22136825
81. Jannetti SA, Zeglis BM, Zalutsky MR, Reiner T. Poly(ADP-Ribose)Polymerase (PARP) Inhibitors and Radiation Therapy. Front Pharmacol (2020) 11:170. doi: 10.3389/fphar.2020.00170
82. de Haan R, van Werkhoven E, van den Heuvel MM, Peulen HMU, Sonke GS, Elkhuizen P, et al. Study Protocols of Three Parallel Phase 1 Trials Combining Radical Radiotherapy With the PARP Inhibitor Olaparib. BMC Cancer (2019) 19(1):901. doi: 10.1186/s12885-019-6121-3
83. Mao Y, Huang X, Shuang Z, Lin G, Wang J, Duan F, et al. PARP Inhibitor Olaparib Sensitizes Cholangiocarcinoma Cells to Radiation. Cancer Med (2018) 7(4):1285–96. doi: 10.1002/cam4.1318
84. Maréchal A, Zou L. RPA-Coated Single-Stranded DNA as a Platform for Post-Translational Modifications in the DNA Damage Response. Cell Res (2015) 25(1):9–23. doi: 10.1038/cr.2014.147
85. Gao M, Guo G, Huang J, Hou X, Ham H, Kim W, et al. DOCK7 Protects Against Replication Stress by Promoting RPA Stability on Chromatin. Nucleic Acids Res (2021) 49(6):3322–37. doi: 10.1093/nar/gkab134
86. Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, et al. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell (2013) 155(5):1088–103. doi: 10.1016/j.cell.2013.10.043
87. Liu JS, Kuo SR, Melendy T. DNA Damage-Induced RPA Focalization is Independent of Gamma-H2AX and RPA Hyper-Phosphorylation. J Cell Biochem (2006) 99(5):1452–62. doi: 10.1002/jcb.21066
88. DeVeaux LC, Müller JA, Smith J, Petrisko J, Wells DP, DasSarma S. Extremely Radiation-Resistant Mutants of a Halophilic Archaeon With Increased Single-Stranded DNA-Binding Protein (RPA) Gene Expression. Radiat Res (2007) 168(4):507–14. doi: 10.1667/RR0935.1
89. Andrade MJ, Van Lonkhuyzen DR, Upton Z, Satyamoorthy K. RPA Facilitates Rescue of Keratinocytes From UVB Radiation Damage Through Insulin-Like Growth Factor-I Signalling. J Cell Sci (2021) 134(12):jcs255786. doi: 10.1242/jcs.255786
90. Pedersen H, Anne Adanma Obara E, Elbæk KJ, Vitting-Serup K, Hamerlik P. Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells. Int J Mol Sci (2020) 21(5):1588. doi: 10.3390/ijms21051588
91. Inano S, Sato K, Katsuki Y, Kobayashi W, Tanaka H, Nakajima K, et al. RFWD3-Mediated Ubiquitination Promotes Timely Removal of Both RPA and RAD51 From DNA Damage Sites to Facilitate Homologous Recombination. Mol Cell (2017) 66(5):622–34.e8. doi: 10.1016/j.molcel.2017.04.022
92. Bagge J, Oestergaard VH, Lisby M. Functions of TopBP1 in Preserving Genome Integrity During Mitosis. Semin Cell Dev Biol (2021) 113:57–64. doi: 10.1016/j.semcdb.2020.08.009
93. Frattini C, Promonet A, Alghoul E, Vidal-Eychenie S, Lamarque M, Blanchard MP, et al. TopBP1 Assembles Nuclear Condensates to Switch on ATR Signaling. Mol Cell (2021) 81(6):1231–45.e8. doi: 10.1016/j.molcel.2020.12.049
94. Wang J, Chen J, Gong Z. TopBP1 Controls BLM Protein Level to Maintain Genome Stability. Mol Cell (2013) 52(5):667–78. doi: 10.1016/j.molcel.2013.10.012
95. Liu K, Graves JD, Lee YJ, Lin FT, Lin WC. Cell Cycle-Dependent Switch of TopBP1 Functions by Cdk2 and Akt. Mol Cell Biol (2020) 40(8):e00599-19. doi: 10.1128/MCB.00599-19
96. Liu K, Graves JD, Lin FT, Lin WC. Overexpression of TopBP1, a Canonical ATR/Chk1 Activator, Paradoxically Hinders ATR/Chk1 Activation in Cancer. J Biol Chem (2021) 296:100382. doi: 10.1016/j.jbc.2021.100382
97. Menolfi D, Zha SATM. ATR and DNA-PKcs Kinases-the Lessons From the Mouse Models: Inhibition ≠ Deletion. Cell Biosci (2020) 10:8. doi: 10.1186/s13578-020-0376-x
98. Li F, Kozono D, Deraska P, Branigan T, Dunn C, Zheng XF, et al. CHK1 Inhibitor Blocks Phosphorylation of FAM122A and Promotes Replication Stress. Mol Cell (2020) 80(3):410–22.e6. doi: 10.1016/j.molcel.2020.10.008
99. Wang M, Liu G, Shan GP, Wang BB. In Vivo and In Vitro Effects of ATM/ATR Signaling Pathway on Proliferation, Apoptosis, and Radiosensitivity of Nasopharyngeal Carcinoma Cells. Cancer Biotherapy Radiopharmaceuticals (2017) 32(6):193–203. doi: 10.1089/cbr.2017.2212
100. Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL, et al. Radiosensitization by the ATR Inhibitor AZD6738 Through Generation of Acentric Micronuclei. Mol Cancer Ther (2017) 16(1):25–34. doi: 10.1158/1535-7163.MCT-16-0239
101. Šalovská B, Janečková H, Fabrik I, Karlíková R, Čecháková L, Ondrej M, et al. Radio-Sensitizing Effects of VE-821 and Beyond: Distinct Phosphoproteomic and Metabolomic Changes After ATR Inhibition in Irradiated MOLT-4 Cells. PloS One (2018) 13(7):e0199349. doi: 10.1371/journal.pone.0199349
102. Patties I, Kallendrusch S, Böhme L, Kendzia E, Oppermann H, Gaunitz F, et al. The Chk1 Inhibitor SAR-020106 Sensitizes Human Glioblastoma Cells to Irradiation, to Temozolomide, and to Decitabine Treatment. J Exp Clin Cancer Res (2019) 38(1):420. doi: 10.1186/s13046-019-1434-2
103. Lücking U, Wortmann L, Wengner AM, Lefranc J, Lienau P, Briem H, et al. Damage Incorporated: Discovery of the Potent, Highly Selective, Orally Available ATR Inhibitor BAY 1895344 With Favorable Pharmacokinetic Properties and Promising Efficacy in Monotherapy and in Combination Treatments in Preclinical Tumor Models. J medicinal Chem (2020) 63(13):7293–325. doi: 10.1021/acs.jmedchem.0c00369
104. Bonilla B, Hengel SR, Grundy MK, Bernstein KA. RAD51 Gene Family Structure and Function. Annu Rev Genet (2020) 54:25–46. doi: 10.1146/annurev-genet-021920-092410
105. Zeman MK, Cimprich KA. Causes and Consequences of Replication Stress. Nat Cell Biol (2014) 16(1):2–9. doi: 10.1038/ncb2897
106. Bhat KP, Cortez D. RPA and RAD51: Fork Reversal, Fork Protection, and Genome Stability. Nat Struct Mol Biol (2018) 25(6):446–53. doi: 10.1038/s41594-018-0075-z
107. Wang D, Zhang KF, Du G, Wang J, Zhao J. Berberine Enhances the Radiosensitivity of Osteosarcoma by Targeting Rad51 and Epithelial-Mesenchymal Transition. J Cancer Res Ther (2020) 16(2):215–21. doi: 10.4103/jcrt.JCRT_293_19
108. Liu G, Lim D, Cai Z, Ding W, Tian Z, Dong C, et al. The Valproate Mediates Radio-Bidirectional Regulation Through RFWD3-Dependent Ubiquitination on Rad51. Front Oncol (2021) 11:646256. doi: 10.3389/fonc.2021.646256
109. Traver G, Sekhar KR, Crooks PA, Keeney DS, Freeman ML. Targeting NPM1 in Irradiated Cells Inhibits NPM1 Binding to RAD51, RAD51 Foci Formation and Radiosensitizes NSCLC. Cancer Lett (2021) 500:220–7. doi: 10.1016/j.canlet.2020.12.023
110. Kaur E, Agrawal R, Sengupta S. Functions of BLM Helicase in Cells: Is It Acting Like a Double-Edged Sword? Front Genet (2021) 12:634789. doi: 10.3389/fgene.2021.634789
111. Hambarde S, Tsai CL, Pandita RK, Bacolla A, Maitra A, Charaka V, et al. EXO5-DNA Structure and BLM Interactions Direct DNA Resection Critical for ATR-Dependent Replication Restart. Mol Cell (2021) 81(14):2989–3006.e9. doi: 10.1016/j.molcel.2021.05.027
112. Ellis N, Zhu J, Yagle MK, Yang WC, Huang J, Kwako A, et al. RNF4 Regulates the BLM Helicase in Recovery From Replication Fork Collapse. Front Genet (2021) 12:753535. doi: 10.3389/fgene.2021.753535
113. Shorrocks AK, Jones SE, Tsukada K, Morrow CA, Belblidia Z, Shen J, et al. The Bloom Syndrome Complex Senses RPA-Coated Single-Stranded DNA to Restart Stalled Replication Forks. Nat Commun (2021) 12(1):585. doi: 10.1038/s41467-020-20818-5
114. Du X, Zhang C, Yin C, Wang W, Yan X, Xie D, et al. High BLM Expression Predicts Poor Clinical Outcome and Contributes to Malignant Progression in Human Cholangiocarcinoma. Front Oncol (2021) 11:633899. doi: 10.3389/fonc.2021.633899
115. Ruan Y, Xu H, Ji X, Zhao J. BLM Interaction With EZH2 Regulates MDM2 Expression and is a Poor Prognostic Biomarker for Prostate Cancer. Am J Cancer Res (2021) 11(4):1347–68.
116. Flanagan M, Cunniff CM. Bloom Syndrome. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Gripp KW, editors. GeneReviews® Seattle (WA): University of Washington (2006).
117. Rosenthal AS, Dexheimer TS, Nguyen G, Gileadi O, Vindigni A, Simeonov A, et al. Discovery of ML216, a Small Molecule Inhibitor of Bloom (BLM) Helicase. In: Probe Reports From the NIH Molecular Libraries Program. Bethesda (MD: National Center for Biotechnology Information (US (2010).
118. Wang L, Zhan L, Zhao Y, Huang Y, Wu C, Pan T, et al. The ATR-WEE1 Kinase Module Inhibits the MAC Complex to Regulate Replication Stress Response. Nucleic Acids Res (2021) 49(3):1411–25. doi: 10.1093/nar/gkaa1082
119. Busch CJ, Kröger MS, Jensen J, Kriegs M, Gatzemeier F, Petersen C, et al. G2-Checkpoint Targeting and Radiosensitization of HPV/p16-Positive HNSCC Cells Through the Inhibition of Chk1 and Wee1. Radiother Oncol (2017) 122(2):260–6. doi: 10.1016/j.radonc.2016.11.017
120. Cuneo KC, Morgan MA, Davis MA, Parcels LA, Parcels J, Karnak D, et al. Wee1 Kinase Inhibitor AZD1775 Radiosensitizes Hepatocellular Carcinoma Regardless of TP53 Mutational Status Through Induction of Replication Stress. Int J Radiat Oncol Biol Phys (2016) 95(2):782–90. doi: 10.1016/j.ijrobp.2016.01.028
121. Ma H, Lian R, Wu Z, Li X, Yu W, Shang Y, et al. MiR-503 Enhances the Radiosensitivity of Laryngeal Carcinoma Cells via the Inhibition of WEE1. Tumour Biol (2017) 39(10):1010428317706224. doi: 10.1177/1010428317706224
122. Wang Q, Chen Y, Lu H, Wang H, Feng H, Xu J, et al. Quercetin Radiosensitizes Non-Small Cell Lung Cancer Cells Through the Regulation of miR-16-5p/WEE1 Axis. IUBMB Life (2020) 72(5):1012–22. doi: 10.1002/iub.2242
123. Kong A, Mehanna H. WEE1 Inhibitor: Clinical Development. Curr Oncol Rep (2021) 23(9):107. doi: 10.1007/s11912-021-01098-8
124. Akter J, Katai Y, Sultana P, Takenobu H, Haruta M, Sugino RP, et al. Loss of P53 Suppresses Replication Stress-Induced DNA Damage in ATRX-Deficient Neuroblastoma. Oncogenesis (2021) 10(11):73. doi: 10.1038/s41389-021-00363-6
125. Benedict B, van Harn T, Dekker M, Hermsen S, Kucukosmanoglu A, Pieters W, et al. Loss of P53 Suppresses Replication-Stress-Induced DNA Breakage in G1/S Checkpoint Deficient Cells. Elife (2018) 7:e37868. doi: 10.7554/eLife.37868
126. Kataoka Y, Iimori M, Fujisawa R, Morikawa-Ichinose T, Niimi S, Wakasa T, et al. DNA Replication Stress Induced by Trifluridine Determines Tumor Cell Fate According to P53 Status. Mol Cancer Res (2020) 18(9):1354–66. doi: 10.1158/1541-7786.MCR-19-1051
127. Zhang J, Shen L, Sun LQ. The Regulation of Radiosensitivity by P53 and its Acetylation. Cancer Lett (2015) 363(2):108–18. doi: 10.1016/j.canlet.2015.04.015
128. Lamarche BJ, Orazio NI, Weitzman MD. The MRN Complex in Double-Strand Break Repair and Telomere Maintenance. FEBS Lett (2010) 584(17):3682–95. doi: 10.1016/j.febslet.2010.07.029
129. Oh J, Symington LS. Role of the Mre11 Complex in Preserving Genome Integrity. Genes (Basel) (2018) 9(12):589. doi: 10.3390/genes9120589
130. Syed A, Tainer JA. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu Rev Biochem (2018) 87:263–94. doi: 10.1146/annurev-biochem-062917-012415
131. Wang YY, Hung AC, Lo S, Hsieh YC, Yuan SF. MRE11 as a Molecular Signature and Therapeutic Target for Cancer Treatment With Radiotherapy. Cancer Lett (2021) 514:1–11. doi: 10.1016/j.canlet.2021.05.013
132. Fagan-Solis KD, Simpson DA, Kumar RJ, Martelotto LG, Mose LE, Rashid NU, et al. A P53-Independent DNA Damage Response Suppresses Oncogenic Proliferation and Genome Instability. Cell Rep (2020) 30(5):1385–99.e7. doi: 10.1016/j.celrep.2020.01.020
133. Joubert A, Zimmerman KM, Bencokova Z, Gastaldo J, Chavaudra N, Favaudon V, et al. DNA Double-Strand Break Repair Defects in Syndromes Associated With Acute Radiation Response: At Least Two Different Assays to Predict Intrinsic Radiosensitivity? Int J Radiat Biol (2008) 84(2):107–25. doi: 10.1080/09553000701797039
134. Sahlberg SH, Gustafsson AS, Pendekanti PN, Glimelius B, Stenerlöw B. The Influence of AKT Isoforms on Radiation Sensitivity and DNA Repair in Colon Cancer Cell Lines. Tumour Biol (2014) 35(4):3525–34. doi: 10.1007/s13277-013-1465-9
135. Dupré A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, et al. A Forward Chemical Genetic Screen Reveals an Inhibitor of the Mre11-Rad50-Nbs1 Complex. Nat Chem Biol (2008) 4(2):119–25. doi: 10.1038/nchembio.63
136. Shin SH, Yoon MJ, Kim M, Kim JI, Lee SJ, Lee YS, et al. Enhanced Lung Cancer Cell Killing by the Combination of Selenium and Ionizing Radiation. Oncol Rep (2007) 17(1):209–16. doi: 10.3892/or.17.1.209
137. Kuroda S, Fujiwara T, Shirakawa Y, Yamasaki Y, Yano S, Uno F, et al. Telomerase-Dependent Oncolytic Adenovirus Sensitizes Human Cancer Cells to Ionizing Radiation via Inhibition of DNA Repair Machinery. Cancer Res (2010) 70(22):9339–48. doi: 10.1158/0008-5472.CAN-10-2333
138. Shiloh Y, Ziv Y. The ATM Protein Kinase: Regulating the Cellular Response to Genotoxic Stress, and More. Nat Rev Mol Cell Biol (2013) 14(4):197–210. doi: 10.1038/nrm3546
139. Maréchal A, Zou L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harbor Perspect Biol (2013) 5(9):a012716. doi: 10.1101/cshperspect.a012716
140. Choi M, Kipps T, Kurzrock R. ATM Mutations in Cancer: Therapeutic Implications. Mol Cancer Ther (2016) 15(8):1781–91. doi: 10.1158/1535-7163.MCT-15-0945
141. Durant ST, Zheng L, Wang Y, Chen K, Zhang L, Zhang T, et al. The Brain-Penetrant Clinical ATM Inhibitor AZD1390 Radiosensitizes and Improves Survival of Preclinical Brain Tumor Models. Sci Adv (2018) 4(6):eaat1719. doi: 10.1126/sciadv.aat1719
142. Jie X, Fong WP, Zhou R, Zhao Y, Zhao Y, Meng R, et al. USP9X-Mediated KDM4C Deubiquitination Promotes Lung Cancer Radioresistance by Epigenetically Inducing TGF-β2 Transcription. Cell Death Differ (2021) 28(7):2095–111. doi: 10.1038/s41418-021-00740-z
143. Bian L, Meng Y, Zhang M, Guo Z, Liu F, Zhang W, et al. ATM Expression Is Elevated in Established Radiation-Resistant Breast Cancer Cells and Improves DNA Repair Efficiency. Int J Biol Sci (2020) 16(7):1096–106. doi: 10.7150/ijbs.41246
144. Ayars M, Eshleman J, Goggins M. Susceptibility of ATM-Deficient Pancreatic Cancer Cells to Radiation. Cell Cycle (Georgetown Tex) (2017) 16(10):991–8. doi: 10.1080/15384101.2017.1312236
145. Kim N, Kim SH, Kang SG, Moon JH, Cho J, Suh CO, et al. ATM Mutations Improve Radio-Sensitivity in Wild-Type Isocitrate Dehydrogenase-Associated High-Grade Glioma: Retrospective Analysis Using Next-Generation Sequencing Data. Radiat Oncol (London England) (2020) 15(1):184. doi: 10.1186/s13014-020-01619-y
146. Tang S, Li Z, Yang L, Shen L, Wang Y. A Potential New Role of ATM Inhibitor in Radiotherapy: Suppressing Ionizing Radiation-Activated EGFR. Int J Radiat Biol (2020) 96(4):461–8. doi: 10.1080/09553002.2020.1707325
147. Rainey MD, Charlton ME, Stanton RV, Kastan MB. Transient Inhibition of ATM Kinase is Sufficient to Enhance Cellular Sensitivity to Ionizing Radiation. Cancer Res (2008) 68(18):7466–74. doi: 10.1158/0008-5472.CAN-08-0763
148. Heylmann D, Badura J, Becker H, Fahrer J, Kaina B. Sensitivity of CD3/CD28-Stimulated Versus non-Stimulated Lymphocytes to Ionizing Radiation and Genotoxic Anticancer Drugs: Key Role of ATM in the Differential Radiation Response. Cell Death Dis (2018) 9(11):1053. doi: 10.1038/s41419-018-1095-7
149. Hammond EM, Muschel RJ. Radiation and ATM Inhibition: The Heart of the Matter. J Clin Invest (2014) 124(8):3289–91. doi: 10.1172/JCI77195
150. Riches LC, Trinidad AG, Hughes G, Jones GN, Hughes AM, Thomason AG, et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol Cancer Ther (2020) 19(1):13–25. doi: 10.1158/1535-7163.MCT-18-1394
151. Jucaite A, Stenkrona P, Cselényi Z, De Vita S, Buil-Bruna N, Varnäs K, et al. Brain Exposure of the ATM Inhibitor AZD1390 in Humans-a Positron Emission Tomography Study. Neuro Oncol (2021) 23(4):687–96. doi: 10.1093/neuonc/noaa238
152. Hou H, Sun D, Zhang X. The Role of MDM2 Amplification and Overexpression in Therapeutic Resistance of Malignant Tumors. Cancer Cell Int (2019) 19:216. doi: 10.1186/s12935-019-0937-4
153. Miles X, Vandevoorde C, Hunter A, Bolcaen J. Mdm2/X Inhibitors as Radiosensitizers for Glioblastoma Targeted Therapy. Front Oncol (2021) 11:703442. doi: 10.3389/fonc.2021.703442
154. Gudkov AV, Komarova EA. The Role of P53 in Determining Sensitivity to Radiotherapy. Nat Rev Cancer (2003) 3(2):117–29. doi: 10.1038/nrc992
155. Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of P53 Function Leads to Tumour Regression In Vivo. Nature (2007) 445(7128):661–5. doi: 10.1038/nature05541
156. Dowarha D, Chou RH, Yu C. S100A1 Blocks the Interaction Between P53 and Mdm2 and Decreases Cell Proliferation Activity. PloS One (2020) 15(6):e0234152. doi: 10.1371/journal.pone.0234152
157. Gao J, Xia R, Chen J, Gao J, Luo X, Ke C, et al. Inhibition of Esophageal-Carcinoma Cell Proliferation by Genistein via Suppression of JAK1/2-STAT3 and AKT/MDM2/p53 Signaling Pathways. Aging (2020) 12(7):6240–59. doi: 10.18632/aging.103019
158. Feng FY, Zhang Y, Kothari V, Evans JR, Jackson WC, Chen W, et al. MDM2 Inhibition Sensitizes Prostate Cancer Cells to Androgen Ablation and Radiotherapy in a P53-Dependent Manner. Neoplasia (2016) 18(4):213–22. doi: 10.1016/j.neo.2016.01.006
159. Yi H, Yan X, Luo Q, Yuan L, Li B, Pan W, et al. A Novel Small Molecule Inhibitor of MDM2-P53 (APG-115) Enhances Radiosensitivity of Gastric Adenocarcinoma. J Exp Clin Cancer Res (2018) 37(1):97. doi: 10.1186/s13046-018-0765-8
160. Pan Q, Wang L, Liu Y, Li M, Zhang Y, Peng W, et al. Knockdown of POLQ Interferes the Development and Progression of Hepatocellular Carcinoma Through Regulating Cell Proliferation, Apoptosis and Migration. Cancer Cell Int (2021) 21(1):482. doi: 10.1186/s12935-021-02178-2
161. Koole W, van Schendel R, Karambelas AE, van Heteren JT, Okihara KL, Tijsterman M. A Polymerase Theta-Dependent Repair Pathway Suppresses Extensive Genomic Instability at Endogenous G4 DNA Sites. Nat Commun (2014) 5:3216. doi: 10.1038/ncomms4216
162. Li J, Ko JM, Dai W, Yu VZ, Ng HY, Hoffmann JS, et al. Depletion of DNA Polymerase Theta Inhibits Tumor Growth and Promotes Genome Instability Through the cGAS-STING-ISG Pathway in Esophageal Squamous Cell Carcinoma. Cancers (Basel) (2021) 13(13):3204. doi: 10.3390/cancers13133204
163. Dai CH, Chen P, Li J, Lan T, Chen YC, Qian H, et al. Co-Inhibition of Pol θ and HR Genes Efficiently Synergize With Cisplatin to Suppress Cisplatin-Resistant Lung Cancer Cells Survival. Oncotarget (2016) 7(40):65157–70. doi: 10.18632/oncotarget.11214
164. Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, et al. Homologous-Recombination-Deficient Tumours are Dependent on Polθ-Mediated Repair. Nature (2015) 518(7538):258–62. doi: 10.1038/nature14184
165. Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A. Mammalian Polymerase θ Promotes Alternative NHEJ and Suppresses Recombination. Nature (2015) 518(7538):254–7. doi: 10.1038/nature14157
166. Zhou J, Gelot C, Pantelidou C, Li A, Yücel H, Davis RE, et al. A First-in-Class Polymerase Theta Inhibitor Selectively Targets Homologous-Recombination-Deficient Tumors. Nat Cancer (2021) 2(6):598–610. doi: 10.1038/s43018-021-00203-x
167. Lowery MA, Kelsen DP, Capanu M, Smith SC, Lee JW, Stadler ZK, et al. Phase II Trial of Veliparib in Patients With Previously Treated BRCA-Mutated Pancreas Ductal Adenocarcinoma. Eur J Cancer (2018) 89:19–26. doi: 10.1016/j.ejca.2017.11.004
168. Long DT, Joukov V, Budzowska M, Walter JC. BRCA1 Promotes Unloading of the CMG Helicase From a Stalled DNA Replication Fork. Mol Cell (2014) 56(1):174–85. doi: 10.1016/j.molcel.2014.08.012
169. Cong K, Peng M, Kousholt AN, Lee WTC, Lee S, Nayak S, et al. Replication Gaps are a Key Determinant of PARP Inhibitor Synthetic Lethality With BRCA Deficiency. Mol Cell (2021) 81(15):3128–44.e7. doi: 10.1016/j.molcel.2021.06.011
170. Yi M, Dong B, Qin S, Chu Q, Wu K, Luo S. Advances and Perspectives of PARP Inhibitors. Exp Hematol Oncol (2019) 8:29. doi: 10.1186/s40164-019-0154-9
171. Przetocka S, Porro A, Bolck HA, Walker C, Lezaja A, Trenner A, et al. CtIP-Mediated Fork Protection Synergizes With BRCA1 to Suppress Genomic Instability Upon DNA Replication Stress. Mol Cell (2018) 72(3):568–82.e6. doi: 10.1016/j.molcel.2018.09.014
172. Baert A, Depuydt J, Van Maerken T, Poppe B, Malfait F, Van Damme T, et al. Analysis of Chromosomal Radiosensitivity of Healthy BRCA2 Mutation Carriers and non-Carriers in BRCA Families With the G2 Micronucleus Assay. Oncol Rep (2017) 37(3):1379–86. doi: 10.3892/or.2017.5407
173. Baert A, Depuydt J, Van Maerken T, Poppe B, Malfait F, Storm K, et al. Increased Chromosomal Radiosensitivity in Asymptomatic Carriers of a Heterozygous BRCA1 Mutation. Breast Cancer Res BCR (2016) 18(1):52. doi: 10.1186/s13058-016-0709-1
174. Edwards E, Geng L, Tan J, Onishko H, Donnelly E, Hallahan DE. Phosphatidylinositol 3-Kinase/Akt Signaling in the Response of Vascular Endothelium to Ionizing Radiation. Cancer Res (2002) 62(16):4671–7. doi: 10.1002/cncr.10753
175. Schmidt-Ullrich RK, Contessa JN, Lammering G, Amorino G, Lin PS. ERBB Receptor Tyrosine Kinases and Cellular Radiation Responses. Oncogene (2003) 22(37):5855–65. doi: 10.1038/sj.onc.1206698
176. Huang TT, Brill E, Nair JR, Zhang X, Wilson KM, Chen L, et al. Targeting the PI3K/mTOR Pathway Augments CHK1 Inhibitor-Induced Replication Stress and Antitumor Activity in High-Grade Serous Ovarian Cancer. Cancer Res (2020) 80(23):5380–92. doi: 10.1158/0008-5472.CAN-20-1439
177. Ersahin T, Tuncbag N, Cetin-Atalay R. The PI3K/AKT/mTOR Interactive Pathway. Mol bioSystems (2015) 11(7):1946–54. doi: 10.1039/C5MB00101C
178. Sun R, Chen C, Deng X, Wang F, Song S, Cai Q, et al. IL-11 Mediates the Radioresistance of Cervical Cancer Cells via the PI3K/Akt Signaling Pathway. J Cancer (2021) 12(15):4638–47. doi: 10.7150/jca.56185
179. Yang X, Wang G, You J, Gu R, Xu X, Xu C, et al. High Expression of Cancer-IgG Is Associated With Poor Prognosis and Radioresistance via PI3K/AKT/DNA-PKcs Pathway Regulation in Lung Adenocarcinoma. Front Oncol (2021) 11:675397. doi: 10.3389/fonc.2021.675397
180. Mardanshahi A, Gharibkandi NA, Vaseghi S, Abedi SM, Molavipordanjani S. The PI3K/AKT/mTOR Signaling Pathway Inhibitors Enhance Radiosensitivity in Cancer Cell Lines. Mol Biol Rep (2021) 48(8):1–14. doi: 10.1007/s11033-021-06607-3
181. Chopra SS, Jenney A, Palmer A, Niepel M, Chung M, Mills C, et al. Torin2 Exploits Replication and Checkpoint Vulnerabilities to Cause Death of PI3K-Activated Triple-Negative Breast Cancer Cells. Cell Syst (2020) 10(1):66–81.e11. doi: 10.1016/j.cels.2019.11.001
182. Omeljaniuk WJ, Krętowski R, Ratajczak-Wrona W, Jabłońska E, Cechowska-Pasko M. Novel Dual PI3K/mTOR Inhibitor, Apitolisib (GDC-0980), Inhibits Growth and Induces Apoptosis in Human Glioblastoma Cells. Int J Mol Sci (2021) 22(21):11511. doi: 10.3390/ijms222111511
183. Lecona E, Rodriguez-Acebes S, Specks J, Lopez-Contreras AJ, Ruppen I, Murga M, et al. USP7 is a SUMO Deubiquitinase Essential for DNA Replication. Nat Struct Mol Biol (2016) 23(4):270–7. doi: 10.1038/nsmb.3185
184. Chang HM, Yeh ETH. SUMO: From Bench to Bedside. Physiol Rev (2020) 100(4):1599–619. doi: 10.1152/physrev.00025.2019
185. Galarreta A, Valledor P, Fernandez-Capetillo O, Lecona E. Coordinating DNA Replication and Mitosis Through Ubiquitin/SUMO and CDK1. Int J Mol Sci (2021) 22(16):8796. doi: 10.3390/ijms22168796
186. Deng L, Wu RA, Sonneville R, Kochenova OV, Labib K, Pellman D, et al. Mitotic CDK Promotes Replisome Disassembly, Fork Breakage, and Complex DNA Rearrangements. Mol Cell (2019) 73(5):915–29.e6. doi: 10.1016/j.molcel.2018.12.021
187. Huang Y, Yang X, Lu Y, Zhao Y, Meng R, Zhang S, et al. UBE2O Targets Mxi1 for Ubiquitination and Degradation to Promote Lung Cancer Progression and Radioresistance. Cell Death Differ (2021) 28(2):671–84. doi: 10.1038/s41418-020-00616-8
188. Hu C, Jiang X. The SUMO-Specific Protease Family Regulates Cancer Cell Radiosensitivity. BioMed Pharmacother (2019) 109:66–70. doi: 10.1016/j.biopha.2018.10.071
189. Lee D, Apelt K, Lee SO, Chan HR, Luijsterburg MS, Leung JWC, et al. ZMYM2 Restricts 53BP1 at DNA Double-Strand Breaks to Favor BRCA1 Loading and Homologous Recombination. Nucleic Acids Res (2022) 50(7):3922–43. doi: 10.1093/nar/gkac160
190. Vyas R, Kumar R, Clermont F, Helfricht A, Kalev P, Sotiropoulou P, et al. RNF4 is Required for DNA Double-Strand Break Repair In Vivo. Cell Death Differ (2013) 20(3):490–502. doi: 10.1038/cdd.2012.145
191. Hetz C, Zhang K, Kaufman RJ. Mechanisms, Regulation and Functions of the Unfolded Protein Response. Nat Rev Mol Cell Biol (2020) 21(8):421–38. doi: 10.1038/s41580-020-0250-z
192. Koumenis C, Wouters BG. "Translating" Tumor Hypoxia: Unfolded Protein Response (UPR)-Dependent and UPR-Independent Pathways. Mol Cancer Res (2006) 4(7):423–36. doi: 10.1158/1541-7786.MCR-06-0150
193. Madden E, Logue SE, Healy SJ, Manie S, Samali A. The Role of the Unfolded Protein Response in Cancer Progression: From Oncogenesis to Chemoresistance. Biol Cell (2019) 111(1):1–17. doi: 10.1111/boc.201800050
194. Lai C, Zhang J, Tan Z, Shen LF, Zhou RR, Zhang YY. Maf1 Suppression of ATF5-Dependent Mitochondrial Unfolded Protein Response Contributes to Rapamycin-Induced Radio-Sensitivity in Lung Cancer Cell Line A549. Aging (2021) 13(5):7300–13. doi: 10.18632/aging.202584
195. Carty M, Guy C, Bowie AG. Detection of Viral Infections by Innate Immunity. Biochem Pharmacol (2021) 183:114316. doi: 10.1016/j.bcp.2020.114316
196. Li D, Wu M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct Target Ther (2021) 6(1):291. doi: 10.1038/s41392-021-00687-0
197. Ragu S, Matos-Rodrigues G, Lopez BS. Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes (Basel) (2020) 11(4):409. doi: 10.3390/genes11040409
198. Ivashkiv LB, Donlin LT. Regulation of Type I Interferon Responses. Nat Rev Immunol (2014) 14(1):36–49. doi: 10.1038/nri3581
199. Zhu H, Tang YD, Zhan G, Su C, Zheng C. The Critical Role of PARPs in Regulating Innate Immune Responses. Front Immunol (2021) 12:712556. doi: 10.3389/fimmu.2021.712556
200. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an Innate Immune Sensor for Intracellular DNA. Nat Immunol (2010) 11(11):997–1004. doi: 10.1038/ni.1932
201. Ye Z, Shi Y, Lees-Miller SP, Tainer JA. Function and Molecular Mechanism of the DNA Damage Response in Immunity and Cancer Immunotherapy. Front Immunol (2021) 12:797880. doi: 10.3389/fimmu.2021.797880
202. Lamberti MJ, Mentucci FM, Roselli E, Araya P, Rivarola VA, Rumie Vittar NB, et al. Photodynamic Modulation of Type 1 Interferon Pathway on Melanoma Cells Promotes Dendritic Cell Activation. Front Immunol (2019) 10:2614. doi: 10.3389/fimmu.2019.02614
203. Craig DJ, Nanavaty NS, Devanaboyina M, Stanbery L, Hamouda D, Edelman G, et al. The Abscopal Effect of Radiation Therapy. Future Oncol (2021) 17(13):1683–94. doi: 10.2217/fon-2020-0994
204. Gessani S, Conti L, Del CornГІ M, Belardelli F. Type I Interferons as Regulators of Human Antigen Presenting Cell Functions. Toxins (2014) 6(6):1696–723. doi: 10.3390/toxins6061696
205. Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I Interferon Response and Innate Immune Sensing of Cancer. Trends Immunol (2013) 34(2):67–73. doi: 10.1016/j.it.2012.10.004
206. Wang L, Yang L, Wang C, Zhao W, Ju Z, Zhang W, et al. Inhibition of the ATM/Chk2 Axis Promotes cGAS/STING Signaling in ARID1A-Deficient Tumors. J Clin Invest (2020) 130(11):5951–66. doi: 10.1172/JCI130445
207. Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, et al. Exosomes Maintain Cellular Homeostasis by Excreting Harmful DNA From Cells. Nat Commun (2017) 8:15287. doi: 10.1038/ncomms15287
208. Ma J, Zhang H, Tang K, Huang B. Tumor-Derived Microparticles in Tumor Immunology and Immunotherapy. Eur J Immunol (2020) 50(11):1653–62. doi: 10.1002/eji.202048548
209. Zhao F, Zheng T, Gong W, Wu J, Xie H, Li W, et al. Extracellular Vesicles Package dsDNA to Aggravate Crohn's Disease by Activating the STING Pathway. Cell Death Dis (2021) 12(9):815. doi: 10.1038/s41419-021-04101-z
210. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity (2014) 41(5):843–52. doi: 10.1016/j.immuni.2014.10.019
211. Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, et al. Nuclear cGAS Suppresses DNA Repair and Promotes Tumorigenesis. Nature (2018) 563(7729):131–6. doi: 10.1038/s41586-018-0629-6
212. Chen H, Chen H, Zhang J, Wang Y, Simoneau A, Yang H, et al. cGAS Suppresses Genomic Instability as a Decelerator of Replication Forks. Sci Adv (2020) 6(42):eabb8941. doi: 10.1126/sciadv.abb8941
213. Schaue D, Micewicz ED, Ratikan JA, Xie MW, Cheng G, McBride WH. Radiation and Inflammation. Semin Radiat Oncol (2015) 25(1):4–10. doi: 10.1016/j.semradonc.2014.07.007
214. Diegeler S, Hellweg CE. Intercellular Communication of Tumor Cells and Immune Cells After Exposure to Different Ionizing Radiation Qualities. Front Immunol (2017) 8:664. doi: 10.3389/fimmu.2017.00664
215. Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-Dependent MDSC Mobilization Drives Extrinsic Radiation Resistance. Nat Commun (2017) 8(1):1736. doi: 10.1038/s41467-017-01566-5
216. Du SS, Chen GW, Yang P, Chen YX, Hu Y, Zhao QQ, et al. Radiation Therapy Promotes Hepatocellular Carcinoma Immune Cloaking via PD-L1 Upregulation Induced by cGAS-STING Activation. Int J Radiat Oncol Biol Phys (2022) 112(5):1243–55. doi: 10.1016/j.ijrobp.2021.12.162
217. Gerber DE, Urbanic JJ, Langer C, Hu C, Chang IF, Lu B, et al. Treatment Design and Rationale for a Randomized Trial of Cisplatin and Etoposide Plus Thoracic Radiotherapy Followed by Nivolumab or Placebo for Locally Advanced Non-Small-Cell Lung Cancer (RTOG 3505). Clin Lung Cancer (2017) 18(3):333–9. doi: 10.1016/j.cllc.2016.10.009
218. Bozorgmehr F, Hommertgen A, Krisam J, Lasitschka F, Kuon J, Maenz M, et al. Fostering Efficacy of Anti-PD-1-Treatment: Nivolumab Plus Radiotherapy in Advanced non-Small Cell Lung Cancer - Study Protocol of the FORCE Trial. BMC Cancer (2019) 19(1):1074. doi: 10.1186/s12885-019-6205-0
219. Tuyaerts S, Van Nuffel AMT, Naert E, Van Dam PA, Vuylsteke P, De CaluwГ© A, et al. PRIMMO Study Protocol: A Phase II Study Combining PD-1 Blockade, Radiation and Immunomodulation to Tackle Cervical and Uterine Cancer. BMC Cancer (2019) 19(1):506. doi: 10.1186/s12885-019-5676-3
220. Johansson H, Andersson R, Bauden M, Hammes S, Holdenrieder S, Ansari D. Immune Checkpoint Therapy for Pancreatic Cancer. World J Gastroenterol (2016) 22(43):9457–76. doi: 10.3748/wjg.v22.i43.9457
221. Shinde-Jadhav S, Mansure JJ, Rayes RF, Marcq G, Ayoub M, Skowronski R, et al. Role of Neutrophil Extracellular Traps in Radiation Resistance of Invasive Bladder Cancer. Nat Commun (2021) 12(1):2776. doi: 10.1038/s41467-021-23086-z
222. Kho VM, Mekers VE, Span PN, Bussink J, Adema GJ. Radiotherapy and cGAS/STING Signaling: Impact on MDSCs in the Tumor Microenvironment. Cell Immunol (2021) 362:104298. doi: 10.1016/j.cellimm.2021.104298
223. Bhattacharya S, Srinivasan K, Abdisalaam S, Su F, Raj P, Dozmorov I, et al. RAD51 Interconnects Between DNA Replication, DNA Repair and Immunity. Nucleic Acids Res (2017) 45(8):4590–605. doi: 10.1093/nar/gkx126
224. Dillon MT, Bergerhoff KF, Pedersen M, Whittock H, Crespo-Rodriguez E, Patin EC, et al. ATR Inhibition Potentiates the Radiation-Induced Inflammatory Tumor Microenvironment. Clin Cancer Res (2019) 25(11):3392–403. doi: 10.1158/1078-0432.CCR-18-1821
225. Zhang N, Gao Y, Zeng Z, Luo Y, Jiang X, Zhang J, et al. PARP Inhibitor Niraparib as a Radiosensitizer Promotes Antitumor Immunity of Radiotherapy in EGFR-Mutated non-Small Cell Lung Cancer. Clin Trans Oncol (2021) 23(9):1827–37. doi: 10.1007/s12094-021-02591-z
226. Wang B, Sun L, Yuan Z, Tao Z. Wee1 Kinase Inhibitor AZD1775 Potentiates CD8+Вђ‰T Cell-Dependent Antitumour Activity via Dendritic Cell Activation Following a Single High Dose of Irradiation. Med Oncol (Northwood London England) (2020) 37(8):66. doi: 10.1007/s12032-020-01390-w
Keywords: replication stress, DNA damage repair, radiation therapy, radioresistance, radiosensitizer
Citation: Zhang Y, Wu L, Wang Z, Wang J, Roychoudhury S, Tomasik B, Wu G, Wang G, Rao X and Zhou R (2022) Replication Stress: A Review of Novel Targets to Enhance Radiosensitivity-From Bench to Clinic. Front. Oncol. 12:838637. doi: 10.3389/fonc.2022.838637
Received: 18 December 2021; Accepted: 15 June 2022;
Published: 08 July 2022.
Edited by:
Qiang Zhang, University of Michigan, United StatesReviewed by:
Vijay Menon, Yale University, United StatesWeiwei Wang, First Affiliated Hospital of Zhengzhou University, China
Copyright © 2022 Zhang, Wu, Wang, Wang, Roychoudhury, Tomasik, Wu, Wang, Rao and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rui Zhou, bWltaXJ1aXJ1aTJAMTYzLmNvbQ==; Xinrui Rao, cmFveGlucnVpQGh1c3QuZWR1LmNu