
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 25 February 2022
Sec. Cancer Molecular Targets and Therapeutics
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.812534
 Hong-Tao Wang1,2†
Hong-Tao Wang1,2† Jie Ju1†Shao-Cong Wang1†Yu-Hui Zhang3
Jie Ju1†Shao-Cong Wang1†Yu-Hui Zhang3 Cui-Yun Liu1Tao Wang1Xue Yu1Fei Wang1Xue-Ru Cheng1
Cui-Yun Liu1Tao Wang1Xue Yu1Fei Wang1Xue-Ru Cheng1 Kun Wang1*Zhao-Yang Chen4*
Kun Wang1*Zhao-Yang Chen4*Ferroptosis is a new form of programmed cell death (PCD) characterized by an excess iron accumulation and subsequent unbalanced redox states. Ferroptosis is different from the already reported PCD and has unique morphological features and biochemical processes. Ferroptosis was first elaborated by Brent R. Stockwell’s lab in 2012, in which small molecules erastin and RSL-3 induce PCD in Ras mutant cell lines. Ferroptosis involves various physiological processes and occurrence of disease and especially shows strong potential in cancer treatment. Development of small molecule compounds based on Stockwell’s research was found to kill cancer cells, and some FDA-approved drugs were discovered to result in ferroptosis of cancer cells. Radiotherapy and checkpoint therapy have been widely used as a treatment for many types of cancer. Recently, some papers have reported that chemotherapy, radiotherapy, and checkpoint therapy induce ferroptosis of cancer cells, which provides new strategies for cancer treatment. Nevertheless, the limitless proliferation of tumor cells and the lack of cell death mechanisms are important reasons for drug resistance for tumor therapy. Therefore, we reviewed the molecular mechanism of ferroptosis and sensitivity to ferroptosis of different cancer cells and tumor treatment strategy.
Cell death is the ultimate destination of all cells. It has been thought that cell death is not controlled by genes for a long time. Programmed cell death (PCD) is induced by specific extracellular or intracellular signals and regulated by death-related genes (1). With the deepening of cell death research, the concept of PCD has been continuously enriched and is still being expanded. Apoptosis is the first discovered PCD, in which apoptosis effector caspase is involved in this pathway. In recent years, many forms of PCD have been discovered, including necroptosis, autophagy, pyroptosis, and ferroptosis. Ferroptosis, as a new form of PCD, has gradually come into view and has become a research hotspot in the field of cell death.
Ferroptosis is triggered by the accumulation of excess iron and subsequent reactive oxygen species (ROS) production and is morphologically, biochemically, and genetically distinct from apoptosis, necrosis, and autophagy. Glutathione peroxidase 4 (GPX4) is a protein resistant to ferroptosis, which catalyzes the reduction reaction of lipid peroxide (LOOH) and maintains the balance of redox reaction. The deficiency of GPX4 causes ferroptosis owing to the imbalance of redox states. Iron is an important microelement to support life. Nevertheless, excess iron generates abundant ROS by Fenton reaction, which breaks redox balance and induces ferroptosis. Ferroptosis is involved in multiple physiological and pathological processes, such as intestinal, renal, and cardiac ischemia and reperfusion (I/R), neurodegenerative diseases, hemochromatosis, and cancer (2–6).
Malignant tumors are commonly referred to as “cancers.” According to the statistics of the World Health Organization, about 9.6 million people died of cancer in 2018, and nearly one-sixth of all deaths were caused by cancer worldwide. Malignant tumor is the second leading cause of death in the world and one of the major diseases that seriously threaten human life and health (7). Small molecular compounds erastin and RSL-3 trigger ferroptosis in Ras mutant cancer cell lines and lead to the death of cancer cells (8, 9). These findings provide a new strategy for cancer treatment by pharmacological or genetic intervention related to ferroptosis, which is a significant work in scientific research as well as pharmaceutical research. This paper will mainly describe the mechanism of ferroptosis and the research progress in cancer treatment, especially chemotherapy, radiotherapy, and tumor immunotherapy, which provides new ideas for cancer drug resistance in several aspects.
In 1959, Harry Eagle strived to search for necessary nutrients of cell metabolism and found only deficiency of cysteine inhibiting the growth of human and mouse cells. A cell lacking cysteine presents representative characteristics different from deprivation of other amino acids (10). Glutathione (GSH) synthesis inhibition and cysteine deficiency are certified to be important causes of cell death in the next few years (11). It is not recognized as ferroptosis until recent studies found that both iron chelators and lipophilic antioxidants prevent the occurrence of this kind of cell death (12).
Stockwell’s laboratory screened compounds lethal for HRAS mutant cell lines in a small-molecule pool and, in 2003, identified a compound named “erastin” which is proved to trigger ferroptosis (13). The system Xc-, Na+-independent cystine/glutamate antiporter is a direct target of erastin. Cells treated with erastin inhibit the function of system Xc-, which attenuates cystine uptake and GSH synthesis and leads to ferroptosis. Ferroptosis was finally named in 2012 and described as a regulated cell death characterized by excessive lipid peroxidation and redox active iron (9). Another well-known ferroptosis inducer RSL-3, a GPX4 inhibitor, induces ferroptosis selectively in HRASv12-mutant but not wild-type BJeLR-derived cell lines (8).
The most obvious morphological features of ferroptosis are shrunken mitochondria, increased membrane density, cristae degeneration and breakdown, and outer mitochondrial membrane rupture.
The morphology of the nucleus remains unchanged and without chromatin condensation appearance. However, margination, condensation, and fragmentation of chromatin are the main features during apoptosis (14). Besides, ROS augments peroxidation of membrane lipid and then damages the barrier function of the cytomembrane, but apoptosis involves apoptotic body production and without inflammation emergence (1). Necroptosis relies on a cascade pathway composed of receptor-interacting protein 1/3 (RIPK1/RIPK3) and mixed lineage kinase domain-like pseudokinase (MLKL) activation with the characteristics of membrane rupture and Annexin V-positive membrane bubble production, which is different from ferroptosis (1). Autophagy is another form of PCD characterized by the de novo synthesis of organelles enclosed by a double membrane and fused with a lysosome, which leads to their digestion. The main morphology of autophagy is also not present in ferroptosis (15). In all, the morphology of ferroptosis is sensibly different from that of apoptosis, necroptosis, and autophagy. Therefore, we compared the four different patterns of cell death, as shown in Table 1.
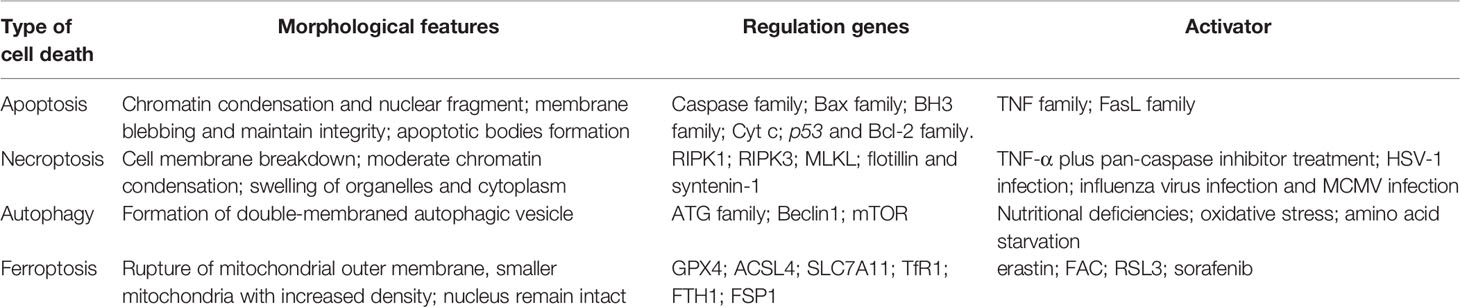
Table 1 Comparison of different types of cell death.
The molecular mechanism of ferroptosis is mainly dependent on the production and elimination of lipid peroxidation, two competing biochemical processes in the cell. Iron and polyunsaturated fatty acids (PUFAs) are used as materials in the lipid peroxidation process to promote ferroptosis. GSH as a substrate of GPX4 clears peroxide and regulates ferroptosis negatively. When cells are unable to remove the excess peroxide effectively through an antioxidative mechanism, the accumulation of peroxidative lipids induces ferroptosis. Many physiological processes including iron metabolism, amino acid metabolism, and lipid metabolism can regulate ferroptosis by influencing cellular redox states.
Iron originated from food is absorbed by intestinal epithelial cells of duodenal mucosa, where iron ion reductase catalyzes ferric iron (Fe3+) and reduces to Fe2+, and Fe2+ is transported into cells by divalent metal transporter 1 (DMT1) (16, 17). Extracellular Fe3+ combined with transferrin is transported into the cell with the help of cell surface receptor transferrin receptor 1 (TfR1). Heat shock protein family B member 1 (HSPB1) suppresses the TfR1 recycling and iron uptake. The deficiency of transferrin and TfR1 in glutamate-free culture medium significantly inhibits ferroptosis. Fe3+ is transported into the endosome and reduced to Fe2+ by STEAP family member 3 (STEAP3) in the cell (18). The reduced Fe2+ is transported into the cytoplasm with the help of DMT1 and becomes the main component of the iron pool (19). Intracellular iron is exported into the extracellular matrix and oxidized Fe2+ to Fe3+ in the meantime by ferroportin (FPN) to maintain normal levels of iron.
Excess iron generates ROS and then triggers ferroptosis. In order to avoid cell death, the excess iron is stocked in ferritin and produces redox-inactive ferritin heteropolymers to sustain the redox balance and protect against ferroptosis. Besides, the stocked iron is released from ferritin in which ferritin is degraded by nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy to induce ferroptosis. NCOA4 interacts with HERC2 ubiquitin E3 ligase and is degraded by the ubiquitin proteasome degradation pathway to ensure a normal concentration. Under low cellular iron conditions, the interaction of NCOA4 and HERC2 is interrupted resulting in the elevation of NCOA4 expression, which increases ferritinophagy and iron levels (20). In all, NCOA4 overexpression increases the degradation of ferritin, which leads to upregulation of the iron concentration and promotes ferroptosis. On the other hand, the downregulation of NCOA4 inhibits degradation of ferritin and attenuates sensitivity of cells to oxidative damage (21) (Figure 1A).
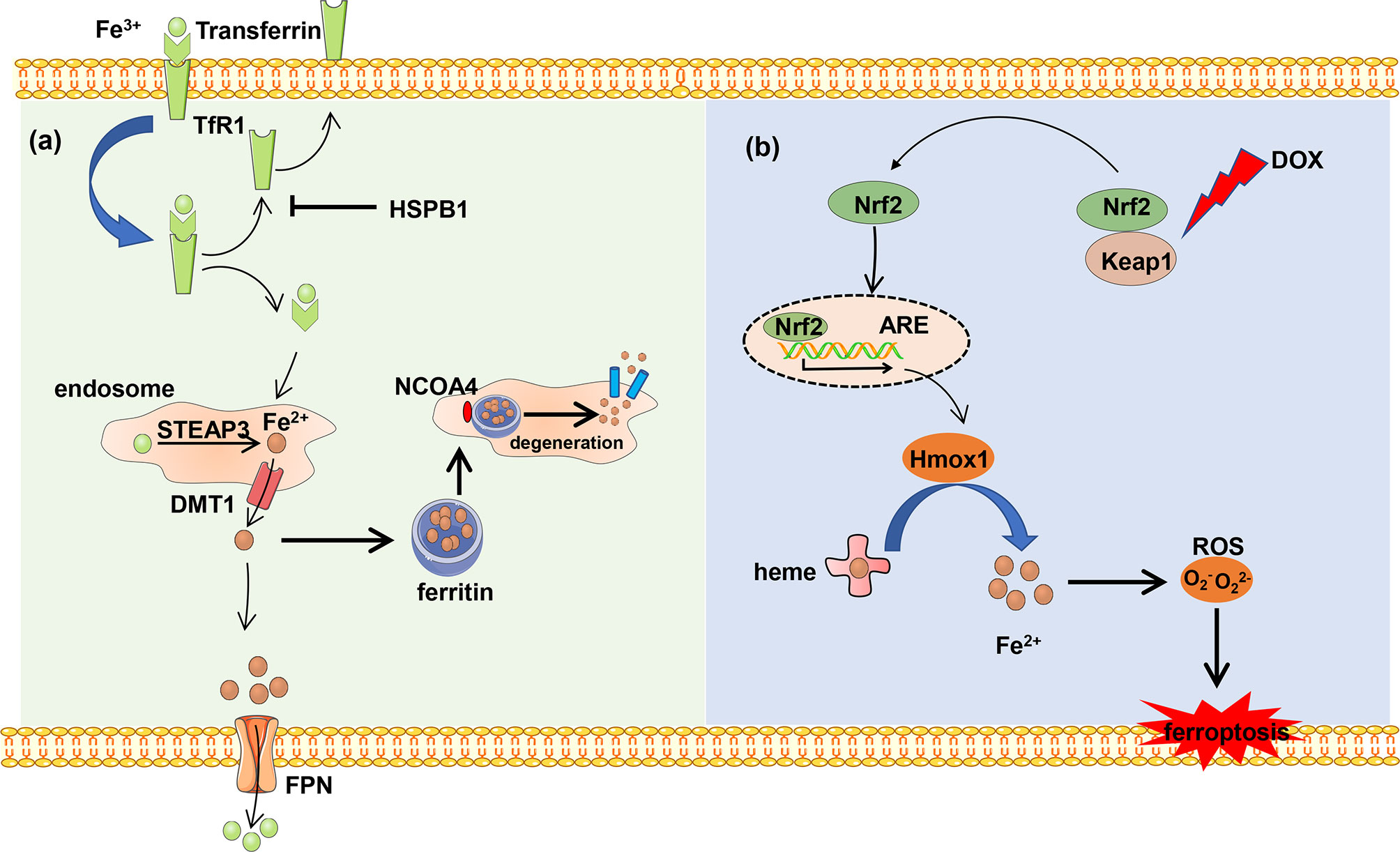
Figure 1 The metabolism of iron. (A) Ferric iron (Fe3+) coupled with transferrin and then was imported into intercellular circumstance by TfR1. Transferrin was recycled and exported out of the cell, which can be blocked by HSPB1. Fe3+ is reduced and forms reduced iron (Fe2+) by DMT1 in the endosome, and Fe2+ is transported into the cytoplasm. Excess Fe2+ is stocked in the ferritin to avoid iron overload. The stocked Fe2+ can be released from ferritin by NCOA4-mediated ferritinophagy. Besides, Fe2+ is exported out of the cell and is oxidized by FPN. (B) DOX induces ferroptosis. The heart-exported DOX activates the Keap1/Nrf2 pathway, and transcription factor Nrf2 activates downstream protein Hmox1 which oxidizes heme and release of iron which results in ferroptosis.
Each heme consists of four pyrrole subunits forming a ring with a ferrous ion at the center. The heme oxygenase enzyme 1 (Hmox1) catalyzes the region-specific hydroxylation of heme and produces biliverdin, carbon monoxide, and Fe2+. In some pathological states such as doxorubicin (DOX)-induced cardiomyopathy and cardiac ischemia/reperfusion (I/R), Hmox1 is significantly upregulated. Non-heme iron is accumulated by degrading heme via the nuclear factor E2-related factor 2 (Nrf2)-mediated upregulation of Hmox1. The excess iron induces ferroptosis and cardiac injury which can be blocked by ferroptosis inhibitors ferrostatin-1 and iron chelation dexrazoxane (Figure 1B).
GSH is a substrate of GPX4 which maintains the redox balance in cells and determines whether cells undergo ferroptosis or not. GPX4 uses 2-molecule GSH as an electron donor to reduce toxic lipid peroxides (L-OOH) to non-toxic fatty alcohols (L-OH), and GSH is oxidized into oxidized glutathione (GSSG) at the same time. GSSG can be reduced by NADPH to GSH and is then involved in redox reaction. GSH is a tripeptide containing the γ-amide bond and sulfhydryl group and composed of glutamic acid, cysteine, and glycine. Cysteine serves as the rate-limiting precursor for GSH. Excess methionine is converted into cysteine through transsulfuration in mammals to initiate cysteine formation. The cystathionine β-synthase enzyme catalyzes serine with the methionine cycle intermediate homocysteine to form cystathionine, which is subsequently cleaved by cystathionine γ-lyase to release cysteine (22). When cystine is deficient, some cells synthesize cysteine through methionine transsulfuration pathways bypassing the system Xc- and therefore resist to ferroptosis caused by system Xc- inhibition (23). It is reported that knockdown of cysteinyl-tRNA synthetase (CARS) leads to enhanced activity of transsulfuration and inhibits ferroptosis induced by erastin, in which the lipid ROS is prevented without altering iron homeostasis (24). However, most cancer cells need specific transporter-system Xc- (composed by SLC3A2 and SLC7A11 connected with disulfide bond) to import cystine which is converted to cysteine through an NADPH-consuming reduction reaction and is used for GSH synthesis (25). Inhibition of system Xc- leads to the imbalance of amino acid metabolism and subsequent ferroptosis. Cystine is directly transferred into the cell through the alanine, serine, cysteine-preferring system (ASC system) when the cell is in a reducing state. ASC system activation inhibits ferroptosis induced by erastin (18) (Figure 2A).
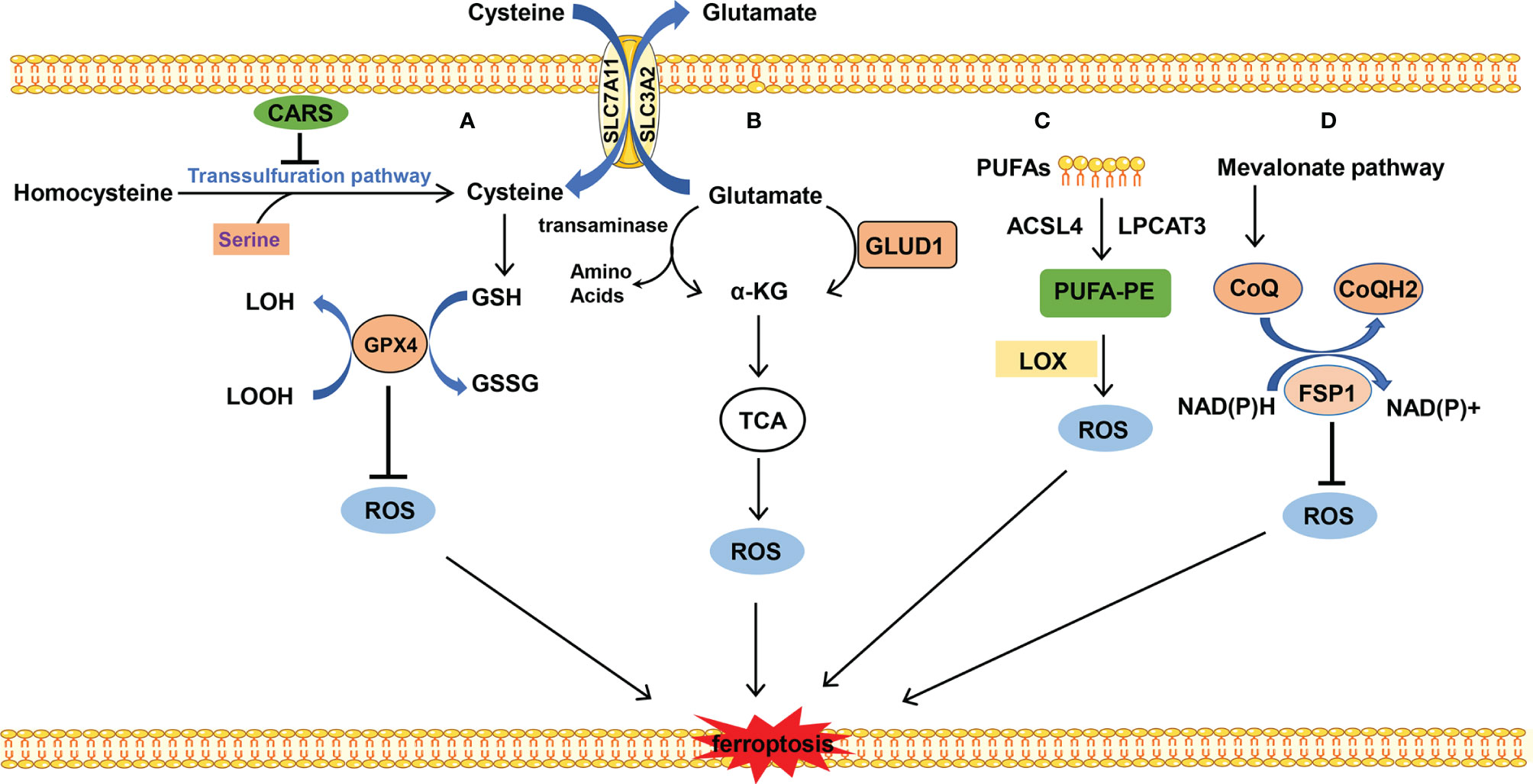
Figure 2 The metabolism of amino acids and lipid. (A) Cysteine is transported into the cell, and glutamate is transported out of the cell by system Xc- at the same time. Cysteine is used to synthesize GSH to maintain the balance of redox states. Besides, cysteine can be synthesized by the transsulfuration pathway, which is blocked by CARS. (B) Glutamate is converted into α-KG by transaminase or GLUD1 pathway and participates in TCA, which results in ROS production. (C) PUFAs which mainly stem from the cellular membrane are catalyzed into PUFA-PE by ACSL4 and LPCAT. PUFA-PE is peroxided by the LOX family. (D) FSP1 depends on the balance of redox. CoQ is synthesized by the mevalonate pathway and plays an important role in the CoQ antioxidant system.
Glutamine is the most abundant amino acid which is used for biosynthesis of nucleotides, as a nitrogen source, and in the tricarboxylic acid cycle as a carbon source. The uptake of glutamate relies on the receptor complex of SLC38A1 and SLC1A5. The inhibition of SLC1A5 by chemical compound L-g-glutamyl-p-nitroanilide (GPNA) or RNA interference blocks ferroptosis, indicating that glutamine is indispensable for ferroptosis (26). The glutamate deamination mediated by glutamate dehydrogenase (GLUD1) or transamination pathway catalyzes glutamate to generate α-ketoglutarate (α-KG). Transaminase inhibitor amino-oxyacetate (AOA) or RNAi of the transaminase GOT1 inhibits ferroptosis. α-KG in combination with dialyzed FBS induces ferroptosis even during co-treatment with AOA in case of amino acid starvation. It is indicated that downstream metabolites of glutaminolysis α-KG play an important role in ferroptosis (27). Besides, high-concentration glutamate inhibits the function of system Xc- and results in ferroptosis, which may explain the neurotoxicity of glutamate when glutamate is accumulated at a high concentration (9) (Figure 2B).
ROS are a group of molecules with partially reduced oxygen, including peroxides, superoxides, singlet oxygen, and free radicals, which are detrimental to DNA or proteins. One of the sources of ROS production is lipid peroxidation. It is reported that lipid peroxides cause cell damage in many ways. Firstly, lipid peroxides are further decomposed into ROS, which further amplifies the lipid peroxidation process. Secondly, lipid peroxidation changes the physical structure of the membrane, such as the thickness and bending degree of the membrane, or by forming holes in the membrane, releasing harmful substances out of the cell. Thirdly, the by-products (aldehydes) of lipid peroxidation cause cell damage, such as malondialdehyde (MDA) and 4-HNE (28, 29). Therefore, the accumulation of lipid peroxides, especially phospholipid peroxides, is considered to be the hallmark event of ferroptosis (30).
The substrate of lipid peroxidation is fatty acid, and polyunsaturated fatty acids (PUFAs) are more prone to be oxidized compared with saturated fatty acids and monounsaturated fatty acids (30), in which the free PUFA is esterified into membrane phospholipids which are further oxidized to induce ferroptosis. This enzymatic reaction process of lipid peroxidation is mediated by long-chain lipidyl coenzyme A ligase 4 (ACSL4) (31), lysophosphatidylcholine acyltransferase 3 (LPCAT3) (32), lipoxygenase (LOX) (33), and so on. ACSL4 converts free long-chain fatty acids into fatty acyl-CoA esters and plays a key role in lipid biosynthesis and fatty acid degradation. LPCAT3 possesses transferring acyl groups and 1-acylglycerophosphocholine O-acyltransferase activity. ACSL4 and LPCAT3 are involved in the synthesis of PUFA-phosphatidylethanolamine (PUFA-PE) in cellular membranes. The former is involved in catalyzing the esterification reaction, especially preferring to esterify the acyl of arachidonic acid. The latter helps esterify the acyl of arachidonic acid to insert it into the membrane phospholipids (32). Subsequently, LOX mediates the peroxidation of membrane phospholipids. Cells with LOX overexpression tend to have stronger lipid peroxidation and are more sensitive to erastin- or RSL3-induced ferroptosis, although to a slightly different degree (34, 35). Besides, it is reported that GPX4 and ACSL4 double-knockout cells are resistant to ferroptosis (31). This process prefers to use free PUFA as substrate rather than phospholipids containing PUFA (36) (Figure 2C).
Besides iron, lipid, and amino acid metabolism, ferroptosis is also regulated by ferroptosis suppressor protein 1 (FSP1), Nrf2, heat shock protein (HSP), and so on. Particularly, FSP1 catalyzes the regeneration of CoQ10 using NAD(P)H and the reduction form of CoQ10 traps lipid peroxyl radicals that mediate lipid peroxidation, which inhibits ferroptosis (37). Nrf2 is an important transcription factor, which activates the antioxidant genes expression (38). HSPB1 regulates iron homeostasis and prevents ROS production caused by the high concentration of intracellular iron (39).
FSP1 is a recently discovered important ferroptosis regulator. Previously, GPX4 was thought of as a central mechanism for ferroptosis inhibition. Some researchers have tried to find other protective mechanisms parallel to GPX4-inhibition-mediated ferroptosis, in other words, whether cells can survive in ferroptosis-inducing conditions and without GPX4 participation. Coincidentally, the two research groups discovered FSP1 almost simultaneously (37, 40). FSP1 is a negative regulator of ferroptosis parallel to GPX4. Mechanistically, the myristoylation of FSP1 mediates the recruitment of FSP1 to lipid droplets and plasma membrane where FSP1 mediates the NADH-dependent reduction of coenzyme Q (CoQ) which functions as a radical-trapping antioxidant and suppresses the generation of lipid peroxides. The mevalonic acid pathway plays an important role in the upstream of CoQ synthesis; small molecular compound FIN56 induces ferroptosis by mevalonic acid pathway inhibition. In addition, supplementation of cells with edibenzoquinone (an analogue of CoQ) protects against the lethality of FIN56 (41) (Figure 2D).
Nrf2 is a transcription factor, closely related with ferroptosis by regulating the intracellular oxidation homeostasis and controlling lipid peroxidation. Nrf2 inhibits ferroptosis by regulating the expression of downstream proteins such as metallothionein 1G (MT-1G), SLC7A11, and Hmox1. It is reported that Nrf2 inhibits ferroptosis and protects against acute lung injury due via regulating SLC7A11 and Hmox1 (42). Besides, the Nrf2/Hmox1 pathway participates in heme degradation and iron release and causes ferroptosis of the heart in doxorubicin-induced cardiomyopathy (4). Activation of Nrf2 is essential for induction of MT-1G expression and ferroptosis inhibition following sorafenib treatment, which is an important mechanism of sorafenib resistance in hepatocellular carcinoma therapy (43).
HSP is expressed constitutively under normal conditions, but overexpression is induced under stress conditions. HSPB1 also called human HSP27 or mouse HSP25 is a member of HSPs and plays a negative role in ferroptosis of cancer cells. HSF1 is a master regulator of HSP expression (44). It is reported that protein kinase C-mediated phosphorylation of HSPB1 inhibits ferroptosis by reducing lipid ROS production. Erastin-induced ferroptosis is enhanced by knockdown of HSP1 and HSPB1. Consistent with this phenomenon, inhibition of HSF1-HSPB1 and phosphorylation of HSPB1 increase the anticancer activity of erastin in vivo (45).
In all, the abnormal metabolism of iron, amino acid, and lipid which are important factors to trigger ferroptosis is shown. Some factors such as FSP1, Nrf2, and HSPB1 recently have proved to participate in ferroptosis.
Eukaryotic cells are organized by membrane-bound organelles which maintain intracellular homeostasis. Stress conditions lead to the dysfunction of cell organelles and augment of cell death. Some cell organelles were reported to involve in ferroptosis regulation such as mitochondria, endoplasmic reticulum (ER) and lysosome. Hereon, we will discuss the specific functions of different organelles in the regulation of ferroptosis.
Mitochondria are also called the powerhouse, which provide energy for the cell and are a site of aerobic respiration. The researchers have found that mitochondria are involved in the regulation of apoptosis. However, the functions of mitochondria in ferroptosis remain unknown. Gao et al. have found that mitochondria play an important role in ferroptosis induced by cysteine deprivation and are dispensable in GPX4 inhibition-induced ferroptosis. It is likely that mitochondria function upstream of GPX4 and promote the exhaustion of GSH under cysteine deprivation conditions. The production of glutaminolysis α-KG replenishes the tricarboxylic acid (TCA) cycle intermediates which are involved in cysteine deprivation-induced ferroptosis. The TCA cycle and electron transport chain (ETC) are the main functions of mitochondria which play roles as a major resource for lipid ROS production in cysteine-deprivation induced ferroptosis (46). The function of mitochondria in ferroptosis regulation is confirmed in DOX-induced cardiomyocyte ferroptosis in which the lipid peroxidation and iron are increased in the mitochondria (4). Besides, mitochondrial ATP–binding cassette (ABC) transporters play an important role in regulating cellular iron metabolism and maintenance of redox status. Deficiency of mitochondrial ABC transporters impairs the activity of mitochondrial ETC, iron overload, and elevation of ROS which results in mitochondrial dysfunction (47).
Lysosome contains a variety of hydrolytic enzymes which hydrolyze a variety of exogenous and endogenous macromolecular substances to maintain intracellular homeostasis by multiple pathways especially autophagy (48). Lysosomal cell death is mainly executed by cathepsin and induced by iron overload or oxidative injury (49). It is reported that ferroptosis differs from lysosomal cell death and autophagy. However, elevation of autophagy and lysosomal activity is related with the induction of ferroptosis (50, 51). The NCOA4-mediated degradation of ferritin promotes ferroptosis, which is related with lysosome (52). STAT3 as an important transcription factor that regulates lysosomal cell death in vitro and in vivo by mediating the expression of cathepsin B, which is indispensable for ferroptosis (53).
The alterations of redox state and calcium levels and failure of secretory proteins post-translational modification activate unfolded protein response (UPR) to defend against ER stress. Recent studies reveal that the ER stress response induced by the ferroptotic agent plays an important role in the cross talk between ferroptosis and apoptosis. Erastin induces ER stress and promotes the expression of the p53-upregulated modulator of apoptosis (PUMA) pathway via transcription factor C/EBP-homologous protein (CHOP) (54). p38 phosphorylates and activates CHOP, which causes an elevated expression of Bim and DR5 and induces apoptosis. PUMA is inactive after treatment with the ferroptic agent while PUMA transforms into an active state after combined treatment with both erastin and TRAIL. This conversion of PUMA from an inactive to active state determines whether necrosis or apoptosis occurs (55).
The treatment of malignant tumors has been accelerating and bringing obvious clinical prospects for patients in recent years. The global expenditure on oncology drugs and the complexity of clinical trial activities are increasing dramatically. Despite the high level of basic and clinical research, tumor therapy remains one of the most challenging areas of research with a significant risk of failure and long development period. At present, surgery, chemotherapy, radiotherapy, and immunotherapy are the main strategies for the treatment of cancer (56). Even though a wide range of clinical antitumor therapies was developed, patients are still subjected to poor efficacy of treatment, recurrence, and metastasis, and eventually death due to treatment failure. How to overcome tumor therapeutic resistance and find new therapy strategy has become a clinical problem to be solved urgently. A recent study found that combined treatment with both ferroptosis inducer erastin and antitumor drugs enhances the antitumor activity of drugs, which provides great clinical value in the treatment of tumor (57–61). All the details are shown in Table 2.
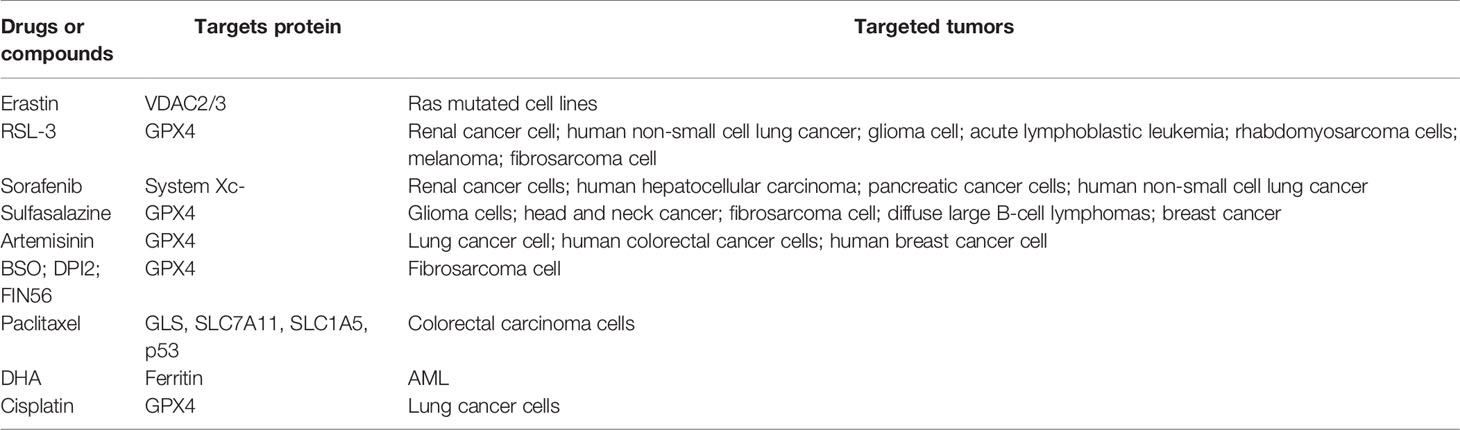
Table 2 Drugs and compounds that inhibit tumor growth.
At present, many small-molecule compounds have been proved to induce the death of tumor cells by inducing ferroptosis. Therefore, ferroptosis inhibitors are expected to be developed into new antitumor small-molecule drugs. According to the different targets of compounds, the ferroptosis inducer can be divided into four categories: (1) system Xc- inhibitor; (2) GPX4 inhibitor; (3) GPX4 and CoQ degradation inducer; and (4) lipid peroxide inducer. It is worth mentioning that some FDA-approved clinical drugs are proved to induce ferroptosis, which extends its clinical application value.
Sorafenib is an oncogenic kinase inhibitor which has been approved as an anticancer drug in the clinical treatment. The cytotoxic effect of sorafenib on hepatocellular carcinoma (HCC) cells is restrained iron chelation deferoxamine. Importantly, sorafenib induction of ferroptosis in cancer cells is unrelated with inhibition of the RAF-MEK-ERK kinase cascade. Lachaier et al. show that sorafenib blocks the system Xc- activity and the synthesis of GSH in a similar way as erastin to induce ferroptosis in different solid tumors (62).
Sulfasalazine (SAS) is a sulfonamide-based antimicrobial agent which is used to treat chronic inflammation of the gut, joints, and retina. A recent study reveals that sulfasalazine inhibits breast cancer cell viability by ferroptosis with the characterization of an abnormal increase in ROS and inhibition of GPX4 and system Xc-. What is more, liproxstatin-1 inhibits the SAS-induced increase in ROS. Importantly, sulfasalazine is more likely to trigger ferroptosis in breast cancer cells, especially in cells with low estrogen receptor expression (63). Inhibition of system Xc- by SAS overcomes the cisplatin resistance of head and neck cancer (HNC) cells by inducing ferroptosis (57, 58).
Artemisinin is the most effective drug for treating malaria resistance. Nowadays, artemisinin and its derivatives have been explored as potential anticancer agents. Dihydroartemisinin (DAT) is a derivative of artemisinin which induces lysosomal degradation of ferritin and increases the cellular free iron level (64). The binding of iron regulatory proteins (IRPs) with mRNA molecules containing iron-responsive element (IRE) sequences is an important iron homeostasis mechanism. DAT impairs IRP/IRE-controlled iron homeostasis and further increases cellular free iron. Besides, DAT promotes ferroptosis of a cohort of cancer cells mediated by GPX4 inhibition, which are originally resistant to ferroptosis (65). Artesunate-induced ferroptosis in Burkitt lymphoma DAUDI and CA46 cells results in ER stress response and activation of the ATF4-CHOP-CHAC1 pathway, which is blocked by liproxstatin-1, ferrostatin-1, and deferoxamine (DFO) (66). A previous study has reported that the antitumor effect of artesunate relies on mRNA upregulation of iron-related genes and induces ferroptosis in an iron-dependent manner (67).
Paclitaxel (PTX) is a natural alkaloid isolated from the bark of Taxus brevifolia. Low-dose PTX is a promising treatment for some cancers, especially colorectal carcinoma cells. The expression of glutaminolysis-related genes GLS, SLC7A11, and SLC1A5 is reduced after low-dose PTX treatment in colorectal carcinoma cells, and the expression of tumor-suppressor genes p53 and p21 is elevated at the same time. Low-dose PTX increases lactate production and decreases the pH of tumor microenvironments, which results in the inhibition of tumor cell growth (68).
Dihydroartemisinin (DHA) is able to inhibit the growth of tumor cells including acute myeloid leukemia (AML). DHA arrests the tumor cell cycle at the G0/G1 phase and inhibits AML cell viability. Besides, DHA induces ferroptosis in an iron-dependent manner by regulating the activity of the AMPK/mTOR/p70S6k signaling pathway and promoting the autophagy degradation of ferritin and leads to an increase in the iron pool and elevates the accumulation of ROS (69).
Cisplatin is a first-line therapy for tumor. Cisplatin was found as an inducer of both ferroptosis and apoptosis among the five chemotherapeutic drugs in A549 and HCT116 cell lines. Cancer cells treated with cisplatin show downregulation of GSH and inactivation of GPX4, which results in initiation of ferroptosis. Combination therapy of cisplatin and erastin significantly improves antitumor efficiency (70). PRLX93936 is an analogue of erastin, which has been tested in clinical trials. Co-treatment of cisplatin and PRLX93936 induces the production of lipid peroxidation and Fe2+ and eventually promotes ferroptosis (71).
Besides the above drugs already used in the clinic, many small molecular ferroptosis compounds still stay in a laboratory research phase. Erastin was first found to induce ferroptosis. The chemosynthesis of more soluble erastin derivatives has been shown to have a more effective treatment effect in animal disease models of fibrosarcoma and diffuse large B-cell lymphoma (9). RSL-3 is a representative of the second class of compounds, which can induce ferroptosis in fibrosarcoma. Mechanically, RSL-3 targets the nucleophilic active site of GPX4 to inhibit the activity of GPX4 and induce ferroptosis (8). FIN56 triggers ferroptosis through a mechanism involving the regulation of GPX4 degradation. FIN56 also binds to and activates squalene synthase, an enzyme involved in isoprenoid biosynthesis, which leads to the lack of CoA and promotes ferroptosis (41). 1,2-Dioxolane is an organic peroxide and induces ferroptosis by influencing iron levels and inactivating GPX4, which is the fourth class of small-molecule drug (72).
Chemotherapy is one of the main therapeutic strategies for malignant tumors, but the phenomenon of multidrug resistance (MDR) has become the main reason for the failure of chemotherapy in tumor patients (73). At present, it is widely believed that MDR in tumors can be divided into two cases. The first case is known as intrinsic resistance, in which the tumor cells and tissues are resistant to the drug before receiving chemotherapy (74). The other is known as acquired resistance, that is, drug resistance in patients after effective chemotherapy (75). In recent years, more and more studies have been focused on how to effectively overcome MDR, and with ferroptosis coming into our sight, we will see the light for overcoming MDR.
Recent studies have demonstrated that restraint of ferroptosis is important for drug resistance. Treatment of lung or breast cancer cells with PTX or DOX activates NRF2 and leads to MDR. The abnormal activation of NRF2 caused by Keap1 mutation and p62 upregulation protects cancer cells against ferroptosis. The activation of NRF2 leads to an increase in downstream protein Fth1 and SLC7A11 upregulation, which causes ferroptosis resistance (76). Generally, cancer cells with high levels of GSH and SLC7A11 show higher MDR (56). Therefore, exploring small-molecule inhibitors targeting Keap1/NRF2 against MDR is of therapeutic significance. Pathways to increase intracellular iron concentration such as elevation of iron uptake by TfR1 overexpression, reduction of iron storage by ferritin knockdown or induction of ferritinophagy, and decrease of iron export enhance ferroptosis sensitivity and inhibit MDR, but on the other, reduction of iron results in ferroptosis resistance (77).
The current strategies of MDR include inhibition of the ABC transporter, development of new drug dosage forms, targeting of the tumor microenvironment, and regulation of ROS. Especially, ROS produced during ferroptosis may be destined to have border applications. A large number of studies have demonstrated that compounds regulating cellular ROS levels can enhance the death of MDR cancer cells and make MDR cancer cells sensitive to certain chemotherapy drugs (78). Besides, some redox-regulating enzymes, including mitochondrial ETC complexes, NADPH oxidases (NOXs), system Xc-, thioredoxin reductases, and Nrf2 play important roles in regulating cellular ROS levels and drug resistance as well as their clinical significance (79). Zhang et al. found that cisplatin significantly increased the expression of system Xc- and subsequently increased in GSH levels in tongue squamous cell carcinoma (TSCC). The upregulation of system Xc- and intercellular GSH levels contributed to cisplatin resistance in TSCC cells. Consistently, system Xc- suppression sensitizes TSCC to cisplatin treatment (80). The GPX4 inhibitor RSL-3 enhances the antitumor efficacy of cisplatin by increasing the accumulation of ROS and augmenting the level of iron in tumor cells to activate ferroptosis (81). Besides, activation of the Nrf2-ARE pathway resists GPX4 inhibition of HNC, and inhibition of Nrf2-ARE pathway sensitizes to ferroptosis in HNC (82).
The development of ferroptosis inhibitors and inducers accelerates the research progress in the mechanism study of ferroptosis and chemosensitivity. Ferroptosis inducer sulfasalazine enhances the antitumor sensitivity of cisplatin in HNC and rectal cancer (57). It is reported that different doses of ferroptosis inducer erastin are slightly different. For example, erastin can induce ferroptosis in the AML cell line at the concentration of 5 μmol/l and enhance the antitumor efficiency of cytosine arabinoside and doxorubicin at 1.5 μmol/l (83). Hangauer et al. reported that drug resistance cell lines are vulnerable to GPX4 inhibition. Inhibition of ATF4/HSPA5/GPX4 and system Xc- elevates the sensitivity of pancreatic ductal carcinoma to cisplatin and gemcitabine (84). The depletion of GSH caused by cisplatin and inactivation of GPX4 plays an important role in cisplatin-induced PCD of the non-small cell lung cancer cell line (70). Inhibition of the STAT3/Nrf2/GPX4 pathway promotes the sensitivity of cisplatin-resistant osteosarcoma to cisplatin (85).
Radiotherapy (RT) is one of the most effective cancer treatment methods; RT is the first-line treatment strategy for about 60% of cancer patients. The main mechanism of RT induced is summarized as follows. Firstly, RT induces oxidative damage by radiolysis of cellular water and stimulation of oxidase in all cellular compartments, including the lipid membrane. The accumulation of lipid peroxidation has been implicated to cause ferroptosis, a new form of PCD (9). However, the hypoxic environment in solid tumors limits the effectiveness of RT, because even though the double-stranded structure of DNA is damaged when exposed to RT, hypoxic conditions repair the ROS-induced double-stranded breaks in DNA. Therefore, the O2 concentration of the tumor microenvironment is elevated in vivo, so as to change the hypoxia state of tumor cells and promote RT sensitization (86). In addition, tumor cells with RT will release microparticles into the extracellular environment, which can enhance the effect of radiotherapy by inducing ferroptosis (87). What is more, RT induces intracellular mitochondria DNA damage, thereby activating the STING1/TMEM173-mediated DNA sensing pathway, leading to autophagy-dependent ferroptosis via lipid peroxidation (88).
Ivanov et al. found that treatment with iron-containing water for a long period before RT accelerated glioma death in rats and elevated the efficiency of radiotherapy due to the combination of apoptosis and ferroptosis, which can be blocked by iron chelator DFO (89). Lei et al. found that IR or deficiency of Keap1 promotes the expression of SLC7A11, which enhances radioresistance by inhibiting ferroptosis. Ferroptosis inducers such as RSL3, erastin, sorafenib, and sulfasalazine or inactivating SLC7A11 and GPX4 sensitizes radioresistant cancer cells (90). Besides, it has been reported that gold nanoparticles (AuNPs) induce the production of ROS under X-ray and UV radiation, which indicated that AuNPs can be used as radiosensitizer potential (90, 91). RGD/P-AuNPs formed by the combination of arginine-glycine-aspartic acid (RGD) tripeptide and polyethylene glycolated gold nanoparticles (P-AuNPs) inhibit the invasion of breast cancer cells after radiotherapy (92). Combination therapy of internalized RGD and RGD-conjugated mesoporous silica-coated gold nanorods with radiotherapy can cooperatively induce the G2/M phase arrest of MDA-MB-231 cells and promote ROS production, which enhances the radiosensitivity of breast cancer cells (93).
The tumor microenvironment includes tumor cells, tumor vascular system, extracellular matrix, and immune cells which are important factors in the therapeutic effect of tumor. Hypoxia, chronic inflammation, and immunosuppression are the main characteristics of the tumor environment. The expression of programmed death-ligand 1 (PD-L1) is upregulated in tumor cells to escape immunological surveillance, which interacts with PD-1 of the T cell surface receptor to trigger immune checkpoint response (94). Therefore, anti-PD-1 and anti-PD-L1 immunotherapy has been the most remarkable promise in treating tumors. However, immunotherapy cannot obtain the expected antitumor effect because the physical barrier composed of the fibroblast and dense extracellular matrix in the tumor microenvironment blocks the drugs delivery (95).
Immunotherapy-activated CD8+ T cells enhance specific lipid peroxidation related with ferroptosis in tumor cells, which benefits with the antitumor effect of immunotherapy. Mechanistically, CD8+ T cell downregulates the expression of system Xc- subunits SLC7A11 and SLC3A2 by releasing interferon γ (IFN-γ), which reduces cystine uptake and promotes lipid peroxidation in tumor cells. Depletion of cystine combines with PD-L1 blockade therapy and synergistically induces ferroptosis, which elevates T cell-mediated antitumor immunity (96). In conclusion, ferroptosis inducer and immune checkpoint inhibitor combination therapy will be very promising in the future. Damage-associated molecular pattern molecules (DAMPs) released from irradiated tumor cells and combined therapy of immune checkpoint with the TGF-β inhibitor transform procarcinogenic M2 macrophage into antitumor M1-polarized macrophage (87). A recent study has found a biomimetic magnetosome composed of a Fe3O4 magnetic nanocluster as core and pre-engineered leukocyte membranes as the cloak. The TGF-β inhibitor is anchored inside the membrane, and the PD-1 antibody is loaded on the membrane surface. The biomimetic magnetosome will enter into the tumor where the PD-1 antibody and TGF-β inhibitor create an immunogenic microenvironment synergistically, which increases the content of H2O2 and promotes the Fenton reaction with irons released from the magnetic nanocluster (97). Sun et al. developed a sorafenib and chlorin e6-co-loaded ROS-responsive nanoparticle (NP-sfb/ce6) which strengthens the antitumor effect. With the irradiation of 660 nm, ROS is generated from chlorin e6 (ce6) and destroys the nanoparticles and, as a consequence, promotes sorafenib release from NP-sfb/ce6. The released sorafenib with lose-dose irradiation attenuates tumor progression by inducing powerful T cell antitumor immunity in local and systemic response, remodeling the tumor microenvironment and disrupting the interaction between CD8+ T cell and diverse immunosuppressive cells (98). These findings illustrate the relationship between ferroptosis and immunotherapy and lay a theoretical foundation of ferroptosis–immunotherapy synergism for the tumor treatment. It meanwhile provides a direction for further exploring the molecular mechanism of ferroptosis to promote the efficacy of immunotherapy.
Ferroptosis is a kind of iron-dependent cell death, so the detection of iron content in tumor cells is an important way to detect ferroptosis. Phen Green SK probes are green permeability dyes used to detect intracellular iron in living cells by flow cytometry or confocal microscopy. Iron reacted with ferrozine to form a violet-colored complex and was determined by spectrophotometry at 560 nm. Serum non-heme iron was measured using iron/TIBC reagent designed by Pointe Scientific, Inc. (Canton, MI, USA), and was determined by the chromogen method (99). Lipid peroxidation is one of the features of ferroptosis, which can be detected by BODIPY 581/591 C-11 or DCFH-DA. BODIPY 581/591 C-11 is a lipophilic dye that accumulates in cell membranes. Light emission at 530 and 590 nm (red fluorescence) will be detected with light excitation at 488 and 568 nm. When the dye is oxidized, the emission light will migrate from 590 to 510 nm (green fluorescence), which can be detected by flow cytometry. DCFH-DA is hydrolyzed by intracellular esterase to produce DCFH which can be oxidized by ROS into green fluorescent DCF and monitored by fluorescence microscopy or flow cytometry (9). 4-Hydroxynonenal and malondialdehyde are endogenous products derived from the peroxidation of ω-6 PUFAs, which can be used as biomarkers of ferroptosis. The thiobarbituric acid colorimetric method and enzyme-linked immunosorbent assay are used to detect the content of MDA and 4-hydroxynonenal in the cells, tissues, and serum (4). Besides, the expression of some gene changes after ferroptosis. For example, the expression of ACSL4, PTGS2, and TfR1 upregulates but that of GPX4 and FtH1 downregulates in ferroptic cells (100). Although these genes are well known to be closely associated with ferroptosis, further research is needed to discover more specific biomarkers and testing tools that will be helpful in providing meaningful iron guidance to clinicians.
Ferroptosis is initially discovered as a form of PCD in 2012 and plays important roles in cancer treatment. Based on currently acknowledged pathways such as system Xc-, GPX4, FSP1, and iron metabolism pathway, a number of ferroptosis inducers are used in the fundamental research of cancer treatment. Ferroptosis inducers are better to combine with conventional therapy compared with sole application of ferroptosis inducers in tumor treatment, which significantly enhances therapeutic effects.
Although ferroptosis-based cancer therapy has made great progress, it still faces several challenges, especially with regard to regulatory mechanisms and potential applications in clinical oncology. The exact mechanism of ferroptosis is unclear and remains to be investigated, for example, whether ferroptosis is triggered by lipid peroxidation or the by-products of lipid peroxidation. What is more, ferroptosis is involved in the occurrence and development of many diseases, but whether ferroptosis is a pathogenic factor of these diseases remains to be studied. In addition, little study is focused on the physiological function of ferroptosis during growth and development. Studying the role of ferroptosis in growth and development will be a benefit for cancer treatment.
In addition, there are still unaddressed questions regarding the potential application of ferroptosis in cancer treatment, especially in the area of translational drugs. First is whether the ferroptosis inhibitors or inducers can be applied in vivo safely. Fortunately, sorafenib, sulfasalazine, and artemisinin mentioned in previous sections are currently being used in the clinical treatment of different diseases, but their application in clinical practice of tumor treatment requires further investigation. The sensitivity of different tumor cells to ferroptosis varies because of the expression levels of genes associated with ferroptosis. Therefore, it is important for patients to consider the type of tumor to be treated with ferroptosis-related drugs. Given that pharmacokinetic and physicochemical properties are important for the druggability of compounds, the current laboratorial ferroptosis inducers lack these first-class properties, which restricts its clinical application. Although the combination of ferroptosis with other treatments has been shown to improve the clinical outcomes of cancers, most of this is animal experiment and an optimal combination strategy involving ferroptosis and other treatments has not yet been identified. Although the combination therapy of ferroptosis with other treatments improves the clinical outcomes of cancers, most of this is animal experiment and an optimal combination strategy involving ferroptosis and other treatments needs further exploration.
Ferroptosis is a newly discovered form of PCD, and the study of its mechanism and application in diseases is developing rapidly. From the perspective of mechanism, the core of ferroptosis is the excessiveness of iron and the generation of lipid peroxides. Previous studies considered GPX4 as the core molecule of ferroptosis, but it was recently found that FSP1 was also involved in ferroptosis as a negative regulatory factor paralleled to GPX4, which greatly enriched the comprehension of ferroptosis. It may extend more ferroptosis regulators according to FSP1 regulation (101).
Although great progress has been made in tumor biology and therapeutics, there is still a long way to go before the battle against cancer is won. It is worth mentioning that not all cancer cells are sensitive to ferroptosis. Therefore, study of the ferroptosis sensitivity to cancer cells originating from different tissues will benefit clinical practice to a large extent. Ferroptosis is expected to be a new therapeutic target and has attracted more and more attention in the field of tumor biology and tumor therapy. Ferroptosis is a double-edged sword, and thus the potential cytotoxic effect of key proteins and pathways related with the ferroptosis inducer or inhibitor should be studied to ensure the tumor to trigger Fenton reaction. Further research into the mechanism of ferroptosis provides more potential therapeutic targets for clinical applications, which may lead to the development of new drugs and new therapeutic approaches. The dependence of tumor cells on iron makes them sensitize on iron overload and ROS accumulation. Targeting ferroptosis as new antitumor treatment strategy has important significance. What is more, a cross-disciplinary combination of technologies leads to better antitumor therapies. For example, ferroptosis-related nanometer materials or ferroptosis inducer and chemotherapy or radiotherapy or immune checkpoint combination therapy is used for tumor treatment. Above all, as the study of iron death deepened, the drugs and nanomaterials targeting ferroptosis of tumor cells will have a good prospect, which will become an important field in cancer treatment.
KW put forward the theme of ferroptosis and cancers. JJ and HW developed the scope of the review. SW, TW, XY, FW, and XC carried out the PubMed searches. JJ and HW summarized the relevant papers, accomplished the figures, and wrote the manuscript. KW and CL compiled and approved the final version of the manuscript. Z-YC prepares and presents the published work by those from the original review article. Y-HZ takes the responsibility of preparation, creation or presentation of the published work, especially data presentation. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81770275, 81770406, 81873472, 82070313) and Taishan Scholar Program of Shandong Province and Qingdao Public Domain Science and Technology Support Plan Project (19-6-1-6-nsh).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. D’Arcy MS. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. J Cell Biol Int (2019) 43:582–92. doi: 10.1002/cbin.11137
2. Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, et al. Ischemia-Induced ACSL4 Activation Contributes to Ferroptosis-Mediated Tissue Injury in Intestinal Ischemia/Reperfusion. J Cell Death Differ (2019) 26:2284–99. doi: 10.1038/s41418-019-0299-4
3. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. J Nat Cell Biol (2014) 16:1180–91. doi: 10.1038/ncb3064
4. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a Target for Protection Against Cardiomyopathy. J Proc Natl Acad Sci USA (2019) 116:2672–80. doi: 10.1073/pnas.1821022116
5. Do Van B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, et al. Ferroptosis, a Newly Characterized Form of Cell Death in Parkinson’s Disease That is Regulated by PKC. J Neurobiol Dis (2016) 94:169–78. doi: 10.1016/j.nbd.2016.05.011
6. Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of Ferroptosis in Murine Models of Hemochromatosis. J Hepatol (2017) 66:449–65. doi: 10.1002/hep.29117
7. Cortes J, Perez-Garcia JM, Llombart-Cussac A, Curigliano G, El Saghir NS, Cardoso F, et al. Enhancing Global Access to Cancer Medicines. J CA Cancer J Clin (2020) 70:105–24. doi: 10.3322/caac.21597
8. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. J Cell (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
9. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. J Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
10. Eagle H. Amino Acid Metabolism in Mammalian Cell Cultures. J Sci (1959) 130:432–7. doi: 10.1126/science.130.3373.432
11. Yonezawa M, Back SA, Gan X, Rosenberg PA, Volpe JJ. Cystine Deprivation Induces Oligodendroglial Death: Rescue by Free Radical Scavengers and by a Diffusible Glial Factor. J J Neurochem (1996) 67:566–73. doi: 10.1046/j.1471-4159.1996.67020566.x
12. Wang H, Li J, Follett PL, Zhang Y, Cotanche DA, Jensen FE, et al. 12-Lipoxygenase Plays a Key Role in Cell Death Caused by Glutathione Depletion and Arachidonic Acid in Rat Oligodendrocytes. J Eur J Neurosci (2004) 20:2049–58. doi: 10.1111/j.1460-9568.2004.03650.x
13. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of Genotype-Selective Antitumor Agents Using Synthetic Lethal Chemical Screening in Engineered Human Tumor Cells. J Cancer Cell (2003) 3:285–96. doi: 10.1016/S1535-6108(03)00050-3
14. Xu X, Lai Y, Hua ZC. Apoptosis and Apoptotic Body: Disease Message and Therapeutic Target Potentials. J Biosci Rep (2019) 39:BSR20180992. doi: 10.1042/BSR20180992
15. Chang KC, Liu PF, Chang CH, Lin YC, Chen YJ, Shu CW. The Interplay of Autophagy and Oxidative Stress in the Pathogenesis and Therapy of Retinal Degenerative Diseases. J Cell Biosci (2022) 12:1. doi: 10.1186/s13578-021-00736-9
16. McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, et al. An Iron-Regulated Ferric Reductase Associated With the Absorption of Dietary Iron. J Sci (2001) 291:1755–9. doi: 10.1126/science.1057206
17. Rouault TA. The Intestinal Heme Transporter Revealed. J Cell (2005) 122:649–51. doi: 10.1016/j.cell.2005.08.027
18. Doll S, Conrad M. Iron and Ferroptosis: A Still Ill-Defined Liaison. J IUBMB Life (2017) 69:423–34. doi: 10.1002/iub.1616
19. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is Induced Following Siramesine and Lapatinib Treatment of Breast Cancer Cells. J Cell Death Dis (2016) 7:e2307. doi: 10.1038/cddis.2016.208
20. Quiles Del Rey M, Mancias JD. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. J Front Neurosci (2019) 13:238. doi: 10.3389/fnins.2019.00238
21. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative Proteomics Identifies NCOA4 as the Cargo Receptor Mediating Ferritinophagy. J Nature (2014) 509:105–9. doi: 10.1038/nature13148
22. Zhu J, Berisa M, Schworer S, Qin W, Cross JR, Thompson CB. Transsulfuration Activity Can Support Cell Growth Upon Extracellular Cysteine Limitation. J Cell Metab (2019) 30:865–876 e865. doi: 10.1016/j.cmet.2019.09.009
23. Chio IIC, Tuveson DA. ROS in Cancer: The Burning Question. J Trends Mol Med (2017) 23:411–29. doi: 10.1016/j.molmed.2017.03.004
24. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA Synthetase (CARS) Induces the Transsulfuration Pathway and Inhibits Ferroptosis Induced by Cystine Deprivation. J Cell Death Differ (2016) 23:270–8. doi: 10.1038/cdd.2015.93
25. Combs JA, DeNicola GM. The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. J Cancers (Basel) (2019) 11:678. doi: 10.3390/cancers11050678
26. McGivan JD, Bungard CI. The Transport of Glutamine Into Mammalian Cells. J Front Biosci (2007) 12:874–82. doi: 10.2741/2109
27. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. J Mol Cell (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
28. Gaschler MM, Stockwell BR. Lipid Peroxidation in Cell Death. J Biochem Biophys Res Commun (2017) 482:419–25. doi: 10.1016/j.bbrc.2016.10.086
29. Feng H, Stockwell BR. Unsolved Mysteries: How Does Lipid Peroxidation Cause Ferroptosis? J PloS Biol (2018) 16:e2006203. doi: 10.1371/journal.pbio.2006203
30. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. J Nat Chem Biol (2017) 13:81–90. doi: 10.1038/nchembio.2238
31. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. J Nat Chem Biol (2017) 13:91–8. doi: 10.1038/nchembio.2239
32. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. J ACS Chem Biol (2015) 10:1604–9. doi: 10.1021/acschembio.5b00245
33. Shah R, Shchepinov MS, Pratt DA. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. J ACS Cent Sci (2018) 4:387–96. doi: 10.1021/acscentsci.7b00589
34. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. J Proc Natl Acad Sci USA (2016) 113:E4966–4975. doi: 10.1073/pnas.1603244113
35. Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress Into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. J Cell Metab (2008) 8:237–48. doi: 10.1016/j.cmet.2008.07.005
36. Kuhn H, Banthiya S, van Leyen K. Mammalian Lipoxygenases and Their Biological Relevance. J Biochim Biophys Acta (2015) 1851:308–30. doi: 10.1016/j.bbalip.2014.10.002
37. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. J Nat (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1707-0
38. Song C, Li D, Zhang J, Zhao X. Role of Ferroptosis in Promoting Cardiotoxicity Induced by Imatinib Mesylate via Down-Regulating Nrf2 Pathways In Vitro and In Vivo. J Toxicol Appl Pharmacol (2021) 435:115852. doi: 10.1016/j.taap.2021.115852
39. Sha W, Hu F, Xi Y, Chu Y, Bu S. Mechanism of Ferroptosis and Its Role in Type 2 Diabetes Mellitus. J J Diabetes Res (2021) 2021:9999612. doi: 10.1155/2021/9999612
40. Bersuker K, Hendricks J, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. J Nat (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1705-2
41. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. J Nat Chem Biol (2016) 12:497–503. doi: 10.1038/nchembio.2079
42. Dong H, Qiang Z, Chai D, Peng J, Xia Y, Hu R, et al. Nrf2 Inhibits Ferroptosis and Protects Against Acute Lung Injury Due to Intestinal Ischemia Reperfusion via Regulating SLC7A11 and HO-1. J Aging (Albany NY) (2020) 12:12943–59. doi: 10.18632/aging.103378
43. Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1G Facilitates Sorafenib Resistance Through Inhibition of Ferroptosis. J Hepatol (2016) 64:488–500. doi: 10.1002/hep.28574
44. Wu C. Heat Shock Transcription Factors: Structure and Regulation. J Annu Rev Cell Dev Biol (1995) 11:441–69. doi: 10.1146/annurev.cb.11.110195.002301
45. Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, et al. HSPB1 as a Novel Regulator of Ferroptotic Cancer Cell Death. J Oncogene (2015) 34:5617–25. doi: 10.1038/onc.2015.32
46. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of Mitochondria in Ferroptosis. J Mol Cell (2019) 73:354–63.e353. doi: 10.1016/j.molcel.2018.10.042
47. Kumar V, Santhosh Kumar TR, Kartha CC. Mitochondrial Membrane Transporters and Metabolic Switch in Heart Failure. J Heart Fail Rev (2019) 24:255–67. doi: 10.1007/s10741-018-9756-2
48. Kroemer G, Jaattela M. Lysosomes and Autophagy in Cell Death Control. J Nat Rev Cancer (2005) 5:886–97. doi: 10.1038/nrc1738
49. Aits S, Jaattela M. Lysosomal Cell Death at a Glance. J J Cell Sci (2013) 126:1905–12. doi: 10.1242/jcs.091181
50. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy Promotes Ferroptosis by Degradation of Ferritin. J Autophagy (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
51. Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, et al. An Essential Role for Functional Lysosomes in Ferroptosis of Cancer Cells. J Biochem J (2016) 473:769–77. doi: 10.1042/BJ20150658
52. Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang D, et al. Ferroptosis is a Lysosomal Cell Death Process. J Biochem Biophys Res Commun (2018) 503:1550–6. doi: 10.1016/j.bbrc.2018.07.078
53. Sargeant TJ, Lloyd-Lewis B, Resemann HK, Ramos-Montoya A, Skepper J, Watson CJ. Stat3 Controls Cell Death During Mammary Gland Involution by Regulating Uptake of Milk Fat Globules and Lysosomal Membrane Permeabilization. J Nat Cell Biol (2014) 16:1057–68. doi: 10.1038/ncb3043
54. Lee YS, Lee DH, Choudry HA, Bartlett DL, Lee YJ. Ferroptosis-Induced Endoplasmic Reticulum Stress: Cross-Talk Between Ferroptosis and Apoptosis. J Mol Cancer Res (2018) 16:1073–6. doi: 10.1158/1541-7786.MCR-18-0055
55. Yoshida GJ. The Interplay Between Apoptosis and Ferroptosis Mediated by ER Stress. J Apoptosis (2020) 25:784–5. doi: 10.1007/s10495-020-01641-1
56. Wu Y, Yu C, Luo M, Cen C, Qiu J, Zhang S, et al. Ferroptosis in Cancer Treatment: Another Way to Rome. J Front Oncol (2020) 10:571127. doi: 10.3389/fonc.2020.571127
57. Roh JL, Kim EH, Jang HJ, Park JY, Shin D. Induction of Ferroptotic Cell Death for Overcoming Cisplatin Resistance of Head and Neck Cancer. J Cancer Lett (2016) 381:96–103. doi: 10.1016/j.canlet.2016.07.035
58. Ma MZ, Chen G, Wang P, Lu WH, Zhu CF, Song M, et al. Xc- Inhibitor Sulfasalazine Sensitizes Colorectal Cancer to Cisplatin by a GSH-Dependent Mechanism. J Cancer Lett (2015) 368:88–96. doi: 10.1016/j.canlet.2015.07.031
59. Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Maziere JC, Chauffert B, et al. Iron-Dependent Cell Death of Hepatocellular Carcinoma Cells Exposed to Sorafenib. J Int J Cancer (2013) 133:1732–42. doi: 10.1002/ijc.28159
60. Roh JL, Kim EH, Jang H, Shin D. Nrf2 Inhibition Reverses the Resistance of Cisplatin-Resistant Head and Neck Cancer Cells to Artesunate-Induced Ferroptosis. J Redox Biol (2017) 11:254–62. doi: 10.1016/j.redox.2016.12.010
61. Chen Y, Mi Y, Zhang X, Ma Q, Song Y, Zhang L, et al. Dihydroartemisinin-Induced Unfolded Protein Response Feedback Attenuates Ferroptosis via PERK/ATF4/HSPA5 Pathway in Glioma Cells. J J Exp Clin Cancer Res (2019) 38:402. doi: 10.1186/s13046-019-1413-7
62. Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, et al. Sorafenib Induces Ferroptosis in Human Cancer Cell Lines Originating From Different Solid Tumors. J Anticancer Res (2014) 34:6417–22. doi: 10.3892/or.2019.7189
63. Yu H, Yang C, Jian L, Guo S, Chen R, Li K, et al. Sulfasalazineinduced Ferroptosis in Breast Cancer Cells is Reduced by the Inhibitory Effect of Estrogen Receptor on the Transferrin Receptor. J Oncol Rep (2019) 42:826–38.
64. Zhu S, Yu Q, Huo C, Li Y, He L, Ran B, et al. Ferroptosis: A Novel Mechanism of Artemisinin and its Derivatives in Cancer Therapy. J Curr Med Chem (2021) 28:329–45. doi: 10.2174/0929867327666200121124404
65. Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, Jiang X. Artemisinin Compounds Sensitize Cancer Cells to Ferroptosis by Regulating Iron Homeostasis. J Cell Death Differ (2020) 27:242–54. doi: 10.1038/s41418-019-0352-3
66. Wang N, Zeng GZ, Yin JL, Bian ZX. Artesunate Activates the ATF4-CHOP-CHAC1 Pathway and Affects Ferroptosis in Burkitt’s Lymphoma. J Biochem Biophys Res Commun (2019) 519:533–9. doi: 10.1016/j.bbrc.2019.09.023
67. Ooko E, Saeed ME, Kadioglu O, Sarvi S, Colak M, Elmasaoudi K, et al. Artemisinin Derivatives Induce Iron-Dependent Cell Death (Ferroptosis) in Tumor Cells. J Phytomed (2015) 22:1045–54. doi: 10.1016/j.phymed.2015.08.002
68. Lv C, Qu H, Zhu W, Xu K, Xu A, Jia B, et al. Low-Dose Paclitaxel Inhibits Tumor Cell Growth by Regulating Glutaminolysis in Colorectal Carcinoma Cells. J Front Pharmacol (2017) 8:244. doi: 10.3389/fphar.2017.00244
69. Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA Inhibits Proliferation and Induces Ferroptosis of Leukemia Cells Through Autophagy Dependent Degradation of Ferritin. J Free Radic Biol Med (2019) 131:356–69. doi: 10.1016/j.freeradbiomed.2018.12.011
70. Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: A Novel Anti-Tumor Action for Cisplatin. J Cancer Res Treat (2018) 50:445–60. doi: 10.4143/crt.2016.572
71. Liang Z, Zhao W, Li X, Wang L, Meng L, Yu R. Cisplatin Synergizes With PRLX93936 to Induce Ferroptosis in non-Small Cell Lung Cancer Cells. J Biochem Biophys Res Commun (2021) 569:79–85. doi: 10.1016/j.bbrc.2021.06.088
72. Abrams RP, Carroll WL, Woerpel KA. Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. J ACS Chem Biol (2016) 11:1305–12. doi: 10.1021/acschembio.5b00900
73. Perez-Tomas R. Multidrug Resistance: Retrospect and Prospects in Anti-Cancer Drug Treatment. J Curr Med Chem (2006) 13:1859–76. doi: 10.2174/092986706777585077
74. Nikolaou M, Pavlopoulou A, Georgakilas AG, Kyrodimos E. The Challenge of Drug Resistance in Cancer Treatment: A Current Overview. J Clin Exp Metastasis (2018) 35:309–18. doi: 10.1007/s10585-018-9903-0
75. Cao Y. Adipocyte and Lipid Metabolism in Cancer Drug Resistance. J J Clin Invest (2019) 129:3006–17. doi: 10.1172/JCI127201
76. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the P62-Keap1-NRF2 Pathway Protects Against Ferroptosis in Hepatocellular Carcinoma Cells. J Hepatol (2016) 63:173–84. doi: 10.1002/hep.28251
77. Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. J Cell Chem Biol (2020) 27:420–35. doi: 10.1016/j.chembiol.2020.02.005
78. Gao M, Deng J, Liu F, Fan A, Wang Y, Wu H, et al. Triggered Ferroptotic Polymer Micelles for Reversing Multidrug Resistance to Chemotherapy. J Biomater (2019) 223:119486. doi: 10.1016/j.biomaterials.2019.119486
79. Cui Q, Wang JQ, Assaraf YG, Ren L, Gupta P, Wei L, et al. Modulating ROS to Overcome Multidrug Resistance in Cancer. J Drug Resist Updat (2018) 41:1–25. doi: 10.1016/j.drup.2018.11.001
80. Zhang P, Wang W, Wei Z, Xu LI, Yang X, Du Y. xCT Expression Modulates Cisplatin Resistance in Tca8113 Tongue Carcinoma Cells. J Oncol Lett (2016) 12:307–14. doi: 10.3892/ol.2016.4571
81. Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. J Front Pharmacol (2018) 9:1371. doi: 10.3389/fphar.2018.01371
82. Polewski MD, Reveron-Thornton RF, Cherryholmes GA, Marinov GK, Cassady K, Aboody KS. Increased Expression of System Xc- in Glioblastoma Confers an Altered Metabolic State and Temozolomide Resistance. J Mol Cancer Res (2016) 14:1229–42. doi: 10.1158/1541-7786.MCR-16-0028
83. Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, et al. The Ferroptosis Inducer Erastin Enhances Sensitivity of Acute Myeloid Leukemia Cells to Chemotherapeutic Agents. J Mol Cell Oncol (2015) 2:e1054549. doi: 10.1080/23723556.2015.1054549
84. Chen MS, Wang SF, Hsu CY, Yin PH, Yeh TS, Lee HC, et al. CHAC1 Degradation of Glutathione Enhances Cystine-Starvation-Induced Necroptosis and Ferroptosis in Human Triple Negative Breast Cancer Cells via the GCN2-eIF2alpha-ATF4 Pathway. J Oncotarget (2017) 8:114588–602. doi: 10.18632/oncotarget.23055
85. Liu Q, Wang K. The Induction of Ferroptosis by Impairing STAT3/Nrf2/GPx4 Signaling Enhances the Sensitivity of Osteosarcoma Cells to Cisplatin. J Cell Biol Int (2019) 43:1245–56. doi: 10.1002/cbin.11121
86. Meng L, Cheng Y, Gan S, Zhang Z, Tong X, Xu L, et al. Facile Deposition of Manganese Dioxide to Albumin-Bound Paclitaxel Nanoparticles for Modulation of Hypoxic Tumor Microenvironment To Improve Chemoradiation Therapy. J Mol Pharm (2018) 15:447–57. doi: 10.1021/acs.molpharmaceut.7b00808
87. Wan C, Sun Y, Tian Y, Lu L, Dai X, Meng J, et al. Irradiated Tumor Cell-Derived Microparticles Mediate Tumor Eradication via Cell Killing and Immune Reprogramming. J Sci Adv (2020) 6:eaay9789. doi: 10.1126/sciadv.aay9789
88. Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, Tang D. Mitochondrial DNA Stress Triggers Autophagy-Dependent Ferroptotic Death. J Autophagy (2021) 17:948–60. doi: 10.1080/15548627.2020.1739447
89. Ivanov SD, Semenov AL, Kovan’ko EG, Yamshanov VA. Effects of Iron Ions and Iron Chelation on the Efficiency of Experimental Radiotherapy of Animals With Gliomas. J Bull Exp Biol Med (2015) 158:800–3. doi: 10.1007/s10517-015-2865-1
90. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The Role of Ferroptosis in Ionizing Radiation-Induced Cell Death and Tumor Suppression. J Cell Res (2020) 30:146–62. doi: 10.1038/s41422-019-0263-3
91. Her S, Jaffray DA, Allen C. Gold Nanoparticles for Applications in Cancer Radiotherapy: Mechanisms and Recent Advancements. J Adv Drug Delivery Rev (2017) 109:84–101. doi: 10.1016/j.addr.2015.12.012
92. Wu PH, Onodera Y, Ichikawa Y, Rankin EB, Giaccia AJ, Watanabe Y, et al. Targeting Integrins With RGD-Conjugated Gold Nanoparticles in Radiotherapy Decreases the Invasive Activity of Breast Cancer Cells. J Int J Nanomed (2017) 12:5069–85. doi: 10.2147/IJN.S137833
93. Zhao N, Yang Z, Li B, Meng J, Shi Z, Li P, et al. RGD-Conjugated Mesoporous Silica-Encapsulated Gold Nanorods Enhance the Sensitization of Triple-Negative Breast Cancer to Megavoltage Radiation Therapy. J Int J Nanomed (2016) 11:5595–610. doi: 10.2147/IJN.S104034
94. Chen L, Han X. Anti-PD-1/PD-L1 Therapy of Human Cancer: Past, Present, and Future. J J Clin Invest (2015) 125:3384–91. doi: 10.1172/JCI80011
95. Xu H, Hu M, Liu M, An S, Guan K, Wang M, et al. Nano-Puerarin Regulates Tumor Microenvironment and Facilitates Chemo- and Immunotherapy in Murine Triple Negative Breast Cancer Model. J Biomater (2020) 235:119769. doi: 10.1016/j.biomaterials.2020.119769
96. Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T Cells Regulate Tumour Ferroptosis During Cancer Immunotherapy. J Nature (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
97. Zhang F, Li F, Lu GH, Nie W, Zhang L, Lv Y, et al. Engineering Magnetosomes for Ferroptosis/Immunomodulation Synergism in Cancer. J ACS Nano (2019) 13:5662–73. doi: 10.1021/acsnano.9b00892
98. Sun X, Cao Z, Mao K, Wu C, Chen H, Wang J, et al. Photodynamic Therapy Produces Enhanced Efficacy of Antitumor Immunotherapy by Simultaneously Inducing Intratumoral Release of Sorafenib. J Biomater (2020) 240:119845. doi: 10.1016/j.biomaterials.2020.119845
99. Zhang F, Tao Y, Zhang Z, Guo X, An P, Shen Y, et al. Metalloreductase Steap3 Coordinates the Regulation of Iron Homeostasis and Inflammatory Responses. J Haematol (2012) 97:1826–35. doi: 10.3324/haematol.2012.063974
100. Ju J, Song YN, Wang K. Mechanism of Ferroptosis: A Potential Target for Cardiovascular Diseases Treatment. J Aging Dis (2021) 12:261–76. doi: 10.14336/AD.2020.0323
Keywords: ferroptosis, metabolism, tumor treatment, cancers, drug resistance
Citation: Wang H-T, Ju J, Wang S-C, Zhang Y-H, Liu C-Y, Wang T, Yu X, Wang F, Cheng X-R, Wang K and Chen Z-Y (2022) Insights Into Ferroptosis, a Novel Target for the Therapy of Cancer. Front. Oncol. 12:812534. doi: 10.3389/fonc.2022.812534
Received: 10 November 2021; Accepted: 26 January 2022;
Published: 25 February 2022.
Edited by:
Fabrizio Carta, University of Florence, ItalyReviewed by:
Ilaria D’Agostino, University of Siena, ItalyCopyright © 2022 Wang, Ju, Wang, Zhang, Liu, Wang, Yu, Wang, Cheng, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kun Wang, d2FuZ2s2OTZAcWR1LmVkdS5jbg==; Zhao-Yang Chen, Y2hlbnpoYW95MjAwNjgwOUB5ZWFoLm5ldA==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.