 Shuye Lin1†
Shuye Lin1† Hanli Xu2†Mengdi Pang2Xiaomeng Zhou2,3Yuanming Pan1Lishu Zhang2Xin Guan2Xiaoyue Wang2Bonan Lin2Rongmeng Tian2Keqiang Chen4Xiaochen Zhang2Zijiang Yang2Fengmin Ji2Yingying Huang5Wu Wei5Wanghua Gong6Jianke Ren5
Hanli Xu2†Mengdi Pang2Xiaomeng Zhou2,3Yuanming Pan1Lishu Zhang2Xin Guan2Xiaoyue Wang2Bonan Lin2Rongmeng Tian2Keqiang Chen4Xiaochen Zhang2Zijiang Yang2Fengmin Ji2Yingying Huang5Wu Wei5Wanghua Gong6Jianke Ren5 Ji Ming Wang4
Ji Ming Wang4 Mingzhou Guo3*
Mingzhou Guo3* Jiaqiang Huang1,2,4*
Jiaqiang Huang1,2,4*- 1Cancer Research Center, Beijing Chest Hospital, Capital Medical University, Beijing Tuberculosis and Thoracic Tumor Institute, Beijing, China
- 2College of Life Sciences & Bioengineering, Beijing Jiaotong University, Beijing, China
- 3Department of Gastroenterology and Hepatology, Chinese People’s Liberation Army of China (PLA) General Hospital, Beijing, China
- 4Laboratory of Cancer Immunometabolism, Center for Cancer Research, National Cancer Institute, Frederick, MD, United States
- 5Chinese Academy of Sciences (CAS) Key Laboratory of Computational Biology, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai, China
- 6Basic Research Program, Leidos Biomedical Research, Inc., Frederick, MD, United States
Background: Hepatocellular carcinoma (HCC) is one of the most lethal human tumors with extensive intratumor heterogeneity (ITH). Serine protease 3 (PRSS3) is an indispensable member of the trypsin family and has been implicated in the pathogenesis of several malignancies, including HCC. However, the paradoxical effects of PRSS3 on carcinogenesis due to an unclear molecular basis impede the utilization of its biomarker potential. We hereby explored the contribution of PRSS3 transcripts to tumor functional heterogeneity by systematically dissecting the expression of four known splice variants of PRSS3 (PRSS3-SVs, V1~V4) and their functional relevance to HCC.
Methods: The expression and DNA methylation of PRSS3 transcripts and their associated clinical relevance in HCC were analyzed using several publicly available datasets and validated using qPCR-based assays. Functional experiments were performed in gain- and loss-of-function cell models, in which PRSS3 transcript constructs were separately transfected after deleting PRSS3 expression by CRISPR/Cas9 editing.
Results: PRSS3 was aberrantly differentially expressed toward bipolarity from very low (PRSS3Low) to very high (PRSS3High) expression across HCC cell lines and tissues. This was attributable to the disruption of PRSS3-SVs, in which PRSS3-V2 and/or PRSS3-V1 were dominant transcripts leading to PRSS3 expression, whereas PRSS3-V3 and -V4 were rarely or minimally expressed. The expression of PRSS3-V2 or -V1 was inversely associated with site-specific CpG methylation at the PRSS3 promoter region that distinguished HCC cells and tissues phenotypically between hypermethylated low-expression (mPRSS3-SVLow) and hypomethylated high-expression (umPRSS3-SVHigh) groups. PRSS3-SVs displayed distinct functions from oncogenic PRSS3-V2 to tumor-suppressive PRSS3-V1, -V3 or PRSS3-V4 in HCC cells. Clinically, aberrant expression of PRSS3-SVs was translated into divergent relevance in patients with HCC, in which significant epigenetic downregulation of PRSS3-V2 was seen in early HCC and was associated with favorable patient outcome.
Conclusions: These results provide the first evidence for the transcriptional and functional characterization of PRSS3 transcripts in HCC. Aberrant expression of divergent PRSS3-SVs disrupted by site-specific CpG methylation may integrate the effects of oncogenic PRSS3-V2 and tumor-suppressive PRSS3-V1, resulting in the molecular diversity and functional plasticity of PRSS3 in HCC. Dysregulated expression of PRSS3-V2 by site-specific CpG methylation may have potential diagnostic value for patients with early HCC.
Introduction
Human primary liver cancer is one of the most lethal tumors with a dismal prognosis, featuring extensive intratumor heterogeneity (ITH) and aggressiveness in the context of genetic and epigenetic aberrations (1–5). Liver hepatocellular carcinoma (HCC or LIHC) accounts for approximately 75-85% of all primary liver cancers. Most HCCs (>90%) develop from chronic inflammation-induced liver cirrhosis contributed by multiple risk factors, such as hepatitis viruses, alcohol consumption, and nonalcoholic fatty liver disease, which trigger the molecular complexity of ITH, increasing HCC phenotypic diversity and therapeutic resistance (3, 5). Regardless of the many approaches developed for the management of HCC in the past decade, its incidence and mortality rate continue to increase worldwide (5).
Large-scale bioinformatics datasets generated with next-generation sequencing technologies reveal a comprehensive landscape of genomic and epigenetic heterogeneity among HCC cell lines and tissue specimens (1, 2, 4, 6, 7). These studies offer invaluable insight into the molecular basis of ITH to categorize HCC into proliferative and nonproliferative subclasses in favor of integrative molecular monitoring of malignant transformation and management of HCC. However, aside from most genetic alterations occurring in passenger genes that may be associated with aging and pollution, most genetic variants, such as driver mutations in TP53, TERT and CTNNB1 detected in HCCs, are not clinically relevant or are not potentially targetable for the existing drugs (3). This gives rise to a growing drive to integrate nongenetic variations into ITH and to distinguish between functional and nonfunctional ITH (7, 8). PremRNA alternative splicing (AS), as a key co and posttranscriptional process, drives nongenetic phenotypic heterogeneity, the disruption of which generates aberrant transcript variants or splice variants (SVs) that contribute to ITH and functional divergence and are thus functionally important to carcinogenesis and oncotherapeutic resistance (9–12).
Proteases play critical roles in multiple biological processes and are associated with a wide variety of pathological conditions, including carcinogenesis (13). As a group of trypsin-family serine proteases, human trypsinogen gene, protease serine 3 (PRSS3), encodes PRSS3, also called mesotrypsinogen (MTG) (14–16). PRSS3 possesses four experimentally validated SVs, referred to as trypsinogen transcript variants 1, 2, 3, and 4 (PRSS3-V1, -V2, -V3 and -V4), encoding PRSS3 isoform 1 (also known as brain form or trypsinogen 4, TRY4) (15, 17), PRSS3-2 (form C or MTG) (14, 18), PRSS3-3 (form B or trypsinogen IV) (19), and PRSS3-4 (new form or trypsinogen 5), respectively (20). In addition to PRSS1 and PRSS2, as the major digestive enzymes in the pancreas, PRSS3 is a minor constituent trypsin isoform but is physiologically critical due to its resistance to common trypsin inhibitors (13, 14, 16). PRSS3 has long been implicated in the pathogenesis of several malignancies and is therefore a promising biomarker and potential therapeutic target for cancer (21–31). However, the functional roles associated with the expression of PRSS3 in cancer development are debatable. On the one hand, PRSS3 was shown to be upregulated in association with cancer metastasis, recurrence and poor prognosis (21–24, 26–31). However, on the other hand, PRSS3 was suggested to be a tumor suppressor gene due to epigenetic silencing (32–36). Although the evidence supports the dual roles of proteases in carcinogenesis depending on cellular sources and the cancer microenvironment (9, 12, 13, 22, 23, 26, 34–36), the underlying molecular basis of PRSS3 for its pro- and antitumorigenic roles shown in different cancer types, even reported in the same type of cancer, such as in esophageal adenocarcinoma (24, 32), lung cancer (29, 35) and liver cancer (21, 36), remains elusive, causing many miscellaneous aliases to PRSS3 to impact its potential target-therapeutic applications (1, 12, 13, 23, 25, 36).
While SVs have emerged as new candidates for diagnostic and prognostic biomarkers and therapeutic targets (9, 10), the expression and function of PRSS3-SVs in cancer development have never been systematically addressed. Here, we hypothesized that the molecular basis of PRSS3 exerts dual roles attributable to its different transcripts. We thereby investigated the functional expression and epigenetic alteration of PRSS3-SVs in relation to HCC heterogeneity. We found divergent expression of PRSS3-SVs in HCC, which were epigenetically dysregulated by site-specific abnormal CpG methylation. We also observed different functionalities and clinical relevance of PRSS3-SVs. Therefore, epigenetic dysregulation of the expression of PRSS3-SVs may integrate the molecular basis of PRSS3 to exert divergent effects on hepatocarcinogenesis.
Materials and Methods
Data Collection
The datasets used for this study are publicly available on the following websites: the Cancer Model Repository (LIMORE) (https://www.picb.ac.cn/limore/home) (6); the Cancer Genome Atlas (TCGA, https://www.cancer.gov/) (38); the Gene Expression Profiling Interactive Analysis (GEPIA, http://gepia.cancer-pku.cn/) (37); the Cancer Cell Line Encyclopedia (CCLE, http://www.broadinstitute.org/ccle) (39); the Cancer Dependency Map (DepMap, https://depmap.org/portal/, DepMap Public 20Q3) (40); and the Broad Genome Data Analysis Center (http://gdac.broadinstitute.org) (41). The expression of PRSS3 protein was analyzed using data obtained from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) Confirmatory/Discovery dataset (http:ualcan.path.uab.edu) (42).
Cell Lines
Human HCC cell lines, including well differentiated (HepG2 and Huh7) and poorly differentiated (SK-Hep-1, SMMC-7721 and LM3) cell lines, were purchased from Cellcook Biotech Co. (Guangzhou, China) and authenticated by STR profiling (Additional files). The cell lines were grown in DMEM (Gibco, Life Technologies, USA) with 10% fetal bovine serum (Gibco, USA), penicillin/streptomycin and glutamine as described previously (36, 43). TransSafe™ Mycoplasma Prevention Reagent was used to prevent mycoplasma contamination (TransGene, China). The cells were split to low density (30% confluence) overnight culture and were then treated with 5 μM 5-aza-2’-deoxycytidine (5-aza-CR) (Sigma–Aldrich, USA) for 96 hours, with the medium exchanged every 24 hours.
Cell Line Construction
The establishment of stable cell lines with PRSS3-V1 overexpression was described previously (36). The OmicsLink™ Expression clones of PRSS3-V2, -V3 and -V4 were purchased from GeneCopoeia (Rockville, MD, USA) (Table S6). The CRISPR/Cas9 bivector lentivirus was custom ordered from GeneChem (Shanghai, China). The sgRNA was GGCACTGAGTGCCTCATCTC. Genomic deletion of PRSS3 transcripts (PRSS3KO) by targeting the common exon 5-8 region in PRSS3High Huh7 cells was performed using the CRISPR/Cas9 system. Puromycin (Puro) (2 μg/ml) was used for selection of the transduced cells. PRSS3KO Huh7 cells were transfected with the PRSS3-V1 to -V4 constructs to establish stable re-expression of PRSS3 transcripts dubbed the PRSS3KO+V cell model. Transfection was performed using Lipofectamine 2000 (Invitrogen, USA) following the manufacturer’s instructions. Stable cell lines with PRSS3-V2, -V3 or -V4 were selected using 0.5 mg/ml G418 (Invitrogen) for 2 weeks.
Cell Viability
HepG2, SK-Hep-1 and Huh7 cells were seeded into 96-well plates at 2 × 103 cells/well. Cell viability was measured every day by using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay kit (KeyGEN Biotech, China). The absorbance at 490 nm was detected using a microplate reader (Thermo Multiskan MK3, Thermo Fisher Scientific Inc., USA) as described (36, 43).
Colony Formation
HCC cells were seeded in 6-well tissue culture plates (100 cells/well) in triplicate. Colonies with more than 50 cells were counted after 2 weeks. The cells were fixed with 75% ethanol for 30 minutes and stained with 0.2% crystal violet (Beyotime Biotech, China) for 20 minutes (36, 43).
Transwell Invasion Assay
A Transwell apparatus was used with 8-μm polyethylene terephthalate membrane filters (Corning Inc., USA). The upper chambers were seeded with 200 µl of serum-free medium containing serum-starved cells (HepG2 and SK-Hep-1: 1 × 104 cells; Huh7: 2 × 104 cells). The lower chambers were filled with 500 µl of 10% FBS-DMEM. After 24 hours, cells that invaded the lower chamber were fixed and stained with 0.2% crystal violet (Beyotime Biotech) as previously described (36). The invaded cell number from experiments in triplicate was counted in five randomly selected fields per chamber under an inverted microscope (Leica, Germany).
RNA Isolation and RT–qPCR
Cells were harvested for RNA isolation using an RNeasy Mini Kit (QIAGEN, USA), and first strand cDNA was synthesized with the Superscript First-Strand Synthesis System (Invitrogen). RT–qPCR was performed using primers as previously described (36). The relative expression level of each mRNA was normalized to β-actin using the 2-ΔΔCt method.
Methylation-Specific qPCR
DNA extraction, bisulfite modification and MSP-PCR were performed as previously described (36, 43). Genomic DNA was extracted from tissues using the QIAamp DNA Mini Kit (Qiagen), followed by quantitative analysis using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA). Bisulfite modification of DNA was performed using a Zymo DNA Methylation Kit (Zymo Research, USA). The positive and negative template controls were the Human Methylated & Nonmethylated DNA Set (Zymo Research). MSP-qPCR was performed by using methylated or unmethylated primer pairs specifically for PRSS3 (36) and β-actin (43). The relative level of methylation and unmethylation of PRSS3 was normalized to β-actin using the 2-ΔΔCt method.
Methylated DNA Immunoprecipitation-qPCR
Genomic DNA was extracted from the HCC cells. The purified DNA was then sonicated into 200~1000 bp fragments. A 10% sonicated DNA sample was kept as an input control. The denatured DNA fragments (input fractions) were incubated with 2 μg anti-5-methylcytidine (5mC) (Active motif, USA) or 2 μg control IgG (Sigma–Aldrich) monoclonal antibodies at 4°C overnight, followed by precipitation using protein A beads. After washing, immunoprecipitated DNA (IP fractions) and the input control fraction were purified by using a QIAquick purification kit (QIAGEN) and analyzed by qPCR using the following primers: F: 5’- CTGTGATGGAGAGGGGGTTC -3’; R: 5’- GAGTAGTGTGCGCATCGGT-3’.
Western Blotting
HCC cells were lysed in RIPA buffer (Beyotime Biotech) containing protease and phosphatase inhibitors (Sigma–Aldrich). Equal amounts of total protein were loaded on and separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and then transferred onto polyvinylidene difluoride membranes using a Bio–Rad Mini PROTEAN 3 system. The membranes were blocked for 1 h in PBS containing 5% milk (v/v) and 0.1% Tween-20 (v/v) and incubated with the indicated primary antibodies against PRSS3 (Cat. ab107430, Abcam) and PRSS3 isoforms (Figure S1C) overnight at 4°C, followed by incubation for 1 h with the appropriate secondary antibodies. Immunoreactive bands were visualized by using the Amersham ECL Western Blotting Detection Kit according to the manufacturer’s instructions. β-Actin served as a loading control.
Tumorigenicity
The animal handling and all in vivo experimental procedures were approved by the Institutional Animal Ethics Committee of Beijing Chest Hospital. Huh7 cells (2 × 106) with or without constructs suspended in 0.1 ml PBS were injected into the subcutaneous flanks of 4-week-old Balb/c female athymic mice (Vital River Laboratories, Beijing, China). The tumor diameters and body weights of nude mice bearing HCC xenograft tumors were measured and documented every 3 days until the animals were sacrificed at day 15. HCC tumor xenografts were isolated and weighed. Tumor volume was calculated by measuring the longest (a) and shortest (b) diameters of the tumor and calculated by the formula: ab2/2.
Statistical Analysis
The data are expressed as the means ± standard deviation (SD) of at least three independent experiments. PRSS3 transcript expression, epigenetic alterations and associated clinicopathological correlations were analyzed by using the two−tailed Student’s t−test, Wilcoxon rank sum test, one−way analysis of variance (ANOVA) with Tukey’s post-hoc test, Spearman rank test and Fisher’s exact test, or χ2 or Fisher’s exact tests. Cancer-related survival was analyzed using the Kaplan–Meier method and was compared using log-rank tests. Statistical significance was considered when P < 0.05. All statistical analyses were performed using SPSS version 23.0 (IBM Corp.).
Results
Aberrant Differential Expression of PRSS3 in HCC Was Attributable to its Transcript Heterogeneity in Human HCC
The RNA-seq data from the Cancer Model Repository (LIMORE) and the DepMap portal revealed that PRSS3 as a whole was differentially expressed in HCC cell lines (Figure S1A, Figure 1A and Table S1). RT–qPCR validation showed that the expression levels of PRSS3 ranged from very low (PRSS3Low) to very high (PRSS3High) compared to human fetal liver L02 cells (Figure 1B). Western blot using an anti-PRSS3 antibody confirmed the differential expression of PRSS3 at protein level (Figure 1C). Comparative analysis using TCGA RNA-seq data from FIREHOSE Broad GDAC showed divergent features of PRSS3 expression in HCC tissues compared to their matched nontumor tissues (n=50) (Table S2 and Figure 1D). This was further evidenced by the analysis of more HCC tissue specimens (tumor =371) (Table 1, Figure 1E). The GEPIA portal combined TCGA with GTEx RNA-seq datasets showed that PRSS3 expression was more varied in HCC tissues (n=369) than in normal controls (n=160) (Figure S1B) (38, 41). Although not statistically significant, the PRSS3 mRNA level was lower but had an extraordinarily wide range in HCC tissues compared to normal tissues, consistent with the analysis of CPTAC data showing that PRSS3 protein was more highly expressed in normal human live tissues than in HCC tissues (Figure 1F). These results suggest that PRSS3, as a differentially expressed gene (DEG), was aberrantly and divergently expressed in HCC.
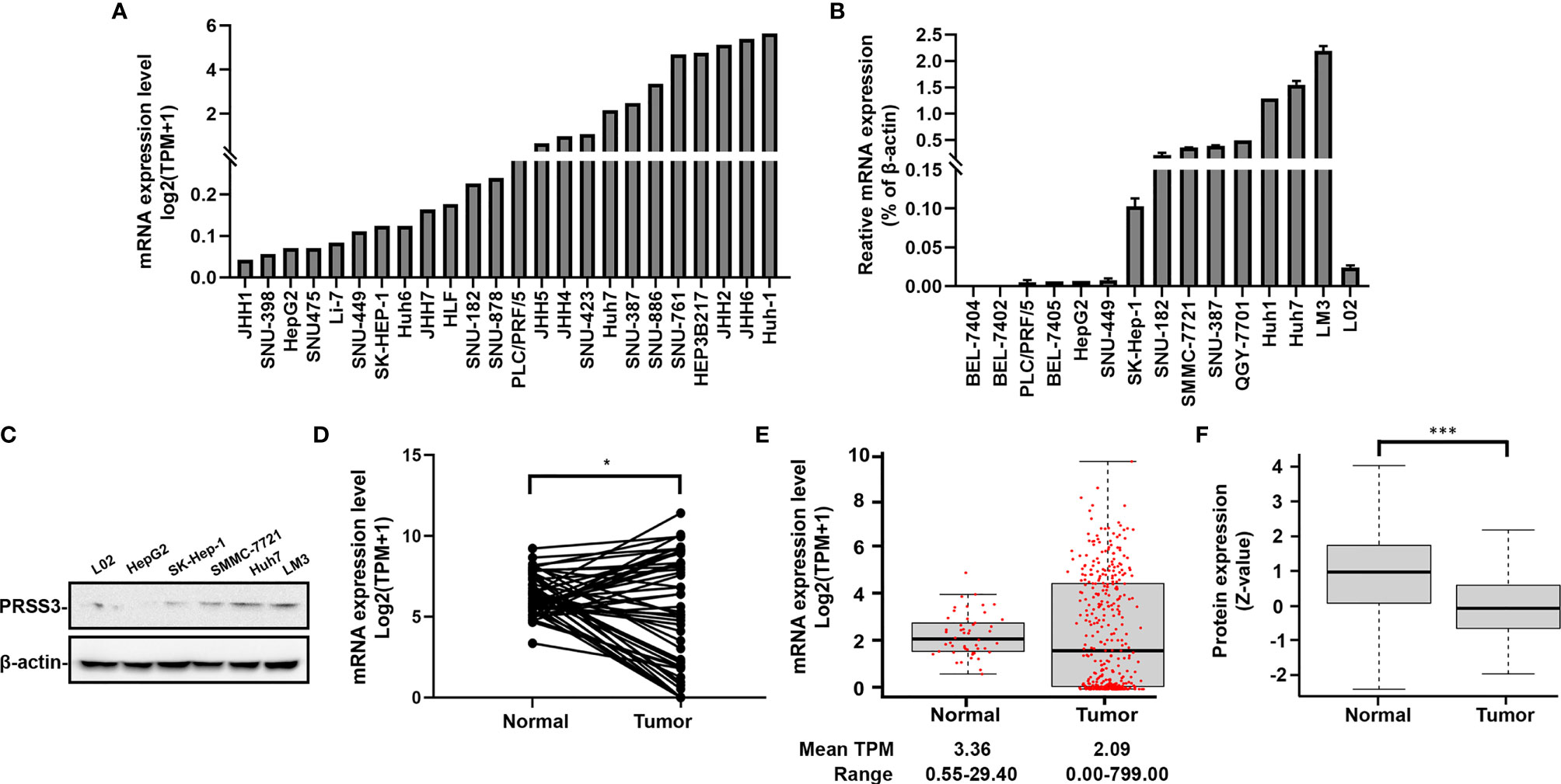
Figure 1 PRSS3 expression in human HCC cell lines and tissues. (A) The mRNA level of PRSS3 expression in 24 HCC cell lines using RNA-seq data extracted from the DepMap website. The expression bar chart is sorted by PRSS3 mRNA expression levels processed on a log2 (TPM+1) scale. TPM: transcripts per million. (B) RT–qPCR analysis of PRSS3 expression in 14 HCC cell lines and the human fetal liver cell line L02. The relative expression of PRSS3 mRNA was normalized to β-actin, presented as the mean ± SD from three independent experiments performed in triplicate. (C) Western blot analysis of PRSS3 in HCC cell lines and L02 cells. (D, E) The RNA-seq data were sourced from FIREHOSE (Table 1). PRSS3 expression in 50 pairs of tumors and matched solid normal tissues from patients with HCC (D) or extensively in 371 tumor samples versus 50 normal tissues from HCC patients (E). * P < 0.05, by Student’s t test. (F) PRSS3 protein expression in human HCC (n = 165) in comparison with normal liver tissues (Normal) (n = 165), based on data from UALCAN portal analysis of CPTAC Confirmatory/Discovery dataset. Z-values represent the standard deviation from the median across samples for a given cancer type. Log2 spectral count ratio values from CPTAC were first normalized within each sample profile and normalized across samples. ***P < 0.001, by the Wilcoxon rank sum test.
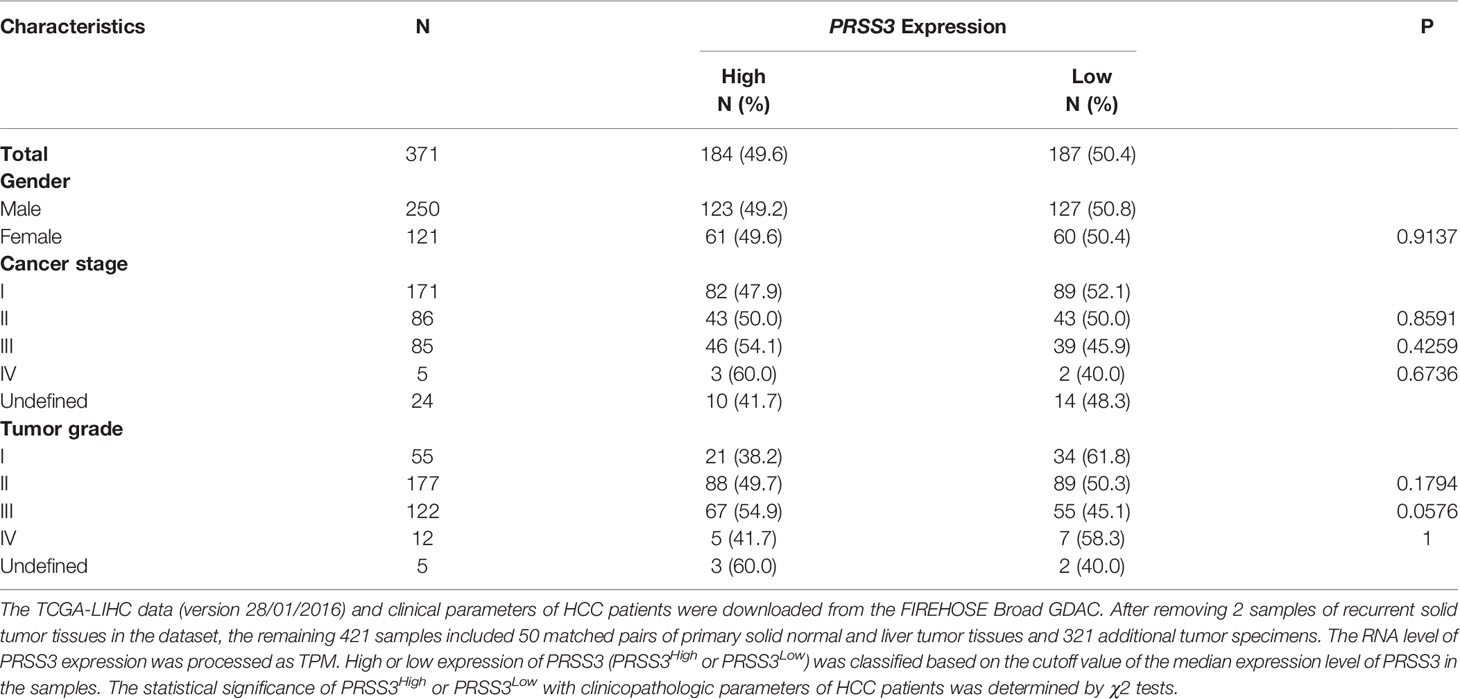
Table 1 Correlation between the mRNA levels of PRSS3 transcripts and clinicopathologic characteristics in patients with HCC.
To explore the molecular basis of the divergent expression of PRSS3 in HCC, we dissected the expression of PRSS3-V1~-V4 in HCC (14, 15, 17–20) (Figure 2A). Analysis of the DepMap data revealed that in 24 HCC cell lines, PRSS3-V2 and/or -V1 were two major transcripts contributing to the expression of PRSS3 because PRSS3-V3 was infrequent and/or poorly expressed, while PRSS3-V4 was absent (Table S1 and Figure 2B). RT–qPCR showed that despite almost undetectable PRSS3-V4 and very low expression of PRSS3-V3 in all cell lines, PRSS3-V1 was expressed at low levels in L02 cells, whereas PRSS3-V1 and -V2 were minimally expressed in HepG2, SK-Hep-1 and SMMC-7721 cells but highly expressed in Huh7 and LM3 cells (Figure 2C). Western blot using antibodies against PRSS3-V1 to -V4 showed that PRSS3-V1 and -V2 were detected in Huh7 and LM3 cells (Figure S1C), in parallel to their mRNA levels. Through comparative analysis of the expression levels of PRSS3 transcripts, including PRSS3 and its four SVs, in 50 paired tissue samples, we found that PRSS3-V2 and -V1 were predominantly present in both normal and tumor tissues (Table S2 and Figure 2D). Figure 2E shows that the expression of PRSS3-SVs (no data available for PRSS3-V4) tended toward bipolarity in 371 HCC tissue samples compared to normal liver tissues, although only PRSS3-V2 expression was significantly decreased. Coexpression analysis of both HCC cell lines and tissues summarized in Table 2 further showed that the highest contributor of PRSS3-SVs to PRSS3High was PRSS3-V2, suggesting its expression dominance in PRSS3High in HCC. Moreover, PRSS3Low also resulted from decreased expression of PRSS3-V2 and/or -V1 because PRSS3-V3 was minimally expressed in HCC and minimally affected the eventual expression of PRSS3, although PRSS3-V3Low was most frequently associated with PRSS3Low. These results thereby revealed disruption of PRSS3 transcripts toward bipolar expression contributing to aberrant and differential expression of PRSS3 in HCC, in which PRSS3-V2 and/or PRSS3-V1 were dominant transcripts leading to PRSS3 expression.
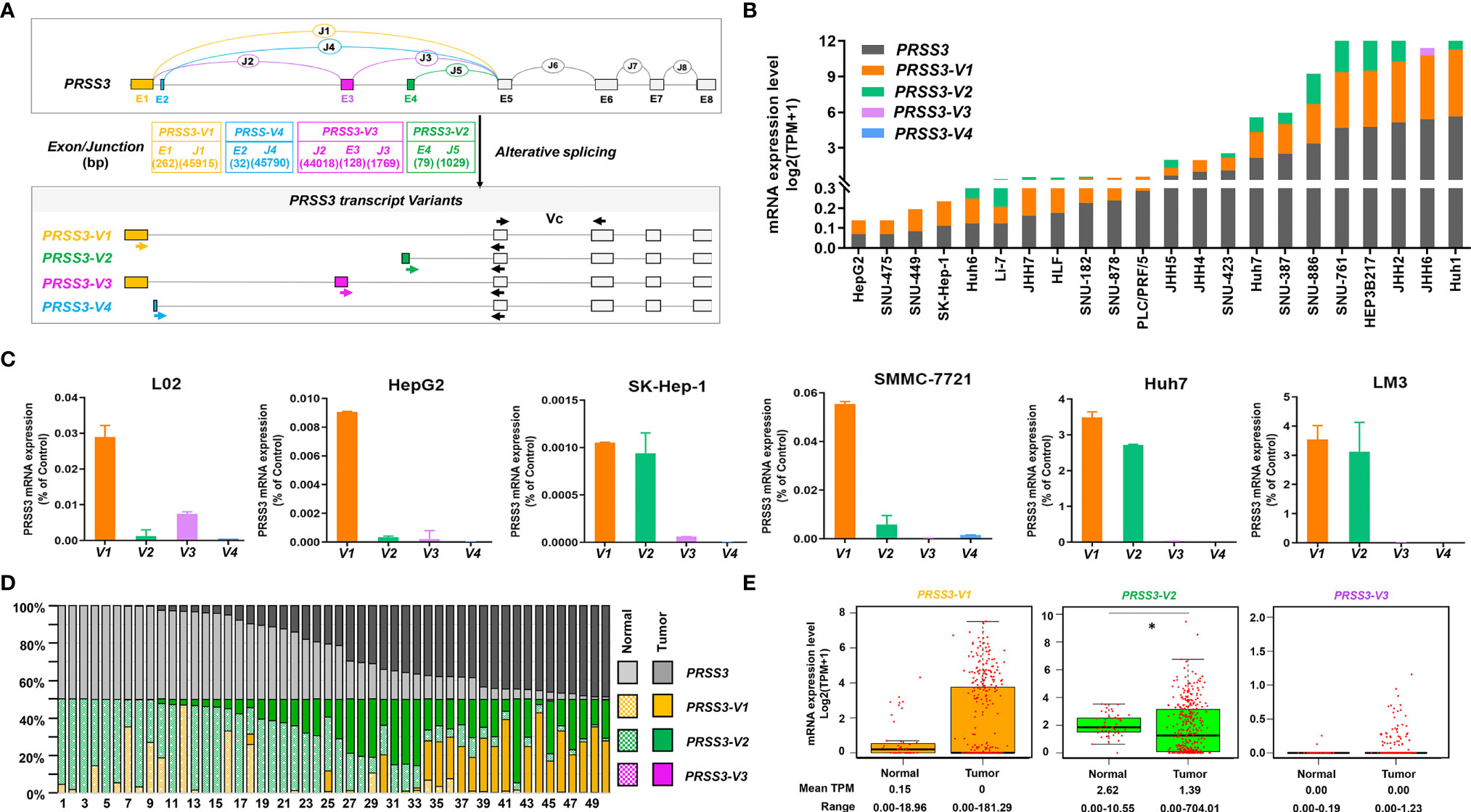
Figure 2 The expression of PRSS3 splice variants in human HCC cells and tissues. (A) A schematic overview of the human PRSS3 gene structure and its splicing variants (SVs) and the designed RT–qPCR primers. The top diagram represents the genomic organization of PRSS3. Alternative splicing within the 5’ region of the PRSS3 gene leads to the creation of PRSS3-V1 ~ -V4. The exons and introns are represented as boxes and lines (not drawn to scale). E1-E8: Exons; J1-J8: Junctions. E5-8: gray boxes common to all four variants. E1 to 4: sequence-specific for PRSS3-V1 (brown), -V4 (blue), -V3 (purple), and -V2 (green), respectively. Arrowheads indicate primer set locations used for amplification of PRSS3-SVs. Forward primers were designed specifically for PRSS3-SVs. Reverse primers were common to all. Vc: RT–qPCR primer set common to PRSS3-SVs. (B) Expression level of PRSS3-SVs in HCC cell lines. Data from the DepMap (Table S1). (C) RT–qPCR of PRSS3 transcripts expressed in the human fetal liver cell line L02 and HCC cell lines. The relative expression level of each mRNA was normalized against β-actin. (D) Comparison of the mRNA expression of PRSS3 and its transcript variants in 50 paired HCC and normal liver tissues (Table S2). The relative percentage of PRSS3 transcripts expressed in each paired sample (TPM scale) was visualized by a 100% stacked bar graph. (E) The mRNA expression of PRSS3 transcripts in HCC tissues (n=371) and normal liver tissues (n=50) based on data from FIREHOSE. The relative transcript level is presented as a log2 (TPM+1) scale. *P < 0.05 by Wilcoxon rank sum test.
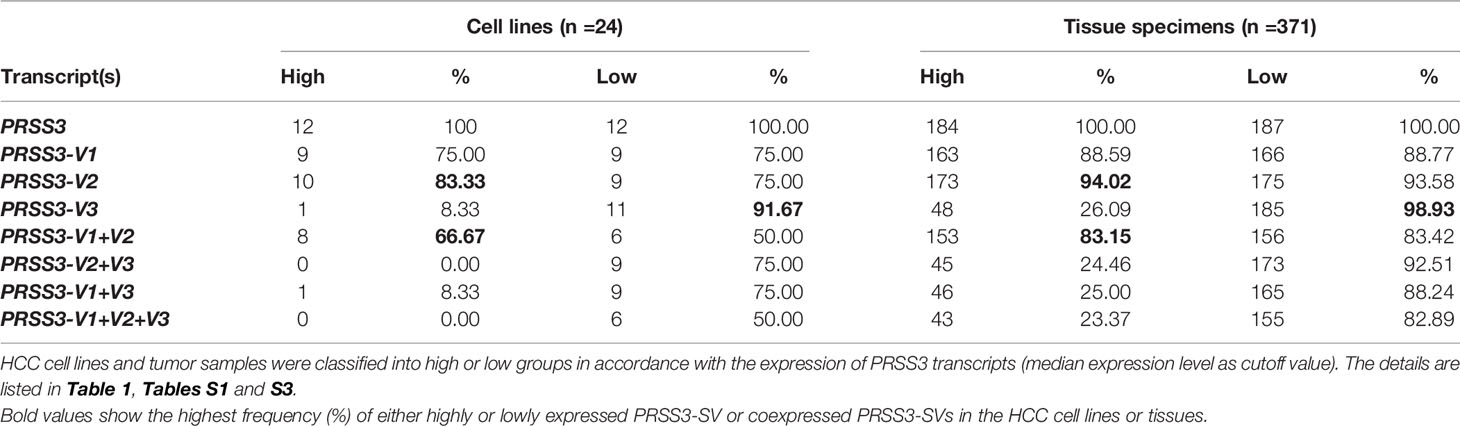
Table 2 Predominance and coexpression of PRSS3 transcripts in HCC cell lines and tissues.
CpG Site-Specific Methylation Regulated the Expression Divergence of PRSS3-SVs in HCC
We previously observed epigenetic silencing of PRSS3 in HCC (32, 34–36). However, methylation in association with the expression of its SVs has never been addressed. We next assessed the contribution of DNA methylation to the expression of PRSS3-SVs based on the data available from DepMap and FIREHOSE (39, 40) for three genomic regions in PRSS3. These were referred to as the promoter region and upstream and extended fragment, respectively (Figure 3A). The promoter region approximately 2400 bp (-1749 to 653 bp) around the TSS shared by PRSS3-V1/3 contains 17 CpG sites (CpGs 1-17), including 5 CpGs (CpGs 2-7) in the 1 kb upstream fragment (-1000 bp to the TSS of PRSS3-V1/3), while the extended fragment includes 6 CpGs (defined as A, B, C, D, E and F) scattering around a broad genomic region approximately 34.5 kb in scale from -170 to 34,654 bp of the TSS of PRSS3-V1/3 but still -10,643 bp upstream of the TSS of PRSS3-V2. The genomic position of each CpG site is shown in Figure 3A relative to the TSS of PRSS3-V1/3 (Table S4).
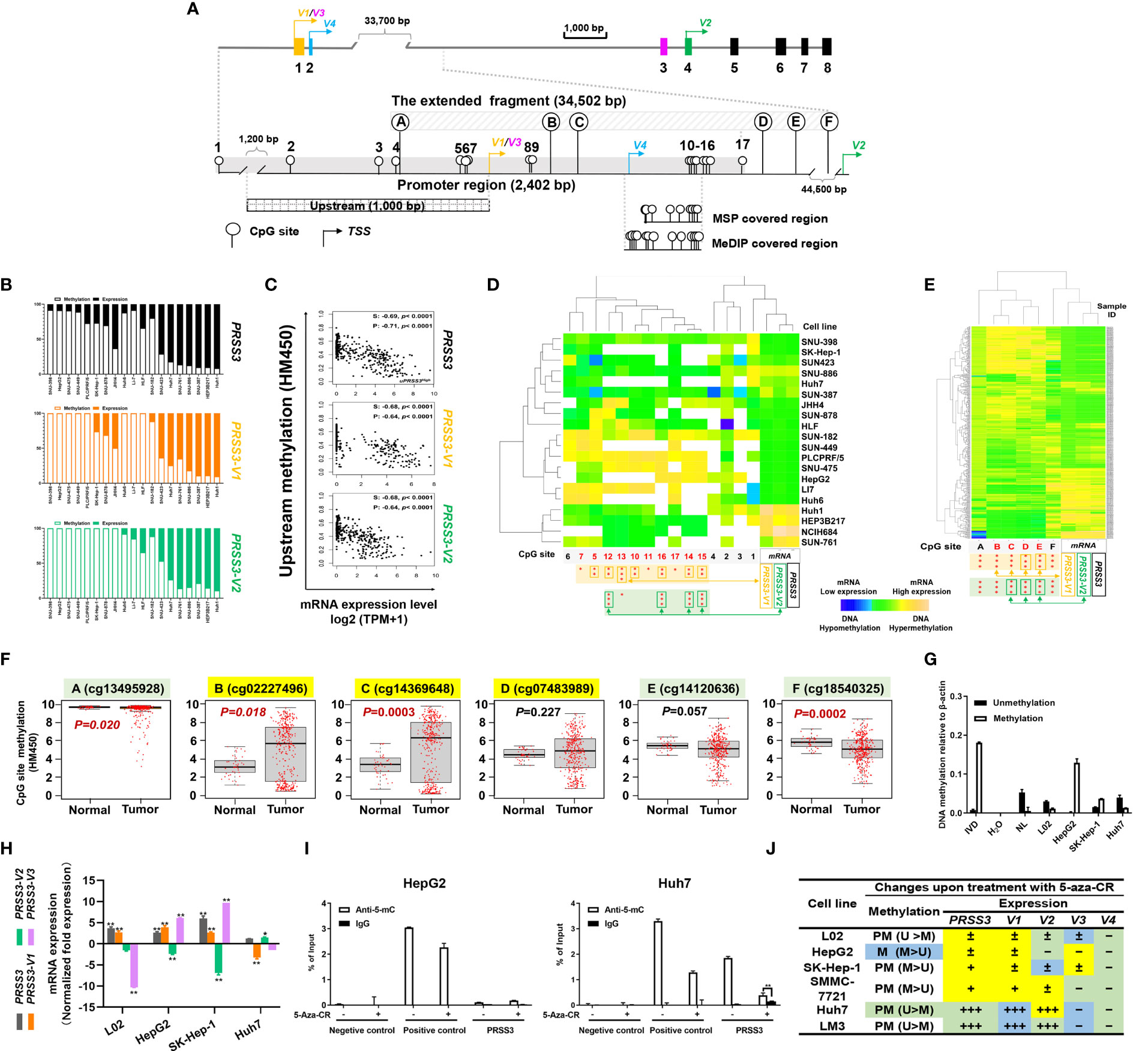
Figure 3 CpG methylation in the regulation of PRSS3 transcript expression in HCC. (A) Schematic of the PRSS3 5’-genomic region including the extended promoter region and upstream and extended fragments. The promoter region (-1749 to 653 bp) shared by PRSS3-V1/3 contains 17 CpG sites (CpGs), including 5 CpGs (CpG sites 2-7) in the 1 kb upstream fragment (-1000 bp from the TSS of PRSS3-V1/3). The extended fragment includes 6 CpGs (defined as A–F) scattering around a broad genomic region approximately 34.5 kb from -170 to 34,654 bp of the PRSS3-V1/3 TSS but -10,643 bp from the TSS of PRSS3-V2. The genomic position of each CpG site is shown relative to the TSS of PRSS3-V1/3 (Table S4). Primer-covered regions for MSP-qPCR and MeDIP-qPCR are shown. (B) 1 kb upstream methylation normalized as a percentage relative to PRSS3 expression in HCC cell lines visualized by a 100% stacked bar graph. (C) Spearman and Pearson correlation analysis of 1 kb upstream methylation associated with PRSS3 expression in human primary liver tumor samples (n=371). (D, E) Clustered heatmap of the correlation between CpG site methylation and PRSS3 transcript expression. Data were visualized by using correlation as a distance function for heatmap cluster analysis of CpG methylation at the promoter in 20 HCC cell lines (D) and at the extended fragment in HCC tissue specimens (n=414) (E). In the heatmap, blue indicates low, green indicates intermediate and yellow indicates high DNA methylation or mRNA values. Rows: CpG sites arranged based on the correlation between the methylation and mRNA expression levels of PRSS3 transcripts. The values of DNA methylation levels were renormalized with mean=0 and standard deviation=1. Columns: HCC cell lines or tissue specimens. The statistical significance of correlation coefficients between CpG sites (red) and mRNA expression of PRSS3 transcripts are shown at the bottom. *P < 0.05, **P < 0.01, ***P < 0.001 (Figures S3, S4 and Table S5). (F) Association analysis of CpG site methylation with PRSS3-SV expression in 414 HCC tissue specimens compared with 41 normal controls (Wilcoxon rank sum test). (G) MS-qPCR of PRSS3 methylation in HCC cell lines and L02 cells. In vitro methylated DNA (IVD) and normal human peripheral lymphocyte DNA (NL) served as positive and negative methylation controls, respectively. (H) RT–qPCR of the expression of PRSS3 transcripts in HCC cell lines and L02 cells upon treatment with the epigenetic reagent 5-aza-CR (5 μM, 96 h). *P < 0.05, **P < 0.01 by Student’s t test. (I) MeDIP-qPCR to analyze 5-mC-enriched genomic DNA associated with the extended promoter region in HCC cell lines and L02 cells after 5-aza-CR treatment. **P < 0.01 by Student’s t test. (J) In the summary table, the differential expression changes of PRSS3 transcripts responding to treatment with 5-aza-CR are visualized with symbols and colors. Methylation was defined as partial methylation (PM) or methylation (M) based on the MSP results. PRSS3 expression: “–”, < 0.001%; “±”, 0.001-0.05%; “+”, > 0.05%; “+++”, > 1%. The fold changes upon 5-aza-CR treatment are shown in color: yellow, upregulation; green, downregulation; blue, no change.
Association analysis demonstrated an inverse association between the upstream methylation and mRNA expression of PRSS3 and its transcripts PRSS3-V1 and -V2 that could distinguish HCC cell lines phenotypically between hypermethylation of PRSS3Low (mPRSS3Low) and hypomethylation of PRSS3High (umPRSS3High) groups (Figure 3B). The pattern of mPRSS3Low versus umPRSS3High was further confirmed in tumor samples showing more similarity between PRSS3 and PRSS3-V2, whereas PRSS3-V1 was more phenotypically defined with mPRSS3Low and umPRSS3High groups (Figure 3C). Together with the intragenic methylation associated with PRSS3 expression shown in our previous study (35), these results support the regulatory effect of DNA methylation on PRSS3 transcripts.
Unsupervised clustering combined with Spearman correlation analysis of CpG site methylation and expression of PRSS3 transcripts in HCC cell lines revealed that among 17 CpGs distributed in the promoter region, methylation occurring at CpG sites 5-17 (-89~653 bp from the TSS of PRSS3-V1/V3) (Table S4) was inversely correlated with the mRNA expression level of PRSS3-V1, while methylation at CpG sites 12-16 (522 to 564 bp to PRSS3-V1 TSS) was highly related to PRSS3-V2 expression (Figure 3D and Figure S2, Table S5). No associative comparison was conducted on PRSS3-V3 and -V4 due to their rare expression in HCC. This result confirms the patterns of mPRSS3Low versus umPRSS3High in HCC cells. However, only CpG site 5 in the upstream was significantly associated with the expression of PRSS3-V1 (Figure 3D, and Figure S2), suggesting CpG site-specific regulation of PRSS3 transcript expression in HCC cells. Moreover, despite a positive association shown at CpG site F, methylation at CpG sites A-E was negatively correlated with PRSS3 expression (Figure 3E and Figure S3A), in which the associative significance of site methylation with PRSS3 and PRSS3-V2 was B, C, D, E but reversed for PRSS3-V1 (Figure S3B). CpG site methylation at the extended fragment of PRSS3 was decreased at site A, increased at B, C and D, and then decreased at E and F in HCC tumors compared to normal controls (Figure 3F). The CpG site methylation in the PRSS3 promoter region from -89 bp (CpG site 5) to 785 bp (CpG site E) to the TSS of PRSS3-V1/3 associated with the expression of PRSS3 transcripts suggests an epigenetic promoter contribution to divergent expression of PRSS3 transcripts in HCC (Figure 3A).
We then examined the methylation-specific effect on PRSS3 expression using qPCR-based assays (Figure 3A). MSP-qPCR showed that PRSS3 was hypermethylated in PRSS3Low cell lines (HepG2, SK-Hep-1) but hypomethylated in PRSS3high Huh7 cells compared to L02 cells (Figure 3G), consistent with previous observations (36). Figure 3H reveals that treatment with the DNA methyltransferase inhibitor 5-aza-CR caused significant upregulation of PRSS3 expression in PRSS3Low cell lines but had no effect on PRSS3High Huh7 cells. Notably, a bipolar expression pattern was observed in PRSS3Low cell lines upon 5-aza-CR treatment, showing significant upregulation of PRSS3-V1 and -V3 opposite to downregulation of PRSS3-V2, eventually integrative to the upregulation of PRSS3, whereas the treatment had no effect on PRSS3high Huh7 cells, actually due to integration between upregulation of PRSS3-V2 and downregulation of PRSS3-V1 and -V3. MeDIP-qPCR further showed that the anti-5-methylcytosine (5-mC) antibody significantly enriched fewer genomic DNA fragments in HepG2 cells but not in Huh7 cells upon 5-aza-CR treatment (Figure 3I), suggesting that 5-aza-CR was effective in the expression of PRSS3 specifically by altering DNA methylation in this promoter region. Although the expression of PRSS3-V3 in L02 or PRSS3-V2 in HepG2 and SK-Hep-1 cells was too low to take into account its decreased significance level, these results are consistent with bioinformatic analysis of HCC cell lines and tissues, as well as our previous report (36), suggesting that methylation occurring at this region is more critical for epigenetically controlling PRSS3 transcript activities in HCC. As shown in the summarized table (Figure 3J), the divergence of PRSS3 transcript expression and their response to 5-aza-CR treatment was negatively associated with site-specific CpG methylation, which eventually determined the expression level of PRSS3 as a whole. These results suggest that differential methylation of the promoter controls the expression of PRSS3-SVs in a site-specific manner in HCC.
PRSS3-V2 Exerts Oncogenic Functions Distinct From the Tumor-Suppressive Effects of PRSS3-V1 and PRSS3-V3 in HCC Cells
The functional role of PRSS3-SVs was assessed by transfecting PRSS3-V1 to -V4 into PRSS3Low HepG2 and SK-Hep-1 cells (defined as V1 to V4) (Figure 4A and Figure S4A). MTT assays showed that ectopic expression of PRSS3-V1 or PRSS3-V3 significantly inhibited HCC cell proliferation in contrast to notably enhancing the effect of ectopic PRSS3-V2 expression or nonfunctional PRSS3-V4 on HCC cell proliferation compared to the vector controls (Figure 4B). Moreover, the results of the clone formation assay showed that overexpression of PRSS3-V1 or PRSS3-V3 remarkably diminished the number of colonies of HCC cells compared with the control group, but PRSS3-V2 overexpression resulted in an increased number of colonies only effectively in HepG2 cells. However, ectopic PRSS3-V4 significantly reduced clone formation in SK-Hep-1 cells but had no effect in HepG2 cells (Figure 4C). Transwell assays further showed an inhibitory effect of PRSS3-V1 or PRSS3-V3 on HCC cell migration, in contrast to PRSS3-V2, which showed an enhanced effect in the cells (Figure 4D). These results suggest a tumor-suppressive effect of PRSS3-V1/V3 versus an oncogenic effect of PRSS3-V2 in HCC cells.
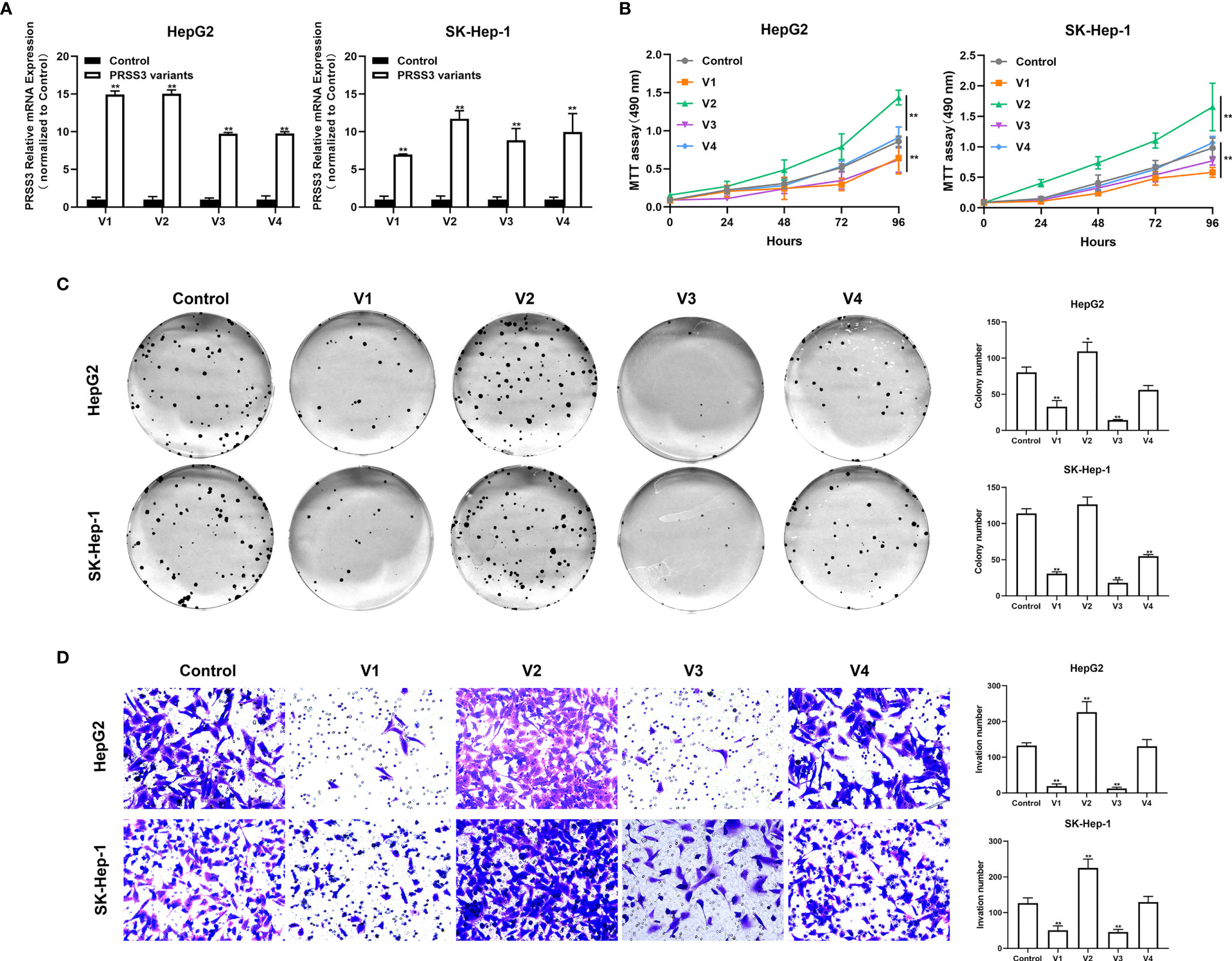
Figure 4 Effects of ectopically expressed PRSS3 transcripts on HCC cell malignancy. The PRSS3 splicing variants were separately transfected into HepG2 and SK-Hep-1 cells to establish stable cell lines with individual overexpression of either PRSS3-V1 to -V4 (V1 to V4) or vector control (Control). (A) The mRNA expression levels of PRSS3 transcripts in the transfected cells were measured by RT–qPCR and quantified relative to the control cells (Student’s t test). (B) Cell viability of HepG2 and SK-Hep-1 cells with ectopic expression of either PRSS3 transcript was detected by MTT assays compared with the vector control (two‐tailed Student’s t‐test). (C) Colony assays showing the colony formation of HepG2 and SK-Hep-1 cells after overgrowing for 2 weeks. Representative images are presented in the left panel; quantitation of the colony numbers is shown on the right. (D) Transwell invasion assay assessing cell invasion capacity following transfection of PRSS3 transcripts. Left panel: representative image at 200× magnification; right panel: quantitation of the migrated cells. One−way ANOVA with Tukey’s post-hoc test was calculated for the transfected cells compared with the vector control in (C, D). *P < 0.05, **P < 0.01, versus control. Data are presented as the mean ± SD of three independent experiments performed in triplicate.
To further define the phenotypic properties of PRSS3-SVs in HCC cells, we established a PRSS3KO+V cell model in which each PRSS3 transcript construct was separately transfected after endogenous PRSS3 was deleted through the CRISPR/Cas9 system (Figure 5A). RT–qPCR showed that all the detected PRSS3 transcripts were effectively deleted, and their constructs were stably expressed in Huh7 cells, designated PRSS3KO+V1 to PRSS3KO+V4, or the vector control (PRSS3KO+V) (Figure 5B), which was further confirmed by Western blot analysis of PRSS3 protein isoforms (Figure S4B). Functional assays, as shown in Figure 5C–E, revealed that PRSS3 deletion in Huh7 cells facilitated cell proliferation, colony formation and migration, which were abolished by re-expression of PRSS3-V1 or PRSS3-V3. Ectopic re-expression of PRSS3-V2 augmented the PRSS3-deletion effects on cell proliferation and, remarkably, on the migration of PRSS3KO Huh7 cells. Unexpectedly, PRSS3-V4 re-expression did not affect Huh7 cell proliferation but resulted in significant inhibition of PRSS3KO Huh7 cell activity. To analyze the functional roles of the PRSS3 variants in tumor growth in vivo, a tumor xenograft assay was performed by injecting PRSS3KO+V cells into nude mice (Figure S5). Consistent with in vitro findings, PRSS3 deletion favored xenograft tumor growth formed by Huh7 cells, with a significant augmentation by re-expression of PRSS3-V2 (PRSS3KO+V2), whereas re-expressing either PRSS3-V1 or PRSS3-V3 (PRSS3KO+V1/3) in the cells caused a marked inhibitory effect on xenograft tumor growth in contrast to a minimal role of PRSS3KO+V4 (Figure 5F). These results exclusively demonstrate the dual roles of PRSS-SVs in HCC cells, and divergent disruption of PRSS3 transcripts may be integrated to establish their functional heterogeneity in HCC cells.
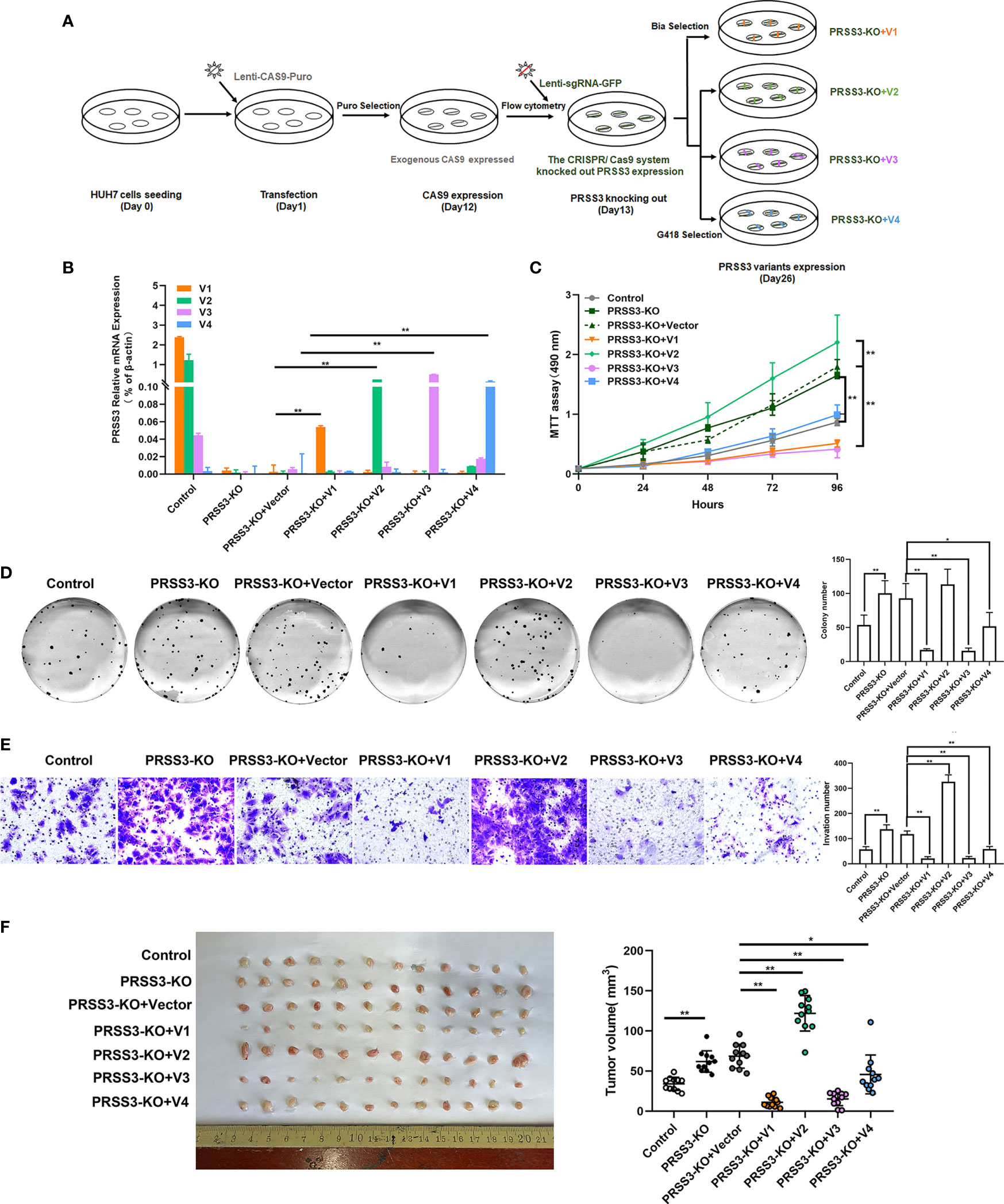
Figure 5 Functional divergence of PRSS3 transcript variants in a gain- and loss-of-function cell model. (A) Schematic of the workflow for the construction of a cell model by endogenous deletion and then ectopic expression of the PRSS3 transcript in Huh7 cells (PRSS3 KO+V cell model). Genomic deletion of PRSS3 transcripts by targeting the common exon 5-8 region in PRSS3High Huh7 cells was performed using the CRISPR/Cas9 system, followed by transfection with PRSS3-V1 to PRSS3-V4 constructs (PRSS3 KO+V1 to PRSS3KO+V4) or vector control (PRSS3KO+C). Puromycin (Puro), blasticidin (Bla) and geneticin (G418) were used for selection of the transduced cells. (B) RT–qPCR analysis of PRSS3 mRNA expression in the transfected cells. The relative mRNA expression of PRSS3 transcripts normalized to β-actin (Student’s t test). (C) MTT assays showed the viability of Huh7 cells (two‐tailed Student’s t‐test). (D) Colony formation of Huh7 cells for 2 weeks. Left panel: representative image; Right panel: The colony numbers counted. (E) Transwell invasion assay assessing the invasion capacity of Huh7 cells upon transfection. Left panel: representative images at 200× magnification; right panel: quantitation of the invaded cells. One−way ANOVA with Tukey’s post-hoc test was calculated for the transfected cells compared with the vector control in (C–F). *P < 0.05, **P < 0.01, versus control. Data are presented as the mean ± SD of three independent experiments performed in triplicate. (F) Effects of PRSS-SVs on HCC tumorigenicity using the PRSS3 KO+V Huh7 cell model. Photographs (left panel) and tumor volumes (right panel) of dissected xenograft tumors from different groups of nude mice (n=12) after sacrifice. *P < 0.05, **P < 0.01, one−way ANOVA with Tukey’s post hoc test.
To explore the possible mechanisms by which the PRSS3 transcript variants exerted the dual effects on hepatocarcinogenesis, potential PRSS3-targeted downstream genes were sorted using network analysis of TCGA-LIHC tissue dataset available from SEEK (http://seek.princeton.edu) (Figure S6A), among which 8 key hub genes were shown in most association with PRSS3 transcripts (except PRSS3-V4) either positively (F2RL1, SMPDL3B, DUOX2, SLC43A3, TMEM45A and VNN1) or negatively (GLUL and NKD1) in the network (Figure S6B), consistent with the validation in HCC cells using the CCLE dataset (Tables S1, S7 and S8). In addition to a heatmap visualizing the differential expression of the hub genes (Figure S6C), Figure S6D shows significant upregulation of F2RL1, SMPDL3B, DUOX2, SLC43A3, GLUL and NKD1, but upregulation of TMEM45A and VNN1 in HCC tissues compared with normal human live tissues. As shown in the summarized table based on the available data from UALCAN (https://www.ualcan.path.uab.edu/), there was a divergent association of the clinical significance between PRSS3 and the hub genes (Figure S6E). For instance, the pattern of PRSS3 downregulation associated with the clinical relevance of HCC patients was similar to that of TMEM45A and VNN1, which are positively co-expressed genes of PRSS3 in HCC patients showing oncogenic effects on cancer-associated events (44, 45). However, GLUL and NKD1, completely opposite to PRSS3, showed increased expression related to clinical relevance, displaying the ability to regulate the invasion and migration of hepatocarcinoma cells (46–49). Importantly, PRSS3/MTG linked to F2RL1 (also known as PAR2), was reported to modulate inflammation and tumorigenesis in several cancer types, such as colon cancer and breast cancer (23, 24). In support of this point, Kaplan–Meier survival analysis showed divergent survival curves for HCC patients with high or low expression of the hub genes (Figure S6F). Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis indicated that these cancer-associated genes may be involved in the cell cycle and senescence through the PRSS3-V1-associated p53 signaling pathway, or via the PI3K-Akt pathway in association with PRSS3-V2/PRSS3, due to their corresponding pathways with more parallel lines (Figure S6G). These data therefore suggest that PRSS3 transcripts are bifunctional, possibly via interplay with different cancer-associated gene pathways.
Epigenetic Alteration of PRSS3-V2 Is Associated With Clinical Relevance in Patients With Early HCC
To further explore the contribution of PRSS3 transcripts to tumor heterogeneity, we used the TCGA dataset to analyze their clinical relevance. We found that the expression of PRSS3 and PRSS3-V2 was similarly downregulated but with a gradually increasing tendency in HCC tumors compared with control tissues, following the progression of tumor stages (Figure 6A) and pathological grades (Figure 6B), in which PRSS3-V2Low was significantly detected in tumors of early HCC patients in contrast to PRSS3-V2High in advanced tumors. Kaplan–Meier (K-M) analysis revealed that PRSS3-V2Low was a favorable factor for the overall survival of HCC patients based on cancer stage (Figure 6C) and grade (Figure 6D), in which PRSS3-V2Low patient groups with low-grade tumors showed significantly favorable outcomes (P=0.011). Moreover, divergent disruption of CpG site methylation (A to F) was shown throughout the clinical progression of tumors but occurred more frequently and significantly in tumors of HCC patients with early-stage (Figure 6E) and lower-grade tumors (Figure 6F). In such tumors, alteration in CpG methylation at site D was most inversely correlated with the expression of PRSS3 and PRSS-V2. Since the region located at site D was shown to be an important regulatory region specifically for epigenetic regulation of PRSS3 transcripts (Figure 3), the data suggest that site-specific epigenetic alteration of PRSS3-V2 in HCC tissues was distinct between mPRSS3-V2Low in early HCC and umPRSS3High in advanced HCC patients, in which early HCC patients with PRSS3-V2Low tumors had better outcomes.
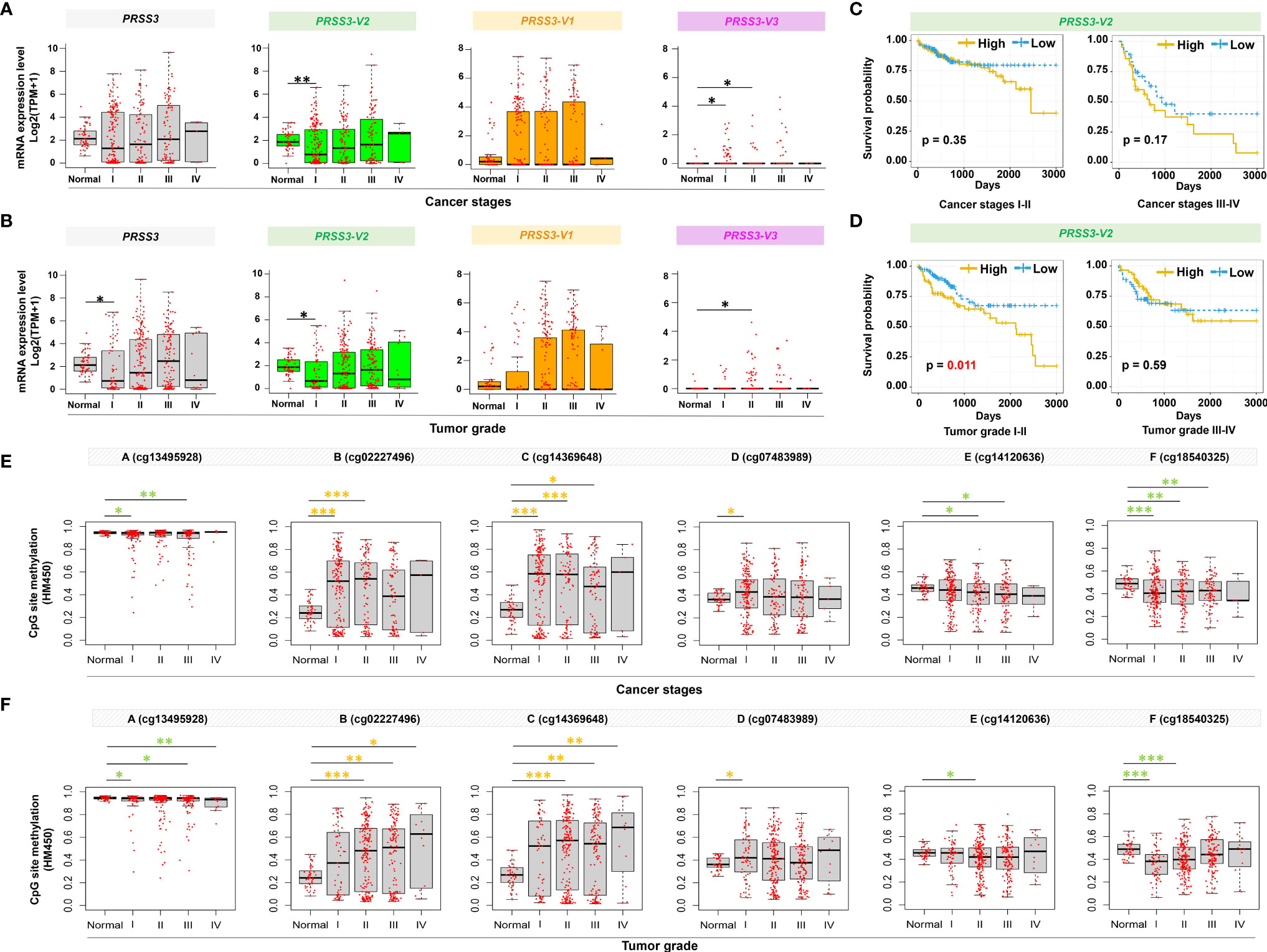
Figure 6 Clinical relevance of epigenetic alteration of PRSS3-SVs in HCC patients. (A, B) Box-and-whisker plot with overlay of individual data points showing mRNA expression of PRSS3 transcripts in HCC tissues (Tumor=371) and normal controls (Normal=50), based on (A) cancer stages (171 stage I, 86 stage II, 85 stage IIII, 5 stage IV) and (B) tumor grades (55 grade I, 177 grade II, 122 grade III, 12 grade IV) (Table 1). (C, D) HCC patients were grouped into PRSS3-V2High and PRSS3-V2Low groups based on the mean value of each transcript in tumors (Table 1). The Kaplan–Meier method was used to determine patient survival, and the log-rank (Mantel–Cox) test was used to compare survival rates. The results of HCC patient survival curves from left to right panels: cancer stages I-II and III-IV (C), tumor grades I-II and tumor grades III-IV (D). (E, F) The association of methylation of CpG sites (A–F) within the extended fragment with different clinical stages (E) and pathological grades (F) in HCC tissue specimens (n=414) in comparison with normal liver control tissues (Normal=41). The data were extracted from the FIREHOSE. Statistical significance was determined by the Wilcoxon rank sum test. Asterisks in green and yellow indicate the changes in hypermethylation and hypomethylation, respectively. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Paradoxical effects of many genes have been observed during tumorigenesis (13, 50, 51). The protease PRSS3 is the first to link the enzyme to prostate cancer, leading to the development of a compound to stop PRSS3 from promoting metastasis (13, 52). Since the high similarity in both sequences and structures to different trypsinogen isoenzymes made it difficult to delineate their functionally associated transcripts distributed in different tissues (13, 16), the protumor (21–31) or antitumor properties of PRSS3 (32–36) were too sophisticated to be deciphered. In this study, we found differentially expressed PRSS3 in HCC due to CpG methylation-mediated epigenetic dysregulation of its splice variants. Different PRSS3-SVs were expressed in HCC, showing a dual role in hepatocarcinogenesis that may increase phenotypic diversity. Our study uncovered epigenetic-mediated PRSS3 transcript variance contributing to the nongenetic phenotypic diversity of HCC (50). To the best of our knowledge, this is the first study of functional dissection of the expression of PRSS3-SVs in cancer and thus has important implications in HCC patient-tailored management.
PRSS3 is a digestive protease with restricted expression in the pancreas. However, the preferential expression of PRSS3-SVs differs in human tissues, suggesting tissue-selective expression. For instance, PRSS3-V2 was exclusively expressed in human pancreatic tissue and fluid encoding MTG (15, 53). Canonical PRSS3-V1 was originally identified in the human brain (17, 53). PRSS3-V3 shares the same TSS with PRSS3-V1 but has a different in-frame exon with a deduced 261-amino acid sequence (formerly named isoform B) (19). PRSS3-V4 was cloned from keratinocytes and shown to participate in keratinocyte terminal differentiation (20). Our study showed the differential expression of PRSS3 as a DEG in HCC across a large expression range that could be used to phenotypically distinguish between PRSS3Low and PRSS3High HCC cells and tissues. Accordingly, we found divergent expression of PRSS3-SVs toward bipolarity following clinical progression from downregulation in early HCC to upregulation in advanced cancer, unveiling the molecular basis of PRSS3 in tissue-selective expression of its splice transcripts in HCC. Despite the infrequent and/or minimal expression of PRSS3-V3 and unexpressed PRSS3-V4, the divergent expression changes of PRSS3-V2 and/or -V1 were major contributors to the transcript heterogeneity of PRSS3 in HCC. Notably, the expression of PRSS3-SVs was dynamically altered following clinical progression from downregulation in early HCC to upregulation in advanced cancer. PRSS3 transcript heterogeneity was further evidenced by its divergent responses to 5-aza-CR treatment of HCC cells, distinguishing between upregulation of PRSS3-V1 or -V3 but downregulation of PRSS3-V2 in PRSS3Low HCC and downregulation of PRSS3-V1 but upregulation of PRSS3-V2 in PRSS3High HCC. The divergent expression of PRSS3 transcripts and their response to 5-aza-CR prompted our consideration of the effects of nongenetic heterogeneity on the chemotherapy response because this well-known anticancer drug has broad clinical applications and mis-splicing regulation, as a nongenetic mechanism is frequently linked to therapy escape (54–56). For precise evaluation of the clinical effectiveness and drug resistance by using a DEG, its functional splice variants, rather than its overall expression, need to be considered. Nevertheless, it was clear that differentially expressed PRSS3 decreased as a whole, which was mainly attributable to its aberrant transcript variance expressed in HCC.
PRSS3 translocates from chromosome 7q34, the loci of PRSS1 and PRSS2, to chromosome 9p11.2, a region frequently containing alterations (13, 57). However, frequent genetic variations occurring in PRSS3 have not yet demonstrated disease-associated PRSS3 variants (https://www.nextprot.org/entry/NX_P35030/medical). Alternative splicing forms a dynamic interactome offering precise therapeutic approaches to correcting cancer-specific defects caused by mis-splicing regulation, in which epigenetics plays an essential role (9, 11, 12, 55, 58–60). Our previous study showed epigenetic silencing of PRSS3 in HCC (36), and we reasoned that epigenetic regulation of PRSS3-SVs contributes to nongenetic heterogeneity in HCC. The different TSSs and start codes in PRSS3 suggest that PRSS3, like the majority of protein-coding genes, tends to be regulated by multiple or alternative promoters, the usage of which provides pretranscriptional control of gene activity to express its different isoforms in a tissue-specific manner (1, 6, 9, 24). Here, we found an extended promoter region covering the upstream and intragenic regions of PRSS3-V1/V3 and PRSS3-V4, providing a site-specific way to regulate the expression of PRSS3-SVs. Both HCC cells and tissues were phenotypically classified as mPRSS3Low and umPRSS3High based on CpG methylation in association with the expression of PRSS3 transcripts. Compared to consistent upstream hypermethylation, site-specific CpG methylation in the intragenic region was found to be more associated with the expression of PRSS3-V1 and PRSS3-V2, suggesting that this extended promoter region played a central role in the regulation of both PRSS3-V1 and PRSS3-V2. Given that epigenetic promoter alterations can change the chromatin accessibility of transcription regulatory elements binding to transcription factors (11, 12, 50, 60–63), the upstream hypermethylation of PRSS3 may impact tissue-specific cis-regulatory modules that may alter the transcriptional activity of PRSS3-SVs in HCC. Dynamic disruption of methylation of different CpG sites within the extended promoter region may affect the occupancy of certain transcriptional regulators or splicing factors, resulting in an alteration in exon skipping to control the expression of PRSS3-V1 or -V3. Meanwhile, site-specific epigenetic control of PRSS3-V2 suggests that the extended promoter may be a distal regulatory region in the regulation of PRSS3-V2 through a very different epigenetic pathway (64). Consistent with this, epigenetic silencing of PRSS3 was found in several cancer types (32–35), and our previous study showed intragenic DNA methylation within the extended promoter region contributing to PRSS3/TRY-4 downregulation in HCC (36). This study was the first to dissect epigenetic heterogeneity in the regulation of PRSS3-SVs, which may provide important implications for understanding epigenetic contributions to the genomic occupancy of transcription factors during transcription, in which many events may appear to be cospliced with distant events (58, 61–63).
Many transcript isoforms can exist per gene (9–11), most of which are thought not to be functionally relevant (65). Recently, comprehensive gain- and loss-of-function studies have shown the functional importance of SVs in tumor heterogeneity by linking genetic variants to individual phenotypes (58–60, 66, 67). PRSS3 appears to be transcribed differentially to display heterogeneous functions in cancer, in which a dual role or contradictory effects reported might be due to MTG (PRSS3-V2) being functionally regarded as PRSS3 (13, 16, 22, 23, 29). We hereby deciphered in vitro and in vivo functional differences among the PRSS3 isoforms by using a constructed Huh7 cell model. Despite PRSS3-V2/MTG-mediated oncogenic effects in HCC in line with the promalignant activities of MTG shown in other cancer types (13, 16, 22, 23, 29), PRSS3-V1/TRY-4 or -V3 were found to be tumor suppressors in HCC cells, while ectopic PRSS3-V4 showed an inhibitory effect on PRSS3ko Huh7 cells. PRSS3ko resulted in protumor effects in Huh7 cells, suggesting a tumor-suppressive role of PRSS3 in HCC that was attributed to the coexpression of PRSS3-V1 and PRSS3-V2, the two isoforms with opposite functionality. This is in line with our previous observations on PRSS3/TRY-4 (36) and may explain some but not all cases of a similar phenotype with well-differentiated and/or low metastatic potential appearing in either PRSS3Low (e.g., HepG2 and SK-Hep1 cells) or PRSS3High (Huh7 cells) live cancer cell lines or a dual role of PRSS3 contradictorily shown in carcinogenesis. To support this, corresponding clinicopathological analysis of HCC specimens compared to the normal tissue controls revealed that PRSS3-V1 and -V2 were main functional components of clinical relevance since PRSS3-V1 and -V2 were bipolarly present in either PRSS3Low or PRSS3High tissues; therefore, their abnormal coexpression could result in functional heterogeneity including insignificant or paradoxical clinical associations. However, a signature pattern of epigenetic regulation of PRSS3 expression by site-specific CpG methylation was dynamically shown from mPRSS3Low to umPRSS3High through clinical progression, better matched to PRSS3-V2, suggesting PRSS3-V2 to be a more prevalent isoform functionally through clinical progression of HCC. Accordingly, significant epigenetic downregulation of PRSS3-V2 was observed in early HCC with favorable patient outcomes. This finding supports an oncogenic role of PRSS3-V2/MTG predominantly in HCC, thus providing early diagnostic and prognostic value for HCC (16, 22, 23, 29). Thus, our study provides additional evidence supporting the hypothesis of functional hepato-heterogeneity attributed to genetic and epigenetic factors (1, 2, 4, 6).
Aberrant expression of SVs in cancer generates functional tumor heterogeneity resulting in cellular phenotype(s) or influencing cell fate determination (1, 4, 7, 8). In this regard, delineation of the heterogeneity of PRSS3 expression and epigenetic regulation is critical for clarifying the molecular basis of PRSS3 transcripts, thus facilitating functional interpretation of the paradoxical effects of PRSS3 in cancer development. Functional classification and experimental dissection of PRSS3-SVs and their response to 5-aza-CR treatment distinct between PRSS3Low and PRSS3High HCC cells (such as Huh7 versus HepG2 cells) may be used as an experimental model for studying PRSS3 splicing-mediated functional heterogeneity during hepatocarcinogenesis. In contrast to permanent genetic mutations, epigenetic disruptions frequently occur in early clinical stages and play an important role in modulating cell malignancy in a progressive and reversible manner. Therefore, delineation of the precise molecular mechanisms underlying epigenetic regulation of PRSS3-SVs could contribute to the molecular phenotypes of HCC.
This study on bioinformatic analysis of RNA sequencing data of PRSS3-SVs and their clinical relevance gave many insignificantly divergent results. For instance, PRSS3Low was shown in 50 paired HCC tissues, consistent with our previous observation (36) and the analyses shown in the TCGA and UALCAN portals (38). However, its decrease was no longer statistically significant in more HCC tissue specimens due to different statistical methods or integration of the RNA-seq data with different median cutoff values for extensively divergent expression of PRSS3-SVs in HCC specimens. Therefore, conventional parameters, such as the median cutoff values, may need to be reevaluated for grouping a DEG with divergent expression levels. Moreover, functional heterogeneity could be caused by the microenvironment enhancing the coexpression diversity of PRSS3-SVs. As a result, further studies with larger sample sizes of paired HCC specimens are warranted to validate our observations. Moreover, due to the lack of commercial antibodies capable of discriminating well among PRSS3 isoforms, the functional pathways corresponding to PRSS3 isoforms could not be precisely distinguished from each other. This may yield inconsistent reports of PRSS3 effects on carcinogenesis, resulting in inconclusive informatics analyses of the molecular mechanisms related to PRSS3 isoforms. Therefore, customized generation of more isoform-specific antibodies will be the subject of our future investigation to explore the molecular mechanisms underlying the dual role of PRSS3 transcript isoforms in cancer development.
Conclusions
In summary, PRSS3 was aberrantly expressed in HCC due to epigenetic dysregulation that was integrated with divergent expression of PRSS3-SVs by site-specific CpG methylation. The effects of oncogenic PRSS3-V2 and tumor-suppressive PRSS3-V1 in HCC cells may increase the molecular diversity and functional plasticity of hepatocarcinogenesis. Epigenetic dysregulation of PRSS3-V2 distinct between mPRSS3-V2Low in early clinical stages and umPRSS3High in advanced tumors has potential diagnostic value for patients with early HCC (Figure 7).
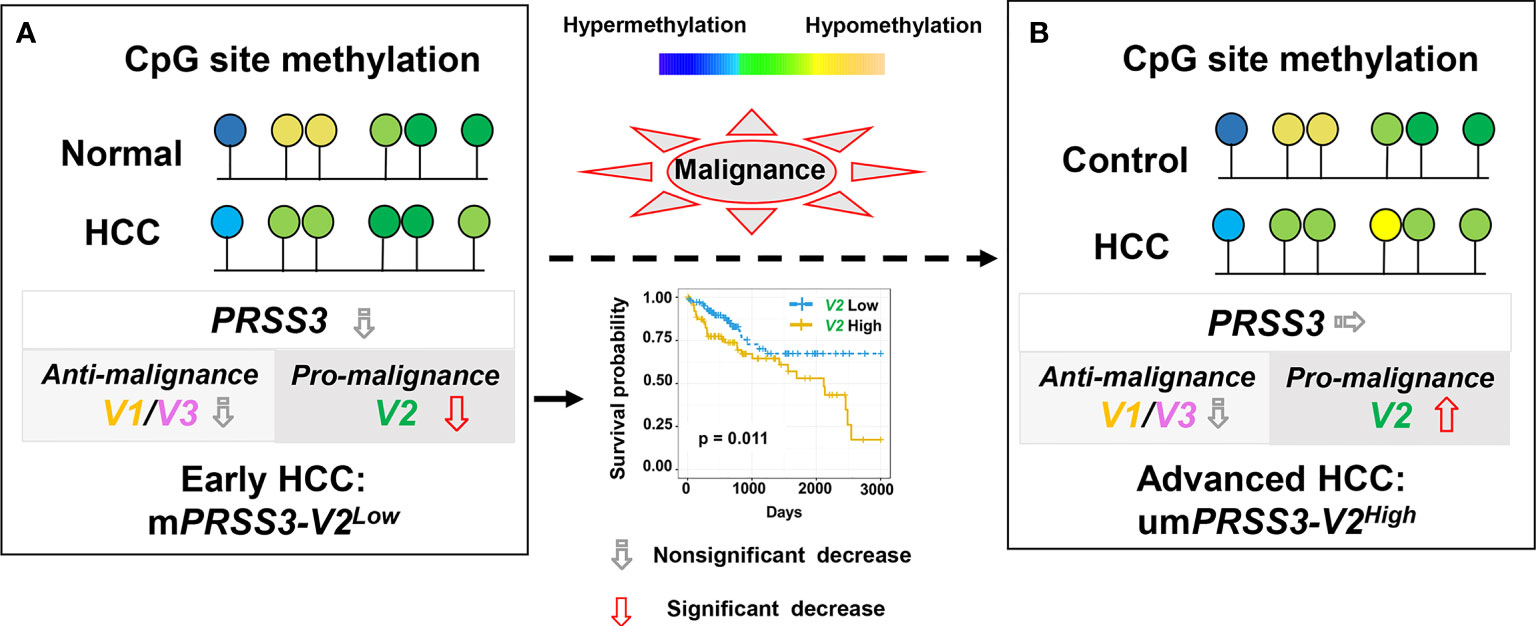
Figure 7 A schematic model for epigenetic dysregulation of PPRSS3 transcripts functionally contributing to hepatocarcinogenesis and its biomarker potential. (A) Epigenetic silencing of PRSS3-SVs by site-specific CpG methylation in the tumors of patients with early HCC, in which mPRSS3Low was a potential biomarker favorable for patient survival. (B) Epigenetic disruption resulted in umPRSS3High in tumors of advanced HCC patients.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
The animal handling protocols and all in vivo experimental procedures were approved by the Institutional Animal Ethics Committee of the Beijing Chest Hospital.
Author Contributions
Conceptualization, SL, HX and JH; Formal analysis, SL, HX, MP, XG, XCZ, LZ, FJ, YH, WW, JR and JH; Funding acquisition, JW, MG and JH; Investigation, SL, HX, XMZ, MP, LZ, XG, YP and ZY; Methodology, SL, XMZ, HX, MP, XW, BL, RT, XCZ, ZY and JH; Resources, KC and WG; Supervision, MG and JH; writing-original draft, SL and JH; writing-review & editing, JW and JH. All authors have reviewed and agreed to the final version of the manuscript.
Funding
This study was funded by the Scientific Research Project of Beijing Educational Committee (Grant No. KM202110025004), the Intramural Research Funding Program from Beijing Tuberculosis and Thoracic Tumor Research Institute/Beijing Chest Hospital, National Key Research and Development Program of China (2018YFA0208902, 2020YFC2002705); Beijing Natural Science Foundation of China (7214242, 7171008), National Science Foundation of China (NSFC Grant No. 81872021, U1604281, 81672138); National Key Scientific Instrument Special Program of China (Grant No. 2011YQ03013405). KC, JH and JMW were also funded in part by Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E and were supported in part by the Intramural Research Program of the NCI, CCR, LCIM, NIH.
Conflict of Interest
Author WG is employed by Basic Research Program, Leidos Biomedical Research, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all our laboratory members for technical help and helpful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.831268/full#supplementary-material
Abbreviations
5-aza-CR, 5 aza-2’-deoxycytidine; CCLE, Cancer Cell Line Encyclopedia; DEG, Differentially expressed gene; DepMap, The Cancer Dependency Map; FPKM, Fragments Per Kilobase Million; GTEx, Genotype-Tissue Expression; GEPIA, Gene Expression Profiling Interactive Analysis; HCC, Hepatocellular carcinoma; LIHC, Liver hepatocellular carcinoma; MeDIP-qPCR, Methylated DNA immunoprecipitation-qPCR; mPRSS3Low lowly expressed PRSS3-SVs with hypermethylation; MS-qPCR, Methylation-specific qPCR; MTG, Trypsin-3 isoform 2/mesotrypsinogen; PRSS3, Serine protease 3 gene; PRSS3, Serine protease 3; PRSS3High or PRSS3Low, Highly or minimally expressed PRSS3; PRSS3-V1~-V4, PRSS3 transcript variants 1, -2, -3 and -4; PRSS3-SVs, Splice transcripts of PRSS3; TCGA, The Cancer Genome Atlas; TPM, Transcripts Per Million; TRY4, Trypsin-3 isoform 1/trypsinogen 4; umPRSS3High, highly expressed PRSS3-SVs with hypomethylation.
References
1. Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell (2017) 169(7):1327–41.e1323. doi: 10.1016/j.cell.2017.05.046
2. Lin DC, Mayakonda A, Dinh HQ, Huang P, Lin L, Liu X, et al. Genomic and Epigenomic Heterogeneity of Hepatocellular Carcinoma. Cancer Res (2017) 77(9):2255–65. doi: 10.1158/0008-5472.Can-16-2822
3. Llovet JM, Montal R, Sia D, Finn RS. Molecular Therapies and Precision Medicine for Hepatocellular Carcinoma. Nat Rev Clin Oncol (2018) 15(10):599–616. doi: 10.1038/s41571-018-0073-4
4. Dhanasekaran R. Deciphering Tumor Heterogeneity in Hepatocellular Carcinoma (HCC)-Multi-Omic and Singulomic Approaches. Semin Liver Dis (2021) 41(1):9–18. doi: 10.1055/s-0040-1722261
5. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
6. Qiu Z, Li H, Zhang Z, Zhu Z, He S, Wang X, et al. A Pharmacogenomic Landscape in Human Liver Cancers. Cancer Cell (2019) 36(2):179–93.e111. doi: 10.1016/j.ccell.2019.07.001
7. Nam AS, Chaligne R, Landau DA. Integrating Genetic and non-Genetic Determinants of Cancer Evolution by Single-Cell Multi-Omics. Nat Rev Genet (2021) 22(1):3–18. doi: 10.1038/s41576-020-0265-5
8. Chan LK, Tsui YM, Ho DW, Ng IO. Cellular Heterogeneity and Plasticity in Liver Cancer. Semin Cancer Biol (2021) S1044-579X(21):00050-X. doi: 10.1016/j.semcancer.2021.02.015
9. Dvinge H, Guenthoer J, Porter PL, Bradley RK. RNA Components of the Spliceosome Regulate Tissue- and Cancer-Specific Alternative Splicing. Genome Res (2019) 29(10):1591–604. doi: 10.1101/gr.246678.118
10. Bonnal SC, López-Oreja I, Valcárcel J. Roles and Mechanisms of Alternative Splicing in Cancer - Implications for Care. Nat Rev Clin Oncol (2020) 17(8):457–74. doi: 10.1038/s41571-020-0350-x
11. Sonneveld S, Verhagen BMP, Tanenbaum ME. Heterogeneity in mRNA Translation. Trends Cell Biol (2020) 30(8):606–18. doi: 10.1016/j.tcb.2020.04.008
12. Zhang J, Zhang YZ, Jiang J, Duan CG. The Crosstalk Between Epigenetic Mechanisms and Alternative RNA Processing Regulation. Front Genet (2020) 11:998. doi: 10.3389/fgene.2020.00998
13. Lópezotín C, Matrisian LM. Emerging Roles of Proteases in Tumour Suppression. Nat Rev Cancer (2007) 7(10):800–8. doi: 10.1038/nrc2228
14. Rinderknecht H, Renner IG, Abramson SB, Carmack C. Mesotrypsin: A New Inhibitor-Resistant Protease From a Zymogen in Human Pancreatic Tissue and Fluid. Gastroenterology (1984) 86(4):681–92. doi: 10.1016/S0016-5085(84)80117-1
15. Nyaruhucha CN, Kito M, Fukuoka SI. Identification and Expression of the cDNA-Encoding Human Mesotrypsin(Ogen), an Isoform of Trypsin With Inhibitor Resistance. J Biol Chem (1997) 272(16):10573–8. doi: 10.1074/jbc.272.16.10573
16. Salameh MA, Radisky ES. Biochemical and Structural Insights Into Mesotrypsin: An Unusual Human Trypsin. Int J Biochem Mol Biol (2013) 4(3):129–39.
17. Wiegand U, Corbach S, Minn A, Kang J, Müller-Hill B. Cloning of the cDNA Encoding Human Brain Trypsinogen and Characterization of its Product. Gene (1993) 136(1-2):167–75. doi: 10.1016/0378-1119(93)90460-k
18. Tóth J, Siklódi E, Medveczky P, Gallatz K, Németh P, Szilágyi L, et al. Regional Distribution of Human Trypsinogen 4 in Human Brain at mRNA and Protein Level. Neurochem Res (2007) 32(9):1423–33. doi: 10.1007/s11064-007-9327-8
19. Cottrell GS, Amadesi S, Grady EF, Bunnett NW. Trypsin IV, a Novel Agonist of Protease-Activated Receptors 2 and 4. J Biol Chem (2004) 279(14):13532–9. doi: 10.1074/jbc.M312090200
20. Nakanishi J, Yamamoto M, Koyama J, Sato J, Hibino T. Keratinocytes Synthesize Enteropeptidase and Multiple Forms of Trypsinogen During Terminal Differentiation. J Invest Dermatol (2010) 130(4):944. doi: 10.1038/jid.2009.364
21. Terada T, Ohta T, Minato H, Nakanuma Y. Expression of Pancreatic Trypsinogen/Trypsin and Cathepsin B in Human Cholangiocarcinomas and Hepatocellular Carcinomas. Hum Pathol (1995) 26(7):746–52. doi: 10.1016/0046-8177(95)90222-8
22. Jiang G, Cao F, Ren G, Gao D, Bhakta V, Zhang Y, et al. PRSS3 Promotes Tumour Growth and Metastasis of Human Pancreatic Cancer. Gut (2010) 59(11):1535–44. doi: 10.1136/gut.2009.200105
23. Hockla A, Miller E, Salameh MA, Copland JA, Radisky DC, Radisky ES. PRSS3/mesotrypsin Is a Therapeutic Target for Metastatic Prostate Cancer. Mol Cancer Res (2012) 10(12):1555–66. doi: 10.1158/1541-7786.Mcr-12-0314
24. Han S, Lee CW, Trevino JG, Hughes SJ, Sarosi GA Jr. Autocrine Extra-Pancreatic Trypsin 3 Secretion Promotes Cell Proliferation and Survival in Esophageal Adenocarcinoma. PloS One (2013) 8(10):e76667. doi: 10.1371/journal.pone.0076667
25. Radisky ES. PRSS3/mesotrypsin in Prostate Cancer Progression: Implications for Translational Medicine. Asian J Androl (2013) 15(4):439–40. doi: 10.1038/aja.2013.14
26. Ghilardi C, Silini A, Figini S, Anastasia A, Lupi M, Fruscio R, et al. Trypsinogen 4 Boosts Tumor Endothelial Cells Migration Through Proteolysis of Tissue Factor Pathway Inhibitor-2. Oncotarget (2015) 6(29):28389–400. doi: 10.18632/oncotarget.4949
27. Ma R, Ye X, Cheng H, Ma Y, Cui H, Chang X. PRSS3 Expression is Associated With Tumor Progression and Poor Prognosis in Epithelial Ovarian Cancer. Gynecol Oncol (2015) 137(3):546–52. doi: 10.1016/j.ygyno.2015.02.022
28. Qian L, Gao X, Huang H, Lu S, Cai Y, Hua Y, et al. PRSS3 is a Prognostic Marker in Invasive Ductal Carcinoma of the Breast. Oncotarget (2017) 8(13):21444–53. doi: 10.18632/oncotarget.15590
29. Ma H, Hockla A, Mehner C, Coban M, Papo N, Radisky DC, et al. PRSS3/Mesotrypsin and Kallikrein-Related Peptidase 5 are Associated With Poor Prognosis and Contribute to Tumor Cell Invasion and Growth in Lung Adenocarcinoma. Sci Rep (2019) 9(1):1844. doi: 10.1038/s41598-018-38362-0
30. Aboulouard S, Wisztorski M, Duhamel M, Saudemont P, Cardon T, Narducci F, et al. In-Depth Proteomics Analysis of Sentinel Lymph Nodes From Individuals With Endometrial Cancer. Cell Rep Med (2021) 2(6):100318. doi: 10.1016/j.xcrm.2021.100318
31. Schaid DJ, McDonnell SK, FitzGerald LM, DeRycke L, Fogarty Z, Giles GG, et al. Two-Stage Study of Familial Prostate Cancer by Whole-Exome Sequencing and Custom Capture Identifies 10 Novel Genes Associated With the Risk of Prostate Cancer. Eur Urol (2021) 79(3):353–61. doi: 10.1016/j.eururo.2020.07.038
32. Keishi Y, Koshi M, Hiroshi I, Masaki M, David S. A Tumor-Suppressive Role for Trypsin in Human Cancer Progression. Cancer Res (2003) 63(20):6575–8.
33. Marsit CJ, Karagas MR, Schned A, Kelsey KT. Carcinogen Exposure and Epigenetic Silencing in Bladder Cancer. Ann N Y Acad Sci (2006) 1076:810–21. doi: 10.1196/annals.1371.031
34. Marsit CJ, Houseman EA, Schned AR, Karagas MR, Kelsey KT. Promoter Hypermethylation is Associated With Current Smoking, Age, Gender and Survival in Bladder Cancer. Carcinogenesis (2007) 28(8):1745–51. doi: 10.1093/carcin/bgm116
35. Marsit CJ, Chinedu O, Hadi D, Kelsey KT. Epigenetic Silencing of the PRSS3 Putative Tumor Suppressor Gene in non-Small Cell Lung Cancer. Mol Carcinog (2010) 44(2):146–50. doi: 10.1002/mc.20125
36. Lin B, Zhou X, Lin S, Wang X, Zhang M, Cao B, et al. Epigenetic Silencing of PRSS3 Provides Growth and Metastasis Advantage for Human Hepatocellular Carcinoma. J Mol Med (Berl) (2017) 95(11):1237–49. doi: 10.1007/s00109-017-1578-5
37. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res (2017) 45(W1):W98–w102. doi: 10.1093/nar/gkx247
38. Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, et al. A Pathology Atlas of the Human Cancer Transcriptome. Science (2017) 357(6352):eaan2507. doi: 10.1126/science.aan2507
39. Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature (2019) 569(7757):503–8. doi: 10.1038/s41586-019-1186-3
40. Ben-David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, et al. Genetic and Transcriptional Evolution Alters Cancer Cell Line Drug Response. Nature (2018) 560(7718):325–30. doi: 10.1038/s41586-018-0409-3
41. Deng M, Brägelmann J, Kryukov I, Saraiva-Agostinho N, Perner S. FirebrowseR: An R Client to the Broad Institute's Firehose Pipeline. Database (Oxf) (2017) 2017:baw160. doi: 10.1093/database/baw160
42. Chen F, Chandrashekar DS, Varambally S, Creighton CJ. Pan-Cancer Molecular Subtypes Revealed by Mass-Spectrometry-Based Proteomic Characterization of More Than 500 Human Cancers. Nat Commun (2019) 10(1):5679. doi: 10.1038/s41467-019-13528-0
43. Lin S, Wang X, Pan Y, Tian R, Lin B, Jiang G, et al. Transcription Factor Myeloid Zinc-Finger 1 Suppresses Human Gastric Carcinogenesis by Interacting With Metallothionein 2a. Clin Cancer Res (2019) 25(3):1050–62. doi: 10.1158/1078-0432.Ccr-18-1281
44. Løvf M, Nome T, Bruun J, Eknaes M, Bakken AC, Mpindi JP, et al. A Novel Transcript, VNN1-AB, as a Biomarker for Colorectal Cancer. Int J Cancer (2014) 135(9):2077–84. doi: 10.1002/ijc.28855
45. Schmit K, Michiels C. TMEM Proteins in Cancer: A Review. Front Pharmacol (2018) 9:1345. doi: 10.3389/fphar.2018.01345
46. Zhang S, Li J, Wang X. NKD1 Correlates With a Poor Prognosis and Inhibits Cell Proliferation by Inducing P53 Expression in Hepatocellular Carcinoma. Tumour Biol (2016) 37(10):14059–67. doi: 10.1007/s13277-016-5173-0
47. Lin YY, Yu MW, Lin SM, Lee SD, Chen CL, Chen DS, et al. Genome-Wide Association Analysis Identifies a GLUL Haplotype for Familial Hepatitis B Virus-Related Hepatocellular Carcinoma. Cancer (2017) 123(20):3966–76. doi: 10.1002/cncr.30851
48. Wang C, Mou L, Chai HX, Wang F, Yin YZ, Zhang XY. Long non-Coding RNA HNF1A-AS1 Promotes Hepatocellular Carcinoma Cell Proliferation by Repressing NKD1 and P21 Expression. BioMed Pharmacother (2017) 89:926–32. doi: 10.1016/j.biopha.2017.01.031
49. Schmidt A, Armento A, Bussolati O, Chiu M, Ellerkamp V, Scharpf MO, et al. Hepatoblastoma: Glutamine Depletion Hinders Cell Viability in the Embryonal Subtype But High GLUL Expression is Associated With Better Overall Survival. J Cancer Res Clin Oncol (2021) 147(11):3169–81. doi: 10.1007/s00432-021-03713-4
50. Li Y, McGrail DJ, Xu J, Mills GB, Sahni N, Yi S. Gene Regulatory Network Perturbation by Genetic and Epigenetic Variation. Trends Biochem Sci (2018) 43(8):576–92. doi: 10.1016/j.tibs.2018.05.002
51. Belluti S, Rigillo G, Imbriano C. Transcription Factors in Cancer: When Alternative Splicing Determines Opposite Cell Fates. Cells (2020) 9(3):760. doi: 10.3390/cells9030760
52. Cohen I, Coban M, Shahar A, Sankaran B, Hockla A, Lacham S, et al. Disulfide Engineering of Human Kunitz-Type Serine Protease Inhibitors Enhances Proteolytic Stability and Target Affinity Toward Mesotrypsin. J Biol Chem (2019) 294(13):5105–20. doi: 10.1074/jbc.RA118.007292
53. Tani T, Kawashima I, Mita K, Takiguchi Y. Nucleotide Sequence of the Human Pancreatic Trypsinogen III cDNA. Nucleic Acids Res (1990) 18(6):1631. doi: 10.1093/nar/18.6.1631
54. Ramesh V, Ganesan K. Integrative Functional Genomic Analysis Unveils the Differing Dysregulated Metabolic Processes Across Hepatocellular Carcinoma Stages. Gene (2016) 588(1):19–29. doi: 10.1016/j.gene.2016.04.039
55. Urbanski LM, Leclair N, Anczuków O. Alternative-Splicing Defects in Cancer: Splicing Regulators and Their Downstream Targets, Guiding the Way to Novel Cancer Therapeutics. Wiley Interdiscip Rev RNA (2018) 9(4):e1476. doi: 10.1002/wrna.1476
56. Duchmann M, Itzykson R. Clinical Update on Hypomethylating Agents. Int J Hematol (2019) 110(2):161–9. doi: 10.1007/s12185-019-02651-9
57. Rowen L, Williams E, Glusman G, Linardopoulou E, Friedman C, Ahearn ME, et al. Interchromosomal Segmental Duplications Explain the Unusual Structure of PRSS3, the Gene for an Inhibitor-Resistant Trypsinogen. Mol Biol Evol (2005) 22(8):1712–20. doi: 10.1093/molbev/msi166
58. Sharma A, Merritt E, Hu X, Cruz A, Jiang C, Sarkodie H, et al. Non-Genetic Intra-Tumor Heterogeneity Is a Major Predictor of Phenotypic Heterogeneity and Ongoing Evolutionary Dynamics in Lung Tumors. Cell Rep (2019) 29(8):2164–74.e2165. doi: 10.1016/j.celrep.2019.10.045
59. Calviello L, Hirsekorn A, Ohler U. Quantification of Translation Uncovers the Functions of the Alternative Transcriptome. Nat Struct Mol Biol (2020) 27(8):717–25. doi: 10.1038/s41594-020-0450-4
60. Gopi LK, Kidder BL. Integrative Pan Cancer Analysis Reveals Epigenomic Variation in Cancer Type and Cell Specific Chromatin Domains. Nat Commun (2021) 12(1):1419. doi: 10.1038/s41467-021-21707-1
61. Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene Body Methylation can Alter Gene Expression and is a Therapeutic Target in Cancer. Cancer Cell (2014) 26(4):577–90. doi: 10.1016/j.ccr.2014.07.028
62. Demircioğlu D, Cukuroglu E, Kindermans M, Nandi T, Calabrese C, Fonseca NA, et al. A Pan-Cancer Transcriptome Analysis Reveals Pervasive Regulation Through Alternative Promoters. Cell (2019) 178(6):1465–77.e1417. doi: 10.1016/j.cell.2019.08.018
63. Gomez L, Odom GJ, Young JI, Martin ER, Liu L, Chen X, et al. Comethdmr: Accurate Identification of Co-Methylated and Differentially Methylated Regions in Epigenome-Wide Association Studies With Continuous Phenotypes. Nucleic Acids Res (2019) 47(17):e98. doi: 10.1093/nar/gkz590
64. Schoenfelder S, Fraser P. Long-Range Enhancer-Promoter Contacts in Gene Expression Control. Nat Rev Genet (2019) 20(8):437–55. doi: 10.1038/s41576-019-0128-0
65. Gallego-Paez LM, Bordone MC, Leote AC, Saraiva-Agostinho N, Ascensão-Ferreira M, Barbosa-Morais NL. Alternative Splicing: The Pledge, the Turn, and the Prestige : The Key Role of Alternative Splicing in Human Biological Systems. Hum Genet (2017) 136(9):1015–42. doi: 10.1007/s00439-017-1790-y
66. eGTEx Project. Enhancing GTEx by Bridging the Gaps Between Genotype, Gene Expression, and Disease. Nat Genet (2017) 49(12):1664–70. doi: 10.1038/ng.3969
Keywords: liver cancer, hepatocellular carcinoma, PRSS3, transcript variant, intratumor heterogeneity, CpG methylation, epigenetics, biomarker
Citation: Lin S, Xu H, Pang M, Zhou X, Pan Y, Zhang L, Guan X, Wang X, Lin B, Tian R, Chen K, Zhang X, Yang Z, Ji F, Huang Y, Wei W, Gong W, Ren J, Wang JM, Guo M and Huang J (2022) CpG Site-Specific Methylation-Modulated Divergent Expression of PRSS3 Transcript Variants Facilitates Nongenetic Intratumor Heterogeneity in Human Hepatocellular Carcinoma. Front. Oncol. 12:831268. doi: 10.3389/fonc.2022.831268
Received: 08 December 2021; Accepted: 16 March 2022;
Published: 11 April 2022.
Edited by:
Hongquan Zhang, Peking University Health Science Centre, ChinaReviewed by:
Haiwei Mou, Cold Spring Harbor Laboratory, United StatesJianxiang Chen, Hangzhou Normal University, China
Copyright © 2022 Lin, Xu, Pang, Zhou, Pan, Zhang, Guan, Wang, Lin, Tian, Chen, Zhang, Yang, Ji, Huang, Wei, Gong, Ren, Wang, Guo and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiaqiang Huang, anFodWFuZ0BianR1LmVkdS5jbg==; Mingzhou Guo, bXpndW9AaG90bWFpbC5jb20=
†These authors share first authorship