 Serine Avagyan
Serine Avagyan Akiko Shimamura
Akiko Shimamura
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 09 March 2022
Sec. Hematologic Malignancies
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.813149
This article is part of the Research Topic Advancing Science for Clinical Care in MDS View all 6 articles
Pediatric myelodysplastic syndromes (MDS) often raise concern for an underlying germline predisposition to hematologic malignancies, referred to as germline predisposition herein. With the availability of genetic testing, it is now clear that syndromic features may be lacking in patients with germline predisposition. Many genetic lesions underlying germline predisposition may also be mutated somatically in de novo MDS and leukemias, making it critical to distinguish their germline origin. The verification of a suspected germline predisposition informs therapeutic considerations, guides monitoring pre- and post-treatment, and allows for family counseling. Presentation of MDS due to germline predisposition is not limited to children and spans a wide age range. In fact, the risk of MDS may increase with age in many germline predisposition conditions and can present in adults who lack classical stigmata in their childhood. Furthermore, germline predisposition associated with DDX41 mutations presents with older adult-onset MDS. Although a higher proportion of pediatric patients with MDS will have a germline predisposition, the greater number of MDS diagnoses in adult patients may result in a larger overall number of those with an underlying germline predisposition. In this review, we present a framework for the evaluation of germline predisposition to MDS across all ages. We discuss characteristics of personal and family history, clinical exam and laboratory findings, and integration of genetic sequencing results to assist in the diagnostic evaluation. We address the implications of a diagnosis of germline predisposition for the individual, for their care after MDS therapy, and for family members. Studies on MDS with germline predisposition have provided unique insights into the pathogenesis of hematologic malignancies and mechanisms of somatic genetic rescue vs. disease progression. Increasing recognition in adult patients will inform medical management and may provide potential opportunities for the prevention or interception of malignancy.
Although historically considered in the context of pediatric myelodysplastic syndromes (MDS), genetic predisposition to hematologic malignancies, including MDS, is now recognized to manifest across the age spectrum (1–4). Germline etiology has been associated with up to 15% of MDS diagnoses (1, 2, 5, 6), a number that undoubtedly will increase with the availability of clinical genetic testing and our understanding of MDS pathobiology. Most germline MDS predisposition conditions were originally characterized in patients with severe early-onset presentations with specific syndromic features. However, the absence of classic physical features or family history of hematologic malignancies does not preclude consideration of germline predisposition, due to clinically cryptic phenotypes or de novo germline mutations.
MDS associated with germline predisposition is now classified separately in the 2016 World Health Organization system (7). Recognition of an underlying germline predisposition has broad implications for both patients and their families. As a hematopoietic stem cell transplantation is the curative therapy for many patients with MDS, donor choices will be influenced by the underlying genetics, which may affect other family members. Lastly, the diagnosis of germline MDS predisposition guides family counseling and family planning and allows appropriate monitoring of affected asymptomatic individuals to maintain health. We present here a framework for evaluating MDS that is applicable to both pediatric and adult patients.
Consider the following cases presenting to an adult clinic: (1) a 27-year-old sibling of a 19-year-old man with MDS is undergoing evaluation as a possible donor for a stem cell transplantation. This sibling is unexpectedly found to have macrocytic anemia and thrombocytopenia. A subsequent bone marrow evaluation reveals multilineage dysplasia with trisomy 8 consistent with MDS; (2) A 57-year-old woman with MDS reports that her 16-year-old grandson was treated for acute myeloid leukemia. Should these cases raise concern for predisposition syndromes? The first case of both a proband and sibling with MDS would be suspicious for an underlying genetic etiology, which, in this case, was a germline GATA2 mutation. The second case of multiple family members with myeloid malignancy should also raise consideration of germline predisposition, despite the adult-onset MDS diagnosis. This family harbored a germline mutation in RUNX1 that manifested across generations.
Approximately 90% of MDS is diagnosed in adults over the age of 60 years, with only 6% diagnosed in those younger than 50 years (8). It is increasingly recognized that patients with conditions previously considered pediatric diseases may first come to medical attention with adult-onset MDS. The diagnosis of MDS may be the first presentation of the underlying germline predisposition or may be the hematologic presentation of a syndrome unrecognized with subtle or non-hematologic manifestations, like short-stature in Shwachman–Diamond syndrome (SDS) (2) or lymphedema in GATA2 deficiency (9). Furthermore, due to high degree of variability of penetrance of a specific phenotype, manifestations of the different aspects of the syndrome may present at different ages. Even within single families, the genotype often poorly correlates with the phenotype in germline predisposition. For example, RUNX1-familial platelet disorder (RUNX1-FPD), a syndrome associated with germline mutations in RUNX1, may present with MDS, AML, T-cell acute lymphoblastic leukemia, or other lymphoid cancers between the ages of 9 and 65 (10). In telomere biology disorders (TBD), MDS and myeloid leukemia were the most commonly diagnosed cancers, with a median age at diagnosis of 53 years (range of 12–71 years old) (11). TERT mutations were identified in 2.7% of adult patients with MDS (12). DDX41 mutations associated with MDS and AML are characterized by presentation in older adulthood, often without any family history (13, 14). Identification of germline mutations in DDX41 in familial malignancies presenting at older ages has shifted the paradigm of germline predisposition affecting only children and young adults. Over 70 families with germline DDX41 mutations have been reported, with a median age of MDS onset of 65 years (range of 41–88 years old), similar to the median age of onset of sporadic MDS in early 70s (15, 16). Thus, predisposition syndromes should be considered in adult patients with MDS to inform medical management.
MDS may be the first presenting clinical feature of a germline predisposition condition. The diagnostic evaluation for a potential genetic predisposition condition integrates the history, laboratory tests, histopathological findings in the bone marrow, and genetic testing. Focusing on the syndromes that span pediatric and adult hematology, we will focus on clues to the diagnosis of germline predisposition. Since many of these syndromes have been described previously, we will not review them in detail here but refer the reader to these excellent references (17–19).
In general, the presenting signs and symptoms of MDS do not differ in patients with germline predisposition and include nonspecific clinical findings of fatigue, pallor, malaise, fevers, bleeding, increased bruising, weight loss, and anorexia. The marrow is often hypocellular in pediatric MDS or MDS in germline predisposition, in contrast to adult-onset MDS, which is more commonly hypercellular. Adult patients with hypocellular MDS have significantly more pronounced cytopenias at presentation compared to those presenting with normocellular or hypercellular marrows (20). It is yet unknown whether the small subset of hypocellular MDS in adults, between 10% and 20% of all MDS (21, 22), are more likely to be related to germline predisposition. For example, germline DDX41-related adult-onset MDS is associated with a hypocellular marrow (14, 16). The duration of specific clinical symptoms may allow insight into the chronicity of cytopenias. In patients with history of neutropenia in childhood and adolescence, information on the length and dose of granulocyte-colony stimulating factor (G-CSF) usage is helpful in the evaluation of a patient with new MDS, as it may suggest germline mutations associated with severe congenital neutropenia (SCN) due to germline ELANE mutations. Prolonged use, particularly with high doses, of G-CSF in SCN is associated with a 22% cumulative incidence of MDS or secondary AML after 15 years of therapy (23). The risk of MDS development is associated with a somatic acquisition of mutations in CSF3R, the receptor for G-CSF (24–26). A history of prior cytopenias or unexplained macrocytosis may also serve as a clinical clue for germline predisposition. Elevated hemoglobin F may be associated with inherited germline predisposition (27) but can also be elevated in MDS and, thus, should be interpreted with caution (28–30).
Past medical history of bleeding, easy bruising, or diagnosis of “chronic immune thrombocytopenia” is a feature associated with mutations in transcription factors important for megakaryopoiesis and platelet function, including RUNX1, ANKRD26, and ETV6 (31–33). Patients with germline mutations in these genes may have quantitative and/or qualitative platelet defects, manifesting with easy bruising, heavy menses in women, or other bleeding symptoms, which may be out of proportion to the platelet count. Amegakaryocytic thrombocytopenia, often with radioulnar synostosis, is associated with germline MECOM mutations, presenting as congenital bone marrow failure in early childhood (34, 35). In rare case reports, mutations in MECOM have been described in association with adult-onset MDS (36). A history of thrombosis has been reported in GATA2 deficiency, related to cerebrovascular accidents, deep vein thrombosis, pulmonary embolism, and portal vein thrombosis (9). Clinical features of GATA2 deficiency may include recurrent infections, atypical mycobacterial infections, warts, pulmonary alveolar proteinosis, or lymphedema. Although not with clear association to vascular dysfunction or immune deficiencies, premature labor and miscarriages have been described in GATA2 deficiency (9, 37) and other germline conditions (38–40).
Past medical history of malignancy should be investigated as part of the evaluation of germline predisposition (3). In RUNX1-FPD, cases of secondary malignancy with MDS after therapy for acute lymphoblastic leukemia (ALL) have been described, particularly in survivors of childhood T-cell ALL (10, 41, 42). A history of excessive toxicities with chemotherapy or radiation may also suggest a genetic predisposition such as Fanconi anemia (FA). A history of multiple malignancies should raise concern for possible germline predisposition. Solid organ cancers are associated with conditions such as FA [squamous cell carcinomas of head and neck, gastrointestinal (GI) and genital tract, and other solid organ cancers], TBD (head/neck and GI) (43), and Diamond–Blackfan anemia (DBA), particularly in adults (44). In the case of FA, patients may have subclinical mild cytopenias or normal blood counts in childhood, only to present with a solid organ cancer in adulthood, and experience severe protracted pancytopenia with chemotherapy or radiation (45). The diagnosis of a germline predisposition in a patient with a past medical history of malignancy guides subsequent cancer monitoring. This is particularly important in syndromes associated with mutations affecting genomic instability such as FA and Li–Fraumeni syndrome (LFS).
A detailed investigation of the past medical history of a patient with MDS may also identify non-hematologic clinical manifestations of germline predisposition (Table 1).
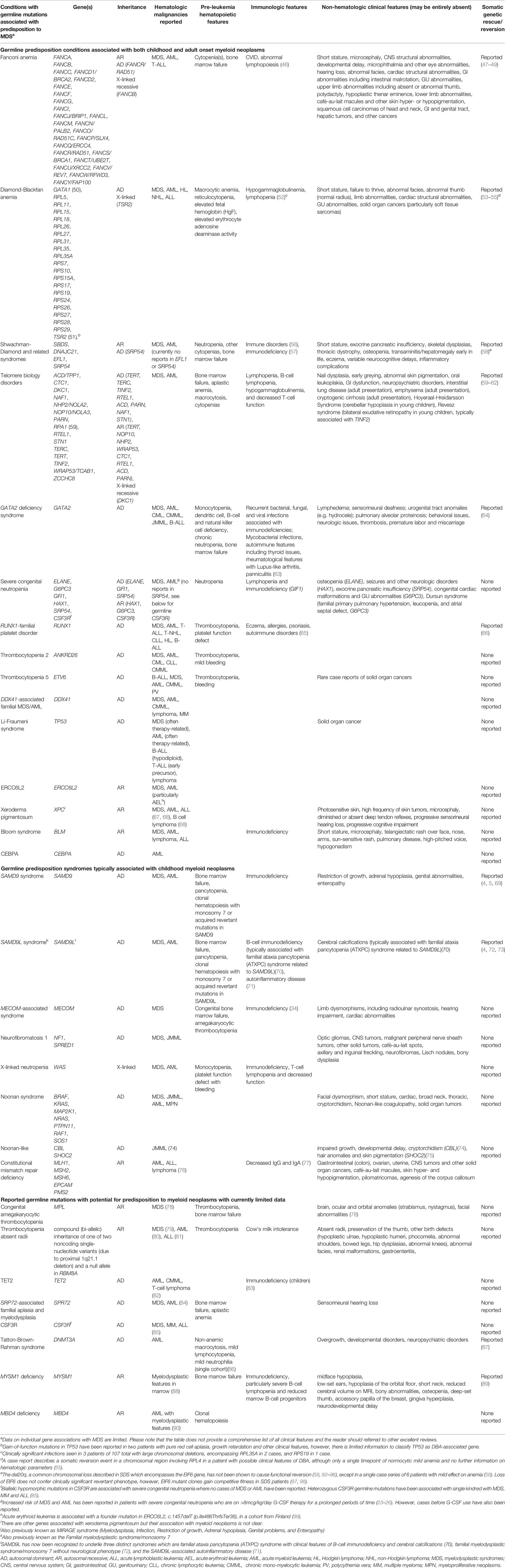
Table 1 Clinical features of and genetic mutations underlying germline conditions with predisposition to myeloid neoplasms.
Growth and Congenital Anomalies. Many germline predisposition conditions are associated with a history of intrauterine growth retardation, failure to thrive in childhood, and short stature. Congenital malformations, including skeletal anomalies, abnormal digits, cardiac structural defects, and gastrointestinal (GI) or genitourinary (GU) abnormalities, are important to recognize, many of which may have been repaired in childhood. Such malformations may be part of early presentations of FA (99), TBD (100), DBA (101, 102), and other germline predisposition (Table 1). A history of hydrocele in infancy in patients with GATA2 deficiency may be one of the earliest manifestations of the lymphatic dysfunction of that syndrome that is often unrecognized.
Immunologic Features. Many predisposition conditions are associated with immunodeficiency, which may manifest with recurrent infections or immune dysregulation, or may be clinically asymptomatic but detected by laboratory testing for lymphocyte counts, lymphocyte subsets (T-, B-, and NK cells), and immunoglobulin levels. GATA2 deficiency syndrome, for instance, is associated with atypical mycobacterial infections and warts (genital and extragenital) due to the decreased natural killer and dendritic cells, as well as B-cell lymphopenia (9, 103). Immunodeficiency has also been described in TBD (104–107) and in germline TET2 deficiency (83), particularly in childhood for the latter. There may also be autoimmune or inflammatory manifestations of germline predisposition conditions that are actively being studied. These include thyroid disease [GATA2 deficiency (9)], eczema and allergies [RUNX1-FPD (10, 65)], psoriasis [RUNX1-FPD (10)], panniculitis, and lupus-like arthritis [GATA2 deficiency (103, 108)], and others listed in Table 1. Laboratory screening of immunological abnormalities is recommended below and should be considered in the workup of MDS patients.
Beyond the Blood. Given the multifaceted functional roles of the underlying genes in germline predisposition, it is not surprising to find tissues other than blood being affected in these conditions. Table 1 lists examples of non-hematologic manifestations of these conditions. A history of extremity edema, typically unilateral and sometimes diagnosed as hemihypertrophy, or genital edema (for example, hydrocele) may be related to germline GATA2 deficiency-associated lymphedema (9, 63). GATA2 is expressed in the lymphatic system (109, 110) and is thought to underlie various forms of lymphatic dysfunction, occasionally with atypical presentations such as swelling on the face or preauricular region (111). Adult-onset non-hematologic manifestations of TBD include a vast range of clinical findings (107). These include interstitial lung disease, particularly pulmonary fibrosis, and liver cirrhosis, often diagnosed and managed by various subspecialists. We inquire about hearing loss of any type and, if clinically indicated, pursue testing.
Family history provides important clues to an underlying germline predisposition; however, a negative family history does not rule out a genetic disorder. Attention should be paid to the hematologic and non-hematologic manifestations of germline predisposition conditions. Given the variable expressivity and penetrance of many of these conditions, with limited genotype–phenotype correlations described to date, clinical manifestations may vary or even be absent between different family members carrying the same genetic predisposition mutation. For example, studies in kindreds with GATA2 deficiency have reported that by age 40 years, 80% of affected individuals presented with either hematologic malignancies or infections (9, 103). However, the prevalence of clinical manifestations of germline predisposition should be interpreted with caution given potential confounding factors such as selection and ascertainment bias, and mechanisms of somatic genetic reversion discussed later.
A negative family history of MDS or other hematologic malignancies should not preclude consideration of an underlying germline genetic etiology of MDS. Silent clinical presentations have been described in a number of germline MDS syndromes. Disease manifestation may present at various ages within a kindred with the same germline genetic mutation (112). Disease anticipation has been described for some of these conditions, with clinical features being more severe and earlier in onset for each subsequent generation (113). Finally, the germline syndrome may be due to a de novo or recessive mutation. Table 1 lists many clinical features variably associated with germline predisposition, which can be used as a general guide; however, absence of these findings does not rule out a germline condition. As the inheritance of these conditions may be autosomal dominant or recessive, or X-linked (Table 1), in addition to the history of immediate family members (parents, grandparents, children, and siblings), querying the history of extended family members may also be illuminating.
It is important to inquire about family members with any cytopenias, need for transfusions, growth factor support, or splenectomy as possible therapy for thrombocytopenia. If there is a history of antecedent cytopenia, the type, duration, and severity of the cytopenia, prior diagnostic evaluation (such as bone marrow results), and responses to any prior treatments should be queried. Inquiring about clinical symptoms of bleeding, frequent infections, and chronic anemia will address any cellular dysfunction. For example, bleeding symptoms disproportionately severe relative to the platelet counts may be a clue to RUNX1-FPD or ANKRD26 mutations. A family history of recurrent infections of any type, but particularly viral (HPV caused warts in GATA2 deficiency), opportunistic infections [herpes zoster reactivation, primary CMV with retinitis/encephalitis, Pneumocystis jirovecii pneumonia, and Candida esophagitis in TBD (104, 107, 114)] or atypical bacterial (mycobacterial infections in GATA2 deficiency), may suggest the presence of concurrent immunodeficiency and should prompt a more careful laboratory evaluation.
We inquire about any family members with previous diagnoses of hematologic malignancies, such as MDS, myeloid or lymphoid leukemia, or lymphoma, and the age at which these diagnoses were made. A diagnosis of a malignancy at an unusually young age should be noted. Germline predisposition, such as those associated with mutations in hematopoietic transcription factors RUNX1, ANKRD26, GATA2, and ETV6, can present with different types of hematopoietic malignancies, including MDS, AML, T-cell ALL, B-cell ALL, and myeloproliferative neoplasms (MPNs). A family history of chemotherapy sensitivity or multiple malignancies also raise suspicion for a potential germline disorder. Solid organ or head and neck cancers may be presenting features of conditions such as FA, TBD, LFS, or DBA. A family history of clinical features associated with genetic predisposition should also raise suspicion.
Finally, a history of consanguinity in the family may raise the possibility of autosomal recessive syndromes. There are rare genetic associations with founder variants in germline predisposition, for example, North African XPC founder mutation associated with xeroderma pigmentosum subtype C (67, 68) and the ERCC6L2 mutation in the Finnish population (98). Thus, asking about ancestral origins may guide the differential diagnoses.
The physical exam may provide additional clues to an underlying germline predisposition in the context of an MDS diagnosis; however, the lack of syndromic features (Table 1) does not rule out an underlying germline predisposition.
Short stature relative to predicted height is one of the most telling physical exam findings suggestive of a predisposition condition in an adult with MDS (Table 1). In a large study of MDS patients who underwent stem cell transplantation, among patients younger than 40 years of age, biallelic SBDS mutations were associated with significantly lower height percentile (2). Skin hypo- and hyperpigmentation are features of TBD and may also be present with FA, where café-au-lait spots are often observed. A careful oral exam, especially in patients with suspected TBD and FA, should be conducted. Suspicious oral lesions should be referred to an otolaryngologist for further evaluation. Patients with FA are at high risk for squamous cell carcinomas (99). Exam of extremities should note any type of thumb abnormalities found in FA (for example, hypoplasia or pouce flottant, “floating” thumb, with associated radius abnormalities), DBA (for example, small, large, or triphalangeal, finger-like, thumb), and MECOM-associated syndrome (radioulnar synostosis). Hand and forearm X-rays may be helpful to diagnose findings not readily apparent on exam if clinical suspicion is high.
In addition to the complete blood count with differential and reticulocyte count at presentation, prior blood count reports are helpful to detect pre-existing cytopenias suggestive of a pre-existing disorder. Liver function tests are helpful in consideration of TBD and FA. To screen for immunological abnormalities, we typically test lymphocytes subsets for absolute counts of CD3+ T-cells, with CD4 and CD8 subpopulations, CD19+ or CD20+ B-cells, and CD16+ or CD56+ NK cells. We also screen for levels of IgG, IgA, and IgM immunoglobulins. Combined cytopenias and lymphocytopenias or lymphocyte dysfunction (low IgM or IgG levels) may flag a potential germline condition. B- or NK cell lymphocytopenias may be present in GATA2 deficiency, and abnormal Ig levels may be present in GATA2 deficiency and TBD. It should be noted that lympocytopenia (low absolute lymphocyte count, ALC, of <1.2 × 109/L) has been described in MDS (115, 116). Here, again, a comparison of the ALC with previous values, if available, could be helpful to distinguish pre-existing abnormalities from those associated with MDS.
Chromosomal breakage testing for FA should be interpreted with caution in patients with MDS, particularly if peripheral blasts are present or if prior cytoreductive therapy has been administered. FA chromosomal breakage test is typically performed on peripheral blood leukocytes exposed to clastogenic agents diepoxybutane (DEB) and mitomycin C (MMC). Increased chromosomal breaks and fusions with radial forms are observed in FA. FA chromosomal breakage testing in the blood may be non-diagnostic due to peripheral blood somatic mosaicism resulting in two distinct populations of cells, where one of the populations harbors the germline mutation, while the other has a somatic genetic rescue that corrects the hypersensitivity to DNA cross-linking agents (47, 48). Skin fibroblasts can help diagnose blood mosaicism. The FA chromosomal breakage tests can be affected by prior chemotherapy or radiation therapy, since the lymphocytes may manifest baseline chromosomal breakage, however, a further increase in chromosomal breakage with the addition of DEB or MMC would support the diagnosis of FA. In such cases, performing FA testing on skin fibroblasts may be helpful. The results of the FA chromosome breakage test should be interpreted with caution and with advice from experts in the field. The diagnosis of FA informs the therapeutic plan, particularly for stem cell transplantation conditioning regimen given the sensitivity to genotoxic agents, making this is a critical part of MDS evaluation.
The telomere length testing for TBD is available clinically using a flow-FISH assay (117); however, interpretation can be challenging in the context of MDS, particularly following prior cytoreductive therapy, which might result in secondary telomere shortening. In untreated MDS patients without excess blasts, telomere lengths less than the first percentile for age in at least three different lymphocyte subsets are highly suspicious for a primary disorder of telomere maintenance (118, 119). It should be stressed that this parameter, although highly predictive in children, is not as well defined in adults, where the telomere length may fall above the first percentile for age even with known underlying genetic mutations related to telomere biology (11). If telomere measurement is available only in a single lymphocyte subset and is less than the first percentile for age, this could be suggestive of an underlying TBD and should be correlated with personal or family history of clinical features of TBD and/or germline genetic lesions. Shortening of telomeres in the granulocytes is a non-specific finding and, in isolation, is insufficient to flag a TBD. The analysis of telomere length in patients with MDS needs further study. Flow-FISH telomere length measurement must be interpreted with caution in a patient with MDS, and the potential influence of excess blasts or other secondary hematologic stressors has not been defined for this assay. In the research setting, the telomere length in MDS measured using real-time quantitative polymerase chain reaction (qPCR) analysis has been described to be shorter than in healthy individuals and is independently related to outcome in MDS after stem cell transplantation (120). The telomere length should be integrated with the clinical context, personal or family history of any TBD-related manifestations, other laboratory findings suggestive of a genetic telomere disorder, and genetic testing (see below).
Additional focused laboratory testing may be considered if clinically indicated. Evaluation of pancreatic insufficiency in SDS or SDS-like syndromes includes a pancreatic isoamylase for anyone older than 3 years old. Abdominal imaging may be helpful to assess for pancreatic lipomatosis or an atretic/abnormal pancreas suggestive of SDS. If a prior history of significant idiopathic macrocytosis or non-immune red cell aplasia is identified, the measurement of erythrocyte adenosine deaminase may be considered for evaluation of DBA.
One of the distinguishing features of pediatric MDS, and MDS associated with predisposition conditions, is hypocellularity of the marrow unlike adult-onset MDS, which is typically hypercellular. Up to 10%–15% of MDS in adults may be hypocellular with bone marrow cellularity of <30%, and it is not yet known if this indicates an association with a germline predisposition. Studies in adult patients have found that MDS with hypocellular marrows is observed in younger patients and is associated with severe cytopenias, when compared to MDS with normocellular or hypercellular marrows (21, 121).
Some germline predisposition conditions are associated with baseline dysplasia (122), which may be confused with MDS. Megakaryocyte dysmorphology is a frequent baseline finding in the absence of MDS in GATA2 deficiency (123), RUNX1-FPD (124), or other germline predisposition conditions (125, 126). Megakaryocyte immunostaining in the bone marrow with CD61 helps highlight atypical megakaryocytes, which may be difficult to identify with hematoxylin and eosin (H&E) stain. Baseline myeloid dysplasia with nuclear hypolobation (Pelger–Huët-like cells) and hypogranulation is often seen without MDS in patients with SDS (127). Baseline mild dysmorphology is also observed in FA, including megaloblastic erythroid precursors, Pelger–Huët-like neutrophils, and hypolobated megakaryocytes, and should be considered in the diagnostic pathology (126). In such cases, serial marrow exams showing progressively increasing dysplasia, high-risk cytogenetic findings (for example, monosomy 7 or deletion of 7q, complex cytogenetic abnormalities), certain patterns of high risk somatic clones [for example, rising mutant TP53 clone size in SDS (96)], or increased blasts would raise concern for MDS.
We routinely perform cytogenetic evaluation and FISH staining on all MDS marrows. Since the proportion of abnormal cells may be small particularly if the marrow is hypocellular, and/or these cells may have a relative disadvantage of growth and missed in cytogenetic testing, FISH testing may be helpful irrespective of cytogenetic findings (128). Complete or partial loss of chromosome 7 is the most common cytogenetic abnormality in pediatric MDS and is the most common cytogenetic abnormality observed in MDS associated with predisposition conditions (129, 130). Monosomy 7/deletion of 7q has been found in up to one-third of MDS and AML associated with FA, SDS, GATA2 deficiency, and SCN, and has been described in MDS associated with RUNX1-FPD, TBD, and ERCC6L2 (129). In a cohort of adolescent patients with monosomy 7 MDS, the prevalence of germline GATA2 deficiency was reported to be 72% (6). Isochromosome 7q [i(7)(q10)] or del(20q) are cytogenetic abnormalities found in SDS, but these clones have not been associated with progression to malignancy (91, 131). Other common cytogenetic abnormalities described with germline predisposition conditions include trisomy 8 with GATA2 deficiency (6) and gains of chromosomes 1q and 3q with FA (126).
The availability of clinical next-generation sequencing (NGS) has revealed the complex landscape of germline mutations in myeloid neoplasms, including MDS. Many germline mutations underlying predisposition conditions are also often found as somatic mutations in myeloid leukemias and may first be picked up on a somatic mutation panel as part of the diagnostic evaluation for MDS. Distinguishing germline from somatic mutations, particularly in consideration of germline predisposition conditions in young patients, has added to the complexity of interpretating the NGS result and carries profound clinical implications discussed below.
Most genetic panels designed for somatic mutation analysis of myeloid malignancies target mutations recurrently found in adult hematologic malignancies. Commercial somatic and germline NGS panels differ with respect to what genes and gene regions are included. Analytic pipelines for germline versus somatic mutations are also distinct. Additionally, many of these somatic genetic panels can miss deletions or duplications in a gene (for example, in GATA2 deficiency and RUNX1-FPD) and changes in non-coding regions like promoter regions (ANKRD26) and introns (GATA2) that may underlie germline predisposition conditions. Indeed, a normal/negative somatic NGS panel does not rule out a germline genetic cause of an MDS diagnosis. In our practice, we utilize a somatic NGS panel of 88 genes to evaluate somatic MDS mutations (132), accompanied by a separate gene panel designed for genetic analysis of germline variants. The identification of variants in genes known to be mutated either somatically or in the germline, such as RUNX1, GATA2, and TP53, warrant further consideration regardless of variant allele frequency, which may be affected by confounding factors such as copy number variants, loss of heterozygosity, or somatic reversion. Approaches to distinguish germline from somatic mutations include examination of non-hematopoietic tissues such as skin fibroblasts and persistence of the variant at a constant appropriate variant allele fraction (VAF) in remission marrows after MDS-directed therapy. For example, we reported a case of an adolescent patient with RUNX1-FPD who presented with B-ALL, who had a personal and family history of mild bleeding symptoms. She was found with a RUNX1 variant on the NGS panel with a VAF of 52%, initially classified as a variant of unknown clinical significance (VUS). Remission marrow showed persistence of this variant at a similar VAF, which was confirmed to be a germline with fibroblast sequencing and positive testing of her father who had long-standing bleeding symptoms (133).
The designation of a VUS indicates lack of sufficient evidence to meet current American College of Medical Genetics (ACMG) requirements for pathogenic/likely pathogenic or benign/likely benign designation. Many germline variants classified as VUS have not been described in the literature or in publicly available databases gene variants such as ClinVar (https://www.ncbi.nlm.nih.gov/clinvar) or gnomAD (https://gnomad.broadinstitute.org). We work closely with experts in specific genes or diseases and colleagues in genetic counseling to analyze these variants with the clinical history, laboratory evaluation, association of the variant within family pedigree phenotypes, variant allele frequencies in control populations, and gene-specific structural and functional data (134). There are ongoing research and collaborative expert panel recommendations such as Clinical Genome Consortium (ClinGen: https://clinicalgenome.org/) for the interpretation and classification of germline variants (135). For specific gene/disease pairs, such as for RUNX1, TP53, and others, guidelines are curated by a Myeloid Malignancy Variant Curation Expert Panel (MM-VCEP) and include additional functional assessment guidelines to enhance the accuracy of variant interpretation (136–138). Variant classification is a highly active area of research, and consultation with an expert in the field can be helpful.
Interpretation of the sequencing results may be confounded by somatic genetic rescue (SGR) (139). In a number of these syndromes, SGR results in the correction of the germline mutation in a single hematopoietic stem and progenitor cell, which then results in a selective growth of the wild-type corrected clone over time, with reduction in the relative presence of the germline mutant cells in the hematopoietic tissues (97). Examples of SGR or reversion reported in literature are listed in Table 1. As such, VAFs of true germline mutations will be lower than the expected heterozygous allele frequencies, which may result in the omission of the variant by the analytic pipeline for germline mutations or mis-attribution to a somatic origin. We have limited understanding whether the remaining abnormal germline clones have the same high risk of malignant transformation. For example, in FA, revertant mosaicism in the blood may improve overall blood counts (47, 140), but still confers a risk for leukemogenesis (141), while in the rare single cases of reversion in GATA2 deficiency (64) and RUNX1-FPD (66), hematologic malignancies have not been reported. A curious example of SGR in hematopoiesis is the case of SAMD9 and SAMD9L syndromes. SAMD9 and SAMD9L are located near each other on chromosome 7q21.3. Gain-of-function (GOF) mutations in these genes are associated with childhood cytopenias and high risk of developing monosomy 7 or 7q− MDS (142). GOF mutations in stem and progenitor cells result in reduced proliferation, and thus, a cellular competitive advantage is gained by a clone that losses the mutant allele. SGR in patients with germline SAMDL/SAMD9L mutations may occur through several mechanisms: gene correction via genetic reversion or copy neutral loss of heterozygosity (uniparental disomy), inactivation of the mutant allele by a somatic loss of function mutation within the mutant allele, or through complete or partial loss of the chromosome harboring the mutant allele (chromosome 7), termed adaptation by aneuploidy (69, 72, 143, 144). The last mechanism generates a monosomy 7 or deletion of 7q clone(s), which may progress to myeloid malignancy. The germline mutations may be detected by sequencing a non-hematopoietic tissue such as a skin fibroblast. Some centers routinely test skin fibroblasts for the evaluation of germline genetic predisposition in patients with MDS.
The recognition of the underlying germline predisposition informs the approach to therapy in MDS. A therapy with curative intent will often involve stem cell transplantation (SCT) where donor selection is a critical part of the workup. Identification of a germline genetic predisposition allows evaluation of potential family member donors to avoid choosing an affected but clinically asymptomatic donor (145). If a pathogenic germline mutation is identified in the proband, targeted sequencing of the suspected germline variant should be evaluated in the potential family donor. As genetic testing of the donor is time sensitive, the sequencing platform that can most rapidly return test results should be utilized. If the workup of the patient reveals abnormal functional test results, for example, short telomeres concerning TBD, without identification of a specific gene mutation, then evaluation of the related donor for that functional test and a bone marrow examination to rule out any significant hematologic abnormalities should be considered. In adult patients with MDS, the diagnosis of a germline MDS predisposition will also inform the decision around SCT versus chemotherapy or other alternative therapeutic measures, as SCT will not only cure the disease at hand but also eliminate the future heightened risk of hematologic malignancy (146).
Beyond donor selection, identification of germline predisposition conditions may change the conditioning regimen used in SCT to reduce non-relapse mortality (NRM). NRM is a significant complication in SCT for many predisposition conditions such as TBD (147) and FA, where there is a clear benefit for reduced intensity conditioning (RIC). For pediatric patients with MDS, SCT often involves myeloablative conditioning (MAC) regimens, although some RIC regimens are also used. A randomized trial in adults with MDS showed comparable relapse outcomes for patients receiving RIC versus MAC (148). Hence, the presence of certain germline predisposition conditions, particularly those associated with DNA-damage repair mechanism or high NRM, may benefit from receiving RIC and not MAC regimens to reduce NRM. Lastly, SCT outcomes may be affected by the underlying germline condition and associated comorbidities (2, 120) and should be considered at the time of transplantation planning.
Caring of patients during and after SCT for MDS may also be influenced by a germline predisposition condition. For example, in a single center cohort of patients undergoing SCT for GATA2 deficiency-related blood disorders, we found an increased risk of thrombotic and neurological complications, compared to patients with GATA2 wild-type MDS or ALL (147). A second cohort of patients with GATA2 deficiency-related MDS who underwent SCT reported four cases of neurological complications and several patients with transplant-associated thrombotic microangiopathy (149). Patients may also experience clinical signs of non-hematologic organ toxicity, which do not correct with SCT, such as GATA2 deficiency-related lymphatic dysfunction, or lung and liver disease in TBD. Attention to post-SCT monitoring is needed for a patient with germline predisposition conditions with an increased risk of solid tumors [for example, FA, LFS, GATA2 deficiency (103), and DBA (150)].
Diagnosis of germline predisposition conditions informs family counseling. In the current era of successful oncofertility programs, young patients undergoing MDS-directed therapy, including SCT, have an opportunity to consider family planning options after their treatment. At the appropriate time, genetic counselors are helpful to engage in discussions about prenatal counseling, preimplantation genetic testing, and other possibilities.
Testing of family members should be discussed to allow informed decision-making and to guide their health maintenance. Testing of siblings, particularly those who may be considered for stem cell donation, is often a first step in this process. Testing of all direct family members (parents, siblings, and children) of the affected patient and interested biologically related members of the extended family should be discussed together with a genetic counselor with expertise in these conditions.
We have gained valuable insight about MDS pathogenesis from pediatric MDS with germline predisposition conditions. Expanding the systematic evaluation for these conditions to adult patients with MDS will guide medical management and improve outcomes with tailored therapies, appropriate management of co-morbidities, and inform donor selection. Hematologists should be alert to potential red flags from personal and family history, physical exam, blood and marrow examination, and somatic NGS results, which are suggestive of genetic predisposition in adult patients with MDS.
Investigations into germline predisposition to hematologic malignancies continue to address unanswered questions of disease penetrance, incidence, mechanisms of somatic genetic rescue, and prognostic markers of MDS risk once a conditions is identified in asymptomatic patients. Adults with germline predisposition to MDS may present without a suggestive childhood history. The identification of older adult-onset MDS for genetic predisposition conditions such as DDX41 or TBD-related MDS has expanded the age spectrum of consideration of germline conditions in MDS. Increasing awareness and evaluation for genetic predisposition to MDS in the diagnostic workup of adult patients will improve patient care, guide therapies, and inform monitoring of toxicities and comorbidities with the goal of improving patient outcomes.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by NIH/NIDDK 1RC2DK 122533 to AS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to acknowledge Christopher Reilly for his critical reading of the manuscript.
1. Keel SB, Scott A, Bonilla MS, Ho PA, Gulsuner S, Pritchard CC, et al. Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients. Haematologica (2016) 101:1343–50. doi: 10.3324/haematol.2016.149476
2. Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic Mutations in Myelodysplastic Syndrome After Stem-Cell Transplantation. N Engl J Med (2017) 376:536–47. doi: 10.1056/NEJMoa1611604
3. Feurstein S, Churpek JE, Walsh T, Keel S, Hakkarainen M, Schroeder T, et al. Germline Variants Drive Myelodysplastic Syndrome in Young Adults. Leuk (2021) 35:2439–44. doi: 10.1038/s41375-021-01137-0
4. Bluteau O, Sebert M, Leblanc T, De Latour RP, Quentin S, Lainey E, et al. A Landscape of Germ Line Mutations in a Cohort of Inherited Bone Marrow Failure Patients. Blood (2018) 131:717–32. doi: 10.1182/blood-2017-09-806489
5. Schwartz JR, Ma J, Lamprecht T, Walsh M, Wang S, Bryant V, et al. The Genomic Landscape of Pediatric Myelodysplastic Syndromes. Nat Commun (2017) 8:1557. doi: 10.1038/s41467-017-01590-5
6. Wlodarski MW, Hirabayashi S, Pastor V, Stary J, Hasle H, Masetti R, et al. Prevalence, Clinical Characteristics, and Prognosis of GATA2-Related Myelodysplastic Syndromes in Children and Adolescents. Blood (2016) 127:1387–97. doi: 10.1182/blood-2015-09-669937
7. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
8. Ma X, Does M, Raza A, Mayne ST. Myelodysplastic Syndromes: Incidence and Survival in the United States. Cancer (2007) 109:1536–42. doi: 10.1002/cncr.22570
9. Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 Deficiency: A Protean Disorder of Hematopoiesis, Lymphatics, and Immunity. Blood (2014) 123:809–21. doi: 10.1182/blood-2013-07-515528
10. Brown AL, Arts P, Carmichael CL, Babic M, Dobbins J, Chong CE, et al. RUNX1-Mutated Families Show Phenotype Heterogeneity and a Somatic Mutation Profile Unique to Germline Predisposed AML. Blood Adv (2020) 4:1131–44. doi: 10.1182/bloodadvances.2019000901
11. Schratz KE, Haley L, Danoff SK, Blackford AL, DeZern AE, Gocke CD, et al. Cancer Spectrum and Outcomes in the Mendelian Short Telomere Syndromes. Blood (2020) 135:1946–56. doi: 10.1182/blood.2019003264
12. Reilly CR, Myllymaki M, Redd RA, Padmanaban S, Karunakaran D, Tesmer VM, et al. The Clinical and Functional Effects of TERT Variants in Myelodysplastic Syndrome. Blood (2021) 138(10):898–911. doi: 10.1182/blood.2021011075
13. Sébert M, Passet M, Raimbault A, Rahme R, Raffoux E, De Fontbrune FS, et al. Germline DDX41 Mutations Define a Significant Entity Within Adult MDS/AML Patients. Blood (2019) 134:1441–4. doi: 10.1182/blood.2019000909
14. Lewinsohn M, Brown AL, Weinel LM, Phung C, Rafidi G, Lee MK, et al. Novel Germ Line DDX41 Mutations Define Families With a Lower Age of MDS/AML Onset and Lymphoid Malignancies. Blood (2016) 127:1017–23. doi: 10.1182/blood-2015-10-676098
15. Cheah JJC, Hahn CN, Hiwase DK, Scott HS, Brown AL. Myeloid Neoplasms With Germline DDX41 Mutation. Int J Hematol (2017) 106:163–74. doi: 10.1007/s12185-017-2260-y
16. Polprasert C, Schulze I, Sekeres MA, Makishima H, Przychodzen B, Hosono N, et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell (2015) 27:658–70. doi: 10.1016/j.ccell.2015.03.017
17. Kennedy AL, Shimamura A. Genetic Predisposition to MDS: Clinical Features and Clonal Evolution. Blood (2019) 133:1071–85. doi: 10.1182/blood-2018-10-844662
18. Brown AL, Churpek JE, Malcovati L, Döhner H, Godley LA. Recognition of Familial Myeloid Neoplasia in Adults. Semin Hematol (2017) 54(2):60–8. doi: 10.1053/j.seminhematol.2016.11.003
19. Godley LA. Inherited Predisposition to Acute Myeloid Leukemia. Semin Hematol (2014) 51(4):306–21. doi: 10.1053/j.seminhematol.2014.08.001
20. Tong WG, Quintas-Cardama A, Kadia T, Borthakur G, Jabbour E, Ravandi F, et al. Predicting Survival of Patients With Hypocellular Myelodysplastic Syndrome: Development of a Disease-Specific Prognostic Score System. Cancer (2012) 118:4462–70. doi: 10.1002/cncr.27420
21. Marisavljević D, Čemerikić V, Rolović Z, Bošković D, Čolović M. Hypocellular Myelodysplastic Syndromes: Clinical and Biological Significance. Med Oncol (2005) 22:169–75. doi: 10.1385/MO:22:2:169
22. Huang TC, Ko BS, Tang JL, Hsu C, Chen CY, Tsay W, et al. Comparison of Hypoplastic Myelodysplastic Syndrome (MDS) With Normo-/Hypercellular MDS by International Prognostic Scoring System, Cytogenetic and Genetic Studies. Leukemia (2008) 22:544–50. doi: 10.1038/sj.leu.2405076
23. Rosenberg PS, Zeidler C, Bolyard AA, Alter BP, Bonilla MA, Boxer LA, et al. Stable Long-Term Risk of Leukaemia in Patients With Severe Congenital Neutropenia Maintained on G-CSF Therapy: Short Report. Br J Haematol (2010) 150:196–9. doi: 10.1111/j.1365-2141.2010.08216.x
24. Dong F, Brynes RK, Tidow N, Welte K, Lowenberg B, Touw IP. Mutations in the Gene for the Granulocyte Colony-Stimulating–Factor Receptor in Patients With Acute Myeloid Leukemia Preceded by Severe Congenital Neutropenia. N Engl J Med (1995) 333:487–93. doi: 10.1056/NEJM199508243330804
25. Dong F, Dale DC, Bonilla MA, Freedman M, Fasth A, Neijens HJ, et al. Mutations in the Granulocyte Colony-Stimulating Factor Receptor Gene in Patients With Severe Congenital Neutropenia. Leukemia (1997) 11:120–5. doi: 10.1038/sj.leu.2400537
26. Germeshausen M, Ballmaier M, Welte K. Incidence of CSF3R Mutations in Severe Congenital Neutropenia and Relevance for Leukemogenesis: Results of a Long-Term Survey. Blood (2007) 109:93–9. doi: 10.1182/blood-2006-02-004275
27. Alter BP, Rosenberg PS, Day T, Menzel S, Giri N, Savage SA, et al. Genetic Regulation of Fetal Haemoglobin in Inherited Bone Marrow Failure Syndromes. Br J Haematol (2013) 162:542–6. doi: 10.1111/bjh.12399
28. Kudo S, Harigae H, Watanabe N, Takasawa N, Kimura J, Kameoka JI, et al. Increased HbF Levels in Dyserythropoiesis. Clin Chim Acta (2000) 291:83–7. doi: 10.1016/S0009-8981(99)00186-2
29. Reinhardt D, Haase D, Schoch C, Wollenweber S, Hinkelmann E, Heyden WV, et al. Hemoglobin F in Myelodysplastic Syndrome. Ann Hematol (1998) 76:135–8. doi: 10.1007/s002770050377
30. Choi JW, Kim Y, Fujino M, Ito M. Significance of Fetal Hemoglobin-Containing Erythroblasts (F Blasts) and the F Blast/F Cell Ratio in Myelodysplastic Syndromes. Leukemia (2002) 16:1478–83. doi: 10.1038/sj.leu.2402536
31. Sood R, Kamikubo Y, Liu P. Role of RUNX1 in Hematological Malignancies. Blood (2017) 129(15):2070–82. doi: 10.1182/blood-2016-10-687830
32. Marquez R, Hantel A, Lorenz R, Neistadt B, Wong J, Churpek JE, et al. A New Family With a Germline ANKRD26 Mutation and Predisposition to Myeloid Malignancies. Leuk Lymphoma (2014) 55:2945–6. doi: 10.3109/10428194.2014.903476
33. Hock H, Shimamura A. ETV6 in Hematopoiesis and Leukemia Predisposition. Semin Hematol (2017) 54:98–104. doi: 10.1053/j.seminhematol.2017.04.005
34. Germeshausen M, Ancliff P, Estrada J, Metzler M, Ponstingl E, Rutschle H, et al. MECOM-Associated Syndrome: A Heterogeneous Inherited Bone Marrow Failure Syndrome With Amegakaryocytic Thrombocytopenia. Blood Adv (2018) 2:586–96. doi: 10.1182/bloodadvances.2018016501
35. Niihori T, Ouchi-Uchiyama M, Sasahara Y, Kaneko T, Hashii Y, Irie M, et al. Mutations in MECOM, Encoding Oncoprotein EVI1, Cause Radioulnar Synostosis With Amegakaryocytic Thrombocytopenia. Am J Hum Genet (2015) 97:848–54. doi: 10.1016/j.ajhg.2015.10.010
36. Ripperger T, Hofmann W, Koch JC, Shirneshan K, Haase D, Wulf G, et al. MDS1 and EVI1 Complex Locus (MECOM): A Novel Candidate Gene for Hereditary Hematological Malignancies. Haematologica (2018) 103:e55–8. doi: 10.3324/haematol.2017.178723
37. Fu YY, Ren CE, Qiao PY, Meng YH. Uterine Natural Killer Cells and Recurrent Spontaneous Abortion. Am J Reprod Immunol (2021) 86:e13433. doi: 10.1111/aji.13433
38. Alter BP, Kumar M, Lockhart LL, Sprinz PG, Rowe TF. Pregnancy in Bone Marrow Failure Syndromes: Diamond-Blackfan Anaemia and Shwachman-Diamond Syndrome. Br J Haematol (1999) 107:49–54. doi: 10.1046/j.1365-2141.1999.01668.x
39. Faivre L, Meerpohl J, Da Costa L, Marie I, Nouvel C, Gnekow A, et al. High-Risk Pregnancies in Diamond-Blackfan Anemia: A Survey of 64 Pregnancies From the French and German Registries Disorders of Erythropoiesis • Brief Report. Haematologica (2006) 91(4):530–3.
40. Gansner JM, Achebe MM, Gray KJ, Yefidoff-Freedman R, Labovitis E, Parnes A, et al. Pregnancy Outcomes in Inherited Bone Marrow Failure Syndromes. Blood (2017) 130:1671–4. doi: 10.1182/blood-2017-08-798462
41. Antony-Debré I, Duployez N, Bucci M, Geffroy S, Micol JB, Renneville A, et al. Somatic Mutations Associated With Leukemic Progression of Familial Platelet Disorder With Predisposition to Acute Myeloid Leukemia. Leukemia (2016) 30:999–1002. doi: 10.1038/leu.2015.236
42. Latger-Cannard V, Philippe C, Bouquet A, Baccini V, Alessi MC, Ankri A, et al. Haematological Spectrum and Genotype-Phenotype Correlations in Nine Unrelated Families With RUNX1 Mutations From the French Network on Inherited Platelet Disorders. Orphanet J Rare Dis (2016) 11:49. doi: 10.1186/s13023-016-0432-0
43. Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in Dyskeratosis Congenita. Blood (2009) 113:6549–57. doi: 10.1182/blood-2008-12-192880
44. Vlachos A, Rosenberg PS, Atsidaftos E, Kang J, Onel K, Sharaf RN, et al. Increased Risk of Colon Cancer and Osteogenic Sarcoma in Diamond-Blackfan Anemia. Blood (2018) 132:2205–8. doi: 10.1182/blood-2018-05-848937
45. Lach FP, Singh S, Rickman KA, Ruiz PD, Noonan RJ, Hymes KB, et al. Esophageal Cancer as Initial Presentation of Fanconi Anemia in Patients With a Hypomorphic FANCA Variant. Cold Spring Harb Mol Case Stud (2020) 6(6):a005595. doi: 10.1101/mcs.a005595
46. Sekinaka Y, Mitsuiki N, Imai K, Yabe M, Yabe H, Mitsui-Sekinaka K, et al. Common Variable Immunodeficiency Caused by FANC Mutations. J Clin Immunol (2017) 37:434–44. doi: 10.1007/s10875-017-0396-4
47. Waisfisz Q, Morgan N, Savino M, et al. Spontaneous Functional Correction of Homozygous Fanconi Anaemia Alleles Reveals Novel Mechanistic Basis for Reverse Mosaicism. Nat Genet (1999) 22:379–83. doi: 10.1038/11956
48. Gross M, Hanenberg H, Lobitz S, Friedl R, Herterich S, Dietrich R, et al. Reverse Mosaicism in Fanconi Anemia: Natural Gene Therapy via Molecular Self-Correction. In: Cytogenetic and Genome Research, vol. 98. (2002). p. 126–35. S. Karger AG.
49. Ramírez MJ, Pujol R, Trujillo-Quintero JP, Minguillon J, Bogliolo M, Rio P, et al. Natural Gene Therapy by Reverse Mosaicism Leads to Improved Hematology in Fanconi Anemia Patients. Am J Hematol (2021) 96:989–99. doi: 10.1002/ajh.26234
50. Ludwig LS, Gazda HT, Eng JC, Eichhorn SW, Thiru P, Ghazvinian R, et al. Altered Translation of GATA1 in Diamond-Blackfan Anemia. Nat Med (2014) 20:748–53. doi: 10.1038/nm.3557
51. Iskander D, Roberts I, Rees C, Szydlo R, Alikian M, Neale M, et al. Impaired Cellular and Humoral Immunity is a Feature of Diamond-Blackfan Anaemia; Experience of 107 Unselected Cases in the United Kingdom. Br J Haematol (2019) 186:321–6. doi: 10.1111/bjh.15915
52. Venugopal P, Moore S, Lawrence DM, George AJ, Hannan RD, Bray SCE, et al. Self-Reverting Mutations Partially Correct the Blood Phenotype in a Diamond Blackfan Anemia Patient. Haematologica (2017) 102:e506–9. doi: 10.3324/haematol.2017.166678
53. Garelli E, Quarello P, Giorgio E, Carando A, Menegatti E, Mancini C, et al. Spontaneous Remission in a Diamond-Blackfan Anaemia Patient Due to a Revertant Uniparental Disomy Ablating a de novo RPS19 mutation. Br J Haematol (2019) 185:994–8. doi: 10.1111/bjh.15688
54. Jongmans MCJ, Diets IJ, Quarello P, Garelli E, Kuiper RP, Pfundt R. Somatic Reversion Events Point Towards RPL4 as a Novel Disease Gene in a Condition Resembling Diamond-Blackfan Anemia. Haematologica (2018) 103:e607–9. doi: 10.3324/haematol.2018.200683
55. Gripp KW, Curry C, Olney AH, Sandoval C, Fisher J, Chong JXL, et al. Diamond-Blackfan Anemia With Mandibulofacial Dystostosis Is Heterogeneous, Including the Novel DBA Genes TSR2 and RPS28. Am J Med Genet (2014) A 164A:2240–9. doi: 10.1002/ajmg.a.36633
56. Furutani E, Shah AS, Zhao Y, Andorsky D, Dedeoglu F, Geddis A, et al. Inflammatory Manifestations in Patients With Shwachman–Diamond Syndrome: A Novel Phenotype. Am J Med Genet Part A (2020) 182:1754–60. doi: 10.1002/ajmg.a.61593
57. Khan S, Hinks J, Shorto J, Schwarz MJ, Sewell WAC. Some Cases of Common Variable Immunodeficiency may be Due to a Mutation in the SBDS Gene of Shwachman-Diamond Syndrome. Clin Exp Immunol (2008) 151:448–54. doi: 10.1111/j.1365-2249.2007.03556.x
58. Nacci L, Valli R, Maria Pinto R, Zecca M, Cipolli M, Morini J, et al. Parental Origin of the Deletion Del(20q) in Shwachman-Diamond Patients and Loss of the Paternally Derived Allele of the Imprinted L3MBTL1 Gene. Genes Chromosom Cancer (2017) 56:51–8. doi: 10.1002/gcc.22401
59. Sharma R, Sahoo SS, Honda M, Granger SL, Goodings C, Sanchez L, et al. Gain-Of-Function Mutations in RPA1 Cause a Syndrome With Short Telomeres and Somatic Genetic Rescue. Blood (2021) 139(7):1039–51. doi: 10.1182/blood.2021011980
60. Maryoung L, Yue Y, Young A, Newton CA, Barba C, Van Oers NSC, et al. Somatic Mutations in Telomerase Promoter Counterbalance Germline Loss-of-Function Mutations. J Clin Invest (2017) 127:982–6. doi: 10.1172/JCI91161
61. Gutierrez-Rodrigues F, Donaires FS, Pinto A, Vicente A, Dillon LW, Cle DV, et al. Pathogenic TERT Promoter Variants in Telomere Diseases. Genet Med (2019) 21:1594–602. doi: 10.1038/s41436-018-0385-x
62. Jongmans MCJ, Verwiel ETP, Heijdra Y, Vulliamy T, Kamping EJ, Hehir-Kwa JY, et al. Revertant Somatic Mosaicism by Mitotic Recombination in Dyskeratosis Congenita. Am J Hum Genet (2012) 90:426–33. doi: 10.1016/j.ajhg.2012.01.004
63. Collin M, Dickinson R, Bigley V. Haematopoietic and Immune Defects Associated With GATA2 Mutation. Br J Haematol (2015) 169:173–87. doi: 10.1111/bjh.13317
64. Catto LFB, Borges G, Pinto AL, Cle DV, Chahud F, Santana BA, et al. Somatic Genetic Rescue in Hematopoietic Cells in GATA2 Deficiency. Blood (2020) 136:1002–5. doi: 10.1182/blood.2020005538
65. Sacco K, Laky K, Li M, Merguerian M, Craft K, Cunningham L, et al. Germline RUNX1 Deficiency Predisposes to Allergy and Autoimmunity. J Allergy Clin Immunol (2021) 147:AB68. doi: 10.1016/j.jaci.2020.12.266
66. Glembotsky AC, Marin Oyarzun CP, de Luca G, Marzac C, Auger N, Goette NP, et al. First Description of Revertant Mosaicism in Familial Platelet Disorder With Predisposition to Acute Myelogenous Leukemia: Correlation With the Clinical Phenotype. Haematologica (2020) 105:535–9. doi: 10.3324/haematol.2020.253070
67. Oetjen KA, Levoska MA, Tamura D, Ito S, Douglas D, Khan SG, et al. Predisposition to Hematologic Malignancies in Patients With Xeroderma Pigmentosum. Haematologica (2020) 105:1–11. doi: 10.3324/haematol.2019.223370
68. Sarasin A, Quentin S, Droin N, Sahbatou M, Saada V, Auger N, et al. Familial Predisposition to TP53/complex Karyotype MDS and Leukemia in DNA Repair-Deficient Xeroderma Pigmentosum. Blood (2019) 133:2718–24. doi: 10.1182/blood-2019-01-895698
69. Shima H, Koehler K, Nomura Y, Sugimoto K, Satoh A, Ogata T, et al. Two Patients With MIRAGE Syndrome Lacking Haematological Features: Role of Somatic Second-Site Reversion SAMD9 Mutations. J Med Genet (2018) 55:81–5. doi: 10.1136/jmedgenet-2017-105020
70. Chen DH, Below JE, Shimamura A, Keel SB, Matsushita M, Wolff J, et al. Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am J Hum Genet (2016) 98:1146–58. doi: 10.1016/j.ajhg.2016.04.009
71. de Jesus AA, Hou Y, Brooks S, Malle L, Biancotto A, Huang Y, et al. Distinct Interferon Signatures and Cytokine Patterns Define Additional Systemic Autoinflammatory Diseases. J Clin Invest (2020) 130:1669–82. doi: 10.1172/JCI129301
72. Pastor VB, Sahoo SS, Boklan J, Schwabe GC, Saribeyoglu E, Strahm B, et al. Constitutional SAMD9L Mutations Cause Familial Myelodysplastic Syndrome and Transient Monosomy 7. Haematologica (2018) 103:427–37. doi: 10.3324/haematol.2017.180778
73. Tesi B, Davidsson J, Voss M, Rahikkala E, Holmes TD, Chiang SCC, et al. Gain-Of-Function SAMD9L Mutations Cause a Syndrome of Cytopenia, Immunodeficiency, MDS, and Neurological Symptoms. Blood (2017) 129:2266–79. doi: 10.1182/blood-2016-10-743302
74. Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, et al. Germline CBL Mutations Cause Developmental Abnormalities and Predispose to Juvenile Myelomonocytic Leukemia. Nat Genet (2010) 42:641. doi: 10.1038/ng.641
75. Cordeddu V, Di Schiavi E, Pennacchio LA, Ma’ayan A, Sarkozy A, Fodale V, et al. Mutation of SHOC2 Promotes Aberrant Protein N-Myristoylation and Causes Noonan-Like Syndrome With Loose Anagen Hair. Nat Genet (2009) 41:1022–6. doi: 10.1038/ng.425
76. Tabori U, Hansford JR, Achatz MI, Kratz CP, Plon SE, Frebourg T, et al. Clinical Management and Tumor Surveillance Recommendations of Inherited Mismatch Repair Deficiency in Childhood. Clin Cancer Res (2017) 23:e32–7. doi: 10.1158/1078-0432.CCR-17-0574
77. Wimmer K, Kratz CP, Vasen HFA, Caron O, Colas C, Entz-Werle N, et al. Diagnostic Criteria for Constitutional Mismatch Repair Deficiency Syndrome: Suggestions of the European Consortium “Care for CMMRD” (C4CMMRD). J Med Genet (2014) 51:355–65. doi: 10.1136/jmedgenet-2014-102284
78. Germeshausen M, Ballmaier M. CAMT-MPL: Congenital Amegakaryocytic Thrombocytopenia Caused by MPL Mutations - Heterogeneity of a Monogenic Disorder - A Comprehensive Analysis of 56 Patients. Haematologica (2021) 106:2439–48. doi: 10.3324/haematol.2020.257972
79. Jameson-Lee M, Chen K, Ritchie E, Shore T, Al-Khattab O, Gergis U. Acute Myeloid Leukemia in a Patient With Thrombocytopenia With Absent Radii: A Case Report and Review of the Literature. Hematol Oncol Stem Cell Ther (2018) 11:245–7. doi: 10.1016/j.hemonc.2017.02.001
80. Go RS, Johnston KL. Acute Myelogenous Leukemia in an Adult With Thrombocytopenia With Absent Radii Syndrome. Eur J Haematol (2003) 70:246–8. doi: 10.1034/j.1600-0609.2003.00054.x
81. Camitta BM, Rock A. Acute Lymphoidic Leukemia in a Patient With Thrombocytopenia/Absent Radii (Tar) Syndrome. Am J Pediatr Hematol Oncol (1993) 15:335–7.
82. Duployez N, Goursaud L, Fenwarth L, Bories C, Marceau-Renaut A, Boyer T, et al. Familial Myeloid Malignancies With Germline TET2 Mutation. Leukemia (2020) 34:1450–3. doi: 10.1038/s41375-019-0675-6
83. Spegarova JS, Lawless D, Mohamad SMB, Engelhardt KR, Doody G, Shrimpton J, et al. Germline TET2 Loss of Function Causes Childhood Immunodeficiency and Lymphoma. Blood (2020) 136:1055–66. doi: 10.1182/blood.2020005844
84. Kirwan M, Walne AJ, Plagnol V, Velangi M, Ho A, Hossain U, et al. Exome Sequencing Identifies Autosomal-Dominant SRP72 Mutations Associated With Familial Aplasia and Myelodysplasia. Am J Hum Genet (2012) 90:888–92. doi: 10.1016/j.ajhg.2012.03.020
85. Trottier AM, Druhan LJ, Kraft IL, Lance A, Feurstein S, Helgeson M, et al. Heterozygous Germ Line CSF3R Variants as Risk Alleles for Development of Hematologic Malignancies. Blood Adv (2020) 4:5269–84. doi: 10.1182/bloodadvances.2020002013
86. Tovy A, Rosas C, Gaikwad AS, Medrano G, Zhang L, Reyes JM, et al. Perturbed Hematopoiesis in Individuals With Germline DNMT3A Overgrowth Tatton-Brown-Rahman Syndrome. Haematologica (2021). doi: 10.3324/haematol.2021.278990
87. Tovy A, Reyes JM, Gundry MC, Brunetti L, Lee-Six H, Petljak M, et al. Tissue-Biased Expansion of DNMT3A-Mutant Clones in a Mosaic Individual Is Associated With Conserved Epigenetic Erosion. Cell Stem Cell (2020) 27:326–335.e4. doi: 10.1016/j.stem.2020.06.018
88. Bahrami E, Witzel M, Racek T, Puchałka J, Hollizeck S, Greif-Kohistani N, et al. Myb-Like, SWIRM, and MPN Domains 1 (MYSM1) Deficiency: Genotoxic Stress-Associated Bone Marrow Failure and Developmental Aberrations. J Allergy Clin Immunol (2017) 140:1112–9. doi: 10.1016/j.jaci.2016.10.053
89. Le Guen T, Touzot F, Andre-Schmutz I, Lagresle-Peyrou C, France B, Kermasson L, et al. An In Vivo Genetic Reversion Highlights the Crucial Role of Myb-Like, SWIRM, and MPN Domains 1 (MYSM1) in Human Hematopoiesis and Lymphocyte Differentiation. J Allergy Clin Immunol (2015) 136:1619–1626.e5. doi: 10.1016/j.jaci.2015.06.008
90. Sanders MA, Chew E, Flensburg C, Zeilemaker A, Miller SE, Al Hinai AS, et al. MBD4 Guards Against Methylation Damage and Germ Line Deficiency Predisposes to Clonal Hematopoiesis and Early-Onset AML. Blood (2018) 132:1526–34. doi: 10.1182/BLOOD-2018-05-852566
91. Pressato B, Valli R, Marletta C, Mare L, Montalbano G, Lo CF, et al. Deletion of Chromosome 20 in Bone Marrow of Patients With Shwachman-Diamond Syndrome, Loss of the EIF6 Gene and Benign Prognosis. Br J Haematol (2012) 157:503–5. doi: 10.1111/j.1365-2141.2012.09033.x
92. Maserati E, Minelli A, Pressato B, Valli R, Crescenzi B, Stefanelli M, et al. Shwachman Syndrome as Mutator Phenotype Responsible for Myeloid Dysplasia/Neoplasia Through Karyotype Instability and Chromosomes 7 and 20 Anomalies. Genes Chromosom Cancer (2006) 45:375–82. doi: 10.1002/gcc.20301
93. Maserati E, Pressato B, Valli R, Minelli A, Sainati L, Patitucci F, et al. The Route to Development of Myelodysplastic Syndrome/Acute Myeloid Leukaemia in Shwachman-Diamond Syndrome: The Role of Ageing, Karyotype Instability, and Acquired Chromosome Anomalies. Br J Haematol (2009) 145:190–7. doi: 10.1111/j.1365-2141.2009.07611.x
94. Valli R, Pressato B, Marletta C, Mare L, Montalbano G, Lo CF, et al. Different Loss of Material in Recurrent Chromosome 20 Interstitial Deletions in Shwachman-Diamond Syndrome and in Myeloid Neoplasms. Mol Cytogenet (2013) 6:56. doi: 10.1186/1755-8166-6-56
95. Crescenzi B, La Starza R, Sambani C, Parcharidou A, Pierini V, Nofrini V, et al. Totipotent Stem Cells Bearing Del(20q) Maintain Multipotential Differentiation in Shwachman Diamond Syndrome. Br J Haematol (2009) 144:116–9. doi: 10.1111/j.1365-2141.2008.07448.x
96. Kennedy AL, Myers KC, Bowman J, Gibson CJ, Camarda ND, Furutani E, et al. Distinct Genetic Pathways Define Pre-Malignant Versus Compensatory Clonal Hematopoiesis in Shwachman-Diamond Syndrome. Nat Commun (2021) 12:1–15. doi: 10.1038/s41467-021-21588-4
97. Tan S, Kermasson L, Hilcenko C, Kargas V, Traynor D, Boukerrou AZ, et al. Somatic Genetic Rescue of a Germline Ribosome Assembly Defect. Nat Commun (2021) 12:1–17. doi: 10.1038/s41467-021-24999-5
98. Douglas SPM, Siipola P, Kovanen PE, Pyorala M, Kakko S, Savolainen ER, et al. ERCC6L2 Defines a Novel Entity Within Inherited Acute Myeloid Leukemia. Blood (2019) 133:2724–8. doi: 10.1182/blood-2019-01-896233
99. Anemia Research Fund, F. Fanconi Anemia Clinical Care Guidelines_5th Edition 2020_Web. Fanconi Anemia Research Fund (2020).
100. Savage SA, Alter BP. Dyskeratosis Congenita. Hematol Oncol Clinics North America (2009) 23:215–31. doi: 10.1016/j.hoc.2009.01.003
101. Vlachos A, Osorio DS, Atsidaftos E, Kang J, Lababidi ML, Seiden HS, et al. Increased Prevalence of Congenital Heart Disease in Children With Diamond Blackfan Anemia Suggests Unrecognized Diamond Blackfan Anemia as a Cause of Congenital Heart Disease in the General Population: A Report of the Diamond Blackfan Anemia Registry. Circ Genomic Precis Med (2018) 11:e002044. doi: 10.1161/CIRCGENETICS.117.002044
102. Lipton JM, Ellis SR. Diamond Blackfan Anemia: Diagnosis, Treatment and Molecular Pathogenesis. Hematol/Oncol Clin North Am (2009) 23(2):261–82. doi: 10.1016/j.hoc.2009.01.004
103. Donadieu J, Lamant M, Fieschi C, De Fontbrune FS, Caye A, Ouachee M, et al. Natural History of GATA2 Deficiency in a Survey of 79 French and Belgian Patients. Haematologica (2018) 103:1278–87. doi: 10.3324/haematol.2017.181909
104. Jyonouchi S, Forbes L, Ruchelli E, Sullivan KE. Dyskeratosis Congenita: A Combined Immunodeficiency With Broad Clinical Spectrum - A Single-Center Pediatric Experience. Pediatr Allergy Immunol (2011) 22:313–9. doi: 10.1111/j.1399-3038.2010.01136.x
105. Khan S, Pereira J, Darbyshire PJ, Holding S, Dore PC, Sewell WAC, et al. Do Ribosomopathies Explain Some Cases of Common Variable Immunodeficiency? Clin Exp Immunol (2011) 163:96–103. doi: 10.1111/j.1365-2249.2010.04280.x
106. Knudson M, Kulkarni S, Ballas ZK, Bessler M, Goldman F. Association of Immune Abnormalities With Telomere Shortening in Autosomal-Dominant Dyskeratosis Congenita. Blood (2005) 105:682–8. doi: 10.1182/blood-2004-04-1673
107. Schratz KE. Extrahematopoietic Manifestations of the Short Telomere Syndromes. Hematol Am Soc Hematol Educ Progr (2020) 2020:115–22. doi: 10.1182/hematology.2020000170
108. Polat A, Dinulescu M, Fraitag S, Nimubona S, Toutain F, Jouneau S, et al. Skin Manifestations Among GATA2-Deficient Patients. Br J Dermatol (2018) 178:781–5. doi: 10.1111/bjd.15548
109. Kazenwadel J, Betterman KL, Chong CE, Stokes PH, Lee YK, Secker GA, et al. GATA2 Is Required for Lymphatic Vessel Valve Development and Maintenance. J Clin Invest (2015) 125:2879–994. doi: 10.1172/JCI78888
110. Shin M, Nozaki T, Idrizi F, Isogai S, Ogasawara K, Ishida K, et al. Valves Are a Conserved Feature of the Zebrafish Lymphatic System. Dev Cell (2019) 51:374–386.e5. doi: 10.1016/j.devcel.2019.08.019
111. Wlodarski MW, Collin M, Horwitz MS. GATA2 Deficiency and Related Myeloid Neoplasms. Semin Hematol (2017) 54:81–6. doi: 10.1053/j.seminhematol.2017.05.002
112. DiNardo CD, Bannon SA, Routbort M, Franklin A, Mork M, Armanios M, et al. Evaluation of Patients and Families With Concern for Predispositions to Hematologic Malignancies Within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk (2016) 16(7):417–28.e2. doi: 10.1016/j.clml.2016.04.001
113. Churpek JE, Garcia JS, Madzo J, Jackson SA, Onel K, Godley LA. Identification and Molecular Characterization of a Novel 3′ Mutation in RUNX1 in a Family With Familial Platelet Disorder. Leuk Lymphoma (2010) 51:1931–5. doi: 10.3109/10428194.2010.503821
114. Wagner CL, Hanumanthu VS, Conover Talbot C, Abraham RS, Hamm D, Gable DL, et al. Short Telomere Syndromes Cause a Primary T Cell Immunodeficiency. J Clin Invest (2018) 128:5222–34. doi: 10.1172/JCI120216
115. Silzle T, Blum S, Schuler E, Kaivers J, Rudelius M, Hildebrandt B, et al. Lymphopenia at Diagnosis is Highly Prevalent in Myelodysplastic Syndromes and has an Independent Negative Prognostic Value in IPSS-R-Low-Risk Patients. Blood Cancer J (2019) 9:63. doi: 10.1038/s41408-019-0223-7
116. Saeed L, Patnaik MM, Begna KH, Al-Kali A, Litzow MR, Hanson CA, et al. Prognostic Relevance of Lymphocytopenia, Monocytopenia and Lymphocyte-to-Monocyte Ratio in Primary Myelodysplastic Syndromes: A Single Center Experience in 889 Patients. Blood Cancer J (2017) 7:e550. doi: 10.1038/bcj.2017.30
117. Baerlocher GM, Vulto I, de Jong G, Lansdorp PM. Flow Cytometry and FISH to Measure the Average Length of Telomeres (Flow FISH). Nat Protoc (2006) 1:2365–76. doi: 10.1038/nprot.2006.263
118. Alter BP, Giri N, Savage SA, Rosenberg PS. Telomere Length in Inherited Bone Marrow Failure Syndromes. Haematologica (2015) 100:49–54. doi: 10.3324/haematol.2014.114389
119. Alder JK, Hanumanthu VS, Strong MA, DeZern AE, Stanley SE, Takemoto CM, et al. Diagnostic Utility of Telomere Length Testing in a Hospital-Based Setting. Proc Natl Acad Sci U S A (2018) 115:E2358–65. doi: 10.1073/pnas.1720427115
120. Myllymäki M, Redd R, Reilly CR, Saber W, Spellman SR, Gibson CJ, et al. Short Telomere Length Predicts Nonrelapse Mortality After Stem Cell Transplantation for Myelodysplastic Syndrome. Blood (2020) 136:3070–81. doi: 10.1182/blood.2020005397
121. Hyun SY, Kong JH, Shim KY, Lee JI. Hypocellular Myelodysplastic Syndrome in the Hypomethylating Agent Era. Ann Oncol (2014) 25:iv336. doi: 10.1093/annonc/mdu339.33
122. Galera P, Dulau-Florea A, Calvo KR. Inherited Thrombocytopenia and Platelet Disorders With Germline Predisposition to Myeloid Neoplasia. Int J Lab Hematol (2019) 41(Suppl 1):131–41. doi: 10.1111/ijlh.12999
123. Ganapathi KA, Townsley DM, Hsu AP, Arthur DC, Zerbe CS, Cuellar-Rodriguez J, et al. GATA2 Deficiency-Associated Bone Marrow Disorder Differs From Idiopathic Aplastic Anemia. Blood (2015) 125:56–70. doi: 10.1182/blood-2014-06-580340
124. Chisholm KM, Denton C, Keel S, Geddis AE, Xu M, Appel BE, et al. Bone Marrow Morphology Associated With Germline RUNX1 Mutations in Patients With Familial Platelet Disorder With Associated Myeloid Malignancy. Pediatr Dev Pathol (2019) 22:315–28. doi: 10.1177/1093526618822108
125. Zhang MY, Churpek JE, Keel SB, Walsh T, Lee MK, Loeb KR, et al. Germline ETV6 Mutations in Familial Thrombocytopenia and Hematologic Malignancy. Nat Genet (2015) 47(2):180–5. doi: 10.1038/ng.3177
126. Cioc AM, Wagner JE, MacMillan ML, DeFor T, Hirsch B. Diagnosis of Myelodysplastic Syndrome Among a Cohort of 119 Patients With Fanconi Anemia: Morphologic and Cytogenetic Characteristics. Am J Clin Pathol (2010) 133:92–100. doi: 10.1309/AJCP7W9VMJENZOVG
127. Myers KC, Furutani E, Weller E, Siegele B, Galvin A, Arsenault V, et al. Clinical Features and Outcomes of Patients With Shwachman-Diamond Syndrome and Myelodysplastic Syndrome or Acute Myeloid Leukaemia: A Multicentre, Retrospective, Cohort Study. Lancet Haematol (2020) 7:e238–46. doi: 10.1016/S2352-3026(19)30206-6
128. Furutani E, Liu S, Galvin A, Steltz S, Malsch MM, Loveless SK, et al. Hematologic Complications With Age in Shwachman-Diamond Syndrome. Blood Adv (2022) 6:297–306. doi: 10.1182/bloodadvances.2021005539
129. Wlodarski MW, Sahoo SS, Niemeyer CM. Monosomy 7 in Pediatric Myelodysplastic Syndromes. Hematol Oncol Clin North Am (2018) 32:729–43. doi: 10.1016/j.hoc.2018.04.007
130. Pezeshki A, Podder S, Kamel R, Corey SJ. Monosomy 7/Del (7q) in Inherited Bone Marrow Failure Syndromes: A Systematic Review. Pediatr Blood Cancer (2017) 64:e26714. doi: 10.1002/pbc.26714
131. Dror Y. Shwachman-Diamond Syndrome. Pediatr Blood Cancer (2005) 45:892–901. doi: 10.1002/pbc.20478
132. Kluk MJ, Lindsley RC, Aster JC, Lindeman NI, Szeto D, Hall D, et al. Validation and Implementation of a Custom Next-Generation Sequencing Clinical Assay for Hematologic Malignancies. J Mol Diagn (2016) 18:507–15. doi: 10.1016/j.jmoldx.2016.02.003
133. Six KA, Gerdemann U, Brown AL, Place AE, Cantor AB, Kutny MA, et al. B-Cell Acute Lymphoblastic Leukemia in Patients With Germline RUNX1 Mutations. Blood Adv (2021) 5:3199–202. doi: 10.1182/bloodadvances.2021004653
134. Furutani E, Shimamura A. Germline Genetic Predisposition to Hematologic Malignancy. J Clin Oncol (2017) 35:1018–28. doi: 10.1200/JCO.2016.70.8644
135. Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN Maintains Haematopoietic Stem Cells and Acts in Lineage Choice and Leukaemia Prevention. Nature (2006) 441:518–22. doi: 10.1038/nature04747
136. Luo X, Feurstein S, Mohan S, Porter CC, Jackson SA, Keel S, et al. ClinGen Myeloid Malignancy Variant Curation Expert Panel Recommendations for Germline RUNX1 Variants. Blood Adv (2019) 3:2962–79. doi: 10.1182/bloodadvances.2019000644
137. Wu D, Luo X, Feurstein S, Kesserwan C, Mohan S, Pineda-Alvarez DE, et al. How I Curate: Applying American Society of Hematology-Clinical Genome Resource Myeloid Malignancy Variant Curation Expert Panel Rules for RUNX1 Variant Curation for Germline Predisposition to Myeloid Malignancies. Haematologica (2020) 105:870–87. doi: 10.3324/haematol.2018.214221
138. Fortuno C, Lee K, Olivier M, Pesaran T, Mai PL, de Andrade KC, et al. Specifications of the ACMG/AMP Variant Interpretation Guidelines for Germline TP53 Variants. Hum Mutat (2021) 42:223–36. doi: 10.1002/humu.24152
139. Revy P, Kannengiesser C, Fischer A. Somatic Genetic Rescue in Mendelian Haematopoietic Diseases. Nat Rev Genet (2019) 20:582–98. doi: 10.1038/s41576-019-0139-x
140. Mankad A, Taniguchi T, Cox B, Akkari Y, Rathbun RK, Lucas L, et al. Natural Gene Therapy in Monozygotic Twins With Fanconi Anemia. Blood (2006) 107:3084–90. doi: 10.1182/blood-2005-07-2638
141. Gregory JJ, Wagner JE, Verlander PC, Levran O, Batish SD, Eide CR, et al. Somatic Mosaicism in Fanconi Anemia: Evidence of Genotypic Reversion in Lymphohematopoietic Stem Cells. Proc Natl Acad Sci U S A (2001) 98:2532–7. doi: 10.1073/pnas.051609898
142. Sahoo SS, Pastor VB, Goodings C, Voss RK, Kozyra EJ, Szvetnik A, et al. Clinical Evolution, Genetic Landscape and Trajectories of Clonal Hematopoiesis in SAMD9/SAMD9L Syndromes. Nat Med (2021) 27:1806–17. doi: 10.1038/s41591-021-01511-6
143. Buonocore F, Kuhnen P, Suntharalingham JP, Del Valle I, Digweed M, Stachelscheid H, et al. Somatic Mutations and Progressive Monosomy Modify SAMD9-Related Phenotypes in Humans. J Clin Invest (2017) 127:1700–13. doi: 10.1172/JCI91913
144. Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 Mutations Cause a Novel Multisystem Disorder, MIRAGE Syndrome, and are Associated With Loss of Chromosome 7. Nat Genet (2016) 48:792–7. doi: 10.1038/ng.3569
145. Galera P, Hsu AP, Wang W, Droll S, Chen R, Schwartz JR, et al. Donor-Derived MDS/AML in Families With Germline GATA2 Mutation. Blood (2018) 132(18):1994–8. doi: 10.1182/blood-2018-07-861070
146. Churpek JE, Godley LA. How I Diagnose and Manage Individuals at Risk for Inherited Myeloid Malignancies. Blood (2016) 128:1800–13. doi: 10.1182/blood-2016-05-670240
147. Hofmann I, Avagyan S, Stetson A, Guo D, Al-Sayegh H, London WB. Comparison of Outcomes of Myeloablative Allogeneic Stem Cell Transplantation for Pediatric Patients With Bone Marrow Failure, Myelodysplastic Syndrome and Acute Myeloid Leukemia With and Without Germline GATA2 Mutations. Biol Blood Marrow Transplant (2020) 26:1124–30. doi: 10.1016/j.bbmt.2020.02.015
148. Pulsipher MA. Reduced Intensity for Myelodysplastic Syndrome: Worth the Gamble? J Clin Oncol (2017) 35:2106–8. doi: 10.1200/JCO.2017.72.8048
149. Bortnick R, Wlodarski M, de Haas V, De Moerloose B, Dworzak M, Hasle H, et al. Hematopoietic Stem Cell Transplantation in Children and Adolescents With GATA2-Related Myelodysplastic Syndrome. Bone Marrow Transplant (2021) 56:2732–41. doi: 10.1038/s41409-021-01374-y
Keywords: germline, inherited bone marrow failure syndromes, predisposition syndromes, myelodysplastic syndrome, hematologic malignancy
Citation: Avagyan S and Shimamura A (2022) Lessons From Pediatric MDS: Approaches to Germline Predisposition to Hematologic Malignancies. Front. Oncol. 12:813149. doi: 10.3389/fonc.2022.813149
Received: 11 November 2021; Accepted: 26 January 2022;
Published: 09 March 2022.
Edited by:
Amy DeZern, Johns Hopkins Medicine, United StatesReviewed by:
Marcin Wlodarski, University of Freiburg Medical Center, GermanyCopyright © 2022 Avagyan and Shimamura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Akiko Shimamura, YWtpa28uc2hpbWFtdXJhQGNoaWxkcmVucy5oYXJ2YXJkLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.