
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 20 December 2022
Sec. Molecular and Cellular Oncology
Volume 12 - 2022 | https://doi.org/10.3389/fonc.2022.1099280
This article is part of the Research Topic Differential Diagnoses of Thyroid Neoplasms: Molecular and Histological Features and the Impact on Follow-Up View all 10 articles
 Giusy Elia1†
Giusy Elia1† Armando Patrizio2†
Armando Patrizio2† Francesca Ragusa1
Francesca Ragusa1 Sabrina Rosaria Paparo3
Sabrina Rosaria Paparo3 Valeria Mazzi1
Valeria Mazzi1 Eugenia Balestri1
Eugenia Balestri1 Chiara Botrini1
Chiara Botrini1 Licia Rugani1
Licia Rugani1 Salvatore Benvenga4,5,6
Salvatore Benvenga4,5,6 Gabriele Materazzi1
Gabriele Materazzi1 Claudio Spinelli1
Claudio Spinelli1 Alessandro Antonelli1*
Alessandro Antonelli1* Poupak Fallahi3
Poupak Fallahi3 Silvia Martina Ferrari7
Silvia Martina Ferrari7Poorly differentiated thyroid cancer (PDTC) and anaplastic thyroid cancer (ATC) have a worse prognosis with respect to well differentiated TC, and the loss of the capability of up-taking 131I is one of the main features characterizing aggressive TC. The knowledge of the genomic landscape of TC can help clinicians to discover the responsible alterations underlying more advance diseases and to address more tailored therapy. In fact, to date, the antiangiogenic multi-targeted kinase inhibitor (aaMKIs) sorafenib, lenvatinib, and cabozantinib, have been approved for the therapy of aggressive radioiodine (RAI)-resistant papillary TC (PTC) or follicular TC (FTC). Several other compounds, including immunotherapies, have been introduced and, in part, approved for the treatment of TC harboring specific mutations. For example, selpercatinib and pralsetinib inhibit mutant RET in medullary thyroid cancer but they can also block the RET fusion proteins-mediated signaling found in PTC. Entrectinib and larotrectinib, can be used in patients with progressive RAI-resistant TC harboring TRK fusion proteins. In addition FDA authorized the association of dabrafenib (BRAFV600E inhibitor) and trametinib (MEK inhibitor) for the treatment of BRAFV600E-mutated ATC. These drugs not only can limit the cancer spread, but in some circumstance they are able to induce the re-differentiation of aggressive tumors, which can be again submitted to new attempts of RAI therapy. In this review we explore the current knowledge on the genetic landscape of TC and its implication on the development of new precise therapeutic strategies.
Thyroid cancer (TC) is a highly diffuse endocrine tumor affecting especially the female gender with a low death rate but increasing worldwide (1–4). TCs classification is based on the cells of origin with an incidence that changes according to the different histotypes. The differentiated TC (DTC) is the most common tumor, which arises from thyroid follicular cells, and represents with papillary TC (PTC), and follicular TC (FTC) about 85–95% of all TCs. Hürthle cells TC and poorly differentiated TC (PDTC) account for 2–5% of all TCs, and the anaplastic TC (ATC) comprises about 1.7% of all cases of TC. Medullary TC (MTC) arises from para-follicular C cells of neuroendocrine origin, accounting for 3–5% of all TCs (5).
Patients with PDTC and ATC have a worse prognosis with respect to well differentiated TC (WDTC), and a lower overall survival (OS) rate with a mean survival of about 3.2 years and 6 months, respectively (6). High rate of disease relapse is registered in PDTC patients, who report frequent local invasion of the disease at the level of trachea and/or esophagus, and also distant progression to the liver, lungs, bone and brain (7–9).
Some PDTC tumors are characterized by refractoriness to T4-mediated TSH suppression or to the therapy with radioiodine (RAI) (7).
ATC is a very aggressive cancer usually originating from DTCs or PDTCs, and is characterized by a quickly growth that can vary from days to several weeks; it is often associated to dysphagia, acute hoarseness, dyspnea, and/or neck pain (7, 10).
Thyroid ultrasound (US) helps in stratifying the risk of malignancy of thyroid nodules, that according to their morphological features (shape, size, echogenicity, margins, the presence of microcalcifications) can be further examined by the fine needle aspiration cytology (FNAC) (11).
The criteria defining the risk of malignancy for biopsied nodules and their subsequent clinical management follow the Bethesda classification system (12).
However, it is often challenging make the right therapeutic decision with indeterminate thyroid nodules, and molecular testing of genetic mutations related to TC can improve the risk stratification supporting the decision-making process in order to avoid unnecessary invasive procedure, such as surgery, and predicting possible adverse clinical outcomes in the post-operative phase (11, 12).
Some of the genetic TC alterations are called “driver” mutations that promote the normal cell transition into cancerous one, whereas the “passenger” mutations are the consequences of carcinogenesis and of loss of differentiation (13, 14). About 90% of alterations are mutually exclusive activating oncogene BRAF (~60%), RAS (~13%), and rearrangements [ALK, RET, and NTRK genes (~5%)]; whereas the other 10% are loss-of-function of tumor suppressor genes (including PPARγ, PTEN, and TP53) (15–17). The Cancer Genome Atlas (TCGA) documented aberrations of genes in 97% of PTCs, including driver genes CHEK2, EIF1AX, and PPM1D, members of the phosphoinositide 3-kinase (PI3K) pathway and other gene fusions (17), however 3% of PTCs (called “dark matter”) still are genetically undefined (18). The molecular mechanisms that guide the progression to a more aggressive pattern are not largely elucidated (19). The genetic alterations per tumor found in ATC are higher in comparison to PTC and FTC (16); and according to TCGA, PDTC has also a higher mutational burden compared to PTC, but lower than ATC. Genomic instability in PDTC and ATC involve both somatic driver mutations and gene fusions (17, 20). Parallel sequencing studies have been carried out on both PDTCs and ATCs, in order to study their molecular features and discovering the differences between these two types of tumors. Elevated frequencies of TERT promoter, TP53, PTEN and PIK3CA mutations have been observed in ATCs with respect to PDTCs; ATCs also have NF1, NF2, ATM, CDKN2A, CDKN2B and RB1 mutations. Instead, PDTCs showed a higher frequencies of gene fusions (RET, ALK, NTRK1, NTRK3) (21). Recently, next-generation sequencing (NGS) studies, have revealed molecular clues underlying the progression of DTC to PDTC and ATC (15, 16).
Most of the TC primary driver oncogenes activate the mitogen activated protein kinase (MAPK) and the PI3K pathways (22–24); the alterations involving these pathways are the most found in ATC and PDTC (20). BRAFV600E and RAS-like mutations, including three highly homologous isoforms (NRAS, KRAS and HRAS) are the most common found in TC.
According to the TCGA, BRAFV600E is the most frequent driver mutations associated with PTC (1, 17, 25); found in 25% of ATC, and associated with tumor aggressiveness, and a bad prognosis (26, 27). Moreover, it is related to an absent or reduced expression of various genes, such as those encoding thyroid-peroxidase, the sodium-iodide symporter, Tg, TSH receptor, and pendrin genes (SLC26A4) (1, 28, 29). Therefore, it is suggested as a predictive marker of PTC persistence or recurrence, decreased efficacy of RAI therapy (30), and reduction of the OS (13, 31–33).
Mutated BRAF PTC has been related to different clinical-pathological conditions with a negative prognostic impact, and a more aggressive behaviour (extra-thyroidal extension, lymph node metastases, advanced disease stage) (1, 34, 35). Other studies showed no kind of correlation between BRAFV600E and any of the PTC aggressiveness features (13, 36). However, the detection of BRAFV600E in FNAC improves the diagnostic accuracy of PTC reducing also false-negative results (13).
RAS genes mutations are mainly found in FTC and in follicular variant PTC (FVPTC) (30-45%), in PDTC (20-40%), in a less percentage of ATC (10-20%), and also in benign follicular thyroid adenoma (20-25%), while rarely in classical PTC (1, 37). These mutations are more commonly related to indolent behaviour, follicular growth, encapsulation, and a lower incidence of nodal metastasis (11, 38). However, RAS mutations are believed to worsen TC prognosis and life expectancy inducing the passage from a WDTC to a de-differentiated type, the development of distant metastases, and recurrence (13). Furthermore, the de-differentiation effect has been supported by the chromosome instability because of mutant RAS (1, 39, 40).
Association with clinical-pathological manifestation is controversial (41, 42). Disease-specific death risk is 2.9 times higher in subjects harbouring RAS mutation with respect to those without RAS mutation (43). The detection of RAS mutation in FNAC has an important clinical meaning for indeterminate nodules, with a predictive value for cancer ranging between 74% and 88% (13).
The gradual passage or de-differentiation of WDTC to ATC it has been hypothesized to be induced by the accumulation of genetic alterations, particularly of BRAF or RAS mutations (6, 44, 45).
Point mutations have been also identified in TERT promoter, resulting in a telomerase activation that is up-regulated in 80-90% of TC; whereas it is not present in normal thyroid cells (1, 46, 47).
Duan et al. studies found that: 1) ATC with PTC components is typically characterised by a BRAF mutation, and at least one late mutation event (TP53, TERT, or PIK3CA); 2) RET fusion is more frequently associated to PDTC with PTC components. In subjects with PDTC/ATC a worse OS is related to TERT and concurrent PIK3CA mutations (6). The prognostic effect related to TERT promoter mutations was not present when BRAF mutation occurred separately, showing that the co-existence of both mutations is determinant for tumor aggressiveness (40).
Other alterations mostly found in PDTC (10-14%) than in ATC (3-5%) are the genes fusions (48). RET represents the most frequent genetic fusion, especially RET/PTC1 and RET/PTC3; NTRK, ALK and BRAF fusions are unusual (20, 48). Post-radiation exposure TC, and children, reporting or not a previously irradiation history, display a high frequency of RET/PTC1 and RET/PTC3 rearrangements. RET/PTC3 is related with the tendency for aggressive behaviour and advanced stage, higher rates of extra-thyroidal extension and lymph node metastases (13). It seems that RET/PTC is a leading mutation in thyroid carcinogenesis (49–51), it is especially related to the classic PTC subtype (51). However, according to TCGA, RET/PTC is considered a primary genetic event in only 6.8% of the PTC cohort (1, 17). The diagnostic and predictive value of RET/PTC is controversial; in fact, in cases of indeterminate cytology it is still not routinely examined by molecular testing (13, 52, 53).
As regard the NTRK1/3 rearrangements, their encoded protein is constitutively active, causing the activation of the pro-oncogenic pathways PI3K/AKT, phospholipase C (PLC-γ), and MAPK (1, 7, 54).
Also ALK, a transmembrane tyrosine kinase, when activated can trigger downstream signalling pathways, including MAPK, JAK/STAT, and PI3K/AKT. ALK gene alterations lead to disease progression and aggressiveness, and they are more detected in PDTCs, ATCs than in PTCs (1, 55).
The mechanism of age-associated genetic alterations is not still fully understood, however chromosomal rearrangements are strongly related to the exposition to ionizing radiation, while BRAFV600E point mutations may be associated to excess dietary iodine intake or exposure to chemical disruptors in volcanic regions (56, 57). DNA fragility and impaired repair mechanism are both implicated in radiation-induced genetic damaged or stocastic oncogenic fusion (58–60). Young children might develop more frequently uncoupled double-stranded breaks and translocation with partner genes because they seem to be more vulnerable to the ionizing radiation activity and to the loss of the DNA repair capacity (60).
Only 2.3–2.5% of FTCs display microsatellite instability (MSI), which derived from persistent oxidative stress and subsequent impairment of DNA mismatch repair gene(s) encoding MutL-homolog DNA mismatch repair enzymes PMS1, PMS2, and MLH1, MLH3 (61–63). Since tumors harboring MSI are susceptible to anti-programmed cell death ligand 1 (PD-L1) immunotherapy, additional efforts are needed to clarify the role of mismatch repair gene deficiency in TC (64).
Epigenetic alterations influence gene expression: hyper-methylation of gene promoter sequences lead to heritable inhibition of transcription, while unmethylation results in increased gene transcription (65). Thyroid-specific tumor suppressor genes can promote cell de-differentiation if wrongly methylated during the first steps of tumorigenesis. If cell lines, with TSHR gene silenced by hyper-methylation, are treated with a demethylating agent, they can in part restore TSHR expression and subsequent TSH-induced iodine uptake and effectiveness of RAI (66–68). Other tumor suppressor genes silenced by aberrant methylation are genes encoding cyclin-dependent kinase inhibitors p15INKa and p16INK4b (69), RASSF1A (70), ECAD, RARβ-2, NIS-I, DAPK, ATM, SLC26A, SLC5A8, and TIMP3 (71, 72). It has been suggested that the latter four are associated with aggressive features (18).
Several molecular driven therapies have been evaluated in aggressive TC (Table 1) (73). Systemic treatments for unresponsive metastatic non-anaplastic follicular cell derived TC include the antiangiogenic multitargeted kinase inhibitor (aaMKIs) sorafenib and lenvatinib (7, 23, 74–77). The Food And Drug Administration (FDA) authorized these aaMKIs, because they can improve progression-free survival as emerged from phase III randomized double blinded crossover clinical trials. Although non tested in a “head-to-head” trial, lenvatinib showed a longer progression-free median survival (18.3 months vs 3.6 months of the placebo group, p < 0.001) compared to sorafenib and to placebo group (10.8 months vs 5.8 months respectively, p < 0.0001), becoming the first-choice agent among oral aaMKIs (78, 79). Most patients demonstrated disease stabilization or minor/partial responses, which lasted mean period of 12-24 months (78). Lately, also Cabozantinib, an aaMKI previously approved by FDA for the treatment of MTC, has been authorized in case of failure of first-line therapy with lenvatinib and sorafenib, since it improves progression free survival as a second-line agent (80). These compounds do not require mutation profiling of the tumors and they can be also administered when specific targetable mutation (eg, NTRK, ALK, RET, or BRAF) have not been identified. As they target primarily the angiogenic vascular endothelial growth factor receptor (VEGFR) signaling, the side effects may include fatigue, hypertension, diarrhea, hand-foot skin reaction and other rashes, thyroid dysfunctions, hepatotoxicity, renal toxicity and fistula formation in the gastrointestinal tract and/or in other locations.
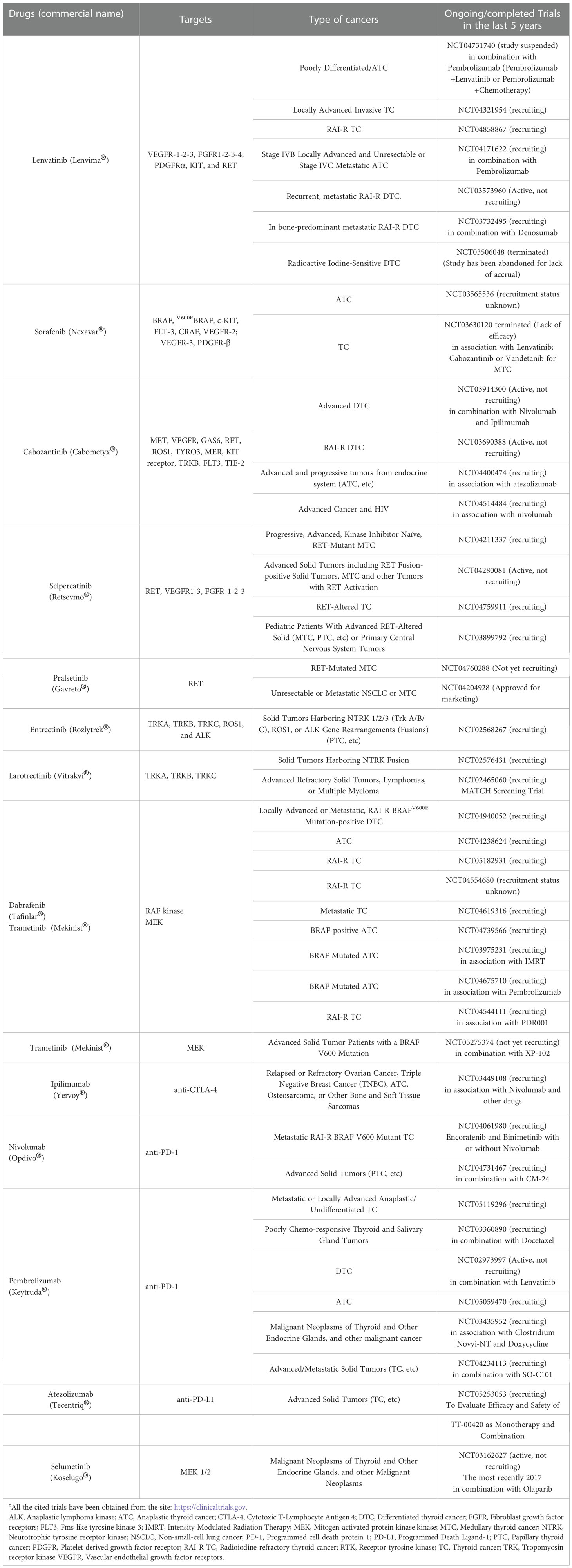
Table 1 FDA-approved therapies for thyroid cancer.
On the other hand, if specific driver mutations are identified (eg, NTRK, ALK, RET, BRAF), new mutation-specific kinase inhibitor should be considered which have been FDA-approved, specifically for TCs or for any tumor type harboring the same molecular target (7, 23, 81, 82). For this reason, these compounds require the tumor mutation profiling to prove their pertinence to a specific patient. For example, selpercatinib and pralsetinib inhibit mutant RET in MTC but they can also block the RET fusion proteins-mediated signaling found in PTC and other types of tumor (such as lung cancer) as documented by enduring high partial response and several complete responses rates in Phase III trials (83, 84). These RET inhibitors appear also to be better tolerated than the aaMKIs. However, emerging over time new RET mutations can cause therapeutic resistance by blocking drugs access to the active site or through other mechanisms (85, 86). The clinical trials performed for TRK inhibitors, entrectinib and larotrectinib, have documented activity also for TC (87, 88) and they can be used in some patients with progressive RAI-resistant TC harboring TRK fusion proteins. In addition, the FDA, according to a small cohort study in which ~50% of patients had partial responses to therapy, authorized the association of dabrafenib (BRAFV600E inhibitor) and trametinib (MEK inhibitor) for the treatment of BRAFV600E-mutated ATC (89, 90). A subgroup of patients of that cohort displayed a prolonged responses of several years (89). Based on these data, it is recommended to obtain rapid BRAFV600E testing in all patients with ATC (91). Regarding BRAFV600-mutant PTC, off-label administration of a BRAF inhibitor could be considered especially for whom aaMKI therapy is contraindicate. Furthermore, BRAFV600E inhibitors have showed promising results for advanced DTC in phase II studies (92, 93). Ultimately, the activity of FDA-approved immune checkpoint inhibitors (such as anti-PD1 and anti-PDL1) is also routinely tested in TC samples and predictors of response are the detection of MSI and high mutational burden (24).
The loss of the capability of up-taking 131I is one of the main features characterizing aggressive TC. The cancer therapy with RAI is based in the exploiting of Na/I symporter (NIS). NIS is primarily regulated by TSH through the cAMP pathway, and it is necessary to transport the iodide against a concentration gradient in thyroid follicular cells to synthetize thyroid hormone (94). This mechanism is lost in case of NIS downregulation or loss of function.
RAI refractoriness can be defined by different scenarios, such as the absence of RAI uptake at the initial whole body scan (WBS) or in metastatic lesions, or the loss of the capacity to uptake RAI after a previous WBS showing avidly uptake RAI metastases; a progression of the disease in a subject who has previously received RAI, or a cumulative activity of 600 mCi of 131I; the presence of locally advanced disease that cannot be treated by surgery or evaluated by RAI uptake (95, 96). Genetic and epigenetic alterations in the RTK/BRAF/MAPK/ERK and PI3K-AKT-mTOR pathways underly the diminished NIS signalling/activity that lead to RAI refractoriness and to a more aggressive behaviour (97): their identification can be useful to investigate new compounds able to act against these aberrant molecular mechanisms overcoming the standard cancer therapy resistance.
In vivo studies in mice focused on the disruption of BRAFV600E-driven MAPK signaling and found an increase of the iodine uptake (29). According to these findings a clinical trial has been conducted on RAI-resistant metastatic TC subjects who had undergone a whole body I124 PET/CT, who were then treated with selumetinib (a MEK inhibitor) for 4 weeks, and subsequently underwent a second scan (98). A partial response has been obtained in approximately 62.5% of the treated subjects, whereas the others had stable disease over a year. It has been observed a difference in the response of the patients according to their mutational status, in fact those harboring RAS mutations responded more frequently with respect to those with the BRAFV600E mutations.
Other studies have been carried out by using different drugs including BRAFV600E, TRK, and RET inhibitors in selected patients according to their genomic tests (58, 99–101).
A study enrolled non-genomically identified patients for first RAI therapy after surgery, who were randomly assigned in a “receiving selumetinib group” and in a “no selumetinib group” and benefits in response rates between the groups were not reported (102). The redifferentiation approach could be in the future a useful strategy to delay long-term treatment with kinase inhibitors using RAI therapy.
These results suggest the use of genomic tests for treatment decisions (24).
De-differentiated TC and ATC have a worse prognosis with respect to WDTC and the loss of the capability of up-taking 131I is one of the main features characterizing de-differentiated and aggressive TC. The knowledge of the genomic landscape of TC can help clinicians to discover the responsible alterations underlying more advance diseases and to address more tailored therapy (103–109). In fact, to date, the aaMKIs sorafenib, lenvatinib, and cabozantinib, have been approved for the therapy of aggressive RAI-resistant PTC or FTC. Several other compounds, including immunotherapies, have been introduced and, in part, approved for the treatment of TC harboring specific mutations. For example, selpercatinib and pralsetinib inhibit mutant RET in MTC but they can also block the RET fusion proteins-mediated signaling found in PTC. Entrectinib and larotrectinib, can be used in some patients with progressive RAI-resistant TC harboring TRK fusion proteins. In addition FDA authorized the association of dabrafenib and trametinib for the treatment of BRAFV600E-mutated ATC (89, 90).
Tyrosine kinase inhibitors drugs can act against different altered pathways implicated in the pathogenetic process of aggressive TC. However, patients can’t have a good therapeutic response to the therapies with activation of other pathways able to evade the drugs antitumoral effect. Moreover, patients can experience important side effects that can lead to the interruption of the therapy.
New therapies strategies are under investigations, with drugs against immune checkpoint inhibitors.
A good therapy strategy is knowing the molecular pattern of each patient that could aid in the choice of right therapies avoiding the administration of ineffective drugs. A personalized therapy is the challenge of the precision medicine. This challenge can be largely support by in vitro drug tests performed on primary tumor cells obtained from patients, that reflect the in vivo behavior with a predictive positive value of 60%, and negative predictive value of 90% (110–112). Furthermore, in vitro studies can be performed in cells obtained by using the non-invasive technique of FNAC, without the use of surgery (113–115).
Therefore, additional studies about molecular implications involved in the development of aggressive cancer, as well as about each individual patients response to chemotherapeutics will pave the way in the battle against thyroid aggressive cancer.
GE, AP, SB, GM, CS, AA, PF, SMF conceived the paper. GE, AP, SMF specifically wrote the paper and controlled references. All authors reviewed and approved the final version of the manuscript. GE and AP equally contributed as first authors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Elia G, Ferrari SM, Ragusa F, Paparo SR, Mazzi V, Ulisse S, et al. Advances in pharmacotherapy for advanced thyroid cancer of follicular origin (PTC, FTC). new approved drugs and future therapies. Expert Opin Pharmacother (2022) 23:599–610. doi: 10.1080/14656566.2022.2030704
2. Hernandez-Prera JC. The evolving concept of aggressive histological variants of differentiated thyroid cancer. Semin Diagn Pathol (2020) 37:228–33. doi: 10.1053/j.semdp.2020.03.002
3. Antonelli A, Ferri C, Fallahi P. Thyroid cancer in patients with hepatitis c infection. JAMA (1999) 281:1588. doi: 10.1001/jama.281.17.1588
4. Ferrari SM, Fallahi P, Elia G, Ragusa F, Ruffilli I, Paparo SR, et al. Thyroid autoimmune disorders and cancer. Semin Cancer Biol (2020) 64:135–46. doi: 10.1016/j.semcancer.2019.05.019
5. Fugazzola L, Elisei R, Fuhrer D, Jarzab B, Leboulleux S, Newbold K, et al. 2019 European Thyroid association guidelines for the treatment and follow-up of advanced radioiodine-refractory thyroid cancer. Eur Thyroid J (2019) 8:227–45. doi: 10.1159/000502229
6. Duan H, Li Y, Hu P, Gao J, Ying J, Xu W, et al. Mutational profiling of poorly differentiated and anaplastic thyroid carcinoma by the use of targeted next-generation sequencing. Histopathology (2019) 75:890–9. doi: 10.1111/his.13942
7. Cabanillas ME, Ryder M, Jimenez C. Targeted therapy for advanced thyroid cancer: Kinase inhibitors and beyond. Endocr Rev (2019) 40:1573–604. doi: 10.1210/er.2019-00007
8. Xu B, Ibrahimpasic T, Wang L, Sabra MM, Migliacci JC, Tuttle RM, et al. Clinicopathologic features of fatal non-anaplastic follicular cell-derived thyroid carcinomas. Thyroid (2016) 26:1588–97. doi: 10.1089/thy.2016.0247
9. Ibrahimpasic T, Ghossein R, Carlson DL, Chernichenko N, Nixon I, Palmer FL, et al. Poorly differentiated thyroid carcinoma presenting with gross extrathyroidal extension: 1986-2009 memorial Sloan-Kettering cancer center experience. Thyroid (2013) 23:997–1002. doi: 10.1089/thy.2012.0403
10. Ferrari SM, Elia G, Ragusa F, Ruffilli I, La Motta C, Paparo SR, et al. Novel treatments for anaplastic thyroid carcinoma. Gland Surg (2020) 9(Suppl 1):S28–42. doi: 10.21037/gs.2019.10.18
11. Semsar-Kazerooni K, Morand GB, Payne AE, da Silva SD, Forest VI, Hier MP, et al. Mutational status may supersede tumor size in predicting the presence of aggressive pathologic features in well differentiated thyroid cancer. J Otolaryngol Head Neck Surg (2022) 51:9. doi: 10.1186/s40463-022-00559-9
12. Melo-Uribe MA, Sanabria Á, Romero-Rojas A, Pérez G, Vargas EJ, Abaúnza MC, et al. The Bethesda system for reporting thyroid cytopathology in Colombia: Correlation with histopathological diagnoses in oncology and non-oncology institutions. J Cytol (2015) 32:12–6. doi: 10.4103/0970-9371.155224
13. Niciporuka R, Nazarovs J, Ozolins A, Narbuts Z, Miklasevics E, Gardovskis J. Can we predict differentiated thyroid cancer behavior? Role of genetic and molecular markers. Medicina (Kaunas) (2021) 57:1131. doi: 10.3390/medicina57101131
14. Khatami F, Tavangar SM. A review of driver genetic alterations in thyroid cancers. Iran J Pathol (2018) 13:125–35.
15. Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest (2016) 126:1052–66. doi: 10.1172/JCI85271
16. Pozdeyev N, Gay LM, Sokol ES, Hartmaier R, Deaver KE, Davis S, et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res (2018) 24:3059–68. doi: 10.1158/1078-0432.CCR-18-0373
17. Cancer genome atlas research network. Integrated genomic characterization of papillary thyroid carcinoma. Cell (2014) 159:676–90. doi: 10.1016/j.cell.2014.09.050
18. Singh A, Ham J, Po JW, Niles N, Roberts T, Lee CS. The genomic landscape of thyroid cancer tumourigenesis and implications for immunotherapy. Cells (2021) 10:1082. doi: 10.3390/cells10051082
19. Yoo SK, Song YS, Lee EK, Hwang J, Kim HH, Jung G, et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid cancer. Nat Commun (2019) 10:2764. doi: 10.1038/s41467-019-10680-5
20. Prete A, Matrone A, Gambale C, Torregrossa L, Minaldi E, Romei C, et al. Poorly differentiated and anaplastic thyroid cancer: Insights into genomics, microenvironment and new drugs. Cancers (Basel) (2021) 13:3200. doi: 10.3390/cancers13133200
21. Macerola E, Poma AM, Vignali P, Basolo A, Ugolini C, Torregrossa L, et al. Molecular genetics of follicular-derived thyroid cancer. Cancers (Basel) (2021) 13:1139. doi: 10.3390/cancers13051139
22. Fagin JA, Wells SA Jr. Biologic and clinical perspectives on thyroid cancer. N Engl J Med (2016) 375:2307. doi: 10.1056/NEJMc1613118
23. Ringel MD. New horizons: Emerging therapies and targets in thyroid cancer. J Clin Endocrinol Metab (2021) 106:e382–8. doi: 10.1210/clinem/dgaa687
24. Sipos JA, Ringel MD. Molecular testing in thyroid cancer diagnosis and management. Best Pract Res Clin Endocrinol Metab (2022) 101680. doi: 10.1016/j.beem.2022.101680
25. Zoghlami A, Roussel F, Sabourin JC, Kuhn JM, Marie JP, Dehesdin D, et al. BRAF mutation in papillary thyroid carcinoma: predictive value for long-term prognosis and radioiodine sensitivity. Eur Ann Otorhinolaryngol Head Neck Dis (2014) 131:7–13. doi: 10.1016/j.anorl.2013.01.004
26. Fallahi P, Ferrari SM, Galdiero MR, Varricchi G, Elia G, Ragusa F, et al. Molecular targets of tyrosine kinase inhibitors in thyroid cancer. Semin Cancer Biol (2022) 79:180–96. doi: 10.1016/j.semcancer.2020.11.013
27. Perri F, Pezzullo L, Chiofalo MG, Lastoria S, Di Gennaro F, Scarpati GD, et al. Targeted therapy: a new hope for thyroid carcinomas. Crit Rev Oncol Hematol (2015) 94:55–63. doi: 10.1016/j.critrevonc.2014.10.012
28. Durante C, Puxeddu E, Ferretti E, Morisi R, Moretti S, Bruno R, et al. BRAF mutations in papillary thyroid carcinomas inhibit genes involved in iodine metabolism. J Clin Endocrinol Metab (2007) 92:2840–3. doi: 10.1210/jc.2006-2707
29. Chakravarty D, Santos E, Ryder M, Knauf JA, Liao XH, West BL, et al. Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J Clin Invest (2011) 121:4700–11. doi: 10.1172/JCI46382
30. Ge J, Wang J, Wang H, Jiang X, Liao Q, Gong Q, et al. The BRAF V600E mutation is a predictor of the effect of radioiodine therapy in papillary thyroid cancer. J Cancer (2020) 11:932–9. doi: 10.7150/jca.33105
31. Zhang Q, Liu SZ, Zhang Q, Guan YX, Chen QJ, Zhu QY. Meta-analyses of association between BRAF(V600E) mutation and clinicopathological features of papillary thyroid carcinoma. Cell Physiol Biochem (2016) 38:763–76. doi: 10.1159/000443032
32. Liu C, Chen T, Liu Z. Associations between BRAF(V600E) and prognostic factors and poor outcomes in papillary thyroid carcinoma: a meta-analysis. World J Surg Oncol (2016) 14:241. doi: 10.1186/s12957-016-0979-1
33. Xing M, Alzahrani AS, Carson KA, Viola D, Elisei R, Bendlova B, et al. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA (2013) 309:1493–501. doi: 10.1001/jama.2013.3190
34. Xing M, Westra WH, Tufano RP, Cohen Y, Rosenbaum E, Rhoden KJ, et al. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J Clin Endocrinol Metab (2005) 90:6373–9. doi: 10.1210/jc.2005-0987
35. Wang Y, Ji M, Wang W, Miao Z, Hou P, Chen X, et al. Association of the T1799A BRAF mutation with tumor extrathyroidal invasion, higher peripheral platelet counts, and over-expression of platelet-derived growth factor-b in papillary thyroid cancer. Endocr Relat Cancer (2008) 15:183–90. doi: 10.1677/ERC-07-0182
36. Yan C, Huang M, Li X, Wang T, Ling R. Relationship between BRAF V600E and clinical features in papillary thyroid carcinoma. Endocr Connect (2019) 8:988–96. doi: 10.1530/EC-19-0246
37. Xing M. Clinical utility of RAS mutations in thyroid cancer: A blurred picture now emerging clearer. BMC Med (2016) 14:12. doi: 10.1186/s12916-016-0559-9
38. Patel SG, Carty SE, McCoy KL, Ohori NP, LeBeau SO, Seethala RR, et al. Preoperative detection of RAS mutation may guide extent of thyroidectomy. Surgery (2017) 161:168–75. doi: 10.1016/j.surg.2016.04.054
39. Saavedra HI, Knauf JA, Shirokawa JM, Wang J, Ouyang B, Elisei R, et al. The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene (2000) 19:3948–54. doi: 10.1038/sj.onc.1203723
40. Marotta V, Sciammarella C, Colao A, Faggiano A. Application of molecular biology of differentiated thyroid cancer for clinical prognostication. Endocr Relat Cancer (2016) 23:R499–515. doi: 10.1530/ERC-16-0372
41. Vuong HG, Duong UN, Altibi AM, Ngo HT, Pham TQ, Tran HM, et al. A meta-analysis of prognostic roles of molecular markers in papillary thyroid carcinoma. Endocr Connect (2017) 6:R8–17. doi: 10.1530/EC-17-0010
42. Howell GM, Hodak SP, Yip L. RAS mutations in thyroid cancer. Oncologist (2013) 18:926–32. doi: 10.1634/theoncologist.2013-0072
43. Pak K, Suh S, Kim SJ, Kim IJ. Prognostic value of genetic mutations in thyroid cancer: A meta-analysis. Thyroid (2015) 25:63–70. doi: 10.1089/thy.2014.0241
44. Oishi N, Kondo T, Ebina A, Sato Y, Akaishi J, Hino R, et al. Molecular alterations of coexisting thyroid papillary carcinoma and anaplastic carcinoma: identification of TERT mutation as an independent risk factor for transformation. Mod Pathol (2017) 30:1527–37. doi: 10.1038/modpathol.2017.75
45. Nikiforov YE. Genetic alterations involved in the transition from well-differentiated to poorly differentiated and anaplastic thyroid carcinomas. Endocr Pathol (2004) 15:319–27. doi: 10.1385/ep:15:4:319
46. Karapanou O, Simeakis G, Vlassopoulou B, Alevizaki M, Saltiki K. Advanced RAI-refractory thyroid cancer: an update on treatment perspectives. Endocr Relat Cancer (2022) 29:R57–66. doi: 10.1530/ERC-22-0006
47. Antonelli A, Ferrari SM, Elia G, Patrizio A, Fallahi P. Metastases free thyroid cancer patients harbouring TERT mutations may benefit from a more intensive treatment and follow-up. Gland Surg (2019) 8:298–300. doi: 10.21037/gs.2019.05.05
48. Yakushina VD, Lerner LV, Lavrov AV. Gene fusions in thyroid cancer. Thyroid (2018) 28:158–67. doi: 10.1089/thy.2017.0318
49. Marotta V, Guerra A, Sapio MR, Vitale M. RET/PTC rearrangement in benign and malignant thyroid diseases: A clinical standpoint. Eur J Endocrinol (2011) 165:499–507. doi: 10.1530/EJE-11-0499
50. Tallini G, Santoro M, Helie M, Carlomagno F, Salvatore G, Chiappetta G, et al. RET/PTC oncogene activation defines a subset of papillary thyroid carcinomas lacking evidence of progression to poorly differentiated or undifferentiated tumor phenotypes. Clin Cancer Res (1998) 4:287–94.
51. Lam AK, Montone KT, Nolan KA, Livolsi VA. Ret oncogene activation in papillary thyroid carcinoma: Prevalence and implication on the histological parameters. Hum Pathol (1998) 29:565–8. doi: 10.1016/s0046-8177(98)80004-x
52. Romei C, Elisei R. RET/PTC translocations and clinico-pathological features in human papillary thyroid carcinoma. Front Endocrinol (Lausanne) (2012) 3:54. doi: 10.3389/fendo.2012.00054
53. Nikiforov YE. RET/PTC rearrangement in thyroid tumors. Endocr Pathol (2002) 13:3–16. doi: 10.1385/ep:13:1:03
54. Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol (2018) 19:705–14. doi: 10.1016/S1470-2045(18)30119-0
55. Ratajczak M, Gaweł D, Godlewska M. Novel inhibitor-based therapies for thyroid cancer-an update. Int J Mol Sci (2021) 22:11829. doi: 10.3390/ijms222111829
56. Nikiforov YE, Nikiforova MN. Molecular genetics and diagnosis of thyroid cancer. Nat Rev Endocrinol (2011) 7:569–80. doi: 10.1038/nrendo.2011.142
57. Ferrari SM, Fallahi P, Antonelli A, Benvenga S. Environmental issues in thyroid diseases. Front Endocrinol (Lausanne) (2017) 8:50. doi: 10.3389/fendo.2017.00050
58. Lee YA, Lee H, Im SW, Song YS, Oh DY, Kang HJ, et al. NTRK and RET fusion-directed therapy in pediatric thyroid cancer yields a tumor response and radioiodine uptake. J Clin Invest (2021) 131:e144847. doi: 10.1172/JCI144847
59. Picarsic JL, Buryk MA, Ozolek J, Ranganathan S, Monaco SE, Simons JP, et al. Molecular characterization of sporadic pediatric thyroid carcinoma with the DNA/RNA ThyroSeq v2 next-generation sequencing assay. Pediatr Dev Pathol (2016) 19:115–22. doi: 10.2350/15-07-1667-OA.1
60. Roukos V, Misteli T. The biogenesis of chromosome translocations. Nat Cell Biol (2014) 16:293–300. doi: 10.1038/ncb2941
61. Ravi N, Yang M, Gretarsson S, Jansson C, Mylona N, Sydow SR, et al. Identification of targetable lesions in anaplastic thyroid cancer by genome profiling. Cancers (Basel) (2019) 11:402. doi: 10.3390/cancers11030402
62. Paulsson JO, Backman S, Wang N, Stenman A, Crona J, Thutkawkorapin J, et al. Whole-genome sequencing of synchronous thyroid carcinomas identifies aberrant DNA repair in thyroid cancer dedifferentiation. J Pathol (2020) 250:183–94. doi: 10.1002/path.5359
63. Genutis LK, Tomsic J, Bundschuh RA, Brock PL, Williams MD, Roychowdhury S, et al. Microsatellite instability occurs in a subset of follicular thyroid cancers. Thyroid (2019) 29:523–9. doi: 10.1089/thy.2018.0655
64. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357:409–13. doi: 10.1126/science.aan6733
65. Baylin SB. Tying it all together: epigenetics, genetics, cell cycle, and cancer. Science (1997) 277:1948–9. doi: 10.1126/science.277.5334.1948
66. Xing M, Usadel H, Cohen Y, Tokumaru Y, Guo Z, Westra WB, et al. Methylation of the thyroid-stimulating hormone receptor gene in epithelial thyroid tumors: a marker of malignancy and a cause of gene silencing. Cancer Res (2003) 63:2316–21.
67. Kartal K, Onder S, Kosemehmetoglu K, Kilickap S, Tezel YG, Kaynaroglu V. Methylation status of TSHr in well-differentiated thyroid cancer by using cytologic material. BMC Cancer (2015) 15:824–30. doi: 10.1186/s12885-015-1861-1
68. Kim WG, Zhu X, Kim DW, Zhang L, Kebebew E, Cheng SY. Reactivation of the silenced thyroid hormone receptor β gene expression delays thyroid tumor progression. Endocrinology (2013) 154:25–35. doi: 10.1210/en.2012-1728
69. Boltze C, Zack S, Quednow C, Bettge S, Roessner A, Schneider-Stock R. Hypermethylation of the CDKN2/p16INK4A promotor in thyroid carcinogenesis. Pathol Res Pract (2003) 199:399–404. doi: 10.1078/0344-0338-00436
70. Shou F, Xu F, Li G, Zhao Z, Mao Y, Yang F, et al. RASSF1A promoter methylation is associated with increased risk of thyroid cancer: a meta-analysis. Onco Targets Ther (2017) 10:247–57. doi: 10.2147/OTT.S124417
71. Hu S, Liu D, Tufano RP, Carson KA, Rosenbaum E, Cohen Y, et al. Association of aberrant methylation of tumor suppressor genes with tumor aggressiveness and BRAF mutation in papillary thyroid cancer. Int J Cancer (2006) 119:2322–9. doi: 10.1002/ijc.22110
72. Xing M, Tokumaru Y, Wu G, Westra WB, Ladenson PW, Sidransky D. Hypermethylation of the pendred syndrome gene SLC26A4 is an early event in thyroid tumorigenesis. Cancer Res (2003) 63:2312–5.
73. Available at: https://clinicaltrials.gov.
74. Haddad RI, Nasr C, Bischoff L, Busaidy NL, Byrd D, Callender G, et al. NCCN guidelines insights: Thyroid carcinoma, version 2.2018. J Natl Compr Canc Netw (2018) 16:1429–40. doi: 10.6004/jnccn.2018.0089
75. Ferrari SM, Politti U, Spisni R, Materazzi G, Baldini E, Ulisse S, et al. Sorafenib in the treatment of thyroid cancer. Expert Rev Anticancer Ther (2015) 15:863–74. doi: 10.1586/14737140.2015.1064770
76. Ferrari SM, Ruffilli I, Centanni M, Virili C, Materazzi G, Alexopoulou M, et al. Lenvatinib in the therapy of aggressive thyroid cancer: State of the art and new perspectives with patents recently applied. Recent Pat Anticancer Drug Discovery (2018) 13:201–8. doi: 10.2174/1574892813666180220110729
77. Ferrari SM, Elia G, Ragusa F, Paparo SR, Mazzi V, Miccoli M, et al. Lenvatinib: an investigational agent for the treatment of differentiated thyroid cancer. Expert Opin Investig Drugs (2021) 30:913–21. doi: 10.1080/13543784.2021.1972971
78. Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med (2015) 372:621–30. doi: 10.1056/NEJMoa1406470
79. Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet (2014) 384:319–28. doi: 10.1016/S0140-6736(14)60421-9
80. Brose MS, Robinson B, Sherman SI, Krajewska J, Lin CC, Vaisman F, et al. Cabozantinib for radioiodine-refractory differentiated thyroid cancer (COSMIC-311): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol (2021) 22:1126–38. doi: 10.1016/S1470-2045(21)00332-6
81. Lubitz CC, Sadow PM, Daniels GH, Wirth LJ. Progress in treating advanced thyroid cancers in the era of targeted therapy. Thyroid (2021) 31:1451–62. doi: 10.1089/thy.2020.0962
82. Tiedje V, Fagin JA. Therapeutic breakthroughs for metastatic thyroid cancer. Nat Rev Endocrinol (2020) 16:77–8. doi: 10.1038/s41574-019-0307-2
83. Wirth LJ, Sherman E, Robinson B, Solomon B, Kang H, Lorch J, et al. Efficacy of selpercatinib in RET-altered thyroid cancers. N Engl J Med (2020) 383:825–35. doi: 10.1056/NEJMoa2005651
84. Subbiah V, Hu MI, Wirth LJ, Schuler M, Mansfield AS, Curigliano G, et al. Pralsetinib for patients with advanced or metastatic RET-altered thyroid cancer (ARROW): A multi-cohort, open-label, registrational, phase 1/2 study. Lancet Diabetes Endocrinol (2021) 9:491–501. doi: 10.1016/S2213-8587(21)00120-0
85. Zhu VW, Madison R, Schrock AB, Ou SI. Emergence of high level of MET amplification as off-target resistance to selpercatinib treatment in KIF5B-RET NSCLC. J Thorac Oncol (2020) 15:e124–7. doi: 10.1016/j.jtho.2020.03.020
86. Solomon BJ, Tan L, Lin JJ, Wong SQ, Hollizeck S, Ebata K, et al. RET solvent front mutations mediate acquired resistance to selective RET inhibition in RET-driven malignancies. J Thorac Oncol (2020) 15:541–9. doi: 10.1016/j.jtho.2020.01.006
87. Waguespack SG, Drilon A, Lin JJ, Brose MS, McDermott R, Almubarak M, et al. Efficacy and safety of larotrectinib in patients with TRK fusion-positive thyroid carcinoma. Eur J Endocrinol (2022) 186:631–43. doi: 10.1530/EJE-21-1259
88. Liu SV, Macke LA, Colton BS, Imran SS, Christiansen J, Chow-Maneval E, et al. Response to entrectinib in differentiated thyroid cancer with a ROS1Fusion. JCO Precis Oncol (2017) 1. doi: 10.1200/PO.17.00105. PO.17.00105.
89. Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib plus trametinib in patients with BRAF V600E-mutant anaplastic thyroid cancer: updated analysis from the phase II ROAR basket study. Ann Oncol (2022) 33:406–15. doi: 10.1016/j.annonc.2021.12.014
90. Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol (2018) 36:7–13. doi: 10.1200/JCO.2017.73.6785
91. Shonka DC Jr, Ho A, Chintakuntlawar AV, Geiger JL, Park JC, Seetharamu N, et al. American Head and neck society endocrine surgery section and international thyroid oncology group consensus statement on mutational testing in thyroid cancer: Defining advanced thyroid cancer and its targeted treatment. Head Neck (2022) 44:1277–300. doi: 10.1002/hed.27025
92. Brose MS, Cabanillas ME, Cohen EE, Wirth LJ, Riehl T, Yue H, et al. Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol (2016) 17:1272–82. doi: 10.1016/S1470-2045(16)30166-8
93. Falchook GS, Millward M, Hong D, Naing A, Piha-Paul S, Waguespack SG, et al. BRAF inhibitor dabrafenib in patients with metastatic BRAF-mutant thyroid cancer. Thyroid (2015) 25:71–7. doi: 10.1089/thy.2014.0123
94. Ohno M, Zannini M, Levy O, Carrasco N, di Lauro R. The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol Cell Biol (1999) 19:2051–60. doi: 10.1128/MCB.19.3.2051
95. Schlumberger M, Brose M, Elisei R, Leboulleux S, Luster M, Pitoia F, et al. Definition and management of radioactive iodine-refractory differentiated thyroid cancer. Lancet Diabetes Endocrinol (2014) 2:356–8. doi: 10.1016/S2213-8587(13)70215-8
96. Pitoia F, Jerkovich F, Trimboli P, Smulever A. New approaches for patients with advanced radioiodine-refractory thyroid cancer. World J Clin Oncol (2022) 13:9–27. doi: 10.5306/wjco.v13.i1.9
97. Aashiq M, Silverman DA, Na'ara S, Takahashi H, Amit M. Radioiodine-refractory thyroid cancer: Molecular basis of redifferentiation therapies, management, and novel therapies. Cancers (Basel) (2019) 11:1382. doi: 10.3390/cancers11091382
98. Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med (2013) 368:623–32. doi: 10.1056/NEJMoa1209288
99. Dunn LA, Sherman EJ, Baxi SS, Tchekmedyian V, Grewal RK, Larson SM, et al. Vemurafenib redifferentiation of BRAF mutant, RAI-refractory thyroid cancers. J Clin Endocrinol Metab (2019) 104:1417–28. doi: 10.1210/jc.2018-01478
100. Rothenberg SM, McFadden DG, Palmer EL, Daniels GH, Wirth LJ. Redifferentiation of iodine-refractory BRAF V600E-mutant metastatic papillary thyroid cancer with dabrafenib. Clin Cancer Res (2015) 21:1028–35. doi: 10.1158/1078-0432.CCR-14-2915
101. Groussin L, Theodon H, Bessiene L, Bricaire L, Bonnet-Serrano F, Cochand-Priollet B, et al. Redifferentiating effect of larotrectinib in NTRK-rearranged advanced radioactive-iodine refractory thyroid cancer. Thyroid (2022) 32:594–8. doi: 10.1089/thy.2021.0524
102. Tchekmedyian V, Dunn L, Sherman E, Baxi SS, Grewal RK, Larson SM, et al. Enhancing radioiodine incorporation in BRAF-mutant, radioiodine-refractory thyroid cancers with vemurafenib and the anti-ErbB3 monoclonal antibody CDX-3379: Results of a pilot clinical trial. Thyroid (2022) 32:273–82. doi: 10.1089/thy.2021.0565
103. Baldini E, Tuccilli C, Prinzi N, Sorrenti S, Antonelli A, Gnessi L, et al. Effects of selective inhibitors of aurora kinases on anaplastic thyroid carcinoma cell lines. Endocr Relat Cancer (2014) 21:797–811. doi: 10.1530/ERC-14-0299
104. Baldini E, Tuccilli C, Prinzi N, Sorrenti S, Falvo L, De Vito C, et al. Deregulated expression of aurora kinases is not a prognostic biomarker in papillary thyroid cancer patients. PLos One (2015) 10:e0121514. doi: 10.1371/journal.pone.0121514
105. Gabillard JC, Ulisse S, Baldini E, Sorrenti S, Cremet JY, Coccaro C, et al. Aurora-c interacts with and phosphorylates the transforming acidic coiled-coil 1 protein. Biochem Biophys Res Commun (2011) 408:647–53. doi: 10.1016/j.bbrc.2011.04.078
106. Baldini E, Sorrenti S, D'Armiento E, Di Matteo FM, Catania A, Ulisse S. The urokinase plasminogen activating system in thyroid cancer: clinical implications. G Chir (2012) 33:305–10.
107. Baldini E, Tuccilli C, Pironi D, Catania A, Tartaglia F, Di Matteo FM, et al. Expression and clinical utility of transcription factors involved in epithelial-mesenchymal transition during thyroid cancer progression. J Clin Med (2021) 10:4076. doi: 10.3390/jcm10184076
108. Bulfamante AM, Lori E, Bellini MI, Bolis E, Lozza P, Castellani L, et al. Advanced differentiated thyroid cancer: A complex condition needing a tailored approach. Front Oncol (2022) 12:954759. doi: 10.3389/fonc.2022.954759
109. Baldini E, Presutti D, Favoriti P, Santini S, Papoff G, Tuccilli C, et al. In vitro and In vivo effects of the urokinase plasminogen activator inhibitor WX-340 on anaplastic thyroid cancer cell lines. Int J Mol Sci (2022) 23:3724. doi: 10.3390/ijms23073724
110. Antonelli A, Ferrari SM, Fallahi P, Berti P, Materazzi G, Minuto M, et al. Thiazolidinediones and antiblastics in primary human anaplastic thyroid cancer cells. Clin Endocrinol (Oxf) (2009) 70:946–53. doi: 10.1111/j.1365-2265.2008.03415.x
111. Antonelli A, Ferrari SM, Fallahi P, Berti P, Materazzi G, Marchetti I, et al. Evaluation of the sensitivity to chemotherapeutics or thiazolidinediones of primary anaplastic thyroid cancer cells obtained by fine-needle aspiration. Eur J Endocrinol (2008) 159:283–91. doi: 10.1530/EJE-08-0190
112. Fallahi P, Ferrari SM, Elia G, Ragusa F, Patrizio A, Paparo SR, et al. Primary cell cultures for the personalized therapy in aggressive thyroid cancer of follicular origin. Semin Cancer Biol (2022) 79:203–16. doi: 10.1016/j.semcancer.2020.06.013
113. Ferrari SM, La Motta C, Elia G, Ragusa F, Ruffilli I, Quattrini L, et al. Antineoplastic effect of lenvatinib and vandetanib in primary anaplastic thyroid cancer cells obtained from biopsy or fine needle aspiration. Front Endocrinol (Lausanne) (2018) 9:764. doi: 10.3389/fendo.2018.00764
114. Ferrari SM, Fallahi P, Ruffilli I, Elia G, Ragusa F, Paparo SR, et al. Molecular testing in the diagnosis of differentiated thyroid carcinomas. Gland Surg (2018) 7(Suppl 1):S19–29. doi: 10.21037/gs.2017.11.07
Keywords: aggressive thyroid cancer, genetic mutations, molecular features, RAI refractioness, targeted therapy
Citation: Elia G, Patrizio A, Ragusa F, Paparo SR, Mazzi V, Balestri E, Botrini C, Rugani L, Benvenga S, Materazzi G, Spinelli C, Antonelli A, Fallahi P and Ferrari SM (2022) Molecular features of aggressive thyroid cancer. Front. Oncol. 12:1099280. doi: 10.3389/fonc.2022.1099280
Received: 15 November 2022; Accepted: 06 December 2022;
Published: 20 December 2022.
Edited by:
Augusto Lauro, Sapienza University of Rome, ItalyReviewed by:
Eleonora Lori, Sapienza University of Rome, ItalyCopyright © 2022 Elia, Patrizio, Ragusa, Paparo, Mazzi, Balestri, Botrini, Rugani, Benvenga, Materazzi, Spinelli, Antonelli, Fallahi and Ferrari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Antonelli, YWxlc3NhbmRyby5hbnRvbmVsbGlAdW5pcGkuaXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.