 Anne-Laure Désage1,2
Anne-Laure Désage1,2 Aurélie Swalduz
Aurélie Swalduz Sandra Ortiz-Cuaran
Sandra Ortiz-Cuaran
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 16 February 2022
Sec. Thoracic Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.796832
This article is part of the Research Topic Targeting KRAS in Lung Cancer View all 7 articles
Although KRAS-activating mutations represent the most common oncogenic driver in non-small cell lung cancer (NSCLC), various attempts to inhibit KRAS failed in the past decade. KRAS mutations are associated with a poor prognosis and a poor response to standard therapeutic regimen. The recent development of new therapeutic agents (i.e., adagrasib, sotorasib) that target specifically KRAS G12C in its GDP-bound state has evidenced an unprecedented success in the treatment of this subgroup of patients. Despite providing pre-clinical and clinical efficacy, several mechanisms of acquired resistance to KRAS G12C inhibitors have been reported. In this setting, combined therapeutic strategies including inhibition of either SHP2, SOS1 or downstream effectors of KRAS G12C seem particularly interesting to overcome acquired resistance. In this review, we will discuss the novel therapeutic strategies targeting KRAS G12C and promising approaches of combined therapy to overcome acquired resistance to KRAS G12C inhibitors.
RAS (Rat sarcoma viral oncogene homolog) encodes a membrane-bound protein, initially described in 1960s by Harvey (1) and Kirsten (2) as a retroviral oncogene involved in cell proliferation, differentiation and survival. RAS transforming properties were firstly reported in 1982 in human bladder cancer cell lines (3) and is considered the most frequently mutated oncogene in humans (i.e., 19% of cancer patients harboring a RAS mutation) (4). Notably, the KRAS (Kirsten rat sarcoma viral oncogene) isoform represents 75% of RAS mutant cancers (4).
In particular, the highest frequency of KRAS alterations is identified in pancreatic adenocarcinoma (88%), colon and rectal adenocarcinoma (50%), and in lung adenocarcinoma (32%) (4, 5). In pancreatic carcinoma, KRAS mutations are predominantly found in codon G12, followed by a smaller proportion of mutations in codons Q61 and G13. A similar distribution is observed in non-small cell lung cancer (NSCLC) patients. However, in the latter, the location of mutations beyond the Q61 codon within the KRAS gene is more heterogeneous than in pancreatic adenocarcinoma cases (6).
In non-small cell lung cancer (NSCLC), the clinical significance of KRAS mutated oncogene was firstly demonstrated in 1984 (7). Since this discovery, and based on the successful development of targeted therapies in other oncogenic-driven NSCLC, many attempts to target KRAS in NSCLC were made in past decades. Despite thorough pre-clinical and clinical research, these attempts failed, thus considering KRAS as an “undruggable” alteration. The recent discovery of sotorasib and adagrasib, which specifically target KRAS G12C, provides new therapeutic strategies to improve patient outcomes.
In this literature review, we focus on KRAS mutations in NSCLC and discuss the pre-clinical and clinical development of sotorasib and adagrasib. Herein, we outline the main mechanisms of acquired resistance described to KRAS G12C inhibitors to present and summarize therapeutic strategies to overcome resistance.
RAS encodes a membrane bound protein with a guanosine triphosphatase (GTPase), that is expressed in all mammalian cells (8, 9). RAS protein acts as a molecular switch, cycling between an active guanosine triphosphate (GTP)-bound state and an inactive guanosine diphosphate (GDP)-bound state (10, 11) (Figure 1). The cycling of RAS protein to its active form is promoted by guanine nucleotide exchange factors (GEF) while GTPase activating proteins (GAPs) contribute to maintain RAS in its inactive state through GTP hydrolysis (10, 11) (Figure 1). RAS cycling regulation needs the recruitment of either GEF or GAPs to the inner face of the cell membrane (8). GEF activation, which is necessary to activate RAS, is mostly related to signaling from either tyrosine kinase receptors or G-protein coupled receptors (10). Notably, EGFR (epidermal growth factor receptor) activation is known to induce wild-type RAS activation through the recruitment and the interaction of Grb2 complex (Growth factor receptor-bound protein 2) with SOS protein (Son of sevenless) (12, 13). In a lesser extent, upregulated gene expression or missense mutation might result in aberrant GEF activation (10), thereby promoting RAS activation.
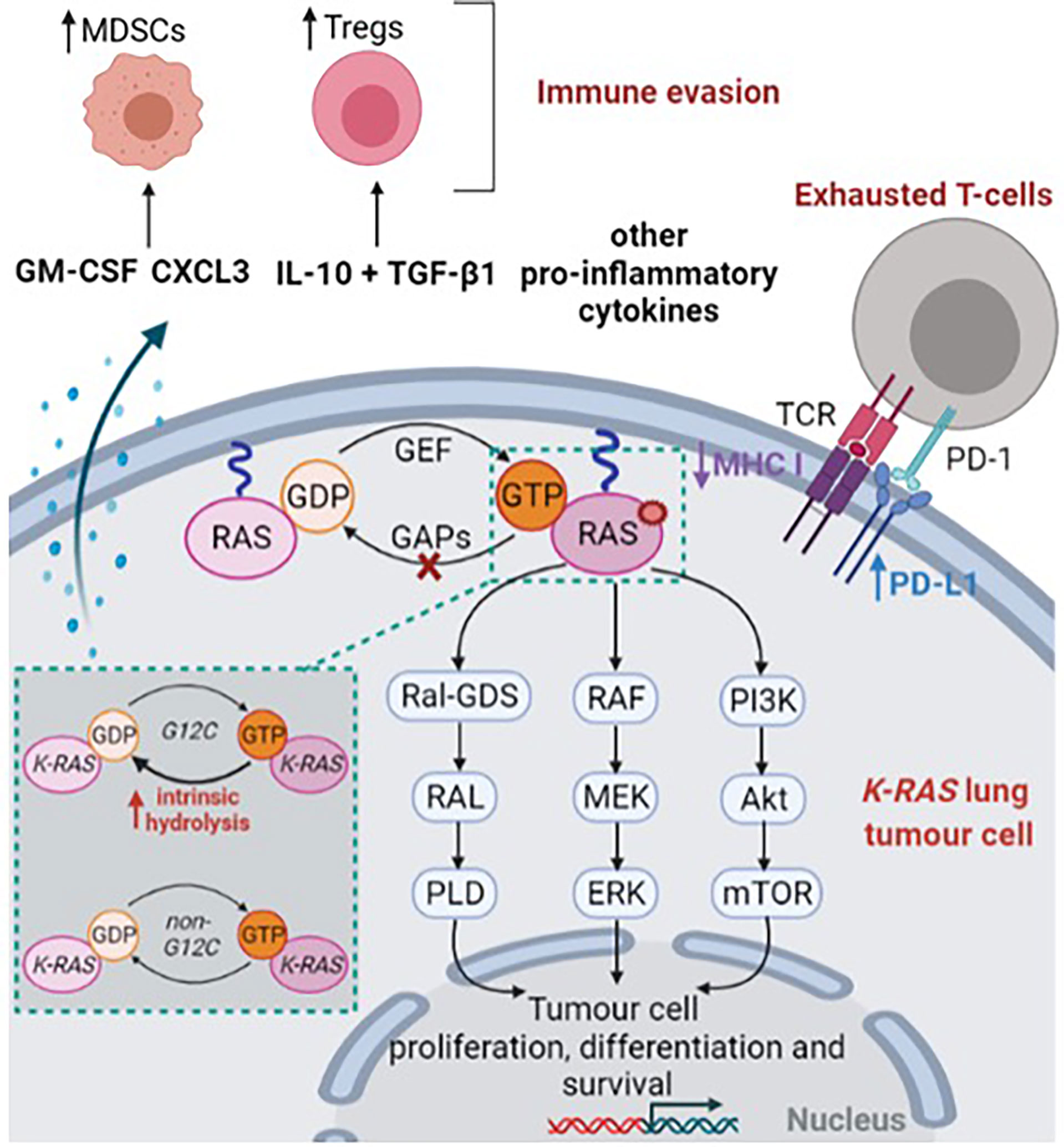
Figure 1 KRAS-mutant lung tumor cell. This figure was created with Biorender.com.
RAS protein is composed of three major domains: (1) the G-domain – a highly conserved domain between RAS isoforms – which contains switch-I (aa 30-38) and switch-II (aa 59-76) loops is responsible for GTP-GDP exchange; (2) the C-terminal domain known as the hypervariable region and associated with significant variations among RAS isoforms and (3) the C-terminal CaaX box responsible for post translational modifications (10, 14–16). Of note, the C-terminal CaaX box is involved in the farnesylation of the cysteine residue; a first-step that ensures RAS localisation in the inner face of the cell membrane (16).
As a result, the binding of GTP to RAS induces a conformational change in switch-I and switch-II loops which, in turn, activates RAS (14). Consequently, activated RAS promotes RAF recruitment (17–20) and PI3K activation (21, 22) contributing to cell proliferation, differentiation and survival (Figure 1). Moreover, Ral GDS pathway (Ral guanine nucleotide dissociation stimulator) was also found as a key RAS effector, involved in vesicle trafficking and cytoskeletal organisation (23, 24) (Figure 1). In the case of somatic activating RAS mutant, RAS presents a reduced ability to hydrolyse GTP or to interact with GAPs, thereby leading to a permanent and constitutive activation of RAS and downstream effectors which promotes tumorigenesis (8, 9, 25) (Figure 1).
KRAS is considered as a key oncogenic factor as it represents 75% of all RAS mutations (4). Indeed, preclinical studies on genetically engineered mouse models assessed that KRAS mutations predisposed to early onset lung cancer (26). The KRAS gene consists of 6 exons on chromosome 12 (8, 27). These point mutations mainly occur at exon 2 and exon 3 (8, 27). Indeed, 96% of KRAS mutations in NSCLC occur at either G12, G13 or Q61 codons (28).
KRAS mutations in NSCLC are often associated with tobacco history (i.e., 6.9% in never smokers vs. 32.3% and 36.9% in former and current smokers respectively) (29). As well, KRAS alterations are more frequent in adenocarcinoma (32%) than squamous cell carcinoma (4%) (4, 29, 30). Notably, the G12C mutation (i.e., mutation from amino acid glycine to cysteine) is the most frequent, as it constitutes 40% of all KRAS mutations in NSCLC (29). Nassar et al. recently outlined that KRAS G12C mutations are found in 13.8% of patients with a NSCLC (31). The G12V and G12D mutations represent 21% and 17% of KRAS mutations in NSCLC, respectively (29). Moreover, KRAS G12C mutations seem to be more frequent in women (i.e., 43.4%; p=0.007) and younger patients (i.e., median age 63.1 years old; p=0.0092) compared with other KRAS mutations (29, 30) and KRAS wild-type (32). Likewise, it was reported that KRAS G12C mutations were more frequent in black and white patients than Asian patients (p<0.001) (31), and are more frequent in current or former smokers (i.e., 41%), while G12D mutations mostly occur in non-smokers (i.e., 56%) (29). As well, KRAS G12C-positive patients present a higher rate of metastasis at diagnosis compared with KRAS wild-type (i.e., 94.4% vs. 88.4%) (32). Despite some reports highlighting higher frequency of brain metastasis in KRAS mutated NSCLC patients (33, 34), KRAS mutations are not considered to specifically drive brain metastasis as is the case for ALK rearrangements.
Notably, co-occurring alterations in non-oncogenic drivers of NSCLC are reported in about one-half of patients harboring KRAS mutations (35). Co-occurring mutations in TP53 (about 40%), STK11 (i.e., serine/threonine kinase 11; 19.8% to 28%) and KEAP1 (i.e., kelch like ECH associated protein 1; 13% to 24%) are the most frequently reported (35–37).
KRAS mutations do not usually coexist in the context of EGFR-, ALK-, or ROS- driven NSCLC. Schmid et al. (38), reported nine cases of metastatic NSCLC which harbor ALK/KRAS co-alterations, a fraction of which (86%) were primary refractory to crizotinib (ALK inhibitor). Although EGFR and KRAS mutations typically occur in a mutually exclusive fashion in lung cancer (39, 40); anecdotal reports show evidence of co-occurrence of KRAS and EGFR mutations in lung cancer patients (41–43).
KRAS G12C and other KRAS mutations more frequently co-occurred with MET amplification in both localised (30) and metastatic (35, 36), treatment-naïve NSCLC. KRAS G12C-positive patients were also found to significantly harbor increased frequency of ERBB2 amplification (p=0.002) or ERBB4 mutations (p=0.025) compared with non- KRAS G12C patients (35).
Besides promoting tumorigenesis through downstream effectors, mutant KRAS cells have been found to interact with the tumor microenvironment (Figure 1). Firstly, KRAS mutations downregulate the expression of major histocompatibility class I molecules in colorectal carcinoma, thereby decreasing priming and cross-presentation to T-cells (44, 45). Similarly, KRAS mutations are associated with higher PD-L1 (programmed cell death ligand 1) expression in NSCLC, thus contributing to exhausted T-cells (46, 47). Notably, the proportion of PD-L1 TPS (i.e., tumor proportion score) ≥50% was reported in a range of 34 (32) to 41% (48) for KRAS G12C patients compared with 20.4% (32) to 25.9% (48) among KRAS wild-type patients. In vitro, downstream effectors of KRAS-mutated NSCLC cell lines, including MAPK and STAT3 signaling pathways were found to promote ectopic PD-L1 expression (49, 50).
As another assessment of promoting immunosuppressive microenvironment, analyses in an in vitro model of KRAS G12V induction highlighted that KRAS-mutant cells enhance the secretion of TGF-β1 and IL-10, thus inducing regulatory T cells (Tregs) (51). Such results were also further confirmed in mouse models with lung tumors (51). Several reports also outlined that pro-inflammatory cytokines such as IL-6, IL-17, IL-8 and IL-22, are highly secreted by KRAS-mutant cells (52–55). Likewise, KRAS mutant cells were found to markedly secrete granulocyte macrophage colony-stimulating factor (GM-CSF) which contributes to the recruitment and activation of myeloid-derived suppressor cells (MDSCs) (53, 56). The recruitment of MDSCs was also found to be promoted by repression of interferon regulatory factor 2 in KRAS colorectal carcinoma, which in turn increased the expression of CXCL3, a chemokine which binds to CXCR2 receptor expressed on MDSCs (57).
Despite some conflicting results (32, 58, 59), KRAS mutations in NSCLC seem to be associated with worse prognosis (60–63). Notably, in a large meta-analysis of 43 observational studies including stage IIIB-IV NSCLC patients treated with either chemotherapy combined with or without bevacizumab and EGFR TKIs, Goulding et al found that KRAS mutations are associated with a significant shorter overall survival (HR=1.71; 95%CI [1.07-2.84]) and progression-free survival (HR=1.18; 95%CI [1.02-1.36]) (61). The objective response rate (RR) was also significantly lower for patients harboring KRAS mutations (RR=0.38; 95%CI [0.16-0.63]) (61). KRAS mutations were also identified as an independent worse prognostic factor in stage I disease (64). On the contrary, in a large cohort of 1039 advanced or metastatic NSCLC patients treated with chemotherapy alone or in combination with immune checkpoint inhibitor, no significant difference was observed in terms of PFS and OS between KRAS wild-type, KRAS G12C and non-KRAS G12C patients (32).
The KRAS mutation sub-type as well as co-occurring mutations seem important to take into account as they might impact prognosis and response to treatment. Indeed, a better disease-free survival for patients with stage I lung adenocarcinoma harboring G12V/G12C mutations (p=0.0271) was reported, compared with other KRAS mutations (64). Conversely, Finn et al. recently reported among a cohort of 1012 patients with a stage I-III lung adenocarcinoma that KRAS G12C mutations are associated with a significant shorter OS and relapse-free survival, compared with other KRAS mutations and wild-type KRAS (30). On the contrary, no significant difference was reported in terms of OS and PFS between KRAS G12C and non- KRAS G12C patients treated with chemotherapy alone or in combination with checkpoint inhibitor as first-line treatment for advanced or metastatic NSCLC (32). Notably, patients with both KRAS and either STK11 (35, 65) or KEAP1 (37) co-mutations were reported to have shorter survival.
Despite thorough pre-clinical and clinical research, KRAS was considered as an elusive target for a long time. Indeed, the picomolar affinity of RAS for GTP and the smooth surface of KRAS with a lack of well-defined hydrophobic pocket prevented either the development of GTP inhibitors or the development of KRAS inhibitors (66). Likewise, inhibitors of KRAS membrane positioning such as farnesyl transferase inhibitors (67, 68) and inhibitors of RAS membrane binding (69) failed to demonstrate clinical efficacy due to adaptative KRAS prenylation by geranylgeranyl transferase (70). In the same way, strategies to inhibit RAS downstream effectors or synthetic lethality approaches failed to show clinical efficacy (68, 71–74) due to either feedback activation of RAS upstream effectors or tumor heterogeneity regarding KRAS synergistic oncogenic dependencies (75).
In 2013, in the seminal work of Ostrem and colleagues, X-ray crystallographic studies revealed a new allosteric pocket beneath the effector switch-II region (76). This specific allosteric pocket was found to be located between the central β-sheet of RAS, and the α-2 (switch-II) and α-3 helices. Notably, switch-II binding pocket was only visible in RAS GDP-bound state (76). In line with these observations, a tethering approach (i.e., a fragment-based drug discovery approach) was used to screen compounds against KRAS G12C in the GDP-state (76).
The identification of early-stage compounds evidenced that direct targeting of KRAS G12C relies on covalent binding to cysteine 12 and switch-II binding pocket region when KRAS G12C is in its inactive GDP-state (76). Consistently with these findings, pre-loading of GTP to KRAS impairs binding of these early-stage compounds to KRAS G12C (76). Of note, these early-stage compounds were shown to decrease cell viability and to induce cell apoptosis in KRAS G12C lung cancer cell lines (76). Overall, these experiments provided new strategies to target KRAS G12C, while sparing wild-type KRAS or other KRAS mutants, based on covalent binding to mutant cysteine 12 residue and switch-II binding pocket.
Based on this promising proof-of-concept, subsequent KRAS G12C inhibitors with higher specificity were developed through structure-based optimization, such as ARS-853 (77, 78) and ARS-1620 (79). Janes et al. demonstrated that ARS-1620 significantly inhibits tumor growth in MIA-PaCa2 pancreatic cancer xenograft models (79).
Subsequent pre-clinical studies allowed to precise the mechanism of action of KRAS G12C inhibitors (77–79). Notably, these studies showed that KRAS G12C inhibitors trap KRAS in its inactive GDP-bound state by reducing its susceptibility to nucleotide exchange factors (77, 78), and that efficacy of KRAS G12C inhibitors requires intact GTPase activity (77, 78). Indeed, in engineered H358 cells (KRAS G12C) that express the A59G mutation – which abrogates RAS GTPase activity – ARS-853 failed to decrease KRAS GTP levels and ERK phosphorylation (77). As well, RAS mutations that increase nucleotide exchange activity (i.e., Y40A, N116H and A146V) reduced susceptibility to KRAS G12C inhibitors in vitro (77). On the contrary, KRAS G12C inhibition was enhanced by SOS1 inhibition (77). Consistently with these observations, Lito and Patricelli highlighted that KRAS G12C inhibition is dependent on the activity of upstream tyrosine kinase receptors (77, 78). Indeed, when cells are treated with EGF (Epidermal Growth Factor), in vitro activity of ARS-853 decreases (77, 78) while EGFR inhibition with either erlotinib, afatinib (78) or gefitinib (77) enhanced the potency of KRAS G12C inhibitors. Finally, it has been reported that KRAS G12C has a high intrinsic hydrolysis rate, compared to other KRAS mutations (80) (Figure 1). This intrinsic property might explain that KRAS G12C undergoes sufficient hydrolysis to enable inhibition by GDP-state selective drugs (72).
More recently, AMG-510 (81) and MRTX849 (82) were reported to have increased KRAS G12C inhibition activity over previous inhibitors, through enhanced interaction with the H95 residue in the α-3 helix of KRAS G12C. These improvements in structure-based design and biopharmaceutical optimization led to the initiation of the first-in-human trial of AMG-510 in 2018 (ClinicalTrials.gov NCT03600883).
In pre-clinical testing, sotorasib was found to impair cell viability in pancreatic and lung adenocarcinoma cell lines, in monolayer and spheroid models, with a high potency and high selectivity, as it had no impact on cell viability of non-KRAS G12C cell lines (Table 1) (83). Sotorasib inhibits ERK phosphorylation in multiple KRAS G12C-mutant in vitro and in vivo models (i.e., xenograft and syngeneic mouse models and patient-derived xenografts) (Table 1) (83). Besides its impact on tumor cell signaling, sotorasib was also found to restore an efficient immune tumor response (84, 86) as evidenced by increased T-cell infiltration and immune effectors including macrophages, CD103+ dendritic cells and CD4+/CD8+ T-cells (Table 1) (83).
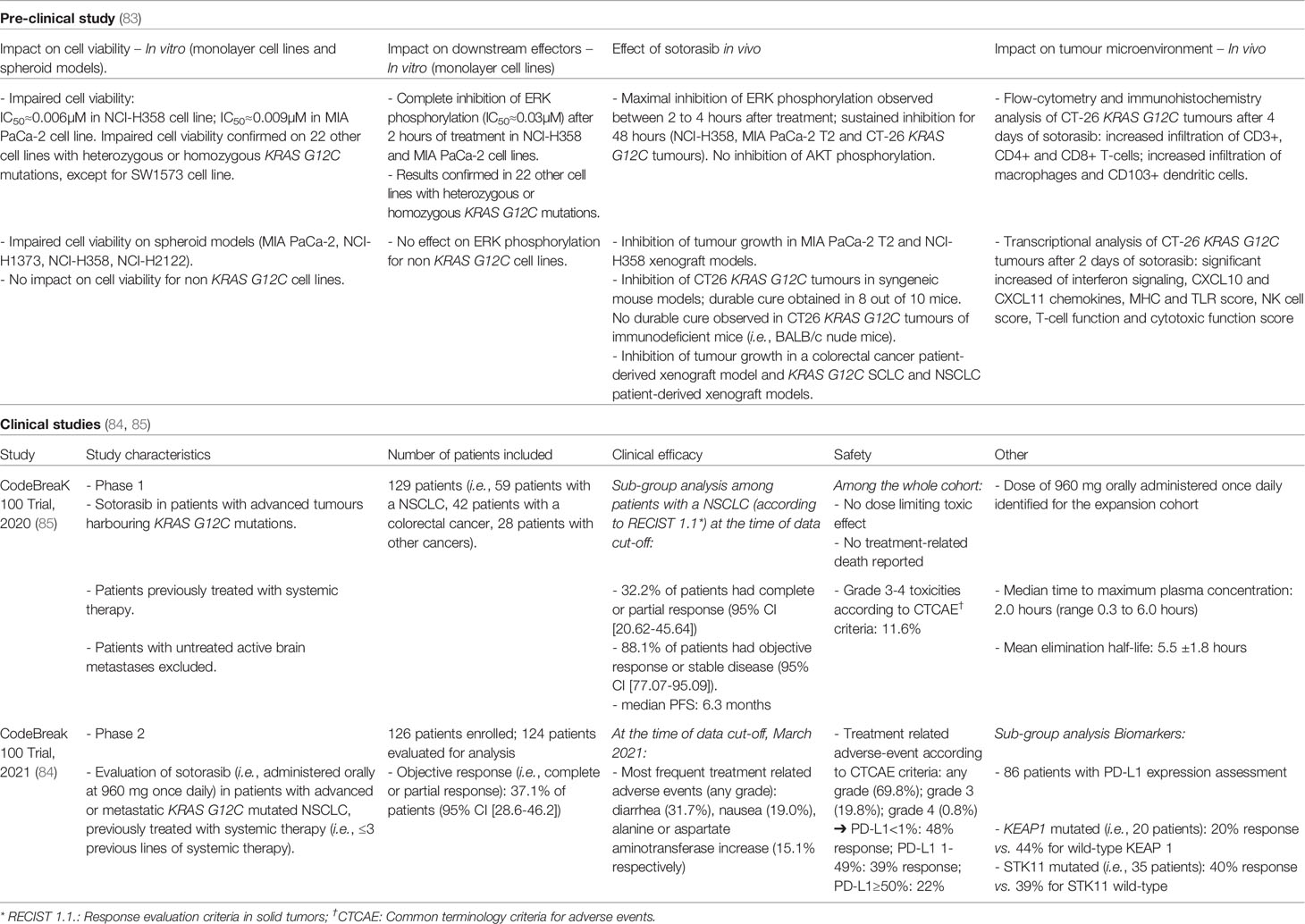
Table 1 Sotorasib: synthesis of pre-clinical and clinical development.
Sotorasib, formerly AMG-510, was the first direct KRAS G12C inhibitor to enter clinical development (85), in a phase I/II first-in-human clinical trial (NSCLC cases in phase I=59 patients enrolled; NSCLC cases in phase II=126 patients enrolled; Table 1) (84, 85). In the phase II trial, sotorasib induced an objective response rate of 37.1% and a disease control rate of 80.6% (84). Among the 124 patients eligible for evaluation, 4 patients presented a complete response (84). Recently, Ramalingam et al. reported the clinical efficacy of sotorasib among patients with stable brain metastasis (n=40) included in the phase I/II CODEBREAK 100 clinical trial. In this sub-group analysis, sotorasib demonstrated clinical efficacy with a median PFS of 5.3 months and a median OS of 8.3 months (87). Sotorasib has a tolerable safety profile: 19.8% of patients experienced grade 3 while 0.8% experienced grade 4 treatment-related adverse events (84). The most frequent treatment related adverse events reported were diarrhea (31.7%), nausea (19%), increase in either alanine aminotransferase (15.1%) or aspartate aminotransferase (15.1%) levels and asthenia (11.1%) (84). In particular, as suggested in a recent report (88), sotorasib might trigger immune-related hepatitis in patients previously treated with immune checkpoint inhibitors. Sub-group analysis of the phase II cohort suggested that sotorasib either maintains or improves quality of life, physical functioning and the severity of key lung cancer-related symptoms, including cough, chest pain and dyspnea (89).
Despite small sub-group sample size, sotorasib demonstrated efficacy across a range of co-occurring mutations including STK11 and TP53, whereas a lower percentage of response was observed among patients harboring a KEAP1 co-mutation (84). Finally, although current limited data available, no predictive biomarkers of response to sotorasib have been yet identified (84, 86). Exploratory analysis of the CODEBREAK 100 trial showed that KRAS G12C patients who harbor a KEAP1 mutation are likely early-progressors (i.e., patients with an event of progressive disease and PFS<3 months) whereas patients with alterations in effectors of cell cycle, WNT pathway and MAPK pathway are more prevalent in the late-progressor group (i.e., patients with an event of progressive disease and PFS≥3 months) (87). Zhao et al. reported among 43 patients treated with sotorasib - including 36 NSCLC patients - that exceptional responders (i.e., defined as a complete response or a partial response lasting more than 12 months) tend to have a lower plasma KRAS G12C allele frequency and a lower tumor burden at baseline compared to other patients (90). Based on the clinical efficacy and safety profile, sotorasib was recently approved by FDA for the treatment of advanced KRAS G12C NSCLC patients following at least one prior systemic therapy.
Adagrasib, formerly MRTX849, is the second irreversible and selective KRAS G12C inhibitor to have entered clinical trials.
Adagrasib was optimized to exhibit a long half-life (≈24 hours) and extensive tissue distribution (91, 92). Similar to sotorasib, adagrasib impairs cell viability of pancreatic and lung adenocarcinoma monolayer and spheroid models harboring KRAS G12C (Table 2) (93). Consistently with pre-clinical findings on sotorasib, adagrasib inhibits ERK phosphorylation while it has no impact on AKT activation (Table 2) (93). Pre-clinical studies revealed that KEAP1 loss might constitute a potential intrinsic resistance to adagrasib as sgRNA targeting KEAP1 were enriched following adagrasib treatment in xenograft models (Table 2) (93). In line with the observations reported for sotorasib, Briere et al. recently outlined, in KRAS G12C syngeneic and genetically engineered mouse models, adagrasib decreases intra-tumor MDSCs while it increases M1-macrophages, dendritic cells and CD4+/CD8+ T-cells (94). Interestingly, the in vivo efficacy of adagrasib was markedly decreased in T-cell deficient nu/nu mice (94).
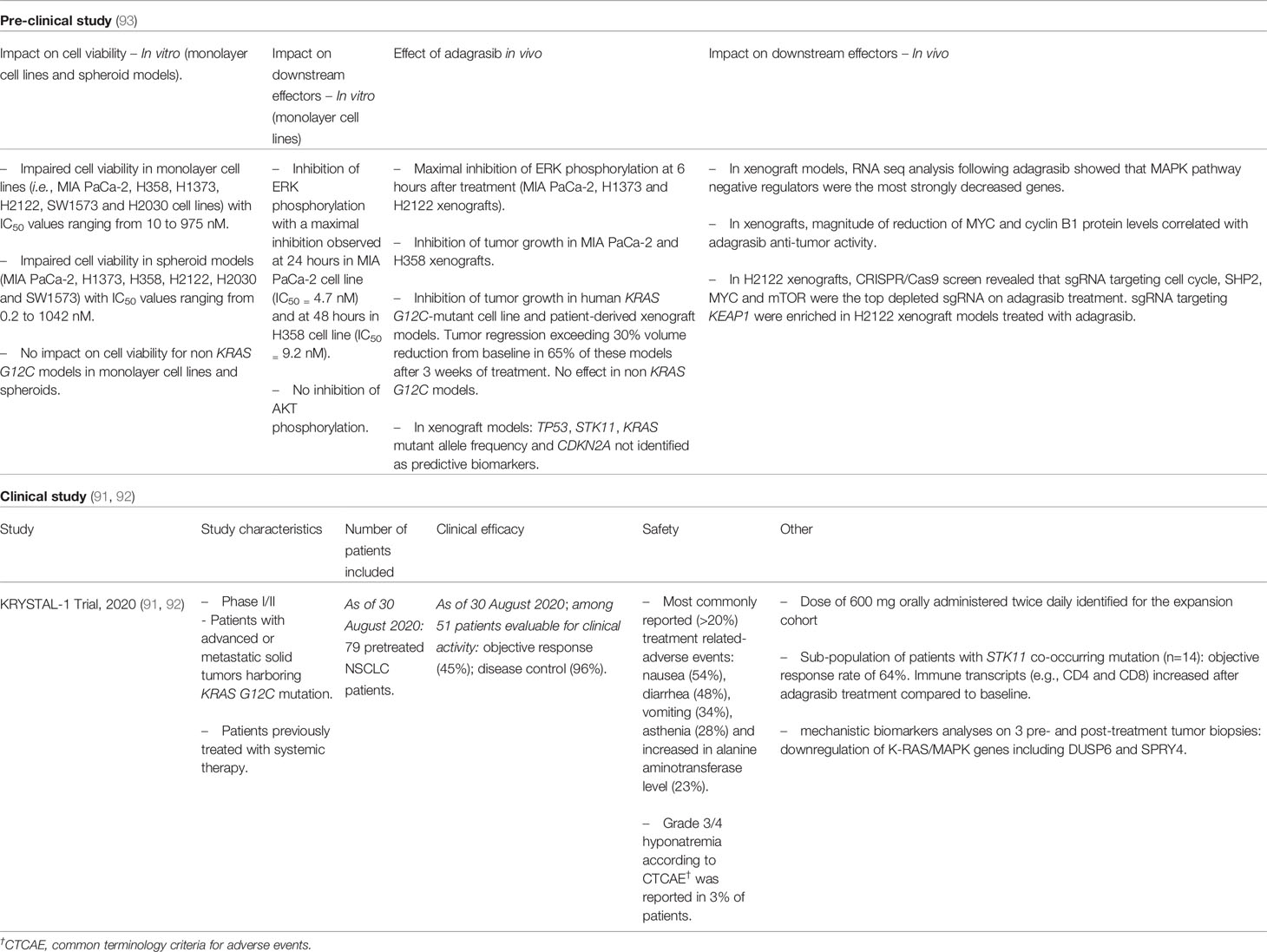
Table 2 Adagrasib: Synthesis of pre-clinical and clinical development.
Adagrasib is currently being tested in the KRYSTAL-1 multi-cohort phase I/II study that enrolled patients with advanced or metastatic solid tumors harboring a KRAS G12C mutation (NCT03785249) (91, 92). A recent preliminary report on the clinical efficacy of adagrasib confirmed an objective response rate and a disease control in respectively 45% and 96% of evaluable patients in the NSCLC cohort (NSCLC patients: n=79; Table 2) (91). Similar to sotorasib, nausea, diarrhea, vomiting, asthenia and increase in alanine aminotransferase level were the most frequent treatment related-adverse events reported (Table 2) (91). Despite small sub-group sample size, adagrasib treatment efficacy is reported regardless of the presence of concurrent mutations in STK11 (92).
The objective response rates obtained for either sotorasib or adagrasib is markedly lower compared to those obtained with osimertinib or alectinib in EGFR and ALK-driven NSCLC respectively (95, 96), suggesting the presence of intrinsic mechanisms of resistance to KRAS G12C inhibitors.
Previous efforts to target the RAS–RAF–MEK pathway have been challenged by early adaptive feedback reactivation of signaling pathways, leading to therapeutic resistance. In KRAS-mutant cancers, trametinib (MEK inhibitor) provokes a compensatory response involving the fibroblast growth factor receptor 1 (FGFR1), resulting in signaling rebound and adaptive drug resistance (97, 98). In KRAS G12C-mutant models, treatment with sotorasib leads to rapid RAS pathway reactivation, followed by a significant rebound of ERK phosphorylation (average of 75% vs baseline levels) (99). In this setting, RAS-MAPK feedback reactivation was driven by intrinsic activation of various receptor tyrosine kinases (RTKs) including EGFR, HER2, FGFR and c-MET, that resulted in stimulation of wild-type RAS (NRAS and HRAS), which is not inhibited by G12C-specific inhibitors but can be counteracted by the combination of KRAS G12C and SHP2 inhibitors (99).
Interestingly, shortly after treatment, some cancer cells are sequestered in a quiescent state with low KRAS activity. The new KRAS G12C is maintained in its active, drug-insensitive state by epidermal growth factor receptor (EGFR) and aurora kinase (AURKA) signaling. In this setting, the synthesis of new KRAS G12C and its trading between the active or inactive states, modulates the divergent early response to KRAS G12C inhibition and allow cells to resume proliferation.
Initial response to KRAS G12C inhibition is also associated with induction of EGFR phosphorylation, following sotorasib treatment in NCI-H358 cells (83), or a marked recovery of ERK phosphorylation in adagrasib-partially sensitive H358 and H2122 cells (93). Furthermore, IGF2 (i.e., insulin-like growth factor 2) mRNA levels were significantly higher in BEAS2BKRAS G12C cells treated with sotorasib, compared to control (100). Similarly, AURKA (Aurora Kinase A) – a cell-cycle regulator – was found to promote early adaptative resistance to ARS-1620 in vitro and in H358-xenograft models (101).
In this section, we outline the mechanisms of acquired resistance, to either sotorasib or adagrasib, described in vitro, in vivo or in the clinical setting. The mechanisms of resistance to KRAS G12C targeted therapies can be classified into: (1) on-target mechanisms; (2) by-pass alterations resulting in abnormal activation of downstream or connecting signaling pathways; and (3) phenotypic transformation (102) (Figure 2).
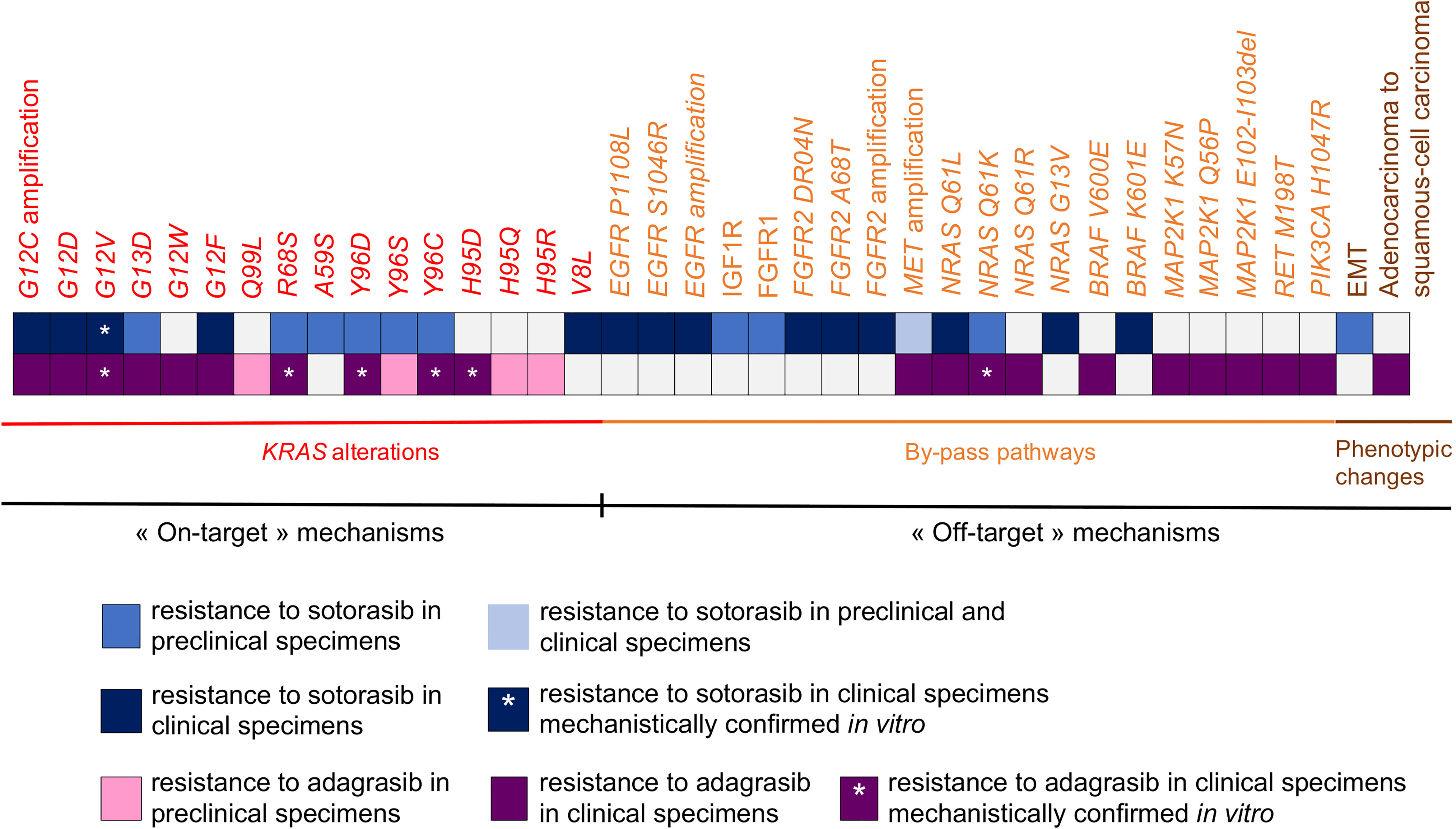
Figure 2 Synthesis of biological mechanisms of acquired resistance to sotorasib and adagrasib described in pre-clinical specimens and clinical specimens.
Secondary mutations in KRAS that confer acquired resistance to KRAS G12C inhibitors are currently described in vitro and in clinical setting (103–105). Similar to the well-known T790M gatekeeper mutation in EGFR-oncogenic driven NSCLC (106), these mutations occur in switch-II binding pocket and thereby alter drug binding (107). Similar to other oncogenic-driven NSCLC (106), acquired KRAS-activating mutations and KRAS amplification mediate acquired resistance through RAS signaling pathway activation (107).
Koga et al. modelled the acquisition of mutations associated with sotorasib or adagrasib resistance, using Ba/F3 cells treated with ENU (i.e., N-ethyl-N-nitrosourea) (103). Secondary KRAS mutations were identified in 87.3% of the 142 resistant clones generated. In sotorasib resistant clones, KRAS G13D was the most frequent secondary mutation (i.e., 23% of KRAS secondary mutations) observed, followed by R68M and A59S (21,2% for each, respectively). In adagrasib-resistant clones, KRAS Q99L was the most frequent secondary mutation (i.e., 52.8% of KRAS secondary mutations), followed by Y96D and R68S (i.e., 15,3% and 13.9% respectively) (103). Of note, although Y96D and A59S were the only mutations shared between sotorasib- and adagrasib-resistant clones, only Y96D mechanistically proved to confer strong cross-resistance to both KRAS G12C inhibitors (103). Cross-resistance of Y96D, but also of Y96S, were also assessed in H358 cells (NSCLC, KRAS G12C) (103). Upon adagrasib treatment, the KRAS Y96D mutation was associated with sustained ERK and AKT activation in several in vitro models including MIA Pa-Ca2 (pancreatic cancer), KRAS G12C/Y96D NSCLC cells and the MGH1138-1 patient-derived model (104). Moreover, a higher level of active GTP-bound KRAS was observed in KRAS G12C/Y96D compared to KRAS G12C despite treatment with adagrasib (104). Secondary mutations that alter drug binding are reported in patients who progressed on adagrasib and were exogenously expressed as double-mutant alleles in Ba/F3 cell lines, to model their mechanistic impact on KRAS G12C-inhibitor sensitivity (105). Notably, switch-II binding pocket mutations, including R68S, H95D/Q/R and Y96C, conferred resistance to adagrasib in Ba/F3 cells compared to the control KRAS G12C allele (105). Consistently with Koga and colleagues, the Y96C mutation caused cross-resistance to both adagrasib and sotorasib in vitro (105).
These mutations were anticipated to structurally alter drug binding (103–105). Indeed, the crystal structure of KRAS G12C binding with sotorasib or adagrasib showed that the residues G13, A59, Q61, R68, Y96 and Q99 face the drug binding pocket (103). In line with these observations, analysis of R68S, H95D/Q/R and Y96C mutant residues were found to disrupt non-covalent binding interactions between KRAS G12C and adagrasib (105). Notably, Y96D was found to abolish the normal direct hydrogen bond with the pyrimidine ring of adagrasib, while it disrupted the water-mediated hydrogen bond between Y96 and the carboxyl-group on sotorasib (104). Y96D also introduced a negatively charged amino-acid which contributes to change the binding pocket towards a substantially more hydrophilic pocket; thereby reducing drug binding (104). Besides disrupting drug binding, secondary mutations such as A59 or Q61 might alter GTPase activity which is normally required for efficient KRAS G12C inhibition (103). Similarly, mutations occurring at codons 13, 59, 61, 117 and 146, that impede GTP hydrolysis or increase GDP-to-GTP nucleotide exchange (80) were also found to promote resistance in vitro to MRTX1257 (i.e., a compound highly related to adagrasib) and sotorasib (103, 105).
These in vitro experiments highlight that several acquired mutations have different sensitivities to KRAS G12C inhibitors. Indeed, G13D and A59S secondary mutations remained partially sensitive to adagrasib while they conferred strong resistance to sotorasib (103). Likewise, Q99L (103) and H95D/Q/R (105) secondary mutations which conferred resistance to adagrasib, remained sensitive to sotorasib. Based on a positive-selection screen for mutations that confer resistance to either sotorasib or MRTX1257, in Ba/F3 cells, Awad et al. identified that mutations occurring at codons 12, 68, 95 and 96 conferred strong resistance to MRTX1257; while mutations occurring at codons 8, 9, 12, 96 and 117 were associated with a strong resistance to sotorasib (105).
Overall, these observations suggest drug-specific binding mechanisms of resistance and indicate for rationales of sequential therapeutic strategies between sotorasib or adagrasib to counteract acquired resistance.
Interestingly, recent reports in the clinical setting support the in vitro findings highlighting that secondary KRAS mutations within drug-binding pocket and activating mutations in KRAS mediate acquired resistance to sotorasib and adagrasib. Tanaka et al. recently reported the case of a 67 year-old woman treated with adagrasib, in the KRYSTAL-1 dose expansion cohort, who developed heterogeneous mechanisms of acquired resistance including KRAS G12C/Y96D, G13D and G12V (104).
In a recent report, genomic and histologic analyses of pre-treatment samples compared with those obtained at progressive disease on adagrasib were conducted among 38 patients enrolled in phase I KRYSTAL-1 trial (i.e., 27 patients with NSCLC, 10 with colorectal cancer and 1 with appendiceal cancer) (105). This analysis revealed that, among patients with putative mechanisms of resistance to adagrasib, 53% had at least one acquired KRAS mutation or KRAS amplification (105). Consistently with in vitro observations, acquired mutations within switch-II binding pocket – Y96C, R68S, H95D – were reported among NSCLC patients. KRAS G12D and G12V activating mutations were reported in 2 NSCLC patients (105). Additional mutation in the G12 codon as well as cis-mutations were also described as potential mechanisms of acquired resistance to adagrasib (104, 105). Indeed, Awad et al. reported an additional mutation in G12, leading to a mutation from cysteine to tryptophane (i.e., G12W mutation) (105). Then, based on the observation of concurrent G12C and G12V in cis on the same strand (i.e., G12F), Tanaka et al. hypothesized that cis mutations resulting in G12C loss and conversion into another KRAS mutation might constitute an additional mechanism of acquired resistance (104). Finally, high-level of KRAS G12C amplification was reported at relapse on adagrasib in one NSCLC patient (105).
As for adagrasib, genomic and histologic analysis of pre-treatment samples compared with those obtained at progressive disease on sotorasib were recently reported among 32 NSCLC patients enrolled in CODEBREAK 100 and CODEBREAK 101 clinical trials (90). Among these, putative mechanisms of acquired resistance were identified in 78% cases. In particular, secondary KRAS mutations were identified in 4 patients (i.e., G12D, G12V, G12F, V8L) while 3 patients presented KRAS amplification (90). The impact of G12V secondary mutation was mechanistically confirmed in KRAS G12V dox-induced H358 cells as it decreased the antiproliferative effect of either sotorasib or adagrasib (90). Finally, loss of KRAS G12C dependency alone or concomitant to secondary KRAS mutations was also found to confer resistance to either adagrasib (105) or sotorasib (90).
Activation of by-pass signaling pathways constitutes another main mechanism of acquired resistance described in oncogenic-driven NSCLC (106), to ensure sustained signaling activation despite therapeutic pressure.
Based on a phospho-RTK array of KRAS G12C NSCLC H23-sensitive and H23-resistant clones to sotorasib, Suzuki et al. showed that MET and HGF (i.e., hepatocyte growth factor) were significantly upregulated in sotorasib-resistant cells (108). Notably, H23 sotorasib-resistant cells had an abnormal MET/CEP7 ratio (i.e., 2.7), indicating that MET amplification promoted acquired resistance in these cells. No acquired KRAS mutations were identified (108). Functional experiments show that MET knockdown restored sensitivity to sotorasib in vitro and reverted sustained ERK phosphorylation (108). RTK activation has been reported with acquisition of epithelial-to-mesenchymal (EMT) features in sotorasib-resistant cells (109). Indeed, in resistant H358 cells that display EMT features, IGF1R (i.e., Insuline-like Growth Factor Receptor 1) activates AKT pathway while FGFR1 (i.e., Fibroblast Growth Factor Receptor 1) promotes ERK rebound activation (109). In line with these observations, IGF1R and FGFR1 were found to mediate acquired resistance in H358 and LU65-sotorasib resistant cells harboring EMT features (109).
In their report, Tanaka et al. showed that several mechanisms of acquired resistance to adagrasib might co-occur in the clinical setting, thereby highlighting the intra-tumoral heterogeneity of resistant tumors. Indeed, cfDNA sequencing and droplet digital PCR (ddPCR) in plasma samples collected at disease progression on adagrasib revealed up to 10 distinct mutations affecting RAS-MAPK pathway effectors (104). In particular, these mutations include activating mutations in NRAS (i.e., Q61L, Q61K and Q61R), BRAF V600E and MAP2K1 (i.e., K57N, Q56P and E102-I103del) (104). The impact of NRAS Q61K secondary mutation on adagrasib sensitivity was mechanistically confirmed in H358-engineered cells and was also found to confer resistance to sotorasib in vitro (90). Moreover, alterations in RET M198T and PIK3CA as well as MET amplification were also detected in 5 NSCLC patients with progressive disease on adagrasib (105). Similar to adagrasib, mutations affecting RAS-MAPK pathway effectors were also identified at acquired resistance to sotorasib (90). In clinical setting, these mutations included NRAS G13V, non V600E- BRAF (i.e., K601E), EGFR (i.e., S1064R and P1108L) and FGFR2 (i.e., A68T and D304N) activating mutations (90). MET and EGFR amplifications were also detected at progressive disease on sotorasib and co-occurred in one patient. Likewise, FGFR2 amplification was detected in one patient (90).
Histologic transformation to either small-cell lung cancer or squamous-cell carcinoma is a well-known biological mechanism of acquired resistance to targeted therapies in NSCLC (110, 111). While there is no current evidence of transdifferentiation to small-cell lung cancer in the context of acquired resistance to KRAS G12C-targeted therapies, Awad et al. reported two cases of phenotypic changes to squamous-cell carcinoma at disease progression on adagrasib (105). Of note, initial KRAS G12C mutation was detected at resistance while no other KRAS acquired mutations occurred in these patients (105). Interestingly, STK11 deletion seems to drive lineage transformation to squamous-cell carcinoma in KRAS mutated lung adenocarcinoma (112).
Epithelial-to-mesenchymal transition represents another non-genomic transcriptional reprogramming that mediates acquired resistance to targeted therapies (111). In the setting of KRAS G12C-mutant NSCLC, the acquisition of EMT features in vitro and in xenograft models was reported to mediate acquired resistance to sotorasib (109). H358 and LU65 cells generated resistant to sotorasib presented features of EMT including E-cadherin downregulation and vimentin upregulation (109). Of note, gene set enrichment analysis of H358 sotorasib-resistant cells showed an enrichment of the EMT transcriptomic signature, compared to their sensitive parental counterparts (109). Likewise, H358 sotorasib-resistant xenograft models presented an induction of vimentin. In both resistant models to sotorasib, no acquired KRAS mutations were detected (109).
Overall, despite the recent development of sotorasib and adagrasib, biological mechanisms of acquired resistance have yet been described in both pre-clinical and clinical specimens (Figure 2).
These findings highlight the diversity of KRAS mutations that emerge in response to KRAS G12C inhibitors, thereby limiting the potential development of efficient next-generation KRAS G12C inhibitors (103–105). Some of these mutations display differential sensitivity to either sotorasib or adagrasib, thus providing a therapeutic rationale at progressive disease (103, 105). In the clinical setting, putative mechanisms of acquired resistance to adagrasib were described in 45% cases, suggesting that other mechanisms might be implicated in resistance to KRAS G12C inhibitors (105). Finally, pre-clinical and clinical observations provide rationale for combined therapy to prevent or delay acquired resistance, since 41% patients were found to have more than one concurrent mechanism of acquired resistance to adagrasib (105). As for adagrasib, concurrent treatment associated alterations at progressive disease on sotorasib were found in 60% cases of patients that present putative mechanisms of acquired resistance (90).
As highlighted in the seminal work of Canon et al., sotorasib might improve therapeutic efficacy of targeted agents including inhibitors of upstream and downstream effectors of KRAS G12C (83). Analysis of synergy scores of sotorasib associated with different targeted therapies provided proof-of concept to enhance therapeutic efficacy to revert or overcome acquired resistance (83). In H358 cells, the highest synergy scores were observed for HER kinases inhibitor (i.e., afatinib), Src Homology Phosphatase 2 inhibitor (i.e., RMC-4550) and MEK inhibitor (i.e., trametinib); the latter achieving the highest synergy score in an NCI-H1373 spheroid model (83). Consistently, a significant reduction in tumor volume was observed in H358 xenograft models treated with sotorasib plus MEK inhibitor, compared to sotorasib or MEK inhibitor monotherapy (83). Interestingly, sotorasib also enhanced therapeutic efficacy of carboplatin in vivo (83).
In line with these observations, adagrasib was found to have enhanced efficacy when associated with other targeted agents (93). Combined therapy of adagrasib with either afatinib or RMC-4550 (SHP2 inhibitor) induced a significant greater anti-tumor efficacy compared to single-agent monotherapy, in xenograft models of NSCLC and oesophageal squamous cell carcinoma (93). Similarly, targeting downstream KRAS effectors such as mTOR and members of the cyclin D family might constitute an efficient approach to enhanced adagrasib efficacy in vivo (93).
In this section, we will focus on reported therapeutic strategies to overcome acquired resistance to either sotorasib or adagrasib.
As already outlined, KRAS G12C/Y96D mutations were found to confer cross resistance to sotorasib and adagrasib, through altered drug binding (103, 104). In this setting, Tanaka and colleagues sought to determine whether structurally and functionally different KRAS G12C inhibitors could target the KRAS Y96D mutation (104). RM-018 is a novel KRAS G12C inhibitor that exploits cyclophilin A to bind and inhibit KRAS G12C in its GTP-bound state (113, 114). In vitro, RM-018 was found to impair cell viability and to markedly decrease RAS-MAPK signaling in cell lines harboring KRAS G12C (113, 114). In vivo, administration of RM-018 in H358 KRAS G12C-NSCLC xenograft models resulted in dose-dependent tumor regression and was well tolerated (113, 114). Moreover, in the context of acquired resistance, RM-018 achieved in vitro efficacy (104). Indeed, no IC50 shift was observed upon RM-018 therapeutic pressure in several models including MIA PaCa-2, H358, Ba/F3 cells and the MGH1138-1 patient-derived model, harboring KRAS G12C/Y96D compared to KRAS G12C parental cells (104). In line with these observations, RM-018 also inhibited ERK activation with a high potency in vitro (104). Taken together, these pre-clinical data provide proof-of-concept that RM-018 could overcome KRAS Y96D-resistance mutation (Figure 3).
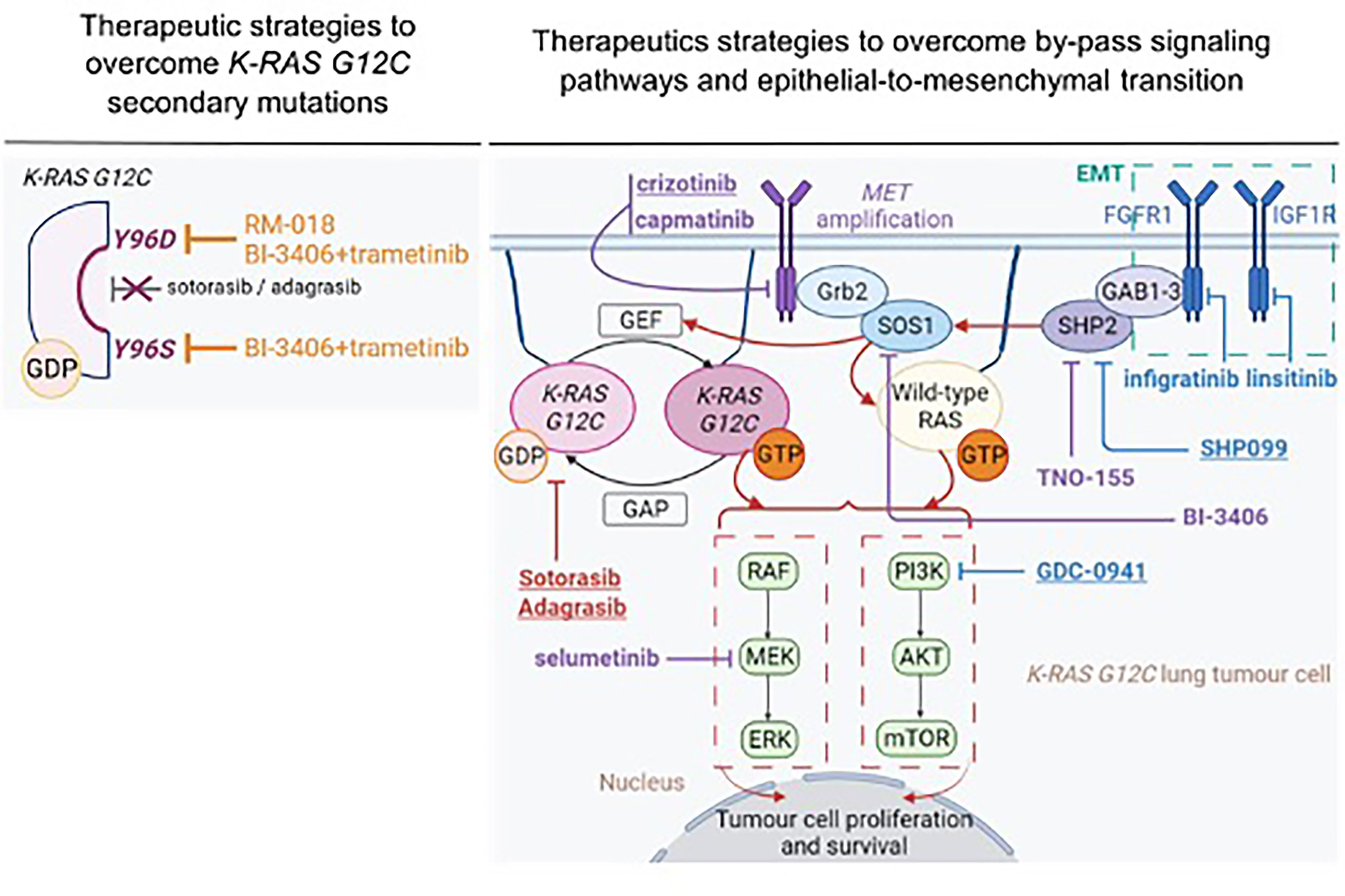
Figure 3 Therapeutic strategies to overcome acquired resistance to sotorasib and adagrasib. Treatment that demonstrated therapeutic efficacy in both in vitro and in vivo setting are underlined. Therapeutic strategies that demonstrated efficacy in pre-clinical specimens in the context of acquired resistance related to MET amplification are represented in purple. Therapeutic strategies that demonstrated efficacy in pre-clinical specimens in the context of acquisition of epithelial-to-mesenchymal features are represented in blue. This figure was created with Biorender.com.
Observations in vitro and in vivo at acquired resistance to KRAS G12C inhibitors highlighted that cancer cells develop adaptative strategies to overcome the selective pressure and survive under treatment. This might result from KRAS secondary mutations or from activation of upstream effectors such as RTKs, in the context of by-pass signaling pathway or epithelial-to-mesenchymal transition. In this setting, several strategies are currently described to overcome acquired resistance based on combined therapy targeting either the specific RTKs involved in the resistance process or upstream and downstream effectors of KRAS G12C (115).
Some pre-clinical studies sought to determine the impact of targeting RTK-mediated acquired resistance to KRAS G12C inhibitors (108, 109) (Figure 3). Combined treatment with sotorasib and crizotinib in H23 sotorasib-resistant cells that acquired MET amplification, was more efficient in inhibiting ERK, AKT and MET activation and inducing apoptosis (i.e., increased cleaved-PARP and BIM expression), compared to single monotherapy (108). Moreover, combination therapy was efficient enough to eliminate RAS-GTP in similar levels as those observed in H23 parental cells. In line with these observations, the IC50 value of combined treatment (IC50 = 0.22 µM) was markedly inferior to those obtained with either sotorasib (IC50 = 69.33µM) or crizotinib (IC50 = 2.68µM) monotherapy, in MET-amplified, sotorasib-resistant cells (108). Notably, combined treatment with sotorasib and crizotinib during four weeks, significantly decreased tumor growth in MET-amplified H23-sotorasib resistant xenograft models compared to monotherapy (108). Interestingly, combined treatment with sotorasib and capmatinib – a MET inhibitor that is more specific of the MET kinase – decreased cell viability with higher potency (i.e., IC50 = 0.07µM) (108) (Figure 3).
MEK inhibitor might constitute an alternative strategy in MET-amplified tumors as pre-clinical experiments highlighted the importance of MAPK pathway in this setting (116). Selumetinib (i.e., MEK inhibitor) also showed interesting in vitro results when associated with sotorasib as assessed by complete inhibition of ERK phosphorylation in H23 MET-amplified sotorasib-resistant cells (108) (Figure 3). Adachi et al. reported in vitro efficacy when targeting IGF1R and FGFR1, RTKs that mediate acquired resistance to KRAS G12C inhibitors through EMT (109). Combined treatment with sotorasib, linsitinib (IGF1R inhibitor) and infigratinib (FGFR1 inhibitor) markedly decreased phosphorylation of AKT, ERK and S6 in H358-sotorasib resistant cells (109). Likewise, cell proliferation was significantly decreased with this triple-combination treatment compared to bitherapies of sotorasib with either linsitinib or infigratinib (109) (Figure 3).
Overall, these pre-clinical studies highlight that targeting the specific RTK that are involved in by-pass signaling pathways might be a promising strategy to overcome acquired resistance to KRAS G12C inhibitors. However, the clinical efficacy of such therapeutic strategies seems difficult to prove in specifically dedicated clinical trials, due to the small proportion of patients who present these alterations. Moreover, mechanisms of acquired resistance to KRAS G12C inhibitors seem highly heterogeneous and might co-occur, thereby providing rationale for the instauration of combination treatments at baseline rather than at disease progression.
SHP2 (Src Homology Phosphatase 2), encoded by PTPN11, is a protein tyrosine phosphatase that interacts with activated RTKs either directly by binding with phosphorylated tyrosine residues or indirectly through scaffolding proteins such as GAB1-3, FRS-2 and IRS1-4 (117–119). SHP2 promotes RAS activation pathway, in particular through SOS1 activation (120), and interacts with SRC kinase (121), thereby promoting its activation and subsequent RAS signaling pathway activation (Figure 3).
Pre-clinical in vivo studies highlight that KRAS-mutant NSCLC depends on SHP2 during carcinogenesis (93, 122, 123). Based on these observations, it has been reported that combined therapy with either sotorasib (83) or adagrasib (93) and RMC-4550 enhanced efficacy compared to single monotherapy. Indeed, RMC-4550 combined with AMG-510 achieved high synergy score in both H358-monolayer cell line and CT-26 KRAS G12C spheroid models (i.e., synergy score 22.8 and 11.7 respectively) (83). RMC-4550 combined with MRTX849 decreased ERK phosphorylation in vitro and in xenograft models of oesophageal cancer (KYSE-410) and NSCLC (H358) (93). Likewise, combined therapy of RMC-4550 plus MRTX849 resulted in significantly greater anti-tumor efficacy in 4 out of 6 KRAS G12C in vivo models, compared to monotherapy (93). Notably, results of phase 1 first-in-human clinical trial showed that RMC-4630 led to a disease control rate of 71% in KRAS G12C mutated NSCLC patients (n=19), as a single-agent monotherapy (124). Likewise, TNO-155 is currently being tested in a phase 1 dose escalation/expansion trial (NCT03114319) in adults with advanced solid tumors alone (125) or in combination with JDQ443 (KRAS G12C inhibitor), in KRAS G12C patients with advanced solid tumors (NCT04699188).
In the setting of acquired resistance to KRAS G12C inhibitors, SHP099 (109) and TNO-155 (108) have demonstrated interesting results to overcome acquired resistance to sotorasib (Figure 3). SHP099 was found to significantly reduce tumor growth in patient-derived orthotopic xenografts and KRAS mutant NSCLC patient-derived xenograft models (122). In EMT-driven, sotorasib-resistant H358 cells, characterized by FGFR1 and IGF1R-ISR1 upregulation, combined therapy with sotorasib, GDC-0941 (PI3K inhibitor) and SHP099 efficiently suppressed PI3K-AKT, MAPK and S6 phosphorylation (109). Similar results were observed in sotorasib-insensitive SW1573 and LU99 KRAS-mutated, mesenchymal-like cells (109). Consistently with these observations, combined treatment with sotorasib, GDC-0941 and SHP099 was found to reduce tumor growth and inhibited AKT and ERK signaling pathways in sotorasib-resistant xenograft models (109). Notably, only the combination of these three treatments reversed acquired resistance to sotorasib in vivo, compared to bitherapies with sotorasib and either GDC-0941 or SHP099 (109) (Figure 3). These results further support that targeting either downstream or upstream effectors of KRAS G12C might not be efficient enough to overcome resistance. TNO-155 is another allosteric SHP2 inhibitor (126) that demonstrated synergistic efficacy with an early-stage compound (82) of adagrasib (i.e., compound 12) in H2122 and H1373 KRAS mutant NSCLC cells (127). Combined therapy with compound 12 and TNO-155 inhibited ERK phosphorylation and impaired cell proliferation in these models (127). Interestingly, combined treatment of sotorasib and TNO-155 in MET-amplified, H23-sotorasib resistant cells inhibited ERK phosphorylation and decreased the rate of RAS-GTP levels, compared to single agent treatment (108) (Figure 3).
SOS1 is a key guanine nucleotide exchange factor for KRAS activation. SOS1 can either bind to KRAS in its GDP-bound state at its catalytic site or binds to KRAS in its GTP-bound state at its allosteric binding site, thereby promoting KRAS activation (128) (Figure 3). SOS1 is also associated in RAS-mutated carcinogenesis (129). SOS1 is a promising target in KRAS-mutated cancers as it plays a central role in feedback MAPK reactivation (130, 131). Notably, activated MAPK kinases were found to selectively phosphorylate the C-terminal tail of SOS1, but not its paralog SOS2, thereby uncoupling Grb2-SOS1 complex with membrane-bound receptor and activating RAS pathway (130, 131). Based on high-throughput screening and structure-based optimization, BI-3406 was recently developed as a potent and selective SOS1 inhibitor (132). BI-3406 bounds specifically to the catalytic site of SOS1, thus blocking SOS1 interaction with GDP-bound KRAS (132). Interestingly, pre-clinical studies on BI-3406 reveal that therapeutic efficacy was not limited to KRAS G12C-mutant models, as it also induced tumor growth inhibition in xenograft models of KRAS G12C (MIA PaCa-2 cells), KRAS G12V (SW620 cells), KRAS G13D (LoVo cells) and KRAS G12S (A549 cells) (132). Moreover, BI-3406 was found to prevent adaptative resistance to MEK inhibition in several KRAS-mutated cancer cell lines, and prevented ERK activation rebound following sotorasib treatment in vitro (132).
Interestingly, BI-3406 demonstrated promising pre-clinical results to overcome acquired resistance to KRAS G12C inhibitors, when combined with treatments that target downstream KRAS G12C effectors. BI-3406 or TNO-155 monotherapy did not restore sensitivity to KRAS G12C inhibitors in Ba/F3 cells harboring KRAS G12C and Y96S or Y96D secondary mutations (103). In contrast, H358 cells (KRAS G12C), modified to express KRAS Y96S or KRAS Y96D resistant mutations, were sensitive to the combination of BI-3406 and trametinib in both monolayer and 3D cell line models (103) (Figure 3). Combined treatment with sotorasib and BI-3406 markedly decreased RAS-GTP levels and inhibited ERK phosphorylation, while AKT activation was less altered, in MET-amplified sotorasib-resistant H23 cells (108) (Figure 3).
Overall, these studies evidence promising pre-clinical rationales to revert or delay the emergence of acquired resistance to sotorasib and adagrasib (Figure 3). Several clinical trials are currently ongoing to evaluate the efficacy of KRAS G12C inhibitors in combination with chemotherapy (133, 134) or with targeted therapies including cetuximab, afatinib, pembrolizumab and SHP2, mTOR, CDK4/6 inhibitors (91, 135, 136). These results would be of interest to evaluate whether combined treatments increased response rate to KRAS G12C inhibitors, thereby contributing to prevent or delay acquired resistance. Of note, clinical trial evaluating combined treatment with mTOR or CDK4/6 inhibitors (135) will be interesting as these alterations have been recently evidenced to promote acquired resistance to sotorasib in clinical setting (90). As PI3K-AKT pathway is less dependent on RAS pathway and less sensitive to KRAS G12C inhibitors, combined strategies targeting upstream effectors (i.e., SOS1 or SHP2) and PI3K-AKT-mTOR pathway seem particularly relevant.
Based on tobacco history, high TMB, high levels of PD-L1 expression and pro-inflammatory microenvironment of KRAS-mutant NSCLC patients, KRAS mutated NSCLC patients are expected to benefit from immune checkpoint inhibitors (ICIs) (137, 138), and this has been demonstrated in KRAS G12C-mutant patients in particular (139–141). In IMMUNOTARGET study, among 271 KRAS mutated patients, where immunotherapy was administered as monotherapy in advanced line, response rate was 26% with higher proportion of long responders (12-months PFS: 25.6%) as compared with other oncogenic-driven NSCLC (142). No significant difference has been observed between KRAS mutation subtypes in term of response or PFS (142) although KRAS G12C patients tend to have higher response rate to ICIs (26.9%) and a longer mPFS (4.0 months) compared with non-KRAS G12C (response rate: 18.8% and mPFS 2.9 months) (141). However, as expected, PFS positively correlated with PD-L1 expression. In the Flatiron database of PD-L1≥50% NSCLC, among patients receiving first-line ICI monotherapy, KRAS mutations (versus KRAS wt) were associated with significant superior survival (mOS,21.1 vs 13.6 months) (137).
In this context, co-occurring mutations might counterbalance clinical benefit of ICIs. While TP53 co-mutation seems to be associated with a better response to ICIs, STK11 and KEAP1 co-mutations seem to impair response to ICIs. Indeed, TP53/KRAS co-mutated patients present higher PD-L1 expression, increased CD8+ TILs (143) and significantly higher mutational load, compared with either STK11 or KEAP1 co-mutated patients. Moreover, patients harboring TP53/KRAS mutations were reported to have prolonged PFS to anti- PD-1 therapy compared to TP53 and KRAS wild-type patients (143). In contrast, STK11 and KEAP1 co-occurring mutations are associated with resistance to ICIs in KRAS-mutant patients, independently of PD-L1 expression (37, 144, 145). Indeed, post-hoc analysis of the IMPOWER 150 Phase III trial showed that PFS and OS were markedly decreased for patients harboring KRAS, STK11 and/or KEAP1 co-mutations treated with carboplatin-paclitaxel combined with either atezolizumab, bevacizumab or atezolizumab and bevacizumab regimen compared to patients with STK11 and KEAP1 wild-type (145). KRAS/STK11 co-mutation might drive intrinsic resistance to PD-1/PD-L1 inhibitors, as it was the only marker associated with PD-L1 negativity among 924 intermediate/high TMB lung adenocarcinoma patients (144). Likewise, KRAS/STK11 co-mutated tumors are usually considered as “cold-tumors” with paucity of CD3+, CD4+ and CD8+ TILs (146). KEAP1 loss downregulates the STING pathway through its interaction with NRF2 (i.e., Nuclear Factor Erythroid 2-like 2) (146). Indeed, KEAP1 loss mediates the degradation of NRF2, a transcriptional factor that is highly involved in cellular antioxidant pathway (147). It was recently reported that patients with high PD-L1 expression (i.e., ≥50%) had significantly higher levels of stromal-SHP2 compared to those with PD-L1<50% (p=0.039) (148). In particular, a significantly higher expression of CD8+/CD4+ T-cells and CD64+ macrophages was observed in stromal compartment of patients with high stromal SHP2 expression compared to those with low stromal SHP2 expression (148). Consistently with these observations, despite small sub-group size, sub-groups analysis showed that patients with high SHP2 expression and PD-L1≥1% had significantly prolonged PFS and OS (148).
In the context of KRAS G12C inhibitors, pre-clinical studies highlighted that either sotorasib or adagrasib have enhanced anti-tumor efficacy when combined with anti- PD-1/PD-L1 therapy. In contrast, although either AMG-510 or anti- PD-1 monotherapy caused tumor complete regression in only one out of ten mice, for each therapy; combined treatment achieved a complete and prolonged response in 9/10 CT-26 KRAS G12C mice (83). Notably, treatment response was maintained 112 days following treatment arrest. Subsequently, mice that were cured by AMG-510 and anti- PD-1 were then rechallenged with tumor inoculum and showed no tumor reformation (83). Consistently with these observations, co-treatment with adagrasib and anti- PD-1 demonstrated complete and prolonged response in vivo, without tumor regrowth after tumor cell inoculation, and increased PFS in KRAS G12C genetically engineered mouse models, compared with single-agent monotherapy (94). Based on these pre-clinical results, several clinical trials are currently ongoing evaluating anti- PD-1/PD-L1 with either sotorasib (NCT04185883, NCT03600883) or adagrasib (NCT03785249, NCT04613596). Besides, the potential impact of targeting immune pathways, several other combinations are on study targeting upstream (EGFR mAb and TKIs, pan-ERRB inhibitors, SHP2 and SOS1 inhibitors) and downstream signaling pathways (PIK3, ERK/RAF, MEK, mTOR and CDK4/6 inhibitors) (Table 3).
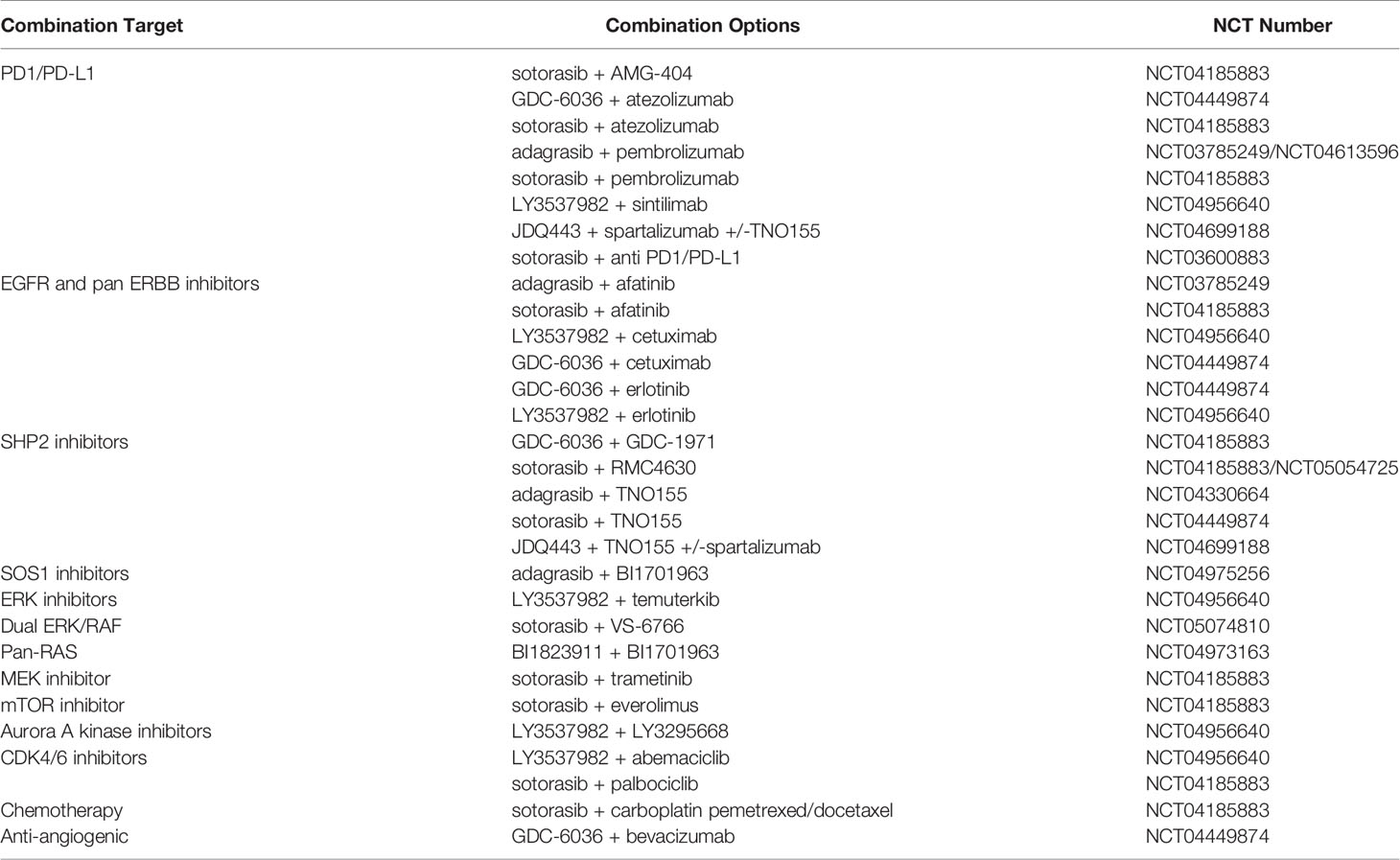
Table 3 Ongoing clinical trials to target mutant KRAS.
Similar other targeted therapies, the biological mechanisms of acquired resistance to KRAS G12C are highly heterogeneous. The development of combine therapeutic approaches, based on SHP2 and SOS1 inhibitors, might be a valuable strategy to target both intrinsic and acquired resistance. Moreover, pre-clinical evidence highlights that combined treatment involving KRAS G12C inhibitors as well as upstream and downstream effectors is usually necessary to achieve therapeutic efficacy. Although promising, these observations rise concerns on the safety profile of such combined treatments in clinical setting. After long decades of considering KRAS as an elusive target in NSCLC, sotorasib and adagrasib as well as emerging KRAS-mutant targeted treatments constitute an unprecedent improvement to efficiently target KRAS-mutant NSCLC. Besides providing new therapeutic strategies to target KRAS-mutant cancer, sotorasib and adagrasib also provide a potential therapeutic rational to overcome KRAS secondary mutations mediating resistance to other therapies in other oncogenic driven NSCLC.
A-LD, AS, SO-C: contributed to this paper with the design. A-LD, CL, AS, SO-C: literature search. A-LD, CL, AS, SO-C: revision, editing and final approval. All authors contributed to the article and approved the submitted version.
A-LD was supported by a 2020–2021 clinical fellowship award for a Master degree from Saint-Etienne Jean Monnet University. CL received a PhD fellowship from La Ligue contre le Cancer (2017–2020). This work was supported by the Institut National du Cancer (INCa) (PRT-K program no. 2018-024 EMT-CoNCEPT).
AS received honoraria from Amgen for participation in Board meetings.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Harvey JJ. An Unidentified Virus Which Causes the Rapid Production of Tumours in Mice. Nature (1964) 204:1104–5. doi: 10.1038/2041104b0
2. Kirsten WH, Mayer LA. Morphologic Responses to a Murine Erythroblastosis Virus. J Natl Cancer Inst (1967) 39:311–35. doi: 10.1093/jnci/39.2.311
3. Reddy EP, Reynolds RK, Santos E, Barbacid M. A Point Mutation Is Responsible for the Acquisition of Transforming Properties by the T24 Human Bladder Carcinoma Oncogene. Nature (1982) 300:149–52. doi: 10.1038/300149a0
4. Prior IA, Hood FE, Hartley JL. The Frequency of Ras Mutations in Cancer. Cancer Res (2020) 80:2969–74. doi: 10.1158/0008-5472.CAN-19-3682
5. Barlesi F, Mazieres J, Merlio J-P, Debieuvre D, Mosser J, Lena H, et al. Routine Molecular Profiling of Patients With Advanced Non-Small-Cell Lung Cancer: Results of a 1-Year Nationwide Programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet (2016) 387:1415–26. doi: 10.1016/S0140-6736(16)00004-0
6. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the Cbioportal. Sci Signal (2013) 6:pl1–1. doi: 10.1126/scisignal.2004088
7. Santos E, Martin-Zanca D, Reddy EP, Pierotti MA, Della Porta G, Barbacid M. Malignant Activation of a K-Ras Oncogene in Lung Carcinoma But Not in Normal Tissue of the Same Patient. Science (1984) 223:661–4. doi: 10.1126/science.6695174
8. Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell (2017) 170:17–33. doi: 10.1016/j.cell.2017.06.009
9. Malumbres M, Barbacid M. RAS Oncogenes: The First 30 Years. Nat Rev Cancer (2003) 3:459–65. doi: 10.1038/nrc1097
10. Vigil D, Cherfils J, Rossman KL, Der CJ. Ras Superfamily GEFs and GAPs: Validated and Tractable Targets for Cancer Therapy? Nat Rev Cancer (2010) 10:842–57. doi: 10.1038/nrc2960
11. Cherfils J, Zeghouf M. Regulation of Small GTPases by GEFs, GAPs, and GDIs. Physiol Rev (2013) 93:269–309. doi: 10.1152/physrev.00003.2012
12. Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell (2000) 103:211–25. doi: 10.1016/S0092-8674(00)00114-8
13. Gale NW, Kaplan S, Lowenstein EJ, Schlessinger J, Bar-Sagi D. Grb2 Mediates the EGF-Dependent Activation of Guanine Nucleotide Exchange on Ras. Nature (1993) 363:88–92. doi: 10.1038/363088a0
14. Vetter IR, Wittinghofer A. The Guanine Nucleotide-Binding Switch in Three Dimensions. Science (2001) 294:1299–304. doi: 10.1126/science.1062023
15. Vögler O, Barceló JM, Ribas C, Escribá PV. Membrane Interactions of G Proteins and Other Related Proteins. Biochim Biophys Acta (2008) 1778:1640–52. doi: 10.1016/j.bbamem.2008.03.008
16. Gao J, Liao J, Yang G-Y. CAAX-Box Protein, Prenylation Process and Carcinogenesis. Am J Transl Res (2009) 1:312–25.
17. Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of Ras.GTP With Raf-1 and Mitogen-Activated Protein Kinase Kinase. Science (1993) 260:1658–61. doi: 10.1126/science.8503013
18. Zhang XF, Settleman J, Kyriakis JM, Takeuchi-Suzuki E, Elledge SJ, Marshall MS, et al. Normal and Oncogenic P21ras Proteins Bind to the Amino-Terminal Regulatory Domain of C-Raf-1. Nature (1993) 364:308–13. doi: 10.1038/364308a0
19. Warne PH, Viciana PR, Downward J. Direct Interaction of Ras and the Amino-Terminal Region of Raf-1 In Vitro. Nature (1993) 364:352–5. doi: 10.1038/364352a0
20. Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras Interacts Directly With the Serine/Threonine Kinase Raf. Cell (1993) 74:205–14. doi: 10.1016/0092-8674(93)90307-C
21. Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH Kinase as a Direct Target of Ras. Nature (1994) 370:527–32. doi: 10.1038/370527a0
22. Sjölander A, Yamamoto K, Huber BE, Lapetina EG. Association of P21ras With Phosphatidylinositol 3-Kinase. Proc Natl Acad Sci USA (1991) 88:7908–12. doi: 10.1073/pnas.88.18.7908
23. Hofer F, Fields S, Schneider C, Martin GS. Activated Ras Interacts With the Ral Guanine Nucleotide Dissociation Stimulator. Proc Natl Acad Sci USA (1994) 91:11089–93. doi: 10.1073/pnas.91.23.11089
24. Kikuchi A, Demo SD, Ye ZH, Chen YW, Williams LT. ralGDS Family Members Interact With the Effector Loop of Ras P21. Mol Cell Biol (1994) 14:7483–91. doi: 10.1128/mcb.14.11.7483-7491.1994
25. Gibbs JB, Sigal IS, Poe M, Scolnick EM. Intrinsic GTPase Activity Distinguishes Normal and Oncogenic Ras P21 Molecules. Proc Natl Acad Sci USA (1984) 81:5704–8. doi: 10.1073/pnas.81.18.5704
26. Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic Activation of the K-Ras Oncogene Causes Early Onset Lung Cancer in Mice. Nature (2001) 410:1111–6. doi: 10.1038/35074129
27. Prior IA, Lewis PD, Mattos C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res (2012) 72:2457–67. doi: 10.1158/0008-5472.CAN-11-2612
28. Wood K, Hensing T, Malik R, Salgia R. Prognostic and Predictive Value in KRAS in Non–Small-Cell Lung Cancer: A Review. JAMA Oncol (2016) 2:805–12. doi: 10.1001/jamaoncol.2016.0405
29. Dogan S, Shen R, Ang DC, Johnson ML, D’Angelo SP, Paik PK, et al. Molecular Epidemiology of EGFR and KRAS Mutations in 3,026 Lung Adenocarcinomas: Higher Susceptibility of Women to Smoking-Related KRAS-Mutant Cancers. Clin Cancer Res (2012) 18:6169–77. doi: 10.1158/1078-0432.CCR-11-3265
30. Finn SP, Addeo A, Dafni U, Thunnissen E, Bubendorf L, Madsen LB, et al. Prognostic Impact of KRAS G12C Mutation in Patients With NSCLC: Results From the European Thoracic Oncology Platform Lungscape Project. J Thorac Oncol (2021) 16:990–1002. doi: 10.1016/j.jtho.2021.02.016
31. Nassar AH, Adib E, Kwiatkowski DJ. Distribution of KRAS G12C Somatic Mutations Across Race, Sex, and Cancer Type. N Engl J Med (2021) 384:185–7. doi: 10.1056/NEJMc2030638
32. Sebastian M, Eberhardt WEE, Hoffknecht P, Metzenmacher M, Wehler T, Kokowski K, et al. KRAS G12C-Mutated Advanced Non-Small Cell Lung Cancer: A Real-World Cohort From the German Prospective, Observational, Nation-Wide CRISP Registry (AIO-TRK-0315). Lung Cancer (2021) 154:51–61. doi: 10.1016/j.lungcan.2021.02.005
33. Zhao N, Wilkerson MD, Shah U, Yin X, Wang A, Hayward MC, et al. Alterations of LKB1 and KRAS and Risk of Brain Metastasis: Comprehensive Characterization by Mutation Analysis, Copy Number, and Gene Expression in Non-Small-Cell Lung Carcinoma. Lung Cancer (2014) 86:255–61. doi: 10.1016/j.lungcan.2014.08.013
34. Macerelli M, Caramella C, Faivre L, Besse B, Planchard D, Polo V, et al. Does KRAS Mutational Status Predict Chemoresistance in Advanced Non-Small Cell Lung Cancer (NSCLC)? Lung Cancer (2014) 83:383–8. doi: 10.1016/j.lungcan.2013.12.013
35. Scheffler M, Ihle MA, Hein R, Merkelbach-Bruse S, Scheel AH, Siemanowski J, et al. K-Ras Mutation Subtypes in NSCLC and Associated Co-Occuring Mutations in Other Oncogenic Pathways. J Thorac Oncol (2019) 14:606–16. doi: 10.1016/j.jtho.2018.12.013
36. Nusrat M, Roszik J, Holla V, Cai T, Hong M, Coker O, et al. Therapeutic Vulnerabilities Among KRAS G12C Mutant (Mut) Advanced Cancers Based on Co-Alteration (Co-Alt) Patterns. J Clin Oncol (2020) 38:3625–5. doi: 10.1200/JCO.2020.38.15_suppl.3625
37. Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, et al. Effects of Co-Occurring Genomic Alterations on Outcomes in Patients With KRAS-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res (2018) 24:334–40. doi: 10.1158/1078-0432.CCR-17-1841
38. Schmid S, Gautschi O, Rothschild S, Mark M, Froesch P, Klingbiel D, et al. Clinical Outcome of ALK-Positive Non-Small Cell Lung Cancer (NSCLC) Patients With De Novo EGFR or KRAS Co-Mutations Receiving Tyrosine Kinase Inhibitors (TKIs). J Thorac Oncol (2017) 12(4):681–8. doi: 10.1016/j.jtho.2016.12.003
39. Pao W, Miller VA. Epidermal Growth Factor Receptor Mutations, Small-Molecule Kinase Inhibitors, and Non-Small-Cell Lung Cancer: Current Knowledge and Future Directions. J Clin Oncol (2005) 23(11):2556–68. doi: 10.1200/JCO.2005.07.799
40. Unni AM, Lockwood WW, Zejnullahu K, Lee-Lin SQ, Varmus H. Evidence That Synthetic Lethality Underlies the Mutual Exclusivity of Oncogenic KRAS and EGFR Mutations in Lung Adenocarcinoma. Elife (2015) 5:e06907. doi: 10.7554/eLife.06907.015
41. Gainor JF, Varghese AM, Ou SH, Kabraji S, Awad MM, Katayama R, et al. ALK Rearrangements Are Mutually Exclusive With Mutations in EGFR or KRAS: An Analysis of 1,683 Patients With Non-Small Cell Lung Cancer. Clin Cancer Res (2013) 19(15):4273–81. doi: 10.1158/1078-0432.CCR-13-0318
42. Choughule A, Sharma R, Trivedi V, Thavamani A, Noronha V, Joshi A, et al. Coexistence of KRAS Mutation With Mutant But Not Wild-Type EGFR Predicts Response to Tyrosine-Kinase Inhibitors in Human Lung Cancer. Br J Cancer (2014) 111:2203–4. doi: 10.1038/bjc.2014.401
43. Ortiz-Cuaran S, Scheffler M, Plenker D, Dahmen L, Scheel AH, Fernandez-Cuesta L, et al. Heterogeneous Mechanisms of Primary and Acquired Resistance to Third-Generation EGFR Inhibitors. Clin Cancer Res (2016) 22:4837–47. doi: 10.1158/1078-0432.CCR-15-1915
44. Koelzer VH, Dawson H, Andersson E, Karamitopoulou E, Masucci GV, Lugli A, et al. Active Immunosurveillance in the Tumor Microenvironment of Colorectal Cancer Is Associated With Low Frequency Tumor Budding and Improved Outcome. Transl Res (2015) 166:207–17. doi: 10.1016/j.trsl.2015.02.008
45. Atkins D, Breuckmann A, Schmahl GE, Binner P, Ferrone S, Krummenauer F, et al. MHC Class I Antigen Processing Pathway Defects, Ras Mutations and Disease Stage in Colorectal Carcinoma. Int J Cancer (2004) 109:265–73. doi: 10.1002/ijc.11681
46. Chen N, Fang W, Lin Z, Peng P, Wang J, Zhan J, et al. KRAS Mutation-Induced Upregulation of PD-L1 Mediates Immune Escape in Human Lung Adenocarcinoma. Cancer Immunol Immunother CII (2017) 66:1175–87. doi: 10.1007/s00262-017-2005-z
47. Li D, Zhu X, Wang H, Li N. Association Between PD-L1 Expression and Driven Gene Status in NSCLC: A Meta-Analysis. Eur J Surg (2017) 43:1372–9. doi: 10.1016/j.ejso.2017.02.008
48. Judd J, Abdel Karim N, Khan H, Naqash AR, Baca Y, Xiu J, et al. Characterization of KRAS Mutation Subtypes in Non-Small Cell Lung Cancer. Mol Cancer Ther (2021) 21:0201. doi: 10.1158/1535-7163.MCT-21-0201
49. Sumimoto H, Takano A, Teramoto K, Daigo Y. RAS-Mitogen-Activated Protein Kinase Signal Is Required for Enhanced PD-L1 Expression in Human Lung Cancers. PloS One (2016) 11:e0166626. doi: 10.1371/journal.pone.0166626
50. Miura Y, Sunaga N, Kaira K, Tsukagoshi Y, Osaki T, Sakurai R, et al. Abstract 4028: Oncogenic KRAS Mutations Induce PD-L1 Overexpression Through MAPK Pathway Activation in Non-Small Cell Lung Cancer Cells. Cancer Res Am Assoc Cancer Res (2016) 76:4028–8. doi: 10.1158/1538-7445.AM2016-4028
51. Zdanov S, Mandapathil M, Abu Eid R, Adamson-Fadeyi S, Wilson W, Qian J, et al. Mutant KRAS Conversion of Conventional T Cells Into Regulatory T Cells. Cancer Immunol Res (2016) 4:354–65. doi: 10.1158/2326-6066.CIR-15-0241
52. Sunaga N, Imai H, Shimizu K, Shames DS, Kakegawa S, Girard L, et al. Oncogenic KRAS-Induced Interleukin-8 Overexpression Promotes Cell Growth and Migration and Contributes to Aggressive Phenotypes of Non-Small Cell Lung Cancer. Int J Cancer (2012) 130:1733–44. doi: 10.1002/ijc.26164
53. Chang SH, Mirabolfathinejad SG, Katta H, Cumpian AM, Gong L, Caetano MS, et al. T Helper 17 Cells Play a Critical Pathogenic Role in Lung Cancer. Proc Natl Acad Sci USA (2014) 111:5664–9. doi: 10.1073/pnas.1319051111
54. Caetano MS, Zhang H, Cumpian AM, Gong L, Unver N, Ostrin EJ, et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-Ras-Mutant Lung Cancer. Cancer Res (2016) 76:3189–99. doi: 10.1158/0008-5472.CAN-15-2840
55. Khosravi N, Caetano MS, Cumpian AM, Unver N, de la Garza Ramos C, Noble O, et al. IL22 Promotes Kras-Mutant Lung Cancer by Induction of a Protumor Immune Response and Protection of Stemness Properties. Cancer Immunol Res (2018) 6:788–97. doi: 10.1158/2326-6066.CIR-17-0655
56. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell (2012) 21:822–35. doi: 10.1016/j.ccr.2012.04.025
57. Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell (2019) 35:559–72.e7. doi: 10.1016/j.ccell.2019.02.008
58. Villaruz LC, Socinski MA, Cunningham DE, Chiosea SI, Burns TF, Siegfried JM, et al. The Prognostic and Predictive Value of KRAS Oncogene Substitutions in Lung Adenocarcinoma. Cancer (2013) 119:2268–74. doi: 10.1002/cncr.28039
59. Shepherd FA, Domerg C, Hainaut P, Jänne PA, Pignon J-P, Graziano S, et al. Pooled Analysis of the Prognostic and Predictive Effects of KRAS Mutation Status and KRAS Mutation Subtype in Early-Stage Resected Non-Small-Cell Lung Cancer in Four Trials of Adjuvant Chemotherapy. J Clin Oncol (2013) 31:2173–81. doi: 10.1200/JCO.2012.48.1390
60. Mascaux C, Iannino N, Martin B, Paesmans M, Berghmans T, Dusart M, et al. The Role of RAS Oncogene in Survival of Patients With Lung Cancer: A Systematic Review of the Literature With Meta-Analysis. Br J Cancer (2005) 92:131–9. doi: 10.1038/sj.bjc.6602258
61. Goulding RE, Chenoweth M, Carter GC, Boye ME, Sheffield KM, John WJ, et al. KRAS Mutation as a Prognostic Factor and Predictive Factor in Advanced/Metastatic Non-Small Cell Lung Cancer: A Systematic Literature Review and Meta-Analysis. Cancer Treat Res (2020) 24:100200. doi: 10.1016/j.ctarc.2020.100200
62. Marabese M, Ganzinelli M, Garassino MC, Shepherd FA, Piva S, Caiola E, et al. KRAS Mutations Affect Prognosis of Non-Small-Cell Lung Cancer Patients Treated With First-Line Platinum Containing Chemotherapy. Oncotarget (2015) 6:34014–22. doi: 10.18632/oncotarget.5607
63. Hames ML, Chen H, Iams W, Aston J, Lovly CM, Horn L. Correlation Between KRAS Mutation Status and Response to Chemotherapy in Patients With Advanced Non-Small Cell Lung Cancer. Lung Cancer (2016) 92:29–34. doi: 10.1016/j.lungcan.2015.11.004
64. Izar B, Zhou H, Heist RS, Azzoli CG, Muzikansky A, Scribner EEF, et al. The Prognostic Impact of KRAS, Its Codon and Amino Acid Specific Mutations, on Survival in Resected Stage I Lung Adenocarcinoma. J Thorac Oncol (2014) 9:1363–9. doi: 10.1097/JTO.0000000000000266
65. El Osta B, Behera M, Kim S, Berry LD, Sica G, Pillai RN, et al. Characteristics and Outcomes of Patients With Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J Thorac Oncol (2019) 14:876–89. doi: 10.1016/j.jtho.2019.01.020
66. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the Undruggable RAS: Mission Possible? Nat Rev Drug Discov (2014) 13:828–51. doi: 10.1038/nrd4389
67. Adjei AA, Mauer A, Bruzek L, Marks RS, Hillman S, Geyer S, et al. Phase II Study of the Farnesyl Transferase Inhibitor R115777 in Patients With Advanced Non–Small-Cell Lung Cancer. J Clin Oncol (2003) 21(9):1760–6. doi: 10.1200/JCO.2003.09.075
68. Papke B, Der CJ. Drugging RAS: Know the Enemy. Science (2017) 355:1158–63. doi: 10.1126/science.aam7622
69. Riely GJ, Johnson ML, Medina C, Rizvi NA, Miller VA, Kris MG, et al. A Phase II Trial of Salirasib in Patients With Lung Adenocarcinomas With KRAS Mutations. J Thorac Oncol (2011) 6:1435–7. doi: 10.1097/JTO.0b013e318223c099
70. Cox AD, Der CJ, Philips MR. Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin Cancer Res (2015) 21:1819–27. doi: 10.1158/1078-0432.CCR-14-3214
71. Jänne PA, van den Heuvel MM, Barlesi F, Cobo M, Mazieres J, Crinò L, et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA (2017) 317:1844–53. doi: 10.1001/jama.2017.3438
72. Kim D, Xue JY, Lito P. Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients. Cell (2020) 183:850–9. doi: 10.1016/j.cell.2020.09.044
73. Goldman JW, Mazieres J, Barlesi F, Dragnev KH, Koczywas M, Göskel T, et al. A Randomized Phase III Study of Abemaciclib Versus Erlotinib in Patients With Stage IV Non-Small Cell Lung Cancer With a Detectable KRAS Mutation Who Failed Prior Platinum-Based Therapy: JUNIPER. Front Oncol (2020) 10:578756. doi: 10.3389/fonc.2020.578756
74. Middleton G, Fletcher P, Popat S, Savage J, Summers Y, Greystoke A, et al. The National Lung Matrix Trial of Personalized Therapy in Lung Cancer. Nature (2020) 583:807–12. doi: 10.1038/s41586-020-2481-8
75. Xie M, Xu X, Fan Y. KRAS-Mutant Non-Small Cell Lung Cancer: An Emerging Promisingly Treatable Subgroup. Front Oncol (2021) 11:672612. doi: 10.3389/fonc.2021.672612
76. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature (2013) 503:548–51. doi: 10.1038/nature12796
77. Lito P, Solomon M, Li L-S, Hansen R, Rosen N. Allele-Specific Inhibitors Inactivate Mutant KRAS G12C by a Trapping Mechanism. Science (2016) 351:604–8. doi: 10.1126/science.aad6204
78. Patricelli MP, Janes MR, Li L-S, Hansen R, Peters U, Kessler LV, et al. Selective Inhibition of Oncogenic KRAS Output With Small Molecules Targeting the Inactive State. Cancer Discov (2016) 6:316–29. doi: 10.1158/2159-8290.CD-15-1105
79. Janes MR, Zhang J, Li L-S, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers With a Covalent G12C-Specific Inhibitor. Cell (2018) 172:578–89.e17. doi: 10.1016/j.cell.2018.01.006
80. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res (2015) 13:1325–35. doi: 10.1158/1541-7786.MCR-15-0203
81. Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J Med Chem (2020) 63:52–65. doi: 10.1021/acs.jmedchem.9b01180
82. Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J Med Chem (2020) 63:6679–93. doi: 10.1021/acs.jmedchem.9b02052
83. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature (2019) 575:217–23. doi: 10.1038/s41586-019-1694-1
84. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for Lung Cancers With KRAS P.G12C Mutation. N Engl J Med (2021) 384:2371–81. doi: 10.1056/NEJMoa2103695
85. Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. KRASG12C Inhibition With Sotorasib in Advanced Solid Tumors. N Engl J Med (2020) 383:1207–17. doi: 10.1056/NEJMoa1917239
86. Hong DS, Bang Y-J, Barlesi F, Durm GA, Falchook GS, Govindan R, et al. 1257o Durability of Clinical Benefit and Biomarkers in Patients (Pts) With Advanced Non-Small Cell Lung Cancer (NSCLC) Treated With AMG 510 (Sotorasib). Ann Oncol (2020) 31:S812. doi: 10.1016/j.annonc.2020.08.1571
87. IASLC. World Conference on Lung Cancer (WCLC 2021) (2021). Available at: https://wclc2021.iaslc.org/.
88. Begum P, Goldin RD, Possamai LA, Popat S. Severe Immune Checkpoint Inhibitor Hepatitis in KRAS G12C-Mutant NSCLC Potentially Triggered by Sotorasib: Case Report. JTO Clin Res Rep (2021) 2(9):100–213. doi: 10.1016/j.jtocrr.2021.100213
89. Spira AI, Wilson FH, Shapiro G, Dooms C, Curioni-Fontecedro A, Esaki T, et al. Patient-Reported Outcomes (PRO) From the Phase 2 CodeBreaK 100 Trial Evaluating Sotorasib in KRAS P.G12C Mutated Non-Small Cell Lung Cancer (NSCLC). J Clin Oncol (2021) 39:9057–7. doi: 10.1200/JCO.2021.39.15_suppl.9057
90. Zhao Y, Murciano-Goroff YR, Xue JY, Ang A, Lucas J, Mai TT, et al. Diverse Alterations Associated With Resistance to KRAS(G12C) Inhibition. Nature (2021) 599:679–83. doi: 10.1038/s41586-021-04065-2
91. Jänne PA, Rybkin II, Spira AI, Riely GJ, Papadopoulos KP, Sabari JK, et al. KRYSTAL-1: Activity and Safety of Adagrasib (MRTX849) in Advanced/Metastatic Non–Small-Cell Lung Cancer (NSCLC) Harboring KRAS G12C Mutation. Eur J Cancer (2020) 138:S1–2. doi: 10.1016/S0959-8049(20)31076-5
92. Riely GJ, Ou S-HI, Rybkin I, Spira A, Papadopoulos K, Sabari JK, et al. 99o_PR KRYSTAL-1: Activity and Preliminary Pharmacodynamic (PD) Analysis of Adagrasib (MRTX849) in Patients (Pts) With Advanced Non–Small Cell Lung Cancer (NSCLC) Harboring KRASG12C Mutation. J Thorac Oncol (2021) 16:S751–2. doi: 10.1016/S1556-0864(21)01941-9
93. Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRASG12C Inhibitor MRTX849 Provides Insight Toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov (2020) 10:54–71. doi: 10.1158/2159-8290.CD-19-1167
94. Briere DM, Li S, Calinisan A, Sudhakar N, Aranda R, Hargis L, et al. The KRASG12C Inhibitor MRTX849 Reconditions the Tumor Immune Microenvironment and Sensitizes Tumors to Checkpoint Inhibitor Therapy. Mol Cancer Ther (2021) 20:975–85. doi: 10.1158/1535-7163.MCT-20-0462
95. Soria J-C, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med (2018) 378:113–25. doi: 10.1056/NEJMoa1713137
96. Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim D-W, et al. Alectinib Versus Crizotinib in Untreated ALK-Positive Non–Small-Cell Lung Cancer. N Engl J Med (2017) 377:829–38. doi: 10.1056/NEJMoa1704795
97. Manchado E, Weissmueller S, Morris JP, Chen C-C, Wullenkord R, Lujambio A, et al. A Combinatorial Strategy for Treating KRAS-Mutant Lung Cancer. Nature (2016) 534:647–51. doi: 10.1038/nature18600
98. Kitai H, Ebi H, Tomida S, Floros KV, Kotani H, Adachi Y, et al. Epithelial-To-Mesenchymal Transition Defines Feedback Activation of Receptor Tyrosine Kinase Signaling Induced by MEK Inhibition in KRAS-Mutant Lung Cancer. Cancer Discov (2016) 6:754–69. doi: 10.1158/2159-8290.CD-15-1377
99. Ryan MB, Fece de la Cruz F, Phat S, Myers DT, Wong E, Shahzade HA, et al. Vertical Pathway Inhibition Overcomes Adaptive Feedback Resistance to KRASG12C Inhibition. Clin Cancer Res (2020) 26:1633–43. doi: 10.1158/1078-0432.CCR-19-3523
100. Kobayashi K, Terai H, Yasuda H, Hamamoto J, Hayashi Y, Takeuchi O, et al. Functional Dissection of the KRAS G12C Mutation by Comparison Among Multiple Oncogenic Driver Mutations in a Lung Cancer Cell Line Model. Biochem Biophys Res Commun (2021) 534:1–7. doi: 10.1016/j.bbrc.2020.11.110
101. Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid Non-Uniform Adaptation to Conformation-Specific KRAS(G12C) Inhibition. Nature (2020) 577:421–5. doi: 10.1038/s41586-019-1884-x
102. Aldea M, Andre F, Marabelle A, Dogan S, Barlesi F, Soria J-C. Overcoming Resistance to Tumor-Targeted and Immune-Targeted Therapies. Cancer Discov (2021) 11:874–99. doi: 10.1158/2159-8290.CD-20-1638
103. Koga T, Suda K, Fujino T, Ohara S, Hamada A, Nishino M, et al. KRAS Secondary Mutations That Confer Acquired Resistance to KRAS G12C Inhibitors, Sotorasib and Adagrasib, and Overcoming Strategies: Insights From In Vitro Experiments. J Thorac Oncol (2021) 16:1321–32. doi: 10.1016/j.jtho.2021.04.015
104. Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical Acquired Resistance to KRASG12C Inhibition Through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov (2021) 11:1913–22. doi: 10.1158/2159-8290.CD-21-0365
105. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired Resistance to KRASG12C Inhibition in Cancer. N Engl J Med (2021) 384:2382–93. doi: 10.1056/NEJMoa2105281
106. Camidge DR, Pao W, Sequist LV. Acquired Resistance to TKIs in Solid Tumours: Learning From Lung Cancer. Nat Rev Clin Oncol (2014) 11:473–81. doi: 10.1038/nrclinonc.2014.104
107. Romero D. Uncovering Adagrasib Resistance. Nat Rev Clin Oncol (2021) 18:541–1. doi: 10.1038/s41571-021-00545-6
108. Suzuki S, Yonesaka K, Teramura T, Takehara T, Kato R, Sakai H, et al. KRAS Inhibitor-Resistance in MET-Amplified KRAS G12C Non-Small Cell Lung Cancer Induced by RAS- and Non-RAS-Mediated Cell Signaling Mechanisms. Clin Cancer Res (2021) 27(20):clincanres.0856.2021. doi: 10.1158/1078-0432.CCR-21-0856
109. Adachi Y, Ito K, Hayashi Y, Kimura R, Tan TZ, Yamaguchi R, et al. Epithelial-To-Mesenchymal Transition Is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C-Mutant Non-Small Cell Lung Cancer. Clin Cancer Res (2020) 26:5962–73. doi: 10.1158/1078-0432.CCR-20-2077
110. Passaro A, Jänne PA, Mok T, Peters S. Overcoming Therapy Resistance in EGFR -Mutant Lung Cancer. Nat Cancer (2021) 2:377–91. doi: 10.1038/s43018-021-00195-8
111. Marine J-C, Dawson S-J, Dawson MA. Non-Genetic Mechanisms of Therapeutic Resistance in Cancer. Nat Rev Cancer (2020) 20:743–56. doi: 10.1038/s41568-020-00302-4
112. Zhang H, Fillmore Brainson C, Koyama S, Redig AJ, Chen T, Li S, et al. Lkb1 Inactivation Drives Lung Cancer Lineage Switching Governed by Polycomb Repressive Complex 2. Nat Commun (2017) 8:14922. doi: 10.1038/ncomms14922
113. Nichols R, Schulze C, Bermingham A, Choy T, Cregg J, Kiss G, et al. A06 Tri-Complex Inhibitors of the Oncogenic, GTP-Bound Form of KRASG12C Overcome RTK-Mediated Escape Mechanisms and Drive Tumor Regressions in Preclinical Models of NSCLC. J Thorac Oncol (2020) 15:S13–4. doi: 10.1016/j.jtho.2019.12.035
114. Schulze CJ, Bermingham A, Choy TJ, Cregg JJ, Kiss G, Marquez A, et al. Abstract PR10: Tri-Complex Inhibitors of the Oncogenic, GTP-Bound Form of KRASG12C Overcome RTK-Mediated Escape Mechanisms and Drive Tumor Regressions In Vivo. Mol Cancer Ther (2019) 18:PR10–0. doi: 10.1158/1535-7163.TARG-19-PR10
115. Dunnett-Kane V, Nicola P, Blackhall F, Lindsay C. Mechanisms of Resistance to KRASG12C Inhibitors. Cancers (2021) 13:E151. doi: 10.3390/cancers13010151
116. Chiba M, Togashi Y, Tomida S, Mizuuchi H, Nakamura Y, Banno E, et al. MEK Inhibitors Against MET-Amplified Non-Small Cell Lung Cancer. Int J Oncol (2016) 49:2236–44. doi: 10.3892/ijo.2016.3736
117. Neel BG, Gu H, Pao L. The ‘Shp’ing News: SH2 Domain-Containing Tyrosine Phosphatases in Cell Signaling. Trends Biochem Sci (2003) 28:284–93. doi: 10.1016/S0968-0004(03)00091-4
118. Vogel W, Lammers R, Huang J, Ullrich A. Activation of a Phosphotyrosine Phosphatase by Tyrosine Phosphorylation. Science (1993) 259:1611–4. doi: 10.1126/science.7681217
119. Chan RJ, Feng G-S. PTPN11 Is the First Identified Proto-Oncogene That Encodes a Tyrosine Phosphatase. Blood (2007) 109:862–7. doi: 10.1182/blood-2006-07-028829
120. Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, et al. RAS Nucleotide Cycling Underlies the SHP2 Phosphatase Dependence of Mutant BRAF-, NF1- and RAS-Driven Cancers. Nat Cell Biol (2018) 20:1064–73. doi: 10.1038/s41556-018-0169-1
121. Zhang SQ, Yang W, Kontaridis MI, Bivona TG, Wen G, Araki T, et al. Shp2 Regulates SRC Family Kinase Activity and Ras/Erk Activation by Controlling Csk Recruitment. Mol Cell (2004) 13:341–55. doi: 10.1016/S1097-2765(04)00050-4
122. Mainardi S, Mulero-Sánchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, et al. SHP2 Is Required for Growth of KRAS-Mutant Non-Small-Cell Lung Cancer In Vivo. Nat Med (2018) 24:961–7. doi: 10.1038/s41591-018-0023-9
123. Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-Driven Cancers Depend on PTPN11/SHP2 Phosphatase. Nat Med (2018) 24:954–60. doi: 10.1038/s41591-018-0024-8
124. Ou SI, Koczywas M, Ulahannan S, Janne P, Pacheco J, Burris H, et al. A12 The SHP2 Inhibitor RMC-4630 in Patients With KRAS-Mutant Non-Small Cell Lung Cancer: Preliminary Evaluation of a First-In-Man Phase 1 Clinical Trial. J Thorac Oncol (2020) 15:S15–6. doi: 10.1016/j.jtho.2019.12.041
125. Brana I, Shapiro G, Johnson ML, Yu HA, Robbrecht D, Tan DS-W, et al. Initial Results From a Dose Finding Study of TNO155, a SHP2 Inhibitor, in Adults With Advanced Solid Tumors. J Clin Oncol (2021) 39:3005–5. doi: 10.1200/JCO.2021.39.15_suppl.3005
126. LaMarche MJ, Acker M, Argintaru A, Bauer D, Boisclair J, Chan H, et al. Identification of TNO155, an Allosteric SHP2 Inhibitor for the Treatment of Cancer. J Med Chem (2020) 63:13578–94. doi: 10.1021/acs.jmedchem.0c01170
127. Liu C, Lu H, Wang H, Loo A, Zhang X, Yang G, et al. Combinations With Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling. Clin Cancer Res (2021) 27:342–54. doi: 10.1158/1078-0432.CCR-20-2718
128. Freedman TS, Sondermann H, Friedland GD, Kortemme T, Bar-Sagi D, Marqusee S, et al. A Ras-Induced Conformational Switch in the Ras Activator Son of Sevenless. Proc Natl Acad Sci USA (2006) 103:16692–7. doi: 10.1073/pnas.0608127103
129. Jeng H-H, Taylor LJ, Bar-Sagi D. Sos-Mediated Cross Activation of Wild-Type Ras by Oncogenic Ras Is Essential for Tumorigenesis. Nat Commun (2012) 3:1168. doi: 10.1038/ncomms2173
130. Rozakis-Adcock M, van der Geer P, Mbamalu G, Pawson T. MAP Kinase Phosphorylation of Msos1 Promotes Dissociation of Msos1-Shc and Msos1-EGF Receptor Complexes. Oncogene (1995) 11:1417–26.
131. Corbalan-Garcia S, Yang SS, Degenhardt KR, Bar-Sagi D. Identification of the Mitogen-Activated Protein Kinase Phosphorylation Sites on Human Sos1 That Regulate Interaction With Grb2. Mol Cell Biol (1996) 16:5674–82. doi: 10.1128/MCB.16.10.5674
132. Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR, et al. BI-3406, a Potent and Selective SOS1–KRAS Interaction Inhibitor, Is Effective in KRAS-Driven Cancers Through Combined MEK Inhibition. Cancer Discov (2021) 11:142–57. doi: 10.1158/2159-8290.CD-20-0142
133. Reck M, Spira A, Besse B, Wolf J, Skoulidis F, Borghaei H, et al. 1416tip CodeBreak 200: A Phase III Multicenter Study of Sotorasib (AMG 510), a KRAS(G12C) Inhibitor, Versus Docetaxel in Patients With Previously Treated Advanced Non-Small Cell Lung Cancer (NSCLC) Harboring KRAS P.G12C Mutation. Ann Oncol (2020) 31:S894–5. doi: 10.1016/j.annonc.2020.08.1730
134. Mok TSK, Lawler WE, Shum MK, Dakhil SR, Spira AI, Barlesi F, et al. KRYSTAL-12: A Randomized Phase 3 Study of Adagrasib (MRTX849) Versus Docetaxel in Patients (Pts) With Previously Treated Non-Small-Cell Lung Cancer (NSCLC) With KRASG12C Mutation. J Clin Oncol (2021) 39:TPS9129–TPS9129. doi: 10.1200/JCO.2021.39.15_suppl.TPS9129
135. Hong DS, Strickler JH, Fakih M, Falchook GS, Li BT, Durm GA, et al. Trial in Progress: A Phase 1b Study of Sotorasib, A Specific and Irreversible KRASG12C Inhibitor, as Monotherapy in Non-Small Cell Lung Cancer (NSCLC) With Brain Metastasis and in Combination With Other Anticancer Therapies in Advanced Solid Tumors (CodeBreaK 101). J Clin Oncol (2021) 39:TPS2669–TPS2669. doi: 10.1200/JCO.2021.39.15_suppl.TPS2669
136. Sabari JK, Park H, Tolcher AW, Ou S-HI, Garon EB, George B, et al. KRYSTAL-2: A Phase I/II Trial of Adagrasib (MRTX849) in Combination With TNO155 in Patients With Advanced Solid Tumors With KRAS G12C Mutation. J Clin Oncol (2021) 39:TPS146–6. doi: 10.1200/JCO.2021.39.3_suppl.TPS146
137. Sun L, Hsu M, Cohen RB, Langer CJ, Mamtani R, Aggarwal C. Association Between KRAS Variant Status and Outcomes With First-Line Immune Checkpoint Inhibitor-Based Therapy in Patients With Advanced Non-Small-Cell Lung Cancer. JAMA Oncol (2021) 7:937–9. doi: 10.1001/jamaoncol.2021.0546
138. Liu C, Zheng S, Jin R, Wang X, Wang F, Zang R, et al. The Superior Efficacy of Anti-PD-1/PD-L1 Immunotherapy in KRAS-Mutant Non-Small Cell Lung Cancer That Correlates With an Inflammatory Phenotype and Increased Immunogenicity. Cancer Lett (2020) 470:95–105. doi: 10.1016/j.canlet.2019.10.027
139. Gadgeel S, Rodriguez-Abreu D, Felip E, Esteban E, Speranza G, Reck M, et al. KRAS Mutational Status and Efficacy in KEYNOTE-189: Pembrolizumab (Pembro) Plus Chemotherapy (Chemo) vs Placebo Plus Chemo as First-Line Therapy for Metastatic Non-Squamous NSCLC. Ann Oncol (2019) 30:xi64–5. doi: 10.1093/annonc/mdz453.002
140. Herbst RS, Lopes G, Kowalski DM, Kasahara K, Wu Y-L, Castro GD, et al. LBA4 Association of KRAS Mutational Status With Response to Pembrolizumab Monotherapy Given as First-Line Therapy for PD-L1-Positive Advanced Non-Squamous NSCLC in Keynote-042. Ann Oncol (2019) 30:xi63–4. doi: 10.1093/annonc/mdz453.001
141. Mazieres J, Tomasini P, Lusque A, Boucekine M, Gautschi O, Cortot AB, et al. 1279p Impact of KRAS Mutations and Subtypes on Efficacy of Immune-Checkpoint Inhibitors (ICI) in Non-Small Cell Lung Cancer (NSCLC). Ann Oncol (2020) 31:S826–7. doi: 10.1016/j.annonc.2020.08.1593
142. Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune Checkpoint Inhibitors for Patients With Advanced Lung Cancer and Oncogenic Driver Alterations: Results From the IMMUNOTARGET Registry. Ann Oncol (2019) 30(8):1321–8. doi: 10.1093/annonc/mdz167
143. Dong Z-Y, Zhong W-Z, Zhang X-C, Su J, Xie Z, Liu S-Y, et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res (2017) 23:3012–24. doi: 10.1158/1078-0432.CCR-16-2554
144. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov (2018) 8:822–35. doi: 10.1158/2159-8290.CD-18-0099
145. West H, Cappuzzo F, Reck M, Mok T, Jotte RM, Nishio M, et al. 1265p IMpower150: A Post Hoc Analysis of Efficacy Outcomes in Patients With KRAS, STK11 and KEAP1 Mutations. Ann Oncol (2020) 31:S817–8. doi: 10.1016/j.annonc.2020.08.1579
146. Skoulidis F, Heymach JV. Co-Occurring Genomic Alterations In Non-Small-Cell Lung Cancer Biology and Therapy. Nat Rev Cancer (2019) 19:495–509. doi: 10.1038/s41568-019-0179-8
147. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, et al. Keap1 Loss Promotes Kras-Driven Lung Cancer and Results in Dependence on Glutaminolysis. Nat Med (2017) 23:1362–8. doi: 10.1038/nm.4407
Keywords: KRAS G12C, non-small cell lung cancer (NSCLC), adagrasib, sotorasib, acquired resistance
Citation: Désage A-L, Léonce C, Swalduz A and Ortiz-Cuaran S (2022) Targeting KRAS Mutant in Non-Small Cell Lung Cancer: Novel Insights Into Therapeutic Strategies. Front. Oncol. 12:796832. doi: 10.3389/fonc.2022.796832
Received: 17 October 2021; Accepted: 07 January 2022;
Published: 16 February 2022.
Edited by:
Matthias Scheffler, University Hospital of Cologne, GermanyReviewed by:
Francesco Facchinetti, Gustave Roussy Cancer Campus, FranceCopyright © 2022 Désage, Léonce, Swalduz and Ortiz-Cuaran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Ortiz-Cuaran, c2FuZHJhLm9ydGl6LWN1YXJhbkBseW9uLnVuaWNhbmNlci5mcg==; Aurélie Swalduz, QXVyZWxpZS5TV0FMRFVaQGx5b24udW5pY2FuY2VyLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.