
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 07 February 2022
Sec. Molecular and Cellular Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.810610
 Yinshuang Chen1Man Yang1Fanyi Meng1Yawen Zhang1Mengmeng Wang1Xuqin Guo1Jie Yang2Hongjian Zhang1Haiyang Zhang1*Jing Sun2*Weipeng Wang1*
Yinshuang Chen1Man Yang1Fanyi Meng1Yawen Zhang1Mengmeng Wang1Xuqin Guo1Jie Yang2Hongjian Zhang1Haiyang Zhang1*Jing Sun2*Weipeng Wang1*SRSF3, an important member of the serine/arginine-rich protein (SRp) family, is highly expressed in various tumors and plays an important role in tumor cell proliferation, migration and invasion. However, it is still unclear whether SRSF3 is involved in tumor angiogenesis. In this study, we first revealed that SRSF3 regulated the expression of numerous genes related to angiogenesis, including proangiogenic SRF. Then, we confirmed that SRSF3 was highly expressed in colorectal cancer (CRC) and was positively correlated with SRF. Mechanistic studies revealed that SRSF3 directly bound to the “CAUC” motif in exon 6 of SRF and induced the exclusion of introns. Knockdown of SRSF3 significantly reduced the secretion of VEGF from CRC cells. Conditioned medium from SRSF3-knockdown CRC cells significantly inhibited the migration, invasion and tube formation of human umbilical vein endothelial cells (HUVECs). In addition, SRF silencing inhibited angiogenesis, while SRF overexpression reversed the antiangiogenic effects of SRSF3 knockdown on tube formation. These findings indicate that SRSF3 is involved in the splicing of SRF and thereby regulates the angiogenesis of CRC, which offers novel insight into antiangiogenic therapy in CRC.
CRC is the third most common cancer in the world, and its mortality rate ranks the second (1). Approximately 86% of advanced CRC patients still relapse within 5 years after surgery, and most of these recurrences are in the form of metastasis (2, 3). Moreover, metastatic CRC is difficult to overcome (4). The blood vessels in tumors can transport oxygen and nutrients to promote tumor growth, so angiogenesis is one of the hallmarks of cancer (5). Moreover, angiogenesis plays a vital role in CRC development and has been used as a potential target for metastatic CRC treatment (6, 7). Recently, antiangiogenic therapy has made a major breakthrough in metastatic CRC treatment, significantly prolonging the survival time of patients (4, 8, 9). For example, monoclonal antibodies cetuximab and panitumumab that block epidermal growth factor (EGFR) (10), bevacizumab, aflibercept, ramoximab and regofenib that block vascular endothelial growth factor (VEGF) and VEGF receptor (11–13) have achieved great therapeutic effects in the clinical treatment of patients with metastatic CRC (14). However, there are still many problems in the antiangiogenic therapy of CRC, such as limited scope of application, drug resistance and poor prognosis (11, 15).
Alternative splicing is a critical mechanism to generate diverse structural and functional proteins (16), and more than 95% of human genes undergo alternative splicing. Alternative splicing occurs in various biological processes related to cancer, including invasion, metastasis and angiogenesis (17). Therefore, aberrant splicing could be used not only as a marker of cancer but also as a potential target for cancer treatment (18). Oncogenic mRNA transcripts are produced by the interaction of splicing factors and pre-mRNA. For example, the serine/arginine-rich (SR) proteins SRSF1 and SRSF5 promote the splicing of vascular endothelial growth factor A (VEGFA) and generate proangiogenic isoforms, which are upregulated in tumors (19). Splicing factor 3a subunit 3 (SF3A3) accelerates the production of platelet-derived growth factor receptor (PDGFR), which is associated with angiogenesis (20). RNA binding motif protein 4 (RBM4) and SRSF1 motivate the production of hypoxia inducible factor 1 subunit alpha (HIF-1α) full length isoform and Δ14 HIF-1α isoform, which are proangiogenic (21). ESRP1 enhances the expression of mesenchymal spliced variant CD44s (standard), which plays an important role in the epithelial-mesenchymal transition (EMT) process of epithelial ovarian cancer (22).
Splicing factor SRSF3, an important member of the serine/arginine-rich protein (SRps) family, contains an N-terminal RNA-binding domain and a downstream SR-rich domain. It binds to RNA and acts as a regulator of alternative splicing for many genes. SRSF3 has been identified as a proto-oncogene and overexpressed in multiple cancers (23–25). For example, SRSF3 acts as a PKM splicer and plays a positive role in cancer-specific energy metabolism in CRC (26). SRSF3 also affects the expression of spliced variant coiled-coil domain containing 50 short (CCDC50S) to contribute to the growth and metastasis of hepatocellular carcinoma (HCC) through the Ras/Foxo4 signaling pathway (27). Recent studies have suggested that SRSF3 is a significant regulator of glioblastoma-related alternative splicing and is directly associated with glioblastoma development, progression, aggressiveness and patient survival. It represents a novel potential therapeutic target to tackle this devastating pathology (28, 29). However, the role of SRSF3 in CRC angiogenesis is still unclear.
In this study, we demonstrated that SRSF3 regulated the expression of numerous genes related to angiogenesis. We subsequently identified the proangiogenic role of SRSF3 in CRC. Furthermore, we showed that SRSF3 regulated serum response factor (SRF) expression by binding to the “CAUC” motif in exon 6 of SRF pre-mRNA and participated in the splicing of SRF. Moreover, we confirmed that SRF had a proangiogenic effect and that SRSF3 promoted the angiogenesis of CRC by splicing SRF.
HCT-116 and HCT-8 cell lines (ATCC, USA) were cultured in DMEM (HyClone, USA) with 10% fetal bovine serum (FBS, Gibco). Human umbilical vein endothelial cells (HUVECs) were a gift from the Institute for Cardiovascular Science of Soochow University and cultured in DMEM/F12 (HyClone, USA) containing 10% FBS. All cells were cultivated at 37°C in a humidified incubator containing 5% CO2.
SRSF3-specific siRNA, SRF-specific siRNA and siRNA control were purchased from GenePharma. The human Flag-tagged SRSF3 expression plasmid was cloned into the pcDNA3.1 (+) vector and synthesized by GENEWIZ. The human Flag-tagged SRF expression plasmid was cloned into the GV492 vector, which was synthesized by GeneChem. Minigene recombinant plasmids expressing SRF exons 5-7 with or without point mutations were synthesized by Synbio Technologies. siRNAs and plasmids were transfected into HCT-116 and HCT-8 cell lines using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s protocol.
Total RNA was isolated from CRC cells using RNAiso Plus (#9109, Takara), and the quantity was measured with a NanoDrop 2000 spectrophotometer (Thermo). cDNA synthesis was performed from 1000 ng of total RNA with an RT-Kit (Thermo) and random primers (TaKaRa) as described by the manufacturer. An equal amount of cDNA was amplified by PCR using premix Taq™ (TaKaRa) and separated on an agarose gel. Signal intensities of ethidium bromide-stained bands were quantified using ImageJ software. Specific mRNA expression was measured by qPCR using SYBR Green (Bio–Rad) operated on the Bio–Rad CFX96-C1000, and the relative RNA amount was calculated by the 2-ΔΔCt method with normalization to GAPDH. Primer sequences are shown in Supplementary Table 1.
Protein samples were extracted from the cells with RIPA buffer (Beyotime) containing a protease and phosphatase inhibitor mixture. The extracted proteins were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred onto 0.45 μm PVDF membranes (Merck Millipore). After blocking for 1.5 h at room temperature with 5% nonfat milk, the membranes were incubated overnight at 4°C with primary antibodies against SRSF3 (ab198291, Abcam, 1:10000), SRF (16821-1-AP, Proteintech, 1:1000), GAPDH (AF0006, Beyotime, 1:1000) and β-actin (AF0003, Beyotime, 1:1000). Following incubation with an appropriate HRP-conjugated secondary antibody for 1 h at room temperature, the proteins on the membranes were detected with an ECL Western Blotting Detection System (Merck Millipore). The intensities of bound antibodies were quantified using ImageJ software.
HCT-116 cells were transfected with SRSF3 siRNA and siRNA control for 48 h, and total RNA was extracted as described above. Then, library construction and RNA sequencing (RNA-seq) were performed by Shanghai OE Biotech, followed by computational analysis. Briefly, the count number of each gene was normalized and then the fold change (Fc) was calculated by using DESeq software. The significance of the difference in the number of reads was tested by using negative binomial distribution test. Genes were identified as significantly differentially expressed with nominal P-value <0.05 and Fc > 2 or < 0.5.
Fifty-five CRC tissue samples were collected by the Second People’s Hospital of Changshu from March 2018 to February 2019. All samples were stained with hematoxylin-eosin and confirmed by two pathologists. None of the patients had received chemotherapy or radiotherapy before surgery. The Ethics Committee of Soochow University approved all aspects of this study, and all patients signed informed consent forms.
Immunohistochemistry (IHC) staining was performed by Servicebio (Wuhan) on human CRC tissues. Briefly, CRC tissues were cut into 4 µL-thick sections, deparaffinized in xylene, hydrated in ethyl alcohol and washed in tap water in an orderly manner. Next, the sections were incubated with SRSF3 and SRF antibodies (Santa Cruz). Finally, the sections were visualized under an inverted microscope (Olympus, Japan). The intensity of staining was reviewed by two independent pathologists. For each section, the stain strength was scored at 0-4, and the staining extent was scored as follows: 0 = 0% of tumor cells were positive staining, 1 = 1%-25% of cells were positive staining, 2 = 26%-50% of cells were positive staining, 3 = 51%-75% of cells were positive staining, or 4 = 76%-100% of cells were positive staining. The expression levels of SRSF3 and SRF were classified as negative (score 0), low (score 1-2), and high (score 3-4), respectively.
HCT-116 or HCT-8 cells were seeded in a 6-well plate (3.5×105 cells/well for transfection with siRNAs or 7×105 cells/well for transfection with plasmids). At 48 h after transfection, the medium in the plate was replaced with fresh medium containing 1% FBS and incubated for 24 h. Then, the conditioned medium (CM) was collected and centrifuged at 800 rpm for 10 min to remove cells and cell debris. The supernatant was stored at -80°C for subsequent ELISAs and HUVEC proliferation, migration, invasion and tube formation assays.
The concentration of VEGF in cell culture medium from HCT-116 or HCT-8 cells was analyzed by using a human VEGF ELISA kit (70-EK183-96, LiankeBio) according to the manufacturer’s instructions. Briefly, 1× washing solution was added to a 96-well microtiter plate at 300 μL per well and soaked for 30 s. Then, 100 μL standards and samples were successively added to the plate after discarding the washing solution. At the same time, 50 μL antibody was added to each well, the microtiter plate was sealed with a sealing membrane, and then the plate was placed in a constant temperature shaker at 25°C for 2 h. Coated wells were washed 6 times with 300 μL washing solution and blocked with 100 μL horseradish peroxidase-labeled streptavidin at 25°C for 45 min. After washing 6 times, 100 μL chromogenic substrate was added to each well and incubated for 5-30 min in a dark place at room temperature. Finally, 100 μL stop solution was added to each well. The OD values of the samples in the plate were measured at a maximum absorption wavelength of 450 nm and a reference wavelength of 570 nm or 630 nm by using an enzyme plate analyzer.
Approximately 5000 HUVECs were seeded into a 96-well plate and incubated with the collected CM in a 37°C incubator with 5% CO2 for 24 h. Then, 10 μL MTT solution (5 mg/mL) was added to each well, and the cells were further cultured in the incubator for 2 h. After that, the medium containing MTT was removed, and 150 μL DMSO was added to each well. Finally, the 96-well plate was placed on a constant temperature shaker (800 rpm) at 37°C for 10 min, and the OD value at 492 nm was measured.
For the migration assay, 3×104 HUVECs in 200 μL serum-free medium were seeded into the upper chamber (8 μm pore size, 24-well, #3422, Corning). Then, 600 μL CM was added to the lower chamber and incubated for 24 h at 37°C with 5% CO2. To assess the invasive capacity of HUVECs, the upper chambers were first coated with 50 μL diluted Matrigel (200 μg/mL, #356234, Corning). Then, 5×104 HUVECs in 200 μL serum-free medium were seeded into the upper chamber, 600 μL CM was added to the lower chamber and incubated in a 37°C incubator with 5% CO2 for 48 h.
After 24 h of incubation for the migration assay or 48 h for the invasion assay, cells that could not migrate and invade were removed with a cotton swab from the upper part of the Transwell, and the inserts were fixed with 4% paraformaldehyde for 30 min at room temperature. Transwell inserts were stained in 600 μL 0.1% crystal violet solution for 20 min, and the stained cells were counted in at least three randomly selected fields at 100× magnification under a microscope to minimize bias.
A 96-well plate was coated with 50 μL Matrigel and incubated for 1 h at 37°C. Then, 2×104 HUVECs in 100 μL CM were seeded into 96-well plates. After the plate was incubated for 10 h at 37°C, the tube structures were photographed using an inverted microscope and analyzed by Image-Pro Plus software. Three independent experiments were required for each treatment.
Protein-A/G beads were first preincubated with 5% BSA of NT2 buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM MgCl2 and 0.05% NP-40, pH 7.4). Then, 3 μg SRSF3 antibody or control rabbit IgG (#A7016, Beyotime) was added and incubated at 4°C overnight to obtain SRSF3 antibody or rabbit IgG grafted beads. The beads were collected by centrifugation at 4°C and washed with NT2 buffer 5 times. HCT-116 cells cultured in 100 mm petri dishes were washed twice with 5 mL ice-cold PBS and then lysed in lysis buffer containing 100 mM KCl, 5 mM MgCl2, 10 mM HEPES (pH 7.0), 0.5% NP-40, 1 mM DTT, 1 mM PMSF and 100 U/mL RNase inhibitor for 30 min. After that, the lysis solution was collected by centrifugation. Subsequently, the cell lysate and the antibody-conjugated beads were incubated at 4°C for 4 h, and the immunoprecipitated beads were collected after washing with NT2 buffer 5 times. The beads were resuspended in NT2 buffer containing 0.3 mg/mL proteinase K and then incubated at 55°C for 30 min with shaking. After incubation, RNAiso Plus (#9109, Takara) was added to extract the immunoprecipitated RNA for PCR assays.
To construct the SRF minigene plasmid, a genomic DNA fragment containing SRF exons 5-7 and a 100 bp flanking sequence was cloned into the pcDNA3.1(+) plasmid. According to the RBPmap website (http://rbpmap.technion.ac.il/), three SRF minigene mutant plasmids were constructed by mutating the sequence of exon 6 to determine the binding sites of the SRSF3 protein and SRF pre-mRNA. To investigate the effect of SRSF3 on the splicing of SRF minigene plasmids, minigene plasmids were cotransfected with SRSF3 siRNA or overexpression plasmid into HCT-116 cells. The transcripts were amplified by RT–PCR with primers (FP1: TCATCCGTGCCCACAACTGT; FP2: GTTTCAGCAGTTCAGCTCCACC; RP1: CATTCATCTTGGTGCTGTGGG). The PCR products were then separated on agarose gels.
The relationship between SRF expression and clinicopathological features was analyzed by SPSS v26. The Pearson correlation coefficient was used to assess the correlation between SRSF3 expression and the other genes. For two-group analysis, a two-tailed Student’s t test was used to examine group differences. Significance threshold was p < 0.05. Data were analyzed and plotted using GraphPad Prism 8.0.1 (GraphPad Software Inc., USA).
SRSF3-regulated aberrant splicing is usually associated with numerous aspects of human cancers, such as the cell cycle, cytoskeleton, cell proliferation, apoptosis and other functions. To explore the role of SRSF3 in CRC, we first performed RNA-seq on the extracted RNA from HCT-116 cells transfected with SRSF3 siRNA and siRNA control. The RNA-seq results showed that 1152 genes were upregulated and 870 genes were downregulated when SRSF3 was knocked down (Figure 1A). Then, we performed Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis on these downregulated genes (FC>2, P<0.05) and found that the downregulated genes were involved in the VEGF and TGF-β signaling pathways (Figure 1B). Therefore, the genes regulated by SRSF3 were involved in a variety of biological processes of cancer, suggesting that SRSF3 is expected to be a new therapeutic target for CRC.
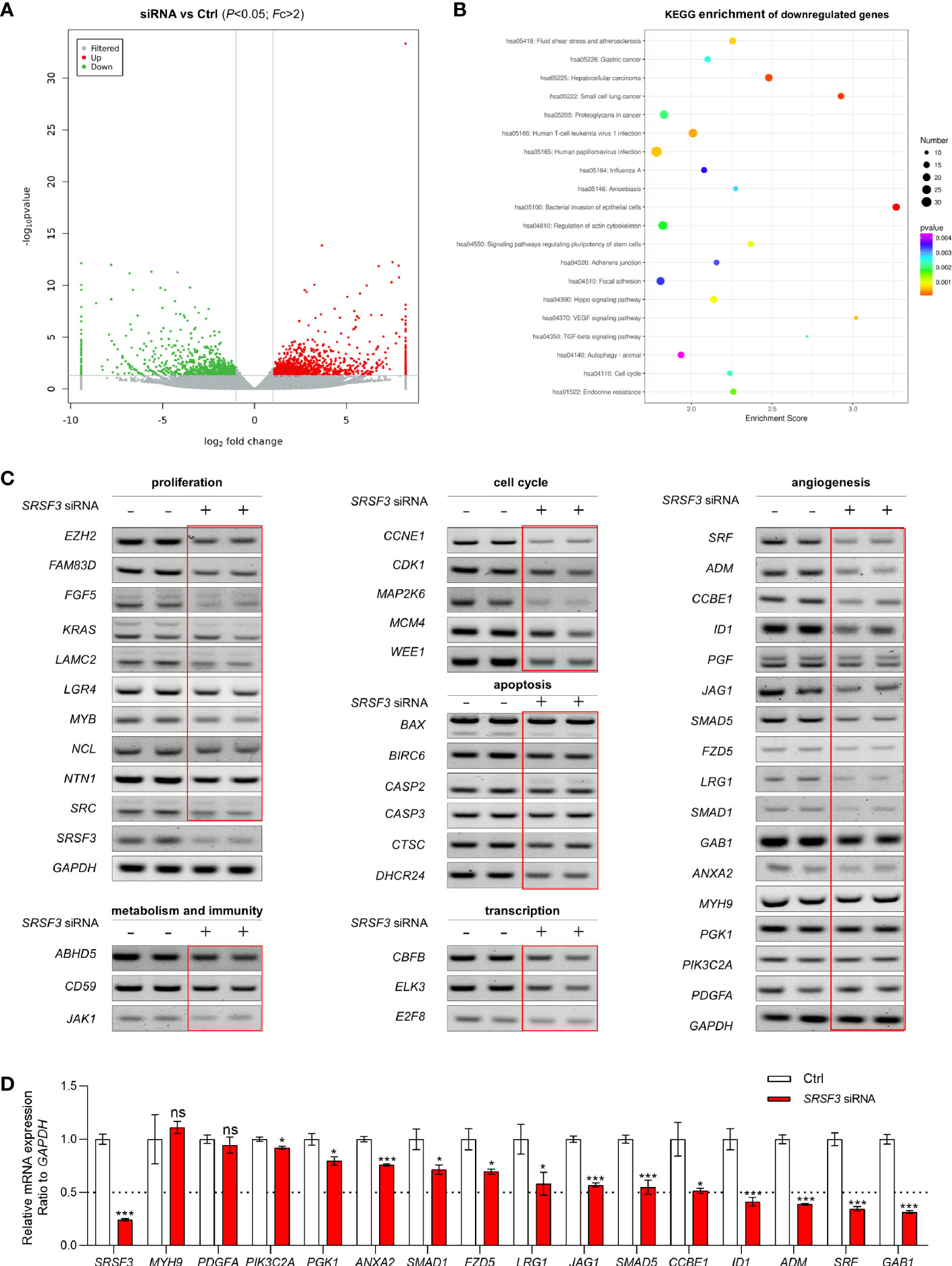
Figure 1 The expression and function of SRSF3-regulated genes in HCT-116 cells. (A) Clustered heatmap for differentially expressed genes regulated by SRSF3. The gene expression was measured by RNA-seq. (B) KEGG analysis of the SRSF3-downregulated genes. (C) RT–PCR assays for SRSF3-regulated genes involved in various biological processes (n=2). (D) qPCR assays for SRSF3-regulated genes related to angiogenesis (n=6). Data represent mean ± SD. Significance was assessed by two-sided t test. ***P < 0.001; *P < 0.05; ns, no significance.
Based on the RNA-seq results, we further screened and verified the expression of genes regulated by SRSF3 using RT–PCR assays. As shown in Figure 1C, the expression of many genes was decreased in the SRSF3-knockdown group, which indicated that SRSF3 regulated the expression of genes related to the cell cycle (MAP2K6, WEE1, CDK1), cell proliferation (NCL, EZH2), apoptosis (CTSC, BIRC6), immune function (CD59, JAK1), metabolism (ABHD5) and transcription factors (ELK3, CBFB, E2F8). Moreover, SRSF3 was positively correlated with genes related to angiogenesis, such as ADM, SRF, ID1 and GAB1. These results were further verified by qPCR assay (Figure 1D).
To further explore the target genes of SRSF3 regulating angiogenesis, the expression correlation between SRSF3 and 16 angiogenesis-related genes based on the TCGA database was analyzed, as shown in Supplementary Table 2. Among these genes, SRF had a good correlation with SRSF3 expression. According to the results of qPCR and WB assays (Figures 2A, B), SRF mRNA and protein expression was significantly inhibited when SRSF3 was knocked down in HCT-116 and HCT-8 cells. Therefore, we concluded that SRSF3 had a positive regulatory effect on the expression of SRF mRNA and protein. SRF was chosen for subsequent experimental studies.
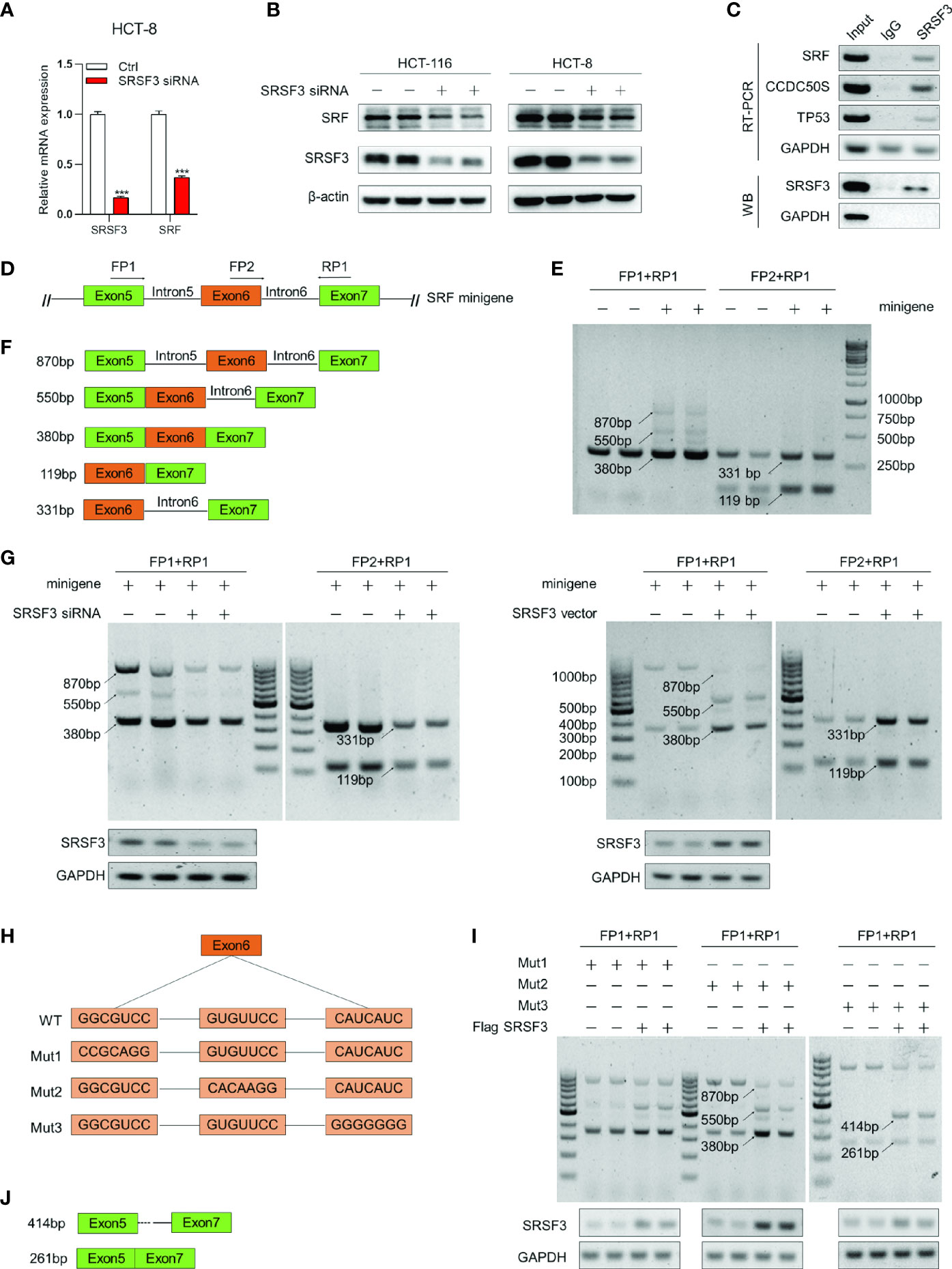
Figure 2 SRSF3-regulated splicing of SRF. (A) qPCR assays for analyzing the effects of SRSF3 knockdown on SRF mRNA expression in HCT-8 cells (n=6). (B) Western blotting to analyze the effects of SRSF3 knockdown on SRF protein expression in HCT-116 and HCT-8 cells (n=2). (C) RIP assays for analyzing the binding of SRSF3 protein with SRF pre-mRNA in HCT-116 cells. GAPDH was used as a negative control, while TP53 and CCDC50S were used as positive controls (upper panels: RT–PCR for SRF mRNA, lower panels: immunoblotting for SRSF3 protein). (D) The schematic diagram of SRF mRNA and minigene. The SRF mRNA contains seven exons, and the predicted binding sites of SRSF3 were enriched on exons 5 to 7. The exons 5–7 were constructed into pcDNA3.1 vectors. Two forward primers (FP1 and FP2) and one reverse primer (RP1) were designed to amplify the transcripts of minigenes. (E) RT–PCR assays for investigating the transcripts of the SRF minigene in HCT-116 cells (n=2). (F) Schematic diagram and sizes of SRF minigene transcripts analyzed by DNA sequencing. (G) RT–PCR assays for investigating the effects of SRSF3 knockdown or overexpression on SRF minigene transcripts in HCT-116 cells (n=2). (H) Schematic diagram of three SRF minigene mutant plasmids. (I) RT–PCR assays for investigating the effects of SRSF3 overexpression on transcripts of SRF minigene mutant plasmids in HCT-116 cells (n=2). (J) Schematic diagram and sizes of SRF Mut3 plasmid transcripts analyzed by DNA sequencing. Data represent mean ± SD. Significance was assessed by two-sided t test. ***P < 0.001.
As an RNA binding protein, SRSF3 can bind to the binding sites on pre-mRNA and then participate in the splicing of target genes (30). Accordingly, we investigated whether SRSF3 could bind to SRF pre-mRNA and regulate SRF pre-mRNA splicing. We performed RIP assay to verify it. In this experiment, CCDC50S and tumor protein 53 (TP53) were chosen as positive controls, which have been identified as SRSF3 target genes (27, 31), while GAPDH was used as a negative control. As shown in Figure 2C, the SRSF3 protein in HCT-116 cells could be pulled down by the bead-antibody (SRSF3) complex. Meanwhile, the PCR results showed that endogenous CCDC50S, TP53 and SRF mRNA could be detected in the SRSF3 protein-pulled down complex, while these could not be detected in the IgG control, as shown in Figure 2C. These results proved that the SRSF3 protein could directly bind to SRF mRNA.
To further explore the mechanism of SRSF3 splicing SRF, a minigene reporter assay was performed, and the design pattern of the SRF minigene is shown in Figure 2D. The SRF minigene plasmid and control plasmid were first transfected into HCT-116 cells. Then, the transcripts were analyzed by RT–PCR using specific primers. As shown in Figure 2E, amplicons of 870 bp, 550 bp and 380 bp were produced by FP1 and RP1, and amplicons of 331 bp and 119 bp were produced by FP2 and RP1. Compared with the control group, two amplicons (870 bp and 550 bp) were only detected in the SRF minigene group, and other amplicons (380 bp, 119 bp and 331 bp) were significantly enhanced. These results proved that the SRF minigene plasmid was successfully constructed. Furthermore, the above PCR products were sequenced, and the splicing patterns are shown in Figure 2F. To determine whether SRSF3 could affect the splicing of SRF pre-mRNA, the SRF minigene was cotransfected with SRSF3 siRNA or expression plasmid into HCT-116 cells. The RT–PCR assay results showed that five transcripts (870 bp, 550 bp, 380 bp, 119 bp and 331 bp) were reduced when SRSF3 was knocked down, while four transcripts were enhanced except 870 bp when SRSF3 was overexpressed (Figure 2G). These results proved that SRSF3 could directly bind to SRF pre-mRNA and participate in the splicing of SRF pre-mRNA, thereby regulating SRF expression.
The RBPmap analysis showed that the potential binding sites of SRSF3 on SRF pre-mRNA were enriched in exon 6 of SRF. To determine the binding sites, we mutated three sequences in exon 6 separately and constructed three SRF minigene mutant plasmids, named Mut1, Mut2 and Mut3 (Figure 2H). Three mutant plasmids were cotransfected with the SRSF3 expression plasmid into HCT-116 cells, and RT–PCR assays were performed to detect the changes in transcripts. As shown in Figure 2I, the group transfected with Mut1 and Mut2 plasmids had the same changes in transcripts as the WT group (the 870 bp transcript was reduced, while the other transcripts were enhanced). This result indicated that the mutant sequences of Mut1 and Mut2 were not the binding sites of SRSF3 and SRF pre-mRNA. Furthermore, transcripts of 414 bp and 261 bp were detected in the transfected Mut3 plasmid group compared with the WT group, but the 550 bp and 380 bp bands were almost undetectable. These results indicated that the mutated sequences of Mut3 changed the splicing mode of SRSF3 to SRF pre-mRNA, and the mutated sequence “CAUC” was the binding site of SRSF3 on SRF pre-mRNA. We also performed DNA sequencing on the 414 bp and 261 bp transcripts, and the splicing patterns are described in Figure 2J. The results showed that SRSF3 no longer binds to exon 6 when the binding sites in exon 6 were mutated, resulting in exon 6 skipping and partial intron retention.
Our previous study demonstrated that SRSF3 expression was enhanced in CRC tissues (32). Moreover, the IHC results showed that SRSF3 also had higher expression in perivascular tumor cells (Figure 3A). We also explored the expression of SRF in CRC. The results of the IHC assay showed that SRF was highly expressed in the endothelial cells around blood vessels (Figure 3A). Moreover, SRSF3 mRNA expression was significantly correlated with SRF in the TCGA database (Figure 3B). In addition, the perivascular expression of SRF in CRC tissues was significantly positively correlated with SRSF3 expression and was associated with lymph node metastasis (Table 1). These results suggested that SRSF3 might be related to CRC angiogenesis and play an important role in the development of CRC.
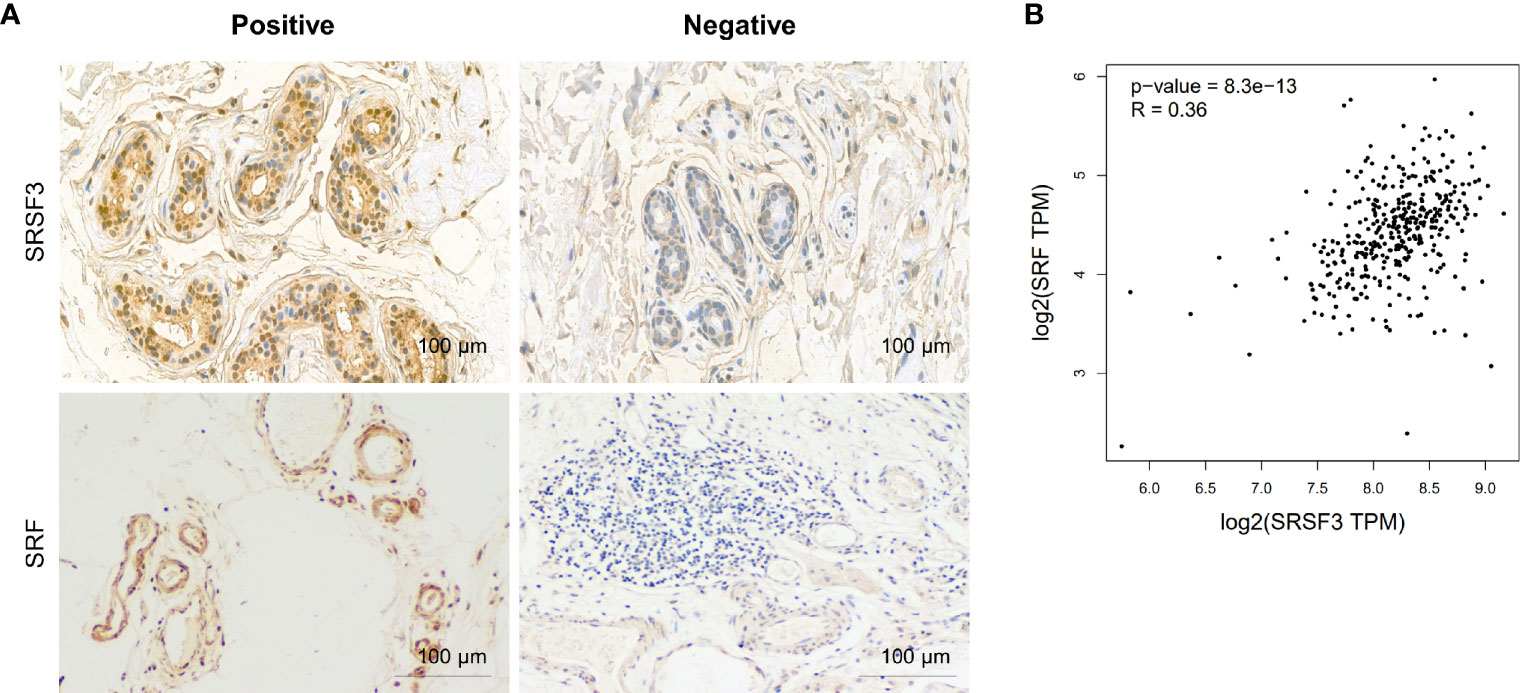
Figure 3 The expression of SRSF3 and SRF in CRC. (A) IHC staining of the SRSF3 and SRF proteins in endothelial cells around blood vessels (n=55). (B) The correlation between SRSF3 and SRF mRNA expression in TCGA database.
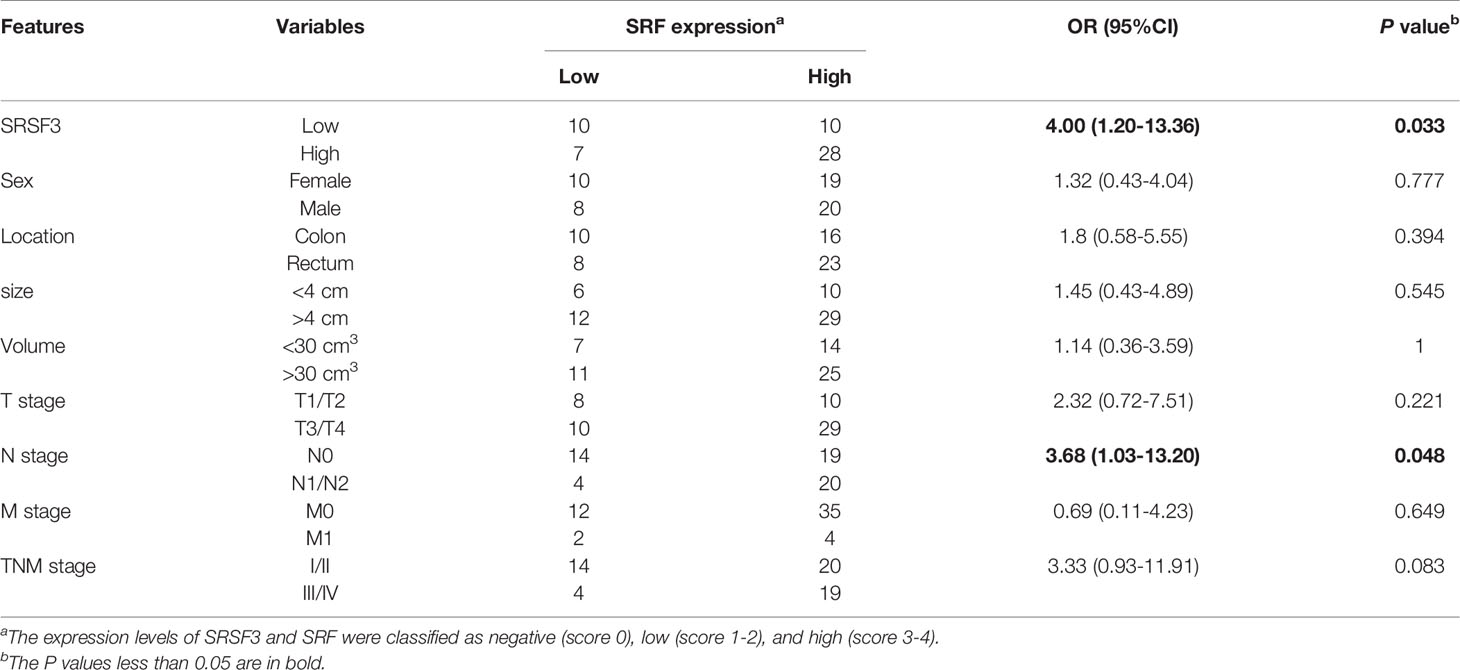
Table 1 The relationship between SRF and clinical characteristics in CRC.
Since SRSF3 regulated angiogenesis-related genes, we wondered whether SRSF3 would affect VEGF secretion and angiogenesis in CRC. Therefore, we first performed ELISA to measure VEGF protein expression in CM from SRSF3-knockdown HCT-116 and HCT-8 cells (Figure 4A). We found that VEGF protein expression in CM from SRSF3-knockdown HCT-116 or HCT-8 cells was significantly decreased compared with that in CM from scrambled control cells (Figure 4B). Next, we used CM from SRSF3-knockdown HCT-116 or HCT-8 cells to explore the effects of SRSF3 on the proliferation, migration, invasion and tube formation of HUVECs. We found that CM had no effect on the proliferation of HUVECs (Figure 4C), but significantly reduced the number of HUVECs that migrated or invaded the Transwell chambers compared with the scrambled control (Figures 4D, E). Meanwhile, the results of the tube formation assay shown in Figure 4F demonstrated that the number of nodes, meshes and branches in SRSF3-knockdown CM was significantly lower than that in the control group. The above results indicated that CM from SRSF3-knockdown HCT-116 or HCT-8 cells significantly inhibited the migration, invasion and tube formation of HUVECs, which verified the important role of SRSF3 in CRC angiogenesis.
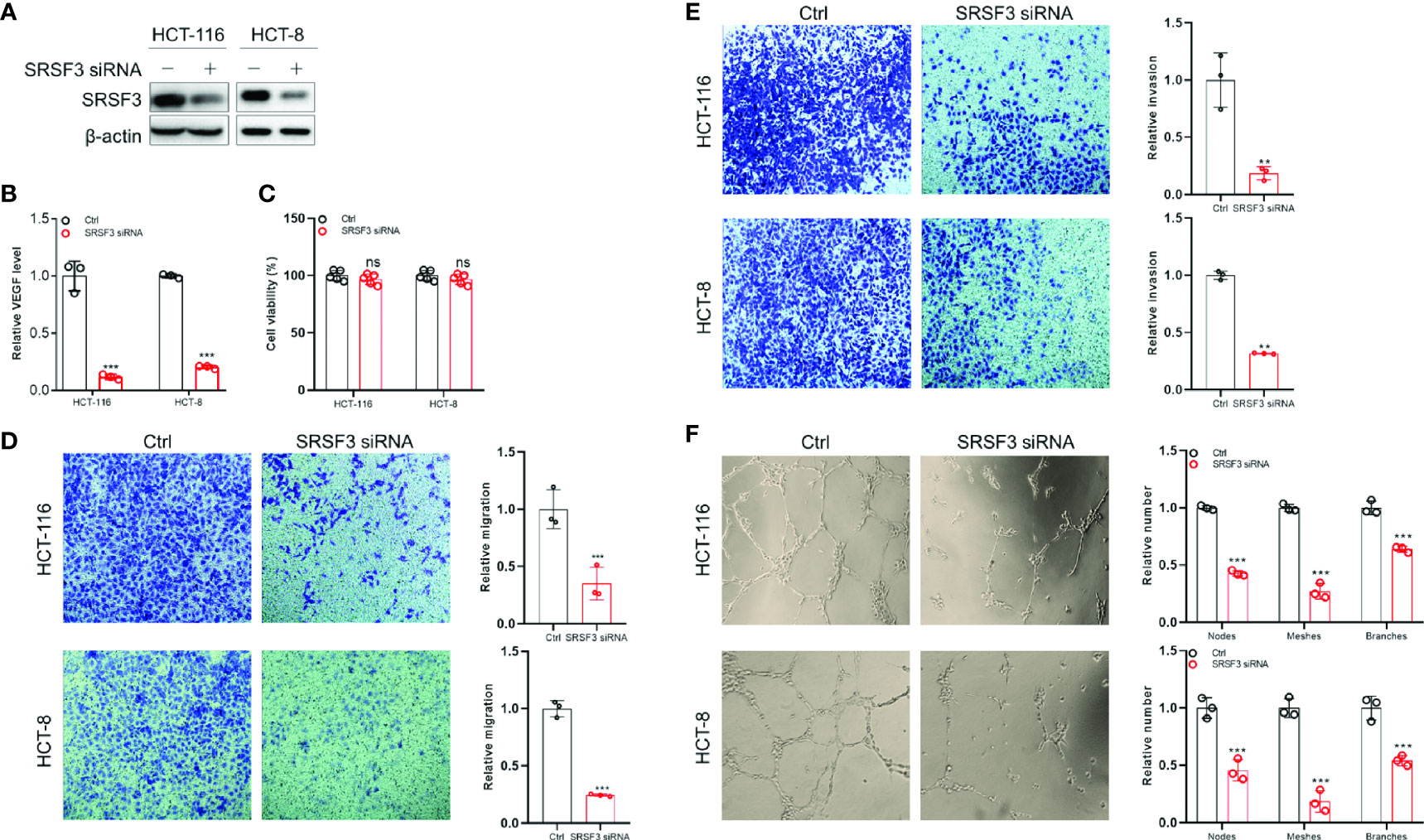
Figure 4 The SRSF3-promoted angiogenesis in CRC. (A) Western blotting for evaluating SRSF3 siRNA in HCT-116 and HCT-8 cells. (B) ELISAs for detecting the effect of SRSF3 siRNA on VEGF secretion in HCT-116 and HCT-8 cells (n=3). (C) MTT assays to investigate the effect of CM from SRSF3-silenced HCT-116 and HCT-8 cells on the proliferation of HUVECs (n=6). (D) Transwell assays for investigating the effect of CM from SRSF3-silenced HCT-116 and HCT-8 cells on the migration of HUVECs (n=3). (E) Transwell assays for investigating the effect of CM from SRSF3-silenced HCT-116 and HCT-8 cells on the invasion of HUVECs (n=3). (F) The effects of CM from SRSF3-silenced HCT-116 and HCT-8 cells on the tube formation of HUVECs (n=3). Data represent mean ± SD. Significance was assessed by two-sided t test. ***P < 0.001; **P < 0.01; ns, no significance.
To investigate the role of SRF in CRC angiogenesis, HCT-116 and HCT-8 cells were first transfected with SRF siRNA or expression plasmid, and then CM was collected. The results of western blot and qPCR assays demonstrated that SRF siRNA and expression plasmids regulated SRF expression at the mRNA and protein levels (Figures 5A, B). As shown in Figure 5C, the ELISA results showed that the secretion of VEGF was significantly decreased in SRF-knockdown CM. We further investigated the effect of SRF-knockdown CM on the proliferation, migration, invasion and tube formation of HUVECs. Consistent with the SRSF3 knockdown results, SRF-knockdown CM had no effect on the proliferation of HUVECs (Figure 5D). However, the number of HUVECs crossing the Transwell chambers was significantly lower than that crossing the scrambled control, regardless of the presence of Matrigel in the chambers (Figures 5E, F). Moreover, SRF-knockdown CM induced HUVECs to develop fewer tubes than the scrambled control (Figure 5G). These results showed that SRF-knockdown CM could significantly inhibit the migration, invasion and tube formation of HUVECs, suggesting that SRF also played an important role in CRC angiogenesis.

Figure 5 The SRF-promoted angiogenesis in CRC. (A) Western blotting assays for verifying SRF siRNA and expression vector in HCT-116 and HCT-8 cells. (B) qPCR assays for verifying SRF siRNA in HCT-116 and HCT-8 cells (n=6). (C) ELISAs to investigate the effects of SRF silencing on VEGF secretion in HCT-116 and HCT-8 cells (n=3). (D) MTT assays for investigating the effects of CM from SRF-knockdown HCT-116 and HCT-8 cells on the proliferation of HUVECs (n=6). (E) Transwell assays to investigate the effects of CM on the migration of HUVECs (n=2). (F) Transwell assays to investigate the effects of CM on the invasion of HUVECs (n=2). (G) The effects of CM on the tube formation of HUVECs (n=3). (H) The effects of CM from HCT-116 and HCT-8 cells transfected with SRSF3 siRNA and SRF expression plasmid on the tube formation of HUVECs (n=3). Data represent mean ± SD. Significance was assessed by two-sided t test. ***P < 0.001; **P < 0.01; ns, no significance.
To explore whether SRSF3 promoted the angiogenesis of CRC by regulating SRF, we performed rescue experiments by cotransfecting SRSF3 siRNA with an SRF expression plasmid into HCT-116 and HCT-8 cells. As shown in Figure 5H, the results showed that the overexpression of SRF diminished the inhibitory effect of SRSF3 knockdown on HUVEC tube formation, indicating that SRSF3 promoted the angiogenesis of CRC by regulating SRF.
The high morbidity and mortality of CRC remain a worldwide challenge. Abnormal angiogenesis is one of the common clinical traits of CRC. In this study, we demonstrated that SRSF3 was highly expressed in CRC tissues and tumor cells around blood vessels and verified that SRSF3 played important roles in the angiogenesis of CRC. Moreover, our data showed that SRSF3 positively regulated SRF expression and consequently promoted CRC angiogenesis by driving the migration, invasion and tube formation of HUVECs.
Alternative splicing is a common process leading to transcript variation and proteome diversity (17), and aberrant splicing is usually the cause of cancer occurrence and development. It has been reported that aberrant splicing leads to an increase in proangiogenic isoforms of VEGFA and prompts tumor angiogenesis (33). For example, aberrant splicing-mediated upregulation of BCL2-like 1 (BCL2L1) antiapoptotic isoform enhances the antiapoptotic ability of cancer (34). Aberrant splicing is commonly caused by mutations or abnormal expression of splicing factors. SRSF3 is a member of the SR protein family with the highest expression in CRC (35), which regulates the alternative splicing of multiple genes and participates in numerous steps of RNA biological metabolism. Previous studies have shown that SRSF3 expression could be used as a biomarker for cancer diagnosis and prognosis (29, 36). In this study, we first performed IHC staining on 55 CRC tissues, and the results showed that SRSF3 had a higher expression in CRC tissues, which was consistent with other studies (35, 37, 38). In addition, SRSF3 was also highly expressed around tumor blood vessels. These results suggested that SRSF3 could have a positive effect on the angiogenesis of CRC. Then, we performed RNA-seq on HCT-116 cells transfected with SRSF3 siRNA or negative control. The results showed that genes regulated by SRSF3 are involved in a variety of biological processes, such as the cell cycle, proliferation, migration, invasion, cell metabolism and immune response. Moreover, the results of RT–PCR and qPCR assays showed that SRSF3 regulated the expression of genes related to angiogenesis, including SRF. We utilized RIP assays and minigene reporter assays to prove that SRSF3 participated in the splicing of SRF pre-mRNA by binding to the “CAUC” motif in exon 6, thereby regulating SRF expression.
SRF, a member of the MADS box superfamily of transcription factors, mediates the transcription of genes related to cell growth, migration, cytoskeleton, and energy metabolism and regulates the expression of cell adhesion factors (39, 40). SRF is highly expressed in gastrointestinal cancers, such as hepatocellular carcinoma, CRC and esophageal cancer. It also regulates the Wnt/β-catenin pathway, promotes the expression of MMP2, MMP9 and E-cadherin/β-catenin, and enhances the proliferation, invasion and angiogenesis of tumor cells, thereby promoting tumor metastasis (41). VEGF is one of the major factors that initiates and regulates angiogenesis (42). Several studies have found that SRF is a downstream mediator of VEGF signal transduction in endothelial cells and is also a key condition for VEGF-induced angiogenesis (43, 44). VEGF can induce SRF expression and nuclear translocation through the MEK-ERK and Rho GTPase signaling pathways and increase the binding activity of SRF to DNA in endothelial cells (45).
Our study demonstrated that SRF played a proangiogenic role in CRC. We performed IHC staining on 55 CRC tissue samples for SRF, and the results showed that SRF was highly expressed not only in CRC tissues but also around tumor blood vessels. Meanwhile, SRF could act as an upstream regulator to affect the expression of VEGF. The ELISA results showed that the expression of VEGF was downregulated in the CM from SRF-knockdown HCT-116 and HCT-8 cells, indicating that there was a mutual regulatory relationship between SRF and VEGF. Moreover, CM from SRF-knockdown HCT-116 and HCT-8 cells significantly inhibited the migration, invasion and tube formation of HUVECs. We also found that the overexpression of SRF reversed the inhibitory effect of SRSF3 knockdown on HUVEC tube formation. Our study suggested that SRF played an important role in the angiogenesis and development of CRC.
In summary, we confirmed that SRSF3 promoted the angiogenesis of CRC by regulating SRF through a series of in vitro experiments. SRSF3 could directly bind to SRF pre-mRNA and participate in the splicing of SRF pre-mRNA, thereby positively regulating SRF expression. In short, our study provides a theoretical basis for SRSF3 as a therapeutic target for CRC and provides a new direction for the treatment of CRC.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by The Ethics Committee of Soochow University. The patients/participants provided their written informed consent to participate in this study.
WW, JS, and HYZ conceived the idea and directed the study. YC, YZ, MY, HYZ, JS, and WW designed the experiments and wrote the manuscript. YC, YZ, MY, JY, and FM performed experiments. YC, YZ, and WW analyzed the data. YC, MY, YZ, HYZ, and WW generated figures. WW, JS, and HYZ critically revised the manuscript for important intellectual content. All authors contributed to the article and approved the submitted version.
This research was supported by the National Natural Science Foundation of China (No. 81773044), Science and Technology Special Project of Clinical Medicine in Jiangsu Province (BL2014046), Social Development Project of Jiangsu Province (BE2019657), Qinglan Project of Jiangsu Province, and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.810610/full#supplementary-material
1. Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal Cancer Statistics, 2020. CA: Cancer J Clin (2020) 70(3):145–64. doi: 10.3322/caac.21601
2. Tauriello DV, Calon A, Lonardo E, Batlle E. Determinants of Metastatic Competency in Colorectal Cancer. Mol Oncol (2017) 11(1):97–119. doi: 10.1002/1878-0261.12018
3. Wrobel P, Ahmed S. Current Status of Immunotherapy in Metastatic Colorectal Cancer. Int J Colorectal Dis (2019) 34(1):13–25. doi: 10.1007/s00384-018-3202-8
4. Modest DP, Pant S, Sartore-Bianchi A. Treatment Sequencing in Metastatic Colorectal Cancer. Eur J Cancer (Oxford Engl 1990) (2019) 109:70–83. doi: 10.1016/j.ejca.2018.12.019
5. Wang R, Ma Y, Zhan S, Zhang G, Cao L, Zhang X, et al. B7-H3 Promotes Colorectal Cancer Angiogenesis Through Activating the NF-κb Pathway to Induce VEGFA Expression. Cell Death Dis (2020) 11(1):55. doi: 10.1038/s41419-020-2252-3
6. Giordano G, Febbraro A, Venditti M, Campidoglio S, Olivieri N, Raieta K, et al. Targeting Angiogenesis and Tumor Microenvironment in Metastatic Colorectal Cancer: Role of Aflibercept. Gastroenterol Res Pract (2014) 2014:526178. doi: 10.1155/2014/526178
7. Cheng WK, Oon CE. How Glycosylation Aids Tumor Angiogenesis: An Updated Review. Biomed Pharmacother = Biomed Pharmacother (2018) 103:1246–52. doi: 10.1016/j.biopha.2018.04.119
8. Battaglin F, Puccini A, Intini R, Schirripa M, Ferro A, Bergamo F, et al. The Role of Tumor Angiogenesis as a Therapeutic Target in Colorectal Cancer. Expert Rev Anticancer Ther (2018) 18(3):251–66. doi: 10.1080/14737140.2018.1428092
9. Marques I, Araújo A, de Mello RA. Anti-Angiogenic Therapies for Metastatic Colorectal Cancer: Current and Future Perspectives. World J Gastroenterol (2013) 19(44):7955–71. doi: 10.3748/wjg.v19.i44.7955
10. Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, et al. Addition of Cetuximab to Chemotherapy as First-Line Treatment for KRAS Wild-Type Metastatic Colorectal Cancer: Pooled Analysis of the CRYSTAL and OPUS Randomised Clinical Trials. Eur J Cancer (Oxford Engl 1990) (2012) 48(10):1466–75. doi: 10.1016/j.ejca.2012.02.057
11. Annett S, Moore G, Short A, Marshall A, McCrudden C, Yakkundi A, et al. FKBPL-Based Peptide, ALM201, Targets Angiogenesis and Cancer Stem Cells in Ovarian Cancer. Br J Cancer (2020) 122(3):361–71. doi: 10.1038/s41416-019-0649-5
12. Arnold D, Fuchs CS, Tabernero J, Ohtsu A, Zhu AX, Garon EB, et al. Meta-Analysis of Individual Patient Safety Data From Six Randomized, Placebo-Controlled Trials With the Antiangiogenic VEGFR2-Binding Monoclonal Antibody Ramucirumab. Ann Oncol (2017) 28(12):2932–42. doi: 10.1093/annonc/mdx514
13. Mody K, Baldeo C, Bekaii-Saab T. Antiangiogenic Therapy in Colorectal Cancer. Cancer J (Sudbury Mass) (2018) 24(4):165–70. doi: 10.1097/PPO.0000000000000328
14. Bupathi M, Ahn DH, Bekaii-Saab T. Spotlight on Bevacizumab in Metastatic Colorectal Cancer: Patient Selection and Perspectives. Gastrointest Cancer (2016) 6:21–30. doi: 10.2147/GICTT.S97740
15. Van Emburgh BO, Arena S, Siravegna G, Lazzari L, Crisafulli G, Corti G, et al. Acquired RAS or EGFR Mutations and Duration of Response to EGFR Blockade in Colorectal Cancer. Nat Commun (2016) 7:13665. doi: 10.1038/ncomms13665
16. Qi F, Li Y, Yang X, Wu YP, Lin LJ, Liu XM. Significance of Alternative Splicing in Cancer Cells. Chin Med J (2020) 133(2):221–8. doi: 10.1097/CM9.0000000000000542
17. Sveen A, Kilpinen S, Ruusulehto A, Lothe RA, Skotheim RI. Aberrant RNA Splicing in Cancer; Expression Changes and Driver Mutations of Splicing Factor Genes. Oncogene (2016) 35(19):2413–27. doi: 10.1038/onc.2015.318
18. Yang Q, Zhao J, Zhang W, Chen D, Wang Y. Aberrant Alternative Splicing in Breast Cancer. J Mol Cell Biol (2019) 11(10):920–9. doi: 10.1093/jmcb/mjz033
19. Biselli-Chicote PM, Oliveira AR, Pavarino EC, Goloni-Bertollo EM. VEGF Gene Alternative Splicing: Pro- and Anti-Angiogenic Isoforms in Cancer. J Cancer Res Clin Oncol (2012) 138(3):363–70. doi: 10.1007/s00432-011-1073-2
20. Zuo ZH, Yu YP, Martin A, Luo JH. Cellular Stress Response 1 Down-Regulates the Expression of Epidermal Growth Factor Receptor and Platelet-Derived Growth Factor Receptor Through Inactivation of Splicing Factor 3A3. Mol Carcinog (2017) 56(2):315–24. doi: 10.1002/mc.22494
21. Chang HL, Lin JC. SRSF1 and RBM4 Differentially Modulate the Oncogenic Effect of HIF-1α in Lung Cancer Cells Through Alternative Splicing Mechanism. Biochim Biophys Acta Mol Cell Res (2019) 1866(12):118550. doi: 10.1016/j.bbamcr.2019.118550
22. Chen L, Yao Y, Sun L, Zhou J, Miao M, Luo S, et al. Snail Driving Alternative Splicing of CD44 by ESRP1 Enhances Invasion and Migration in Epithelial Ovarian Cancer. Cell Physiol Biochem (2017) 43(6):2489–504. doi: 10.1159/000484458
23. Ke H, Zhao L, Zhang H, Feng X, Xu H, Hao J, et al. Loss of TDP43 Inhibits Progression of Triple-Negative Breast Cancer in Coordination With SRSF3. Proc Natl Acad Sci USA (2018) 115(15):E3426–35. doi: 10.1073/pnas.1714573115
24. Su YA, Yang J, Tao L, Nguyen H, He P. Undetectable and Decreased Expression of KIAA1949 (Phostensin) Encoded on Chromosome 6p21.33 in Human Breast Cancers Revealed by Transcriptome Analysis. J Cancer (2010) 1:38–50. doi: 10.7150/jca.1.38
25. Iborra S, Hirschfeld M, Jaeger M, Zur Hausen A, Braicu I, Sehouli J, et al. Alterations in Expression Pattern of Splicing Factors in Epithelial Ovarian Cancer and its Clinical Impact. Int J Gynecol Cancer (2013) 23(6):990–6. doi: 10.1097/IGC.0b013e31829783e3
26. Kuranaga Y, Sugito N, Shinohara H, Tsujino T, Taniguchi K, Komura K, et al. SRSF3, a Splicer of the PKM Gene, Regulates Cell Growth and Maintenance of Cancer-Specific Energy Metabolism in Colon Cancer Cells. Int J Mol Sci (2018) 19(10):3012. doi: 10.3390/ijms19103012
27. Wang H, Zhang CZ, Lu SX, Zhang MF, Liu LL, Luo RZ, et al. A Coiled-Coil Domain Containing 50 Splice Variant Is Modulated by Serine/Arginine-Rich Splicing Factor 3 and Promotes Hepatocellular Carcinoma in Mice by the Ras Signaling Pathway. Hepatol (Baltimore Md) (2019) 69(1):179–95. doi: 10.1002/hep.30147
28. Song X, Wan X, Huang T, Zeng C, Sastry N, Wu B, et al. SRSF3-Regulated RNA Alternative Splicing Promotes Glioblastoma Tumorigenicity by Affecting Multiple Cellular Processes. Cancer Res (2019) 79(20):5288–301. doi: 10.1158/0008-5472.CAN-19-1504
29. Fuentes-Fayos AC, Vázquez-Borrego MC, Jiménez-Vacas JM, Bejarano L, Pedraza-Arévalo S, LL F, et al. Splicing Machinery Dysregulation Drives Glioblastoma Development/Aggressiveness: Oncogenic Role of SRSF3. Brain (2020) 143(11):3273–93. doi: 10.1093/brain/awaa273
30. Lee SC, Abdel-Wahab O. Therapeutic Targeting of Splicing in Cancer. Nat Med (2016) 22(9):976–86. doi: 10.1038/nm.4165
31. Tang Y, Horikawa I, Ajiro M, Robles AI, Fujita K, Mondal AM, et al. Downregulation of Splicing Factor SRSF3 Induces P53β, an Alternatively Spliced Isoform of P53 That Promotes Cellular Senescence. Oncogene (2013) 32(22):2792–8. doi: 10.1038/onc.2012.288
32. Zhang C, Chen Y, Li F, Yang M, Meng F, Zhang Y, et al. B7-H3 is Spliced by SRSF3 in Colorectal Cancer. Cancer Immunol Immunother CII (2020) 70(2):311–21. doi: 10.1007/s00262-020-02683-9
33. Ladomery MR, Harper SJ, Bates DO. Alternative Splicing in Angiogenesis: The Vascular Endothelial Growth Factor Paradigm. Cancer Lett (2007) 249(2):133–42. doi: 10.1016/j.canlet.2006.08.015
34. Boise LH, González-García M, Postema CE, Ding L, Lindsten T, Turka LA, et al. Bcl-X, a Bcl-2-Related Gene That Functions as a Dominant Regulator of Apoptotic Cell Death. Cell (1993) 74(4):597–608. doi: 10.1016/0092-8674(93)90508-N
35. Park WC, Kim HR, Kang DB, Ryu JS, Choi KH, Lee GO, et al. Comparative Expression Patterns and Diagnostic Efficacies of SR Splicing Factors and HNRNPA1 in Gastric and Colorectal Cancer. BMC Cancer (2016) 16:358. doi: 10.1186/s12885-016-2387-x
36. Che Y, Fu L. Aberrant Expression and Regulatory Network of Splicing Factor-SRSF3 in Tumors. J Cancer (2020) 11(12):3502–11. doi: 10.7150/jca.42645
37. Lin JC, Lee YC, Tan TH, Liang YC, Chuang HC, Fann YC, et al. RBM4-SRSF3-MAP4K4 Splicing Cascade Modulates the Metastatic Signature of Colorectal Cancer Cell. Biochim Biophys Acta Mol Cell Res (2018) 1865(2):259–72. doi: 10.1016/j.bbamcr.2017.11.005
38. Wang JL, Guo CR, Sun TT, Su WY, Hu Q, Guo FF, et al. SRSF3 Functions as an Oncogene in Colorectal Cancer by Regulating the Expression of Arhgap30. Cancer Cell Int (2020) 20:120. doi: 10.1186/s12935-020-01201-2
39. Gau D, Roy P. SRF'ing and SAP'ing - the Role of MRTF Proteins in Cell Migration. J Cell Sci (2018) 131(19):jcs218222. doi: 10.1242/jcs.218222
40. Miano JM, Long X, Fujiwara K. Serum Response Factor: Master Regulator of the Actin Cytoskeleton and Contractile Apparatus. Am J Physiol Cell Physiol (2007) 292(1):C70–81. doi: 10.1152/ajpcell.00386.2006
41. Yin J, Lv X, Hu S, Zhao X, Liu Q, Xie H. Overexpression of Serum Response Factor is Correlated With Poor Prognosis in Patients With Gastric Cancer. Hum Pathol (2019) 85:10–7. doi: 10.1016/j.humpath.2018.10.018
42. Gianni-Barrera R, Butschkau A, Uccelli A, Certelli A, Valente P, Bartolomeo M, et al. PDGF-BB Regulates Splitting Angiogenesis in Skeletal Muscle by Limiting VEGF-Induced Endothelial Proliferation. Angiogenesis (2018) 21(4):883–900. doi: 10.1007/s10456-018-9634-5
43. Franco CA, Li Z. SRF in Angiogenesis: Branching the Vascular System. Cell Adhes Migr (2009) 3(3):264–7. doi: 10.4161/cam.3.3.8291
44. Franco CA, Mericskay M, Parlakian A, Gary-Bobo G, Gao-Li J, Paulin D, et al. Serum Response Factor is Required for Sprouting Angiogenesis and Vascular Integrity. Dev Cell (2008) 15(3):448–61. doi: 10.1016/j.devcel.2008.07.019
Keywords: angiogenesis, colorectal cancer, splicing, SRF, SRSF3
Citation: Chen Y, Yang M, Meng F, Zhang Y, Wang M, Guo X, Yang J, Zhang H, Zhang H, Sun J and Wang W (2022) SRSF3 Promotes Angiogenesis in Colorectal Cancer by Splicing SRF. Front. Oncol. 12:810610. doi: 10.3389/fonc.2022.810610
Received: 07 November 2021; Accepted: 17 January 2022;
Published: 07 February 2022.
Edited by:
Almudena Porras, Complutense University of Madrid, SpainReviewed by:
Massimo Pancione, University of Sannio, ItalyCopyright © 2022 Chen, Yang, Meng, Zhang, Wang, Guo, Yang, Zhang, Zhang, Sun and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiyang Zhang, hyzhang2020@suda.edu.cn; Jing Sun, jsun@szhct.edu.cn; Weipeng Wang, wangweipeng@suda.edu.cn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.