 Yanzi Pei
Yanzi Pei Yujie Qian1
Yujie Qian1 Li Tan
Li Tan- 1Center for Medical Research and Innovation, Shanghai Pudong Hospital, Fudan University Pudong Medical Center, and Shanghai Key Laboratory of Medical Epigenetics, Institutes of Biomedical Sciences, Fudan University, Shanghai, China
- 2Department of Anesthesiology, Zhongshan Hospital, Fudan University, Shanghai, China
Ferroptosis is an evolutionarily conserved form of regulated cell death triggered by iron-dependent phospholipid peroxidation. Ferroptosis contributes to the maintenance of tissue homeostasis under physiological conditions while its aberration is tightly connected with lots of pathophysiological processes such as acute tissue injury, chronic degenerative disease, and tumorigenesis. Epigenetic regulation controls chromatin structure and gene expression by writing/reading/erasing the covalent modifications on DNA, histone, and RNA, without altering the DNA sequence. Accumulating evidences suggest that epigenetic regulation is involved in the determination of cellular vulnerability to ferroptosis. Here, we summarize the recent advances on the epigenetic mechanisms that control the expression of ferroptosis-associated genes and thereby the ferroptosis process. Moreover, the potential value of epigenetic drugs in targeting or synergizing ferroptosis during cancer therapy is also discussed.
Introduction
Ferroptosis, which was first proposed by Dixon et al. in 2012, is a novel programmed cell death driven by iron-dependent accumulation of lipid peroxidation (1–7). As an evolutionally-conserved cell death form, ferroptosis plays a critical role in diverse physiological and pathological processes (8). Apparently, understanding the comprehensive molecular mechanisms of ferroptosis has great biological importance and clinical significance. Epigenetic regulation controls gene expression by writing/reading/erasing the covalent modifications on DNA, histone, and RNA, without altering the DNA sequence (9). Accumulating evidence suggests that epigenetic regulation modulates the expression dosages of ferroptosis-associated genes and consequently contributes to the determination of cell sensitivity to ferroptosis. Here we summarize current knowledge on the role of epigenetic regulation in ferroptosis and its implication in cancer therapy.
The Regulatory Circuits of Ferroptosis
Biochemistry of Ferroptosis
Since the ferroptosis was first defined in 2012, the core biochemical components of ferroptosis have been rapidly identified in the past decade.
Lipid Peroxidation
Polyunsaturated fatty acids are susceptible to lipid peroxidation and essential for the ferroptosis process (10). The abundance and localization of polyunsaturated fatty acids determine the level of intracellular lipid peroxidation and furtherly decide the sensitivity of cells to ferroptosis (11). Phosphatidylethanolamines (PEs) containing arachidonic acid are the major phospholipid that peroxides and promotes ferroptosis (7, 12). Further on, the PE-Coenzyme-A derivatives form and insert into phospholipid, which was defined as a necessary step of pro-ferroptotic signal production (7). In 2015, through the haploid genetic screening, Dixon et al. identified 9 lipid metabolism-associated genes, including the lipid remodeling gene LPCAT3 and fat acid metabolism gene ACSL4, that play essential roles in ferroptosis (5). Doll et al. convincingly proved that ACSL4 is involved in the generation of pro-ferroptotic state (7).
Intracellular Iron
Iron complexes or loosely bound iron structure are essential for the formation of reactive oxygen (13). In eukaryotic cells, the uncoordinated redox-active Fe2+ that was temporally released in the plasma is generally referred as “The Labile Iron Pool” or “free Fe2+” (14). Technically, free Fe2+ catalyzes the formation of hydroxyl radical and hydroxide from H2O2 through “Fenton reaction”. Consistent to the name of “ferroptosis”, both Fenton reaction and iron-dependent enzymes are the formation of reactive oxygen, which causes severe oxidative damage to neighbor cell structure, are key players in ferroptosis (15, 16). Conversely, treatment with iron chelators such as could inhibit ferroptosis. Under normal physiological condition, the level of iron is well regulated by transferrin (extracellular environment) and ferritin (intracellular environment). Apparently, the signaling pathways that alter iron metabolism have potential effect on the regulation of ferroptosis.
Glutathione and GPX4
Glutathione (GSH) metabolism was identified as the first pathway regulating the ferroptosis process. The hydro-sulfuryl structure makes GSH as a commonly considered antioxidant and free radical scavenger in the intracellular environment (17). In 1986, the glutamate-cystine transportation system was identified by Bannai et al. (18). The transporter is composed by one light chain subunit and one heavy chain subunit, which were respectively encoded by SLC7A11 and SLC3A2. System transfers glutamate out of cells and cystine into cells at a ratio of 1:1, and then cystine is reduced into cysteine which participates in the synthesis of GSH (Figure 1) (19). High concentration of extracellular glutamate inhibits cystine uptake through inhibition of Xc- leading to glutathione decrease and oxidative cell death (20, 21). Indeed, the first ferroptosis inducer, erastin, mainly targets on system (1).
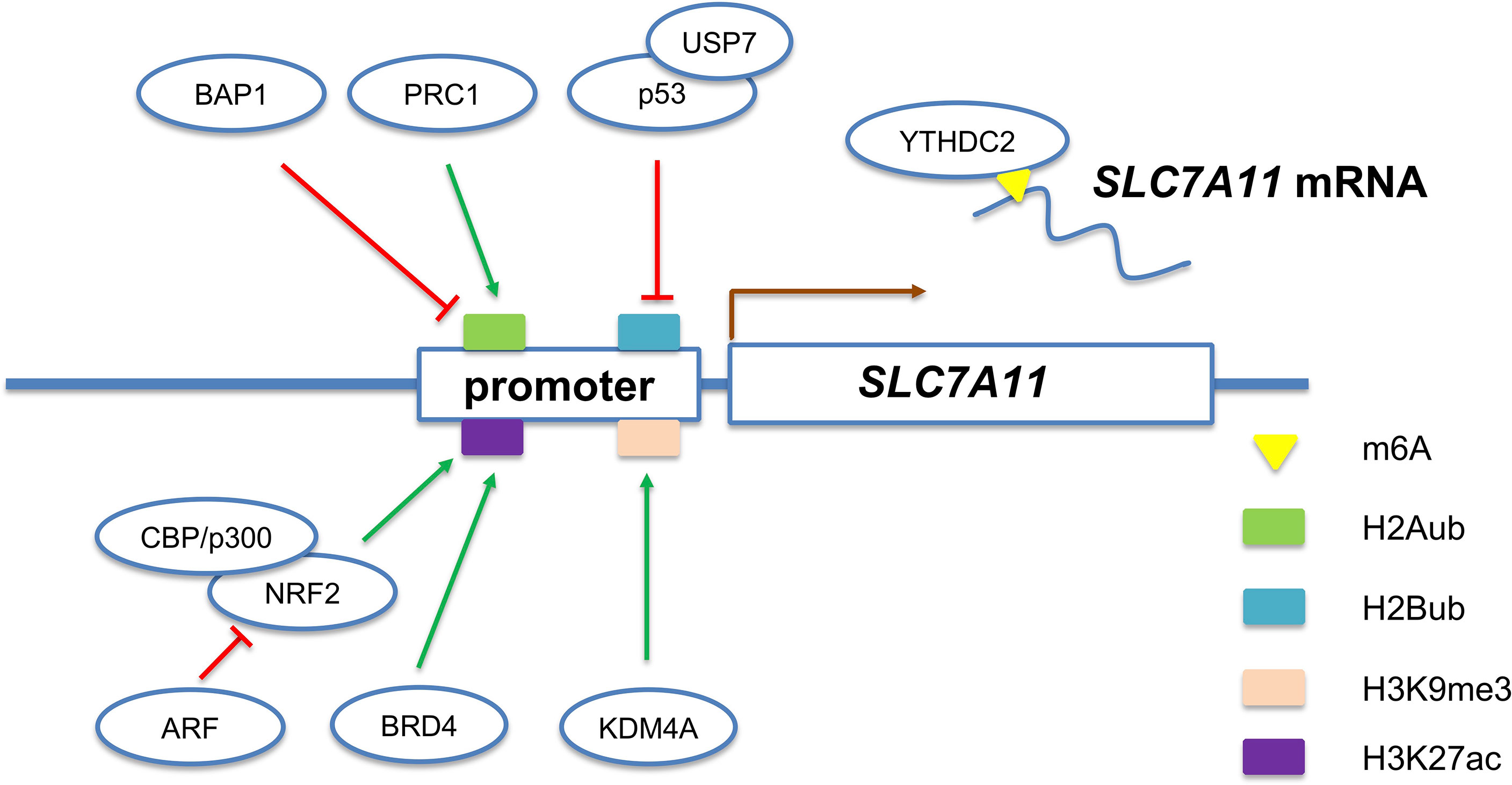
Figure 1 Multiple epigenetic mechanisms regulate the expression of SLC7A11 gene, a representative ferroptosis-associated gene.
The glutathione peroxidases (GPXs) are series of peroxide-degrading enzymes. GPX4 uses GSH as an essential cofactor to prevent lipid peroxide and maintain redox homeostasis (22). In 2008, Seiler et al. identified lipid peroxidation as the key mediator of cell death in glutathione peroxidase 4 (GPX4) knockout cells (23). Thus, people began to consider this type of cell death different from either apoptosis or necrosis. Several small molecule compounds were screened out as ferroptosis inducer (24–27). Among them, RSL3 targets on GPX4 (25).
The FSP1-CoQ10 Pathway
Independent of GPX4 regulatory pathway, the FSP1-CoQ pathway is a novel ferroptosis pathway identified by Doll et al. through an expression cloning approach to identify genes in human cancer cells that are able to complement the loss of GPX4 (6).
Doll et al. revealed that apoptosis-inducing factor mitochondria-associated 2 (AIFM2, renamed as FSP1) overexpression can largely abrogate GPX4 inhibition-induced ferroptosis. A previous study showed that FSP1 functions as a NADP dependent coenzyme Q (CoQ) oxidoreductase in vitro (28). CoQ10 is a mobile lipophilic electron carrier that endogenously synthesizes lipid-soluble antioxidants and acts as a lipophilic free radical-trapping agents (RTAs) in the plasma membrane (29). Intriguingly, FSP1 overexpression fails to suppress ferroptosis in both CoQ2 knockout cells and in cells treated with the CoQ2 inhibitor (6, 30). CoQ2 is the enzyme that catalyzes the first step in CoQ10 biosynthesis, and the soluble analog of CoQ10 is sufficient for suppressing ferroptosis and lipid peroxidation (6, 30). These two latest studies clearly suggest that FSP1 acts parallel to GPX4 to inhibit ferroptosis by regulating the nonmitochondrial CoQ10 antioxidant system.
The DHODH-CoQH2 Pathway
Dihydroorotate dehydrogenase (DHODH), an enzyme essential for the de novo biosynthesis of pyrimidine-based nucleotides, is a known therapeutic target for multiple diseases (31). Furthermore, DHODH inhibitors, including brequinar, leflunomide, and teriflunomide, have been clinically evaluated but failed to receive FDA approval for the treatment of cancer (32–34). Mao et al. identified DHODH as the third anti-ferroptosis pathway, independent of GPX4 and FSP1 (35). Mechanistically, DHODH in the mitochondrial inner membrane regulates the production of CoQH2, a radical-trapping antioxidant in mitochondrial. Importantly, DHODH inhibitor brequinar selectively suppresses GPX4-low tumor growth by inducing ferroptosis, whereas combined treatment with brequinar and sulfasalazine synergistically induces ferroptosis and suppresses GPX4-high tumor growth.
The GCH1-BH4 Pathway
Tetrahydrobiopterin (BH4) is a redox-active cofactor involved in the production of nitric oxide, neurotransmitters, and aromatic amino acids (36). The GCH1-PTS-SPR pathway catalyzes GTP to BH4, and GCH1 is a rate-limiting enzyme in the synthesis of BH4 (37, 38).
Kraft et al. found that the overexpression of GCH1 provide protection to against ferroptosis by abolishing lipid peroxidation (36). GCH1 overexpression exhibits robust protection against RSL3- and imidazole ketone erastin (IKE)-induced ferroptosis and genetic ablation of GPX4-induced ferroptosis but does not protect cells against inducers of apoptosis and is only marginally effective against necroptosis. Those results indicate that GCH1 selectively inhibits ferroptotic cell death (36).
Intriguingly, BH4 loss in cells leads the accumulation of coenzyme A, NADP, and oxidized GSH (GSSG) in cells. Further, the elevation of reduced CoQ10 in cells with GCH1 overexpression have been detected (38). Thus, these results indicate that the GCH1-BH4 pathway acts as an endogenous antioxidant pathway to inhibit ferroptosis through a mechanism independent of the GPX4 signal pathway.
The Signal Pathways of Ferroptosis
Several canonical oncogenic and tumor suppressive pathways have been reported to converge to the ferroptosis process. In general, these pathways alter the ferroptosis sensitivity through modulating the expression levels and enzymatic activities of core ferroptosis executors.
The p53 Pathway
The p53 pathway inhibits cystine uptake and sensitize cells to ferroptosis through repressing SLC7A11 expression (39). Notably, the acetylation–defective mutant p53KR loses the function of inducing cell-cycle arrest but still retains the ability to regulate SLC7A11. Moreover, the spermidine/spermine N1-acetyltransferase 1 (SAT1) gene, which encodes a rate-limiting enzyme in polyamine catabolism, was identified as a transcription target of p53 and promote ferroptosis through conversing spermidine and spermine back to putrescine (40). However, there were also some controversial reports on the function of p53 pathway in ferroptosis. For instance, the dipeptidyl-peptidase-4 (DPP4) can be blocked by p53, resulting in resistance to ferroptosis (41). Alternatively, the wild-type p53 stabilization can delay the induction of ferroptosis in cancer cells upon system inhibition (42). These findings indicate a content-dependent role of p53 in the regulation of ferroptosis.
The KEAP1-NRF2 Pathway
It has been well-established that nuclear factor erythroid 2-related factor 2 (NRF2) pathway plays an essential role in antioxidant response. The correlation between NRF2 pathway and ferroptosis has also been studied (43, 44). NRF2 upregulates system and thereby protects brain tumor cells from ferroptosis (45). Since the NRF2 pathway is commonly activated in diverse malignant tumors, it is likely that aberrant NRF2 activation contributes to protect tumor cells against ferroptosis. A recent work revealed that 3D organoid culture causes ferroptosis and insufficient NRF2 activation leads to the failure of establishment of organoids (46). In addition, the tumor suppressor ARF (CDKN2A) has recently been identified as a binding partner for NRF2 and impacts ferroptosis sensitivity (47). Mechanistically, ARF represses NRF2-induced transcriptional upregulation of SLC7A11 and other antioxidant genes.
The Hippo Pathway
Hippo pathway controls organ size by regulating cell proliferation, apoptosis, and stem cell self-renewal (48). Wu et al. observed that high cell density protects many types of cells against ferroptosis during the in vitro cell culture. Furthermore, they revealed that the cell density-dependent acquisition of ferroptosis resistance is triggered by E-cadherin-mediated activation of intracellular NF2 (also known as merlin) and Hippo signaling pathway (49, 50). Antagonizing this signaling allows the nuclear translocation of proto-oncogenic transcriptional co-activator YAP and promotes ferroptosis by upregulating several ferroptosis modulators, including ACSL4 and TFRC (50). The identification of E-Cad/Hippo/YAP/ACSL4 axis may explain a long term-observed phenotype that mesenchymal cells are more sensitive to ferroptosis than epithelial cells (50). Similarly, epithelial-mesenchymal transition (EMT) increases the vulnerability of cells to ferroptosis, which may be partially attributed to the inactivation of Hippo pathway during EMT (51).
Besides the above three well-established signaling pathways, other canonical development and disease-associated pathways such as RAS-RAF-MAPK and PI3K/Akt also have intersection with the core regulatory circuity of ferroptosis and participate in the regulation of ferroptosis process.
Epigenetic Mechanisms Underlying Ferroptosis
DNA Modification
DNA methylation is the most common epigenetic modification that has been studied in gene regulation. Homocysteine treatment induced DNA methylation of GPX4 gene in nucleus pulposus, leading to ferroptosis sensitivity (52). DNA hypermethylation of CDH1 gene promoter in head and neck cancer cells repressed E-cadherin (encoding by CDH1) expression and increased ferroptosis susceptibility (51). These two cases clearly demonstrate that DNA methylation is involved in the epigenetic silencing of ferroptosis-associated genes. However, whether other ferroptosis-associated genes are also affected by DNA methylation requires further study. Moreover, TET (ten-eleven translocation) proteins could catalyze 5mC oxidation, which in turn initiates the active or passive DNA demethylation (53). It is still unclear whether TET proteins-mediated DNA demethylation also plays a role in the regulation of ferroptosis.
Histone Modifications
Histones form the framework of DNA entangling (54). The tails of four core histones (H2A, H2B, H3 and H4) are proved to undergo chemical modifications, including lysine methylation/acetylation, arginine methylation/citrullination and serine/threonine/tyrosine phosphorylation (55). These chemical modifications alter the interaction between histones and other nuclear proteins including the transcriptional machine, thereby changing the expression of targeted genes.
Histone acetylation marks are written by histone acetyltransferases (HATs), read by bromodomains (BRDs), and erased by histone deacetylases (HDACs). NRF2 activates the transcription of SLC7A11 gene partially through recruiting HATs (CBP and p300) (47, 56). Moreover, the expression of many ferroptosis-associated genes (GPX4, SLC7A11, and SLC3A2) were down-regulated in breast and lung cancer cell lines upon BRD4 knockdown (57). The inhibition of BRD4 also enhances the expression of a histone deacetylase called sirtuin 1 (SIRT1) (57). Additionally, SIRT1 causes epigenetic reprogramming of epithelial-mesenchymal transition (EMT) thus promotes ferroptosis in head and neck cancer (51).
H2A ubiquitination/de-ubiquitination play a critical role in the regulation of SLC7A11 expression and erastin-induced ferroptosis (Figure 1). PRC1 complex, the best-known ubiquitin ligase of H2Aub, is responsible for the establishment of H2A ubiquitination on SLC7A11 promoter (58). In contrast, a nuclear deubiquitinating enzyme named BRCA1-associated protein 1 (BAP1) could decrease the H2A ubiquitination occupancy on the SLC7A11 promoter (59). Interestingly, although H2Aub is generally correlated with gene repression, both BAP1 and PRC1 represses SLC7A11 expression. The weird results indicate that BAP1 and PRC1 coordinately repress SLC7A11 expression through dynamic regulation of H2Aub levels on the SLC7A11 promoter. However, the exact role of H2Aub in SLC7A11 gene expression requires further research. In addition to H2A, mono-ubiquitination of histone H2B on lysine 120 is an epigenetic active marker associated with SLC7A11 expression. Wang et al. revealed that P53-mediated repression of SLC7A11 is dependent on USP7-mediated H2B de-ubiquitination (60).
The di- or tri-methylation of H3K9 are well-established epigenetic marks of heterochromatin and associated with transcriptional silencing (61). Inhibition of SUV39H1 (one of histone H3K9me3 methyltransferases) by small chemical molecules or siRNA upregulates DPP4 expression through reducing the H3K9me3, thereby inducing iron accumulation, lipid peroxidation, and ferroptosis (62). In contrast, KDM3B, a histone H3 lysine 9 demethylase, was reported to prevent erastin-induced ferroptosis of HT-1080 cells (63). Mechanistically, KDM3B knockdown did not change the H3K9 methylation level on the SLC7A11 promoter, while KDM3B cooperates with transcription factor ATF4 to upregulate the expression of SLC7A11. Also, KDM4A, a histone demethylase, was revealed to regulate SLC7A11 transcription by controlling H3K9me3 demethylation in the promoter of SLC7A11 (Figure 1) (64). Besides H3K9, multiple lysine and arginine residues of histones (such as H3R2, H3K4, H3K27, H3K79, H4R3, H4K20, and H2BK5) also undergo methylation/demethylation dynamics and exhibit pleiotropic roles in gene transcription (65). Therefore, it is of great interest to determine whether the histone methylation of other sites and their writers, readers, and erasers also participate in the regulation of ferroptosis-associated genes.
RNA Modifications
N6-methyladenosine (m6A) RNA modification emerges in recent years as a new layer of regulatory mechanism controlling gene expression in eukaryotes (66). The m6A RNA modification is a reversible epigenetic modification that targets on mRNA and noncoding RNAs. The m6A modification regulates gene expression by affecting the fate of the modified RNA molecules (67). Intriguingly, m6A modification has been observed to play a regulatory function in ferroptosis. The m6A reader YTH domain containing 2 (YTHDC2) can bind to SLC7A11 mRNA and thereafter promotes its decay (Figure 1) (68). The main death type of tissue ischemic reperfusion injury has been proved to be ferroptosis (11). Xu et al. revealed that m6A methylase methyltransferase like 14 (METTL14) promotes renal ischemic reperfusion injury (69). Mechanistically, they identified YAP1 mRNA as a target of METTL14 and the translation of m6A-modified YAP1 mRNA was inhibited. However, a recent study revealed that YAP1 activates ACSCL4 gene transcription and thereby promotes ferroptosis (69). These paradoxical results suggest that the role of YAP1 in ferroptosis might be context-dependent and tissue or cell-type specific.
Non-Coding RNAs
The microRNA (miRNA) is a series of single strand noncoding small RNA, which is made up of 20-22 nucleotides. The miRNA can targets the 3’-UTR region of mRNA, triggering mRNA decay or translational inhibition (70). Plenty of miRNAs have been identified to participate in the regulation of the key genes of ferroptosis. For instance, miRNA-17-92 can protect cells from erastin-induced ferroptosis through targeting the ACSL4 axis and down regulating the ACSL4 expression (71). miRNA-4715-3p induces ferroptosis by inhibiting GPX4 expression (72). miRNA-137 targets SLC1A5 to suppress glutamine transportation and induce ferroptosis (73).
The role of long noncoding RNA (lncRNA) in gene regulation is gradually focused during the recent years (74). lncRNA is defined as transcripts of more than 200 nucleotides that are not translated into proteins (75). lncRNAs including HOX transcript antisense RNA and metastasis−associated lung adenocarcinoma transcript 1 are identified in the mechanism of ferroptosis suppress induced by XAV939 treatment (76). The lncRNA P53RRA induces ferroptosis by interacting with Ras GTPase-activating protein-binding protein 1 (G3BP1) and activating p53 pathway, then induce ferroptosis by affecting transcription of several metabolic genes (77). Wang et al. found lncRNA LINC00336 combines with ELAVL1 and inhibit ferroptosis by decreasing intracellular iron and lipid ROS level (78).
Circular RNA (cirRNA) is a type of single-stranded RNA which, unlike linear RNA, forms a covalently closed continuous loop. Circular RNA can regulate gene regulation by directly conjugating mRNA or indirectly transporting miRNAs in the cell (79, 80). In addition, cirRNAs are appealed to involve the ferroptosis regulation. Circular RNA cIARS regulates ferroptosis in HCC cells through interacting with RNA binding protein alkB homolog 5 (ALKBH5) (81). CircABCB10 silencing inhibits the cell ferroptosis and apoptosis by regulating the miR-326/CCL5 axis in rectal cancer (82). Circ_0008035 contributes to cell proliferation and inhibits apoptosis and ferroptosis in gastric cancer via miR-599/EIF4A1 axis (83).
Collectively, these findings demonstrate that epigenetic mechanisms contribute to the regulation of ferroptosis-associated genes (Table 1). However, whether, when, and how those key regulator genes as well as many newly found genes are epigenetically modulated are poorly understood. Therefore, systematic identification of the epigenetic regulatory network underlying ferroptosis is required in future study.

Table 1 Epigenetic regulation of ferroptosis-associated genes.
Targeting Epigenetic Regulation: A New Strategy in the Prevention and Therapy of Ferroptosis-Associated Diseases
As aforementioned, epigenetic mechanisms play a critical role in the regulation of ferroptosis-associated genes, thereby finetuning the cellular response to ferroptotic stress. Therefore, targeting epigenetic regulation represents a promising strategy to enhance or inhibit ferroptosis and has potential application in the prevention and therapy of ferroptosis-associated diseases. Indeed, many epigenetic drugs have been reported to display exciting results in cancer therapy through modulating ferroptosis (Table 2).
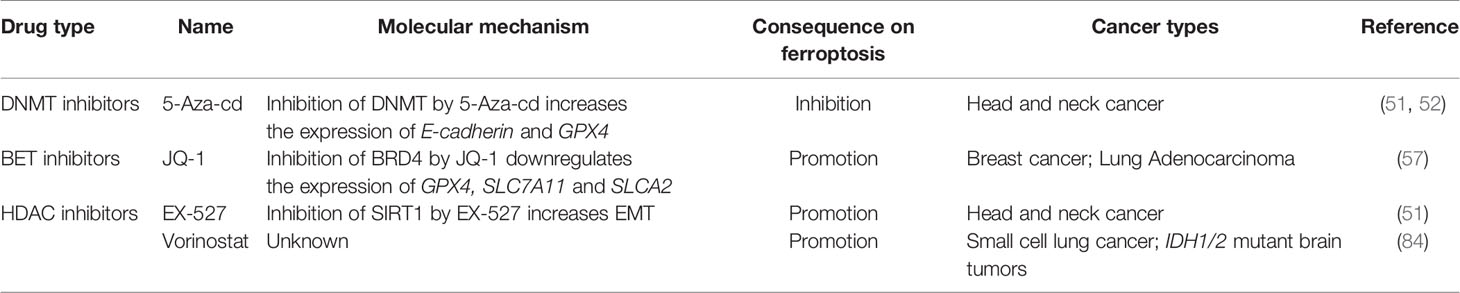
Table 2 Epigenetic drugs that modulate ferroptosis in cancer therapy.
DNMT Inhibitors
DNMT inhibitors have been successfully used in the treatment of certain hematopoietic malignancies (85). Moreover, recent work showed that DNMT inhibitors could enhance the efficiency of immune checkpoint inhibitors (ICI) in cancer immunotherapy (86). As aforementioned, 5-Aza-cd treatment could release the DNA methylation-mediated epigenetic silencing of GPX4 and CDH1 genes, restoring the resistance of cells to ferroptosis (51, 52). Given that DNMT inhibitors have very broad effect on gene expression and genomic stability, it should be careful to distinguish whether their effect on ferroptosis is achieved through the direct regulation of specific ferroptosis-associated gene or the indirect activation of certain signaling pathways due to epigenetic reprogramming.
BET Inhibitors
Bromodomain and extra terminal protein (BET) inhibitors are a class of drugs that prevent the interaction between BET proteins and acetylated histones (87–89). Sui et al. revealed that ferroptosis is involved in JQ1-induced cell death of BRCA and LUAD (57). Moreover, treatment with JQ1 and ferroptosis inducers (RSL3, erastin, or sorafenib) produced a satisfactory anticancer effect, suggesting that the combination of BET inhibitors with ferroptosis inducers may become a new therapeutic modality.
HDAC Inhibitors
Pharmacological inhibition of SIRT1 by EX-527 increases ferroptosis susceptibility by suppressing EMT, while SIRT1 agonists, resveratrol and SRT1720, promote ferroptosis (51). A recent drug screening also identified a class I HDAC inhibitor, Vorinostat, as an inducer of ferroptosis in small cell lung cancer (SCLC) and isocitrate dehydrogenase (IDH1/2)-mutant brain tumors, suggesting an unique vulnerability that is regulated by histone or non-histone acetylation (84).
Discussion
As a new concept introduced in 2012, ferroptosis has attracted tremendous attention in biomedical fields. The existing work about the epigenetic regulation of ferroptosis mainly focused on several key ferroptosis genes. Whether the epigenetic mechanisms affect multiple ferroptosis genes and how these different epigenetic mechanisms corporate with diverse signaling pathways to determine the responsiveness of cells to ferroptosis stimuli remain unknown. Therefore, a systematic study on the epigenetic regulatory network of ferroptosis process is still a blank in this field and requires extensive investigation.
Given that ferroptosis plays a role of surveillance in tumorigenesis and also contributes to the efficiency of multiple cancer therapies [chemotherapy, radiotherapy (90), and immunotherapy (91)], it is rationale to speculate that targeting the epigenetic machines alone or in combination with the traditional therapies will be promising strategies for cancer therapy. Since ferroptosis is closely related to neurodegenerative diseases, ischemia-reperfusion injury of organ, neurotoxicity, and others (11, 92–95), it is also of great interest to explore the epigenetic mechanisms underlying the altered ferroptosis sensitivity under different pathological processes. The advances in this cross-disciplinary research field may shed light on the treatment of diseases mentioned above by modulating ferroptosis.
Author Contributions
Conceptualization, YP, HW, and LT. Writing—original draft preparation, YP and LT. Writing—review and editing, YQ, HW, and LT. Funding acquisition, LT. All authors have read and agreed to the published version of the manuscript.
Funding
This review was supported by the National Natural Science Foundation of China (81672785, 31871291, and 82073113) and the National Key R&D Project of China (2016YFA0101800). LT was also supported by the innovative research team of high-level local university in Shanghai.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, SJ, declared a shared parent affiliation with the authors to the handling editor at the time of the review.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Dr Pingzhao Zhang for providing us critical advice.
References
1. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
2. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat Cell Biol (2014) 16:1180–91. doi: 10.1038/ncb3064
3. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
4. Li Y, Feng D, Wang Z, Zhao Y, Sun R, Tian D, et al. Ischemia-Induced ACSL4 Activation Contributes to Ferroptosis-Mediated Tissue Injury in Intestinal Ischemia/Reperfusion. Cell Death Differ (2019) 26:2284–99. doi: 10.1038/s41418-019-0299-4
5. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol (2015) 10:1604–9. doi: 10.1021/acschembio.5b00245
6. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 Is a Glutathione-Independent Ferroptosis Suppressor. Nature (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
7. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat Chem Biol (2017) 13:91–8. doi: 10.1038/nchembio.2239
8. Jiang X, Stockwell BR, Conrad M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat Rev Mol Cell Biol (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
9. Dawson MA, Kouzarides T. Cancer Epigenetics: From Mechanism to Therapy. Cell (2012) 150:12–27. doi: 10.1016/j.cell.2012.06.013
10. Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol (2016) 26:165–76. doi: 10.1016/j.tcb.2015.10.014
11. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
12. Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat Chem Biol (2017) 13:81–90. doi: 10.1038/nchembio.2238
13. Halliwell B, Cross CE. Oxygen-Derived Species: Their Relation to Human Disease and Environmental Stress. Environ Health Perspect (1994) 102 Suppl:10. doi: 10.1289/ehp.94102s105
14. Kakhlon O, Cabantchik ZI. The Labile Iron Pool: Characterization, Measurement, and Participation in Cellular Processes(1). Free Radical Biol Med (2002) 33:1037–46. doi: 10.1016/S0891-5849(02)01006-7
15. Dixon SJ, Stockwell BR. The Role of Iron and Reactive Oxygen Species in Cell Death. Nat Chem Biol 10 (2014) 10:9–17. doi: 10.1038/nchembio.1416
16. Doll S, Conrad M. Iron and Ferroptosis: A Still Ill-Defined Liaison. IUBMB Life (2017) 69:423–34. doi: 10.1002/iub.1616
17. Paul BD, Sbodio JI, Snyder SH. Cysteine Metabolism in Neuronal Redox Homeostasis. Trends Pharmacol Sci (2018) 39:513–24. doi: 10.1016/j.tips.2018.02.007
18. Bannai S. Exchange of Cystine and Glutamate Across Plasma Membrane of Human Fibroblasts. J Biol Chem (1986) 261:2256–63. doi: 10.1016/S0021-9258(17)35926-4
19. Koppula P, Zhuang L, Gan B. Cystine Transporter SLC7A11/xCT in Cancer: Ferroptosis, Nutrient Dependency, and Cancer Therapy. Protein Cell (2020) 12:599–620. doi: 10.1007/s13238-020-00789-5
20. Tan S, Wood M, Maher P. Oxidative Stress Induces a Form of Programmed Cell Death With Characteristics of Both Apoptosis and Necrosis in Neuronal Cells. J Neurochem (1998) 71:95–105. doi: 10.1046/j.1471-4159.1998.71010095.x
21. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate Toxicity in a Neuronal Cell Line Involves Inhibition of Cystine Transport Leading to Oxidative Stress. Neuron (1989) 2:1547–58. doi: 10.1016/0896-6273(89)90043-3
22. Ekoue DN, He C, Diamond AM, Bonini MG. Manganese Superoxide Dismutase and Glutathione Peroxidase-1 Contribute to the Rise and Fall of Mitochondrial Reactive Oxygen Species Which Drive Oncogenesis. Biochim Biophys Acta Bioenerg (2017) 1858:628–32. doi: 10.1016/j.bbabio.2017.01.006
23. Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress Into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab (2008) 8:237–48. doi: 10.1016/j.cmet.2008.07.005
24. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of Genotype-Selective Antitumor Agents Using Synthetic Lethal Chemical Screening in Engineered Human Tumor Cells. Cancer Cell (2003) 3:285–96. doi: 10.1016/S1535-6108(03)00050-3
25. Yang WS, Stockwell BR. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem Biol (2008) 15:234–45. doi: 10.1016/j.chembiol.2008.02.010
26. Ko P-J, Woodrow C, Dubreuil MM, Martin BR, Skouta R, Bassik MC, et al. A ZDHHC5-GOLGA7 Protein Acyltransferase Complex Promotes Nonapoptotic Cell Death. Cell Chem Biol (2019) 26:1716–24. doi: 10.1016/j.chembiol.2019.09.014
27. Yang WS, Shimada K, Delva D, Patel M, Ode E, Skouta R, et al. Identification of Simple Compounds With Microtubule-Binding Activity That Inhibit Cancer Cell Growth With High Potency. ACS Med Chem Lett (2012) 3:35–8. doi: 10.1021/ml200195s
28. Marshall KR, Gong M, Wodke L, Lamb JH, Jones DJL, Farmer PB, et al. The Human Apoptosis-Inducing Protein AMID Is an Oxidoreductase With a Modified Flavin Cofactor and DNA Binding Activity. J Biol Chem (2005) 280:30735–40. doi: 10.1074/jbc.M414018200
29. Crane FL. Discovery of Ubiquinone (Coenzyme Q) and an Overview of Function. Mitochondrion (2007) 7 Suppl:S2–7. doi: 10.1016/j.mito.2007.02.011
30. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
31. Madak JT, Bankhead A, Cuthbertson CR, Showalter HD, Neamati N. Revisiting the Role of Dihydroorotate Dehydrogenase as a Therapeutic Target for Cancer. Pharmacol Ther (2019) 195:111–31. doi: 10.1016/j.pharmthera.2018.10.012
32. Fu H, Zhang Z, Dai Y, Liu S, Fu E. Brequinar Inhibits Enterovirus Replication by Targeting Biosynthesis Pathway of Pyrimidines. Am J Transl Res (2020) 12:8247–55.
33. Boyd AS. Leflunomide in Dermatology. J Am Acad Dermatol (2012) 66:673–9. doi: 10.1016/j.jaad.2011.08.025
34. Bar-Or A, Pachner A, Menguy-Vacheron F, Kaplan J, Wiendl H. Teriflunomide and Its Mechanism of Action in Multiple Sclerosis. Drugs (2014) 74:659–74. doi: 10.1007/s40265-014-0212-x
35. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-Mediated Ferroptosis Defence Is a Targetable Vulnerability in Cancer. Nature (2021) 593:586–90. doi: 10.1038/s41586-021-03539-7
36. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis Through Lipid Remodeling. ACS Cent Sci (2020) 6:41–53. doi: 10.1021/acscentsci.9b01063
37. Xu J, Wu Y, Song P, Zhang M, Wang S, Zou M-H. Proteasome-Dependent Degradation of Guanosine 5’-Triphosphate Cyclohydrolase I Causes Tetrahydrobiopterin Deficiency in Diabetes Mellitus. Circulation (2007) 116:944–53. doi: 10.1161/CIRCULATIONAHA.106.684795
38. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic Determinants of Cancer Cell Sensitivity to Canonical Ferroptosis Inducers. Nat Chem Biol (2020) 16:1351–60. doi: 10.1038/s41589-020-0613-y
39. Jiang L, Kon N, Li T, Wang S-J, Su T, Hibshoosh H, et al. Ferroptosis as a P53-Mediated Activity During Tumour Suppression. Nature (2015) 520:57–62. doi: 10.1038/nature14344
40. Ou Y, Wang S-J, Li D, Chu B, Gu W. Activation of SAT1 Engages Polyamine Metabolism With P53-Mediated Ferroptotic Responses. Proc Natl Acad Sci USA (2016) 113:E6806–12. doi: 10.1073/pnas.1607152113
41. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The Tumor Suppressor P53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep (2017) 20:1692–704. doi: 10.1016/j.celrep.2017.07.055
42. Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. P53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep (2018) 22:569–75. doi: 10.1016/j.celrep.2017.12.077
43. Song X, Long D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front Neurosci (2020) 14:267. doi: 10.3389/fnins.2020.00267
44. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 Plays a Critical Role in Mitigating Lipid Peroxidation and Ferroptosis. Redox Biol (2019) 23:101107. doi: 10.1016/j.redox.2019.101107
45. Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 Pathway Promotes Cell Proliferation and Diminishes Ferroptosis. Oncogenesis (2017) 6:e371. doi: 10.1038/oncsis.2017.65
46. Takahashi N, Cho P, Selfors LM, Kuiken HJ, Kaul R, Fujiwara T, et al. 3d Culture Models With CRISPR Screens Reveal Hyperactive NRF2 as a Prerequisite for Spheroid Formation via Regulation of Proliferation and Ferroptosis. Mol Cell (2020) 80:828–44.e6. doi: 10.1016/j.molcel.2020.10.010
47. Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R, et al. NRF2 Is a Major Target of ARF in P53-Independent Tumor Suppression. Mol Cell (2017) 68:224–32.e4. doi: 10.1016/j.molcel.2017.09.009
48. Meng Z, Moroishi T, Guan K-L. Mechanisms of Hippo Pathway Regulation. Genes Dev (2016) 30:1–17. doi: 10.1101/gad.274027.115
49. Yang W-H, Ding C-KC, Sun T, Rupprecht G, Lin C-C, Hsu D, et al. The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep (2019) 28:2501–8.e4. doi: 10.2139/ssrn.3316797
50. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular Interaction Dictates Cancer Cell Ferroptosis via NF2-YAP Signalling. Nature (2019) 572:402–6. doi: 10.1038/s41586-019-1426-6
51. Lee J, You JH, Kim M-S, Roh J-L. Epigenetic Reprogramming of Epithelial-Mesenchymal Transition Promotes Ferroptosis of Head and Neck Cancer. Redox Biol (2020) 37:101697. doi: 10.1016/j.redox.2020.101697
52. Zhang X, Huang Z, Xie Z, Chen Y, Zheng Z, X.a. Wei B, et al. Homocysteine Induces Oxidative Stress and Ferroptosis of Nucleus Pulposus via Enhancing Methylation of GPX4. Free Radical Biol Med (2020) 160:552–65. doi: 10.1016/j.freeradbiomed.2020.08.029
53. Tan L, Shi YG. Tet Family Proteins and 5-Hydroxymethylcytosine in Development and Disease. Dev (Cambridge England) (2012) 139:1895–902. doi: 10.1242/dev.070771
54. Bannister AJ, Schneider R, Kouzarides T. Histone Methylation: Dynamic or Static? Cell (2002) 109:801–6. doi: 10.1016/S0092-8674(02)00798-5
55. Rodríguez-Paredes M, Esteller M. Cancer Epigenetics Reaches Mainstream Oncology. Nat Med (2011) 17:330–9. doi: 10.1038/nm.2305
56. Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by P300/CBP Augments Promoter-Specific DNA Binding of Nrf2 During the Antioxidant Response. Mol Cell Biol (2009) 29:2658–72. doi: 10.1128/MCB.01639-08
57. Sui S, Zhang J, Xu S, Wang Q, Wang P, Pang D. Ferritinophagy Is Required for the Induction of Ferroptosis by the Bromodomain Protein BRD4 Inhibitor (+)-JQ1 in Cancer Cells. Cell Death Dis (2019) 10:331. doi: 10.1038/s41419-019-1564-7
58. Zhang Y, Koppula P, Gan B. Regulation of H2A Ubiquitination and SLC7A11 Expression by BAP1 and PRC1. Cell Cycle (Georgetown Tex) (2019) 18:773–83. doi: 10.1080/15384101.2019.1597506
59. Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 Links Metabolic Regulation of Ferroptosis to Tumour Suppression. Nat Cell Biol (2018) 20:1181–92. doi: 10.1038/s41556-018-0178-0
60. Wang Y, Yang L, Zhang X, Cui W, Liu Y, Sun QR, et al. Epigenetic Regulation of Ferroptosis by H2B Monoubiquitination and P53. EMBO Rep (2019) 20:e47563. doi: 10.15252/embr.201847563
61. Becker JS, Nicetto D, Zaret KS. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet (2016) 32:29–41. doi: 10.1016/j.tig.2015.11.001
62. Wang J, Yin X, He W, Xue W, Zhang J, Huang Y. SUV39H1 Deficiency Suppresses Clear Cell Renal Cell Carcinoma Growth by Inducing Ferroptosis. Acta Pharm Sinica B (2021) 11:406–19. doi: 10.1016/j.apsb.2020.09.015
63. Wang Y, Zhao Y, Wang H, Zhang C, Wang M, Yang Y, et al. Histone Demethylase KDM3B Protects Against Ferroptosis by Upregulating SLC7A11. FEBS Open Bio (2020) 10:637–43. doi: 10.1002/2211-5463.12823
64. Chen M, Jiang Y, Sun Y. KDM4A-Mediated Histone Demethylation of SLC7A11 Inhibits Cell Ferroptosis in Osteosarcoma. Biochem Biophys Res Commun (2021) 550:77–83. doi: 10.1016/j.bbrc.2021.02.137
65. Jambhekar A, Dhall A, Shi Y. Roles and Regulation of Histone Methylation in Animal Development. Nat Rev Mol Cell Biol (2019) 20:625–41. doi: 10.1038/s41580-019-0151-1
66. Chen X-Y, Zhang J, Zhu J-S. The Role of mA RNA Methylation in Human Cancer. Mol Cancer (2019) 18:103. doi: 10.1186/s12943-019-1033-z
67. Huang H, Weng H, Chen J. mA Modification in Coding and Non-Coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell (2020) 37:270–88. doi: 10.1016/j.ccell.2020.02.004
68. Ma L, Chen T, Zhang X, Miao Y, Tian X, Yu K, et al. The M6a Reader YTHDC2 Inhibits Lung Adenocarcinoma Tumorigenesis by Suppressing SLC7A11-Dependent Antioxidant Function. Redox Biol (2021) 38:101801. doi: 10.1016/j.redox.2020.101801
69. Xu Y, Yuan XD, Wu JJ, Chen RY, Xia L, Zhang M, et al. The N6-Methyladenosine mRNA Methylase METTL14 Promotes Renal Ischemic Reperfusion Injury via Suppressing YAP1. J Cell Biochem (2020) 121:524–33. doi: 10.1002/jcb.29258
70. Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol (2018) 141:1202–7. doi: 10.1016/j.jaci.2017.08.034
71. Xiao F-J, Zhang D, Wu Y, Jia Q-H, Zhang L, Li Y-X, et al. miRNA-17-92 Protects Endothelial Cells From Erastin-Induced Ferroptosis Through Targeting the A20-ACSL4 Axis. Biochem Biophys Res Commun (2019) 515:448–54. doi: 10.1016/j.bbrc.2019.05.147
72. Gomaa A, Peng D, Chen Z, Soutto M, Abouelezz K, Corvalan A, et al. Epigenetic Regulation of AURKA by miR-4715-3p in Upper Gastrointestinal Cancers. Sci Rep (2019) 9:16970. doi: 10.1038/s41598-019-53174-6
73. Luo M, Wu L, Zhang K, Wang H, Zhang T, Gutierrez L, et al. miR-137 Regulates Ferroptosis by Targeting Glutamine Transporter SLC1A5 in Melanoma. Cell Death Differ (2018) 25:1457–72. doi: 10.1038/s41418-017-0053-8
74. Ulitsky I, Bartel DP. lincRNAs: Genomics, Evolution, and Mechanisms. Cell (2013) 154:26–46. doi: 10.1016/j.cell.2013.06.020
75. Kopp F, Mendell JT. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell (2018) 172:393–407. doi: 10.1016/j.cell.2018.01.011
76. Yu H, Han Z, Xu Z, An C, Xu L, Xin H. RNA Sequencing Uncovers the Key Long Non-Coding RNAs and Potential Molecular Mechanism Contributing to XAV939-Mediated Inhibition of Non-Small Cell Lung Cancer. Oncol Lett (2019) 17:4994–5004. doi: 10.3892/ol.2019.10191
77. Mao C, Wang X, Liu Y, Wang M, Yan B, Jiang Y, et al. A G3BP1-Interacting lncRNA Promotes Ferroptosis and Apoptosis in Cancer via Nuclear Sequestration of P53. Cancer Res (2018) 78:3484–96. doi: 10.1158/0008-5472.CAN-17-3454
78. Wang M, Mao C, Ouyang L, Liu Y, Lai W, Liu N, et al. Long Noncoding RNA LINC00336 Inhibits Ferroptosis in Lung Cancer by Functioning as a Competing Endogenous RNA. Cell Death Differ (2019) 26:2329–43. doi: 10.1038/s41418-019-0304-y
79. Hentze MW, Preiss T. Circular RNAs: Splicing’s Enigma Variations. EMBO J (2013) 32:923–5. doi: 10.1038/emboj.2013.53
80. Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs Are a Large Class of Animal RNAs With Regulatory Potency. Nature (2013) 495:333–8. doi: 10.1038/nature11928
81. Liu Z, Wang Q, Wang X, Xu Z, Wei X, Li J. Circular RNA Regulates Ferroptosis in HCC Cells Through Interacting With RNA Binding Protein ALKBH5. Cell Death Discov (2020) 6:72. doi: 10.1038/s41420-020-00306-x
82. Xian ZY, Hu B, Wang T, Cai JL, Zeng JY, Zou Q, et al. CircABCB10 Silencing Inhibits the Cell Ferroptosis and Apoptosis by Regulating the miR-326/CCL5 Axis in Rectal Cancer. Neoplasma (2020) 67:1063–73. doi: 10.4149/neo_2020_191024N1084
83. Li C, Tian Y, Liang Y, Li Q. Circ_0008035 Contributes to Cell Proliferation and Inhibits Apoptosis and Ferroptosis in Gastric Cancer via miR-599/EIF4A1 Axis. Cancer Cell Int (2020) 20:84. doi: 10.1186/s12935-020-01168-0
84. Yang H, Zhao L, Gao Y, Yao F, Marti TM, Schmid RA, et al. Pharmacotranscriptomic Analysis Reveals Novel Drugs and Gene Networks Regulating Ferroptosis in Cancer. Cancers (2020) 12:3273. doi: 10.3390/cancers12113273
85. Claus R, Lübbert M. Epigenetic Targets in Hematopoietic Malignancies. Oncogene (2003) 22:6489–96. doi: 10.1038/sj.onc.1206814
86. Perrier A, Didelot A, Laurent-Puig P, Blons H, Garinet S. Epigenetic Mechanisms of Resistance to Immune Checkpoint Inhibitors. Biomolecules (2020) 10:1061. doi: 10.3390/biom10071061
87. Garnier J-M, Sharp PP, Burns CJ. BET Bromodomain Inhibitors: A Patent Review. Expert Opin Ther Patents (2014) 24:185–99. doi: 10.1517/13543776.2014.859244
88. Shi J, Vakoc CR. The Mechanisms Behind the Therapeutic Activity of BET Bromodomain Inhibition. Mol Cell (2014) 54:728–36. doi: 10.1016/j.molcel.2014.05.016
89. Bowry A, Piberger AL, Rojas P, Saponaro M, Petermann E. BET Inhibition Induces HEXIM1- and RAD51-Dependent Conflicts Between Transcription and Replication. Cell Rep (2018) 25:2061–9.e4. doi: 10.1101/374660
90. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The Role of Ferroptosis in Ionizing Radiation-Induced Cell Death and Tumor Suppression. Cell Res (2020) 30:146–62. doi: 10.1038/s41422-019-0263-3
91. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T Cells Regulate Tumour Ferroptosis During Cancer Immunotherapy. Nature (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
92. Devos D, Moreau C, Devedjian JC, Kluza J, Petrault M, Laloux C, et al. Targeting Chelatable Iron as a Therapeutic Modality in Parkinson’s Disease. Antioxid Redox Signaling (2014) 21:195–210. doi: 10.1089/ars.2013.5593
93. Moreau C, Duce JA, Rascol O, Devedjian J-C, Berg D, Dexter D, et al. Iron as a Therapeutic Target for Parkinson’s Disease. Mov Disord Off J Mov Disord Soc (2018) 33:568–74. doi: 10.1002/mds.27275
94. Bulk M, Kenkhuis B, van der Graaf LM, Goeman JJ, Natté R, van der Weerd L. Postmortem T2*- Weighted MRI Imaging of Cortical Iron Reflects Severity of Alzheimer’s Disease. J Alzheimer’s Dis JAD (2018) 65:1125–37. doi: 10.3233/JAD-180317
Keywords: ferroptosis, epigenetic regulation, cancer, DNA methylation, histone modifications, RNA methylation
Citation: Pei Y, Qian Y, Wang H and Tan L (2022) Epigenetic Regulation of Ferroptosis-Associated Genes and Its Implication in Cancer Therapy. Front. Oncol. 12:771870. doi: 10.3389/fonc.2022.771870
Received: 07 September 2021; Accepted: 10 January 2022;
Published: 31 January 2022.
Edited by:
Jian Chen, Guilin Medical University, ChinaCopyright © 2022 Pei, Qian, Wang and Tan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Tan, bGl0YW5AZnVkYW4uZWR1LmNu; Hao Wang, d2FuZy5oYW8yQHpzLWhvc3BpdGFsLnNoLmNu