 Joanna Moes-Sosnowska
Joanna Moes-Sosnowska Joanna Chorostowska-Wynimko
Joanna Chorostowska-Wynimko- Department of Genetics and Clinical Immunology, National Institute of Tuberculosis and Lung Diseases, Warsaw, Poland
Fibroblast growth factor receptor (FGFR) inhibitors (FGFRis) are a potential therapeutic option for squamous non-small cell lung cancer (Sq-NSCLC). Because appropriate patient selection is needed for targeted therapy, molecular profiling is key to discovering candidate biomarker(s). Multiple FGFR aberrations are present in Sq-NSCLC tumors—alterations (mutations and fusions), amplification and mRNA/protein overexpression—but their predictive potential is unclear. Although FGFR1 amplification reliability was unsatisfactory, FGFR mRNA overexpression, mutations, and fusions are promising. However, currently their discriminatory power is insufficient, and the available clinical data are from small groups of Sq-NSCLC patients. Here, we focus on FGFR aberrations as predictive biomarkers for FGFR-targeting agents in Sq-NSCLC. Known and suggested molecular determinants of FGFRi resistance are also discussed.
Introduction
Lung cancer is the most common cause of cancer-related death worldwide (1). Non-small cell lung cancer (NSCLC) accounts for over 75% of all lung cancer cases, and 20–30% are squamous non-small cell lung cancers (Sq-NSCLC). The prognosis of patients with advanced Sq-NSCLC is poor and immune checkpoint inhibitors (pembrolizumab, atezolizumab, or cemiplimab in tumors with PD-L1 expression≥50%; pembrolizumab plus carboplatin/nab-paclitaxel chemotherapy or nivolumab and ipilimumab plus 2 cycles of platinum-based chemotherapy regardless of PD-L1 expression) are the only first line systemic therapies approved by both the FDA (US Food and Drug Administration) and the EMA (European Medicines Agency) (2). However, in a randomized trials, approximately 30-40% of patients responded to first-line checkpoint inhibitors (2, 3). It is important to identify new targeted therapies and reliable predictive molecular biomarkers for Sq-NSCLC. Since, FGFR aberrations have been found in targetable oncogenic pathways (4), the FGFR inhibitors emerged as potential targeted therapy agents with promising therapeutic effects assessed in distinct clinical trials [reviewed in (5–7)]. Accordingly, in April 2019 the first FGFR inhibitor (FGFRi), erdafitinib, was approved by the FDA for patients with locally advanced or metastatic urothelial carcinoma with FGFR2 and FGFR3 genetic alterations. In April 2020, the FGFR1/2/3 inhibitor pemigatinib was granted approval and recently, in May 2021, the pan-FGFR1–4 inhibitor BGJ398 (infigratinib) was approved for the treatment of unresectable locally advanced or metastatic cholangiocarcinoma with FGFR2 fusion or other rearrangements.
Sq-NSCLC is a target for FGFRi therapy because of the high rate of amplifications in fibroblast growth factor receptor 1 (FGFR1). Accordingly, FGFR1 amplification and overexpression are promising predictive markers for FGFRi therapy in Sq-NSCLC. However, most early phase clinical trials with new FGFRis showed only a partial response (8–11).
Appropriate patient selection is needed for targeted therapy. This review focuses on FGFR aberrations as reliable predictive biomarkers for response to FGFRis in Sq-NSCLC.
FGFR Signaling Pathway—Structure and Normal Function of Fibroblast Growth Factor Receptors
FGFR1-4 are located on four chromosomes and consist of 18 exons (start codon in exon 2). The proteins encoded by FGFR genes are members of the fibroblast growth factor family, consisting of 18 ligands acting via four highly conserved tyrosine kinase receptors (FGFR 1–4). Each FGFR consists of an extracellular region, composed of three immunoglobulin-like domains (IgI–IgIII), a single hydrophobic transmembrane domain, and a cytoplasmic tyrosine kinase domain (Figure 1, Supplemental Figure S1 and Table S1). FGFRs have multiple alternative splice isoforms with tissue-specific expression (Figure S1). These are generated by deletion of the IgI domain and/or acid box or by a sequence change in the carboxy-terminal half of the IgIII domain, transforming isoform IIIb into IIIc (FGFR1–3) (12, 13). The extracellular domains interact with fibroblast growth factors (FGFs): FGFR1 binds both acidic and basic FGF; FGFR2 binds acidic, basic, and/or keratinocyte FGFs, depending on the isoform; FGFR3 binds acidic and basic FGFs; and FGFR4 binds acidic FGFs (Supplemental Table S1) (14). FGF ligand binding leads to dimerization of FGFR followed by its activation by sequential autophosphorylation of tyrosine residues (Figure 2). FGFR signaling activates the phospho-inositide-3-kinase (PI3K)/AKT, signal transducer and activator of transcription (STAT), and mitogen activated protein kinase (MAPK) pathways (15). FGFRs participate in the regulation of multiple biological activities, including tissue repair; angiogenesis; and cell proliferation, differentiation, migration, and survival (14–16). The four FGFR proteins are expressed in a number of tissues under normal conditions (Table S1). Aberrations of FGFR1–4 genes are associated with a broad range of developmental disorders, such as craniosynostosis and dwarfing syndromes, and with cancers (15).
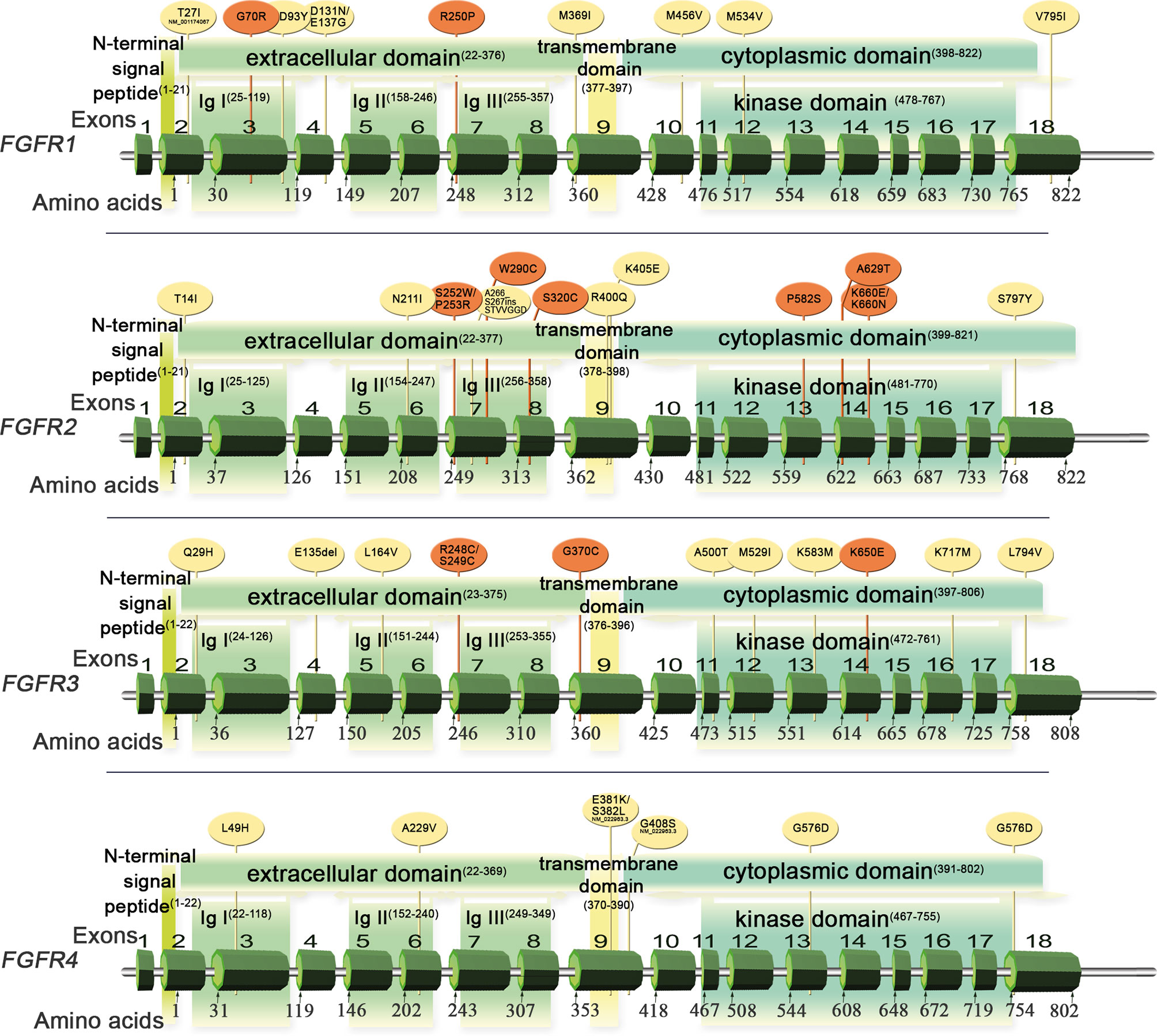
Figure 1 Schematic of FGFR1–4 genes and protein structures with variants detected in Sq-NSCLC tissue (Table 1). Red, pathogenic or likely pathogenic clinical significance; yellow, variants not in the ClinVar database.
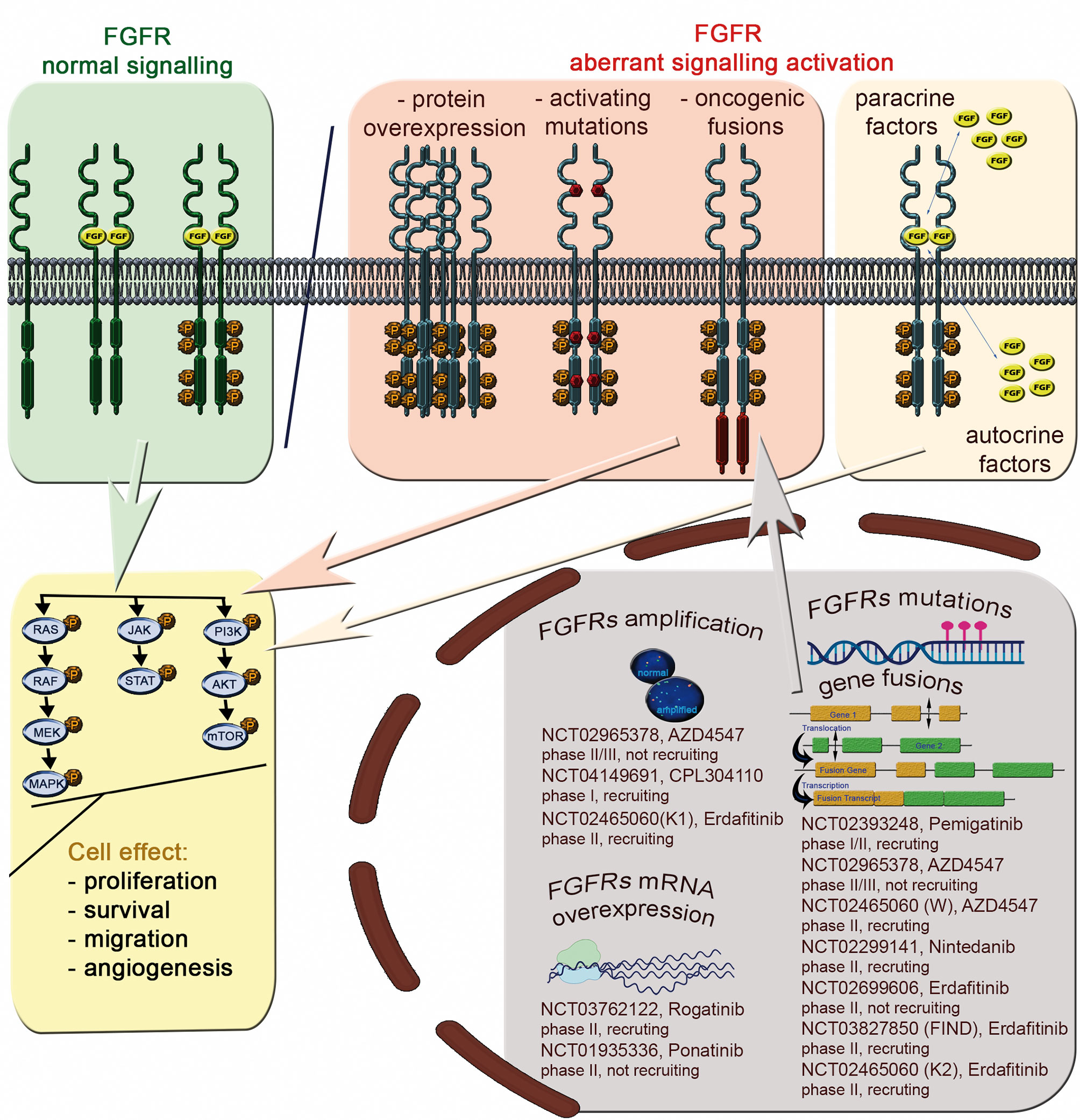
Figure 2 The FGFR signaling pathway and aberrations with potential as predictive biomarkers for FGFRi treatment of Sq-NSCLC.
FGFR1-4 Genetic Aberrations in Cancers
Deregulated FGF/FGFR signaling associated with FGFR aberrations is observed in various cancers (Supplemental Table S2), including lung cancer (Table 1). Activation of effectors of FGFR cancer-related signaling pathways (PI3K/AKT, STAT and MAPK) affects cell proliferation, survival, metabolism, migration, and cell cycle. Furthermore, FGFs released from tumor and stroma may increase autocrine and paracrine signaling and initiate angiogenesis (13) (Figure 2).
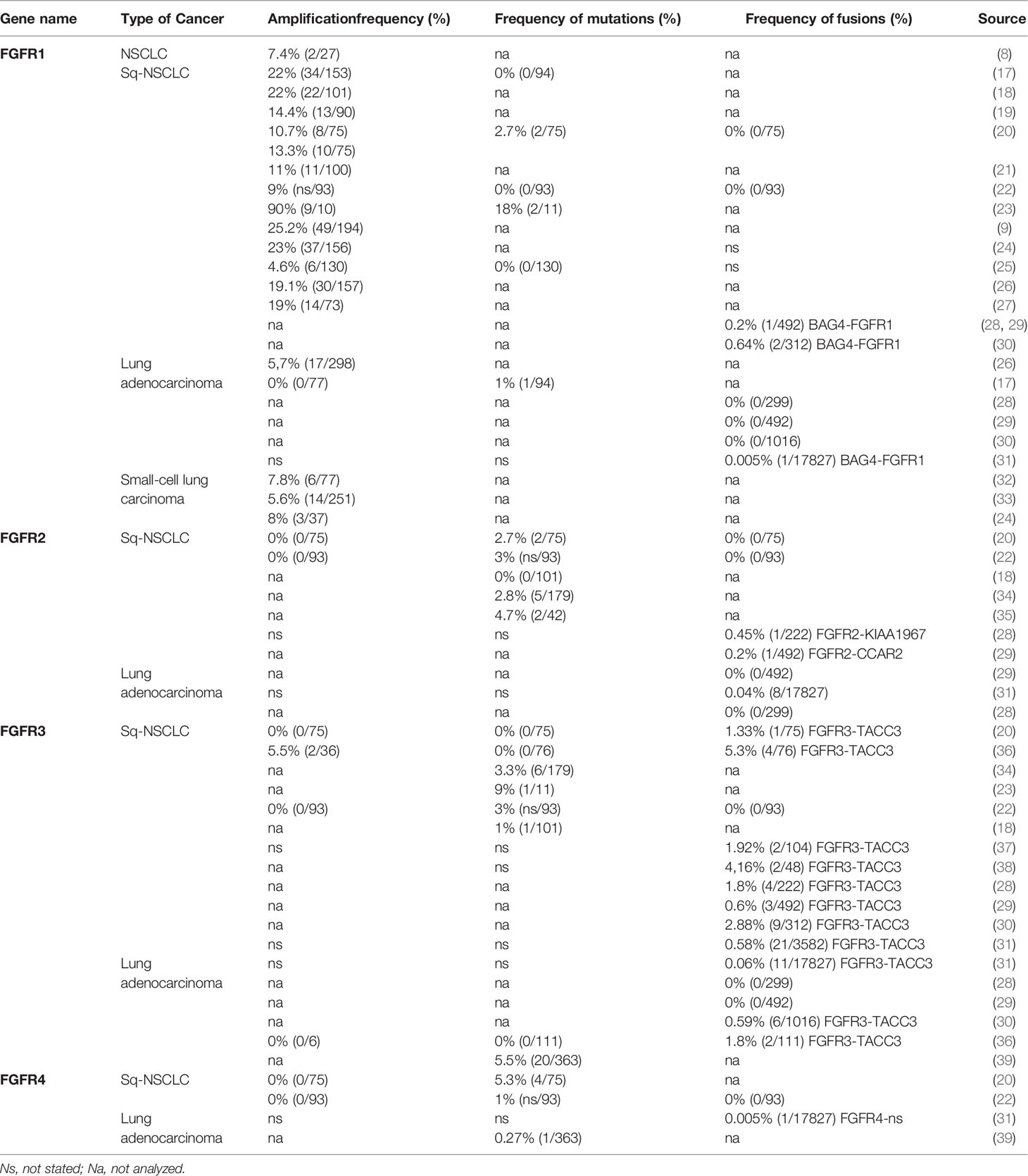
Table 1 Frequencies of FGFR1–4 amplifications, mutations, and fusions in lung cancer.
Preclinical studies have shown that inhibition of FGF/FGFR signaling induces apoptosis in lung cancer cell lines via oxidative stress and impairs tumor growth in xenograft models (40, 41). The FGFR pathway may be altered by gene amplification, mRNA overexpression, and mutation and chromosomal rearrangement (gene fusion). Moreover, alternatively spliced FGFR isoforms participate in oncogenic signaling, promoting tumorigenesis.
FGFR Gene Amplification
FGFR amplification is a main mechanism of FGFR signaling activation and is critical for cancer cell proliferation and survival. It is the most common FGFR alteration in various cancer types (reviewed in (5, 16, 22). FGFR1 amplification is frequent in Sq-NSCLC (4.6–22%, Table 1) and in breast cancer (5–15%), but is less common in other cancer types such as ovarian (5%) and colorectal (2%) carcinomas (4, 18, 22, 42) (Supplemental Table S2).
FGFR1 Amplification in Preclinical In Vitro and In Vivo Studies
It has been proposed to be a biomarker of FGFRi efficacy in advanced Sq-NSCLC, and preclinical in vitro and in vivo studies yielded promising results. For example, FGFRis such as PD173074 (selective FGFR1–3 inhibitor) (17, 43), AZD4547 (a potent inhibitor of FGFR1–3) (19), nintedanib (multikinase inhibitor targeting FGFR, VEGFR, and PDGFR) (20), dovitinib (multitarget inhibitor of RTKs: FLT3/c-Kit, FGFR1–3, and VEGFR1–4) (37), BGJ398 (infigratinib) (44), and ponatinib (AP24534, a multitarget pan-FGFRi) (21, 45, 46) resulted in cell growth inhibition and apoptosis of lung cancer cell lines with FGFR1 amplification (NCI-H520, LK-2, and NCI-H1703 [squamous cell carcinoma] (19–21, 40, 42–46) and NCI-H1581 [large-cell carcinoma] (17, 40, 43, 45, 46). By contrast, a squamous lung carcinoma cell line with the wild-type FGFR1 gene copy number (NCI-H2170) was insensitive to those inhibitors (17, 19, 40, 43). Preclinical in vivo studies based on a primary, FGFR1-amplified Sq-NSCLC xenograft revealed the therapeutic efficacy of some FGFRis. Nintedanib inhibited the growth of tumors formed by FGFR1-amplified NCI-H520 and LK‐2 cells (20); PD173074 shrunk xenografted FGFR1-amplified NCI-H1581 cell tumors; and AZD4547 led to significant tumor growth inhibition (TGI > 94–199%) in four of five FGFR1-amplified patient-derived squamous lung cancer models (47). However, the predictive value of FGFR amplification was not confirmed by other in vitro studies. Two FGFR1 non-amplified cell lines were sensitive to AZD4547 (19). An FGFR1-amplified lung cancer cell line (NCI-H520) was not sensitive to PD173074 or AZD4547 (17, 48), and FGFR1-amplified squamous lung cell lines derived from metastatic sites (HCC95 and SKMES1) were insensitive to ponatinib and AZD4547 (19, 21). Additionally, SKMES1 cells were sensitive to dovitinib (40). Nevertheless, the majority of the promising preclinical evidence resulted in FGFR1 amplification being proposed as a predictive biomarker of FGFR inhibition in advanced Sq-NSCLC.
FGFR1 Amplification in Clinical Trials
Consequently, it has been a key inclusion criterion in phase I/II clinical trials in solid tumors, particularly in Sq-NSCLC. However, most clinical trials in patients with Sq-NSCLC have indicated considerable weakness of FGFR amplification as a biomarker. A complete response (CR) or partial response (PR) to FGFRis has been reported for a few patients; most had stable disease (SD) or disease progression (P). In a phase Ic multicenter study of AZD4547 (NCT00979134), 1 of 24 patients with FGFR-amplified Sq-NSCLC achieved a PR (12 weeks) (49). Similarly, in a phase Ib study, 1 of 13 patients (8%) achieved PR (PFS, 5.4 months), 4 (31%) showed SD and 8 (61%) exhibited P. Changes in tumor size ranged from shrinkage by 35% to growth by 35% (23). Likewise, in a phase II lung-MAP clinical substudy (SWOG S1400D, NCT02965378), 1 of 23 patients (4.3%) with FGFR1 amplification presented a PR (2.9 months) to AZD4547 (11). More promising results were reported in a phase I clinical study of BGJ398 (NCT01004224) (10) and a phase II study of >dovitinib (NCT01861197) (9). In both studies, ~ 11% of patients with FGFR1-amplified Sq-NSCLC achieved PR (4/36 and 3/26, respectively) with a decrease in tumor size of ~ 30–40%, and ~ 40% of patients had SD. The median progression-free survival was 2.9 months and the mean duration of response was 5.2 months (9). A similar outcome was reported by basket- or umbrella-type clinical trials in FGFR1-amplified advanced cancers; Sq-NSCLC had diverse sensitivity to FGFRis; SD was the best response to LY2874455 (a pan-FGFR1–4 inhibitor that occupies the ATP-binding pocket in the kinase domain, NCT01212107) (8), Debio-1347 (CH5183284, an ATP competitive, highly selective inhibitor of FGFR1–3, NCT01948297) (50), and derazantinib (ARQ 087, an ATP-competitive pan-FGFRi with multikinase activity, NCT01752920) (51). Therefore, there is discordance between FGFR1 amplification status and the clinical response to FGFRis in Sq-NSCLC.
FGFR1 Focal Amplification or Arm Amplification
Failure of FGFR1 amplification as a predictive biomarker in lung cancer may be linked to expression of neighboring genes in the 8p11 amplicon. FGFR1 amplification could reflect arm-level 8p11 gain (19, 23, 40) but not focal amplification, as previously claimed (17). Initially, significant correlation between high expression of FGFR1 and its neighboring genes [BAG4, LSM1, NSD3 [WHSC1L1] (19, 40), ASH2L, DDHD2, TACC1, and EIF4EBP1 (19)] and FGFR1 amplification in Sq-NSCLC tissue was shown. However, Paik et al. (23) confirmed that expression of FGFR1 and neighboring genes was variable and lower in tumor samples than in FGFR1-amplified cell lines. FGFR1 expression was low in most tumor samples, but the expression patterns of other genes in the 8p11-12 amplicon were heterogeneous. In some tumors, genes in the 8p11-12 amplicon located closer to the centromere presented high expression, but in others expression levels were uniformly low, or the pattern was inconsistent. There was also no correlation between AZD4547 efficacy and 8p11 amplicon gene expression (23). This implies the importance of other genes in this locus and that FGFR1 amplification alone does not provide adequate sensitivity to reliably predict response to FGFRis.
FGFR1 Copy Number and mRNA Expression
Important insight into the mechanism of failure of FGFR1 amplification as a biomarker is provided by the poor correlation between FGFR1 mRNA expression and its amplification. Only 50% of Sq-NSCLCs with an increased FGFR1 gene copy number (CGN) overexpress FGFR1 mRNA (4). This observation was confirmed by others; 31–46% of FGFR1-amplified Sq-NSCLCs presented high FGFR1 mRNA expression, and no FGFR1 mRNA expression was found in 25% of FGFR1-amplified tumors (9). Similarly, Wynes et al. (21) showed that a high FGFR1 CGN overlapped with an increased FGFR1 mRNA level in 46% of tumors (squamous/mixed NSCLC and NOS). These data imply the existence of mechanisms other than increased GCN regulating FGFR1 transcription.
FGFR1 Amplification Functional Impact - Driver of Cancer or “Passenger” Event
Additionally, it remains unclear whether FGFR1 amplification represents the underlying molecular cause as a driver of cancer, or simply exists as “passenger” event within the overall mutational profile of cancer. Initially, the FGFR1 amplification in Sq-NSCLC was hypothesized as oncogenic driver event. Functional studies revealed that silencing of FGFR1 strongly reduced the viability of the FGFR1-amplified lung cancer cells, inhibited cancer cell growth and clonogenicity, while application of the FGFR inhibitor revealed growth inhibition (p=0.0002) and induced apoptosis (p=0.008) (17, 46). FGFR1 amplification also have indicated poor prognosis, since its association with shorter overall survival and shorter disease-free survival was reported (52). Despite initial evidence, data from in vitro, in vivo and clinical studies (as discussed in dedicated sections) revealed conflicting data. Mainly, poor correlation of FGFR1 amplification with FGFR1 gene and protein expression in Sq-NSCLC cells, and ambiguous response to FGFR1 inhibition affected either due to the FGFR1 expression or presence of other alternations and subsequent activation of alternative signaling pathways, might indicate that FGFR1 amplification is a passenger alteration event rather than a driver alteration. Furthermore, a concurrent amplification of FGFR1 and other genes located in distant loci (on other chromosomes) has been acknowledged in Sq-NSCLC recently (27), specifically the co-amplification of FGFR1 with Defective In Cullin Neddylation 1 Domain Containing 1 gene (DCUN1D1, activation of the focal adhesion kinase and hedgehog signaling pathways) and/or BCL9 Transcription Coactivator gene (BCL9, an alternate activation of Wnt/b-catenin pathway) in the 93% FGFR1-amplified Sq-NSCLC. Still, association of this co-amplification with FGFRi treatment effect has not been examined yet.
FGFR2–4 Amplification
In contrast, the reliability of FGFR2–4 amplification (which is rare in Sq-NSCLC [0–4.7%], Table 1) (11, 20, 22, 36, 53) as a predictive biomarker for Sq-NSCLC has not been investigated. The only phase II clinical study (NCT02965378) of AZD4547 revealed similar inconsistencies in FGFR1 amplification. Two patients with FGFR3-amplified Sq-NSCLC have been reported: one with tumor shrinkage by 25% and the other (with concomitant FGFR1 amplification) with a 10% increase in tumor size (11).
These data imply reasons for the low predictive value of FGFR1 amplification for response to FGFRis. Unfortunately, few Sq-NSCLC patients have been treated with FGFRis in clinical trials, precluding firm conclusions. However, a number of clinical trials of FGFRi are ongoing and are actively enrolling FGFR-amplified patients with advanced Sq-NSCLC (Figure 2).
FGFR mRNA/Protein Overexpression
FGFRs are overexpressed not only in lung cancer but also in other solid tumors, including cancers of the prostate, breast, brain, gastric and head and neck, and sarcoma (54, 55). Increased FGFR1 mRNA levels are frequently observed in Sq-NSCLC. Medium and high tumor FGFR1 mRNA expression was reported in 27–60% of patients with Sq-NSCLC (4, 19, 26), and 55 of 118 (47%) Sq-NSCLC tumors had FGFR1–3 mRNA overexpression (55, 56).
FGFR mRNA Expression
As FGFR1 amplification is a poor predictive biomarker for response to FGFRis in Sq-NSCLC, FGFR expression levels have been evaluated in preclinical and clinical studies. In vitro, a high FGFR1 mRNA level, independently of its amplification, was a significant predictor of sensitivity to AZD4547 in two FGFR1 non-amplified squamous lung cancer cell lines (H226 and LK2) compared to the H1703 and H520 lines, characterized by both high FGFR1 amplification and overexpression (19). Moreover, BGJ398 treatment of FGFR1-silenced H-520 cells showed results similar to BGJ398 or siRNA-FGFR1 treatments alone, indicating that the drug inhibitory effects actually relied on FGFR1 inhibition, while complete silencing of the FGFR1 expression in H-520 cell line lead to significant reduction of cell proliferation, increased the percentage of cells in the G0/1 phase of the cell cycle and resulted in down-regulation of both MAPK and AKT/mTOR pathways (44, 46). Also, the majority of Sq-NSCLC cell lines established from primary or metastatic tumors with FGFR1 expression subsequently transfected by a dominant-negative FGFR1 (dnFGFR1) IIIc-green fluorescent protein chimera or treated with FGFR small-molecule inhibitors (SU5402 and PD166866) showed significantly reduced growth, survival, clonogenicity, and migratory potential. The FGFRi treatment had no effect on nonmalignant bronchoepithelial cell lines (BEAS-2B) expressing very low levels of FGFR1 (57).
In addition, FGFR3, but not FGFR2 or FGFR4, mRNA levels were weakly associated with sensitivity to ponatinib (no correlation to AZD4547) (19, 21). These promising results were confirmed in vivo: significant inhibition of tumor growth by rogaratinib (a potent and selective FGFR1–4 inhibitor) was observed in two patient-derived xenografts (PDX) (LU299–Sq-NSCLC and LXFL1121-NSCLC) overexpressing FGFR1 mRNA but not harboring high FGFR amplification (58). Also, two differentially expressed gene profiles of Sq-NSCL tumors (PDX), sensitive or resistant to dovitinib were observed (40). Notably, FGFR3, FGF3 and FGF19 were the most diversely expressed genes, and significant differences in GRB2, KLB, MAPK1, NRAS, and SOS1 expression were reported. The different expression patterns encompassed genes related to the FGFR pathway, including FGFR1–4 receptors but also other oncogenic genes, which might be linked to tumor sensitivity to FGFRis. The results of phase II clinical studies are inconsistent. Promising results were obtained in 40 advanced Sq-NSCLC patients with FGFR1-3 overexpression (NCT03762122) treated with rogaratinib; two patients (5.6%) achieved PR (in one, for > 16 months) and SD was achieved in 64% (51, 59). However, only 1 of 10 patients (1%) had 6 months PFS in continuation of that study (60). Also, in three Sq-NSCLC patients with enhanced FGFR1 mRNA levels, two responded to ponatinib with SD and one showed P (61). In contrary, in one of three dovitinib responders in the NCT01861197 trial, low FGFR1–3 mRNA levels and a high FGFR1 amplification level in the tumor were reported (9).
FGFRs Protein Expression
FGFR1-3 protein levels are frequently high in NSCLC. Notably, FGFRs expression level differs between histological subtypes. A high FGFR1 protein level was reported in: Sq-NSCLCs - 16 of 171 (9%) (24), 13 of 212 (6%) (26), and in 22 of 267 (8.2%) (36); adenocarcinoma histological subtype- 40 of 114 (35%) (24), 13 of 383 (3.5%) (26), and 40 of 309 (12.9%) (36). High FGFR2 and FGFR3 protein levels in were also reported in both NSCLC histotypes: Sq-NSCLC: 9 (3.4%) and 18 (6.6%), respectively; -adenocarcinoma: 66 (21.8) and 2 (0.6%), respectively (36). Nonetheless, there was no significant correlation between FGFR1 protein expression and gene amplification assessed by FISH or NGS (23, 24), although there was a weak correlation between FGFR1 mRNA and protein levels in Sq-NSCLC (19, 26). The FGFR protein levels have also been investigated as potential biomarkers for response to FGFRi treatment. Some preclinical studies have indicated that FGFRi response associates closely with high FGFR1 protein expression. For example, Wynes et al. (21) showed that FGFR1 mRNA and protein expression, but not GCN, predict FGFR TKI sensitivity in lung cancer cell lines. Similarly, use of rogaratinib against cancer cell lines with high FGFR expression but no FGFR amplification revealed the selective antiproliferative activity of FGFRis (58). On the other hand, two FGFR1-amplified NSCLC PDX (from large cell and adenocarcinoma subtypes) with co-overexpression of FGFR1 protein responded to M6123 (selective FGFR1 antagonist) (26). To our knowledge, besides one Sq-NSCLC patient with FGFR1 amplified tumor, who achieved PR in NCT00979134 study and had no FGFR1 protein overexpression (23) there is luck of clear results for clinical significance of FGFR1 protein expression. Because of the dearth of clinical data, the relationship between FGFR mRNA/protein levels and response to FGFRis remains unclear. On-going trials of FGFRis are enrolling FGFR mRNA- and protein- overexpressing patients with advanced Sq-NSCLC (Figure 2).
FGFR Mutations and Fusions
FGFRs Mutations
FGFR mutations are frequent in human cancers (16, 22), with the highest prevalence in NSCLC (FGFR1 range 0–18%) (17, 20, 23, 62), endometrial carcinoma (FGFR2, range 0–9%) (35, 63), bladder carcinoma (FGFR3, range 8.5–26%) (53, 64), and rhabdomyosarcoma (FGFR4 7.5%) (65) (Table 1 and Supplemental Table S2). Mutations are found in the regions of the gene encoding the extracellular, trans-membrane, and kinase domains of the FGFR receptor (Figure 1 and Tables 2 and 3) but are most frequent outside the kinase domain in exons corresponding to the extracellular domains (Ig-domains) and alternative exons.
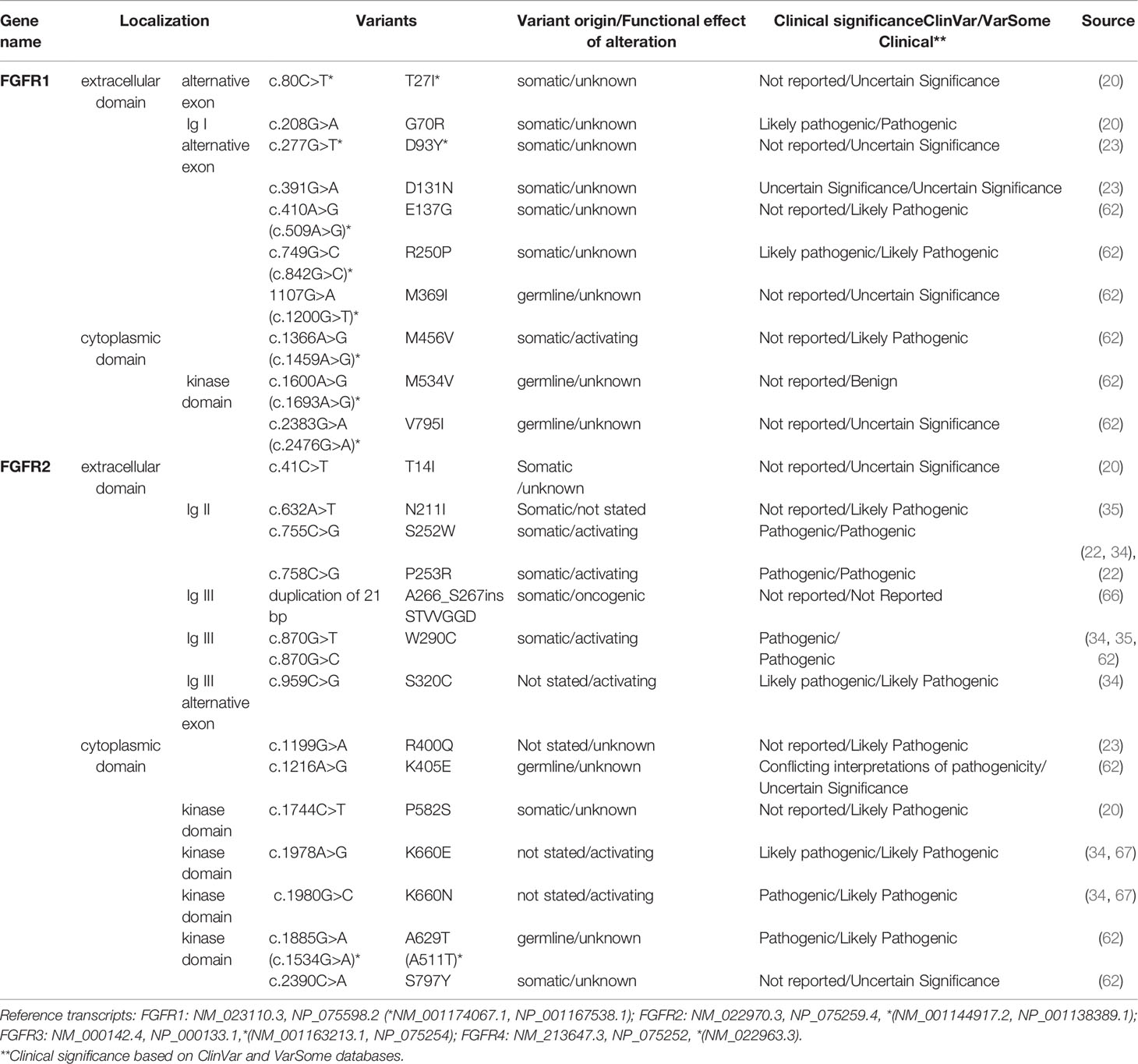
Table 2 FGFR1-2 variants detected in squamous non-small cell lung cancer (Sq-NSCLC). Variant localisation is shown in Figure 2.
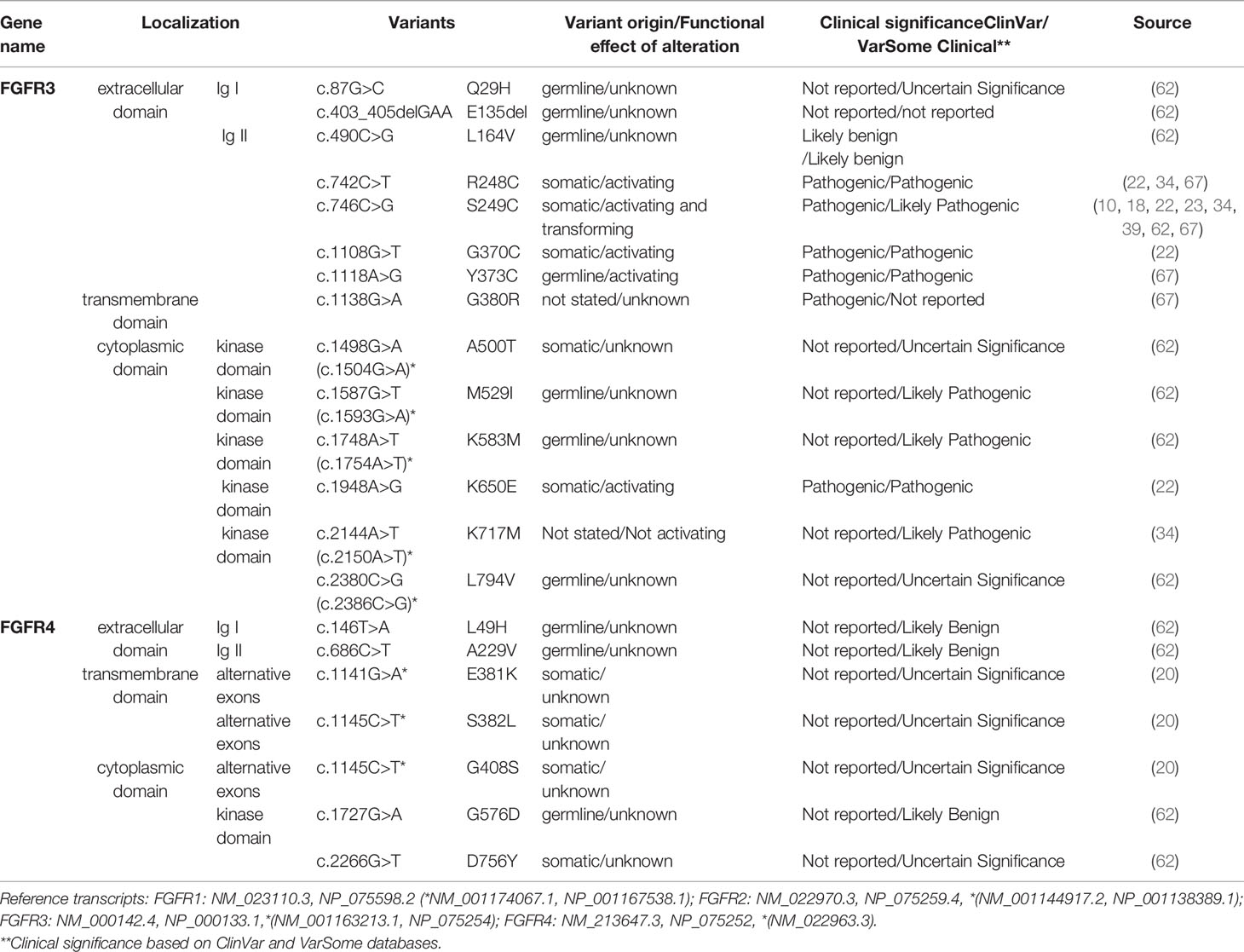
Table 3 FGFR3-4 variants detected in squamous non-small cell lung cancer (Sq-NSCLC). Variant localisation is shown in Figure 2.
This is important because point mutations in extracellular domains lead to receptor stimulation in a ligand-independent manner by obligate receptor dimerization (68). For example, S249C mutation in the extracellular domain (IgIII) of FGFR3 induces constitutive dimerization and receptor activation via modest dimer stabilization in the absence of ligand (69). Similarly, rare FGFR mutations in the transmembrane domain may reduce dimerization efficiency and overall stability, leading to a constitutively active protein (70). Kinase-domain point mutations are typically localized in regulatory elements such as the molecular brake, the A-loop, the kinase hinge, and the DFG-latch. This results in receptor autophosphorylation and constitutive receptor activation in a ligand-independent manner, as exemplified by oncogenic-like FGFR2 mutations (A266_S267ins, 290_291WI>C) (66) and point mutations in FGFR3 (N540, K650) (68).
FGFRs Fusions
FGFR gene fusions originating from chromosomal rearrangement of two genes (through translocation, insertion, inversion or deletion) are found in all common tumor types. The Cancer Genome Atlas study reported FGFR fusions in 8 of 20 tumor types (29). Chromosomal rearrangements involving FGFR genes often lead to protein fusion with the FGFR kinase domain and its subsequent activation (71). FGFR2 gene fusions are most frequent in cholangiocarcinoma, and are also detected, albeit less commonly, in colorectal cancer, hepatocarcinoma and NSCLC (22). FGFR3 fusions are more common in glioblastoma, urothelial carcinoma, and lung cancer, with several dozen fusion partners. FGFR1 and FGFR4 fusions are rare in solid tumors. FGFR1 fusions are typically found in gastrointestinal stromal tumor (GIST), breast cancer, and bladder urothelial carcinoma. Lung cancer develops via a multistep process of tumor biogenesis involving accumulation of inherited or acquired genetic abnormalities. Analysis of the FGFR alterations in Sq-NSCLC showed that 20% of identified variants were in FGFR1-4 (20, 34, 62) (Table 2 and 3); FGFR1 ~ 18% (17, 20, 23, 62), FGFR2 2.5–4.7% (20, 34, 35), FGFR3 0–9% (20, 23, 34), and FGFR4 5.3% (20). In addition, 0.6–3.5% of FGFR aberrations are chromosomal translocations such as FGFR3-TACC3 (3.5%), BAG4-FGFR1 (< 1%), and FGFR2-KIAA1967 (0.3%) (31, 72). Fusions of all four FGFR genes have been found in Sq-NSCLC (30, 31, 67, 71, 73).
Importance of FGFRs Mutations and Fusions as FGFRi Predictive Biomarkers
FGFR mutations and fusions are candidate predictive biomarkers for FGFRis. For instance, it was recently shown that estimated drug sensitivity of FGFR3 S249C was intermediate for all FGFR inhibitors examined in mouse fibroblast cell line (3T3 cells), while different variants at FGFR2 N549 showed different drug sensitivity but similar oncogenicity (74). However, there is little evidence for the predictive power of FGFR variants in Sq-NSCLC. Contradictory results from AZD4547 trials in patients with Sq-NSCLC with the FGFR3 S249C pathogenic variant have been reported. The phase II lung-MAP substudy (SWOG S1400D, NCT02965378) and a phase Ib clinical study (NCT00979134) with AZD4547 FGFRi revealed single Sq-NSCLC patients with FGFR3 S249C who achieved PR (for 1.5 months with ~ 32% tumor shrinkage) (11) or SD (2.6-month progression-free survival [PFS] and 12% tumor shrinkage) (23). No significant benefit of AZD4547 (11) or BGJ398 (infigratinib, NCT01004224) (10) was shown in two other patients carrying the FGFR3 S249C variant (2–4% tumor size decrease). Diverse treatment outcomes were observed in Sq-NSCLCs with other FGFR variants: SD (4.1 months PFS and 20% tumor shrinkage) was reported in one patient (6.6%) with simultaneous FGFR1 missense variants: D93Y and H841Y (23), whereas no response was reported in a patient (1/27, 1.4%) with FGFR3 fusion (11). Additionally, the objective response rate was observed in 1 (5%) patient carrying the FGFR mutation or fusion among 24 patients with NSCLC (13%) treated with erdafitinib (NCT01703481) (75). Molecular profiling revealed 10 (42%) FGFR mutations, 8 (33%) fusions, and 5 (21%) amplifications in that group. FGFR variants and fusions may have predictive utility in other tumor types such as bladder/urothelial cancer, cholangiocarcinoma, and adrenocortical carcinoma (NCT01004224, NCT01752920, NCT02465060) (10, 47, 76, 77). Abnormal protein conformation resulting from FGFR aberrations, and consequently ineffective FGFRi binding, may be responsible for the weak predictive power of FGFR variants and fusions for response to FGFRis (78). Functional analysis can identify potential associations of rare and newly identified variants with FGFR activation, with later confirmation by transforming activity in vitro. Additionally, other factors such as pre-existing mutations of neighboring genes might also influence FGFRi binding. The scarcity of available data, mostly from individual Sq-NSCLC cases as well as a few clinical studies, indicates the need for further research. A number of ongoing clinical trials involve patients with advanced Sq-NSCLC and FGFR mutations and/or translocations (Figure 2).
Potential Markers of FGFRi Resistance
Anti-FGFR therapeutic strategies based on small-molecule chemical tyrosine kinase inhibitors (TKIs) are by far the most extensively explored. However, in clinical and preclinical trials, cancer cells developed diverse resistance mechanisms to FGFRis (5).
FGFRs Mutations
FGFR secondary mutations at the gatekeeper residue and gene fusions are the main mechanisms of resistance to FGF/FGFRis. For instance, the V561M FGFR1 gatekeeper mutation caused resistance to AZD4547 via STAT3 activation and the epithelial-mesenchymal transition in H1581 cells (79) and prolonged exposure of KMS-11 multiple myeloma cells (FGFR3 Y373C) to AZD4547 resulted in a second FGFR3 kinase domain point mutation (V555M), triggering a conformational change and preventing adequate drug binding (80). Additionally, the3T3 cells with FGFR1 N546K and FGFR2 N549D/K were resistant to AZD4547, BGJ398 (infigratinib), erdafitinib, and pemigatinib, and simultaneously they were relatively sensitive to E7090 (a potent selective FGFR1‐3 inhibitor) and futibatinib (selective, irreversible FGFR1–4 inhibitor) (74). The authors also showed that the3T3 cells with concurrent mutations in FGFR3 (S249C and K650M) were greatly resistant to E7090 in comparison to cells with individual mutations. However, such a strong association was not demonstrated for Erdafinib. The FGFR2 mutation V564F in the kinase domain conferred BGJ398 (infigratinib) resistance in cholangiocarcinoma FGFR2 fusion-positive patients (81) and the FGFR2-ASCL5 fusion (Ig2, I-set, a tyrosine kinase domain from FGFR2, and a truncated AMP-binding domain from ACSL5) was reported in post-progression biopsy of a patient with FGFR2-amplified gastric cancer with prior response to the FGFRi LY2874455 (82). FGFR structure analysis has shown that some FGFR gatekeeper mutations lead to steric clashes with FGFRis (78). Consequently, shifting the drug-binding site from the mutation site can overcome resistance to LY2874455, despite FGFR gatekeeper mutations (FGFR1 (V561M), FGFR2 (V564F), FGFR3 (V555M), and FGFR4 (V550M and V550L) (78).
Activation of Multiples Signaling Pathways
PI3K–AKT signaling mediates resistance to FGFRis by directly affecting cell proliferation or activating the mTOR pathway, altering cell metabolism and anti-apoptotic signaling (83). Recently, the dysregulation of RAS/RAF/MAPK pathway through MAPK14 gene (Mitogen-Activated Protein Kinase 14, p38 MAPK) was shown as a driver of resistance to a novel FGFR inhibitor CPL304110 (NCT04149691) (84). Authors revealed that activation of p38 kinase resulted in resistance to CPL304110 in parental cells, while inhibition of p38 MAPK resensitised resistant cells to that FGFRi treatment. Other receptor tyrosine kinases such as hepatocyte growth factor receptor (MET/HGFR) may be upregulated in response to FGFRis, thereby serving as a bypass mechanism for activation of signaling (85, 86). Data from preclinical studies, including cellular models of lung, colorectal, and gastric cancers, indicate that the MET pathway is vital for the growth, survival, and invasive potential of cancers (87). However, little is known of primary resistance to FGFRis. Primary resistance mechanisms may be related to alterations unique to FGFRs and METs. Hepatocyte growth factor (HGF) binding to MET induces activation of various downstream signaling pathways, including the RAS/MAPK and PI3K/AKT pathways, leading to cell proliferation, survival, and migration (87). MET amplification is an important mechanism by which cancers develop resistance to epidermal growth factor receptor (EGFR) inhibitors and FGFRis, and a high MET GCN has been detected in 1–11% of NSCLCs. MET amplification may lead to secondary resistance to EGFR-TKIs in patients with EGFR-mutated lung adenocarcinoma (88). However, MET activation is not observed solely in the presence of amplification, implying other mechanisms of signaling activation (89). These may depend on the molecular aberrations of the MET gene altering MET transcription, MET translation or degradation, or may directly transactivate MET receptor. In 3–4% of NSCLCs, MET splice mutations explained exon 14 skipping, leading to impaired MET degradation (82). Additionally, in vitro models of FGFRi-resistance confirmed a role for AKT and MET activation and clonal expansion of the S266L AKT1 mutation (86). An improved understanding of the complex mechanisms of FGFRi resistance in lung cancer is needed.
Perspectives
Most in vitro studies have demonstrated the potential of FGFR aberrations to predict sensitivity to FGFRis. Unfortunately, clinical data are scarce and discordant.
Important obstacle to patient selection is the availability of archival tumor samples for molecular analysis. Liquid biopsy has emerged as a potential diagnostic approach. Cell-free DNA (cfDNA) analysis is a non-invasive method routinely used in the clinical setting to evaluate secondary resistance mutations to TKIs [reviewed in (90)]. The FGFR alterations detected in cfDNA however, show discordance with tumor molecular profiles. A retrospective analysis of 17 cholangiocarcinoma cases revealed FGFR2-3 alterations in 82.3% of archival tumour samples and 50% of DNA samples (4/5 SNVs, 1/2 amplifications, and 5/13 fusions) (91). Likewise, FGFR2 fusions were detected in the primary tumor and cfDNA in 8 of 12 (67%) patients prior to BGJ398 treatment (NCT02150967) (92). cfDNA has potential for analysis of FGFR2 amplification in gastric and breast cancers. In Sq-NSCLC, cfDNA enabled monitoring of copy number variation and identified a 71% overlap of PIK3CA gain in cfDNA and tumour tissue (phase II clinical trial, GO27912, NCT01493843 (93). cfDNA also has potential for continuous monitoring of the molecular response and resistance mechanisms, as for EGFR TKIs (94). Accordingly, in three patients with cholangiocarcinoma treated with BGJ398 (phase II study, NCT02160041) the FGFR2-BICC1 fusion decreased in cfDNA upon initiation of treatment and increased at the time of radiological progression (81). FGFR2 mutations that potentially confer resistance (V564L, V564F) and activation (N549K) together with alterations in other candidate resistance genes (PTEN and MAP2K1) and a low variant allele frequency of NRAS G12D and BRAF A694T were detected in cfDNA samples from patients on FGFRis who experienced disease progression (91). Notably, because high FGFR mRNA levels were predictive of responsiveness to FGFRis, cell-free RNA (cfRNA) may be an alternative to tissue-based gene-expression analysis. This approach is, however, technically challenging because of the potential for sample contamination with white-blood-cell mRNA. Also, stable normalization transcripts in plasma are unknown, although some results are promising (95).
Discussion
FGFR genomic aberrations facilitate a response to FGFRis, prompting a search for reliable predictive biomarkers. Unfortunately, despite the promising data from preclinical and clinical studies of FGFRis, we are far from identifying specific aberration(s) in FGFRs. FGFR mRNA overexpression, mutations, and fusions show promise, but their discriminatory power is insufficient. Despite the FGFR aberrations in Sq-NSCLC tumors (alterations: mutations and fusions; amplification; mRNA/protein overexpression), the association between the molecular profile and the response to FGFRi treatment is unclear. Small clinical trials of FGFRis in Sq-NSCLC have yielded little data. Moreover, most clinical trials have been of the basket-type design and enrolled small numbers of patients with Sq-NSCLC. Additionally, genomic aberrations other than FGFR could affect the response to FGFRis.
In conclusion, identification of clinically relevant predictive biomarkers of the response and/or secondary resistance to FGFRis in Sq-NSCLC patients is challenging. There is an urgent need for basic and clinical research based on squamous lung cancer histology and deep DNA/RNA profiling to provide insight into cancer biology and to promote discovery of candidate biomarker(s).
Author Contributions
All authors were involved in the concept and design. JM-S wrote the manuscript. JC-W critically reviewed the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by STRATEGMED2 grant (STRATEGMED2/266776/17/NCBR/2015, project “CELONKO”).
Conflict of Interest
JC-W disclose grants, personal fees or non-financial support outside the submitted work from: Grifols, AstraZeneca, MSD, BMS, GSK, Novartis, Chiesi, Roche, Boehringer, CSL Behring, CelonPharma, Amgen, Lekam, Mereo, Takeda.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.780650/full#supplementary-material
References
1. Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Piñeros M, Znaor A, et al. Cancer Statistics for the Year 2020: An Overview. Int J Cancer (2021) 149(4):778–89. doi: 10.1002/ijc.33588
2. Reck M, Remon J, Hellmann MD. First-Line Immunotherapy for Non-Small-Cell Lung Cancer. J Clin Oncol (2022) 40(6):586–97. doi: 10.1200/JCO.21.01497
3. Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab Versus Chemotherapy for PD-L1–positive Non–Small-Cell Lung Cancer. N Engl J Med (2016) 375:1823–33. doi: 10.1056/nejmoa1606774
4. Cancer Genome Atlas Research Network. Comprehensive Genomic Characterization of Squamous Cell Lung Cancers. Nature (2012) 489:519–25. doi: 10.1038/nature11404
5. Babina IS, Turner NC. Advances and Challenges in Targeting FGFR Signalling in Cancer. Nat Rev Cancer (2017) 17:318–32. doi: 10.1038/nrc.2017.8
6. Hashemi-Sadraei N, Hanna N. Targeting FGFR in Squamous Cell Carcinoma of the Lung. Target Oncol (2017) 12:741–55. doi: 10.1007/s11523-017-0513-6
7. Marseglia G, Lodola A, Mor M, Castelli R. Fibroblast Growth Factor Receptor Inhibitors: Patent Review (2015-2019). Expert Opin Ther Pat (2019) 29:965–77. doi: 10.1080/13543776.2019.1688300
8. Michael M, Bang Y-J, Park YS, Kang Y-K, Kim TM, Hamid O, et al. A Phase 1 Study of LY2874455, an Oral Selective Pan-FGFR Inhibitor, in Patients With Advanced Cancer. Target Oncol (2017) 12:463–74. doi: 10.1007/s11523-017-0502-9
9. Lim SH, Sun J-M, Choi Y-L, Kim HR, Ahn S, Lee JY, et al. Efficacy and Safety of Dovitinib in Pretreated Patients With Advanced Squamous Non-Small Cell Lung Cancer With FGFR1 Amplification: A Single-Arm, Phase 2 Study: Dovitinib Infgfr1-Amplified SCC. Cancer (2016) 122:3024–31. doi: 10.1002/cncr.30135
10. Nogova L, Sequist LV, Perez Garcia JM, Andre F, Delord J-P, Hidalgo M, et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J Clin Oncol (2017) 35:157–65. doi: 10.1200/JCO.2016.67.2048
11. Aggarwal C, Redman MW, Lara PN Jr, Borghaei H, Hoffman P, Bradley JD, et al. SWOG S1400D (NCT02965378), a Phase II Study of the Fibroblast Growth Factor Receptor Inhibitor AZD4547 in Previously Treated Patients With Fibroblast Growth Factor Pathway-Activated Stage IV Squamous Cell Lung Cancer (Lung-MAP Substudy). J Thorac Oncol (2019) 14:1847–52. doi: 10.1016/j.jtho.2019.05.041
12. Chellaiah AT, McEwen DG, Werner S, Xu J, Ornitz DM. Fibroblast Growth Factor Receptor (FGFR) 3. Alternative Splicing in Immunoglobulin-Like Domain III Creates a Receptor Highly Specific for Acidic FGF/FGF-1. J Biol Chem (1994) 269:11620–7. doi: 10.1016/s0021-9258(19)78170-8
13. Lieu C, Heymach J, Overman M, Tran H, Kopetz S. Beyond VEGF: Inhibition of the Fibroblast Growth Factor Pathway and Antiangiogenesis. Clin Cancer Res (2011) 17:6130–9. doi: 10.1158/1078-0432.CCR-11-0659
14. Beenken A, Mohammadi M. The FGF Family: Biology, Pathophysiology and Therapy. Nat Rev Drug Discov (2009) 8:235–53. doi: 10.1038/nrd2792
15. Ornitz DM, Itoh N. The Fibroblast Growth Factor Signaling Pathway. Wiley Interdiscip Rev Dev Biol (2015) 4:215–66. doi: 10.1002/wdev.176
16. Turner N, Grose R. Fibroblast Growth Factor Signalling: From Development to Cancer. Nat Rev Cancer (2010) 10:116–29. doi: 10.1038/nrc2780
17. Weiss J, Sos ML, Seidel D, Peifer M, Zander T, Heuckmann JM, et al. Frequent and Focal FGFR1 Amplification Associates With Therapeutically Tractable FGFR1 Dependency in Squamous Cell Lung Cancer. Sci Transl Med (2010) 2:62ra93. doi: 10.1126/scitranslmed.3001451
18. Flockerzi FA, Roggia C, Langer F, Holleczek B, Bohle RM. FGFR1 Gene Amplification in Squamous Cell Carcinomas of the Lung: A Potential Favorable Prognostic Marker for Women and for Patients With Advanced Cancer. Virchows Arch (2018) 472:759–69. doi: 10.1007/s00428-017-2282-0
19. Rooney C, Geh C, Williams V, Heuckmann JM, Menon R, Schneider P, et al. Characterization of FGFR1 Locus in sqNSCLC Reveals a Broad and Heterogeneous Amplicon. PloS One (2016) 11:e0149628. doi: 10.1371/journal.pone.0149628
20. Hibi M, Kaneda H, Tanizaki J, Sakai K, Togashi Y, Terashima M, et al. FGFR Gene Alterations in Lung Squamous Cell Carcinoma Are Potential Targets for the Multikinase Inhibitor Nintedanib. Cancer Sci (2016) 107:1667–76. doi: 10.1111/cas.13071
21. Wynes MW, Hinz TK, Gao D, Martini M, Marek LA, Ware KE, et al. FGFR1 mRNA and Protein Expression, Not Gene Copy Number, Predict FGFR TKI Sensitivity Across All Lung Cancer Histologies. Clin Cancer Res (2014) 20:3299–309. doi: 10.1158/1078-0432.CCR-13-3060
22. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR Landscape in Cancer: Analysis of 4,853 Tumors by Next-Generation Sequencing. Clin Cancer Res (2016) 22:259–67. doi: 10.1158/1078-0432.CCR-14-3212
23. Paik PK, Shen R, Berger MF, Ferry D, Soria J-C, Mathewson A, et al. A Phase Ib Open-Label Multicenter Study of AZD4547 in Patients With Advanced Squamous Cell Lung Cancers. Clin Cancer Res (2017) 23:5366–73. doi: 10.1158/1078-0432.CCR-17-0645
24. Elakad O, Lois A-M, Schmitz K, Yao S, Hugo S, Lukat L, et al. Fibroblast Growth Factor Receptor 1 Gene Amplification and Protein Expression in Human Lung Cancer. Cancer Med (2020) 9:3574–83. doi: 10.1002/cam4.2994
25. Sands JM, Nguyen T, Shivdasani P, Sacher AG, Cheng ML, Alden RS, et al. Next-Generation Sequencing Informs Diagnosis and Identifies Unexpected Therapeutic Targets in Lung Squamous Cell Carcinomas. Lung Cancer (2020) 140:35–41. doi: 10.1016/j.lungcan.2019.12.005
26. Bogatyrova O, Mattsson JSM, Ross EM, Sanderson MP, Backman M, Botling J, et al. FGFR1 Overexpression in Non-Small Cell Lung Cancer Is Mediated by Genetic and Epigenetic Mechanisms and Is a Determinant of FGFR1 Inhibitor Response. Eur J Cancer (2021) 151:136–49. doi: 10.1016/j.ejca.2021.04.005
27. Chatziandreou I, Psaraki A, Paschidis K, Lazaris AC, Saetta AA. Evidence for Frequent Concurrent DCUN1D1, FGFR1, BCL9 Gene Copy Number Amplification in Squamous Cell Lung Cancer. Pathol Res Pract (2021) 221(153412):153412. doi: 10.1016/j.prp.2021.153412
28. Wu Y-M, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer Discov (2013) 3:636–47. doi: 10.1158/2159-8290.CD-13-0050
29. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The Landscape of Kinase Fusions in Cancer. Nat Commun (2014) 5:4846. doi: 10.1038/ncomms5846
30. Wang R, Wang L, Li Y, Hu H, Shen L, Shen X, et al. FGFR1/3 Tyrosine Kinase Fusions Define a Unique Molecular Subtype of Non-Small Cell Lung Cancer. Clin Cancer Res (2014) 20:4107–14. doi: 10.1158/1078-0432.CCR-14-0284
31. Qin A, Johnson A, Ross JS, Miller VA, Ali SM, Schrock AB, et al. Detection of Known and Novel FGFR Fusions in Non-Small Cell Lung Cancer by Comprehensive Genomic Profiling. J Thorac Oncol (2019) 14:54–62. doi: 10.1016/j.jtho.2018.09.014
32. Zhang L, Yu H, Badzio A, Boyle TA, Schildhaus H-U, Lu X, et al. Fibroblast Growth Factor Receptor 1 and Related Ligands in Small-Cell Lung Cancer. J Thorac Oncol (2015) 10:1083–90. doi: 10.1097/JTO.0000000000000562
33. Schultheis AM, Bos M, Schmitz K, Wilsberg L, Binot E, Wolf J, et al. Fibroblast Growth Factor Receptor 1 (FGFR1) Amplification Is a Potential Therapeutic Target in Small-Cell Lung Cancer. Mod Pathol (2014) 27:214–21. doi: 10.1038/modpathol.2013.141
34. Liao RG, Jung J, Tchaicha J, Wilkerson MD, Sivachenko A, Beauchamp EM, et al. Inhibitor-Sensitive FGFR2 and FGFR3 Mutations in Lung Squamous Cell Carcinoma. Cancer Res (2013) 73:5195–205. doi: 10.1158/0008-5472.CAN-12-3950
35. Dutt A, Salvesen HB, Chen T-H, Ramos AH, Onofrio RC, Hatton C, et al. Drug-Sensitive FGFR2 Mutations in Endometrial Carcinoma. Proc Natl Acad Sci USA (2008) 105:8713–7. doi: 10.1073/pnas.0803379105
36. Theelen WS, Mittempergher L, Willems SM, Bosma AJ, Peters DD, van der Noort V, et al. FGFR1, 2 and 3 Protein Overexpression and Molecular Aberrations of FGFR3 in Early Stage Non-Small Cell Lung Cancer: FGFR Expression in Early Stage Non-Small Lung Cancer. J Pathol Clin Res (2016) 2:223–33. doi: 10.1002/cjp2.51
37. Kim Y, Hammerman PS, Kim J, Yoon J-A, Lee Y, Sun J-M, et al. Integrative and Comparative Genomic Analysis of Lung Squamous Cell Carcinomas in East Asian Patients. J Clin Oncol (2014) 32:121–8. doi: 10.1200/JCO.2013.50.8556
38. Majewski IJ, Mittempergher L, Davidson NM, Bosma A, Willems SM, Horlings HM, et al. Identification of Recurrent FGFR3 Fusion Genes in Lung Cancer Through Kinome-Centred RNA Sequencing: Kinase Fusion Genes in NSCLC. J Pathol (2013) 230:270–6. doi: 10.1002/path.4209
39. Chandrani P, Prabhash K, Prasad R, Sethunath V, Ranjan M, Iyer P, et al. Drug-Sensitive FGFR3 Mutations in Lung Adenocarcinoma. Ann Oncol (2017) 28:597–603. doi: 10.1093/annonc/mdw636
40. Kim HR, Kang HN, Shim HS, Kim EY, Kim J, Kim DJ, et al. Co-Clinical Trials Demonstrate Predictive Biomarkers for Dovitinib, an FGFR Inhibitor, in Lung Squamous Cell Carcinoma. Ann Oncol (2017) 28:1250–9. doi: 10.1093/annonc/mdx098
41. Giacomini A, Taranto S, Rezzola S, Matarazzo S, Grillo E, Bugatti M, et al. Inhibition of the FGF/FGFR System Induces Apoptosis in Lung Cancer Cells via C-Myc Downregulation and Oxidative Stress. Int J Mol Sci (2020) 21(24):9376. doi: 10.3390/ijms21249376
42. Pearson A, Smyth E, Babina IS, Herrera-Abreu MT, Tarazona N, Peckitt C, et al. High-Level Clonal FGFR Amplification and Response to FGFR Inhibition in a Translational Clinical Trial. Cancer Discov (2016) 6:838–51. doi: 10.1158/2159-8290.cd-15-1246
43. Dutt A, Ramos AH, Hammerman PS, Mermel C, Cho J, Sharifnia T, et al. Inhibitor-Sensitive FGFR1 Amplification in Human Non-Small Cell Lung Cancer. PloS One (2011) 6:e20351. doi: 10.1371/journal.pone.0020351
44. Fumarola C, Cretella D, La Monica S, Bonelli MA, Alfieri R, Caffarra C, et al. Enhancement of the Anti-Tumor Activity of FGFR1 Inhibition in Squamous Cell Lung Cancer by Targeting Downstream Signaling Involved in Glucose Metabolism. Oncotarget (2017) 8:91841–59. doi: 10.18632/oncotarget.19279
45. Gozgit JM, Wong MJ, Moran L, Wardwell S, Mohemmad QK, Narasimhan NI, et al. Ponatinib (AP24534), a Multitargeted Pan-FGFR Inhibitor With Activity in Multiple FGFR-Amplified or Mutated Cancer Models. Mol Cancer Ther (2012) 11:690–9. doi: 10.1158/1535-7163.MCT-11-0450
46. Ren M, Hong M, Liu G, Wang H, Patel V, Biddinger P, et al. Novel FGFR Inhibitor Ponatinib Suppresses the Growth of Non-Small Cell Lung Cancer Cells Overexpressing FGFR1. Oncol Rep (2013) 29:2181–90. doi: 10.3892/or.2013.2386
47. Zhang J, Zhang L, Su X, Li M, Xie L, Malchers F, et al. Translating the Therapeutic Potential of AZD4547 in FGFR1-Amplified Non-Small Cell Lung Cancer Through the Use of Patient-Derived Tumor Xenograft Models. Clin Cancer Res (2012) 18:6658–67. doi: 10.1158/1078-0432.CCR-12-2694
48. Wang L, Šuštić T, Leite de Oliveira R, Lieftink C, Halonen P, van de Ven M, et al. A Functional Genetic Screen Identifies the Phosphoinositide 3-Kinase Pathway as a Determinant of Resistance to Fibroblast Growth Factor Receptor Inhibitors in FGFR Mutant Urothelial Cell Carcinoma. Eur Urol (2017) 71:858–62. doi: 10.1016/j.eururo.2017.01.021
49. Andre F, Ranson M, Dean E, Varga A, van der Noll R, Stockman PK, et al. Abstract LB-145: Results of a Phase I Study of AZD4547, an Inhibitor of Fibroblast Growth Factor Receptor (FGFR), in Patients With Advanced Solid Tumors. Cancer Res (2013) 73 Suppl 8:LB–145-LB-145. doi: 10.1158/1538-7445.am2013-lb-145
50. Voss MH, Hierro C, Heist RS, Cleary JM, Meric-Bernstam F, Tabernero J, et al. Open-Label, Multicenter, Dose-Escalation Study of the Oral Selective FGFR Inhibitor Debio 1347 in Patients With Advanced Solid Tumors Harboring FGFR Gene Alterations. Clin Cancer Res (2019) 25:2699–707. doi: 10.1158/1078-0432.CCR-18-1959
51. Papadopoulos KP, El-Rayes BF, Tolcher AW, Patnaik A, Rasco DW, Harvey RD, et al. A Phase 1 Study of ARQ 087, an Oral Pan-FGFR Inhibitor in Patients With Advanced Solid Tumours. Br J Cancer (2017) 117:1592–9. doi: 10.1038/bjc.2017.330
52. Seo AN, Jin Y, Lee HJ, Sun P-L, Kim H, Jheon S, et al. FGFR1 Amplification Is Associated With Poor Prognosis and Smoking in Non-Small-Cell Lung Cancer. Virchows Arch (2014) 465(5):547–58. doi: 10.1007/s00428-014-1634-2
53. Baldia PH, Maurer A, Heide T, Rose M, Stoehr R, Hartmann A, et al. Fibroblast Growth Factor Receptor (FGFR) Alterations in Squamous Differentiated Bladder Cancer: A Putative Therapeutic Target for a Small Subgroup. Oncotarget (2016) 7:71429–39. doi: 10.18632/oncotarget.12198
54. Ahmad I, Iwata T, Leung HY. Mechanisms of FGFR-Mediated Carcinogenesis. Biochim Biophys Acta (2012) 1823:850–60. doi: 10.1016/j.bbamcr.2012.01.004
55. Schuler M, Cho BC, Sayehli CM, Navarro A, Soo RA, Richly H, et al. Rogaratinib in Patients With Advanced Cancers Selected by FGFR mRNA Expression: A Phase 1 Dose-Escalation and Dose-Expansion Study. Lancet Oncol (2019) 20:1454–66. doi: 10.1016/S1470-2045(19)30412-7
56. Joerger M, Cho BC, Mach N, Caballero C, Soo RA, Wirth M, et al. Early Clinical Experience With the Pan-FGFR Inhibitor Rogaratinib in Patients With Non-Small Cell Lung Cancer Selected Based on FGFR mRNA Expression Levels. J Clin Oncol (2019) 37 Suppl 15:e20661–1. doi: 10.1200/jco.2019.37.15_suppl.e20661
57. Fischer H, Taylor N, Allerstorfer S, Grusch M, Sonvilla G, Holzmann K, et al. Fibroblast Growth Factor Receptor-Mediated Signals Contribute to the Malignant Phenotype of Non-Small Cell Lung Cancer Cells: Therapeutic Implications and Synergism With Epidermal Growth Factor Receptor Inhibition. Mol Cancer Ther (2008) 7(10):3408–19. doi: 10.1158/1535-7163.MCT-08-0444
58. Grünewald S, Politz O, Bender S, Héroult M, Lustig K, Thuss U, et al. Rogaratinib: A Potent and Selective Pan-FGFR Inhibitor With Broad Antitumor Activity in FGFR-Overexpressing Preclinical Cancer Models. Int J Cancer (2019) 145:1346–57. doi: 10.1002/ijc.32224
59. Joerger M, Soo RA, Cho BC, Navarro A, Sayehli CM, Richly H, et al. A Novel mRNA-Based Patient Selection Strategy Identifies Fibroblast Growth Factor Receptor (FGFR) Inhibitor-Sensitive Tumors: Results From Rogaratinib Phase-1 Study. Ann Oncol (2017) 28 Suppl 5:v126–7. doi: 10.1093/annonc/mdx367.013
60. Addeo A, Joerger M, Rothschild S, Eboulet EI, Godar G, Waibel-Pachinger C, et al. Fibroblast Growth Factor Receptor (FGFR) Inhibitor Rogaratinib in Patients With Advanced Pretreated Squamous-Cell Non-Small Cell Lung Cancer Over-Expressing FGFR mRNA: The SAKK 19/18 Phase II Study. J Clin Oncol (2021) 39(15suppl):e21119–9. doi: 10.1200/JCO.2021.39.15_suppl.e21119
61. Ng TL, Yu H, Smith DE, Boyle TA, York ER, Leedy S, et al. Preselection of Lung Cancer Cases Using FGFR1 mRNA and Gene Copy Number for Treatment With Ponatinib. Clin Lung Cancer (2019) 20:e39–51. doi: 10.1016/j.cllc.2018.09.001
62. Li JJ, Yan S, Pan Y, Liu Z, Liu Y, Deng Q, et al. FGFR Genes Mutation Is an Independent Prognostic Factor and Associated With Lymph Node Metastasis in Squamous Non-Small Cell Lung Cancer. Cancer Biol Ther (2018) 19:1108–16. doi: 10.1080/15384047.2018.1480294
63. Pollock PM, Cancer Genome Project, Gartside MG, Dejeza LC, Powell MA, Mallon MA, et al. Frequent Activating FGFR2 Mutations in Endometrial Carcinomas Parallel Germline Mutations Associated With Craniosynostosis and Skeletal Dysplasia Syndromes. Oncogene (2007) 26:7158–62. doi: 10.1038/sj.onc.1210529
64. Gust KM, McConkey DJ, Awrey S, Hegarty PK, Qing J, Bondaruk J, et al. Fibroblast Growth Factor Receptor 3 Is a Rational Therapeutic Target in Bladder Cancer. Mol Cancer Ther (2013) 12:1245–54. doi: 10.1158/1535-7163.mct-12-1150
65. Taylor JG6, Cheuk AT, Tsang PS, Chung J-Y, Song YK, Desai K, et al. Identification of FGFR4-Activating Mutations in Human Rhabdomyosarcomas That Promote Metastasis in Xenotransplanted Models. J Clin Invest (2009) 119:3395–407. doi: 10.1172/JCI39703
66. Tanizaki J, Ercan D, Capelletti M, Dodge M, Xu C, Bahcall M, et al. Identification of Oncogenic and Drug-Sensitizing Mutations in the Extracellular Domain of FGFR2. Cancer Res (2015) 75:3139–46. doi: 10.1158/0008-5472.CAN-14-3771
67. Zhou Z, Liu Z, Ou Q, Wu X, Wang X, Shao Y, et al. Targeting FGFR in Non-Small Cell Lung Cancer: Implications From the Landscape of Clinically Actionable Aberrations of FGFR Kinases. Cancer Biol Med (2021) 18(2):490–501. doi: 10.20892/j.issn.2095-3941.2020.0120
68. Patani H, Bunney TD, Thiyagarajan N, Norman RA, Ogg D, Breed J, et al. Landscape of Activating Cancer Mutations in FGFR Kinases and Their Differential Responses to Inhibitors in Clinical Use. Oncotarget (2016) 7:24252–68. doi: 10.18632/oncotarget.8132
69. Del Piccolo N, Placone J, Hristova K. Effect of Thanatophoric Dysplasia Type I Mutations on FGFR3 Dimerization. Biophys J (2015) 108:272–8. doi: 10.1016/j.bpj.2014.11.3460
70. Sarabipour S, Hristova K. FGFR3 Unliganded Dimer Stabilization by the Juxtamembrane Domain. J Mol Biol (2015) 427:1705–14. doi: 10.1016/j.jmb.2015.02.013
71. Mertens F, Johansson B, Fioretos T, Mitelman F. The Emerging Complexity of Gene Fusions in Cancer. Nat Rev Cancer (2015) 15:371–81. doi: 10.1038/nrc3947
72. Tiseo M, Gelsomino F, Alfieri R, Cavazzoni A, Bozzetti C, De Giorgi AM, et al. FGFR as Potential Target in the Treatment of Squamous Non Small Cell Lung Cancer. Cancer Treat Rev (2015) 41:527–39. doi: 10.1016/j.ctrv.2015.04.011
73. De Luca A, Esposito Abate R, Rachiglio AM, Maiello MR, Esposito C, Schettino C, et al. FGFR Fusions in Cancer: From Diagnostic Approaches to Therapeutic Intervention. Int J Mol Sci (2020) 21(18):6856. doi: 10.3390/ijms21186856
74. Nakamura IT, Kohsaka S, Ikegami M, Ikeuchi H, Ueno T, Li K, et al. Comprehensive Functional Evaluation of Variants of Fibroblast Growth Factor Receptor Genes in Cancer. NPJ Precis Oncol (2021) 5(1):66. doi: 10.1038/s41698-021-00204-0
75. Bahleda R, Italiano A, Hierro C, Mita A, Cervantes A, Chan N, et al. Multicenter Phase I Study of Erdafitinib (JNJ-42756493), Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients With Advanced or Refractory Solid Tumors. Clin Cancer Res (2019) 25:4888–97. doi: 10.1158/1078-0432.CCR-18-3334
76. Mazzaferro V, El-Rayes BF, Droz dit Busset M, Cotsoglou C, Harris WP, Damjanov N, et al. Derazantinib (ARQ 087) in Advanced or Inoperable FGFR2 Gene Fusion-Positive Intrahepatic Cholangiocarcinoma. Br J Cancer (2019) 120:165–71. doi: 10.1038/s41416-018-0334-0
77. Chae YK, Hong F, Vaklavas C, Cheng HH, Hammerman P, Mitchell EP, et al. Phase II Study of AZD4547 in Patients With Tumors Harboring Aberrations in the FGFR Pathway: Results From the NCI-MATCH Trial (EAY131) Subprotocol W. J Clin Oncol (2020) 38:2407–17. doi: 10.1200/JCO.19.02630
78. Wu D, Guo M, Min X, Dai S, Li M, Tan S, et al. LY2874455 Potently Inhibits FGFR Gatekeeper Mutants and Overcomes Mutation-Based Resistance. Chem Commun (Camb) (2018) 54:12089–92. doi: 10.1039/c8cc07546h
79. Ryan MR, Sohl CD, Luo B, Anderson KS. The FGFR1 V561M Gatekeeper Mutation Drives AZD4547 Resistance Through STAT3 Activation and EMT. Mol Cancer Res (2019) 17:532–43. doi: 10.1158/1541-7786.MCR-18-0429
80. Chell V, Balmanno K, Little AS, Wilson M, Andrews S, Blockley L, et al. Tumour Cell Responses to New Fibroblast Growth Factor Receptor Tyrosine Kinase Inhibitors and Identification of a Gatekeeper Mutation in FGFR3 as a Mechanism of Acquired Resistance. Oncogene (2013) 32:3059–70. doi: 10.1038/onc.2012.319
81. Goyal L, Saha SK, Liu LY, Siravegna G, Leshchiner I, Ahronian LG, et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients With FGFR2 Fusion–Positive Cholangiocarcinoma. Cancer Discov (2017) 7:252–63. doi: 10.1158/2159-8290.cd-16-1000
82. Kim SY, Ahn T, Bang H, Ham JS, Kim J, Kim ST, et al. Acquired Resistance to LY2874455 in FGFR2-Amplified Gastric Cancer Through an Emergence of Novel FGFR2-ACSL5 Fusion. Oncotarget (2017) 8:15014–22. doi: 10.18632/oncotarget.14788
83. Datta J, Damodaran S, Parks H, Ocrainiciuc C, Miya J, Yu L, et al. Akt Activation Mediates Acquired Resistance to Fibroblast Growth Factor Receptor Inhibitor BGJ398. Mol Cancer Ther (2017) 16:614–24. doi: 10.1158/1535-7163.mct-15-1010
84. Zarczynska I, Gorska-Arcisz M, Cortez AJ, Kujawa KA, Wilk AM, Skladanowski AC, et al. P38 Mediates Resistance to FGFR Inhibition in Non-Small Cell Lung Cancer. Cells (2021) 10(12):3363. doi: 10.3390/cells10123363
85. Kim S-M, Kim H, Yun MR, Kang HN, Pyo K-H, Park HJ, et al. Activation of the Met Kinase Confers Acquired Drug Resistance in FGFR-Targeted Lung Cancer Therapy. Oncogenesis (2016) 5:e241. doi: 10.1038/oncsis.2016.48
86. Gimenez-Xavier P, Pros E, Aza A, Moran S, Tonda R, Esteve-Codina A, et al. Deep Analysis of Acquired Resistance to FGFR1 Inhibitor Identifies MET and AKT Activation and an Expansion of AKT1 Mutant Cells. Oncotarget (2018) 9:31549–58. doi: 10.18632/oncotarget.25862
87. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in Cancer: Rationale and Progress. Nat Rev Cancer (2012) 12:89–103. doi: 10.1038/nrc3205
88. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science (2007) 316:1039–43. doi: 10.1126/science.1141478
89. Sacco JJ, Clague MJ. Dysregulation of the Met Pathway in Non-Small Cell Lung Cancer: Implications for Drug Targeting and Resistance. Transl Lung Cancer Res (2015) 4:242–52. doi: 10.3978/j.issn.2218-6751.2015.03.05
90. Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating Liquid Biopsies Into the Management of Cancer. Nat Rev Clin Oncol (2017) 14:531–48. doi: 10.1038/nrclinonc.2017.14
91. Moss TJ, Rodon Ahnert J, Oakley HD, Kahle M, Karp DD, Pant S, et al. Baseline cfDNA Characteristics and Evolution of cfDNA Profile During Treatment With Selective FGFR Inhibitor TAS-120. J Clin Oncol (2019) 37 Suppl 15:3056–6. doi: 10.1200/jco.2019.37.15_suppl.3056
92. Makawita S, Kelley RK, Roychowdhury S, Weiss KH, Abou-Alfa GK, Macarulla T, et al. Circulating Free DNA (cfDNA) and Tissue Next-Generation Sequencing Analysis in a Phase II Study of Infigratinib (BGJ398) for Cholangiocarcinoma With FGFR2 Fusions. J Clin Oncol (2020) 38 Suppl 4:579–9. doi: 10.1200/jco.2020.38.4_suppl.579
93. Chen X, Chang C-W, Spoerke JM, Yoh KE, Kapoor V, Baudo C, et al. Low-Pass Whole-Genome Sequencing of Circulating Cell-Free DNA Demonstrates Dynamic Changes in Genomic Copy Number in a Squamous Lung Cancer Clinical Cohort. Clin Cancer Res (2019) 25:2254–63. doi: 10.1158/1078-0432.CCR-18-1593
94. Macías M, Alegre E, Alkorta-Aranburu G, Patiño-García A, Mateos B, Andueza MP, et al. The Dynamic Use of EGFR Mutation Analysis in Cell-Free DNA as a Follow-Up Biomarker During Different Treatment Lines in Non-Small-Cell Lung Cancer Patients. Dis Markers (2019) 2019:7954921. doi: 10.1155/2019/7954921
Keywords: fibroblast growth factor receptor, FGFR inhibitors, squamous non-small cell lung cancer (Sq-NSCLC), molecular biomarkers, targeted therapy, FGFR1, FGFR2, FGFR3
Citation: Moes-Sosnowska J and Chorostowska-Wynimko J (2022) Fibroblast Growth Factor Receptor 1-4 Genetic Aberrations as Clinically Relevant Biomarkers in Squamous Cell Lung Cancer. Front. Oncol. 12:780650. doi: 10.3389/fonc.2022.780650
Received: 21 September 2021; Accepted: 04 March 2022;
Published: 25 March 2022.
Edited by:
Massimo Broggini, Mario Negri Pharmacological Research Institute (IRCCS), ItalyReviewed by:
Torsten Gerriet Blum, HELIOS Klinikum Emil von Behring, GermanyErnest Nadal, Catalan Institute of Oncology, Spain
Copyright © 2022 Moes-Sosnowska and Chorostowska-Wynimko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joanna Chorostowska-Wynimko, ai5jaG9yb3N0b3dza2FAZ21haWwuY29t