 Wei Liu
Wei Liu Patric Teodorescu
Patric Teodorescu Stephanie Halene
Stephanie Halene Gabriel Ghiaur
Gabriel Ghiaur
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Oncol. , 16 March 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.815037
This article is part of the Research Topic Advancing Science for Clinical Care in MDS View all 6 articles
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal bone-marrow diseases with ineffective hematopoiesis resulting in cytopenias and morphologic dysplasia of hematopoietic cells. MDS carry a wide spectrum of genetic abnormalities, ranging from chromosomal abnormalities such as deletions/additions, to recurrent mutations affecting the spliceosome, epigenetic modifiers, or transcription factors. As opposed to AML, research in MDS has been hindered by the lack of preclinical models that faithfully replicate the complexity of the disease and capture the heterogeneity. The complex molecular landscape of the disease poses a unique challenge when creating transgenic mouse-models. In addition, primary MDS cells are difficult to manipulate ex vivo limiting in vitro studies and resulting in a paucity of cell lines and patient derived xenograft models. In recent years, progress has been made in the development of both transgenic and xenograft murine models advancing our understanding of individual contributors to MDS pathology as well as the complex primary interplay of genetic and microenvironment aberrations. We here present a comprehensive review of these transgenic and xenograft models for MDS and future directions.
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal bone-marrow diseases which have in common ineffective hematopoiesis resulting in cytopenias and morphologic dysplasia of hematopoietic cells. MDS is the most common myeloid malignancy in the United States, with a median age at diagnosis of 72 years (1). The diagnosis needs to be supported by the presence of persistent cytopenias (otherwise unexplained) of at least one lineage and morphologic dysplasia of hematopoietic elements or the presence of certain genetic aberrations [del(5q)]. Genetic evidence of clonal hematopoiesis can also contribute to the diagnosis, but as of today, this is not required (Hasserjian, Pathobiology 2019).
The spectrum of genetic abnormalities identified in MDS is wide, ranging from chromosomal abnormalities such as deletions/additions (del(5q), del(7q)), to specific mutations affecting the spliceosome (SF3B1, SRSF2), epigenetic changes (TET2, ASXL1, DNMT3A) or transcription factors (RUNX1, ETV6). While the mechanism by which some of these mutations lead to disease are not fully understood, a number of them have important implications for diagnosis and prognosis (i.e., SF3B1 – ring sideroblasts), and can guide initial treatment (del(5q) – lenalidomide, SF3B1 – luspatercept).
The course of the disease is variable, correlating with the risk-category. Thus, low-risk MDS has an indolent course, characterized by low-grade cytopenias, not requiring treatment for a long time. On the other hand, high-risk MDS is an aggressive disease characterized by profound cytopenias requiring urgent treatment and increased progression to AML. To some extent, high, and low risk MDS appear as two biologically distinct entities. To this end, they also display different combinations of somatic mutations. For instance, SF3B1 are more likely to segregate with low-risk disease (particularly SF3B1K700E), whereas other mutations (i.e., ASXL1, RUNX1, TP53, EZH2, ETV6 and SF3B1K666N) are usually associated in high-risk MDS.
Currently, the therapeutic options in MDS are limited and include supportive care (blood transfusions, antibiotics) and several pharmacologic interventions. Erythropoietin and hypomethylating agents, such as Azacitidine and Decitabine have been the main therapeutic interventions for many years, and immunomodulating agents, such as Lenalidomide are beneficial for patients with del(5q). Most recently, luspatercept (a TGFβ-pathway activin receptor trap) was approved for the treatment of transfusion-dependent MDS with ring-sideroblasts. Nevertheless, none of these treatment options are curative, and in the absence of bone marrow transplantation patients eventually succumb to cytopenias-related complications (infections, hemorrhage) or progression to AML. The exact mechanism by which specific therapeutic interventions interact with downstream consequences of various mutations is currently unknown (2).
As opposed to AML, research in MDS has been hindered by the lack of preclinical models. First of all, the complex molecular landscape of the disease poses a unique challenge when creating transgenic mouse-models. In addition, primary MDS cells are difficult to manipulate ex vivo resulting in a paucity of cell lines and patient derived xenograft models. While high-risk MDS models are probably closer to AML models, the aforementioned challenges are especially difficult to overcome when modeling low-risk MDS. However, in recent years, progress has been made to develop both transgenic and xenograft strategies, with some models reproducing the disease more closely than others.
Murine models offer sophisticated tools to dissect unique aspects of mammalian hematopoiesis from phenotype – function relations in various compartments to complex interactions between hematopoietic cells and their microenvironment during ontogeny. More so, breeding strategies and the development of pure genetic backgrounds has allowed the scientific community to isolate and clearly define the impact of genetic alterations on hematopoiesis.
In 2002 the Hematopathology subcommittee of the Mouse Models of Human Cancers Consortium devised criteria for MDS in mouse models to allow investigators to diagnose lesions as well-defined entities according to universally accepted criteria. Using peripheral blood findings, cytologic features of hematopoietic tissues, histopathology, immunophenotyping, genetic features, and clinical course, they distinguished nonlymphoid leukemias, nonlymphoid hematopoietic sarcomas, myeloid dysplasia, and non-reactive myeloid proliferations (3). These criteria have allowed the uniform evaluation of models and have been particularly useful in mouse models of MDS (Figure 1A).
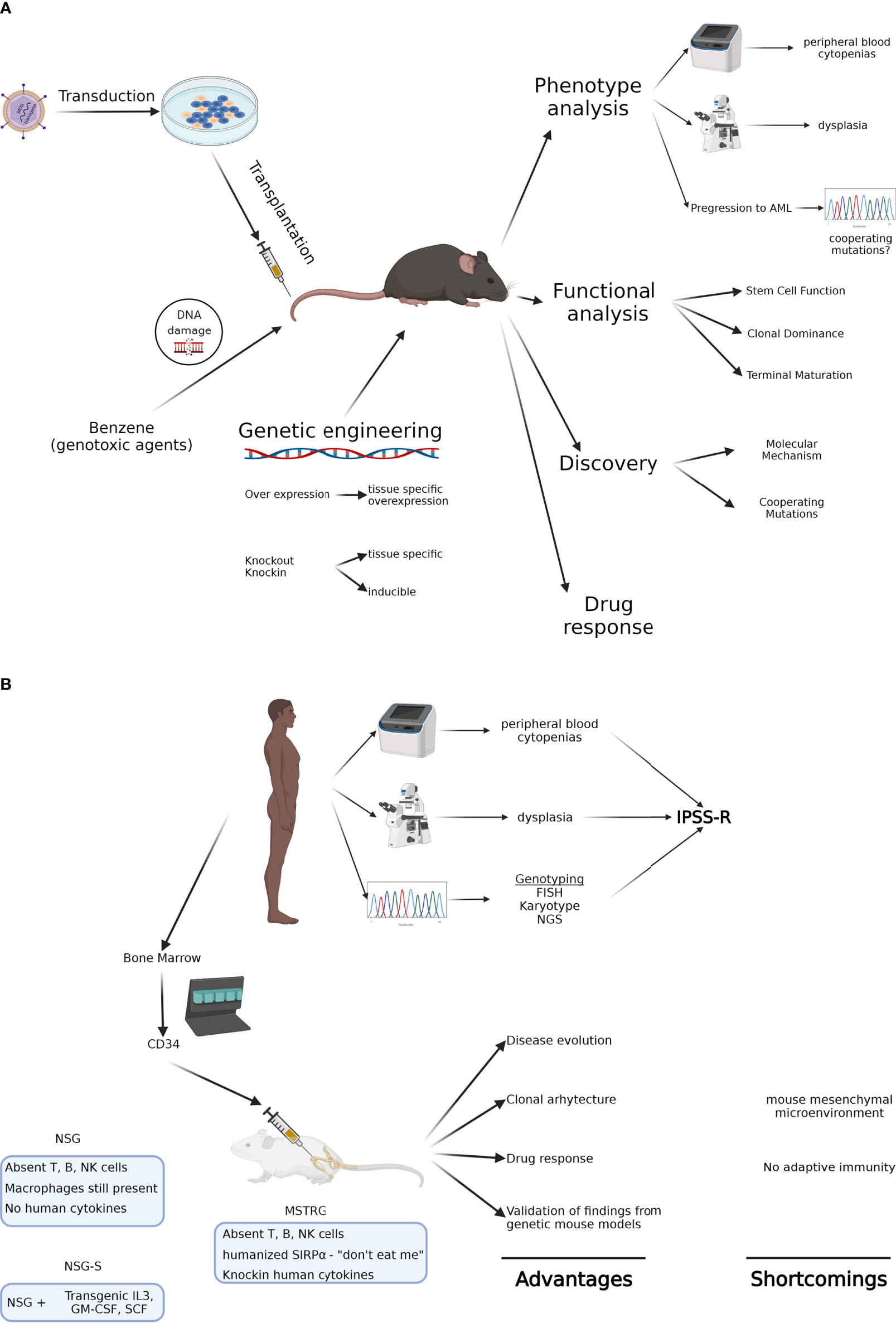
Figure 1 (A) Genetic mouse models of MDS. Using various approaches, these animals allow modeling of MDS in immune competent hosts and in the presence of the endogenous, often unmutated microenvironment. They also provide the analytical tools to study how various mutations impact stem cell function and clonal dominance. Their major shortcoming is that they don’t capture the genetic heterogeneity of MDS. (B) Xenograft mouse models of MDS. Using patient-derived MDS cells, these animals allow modeling of genetically complex disease and the study of clonal architecture and clonal evolution. Most recent humanized immunodeficient mice can even model erythroid maturation, though limited generation of neutrophils and platelets are thus far a major limitation. Created with BioRender.com.
Initial efforts to model MDS in mice used exposure to genotoxic agents to create the disease (4–6). These models provided insight into adaptive physiological processes that are deployed in the setting of bone marrow failure from MDS. Even though this strategy resulted in complex genetic diseases, it was not suitable to explore the contribution of individual gene mutations to the development of MDS.
Gene expression analysis resulting from microarray and more recently RNA sequencing studies hinted towards recurrent molecular patterns present in patients with MDS. Effort to reproduce these patterns in mice took advantage of either “transduction-transplantation” approaches or transgenic mouse technology to create murine models of MDS. In 2010 Beachy et al. provided a comprehensive review of mouse models engineered to replicate alterations in gene expression identified in patients with MDS, replicating some but not all aspects of MDS (7). These models included combinations of Pten/Ship deletions, Evi1 overexpression, Npm1 deletion, Dido deletion, Nup98-Hoxd13 fusion, SALL4B overexpression, co-expression of BCL-2 and mutant NRAS (NrasD12), over-expression of mutant Runx1, deletion of Arid4a, and knock-in of mutant Polg. Though an in depth discussion of each of these mice is beyond the scope of this limited review, we will highlight three of these mouse models to capture the diverse biology of MDS.
Overexpression of Nup98-Hoxd13 fusion under control of the Vav1 promoter generated perhaps one of the most used transgenic mouse models of MDS, NHD13. NUP98 encodes a component of the nuclear pore complex that mediates nucleo-cytoplasmic transport of RNA and protein. Translocations of this gene have been identified in various hematologic malignancies (MDS, AML, CML, pre-T LBL) and frequently the partner genes encode homeodomain proteins belonging to the group of HOX genes. As a result of expression of the NUP98-HOXD13 fusion gene in all hematopoietic tissues, NHD13 mice developed MDS with peripheral leukopenia, neutropenia, and anemia, while the BM was normo- or hypercellular. More than half of the mice eventually progressed to acute leukemia within 18 months (8).
SALL4 is a gene encoding a zinc-finger transcription factor and has two isoforms – SALL4A and SALL4B. Its constitutive expression may play a role in AML pathogenesis. Transgenic mice constitutively expressing human SALL4B developed MDS-like features such as increased number of immature blasts, atypical and dysplastic WBCs, with hyper-segmented neutrophils and pseudo-Pelger-Huet-like cells. Dysplasia was also present in the other lineages, with binucleate erythroid precursors and hypo-lobated megakaryocytes, as well as giant platelets. In addition, around 50% of these mice eventually progressed to AML. These changes were attributed to activation of the Wnt/ß-catenin pathway by the constitutively expressed SALL4B (9).
Even though genomic alterations are by far the most studied events in the pathogenesis of MDS, there is increasing interest in understanding if/how epigenetic events may contribute to disease homeostasis. Such events may alter energy metabolism or the microenvironment of hematopoietic cells and thus promote disease initiation and maintenance. For instance, ablation of the proof reading function of DNA polymerase gamma (POLG), which is responsible for the replication of mitochondrial (mt)DNA, resulted in mtDNA mutations. As a result, Polg deleted mice showed various features of accelerated aging. Starting at the age of 6 months, mutant mice showed progressive macrocytic anemia suggestive of MDS, which worsened rapidly after the age of 11 months. In addition, both B and T lymphocyte counts were decreased. However, none of these mice progressed to acute leukemia (10).
Over the last 15 years, there has been clear evidence that genetic mutations in several conserved pathways are frequent in MDS and amenable to modeling in mice (Table 1). Of these, mutations in transcription factors (e.g. RUNX1, ETV6, BCOR), epigenetic modifiers (DNMT3A, TET2, EZH2, ASXL1), and most recently splicing factors (SRSF2, U2AF1, SF3B1 and ZRSR2) have been already reported in mouse models that offer insight into MDS biology. Mutations in genes belonging to these categories alone are generally insufficient to reproduce all clinical and biological features of human MDS. These models and the mechanistic understanding they have provided now offer the opportunity to recreate the genetic heterogeneity and understand the mutations’ synergy in MDS either in silico or by generating models that combine genetic mutations that frequently co-occur in patients.

Table 1 Mouse models of MDS.
RUNX1 or AML1 encodes a transcription factor with location on chromosome 21q22 and is the most frequent target for chromosomal translocation in leukemia. Mice transplanted with bone-marrow cells infected with a retroviral vector harboring mutant RUNX1 developed MDS-RAEB or MDS/AML with high penetrance. Two types of mutations were used – one located in the Runt homology domain (RUNX1D171N) and the other one causing a frameshift leading to a C-terminal truncation (RUNX1S291fs). While the first one led to leukocytosis and hepatosplenomegaly in mice, the latter caused leukopenia. Interestingly, in both cases multilineage dysplasia was present, with Howell-Jolly bodies, red cell polychromasia, and poikilocytosis. Giant erythroblasts, karyorrhexis, and macrocytosis were also detected. Pseudo-Pelger-Huët anomaly, hyper-segmented neutrophils, and giant platelets were also seen in mice harboring the RUNX1D171N mutation. As a result, MDS/AML was diagnosed in the vast majority of mice in both groups, while a small number of animals classified as MDS-RAEB. The differences between the 2 phenotypes are likely due to the higher expression of Evi1 found only in the RUNX1D171N mice. This is supported by the fact that co-transduction of BM cells with Evi1 and RUNX1D171N, but not RUNX1S291fs, rapidly reproduced the phenotype seen in the transgenic models (23).
EVI1 is a proto-oncogene that has been associated with hematologic malignancies in both mice and men. The transgenic Evi1 mouse showed defects in erythroid hematopoiesis with a reduction in CFU-E derived colonies, but without differences in CFU-G, CFU-GM and CFU-MK derived colonies. More so, there were no other phenotypes observed in the peripheral blood, bone marrow, and spleen of these mice. In the transgenic line harboring the highest number of transgene copies, the phenotype was more severe and showed an important reduction in spleen size, complete absence of the red pulp and erythroblasts, and neutrophil infiltration. Neutrophil infiltration and reduction of erythroblasts were also seen in the bone marrow of these mice. Although reticulocyte counts were decreased, the number of circulating RBCs was normal. Interestingly, in this transgenic line, the Evi1 transgene is X-linked, thus the described phenotype is only present in males (35).
Up to 30% of patients with MDS show alterations in the Ten-Eleven-Translocation-2 (TET2) gene, which is also involved in other myeloid malignancies, such as MPN, CMML, and AML. The TET gene family epigenetically regulates gene expression by opposing methylation-driven gene silencing, thus possibly acting as a tumor suppressor gene. Several genetically engineered Tet2-/- mice models were generated revealing that Tet2 deletion was sufficient to initiate myeloid and lymphoid malignancies in mice (36–39). They developed leukocytosis with monocytosis and neutrophilia as early as two months of age. During aging, the BM, spleen and liver became infiltrated with erythroblasts and mature myeloid cells. Based on the Bethesda criteria, the phenotype was heterogeneous and best defined as MDS with erythroid predominance, CMML, or myeloid leukemia with maturation (37). They showed hematopoietic stem cell expansion and myeloid and lymphoid transformation (36, 38, 39).
Mice deleted for another epigenetic modifier gene, Dnmt3a, revealed an aberrant phenotype affecting all hematopoietic cell lineages. DNMT3A is a methyltransferase frequently mutated in myeloid malignancies such as MPN, MDS and AML. The knockout mice displayed marked myeloid and erythroid dysplasia in their peripheral blood with increased myeloid cells. Bone marrow cellularity was also increased displaying multilineage dysplasia with impaired erythroid maturation. Spleen and liver showed myeloid infiltration with increased blasts, dysplastic megakaryopoiesis, and erythrophagocytosis. These findings were consistent with a diagnosis of MDS/MPN with extramedullary hematopoiesis (40).
ASXL1, a member of the Polycomb group, is altered in various myeloid malignancies (MDS, MPN, CMML, JMML, AML) and generally associated with worse prognosis. The knockout (KO) of both alleles of the gene in mice led to severe developmental abnormalities, such as dwarfism and anophthalmia, and an 80% embryonic lethality. The few surviving mice exhibited multiple cytopenias and dysplastic features such as presence of hyper- and hypo-segmented neutrophils, pseudo-Pelger-Huët anomaly, increased numbers of polychromatophilic RBCs and Howell-Jolly bodies. The BM of these mice was normo- or hypercellular, with myeloid hyperplasia and erythroid hypoplasia, and micromegakaryocytes with hypolobated nuclei. Furthermore, the spleens were small due to reduced red pulp and smaller lymphoid aggregates in the white pulp. As the majority of ASXL1 mutations in patients are heterozygous, Asxl1 haploinsufficient mice were also developed. Heterozygous KO mice recapitulated the phenotype and also showed hyper- and hypo-segmented neutrophils, pseudo-Pelger-Huët anomaly, frequent apoptotic, and hypogranulated neutrophils and increased polychromatophilic RBCs. The spleen architecture was also disrupted, and the BM showed an increased proportion of myeloid cells and a decrease in erythroid islands. The phenotype was more pronounced with age with some mice developing profound anemia, thrombocytopenia, leukopenia, and in some cases leukocytosis and monocytosis, suggesting disease progression with aging (27).
After the discovery of recurrent mutations in key factors of the splicing machinery in greater than 50% of patients with MDS and in a subset of patients with MDS/MPN overlap syndromes such as CMML and AML in SRSF2, U2AF1, SF3B1 and ZRSR2 by Yoshida et al. and others (41, 42), several groups generated inducible knockin [Srsf2 (28, 43), Sf3b1 (30, 44, 45)], transgenic [U2af1 (29, 46)], or knockout [Zrsr2 (47)] mouse models. These mouse models replicated the most common mutations identified in patients. All models phenocopied aspects of MDS but to varying degrees. One important question raised was whether induction of mutations provided mutant stem cells with a competitive advantage as seen in patients in whom splicing factor mutations are almost always a part of the dominant clone. Surprisingly, most models instead showed a competitive disadvantage (Kim, Obeng, Shirai, Mupo, Seiler) for long-term hematopoietic stem cells (LT-HSCs) even though some showed robust initial engraftment when mutant cells were engrafted into irradiated recipient mice to prove the cell-intrinsic nature of the defect (44). Analysis of expression of the mutant versus the wildtype splicing factor transcript in the obligatory heterozygous models revealed expression levels closer to 30%, dependent on the technology used to derive these inducible models. A second Srsf2 P95H mutant model generated in 2018 by the Walkley group achieved expression of mutant Srsf2 closer to 50% as found in patients and exhibited the selective advantage of mutant HSCs expected in MDS. For comprehensive review of the U2af1, Srsf2, and Sf3b1 mutant mouse models we refer the reader to a comprehensive review (48). Interestingly, both Srsf2 mutant and Zrsr2 knockout mouse models showed stronger phenotypes resembling MDS than U2af1 or Sf3b1 mutant mice, which could be attributed to the binding preferences of SRSF2 and ZRSR2. Exons bound by SRSF2 and introns bounds by ZRSR2 as part of the minor (or U12) spliceosome (49) are more highly preserved between human and mouse (50–52) than intronic sequences bound by U2AF1 or SF3B1, resulting in higher overlap of specific genes affected by the respective mutant splicing factors.
Since generation of single mutant mice, compound mutant mice have been generated, providing better understanding of the progression from clonal hematopoiesis to MDS and to AML. Examples include SRSF2/IDH2 co-mutant mice that exhibited profound myelodysplasia and rapid progression to AML likely via reduced expression of INTS3, a member of the integrator complex (53). Similarly, NHD13 MDS mice exhibited accelerated progression to acute leukemia when combined with the Vav1 driven IDH2R140Q transgene, though the phenotype of their leukemia resembled early T-cell precursor ALL rather than AML (54). In contrast, SRSF2/TP53 co-mutant mice did not show increased progression to AML (43) even though loss of TP53 accelerated progression to AML of NHD13 MDS mice (55). On the other hand, U2AF1/RUNX1 co-mutant mice showed normal survival and did not progress to AML unless exposed to alkylating agents (56).
The deletion of the long arm of chromosome 5 [del(5q)] is the most common karyotype abnormality in de novo MDS and defines its own subtype of MDS. While the underlying mechanism of pathogenesis is not completely understood, the loss of several genes located within the deleted region has been identified as possible disease initiating events. Among these, RPS14 (a ribosomal protein) (57), CSNK1A1 (a serine/threonine kinase) (58), and two microRNAs – miR-145 and miR-146a (59) are likely to play a role in the phenotype of del(5q) MDS. In mice, the genes equivalent to the ones located in the 5q region in humans, are located on chromosome 18. Therefore, a mouse model was generated with a deletion of the cd74-nid67 region on chromosome 18 to better understand the pathogenesis of the human 5q- syndrome. These mice developed pronounced macrocytic anemia, thrombocytopenia, and granulocytopenia. They also had a hypocellular bone marrow with a deficit in the hematopoietic progenitor populations (31). Further analysis of the bone marrow compartment showed an accumulation of TP53 protein, cell cycle arrest and increased apoptosis. However, when crossed with TP53-/- mice, the phenotype was almost completely reversed, except for the low RBC counts (present in the original TP53-/- mice as well) and the macrocytosis. More so, activation of the TP53 pathway was associated with loss of RPS14 and increased ribosomal stress (60).
The bone-marrow microenvironment was shown to play an important role in different stages of MDS. Although its most prominent contribution is probably in disease maintenance and progression, several studies have shown that deleting particular genes in the microenvironment can actually initiate MDS.
Dicer1 is an RNase III endonuclease involved in the processing of RNA and in microRNA biogenesis. The deletion of this gene in mouse osteoprogenitor cells in the Osx-GFP-Cre+Dicer1fl/fl mouse model resulted in impaired osteoblastic differentiation and decreased calcified matrix deposition (61). This alteration of the bone marrow niche led to myelodysplasia in these mice. Leukopenia was present in all cases, while some animals also displayed profound anemia and thrombocytopenia. While the BM was normo- or hyper-cellular, no differences were found in the hematopoietic stem and progenitor cells. However, bone marrow of these mice showed dysplastic features such as hyper-segmented nuclei in neutrophils, giant platelets, and micro-megakaryocytes with hypo-lobulated, hyperchromatic nuclei. Consistent with human MDS, B-cells and B-cell progenitors were reduced in the BM in favor of an increased frequency of myeloid cells. In addition, a small percentage of these mice progressed to either AML or myeloid sarcomas. The essential role of the microenvironment in disease initiation in this model was highlighted by reciprocal transplantation experiments in which the disease could not be reproduced in wild type recipient mice. Microarray analysis of the transcriptome of Dicer-/- osteolineage cells suggested that a significant downregulation of the Schwachman-Diamond-Bodian syndrome gene (SBDS) might be responsible for the observed phenotype (32). This hypothesis was further strengthened by the fact that deletion of the SBDS gene in the same compartment in mice led to a similar phenotype by activating the p53-S100A8/9-TLR inflammatory signaling axis, thus driving genotoxic stress (33).
The role of the bone marrow microenvironment in MDS maintenance was also clearly demonstrated in NHD13 mouse models. Even though transgene expression is restricted to hematopoietic cells via the Vav1 promoter-enhancer, these mice showed characteristic MDS-induced alteration of the bone marrow microenvironment including increased endothelial cells and dysfunctional mesenchymal and osteoblastic cellular populations (62, 63). They are, thus, a model to study the interactions between the mutant hematopoietic clone and the surrounding microenvironment and the role of various chemo-/cytokines in the MDS phenotype.
Although the mouse models described above present with several phenotypic features of MDS and allow for in depth characterization of gene function and characterization of mutations, they still have obvious limitations with respect to their ability to recapitulate human MDS. While many gene functions are preserved between mammalian species, some are not and their targets may differ greatly when comparing human and murine species. This has for example become particularly obvious in murine models of splicing factor mutations, affecting 50% of patients with MDS; hallmarks of splicing factor mutant MDS, such as ring sideroblasts in SF3B1 mutant MDS, are absent in Sf3b1 mutant mice (30). Even in monogenic bone marrow failure disorders such as Fanconi Anemia, mouse models fail to recapitulate the human phenotype (64–66). In addition, murine models generally do not replicate genetic complexity or clonal evolution encountered in patients, especially under treatment pressures, making it imperative to study these diseases in primary human cells (Figure 1B).
The study of primary human MDS poses a particular challenge that continues despite several critical improvements over the past several years. The MDS hematopoietic stem cell is defective, reminiscent of stem cell dysfunction in the inherited bone marrow failure syndromes such as Fanconi Anemia. While a few human cell lines have been successfully generated from patients whose bone marrow failure has transformed to leukemia, cell lines that recapitulate the bone marrow failure state are rare and limited in their genetic diversity [reviewed in (67)]. Drexler et al. reviewed 31 candidate MDS cell lines and classified them into three categories: (1) false (cross-contaminated) cell lines and non-malignant cell lines; (2) malignant cell lines established at the AML/MDS leukemic phase but not MDS phase; and (3) MDS cell lines established during the MDS phase (67). Among these cell lines, three cell lines were established during the MDS phase of the diseases. In 1991, the MDS92 cell line was derived from the bone marrow of a 52-year-old male with RARS which developed into RAEB, but prior to leukemic transformation. This cell line carries a complex karyotype, including 5q- and -7, as well as a codon 12 mutation in NRAS. The MDS92 cell line is cytokine-dependent (68, 69); a blastic MDS-L subline was derived in 2000 (70) and shown to be responsive to lenalidomide (71). In 1994, the M-TAT cell line was established from the peripheral blood of a 3-year-old male at relapse of RAEB-T. This cell line is also cytokine-dependent for growth and responds to various cytokines with differentiation down the erythroid or megakaryocytic lineages (72). The TER-3 cell line was established in 2002 from a male patient’s bone marrow at time of progression from RA to RAEB. The complex karyotype included monosomies 7 and 20 among other aberrations. Like M-TAT, this cell line is constitutively cytokine-dependent with potential to differentiate towards the erythroid and megakaryocytic lineages (73).
Given the limited growth potential of primary MDS cells in vitro and the limited number and fidelity of MDS cell lines, models are necessary that allow propagation of primary, patient- derived MDS to allow study of the highly heterogeneous disease. The immunodeficient murine host has become the ideal host to study human tumors in vivo, from an ethical, practical, and cost standpoint.
Historically, direct transplantation of human cells into immune competent mice failed to give positive outgrowth due to immune rejection by the recipient mice; sublethally irradiated mice died without reconstitution of hematopoiesis (74). Since then immunodeficient mouse models have undergone a long evolution from a host lacking murine T- and B-cells to multi-lineage immune-deficient models (T-, B- and NK-cells) with additional modifications; identification and introduction of mutations that enhance recognition of self across the murine-human barrier (SIRPalpha); adaptation of the murine host to express human proteins essential for human cell survival and differentiation, such as cytokines; and introduction of human cellular systems, such as mesenchymal stromal cells and entire ossicles. A detailed review of these mouse models is beyond the scope of this review and provided in detail by Martinov et al. (75). Here we seek to provide a brief summary as it pertains specifically to myeloid malignancies and in particular MDS (Table 2).
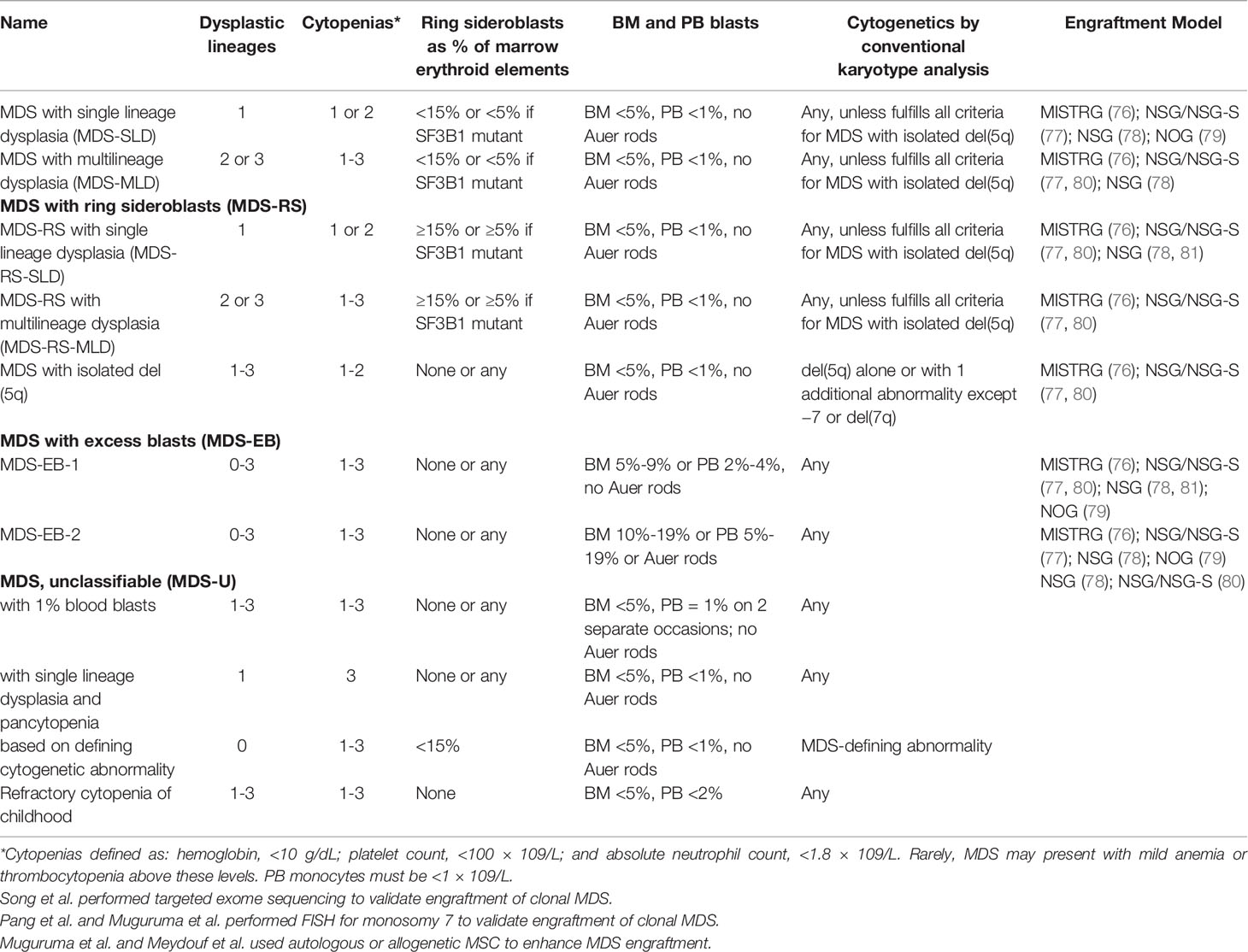
Table 2 PDX mouse models of MDS.
In the late 1970s the first attempt at PDX models of human acute myeloid leukemia (AML) employed subcutaneous implantation of patient AML cells into thymectomized, irradiated mice. In these T-cell deficient, B-cell competent mice, AML cells could be grown as discrete tumors under the skin. However the tumors started regressing 6 days after inoculation without lasting tumor cells (82). Over the next decade two physiologically more relevant models were the bnx mouse which was generated via combination of three mutations, beige, nude and xid, resulting in deficiency of T-, NK- and so-called lymphokine activated killer cells (83), and the severe combined immunodeficiency (SCID) mouse that carries a single point mutation in the protein kinase DNA-activated catalytic polypeptide (Prkdc) gene with impaired T- and B-lymphocyte development but intact NK-cell function and innate immunity (84, 85). While normal human hematopoietic cells (86) and acute lymphoblastic leukemia (ALL) cells (87) could be successfully transplanted into bnx or SCID mice (88), primary AML cells still failed to reliably engraft in either mouse model (89). Interestingly, if mice were treated with the cytokine granulocyte-macrophage stimulating factor (GM-CSF) and human mast cell growth factor (MGF) human undifferentiated blast cells were identifiable in the murine bone marrow (90). These studies suggested that remnant immunity in the murine host compromised lasting engraftment of human myeloid cells and that the murine environment lacked factors relevant to human cell survival and proliferation. However, these changes were not introduced until 2003.
In the following years, mouse models that combined T-, B- and NK-cell deficiency, such as the nonobese diabetic (NOD)-severe combined immunodeficiency (NOD/SCID) mice became the main strain used for xenograft studies and supported engraftment of ALL and a subset of AMLs (91–94). Discovery that a polymorphism in the Sirpa gene in the NOD mouse strain encodes a variant of the SIRPa receptor that cross-reacts with the human CD47 ligand in part explained the improved engraftment levels. Human cells can engage the CD47/SIRPa “don’t eat me” signal in NOD mice and are thereby at least partially protected from phagocytosis by the murine host macrophages.
In regards to primary human MDS cells, in 2002, Nilsson et al. injected 5q- deficient hematopoietic cells obtained from seven MDS patients into NOD/SCID mice. Mice engrafted with cells from one of these patients showed 12% human (CD45+) engraftment and CD45+CD15+ cells proved to carry deletion of 5q. The mice did not exhibit symptoms and engraftment from 6 additional patients were unsuccessful (95). In another study, Benito et al. transplanted bone marrow cells from MDS patients into sub-lethally irradiated NOD/SCID mice. Human CD45+ cells were present in bone marrow and spleen of these mice, albeit with significant delay when compared with healthy donor cell engraftment. Of note, no clonal precursors were found in these grafts validating the poor reconstitution of MDS HSC derived hematopoiesis compared to remnant healthy HSC derived hematopoiesis in the NOD/SCID environment (96).
Despite the improvement afforded by the Nod/Scid model, it became clear that a more permissible host had to be identified to allow MDS HSCs to engraft. One approach was to eliminate residual NK cell activity in the NOD/SCID mouse by elimination or inactivation of the β2-microglobulin (β2m) gene (97–99) or by antibody-mediated elimination of NK cells (100, 101). Indeed, Thanopoulou et al. (102) reported that MDS cells from 4 of 7 MDS patients that included all MDS subtypes engrafted in NOD/SCID β2-microglobulin-deficient mice (NOD/SCID-β2mnull). These grafts also taught the scientific community about the biology of MDS: in contrast to healthy donor transplants that first give rise to erythroid lineage predominance followed by dominance of lymphoid engraftment, most MDS transplants were myeloid predominant. Importantly, in 4 out of 4 MDS cases, the reconstituted cells carried the same cytogenetic abnormalities, namely trisomy 8 and 5q-, as the original samples. While these steps represented great advances, overall engraftment levels remained low, < 1%. Another methodology to abrogate NK cells in the Nod/Scid background was to delete the IL2 receptor common gamma chain (IL2rγ-/-) resulting in the since widely used NOD-scid-IL2rγ-/- (NSG) mice (103). Despite these improvements, engraftment of low risk MDS in NSG mice remained largely unsuccessful, while human AML samples showed robust engraftment (104).
Clearly, combined abrogation of adaptive and partially innate immunity proved insufficient for MDS engraftment suggesting that signals that could support human hematopoiesis were missing. Feurig-Buske et al. compared MDS engraftment in NOD/SCID-β2mnull mice to NOD/SCID-β2m-/- mice expressing human cytokines, specifically human interleukin-3 (IL-3), GM-CSF and Steel factor (SF) (NOD/SCID-β2mnull-3/GM/SF) from a transgene that previously provided excellent engraftment to AML (Feurig-Buske M, et al., Leukemia 2003). Cytokine expression led to reconstitution from all 7 MDS samples tested but with long-term engraftment documented only from 2. Constitutive expression of huIL3, huGM-CSF and huSCF, improved engraftment of primary human AML (105) and enhanced normal human myelopoiesis (106). Nevertheless, engraftment of MDS remained a challenge. In fact, of the MDS cell lines described above, only the MDS-L subline has been engrafted in immunodeficient mice, specifically in NSGS mice (NSG transgenic expressing huIL3, huGM-CSF and huSCF) (107) and employed in drug testing (108–110). While some groups provided evidence that NSGS mice (~5-35%) consistently showed higher engraftment of MDS than NSG mice (~1-9%) (80) several groups have since confirmed that while transient engraftment is enhanced, the constitutive nature of expression of these cytokines is detrimental to normal and MDS HSCs (111, 112).
As an alternative to transgenic cytokine expression, several groups tested co-transplantation of mesenchymal stromal cells (MSCs), either derived from immortalized cell lines or from autologous or allogeneic BM derived MSC cultures. Such studies showed mixed results. In one study cells from 6 MDS patients were transplanted into NOD/SCID-β2mnull mice along with human stroma-derived cell lines HS5 and HS27a with improved but low engraftment levels (0.71-4.44%) (113). Bone marrow CD34 cells from six patient were intrafemorally injected into NOG mice along with human MSCs, resulting in human CD45+ percentages in mouse BM between ~2% and 89% interestingly at the expense of murine hematopoiesis (79). In another study co-injection of autologous MSCs improved engraftment in NSG mice from 1/7 patient samples to 14/20 patient samples with engraftment levels ranging from 1%-22% (80). Interestingly, MSCs were detectable only about a week post transplantation suggesting that their effect on MDS HSC engraftment was transient. The exact mechanism by which MSCs support MDS HSC engraftment remains to be determined; an MSC effect could not be confirmed in several carefully performed studies in NSG or NSGS mice (77, 114).
The most recently advanced strategy to add human microenvironment for human HSPCs engraftment is using human bone organoids (ossicles). Ossicles are created by seeding of human BM-derived MSCs onto a 3D scaffold composed of extracellular matrix (115) or generated by co-injection of MSCs and matrix material under mouse skin; they have provided a favorable niche for normal and leukemic cells (116).
Routinely stem cell engraftment requires elimination of cells occupying the stem and progenitor cell niche. This is typically achieved via irradiation with doses adjusted to the host’s tolerability. Alternatives include conditioning with busulfan (117, 118), attractive in mice with the scid mutation that confers a DNA repair defect that sensitizes host tissues to irradiation. Other alternatives that can be cost-prohibitive include administration of antibody-drug conjugates (119). An attractive option would be a murine host that carries a HSPC defect that would allow normal hematopoiesis at steady state but a competitive disadvantage when challenged with human cells. Indeed, mice bearing mutations in the receptor tyrosine kinase Kit (KitW/Wv), the stem cell factor receptor required for normal hematopoiesis, allow long-term engraftment of injected wildtype HSPCs without conditioning (120). When introduced into immunodeficient hosts (Rag2−/−γc−/−) various mutations in Kit allow engraftment without irradiation (121) and engraftment across the human-mouse barrier (122). Strikingly, these mice displayed robust reconstitution of human erythropoiesis and thrombopoiesis with terminal maturation in the bone marrow (123). Nevertheless, introduction of the KitW41/W41 mutation into NSG mice was insufficient to allow robust MDS engraftment (124).
Abrogation of murine T-, B-, and NK cells via abrogation of IL-2Rγ (Il2rg gene deletion or truncation), abrogation of V(D)J recombination via the Prkdcscid mutation, or deletion of recombination activating genes (RAG)-1 or RAG-2 (125, 126) in combination with either the Sirpa polymorphism encountered in the NOD strain or via introduction of the human SIRPA gene results in optimal immunosuppression. Transgenic expression of critical cytokines provided at least transient engraftment of MDS HSCs. In 2014 Rongvaux et al. presented a novel mouse model with knockin of critical cytokines that lacked cross-reactivity between human and mouse (127). Knockin provided two advantages: 1) expression of human cytokines was regulated by the endogenous murine regulatory elements resulting in physiologic expression of human cytokines and 2) deletion of the murine cytokine provided a competitive disadvantage for the murine host HSPCs, “opening” up the niche for human xenografted cells. “MISTRG” mice, so named for the cytokines replaced and the immunodeficient background strain express human macrophage colony-stimulating factor (M-CSF), IL-3 and GM-CSF, and thrombopoietin (THPO) in the Rag2−/−, IL2Rγ−/− background. To provide phagocytic cross-tolerance human signal regulatory protein alpha (SIRPα) was introduced as transgene (127) and later also knocked in (128). MISTRG mice have proved to be the most promising host for engraftment of MDS patient samples to date (76). Cells from all MDS risk groups efficiently engrafted in this strain; engraftment levels were significantly higher especially for low-risk MDS. Unlike in NSG mice, increasing numbers of CD34+ cells resulted in increasing engraftment levels while engraftment levels in NSG mice remained low, suggesting a lower threshold for engraftment in MISTRG mice. While in NSG mice engraftment in female recipients was up to 11-fold higher than in male recipients (129), engraftment in MISTRG mice was not affected by the sex of the recipient mice. CD34+ cells from MDS bone marrow produced myeloid predominant grafts that engrafted long-term and were transplantable into secondary recipients. Additionally, engrafted MDS cells in MISTRG mice gave rise to erythroid and megakaryocytic lineages and replicated MDS heterogeneity, myeloid dysplasia, and clonal complexity and evolution. MDS PDXs replicated drug treatment responses such as cellular differentiation in response to treatment with inhibitors of mutant IDH2 (76). A shortcoming of MISTRG as for other models was the absence of mature neutrophils, red blood cells, and platelets in circulation, limiting full assessment of the myeloid lineage maturation. This was overcome by humanization of the murine liver, resulting in abrogation of murine complement expression and enhanced red cell survival in circulation (130).
In summary, immunodeficient mouse models have undergone rapid evolution over the past decade from bi-lineage immunodeficiency to an intricate combination of adaptive and innate immune tolerance. In addition, humanization of cytokines and growth factors that are critical for human hematopoietic stem and progenitor cell survival and differentiation have transformed the murine environment into one favoring human cell engraftment.
Over the last decade, we have seen significant therapeutic advances in AML resulting in a number of drugs being either FDA approved or very advanced in their clinical development. Unfortunately, drug development in MDS is still lagging behind. This may be due to unique biological features of MDS compared to AML, particularly the more complex mutational architecture and perhaps higher interdependency between the dysplastic clones and their surrounding immune and stromal microenvironment. Nevertheless, the lack of preclinical models of MDS compared to AML certainly contributed to the slow start of drug development efforts in this disease. Fortunately, the increased accessibility of transgenic technologies coupled with our improved xenograft tools have closed the gap in providing the much needed preclinical models of MDS. While we have come a long way, some challenges remain.
In regard to the transgenic mouse models, we are just beginning to explore the cell intrinsic interactions between various mutations found in patients with MDS. Given the genetic complexity of this disease, modeling all potential interactions is a daunting task. Could information provided by the phenotype of single mutations be integrated to generate algorithms that predict how the combination of mutations would behave? Even if successful, the cell extrinsic interactions between various mutant clones, these clones and residual “unmutated” hematopoiesis or the “unmutated” immune and mesenchymal microenvironment may not be accurately addressed by the current transgenic mouse models. It is important to remember that even if “unmutated”, the residual hematopoiesis and the immune and stromal microenvironment of patients with MDS are not wild type. Can we use transgenic mice to model these interactions?
In regard to xenograft models, they all rely on hosts that have no adaptive and partially compromised innate immunity and once engrafted with human cells represent xenogeneic immune chimera. Thus, at this point, it remains impossible to study the role of immune senescence for instance in MDS homeostasis. Could adaptive transfer of human B and T cells into some of the most advanced xenograft models (i.e. MISTRG) model these interactions, even for a short period of time? Cytopenias are the root cause of much of the morbidity and mortality and lack of quality of life experienced in MDS. We have made significant progress towards generating xenograft models that allow full maturation of several human hematopoietic cell lineages. Nevertheless, these models continue to imperfectly recreate full maturation and functionality of neutrophils and platelets and in part red blood cells.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
WL and PT contributed equally to generating the first draft of the manuscript. SH and GG contributed equally to designing and coordinating the completion of this project. All authors read and approved the final version of the manuscript.
GG was supported by the following grants: R01 CA253981, 1P01CA225618, ASH Bridge award and P30 CA006973-57S2. SH was supported by the following grants: R01 CA253981, R01DK102792, The Frederick A. DeLuca Foundation, Vera and Joseph Dresner Foundation, and The U.S. Department of Defense Peer Reviewed Cancer Research Program, Expansion Award (CA171025, W81XWH1810138).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. DeZern AE. Treatments Targeting MDS Genetics: A Fool’s Errand? Hematology (2018) 2018(1):277–85. doi: 10.1182/asheducation-2018.1.277
2. Teodorescu P, Pasca S, Dima D, Tomuleasa C, Ghiaur G. Targeting the Microenvironment in MDS: The Final Frontier. Front Pharmacol (2020) 11:1044. doi: 10.3389/fphar.2020.01044
3. Kogan SC, Ward JM, Anver MR, Berman JJ, Brayton C, Cardiff RD, et al. Bethesda Proposals for Classification of Nonlymphoid Hematopoietic Neoplasms in Mice. Blood (2002) 100(1):238–45. doi: 10.1182/blood.V100.1.238
4. Mahgoub N, Taylor BR, Le Beau MM, Gratiot M, Carlson KM, Atwater SK, et al. Myeloid Malignancies Induced by Alkylating Agents in Nf1 Mice. Blood (1999) 93(11):3617–23. doi: 10.1182/blood.V93.11.3617.411a49_3617_3623
5. Das M, Chaudhuri S, Law S. Benzene Exposure–an Experimental Machinery for Induction of Myelodysplastic Syndrome: Stem Cell and Stem Cell Niche Analysis in the Bone Marrow. J Stem Cells (2012) 7(1):43–59.
6. Li W, Schnatter AR. Benzene Risk Assessment: Does New Evidence on Myelodysplastic Syndrome Justify a New Approach? Crit Rev Toxicol (2018) 48(6):417–32. doi: 10.1080/10408444.2018.1437389
7. Beachy SH, Aplan PD. Mouse Models of Myelodysplastic Syndromes. Hematol Oncol Clin North Am (2010) 24(2):361–75. doi: 10.1016/j.hoc.2010.02.002
8. Slape C, Lin YW, Hartung H, Zhang Z, Wolff L, Aplan PD. NUP98-HOX Translocations Lead to Myelodysplastic Syndrome in Mice and Men. J Natl Cancer Inst Monogr (2008) 39):64–8. doi: 10.1093/jncimonographs/lgn014
9. Ma Y, Cui W, Yang J, Qu J, Di C, Amin HM, et al. SALL4, a Novel Oncogene, is Constitutively Expressed in Human Acute Myeloid Leukemia (AML) and Induces AML in Transgenic Mice. Blood (2006) 108(8):2726–35. doi: 10.1182/blood-2006-02-001594
10. Chen ML, Logan TD, Hochberg ML, Shelat SG, Yu X, Wilding GE, et al. Erythroid Dysplasia, Megaloblastic Anemia, and Impaired Lymphopoiesis Arising From Mitochondrial Dysfunction. Blood (2009) 114(19):4045–53. doi: 10.1182/blood-2008-08-169474
11. Moody JL, Xu L, Helgason CD, Jirik FR. Anemia, Thrombocytopenia, Leukocytosis, Extramedullary Hematopoiesis, and Impaired Progenitor Function in Pten+/-SHIP-/- Mice: A Novel Model of Myelodysplasia. Blood (2004) 103(12):4503–10. doi: 10.1182/blood-2003-09-3262
12. Buonamici S, Li D, Chi Y, Zhao R, Wang X, Brace L, et al. EVI1 Induces Myelodysplastic Syndrome in Mice. J Clin Invest (2004) 114(5):713–9. doi: 10.1172/JCI21716
13. Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, et al. Role of Nucleophosmin in Embryonic Development and Tumorigenesis. Nature (2005) 437(7055):147–53. doi: 10.1038/nature03915
14. Sportoletti P, Grisendi S, Majid SM, Cheng K, Clohessy JG, Viale A, et al. Npm1 is a Haploinsufficient Suppressor of Myeloid and Lymphoid Malignancies in the Mouse. Blood (2008) 111(7):3859–62. doi: 10.1182/blood-2007-06-098251
15. Fütterer A, Campanero MR, Leonardo E, Criado LM, Flores JM, Hernández JM, et al. Dido Gene Expression Alterations are Implicated in the Induction of Hematological Myeloid Neoplasms. J Clin Invest (2005) 115(9):2351–62. doi: 10.1172/JCI24177
16. Lin Y-W, Slape C, Zhang Z, Aplan PD. NUP98-HOXD13 Transgenic Mice Develop a Highly Penetrant, Severe Myelodysplastic Syndrome That Progresses to Acute Leukemia. Blood (2005) 106(1):287–95. doi: 10.1182/blood-2004-12-4794
17. Chul Won C, Yang Jo C, Christopher S, Peter DA. Impaired Differentiation and Apoptosis of Hematopoietic Precursors in a Mouse Model of Myelodysplastic Syndrome. Haematologica (2008) 93(9):1394–7. doi: 10.3324/haematol.13042
18. Chung YJ, Choi CW, Slape C, Fry T, Aplan PD. Transplantation of a Myelodysplastic Syndrome by a Long-Term Repopulating Hematopoietic Cell. Proc Natl Acad Sci (2008) 105(37):14088–93. doi: 10.1073/pnas.0804507105
19. Choi CW, Chung YJ, Slape C, Aplan PD. A NUP98-HOXD13 Fusion Gene Impairs Differentiation of B and T Lymphocytes and Leads to Expansion of Thymocytes With Partial TCRB Gene Rearrangement. J Immunol (2009) 183(10):6227–35. doi: 10.4049/jimmunol.0901121
20. Omidvar N, Kogan S, Beurlet S, le Pogam C, Janin A, West R, et al. BCL-2 and Mutant NRAS Interact Physically and Functionally in a Mouse Model of Progressive Myelodysplasia. Cancer Res (2007) 67(24):11657–67. doi: 10.1158/0008-5472.CAN-07-0196
21. Wu M-Y, Eldin KW, Beaudet AL. Identification of Chromatin Remodeling Genes Arid4a and Arid4b as Leukemia Suppressor Genes. J Natl Cancer Inst (2008) 100(17):1247–59. doi: 10.1093/jnci/djn253
22. Lavau C, Du C, Thirman M, Zeleznik-Le N. Chromatin-Related Properties of CBP Fused to MLL Generate a Myelodysplastic-Like Syndrome That Evolves Into Myeloid Leukemia. EMBO J (2000) 19(17):4655–64. doi: 10.1093/emboj/19.17.4655
23. Watanabe-Okochi N, Kitaura J, Ono R, Harada H, Harada Y, Komeno Y, et al. AML1 Mutations Induced MDS and MDS/AML in a Mouse BMT Model. Blood (2008) 111(8):4297–308. doi: 10.1182/blood-2007-01-068346
24. Cao Q, Gearhart MD, Gery S, Shojaee S, Yang H, Sun H, et al. BCOR Regulates Myeloid Cell Proliferation and Differentiation. Leukemia (2016) 30(5):1155–65. doi: 10.1038/leu.2016.2
25. Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, et al. DNMT3A Mutations Promote Anthracycline Resistance in Acute Myeloid Leukemia via Impaired Nucleosome Remodeling. Nat Med (2016) 22(12):1488–95. doi: 10.1038/nm.4210
26. Mochizuki-Kashio M, Aoyama K, Sashida G, Oshima M, Tomioka T, Muto T, et al. Ezh2 Loss in Hematopoietic Stem Cells Predisposes Mice to Develop Heterogeneous Malignancies in an Ezh1-Dependent Manner. Blood (2015) 126(10):1172–83. doi: 10.1182/blood-2015-03-634428
27. Wang J, Li Z, He Y, Pan F, Chen S, Rhodes S, et al. Loss of Asxl1 Leads to Myelodysplastic Syndrome–Like Disease in Mice. Blood (2014) 123(4):541–53. doi: 10.1182/blood-2013-05-500272
28. Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell (2015) 27(5):617–30. doi: 10.1016/j.ccell.2015.04.006
29. Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell (2015) 27(5):631–43. doi: 10.1016/j.ccell.2015.04.008
30. Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell (2016) 30(3):404–17. doi: 10.1016/j.ccell.2016.08.006
31. Barlow JL, Drynan LF, Hewett DR, Holmes LR, Lorenzo-Abalde S, Lane AL, et al. A P53-Dependent Mechanism Underlies Macrocytic Anemia in a Mouse Model of Human 5q- Syndrome. Nat Med (2010) 16(1):59–66. doi: 10.1038/nm.2063
32. Raaijmakers MHGP, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone Progenitor Dysfunction Induces Myelodysplasia and Secondary Leukaemia. Nature (2010) 464(7290):852–7. doi: 10.1038/nature08851
33. Zambetti NA, Ping Z, Chen S, Kenswil KJG, Mylona MA, Sanders MA, et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-Leukemia. Cell Stem Cell (2016) 19(5):613–27. doi: 10.1016/j.stem.2016.08.021
34. Basiorka AA, McGraw KL, Eksioglu EA, Chen X, Johnson J, Zhang L, et al. The NLRP3 Inflammasome Functions as a Driver of the Myelodysplastic Syndrome Phenotype. Blood (2016) 128(25):2960–75. doi: 10.1182/blood-2016-07-730556
35. Louz D, van den Broek M, Verbakel S, Vankan Y, van Lom K, Joosten M, et al. Erythroid Defects and Increased Retrovirally-Induced Tumor Formation in Evi1 Transgenic Mice. Leukemia (2000) 14(11):1876–84. doi: 10.1038/sj.leu.2401887
36. Ko M, Bandukwala HS, An J, Lamperti ED, Thompson EC, Hastie R, et al. Ten-Eleven-Translocation 2 (TET2) Negatively Regulates Homeostasis and Differentiation of Hematopoietic Stem Cells in Mice. Proc Natl Acad Sci USA (2011) 108(35):14566–71. doi: 10.1073/pnas.1112317108
37. Li Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, et al. Deletion of Tet2 in Mice Leads to Dysregulated Hematopoietic Stem Cells and Subsequent Development of Myeloid Malignancies. Blood (2011) 118(17):4509–18. doi: 10.1182/blood-2010-12-325241
38. Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell (2011) 20(1):11–24. doi: 10.1016/j.ccr.2011.06.001
39. Quivoron C, Couronné L, Della Valle V, Lopez CK, Plo I, Wagner-Ballon O, et al. TET2 Inactivation Results in Pleiotropic Hematopoietic Abnormalities in Mouse and is a Recurrent Event During Human Lymphomagenesis. Cancer Cell (2011) 20(1):25–38. doi: 10.1016/j.ccr.2011.06.003
40. Guryanova OA, Lieu YK, Garrett-Bakelman FE, Spitzer B, Glass JL, Shank K, et al. Dnmt3a Regulates Myeloproliferation and Liver-Specific Expansion of Hematopoietic Stem and Progenitor Cells. Leukemia (2016) 30(5):1133–42. doi: 10.1038/leu.2015.358
41. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature (2011) 478(7367):64–9. doi: 10.1038/nature10496
42. Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, et al. Recurrent Mutations in the U2AF1 Splicing Factor in Myelodysplastic Syndromes. Nat Genet (2012) 44(1):53–7. doi: 10.1038/ng.1031
43. Smeets MF, Tan SY, Xu JJ, Anande G, Unnikrishnan A, Chalk AM, et al. Srsf2(P95H) Initiates Myeloid Bias and Myelodysplastic/Myeloproliferative Syndrome From Hemopoietic Stem Cells. Blood (2018) 132(6):608–21. doi: 10.1182/blood-2018-04-845602
44. Mupo A, Seiler M, Sathiaseelan V, Pance A, Yang Y, Agrawal AA, et al. Hemopoietic-Specific Sf3b1-K700E Knock-in Mice Display the Splicing Defect Seen in Human MDS But Develop Anemia Without Ring Sideroblasts. Leukemia (2017) 31(3):720–7. doi: 10.1038/leu.2016.251
45. Kon A, Yamazaki S, Nannya Y, Kataoka K, Ota Y, Nakagawa MM, et al. Physiological Srsf2 P95H Expression Causes Impaired Hematopoietic Stem Cell Functions and Aberrant RNA Splicing in Mice. Blood (2018) 131(6):621–35. doi: 10.1182/blood-2017-01-762393
46. Fei DL, Zhen T, Durham B, Ferrarone J, Zhang T, Garrett L, et al. Impaired Hematopoiesis and Leukemia Development in Mice With a Conditional Knock-in Allele of a Mutant Splicing Factor Gene U2af1. Proc Natl Acad Sci USA (2018) 115(44):E10437–46. doi: 10.1073/pnas.1812669115
47. Inoue D, Polaski JT, Taylor J, Castel P, Chen S, Kobayashi S, et al. Minor Intron Retention Drives Clonal Hematopoietic Disorders and Diverse Cancer Predisposition. Nat Genet (2021) 53(5):707–18. doi: 10.1038/s41588-021-00828-9
48. Xu JJ, Smeets MF, Tan SY, Wall M, Purton LE, Walkley CR. Modeling Human RNA Spliceosome Mutations in the Mouse: Not All Mice Were Created Equal. Exp Hematol (2019) 70:10–23. doi: 10.1016/j.exphem.2018.11.001
49. Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, et al. Aberrant Splicing of U12-Type Introns Is the Hallmark of ZRSR2 Mutant Myelodysplastic Syndrome. Nat Commun (2015) 6:6042. doi: 10.1038/ncomms7042
50. Burge CB, Padgett RA, Sharp PA. Evolutionary Fates and Origins of U12-Type Introns. Mol Cell (1998) 2(6):773–85. doi: 10.1016/S1097-2765(00)80292-0
51. Batzoglou S, Pachter L, Mesirov JP, Berger B, Lander ES. Human and Mouse Gene Structure: Comparative Analysis and Application to Exon Prediction. Genome Res (2000) 10(7):950–8. doi: 10.1101/gr.10.7.950
52. Olthof AM, Hyatt KC, Kanadia RN. Minor Intron Splicing Revisited: Identification of New Minor Intron-Containing Genes and Tissue-Dependent Retention and Alternative Splicing of Minor Introns. BMC Genomics (2019) 20(1):686. doi: 10.1186/s12864-019-6046-x
53. Yoshimi A, Lin KT, Wiseman DH, Rahman MA, Pastore A, Wang B, et al. Coordinated Alterations in RNA Splicing and Epigenetic Regulation Drive Leukaemogenesis. Nature (2019) 574(7777):273–7. doi: 10.1038/s41586-019-1618-0
54. Goldberg L, Negi V, Chung YJ, Onozawa M, Zhu YJ, Walker RL, et al. Mutant Idh2 Cooperates With a NUP98-HOXD13 Fusion to Induce Early Immature Thymocyte Precursor ALL. Cancer Res (2021) 81(19):5033–46. doi: 10.1158/0008-5472.CAN-21-1027
55. Xu H, Menendez S, Schlegelberger B, Bae N, Aplan PD, Gohring G, et al. Loss of P53 Accelerates the Complications of Myelodysplastic Syndrome in a NUP98-HOXD13-Driven Mouse Model. Blood (2012) 120(15):3089–97. doi: 10.1182/blood-2012-01-405332
56. Zhen T, Liang Fei D, Zhao L, Lopez G, Varmus H, Liu PP. Runx1 Deficiency and MDS-Associated U2af1 Mutation Cooperate for Leukemia Development in a New Mouse Model. Blood (2016) 128(22):964–4. doi: 10.1182/blood.V128.22.964.964
57. Boultwood J, Pellagatti A, Cattan H, Lawrie CH, Giagounidis A, Malcovati L, et al. Gene Expression Profiling of CD34+ Cells in Patients With the 5q- Syndrome. Br J Haematol (2007) 139(4):578–89. doi: 10.1111/j.1365-2141.2007.06833.x
58. Schneider RK, Adema V, Heckl D, Jaras M, Mallo M, Lord AM, et al. Role of Casein Kinase 1A1 in the Biology and Targeted Therapy of Del(5q) MDS. Cancer Cell (2014) 26(4):509–20. doi: 10.1016/j.ccr.2014.08.001
59. Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, et al. Identification of miR-145 and miR-146a as Mediators of the 5q- Syndrome Phenotype. Nat Med (2010) 16(1):49–58. doi: 10.1038/nm.2054
60. McGowan KA, Li JZ, Park CY, Beaudry V, Tabor HK, Sabnis AJ, et al. Ribosomal Mutations Cause P53-Mediated Dark Skin and Pleiotropic Effects. Nat Genet (2008) 40(8):963–70. doi: 10.1038/ng.188
61. Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, et al. Bone Progenitor Dysfunction Induces Myelodysplasia and Secondary Leukaemia. Nature (2010) 464:852-7.
62. Balderman SR, Li AJ, Hoffman CM, Frisch BJ, Goodman AN, LaMere MW, et al. Targeting of the Bone Marrow Microenvironment Improves Outcome in a Murine Model of Myelodysplastic Syndrome. Blood (2016) 127(5):616–25. doi: 10.1182/blood-2015-06-653113
63. Weidner H, Rauner M, Trautmann F, Schmitt J, Balaian E, Mies A, et al. Myelodysplastic Syndromes and Bone Loss in Mice and Men. Leukemia (2017) 31(4):1003–7. doi: 10.1038/leu.2017.7
64. Bakker ST, van de Vrugt HJ, Rooimans MA, Oostra AB, Steltenpool J, Delzenne-Goette E, et al. Fancm-Deficient Mice Reveal Unique Features of Fanconi Anemia Complementation Group M. Hum Mol Genet (2009) 18(18):3484–95. doi: 10.1093/hmg/ddp297
65. Parmar K, D’Andrea A, Niedernhofer LJ. Mouse Models of Fanconi Anemia. Mutat Res (2009) 668(1-2):133–40. doi: 10.1016/j.mrfmmm.2009.03.015
66. Dubois EL, Guitton-Sert L, Béliveau M, Parmar K, Chagraoui J, Vignard J, et al. A Fanci Knockout Mouse Model Reveals Common and Distinct Functions for FANCI and FANCD2. Nucleic Acids Res (2019) 47(14):7532–47. doi: 10.1093/nar/gkz514
67. Drexler HG, Dirks WG, MacLeod RAF. Many are Called MDS Cell Lines: One Is Chosen. Leuk Res (2009) 33(8):1011–6. doi: 10.1016/j.leukres.2009.03.005
68. Tohyama K, Tsutani H, Ueda T, Nakamura T, Yoshida Y. Establishment and Characterization of a Novel Myeloid Cell Line From the Bone Marrow of a Patient With the Myelodysplastic Syndrome. Br J Haematol (1994) 87(2):235–42. doi: 10.1111/j.1365-2141.1994.tb04904.x
69. Tohyama K, Tohyama Y, Nakayama T, Ueda T, Nakamura T, Yoshida Y. A Novel Factor-Dependent Human Myelodysplastic Cell Line, MDS92, Contains Haemopoietic Cells of Several Lineages. Br J Haematol (1995) 91(4):795–9. doi: 10.1111/j.1365-2141.1995.tb05391.x
70. Nakamura S, Ohnishi K, Yoshida H, Shinjo K, Takeshita A, Tohyama K, et al. Retrovirus-Mediated Gene Transfer of Granulocyte Colony-Stimulating Factor Receptor (G-CSFR) cDNA Into MDS Cells and Induction of Their Differentiation by G-CSF. Cytokines Cell Mol Ther (2000) 6(2):61–70. doi: 10.1080/13684730050515787
71. Matsuoka A, Tochigi A, Kishimoto M, Nakahara T, Kondo T, Tsujioka T, et al. Lenalidomide Induces Cell Death in an MDS-Derived Cell Line With Deletion of Chromosome 5q by Inhibition of Cytokinesis. Leukemia (2010) 24(4):748–55. doi: 10.1038/leu.2009.296
72. Minegishi N, Minegishi M, Tsuchiya S, Fujie H, Nagai T, Hayashi N, et al. Erythropoietin-Dependent Induction of Hemoglobin Synthesis in a Cytokine-Dependent Cell Line M-TAT. J Biol Chem (1994) 269(44):27700–4. doi: 10.1016/S0021-9258(18)47042-1
73. Mishima Y, Terui Y, Mishima Y, Katsuyama M, Mori M, Tomizuka H, et al. New Human Myelodysplastic Cell Line, TER-3: G-CSF Specific Downregulation of Ca2+/calmodulin-Dependent Protein Kinase IV. J Cell Physiol (2002) 191(2):183–90. doi: 10.1002/jcp.10095
74. Dick JE, Lapidot T, Pflumio F. Transplantation of Normal and Leukemic Human Bone Marrow Into Immune-Deficient Mice: Development of Animal Models for Human Hematopoiesis. Immunol Rev (1991) 124(1):25–43. doi: 10.1111/j.1600-065X.1991.tb00614.x
75. Martinov T, McKenna KM, Tan WH, Collins EJ, Kehret AR, Linton JD, et al. Building the Next Generation of Humanized Hemato-Lymphoid System Mice. Front Immunol (2021) 12:643852. doi: 10.3389/fimmu.2021.643852
76. Song Y, Rongvaux A, Taylor A, Jiang T, Tebaldi T, Balasubramanian K, et al. A Highly Efficient and Faithful MDS Patient-Derived Xenotransplantation Model for Pre-Clinical Studies. Nat Commun (2019) 10(1):366. doi: 10.1038/s41467-018-08166-x
77. Rouault-Pierre K, Mian SA, Goulard M, Abarrategi A, Di Tulio A, Smith AE, et al. Preclinical Modeling of Myelodysplastic Syndromes. Leukemia (2017) 31(12):2702–8. doi: 10.1038/leu.2017.172
78. Pang WW, Pluvinage JV, Price EA, Sridhar K, Arber DA, Greenberg PL, et al. Hematopoietic Stem Cell and Progenitor Cell Mechanisms in Myelodysplastic Syndromes. Proc Natl Acad Sci USA (2013) 110:3011–6.
79. Muguruma Y, Matsushita H, Yahata T, Yumino S, Tanaka Y, Miyachi H, et al. Establishment of a Xenograft Model of Human Myelodysplastic Syndromes. Haematologica (2011) 96(4):543–51. doi: 10.3324/haematol.2010.027557
80. Medyouf H, Mossner M, Jann J-C, Nolte F, Raffel S, Herrmann C, et al. Myelodysplastic Cells in Patients Reprogram Mesenchymal Stromal Cells to Establish a Transplantable Stem Cell Niche Disease Unit. Cell Stem Cell (2014) 14(6):824–37. doi: 10.1016/j.stem.2014.02.014
81. Meunier M, Dussiau C, Mauz N, Alary AS, Lefebvre C, et al. Molecular Dissection of Engraftment in a Xenograft Model of Myelodysplastic Syndromes. Oncotarget (2018) 9:14993–5000.
82. Franks CR, Bishop D, Balkwill FR, Oliver RT, Spector WG. Growth of Acute Myeloid Leukaemia as Discrete Subcutaneous Tumours in Immune-Deprived Mice. Br J Cancer (1977) 35(5):697–700. doi: 10.1038/bjc.1977.107
83. Andriole GL, Mulé JJ, Hansen CT, Linehan WM, Rosenberg SA. Evidence That Lymphokine-Activated Killer Cells and Natural Killer Cells are Distinct Based on an Analysis of Congenitally Immunodeficient Mice. J Immunol (1985) 135(5):2911–3.
84. Bosma GC, Custer RP, Bosma MJ. A Severe Combined Immunodeficiency Mutation in the Mouse. Nature (1983) 301(5900):527–30. doi: 10.1038/301527a0
85. Bosma MJ, Carroll AM. The SCID Mouse Mutant: Definition, Characterization, and Potential Uses. Annu Rev Immunol (1991) 9(1):323–50. doi: 10.1146/annurev.iy.09.040191.001543
86. Kamel-Reid S, Dick JE. Engraftment of Immune-Deficient Mice With Human Hematopoietic Stem Cells. Science (1988) 242(4886):1706–9. doi: 10.1126/science.2904703
87. Kamel-Reid S, Letarte M, Sirard C, Doedens M, Grunberger T, Fulop G, et al. A Model of Human Acute Lymphoblastic Leukemia in Immune-Deficient SCID Mice. Science (1989) 246(4937):1597–600. doi: 10.1126/science.2595371
88. Lapidot T, Pflumio F, Doedens M, Murdoch B, Williams DE, Dick JE. Cytokine Stimulation of Multilineage Hematopoiesis From Immature Human Cells Engrafted in SCID Mice. Science (1992) 255(5048):1137–41. doi: 10.1126/science.1372131
89. Cesano A, Hoxie JA, Lange B, Nowell PC, Bishop J, Santoli D. The Severe Combined Immunodeficient (SCID) Mouse as a Model for Human Myeloid Leukemias. Oncogene (1992) 7(5):827–36.
90. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A Cell Initiating Human Acute Myeloid Leukaemia After Transplantation Into SCID Mice. Nature (1994) 367(6464):645–8. doi: 10.1038/367645a0
91. Baersch G, Mollers T, Hotte A, Dockhorn-Dworniczak B, Rube C, Ritter J, et al. Good Engraftment of B-Cell Precursor ALL in NOD-SCID Mice. Klin Padiatr (1997) 209(4):178–85. doi: 10.1055/s-2008-1043947
92. Nijmeijer BA, Mollevanger P, van Zelderen-Bhola SL, Kluin-Nelemans HC, Willemze R, Falkenburg JH. Monitoring of Engraftment and Progression of Acute Lymphoblastic Leukemia in Individual NOD/SCID Mice. Exp Hematol (2001) 29(3):322–9. doi: 10.1016/S0301-472X(00)00669-X
93. Lumkul R, Gorin NC, Malehorn MT, Hoehn GT, Zheng R, Baldwin B, et al. Human AML Cells in NOD/SCID Mice: Engraftment Potential and Gene Expression. Leukemia (2002) 16(9):1818–26. doi: 10.1038/sj.leu.2402632
94. Pearce DJ, Taussig D, Zibara K, Smith LL, Ridler CM, Preudhomme C, et al. AML Engraftment in the NOD/SCID Assay Reflects the Outcome of AML: Implications for Our Understanding of the Heterogeneity of AML. Blood (2006) 107(3):1166–73. doi: 10.1182/blood-2005-06-2325
95. Nilsson L, Åstrand-Grundström I, Anderson K, Arvidsson I, Hokland P, Bryder D, et al. Involvement and Functional Impairment of the CD34+CD38–Thy-1+ Hematopoietic Stem Cell Pool in Myelodysplastic Syndromes With Trisomy 8. Blood (2002) 100(1):259–67. doi: 10.1182/blood-2001-12-0188
96. Benito AI, Bryant E, Loken MR, Sale GE, Nash RA, John Gass M, et al. NOD/SCID Mice Transplanted With Marrow From Patients With Myelodysplastic Syndrome (MDS) Show Long-Term Propagation of Normal But Not Clonal Human Precursors. Leuk Res (2003) 27(5):425–36. doi: 10.1016/S0145-2126(02)00221-7
97. Christianson SW, Greiner DL, Hesselton RA, Leif JH, Wagar EJ, Schweitzer IB, et al. Enhanced Human CD4+ T Cell Engraftment in Beta2-Microglobulin-Deficient NOD-Scid Mice. J Immunol (1997) 158(8):3578–86.
98. Kollet O, Peled A, Byk T, Ben-Hur H, Greiner D, Shultz L, et al. β2 Microglobulin-Deficient (B2mnull) NOD/SCID Mice Are Excellent Recipients for Studying Human Stem Cell Function. Blood (2000) 95(10):3102–5. doi: 10.1182/blood.V95.10.3102
99. Glimm H, Eisterer W, Lee K, Cashman J, Holyoake TL, Nicolini F, et al. Previously Undetected Human Hematopoietic Cell Populations With Short-Term Repopulating Activity Selectively Engraft NOD/SCID-β2 Microglobulin–Null Mice. J Clin Invest (2001) 107(2):199–206. doi: 10.1172/JCI11519
100. Yoshino H, Ueda T, Kawahata M, Kobayashi K, Ebihara Y, Manabe A, et al. Natural Killer Cell Depletion by Anti-Asialo GM1 Antiserum Treatment Enhances Human Hematopoietic Stem Cell Engraftment in NOD/Shi-Scid Mice. Bone Marrow Transplant (2000) 26(11):1211–6. doi: 10.1038/sj.bmt.1702702
101. Hogan CJ, Shpall EJ, Keller G. Differential Long-Term and Multilineage Engraftment Potential From Subfractions of Human CD34+ Cord Blood Cells Transplanted Into NOD/SCID Mice. Proc Natl Acad Sci USA (2002) 99(1):413–8. doi: 10.1073/pnas.012336799
102. Thanopoulou E, Cashman J, Kakagianne T, Eaves A, Zoumbos N, Eaves C. Engraftment of NOD/SCID-β2 Microglobulin Null Mice With Multilineage Neoplastic Cells From Patients With Myelodysplastic Syndrome. Blood (2004) 103(11):4285–93. doi: 10.1182/blood-2003-09-3192
103. Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, et al. Human Lymphoid and Myeloid Cell Development in NOD/LtSz-Scid IL2Rγnull Mice Engrafted With Mobilized Human Hemopoietic Stem Cells. J Immunol (2005) 174(10):6477–89. doi: 10.4049/jimmunol.174.10.6477
104. Martin MG, Welch JS, Uy GL, Fehniger TA, Kulkarni S, Duncavage EJ, et al. Limited Engraftment of Low-Risk Myelodysplastic Syndrome Cells in NOD/SCID Gamma-C Chain Knockout Mice. Leukemia (2010) 24(9):1662–4. doi: 10.1038/leu.2010.156
105. Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M, et al. AML Xenograft Efficiency is Significantly Improved in NOD/SCID-IL2RG Mice Constitutively Expressing Human SCF, GM-CSF and IL-3. Leukemia (2010) 24(10):1785–8. doi: 10.1038/leu.2010.158
106. Miller PH, Cheung AMS, Beer PA, Knapp DJHF, Dhillon K, Rabu G, et al. Enhanced Normal Short-Term Human Myelopoiesis in Mice Engineered to Express Human-Specific Myeloid Growth Factors. Blood (2013) 121(5):e1–4. doi: 10.1182/blood-2012-09-456566
107. Li L, Sheng Y, Li W, Hu C, Mittal N, Tohyama K, et al. β-Catenin Is a Candidate Therapeutic Target for Myeloid Neoplasms With Del(5q). Cancer Res (2017) 77(15):4116–26. doi: 10.1158/0008-5472.CAN-17-0202
108. Hyoda T, Tsujioka T, Nakahara T, Suemori S-I, Okamoto S, Kataoka M, et al. Rigosertib Induces Cell Death of a Myelodysplastic Syndrome-Derived Cell Line by DNA Damage-Induced G2/M Arrest. Cancer Sci (2015) 106(3):287–93. doi: 10.1111/cas.12605
109. Tsujioka T, Yokoi A, Itano Y, Takahashi K, Ouchida M, Okamoto S, et al. Five-Aza-2′-Deoxycytidine-Induced Hypomethylation of Cholesterol 25-Hydroxylase Gene Is Responsible for Cell Death of Myelodysplasia/Leukemia Cells. Sci Rep (2015) 5(1):16709. doi: 10.1038/srep16709
110. Fang J, Liu X, Bolanos L, Barker B, Rigolino C, Cortelezzi A, et al. A Calcium- and Calpain-Dependent Pathway Determines the Response to Lenalidomide in Myelodysplastic Syndromes. Nat Med (2016) 22(7):727–34. doi: 10.1038/nm.4127
111. Nicolini FE, Cashman JD, Hogge DE, Humphries RK, Eaves CJ. NOD/SCID Mice Engineered to Express Human IL-3, GM-CSF and Steel Factor Constitutively Mobilize Engrafted Human Progenitors and Compromise Human Stem Cell Regeneration. Leukemia (2004) 18(2):341–7. doi: 10.1038/sj.leu.2403222
112. Sippel TR, Radtke S, Olsen TM, Kiem H-P, Rongvaux A. Human Hematopoietic Stem Cell Maintenance and Myeloid Cell Development in Next-Generation Humanized Mouse Models. Blood Adv (2019) 3(3):268–74. doi: 10.1182/bloodadvances.2018023887
113. Kerbauy DMB, Lesnikov V, Torok-Storb B, Bryant E, Deeg HJ. Engraftment of Distinct Clonal MDS-Derived Hematopoietic Precursors in NOD/SCID-β2-Microglobulin-Deficient Mice After Intramedullary Transplantation of Hematopoietic and Stromal Cells. Blood (2004) 104(7):2202–3. doi: 10.1182/blood-2004-04-1518
114. Maria K, Xiaochuan S, Chenghui Z, Cedric Dos S, Georges Habineza N, Anthony S, et al. Cytokines Increase Engraftment of Human Acute Myeloid Leukemia Cells in Immunocompromised Mice But Not Engraftment of Human Myelodysplastic Syndrome Cells. Haematologica (2018) 103(6):959–71. doi: 10.3324/haematol.2017.183202
115. Sommerkamp P, Mercier FE, Wilkinson AC, Bonnet D, Bourgine PE. Engineering Human Hematopoietic Environments Through Ossicle and Bioreactor Technologies Exploitation. Exp Hematol (2021) 94:20–5. doi: 10.1016/j.exphem.2020.11.008
116. Reinisch A, Thomas D, Corces MR, Zhang X, Gratzinger D, Hong WJ, et al. A Humanized Bone Marrow Ossicle Xenotransplantation Model Enables Improved Engraftment of Healthy and Leukemic Human Hematopoietic Cells. Nat Med (2016) 22(7):812–21. doi: 10.1038/nm.4103
117. Hayakawa J, Hsieh MM, Uchida N, Phang O, Tisdale JF. Busulfan Produces Efficient Human Cell Engraftment in NOD/LtSz-Scid Il2rγnull Mice. Stem Cells (2009) 27(1):175–82. doi: 10.1634/stemcells.2008-0583
118. Choi B, Chun E, Kim M, Kim S-T, Yoon K, Lee K-Y, et al. Human B Cell Development and Antibody Production in Humanized NOD/SCID/IL-2rγnull (NSG) Mice Conditioned by Busulfan. J Clin Immunol (2011) 31(2):253–64. doi: 10.1007/s10875-010-9478-2
119. Srikanthan MA, Humbert O, Haworth KG, Ironside C, Rajawat YS, Blazar BR, et al. Effective Multi-Lineage Engraftment in a Mouse Model of Fanconi Anemia Using Non-Genotoxic Antibody-Based Conditioning. Mol Ther Methods Clin Dev (2020) 17:455–64. doi: 10.1016/j.omtm.2020.02.001
120. Migliaccio AR, Carta C, Migliaccio G. In Vivo Expansion of Purified Hematopoietic Stem Cells Transplanted in Nonablated W/Wv Mice. Exp Hematol (1999) 27(11):1655–66. doi: 10.1016/S0301-472X(99)00110-1
121. Waskow C, Madan V, Bartels S, Costa C, Blasig R, Rodewald H-R. Hematopoietic Stem Cell Transplantation Without Irradiation. Nat Methods (2009) 6(4):267–9. doi: 10.1038/nmeth.1309
122. Cosgun KN, Rahmig S, Mende N, Reinke S, Hauber I, Schafer C, et al. Kit Regulates HSC Engraftment Across the Human-Mouse Species Barrier. Cell Stem Cell (2014) 15(2):227–38. doi: 10.1016/j.stem.2014.06.001
123. Yurino A, Takenaka K, Yamauchi T, Nunomura T, Uehara Y, Jinnouchi F, et al. Enhanced Reconstitution of Human Erythropoiesis and Thrombopoiesis in an Immunodeficient Mouse Model With KitWv Mutations. Stem Cell Rep (2016) 7(3):425–38. doi: 10.1016/j.stemcr.2016.07.002
124. Mian SA, Abarrategi A, Kong KL, Rouault-Pierre K, Wood H, Oedekoven CA, et al. Ectopic Humanized Mesenchymal Niche in Mice Enables Robust Engraftment of Myelodysplastic Stem Cells. Blood Cancer Discov (2021) 2(2):135–45. doi: 10.1158/2643-3230.BCD-20-0161
125. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-Deficient Mice Have No Mature B and T Lymphocytes. Cell (1992) 68(5):869–77. doi: 10.1016/0092-8674(92)90030-G
126. Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, et al. RAG-2-Deficient Mice Lack Mature Lymphocytes Owing to Inability to Initiate V(D)J Rearrangement. Cell (1992) 68(5):855–67. doi: 10.1016/0092-8674(92)90029-C
127. Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, et al. Development and Function of Human Innate Immune Cells in a Humanized Mouse Model. Nat Biotechnol (2014) 32(4):364–72. doi: 10.1038/nbt.2858
128. Deng K, Pertea M, Rongvaux A, Wang L, Durand CM, Ghiaur G, et al. Broad CTL Response is Required to Clear Latent HIV-1 Due to Dominance of Escape Mutations. Nature (2015) 517(7534):381–5. doi: 10.1038/nature14053
129. Notta F, Doulatov S, Dick JE. Engraftment of Human Hematopoietic Stem Cells is More Efficient in Female NOD/SCID/IL-2Rgc-Null Recipients. Blood (2010) 115(18):3704–7. doi: 10.1182/blood-2009-10-249326
Keywords: humanized mouse models, immunodeficient mouse models, transgenic mouse models, xenograft animal model, myelodysplastic syndromes (MDS)
Citation: Liu W, Teodorescu P, Halene S and Ghiaur G (2022) The Coming of Age of Preclinical Models of MDS. Front. Oncol. 12:815037. doi: 10.3389/fonc.2022.815037
Received: 14 November 2021; Accepted: 21 February 2022;
Published: 16 March 2022.
Edited by:
Maria Figueroa, University of Miami, United StatesReviewed by:
Luisa Cimmino, University of Miami, United StatesCopyright © 2022 Liu, Teodorescu, Halene and Ghiaur. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie Halene, c3RlcGhhbmllLmhhbGVuZUB5YWxlLmVkdQ==; Gabriel Ghiaur, Z2doaWF1cjFAamhtaS5lZHU=
†These authors have contributed equally to this work
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.