
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 19 May 2022
Sec. Genitourinary Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.837127
This article is part of the Research TopicAdvances in Prostate Cancer: Model Systems, Molecular and Cellular Mechanisms, Early Detection, and TherapiesView all 21 articles
 Christopher J. McNally1
Christopher J. McNally1 Joanne Watt2Mary Jo Kurth2John V. Lamont2
Joanne Watt2Mary Jo Kurth2John V. Lamont2 Tara Moore1Peter Fitzgerald2Hardev Pandha3,4
Tara Moore1Peter Fitzgerald2Hardev Pandha3,4 Declan J. McKenna1†
Declan J. McKenna1† Mark W. Ruddock2*†
Mark W. Ruddock2*†Background: Almost 50,000 men in the United Kingdom (UK) are diagnosed each year with prostate cancer (PCa). Secondary referrals for investigations rely on serum prostate-specific antigen (PSA) levels and digital rectal examination. However, both tests lack sensitivity and specificity, resulting in unnecessary referrals to secondary care for costly and invasive biopsies.
Materials and Methods: Serum samples and clinical information were collected from N = 125 age-matched patients (n = 61 non-PCa and n = 64 PCa) and analyzed using Biochip Array Technology on high-sensitivity cytokine array I (IL-2, IL-4, IL-6, IL-8, IL-10, IL-1α, IL-1β, TNFα, MCP-1, INFγ, EGF, and VEGF), cerebral array II (CRP, D-dimer, neuron-specific enolase, and sTNFR1), and tumor PSA oncology array (fPSA, tPSA, and CEA).
Results: The data showed that 11/19 (68.8%) markers were significantly different between the non-PCa and the PCa patients. A combination of EGF, log10 IL-8, log10 MCP-1, and log10 tPSA significantly improved the predictive potential of tPSA alone to identify patients with PCa (DeLong, p < 0.001). This marker combination had an increased area under the receiver operator characteristic (0.860 vs. 0.700), sensitivity (78.7 vs. 68.9%), specificity (76.5 vs. 67.2%), PPV (76.2 vs. 66.7%), and NPV (79.0 vs. 69.4%) compared with tPSA.
Conclusions: The novel combination of serum markers identified in this study could be employed to help triage patients into “low-” and “high-risk” categories, allowing general practitioners to improve the management of patients in primary care settings and potentially reducing the number of referrals for unnecessary, invasive, and costly treatments.
Prostate cancer (PCa) is very common, with almost 50,000 men diagnosed each year in the UK (1) and 240,000 in the US (2). Annually, PCa kills almost 35,000 men in the US (2). Tumors of the prostate are likely to be localized, clinically unapparent, and with International Society of Urological Pathology (ISUP) grade grouping 1 (3, 4). Slow-growing, non-significant PCa may not cause serious harm (5) and often does not require any intervention. However, clinically significant prostate cancers require urgent treatment, as they have the potential to metastasize and cause a serious disease.
Patients with PCa are usually asymptomatic, and the presenting symptoms are not specific and are often observed in men with benign prostate enlargement (BPE), one of the most frequently reported age-related diseases in men over 60 years. The symptoms include painful or burning sensation during urination, frequent urination (particularly at night—nocturia), difficulty stopping and starting urination, sudden erectile dysfunction, blood in the urine (hematuria) or semen, bone pain, and weight loss.
The risk factors for PCa include patient age (>50 years), ethnicity (African-American ethnicity and other minority ethnicities have a greater risk of progression and are more likely to develop aggressive cancer than Caucasian men), obesity (patients who are obese have a higher risk of PCa), and family history (blood relative, e.g., parent) (6). The complications of PCa and subsequent treatment include metastatic spread of the disease, urinary incontinence, and erectile dysfunction (7).
The gold standard for diagnosing PCa is histological assessment of prostate tissue obtained by transrectal ultrasound-guided systematic (TRUS) core needle biopsy. The most common scale used to evaluate the grade of PCa is the Gleason score (8). The higher the Gleason score, the more likely that the cancer will grow and spread quickly (9).
Screening patients for PCa remains controversial and is not recommended due to the potential for overtreatment (10). Data presented by the Surveillance, Epidemiological, and End Results registry have estimated that screening for PCa, using prostate-specific antigen (PSA) alone, resulted in an increase of 28% of patients being over-diagnosed in the US (11). Furthermore, the European Randomised Study of Screening for Prostate Cancer trial also estimated that, when PSA is used alone as a screening tool for PCa, almost 50% of patients were over-diagnosed (12).
Although advances in PCa management have been made, an elevated PSA and/or abnormal digital rectal examination (DRE; nodular, indurated, and/or asymmetry) would still normally warrant a referral for investigation (13). An abnormal DRE is the second most common finding that initiates further investigation for malignancy (14–16). As a result, many patients with elevated PSA/abnormal DRE are referred to secondary care for invasive and costly procedures (17). These are often unnecessary as almost 75% of patients who are referred for further investigation have a negative biopsy (18). In addition, some 2.5 to 3% of patients are admitted to a hospital within a week of their TRUS procedure with a serious infection (urinary tract infections and/or bacterial prostatitis). This could be avoided with better decision-making in primary care but requires more biological information on the patient’s disease to be available to their GP.
Currently, no biomarker or biomarker combinations that have the sensitivity and specificity to replace PSA have been identified (19). Therefore, improved approaches are required to differentiate between men who have a prostate disease that require treatment or surveillance and those who do not. The symptoms and PSA results are not an accurate indicator of disease. Indeed no level of PSA is truly diagnostic (20)—for example, a patient could have a PSA >10 ng/ml and not have any cancer, whereas another patient with a PSA <1 ng/ml could have aggressive cancer. Therefore, there is an urgent need for new tests which can at least stratify patients and, if possible, be diagnostic. However, it is very unlikely, given the heterogeneous nature of PCa, that a single biomarker will prove to be diagnostic.
The effective management of PCa requires an accurate diagnosis. However, the challenge for the clinician is to differentiate benign conditions (BPE) from PCa, which presents with similar symptoms. The PSA test exhibits a negative benefit-to-harm ratio based on population estimates (12). Therefore, biomarkers that would contribute to the sensitivity and specificity of PSA could offer the clinician additional information so that a more informed management decision could be made on whether to refer a patient to secondary care for further investigations or to manage the patient in primary care.
The aim of the study was to investigate the levels of serum markers in patients who present to primary care with PCa-like symptoms so as to identify markers that could be used to improve the triage of patients into low- and high-risk categories, thereby enhancing patient management.
One hundred twenty-five patients were included in the study. The patient cohort consisted of two independent patient sample sets.
The first set of patients (N = 33; n = 10 non-PCa and n = 23 PCa) were recruited by Royal Surrey County Hospital (NHS Foundation Trust) between 2015 and 2018 (Diagnosis of Clinically Significant Prostate Cancer; Royal Surrey County Hospital, Research Development and Innovations Department, The Royal Surrey County Hospital, Leggett Building, Daphne Jackson Road, Guildford, Surrey GU2 7WG, 15/LO/0218). The inclusion criteria included (i) men >18 years referred by their GP to investigate the cause of (ii) an abnormal PSA test. The exclusion criteria included (i) an active urine infection, confirmed by urine dipstick testing or midstream urine microscopy, (ii) men with a PSA <4 and >20 ng/ml, (iii) men already diagnosed with PCa, (iv) men with a prior or concurrent malignancy (apart from basal cell carcinoma of the skin), and (v) men who cannot give informed consent (Supplementary 1). Blood (24 ml) and urine (20–30 ml) were collected after a prostatic examination, along with a detailed clinical history. The study complied with the Declaration of Helsinki, and written informed consent was obtained from all participants.
The second patient cohort (N = 92; n = 54 non-PCa and n = 38 PCa) was obtained from Discovery Life Sciences (DLS; CA, USA). The patient samples were de-identified and publicly available and were thus exempt from the requirement of the Institutional Review Board (IRB) approval (exempt category 4, IRB/EC). However, the DLS samples were procured pursuant to informed consent provided by the individual under approved protocols 45 CFR 46.116. Serum (1 ml) with clinical history was obtained for each DLS patient. The samples were selected from treatment-naive patients based on ICD-10 codes for prostate-related conditions.
Prostate cancer was confirmed by a histological examination of prostate biopsies from both sample sets. The Gleason scores assigned by the pathologists are described in Table 1. The non-PCa group included patients with confirmed benign prostatic hyperplasia (BPH; n = 30/61, 49.2%). All patients were treatment-naïve at the time of prostate biopsy.
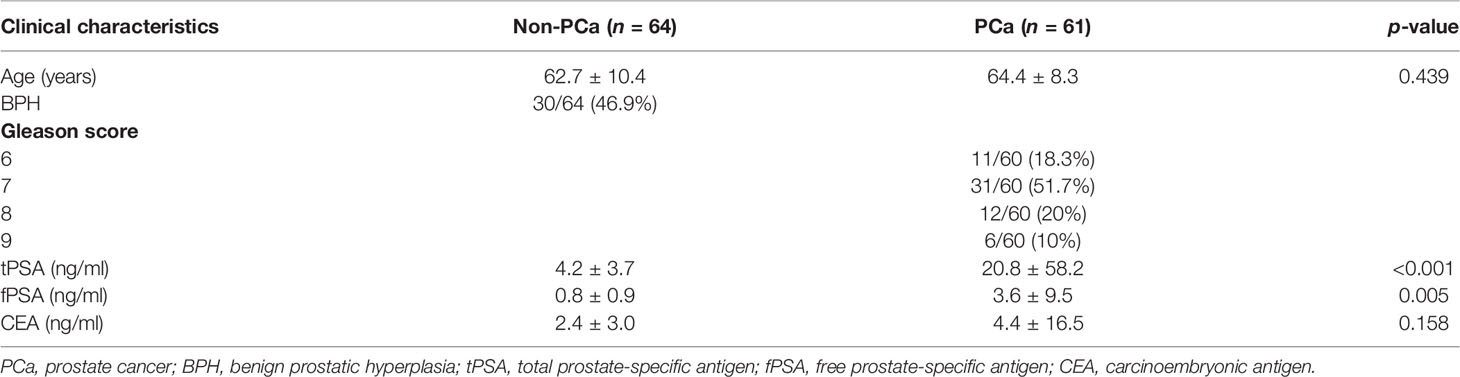
Table 1 Clinical and pathological characteristics of the patients. Data shown as mean ± SD or n/total (%), Wilcoxon rank-sum test; p < 0.05 was considered significant.
Both patient cohorts were combined (N = 125) and separated into two groups, depending on the pathology reports: non-PCa (n = 64/125, 51.2%) and PCa (n = 61/125, 48.8%).
Clinical factors were not available for all patients. However, where data was available, the most common presenting symptoms included the following: lower urinary tract symptoms (LUTS), urine retention, urgency, nocturia, lower back pain, and microscopic hematuria. For many of the patients, there was no previous history of benign disease prior to their PCa diagnosis.
Smoking history and details on alcohol consumption (units/week) were also available for a limited number of patients. Many PCa patients were former smokers. Where data was available, the number of cigarettes smoked per day ranged from 10 to 25. Pack-year data was not available. The alcohol consumption ranged from 1 to 48 units/week (where data was available).
Medications were also noted for a limited number of patients; where data was available, the most common drugs that the patients were prescribed with included sertraline, loratadine, omeprazole, aspirin, tamsulosin, simvastatin, losartan, atorvastatin, imvastatin, bendroflumethiazide, citalopram, sildenafil, fluoxetine, ranitidine, metformin, and bisoprolol.
Patient blood and urine samples were stored in duplicate at -80°C prior to analysis by Randox Laboratory Clinical Services, Antrim, UK, by scientists blinded to the patients’ data. In total, 19 biomarkers were investigated by Biochip Array Technology (BAT) (Randox Laboratories Ltd., Crumlin, UK) (21) using the Evidence Investigator analyzer (Randox Laboratories Ltd., Crumlin, UK) and following the manufacturer’s instructions. The limits of detection (LOD) for the markers on the biochip arrays were as follows: EGF, 2.5 pg/ml; IFNγ, 2.1 pg/ml; IL-1α 0.9, pg/ml; IL-1β, 1.3 pg/ml; IL-2, 4.9 pg/ml; IL-4, 3.5 pg/ml; IL-6, 0.4 pg/ml; IL-8, 2.3 pg/ml; IL-10, 1.1 pg/ml; MCP-1, 25.5 pg/ml; TNFα, 3.7 pg/ml; VEGF, 10.8 pg/ml; CRP, 0.67 mg/L; D-dimer, 2.1 ng/ml; neuron-specific enolase (NSE), 0.26 ng/ml; sTNFR1, 0.24 ng/ml; CEA, 0.29 ng/ml; fPSA, 0.02 ng/ml; and tPSA, 0.045 ng/ml. The biomarkers below the LOD were recorded as 90% of the LOD (22).
Statistical analyses were undertaken using R version 4.0.5 (23). Wilcoxon rank-sum test was used to identify differentially expressed markers. Markers with p <0.05 were considered significant. The ability of the markers to predict PCa was further investigated using logistic LASSO regression following a cross-validation testing of several models. For marker and marker combinations, areas under the receiver operator characteristic (AUROC) (and 95% CI), sensitivity (and 95% CI), specificity (and 95% CI), positive predictive value (PPV), and negative predictive value (NPV) were calculated to identify models that differentiated between the two diagnostic groups (non PCa vs. PCa). DeLong test was used to compare AUROCs for the model and tPSA; p <0.05 was considered significant.
The clinical and pathological characteristics of the patients involved in the study are described in Table 1. Both tPSA and fPSA were significantly elevated in the PCa group. However, CEA was not significantly different.
From the marker results obtained using the biochip arrays, 11/16 (68.8%) markers were significantly different between the non-PCa and the PCa patient groups (Table 2). Of these, 7/16 (43.8%) markers were elevated in the PCa patients vs. non-PCa, 4/16 (25%) were lower in the PCa vs. non-PCa, and 5/16 (31.2%) were not significantly different between either group.
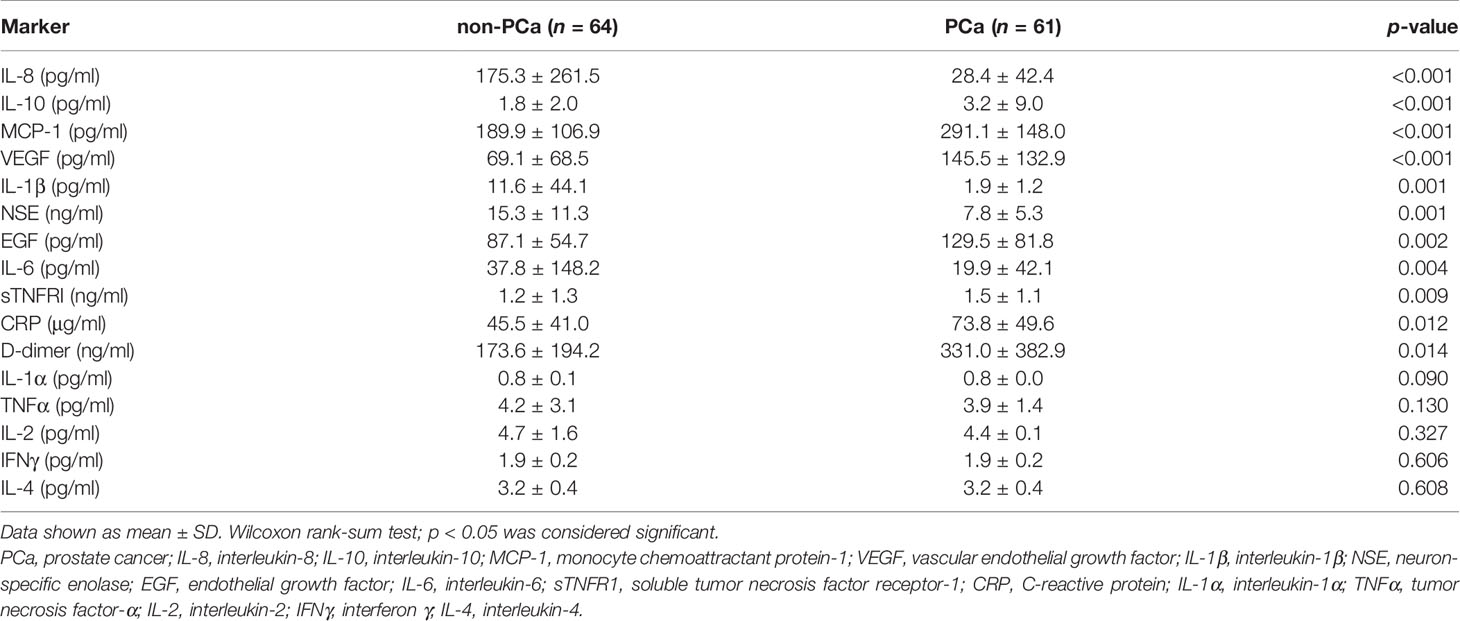
Table 2 The analysis showed that 11/16 (68.8%) serum markers were significantly different between the non-PCa and the PCa patient groups.
Logistic LASSO regression identified a model for a combination of markers that demonstrated higher sensitivity and specificity vs. tPSA alone (Table 3). The four markers selected by LASSO regression to identify patients with PCa included EGF, IL-8, MCP-1, and tPSA (Figure 1A). As some of the data was not normally distributed, log10 transformation was applied to IL-8, MCP-1, and tPSA in the model.
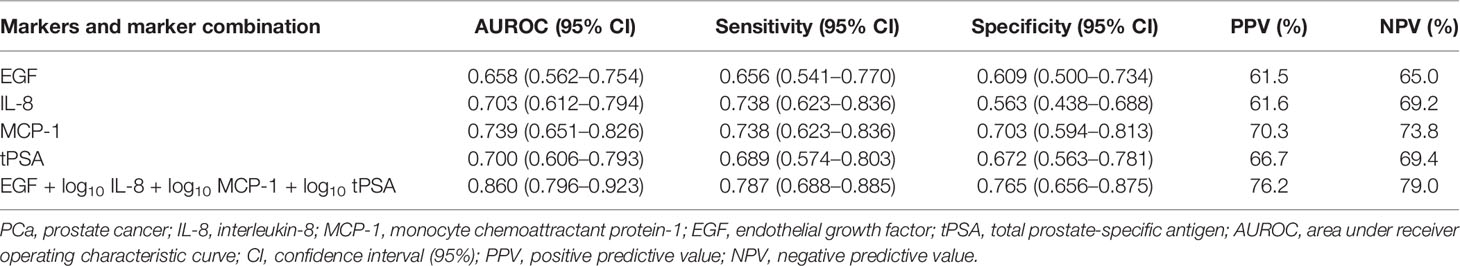
Table 3 Individual analytes and model EGF, IL-8, MCP-1, and tPSA AUROC, sensitivity, specificity, PPV, and NPV for non-PCa vs. PCa.
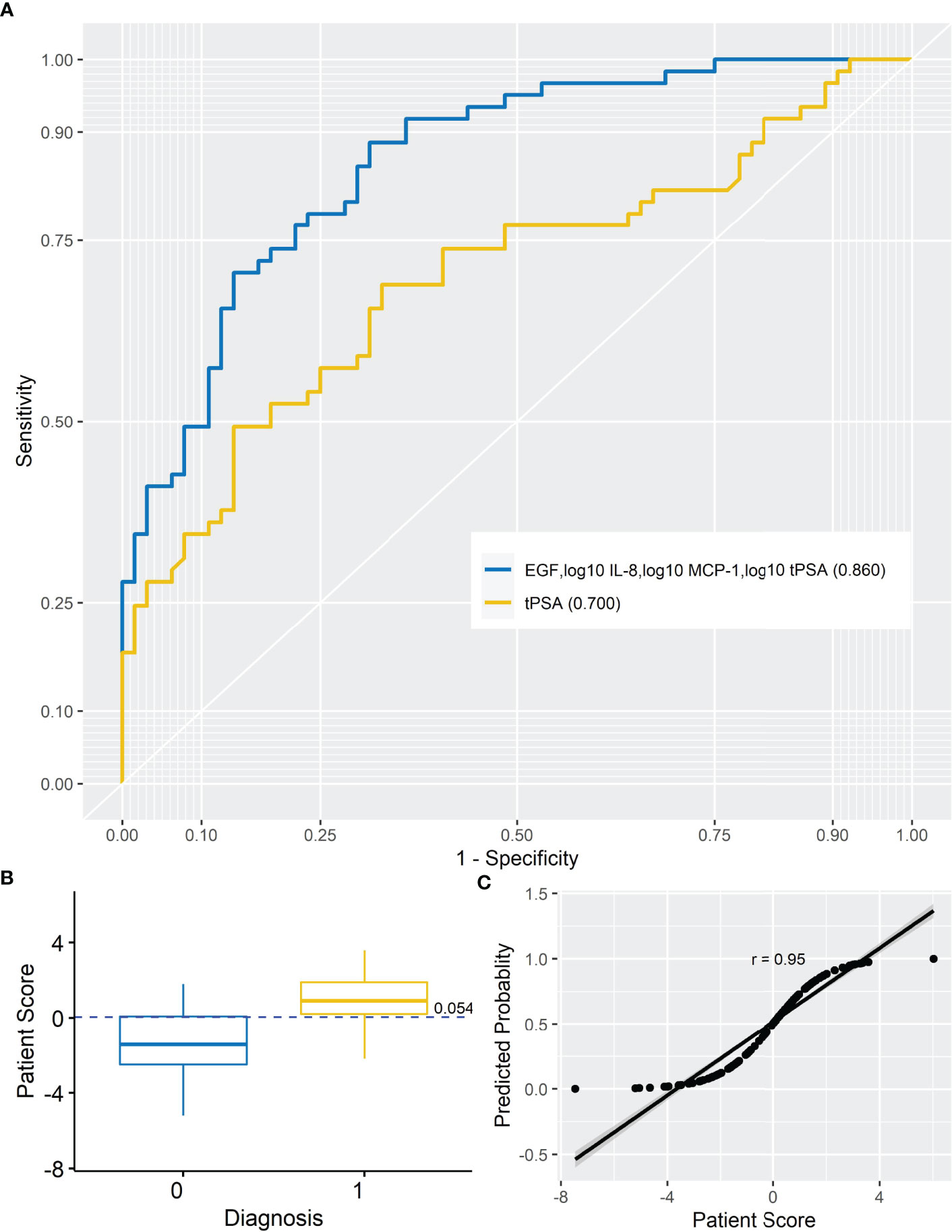
Figure 1 Prostate cancer model. (A) AUROC for analyte model (AUROC, 0.860) and tPSA (AUROC, 0.700). When the AUROC for the model (EGF, log10 IL-8, log10 MCP-1, and log10 tPSA) was compared with the AUROC for tPSA, the model significantly improved upon tPSA alone (DeLong, p < 0.001) at differentiating non-PCa from PCa patients. (B) Simple box plot of patient score by diagnosis [non-PCa (0) and PCa (1); mean ± SD] for the model at a cutoff of 0.054. (C) Simple scatter with fit line for predicted probability by patient score for the marker model (r = 0.95). AUROC, area under receiver operating characteristic; IL-8, interleukin-8; MCP-1, monocyte chemoattractant protein-1; EGF, endothelial growth factor; tPSA, total prostate-specific antigen.
When comparing the new model identified by LASSO to tPSA on its own, the number of false positives was reduced from 21/64 (32.8%) to 15/64 (23.4%), and the number of false negatives increased from 11/61 (18.0%) to 13/61 (21.3%) (Table 4).
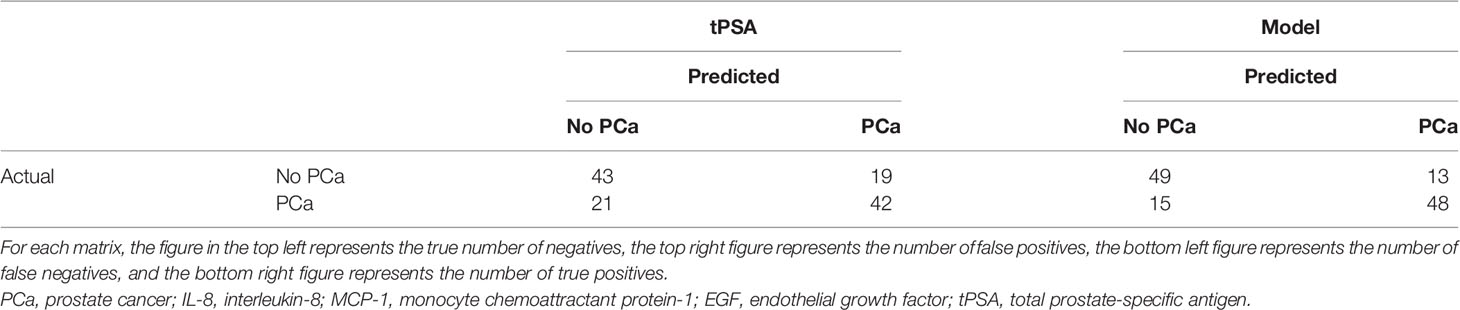
Table 4 Confusion matrices comparing tPSA and the model EGF, IL-8, MCP-1, and tPSA.
The risk of PCa was based on the following marker combination: EGF, log10 IL-8, log10 MCP-1, and log10 tPSA. In this dataset, a cutoff of 0.054 (as shown in Figure 1B) was applied to achieve the highest sensitivity and specificity for identifying patients with PCa; PRS <0.054—patients are negative for PCa, whereas PRS ≥0.054—patients would be positive for PCa. It should be noted that the PRS would be used in combination with clinical risk factors when triaging patients. Thus, patients with a positive risk score and positive clinical risk factors (e.g., painful or burning sensation during urination, frequent urination, difficulty starting or stopping urination, sudden erectile dysfunction, and blood in urine or semen) could be prioritized for urgent referral for further investigations. Patients who were positive for clinical risk factors and negative for marker risk (PRS) could potentially be managed in primary care or referred for investigation as necessary. Importantly, this type of combined measurement approach is recommended for risk stratification methods by the National Institute for Health and Care Excellence (NICE 2019) guidelines for PCa.
To test the linearity of the model, predicted probability was plotted against patient score (Figure 1C). The high correlation between the predicted probability and patient score (r = 0.95) would suggest confidence in the model.
In this study, we investigated 19 serum markers involved in PCa (Supplementary 2). The results showed 11/16 (68.8%) cytokines that were significantly different between the non-PCa vs. PCa groups. Seven of these markers were elevated in the PCa group, whereas 4 markers were elevated in the non-PCa group. In the PCa group, 2/3 (66.6%) cancer markers (fPSA and tPSA) were also elevated.
The serum levels of IL-10, EGF, VEGF, MCP-1, sTNFR1, CRP, and D-dimer were significantly higher in the PCa patients. Of these serum markers, MCP-1 had the highest AUROC for detecting PCa (MCP-1 0.739 vs. tPSA 0.700). MCP-1 (CCL2) is a member of the chemokine family that acts as a paracrine and autocrine factor to promote PCa growth and invasion (24). MCP-1 is also a potent chemotactic factor regulating stromal–epithelial cells in PCa (25). Unsurprisingly, the angiogenic factors VEGF and EGF were also elevated in patients with PCa. However, in other studies, VEGF has been shown to have no significant prognostic or predictive value for expression for localized or advanced PCa (26). In contrast, EGF modulates PCa invasiveness by regulating the urokinase-type plasminogen activity (27). Inhibition of the EGF receptor may prevent tumor cell dissemination (28).
CRP is a general marker for inflammation, although it does not differentiate benign from malignant disease (29). However, IL-10, which was also elevated in our PCa patients, has anti-inflammatory and anti-angiogenic properties (30). Therefore, it was unsurprising that both markers were elevated in the PCa patients.
The thrombotic factor D-dimer has been detected in patients with PCa. However, the relationship between PCa and the coagulation disorder remains unknown (31). Nonetheless, high plasma levels of D-dimer are associated with an increased risk of PCa mortality (32). Similarly, sTNFR1 has been identified in men with PCa. Furthermore, sTNFR1 has been shown to be a potential biomarker for identifying PCa when compared with PSA alone (AUROC 0.97) (33). However, as this was a small study, the authors acknowledged that the results need to be assessed in a much larger patient cohort. In our study, sTNFR1 had an AUROC of 0.635 for PCa.
Prostate cancer is an inflammatory disease; however, we found that 4/11 (36.4%) inflammatory markers (IL-8, IL-1β, NSE, and IL-6 levels) were significantly lower in the PCa patients.
The circulating IL-8 serum levels have not been shown to be a significant predictor of diagnosis, aggressiveness, or prognosis for PCa (34). However, increased circulating IL-8 serum levels have been detected in patients with an underlying inflammatory disease (34). In our study, IL-8 was identified as a marker that could differentiate non-PCa from PCa, potentially by identifying patients with inflammatory disease, i.e., BPH. In addition, IL-1β is elevated in patients with chronic prostatitis, chronic pelvic pain syndrome, and BPH (35, 36). Furthermore, elevated IL-6 has also been reported in men with BPH, LUTS, and erectile disfunction (37). Therefore, it was not surprising that these three markers were elevated in the non-PCa patient group; almost 50% of non-PCa patients had a diagnosis of BPH.
Higher levels of NSE have been observed in non-PCa patients (38), albeit higher levels of NSE have also been observed in patients with metastatic disease (39). In our study, 42/64 (65.6%) PCa patients had a Gleason score ≤7; no information was available on metastatic disease, and there was no significant difference in the NSE levels by Gleason score (data not shown). However, elevated serum NSE has been suggested to correlate with prognosis in advanced PCa (38). The PCa patients in our study were treatment-naïve, and only 6/64 (9.4%) patients presented with a Gleason score ≥9.
In our study, the serum levels of IL-1α, TNFα, IL-2, IL-4, and IFNγ were not significantly different between the non-PCa and the PCa groups.
The results demonstrated that no single marker significantly outperformed tPSA. However, a combination of EGF, log10 IL-8, log10 MCP-1, and log10 tPSA significantly improved the predictive potential of tPSA alone to identify patients with PCa. This marker combination had an increased AUROC (0.860 vs. 0.700), sensitivity (78.7 vs. 68.9%), specificity (76.5 vs. 67.2%), PPV (76.2 vs. 66.7%), and NPV (79.0 vs. 69.4%) compared with tPSA.
Using this marker combination in this patient dataset reduced the number of false positives from 21/64 (32.8%) to 15/64 (23.4%); however, the number of false negatives increased from 11/61 (18.0%) to 13/61 (21.3%) compared with tPSA. Thus, an additional 9.4% (6/64) of patients were correctly assigned as non-PCa using the marker combination. If the management of these patients was based solely on their tPSA results, n = 7 patients could have potentially undergone unnecessary and invasive investigations. An additional 3.3% (2/61) of patients were incorrectly assigned as PCa.
Evidence suggests that the use of multiple markers to differentiate non-clinically significant from clinically significant disease is an important strategy for reducing unnecessary referrals for further investigation (40). Integrating inflammatory serum biomarkers into a risk calculator may provide additional information for detecting and managing PCa risk (40). The predictive value of inflammatory markers for PCa diagnosis has been evaluated in primary care (41). Our data demonstrate the value of measuring multiple markers in this heterogenous pathophysiology in combination with tPSA. The main limitations of this feasibility study included the following: (1) the small number of participants in each patient cohort and (2) the limited patient information [demographics, behaviors, medications, socioeconomic data, and clinicopathological data (e.g., DRE)]. Nevertheless, these results warrant further investigation in a larger cohort, and this will help validate the model.
It is worth noting that other combination models are being investigated elsewhere, including the Stockholm-3 risk-based model (42), the 4kscore (43), the European Randomised Study of Screening for Prostate Cancer risk calculator (44), the Prostate Cancer Prevention Trial (45), and the Irish Prostate Cancer Risk Calculator (46). The work described in this study is therefore an important addition to the global research effort to identify combinations of biological and clinical measurements to inform evidence-based decision-making in PCa patients.
This study demonstrated that a novel serum marker combination of EGF, log10 IL-8, log10 MCP-1, and log10 tPSA significantly improved the predictive potential of tPSA alone to identify patients with PCa. Application of this serum marker combination could provide clinicians with valuable information to help triage their patients into low- and high-risk categories. Improved risk category stratification of patients would enable better management of men who present at primary care with prostate-cancer-like symptoms. In turn, the utilization of this novel combination of markers could potentially reduce the number of patients that are referred to secondary care for unnecessary, costly, and invasive procedures. However, it should be noted that this is a preliminary study and the markers identified would need to be validated in a larger patient cohort.
The raw data supporting the conclusions of this article will be made available by the authors without undue reservation.
The studies involving human participants were reviewed and approved by cohort (1): Ethics was granted by the NHS, Health Research Authority, NRES Committee South East Coast—Brighton and Sussex, Health Research Authority, Skipton House, 80 London Road, London SE1 6LH, UK (REC Reference 15/LO/0218). The second patient cohort was obtained from Discovery Life Sciences (DLS), California, USA. The patient samples are de-identified and publicly available and are thus exempt from the requirement of the Institutional Review Board Ethics Committee (IRB/EC) approval (Exempt Category 4, IRB/EC). All DLS biospecimens are collected under IRB/EC-approved protocols. The samples are procured pursuant to the informed consent provided by the individual, following the general guidelines for informed consent found in the Code of Federal Regulations 45 CFR 46.116. The patients/participants provided their written informed consent to participate in this study.
The authors confirm contribution to the paper as follows: study conception and design: CM, MJK, JL, TM, PF, DM, and MR; data collection: CM, JW, HP, DM, and MR; analysis and interpretation of results: CM, JW, MJK, JL, PF, HP, DM, and MR; and draft manuscript preparation: CM, JW, MJK, TM, HP, DM, and MR. All authors contributed to the article and approved the submitted version.
The authors declare that this study received funding from Randox Laboratories Ltd as part of the Randox Laboratories Ltd – Ulster University PhD Academy Studentship. Randox had the following involvement in the study: analysis of patient samples, statistical analysis, supervision of the project, preparation of the manuscript, and the decision to publish.
JW, MJK, JL, and MR are employees of Randox Laboratories Ltd. but hold no shares in the company. PF is the managing director and owner of Randox Laboratories Ltd. A patent has been filed to protect the biomarker combination disclosed in the manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.837127/full#supplementary-material
1. Cancer Research UK. Prostate Cancer Statistics . Available at: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/prostate-cancer.
2. Cancer.Net. Prostate Cancer: Statistics (2019). Available at: https://www.cancer.net/cancer-types/prostate-cancer/statistics.
3. Popiolek M, Rider JR, Andrén O, Andersson SO, Holmberg L, Adami HO, et al. Natural History of Early, Localized Prostate Cancer: A Final Report From Three Decades of Follow-Up. Eur Urol (2013) 63(3):428–35. doi: 10.1016/j.eururo.2012.10.002
4. Montironi R, Santoni M, Mazzucchelli R, Burattini L, Berardi R, Galosi AB, et al. Prostate Cancer: From Gleason Scoring to Prognostic Grade Grouping. Expert Rev Anticancer Ther (2016) 16(4):433–40. doi: 10.1586/14737140.2016.1160780
5. Prostate Cancer - Symptoms and Causes - Mayo Clinic . Available at: https://www.mayoclinic.org/diseases-conditions/prostate-cancer/symptoms-causes/syc-20353087 (Accessed June 10, 2021).
6. Hamilton W, Sharp DJ, Peters TJ, Round AP. Clinical Features of Prostate Cancer Before Diagnosis: A Population-Based, Case-Control Study. Br J Gen Pract (2006) 56(531):756–62.
7. Simoneau AR. Treatment- and Disease-Related Complications of Prostate Cancer. Rev Urol (2006) 8 Suppl 2(Suppl 2):S56–67.
8. Rubin MA, Dunn R, Kambham N, Misick CP, O’Toole KM. Should a Gleason Score be Assigned to a Minute Focus of Carcinoma on Prostate Biopsy? Am J Surg Pathol (2000) 24(12):1634–40. doi: 10.1097/00000478-200012000-00007
9. Epstein JI. Prostate Cancer Grading: A Decade After the 2005 Modified System. Mod Pathol (2018) 31(S1):47–63. doi: 10.1038/modpathol.2017.133
10. Stark JR, Mucci L, Rothman KJ, Adami HO. Screening for Prostate Cancer Remains Controversial. BMJ (2009) b3601: 339. doi: 10.1136/bmj.b3601
11. Etzioni R, Gulati R, Cooperberg M, Penson D, Weiss N, Thompson I. Limitations of Basing Screening Policies on Screening Trials: The US Preventive Services Task Force and Prostate Cancer Screening. Med Care (2013) 51(4):295–300. doi: 10.1097/MLR.0b013e31827da979
12. Alberts AR, Schoots IG, Roobol MJ. Prostate-Specific Antigen-Based Prostate Cancer Screening: Past and Future. Int J Urol (2015) 22(6):524–32. doi: 10.1111/iju.12750
13. Palmerola R, Smith P, Elliot V, Reese CT, Mahon FB, Harpster LE, et al. The Digital Rectal Examination (DRE) Remains Important-Outcomes From a Contemporary Cohort of Men Undergoing an Initial 12-18 Core Prostate Needle Biopsy. Can J Urol (2012) 19(6):6542–7.
14. Izawa JI, Babaian RJ. Prostate Cancer: Detection and Biopsy Strategies. Prostate Cancer Sci Clin Pract (2003) 2003:129–36. doi: 10.1016/B978-012286981-5/50016-1
15. Galosi AB, Palagonia E, Scarcella S, Cimadamore A, Lacetera V, Delle Fave RF, et al. Detection Limits of Significant Prostate Cancer Using Multiparametric MR and Digital Rectal Examination in Men With Low Serum PSA: Up-Date of the Italian Society of Integrated Diagnostic in Urology. Arch Ital Di Urol E Androl (2021) 93(1):92–100. doi: 10.4081/aiua.2021.1.92
16. Fandella A, Scattoni V, Galosi A, Pepe P, Fiorentino M, Gaudiano C, et al. Italian Prostate Biopsies Group: 2016 Updated Guidelines Insights. Anticancer Res (2017) 37(2):413–24. doi: 10.21873/ANTICANRES.11333
17. Young SM, Bansal P, Vella ET, Finelli A, Levitt C, Loblaw A. Guideline for Referral of Patients With Suspected Prostate Cancer by Family Physicians and Other Primary Care Providers. Can Fam Physician (2015) 61(1):33–9.
18. Prostate Cancer Diagnosis and Management Guidance NICE . Available at: https://www.nice.org.uk/guidance/ng131/chapter/Recommendations#assessment-and-diagnosis.
19. Mcnally CJ, Ruddock MW, Moore T, Mckenna DJ. Biomarkers That Differentiate Benign Prostatic Hyperplasia From Prostate Cancer: A Literature Review. Cancer Manag Res (2020) 12:5225–41. doi: 10.2147/CMAR.S250829
20. Duffy MJ. Biomarkers for Prostate Cancer: Prostate-Specific Antigen and Beyond. Clin Chem Lab Med (2020) 58(3):326–39. doi: 10.1515/cclm-2019-0693
21. FitzGerald SP, Lamont JV, McConnell RI, Benchikh EO. Development of a High-Throughput Automated Analyzer Using Biochip Array Technology. Clin Chem (2005) 51(7):1165–76. doi: 10.1373/clinchem.2005.049429
22. Kurth MJ, McBride WT, McLean G, Watt MJ, Domanska A, Lamont JV, et al. Acute Kidney Injury Risk in Orthopaedic Trauma Patients Pre and Post Surgery Using a Biomarker Algorithm and Clinical Risk Score. Sci Rep (2020) 10(1):20005–5. doi: 10.1038/s41598-020-76929-y
24. Lu Y, Cai Z, Galson DL, Xiao G, Liu Y, George DE, et al. Monocyte Chemotactic Protein-1 (MCP-1) Acts as a Paracrine and Autocrine Factor for Prostate Cancer Growth and Invasion. Prostate (2006) 66(12):1311–8. doi: 10.1002/pros.20464
25. Loberg RD, Day LL. Lauren N St. MCP-1 is a Potent Regulator of Prostate Cancer Cell Chemoaxataxis and Growth. Via Stimulation CCR2 CCRL1 Cancer Res (2006) 66(8 Supplement):578–86. doi: 10.1593/neo.06280
26. Pan L, Baek S, Edmonds PR, Roach M, Wolkov H, Shah S, et al. Vascular Endothelial Growth Factor (VEGF) Expression in Locally Advanced Prostate Cancer: Secondary Analysis of Radiation Therapy Oncology Group (RTOG) 8610. Radiat Oncol (2013) 8(1):1–11. doi: 10.1186/1748-717X-8-100
27. Festuccia C, Angelucci A, Gravina GL, Biordi L, Millimaggi D, Muzi P, et al. Epidermal Growth Factor Modulates Prostate Cancer Cell Invasiveness Regulating Urokinase-Type Plasminogen Activator Activity. EGF-receptor Inhibition May Prevent Tumor Cell Dissemination Thromb Haemost (2005) 93(5):964–75. doi: 10.1160/TH04-09-0637
28. Sigismund S, Avanzato D, Lanzetti L. Emerging Functions of the EGFR in Cancer. Mol Oncol (2018) 12(1):3–20. doi: 10.1002/1878-0261.12155
29. Santotoribio JD, Jimenez-Romero ME. Serum Biomarkers of Inflammation for Diagnosis of Prostate Cancer in Patients With Nonspecific Elevations of Serum Prostate Specific Antigen Levels. Transl Cancer Res (2019) 8(1):273–8. doi: 10.21037/tcr.2019.01.31
30. Shao N, Xu B, Mi YY, Hua LX. IL-10 Polymorphisms and Prostate Cancer Risk: A Meta-Analysis. Prostate Cancer Prostatic Dis (2011) 14(2):129–35. doi: 10.1038/pcan.2011.6
31. Çalışkan S, Sungur M. Fibrinogen and D-Dimer Levels in Prostate Cancer: Preliminary Results. Prostate Int (2017) 5(3):110–2. doi: 10.1016/j.prnil.2017.05.001
32. Ay C, Dunkler D, Pirker R, Thaler J, Quehenberger P, Wagner O, et al. High D-Dimer Levels are Associated With Poor Prognosis in Cancer Patients. Haematologica (2012) 97(8):1158–64. doi: 10.3324/haematol.2011.054718
33. Chadha K, Miller A, Nair B, Schwartz S, Trump D, Underwood W. New Serum Biomarkers for Prostate Cancer Diagnosis. Clin Cancer Investig J (2014) 4(1):72. doi: 10.4103/2278-0513.125802
34. Roumeguère T, Legrand F, El RE, Kaitouni MI, Albisinni S, Rousseau A, et al. A Prospective Clinical Study of the Implications of IL-8 in the Diagnosis, Aggressiveness and Prognosis of Prostate Cancer. Futur Sci OA (2018) 4(2):266. doi: 10.4155/fsoa-2017-0084
35. Nadler RB, Koch AE, Calhoun EA, Campbell PL, Pruden DL, Bennett CL, et al. IL-1β and TNF-α in Prostatic Secretions Are Indicators in the Evaluation of Men With Chronic Prostatitis. J Urol (2000) 164(1):214–8. doi: 10.1016/S0022-5347(05)67497-6
36. Ashok A, Keener R, Rubenstein M, Stookey S, Bajpai S, Hicks J, et al. Consequences of Interleukin 1β-Triggered Chronic Inflammation in the Mouse Prostate Gland: Altered Architecture Associated With Prolonged CD4+ Infiltration Mimics Human Proliferative Inflammatory Atrophy. Prostate (2019) 79(7):732–45. doi: 10.1002/pros.23784
37. Urios A, Ordoño F, García-García R, Mangas-Losada A, Leone P, José Gallego J, et al. Tadalafil Treatment Improves Inflammation, Cognitive Function, And Mismatch Negativity Of Patients With Low Urinary Tract Symptoms And Erectile Dysfunction. Sci Rep (2019) 9(1):1–9. doi: 10.1038/s41598-019-53136-y
38. Kamiya N, Akakura K, Suzuki H, Isshiki S, Komiya A, Ueda T, et al. Pretreatment Serum Level of Neuron Specific Enolase (NSE) as a Prognostic Factor in Metastatic Prostate Cancer Patients Treated With Endocrine Therapy. Eur Urol (2003) 44(3):309–14. doi: 10.1016/S0302-2838(03)00303-8
39. Muoio B, Pascale M, Roggero E. The Role of Serum Neuron-Specific Enolase in Patients With Prostate Cancer: A Systematic Review of the Recent Literature. Int J Biol Markers (2018) 33(1):10–21. doi: 10.5301/ijbm.5000286
40. Jalali A, Kitching M, Martin K, Richardson C, Murphy TB, FitzGerald SP, et al. Integrating Inflammatory Serum Biomarkers Into a Risk Calculator for Prostate Cancer Detection. Sci Rep (2021) 11(1):2525. doi: 10.1038/S41598-021-81965-3
41. Watson J, Salisbury C, Banks J, Whiting P, Hamilton W. Predictive Value of Inflammatory Markers for Cancer Diagnosis in Primary Care: A Prospective Cohort Study Using Electronic Health Records. Br J Cancer (2019) 120(11):1045–51. doi: 10.1038/S41416-019-0458-X
42. Eklund M, Nordström T, Aly M, Adolfsson J, Wiklund P, Brandberg Y, et al. The Stockholm-3 (STHLM3) Model can Improve Prostate Cancer Diagnostics in Men Aged 50-69 Yr Compared With Current Prostate Cancer Testing. Eur Urol Focus (2018) 4(5):707–10. doi: 10.1016/J.EUF.2016.10.009
43. Punnen S, Pavan N, Parekh DJ. Finding the Wolf in Sheep’s Clothing: The 4kscore Is a Novel Blood Test That Can Accurately Identify the Risk of Aggressive Prostate Cancer. Rev Urol (2015) 17(1):3.
44. Schröder FH, Hugosson J, Roobol MJ, Tammela TLJ, Ciatto S, Nelen V, et al. Screening and Prostate-Cancer Mortality in a Randomized European Study. N Engl J Med (2009) 18:1320–8. doi: 10.1056/NEJMoa0810084
45. Thompson IM, Ankerst DP, Chi C, Goodman PJ, Tangen CM, Lucia MS, et al. Assessing Prostate Cancer Risk: Results From the Prostate Cancer Prevention Trial. JNCI J Natl Cancer Inst (2006) 98(8):529–34. doi: 10.1093/JNCI/DJJ131
Keywords: algorithm, EGF, fPSA, IL-8, marker, MCP-1, prostate cancer, tPSA
Citation: McNally CJ, Watt J, Kurth MJ, Lamont JV, Moore T, Fitzgerald P, Pandha H, McKenna DJ and Ruddock MW (2022) A Novel Combination of Serum Markers in a Multivariate Model to Help Triage Patients Into “Low-” and “High-Risk” Categories for Prostate Cancer. Front. Oncol. 12:837127. doi: 10.3389/fonc.2022.837127
Received: 16 December 2021; Accepted: 14 April 2022;
Published: 19 May 2022.
Edited by:
Antonina Mitrofanova, The State University of New Jersey, United StatesReviewed by:
Gian Maria Busetto, University of Foggia, ItalyCopyright © 2022 McNally, Watt, Kurth, Lamont, Moore, Fitzgerald, Pandha, McKenna and Ruddock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark W. Ruddock, bWFyay5ydWRkb2NrQHJhbmRveC5jb20=
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.